Embed Size (px)
Citation preview

Secondary Structure in Denatured DNAis Responsiblefor Its Reaction with Antinative
DNAAntibodies of Systemic Lupus Erythematosus Sera
B. DAVID STOLLARand MICHAEL PAPALIAN, Department of Biochemistryand Pharmacology, Tufts University School of Medicine,Boston, Massachusetts 02111
A B S T R A C T Experiments were designed to deter-mine the basis for the strong competitive reaction ofdenatured DNA with systemic lupus erythematosus(SLE) antinative DNAantibodies. Secondary structurein denatured DNAwas reflected in hyperchromicityupon heating and in multiphase kinetics of its digestionby Si nuclease. Partial digestion by Si nucleasecompletely eliminated the ability of denatured DNAto react with antidenatured DNAantibodies, but notits ability to react with SLE sera. Sl nuclease-resistantcores were isolated from extensively digested de-natured DNA. These cores had secondary structure,including some stable fold-back helical regions. Thecores, from 20 to several hundred base pairs in size,competed with native DNAfor binding by SLE sera.Other experiments measured reactions of denaturedDNA under conditions that affected its secondarystructure content. Its competitive activity decreasedas temperature was increased from 00 to 37°C, whereasthe activity of native DNA was not altered in thistemperature range. With DNApieces of 90-110 basepairs, native fragments were much more effective thanthe denatured fragments, in which stable helicalstructure is less likely to occur than in high molecularweight denatured DNA. Competitive assays withmononucleotides, oligonucleotides, homopolymers, andRNA-DNAhybrids also indicated that two strands ofpolydeoxyribonucleotide were required for optimalreactions with these SLE serum antibodies. Theantibodies can measure stable helical regions indenatured DNA; they may also stabilize short helicalregions that occur in an equilibrium of conforma-tional forms.
INTRODUCTION
Sera of patients with systemic lupus erythematosus(SLE)1 contain a variety of antinucleic acid antibodies.
Received for publication 27 December 1979 and in revisedform 10 March 1980.
210
There has been considerable interest in defining thespecificities of these antibodies, since questions havearisen whether anti-DNA assays are reliable for diagno-sis and monitoring of disease activity, and whetherantibodies with a particular kind of specificity may be es-pecially significant for pathogenesis and diagnosis (1-7).
Anti-DNA antibody specificity has been divided onthe basis of reactivity with either denatured DNAornative DNA. One class of antibody clearly reacts withdenatured DNA only, and recognizes the purine orpyrimidine bases or base sequences, which are notavailable for reaction in the helical native structure.The binding of radioactive denatured DNAby theseantibodies is competed for by unlabeled denaturedDNAbut not at all by native DNA (8). Similar anti-bodies can be induced in experimental animals byimmunization with denatured DNA-methylated bo-vine serum albumin complexes (9), or with nucleoside-or nucleotide-protein conjugates (10).
A second type of antibody in SLE serum reacts withnative DNA. These antibodies bind closed circularbacteriophage DNA (11) or completely helical poly-(dAT) (12). They react with circular helical DNAin situin immunofluorescence assays with Crithidia luciliae(13), and they bind DNA treated with S1 nucleaseto remove single-stranded regions (14). Interestingly,however, the binding of native DNAby these anti-bodies is usually inhibited by denatured DNA(8, 15),and this inhibition may be seen with denatured DNAconcentrations that are nearly the same as thoserequired for native DNA itself. Similarly, denaturedDNAinhibits the precipitation of native DNA(16) orpassive hemagglutination of native DNA-coated eryth-rocytes (17) by most SLE sera.
IAbbreviations used in this paper: PBS, phosphate-buf-fered saline (0.14 MNaCl, 0.01 Mphosphate, pH, 7.2); SLE,systemic lupus erythematosus.
J. Clin. Invest. X) The American Society for Clinical Investigation, Inc. 0021-9738/80/0810210/10 $1.00Volume 66 August 1980 210-219

The finding that a number of SLE sera react withdeterminants that are present on both native anddenatured DNAraises the question whether the anti-bodies truly recognize the secondary helical structureof DNA. Three possibilities raised by these observa-tions are, first, that the reaction is due to exposureof the bases in local single-stranded regions of the DNA;second, that the reaction is with the sugar-phosphatebackbone of one polydeoxyribonucleotide strand andthat such a structure is accessible in both the nativeand denatured DNA(it should be noted that antibodiesto such a determinant, even in combining with de-natured DNA, would differ from those that recognizebases or base sequences; and third, that the antibodiesreact with a determinant that does involve the back-bone of both strands, and that denatured DNAcom-petes for binding because there is in fact a significantamount of helical secondary structure (either stable ortransient) in denatured DNA (18). Such antibodieswould resemble the experimentally induced anti-bodies to double-stranded RNAor RNA-DNAhybrids,which recognize specific helical shape (19).
Experiments described in this article were designedto test these possibilities. The results indicate thatoptimal reaction with antinative DNAantibody doesrequire secondary structure, and that the antibodiesmeasure localized helical regions in denatured DNArather than localized denatured regions in native DNA.
METHODS
Antigens. Calf thymus DNA, obtained from WorthingtonBiochemicals Corp., Freehold, N. J., was purified further asdescribed (20). For denaturation, DNAwas heated at 100°Cfor 10 min and quickly chilled in an ice-water bath. Poly(dA),poly(dT), poly(dI), and poly(A) *poly(dT) were purchasedfrom P-L Biochemicals Inc., Milwaukee, Wis. [3H]Thymidine-labeled DNAwas prepared from a partially thymine-requiringmutant, B3, as described (21), but with a pulse of 500 ,aCi of[3H]thymidine, rather than 60 ,tCi of ['4C]thymidine, duringlog-phase growth. The labeled DNA was treated with SInuclease (Miles Laboratories, Inc., Elkhart, Ind.) in a reactionmixture containing 48 jig of DNAin 1.5 ml of 0.03 Msodiumacetate, pH 5, 0.01 mMZnCl2, and 15 ,ul of enzyme (4,000U). The mixture was incubated at 37°C for 20 min, and thereaction was then stopped by the addition of 2.5 ml of0.06 M sodium phosphate and 0.03 M EDTA, pH 8. Thetreated DNAwas diluted further to 1 ,g/ml for addition to theradioimmunoassay mixtures.
Helical DNAfragments of 90-110 base pairs were preparedas described previously (20). Oligonucleotides averaging 20residues in length were prepared from a pancreatic DNasedigest of calf thymus DNA. 50 ,mg of DNase was added to35 ml of DNA (1.3 mglml with 5 mMMgCl2). The reactionproceeded until 25% hyperchromicity was reached, and wasstopped with EDTA (10 mMfinal concentration). Digestionproducts were applied to a 15-ml DEAE-cellulose column,which was then washed extensively with 0.25 M NaCl in0.05 M cacodylate buffer, pH 6.8. Larger oligonucleotideswere then eluted with 0.6 M NaCl in the cacodylate buffer.
Average chain length was determined from total and terminalphosphorus measurements (22).
Analytical SI nuclease digestion of denatured DNA. 10,ul of SI nuclease (2,700 U) was added to 2 ml of a solutionof 100 ,ug/ml of denatured DNAin 0.03 Macetate, pH 5, with0.015 mMZnCl2; and the change in absorbance was monitoredat 260 nm at room temperature. When the rate of increaseslowed markedly, the temperature was raised to 37°C and theabsorbance monitored during further incubation.
Preparative SI nuclease digestion of denatured DNA. 50,ul (13,500 U) of S1 nuclease was added to 10 ml of de-natured calf thymus DNA (1.8 mg/ml) in 0.03 M sodiumacetate, pH 5, with 0.05 mMZnCl2. The mixture was incubatedat room temperature. Whenthe rate of increase in absorbanceat 260 nm slowed, 10-25-tI increments of enzyme wereadded until hyperchromicity reached 27%; a total of 110 Alof enzyme was used over a 5-h period. The reaction wasstopped by addition of EDTA to 10 mM. The digestionproducts were applied to a 2.5 x 40-cm Bio-Gel P-60 column(Bio-Rad Laboratories, Richmond, Calif.) and washed throughwith phosphate-buffered saline (PBS). The material emergingat the void volume was diluted threefold with water andapplied to a 1.0-ml column of DEAE-cellulose equilibratedwith PBS. The column was washed with PBS and then withsolutions of 0.1, 0.2, 0.3, 0.5, 0.75, and 1.0 M NaCl in PBS.Eluted nucleic acids were dialyzed against a 1:2 dilution ofPBS in water.
Polyacrylamide gel electrophoresis. Samples of 1-2 ugof DNA fragment were loaded onto 10% polyacrylamideslab gels (12 x 16 x 0.16 cm) with a 1-cm, 4% stacking gel.The acrylamide:bisacrylamide ratio wvas 19:1. After electro-phoresis at 50 mA for 3.5 h, gels were stained with 1 ,ug/mlethidium bromide for 30 min, rinsed with water, and photo-graphed under short wave UV light.
Thermal denaturation curves. Calf thymus DNAor DNAfragments were heated in stoppered l-ml silica cuvettes in aZeiss PM6spectrophotometer with a thermo-electric tempera-ture control unit (Carl Zeiss, Inc., New York, N. Y.). Absorb-ance at 260 nm was recorded after equilibration.
Double - antibody radioimmunoassays. Double - antibodyradioimmunoassays (20) or ammonium sulfate radioimmuno-assays (21) were performed as described previously. SLE serawere those used in a previous study (20). Rabbit antinucleo-side antibodies were prepared as described by Erlanger andBeiser (10). Rabbit antibodies to double-stranded RNAandRNA-DNAhybrids were described previously (19).
RESULTS
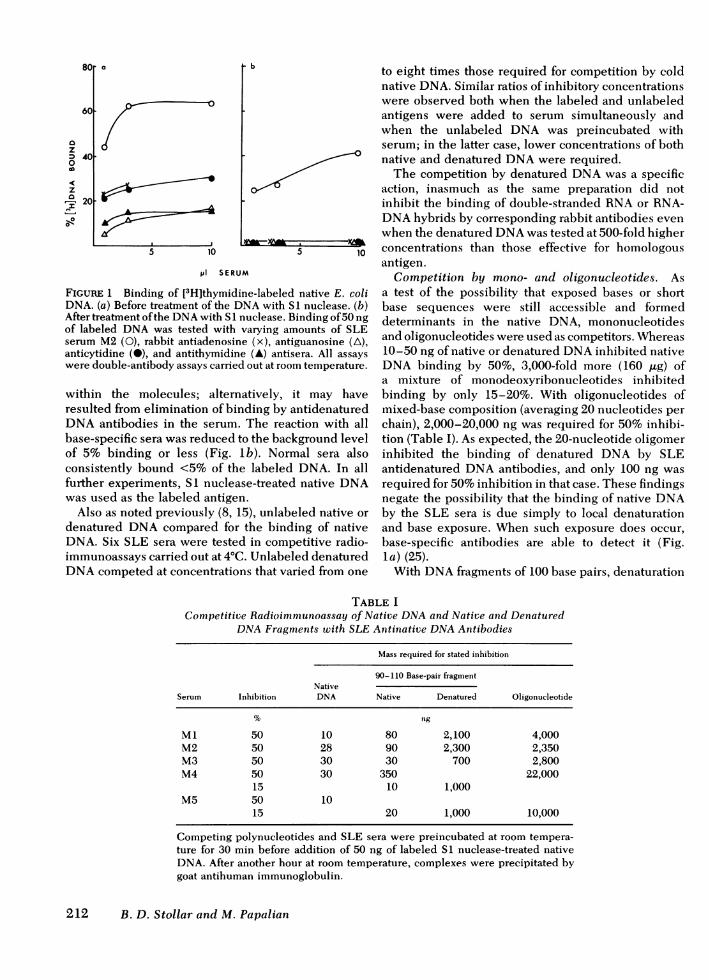
DNAbinding and competition. Initial experimentswere performed to ensure that the antibodies beingmeasured were comparable to those studied previously(8, 15, 16). The DNAused for binding was [3H]thymi-dine-labeled Escherichia coli DNA, prepared from apartial thymine-requiring mutant by the procedure ofMarmur (23). Without further treatment, this DNAwasstill bound by base-specific antibodies to denaturedDNAas well as by SLE sera (Fig. la). After treatmentof the DNA with S1 nuclease, which specificallydigests single-stranded regions (24), most of thebinding by SLE serum was retained (Fig. lb). Thereduction that did occur in binding by SLE serum mayhave resulted from a decrease in molecular weight ofthe DNA if the S1 nuclease cleaved nicked regions
Denatured DNAand SLE Antinative DNAAntibodies 211
80r a
60
0z
) 400co
z020I
0
5 10 5 10
pl SERUM
FIGURE 1 Binding of [3H]thymidine-labeled native E. coliDNA. (a) Before treatment of the DNAwith SI nuclease. (b)After treatment of the DNAwith S1 nuclease. Binding of 50 ngof labeled DNAwas tested with varying amounts of SLEserum M2 (0), rabbit antiadenosine (x), antiguanosine (A),anticytidine (0), and antithymidine (A) antisera. All assayswere double-antibody assays carried out at room temperature.
within the molecules; alternatively, it may haveresulted from elimination of binding by antidenaturedDNAantibodies in the serum. The reaction with allbase-specific sera was reduced to the background levelof 5% binding or less (Fig. lb). Normal sera alsoconsistently bound <5% of the labeled DNA. In allfurther experiments, S1 nuclease-treated native DNAwas used as the labeled antigen.
Also as noted previously (8, 15), unlabeled native ordenatured DNAcompared for the binding of nativeDNA. Six SLE sera were tested in competitive radio-immunoassays carried out at 4°C. Unlabeled denaturedDNAcompeted at concentrations that varied from one
to eight times those required for competition by coldnative DNA. Similar ratios of inhibitory concentrationswere observed both when the labeled and unlabeledantigens were added to serum simultaneously andwhen the unlabeled DNA was preincubated withserum; in the latter case, lower concentrations of bothnative and denatured DNAwere required.
The competition by denatured DNAwas a specificaction, inasmuch as the same preparation did notinhibit the binding of double-stranded RNAor RNA-DNAhybrids by corresponding rabbit antibodies evenwhen the denatured DNAwas tested at 500-fold higherconcentrations than those effective for homologousantigen.
Competition by mono- and oligonucleotides. Asa test of the possibility that exposed bases or shortbase sequences were still accessible and formeddeterminants in the native DNA, mononucleotidesand oligonucleotides were used as competitors. Whereas10-50 ng of native or denatured DNAinhibited nativeDNA binding by 50%, 3,000-fold more (160 jig) ofa mixture of monodeoxyribonucleotides inhibitedbinding by only 15-20%. With oligonucleotides ofmixed-base composition (averaging 20 nucleotides perchain), 2,000-20,000 ng was required for 50% inhibi-tion (Table I). As expected, the 20-nucleotide oligomerinhibited the binding of denatured DNA by SLEantidenatured DNAantibodies, and only 100 ng wasrequired for 50% inhibition in that case. These findingsnegate the possibility that the binding of native DNAby the SLE sera is due simply to local denaturationand base exposure. When such exposure does occur,base-specific antibodies are able to detect it (Fig.la) (25).
With DNAfragments of 100 base pairs, denaturation
TABLE ICompetitive Radioimmunoassay of Native DNAand Native and Denatured
DNAFragments with SLE Antinative DNAAntibodies
Mass required for stated inhibition
90-110 Base-pair fragmentNative
Serum Inhibition DNA Native Denatured Oligonucleotide
% ng
Ml 50 10 80 2,100 4,000M2 50 28 90 2,300 2,350M3 50 30 30 700 2,800M4 50 30 350 22,000
15 10 1,000M5 50 10
15 20 1,000 10,000
Competing polynucleotides and SLE sera were preincubated at room tempera-ture for 30 min before addition of 50 ng of labeled Si nuclease-treated nativeDNA. After another hour at room temperature, complexes were precipitated bygoat antihuman immunoglobulin.
212 B. D. Stollar and M. Papalian
0
-A
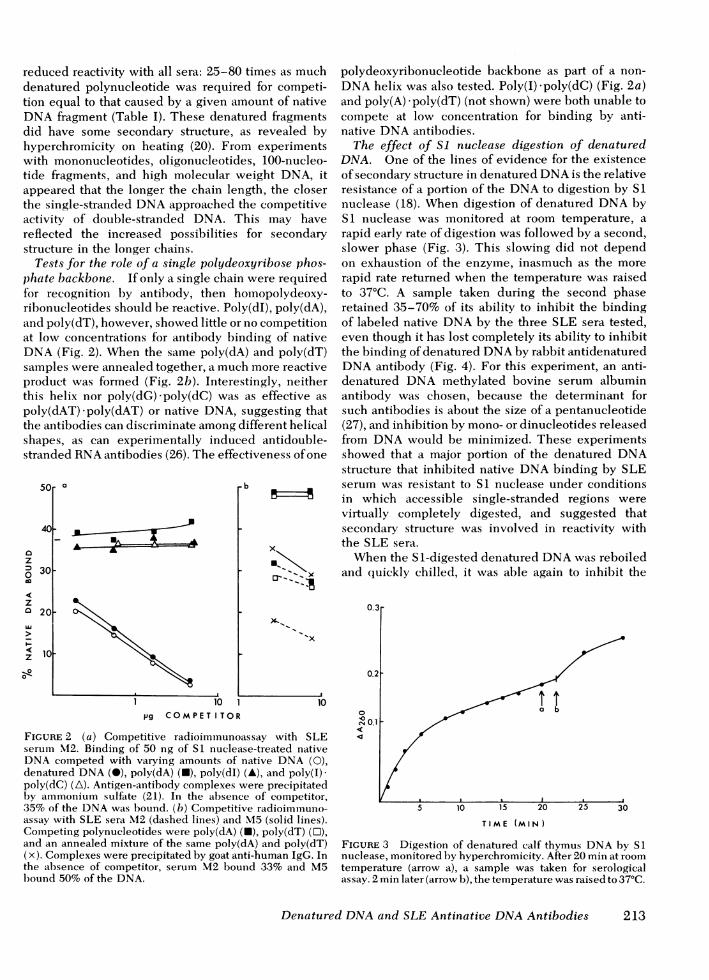
reduced reactivity with all sera: 25-80 timedenatured polynucleotide was required fotion equal to that caused by a given amounDNA fragment (Table I). These denatureddid have some secondary structure, as rehyperchromicity on heating (20). From exwith mononucleotides, oligonucleotides, 11tide fragments, and high molecular weighappeared that the longer the chain length,the single-stranded DNAapproached the ciactivity of double-stranded DNA. Thisreflected the increased possibilities forstructure in the longer chains.
Tests for the role of a single polydeoxyriiphate backbone. If only a single chain werfor recognition by antibody, then homolribonucleotides should be reactive. Poly(dI)and poly(dT), however, showed little or no c(at low concentrations for antibody bindingDNA(Fig. 2). When the same poly(dA) an(samples were annealed together, a much moproduct was formed (Fig. 2b). Interesting]this helix nor poly(dG) poly(dC) was as epoly(dAT) -poly(dAT) or native DNA, suggthe antibodies can discriminate among differshapes, as can experimentally induced astranded RNAantibodies (26). The effectivei
40
azm0
z0
z
0-
50r a
301
20j
10o
10 1pg COMPET TOR
Zs as much polydeoxyribonucleotide backbone as part of a non-r competi- DNAhelix was also tested. Poly(I) -poly(dC) (Fig. 2a)It of native and poly(A) -poly(dT) (not shown) were both unable tofragments compete at low concentration for binding by anti-
vealed by native DNAantibodies.:periments The effect of Si nuclease digestion of denatured00-nucleo- DNA. One of the lines of evidence for the existenceit DNA, it of secondary structure in denatured DNAis the relativethe closer resistance of a portion of the DNAto digestion by SI
ompetitive nuclease (18). When digestion of denatured DNAbymay have Si nuclease was monitored at room temperature, asecondary rapid early rate of digestion was followed by a second,
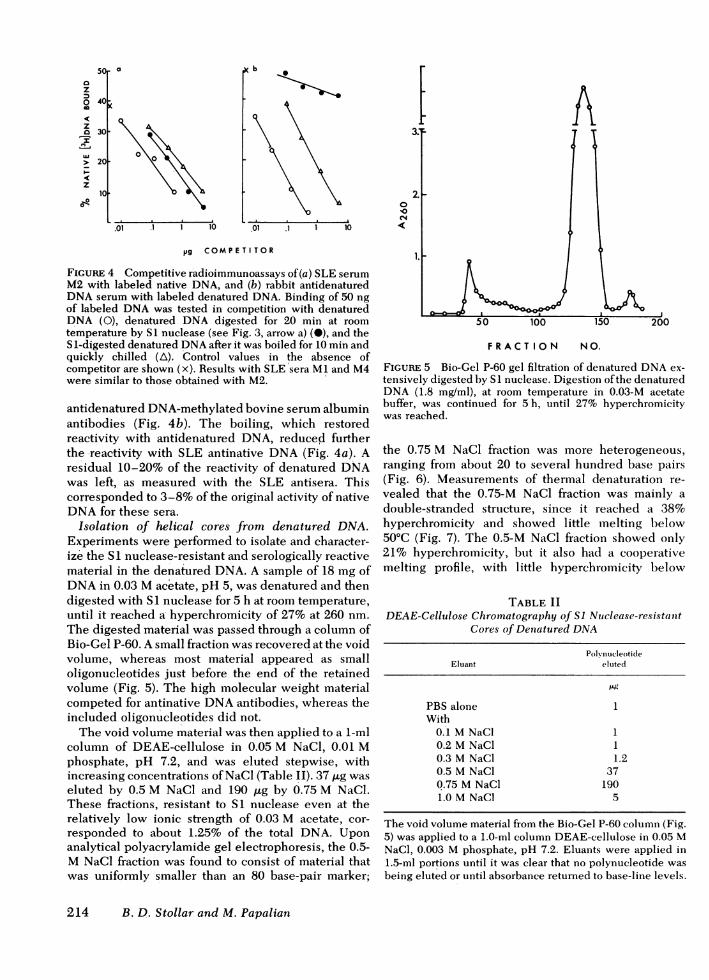
slower phase (Fig. 3). This slowing did not dependbose phos- on exhaustion of the enzyme, inasmuch as the moree required rapid rate returned when the temperature was raisedpolydeoxy- to 37°C. A sample taken during the second phase1, poly(dA), retained 35-70% of its ability to inhibit the bindingompetition of labeled native DNAby the three SLE sera tested,t of native even though it has lost completely its ability to inhibitd poly(dT) the binding of denatured DNAby rabbit antidenaturedore reactive DNAantibody (Fig. 4). For this experiment, an anti-ly, neither denatured DNA methylated bovine serum albuminffective as antibody was chosen, because the determinant foresting that such antibodies is about the size of a pentanucleotide^ent helical (27), and inhibition by mono- or dinucleotides releasedLntidouble- from DNAwould be minimized. These experimentsness of one showed that a major portion of the denatured DNA
structure that inhibited native DNAbinding by SLEserum was resistant to SI nuclease under conditionsin which accessible single-stranded regions werevirtually completely digested, and suggested thatsecondary structure was involved in reactivity with
x the SLE sera.Whenthe SI-digested denatured DNAwas reboiled
m_ -s_x and quickly chilled, it was able again to inhibit the
10
FIGURE 2 (a) Competitive radioimmunoassay with SLEserum M2. Binding of 50 ng of SI nuclease-treated nativeDNA competed with varying amounts of native DNA (0),denatured DNA(i), poly(dA) (N), poly(dI) (A), and poly(I)-poly(dC) (A). Antigen-antibody complexes were precipitatedby aimmonium sulfate (21). In the absence of competitor,35% of the DNAwas bound. (b) Competitive radioimmuno-assay with SLE sera M2 (dashed lines) and M5 (solid lines).Competing polynucleotides were poly(dA) (N), poly(dT) (O),and an annealed mixture of the same poly(dA) and poly(dT)(x). Complexes were precipitated by goat anti-human IgG. Inthe absence of competitor, serum M2 bound 33% and M5bound 50% of the DNA.
00
TIME (MIN)
FIGURE 3 Digestion of denatured calf thymus DNAby Slnuclease, monitored by hyperchromicity. After 20 min at roomtemperature (arrow a), a sample was taken for serologicalassay. 2 min later (arrow b), the temperature was raised to 37°C.
Denatured DNAand SLE Antinative DNAAntibodies
4
L -__-
m-
I
213
50r 00z
o 4C
za 30
> 20
z
0
i.01 .1 1 10 .01
pg COMPETITOR
0 f0'0cm
.1 1 10
FIGURE 4 Competitive radioimmunoassays of(a) SLE serumM2 with labeled native DNA, and (b) rabbit antidenaturedDNAserum with labeled denatured DNA. Binding of 50 ngof labeled DNAwas tested in competition with denaturedDNA (0), denatured DNA digested for 20 min at roomtemperature by SI nuclease (see Fig. 3, arrow a) (M), and theS1-digested denatured DNAafter it was boiled for 10 min andquickly chilled (A). Control values in the absence ofcompetitor are shown (x). Results with SLE sera MI and M4were similar to those obtained with M2.
antidenatured DNA-methylated bovine serum albuminantibodies (Fig. 4b). The boiling, which restoredreactivity with antidenatured DNA, reduced furtherthe reactivity with SLE antinative DNA(Fig. 4a). Aresidual 10-20% of the reactivity of denatured DNAwas left, as measured with the SLE antisera. Thiscorresponded to 3-8% of the original activity of nativeDNAfor these sera.
Isolation of helical cores from denatured DNA.Experiments were performed to isolate and character-ize the SI nuclease-resistant and serologically reactivematerial in the denatured DNA. A sample of 18 mg ofDNAin 0.03 Macetate, pH 5, was denatured and thendigested with SI nuclease for 5 h at room temperature,until it reached a hyperchromicity of 27% at 260 nm.The digested material was passed through a column ofBio-Gel P-60. A small fraction was recovered at the voidvolume, whereas most material appeared as smalloligonucleotides just before the end of the retainedvolume (Fig. 5). The high molecular weight materialcompeted for antinative DNAantibodies, whereas theincluded oligonucleotides did not.
The void volume material was then applied to a 1-mlcolumn of DEAE-cellulose in 0.05 M NaCl, 0.01 Mphosphate, pH 7.2, and was eluted stepwise, withincreasing concentrations of NaCl (Table II). 37 ,tg waseluted by 0.5 M NaCl and 190 gsg by 0.75 M NaCl.These fractions, resistant to S1 nuclease even at therelatively low ionic strength of 0.03 M acetate, cor-responded to about 1.25% of the total DNA. Uponanalytical polyacrylamide gel electrophoresis, the 0.5-MNaCl fraction was found to consist of material thatwas uniformly smaller than an 80 base-pair marker;
A
FRACTION N 0.
FIGURE 5 Bio-Gel P-60 gel filtration of denatured DNAex-tensively digested by S1 nuclease. Digestion of the denaturedDNA (1.8 mg/ml), at room temperature in 0.03-M acetatebuffer, was continued for 5 h, until 27% hyperchromicitywas reached.
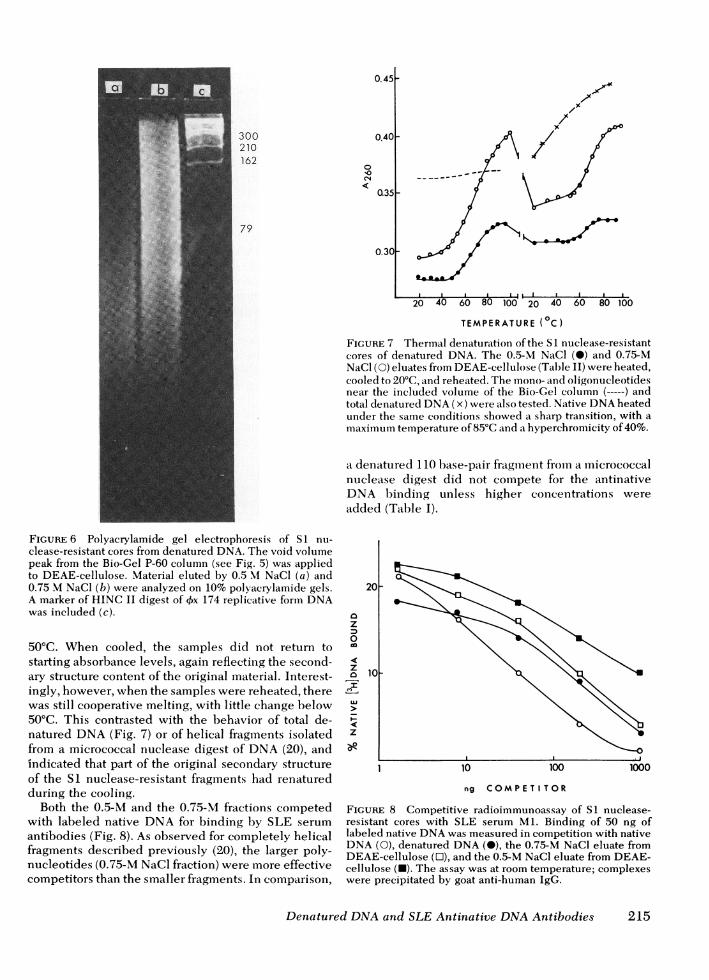
the 0.75 M NaCl fraction was more heterogeneous,ranging from about 20 to several hundred base pairs(Fig. 6). Measurements of thermal denaturation re-vealed that the 0.75-M NaCl fraction was mainly adouble-stranded structure, since it reached a 38%hyperchromicity and showed little melting below50°C (Fig. 7). The 0.5-M NaCl fraction showed only21% hyperchromicity, but it also had a cooperativemelting profile, with little hyperchromicity below
TABLE IIDEAE-Cellulose Chromatography of SI Ntuclease-resistant
Cores of Denatured DNA
PolknucleotideEluant eluted
PBS alone 1With
0.1 MNaCl 10.2 MNaCl 10.3 M NaCl 1.20.5 M NaCl 370.75 MNaCl 1901.0 MNaCl 5
The void volume material from the Bio-Gel P-60 column (Fig.5) was applied to a 1.0-ml column DEAE-cellulose in 0.05 MNaCl, 0.003 M phosphate, pH 7.2. Eluants were applied in1.5-ml portions until it was clear that no polynucleotide wasbeing eluted or until absorbance returned to base-line levels.
214 B. D. Stollar and M. Papalian
0.45F
00
CN4
0Q35
0.30-
20 40 60 80 100- 20 40 60 80 100
TEMPERATURE(0C)
FIGURE 7 Thermal denaturation ofthe SI nuclease-resistantcores of denatured DNA. The 0.5-M NaCl (-) and 0.75-MNaCl (0) eluates from DEAE-cellulose (Table II) were heated,cooled to 20°C, and reheated. The mono- and oligonucleotidesnear the included volume of the Bio-Gel column (-----) andtotal denatured DNA(x) were also tested. Native DNAheatedunder the same conditions showed a sharp transition, with amaximum temperature of 85°C and a hyperchromicity of 40%.
a denatured 110 base-pair fragment from a micrococcalnuclease digest did not compete for the antinativeDNA binding unless higher concentrations were
added (Table I).
FIGURE 6 Polyacrylamide gel electrophoresis of SI nu-
clease-resistant cores from denatured DNA. The void volumepeak from the Bio-Gel P-60 column (see Fig. 5) was appliedto DEAE-cellulose. Material eluted by 0.5 M NaCl (a) and0.75 MNaCl (b) were analyzed on 10% polyacrylamide gels.A marker of HINC II digest of Ox 174 replicative form DNAwas included (c).
50°C. When cooled, the samples did not return tostarting absorbance levels, again reflectiing the second-ary structure content of the original material. Interest-ingly, however, when the samples were reheated, therewas still cooperative melting, with little change below50°C. This contrasted with the behavior of total de-natured DNA(Fig. 7) or of helical fragments isolatedfrom a micrococcal nuclease digest of DNA(20), andindicated that part of the original secondary structureof the SI nuclease-resistant fragments had renaturedduring the cooling.
Both the 0.5-M and the 0.75-M fractions competedwith labeled native DNA for binding by SLE serum
antibodies (Fig. 8). As observed for completely helicalfragments described previously (20), the larger poly-nucleotides (0.75-M NaCl fraction) were more effectivecompetitors than the smaller fragments. In comparison,
20h
az
0
z0
I
I-
z
9
10-
10 100 1000
ng COMPETI TOR
FIGURE 8 Competitive radioimmunoassay of SI nuclease-resistant cores with SLE serum Ml. Binding of 50 ng oflabeled native DNAwas measured in competition with nativeDNA(0), denatured DNA(-), the 0.75-M NaCl eluate fromDEAE-cellulose (0), and the 0.5-M NaCl eluate from DEAE-cellulose (-). The assay was at room temperature; complexeswere precipitated by goat anti-human IgG.
Denatured DNAand SLE Antinative DNAAntibodies
300210162
79
0.40F
I
I
215

The effect of temperature. Secondary structure,which is known to occur in denatured DNA, is unfoldedgradually with increasing temperature in the range of0°-37°C in 0.15 M NaCl. Completely helical DNAisnot altered in this temperature range, but melts sharplyat higher temperatures. In view of this property, theability of denatured DNA to compete with labelednative DNAfor antibody was tested at various tempera-tures. In preliminary experiments it was found that thelabeled native DNAwas bound in the assay when allsteps were carried out at 40, 200, or 370C, and that thesame amount of serum could be used at each tempera-ture. In typical experiments, control binding valuesat 40 and 370C were respectively, 32 and 30%, with 1A1l of serum M5, and 35 and 44%with 2 ,ul of serum M2.
With four sera, competition by denatured DNAwasdecreased at the higher temperatures (Table III). Athree- to sixfold higher concentration was requiredat 37°C than at 4°C. Native DNAwas equally effectiveat both temperatures. With one serum (M4), the sameamount of denatured DNA was effective at 40 and370C, but at both temperatures this was eight timesthe amount of native DNA required for identicalcompetition. In all cases, therefore, denatured DNAhadonly 10-30% of the reactivity of native DNAat 37°C;at this temperature about 30-40% of the secondarystructure in denatured DNAwas melted (Fig. 7).
DISCUSSION
The reaction of SLE serum antibodies with nativeDNAhas had a puzzling aspect, for the reaction can be
TABLE IIITemperature Dependence of the Competition of Denatured
DNAfor Antinative DNAAntibodies
DNArequired for50% competition
37'CSerum DNA 4°C 20'C 37°C 4°C
ng
Ml Denatured 60 100 250 4.1Native 65 70 1.1
M2 Denatured 70 170 450 6.4Native 55 75 1.4
M3 Denatured 35 85 210 6.0Native 50 50 1.0
M4 Denatured 1,000 800 800 0.8Native 120 100 0.8
M5 Denatured 120 330 2.8Native 25 37 1.5
Competing and labeled DNAwere added simultaneously toheated diluted serum. Control binding without competingDNAwas 30-60% at all temperatures.
inhibited by denatured DNA. This has raised questionswhether the antibodies do in fact recognize secondarystructure of helical DNA or, alternatively, detectdenatured regions in apparently native DNA. Severalapproaches have been taken to support the conclusionthat the antibodies react with native DNA. Theseinclude the use of closed circular DNA(11), syntheticpoly(dAT) *poly(dAT) (12), or intracellular circularDNA(13) as test antigens that should be completelyhelical. With other DNAsamples, treatments such asabsorption by various column materials (6) or digestionby S 1 nuclease (14) are used to remove single-strandedregions of DNA. Still, it may be argued that subsequentexposure to the test serum itself may create single-stranded regions by action of serum nucleases. Also,hydrogen-exchange experiments indicate that DNAisin a dynamic equilibrium, in which bases are locallyunpaired and exposed to solvent for a significantfraction of time (with an equilibrium constant foropening of about 0.001-0.01 at neutral pH and 0°C)(28). It is possible, therefore, that the reaction of nativeDNAcould depend on local denaturation and exposureof bases.
The ability of denatured DNAto react with antina-tive DNAantibodies, however, could depend on thepresence of secondary structure in the denatured DNA.This could include base-paired helical regions of vary-ing length and stability. Such structures have beenisolated from single-stranded DNA of small viruses(29-32). Hairpin loops of 44 and 60 base pairs wereisolated from M13 virus DNA(30); similar helical seg-ments have been identified and sequenced in phage G4viral DNA(31). These structures were isolated first ascores of single-stranded DNAthat resist digestion by S 1nuclease or other enzymes specific for unpaired nucleicacids. S 1 nuclease-resistant cores with fold-back helicalstructure also occur in more complex samples such asbacterial and mammalian DNA(18, 33, 34). In a situa-tion that may parallel the ability of antibodies torecognize secondary structure in denatured DNA, du-plex structure within single-stranded OX 174 DNAappears to be responsible for recognition and cleavageby restriction endonucleases (35). In addition to thesestable cores, many other regions with less stablesecondary structure would be present in high molecu-lar weight DNAat low temperature.
In view of the above considerations, it may not bepossible to identify conclusively the reactive struc-ture simply by characterizing the test antigen as na-tive or denatured. The experiments reported in thisarticle, therefore, were designed to determine whetherthere is a relationship between changes in the amountof secondary structure present and the degree of reac-tivity. Additional experiments were done to isolate theS 1 nuclease-resistant cores from calf DNAand to deter-
216 B. D. Stollar and M. Papalian

mine whether they contained base-paired structure andcould react with antinative DNAantibodies.
The failure of mononucleotides to compete, evenat very high concentrations, indicates that the anti-bodies do not recognize single bases; even with oligo-nucleotides averaging 20 residues in length, competi-tive binding required concentrations some 400-foldhigher than those effective with helical DNA. Mostsignificant in this regard, helical fragments of 100 basepairs were effective competitors, whereas the samefragments, when denatured, were much less effec-tive (Table I). There was some residual binding ac-tivity of the denatured fragments. This could mean thata single polydeoxyribonucleotide backbone can bindto these antibodies. SLE sera are known to containheterogeneous antibody populations (17), and somemay in fact bind preferentially to the single back-bone structure. In competition for antibodies thatbind labeled native DNA, however, the single-chainDNA fragments were always much weaker than thehelical forms. This was true also for poly(dA) or poly-(dT) in comparison with poly(dA) poly(dT). Hybridhelices containing one polydeoxyribonucleotidestrand, such as poly(A) poly(dT) or poly(I) -poly(dC),also showed no reactivity when tested at concentrationsat which poly(dA) *poly(dT) was reactive. These resultssupport the conclusion that, although these populationsof antinative DNAantibodies may bind one chain withlow energy, they show much higher binding energywith (i.e., specificity for) helical structures.
Even in the denatured 100-base-pair fragments,secondary structure may be responsible for reactivitywith these antibodies. The denatured fragments doshow hyperchromicity upon heating (20) and there-fore do have secondary structure, but they are unlikelyto have long stable helical segments. There may be anequilibrium in which portions of the fragments spend asmall but finite fraction of time in a helical form, whichis then stabilized by interaction with antibody. Thecompetition by higher concentrations of the denaturedfragments may reflect, in part, such a conformationalequilibrium. A similar analysis has been applied ingreater detail for the reactions of native and unfoldedprotein with conformation-dependent antiprotein anti-bodies (36).
The effect of incubation temperature on reactivityalso indicated that secondary structure in denaturedDNAwas important for reaction with the SLE antina-tive DNAantibodies. Native DNAwas equally reac-tive at 40 and 37°C, over which range its secondarystructure was not appreciably altered. In contrast, asdenatured DNAwas gradually unfolded between 40and 37°C, its reactivity decreased three- to sixfold withfour sera; with the fifth serum, it was only one-eighthas reactive as native DNAat either temperature. At
37°C, native DNAwas from 3.5-9 times as effective asdenatured DNAwith these five sera. Residual activityat 37°C may have reflected the weak binding of a singlepolydeoxyribonucleotide backbone or a conforma-tional equilibrium, as discussed above. In this case,however, there was still a possibility that stable helicalstructure was present within the remaining secondarystructure evident in the thermal denaturation profile(Fig. 7). Experiments with SI nuclease digestiondemonstrated that at least part of the serological re-activity of denatured DNA depended on relativelystable helical secondary structure. With this enzyme,an early rapid rate of digestion probably reflected thehydrolysis of easily accessible, unpaired single strands;more highly folded regions were digested more slowly.When the enzyme had eliminated virtually all regionsrecognized by antidenatured DNA antibodies, theproducts still competed for SLE antinative DNAanti-bodies. Boiling the products restored their reactivitywith the antidenatured DNAand reduced further theirreaction with antinative DNA. The latter was noteliminated completely, however: residual activityreflected either the single backbone contribution tobinding energy or the presence of readily renaturablestructures in the DNA. In further exploration of thesepossibilities, a larger sample of DNA was digestedmore extensively by SI nuclease and resistant coreswere isolated by gel filtration. This preparative scaledigestion was done at relatively low ionic strength,so that only regions of relatively stable secondary struc-ture should remain resistant to prolonged digestion.The resistant cores were heterogeneous in size, rang-ing from about 20 to several hundred base pairs insize. Thermal denaturation experiments indicated theyhad helical structure that melted cooperatively. Someof the denatured material renatured readily upon cool-ing, as reflected in the cooperative melting observedduring a second heating cycle (Fig. 7); this was unlikethe curve seen with total denatured DNA (Fig. 7)or of fragments that were not selected on the basis ofresistance to S1 nuclease (20). The renaturable materialcorresponds to snap-back hairpin structures. The re-mainder of the S1 nuclease-resistant structure corre-sponds to base-paired segments that are separable athigh temperature and do not renature rapidly be-cause they do not have a connecting loop. Identicalproducts of S1 nuclease digestion of mammalian DNAhave been reported by Lin and Lee (34), who usedaqueous dioxane to select the more stable helical re-gions. The isolated fold-back helical structures com-peted effectively for antinative DNAantibodies.
The experiments described in this article supportthe conclusion that optimal reactions of SLE serumantibodies that bind native DNAdepend on helicalstructure, and that the aintibodies detect secondary
Denatured DNAand SLE Antinative DNAAntibodies 217

structure in denatured DNArather than denatured re-gions in native DNA. The conclusions are based ontests of competition by mono- and oligonucleotides,on measurements of quantitative changes in reactivityin relation to changes in secondary structure, and onthe isolation of reactive helical structures from dena-tured DNA. These antibodies are clearly distinct frombase-specific antidenatured DNAantibodies of SLEsera. Exploration of the question of whether antibodiesof different specificities have different pathogeneticimportance or diagnostic value does have a valid im-munochemical basis.
ACKNOWLEDGMENTS
We gratefully acknowledge the technical assistance of M.Ristic.
This work was supported by grant AI-14534 from the Na-tional Institutes of Health.
REFERENCES
1. Pincus, T., P. H. Schur, J. A. Rose, J. L. Decker, and N.Talal. 1969. Measurement of serum DNAbinding ac-tivity in lupus erythematosus. N. Engl. J. Med. 281:701-705.
2. Hughes, G. R. V., S. A. Cohen, and C. L. Christian.1971. Anti-DNA activity in systemic lupus erythematosus:a diagnostic and therapeutic guide. Ann. Rheum. Dis. 30:259-264.
3. Hasselbacher, P., and E. C. Leroy. 1974. Serum DNAbinding activity in healthy subjects and in rheumaticdisease. Arthritis Rheum. 17: 63-71.
4. Bell, C., N. Talal, and P. Schur. 1975. Antibodies to DNAin patients with rheumatoid arthritis and juvenile rheuma-toid arthritis. Arthritis Rheum. 18: 535-540.
5. Adler, M. K., A. Baumgarten, and N. J. Siegel. 1975. Prog-nostic significance of DNAbinding capacity in patientswith lupus nephritis. Ann. Rheum. Dis. 34: 444-450.
6. Locker, J. D., M. E. Medof, R. M. Bennett, and S. Suk-hupunyaraska. 1977. Characterization of DNAused toassay sera for anti-DNA antibodies; determination of thespecificities of anti-DNA antibodies in SLE and non-SLE rheumatic disease states.J. Immunol. 118: 694-701.
7. Lightfoot, R. W., Jr., and G. R. V. Hughes. 1976. Sig-nificance of persisting serological abnormalities in SLE.Arthritis Rheum. 5: 836-843.
8. Stollar, B. D. 1975. The detection of anti-DNA and DNA/anti-DNA complexes. Scand. J. Rheumatol. Supp. 11:7-11.
9. Plescia, 0. J., W. Braun, and N. C. Palczuk. 1964. Produc-tion of antibodies to denatured deoxyribonucleic acid(DNA). Proc. Natl. Acad. Sci. U. S. A. 52: 279-285.
10. Erlanger, B. F., and S. M. Beiser. 1964. Antibodies specificfor ribonucleosides and ribonucleotides and their reactionwith DNA. Proc. Natl. Acad. Sci. U. S. A. 52: 68-74.
11. Aarden, L. A., F. Lakmaker, and E. R. deGroot. 1976.Immunology of DNA. IV. Quantitative aspects of the Farrassay. J. Immunol. Methods. 11: 153-163.
12. Steinman, C. R., U. Deesomchok, and H. Spiera. 1976.Detection of anti-DNA antibody using synthetic antigens.
J. Clin. Invest. 57: 1330-1341.13. Aarden, L. A., E. R. deGroot, and T. E. W. Feltkamp.
1975. Immunology of DNA. III. Crithidia luciliae, asimple substrate for the determination of anti-ds DNAwith the immunofluorescent technique. Ann. N. Y. Acad.Sci. 254: 505-515.
14. Izui, S., P. H. Lambert, and P. A. Miescher. 1976. De-termination of anti-DNA antibodies by a modified 12I-labelled DNA-binding test. Clin. Exp. Immunol. 26:425-430.
15. Picazo, J. J., and E. M. Tan. 1975. Specificities of anti-bodies to native DNA. Scand. J. Rheumatol. Supp. 11:35-41.
16. Arana, R., and M. Seligmann. 1967. Antibodies to nativeand denatured deoxyribonucleic acid in systemic lupuserythematosus.J. Clin. Invest. 46: 1867-1882.
17. Koffler, D., R. Carr, V. Agnello, R. Thobum, and H. G.Kunkel. 1971. Antibodies to polynucleotides in humansera: antigenic specificity and relation to disease. J.Exp. Med. 134: 294-312.
18. Bagchi, B., D. N. Misra, S. Basu, and N. N. Das Gupta.1969. Conformation of denatured and renatured DNA.Biochem. Biophys. Acta. 182: 551-561.
19. Stollar, B. D. 1975. The specificity and application of anti-bodies to helical nucleic acids. Crit. Rev. Biochem. 3:45-69.
20. Papalian, M., E. Lafer, R. Wong, and B. D. Stollar. 1980.The reaction of SLE antinative DNA antibodies withnative DNA fragments from 20 to 1,200 base pairs. J.Clin. Invest. 65: 469-477.
21. Stollar, B. D. 1977. Quantitative microcomplement fixa-tion and radioactive antigen binding assays for measuringanti-DNA antibodies. Ann. Rheum. Dis. 36(Suppl.):102-107.
22. Seaman, E. 1968. Chain length determination by end tototal phosphorus. Methods Enzymol. 12(Pt. B): 218-220.
23. Marmur, J. 1961. A procedure for the isolation of deoxy-ribonucleic acid from microorganisms. J. Mol. Biol. 3:208-218.
24. Ando, T. 1966. A nuclease specific for heat-denaturedDNAisolated from a product of Aspergillus oryzae. Bio-chim. Biophys. Acta. 114: 158-168.
25. Seaman, E., L. Levine, and H. Van Vanukis. 1966. Anti-bodies to the methylene blue sensitized photooxidationproduct in deoxyribonucleic acid. Biochemistry. 5:1216-1223.
26. Johnston, M. I., and B. D. Stollar. 1978. Antigenicstructure of double-stranded RNAanalogues having vary-ing activity in interferon induction. Biochemistry. 17:1959-1964.
27. Wakizaka, A., and E. Okuhara. 1975. Immunologicalstudies on nucleic acids: an investigation of the anti-genic determinants of denatured deoxyribonucleic acid(DNA) reactive with rabbit anti-DNA antisera by aradioimmunoassay technique. Immunochemistry. 12:843-845.
28. Teitelbaum, H., and S. W. Englander. 1975. Open statesin native polynucleotides. II. Hydrogen exchange study ofcytosine-containing double helices. J. Mol. Biol. 92:79-92.
29. Schaller, H., H. Voss, and S. Gucker. 1969. Structure of theDNAof bacteriophage fd. II. Isolation and characteriza-tion of a DNAfraction with double strand-like proper-ties. J. Mol. Biol. 44: 445-458.
30. Niyogi, S. K., and S. Mitra. 1977. Isolation and char-acterization of naturally occurring hairpin structures insingle-stranded DNAof coliphage M13. Biochem. Bio-phys. Res. Commun. 79: 1037-1044.
31. Fiddes, J. C., B. G. Barrell, and G. N. Godson. 1978.
218 B. D. Stollar and M. Papalian

Nucleotide sequences of the separate origins of synthesisof bacteriophage G4 viral and complementary DNAstrands. Proc. Natl. Acad. Sci. U. S. A. 75: 1081-1085.
32. Shishido, K., and Y. Ikeda. 1971. Isolation of double-helical regions rich in guanine-cytosine base pairing frombacteriophage fl DNA. Biochem. Biophys. Res. Commun.42: 482-489.
33. Shishido, K., and T. Ando. 1972. Estimation of the double-helical content in various single-stranded nucleic acids bytreatment with a single strand-specific nuclease. Bio-chim. Biophys. Acta. 287: 477-484.
34. Lin, H. J., and C. L. H. Lee. 1979. Isolation of foldbackDNAutilizing nuclease SI digestion in aqueous dioxane.Anal. Biochem. 96: 144-151.
35. Blakesley, R. W., J. B. Dodgson, I. F. Nes, and R. D.Wells. 1977. Duplex regions in "single-stranded" Ox 174DNA are cleaved by a restriction endonuclease fromHaemophilus aegyptius. J. Biol. Chem. 252: 7300-7306.
36. Furie, B., A. N. Schechter, D. H. Sachs, and C. B. An-finsen. 1975. An immunological approach to the conforma-tional equilibrium of staphylococcal nuclease. J. Mol.Biol. 92: 497-506.
Denatured DNAand SLE Antinative DNAAntibodies 219





























