Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Zur Kenntnis der Stereochemie säurekatalysierterPolyencyclisationen
Author(s): Gut, Michel
Publication Date: 1960
Permanent Link: https://doi.org/10.3929/ethz-a-000150339
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2962
Zur Kenntnis der Stereochemie
säurekatalysierter Polyencyclisationen
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung
der Würde eines Doktors der technischen Wissenschaften
genehmigte
PROMOTION SARBE IT
vorgelegt von
MICHEL GUT
dipl. Ing.-Chem. E. T. H.
von Affoltern a./A.
Referent: Herr Prof. Dr. O. Jeger
Korreferent: Herr Prof. Dr. A. Eschenmoser
Juris-Verlag Zürich
i960
Leer - Vide - Empty

A mes chers parents
avec toute ma reconnaissance
Leer - Vide - Empty

Meinen hochverehrten Lehrern,
Herrn Prof. Dr. L.Ruzicka
und
Herrn Prof. Dr. V.Pr elog
bin ich für ihr stetes Wohlwollen und die Unterstützung der vorliegenden Arbeit zu
grossem Dank verpflichtet.
Herrn Prof. Dr. A. Eschenmoser,
unter dessen Leitung ich die vorliegende Promotionsarbeit ausführte, möchte ich
für die vielen wertvollen Ratschläge und die unermüdliche Hilfe, die er mir immer
zuteil werden liess, meinen aufrichtigsten Dank aussprechen.
Ferner möchte ich auch Herrn Dr. P.Stadler für die vielen Anregungen
und die stets kameradschaftliche Hilfsbereitschaft herzlich danken.
Leer - Vide - Empty

- 7 -
INHALTSVERZEICHNIS
Einleitung 9
THEORETISCHER TEIL 10
A. Literaturbesprechung 10
1. Allgemeines über säurekatalysierte Cyclisationen von
Polyenverbindungen 10
2. Literaturbeispiele 13
B. Eigene Arbeiten 17
1. Darstellung der 7,ll-Dimethyl-2(trans),6(cis), 10-dodecatrien-
1-säure 17
2. Darstellung der cx-Monoeyclo-nor-farnesylsäure 20
3. Darstellung der racemischen und optisch aktiven
v-Monocyclo-nor-farnesylsäure 22
4. Cyclisationsversuche 24
5. Relative Cyclisationsgeschwindigkeiten der 7,11-Dimethyl-
2(trans),6(trans), 10-dodecatrien-l-säure und der
cx-Monocyclo-nor-farnesylsäure 28
C. Schlussfolgerungen 31
EXPERIMENTELLER TEIL 33
7, ll-Dimethyl-2(trans), 6(cis), 10-dodecatrien-l-säure 33
cx-Monocyclo-nor-farnesylsäure 41
(±)-v-Monocyclo-nor-farnesylsäure 45
(+)-Y-Monocyclo-nor-farnesylsäure 49
Relative Cyclisationsgeschwindigkeiten 53
Zusammenfassung 56
Literaturverzeichnis 57
Leer - Vide - Empty

- 9 -
Einleitung
In den letzten Jahren ist die Biosynthese des Cholesterins experimentell weit-1 2)
gehend abgeklärt worden ''.
Eines der bedeutsamsten Ergebnisse dieser Untersuchungen ist ohne Zweifel
die Feststellung >>>>>> >f dass Squalen durch enzymatische Cyclisation in
Lanosterin verwandelt und letzteres durch nachträgliche Umwandlung in Cholesterin
übergeführt wird .
Dieser Uebergang Squalen-Lanosterin lässt sich im Rahmen der heutigen, gülti¬
gen Vorstellungen über säurekatalysierte Cyclisationen gut verstehen, wenn man da¬
bei von gewissen arbiträren Annahmen über den Reaktionsmechanismus ausgeht (Kat¬
ionische Cyclisation, Wagner-Meerwein'sehe Umlagerungen). Durch Uebertra-
gung dieser Annahmen auf die übrigen pentaeyclischen Triterpene gelingt es, alle bis12)
heute bekannten Verbindungen dieser Klasse formal von Squalen abzuleiten '.
Diese Tatsache, sowie die Möglichkeit der Formulierung ähnlicher Zusammen¬
hänge in der Reihe der niedrigeren Terpene, haben zur Aufstellung der biogenetischen13 14 54T
Isoprenregel geführt ' ' '. Obwohl die Gültigkeit dieser Regel für die Biosynthese
der pflanzlichen Triterpene aus Squalen noch nicht gesichert ist ', hat sie sich als
hervorragendes Werkzeug bei der strukturellen Aufklärung und der mechanistischen
17 18 19 20 21 22)
Deutung der bis jetzt bekannten, sowie neu entdeckten Terpene >>»>»'
aus aliphatischen Vorläufern erwiesen.
•* u a , 1 a ,•
11,12,13,14,23,24,25,26). ,
Da nun eine weitgehende formale Analogie>>>>>>>'
zwischen
dem strukturellen Ergebnis dieser enzymgesteuerten Cyclisationen und der in der Che¬
mie der Terpene schon lange bekannten säurekatalysierten Cyclisationen von Polyenen
besteht, ist eine möglichst eingehende Kenntnis des Mechanismus dieser letzteren Re¬
aktion von Interesse.
Die strukturellen Voraussetzungen solcher säurekatalysierten Cyclisationen dür¬
fen als bekannt betrachtet werden; sie bildeten das Thema von Arbeiten, die vor allem
27 28 29)
von H .Schinz und Mitarbeitern durchgeführt wurden ' ' '. Bezüglich der Fra-
OE oA Ol 00 Oo 0^\
ge des sterischen Verlaufes ''' ' ' ' sind am hiesigen Laboratorium einge¬
hende Untersuchungen begonnen worden. Die vorliegende Arbeit stellt, als Fortsetzung
der Untersuchungen von P.Stadler et al. ' 'über den sterischen Verlauf säure¬
katalysierter Cyclisationen in der Reihe der Nor-farnesylsäuren, einen weiteren Bei¬
trag in dieser Richtung dar.
*) Vergleiche jedoch die Biosynthese der Sojasapogenole aus markiertem Mevalo-
lacton!5).

- 10 -
THEORETISCHER TEIL
A. LITERATURBESPRECHUNG
1. Allgemeines über säurekatalysierte Cyclisationen von
t, , w j
13,14,35,36)
Polyenverbindungen' ' » '
Bei säurekatalysierten, in vitro durchgeführten Cyclisationen von Polyenverbin¬
dungen müssen, je nach Reaktionsbedingungen und konstitutionellen Faktoren, eine131
Mehrzahl von Mechanismen angenommen werden '. Von diesen können auf Grund ih¬
res verschiedenen stereochemischen Ergebnisses zwei extreme Typen unterschieden
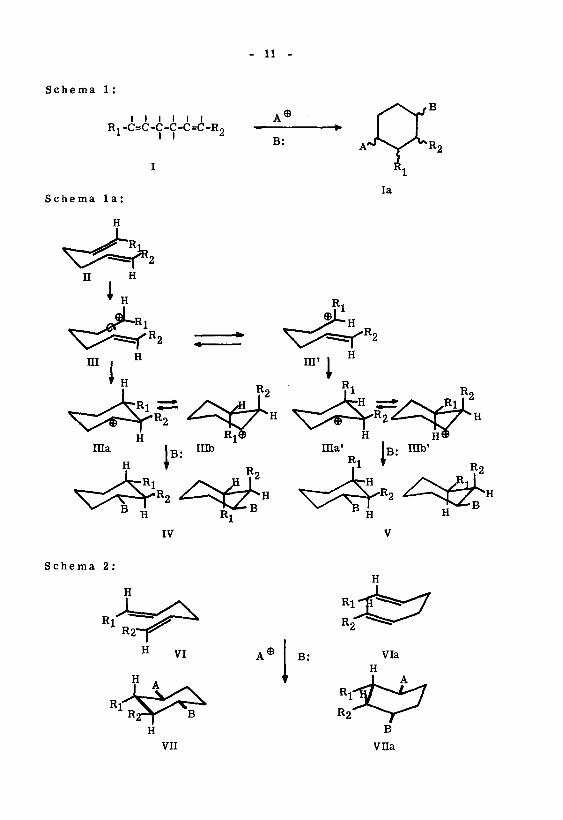
werden13'35).Im einen Fall handelt es sich um einen potentiell stereounspezifischen, sterisch
kontrollierten Reaktionsverlauf mit "formalen" trans- oder eis-Additionen. Man kann
dabei annehmen, dass die im Verlauf der Reaktion auftretenden kationischen Zwischen¬
produkte (z.B. klassische Carboniumkationen) konstellativ schneller aequilibrieren
(Schema la: III^III', ina==nib, IIIa'=IIIb'), als dass sie mit der nächsten Doppel¬
bindung (III—«-lila, in'—lila') oder mit einer externen Base (B: in Iüa/IIIb—-IV,
üla'/inb'——V) weiter reagieren. So können bei einer Cyclisation vom Typus I—-la
(Schema 1) alle acht möglichen Isomeren gebildet werden.
Der andere Reaktionstyp zeichnet sich durch eine Stereospezifität aus, welche
als Folge von stereoelektronisch kontrollierten, antiplanaren trans-Additionen an den
beteiligten Doppelbindungen betrachtet werden kann. Es kann angenommen werden,
dass solche Reaktionen über konfigurativ stabile, kationische Zwischenprodukte (z. B.
nicht klassische Carboniumionen) verlaufen, deren konstellative Aequilibrierung (Sche¬
ma la: III=ni', nia=IIIb, IIIa'==nib') langsamer verläuft als der Additionsschritt
mit der nächsten Doppelbindung (m—«-nia, in'—lila1) oder externen Base (Illa/lIIb
—IV, Ula'/nib'——V). Das sterische Ergebnis' bei diesem Reaktionsverlauf ist mit
dem generellen Schema der antiplanaren Addition vereinbar, einem Schema, das einer¬
seits im Falle von anderen elektrophüen Additionsreaktionen experimentell fundiert
ist ' und das anderseits mit Hilfe stereoelektronischer Betrachtungen theoretisch be-37)
gründet werden kann '. Dieses Schema verlangt, dass die vier am Additionsvorgang
beteiligten Zentren coplanar in einer Reaktionsebene bleiben. Angewandt auf den Reak-
üonsvorgang heisst das: Von den möglichen Konstellationen einer cyclisierenden Mole¬
kel sind deren zwei in dem Sinne ausgezeichnet, dass einzig in ihnen durchwegs anti-
planare Additionsschritte geometrisch möglich sind. Diese beiden Konstellationen
11 -
Sehe ma 1 :
I I I I I I
l ii I
B:
Schema la:
H
la
Ri
*1»j'
Rn
m I m' I
H Rl® H H®
JB- IIIb raa' Iß:ma
R-,
IV V
«1
Schema 2 :
Ri"
H
Rl
R2'
HVI Via
«i^ ^R2JJ^ B
H
VII
B
VHa
- 12 -
S chema 3 :
COAcOH/Ac20/H2S04
Zi-temp,
vra
CÛOAc
IX
XI OH
œ~X
Schema 4 :
COOH
1) HCOOH/H2SO4/20°2) OHe
XII
OOH
ceCOOH
xra
çcCOOHXIV XV

- 13 -
können sinngemäss als Sessel- VI, bzw. als Wannenfaltung Via bezeichnet werden.
Das Charakteristikum dieser beiden Ringschlusskonstellationen Vü/VIIa ist die
primär equatoriale und - bezüglich der neuen gebildeten Ringbindung - antiplana-
re Lage der Addenden A und B im Cyclisationsprodukt Vu bzw. Vila. Gemäss50)
den für solche Cyclisationen in Tab. 1 ' aufgeführten Korrelationen wäre die
Bildung von nur 2 Isomeren zu erwarten. Es ist denkbar, dass bei Cyclisationen
des ersten Typus (potentiell stereounspezifischer Reaktionsverlauf) die kationischen
Zwischenprodukte konstellativ wohl aequilibrieren, die nachfolgenden Additions¬
schritte aber sterisch (z. B. durch sterische Hinderung) derart kontrolliert sein
können, dass bevorzugt solche Endprodukte entstehen, die mit dem Ergebnis eines
antiplanaren Reaktionsverlaufes formal übereinstimmen.
Tabelle 1
Konfiguration von I Relative Konfiguration von Ia bei Cyclisation
über Sesselfaltung über Wannenfaltung
trans-trans trans-trans -trans trans-cis-trans
cis-trans cis-cis-trans cis-trans-trans
trans-cis trans-cis-cis trans-trans-cis
cis-cis cis-trans-cis cis-cis-cis
2. Literaturbeispiele
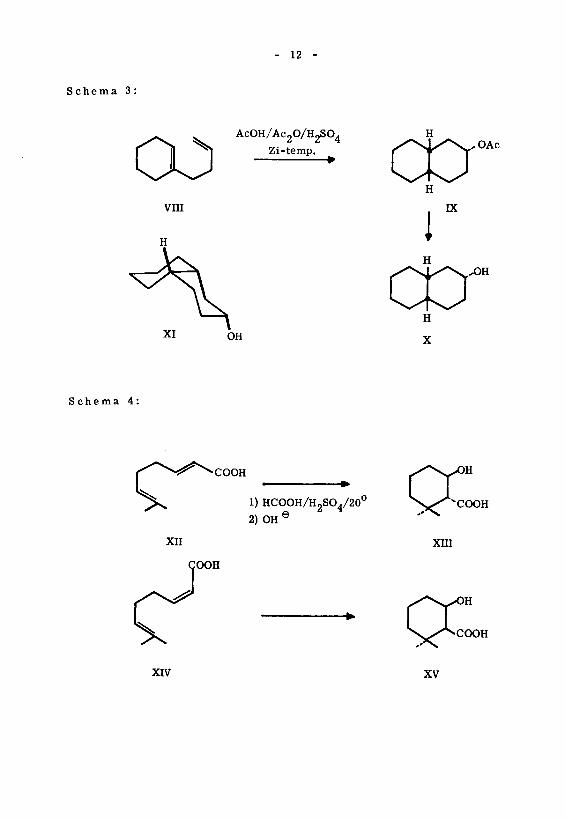
Die ältesten Arbeiten über den sterischen Verlauf von Cyclisationen stammen40)
von R. P. Linstead und Mitarbeitern '. Vom stereochemischen Standpunkt aus41)
ist die Cyclisation des Buten-3-yl-cyclohexens(l) '(VIII) interessant. Es wurde un¬
ter relativ milden Reaktionsbedingungen ein bicyclisches Reaktionsprodukt erhalten,
welches ein Derivat des cis-Decalins X ist. Das sterische Ergebnis dieser Cyclisa¬
tion lässt auf einen antiplanaren Reaktionsablauf der Ringbildung schliessen(XI). Be¬
merkenswert ist, dass die Reaktion entgegen der Regel von Mar kownikow ver¬
läuft.
- 14
Schema 5 :
COOH
19
xvnCOOH
OOH
22
XX H
xvin
XK XXI
cis-Norgeranium-Ri=R. ^ = _COOH
trans-Norggeranium-Ri = _COOH; ^ = _H
Schema 6 :
XXII
Ro
xxm
OOH
XXIV
1) HCOOH/H2SO4/10°2)OHe
COOH
XXV
COOH
XXVI xxvn

- 15 -
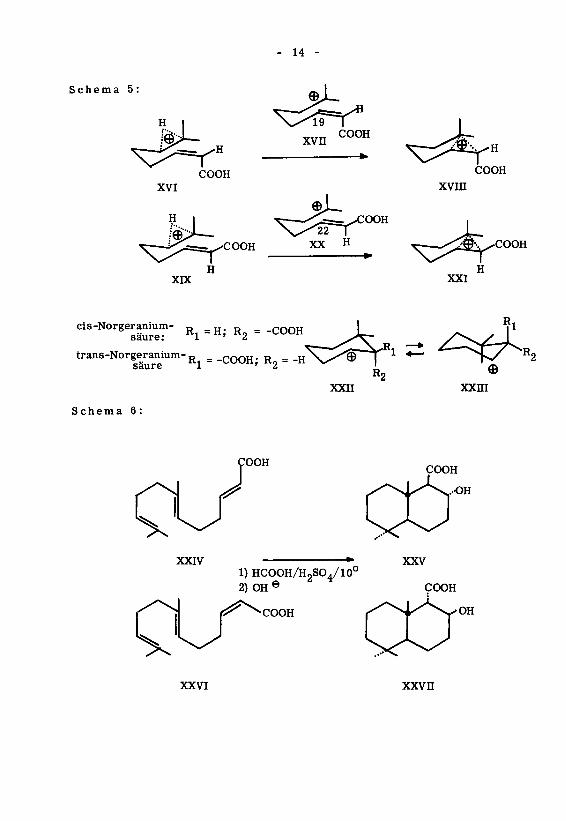
Die Cyclisation der 7-Methyl-2,6-octadiensäuren (XIl/XIV) verläuft ebenfallslfi S1 ^2)
stereoselektiv ' ' '. Die beiden stereoisomeren Norgeraniumsäuren XIl/XIV lie¬
ferten bei der säurekatalysierten Cyclisation isomere cyclische Hydroxysäuren Xin/
XV, die sich nur durch verschiedene relative Konfiguration der Säuregruppe unter¬
scheiden. Dieses Ergebnis entspricht den in Tabelle 1 angegebenen Korrelationen.
Das Resultat lässt nicht eindeutig darauf schliessen, ob ein direkter Uebergang XVI —•-
XVin, XIX—»XXI besteht, oder ob zuerst intermediär das klassische Kation XVII
oder XX gebildet wird, was in Anbetracht der Stabilität von tertiären Kationen '
durchaus möglich wäre. Ebenso bleibt die Frage offen, ob die equatoriale Lage der
Hydroxylgruppe eine Folge der antiplanaren Addition ist (direkte Addition der exter¬
nen Base am überbrückten Ion XVin/XXI), oder ob die aus sterischen Gründen be¬
vorzugte, equatoriale Addition an einem klassischen Kation XXII erfolgt und zwar
schneller, als die Einstellung des Konstellationsgleichgewichtes XXII—•-XXIII '.
Des weiteren wurde in letzter Zeit die Cyclisation der 7,ll-Dimethyl-2(trans),
6(trans),10-dodecatrien-l-säure (XXIV) und der 7,ll-Dimethyl-2(cis),6(trans), 10-
dodecatrien-1-säure (XXVI) von P.Stadler et al. ' ^studiert. Dire Cyclisation
mit Ameisensäure/Schwefelsäure führt zu trans-Dekalinderivaten XXV/XXVH, die
sich lediglich durch die Konfiguration der Säuregruppe unterscheiden ' '. Sie stellt
einen in hohem Masse stereospezifischen Vorgang dar, dessen Ergebnis formal das Re¬
sultat einer antiplanaren Addition XXVin—XXIX, XXX—»XXXI sein könnte. Ueber
den Reaktionsmechanismus lässt sich indessen wenig aussagen, insbesondere nicht
über den Mechanismus der Addition an der mittleren Doppelbindung. Was hingegen
feststeht ist, dass der zweite Ringschluss über die energetisch günstigere Sesselfal¬
tung XXXIl/XXXin vor sich geht, denn die Konstellation der Carboxylgruppe lässt
sich mit der jeweiligen Ringschlusskonstellation eindeutig in Beziehung setzen. Was
die equatoriale Lage der Hydroxylgruppe anbetrifft, so kann sie nicht unbedingt als
Beweis für einen antiplanaren Additionsmechanismus gelten, da auch bei der Ausbil¬
dung eines klassischen Kations XXXIV die Anlagerung der externen Base aus steri¬
schen Gründen ebenfalls equatorial erfolgen würde.
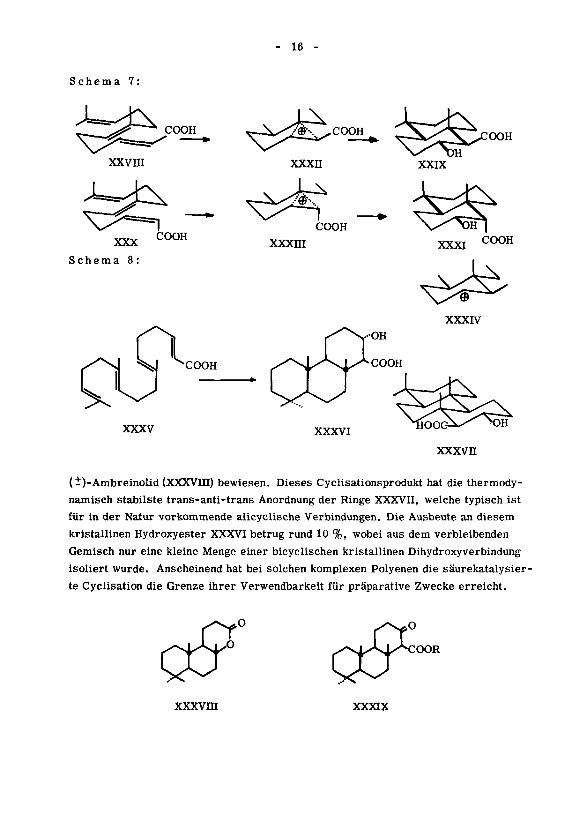
Die kürzlich von J.Meier (unpublizierte Resultate) durchgeführte Cyclisa¬
tion mit Ameisensäure/Schwefelsäure des 7, ll,15-Trimethyl-2,6,10,14-hexadeca-
trien-1-säuremethylesters (XXXV) ergab ein Gemisch, aus welchem der tricyclische
Hydroxyester XXXVI isoliert wurde. Durch Oxydation zum Ketoester XXXIX und Re¬
duktion desselben konnte die Konfiguration der Hydroxylgruppe bewiesen werden. Aus
Analogiegründen (siehe das Resultat der Cyclisation der 7,ll-Dimethyl-2,6,10-dodeca-35 36)
trien-1-säuren (XXIV/XXVI) ' ' wurde die Carbomethoxygruppe equatorial ange¬
nommen. Die Stereochemie des ganzen Ringsystems wurde durch Ueberführung ins
16 -
Schema 7:
COOH /©••. ^COOH OOH
xxvin xxxn
«
XXIX
XXX
Schema 8 :
COOHCOOH
XXXIII XXXICOOH
COOH
XXXV XXXVI
(-)-Ambreinolid (XXXVIII) bewiesen. Dieses Cyclisationsprodukt hat die thermody-
namisch stabilste trans-anti-trans Anordnung der Ringe XXXVH, welche typisch ist
für in der Natur vorkommende alicyclische Verbindungen. Die Ausbeute an diesem
kristallinen Hydroxyester XXXVI betrug rund 10 %, wobei aus dem verbleibenden
Gemisch nur eine kleine Menge einer bicyclischen kristallinen Dihydroxyverbindung
isoliert wurde. Anscheinend hat bei solchen komplexen Polyenen die säurekatalysier¬
te Cyclisation die Grenze ihrer Verwendbarkeit für präparative Zwecke erreicht.
O
et^\L>sl/^COOR
xxxvni xxxix
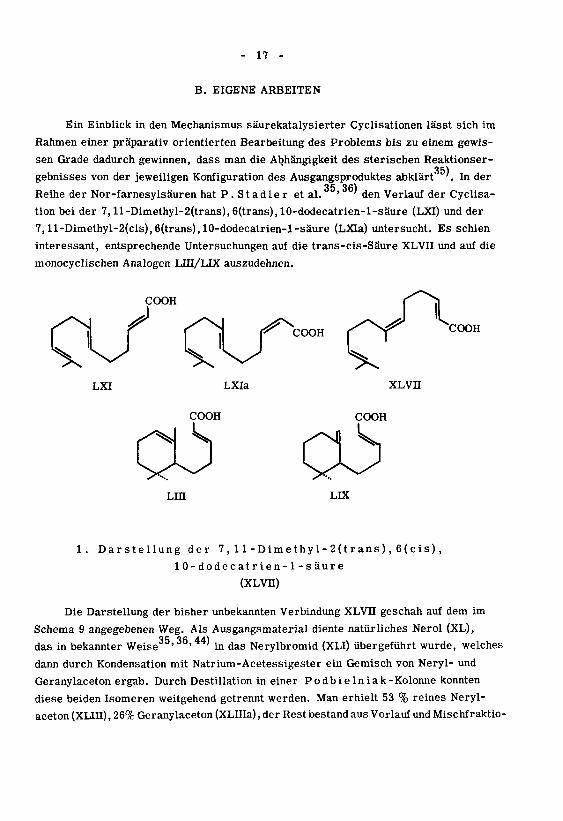
- 17 -
B. EIGENE ARBEITEN
Ein Einblick in den Mechanismus säurekatalysierter Cyclisationen lässt sich im
Rahmen einer präparativ orientierten Bearbeitung des Problems bis zu einem gewis¬
sen Grade dadurch gewinnen, dass man die Abhängigkeit des sterischen Reaktionser-
35)gebnisses von der jeweiligen Konfiguration des Ausgangsproduktes abklärt '. In der
Reihe der Nor-farnesylsäuren hat P .St ad le r et al. ' ' den Verlauf der Cyclisa-
tionbei der 7, ll-Dimethyl-2(trans),6(trans),10-dodecatrien-l-säure (LXI) und der
7,ll-Dimethyl-2(cis),6(trans),10-dodecatrien-l-säure (LXIa) untersucht. Es schien
interessant, entsprechende Untersuchungen auf die trans-cis-Säure XLVII und auf die
monocyclischen Analogen Lin/LIX auszudehnen.
COOH
COOH C^LXI LXIa XLVH
COOH COOH
Lin LDC
1. Darstellung der 7,11 -Dimethyl- 2 (trans), 6(cis),
10-dodecatrien-l-säure
(XLVH)
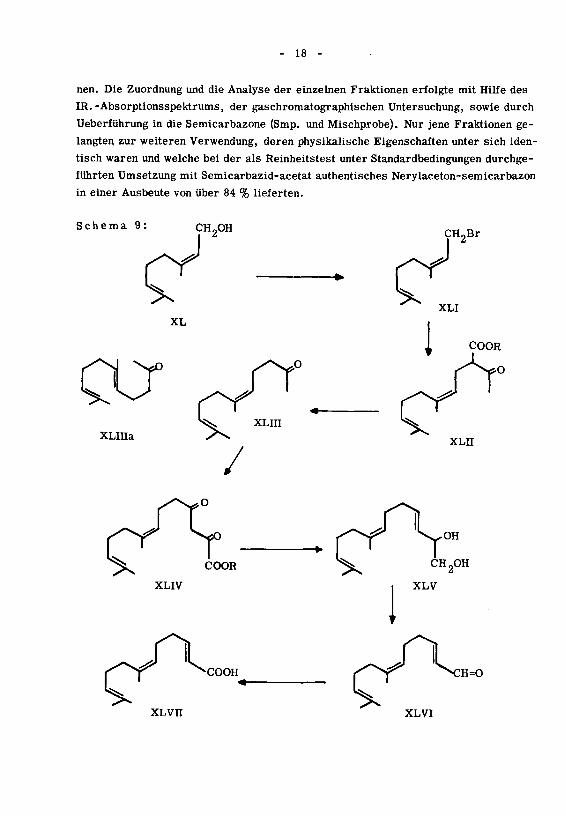
Die Darstellung der bisher unbekannten Verbindung XLVII geschah auf dem im
Schema 9 angegebenen Weg. Als Ausgangsmaterial diente natürliches Nerol (XL),
das in bekannter Weise ''' in das Nerylbromid (XLI) übergeführt wurde, welches
dann durch Kondensation mit Natrium-Acetessigester ein Gemisch von Neryl- und
Geranylaceton ergab. Durch Destillation in einer Podbielniak-Kolonne konnten
diese beiden Isomeren weitgehend getrennt werden. Man erhielt 53 % reines Neryl-
aceton (XLin), 26% Geranylaceton (XLIIIa), der Rest bestand aus Vorlauf und Mischfraktio-
- 18 -
nen. Die Zuordnung und die Analyse der einzelnen Fraktionen erfolgte mit Hilfe des
IR.-Absorptionsspektrums, der gaschromatographischen Untersuchung, sowie durch
Ueberführung in die Semicarbazone (Smp. und Mischprobe). Nur jene Fraktionen ge¬
langten zur weiteren Verwendung, deren physikalische Eigenschaften unter sich iden¬
tisch waren und welche bei der als Reinheitstest unter Standardbedingungen durchge¬
führten Umsetzung mit Semicarbazid-acetat authentisches Nerylaceton-semicarbazon
in einer Ausbeute von über 84 % lieferten.
Schema 9:CH2OH CH2Br
XL
XLinaXLII
COOR
XLIV
C^-COOHXLVIT
er V>s CH2°H
i XLV
|f^ S:h=o
XLVI
- 19 -
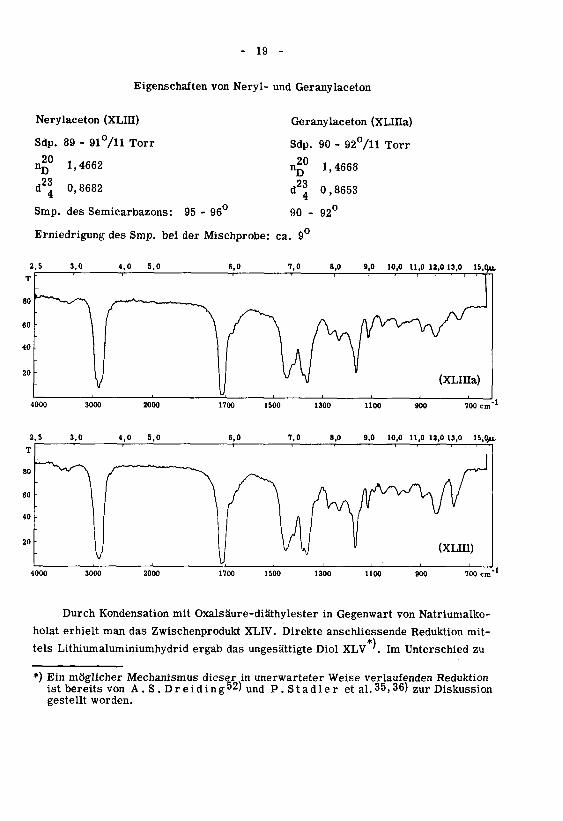
Eigenschaften von Neryl- und Geranylaceton
Nerylaceton (XLIH)
Sdp. 89 - 91°/n Torr
n£° 1,4662
d2^ 0,8682
Geranylaceton (XLIIIa)
Sdp. 90 - 92°/H Torr
n^0 1,4668
d2^ 0,8653
Smp. des Semicarbazons: 95 - 96° 90 - 92°
Erniedrigung des Smp. bei der Mischprobe: ca. 9°
8,0 9,0 10,0 11,0 18,0 13,0 15,0*1
4000 3000 2000 700 cm"
2,5 3,0 4,0 5,0 7,0 8,0 9,0 10,0 11,0 12,0 13,0 15,0^
2000 1700 1500 1100 700 cm"
Durch Kondensation mit Oxalsäure-diäthylester in Gegenwart von Natriumalko-
holat erhielt man das Zwischenprodukt XLIV. Direkte anschliessende Reduktion mit-
tels Lithiumaluminiumhydrid ergab das ungesättigte Diol XLV '. Im Unterschied zu
*) Ein möglicher Mechanismus dieser in unerwarteter Weise verlaufenden Reduktion
ist bereits von A. S .
Dr eiding*2) Und P.Stadler et al. 35,36) ZUr Diskussion
gestellt worden.

- 20 -
früheren Synthesen ' ' wurde die Glykolspaltung mit Natriumperjodat ausgeführt ';
diese verlief mit annähernd gleicher Ausbeute wie die früher angewandte Spaltung mit
Bleitetraacetat. Der so erhaltene Aldehyd XLVI wurde durch Ueberführung in das Se-
micarbazon und 2,4-Dinitrophenylhydrazon charakterisiert. Die Mischprobe dieser Deri¬
vate mit dem Semicarbazon, bzw. 2,4-Dinitrophenylhydrazon des 7,ll-Dimethyl-2
(trans), 6(trans), 10-dodecatrien-l-al zeigte Depressionen des Smp. von 4° bzw.
4,5°, was darauf hinwies, dass im Verlauf der Synthese keine Isomerisierung der
mittleren Doppelbindung stattgefunden hatte. Durch Oxydation mit Silberoxyd erhielt
man die Säure XLVII, welche über ihr S-Benzyl-isothiuroniumsalz vom Smp. 145, 5
bis 146 gereinigt wurde. Eine Mischprobe mit dem entsprechenden Derivat der 7,11-
Dimethyl-2(trans),6(trans),10-dodecatrien-l-säure (LXI) ' 'zeigte bei sehr lang¬
samem Erhitzen eine Depression des Smp. von 3. Durch rasches Zersetzen dieses
Salzes in einer Pufferlösung von pH 1 unterhalb 10 wurde die reine 7,11-Dimethyl-
2(trans),6(cis),10-dodecatrien-l-säure (XLVII) erhalten. Das IR.-Absorptionsspek¬
trum unterschied sich gegenüber dem der 7,ll-Dimethyl-2(trans),6(trans),10-dodeca-OR 9,fi\ —1
trien-1-säure (LXI) ' 'nur durch eine schwache Bande bei 1075 cm und eine
breitere Bande bei 1425 cm".Die mittlere Doppelbindung muss in dieser Verbin¬
dung die ursprüngliche eis-Konfiguration ' des Nerylacetons (XLIII) aufweisen, weil
bei Anwendung der eben beschriebenen Aufbaumethode keine Gelegenheit zur IsOmeri-oe oo A'i AK\
sation bestand. Frühere Erfahrungen ' ' ' 'zeigten, dass die zur Säuregruppe
konjugierte Doppelbindung trans-Konfiguration besitzt.
2. Darstellung der tx -Monocyclo-nor-farnesylsäure (LIII)
Die ebenfalls unbekannte Verbindung LIII wurde gemäss Schema 10 ähnlich der
Synthese der 7, ll-Dimethyl-2(trans),6(cis),10-dodecatrien-l-säure (XLVII) herge¬
stellt. Ausgehend von Dihydro-<x-jonon (XLIX) wurde durch Kondensation mit Oxal-
säure-diäthylester und direkt anschliessende Reduktion mit Lithiumaluminiumhydrid das
Diol LI erhalten. Die Glykolspaltung mit Natriumperjodat ergab den Aldehyd LH, welcher
durch Oxydation mit Silberoxyd in die Säure LUI übergeführtwurde. Die Reinigung dersel¬
ben erfolgte durch Ueberführen in das S-Benzyl-isothiuroniumsalz von Smp. 153 - 154 .
Ueber die Lage der zwei Doppelbindungen kann Folgendes ausgesagt werden:
*) Die beiden grössten Substituenten liegen eis bezüglich der mittleren Doppelbindung.
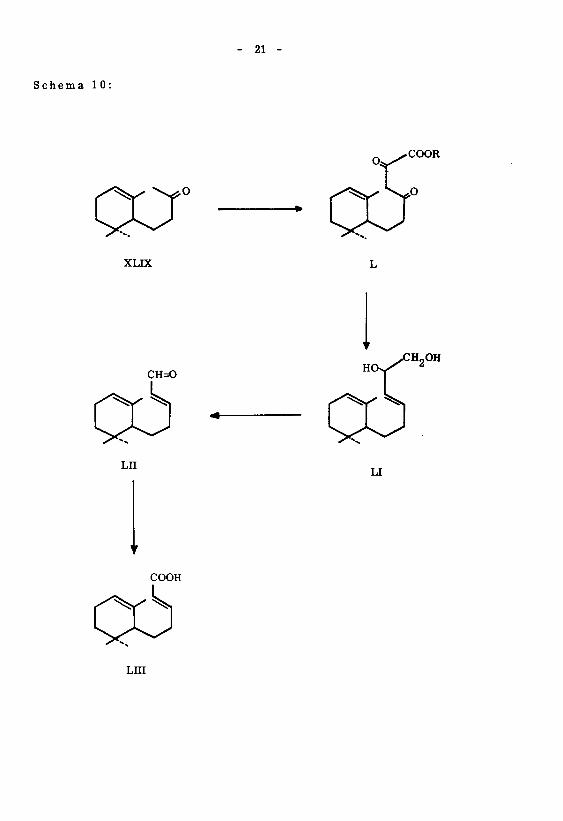
Schema 10:
21
O
XLIX
CH=0
LII
COOH
Lni
Y
OH
LI

- 22 -
Durch das Ausgangsprodukt, Dihydro-cx-jonon (XL1X), welches durch IR.-Ab¬
sorptionsspektrum und durch Ueberführen in das Semicarbazon charakterisiert wurde,
ist die Lage der endocyclischen Doppelbindung gegeben. Sie konnte sich im Verlauf
48)der Synthese nicht geändert haben, da eine Isomerisierung durch Säure ausgeschal¬
tet wurde (gleiche Herstellungsmethoden wie bei der 7, ll-Dimethyl-2(trans), 6(cis),
10-dodecatriensäure (XLVII).
Die zur Säuregruppe konjugierte Doppelbindung besitzt aus den bereits erwähn¬
ten Gründen (siehe Seite 20) trans Konfiguration.
3. Darstellung der racemischen und optisch aktiven
Monocyclo-nor-farnesylsäure
(LIX)
Zur Synthese dieser ebenfalls noch nicht beschriebenen Säuren wurde wiederum
derselbe Weg (vergl. Schema 11) beschritten, wie bei den zwei anderen Säuren XLVII/
Lin. Als Ausgangsmaterial für die Darstellung der racemischen Säure diente durch
wiederholte Umkristallisation gereinigtes, synthetisches (racemisches) Dihydro- j-
jonon-semicarbazon (LIV) vom Smp. 190,5 - 191,5 .Die Zersetzung dieses Semi-
carbazons erfolgte unter Bedingungen, von denen bekannt ist, dass sie keine Isome-
43 47)risierung der Methylendoppelbindung im Ring verursachen '
'. Die gaschromato-
graphische Prüfung des so erhaltenen (t)-Dihydro-^-jonon LV zeigte eine Reinheit
von ca. 95 % an.
Durch Kondensation mit Oxalsäure-diäthylester und sofort anschliessende Re¬
duktion durch Lithiumaluminiumhydrid wurde das Diol LVII erhalten. Dieses wurde
durch Glykolspaltung mit Natriumperjodat in den Aldehyd LVin übergeführt. Oxyda¬
tion mit Silberoxyd im alkalischen Milieu ergab die Säure LIX im rohen Zustand,
welche über das S-Benzyl-isothiuroniumsalz vom Smp. 155 - 156° gereinigt wurde.
Man erhielt so ein Produkt vom Sdp. 102°/0, 02 Torr.
Die zur Säuregruppe konjugierte Doppelbindung dieser Verbindung LIX weist
aus den erwähnten Gründen (siehe Seite 20) trans Konfiguration auf.
In Uebereinstimmung mit der angenommenen Konstitution zeigt das von der
Säure LIX in Nujol aufgenommene IR.-Absorptionsspektrum eine bei der o< -Mono¬
cyclo-nor-farnesylsäure (Lin) nicht vorhandene Bande bei 890 cm (CHg-waggingSchwingung einer RjR2C=CH2 Gruppe).
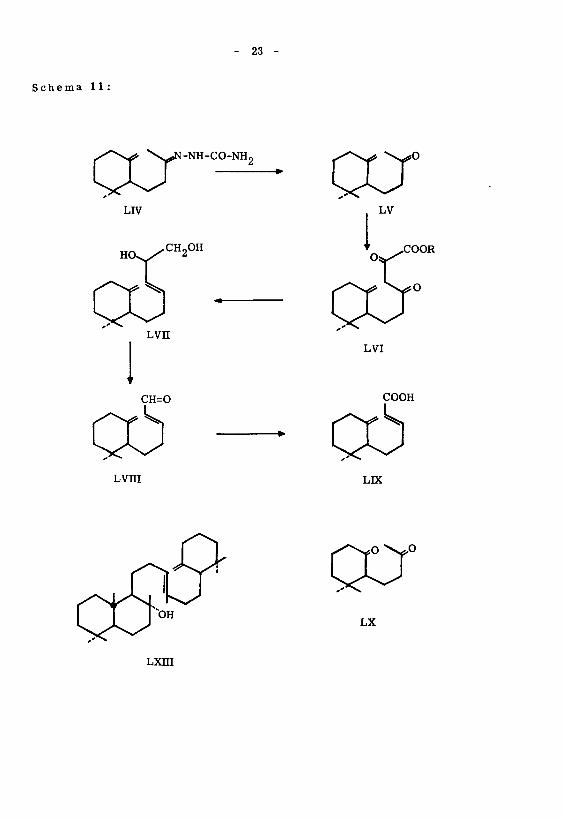
- 23 -
Schema 11:
CCr•NH-CO-NH.,
LIV
HO^/CH2°H
Lvn
CH=0
axLV
LVI
COOH
Lvni LK
Lxni
o\*o
LX

- 24 -
Als Ausgangsmaterial zur Synthese der optisch aktiven y-Monocyclo-nor-farne-
sylsäure (LIX) benutzte man (+)-Dihydro-')f-jonon (LV), welches neben dem Diketon LX49)
durch Oxydation des Ambreins (LXIII) mit Kaliumpermanganat' erhalten wurde.
Eine sorgfältige Destillation erlaubte dieses Diketon LX als Nachlauf abzutrennen.
Das verwendete Dihydro- f -jonon (LV) wies eine spezifische Drehung 'von [«]D =
+ 8,05° auf. Die Synthese der Säure erfolgte auf demselben Weg wie bei der Darstel¬
lung der racemischen ^-Monocyclo-nor-farnesylsäure (LDC) (vergl. Schema 11). In¬
folge Materialmangel musste von einer Reinigung über das S-Benzyl-isothiuronium-
salz abgesehen werden. Das IR.-Absorptionsspektrum war in allen Punkten identisch
mit dem der optisch inaktiven Y-Monocyclo-nor-farnesylsäure (LIX). Die Säure zeig-*) o
te eine spezifische Drehung'von [«*]n = + 6, 07 .
**)
4. Cyclisationsversuche'
Cyclisation der 7,ll-Dimethyl-2(trans),6(cis),10-dodecatrien-l-säure (XLVII).
1) H,SO./HCOOHCOOH —
2) OHe /70°/l/2 Std.
XLVIII
XLVII
Zur Cyclisation verwendete man eine auf 0° abgekühlt Mischung von Ameisen¬
säure und Schwefelsäure (Volumenverhältnis 10:1), welche schon früher von mehre-
ren Autoren benutzt wurde ' ' '. Um eine eventuelle säurekatalysierte Polyme¬
risation möglichst zu unterdrücken, wurde die 7,ll-Dimethyl-2, 6,10-dodecatrien-l-
*) Die Messungen der Drehung wurden in absolutem Dioxan ausgeführt.**) Alle Cyclisationen wurden unter analogen Verhältnissen durchgeführt wie diejeni¬
gen von P.Stadler et al.35,36).

- 25 -
säure (XLVII) durch eine Kapillare möglichst langsam in das Cyclisationsgemisch
eingetropft und durch intensives Rühren mit einem Vibromischer für eine sofortige
Verteilung gesorgt. Die direkte alkalische Verseifung des so erhaltenen Cyclisations-
produktes lieferte in 59%-iger Ausbeute die gut kristallisierende Säure XLVin,
Smp. 233°, deren Konstitution bereits bekannt war' '.
Beim Chromatographieren der Mutterlauge konnte ausser einer kleinen Menge
unreiner Hydroxysäure XLVIII (Smp. 193
isoliert werden.
194 ) kein anderes kristallines Produkt
Cyclisation der oc-Monocyclo-nor-farnesylsäure (LHI)
COOH
Lin
1) H2S04/HCOOH
2) OHe /70°/l/2 Std.
XLVin
Zur Cyclisierung wurde das bereits erwähnte Gemisch aus Ameisensäure-Schwe¬
felsäure im Volumenverhältnis 10:1 verwendet und die Cyclisation im übrigen unter
den genau gleichen Bedingungen durchgeführt wie bei der 7,ll-Dimethyl-2(trans),
6(cis), 10-dodecatrien-l-säure (XLVII). Man erhielt in 56 %-iger Ausbeute wiederum
die bicyclische Hydroxysäure XLVin vom Smp. 233 - 234°. Die Identifikation geschah
durch Smp., Mischprobe mit authentischer Säure XLVIII ' 'und durch IR. -Absorp¬
tionsspektrum.
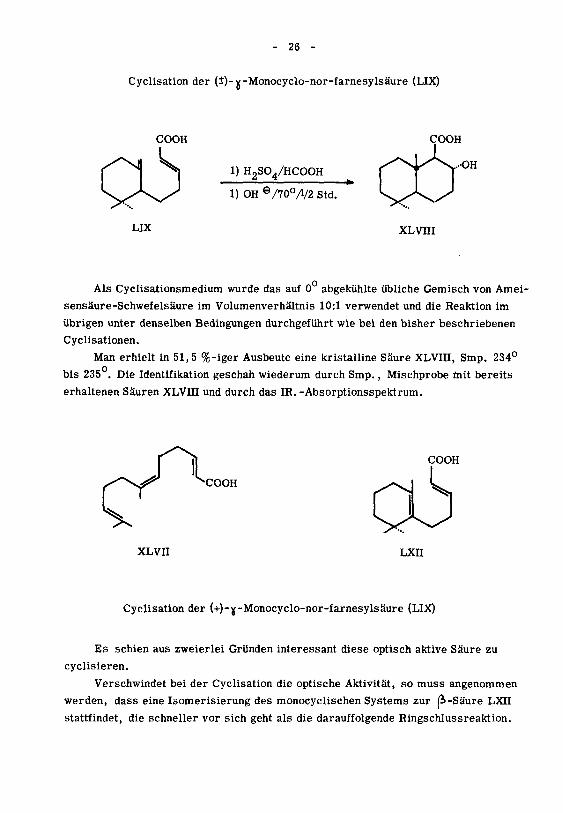
- 26 -
Cyclisation der (i)-Y-Monocyclo-nor-farnesylsäure (LIX)
COOH
LIX
COOH
1) H2S04/HCOOH
1) OH e/70o/l/2 Std.
XLVHI
Als Cyclisationsmedium wurde das auf 0 abgekühlte übliche Gemisch von Amei¬
sensäure-Schwefelsäure im Volumenverhältnis 10:1 verwendet und die Reaktion im
übrigen unter denselben Bedingungen durchgeführt wie bei den bisher beschriebenen
Cyclisationen.
Man erhielt in 51,5 %-iger Ausbeute eine kristalline Säure XLVHI, Smp. 234°
bis 235.Die Identifikation geschah wiederum durch Smp., Mischprobe mit bereits
erhaltenen Säuren XLVHI und durch das IR. -Absorptionsspektrum.
COOH
COOH
XLVII LXn
Cyclisation der (+)-Y-Monocyclo-nor-farnesylsäure (LIX)
Es schien aus zweierlei Gründen interessant diese optisch aktive Säure zu
cyclisieren.
Verschwindet bei der Cyclisation die optische Aktivität, so muss angenommen
werden, dass eine Isomerisierung des monocyclischen Systems zur (i-Säure LXII
stattfindet, die schneller vor sich geht als die darauffolgende Ringschlussreaktion.
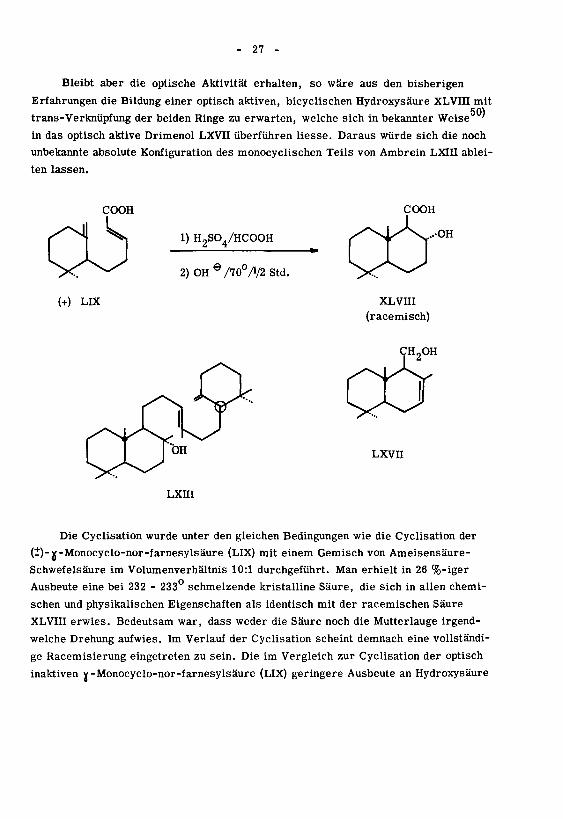
- 27 -
Bleibt aber die optische Aktivität erhalten, so wäre aus den bisherigen
Erfahrungen die Bildung einer optisch aktiven, bicyclischen Hydroxysäure XLVin mit50)
trans-Verknüpfung der beiden Ringe zu erwarten, welche sich in bekannter Weise '
in das optisch aktive Drimenol LXVII überführen liesse. Daraus würde sich die noch
unbekannte absolute Konfiguration des monocyclischen Teils von Ambrein LXIII ablei¬
ten lassen.
COOH COOH
CO 1) H2S04/HCOOH
2) OH® /70°/V2 Std.
(+) LIX XLVm
(racemisch)
CH2OH
LXVII
LXHI
Die Cyclisation wurde unter den gleichen Bedingungen wie die Cyclisation der
(i)-jf-Monocyclo-nor-farnesylsäure (LIX) mit einem Gemisch von Ameisensäure-
Schwefelsäure im Volumenverhältnis 10:1 durchgeführt. Man erhielt in 26 %-iger
Ausbeute eine bei 232 - 233 schmelzende kristalline Säure, die sich in allen chemi¬
schen und physikalischen Eigenschaften als identisch mit der racemischen Säure
XLVIII erwies. Bedeutsam war, dass weder die Säure noch die Mutterlauge irgend¬
welche Drehung aufwies. Im Verlauf der Cyclisation scheint demnach eine vollständi¬
ge Racemisierung eingetreten zu sein. Die im Vergleich zur Cyclisation der optisch
inaktiven v-Monocyclo-nor-farnesylsäure (LIX) geringere Ausbeute an Hydroxysäure
- 28 -
XLVTII von 26 % ist darauf zurückzuführen, dass die optisch aktive Säure LIX aus
Materialmangel nicht über das S-Benzyl-isothiuroniumsalz gereinigt wurde. Erfah-
rungsgemäss (siehe P.Stadler et al. ' ') zeigen Cyclisationen in dieser Reihe
von Säuren, welche nicht über dieses Salz gereinigt wurden, eine um 20 - 30 % ge¬
ringere Ausbeute an Hydroxysäure XLVIII.
COOH
LXI Lm
5. Relative Cy clisationsgeschwindigkeiten der 7,11 -Dimethy 1-
2 (tr ans), 6 (tr ans) ,1 0-dodecatr ien-1 - s äur e (LXI) und
der ot-Monocyclo-nor-farnesylsäure (LIII)
Im Verlauf dieser Arbeit tauchte die Frage auf, ob dieot-Monocyclo-nor-farne-
sylsäure (LIII) nicht ein Zwischenprodukt der Cyclisation der 7, ll-Dimethyl-2(trans),
6(trans),10-dodecatrien-l-säure (LXI) sein könnte. Es schien daher interessant, die
beiden Cyclisationen kinetisch zu verfolgen. Wir nützten zu diesem Zweck den Um¬
stand aus, dass die Endprodukte der Cyclisationen, die Hydroxysäure XLVIII, sich
aus Dioxan-Hexan gut kristallisieren lässt, während die uncyclisierten Säuren in der
Mutterlauge in Lösung bleiben. Ein Modellversuch zeigte, dass 5 mg der bicyclischen
Hydroxysäure XLVHI gut aus 96 mg uncyclisierter Säure LXI isoliert werden können.
Alle Cyclisationen wurden unter identischen Versuchsbedingungen durchgeführt. In
gewissen Zeitabständen wurde V5 der Gesamtvolumenmenge herausgenommen und die
Reaktionslösung unter denselben Bedingungen aufgearbeitet. Durch Kristallisation
aus Dioxan-Hexan erhielt man die cyclisierte Säure XLVIII. Die Resultate der Cycli¬
sationen ergeben die Figur 1. Wie man daraus ersehen kann, ist dieot-Monocyclo-
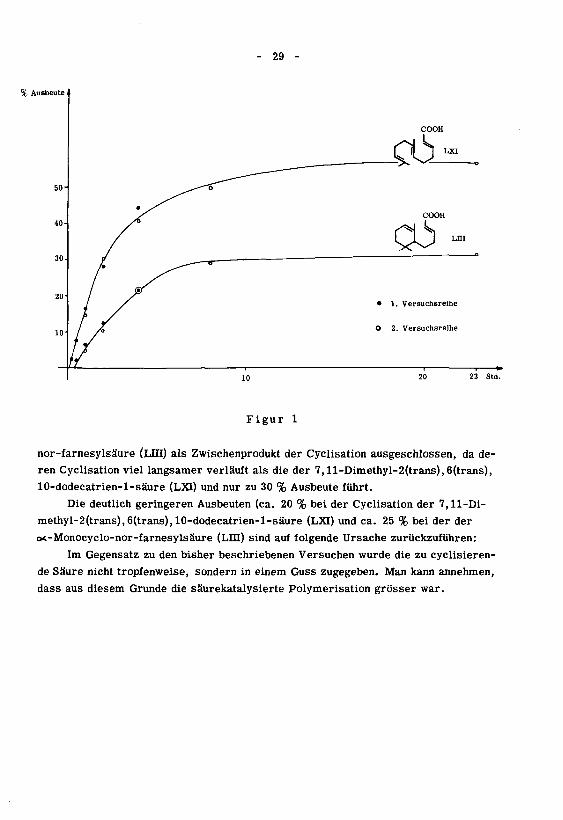
- 29
% Ausbeute .
• 1. Versuchsreihe
O 2. Versuchsreihe
Figur 1
nor-farnesylsäure (LIII) als Zwischenprodukt der Cyclisation ausgeschlossen, da de¬
ren Cyclisation viel langsamer verläuft als die der 7,ll-Dimethyl-2(trans),6(trans),
10-dodecatrien-l-säure (LXI) und nur zu 30 % Ausbeute führt.
Die deutlich geringeren Ausbeuten (ca. 20 % bei der Cyclisation der 7,11-Di-
methyl-2(trans),6(trans), 10-dodecatrien-l-säure (LXI) und ca. 25 % bei der der
ot-Monocyclo-nor-farnesylsäure (LUI) sind auf folgende Ursache zurückzuführen:
Im Gegensatz zu den bisher beschriebenen Versuchen wurde die zu cyclisieren-
de Säure nicht tropfenweise, sondern in einem Guss zugegeben. Man kann annehmen,
dass aus diesem Grunde die säurekatalysierte Polymerisation grösser war.
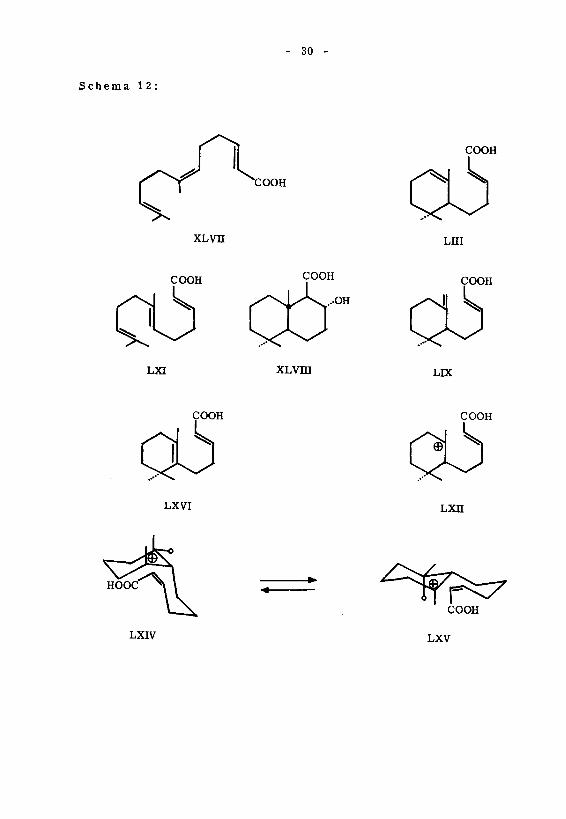
- 30 -
S chema 1 2 :
COOH
XLVU
LXI
COOH
CÔ'XLvni
COOH
LUI
ÇOOH
LK
ÇOOH
LXVI
HOOC ^ \
LXIV
COOH
lxh
COOH
LXV

- 31 -
C. SCHLUSSFOLGERUNGEN
Die in der vorliegenden Arbeit durchgeführten Cyclisationen der 7,11-Dimethyl-
2(trans),6(cis), 10-dodecatrien-l-säure (XLVII), der ot-Monocyclo-nor-farnesylsäu-
re (Lin) und der racemischen und optisch aktiven tf-Monocyclo-nor-farnesylsäure (LIX)
ergaben als Hauptprodukt der Cyclisation immer dasselbe Derivat des trans-
Dekalins, die Hydroxysäure XLVHI, deren Konstitution in der Arbeit von P. Stad-
1er et al. ' bewiesen worden war.
Dieses eher überraschende Resultat hat bezüglich der Frage des Reaktionsab¬
laufes säurekatalysierter Cyclisationen in dieser Reihe folgende Konsequenzen:
Die ausbeutemässig am selektivsten verlaufende Cyclisation der 7,11-Dimethyl-
2(trans),6(trans), 10-dodecatrien-l-säure (LXI) ergibt als Hauptprodukt eine bicycli-50 51}
sehe Hydroxysäure XLVHI mit trans-Verknüpfung der beiden Dekalinringe ''.
Zur Abklärung der Frage, ob diese trans-Verknüpfung als Stütze für die Annahme
eines antiplanaren Additionsablaufes an der mittleren Doppelbindung des cyclisieren-
den Molekels LXI gelten darf, wurde der Versuch mit der isomeren cis-Säure XLVH
durchgeführt. Die Tatsache, dass bei dieser Cyclisation als Hauptprodukt dieselbe
Hydroxysäure XLVHI gebildet wird, zeigt, dass eine trans-Verknüpfung des Deka¬
linringes nicht als Kriterium für die Annahme eines antiplanaren Reaktionsverlaufes
bei der Cyclisation der trans-trans-Säure LXI gewertet werden kann.
Sowohl die e*-Monocyclo-nor-farnesylsäure (LIII) als auch die v-Monocyclo-
nor-farnesylsäure (LIX) sind nicht Zwischenprodukte auf dem Weg 7,11-Dimethyl-
2(trans),6(trans), 10-dodecatrien-l-säure (LXI)—»-Hydroxysäure XLVHI, weil aus¬
beutemässig ihre Cyclisation schlechter verläuft als die der trans-trans-Säure LXI.
Ausserdem ist die Bildungsgeschwindigkeit der Hydroxysäure XLVTH aus der ot -Mo-
nocyclo-nor-farnesylsäure (LHI) kleiner als die derselben Säure XLVHI aus der
trans-trans-Säure LXI.
Der sterische Verlauf der in der vorliegenden Arbeit besprochenen Cyclisatio-50 511
nen, zusammen mit dem Resultat aus früheren Cyclisationen ' ' in der gleichen
Reihe, lassen sich unter einem einheitlichen Gesichtspunkt verstehen, wenn man an¬
nimmt, dass alle diese Cyclisationen über dasselbe Carboniumkation LXIV/LXV ver¬
laufen und dass dieses Kation LXn konstellativ schneller aequilibriertLXIV=LXV,
als dass es mit der nächsten Doppelbindung reagiert. In diesem Fall wäre die spezi¬
fische Bildung eines Dekalinringes mit trans-Verknüpfung das Ergebnis einer forma¬
len trans-Addition an der mittleren Doppelbindung, welche von der axialen Methyl¬
gruppe (vergl. Formel LXV) sterisch kontrolliert ist.

- 32 -
Ueberdies scheint im Falle der optisch aktiven ^-Monocyclo-nor-farnesylsäure(LUI) das entstehende Carboniumkation LXV im Gleichgewicht mit der ß -Form LXVI
dieser Säure zu stehen, wie die totale Racemisierung bei deren Cyclisation es zeigt.

- 33 -
EXPERIMENTELLER TEIL
Nerylaceton (XLIII)
Als Ausgangsmaterial diente natürliches Nerol (XL) (Firmenich & Co., Genf),nr O O A A\
welches in bekannter Weise ' ' ' in das Nerylbromid (XLI) übergeführt wurde
und durch Kondensation mit Natrium-Acetessigester das rohe Nerylaceton (XLIII)50 51)
ergab ''. Die 930 g Rohprodukt XLIII wurden zweimal vordestilliert. Man erhielt
623 g, die einer besseren Fraktionierung in einer Podbielniak-Kolonne unter¬
worfen wurden. Destillationsdauer: 240 Std., Druck in der Destillationsblase 35,5
bis 54,5 Torr, Temp, in der Destillationsblase 157 - 195,Druck im Kolonnenkopf
7,5 Torr, Temp, im Kolonnenkopf 88 - 149° (unkor.), Rücklaufverhältnis 1:120 bis
1:100. Es wurden im ganzen 43 Fraktionen genommen. Von den folgenden Fraktionen,*)
die alle nochmals destilliert wurden, sind die Semicarbazone hergestellt und das
IR.-Absorptionsspektrum aufgenommen worden, ausserdem wurden sie gaschroma-
tographisch untersucht.
Semicarbazone, Gaschromatogramm bei Frakt. : 8,11,16,21,25,29,32,35,42.
IR.-Absorptionsspektrum bei Frakt. : 8, 16,21 42.
Von allen diesen Fraktionen wurde der Brechungsindex bestimmt. Von den
Fraktionen 21 und 42, welche bei der als Reinheitstest unter Standardbedingungen
durchgeführten Umsetzung mit Semicarbazid-acetat Semicarbazon in einer Ausbeute
von über 90 % lieferten, ist eine Analyse gemacht und die Dichte bestimmt worden.
Fraktion 8: Sdp. 120 - 125°/H Torr, n£° 1,4654
Semicarbazon: Smp. 87 - 90° (78 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Linalylaceton zeigt eine De¬
pression des Smp. von 19,5
IR.-Absorptionsspektrum: Banden u.a. bei 2930, 1720, 1450, 1375,
1365, 1165, 875 cm"1.
Gaschromatogramm: eine Substanz (pV 379)
*) Die Semicarbazone wurden unter identischen Bedingungen hergestellt: Eine gewo¬
gene Menge des Ketons (422 - 429 mg) wurde mit je 8 ml einer methanolischen
Lösung von Semicarbazid-acetat versetzt (20 g Semicarbazidhydrochlorid mit 40 g
NatriumacetatflüssigverriebenundmitlOOmlMethanol extrahiert) und 20 Std. bei
Zimmertemperatur stehen gelassen. Anschliessend erhitzte man 2 Std. auf 60° und
liess im Kühlschrank erkalten. Man kristallisierte zweimal aus Methanol-Wasser.

- 34 -
Fraktion 11: Sdp. 120 - 124°/U Torr, n£° 1,4656
Semicarbazon: Smp. 86,5 - 89° (73 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Linalylaceton zeigt eine De¬
pression des Smp. von 19,5
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt keine De¬
pression des Smp.
Gaschromatogramm: eine Substanz (pV 381)
Fraktion 16: Sdp. 120 - 124°/H Torr, n£° 1,4662
Semicarbazon: Smp. 89,5 - 90,5° (84 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Linalylaceton zeigt eine De¬
pression des Smp. von 23.
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt keine De¬
pression des Smp.
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt eine De¬
pression des Smp. von 12,5 .
IR. -Absorptionsspektrum: identisch mit Fraktion 8/21.
Gaschromatogramm: eine Substanz (pV 375)
Fraktion 21: Diese Fraktion ist die Analysenfraktion des Nerylacetons.
Sdp. 120 - 125%1 Torr, n^0 1,4662, d2^ 0,86814
Analyse: C13H220 Ber. C 80,35 H 11,41 %
Gef. C 80,50 H 11,61 %
Semicarbazon: Smp. 95 - 96° (91 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt eine De¬
pression des Smp. von 9.
Analyse: C14H25NgO Ber. C 66,89 H 10,03 %
Gef. C 66,83 H 9,80%
IR.-Absorptionsspektrum: identisch mit Fraktion 8/16.
Gaschromatogramm: eine Substanz (pV 385)
Fraktion 25: Sdp. 120 - 125°/H Torr, n^0 1,4665
Semicarbazon: Smp. 90 - 92° (87 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt keine De¬
pression des Smp.
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt eine De¬
pression des Smp. von 12.
Gaschromatogramm: eine Substanz (pV 360).

- 35 -
Fraktion 29: Sdp. 123 - 124°/H Torr, n^° 1,4666
Semicarbazon: Smp. 90 - 91° (86 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt keine De¬
pression des Smp.
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt eine
Depression des Smp. von 8.
IR.-Absorptionsspektrum: identisch mit Fraktion 8/16/21
Gaschromatogramm: eine Substanz (pV 359)
Fraktion 32: (Mischfraktion)
Sdp. 124 - 127°/11 Torr, n£° 1,4670
Semicarbazon: Smp. 77, 5 - 81° (30 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt keine
Depression des Smp.
Gaschromatogramm: zwei Substanzen (pV 359 und 381).
20,Fraktion 35: Sdp. 124 - 127°/H Torr, n^01,4670
Semicarbazon: 90 - 92° (82 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt eine De¬
pression des Smp. von 21 .
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt keine
Depression des Smp.
Gaschromatogramm: eine Substanz (pV 384).
Fraktion 42: Diese Fraktion ist die Analysenfraktion des Geranylacetons.
Sdp. 124 - 127°/H Torr, np0 1,4666, d2]j 0,86532
Semicarbazon: Smp. 90 - 92° (94, 5 % Ausbeute)
Die Mischprobe mit dem Semicarbazon von Nerylaceton zeigt eine De¬
pression des Smp. von 13.
Die Mischprobe mit dem Semicarbazon von Geranylaceton zeigt keine
Depression des Smp.
Analyse: CjgH^O Ber. C 80,35 H 11,41 %
Get. C 80,25 H 11,33 %
IR.-Absorptionsspektrum: zwei schwache Banden bei 1085 und 1320 cm,
die im IR. -Absorptionsspektrum der Fraktion 21 (Analysenfraktion des
Nerylacetons) vorkommen, sind nicht mehr vorhanden, sonst ist es iden¬
tisch.
Gaschromatogramm: eine Substanz (pV 401).

- 36 -
Die Mischproben der Semicarbazone, die IR.-Absorptionsspektren und die gas-
chromatographischen Untersuchungen erlaubten eine eindeutige Einteilung der einzel¬
nen Fraktionen:
Die Fraktionen 8 bis 15 sind Vorlauffraktionen.
Die Fraktionen 16 bis 29 sind reine Hauptfraktionen des Nerylacetons.
Die Fraktionen 30 bis 34 sind Uebergangsfraktionen zwischen Neryl- und Gera-
nylaceton.
Die Fraktionen 35 bis 43 sind reine Geranylaceton-Fraktionen.
8,12-Dimethyl-3,7,ll-tridecatrien-l,2-diol (XLV)
In einem Kolben wurden 7,1 g blank geschnittenes Natrium und 194 ml über Li¬
thiumaluminiumhydrid destillierter Aether vorgelegt. Der Kolben wurde gut mit
Stickstoff durchgespült. Man liess darauf unter Rühren mit einem Vibromischer 30,6 ml abs.
Aethanol so zutropfen, dass eine lebhafte Wasserstoffentwicklung auftrat. Nachdem alles
eingetropft war, bildete sich mit der Zeit eine weisse Kruste von Natriumalkoholat. Um
diese aufzulösen, begann man die 45,5 g Oxalsäure-diäthylester langsam eintropfen
zu lassen. Sofort setzte die Wasserstoffentwicklung wieder ein, wobei die Farbe sich
gegen gelb-grün änderte. Nachdem alles Natrium aufgelöst und die ganze Menge
Oxalester eingetropft war, rührte man noch eine halbe Stunde und liess darauf in
einem Guss 50 g Nerylaceton (XLin) (Fraktionen 23 - 28) einfliessen. Dabei trat eine
leichte Erwärmung des Aethers auf, und die Farbe der Lösung wurde rot. Unter
leichtem Rührem liess man über Nacht stehen. Zur Aufarbeitung goss man das Re¬
aktionsgemisch in Eiswasser, welchem vorher die theoretische, auf das verwendete
Natrium berechnete Menge Schwefelsäure (15,8 g) und 98 ml einer ges. Lösung von
primärem Natriumphosphat zugefügt worden war. Dabei resultierte ein pH von 3. Man
zog mit Aether aus. Die Aetherlösungen wurden mit eiskalter Kochsalzlösung neutral
gewaschen und mit Natriumsulfat getrocknet. Nach dem Abdampfen des Aethers wur¬
de der dunkelrote Rückstand solange im Wasserstrahlvakuum auf dem Wasserbad er¬
hitzt, bis 15 Torr erreicht wurden. Ausbeute 85 g XLIV, die sofort der Reduktion
mit Lithiumaluminiumhydrid unterworfen wurden.
In einem gut mit Stickstoff durchgespülten Kolben mit Rührer, Rückflusskühler
und Tropftrichter wurden 19, 5 g Lithiumaluminiumhydrid (Metal Hydride Inc., 95 %
rein) vorgelegt und in 291 ml abs. Aether suspendiert. Zu dieser Suspension liess
man im Verlauf von 7 Stunden eine Lösung von 80 g Kondensationsprodukt XLIV in

- 37 -
388 ml Aether langsam unter Rühren zutropfen. Nach Zugabe von weiteren 291 ml
Aether liess man über Nacht stehen. Zur Zersetzung des Reaktionsgemisches wur¬
den unter starkem Rühren 78 g ges. Seignettesalzlösung in die mit einer Eis-Koch¬
salzmischung gekühlte Reaktionslösung eingetropft. Man verdünnte diese mit 2 1
Aether und filtrierte über Cellit, welches selbst mehrmals mit Aether gewaschen
wurde. Die Aetherauszüge wurden einmal mit einer Pufferlösung von Natriumacetat-
Eisessig (pH 3, 5) durchgewaschen, mit Wasser neutral gewaschen und mit Natrium¬
sulfat getrocknet. Das nach Abdampfen des Aethers erhaltene Oel wurde im Hochva¬
kuum fraktioniert. Man erhielt 24,5 g (40 % d.Th.) eines gelblich gefärbten Oeles
XLV, Sdp. 156°/0,17 - 0,15 Torr, n^° 1,4951, d2^ 0,9411. IR.-Absorptionsspek¬
trum: Banden u.a. bei 3350, 2940, 1455, 1380 cm"1.
Analyse: C15H2602 Ber. C 75,58 H 11,00 %
Gef. C 75,18 H 11,09 %
7,ll-Dimethyl-2(trans),6(cis),10-dodecatrien-l-al (XLVI)
24, 5 g Diol XLV in 675 ml Methanol gelöst, wurden in einem Kolben auf 25° er¬
wärmt. Zu dieser Lösung liess man in einem Guss eine Lösung von 33,0 g Natrium-
perjodat in 179 ml Wasser einfHessen. Sofort trat eine voluminöse weisse Fällung
auf. Unter Rühren hielt man während 2V2 Std. die Temperatur konstant auf 34 - 37°,
dann während weiteren 2 Std. auf 27 - 37°.
Man engte die Reaktionsmischung im Vakuum, bei einer Wasserbadtemperatur
von 45°, möglichst stark ein, verdünnte dann mit 400 ml ges. Kochsalzlösung und
zog mit Aether aus. Die Aetherlösungen wurden mit Kochsalzlösung zweimal durch¬
gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des Aethers wurde der
farblose Rest zuerst zweimal vordestilliert, man erhielt 10,61 g, Sdp. 135 /0,06
Torr, n^0 1,4890, UV.-Absorptionsspektrum: A.max
= 218 mu (log 4,16),
die nochmals fraktioniert wurden:
Fraktion Sdp./0,03 Torr
1
2
3
74 - 77°
77 - 80°
über 80°
gn20
nD X. = 218 mu
maxr
log6 =
1,26 1,4872 4,156
7,55 1,4889 4,106
0,73 1,4891 4,095

- 38 -
Zur Analyse gelangte Fraktion 2:
Sdp. 77 - 80°/0,03 Torr, n^0 1,4889, A2\ 0,8832
UV.-Absorptionsspektrum: X = 218 mu (log 6 4,10)
IR.-Absorptionsspektrum: Banden u.a. bei 2930, 2720, 1725, 1690, 1640, 1455,
1380, 830 cm"1.
Analyse: C14H„20 Ber. C 80,50 H 10,75 %
Gef. C 80,27 H 10,60%
2,4-Dinitrophenylhydrazon, fünfmal aus Chloroform-Methanol umkristallisiert,
Smp. 96°.
UV.-Absorptionsspektrum: X. = 244,5 und 375 mju
(log 4,32 4,58)
Die Mischprobe mit dem 2,4-Dinitrophenylhydrazon des 7, ll-Diniethyl-2(trans),
6(cis),10-dodecatrien-l-al ergab keine Depression des Smp.
Die Mischprobe mit dem 2,4-Dinitrophenylhydrazon des 7,ll-Dimethyl-l(trans),
6(trans),10-dodecatrien-l-al ergab eine Depression des Smp. von 4,5 .
Analyse: C2()H2604N4 Ber. C 62,16 H 6,78 N 14,50%
Gef. C 62,46 H 6,97 N 14,50%
Semicarbazon: viermal aus Methanol-Wasser umkristallisiert, Smp. 114,5 - 115.
UV.-Absorptionsspektrum: X = 262,5 mu (log 6 4,59).
Die Mischprobe mit dem Semicarbazon des 7, ll-Dimethyl-2(trâns), 6(cis), 10-dodeca-
trien-1-al ergab keine Depression des Smp.
Die Mischprobe mit dem Semicarbazon des 7, ll-Dimethyl-2(trans),6(trans), 10-do-
decatrien-1-al ergab eine Depression des Smp. von 4.
Analyse: C15H25N30 Ber. C 68,40 H 9,57 N15,96%
Gef. C 68,28 H 9,38 N 16,12%
7,ll-Dimethyl-2(trans),6(cis),10-dodecatrien-l-säure (XLVII)
Zu einem Gemisch von 8 g Aldehyd XLVI (Fraktion 2 und 3), 18,15 g Silberoxyd
in 29 ml Methanol und 29 ml Wasser Hess man im Verlauf von 1 Std. eine Lösung von
5,32 g Natriumhydroxyd in 14,3 ml Wasser langsam zutropfen, wobei die Temperatur
ständig zwischen -10 und 0° gehalten wurde. Anschliessend wurde U/2 Std. bei Zimmer¬
temperatur gerührt. Man liess anschliessend über Nacht unter leichtem Rühren mit
feinem Vibromischer stehen.

- 39 -
Die Reaktionslösung wurde mit Wasser verdünnt, und man filtrierte vom Silber
ab. Der Niederschlag wurde dreimal mit Wasser und einmal mit 1-n. Natronlauge
gewaschen. Die Reaktionslösung und das Waschwasser extrahierte man mit Aether,
wobei im Aether der Neutralteil zurückblieb. Um jede Spur der Säure aus den Aether-
auszügen zu entfernen, wurden diese einmal mit 50 ml 1-n. Natronlauge durchge¬
waschen. Man versetzte den alkalischen Teil mit 150 ml ges. primärer Natriumphos¬
phatlösung und säuerte mit eiskalter Salzsäure bis pH 1 - 2 an. Man extrahierte mit
Aether. Die Aetherlösungen wurden neutral gewaschen und mit Natriumsulfat getrock¬
net. Nach Abdampfen des Aethers blieben 8,8 g zurück, die fraktioniert wurden. Man
erhielt zwei Fraktionen: Sdp. 135 - 138°/0,12 Torr und 138, 5 - 141°/0,1 Torr mit
20demselben Brechungsindex nn 1,4848. UV. -Absorptionsspektrum: A. = 210 mju
(log £ 4,19), die über ihr S-Benzyl-isothiuroniumsalz gereinigt wurden.
S-Benzyl-isothiuroniumsalz: 5,96 g der rohen Säure wurden in 59 ml Methanol ge¬
löst und tropfenweise mit 1-n. Natronlauge auf Phenolphtalein neutralisiert (25,5 ml).
Dann gab man in einem Guss 7, 95 g S-Benzyl-isothiuroniumhydrochlorid in 42 ml
Methanol hinzu, wobei sofort ein voluminöser Niederschlag ausfiel. Man erwärmte
15 Min. auf 60° und liess anschliessend im Kühlschrank erkalten. Die weissen, glän¬
zenden Plättchen wurden dreimal aus Methanol-Wasser umkristallisiert, Smp. 145, 5
bis 146°, Ausbeute 3,29 g (32 % d. Th.). Zur Analyse wurden 0,14 g noch fünfmal
umkristallisiert, Smp. 146.
Analyse: C22H3202N2S Ber. C 68,01 H 8,35 %
Gef. C 68,13 H 8,18 %
Die Mischprobe mit dem S-Benzyl-isothiuroniumsalz der 7, ll-Dimethyl-2(trans),
6(trans),10-dodecatrien-l-säure (LXI) ergab eine Depression des Smp. von 3°.
Um das Salz zu spalten, suspendierte man die 3,29 g S-Benzyl-isothiuronium¬
salz in Aether und fügte 105 ml eisgekühlte, primäre Natriumphosphatlösung hinzu,
die vorher mit Salzsäure auf pH 1 - 2 gebracht worden war. In einem Scheidetrichter
wurde nun solange geschüttelt, bis alles zersetzt war. Die ätherischen Auszüge
wurden nochmals mit der gleichen Menge Pufferlösung behandelt, dann mit Eiswas¬
ser neutral gewaschen und mit Natriumsulfat getrocknet. Das nach dem Abdampfen
des Aethers zurückbleibende Oel wurde im Hochvakuum destilliert. Dabei erhielt man
als Mittelfraktion 1,3 g eines farblosen Oeles vom Sdp. 145°/0,05 Torr, nn 1,4919,
d £ 0,9380, pKMCS = 7,09 (Aequivalentgewicht: Ber. 222, Gef. 226).
UV.-Absorptionsspektrum: X = 210 mu (log E 4,31)
IR.-Absorptionsspektrum: Banden u.a. bei 2930, 1695, 1650, 680 cm".

- 40 -
Gegenüber dem IR.-Absorptionsspektrum der 7,ll-Dimethyl-2(trans),6(trans),or oc\
10-dodecatrien-l-säure (LXI) ' 'sind folgende Unterschiede zu bemerken: Bande
bei 1425 cm ist breiter, eine Bande bei 1070 cm ist bei der trans-Säure LXI
nicht vorhanden.
Analyse: C14H2202 Ber. C 74,63 H 9,97%
Gef. C 75,59 H 9,99 %
Cyclisation der 7, 11-Dimethy 1-2(tr ans), 6(cis)-1 0-dodecatr ien-
1-säure (XLVII)
In einem mit trockenem Stickstoff gut durchgespülten Kolben wurden 20, 4 ml
konz. Ameisensäure (Merck, analysenrein) vorgelegt und auf 0 abgekühlt. Unter
leichtem Rühren mit einem Vibromischer liess man darauf 2,04 ml konz. Schwefel¬
säure (Merck, analysenrein) einfliessen. Dann wurden innert l!/2 Std. 0,4712 g Säure
XLVII (aus der Mittelfraktion) langsam durch eine Kapillare eingetropft, wobei die
Reaktionslösung stark gerührt wurde. Nachdem alles eingetropft war, liess man ohne
zu rühren über Nacht bei 0 stehen (im ganzen 15 Std.).
Aufarbeitung: Die hellrote Reaktionslösung wurde auf Eis gegossen und hierauf
mit Aether extrahiert. Die Aetherauszüge wurden einmal mit Eiswasser gewaschen,
dann viermal mit je 50 ml 2-n. Kalilauge extrahiert, schliesslich wurden sie mit
Wasser neutral gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des
Aethers blieben 0, 026 g Neutralteil zurück, die nicht weiter untersucht wurden.
Der Kalilauge-Auszug wurde V2 Std. auf 70 erhitzt, dann erkalten gelassen
und mit eiskalter Salzsäure auf pH 1 angesäuert, wobei ein flockiger Niederschlag
ausfiel. Man extrahierte mit Aether. Die Aetherlösungen wurden mit Eiswasser neu¬
tral gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des Aethers wur¬
de der Rückstand aus Dioxan-Hexan umkristallisiert. Nach einmaligem Umkristalli¬
sieren wurden 0,2906 g (55,6 % d.Th.) Kristalle erhalten vom Smp. 233° (XLVin).
Die Mischprobe mit der Hydroxysäure XLVHI, cyclisiert aus der 7,11-Dimethyl-
2(trans), 6(trans), 10-dodecatrien-l-säure (LXI) ' '
ergab keine Depression des
Smp. Für das IR.-Absorptionsspektrum wurde ein Teil bei 135°/0,001 Torr sublimiert
(Smp. 234 - 235°).
Das in Nujol aufgenommene IR.-Absorptionsspektrum war vollkommen identisch
mit demjenigen der Hydroxysäure XLVIII, cyclisiert aus der 7,ll-Dimethyl-2(trans),
6(trans), 10-dodecatrien-l-säure (LXI)35,36^.

- 41 -
Aus der Mutterlauge konnten noch 0,043 g Kristalle gewonnen werden, die
zweimal aus Dioxan-Hexan umkristallisiert und bei 135°/0, 002 Torr sublimiert
wurden. Smp. 232 - 234. Zusammen mit den zuerst erhaltenen Kristallen ergibt sich
eine Ausbeute von 59 % d. Th.
Die 0,126 g Restmutterlauge wurden an Silicagel chromatographiert (vierzigfa¬
che Menge). Aus den Benzol-Aether 1:1 Eluaten konnten zwei kristalline Fraktionen
erhalten werden. Sie wurden aus Dioxan-Hexan umkristallisiert. Smp. nach dreima¬
ligem Umkristallisieren 193 - 194 . Die Mischprobe mit der zuerst erhaltenen Hydr-
oxysäure (aus dem ersten Umkristallisieren) zeigte keine Depression.
Kondensation von Dihy dr o-<x-jonon (XLIX) mit Oxals äur e-di-
äthylester
Das bei dieser Synthese verwendete Dihydro-o<-jonon (XLIX) ist durch Ueber-
führen in das Semicarbazon vom konstanten Smp. 161 und IR. -Absorptionsspektrum
(Banden u.a. bei 2940, 2920, 1715, 1775, 1450, 1375 cm"1) charakterisiert worden.
Es siedete bei 87 - 90°/0,5 Torr, nD° 1,478853\In einem Kolben wurden 27,6 g Natrium in 800 ml abs. Aether vorgelegt. Un¬
ter Rühren liess man darauf portionsweise 118 ml abs. Aethanol so einfliessen,
dass eine lebhafte Wasserstoffentwicklung auftrat. Hatte diese nachgelassen, fügte
man ebenfalls portionsweise insgesamt 175 g Oxalsäure-diäthylester hinzu. Dabei
resultierte eine homogene, gelbrote Lösung. War alles Natrium aufgelöst, fügte man
in einem Guss 189 g Dihydro-ot-jonon (XLIX) zu, wobei leichtes Sieden des Aethers
eintrat. Man rührte noch 1 Std. weiter und liess über Nacht bei Zimmertempera¬
tur stehen.
Zur Aufarbeitung wurde das Reaktionsgemisch mit 2 1 Wasser verdünnt und mit
verdünnter Phosphorsäure auf pH 2 angesäuert. Man zog mit Aether aus. Die ätheri¬
schen Lösungen wurden mit Kochsalzlösung neutral gewaschen und mit Natriumsulfat
getrocknet. Nach dem Abdampfen des Lösungsmittels erhitzte man den Rückstand
zwecks Entfernung leichtflüchtiger Anteile im Wasserstrahlvakuum während 30 Mi¬
nuten auf 90 - 95°. Ausbeute: 275,0 g, die sofort der Reduktion mit Lithiumalumi¬
niumhydrid unterworfen wurden.

- 42 -
Diol LI
In einem gut mit trockenem Stickstoff durchgespülten Kolben suspendierte man
76 g Lithiumaluminiumhydrid (Metal Hydride, Inc., 95 % rein) in 1500 ml abs. Aether.
Zu dieser Suspension liess man im Verlauf von 5 Std. unter Rühren mit einem Vibro-
mischer die Lösung von 275 g Kondensationsprodukt in 1 1 Aether zutropfen. Man
liess anschliessend während der Nacht bei Zimmertemperatur stehen.
Die Zersetzung des Reaktionsgemisches erfolgte unter Kühlung durch tropfen¬
weise Zugabe von 20 %-iger Weinsäurelösung (pH 5-6). Dabei trennte sich die Aether¬
schicht vom Aluminiumhydroxyd. Man dekantierte die Aetherschicht ab. Das Alumini¬
umhydroxyd selber wurde durch weitere Zugabe von Weinsäure in Lösung gebracht.
Diese wurde mehrmals mit frischem Aether extrahiert. Die vereinigten Aetheraus-
züge wurden mit Wasser neutral gewaschen und mit Natriumsulfat getrocknet. Nach
Abdampfen des Lösungsmittels blieben 205 g Rohprodukt zurück, die destilliert wur¬
den.
Man erhielt 144 g eines schwach gelb gefärbten Oeles LI, Sdp. 152 - 154°/0,08 - 0,1
Torr (62 % d.Th.).
Analyse: C15H2g02 Ber. C 75,58 H 11,00 %
Get. C 75,63 H 10,86%
tx-Monocy clo-nor-f arnesal (LII)
Zu einer Lösung von 143 g Diol LI in 3 1 Methanol wurden unter starkem Rühren
eine Lösung von 201 g Natriumperjodat in 1200 ml Wasser zugegeben. Sofort trat ei¬
ne voluminöse, weisse Fällung auf und die Temperatur stieg bis auf 38.Man rührte
noch 4 Std. weiter bei einer Temperatur von 35.Zur Aufarbeitung filtrierte man
vom ausgeschiedenen Salz ab und wusch letzteres mehrmals mit wenig Methanol aus.
Das Filtrat versetzte man mit 1 1 ges. Kochsalzlösung und extrahierte dieses mit
Aether. Die Aetherauszüge wurden mit Kochsalzlösung neutral gewaschen und mit
Natriumsulfat getrocknet. Nach Abdampfen des Aethers blieben 119 g Rohprodukt
zurück, die destilliert wurden. Man erhielt 60 g einer zwischen 110 - 115 /0,4 Torr
20siedenden Flüssigkeit nn 1,5002, die nochmals fraktioniert wurden. Als reinen Al¬
dehyd wurde die bei 109 - 113°/0,2 Torr siedende Fraktion betrachtet, n^0 1,4998.
Man erhielt 54 g von LII (44 % d.Th.).
UV.-Absorptionsspektrum: X = 222 mji (log £ 4,15)

- 43 -
IR.-Absorptionsspektrum: Banden u.a. bei 2620, 1685, 1634, 1387, 1366,
820 cm .
Analyse: C14H220 Ber. C 81,50 H 10,75 %
Gef. C 79,97 H 10,66 %
«-Monocyclo-nor-farnesylsäure (LIU)
Zueinemauf-5 vorgekühlten Gemisch von 92,7g Silberoxyd, 41,2g AldehydLII
in 150 ml Wasser und 150 ml Methanol wurde unter Rühren mit einem Vibromischer
innert!/4Std. eine Lösungvon 28 g Natriumhydroxyd in 75 ml Wasser zugetropft. Tempe¬
ratur des Reaktionsgemisches - 2 bis 6°. Anschliessend wurde bei Zimmertemperatur
noch2Std. gerührt. Man Hess über Nacht stehen. Zur Aufarbeitung wurde vom festen
Rückstand abfiltriert und dieser mehrmals mit Wasser gewaschen. Der Neutralteil wurde
durch Extraktion mit Aether abgetrennt. Hierauf versetzte man den alkalischen Teil mit
500 ml ges. primärer Natriumphosphatlösung und säuerte mit verdünnter Phosphor¬
säure an (pH 2). Die flockig ausfallende Säure wurde mit Aether extrahiert. Die Aether-
auszüge wurden mit Kochsalzlösung neutral gewaschen und mit Natriumsulfat getrock¬
net. Nach Abdampfen des Aethers wurde der Rückstand fraktioniert. Man erhielt als
Hauptfraktion 31 g (70 % d.Th.), Sdp. 144 - 148°/0,4 Torr, n£° 1,4928, welche an¬
schliessend über das S-Benzyl-isothiuroniumsalz gereinigt wurden.
S-Benzyl-isothiuroniumsalz: 17,6 g der rohen Säure wurden in 174 ml Methanol ge¬
löst und tropfenweise mit 1-n. Natronlauge auf Phenolphtalein neutralisiert (73,3 ml).
Dazu gab man in einem Guss eine Lösung von 23, 5 g S-Benzyl-isothiuroniumhydro-
chlorid in 125 ml Methanol. Sofort trat eine voluminöse Fällung auf. Man erwärmte
während 20 Minuten auf 60° und liess anschliessend im Kühlschrank erkalten. Der
Niederschlag wurde aus Methanol-Wasser solange umkristallisiert, bis der Schmelz¬
punkt konstant blieb; Smp. 153 - 154°, Ausbeute 4,4 g (14,8 % d.Th.). Zur Analyse
wurden 0,2 g nochmals umkristallisiert, Smp. 153.
Analyse: C22H3202N2S Ber. C 68,01 H 8,30 %
Gef. C 68,10 H 8,22 %
Zur Spaltung des Salzes wurden 4,39 g des S-Benzyl-isothiuroniumsalzes in
Aether suspendiert. Dazu fügte man 136 ml einer eiskalten, primären Natriumphos¬
phatlösung, welche vorher mit Salzsäure auf pH 1 gebracht worden war. Man schüttel-

- 44 -
te so lange, bis alles in Lösung gegangen war. Nachdem die ätherische Lösung noch¬
mals mit der gleichen Menge Pufferlösung behandelt worden war, wurden die Aether-
auszüge mit Eiswasser neutral gewaschen und mit Natriumsulfat getrocknet. Nach
Abdampfen des Aethers blieben 2, 723 g Säure zurück, die im Hochvakuum fraktioniert
wurden. Dabei erhielt man als Mittelfraktion 2,2 g eines farblosen Oeles, Sdp. 122,5 /
0,05 Torr, n£° 1,5039, d2^ 0,9914.
UV.-Absorptionsspektrum: A = 210 mu (log £ 4,03)
pK*g = 7,05 (Aequivalentgewicht: Ber. 222, Gef. 221)
Zur Analyse und für die Aufnahme eines IR. -Absorptionsspektrums wurde ein
Teil nochmals destilliert.
IR.-Absorptionsspektrum: Banden u.a. bei 2920, 1700, 1650, 1425, 1390,
1365 cm"1.
Analyse: C14H2202 Ber. C 75,63 H 9,97%
Gef. C 75,53 H 9, 92%
Cyclisation der o«.-Monocyclo-nor-farnesylsäure (Lin)
In einem mit trockenem Stickstoff gut durchgespülten Kolben wurden 18,7 ml
konz. Ameisensäure (Merck, analysenrein) vorgelegt und auf 0 abgekühlt. Dieser
fügte man unter Rühren mit einem Vibromischer 1,87 ml konz. Schwefelsäure (Merck,
analysenrein) zu. Darauf liess man innert l!/2 Std. 0,428 g Säure Lin durch eine
Kapillare zutropfen, wobei stark gerührt wurde. War alles eingetropft, liess man über
Nacht (15 Std.) bei 0° stehen.
Aufarbeitung: Das Reaktionsprodukt wurde auf Eis gegossen und anschliessend mit
Aether extrahiert. Die Aetherlösungen wurden einmal mit Eiswasser gewaschen,
dann viermal mit je 50 ml 2-n. Kalilauge extrahiert, schliesslich neutral gewaschen
und mit Natriumsulfat getrocknet. Nach Abdampfen des Aethers blieben 0,04 g Neu¬
tralteil zurück, die man nicht weiter untersuchte. Der alkalische Teil wurde während
!/2 Std. auf 70 erwärmt, dann abgekühlt und mit eiskalter, verdünnter Salzsäure auf
pH 2 angesäuert. Die Hydroxysäure wurde mit Aether extrahiert. Die Aetherauszüge
wurden neutral gewaschen und mit Natriumsulfat getrocknet, der Aether anschliessend
abgedampft. Zurück blieb ein fester Rest, der aus Dioxan-Hexan umkristallisiert
wurde. Nach einmaligem Umkristallisieren wurden 0,265 g (56 % d.Th.) vom Smp.
233 - 234° erhalten.
Die Mischprobe mit der Hydroxysäure, cyclisiert aus der 7,ll-Dimethyl-2(trans),50 51)
6(trans),10-dodecatrien-l-säure (LXI) ' '
ergab keine Depression des Smp.

- 45 -
Für das IR.-Absorptionsspektrum wurde ein Teil sublimiert: 130°/0,01 Torr,
Smp. 233 - 234°.
Das in Nujol aufgenommene IR.-Absorptionsspektrum war vollkommen identisch
mit demjenigen der Hydroxysäure, cyclisiert aus der 7,ll-Dimethyl-2(trans),6(trans),
10-dodecatrien-l-säure (LXI) ' ' und aus der 7,ll-Dimethyl-2(trans), 6(cis), 10-
dodecatrien-1-säure (XLVII).
Aus der Mutterlauge konnten 0,036 g Kristalle isoliert werden, die nach zwei¬
maligem Umkristallisieren und Sublimieren einen Smp. von 225 - 227° zeigten.
Die Mischprobe dieser Kristalle mit der Hydroxysäure, cyclisiert aus der 7,11-
Dimethyl-2(trans),6(trans), 10-dodecatrien-l-säure (LXI) ' ' ergab keine Depres¬
sion des Smp. Die 0,093 g Mutterlauge wurden nicht weiter untersucht.
Spaltung von (t)Dihydro- jf-jonon-semicarbazon (LIV)
20 g des Semicarbazons LIV (Smp. 190,5 - 191,5°) wurden in 500 ml Chloro¬
form gelöst, dazu gab man eine Lösung von 30 g Phthalsäureanhydrid in 200 ml heis-
sem Wasser. Man kochte unter Stickstoff milde während 2 Std. am Rückfluss, Hess
dann zuerst bei Zimmertemperatur, anschliessend im Eiswasser erkalten. Die Reak¬
tionsmischung wurde auf 100 ml 10 %-ige, wässerige Natronlauge gegossen. Man zog
mit Chloroform aus, wusch die Auszüge neutral und trocknete sie mit Calciumchlorid.
Das Lösungsmittel wurde im Vakuum bei einer Wasserbadtemperatur von 40 C ent¬
fernt. Beim Eindampfen fiel unzersetztes Semicarbazon aus, das abfiltriert wurde
(0,535 g). Man erhielt 14,89 g Rohprodukt (ein hellgelbes Oel), das im Vakuum frak¬
tioniert wurde. Bei einem Druck von 10 Torr gingen bei einem Sdp. von 115 - 116
2012,55 güber (81,5% d.Th.), nQ 1,4 755. Die gaschromatographische Untersuchung zeigte
eine geringe Verunreinigung von ca. 5 %.
Kondensation des (Î)-Dihy dr o-y-jonons (LV) mit Oxalsäure-di-
äthylester
In einem mit Stickstoff gut gespülten Kolben wurden 1,44 g Natrium und 39 ml
abs. Aether vorgelegt. Unter Rühren mit einem Vibromischer liess man 6,5 ml abs. Ae-
thanol so zutropfen, dass eine lebhafte WasserStoffentwicklung auftrat. Wurde sie geringer,
so liess man in Portionen 9,1g Oxalsäure-diäthylester zufHessen, dabei resultierte eine

- 46 -
gelb-grüne Lösung. War alles Natrium gelöst, rührte man noch 1/4 Std. und liess darauf in
einem Guss 10 g Dihydro-tf-jonon (LV) einfliessen. Es trat keine spürbare Erwär¬
mung auf, nur die Farbe änderte sich gegen rot. Man liess über Nacht unter leich¬
tem Rühren bei Zimmertemperatur stehen. Zur Aufarbeitung wurde das Reaktions¬
gemisch auf eine eiskalte Mischung von 20 ml primäre Natriumphosphatlösung und
3,5 g Schwefelsäure gegossen (pH 2). Man zog mit Aether aus. Die Aetherauszüge
wurden neutral gewaschen und mit Natriumsulfat getrocknet. Der Aether wurde bei
einer Wasserbadtemperatur von 30 - 45 entfernt, zurück blieben 17 g LVI, die so¬
fort der Reduktion mit Lithiumaluminiumhydrid unterworfen wurden.
Diol LVII
In einem mit trockenem Stickstoff gut durchgespülten Kolben wurden 4 g Lithium¬
aluminiumhydrid (Metal Hydride Inc., 95 % rein) und 58 ml abs. Aether vorgelegt.
Zu dieser Suspension liess man unter starkem Umrühren im Verlauf von 6 Std. eine
Lösung von 17 g des Kondensationsproduktes LVI in 68 ml abs. Aether so zutropfen,
dass immer ein leichtes Sieden zu beobachten war. Nachdem alles eingetropft war,
fügte man noch 58 ml abs. Aether zu und liess über Nacht stehen.
Die Zersetzung des Reaktionsgemisches erfolgte unter Kühlung mit einer Eis-
Kochsalzmischung durch Zutropfen von 16 ml ges. Seignettesalzlösung. Nachdem al¬
les zersetzt war, fügte man noch 300 ml Aether zu, dekantierte vom festen Alumini¬
umhydroxyd ab und filtrierte die fast klare Aetherlösung durch Cellit. Der Nieder¬
schlag selbst wurde mehrmals mit frischem Aether extrahiert. Die Aetherauszüge
wurden neutral gewaschen und mit Natriumsulfat getrocknet. Der nach dem Abdamp¬
fen des Aethers erhaltene Rückstand wurde im Hochvakuum fraktioniert.
Man erhielt 5,65 g (46 % d.Th.) LVII, Sdp. 140 - 142°/0,06 Torr, n£° 1,5033.
(i)- y-Monocyclo-nor-f arnesal (LVIH)
In einem Kolben wurden 5,65 g Diol LVII in 115,5 ml Methanol gelöst. Man er¬
wärmte auf 25 und liess unter Umrühren in einem Guss eine Lösung von 7,6 g Natriumperjo-
datin41 ml Wasser einfliessen. Sofort trat eine voluminöse weisse Fällung auf. Man rührte
noch während 31/2 Std., bei einer Temperatur von 35 - 40°, liess anschliessend lang¬
sam erkalten und verdünnte mit ges. Kochsalzlösung. Die Reaktionslösung wurde im

- 47 -
Vakuum zwecks Entfernung des Methanols möglichst stark eingeengt (Wasserbad-Tem¬
peratur 60 j und anschliessend mit Aether extrahiert. Die Aetherauszüge wurden neu¬
tral gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des Aethers blieben
5 g zurück, die im Hochvakuum destilliert wurden. Man erhielt als Hauptfraktion
0,837 g einer bei 92°/0,03 Torr siedenden Flüssigkeit LVTII, n^° 1,4995. UV.-Ab¬
sorptionsspektrum: *max = 233 mu (log £ 4,05). Der Vorlauf dieser Destillation
wurde nochmals fraktioniert.
Fraktion Sdp. Torr g nD max= 223 mji, logE=
1 bis 88° 0,07 1,322 1,4995 4,058
2 88 - 102° 0,05 0,784 1,4978 4,045
3 110 - 130° 0,04 0,258 1,5011 3,788
Für die weitere Oxydation wurde die Hauptfraktion der ersten Destillation plus,
Fraktionen 1 und 2 dieser Vorlaufdestillation verwendet.
( + )- y-Monocy clo-nor-f arnesylsäure (LIX)
In einem Kolben wurden 2, 79 g Aldehyd LVHI, 6,35 g frisch zubereitetes Silber¬
oxyd, 11,5 ml Methanol und 10 ml Wasser vorgelegt und auf 0° gekühlt. Darauf liess man
unter Rühren innerhalb von 3/4 Std. eine Lösung von 1,86 g Natriumhydroxyd in 5 ml
Wasser zutropfen, wobei die Temperatur konstant zwischen 0 und 2 gehalten wurde.
Anschliessend liess man über Nacht bei Zimmertemperatur stehen.
Aufarbeitung: Man filtrierte vom festen Material ab und wusch dieses mehrmals mit
Wasser und einmal mit 1-n. Natronlauge. Die Reaktionslösung und das Waschwasser
wurden mit Aether extrahiert. Nachdem man diese Auszüge mit Wasser neutral ge¬
waschen und getrocknet hatte, wurde der Aether abgedampft. Es blieben 0,457 g Neu¬
tralteil zurück, die nicht untersucht wurden. Der alkalische Teil wurde mit 54 ml ges.
primärer Natriumphosphatlösung versetzt und mit kalter Salzsäure auf pH 2 angesäuert.
Die Extraktion der dabei ausfallenden Säure erfolgte mit Aether. Die Aetherauszüge
wurden neutral gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des
Aethers blieben 2,13 g zurück, die im Hochvakuum destilliert wurden. Man erhielt
1,126 g (37 % d.Th.) einer bei 122 - 126°/0,045 Torr siedenden Flüssigkeit LIX,20
nD 1,4928. Die Säure wurde über ihr S-Benzyl-isothiuroniumsalz gereinigt.

- 48 -
S-Benzyl-isothiuroniumsalz: 1,126 g Säure wurden in 11 ml Methanol gelöst und mit
1-n. Natronlauge auf Phenolphtalein neutralisiert (4,8 ml). Dazu gab man in einem
Guss eine Lösung von 1,519 g S-Benzyl-isothiuronium-hydrochlorid in 8 ml Methanol.
Die voluminöse, weisse Fällung wurde aus Methanol-Wasser umkristallisiert. Nach
viermaligem Umkristallisieren wurde ein Smp. von 157 - 158 erreicht. Ausbeute
0,661 g(36%d.Th.).
Analyse: C22H3202N2S Ber. C 68,01 H 8,30 %
Gef. C 68,20 H 8,38 %
Zur Spaltung des Salzes wurden 0, 661 g Salz in Aether suspendiert. Zu diesem
fügte man 22 ml ges. primäre Natriumphosphatlösung, die vorher mit Salzsäure auf
ein pH von 1 gebracht worden war. Es wurde nun solange geschüttelt, bis alles Salz
in Lösung gebracht war. Der Aetherauszug wurde nochmals mit der gleichen Menge
Pufferlösung behandelt, neutral gewaschen und mit Natriumsulfat getrocknet. Der
nach dem Abdampfen des Aethers erhaltene ölige Rest wurde in einem Kugelrohr de¬
stilliert.
Man erhielt 0,316 g, Sdp. 102°/°, 02 Torr, n^0 1,5017, LIX.
UV.-Absorptionsspektrum: X = 213 mu (log £ 4,18)
IR.-Absorptionsspektrum: Banden u.a. bei 2980, 1700, 1655, 1425, 1385, 1375
cm,und als neue Bande, die bei
der ot-Monocyclo-nor-farnesylsäure (LIIÏ) nicht vorhanden war, eine Bande bei
890 cm"1.
Analyse: C14H2202 Ber. C 75,63 H 9,99 %
Gef. C 75,59 H 9,98 %
Cyclisation der (±) -
f -Monocy c lo-nor-f ar nesy ls äur e (LIX)
In einem mit trockenem Stickstoff gut durchgespülten Kolben wurden 12, 6 ml
Ameisensäure (Merck, analysenrein) vorgelegt und auf 0 abgekühlt. Zu dieser fügte
man unter leichtem Rühren mit einem Vibromischer 1,26 ml konz. Schwefelsäure
(Merck, analysenrein). Unter starkem Rühren wurden 0,268 g Säure LIX innert 1 Std.
durch eine Kapillare eingetropft. Anschliessend liess man ohne zu rühren über Nacht
bei 0 stehen (im ganzen 15 Std.).
Aufarbeitung: Die Reaktionslösung wurde auf Eis gegossen und anschliessend mit
Aether extrahiert. Die Aetherauszüge wurden einmal mit Wasser gewaschen, dann

- 49 -
viermal mit je 50 ml 2-n. Kalilauge extrahiert, neutral gewaschen und mit Natrium¬
sulfat getrocknet. Nach Abdampfen des Aethers blieben als Neutralteil 0,015 g zurück,
die nicht weiter untersucht wurden.
Der alkalische Auszug wurde während !/2 Std. auf 65 - 70° erhitzt, anschliessend
auf 10 abgekühlt und mit verd. Salzsäure bis pH 1 angesäuert. Die ausgefallene Säure
wurde mit Aether extrahiert. Die Aetherlösungen wurden neutral gewaschen und ge¬
trocknet. Der nach dem Verdampfendes Aethers erhaltene Rest wurde aus Dioxan-
Hexan umkristallisiert. Man erhielt 0,153gvonXLVIII(51,5% d.Th.), Smp. 234-235°,Die Mischprobe mit der Hydroxysäure, cyclisiert aus der 7, ll-Dimethyl-2(trans),
or 9ß\
6(trans), 10-dodecatrien-l-säure (LXI) '
zeigte keine Depression des Smp. Für
das IR.-Absorptionsspektrumwurde ein Teil bei 120°/0,05 Torr sublimiert, Smp. 235°.
Das in Nujol aufgenommene IR. -Absorptionsspektrum zeigte dieselben Banden
und war vollkommen identisch mit demjenigen der Hydroxysäure, cyclisiert aus der
ot-Monocyclo-nor-farnesylsäure (LUI) und aus der 7,ll-Dimethyl-2(trans), 6(trans),
10-dodecatrien-l-säure (LXI)35'36*.Aus der Mutterlauge konnten 0,012 g Kristalle gewonnen werden, die nach dem
Sublimieren im Hochvakuum einen Smp. von 229,5 - 230° zeigten. Von diesen wurde
keine Mischprobe gemacht. Die 0,122 g Mutterlauge wurden nicht weiter untersucht.
( + )-Dihy dr o-](-j onon (LV)
49)Das durch Oxydation des Ambreins mit Kaliumpermanganat
' erhaltene optisch
aktive Dihydro- jj-jonon (LV) wurde im Hochvakuum fraktioniert. Die zwischen 70 bis
115°/0,2 - 0,15 Torr siedende Fraktion wurde nochmals destilliert. Als Rückstand
blieb im Kolben ungespaltenes Ambrein zurück. Die zwischen 100 - 105 /0,2 - 0,19
Torr siedende Fraktion war reines Dihydro-jj-jonon (LV), nQ 1,4734, t<*JD = +8,05 .
Die gaschromatographische Untersuchung zeigte nur eine einzige Substanz, das UV.-
Absorptionsspektrum war leer.
20le rraKiion ^,40 g), r
Diketon LX. Sie wurde nicht weiter verwendet
Die zwischen 87 - 92°/u,18 Torr siedendeFraktion (4,45 g),
n£1,4731 ist das

- 50 -
Kondensation von (+)-Dihy dr o-j-jonon (LV) mit Oxalsäure-di-
äthylester
In einem mit trockenem Stickstoff gut gespülten Kolben wurden 0,865 g blankes
Natrium und 23,5 ml abs. Aether vorgelegt. Dazu liess man unter Rühren mit einem Vibro-
mischer 3,7 ml abs. Aethanolsozutropfen, dass eine lebhafte Wasserstoffentwicklung
auftrat. Wurde sie geringer, und war alles Aethanol eingetropft, so liess man in kleinen
Portionen 5,46 g Oxalsäure-diäthylester einfliessen. Die Farbe wurde dabei gelb-grün.
Nachdem alles Natrium aufgelöst war, rührte man noch 4 Std. und liess anschlies¬
send 6 g Dihydro- K -jonon (LV) in einem Guss einfliessen, dabei änderte sich die
Farbe gegen rot. Man liess über Nacht bei Zimmertemperatur stehen.
Aufarbeitung: Die dunkelrote Reaktionslösung wurde auf eine eisgekühlte Mischung
von 15 ml ges. primäre Natriumphosphatlösung und 2,5 g konz. Schwefelsäure gegos¬
sen (pH = 2). Man zog mit Aether aus. Die Auszüge wurden neutral gewaschen und
mit Natriumsulfat getrocknet. Der Aether wurde zuerst auf dem Wasserbad, der letz¬
te Rest im Vakuum entfernt. Zurück blieben 7 g LVI, die der Reduktion mit Lithium¬
aluminiumhydrid unterworfen wurden.
Diol LVIII
In einem mit trockenem Stickstoff gut gespülten Kolben wurden 2,4 g Lithium¬
aluminiumhydrid (Metal Hydride Inc., 95 % rein) in 35 ml abs. Aether suspendiert.
Dazu liess man im Verlauf von 6 Std. unter starkem Umrühren die 7,4 g Kondensa¬
tionsprodukt LVI in 41 ml Aether so zutropfen, dass immer ein leichtes Sieden des
Aethers zu beobachten war. Nachdem alles eingetropft war, fügte man noch 41 ml Ae¬
ther hinzu und liess über Nacht bei Zimmertemperatur stehen. Unter Kühlung mit
einer Eis-Kochsalzmischung liess man zur Reaktionslösung 9, 5 ml Seignettesalz-
lösung eintropfen. Man dekantierte vom ausgeschiedenen Aluminiumhydroxyd ab und
filtrierte die noch leicht trübe Aetherlösung durch Natriumsulfat. Der Niederschlag
selbst wurde fünfmal mit Aether gewaschen. Die Aetherlösungen wurden mit Eiswasser
gewaschen und mit Natriumsulfat getrocknet. Den Aether entfernte man im Vakuum.
Der ölige, farblose Rest wurde im Hochvakuum fraktioniert. Man erhielt 2,13 g eines
bei 132 - 138°/0,06 Torr siedenden Oeles LVII, n^° 1,5013.

- 51 -
( + )-v-Monocyclo-nor-farnesal (LVHI)
2,8 g Diol LVII wurden in 77 ml Methanol gelöst. Unter starkem Rühren gab man
in einem Guss 3, 78 g Natriumperjodat in 20 ml Wasser hinzu. Während 2*/2 Std. er¬
hitzte man auf 42,dann während 1V2 Std. auf 35
.Anschliessend wurde das Methanol
im Vakuum entfernt. Der Rest wurde mit ca. 250 ml ges. Kochsalzlösung verdünnt
und mit Aether ausgezogen. Die Aetherextrakte wurden neutral gewaschen und mit
Natriumsulfat getrocknet. Den Aether entfernte man auf dem Wasserbad, der ölige
Rest wurde im Hochvakuum destilliert.
Die bei 91 - 94°/0,17 Torr siedende Fraktion (0,254 g), n^0 1,4940, UV.-Ab¬
sorptionsspektrum: X = 223 mu (log £ 3,98), und die zwischen 94 - 97°/0,15III 3.X c\n
bis 0,12 Torr siedende Fraktion (1,12 g), nD 1,4964, UV.-Absorptionsspektrum:
\ = 223 mu (log £ 4,01), wurden für die Weiteroxydation verwendet (Gesamt¬
ausbeute 56 % d.Th.).
( +)-v-Monocyclo-nor-farnesylsäure (LIX)
In einem Kolben wurden 1,368 g Aldehyd LVin, 3,12 g Silberoxyd, 5 ml Wasser
und 5 ml Methanol vorgelegt. Man kühlte auf 0° ab und Hess im Verlauf von 1 Std.
eine Lösung von 0,93 g Natriumhydroxyd in 2, 5 ml Wasser eintropfen, wobei ständig ge¬
rührt wurde. War alles eingetropft, liess man über Nacht bei Zimmertemperatur
stehen.
Aufarbeitung: Man filtrierte vom festen Material ab. Der Niederschlag wurde viermal
mit 20 ml 2-n. Natronlauge gewaschen. Man extrahierte den alkalischen Teil mit
Aether. Die Aetherauszüge wurden mit Wasser neutral gewaschen und mit Natrium¬
sulfat getrocknet. Nach dem Abdampfen des Lösungsmittels blieben 0,011 g Neutral¬
teil zurück, die nicht untersucht wurden.
Den alkalischen Teil versetzte man mit 27 ml ges. primärer Natriumphosphat¬
lösung und säuerte mit kalter verd. Salzsäure bis pH 2 an. Man zog mit Aether aus.
Die Aetherlösungen wurden neutral gewaschen und getrocknet. Nach Entfernung des
Aethers wurde der Rückstand in einem Kragenkolben destilliert. Man erhielt 0, 227 g
von LIX, einer zwischenl20-122°/0,02Torr siedenden Flüssigkeit, n^°l,952, UV.-
Absorptionsspektrum: \ = 208 mu (log £ 3,85), IR.-Absorptionsspektrum:
Banden u.a. bei 2930, 1700, 1650, 890 cm,identisch mit dem IR.-Absorptions¬
spektrum der optisch nicht aktiven jj-Monocyclo-nor-farnesylsäure (LIX),[<x.lD =
+ 6,07°.

- 52 -
Cyclisation der ( + ) -
$ -Monocy c lo- nor -f arnesy Is Sure (LIX)
In einem mit trockenem Stickstoff gut gespülten Kolben wurden 9, 9 ml konz.
Ameisensäure (Merck, analysenrein) vorgelegt und auf 0 gekühlt. Unter leichtem
Rühren mit einem Vibromischer liess man dazu 0,99 ml konz. Schwefelsäure (Merck,
analysenrein) einfliessen. Unter starkem Rühren wurden 0, 205 g Säure LIX innert
3/4 Std. durch eine Kapillare eintropfen gelassen. Nachdem alles eingetropft war, liess
man ohne zu rühren über Nacht bei 0 stehen (im ganzen 15 Std.).
Aufarbeitung: Die hellrote Lösung wurde auf Eis gegossen. Der Neutralteil wurde
durch Extraktion mit Aether abgetrennt. Der Aetherauszug wurde einmal mit Eis¬
wasser durchgewaschen, dann viermal mit 50 ml 2-n. Kalilauge extrahiert, schliess¬
lich neutral gewaschen und mit Natriumsulfat getrocknet. Nach Abdampfen des Aethers
blieben 0,024 g Neutralteil zurück, die nicht weiter untersucht wurden. Der alkalische
Auszug wurde l/2 Std. auf 65 - 70° erhitzt, dann abgekühlt und mit eiskalter, verd.
Salzsäure bis pH 1 angesäuert. Der flockige Niederschlag wurde mit Aether extrahiert.
Die Aetherlösungen wurden neutral gewaschen und mit Natriumsulfat getrocknet. Der
nach dem Abdampfen des Aethers erhaltene Rückstand wurde aus Dioxan-Hexan um¬
kristallisiert. Man erhielt 0,059 g Kristalle XLVIII (26 % d.Th.), Smp. 232 - 233°,
MD = o°.
Die Mischprobe mit der Hydroxysäure, cyclisiert aus der optisch inaktiven y-
Monocyclo-nor-farnesylsäure (LIX) zeigte keine Depression des Smp.
Aus der Mutterlauge wurden 0, 026 g schmierige Kristalle erhalten. Sie wurden
sublimiert bei 160°/0,5 Torr; Smp. 228 - 230°, [<x.)D = 0?
Die 0,111 g der Mutterlauge zeigten ebenfalls keine Drehung. Sie wurden nicht
weiter untersucht.
Versuch zur Untersuchung der Trennungsmöglichkeit der cycli-
sierten von der uncyclisierten Säure:
fin Hinblick auf die kinetische Untersuchung der Cyclisationsgeschwindigkeit
von Säuren war es wichtig zu wissen, wie klein die Menge cyclisierter Säure ist, die
sich aus der uncyclisierten Säure isolieren lässt.
Annahme: Es wurden ein Gramm Säure zur Cyclisation gebracht. Aus der gesamten
Menge der Cyclisationsmischung 3,98 ml Schwefelsäure + 39,8 ml Ameisensäure
wurde lfi herausgenommen = 8,73 ml. In den 8, 73 ml haben wir 0,2 g uncyclisierte
Säure. Bei 97,5 % uncyclisierter Säure haben wir 0,005 g Hydroxysäure und 0,1955 g
aliphatische Säure.

- 53 -
Versuch: 0,196 g 7,ll-Dimethyl-2(trans), 6(trans),10-dodecatrien-l-säure (LXI) (97,5%)
und 0, 0053 g Hydroxysäure XLVm wurden in Aether gelöst. Der Aether wurde ver¬
dampft und der ölige Rest aus Dioxan-Hexan umkristallisiert.
Man erhielt 0,0050 g feste, kristalline Hydroxysäure XLVIII vom Smp. 233°
(vorher 234-235°) und 0,1885 gflüssige 7,ll-Dimethyl-2,6,10-dodecatrien-l-säure (LXI).
Untersuchung der relativen C yclisationsgeschwindigkeit der
7, ll-Dimethyl-2(trans),6(trans), 10-dodecatrien-l-säure (LXI)
und der oc -Monocyclo-nor-farnesylsäure (LUI)
Um einheitliche Versuchsbedingungen zu schaffen und damit überhaupt die Mög¬
lichkeit eines Vergleiches der Cyclisationsgeschwindigkeiten zu geben, wurden bei
jedem der vier Versuche folgende Reaktionsbedingungen genau eingehalten:
In einem gut mit trockenem Stickstoff gespülten Kolben wurden 21,62 ml konz.
Ameisensäure (Merck, analysenrein) vorgelegt und unter leichtem Rühren mit einem
Vibromischer auf 0° abgekühlt. Dazu gab man 2,16 ml konz. Schwefelsäure (Merck,
analysenrein), wobei eine leichte Erwärmung auftrat (bis + 5 ). Nachdem alles wie¬
der auf 0 abgekühlt war, liess man in einem Guss ca. 0, 5 g Säure (genaues Gewicht bei
jedem Versuch angegeben) einfliessen. Anschliessend rührte man während 5 Minuten
bei 0,dann während 25 Minuten bei 10
. Nach dieser Zeit wurde der Vibromischer
abgestellt und die Reaktionslösung während der ganzen übrigen Dauer der Cyclisation
konstant auf der Temperatur von 10 gehalten.
Für die Untersuchung des Fortschreitens der Cyclisation wurden nach bestimm¬
ten Zeitabständen Proben von je 1/5 des Gesamtvolumens mit einer geeichten Pipette
herausgenommen und auf 10 g Eis gegossen. Man extrahierte zweimal mit je 40 ml
Aether. Die Aetherextrakte wurden anschliessend viermal mit je 10 ml 2-n. Kalilauge
ausgezogen. Im Aether blieb der Neutralteil zurück, der nicht weiter untersucht wur¬
de. Der alkalische Teil wurde auf dem Wasserbad während 1/2 Std. bei 65 - 70 er¬
wärmt, anschliessend erkalten gelassen und mit verd., eiskalter Salzsäure auf pH 1
angesäuert. Man zog mit Aether aus. Die Aetherextrakte wurden mit Eiswasser neu¬
tral gewaschen und mit Natriumsulfat getrocknet. Der Aether wurde vollständig ab¬
gedampft; der, je nach der Cyclisationszeit, ölige oder feste Rückstand wurde leicht
mit etwas Hexan angespritzt. Die feste, kristalline Hydroxysäure wurde abfiltriert
und der prozentuale Anteil von cyclisierter Säure bestimmt.
Die für die Cyclisation verwendete 7, ll-Dimethyl-2(trans),6(trans), 10-dodeca¬
trien-l-säure (LXI) wurde über das S-Benzyl-isothiuroniumsalz gereinigt, Smp. 151
- 54 -
bis 152°, und hatte einen Sdp. von 135°/0,03 - 0,025 Torr, n^0 1,4901.
0,4972 g 0,5031g
Temp. Ausb. Temp.
°C % °C
10
10
10 14,0 11
10 28,5 10
11 40,0 10
49,5 10
56,0 13
Die für die Cyclisation verwendete ex.-Monocyclo-nor-farnesyl-säure (LUI) wur¬
de über das S-Benzyl-isothiuroniumsalz gereinigt, Smp. 138,und hatte einen Sdp.
von 134 - 137°/0,03 Torr, n^0 1,4933.
Es wurden cyclisiert: 0,4987 g 0,4039 g
Es wurden cyclisiert:
Zeit nach Ausl
Std. %
V4 1,8
V2 7,0
1 15,8
2 27,5
4 43,5
8
23
Zeit nach Ausb. Temp. Ausb. Ter
Std. % °C % °C
V4 0 10
V2 1,6 10
1 5,8 10 4,2 10
2 11,7 10 9,6 10
4 21,0 11 21,0 11
8 29,0 10
23 31,0 15

- 55 -
Die Schmelzpunkte sind korrigiert, die Siedepunkte nicht. Sämtliche UV. -Ab¬
sorptionsspektren sind in Feinsprit aufgenommen worden.
Die Analysen wurden in der mikroanalytischen Abteilung des organisch-chemi¬
schen Institutes der Eidgenössischen Technischen Hochschule unter der Leitung von
Herrn W. Manser ausgeführt, wofür an dieser Stelle bestens gedankt sei.
Fräulein V. Klopf stein möchte ich für die Aufnahme der IR.-Absorptions¬
spektren im Laboratorium von Herrn Prof. Dr. H.H. Gunthar d herzlich danken.
Die UV. -Absorptionsspektren wurden in verdankenswerter Weise im Labora¬
torium von Herrn P.-D. Dr. E . Heilbr onner aufgenommen.
Die Bestimmung der pKî.r„-Werte verdanke ich Herrn Dr. W. Simon.

- 56 -
Zusammenfassung
Als Fortsetzung der am hiesigen Institut begonnenen Arbeiten über die säure¬
katalysierte Cyclisation von Polyenen wurden die 7, ll-Dimethyl-2(trans), 6(cis), 10-
dodecatrien-l-säure, die ot-Monocyclo-nor-farnesylsäure, die optisch aktive und
racemische y-Monocyclo-nor-farnesylsäuren synthetisiert und der Verlauf ihrer
säurekatalysierten Cyclisation untersucht.
Alle diese Ringschlussreaktionen führen zur selben bicyclischen Hydroxysäure
mit trans-Verknüpfung des Dekalinringes, welche bereits bei der Cyclisation der
7, ll-Dimethyl-2(trans),6(trans), 10-dodecatrien-l-säure erhalten worden war.
Durch Vergleich der entsprechenden Cyclisationsgeschwindigkeiten konnte ge¬
zeigt werden, dass die monocyclische ex-Nor-farnesylsäure nicht ein Zwischenpro¬
dukt bei der Cyclisation der 7, ll-Dimethyl-2(trans),6(trans), 10-dodecatrien-l-säure
ist.

- 57 -
Literaturverzeichnis
1) CIBA Foundation Symposium on the Biosynthesis of Terpenes and Sterols, 1959.
(J.A.Churchill Ltd. London).
2)J.W.Cornforth,R.H.Cornforth,M.G.Horning,A.Pelter,G.
Popjak, Tetrahedron, ji, 311 (1959).
3) R. B. Woodward, K. Bloch, J. Amer.Chem.Soc., 75, 2023 (1953).
4)W.G.Dauben,S.Abraman,S.Hotta,I.L.Chaikoff,H.L.Bradlow,
A.H.Soloway, J. Amer.Chem.Soc., ft», 3038(1953).
5) R. B. Clayton, K. Bloch, J.Biol.Chem., £18, 305, 319 (1956).
6) T. T. Tchen, K. Bloch, J.Biol.Chem., 226, 921 (1957).
7) W. Voser, H. V. Mijovié, H. Heusser, O. Jeger, L. Ruzicka, Helv.,
35, 2414 (1952).
8) R. G. Langdon, K. Bloch, J.Biol. Chem., 200, 129, 135 (1953).
9)K.Bloch,A.Maugdal, unpublished work, CIBA Foundation Symposium
on the Biosynthesis of Terpenes and Sterols, 4 (1959).
10) K. Bloch, Vitamins and Hormones, lj), 119 (1957).
11) K. Bloch, N. H. Little, J.Biol.Chem., 162, 441 (1946).
12) L. Ruzicka, A. Eschenmoser, H. Heusser, Exper., £, 362 (1953).
13) A .E schenmoser
,L. Ruzicka, O. Jeger ,
D . Arigoni, Helv., 3Q, 1890
(1955).
14) L. Ruzicka in A. Todd, Perspectives in Organic Chemistry, 1956, 265.
15) D. Arigoni, Exp., 14, 153 (1958).
16) G. Gamboni.H.Schinz, Helv., 39, 1311 (1956).
17) D. Arigoni, CIBA Foundation Symposium on the Biosynthesis of Terpenes and
Sterols, 231 (1959).
18) K. Schaffner, L.Cagliotti.D. Arigoni, O. Jeger, Helv., 41_(1958).
19) J. J. Britt, D. Arigoni, Proc. Chem. Soc, 1958, 224.
20) G. Cainelli, J. J. Britt, D. Arigoni, O. Jeger, Helv., 41, 2053 (1958).
21) F. N. Lamey ,M. V. Leeping, Proc. Chem. Soc, 1958, 342.
22) D. H. Barton, K. Overton, J.Chem.Soc, 1955, 2639.
23)A.Mondon, Angew. Chem., 65, 333(1953).
24) E. Wenkert, Chem. Ind., 1955, 282.
25) G. Stork, A. W. Burgstahler ,J. Amer.Chem.Soc., T7> 5068(1955).
26) K. Bloch, D.Egreen, Currents in Biochem. Research, 1956, 474.
27)R.Helg,F.Zobrist, A.Lauchenauer, K.Brack,A.Caliezi,D.
Stauffacher,E.Zweifel,H.Schinz, Helv., 39, 1269 (1956).

- 58 -
28)C.Daesslé,H.Favre,H.Schinz, Helv., 40, 2270, 2278 (1957).
29) L.Ré.H.Schinz, Helv., 41, 1695,1717(1958).
30) G. Stork, H. Conroy ,J. Amer.Chem.Soc., 73, 4748 (1951).
31) G. Gamboni, H. Schinz, A. Eschenmoser, Helv., 37, 964 (1954).
32) R. Helg, H. Schinz, Helv., 35, 2406 (1952).
33)R.P.Linstead,A.B.L.Wang,J.H.Williams,K.D.Errington,
J.Chem.Soc, 1937, 1136.
34)R.P.Linstead,A.F.Millidge,A.L.Walpole, J.Chem.Soc, 1937,
1140.
35) P.A.Stadler,A.Nechvatal,A. J.Frey,A.Eschenmoser, Helv.,
40, 1373 (1957).
36) P. A. Stadler,Diss. ETH, Nr. 2687.
37) E. Y. Corey, Exper., 9, 329 (1953).
E.Y.Corey, J. Amer. Chem. Soc, 76, 175(1954).
38) G. H. Alt, D.H. R. Bar ton, J.Chem.Soc, 1954, 4284.
39) A.Eschenmoser,D.Felix,M. Gut, J. Meier, P. Stadler, CIBA
Foundation Symposium on the Biosynthesis of Terpenes and Sterols, 219 (1959).
40)R.P.Linstead,A.F.Millidge,A.L.Walpole, J.Chem.Soc, 1937,
1140.
D.C.Hibbit,R.P.Linstead, J.Chem.Soc, 1936, 470.
41) R. P. Linstead, A.B.L. Wang, J. H. Williams, K.D. Errington,
J.Chem.Soc, 1937, 1136.
42) G. Gamboni, Diss. ETH, Nr. 2554.
43) J.Meier, Diss. ETH, Nr. 2806.
44) A.Mondon, B., JÎ8, 724(1955).
45) A. Caliezi, H. Schinz, Helv., 32, 2556 (1949).
46) A. Eschenmossr,Habilitationsschrift ETH, 1956.
47)H.Stoll, Helv., ^8, 1587 (1955).
48) L.Ruzicka.C.F. Seidel, H. Schinz, C.H.Tavel, Helv., 31, 273
(1948).
C.F. Seidel, H. Schinz, L.Ruzicka, Helv., 32, 1739 (1949).
P. Bächli, C. F. Seidel, H. Schinz, L.Ruzicka, Helv., J32, 1744 (1949).
49) E.Lederer,F. Marx, D. Mercier, G. Perot, Helv., 29, 1354 (1946).
50) P.A.Stadler,A.Eschenmoser,H.Schinz,G.Stork, Helv., 40,
2191 (1957).

- 59 -
51) Palfray, Halasz, Rovira, Compt. rend., 210, 765 (1940).
Palfray, Sabetay, Bull. Soc. chim., [ö]4, 950(1937).
Hill, J.Amer.Chem.Soc, J50, 2678(1928).
52) A. S. Dreiding, J. A. Hartman, J. Amer.Chem.Soc., ^75, 939 (1953).
53) L. Ruzicka, G. Büchi, O. Jeger, Helv., jtt, 293 (1948).
54) L. Ruzicka, Proc. Chem. Soc, 1959, 344.

Curriculum vitae
Ich wurde am 25. Juni 1931 in Saint-Cloud bei Paris als Sohn des Walter Gut
und der Madeleine geb. Sicre geboren. Ich besuchte die Primarschule in verschie¬
denen Städten von Frankreich. Als meine Eltern im Jahre 1942 in die Schweiz, nach
Brig kamen, besuchte ich im dortigen Kollegium zuerst eine Spezialschule zum Er¬
lernen der deutschen Sprache, dann während eines Jahres das Progymnasium.
Meine Eltern übersiedelten im Jahre 1945 nach Zürich und ich trat dort in die
erste Klasse des kantonalen Realgymnasiums ein, wo ich im Herbst 1951 die Maturi¬
tätsprüfung (Typus B) bestand. Anschliessend immatrikulierte ich mich an der Abtei¬
lung für Chemie der Eidgenössischen Technischen Hochschule in Zürich und schloss
im August 1956 das Studium mit dem Diplom als Ingenieur-Chemiker ab. Seither ar¬
beitete ich unter LeitungvonHerrn Prof. Dr. L.Ruzicka, Herrn Prof. Dr. V.Pre-
log und Herrn Prof. Dr. A. Eschenmoser unter anderem an der vorliegenden
Promotionsarbeit.
Zürich, im Juli 1959 Michel Gut





























