Embed Size (px)
Citation preview

Review: An update on inflammatory andautoimmune myopathies_ 226..242
M. C. Dalakas*†
*Department of Neurosciences, Imperial College, London, UK, and †Department of Neurology Thomas JeffersonUniversity, Philadelphia, PA, USA
M. C. Dalakas (2011) Neuropathology and Applied Neurobiology 37, 226–242An update on inflammatory and autoimmune myopathies
The review provides an update on the diagnosis of themain subtypes of inflammatory myopathies includingdermatomyositis (DM), polymyositis (PM), necrotizingautoimmune myositis (NAM) and sporadic inclusion bodymyositis (sIBM). The fundamental aspects on musclepathology and the unique pathomechanisms of eachsubset are outlined and the diagnostic dilemmas concern-ing the distinction of PM from sIBM and NAM areaddressed. Dermatomyositis is a complement-mediatedmicroangiopathy leading to destruction of capillaries,hypoperfusion and inflammatory cell stress on the perifas-cicular regions. NAM, is an increasingly recognizedsubacute myopathy triggered by statins, viral infections,
cancer or autoimmuity with macrophages as the finaleffector cells causing fibre injury. In PM and sIBM cyto-toxic CD8-positive T cells clonally expand in situ andinvade major histocompatibility-I-expressing musclefibres. The pathology of sporadic inclusion body myositis iscomplex because, in addition to the inflammatory mecha-nisms, there are degenerative features characterized byvacuolization and the accumulation of stressor andamyloid-related misfolded proteins. Inducible pro-inflammatory molecules, such as interleukin 1-b, mayenhance the accumulation of stressor proteins. Theprinciples for more effective treatment strategies arediscussed.
Keywords: dermatomyositis, inclusion body myositis, muscle immunopathology, necrotizing myositis, polymyositis
Introduction
Inflammatory myopathies (IM) constitute a heteroge-neous group of subacute, chronic or sometimes acuteacquired muscle diseases, which have in common thepresence of moderate to severe muscle weakness andinflammation on muscle biopsy [1,2]. Because these dis-eases represent the largest group of acquired and poten-tially treatable myopathies both in children and adults,early recognition is clinically important [1–8].
Based on distinct clinical, immunopathological, histo-logical and prognostic criteria, as well as different degree ofresponse to therapies, the most common IM seen in prac-
tice can be separated into four distinct subsets: polymyosi-tis (PM), dermatomyositis (DM), necrotizing autoimmunemyositis (NAM) and sporadic inclusion body myositis(sIBM) [7,8]. The disorders have primarily an autoimmunepathogenesis, mediated either by cytotoxic T cells, as in PMand sIBM, by a complement-mediated microangiopathy asin DM, or by macrophages and possibly autoantibodies asin NAM [1–8]. In sIBM, the autoimmune features coexistwith degenerative, highlighted by vacuolization andintrafibre deposition of amyloid and related moleculessimilar to those seen in Alzheimer’s disease; as a result,sIBM is a complex but fascinating disorder with a uniquecombination of autoimmune inflammation and degenera-tion [9]. This review is aimed to provide a balanced updateon the main clinicopathologic features of IM includingfundamental aspects on disease mechanisms, immunopa-thology and response to therapies.
Correspondence: Marinos C. Dalakas, Director NeuromuscularDivision, Thomas Jefferson University, 900 Walnut street, suite200, Philadelphia, PA 19107, USA. Tel: +1 215 955 7952; Fax:+1 215 955 9976; E-mail: [email protected]
226 © 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society
Neuropathology and Applied Neurobiology (2011), 37, 226–242 doi: 10.1111/j.1365-2990.2010.01153.x

Main clinical features
Dermatomyositis affects both children and adults, andwomen more often than men, whereas PM is seen after thesecond decade of life and very rarely in childhood. sIBM ismore frequent in men than women and more likely to affectpersons over the age of 50 years comprising the most com-monly acquired myopathy in this age group [10,11]; in con-trast, PM is the least common while DM is the most frequentIM in children. DM differs from the others because of adistinct red or heliotrope (blue-purple discoloration) rashaccompanying or, more often, preceding muscle weakness.The rash is most prominent on the upper eyelids, face,upper trunk, knuckles and sometimes knees, elbows, ante-rior chest (often in a V sign) or shoulders (shawl sign) [1,2].
All forms have in common a myopathy characterized bymuscle weakness, which usually develops subacutely (weeksto months, as in PM and DM); acutely, even in days, as inNAM; or insidiously over years, as in sIBM, simulating thetempo of a limb girdle muscular dystrophy leading eventu-ally to wheelchair confinement. The patients have difficultyperforming tasks requiring the use of proximal muscles,such as getting up from a chair, climbing steps or liftingobjects; sIBM patients however may experience early diffi-culties with distal muscles such as inability to hold certainobjects, turning on keys, shaking hands or buttoning a shirt.Falling is common among sIBM patients because of earlyinvolvement of the quadriceps muscle that causes bucklingof the knees and weakness of foot extensors [1–3,9–11].Facial muscles are affected mostly in sIBM but rarely in theother forms. The pharyngeal and neck flexor muscles areoften involved, but more severely in sIBM, causing dysph-agia and difficulty in holding up the head. In advanceddisease, respiratory muscles may also be affected, espe-cially in NAM and aggressive forms of DM or PM. Severeweakness is almost always associated with muscularwasting. Sensation remains normal. The tendon reflexesare preserved but may be absent in severely weakened oratrophied muscles, especially in sIBM, where atrophy ofthe quadriceps and the distal muscle is common [1–5].
All forms of myositis can be seen with increased fre-quency in patients with other systemic autoimmune, viralor connective tissue diseases. Up to 10% of patients haveinterstitial lung disease and anti-Jo-1 antibodies, directedagainst histidyl-transfer RNA synthetase; the presence ofanti-Jo-1 antibodies denotes a high probability of havingor developing interstitial lung disease. In sIBM, the coex-istence of another autoimmune disease or autoantibodies
can be seen in up to 33% of the patients, while 75% ofthem have the HLA-B8-DR3 haplotype and increasedfrequency of DRb10301 and DQb10201 alleles, similarto those seen in other autoimmune diseases [12–14].Familial aggregation of sIBM with the typical clinicalphenotype of sIBM, and with histological and immunopa-thological features identical to the sporadic form has beenobserved, a phenomenon also seen in other autoimmunedisorders [15]. An increased incidence of malignancies isseen in patients with DM (particularly over 50 years ofage), but not in PM or sIBM, requiring frequent malignancywork-up and vigilance at least for the first 3 years [1–6].
Diagnosis
The diagnosis of DM is relatively easy when the typicalskin changes are apparent. The diagnosis of PM or NAM isone of exclusion and based on finding a steadily progres-sive myopathy of acute or subacute onset, in adults whodo not have a rash, involvement of the extraocular andfacial muscles, family history of a neuromuscular disease,history of exposure to myotoxic drugs or toxins, endo-crinopathy, neurogenic disease, dystrophy, biochemicalmuscle disorder or sIBM, as determined by muscle pathol-ogy and histochemistry [1–7]. The diagnosis of sIBMshould be considered in an adult who has slow onsetdisease that involves distal muscles, especially foot exten-sors and deep finger flexors, and should be always sus-pected when a patient with presumed PM did not respondto therapy [1–11,16].
The diagnosis is established or confirmed by elevatedlevels of serum muscle enzymes, electromyographic find-ings and, definitively, by the muscle biopsy. The serum CK,in the presence of active disease, can be elevated by asmuch as 50 times above normal and usually parallelsdisease activity; it can be however normal in active DMand sIBM. In NAM, the CK is very high, usually around8–15 000. Along with the serum CK, aldolase, glutamateoxaloacetate transaminase, glutamate pyruvate tran-saminase and lactate dehydrogenase may be elevatedleading sometimes to an erroneous interpretation ofliver disease. Needle electromyography shows myopathicmotor unit potentials characterized by short-duration,low-amplitude polyphasic units on voluntary activation,and increased spontaneous activity with fibrillations,complex repetitive discharges and positive sharp waves[1,2]. This electrophysiological pattern is not specific asit also occurs in a variety of acute, toxic and active
Inflammatory myopathies 227
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

myopathic processes. For definitive diagnosis, a musclebiopsy is essential as detailed below.
Diagnostic muscle biopsy
The following principles on muscle pathology are funda-mental for maximum diagnostic information [4,17].
Proper choice of biopsied muscle A clinically moderatelyweak muscle offers the best chance for a positive biopsy.Magnetic resonance imaging may be at times helpful.Open biopsy, as opposed to needle biopsy, is preferablebecause it offers a larger sample, better suited for findinginflammation considering the sampling variability andthe multifocality of the inflammatory pattern.
Proper processing of muscle The biopsy specimens shouldbe used for cryostat sections that allow enzymatic his-tochemical stains and standardized immunocytochemistryto search for major histocompatibility complex-I (MHC-I) orcomplement and identify lymphocytic subsets or aberrantlyaccumulated molecules. Electron microscopic studies arenot routinely required, but in certain circumstances, or forresearch purposes, can be problem-solving.
Proper interpretation When the typical features of PM,DM, NAM or sIBM are present, there are no diagnosticconcerns. Difficulties arise when the typical changes fallshort of being pathognomonic, requiring clinicopatho-logic correlations, special expertise or even a repeat biopsy.
Specific histological findings
The muscle biopsy shows features distinct for each subsetand remains the most sensitive diagnostic tool.
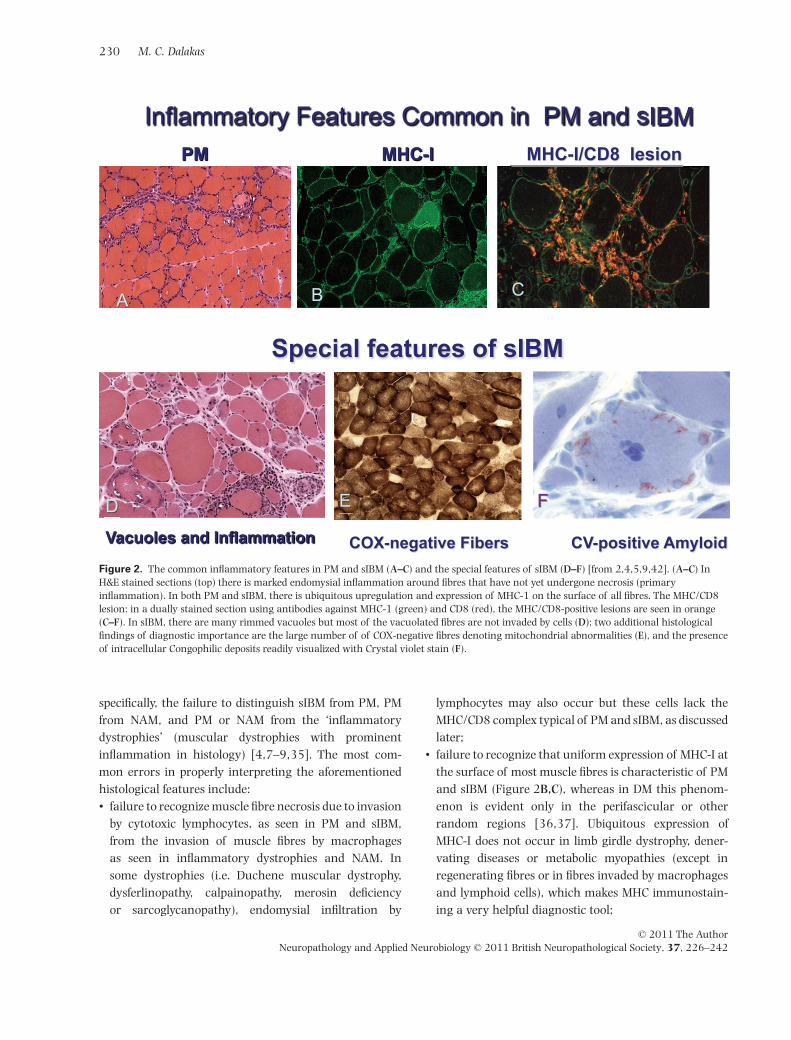
In DM the inflammation is perivascular or at theperiphery of the fascicle and is often associated withperifascicular atrophy (Figure 1A) [1–7]. The presence ofperifascicular atrophy, even in the absence of inflamma-tion, should raise the suspicion of DM. In NAM, there arenecrotic fibres invaded by macrophages; T cells are char-acteristically absent (Figure 1D–F); MHC-I is not upregu-lated (except in necrotic fibres) although overexpressionof MHC-I has been reported in statin-induced NAM [18];in some cases, there may be complement deposits onthickened vessels, as discussed later. In PM and sIBM, theinflammation is in multiple foci within the endomysialparenchyma (Figure 2A–C) and consists predominantly ofCD8+ T cells that invade healthy muscle fibres expressing
the MHC-I antigen, which is ubiquitously upregulated onthe surface of most fibres (Figure 2B,C). The MHC/DC8complex is characteristic of PM and sIBM (Figure 2C)[2,5–9]. In sIBM, in addition to inflammation, there arerimmed-vacuoles (Figure 2D) and tiny amyloid depositsin a variable number of fibres, usually in or near thevacuoles, identified with Congo-red or Crystal violet stains(Figure 2D–F) [4,9], but easily visualized with Texas-redfluorescent optics [19]. The vacuoles contain 12–16 nmfilamentous masses, reported to be identical to the pairedhelical filaments found in the brains of Alzheimer’sdisease [20–22]. The blue granules lining the vacuoles,correspond to whorls of cytomembranes or myelin figures,detectable by electron microscopy [17]. Ragged-red fibreswith mitochondrial excess and multiple mitochondrialDNA deletions, as well as cytochrome oxidase-negativefibres (Figure 2E), are frequent in most examined cases[9,16]. A number of degeneration-associated and stressormolecules accumulate within the myofibres of sIBMmuscles, including beta-amyloid and its precursor protein,alpha-chymotrypsin, phosphorylated tau, best detectedwith antibodies to SMI-31 or the transporter protein p62/SQSTMI, ubiquitin, apolipoprotein E, prion protein andothers [20–22]. These accumulations, which also includea number of nuclear-related proteins, such as TDP-43 andVCP, do not appear specific for sIBM because they are alsofound in other vacuolar myopathies especially myofibril-lar, hereditary sIBM or even in chronic neurogenic condi-tions such as the postpolio syndrome [23–29]. Currently,there is no one molecule that could serve as a biomarkerunique for sIBM. The noted immunoreactivity for TDP-43and SMI-31 [30,31] that identify phosphorylated tau arenot sIBM-specific [7,8,29]; the recently reported immu-noreactivity for p62 [22] is interesting and seems morespecific than SMI-31, but it has not yet been tested inother vacuolar or myofibrillar myopathies. A diagnosticmarker is not necessary for the histologically typicalsIBM but urgently needed for the cases of probable sIBMthat lack vacuoles and Congophilic amyloid deposits, asdiscussed below, and for distinguishing sIBM from non-inflammatory vacuolar myopathies.
Dilemmas in the histological diagnosis ofPM and sIBM
The combination of endomysial inflammation with red-rimmed vacuoles, the ubiquitous MHC-I expression withCD8+ cells along with the presence of COX-negative fibres
228 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

and Congophilic deposits, are diagnostic of sIBM [9]. Diag-nostic concerns arise in patients who have the distinctclinical phenotype of sIBM but their biopsy does not showvacuoles but only inflammation characterized by MHC-Iexpression and CD8+ cells. Such cases labelled ‘PM/sIBM’,‘probable sIBM’ or ‘clinical sIBM’ comprise up to 15% ofsIBM patients [32] and dictate close clinicopathologic cor-relations to avoid the misdiagnosis of PM; a careful view ofthese biopsies, however, shows a large number of COX-negative fibres (Figure 2E) and signs of chronicity (largefibres, splitting, increase connective tissue) that denoteprobable sIBM. A repeat biopsy from another site is oftenhelpful in an effort to unravel the presence of vacuoles andCongo red-positive deposits. The aforementioned histo-biomarkers proposed for the typical sIBM have not been
tested in such cases. Two other distal myopathies that maybe confused with sIBM, are the sporadic form of ‘myofibril-lar myopathy’ [33] and the hereditary sIBM due to GNEmutations [34]. The biopsy in these cases lacks markersof immune inflammation (MHC/CD8) but demonstratevacuoles with all the stressor-related molecules seen insIBM. These cases require high degree of suspicion, carefulsearch for desmin and other myofibrillar deposits, clinico-pathologic correlations and molecular genetic studies.
Pitfalls in muscle histology leadingto misdiagnosis
The most common cause of a clinical misdiagnosis of IMis an erroneous interpretation of the biopsy and, more
Figure 1. The main histopathologic features of DM and NAM. (A–C) Dermatomyositis. (A) Prominent perifascicular atrophy with scatteredperivascular inflammation, are characteristic of DM (H&E.). (B) Imunostaining with the lectin Ulex Europoeaus, which identifies endothelialcell, shows marked reduction of capillaries with dilatation of the lumen of the remaining capillaries in DM compared with a myopathiccontrol muscle (B,C) [from 1,2,4,42]. (D–F) Necrotizing myositis. Trichrome stain of cross and longitudinal sections show necrotic fibresinvaded by macrophages (D,E), best visualized with Acid phosphatase reaction (F). Note the total absence of T cells.
Inflammatory myopathies 229
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242
specifically, the failure to distinguish sIBM from PM, PMfrom NAM, and PM or NAM from the ‘inflammatorydystrophies’ (muscular dystrophies with prominentinflammation in histology) [4,7–9,35]. The most com-mon errors in properly interpreting the aforementionedhistological features include:• failure to recognize muscle fibre necrosis due to invasion
by cytotoxic lymphocytes, as seen in PM and sIBM,from the invasion of muscle fibres by macrophagesas seen in inflammatory dystrophies and NAM. Insome dystrophies (i.e. Duchene muscular dystrophy,dysferlinopathy, calpainopathy, merosin deficiencyor sarcoglycanopathy), endomysial infiltration by
lymphocytes may also occur but these cells lack theMHC/CD8 complex typical of PM and sIBM, as discussedlater;
• failure to recognize that uniform expression of MHC-I atthe surface of most muscle fibres is characteristic of PMand sIBM (Figure 2B,C), whereas in DM this phenom-enon is evident only in the perifascicular or otherrandom regions [36,37]. Ubiquitous expression ofMHC-I does not occur in limb girdle dystrophy, dener-vating diseases or metabolic myopathies (except inregenerating fibres or in fibres invaded by macrophagesand lymphoid cells), which makes MHC immunostain-ing a very helpful diagnostic tool;
Figure 2. The common inflammatory features in PM and sIBM (A–C) and the special features of sIBM (D–F) [from 2,4,5,9,42]. (A–C) InH&E stained sections (top) there is marked endomysial inflammation around fibres that have not yet undergone necrosis (primaryinflammation). In both PM and sIBM, there is ubiquitous upregulation and expression of MHC-1 on the surface of all fibres. The MHC/CD8lesion: in a dually stained section using antibodies against MHC-1 (green) and CD8 (red), the MHC/CD8-positive lesions are seen in orange(C–F). In sIBM, there are many rimmed vacuoles but most of the vacuolated fibres are not invaded by cells (D); two additional histologicalfindings of diagnostic importance are the large number of of COX-negative fibres denoting mitochondrial abnormalities (E), and the presenceof intracellular Congophilic deposits readily visualized with Crystal violet stain (F).
230 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

• failure to recognize that the pathological involvementmay be spotty and a given biopsy may not contain con-vincing pathological changes (‘skip areas’) necessitatingat times the need for a repeat biopsy [4];
• failure to realize that changes typical of perifascicularatrophy are diagnostic of DM, even if there are noinflammatory cell infiltrates; the lack of inflammatorycell infiltrates in these cases may lead to the conclusionof ‘non-specific abnormalities’;
• failure to recognize that up to 15% of biopsies frompatients with typical clinical features of sIBM demon-strate only the inflammatory pattern seen in PM,without vacuoles or amyloid deposits. As mentionedabove, these patients do not have PM but probably sIBM,often referred to as ‘PM/sIBM’, ‘clinical sIBM’ or ‘prob-able sIBM’ [32];
• failure to assess blood vessel pathology or, if needed,seek for complement deposits that can be useful in somecases of DM and NAM; and
• failure to appreciate the presence of other inflammatorycells that might be diagnostic for certain disorders. Forexample, multinucleated giant cells among elongatedinflammatory (‘epithelioid’) cells, macrophages orlymphocytes are characteristic of granulomatousmyopathy. Prominent presence of eosinophilic polymor-phonuclear leucocytes in muscle (or fascia) can occurwith systemic hypereosinophilic syndrome, systemiceosinophilia, due to parasitic infection, vasculitis, toxicfactors or eosinophilic myositis [4]. Certain patientswith genetically defined calpain-gene mutations haveeosinophilic infiltrates in their muscle [38].
Aetiology and pathogenesis
Immunopathology of DM
The primary antigenic target in DM is the vascular endot-helium of the endomysial capillaries and to a lesser extentof larger blood vessels. The disease begins when putativeantibodies directed against endothelial cells activatecomplement C3 that subsequently forms C3b and C4bfragments and lead to the formation of C5b-9 (MAC), thelytic componement of the complement pathway [39,40].MAC, C3b and C4b are detected early in the patients’serum [41] and deposited on capillaries before inflam-matory or structural changes are seen in the muscle(Figure 3A). Sequentially, the complement-mediatedalterations begin with swollen endothelial cells followed
by vacuolization and necrosis of capillaries, perivascularinflammation and ischaemic muscle fibre damage [1–8].The characteristic perifascicular atrophy is probably areflection of the normally occurring relative hypoperfu-sion in the perifascicular zones [1–8]. Finally, thereis marked reduction in the number of capillaries permuscle fibre with compensatory dilatation of the lumen ofthe remaining capillaries [1–7,35,42] (Figures 1B,C and3A). The release of cytokines and chemokines related tocomplement activation, upregulate VCAM-I and ICAM-Ion the endothelial cells [43–48], and facilitate theirexit through the blood vessel wall to the perimysialand endomysial spaces (Figure 3A). Immunophenotypicanalysis of the lymphocytic infiltrates demonstrates B cellsand CD4+ cells in the perimysial and perivascular regionsand plasmacytoid dendritic cells in the perifasciscularregions supporting the view that a humoral mediatedmechanism plays the major role in the disease. The peri-fascicular regions of DM contain many regenerating ordegenerating fibres as they are in a stage of continuousremodelling. As a result, they stain with alkaline phos-phatase, desmin, NCAM, with the autoantibody againstchromatin remodeler Mi-2, as shown recently [49], andwith a variety of antibodies against immune or stressormolecules, including transforming growth factor (TGF)-b, MHC-I, aB-crystallin, cathepsins, amyloid precursorprotein, STAT-1 (triggered by interferon-g) or myxovirusresistance MxA protein (triggered by a/b-interferon)[50,51] (Figure 3A). The proposed theory that the perifas-cicular myofibres may be primarily injured by chronicoverproduction of a/b-interferon-inducible proteinssuch as MxA [51] because they stain with this protein is,accordingly, not pertinent as the perifascicular fibresdisplay not only MxA but all the markers of regenerationand tissue remodelling. Most importantly, such a theorydoes not explain the reduced number of capillariesthroughout the fascicle that occurs very early in thedisease process, or the early activation and deposition ofMAC on the capillaries that precede perifascicular atrophy.Further, a/b-interferon-inducible genes lack specificity oruniqueness for DM because they are also overexpressed inPM [52] and other connective tissue diseases; additionally,there is no evidence that a/b-interferon molecules exerttoxicity to human muscle fibres in vitro.
Using gene arrays, a number of adhesion molecules,cytokines and chemokine genes as well as genes associ-ated with ischaemia and degeneration have been foundupregulated in the muscles of DM patients [53,54] A
Inflammatory myopathies 231
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242
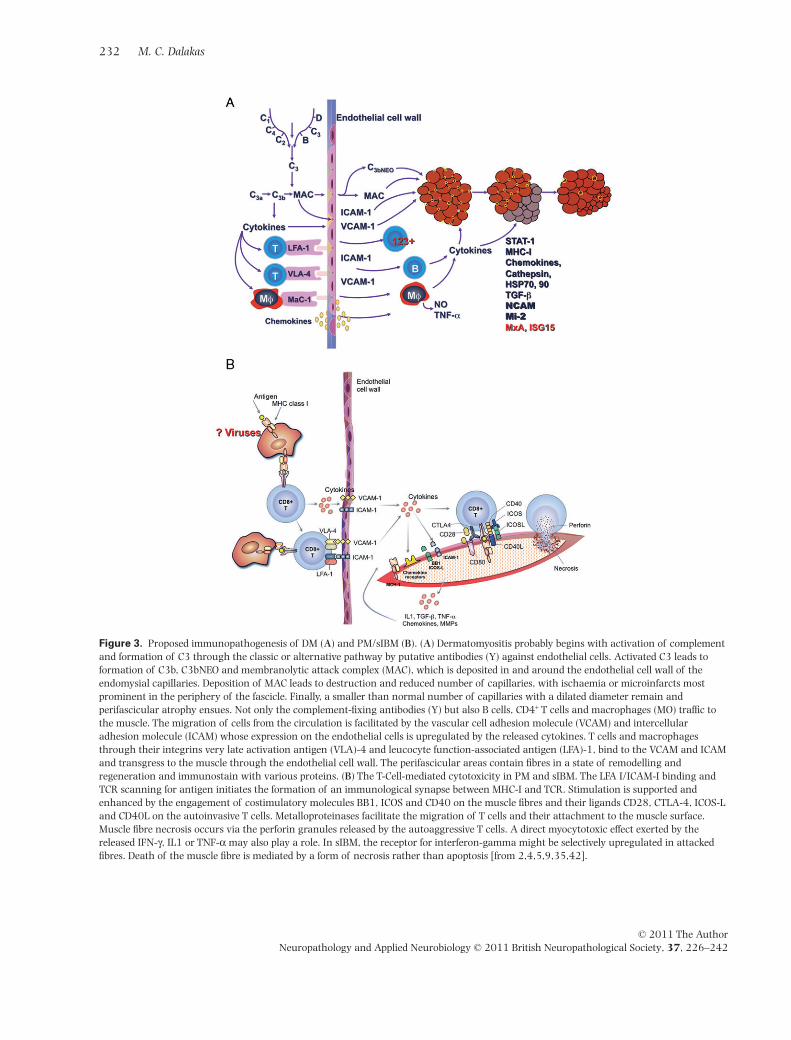
Figure 3. Proposed immunopathogenesis of DM (A) and PM/sIBM (B). (A) Dermatomyositis probably begins with activation of complementand formation of C3 through the classic or alternative pathway by putative antibodies (Y) against endothelial cells. Activated C3 leads toformation of C3b, C3bNEO and membranolytic attack complex (MAC), which is deposited in and around the endothelial cell wall of theendomysial capillaries. Deposition of MAC leads to destruction and reduced number of capillaries, with ischaemia or microinfarcts mostprominent in the periphery of the fascicle. Finally, a smaller than normal number of capillaries with a dilated diameter remain andperifascicular atrophy ensues. Not only the complement-fixing antibodies (Y) but also B cells, CD4+ T cells and macrophages (MO) traffic tothe muscle. The migration of cells from the circulation is facilitated by the vascular cell adhesion molecule (VCAM) and intercellularadhesion molecule (ICAM) whose expression on the endothelial cells is upregulated by the released cytokines. T cells and macrophagesthrough their integrins very late activation antigen (VLA)-4 and leucocyte function-associated antigen (LFA)-1, bind to the VCAM and ICAMand transgress to the muscle through the endothelial cell wall. The perifascicular areas contain fibres in a state of remodelling andregeneration and immunostain with various proteins. (B) The T-Cell-mediated cytotoxicity in PM and sIBM. The LFA I/ICAM-I binding andTCR scanning for antigen initiates the formation of an immunological synapse between MHC-I and TCR. Stimulation is supported andenhanced by the engagement of costimulatory molecules BB1, ICOS and CD40 on the muscle fibres and their ligands CD28, CTLA-4, ICOS-Land CD40L on the autoinvasive T cells. Metalloproteinases facilitate the migration of T cells and their attachment to the muscle surface.Muscle fibre necrosis occurs via the perforin granules released by the autoaggressive T cells. A direct myocytotoxic effect exerted by thereleased IFN-g, IL1 or TNF-a may also play a role. In sIBM, the receptor for interferon-gamma might be selectively upregulated in attackedfibres. Death of the muscle fibre is mediated by a form of necrosis rather than apoptosis [from 2,4,5,9,35,42].
232 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

biologically relevant protein, however, discovered by genearrays, is the KAL-1 adhesion molecule because it is sig-nificantly downregulated in DM patients who improvedafter therapy [54]. The KAL-I is upregulated in vitro byTGF-b and may have a role in inducing fibrosis [54,55]. Itwas suggested that in childhood DM, the upregulation ofa/b and g interferon-inducible genes might be a sign of avirus-driven autoimmune dysregulation [53]; however,no viruses have been amplified from these patients’muscles [56,57].
Immunopathology of PM and sIBM
In PM and sIBM there is evidence of an antigen-directedcytotoxicity mediated by CD8+ T cells invading MHC-I-antigen expressing muscle fibres [36,37,58–63]. Theimmune components associated with this process areidentical in both PM and sIBM, in spite of poor response toimmunotherapies of the latter, and include the following(depicted in Figure 3B):
Activated, cytotoxic CD8+ T cells• The T cells invading muscle fibres are activated express-
ing ICAM-I, MHC-I, CD45RO and inducible costimulator(ICOS) on their surface [3,4,64,65], suggesting anactive participation, rather than a bystander effect. InsIBM, the T-cell invasion of non-necrotic fibres is foundearly and in higher frequency than the Congo-red-positive fibres [66], suggesting that inflammationprecedes the accumulation of amyloid and stressormolecules.
• The CD8+ cells surround healthy, but MHC-I-classexpressing, non-necrotic muscle fibres that eventuallyinvade. The CD8/MHC-I lesion is characteristic of sIBMand PM [2,4,46,62,63] because it does not occur ininflammatory dystrophies or non-immune myopathies[67].
• By immunoelectron microscopy, CD8+ cells and mac-rophages send spike-like processes into non-necroticmuscle fibres, which traverse the basal lamina andfocally displace or compress the muscle fibres [3,58–61].
• The CD8+ T cells, contain perforin and granzyme gran-ules, which are vectorially directed towards the surfaceof the muscle fibre inducing necrosis upon release;further, the perforin-positive CD8+ T cells are autoinva-sive and express costimulatory molecules [65,67,68].These cells are also cytotoxic in vitro when exposed to
autologous myotubes [69]. Consequently, the majorcytotoxic effector mechanism in PM and sIBM is the per-forin pathway; in contrast, the Fas-Fas-L-dependentapoptotic process is not functionally involved [70,71].
Formation of Immunological synapses between the MHC-I-expressing muscle fibres and cytotoxic CD8+ T cells Musclefibres normally do not express detectable amounts of MHCclass I or II antigens. In PM and sIBM however, widespreadoverexpression of MHC is an early event that can be seeneven in areas remote from the inflammation [36,37](Figure 2B,C). Because the MHC-I is induced in vitro bycytokines and chemokines such as IFN-g or TNF-a[72–74], and several of these cytokines are increased inPM and sIBM muscles [44,45,48,72–77], it is likely thatthe upregulation of MHC-I is related to continuous over-expression of cytokines. In other chronic myopathiesincluding the inflammatory dystrophies, and in contrastto PM and sIBM, the muscle fibres do not express theMHC-I antigen or the costimulatory molecules in a ubiq-uitous and consistent pattern, while the few T cells foundin the proximity to the muscle fibres do not release cyto-toxic granules, as in PM and sIBM [67].
The MHC-I-expressing muscle fibres form immunologi-cal synapses with the CD8+ cells, as supported by thefollowing:• Rearrangement of the TCR gene of the endomysial T cells.
The T cells recognize an antigen via the T cell receptors(TCRs), a heterodimer of two a and b chains; the part ofthe TCR that recognizes an antigen is the CDR3 region,which is encoded by specific genes. In patients with PMand sIBM, but not in those with DM or dystrophies, onlycertain T cells of specific TCRa and TCRb families arerecruited to the muscle from the circulation [78–80]Cloning and sequencing of the amplified endomysial orautoinvasive TCR gene families has demonstrated arestricted use of certain gene families with conservedamino acid sequence in the CDR3 region indicating thatthese cells are specifically selected by antigens andclonally expanded in situ probably driven by local anti-gen(s) [78–82]. This conclusion has been further con-firmed by combining spectratyping with molecularlaser-assisted microdissection and by demonstratingthat the clonally expanded TCR families persist over timeeven in different muscles [80–86]. In an important caseof PM, a single clone of g/d T cells was the primarycytotoxic effectors that recognized genuine muscle anti-gens [87–89].
Inflammatory myopathies 233
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

• Presence of Costimulatory molecules for synapseformation.The muscle fibres can present antigenic peptides toCD8+ cells via the MHC-I molecule, thereby serving asantigen-presenting cells, because they possess the B7family of costimulatory molecules [B7-1, B7-2, BB1 orICOS-L (inducible-costimulator-ligand)] and bind totheir respective counter-receptors CD28, CTLA-4 orICOS on the autoinvasive CD8+ T cells [63,64,90](Figure 3B). Because both BB1 and ICOS-L are func-tional molecules induced by interferon-g or TNF-a onhuman myoblasts, the BB1/CD28 and ICOS/ICOS-Linteractions in PM and sIBM would participate inantigen presentation, clonal expansion and costimula-tion of T cells. The upregulation of these moleculesoffers the potential for therapeutic manipulationsbecause of the availability of humanized monoclonalantibodies against CD28, CTLA4 or CD40L.
• Upregulation of cytokines, cytokine signalling, chemok-ines and metalloproteinases.Cytokines and chemokines are essential in enhancingT-cell activation and inducting costimulatory molecules.Various cytokines, including interleukins (IL-1, IL-2,IL-6 and IL-10), TNF-a, INF-g, signal transducer andactivation of transcription (STAT), TGF-b, and chemok-ines such as MCP-1, MIP-1a, IP-10, Mig and its receptorCXCR3, are variably overexpressed on the muscle fibresand on some autoinvasive CD8+ cells [44,45,47,48,72–77]. The muscle, in vivo and in vitro, has the potentialto secrete pro-inflammatory cytokines upon cytokinestimulation, such as INF-g, in an auto-amplificatorymechanism that may facilitate the recruitment of acti-vated T cells to the muscle contributing to the self-sustaining nature of endomysial inflammation anddisease chronicity [44,45,48,72–77,91] (Figure 3B).Various adhesion and extracellular matrix molecules
such as VCAM, ICAM and thrombospondins are alsoupregulated in the tissues of patients with PM and sIBM[92,93] and may enhance the inflammatory response.The metalloproteinases MMP-9 and MMP-2 are also over-expressed playing a role in T-cell adhesion, transmigrationand cytotoxicity [92].
Other immune cells support the autoimmune process in PMand sIBM Myeloid dendritic cells, potent cells in antigenpresentation, are abundantly found within the endomy-sial infiltrates of all IM including PM, DM and sIBM[94,95]. These cells may present local antigens to T cells
and B cells and contribute to muscle fibre injury. A largenumber of plasma cells and clonally expanded B cells arealso found within the sIBM, PM and DM muscles [95].Their presence, frequently noted in the targeted tissues inseveral autoimmune disorders, simply denotes that differ-ent effector mechanisms of T cells and B cells concurrentlyplay an active role in the autoimmune process. Thesuggestion of in situ production of autoantibodies [95]remains unproven.
Viral triggers Although several viruses have been impli-cated in chronic and acute myositis [96,97], sensitive PCRstudies have repeatedly failed to confirm the presence ofsuch viruses in these patients’ muscle [56,98], suggestingthat it is unlikely, although not impossible, for viruses toreplicate in the muscles of patients with PM, DM andsIBM. The best evidence of a viral connection is with theretroviruses. Monkeys infected with the simian immuno-deficiency virus and humans infected with HIV andhuman T-cell lymphotropic virus develop PM or sIBM[99–109]. In HIV- or human T-cell lymphotropic virus-1-infected individuals, an IM may occur as the initial mani-festation of the infection or later in the disease course[100–109]. In these patients, sIBM has been observed in alarge number of patients before the age of 50 years, butseveral years after the first manifestations of the retroviralinfection, suggesting that this association is not fortu-itous. The predominant endomysial cells are CD8+ cyto-toxic T cells which, along with macrophages, invade orsurround MHC-I-antigen expressing non-necrotic musclefibres [105–109]. Viral antigens are not detected withinthe muscle fibres but only in occasional endomysial mac-rophages. Molecular immunology studies using tetramershave shown that among the autoinvasive T cells there areretrovirally specific CD8+ cells that clonally expand in situ[108,109]. Collectively, in retrovirally associated sIBMthere is no evidence of viral replication within the muscle,but the chronic infection triggers an situ persistent inflam-matory response with viral-specific T cells that change thelocal milieu leading finally to sIBM.
Degenerative pathomechanisms in sIBM
Inclusion body myositis is a complex disorder because inaddition to the immunopathogenic events describedabove, there is an equally strong degenerative process asevidenced by the presence of rimmed vacuoles (almostalways in fibres not invaded by T cells (Figure 2D)), and
234 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

intracellular deposition of b-amyloid and related mol-ecules, similar to those seen in neurodegenerative diseases[21]. These proteins can be found inside of inclusions/vacuoles or free within the cytoplasm. One line of evi-dence suggests that the proteasome machinery ismalfunctioning or overloaded with aberrant proteins,which may explain why these proteins accumulate in thecytoplasm of muscle fibres [110,111]. The b-secretase, amajor enzyme relevant for processing of APP, has beenoverexpressed in sIBM muscle fibres [27,111], and mayexplain why processing of APP may be shifted towards thegeneration of b-amyloid. Apart from direct toxicity ofmonomers or oligomers of aberrant proteins, generationof free radicals like NO and other members relevant foroxidative stress, could mediate cell damage. Autophagicprocessing is, apart from the ubiquitin/proteasomesystem, a major mechanism relevant to degradation ofintracellular proteins. The vacuoles have autophagicproperties [112] while the light chain protein 3, a majormarker of autophagy, is present in intracellular vacuolesand co-localize with APP/b-amyloid [112] suggestingthat autophagic processing of APP/b-amyloid is relevantto sIBM pathology. However, it is unclear whether thisautophagic activity is beneficial or detrimental for musclefibres as it can be associated with a cell death programme.Because lysosomal processing of peptides can lead toantigen presentation via MHC-2 [113], macroautophagymay present a direct association to inflammatory mecha-nisms and be relevant to antigen presentation of immunecells that attack the muscle fibres. The contention that therimmed vacuoles are a consequence of the myonuclearbreak down based partly on the observation that theyexpress myonuclear molecules [30,31] lacks any mecha-nistic support and remains largely theoretical.
Amyloid and degeneration-related molecules insIBM: an interrelationship with inflammation
The aforementioned accumulations, although extensivelystudied in sIBM, do not seem to be unique to this disease,because they are also observed in myofibrillar and othervacuolar myopathies [24–29]. What appears unique tosIBM however, compared with other chronic vacuolarmyopathies, is the concomitant accumulation of thesemolecules with a strong primary inflammatory responseand the overexpression of pro-inflammatory mediatorsand MHC-I on all fibres, vacuolated or not. Regardless ofwhether in sIBM the primary event is an inflammatory or
protein dysregulation process, the unique coexistence ofthe two processes and co-localization of APP with cytok-ines (Figure 4A), has led our laboratory to explore aninterrelationship between inflammation and degenerationto understand what drives each process and design tar-geted therapeutic strategies.
Inflammation may trigger or enhance accumulation ofb-amyloid The main inflammatory molecules CCL-3,CCL-4, CXCL-9, IFN-g and IL-1b are expressed to a higherdegree in sIBM muscle compared with PM or DM [91]. InsIBM, but not in PM or DM, there is also a significantcorrelation between the mRNA expression of APP, as a keyrelevant degenerative marker, with the inflammatorymediators IFN-g and CXCL-9, which also co-localize withAPP/b-amyloid proteins [91,114]. Further, exposure ofmuscle cells to pro-inflammatory cytokines IL-1b andIFN-g induces an overexpression of APP with subsequentaccumulation of protein aggregates. It is possible that acontinuous stimulation of inflammatory factors may, aftera long period induce a higher basal expression of APP andan increased sensitivity to de novo pro-inflammatory cytok-ines that trigger a self-perpetuating cycle [91,114,115](Figure 4B). The association between inflammation andaccumulation of b-amyloid in skeletal muscle has recentlybeen shown in a mouse model of sIBM, where LPS-inducedinflammation enhanced the accumulation of proteinssuch as tau and b-amyloid [116].
aB-crystallin and APP are early cell stress markers Almost10 years ago, aB-crystallin has been demonstrated inhealthy-appearing muscle fibres in sIBM muscle [117].aB-crystallin is a heat-shock protein, which can chaper-one proteins in skeletal muscle and is associated with cellstress or b-amyloid clearance. In a quantitative assess-ment of multi-labelling and serial immunohistochemistry,a positive correlation between aB-crystallin and b-amyloid-associated markers was found in sIBM muscles[118]. The normal-appearing muscle fibres that were posi-tive for aB-crystallin were often double-positive for APP,while APP/b-amyloid co-localized with markers of cellstress and regeneration/degeneration. In sIBM therefore,aB-crystallin may be an early event associated with astress-response that precedes accumulation of b-amyloid;this view was supported by in vitro studies, which demon-strated that accumulation of b-amyloid, upon pro-inflammatory cell stress, was preceded by upregulation ofAPP and aB-crystallin [118].
Inflammatory myopathies 235
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242
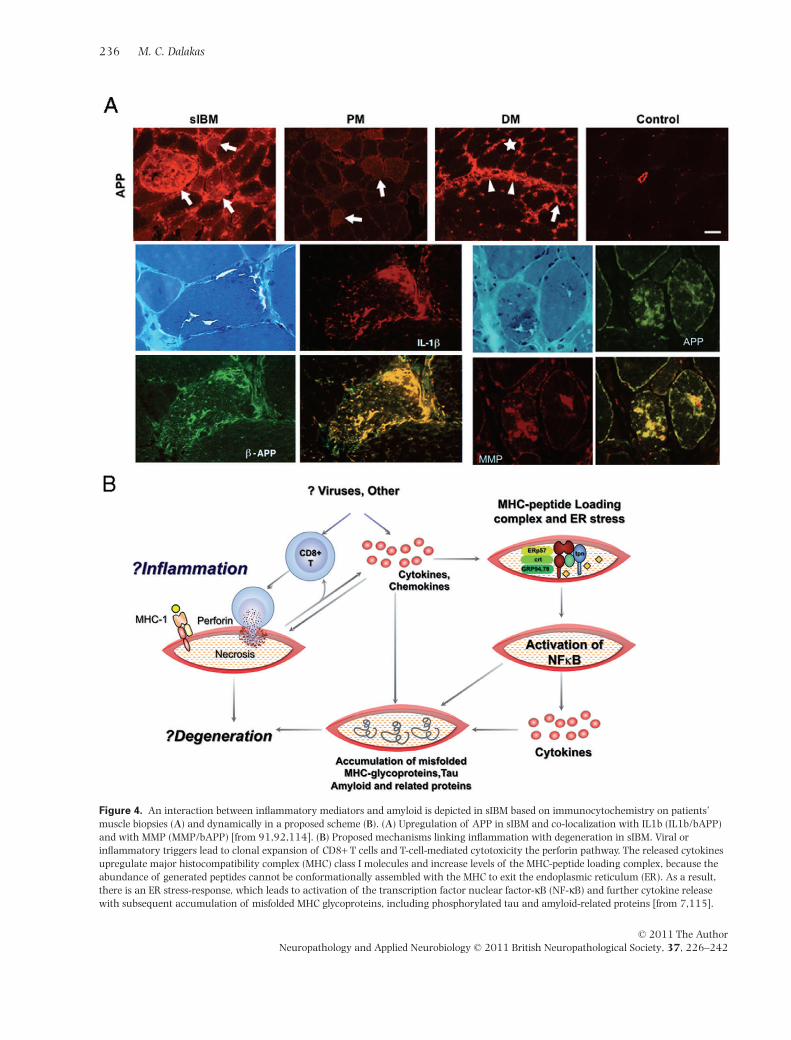
Figure 4. An interaction between inflammatory mediators and amyloid is depicted in sIBM based on immunocytochemistry on patients’muscle biopsies (A) and dynamically in a proposed scheme (B). (A) Upregulation of APP in sIBM and co-localization with IL1b (IL1b/bAPP)and with MMP (MMP/bAPP) [from 91,92,114]. (B) Proposed mechanisms linking inflammation with degeneration in sIBM. Viral orinflammatory triggers lead to clonal expansion of CD8+ T cells and T-cell-mediated cytotoxicity the perforin pathway. The released cytokinesupregulate major histocompatibility complex (MHC) class I molecules and increase levels of the MHC-peptide loading complex, because theabundance of generated peptides cannot be conformationally assembled with the MHC to exit the endoplasmic reticulum (ER). As a result,there is an ER stress-response, which leads to activation of the transcription factor nuclear factor-kB (NF-kB) and further cytokine releasewith subsequent accumulation of misfolded MHC glycoproteins, including phosphorylated tau and amyloid-related proteins [from 7,115].
236 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

NAM
These patients present with high CK, in the thousands,moderate to severe muscle weakness of acute or subacuteonset and with histological features of muscle fibrenecrosis mediated by macrophages as the main effectorcell (Figure 1D–F). There are no T-cell infiltrates orMHC-I expression as seen in PM and sIBM. A number ofpatients have deposition of complement on blood vessels[119,120]. Of interest, antibodies against signal recogni-tion particles [121] or 100–200 kD proteins have beenrecently identified [120]. The cause of NAM is multifacto-rial. Some patients have cancer or an active viral infection(i.e. HIV); others have been exposed to statins that caninduce both, a toxic as well as an autoimmune necrotizingmyositis, with upregulation of MHC-1 in some patients,which responds to immunotherapy [18,122]; others mayhave a smoldering underlying autoimmune process; andstill others have no other disease or apparent exposure toexogenous agents. It is likely, based on the recent studies[120], that NAM is an antibody-mediated disease, assuggested by the presence of specific antibodies and com-plement deposits; the recruitment of macrophages mayrepresent an antibody-dependent cell-mediated cytotoxic-ity process [8,9]. More studies are needed to understandthe pathogenesis and course of this largely overlooked dis-order because it is potentially treatable.
Therapeutic strategies in DM, PM and NAM andchallenges in sIBM
As the specific target antigens in DM, PM and sIBM areunknown, the presently used immunosuppressive thera-pies are not selectively targeting either the autoreactive Tcells or the complement-mediated process on intramuscu-lar blood vessels. Instead, they are inducing a non-specificimmunosuppression or immunomodulation. Further-more, many of these therapies are empirical. Based onexperience, but not controlled studies, the majority ofpatients with PM and DM respond to corticosteroids tosome degree and for a period of time [123–125]. IVIgtested in a controlled study is effective in DM as a second-,and at times, first-line therapy. IVIg appears also effectivein PM and NAM. Immunosuppressants are used assteroid-sparing agents but their efficacy remains unclear[124]. New agents in the form of monoclonal antibodiesor fusion proteins that target cytokines, adhesion mol-ecules, T-cell transduction or transmigration molecules
and B cells or their activation factors, are emerging aspromising immunotherapeutic drugs [123,124]. Amongthem, Rituximab, a B cell-depleting agent, has beenhelpful in some cases of DM and NAM [124].
In contrast to PM and DM, there is currently no effectivetreatment for sIBM. Prednisone, cyclosporine, azathio-prine, methotrexate, total body irradiation and IFN-b havegenerally failed justifying the contention that it could bemore of a degenerative disease rather than autoimmune.A number of patients with sIBM, however, may respond tocommon immunotherapeutic agents early on to somedegree and for a period of time [8,9,126]; up to 25% ofpatients in a controlled study have also respondedtransiently to IVIg [127]. These benefits are arguablylimited and short-lived; sIBM remains a steadily progres-sive disease. This pattern of transient therapeutic responseresembles the one seen in various other autoimmune dis-eases where immune and degenerative features coexistfrom the outset. Primary progressive multiple sclerosisis a classic example as many of these patients partiallyrespond for a period of time but afterwards they becomeresistant to all therapies. The lack of treatment efficacy insIBM may be due to a number of reasons [7,9]: (i) therapyis always initiated late when the degenerative cascadehas already begun, due to insidious onset and very slowdisease progression. It is striking that even patients withminimal clinical weakness already exhibit muscle atrophyand extensive pathology as revealed by histology ormuscle imaging. The observation that aB-crystallin is,along with pro-inflammatory markers, an early eventassociated with cell stress-response that precedes accu-mulation of b-amyloid, supports the view that anti-inflammatory therapy may arrest progression if initiatedearly, as recently shown [128]; (ii) the production of pro-inflammatory mediators by the muscle fibres themselvesmay pose a problem in arresting the process because thestandard immunosuppressants may not be able to sup-press the factors that trigger the continuous production ofcytokines by the muscle fibres themselves; and (iii) thenoted interrelationship between inflammatory mediatorsand degeneration suggests that successful suppression ofendomysial inflammation may have an effect on somedegeneration-associated molecules with resulting short-term clinical stability. On this basis, Alemtuzumab(Campath), a T-cell-depleting monoclonal antibody wasused in a small study. This proof-of-principle, uncontrolledstudy, showed that depletion of T cells from the peripheryalso caused reduction of T cells in the muscle and suppres-
Inflammatory myopathies 237
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

sion of some degeneration-associated molecules resultingin a 6-month disease stability period [129]. This novelstudy highlights that new anti-lymphocyte therapies, ifproven safe for long-term therapy, may have an effect notonly on inflammatory mediators but also in halting degen-eration. Other similar candidate agents may be Rapamy-cin and the IL1b antagonists, because they concurrentlysuppress inflammation and stressor molecules. This is alsoapplicable in Alzheimer’s disease where these very same‘degenerative’ molecules are abundant in the patients’brains. In support of the neuroinflammatory concept,the main drug that currently shows some promise inAlzheimer’s disease is IVIg [130].
References
1 Dalakas MC. Polymyositis, dermatomyositis, andinclusion-body myositis. N Engl J Med 1991; 325:1487–98
2 Dalakas MC, Hohlfeld R. Polymyositis and dermatomyo-sitis. Lancet 2003; 362: 971–82
3 Engel AG, Hohlfeld R, Banker BQ. The polymyositis anddermatomyositis syndrome. In Myology. Eds AG Engel, CFranzini-Armstrong. New York: McGraw-Hill, 2006;1335–83
4 Dalakas MC, Karpati G. The inflammatory myopathies.In Disorders of Voluntary Muscle 8th edn. Eds G Karpati,D Hilton-Jones, K Bushby, RC Griggs. Cambridge:Cambridge University Press, 2010; 427–52
5 Dalakas MC. Polymyositis, dermatomyositis and inclu-sion body myositis. Harrison’s Principles of InternalMedicine 17th edn. Eds AS Fauci, E Braunwald, DLKasper, SL Hauser, DL Longo, JL Jameson, J Loscalzo.New York, NY: McGraw-Hill, 2008; 2696–703
6 Mastaglia FL, Phillips BA. Idiopathic inflammatory myo-pathies: epidemiology, classification and diagnostic cri-teria. Rheum Dis Clin North Am 2002; 28: 723–41
7 Dalakas MC. Inflammatory muscle diseases: a criticalreview on pathogenesis and therapies. Curr Opin Phar-macol 2010; 10: 346–52
8 Schmidt J, Dalakas MC. Pathomechanisms of inflamma-tory myopathies: recent advances and implications fordiagnosis and therapies. Expert Opin Med Diagn 2010; 4:241–50
9 Dalakas MC. Sporadic inclusion body myositis –diagnosis, pathogenesis and therapeutic strategies. NatClin Pract Neurol 2006; 2: 437–47
10 Sekul EA, Dalakas MC. Inclusion body myositis: newconcepts. Semin Neurol 1993; 13: 256–63
11 Needham M, Mastaglia FL. Inclusion body myositis:current pathogenetic concepts and diagnostic andtherapeutic approaches. Lancet Neurol 2007; 6: 620–31
12 Koffman BM, Sivakumar K, Simonis T, Stroncek D,Dalakas MC. HLA allele distribution distinguishessporadic inclusion body myositis from hereditary inclu-sion body myopathies. J Neuroimmunol 1988; 84:139–42
13 Badrising UA, Schreuder GM, Giphart MJ, Geleijns K,Verschuuren JJ, Wintzen AR, Maat-Schieman ML, vanDoorn P, van Engelen BG, Faber CG, Hoogendijk JE, deJager AE, Koehler PJ, de Visser M, van Duinen SG. DutchIBM Study Group. Associations with autoimmunedisorders and HLA class I and II antigens in inclusionbody myositis. Neurology 2004; 63: 2396–8
14 Mastaglia FL, Needham M, Scott A, James I, Zilko P, DayT, Kiers L, Corbett A, Witt CS, Allcock R, Laing N,Garlepp M, Christiansen FT. Sporadic inclusion bodymyositis: HLA DRB1 allele interactions influence diseaserisk and clinical phenotype. Neuromuscul Disord 2009;19: 763–5
15 Sivakumar K, Semino-Mora C, Dalakas MC. An inflam-matory, familial, inclusion body myositis with autoim-mune features and a phenotype identical to sporadicinclusion body myositis: studies in 3 families. Brain1997; 120: 653–61
16 Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G,Mendell JR, Rowland LP. Inclusion body myositis andmyopathies. Ann Neurol 1995; 38: 705–13
17 Karpati G, Carpenter S. Pathology of the inflammatorymyopathies. Baillieres Clin Neurol 1993; 2: 527–56
18 Needham M, Fabian V, Knezevic W, Panegyres P, ZilkoP, Mastaglia FL. Progressive myopathy with upregula-tion of MHC-I associated with statin therapy. Neuromus-cul Disord 2007; 17: 194–200
19 Askanas V, Engel WK, Alvarez RB. Enhanced detectionof Congo-red-positive amyloid deposits in muscle fibersof inclusion body myositis and brain of Alzheimerdisease using fluorescence technique. Neurology 1993;43: 1265–7
20 Askanas V, Engel WK, Alvarez RB, Glenner GG.b-Amyloid protein immunoreactivity in muscle ofpatients with inclusion-body myositis. Lancet 1992;339: 560–1
21 Askanas V, Engel WK, Nogalska A. Inclusion bodymyositis: a degenerative muscle disease associated withintra-muscle fiber multi-protein aggregates, proteasomeinhibition, endoplasmic reticulum stress and decreasedlysosomal degradation. Brain Pathol 2009; 19: 493–506
22 Nogalska A, Terracciano C, D’Agostino C, King Engel W,Askanas V. p62/SQSTM1 is overexpressed and promi-nently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiatingit from polymyositis and dermatomyositis. Acta Neuro-pathol 2009; 118: 407–13
23 De Bleecker JL, Ertl BB, Engel AG. Patterns of abnormalprotein expression in target formations and unstruc-tured cores. Neuromuscul Disord 1996; 6: 339–49
238 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

24 Fidzianska A, Rowinska-Marcinska K, Hausmanowa-Petrusewicz I. Coexistence of X-linked recessive Emery-Dreifuss muscular dystrophy with inclusion bodymyositis-like morphology. Acta Neuropathol 2004; 104:197–203
25 Selcen D, Ohno K, Engel AG. Myofibrillar myopathy:clinical, morphological and genetic studies in 63patients. Brain 2004; 127: 439–51
26 Ferrer I, Martin B, Castano JG, Lucas JJ, Moreno D, OliveM. Proteasomal expression, induction of immunopro-teasome subunits, and local MHC class I presentation inmyofibrillar myopathy and inclusion body myositis.J Neuropathol Exp Neurol 2004; 63: 484–98
27 Ferrer I, Carmona M, Blanco R. Involvement of clusterinand the aggresome in abnormal protein deposits in myo-fibrillar myopathies and Inclusion Body Myositis. BrainPathol 2005; 15: 101–8
28 Semino-Mora C, Dalakas MC. Rimmed vacuoles withb-amyloid and ubiquitinated filamentous deposits in themuscles of patients with long-standing denervation(post-poliomyelitis muscular atrophy): similarities withinclusion body myositis. Hum Pathol 1998; 29: 1128–33
29 Weihl CC, Temiz P, Miller SE, Watts G, Smith C, FormanM, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumu-lation in inclusion body myopathy muscle suggests acommon pathogenic mechanism with frontotemporaldementia. J Neurol Neurosurg Psychiatry 2008; 79:1186–9
30 Salajegheh M, Pinkus JL, Nazareno R, Amato AA,Parker KC, Greenberg SA. Nature of ‘Tau’ immunoreac-tivity in normal myonuclei and inclusion body myositis.Muscle Nerve 2009; 40: 520–8
31 Salajegheh M, Pinkus JL, Taylor JP, Amato AA,Nazareno R, Baloh RH, Greenberg SA. Sarcoplasmicredistribution of nuclear TDP-43 in inclusion bodymyositis. Muscle Nerve 2009; 40: 19–31
32 Chahin N, Engel AG. Correlation of muscle biopsy,clinical course, and outcome in PM and sporadic IBM.Neurology 2008; 70: 418–24
33 Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: mal-functioning desmin causes skeletal and cardiac muscledisease. J Clin Invest 2009; 119: 1806–13
34 Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M,Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G,Bradley WG, Baumbach L, Lancet D, Asher EB, Beck-mann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosaminekinase gene is mutated in recessive hereditary inclusionbody myopathy. Nat Genet 2001; 29: 83–7
35 Dalakas MC. Mechanisms of disease: signaling path-ways and immunobiology of inflammatory myopathies.Nat Clin Pract Rheumatol 2006; 2: 219–27
36 Karpati G, Pouliot Y, Carpenter S. Expression of immu-noreactive major histocompatability complex productsin human skeletal muscles. Ann Neurol 1988; 23: 64–72
37 Emslie-Smith AM, Arahata K, Engel AG. Major histo-compatibility complex class I antigen expression,immunologicalization of interferon subtypes, and Tcell-mediated cytotoxicity in myopathies. Hum Pathol1989; 20: 224–31
38 Krahn M, Lopez de Munain A, Streichenberger N,Bernard R, Pécheux C, Testard H, Pena-Segura JL, YoldiE, Cabello A, Romero NB, Poza JJ, Bouillot-Eimer S,Ferrer X, Goicoechea M, Garcia-Bragado F, Leturcq F,Urtizberea JA, Lévy N. CAPN3 mutations in patientswith idiopathic eosinophilic myositis. Ann Neurol 2006;59: 905–11
39 Kissel JT, Mendell JR, Rammohan KW. Microvasculardeposition of complement membrane attack complex indermatomyositis. N Engl J Med 1986; 314: 329
40 Emslie-Smith AM, Engel AG. Microvascular changes inearly and advanced dermatomyositis: a quantitativestudy. Ann Neurol 1990; 27: 343–56
41 Basta M, Dalakas MC. High-dose intravenous immuno-globulin exerts its beneficial effect in patients withdermatomyositis by blocking endomysial deposition ofactivated complement fragments. J Clin Invest 1994; 94:1729–35
42 Dalakas MC. Immunopathogenesis of inflammatorymyopathies. Ann Neurol 1995; 37: 74–86
43 Stein DP, Dalakas MC. Intercellular adhesionmolecule-1 expression is upregulated in patients withdermatomyositis (DM). Ann Neurol 1993; 34: 268
44 Lundberg I, Brengman JM, Engel AG. Analysis of cytok-ine expression in muscle in inflammatory myopathies,Duchenne’s dystrophy and non-weak controls. J Neu-roimmunol 1995; 63: 9–16
45 Tews DS, Goebel HH. Cytokine expression profiles inidiopathic inflammatory myopathies. J Neuropathol ExpNeurol 1996; 55: 342–7
46 Confalonieri P, Bernasconi P, Megna P, Galbiati S, Cor-nelio F, Mantegazza R. Increased expression of beta-chemokines in muscle of patients with inflammatorymyopathies. J Neuropathol Exp Neurol 2000; 59: 164–9
47 De Bleecker JL, De Paepe B, Vanwalleghem IE, SchröderJM. Differential expression of chemokines in inflamma-tory myopathies. Neurology 2002; 58: 1779–85
48 Figarella-Branger D, Civate M, Bartoli C, Pellissier JF.Cytokines, chemokines, and cell adhesion molecules ininflammatory myopathies. Muscle Nerve 2003; 28:659–82
49 Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermato-myositis autoantigen Mi-2 in regenerating muscle.Arthritis Rheum 2009; 60: 3784–93
50 Gallardo E, de Andrés I, Illa I.. Cathepsins are upregu-lated by IFN-gamma/STAT1 in human muscle culture: apossible active factor in dermatomyositis. J NeuropatholExp Neurol 2001; 60: 847–55
51 Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanou-dou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg
Inflammatory myopathies 239
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

HR, Ericsson M, Park P, Amato AA. Interferon-alpha/beta-mediated innate immune mechanisms in dermato-myositis. Ann Neurol 2005; 57: 664–78
52 Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, PinkusJL, Beggs AH, Amato AA, Greenberg SA. Type Iinterferon-inducible gene expression in blood is presentand reflects disease activity in dermatomyositis andpolymyositis. Arthritis Rheum 2007; 56: 3784–92
53 Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D,Bremer EG, Krasnoselska-Riz I, Kumar A, Pachman LM.Gene expression profiling in DQA1*0501+ children withuntreated dermatomyositis: a novel model of pathogen-esis. J Immunol 2002; 168: 4154–63
54 Raju R, Dalakas MC. Gene expression profile in themuscles of patients with inflammatory myopathies:effect of therapy with IVIg and biological validation ofclinically relevant genes. Brain 2005; 128: 1887–96
55 Amemiya K, Semino-Mora C, Granger RP, Dalakas MC.Downregulation of TGF-b1 mRNA and protein in themuscles of patients with inflammatory myopathies aftertreatment with high-dose intravenous immunoglobulin.Clin Immunol 2000; 94: 99–104
56 Leff RL, Love LA, Miller FW, Greenberg SJ, Klein EA,Dalakas MC, Plotz PH. Viruses in the idiopathic inflam-matory myopathies: absence of candidate viral genomesin muscle. Lancet 1992; 339: 1192–5
57 Leon-Monzon M, Dalakas MC. Absence of persistentinfection with enteroviruses in muscles of patientswith inflammatory myopathies. Ann Neurol 1992; 32:219–22
58 Arahata K, Engel AG. Monoclonal antibody analysis ofmononuclear cells in myopathies. I: quantitation ofsubsets according to diagnosis and sites of accumula-tion and demonstration and counts of muscle fibresinvaded by T cells. Ann Neurol 1984; 16: 193–208
59 Arahata K, Engel AG. Monoclonal antibody analysis ofmononuclear cells in myopathies. IV: cell-mediated cyto-toxicity and muscle fiber necrosis. Ann Neurol 1988; 23:168–73
60 Engel AG, Arahata K. Monoclonal antibody analysis ofmononuclear cells in myopathies. II: phenotypes ofautoinvasive cells in polymyositis and inclusion bodymyositis. Ann Neurol 1984; 16: 209–15
61 Engel AG, Arahata K. Mononuclear cells in myopathies:quantitation of functionally distinct subsets, recogni-tion of antigen-specific cell-mediated cytotoxicity insome diseases, and implications for the pathogenesis ofthe different inflammatory myopathies. Hum Pathol1986; 17: 702–21
62 Hohlfeld R, Engel AG. The immunobiology of muscle.Immunol Today 1994; 15: 269–74
63 Wiendl H, Hohlfeld R, Kieseier BC. Immunobiology ofmuscle: advances in understanding an immunologicalmicroenvironment. Trends Immunol 2005; 26: 373–80
64 Wiendl H, Mitsdoerffer M, Schneider D, Melms A, Loch-muller H, Hohlfeld R, Weller M. Muscle fibres and cul-tured muscle cells express the B7.1/2-related inducibleco-stimulatory molecule, ICOSL: implications for thepathogenesis of inflammatory myopathies. Brain 2003;126: 1026–35
65 Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregu-lated inducible co-stimulator (ICOS) and ICOS-ligand ininclusion body myositis muscle: significance for CD8+ Tcell cytotoxicity. Brain 2004; 127: 1182–90
66 Pruitt JN 2nd, Showalter CJ, Engel AG. Sporadic inclu-sion body myositis: counts of different types of abnormalfibers. Ann Neurol 1996; 39: 139–43
67 Confalonieri P, Oliva L, Andreetta F, Lorenzoni R,Dassi P, Mariani E, Morandi L, Mora M, Cornelio F,Mantegazza R. Muscle inflammation and MHC class Iup-regulation in muscular dystrophy with lack of dys-ferlin: an immunopathological study. J Neuroimmunol2003; 142: 130–6
68 Goebels N, Michaelis D, Engelhardt M, Huber S, BenderA, Pongratz D, Johnson MA, Wekerle H, Tschopp J, JenneD, Hohlfeld R. Differential expression of perforin inmuscle-infiltrating T cell in polymyositis and dermato-myositis. J Clin Invest 1996; 97: 2905
69 Hohlfeld R, Engel AG. Coculture with autologous myo-tubes of cytotoxic T cells isolated from muscle in inflam-matory myopathies. Ann Neurol 1991; 29: 498–507
70 Behrens L, Bender A, Johnson MA, Hohlfeld R. Cyto-toxic mechanisms in inflammatory myopathies: co-expression of Fas and protective Bcl-2 in muscle fibresand inflammatory cells. Brain 1997; 120: 929
71 Schneider C, Gold R, Dalakas MC, Schmied M, Lass-mann H, Toyka KV, Hartung HP. MHC class I mediatedcytotoxicity does not induce apoptosis in muscle fibersnor in inflammatory T cells: studies in patients withpolymyositis, dermatomyositis, and inclusion body myo-sitis. J Neuropath Exp Neurol 1996; 55: 1205–9
72 Mantegazza R, Hughes SM, Mitchell D, Travis M, BlauHM, Steinman L. Modulation of MHC class II antigenexpression in human myoblasts after treatment withIFN-gamma. Neurology 1991; 41: 1128
73 Bao SS, King NJ, dos Remedios CG. Elevated MHC class Iand II antigens in cultured human embryonic myoblastsfollowing stimulation with gamma-interferon. ImmunolCell Biol 1990; 68: 235–41
74 Hohlfeld R, Engel AG. Induction of HLA-DR expressionon human myoblasts with interferon-gamma. Am JPathol 1990; 136: 503
75 Roy R, Dansereau G, Tremblay JP, Belles-Isles M, HuardJ, Labrecque C, Bouchard JP. Expression of major histo-compatibility complex antigens on human myoblasts.Transplant Proc 1991; 23: 799
76 De Bleecker JL, Meire VI, Declercq W, Van Aken HE.Immunolocalization of tumor necrosis factor-alpha andits receptors in inflammatory myopathies. NeuromuscDisord 1999; 9: 239
240 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

77 Raju R, Vasconcelos O, Granger R, Dalakas MC. Expres-sion of IFN-gamma-inducible chemokines in inclusionbody myositis. J Neuroimmunol 2003; 141: 125–31
78 Bender A, Ernst N, Iglesias A, Dornmair K, Wekerle H,Hohlfeld R. T cell receptor in polymyositis: clonal expan-sion of autoaggressive CD81 T cells. J Exp Med 1995;181: 1863–8
79 Mantegazza R, Andreetta F, Bernasconi P, Baggi F,Oksenberg JR, Simoncini O, Mora M, Cornelio F, Stein-man L. Analysis of T cell receptor repertoire of muscleinfiltrating T lymphocytes in polymyositis. J Clin Invest1993; 91: 2880–6
80 O’Hanlon TP, Dalakas MC, Plotz PH, Miller FW. Pre-dominant TCR-alpha-beta variable and joining geneexpression by muscle-infiltrating lymphocytes in theidiopathic inflammatory myopathies. J Immunol 1994;152: 2569–76
81 Benveniste O, Herson S, Salomon B, Dimitri D,Trébeden-Nègre H, Jean L, Bon-Durand V, Antonelli D,Klatzmann D, Boyer O. Long-term persistence ofclonally expanded T cells in patients with polymyositis.Ann Neurol 2004; 56: 867–72
82 Fyhr IM, Moslemi AR, Mosavi AA, Lindberg C,Tarkowski A, Oldfors A. Oligoclonal expansion ofmuscle infiltrating T cells in inclusion body myositis.J Neuroimmunol 1997; 79: 185–9
83 Bender A, Behrens L, Engel AG, Hohlfeld R. T-cell het-erogeneity in muscle lesions of inclusion body myositis.J Neuroimmunol 1998; 84: 86–91
84 Nishio J, Suzuki M, Miyasaka N, Kohsaka H. Clonalbiases of peripheral CD8 T cell repertoire directly reflectlocal inflammation in polymyositis. J Immunol 2001;167: 4051–8
85 Amemiya K, Granger RP, Dalakas MC. Clonal restrictionof T-cell receptor expression by infiltrating lymphocytesin inclusion body myositis persists over time. Studies inrepeated muscle biopsies. Brain 2000; 123: 2030–9
86 Hofbauer M, Wiesener S, Babbe H, Roers A, Wekerle H,Dornmair K, Hohlfeld R, Goebels N. Clonal tracking ofautoaggressive T cells in polymyositis by combininglaser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci USA 2003; 100:4090–5
87 Hohlfeld R, Engel AG, Kunio LI, Harper MC. Polymyositismediated by T lymphocytes that express the gamma/delta receptor. N Engl J Med 1991; 324: 877–81
88 Puschke G, Ruegg D, Hohlfeld R, Engel AG. Autoaggres-sive myocytotoxic T-lymphocytes expressing an unusualgamma delta T cell receptor. J Exp Med 1992; 176:1785–9
89 Wiendl H, Malotka J, Holzwarth B, Weltzien HU,Wekerle H, Hohlfeld R, Dornmair K. An autoreactivegamma delta TCR derived from a polymyositis lesion.J Immunol 2002; 169: 515–21
90 Murata K, Dalakas MC. Expression of the costimulatorymolecule BB-1, the ligands CTLA-4 and CD28 and their
mRNA in inflammatory myopathies. Am J Pathol 1999;155: 453–60
91 Schmidt J, Barthel K, Wrede A, Salajegheh M, Bähr M,Dalakas MC. Interrelation of inflammation and APP insIBM: IL-1 beta induces accumulation of beta-amyloidin skeletal muscle. Brain 2008; 131: 1228–4050
92 Choi YC, Dalakas MC. Expression of matrix metallopro-teinases in the muscle of patients with inflammatorymyopathies. Neurology 2000; 54: 65–71
93 De Bleecker JL, Engel AG. Expression of cell adhesionmolecules in inflammatory myopathies and Duchennedystrophy. J Neuropathol Exp Neurol 1994; 53: 369–76
94 Greenberg SA, Pinkus GS, Amato AA, Pinkus JL.Myeloid dendritic cells in inclusion-body myositis andpolymyositis. Muscle Nerve 2007; 35: 17–23
95 Bradshaw EM, Orihuela A, McArdel SL, Salajegheh M,Amato AA, Hafler DA, Greenberg SA, O’Connor KC. Alocal antigen-driven humoral response is present inthe inflammatory myopathies. J Immunol 2007; 178:547–56
96 Chou SM. Inclusion body myositis: a possible chronicpersistent mumps myositis? Hum Pathol 1986; 17:765–76
97 Dalakas MC. Inflammatory, immune, and viral aspectsof inclusion-body myositis. Neurology 2006; 66: 33–8
98 Nishino H, Engel AG, Rima BK. Inclusion body myositis:the mumps virus hypothesis. Ann Neurol 1989; 25:260–4
99 Dalakas MC, London WT, Gravell M, Sever JL. Polymyo-sitis in an immunodeficiency disease in monkeys inducedby a type D retrovirus. Neurology 1986; 36: 569–72
100 Dalakas MC, Pezeshkpour GH, Gravell M, Sever JL. Poly-myositis in patients with AIDS. JAMA 1986; 256:2381–3
101 Dalakas MC, Pezeshkpour GH. Neuromuscular diseasesassociated with human immunodeficiency virus infec-tion. Ann Neurol 1989; 23 (Suppl.): 38–48
102 Morgan OSC, Rodgers-Johnson P, Mora C, Char G.HTLV-1 and polymyositis in Jamaica. Lancet 1989; 2:1184–7
103 Illa I, Nath A, Dalakas MC. Immunocytochemical andvirological characteristics of HIV-associated inflamma-tory myopathies: similarities with seronegative poly-myositis. Ann Neurol 1991; 29: 474–81
104 Leon-Monzon M, Dalakas MC. Interleukin-1 (IL-1) istoxic to human muscle. Neurology 1994; 44: 132
105 Leon-Monzon M, Lamperth L, Dalakas MC. Searchfor HIV proviral DNA and amplified sequences inthe muscle biopsies of patients with HIV-polymyositis.Muscle Nerve 1993; 16: 408–13
106 Cupler EJ, Leon-Monzon M, Miller J, Semino-Mora C,Anderson TL, Dalakas MC. Inclusion body myositis inHIV-I and HTLV-I infected patients. Brain 1996; 19:1887–93
107 Saito M, Higuchi I, Saito A, Izumo S, Usuku K, BanghamCR, Osame M. Molecular analysis of T cell clonotypes in
Inflammatory myopathies 241
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242

muscle-infiltrating lymphocytes from patients withhuman T lymphotropic virus type 1 polymyositis. J InfectDis 2002; 186: 1231–41
108 Ozden S, Cochet M, Mikol J, Teixeira A, Gessain A, PiqueC. Direct evidence for a chronic CD8+-T-cell-mediatedimmune reaction to tax within the muscle of a humanT-cell leukemia/lymphoma virus type 1-infected patientwith sporadic inclusion body myositis. J Virol 2004; 78:10320–7
109 Dalakas MC, Rakocevic G, Shatunov A, Goldfarb L,Salajegheh M. IBM with HIV infection: 4 new caseswithclonal expansion of viral-specific T cells. AnnNeurology 2007; 61: 466–75
110 Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW,Askanas V. Proteasome inhibition and aggresome for-mation in sporadic inclusion-body myositis and inamyloid-beta precursor protein-overexpressing culturedhuman muscle fibers. Am J Pathol 2005; 167: 517–26
111 Vattemi G, Engel WK, McFerrin J, Buxbaum JD, Pas-torino L, Askanas V. Presence of BACE1 and BACE2 inmuscle fibres of patients with sporadic inclusion-bodymyositis. Lancet 2001; 358: 1962–4
112 Lünemann JD, Schmidt J, Schmid D, Barthel K, WredeA, Dalakas MC, Münz C. Beta-amyloid is a substrate ofautophagy in sporadic inclusion body myositis. AnnNeurol 2007; 61: 476–83
113 Schmid D, Münz C. Localization and MHC class II pre-sentation of antigens targeted for macroautophagy.Methods Mol Biol 2008; 445: 213–25
114 Dalakas MC. Molecular immunology and genetics ofinflammatory muscle diseases. Arch Neurol 1998; 55:1509–12
115 Dalakas MC. Molecular links between inflamation anddegeneration: lessons on ‘Neuroinflammation’ usingIBM as a model. Ann Neurology 2008; 64: 1–3
116 Kitazawa M, Trinh DN, LaFerla FM. Inflammationinduces tau pathology in inclusion body myositis modelvia glycogen synthase kinase-3beta. Ann Neurol 2008;64: 15–24
117 Banwell BL, Engel AG. AlphaB-crystallin immunolocal-ization yields new insights into inclusion body myositis.Neurology 2000; 54: 1020–1
118 Muth IE, Barthel K, Bähr M, Dalakas MC, Schmidt J.Proinflammatory cell stress in sporadic inclusion bodymyositis muscle: overexpression of alphaB-crystallin isassociated with amyloid precursor protein and accumu-lation of beta-amyloid. J Neurol Neurosurg Psychiatry2009; 80: 1344–9
119 Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathywith antibodies to the signal recognition particle:clinical and pathological features. J Neurol NeurosurgPsychiatry 2002; 73: 420–8
120 Christofer-Stine L, Casciola-Rosen LA, Hong G, ChungT, Corse AM, Mammen AL. A novel autoantibodyrecognizing 200 and 100 kDa proteins is associatedwith an immune-mediated necrotizing myopathy. ArthrRheum 2010; 62: 2757–66
121 Hengstman GJ, van Engelen BG, van Venrooij WJ.Myositis specific autoantibodies: changing insights inpathophysiology and clinical associations. Curr OpinRheumatol 2004; 16: 692–9
122 Dalakas MC. Toxic and drug-induced myopathies. JNeurol Neurosurg Psychiatry 2009; 80: 832–8
123 Dalakas MC. Therapeutic targets in patients with inflam-matory myopathies: present approaches and a look tothe future. Neuromuscular Disord 2006; 16: 223–36
124 Dalakas MC. Immunotherapy of myositis: issues, con-cerns and future prospects. Nat Rev Rheumatol 2010; 6:129–37
125 Mastalgia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflam-matory Myopathies: clinical, diagnostic and therapeuticaspects. Muscle Nerve 2003; 27: 407–25
126 Needham M, Mastaglia FL. Sporadic inclusion bodymyositis: a continuing puzzle. Neuromuscul Disord 2008;18: 6–16
127 Dalakas MC, Sekul EA, Cupler EJ, Sivakumar K. TheEfficacy of High Dose Intravenous Immunoglobulin(IVIg) in Patients With Inclusion-Body Myositis (IBM).Neurology 1997; 48: 712–16
128 Layzer R, Lee HS, Iverson D, Margeta M. Dermatomyo-sitis with inclusion body myositis pathology. MuscleNerve 2009; 40: 469–71
129 Dalakas MC, Rakocevic G, Schmidt J, Salajegheh M,McElroy B, Harris-Love MO, Shrader JA, Levy EW,Dambrosia J, Kampen RL, Bruno DA, Kirk AD. Effect ofAlemtuzumab (CAMPATH 1-H) in patients withinclusion-body myositis. Brain 2009; 132: 1536–44
130 Hughes RA, Dalakas MC, Cornblath DR, Latov N,Weksler ME, Relkin N. Clinical applications of intrave-nous immunoglobulins in neurology. Clin Exp Immunol2009; 158 (Suppl. 1): 34–42
Received 4 October 2010Accepted after revision 21 October 2010
Published online Article Accepted on 14 December 2010
242 M. C. Dalakas
© 2011 The AuthorNeuropathology and Applied Neurobiology © 2011 British Neuropathological Society, 37, 226–242





























