Embed Size (px)
Citation preview

Who Technical consulTaTion on ViTamin a in neWborn healTh: mechanisTic sTudies
REPORT


Who Technical consulTaTion on ViTamin a in neWborn healTh: mechanisTic sTudies
GENEVA, SWITZERLAND1–3 DECEMBER 2009
REPORT

WHO Library Cataloguing-in-Publication Data
Report: WHO technical consultation on vitamin A in newborn health: mechanistic studies, Geneva, Switzerland, 1–3 December 2009.
1.Vitamin A – administration and dosage. 2.Vitamin A deficiency – prevention and control. 3.Infant, Newborn. 4.Infant nutrition. I.World Health Organization.
ISBN 978 92 4 150316 7 (NLM classification: WD 110)
© World Health Organization 2012
All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publica-tions – whether for sale or for noncommercial distribution – should be addressed to WHO Press through the WHO web site (http://www.who.int/about/licensing/copyright_form/en/index.html).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization con-cerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate bor-der lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of propri-etary products are distinguished by initial capital letters.
All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either expressed or implied. The responsibility for the interpreta-tion and use of the material lies with the reader. In no event shall the World Health Organiza-tion be liable for damages arising from its use.
Designed by minimum graphics
Suggested citation Report: WHO technical consultation on vitamin A in newborn health: mechanistic studies. Geneva, World Health Organization, 2012.

iii
conTenTs
CONTENTS
Acknowledgements iv
Financial support iv
Introduction 1
Objectives and scope of the technical consultation 2
Management of conflicts of interest 3
Presentations made at the technical consultation 4
Newborn vitamin A supplementation: current situation 4
Recommendations from the WHO Technical Consultation on Neonatal Vitamin A Supplementation Research Priorities (December 2008) 5
Vitamin A supplementation in newborns: a conceptual framework for biological plausibility for reducing mortality using bronchopulmonary dysplasia as an example 8
Possible effects of vitamin A supplementation at birth on immune function 9
Metabolism of vitamin A in early life 12
Mammalian models for understanding mechanisms of retinol and retinol actions 15
Causes of mortality in the neonatal period and first half of infancy 18
References 26
Annex 1. List of participants 27
Annex 2. Background papers 31
A2.1: Vitamin A supplementation in newborns: a conceptual framework for biological plausibility 33
A2.2: Metabolism of vitamin A in early life 47
A2.3: Vitamin A deficiency and the neonatal immune system: possible effects of vitamin A supplementation at birth 77
A2.4: Mammalian models for understanding mechanisms of retinol and retinoid actions 93

iv
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
acknoWledgemenTs
The technical consultation on vitamin A in neonatal health was coordinated by Dr Lisa Rogers under the supervision of Dr Juan Pablo Peña-Rosas. Spe-cial thanks are due to Dr Mathilde Savy for assistance in drafting this report. Thanks are also due to Dr Rajiv Bahl and Dr Jose Martines for their technical support. Ms Grace Rob and Mrs Paule Pillard from the Micronutrients Unit, Department of Nutrition for Health and Development, provided logistic sup-port.
The World Health Organization (WHO) also gratefully acknowledges the technical input of the participants attending the meeting.
Financial supportWHO thanks the Bill & Melinda Gates Foundation for providing financial support for the meeting and the publication of this report.

1
inTroducTion
INTRODUCTION
Vitamin A deficiency among the world’s poor and underprivileged popu-lations is a considerable public health problem, as it can lead to blindness, decreased immune function and ultimately death. Its causes include poverty, infections and lack of access to traditional foods that historically have pro-vided adequate provitamin A.
The term vitamin A includes all compounds with retinol activity and reti-noids that exhibit activity similar to retinol. Preformed vitamin A is derived from animal tissues (retinol and retinyl esters) and is efficiently absorbed in humans (70–90%). Provitamin A carotenoids are cleaved to form retinal; however, their absorption is less efficient in humans (20–50%).
The World Health Organization (WHO) Technical Consultation on Neo-natal Vitamin A Supplementation Research Priorities, held in December 2008, recognized the need to study the biological mechanisms underpinning the potential effects of vitamin A in the first days of life. Following this con-sultation, a grant proposal submitted to the Bill & Melinda Gates Foundation by the WHO, working alongside experts, received a favourable response for research in the following areas related to the mechanisms of action of vitamin A in neonates:
n absorption, transport, distribution and storage of vitamin A following high-dose (50 000 IU) vitamin A supplementation in early life;
n effect of neonatal vitamin A supplementation on: — organ maturation; — innate and adaptive immune responses.
Following a call for expressions of interest, a formal request for proposals, and extensive internal and external reviews, WHO selected the two top-ranked human studies (to be conducted in South Asia and in Africa) and the top-ranked animal study. The Department of Nutrition for Health and Develop-ment (NHD) convened another technical consultation in December 2009 with key experts to review in depth the current knowledge on: role of vitamin A in immunology; metabolism of vitamin A in early life; animal models for the study of the mechanisms of action of vitamin A; and biological plausibil-ity of vitamin A supplementation in reducing neonatal mortality. The con-sultation participants also discussed the proposed studies, including their methodology. Four background papers were commissioned to review current knowledge on these topics.

2
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
objecTiVes and scope of The Technical consulTaTion
The technical consultation reviewed:
n the research questions and the priority research agenda identified by the 2008 WHO Technical Consultation on Neonatal Vitamin A Supplementa-tion Research Priorities;
n existing studies on possible mechanisms of the impact of vitamin A on newborn health;
n current knowledge on metabolism of vitamin A and its role in infant immunity;
n the most appropriate models for studying the mechanisms of action of reti-nol and retinoids;
n the protocols of the proposed studies to standardize methods and out-comes, where possible.
The expected outputs were:
n summarizing the current state of knowledge on the role of vitamin A in neonates and priority research gaps;
n recommendations to finalize the mechanistic study protocols and method-ology for answering the proposed research questions.

3
managemenT of conflicTs of inTeresT
As per the WHO Basic documents, all participating experts submitted a Dec-laration of Interests Form prior to, and verbally declared potential conflicts of interest at the beginning of, the consultation. The potential conflicts of inter-est that were declared are summarized below.
Professor Keith P. West Jr received non-monetary research support from DSM Ltd in the form of micronutrient supplements and periodic potency test-ing for an intervention trial in Bangladesh.
Professor Andrew M. Prentice received research support from the Bill & Melinda Gates Foundation for a landscape analysis.
INTRODUCTION

4
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
presenTaTions made aT The Technical consulTaTion
n Newborn vitamin A supplementation: current situation (Presented by Keith P. West Jr)
Meta-analyses of studies conducted in the 1980s and 1990s have shown that vitamin A supplementation in children 6–72 months of age reduces child mortality by 23–34%, and treatment of severe measles with vitamin A reduces case fatality by 50–80%. Infants are born with only about a 2-week supply of vitamin A, stored in the liver, and in undernourished settings, serum retinol is likely to remain low during the first half of infancy. While in developed countries breastfeeding is usually adequate to meet the vitamin A needs of the young infant, infants in many developing countries likely consume inadequate amounts of vitamin A in the first 6 months, partly due to lower vitamin A con-centrations in breast milk. Given this situation, along with frequent postnatal infections and high rates of infant mortality, can neonatal vitamin A supple-mentation reduce the frequency or severity of infectious illness and mortality in underdeveloped countries? If so, what are the mechanisms of action?
Results of studies are conflicting and a meta-analysis conducted in 2009 (1) did not find convincing evidence for a beneficial effect of vitamin A supple-mentation on morbidity or mortality in infancy. This has raised interest in the issue with WHO currently coordinating three newborn vitamin A trials (see Introduction), this technical consultation and research to explore plausi-ble mechanisms by which newborn vitamin A may reduce infant mortality. A Bangladeshi clinical trial assessing the impact of vitamin A in neonatal sepsis and necrotizing enterocolitis is also nearing recruitment. In addition, literature on the role of vitamin A metabolites in immune function is rapidly expanding.
Assuming that newborn vitamin A supplementation can reduce infant mortality, what biologically plausible mechanisms can explain this effect? What population characteristics can predict it? Or conversely, what are the reasons for not having an effect or potential interactions? At this time, it is hypothesized that there is an effect, which may be due to innate, adaptive and regulatory mechanisms.
Summary of discussion on the presentation
Participants questioned the reason for using the 50 000 IU dose of vitamin A, which was said to be a guess, and whether alternative doses needed to be studied.

5
The common use of serum retinol levels as an indicator of vitamin A status was also questioned, and the best time to give vitamin A supplements to neo-nates. This may be just before discharge, as infants may not come back to the hospital for it at a later time.
Interest was also expressed in the use of birth weight to explain the dif-ferences in outcomes of the various vitamin A supplementation trials. Body weight was not considered to be an explanatory factor.
n Recommendations from the WHO Technical Consultation on Neonatal Vitamin A Supplementation Research Priorities (December 2008)(Presented by Lisa M. Rogers)
Vitamin A supplementation has been promoted as an essential child survival intervention in children 6–59 months of age, but studies have not shown similar benefit in infants 1–5 months of age. Current interest in vitamin A supplemen-tation in the neonatal period (0–28 days) has been sparked by three trials (Indo-nesia, India and Bangladesh) showing a reduction (15–64%) and three trials (Nepal, Zimbabwe and Guinea-Bissau) showing no effect on infant mortality.
A systematic review commissioned by WHO in 2008 concluded that vita-min A supplementation in the neonatal period was not associated with a reduced risk of infant morbidity and mortality or of an increase in adverse effects; however, it identified several issues that needed further research. These were discussed at the WHO Technical Consultation on Neonatal Vitamin A Supplementation Research Priorities, held on 4–5 December 2008, in Geneva, Switzerland, where research needs and gaps in the use of vitamin A supple-ments for neonates were identified and prioritized.
Research questions identified
n Does newborn vitamin A supplementation:— reduce infant mortality in low human immunodeficiency virus (HIV)-
prevalence settings with high infant mortality when given in the first 48 hours of birth?
— have differential effects on infant mortality in Asia and Africa?— have an impact on vitamin A status at 3 and 6 months of age?
n Is the effect of newborn vitamin A supplementation on infant mortal-ity modified by timing of supplementation (first 24 hours, 48 hours, 72 hours, first week), maternal vitamin A status, gender, subsequent vaccines received, birth weight, gestation, season of supplementation, and/or time of initiation of breastfeeding?
n What biological mechanisms underpin the postulated beneficial effects of neonatal vitamin A supplementation?
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

6
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
n What approaches are feasible to deliver interventions such as neonatal vita-min A supplementation?
n Is neonatal vitamin A supplementation safe and beneficial in those who are not vitamin A deficient?
Priority research agenda
n Randomized placebo-controlled trials to determine the effect of neonatal vitamin A supplementation given within the first 2 days after birth on mor-tality in the first 6 months of life:— at least two trials in Africa and one trial in Asia;— trials should be conducted in settings with high infant mortality;— trials should be individually powered to answer the research question.
n A pooled analysis of all published trials on the effects of neonatal vitamin A supplementation, stratified by:— timing of supplementation (first 24 hours, 48 hours, 72 hours, first
week);— season of supplementation;— gender;— vaccines received during follow-up;— birth weight and gestational age;— maternal vitamin A status;— time of initiation of breastfeeding.
n Mechanistic studies exploring the possible beneficial effects of vitamin A supplementation in the first days of life, particularly on immune response and/or organ maturation.
n Operational research on how to reach most babies in developing countries within 2 days, but not exclusively in the context of neonatal vitamin A sup-plementation.
Recommendations for design of priority studies: biological mechanisms (PICOT format)
n Evidence – there is no knowledge on vitamin A metabolism in the first days of life and effects of early supplementation on organ maturation and immune responses in humans.
n Population – animal model: newborn piglets stratified by sex and maternal vitamin A status (as determined by liver vitamin A levels).
n Intervention – vitamin A supplementation (0, 25 000 or 50 000 IU) within the first 2 days of birth.
n Comparison – unsupplemented controls.n Outcomes:
— absorption of vitamin A;

7
— serum metabolites (retinoic acid, retinyl glucuronide, retinoyl glucuro-nide);
— vitamin A distribution in the essential organs;— organ maturation;— immunological responses.
n Timing – first days of life.
Future studies
The Bill & Melinda Gates Foundation has approved a proposal submitted by the WHO Departments of Nutrition for Health and Development (NHD) and Child and Adolescent Health (CAH), working alongside key experts, for funding the conduct of (1) three large randomized controlled trials (RCTs) in neonates in low- and middle-income countries with a high infant mortality rate, and a likelihood of a high prevalence of vitamin A deficiency and a low prevalence of HIV (two in sub-Saharan Africa and one in south Asia), and (2) animal and human mechanistic studies on biological mechanisms underpin-ning possible beneficial effects of neonatal vitamin A supplementation.
Study sites have been chosen in India (n = 40 000), Ghana (n = 32 000) and the United Republic of Tanzania (n = 32 000). Neonates will be given either 50 000 IU vitamin A orally as a single dose within 48 hours of birth or a placebo that is identical in appearance, consistency and taste. Outcomes will include mortality in the first 6 months, neonatal mortality (0–28 days), risk of hospital admissions in the first 6 months, adverse events within 72 hours of supplementation, and vitamin A status at 2 weeks and 3 months of age.
The mechanistic studies are aimed at understanding the biological mecha-nisms through which vitamin A supplementation given at birth can potential-ly impact infant survival, that is, how a large dose of vitamin A given within 48 hours of birth:
n is absorbed, transported and distributed in body tissues;n affects newborn vitamin A stores (magnitude and duration of impact);n affects organ maturation;n affects innate and adaptive immune responses.
Summary of discussion on the presentation
Participants asked about affect of vitamin A on organ maturation, particu-larly lung development, and the spleen, thymus or other organs involved in immune system. Other issues discussed included the meaning of the word ‘maturation’ and how to define that in a premature or malnourished infant.
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

8
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
n Vitamin A supplementation in newborns: a conceptual framework for biological plausibility for reducing mortality using bronchopulmonary dysplasia as an example(Presented by Jayant P. Shenai)
Bronchopulmonary dysplasia (BPD) occurs more frequently in neonates with a lower gestational age and birth weight, and survival of neonates with BPD increased markedly between 1975 and 2005 (data source: Vanderbilt Chil-dren’s Hospital, Nashville, Tennessee, USA). Infants with a birth weight 500–750 g had <20% chance of survival in 1975, while in 2005 the survival rate was close to 80%, leading to an increase in the number of neonates with chronic lung disease, especially BPD.
Most preterm infants are born with low concentrations of retinol in plasma. Clinical studies measuring plasma concentrations of vitamin A have shown consistent low values at lower gestational ages. Liver reserves of vitamin A are also reduced in lower-birth-weight infants. The pathology of vitamin A deficiency generally appears in the following sequence: oral cavity, respiratory tract, genitourinary tract, eyes and skin. BPD develops in immature lungs when there is tissue injury and impaired tissue repair. Tissue injury can be caused by barotraumas/volutrauma, oxygen toxicity, surfactant deficiency, pulmonary oedema, proteolysis or inflammation. Tissue repair may be aided by nutrients such as vitamin A, antioxidants, eicosanoids, growth factors, peptide hormones and extracellular matrix components.
A clinical trial conducted at Vanderbilt Children’s Hospital (2) showed a reduction in BPD in children supplemented with vitamin A (9/20) versus controls (17/20). Supplementation with vitamin A also reduced the need for mechanical ventilation, supplemental oxygen, days in the intensive care unit and the prevalence of airway infections and retinopathy of prematurity. Another clinical trial in extremely-low-birth-weight infants also showed a reduction in BPD in children supplemented with vitamin A (163/346) ver-sus controls (193/347) (3). Vanderbilt Children’s Hospital currently provides vitamin A supplementation to all infants born under 31 weeks’ gestation with a birth weight <1250 g, who have appropriate growth for gestational age, are <24 hours postnatal age, on mechanical ventilation at 12 hours (for infants 1000–1250 g) or independent of ventilatory status (for infants <1000 g) and who do not have congenital anomalies.
See Annex 2 for details of vitamin A biochemical markers and the sched-ule, doses and forms of administration of vitamin A along with post-admin-istration monitoring activities carried out at Vanderbilt Children’s Hospital.
Summary of discussion on the presentation
The discussion focused on the use of retinol-binding protein (RBP) for assess-ing vitamin A status and surfactants as a standard of practice at Vanderbilt

9
Children’s Hospital. Changes in RBP with normal retinol concentrations, and no changes in RBP response to vitamin A administration in preterm infants, were also discussed and participants commented on intramuscular vitamin A administration, its dose, whether it is fat soluble or water miscible, possibility of increased intracranial pressure and field study conditions.
Dr Shenai explained that for measuring septation, a camera takes continu-ous pictures to see the septa grow and develop. Tissue presents bubbles under normal conditions and is not bubbly under abnormal conditions.
n Possible effects of vitamin A supplementation at birth on immune function(Presented by Charles B. Stephensen)
This presentation focuses on immune function of full-term, exclusively breast-fed neonates. It is assumed that infants are born with low vitamin A stores, even if mothers have good vitamin A status, and that mothers may also be vitamin A deficient during breastfeeding, thus the vitamin A intake of infants from breast milk may be inadequate.
The infant immune system is hyporesponsive to pathogen challenge, at least when judged relative to the adult immune system. This lower responsive-ness may allow the neonatal system to tolerate new gut flora during initial colonization after birth without excessive inflammation, which could be dam-aging to the infant. Immune “anergy” (with a type 2 T helper (Th2) cell bias) lasts ~4 months and the whole process of immune system maturation takes ~1 year. Breastfeeding for at least 6 months could help to obtain this maturation.
Innate immunity is provided by the epithelium, granulocytes, monocytes, natural killer (NK) cells and antigen-presenting cells (APCs), which link the innate to adaptive immune system by presenting antigen to T-lymphocytes. Vitamin A is required for epithelial differentiation, with deficiency lead-ing to squamous metaplasia, decreased duodenal goblet cell formation and increased bacterial translocation from the gut (rats). Newborns have been described as having neutrophil “impairment”, characterized by decreased adherence and chemotaxis, and a decreased bacterial killing and granular protein content, leading to an increased risk of invasive bacterial infection. Vitamin A is required for the development of neutrophils, and vitamin A defi-ciency impairs chemotaxis and bacterial killing by neutrophils.
One potential mechanism by which vitamin A deficiency may influence immune function is through impairment of the mucosal epithelial barrier and granulocyte function. When granulocyte function is diminished in newborns there is an increased risk of invasive bacterial infection in vitamin A-deficient infants. Vitamin A supplementation may improve survival in the short term, but the long-term effects are not known. Vitamin A deficiency also affects NK cells number and function by decreasing the abundance of NK cells and
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

10
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
impairing their cytotoxic activity. All these impairments could lead to an increased risk of viral and other infections in vitamin A-deficient infants.
Vitamin A deficiency may also affect macrophages in other ways including altered toll-like receptor (TLR) signalling, thus increasing the risk of infec-tions by impairing immune responses. “Impaired” macrophage function in neonates can increase the risk of intracellular infections (e.g. tuberculo-sis). However, infants are normally protected by passive immunity from the mother and this “impaired” immune function may not necessarily be a prob-lem. “Impairment” may prevent pathological inflammation in newborns. Vitamin A supplementation may reinforce some of the “impairments” of macrophage function (e.g. decreased interleukin-12 production) that may be reversed by proinflammatory effects of vitamin A deficiency.
Unactivated dendritic cells (a type of APC) wait in tissues for something to happen (i.e. exposure to pathogenic organisms). In general, APCs from neonates are less effective in presenting antigen than are adult APCs. How-ever, when activated by exposure to microorganisms, dendritic cells become functional APCs. In newborns, as in adults, APCs are capable of presenting antigen to T-cells when appropriately activated. Under strong inflammatory activation (e.g. by bacillus Calmette-Guérin (BCG), measles, or oral polio vaccines), dendritic cells will differentiate, process and present antigens to T-cells. Another potential mechanism by which vitamin A deficiency may influence immune function is by counteracting the normally hyporesponsive state of neonatal APCs. Thus vitamin A deficiency may lead to an increased development of “inflammatory” T-cells and supplementation may diminish or counteract this proinflammatory effect, which could be deleterious to the infant because hyporesponsive APCs may impair clearance of pathogenic organisms. Again, this “impaired” immune function (see above) may not necessarily be a problem as the supplementation may reinforce some of the “impairments” by preventing dendritic cell maturation. However, retinoic acid plus an inflammatory stimulus may lead to dendritic cell maturation and then “mature” antigen presentation. Therefore, vitamin A supplements giv-en along with BCG, measles or oral polio vaccines may promote rather than impair responses to such vaccines by promoting dendritic cell development. This may not happen with alum-adjuvanted vaccines.
Adaptive immunity is provided by T-cells, B-cells and antibody response, lymphocyte recirculation and mucosal responses. Thymic production of “new” T-cells peaks in the first few months of life. Vitamin A deficiency may impair thymic development resulting in long-term loss of T-cell diversity, thus potentially diminishing efficacy of adaptive immune response to future infec-tions, but supplementation at birth may improve long-term survival because of the high activity of the thymus in first few months of life.
Neonatal T-cells are less responsive via T-cell receptors (TCRs) than adult

11
T-cells. There is a lower expression of TCR complex proteins and lower expres-sion of co-stimulation receptors. More robust co-stimulation during antigen presentation corrects this low response. Lower expression of cell-adhesion molecules, causing less avid interaction of T-cells with dendritic cells, also occurs. In addition, more neonatal than adult T-cells follow the “default” path to Th2 development (this can also be seen as a failure to develop a strong Th1 phenotype, which is common in adults); however, appropriate vaccine adju-vants have been found to overcome this Th2 bias.
Retinoic acid is a key regulator of mucosal immunity to pathogens and tol-erance to commensal gut flora. The “impaired” innate and adaptive immune responses of neonates may allow for the establishment of gut flora that is “tol-erated”. Development of regulatory T-cells (Treg) during this period depends on vitamin A (see Annex 2 for details). Vitamin A supplementation at birth reinforces Th2 and Treg development in response to colonization of the gut with commensal flora, which may prevent “damaging” inflammation in the short term, promotes Th2 responses against mucosal pathogens, and allows for the development of Tregs for long-term “tolerance” of gut flora and pre-vention of damaging inflammation, and disruption of barrier and systemic inflammation.
Thus neonatal vitamin A supplementation (in those at risk of vitamin A deficiency) may decrease risk of mortality “later” in life through:
n reinforcing the “anti-inflammatory tone” of the immune system during the period of maternal immunological protection;
n establishing an adequate commensal relationship with gut flora by mini-mizing inflammation in the short term and allowing development of Treg populations and other aspects of mucosal immunity, through decreased chronic inflammation and/or decreased risk of invasive bacterial infec-tions;
n allowing adequate T-cell development in the thymus.
Summary of discussion on the presentation
Comments focused on gut flora development with vitamin A supplementa-tion. Is there a point of conversion for commensal flora in pathogens and the response of neonates to sepsis? There was some discussion on differences in vitamin A content in colostrum of malnourished and well-nourished women, indicating that concentrations are satisfactory in colostrum, at the expense of the mother, but this may not be true in the worst of situations. Prelacteal introduction (honey, sugar) in Bangladesh that could push back breastfeeding for 2–3 days, which could have a profound effect, was also mentioned.
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

12
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
n Metabolism of vitamin A in early life(Presented by A. Catharine Ross)
Despite decades of research, information on vitamin A metabolism, in partic-ular regulation, is still incomplete, while that on neonatal vitamin A metabo-lism is fragmentary. At birth, vitamin A levels in plasma and tissues are low in animals and humans, and the early months of life are considered critical for building up vitamin A stores to a level that will be sufficient for the prevention of vitamin A deficiency in the post-weaning period.
Dietary and supplemental vitamin A (retinol or retinyl esters) is essential-ly inert until metabolized into its active forms. Retinal is critical for vision, while serving as a transient intermediate in other tissues. Retinoic acid (RA) signalling is critical for cell differentiation in all tissues. Vitamin A metab-olism is highly dependent on proteins that function as chaperones, such as nuclear retinoid receptors (Table A2.2.1), cellular retinoid-binding proteins and enzymes that catalyse the metabolic transformation of retinoids. In utero, factors involved in retinoid metabolism are already expressed. In studying embryonic development, we have learned that limiting the production of RA appears to be as important as generating it, RA needs to be in the right place at the right time and that there is functional redundancy in the retinoid meta-bolic system; few factors are absolutely essential for development.
Vitamin A absorption, storage, transport and elimination depend on adequate functioning of the pancreas, intestine, liver and kidneys, as well as the metabolism of retinol to retinal and RA in appropriate target organs. RA is metabolized rapidly, usually within hours, and a constant resupply is necessary to maintain tissue levels. It is hypothesized that organs such as the intestinal mucosa, respiratory system and immune system that continue to undergo significant maturation in the postnatal period have a higher require-ment for RA, and thus for vitamin A, compared with less rapidly turning over tissues. Conversely, the proper metabolism of vitamin A is likely to depend on adequate organ maturation.
Comparison of the levels of retinol and its transport proteins, RBP and transthyretin (TTR), in cord blood or newborn plasma with maternal plasma have consistently shown lower levels in neonates, even in studies in industri-alized countries where vitamin A deficiency is unlikely. Levels are lower still in preterm neonates. Even in countries not known for vitamin A deficiency, a substantial proportion of infants are born with plasma retinol concentrations <0.70 µmol/L, a value often considered indicative of vitamin A deficiency in older children. But in neonates, this value is not so unusual and its use as an indicator of deficiency needs further validation.
The neonatal liver contains little vitamin A and thus the accumulation of retinyl esters in the hepatic stellate cells (HSCs) has barely begun at birth.

13
However, HSCs are abundant in neonatal liver, suggesting that a lack of this storage cell is not an explanation of the low neonatal liver vitamin A con-tent. The liver concentration of vitamin A, a “gold standard” for the assess-ment of vitamin A status, is often deficient in adults if the level is <20 µg/g liver (70 nmol/g liver), but whether this value is meaningful in neonates is yet unproven.
The amount of vitamin A that neonates receive from colostrum and milk depends significantly on the mother’s vitamin A nutritional status. Overall, breast milk vitamin A levels reflect the mother’s recent diet or supplementa-tion status more than it does her long-term stores as indicated by liver vitamin A concentrations. Nearly all vitamin A in milk is present as retinyl ester, which must be digested to retinol in the lumen of the intestine prior to absorption. Digestion depends on adequate pancreatic function and adequate lipid intake for micellization of the retinol and other products of lipid digestion. Although the exocrine pancreas is immature at birth, neonates are able to digest and utilize milk, a high-fat food. Lipases in milk (e.g. lingual lipase) facilitate triglyceride hydrolysis; however, the enzymology of retinyl ester hydrolysis is still not completely understood. Although direct studies in neonates are lacking, luminal digestion of retinyl ester is not likely to be rate-limiting for vitamin A utilization and neonatal adaptations leading to larger chylomicrons (CMs) should facilitate lipid and vitamin A absorption. Retinol then under-goes intestinal uptake, re-esterification within enterocytes, requiring cellular RBP (CRBP) II and lecithin retinol acyltransferase (LRAT). The retinyl esters are then packaged into the lipid core of the nascent CMs that transport dietary fat. The CM particle is released into the intestinal lymphatic system within the lamina propria, then transported through the lymph ducts to the venous blood stream. Enterocyte maturation is dependent on vitamin A, as there is evidence that RA is important for adaptation of the gut following bowel resec-tion, including an increase in crypt cell proliferation and villus height, leading to an increased area of the absorptive mucosal surface.
In summary, postprandial vitamin A metabolism appears to be adequate in neonates for uptake of vitamin A.
Hepatic metabolism
The liver does not obtain full maturity until 2 years after birth. Preterm infants are at special risk of hepatic decompensation because their immatu-rity results in a delay in achieving normal detoxifying and synthetic function. Postnatal development includes enlargement of the hepatocytes, expansion and multiplication of the liver lobules, and pronounced changes in sinusoidal structure. Vitamin A is stored in the adult liver primarily as retinyl esters in lipid droplets within perisinusoidal HSCs, which are abundant in human
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

14
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
liver <1 month of age, but then decrease by 6 months. Depletion of vitamin A from the liver appears to be a slow process; however, the HSC vitamin A con-tent in the postprandial period can increase rapidly following repletion with a large dose of vitamin A. LRAT activity in the liver is sensitive to vitamin A status, declining at the same rate as plasma retinol declines during vitamin A deficiency. However, LRAT activity in the intestine is not regulated in this manner and does not fall during vitamin A deficiency. The LRAT activity in the intestine seems to be maintained and ready to absorb and esterify vitamin A whenever it becomes available.
RBP accumulates in the liver during vitamin A deficiency and there is a rapid release of holo-RBP into the plasma after administration of even a small amount of vitamin A, and this is the biological rationale for the relative dose response (RDR) test. The rate of RBP synthesis also affects plasma retinol levels. Plasma RBP levels are sensitive to the individual’s nutrient status with respect to protein, calories and other micronutrients such as zinc which affect secretion. RBP synthesis in the liver is also reduced in states of inflammation, even when liver vitamin A is not depleted. Information is lacking on the rate of RBP synthesis in liver of neonates, and therefore it is not known whether RBP accumulates in the neonatal liver as it does in the liver of older animals with similar low hepatic vitamin A stores. The liver also plays a major role in eliminating vitamin A and its metabolites through processes of oxidation and conjugation. It is unknown whether conjugation of retinoid metabolites is slow to develop in the liver of neonates as has been demonstrated for bilirubin conjugation. In most peripheral tissues, retinyl esters are the most abundant form of vitamin A. The uptake of retinol from plasma into cells appears to be mediated by STRA6, a receptor for RBP, and possibly a number of other pathways yet to be identified.
Based on our current understanding of vitamin A metabolism in general, it is anticipated that vitamin A given as an oral supplement will undergo rapid metabolism in the postprandial period, not only in the intestine but also in the liver and some extrahepatic organs. The absorption of vitamin A is consid-ered to be generally efficient in neonates that have no problems with fat diges-tion or absorption and who are consuming adequate dietary fat along with the dose of vitamin A. Since LRAT activity is also high in the small intestine and is not reduced during deficiency, it seems that vitamin A is readily absorbed when administered along with an appropriate amount of fat. It is also antici-pated that the CM remnant vitamin A uptake into the liver occurs at a normal rate regardless of vitamin A status; however, the transfer of retinol to the HSC and its use for retinyl ester formation might be low in the vitamin A-deficient state. A portion of newly absorbed retinol will probably appear in plasma as holo-RBP. The RDR test may be able to provide information on liver vitamin A adequacy, but we do not know enough about the basic regulation of RBP

15
synthesis and secretion in the liver of neonates to know whether the RDR test can be interpreted in the same way for neonates as for older children.
Summary of discussion on the presentation
Comments were made about STRA6 in the eye and lung and about the impor-tance or need for continued instead of single dose (50 000 IU) vitamin A supplementation. STRA6, identified as a receptor for RBP (4), is expressed in the retinal pigment epithelium. It is believed to work by first taking up reti-nol, which is then esterified by LRAT to form a storage pool of retinyl ester. The retinyl ester can be converted to retinal for vision. In the lung, STRA6 is expressed in a developmentally regulated manner, and its expression is increased by retinoic acid (5). STRA6 and LRAT may be important in the lung, analogous to the eye, for creating a storage pool of vitamin A that can be drawn on to provide retinol for later use.
Additional comments were made regarding feeding after supplementation. The sooner the baby can be given supplements, the sooner we can suggest breastfeeding.
n Mammalian models for understanding mechanisms of retinol and retinol actions(Presented by Sherry Tanumihardjo)
Animal models are very useful for the study of vitamin A, but selection of the appropriate model is important and should be based on the specific needs of the question being asked, budget and availability of facilities. The following models are commonly used to study vitamin A metabolism:
n Mice: Useful for the study of RBPs, immune function and cancer; however, they are not a good model for carotenoids (mice do not absorb carotenoids intact) or vitamin A deficiency (mice are not easily depleted). Knockout models have been widely used for RBP studies, and it has been found that mice lacking RBP do not maintain normal vision because of impaired vita-min A circulation in the body. This model has also shown that CM-derived retinyl esters are important for meeting target tissue needs.
n Rats: Useful for the study of vitamin A deficiency since they are rapidly depleted (35 days), but are not good for the study of carotenoids (rats do not absorb carotenoids intact), immune function or cancer.
n Gerbils: Excellent model for the study of carotenoids (gerbils absorb carote-noids partially intact), but they do not absorb lutein or zeaxanthin. Gerbils are not a good model for vitamin A deficiency, immune function or cancer.
n Swine: Excellent model for vitamin A deficiency (can be depleted) and lac-tation (sows produce a lot of milk, 9–13 L). Swine are not good models for the study of carotenoids, immune function or cancer. Swine have gastroin-
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

16
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
testinal tracts and digestive physiology that are similar to the human, and piglets and human infants have anatomical similarities. The test doses of vitamin A used in swine are similar to humans and one can collect swine liver to assay vitamin A directly. An additional benefit of the use of swine is that a sufficient number of piglets are available in each litter for testing a variety of treatments.
n Non-human primates: Good model for studying hypervitaminosis A and age-related macular degeneration, and can be a good model for studying carotenoid metabolism, depending on the species. Monkeys that are given vitamin A every day in their chow have been found to have too much vita-min A in their liver, similar to someone who is taking vitamin A supple-ments daily. If the mother receives vitamin A during pregnancy, her babies will have high liver vitamin A reserves at birth that are sometimes close to the upper limits.
In studying the various outcomes of vitamin A interventions, the modified relative dose response (MRDR) test is often used. It is more sensitive than serum retinol concentrations, only requires a single blood sample, uses high-performance liquid chromatography (HPLC) for analysis and it is easy to interpret the results. As the liver is depleted of vitamin A during times of low dietary intake, apo-RBP begins to accumulate. After a challenge dose of either retinyl ester or dehydroretinyl ester, the retinol or dehydroretinol binds to the accumulated RBP and is transported out into the serum.
High doses of retinyl ester are commonly provided to at-risk populations in areas where vitamin A deficiency is a problem. For women, 400 000 IU given as two doses of 200 000 IU at least 1 day apart and within 6 weeks postpartum are being recommended. Vitamin A supplementation programmes have been highly successful in addressing vitamin A deficiency but are not without risk. The doses administered are at toxic levels (200 000 IU retinyl ester is 85 times the recommended daily allowance (RDA) and 400 000 IU retinyl ester is 172 times the RDA). Acute toxicity may occur at dosages >100 times the adult RDA.
A study of high-dose vitamin A supplementation in Ghanaian women determined the length of time mothers are protected postpartum against vita-min A depletion after receiving either 400 000 IU vitamin A in two divided doses or one 200 000 IU and a placebo dose 24 hours apart. Mean baseline serum retinol concentrations and MRDR values were 1.4 ± 0.5 μmol/L and 0.048 ± 0.037 μmol/L, respectively. Using a repeated measures analysis of variance (ANOVA) with fixed effects, the post-treatment MRDR values at 1, 3 and 5 months after treatment were significantly lower than baseline (P < 0.0001), and there were no two- or three-factor interactions. Furthermore, all women were protected out to 5 months when treated either with 200 000 or 400 000 IU vitamin A.

17
Questions still remain regarding whether malnourished individuals receiving vitamin A supplements have a similar capacity for regulating retinol and retinoic acid, and whether hepatic vitamin A that is efficiently stored by supplemented mothers can be readily mobilized for long-term secretion into breast milk to benefit the breastfed infant.
The recommended intake of vitamin A for infants ranges from 350 μg/day (WHO) to 400–500 μg/day (USA). Assuming an average milk intake of 700 mL/day, a 200 000 IU vitamin A dose given to a breastfeeding mother would provide 5250 μg vitamin A to the breastfed infant over the first 48 hours after administration. A 400 000 IU vitamin A dose given to a breastfeeding mother would provide 10 750 μg vitamin A to the breastfed infant over the first 48 hours after administration. In unsupplemented mothers, only 710 μg vitamin A would be provided to the breastfed infant over 48 hours. Assuming a 50% hepatic storage factor for a 3 kg infant, the high and low dose given to the mother may elevate the vitamin A status of the infant from poor to ade-quate (≥0.07 μmol/g). The nursing infants of these mothers would be expected to increase their liver reserves of vitamin A by a mean of 0.08 mmol/g for the low dose and 0.16 mmol/g for the high dose of vitamin A. This is only a pre-diction though, and the actual increase in vitamin liver reserves in the infant and the extent to which maternal vitamin A supplementation improves infant liver stores of vitamin A are not known.
Swine studies have revealed that derivative retinoid metabolites are observed in serum following vitamin A supplementation, serving as an anti-toxic mechanism. Large doses of vitamin A did not result in long-term milk enrichment and liver reserves were not different between piglets from low- and high-dosed sows. There was no added benefit to the infant when the larger (400 000 IU versus 200 000 IU) maternal dose was used and there was no added benefit to infant when 100 000 IU vitamin A was administered orally over 50 000 IU.
Summary of discussion on the presentation
An important concern in using animal models for the study of vitamin A is the selection of the right model. Dr Tanumihardjo indicated that selection depends on the question. Swine is a good model for vitamin A but not for carotenoids. The issue about the right dose to be tested was raised again, indi-cating that perhaps 25 000 IU vitamin A is enough. Most participants agreed at this point that this is a working question. There was some concern about the use of retinyl acetate in experiments. Apparently it is not different from palmitate, but all participants agreed that the same chemical form of vitamin A should be used in the currently proposed studies to facilitate comparisons between future studies.
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

18
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
n Causes of mortality in the neonatal period and first half of infancy(Presented by Mathilde Savy and Andrew M. Prentice)
Background
Of the 130 million babies born every year worldwide, ~5–6 million die in the first year of life (infancy), and >4 million die in the neonatal period. Three-quarters of the neonatal deaths occur in the first week of life, and >1 million occur in the first 24 hours. Virtually all (99%) of the neonatal deaths occur in low- and middle-income countries, particularly in South-East Asia (36%) and Africa (28%). The average neonatal mortality rate in these regions is 38 and 44 per 1000 live births, respectively.
The counting and identification of causes of death is essential to help design public health strategies. However, live births and deaths of newborns are largely underreported in poor communities, particularly stillbirths or deaths occurring shortly after birth. Information on the causes of death in these areas is also very limited, and often has to be estimated from incomplete data. As later deaths decline at a proportionately faster rate, the design and prior-itization of interventions to reduce the total burden of preventable deaths in low-income countries requires more robust data on the neonatal period.
Among the interventions demonstrated to enhance maternal and child survival (6), vitamin A supplementation in children 6–59 months of age has been recognized as a cost-effective intervention, significantly reducing all-cause mortality in this age group. Although the neonatal period is a critical window where infection rates, nutritional deficiencies and mortality rates are high, the issue of whether such intervention in newborns would also be ben-eficial for early-life survival remains unresolved.
This report focuses on the potential impact of neonatal vitamin A supple-mentation on infant survival. The report will:
n present and analyse neonatal and infant mortality trends over time;n describe the main causes of mortality in children <5 years of age, infants
and neonates globally and by region;n identify the number of deaths that could be potentially avoidable by neona-
tal vitamin A supplementation.
Definitions
n Neonatal mortality: the number of deaths occurring within 0–28 days of birth. This may be subdivided into early, i.e. death occurring within the first 7 days of life, and late neonatal mortality, i.e. death occurring between 7 and 28 days of life.
n Perinatal mortality: the perinatal period begins at 22 completed weeks of

19
gestation and ends 7 completed days after birth. Perinatal mortality there-fore refers to deaths that occur in the early neonatal period, plus fetal deaths or stillbirths.
n Infant mortality: deaths occurring in the first year of life (including neona-tal deaths but not stillbirths).
Data sourcesCounting deathsSince vital registration coverage and hospital-based deliveries are both low in developing countries, information on most neonatal and infant deaths derives from community surveys, including the Demographic and Health Surveys (DHS). For countries where there are no reliable population-based data, death rates have to be estimated using statistical modelling. This paper mainly uses data published in WHO and United Nations Children’s Fund (UNICEF) reports, where child mortality data by region or country have been computed. We also used the WHO Statistical Information System (WHOSIS) (7). This database provides a comprehensive summary of the most basic health out-comes, including mortality, for each Member State, including levels and major causes of child and adult mortality. For estimation of numbers of neonatal deaths by country, WHO reported that <5% of estimates come from vital reg-istration (for countries where coverage is high), 75% from DHS data and 20% are derived from estimates of deaths of children <5 years of age adjusted for HIV prevalence. It should be noted that the DHS data generally cover a lim-ited area of a country and may not be representative; nonetheless when aggre-gating data by region, as in this summary, this limitation becomes less critical.
Causes of deathsCauses of death may be collected through vital registration systems with med-ical certification, verbal autopsy, deaths in hospital with certification of cause of death, disease/injury registries or epidemiological research studies.
Data limitations
Data were not available for some countries, or for some years. Latest esti-mates for certain countries dated back to 1997. Although data were readily accessible for newborn, infant and <5 mortality, most reports/databases do not distinguish between the early and late neonatal period. Similarly, it was almost impossible to obtain information on the 1–6 month period, which was pertinent to the question of whether neonatal vitamin A supplementation may improve survival in the period before the first vitamin A supplement is received under the current protocol.
The quality of the data obtained might also have been poor, especially in developing countries where most deaths are not registered, and when they are,
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

20
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
the cause is very often assessed through interviews only. Mortality rates may also cause concern, since reliable population-based data are rarely available, and methods used to produce estimates may vary widely, as may definitions of terms, leading to misclassification and possible over- or under-estimation of mortality rates.
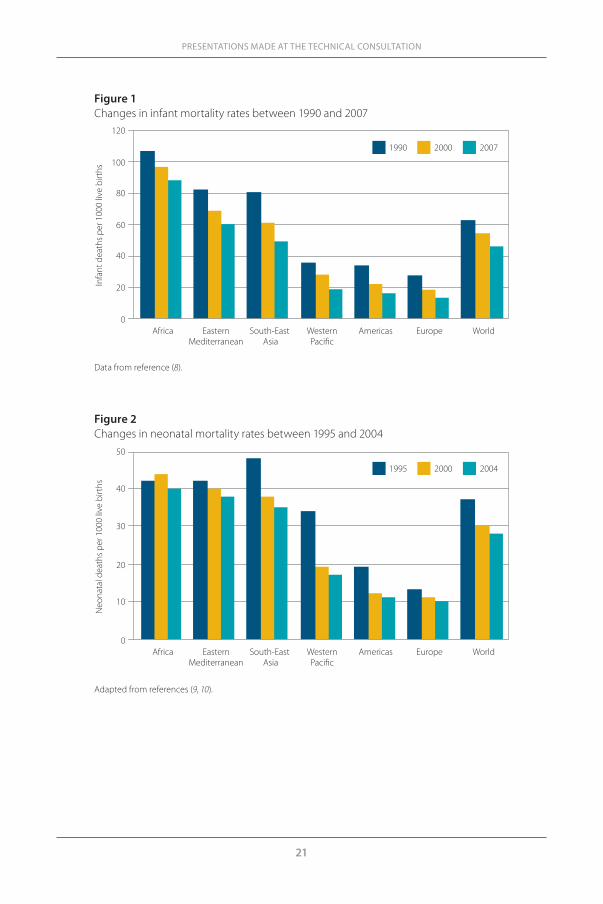
ResultsNeonatal and infant mortality trendsInfant mortality has significantly decreased worldwide and in all regions over recent decades. Globally, the infant mortality rate has fallen from 60 to 40 infant deaths per 1000 live births between 1990 and 2007 (Figure 1). How-ever, in regions such as Africa, Eastern Mediterranean and South-East Asia, mortality rates remains very high (more than 80 deaths per 1000 live births in Africa in 2007). The UN Millennium Development Goal 4 (MDG 4) commit-ment was to reduce childhood mortality by two-thirds between 1990 and 2015 but, at the current rates, this goal will not be reached before 2045.
The neonatal mortality rate has also substantially reduced – from 38 to 28 deaths per 1000 live births between 1995 and 2004 (Figure 2). Decreases in neonatal mortality have been particularly sharp in South-East Asia and the Western Pacific Region between the years 1995 and 2000 (approximately 20% and 44% reduction, respectively). However, in Africa, the decrease in neo-natal mortality is smaller than in the other regions. In sub-Saharan Africa, neonatal mortality is decreasing much more slowly than infant and <5 mor-tality (data not shown). The neonatal mortality rate even increased between 1995 and 2000, and was still as high as 40 deaths per 1000 live births in 2004. Reducing neonatal deaths in high-mortality countries is among the top pri-orities of the United Nations and a core objective of MDG 4.
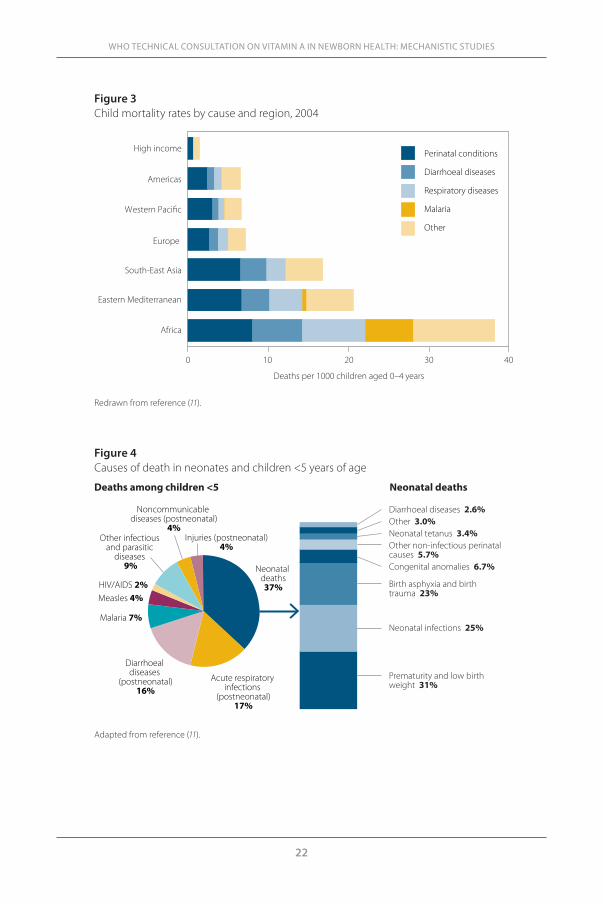
Causes of mortality in different age groupsAs overall <5 mortality declines, perinatal mortality increases as a propor-tion (Figure 3). Perinatal mortality approximately represents one-quarter of the deaths in Africa, and one-third to one-half of the deaths in the Americas, Western Pacific and Europe. In Africa, other major causes of deaths in chil-dren <5 are diarrhoea, respiratory diseases and malaria; the category ‘other’ includes measles, HIV/AIDS, other infections and parasitic diseases, postneo-natal injuries and noncommunicable diseases.
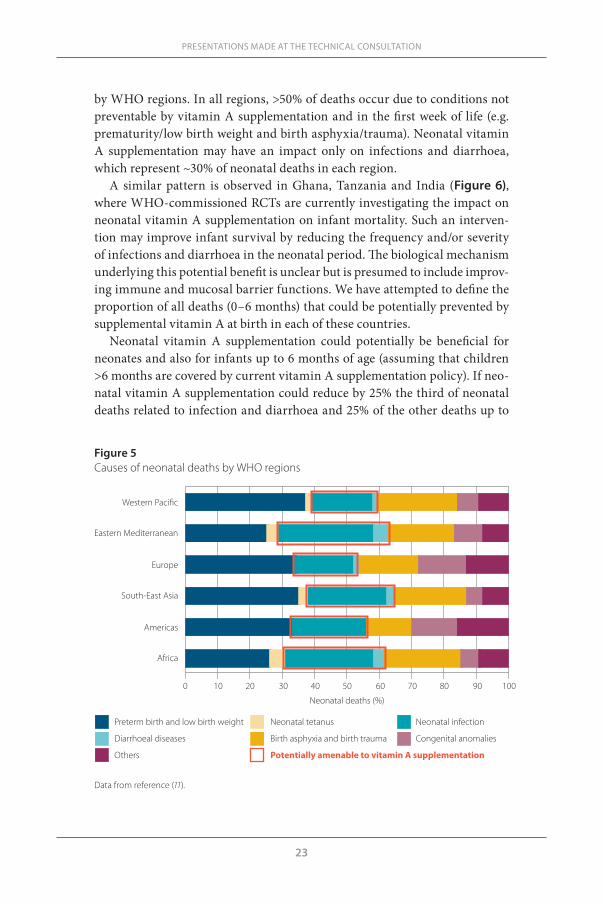
Neonatal deaths account for 37% of deaths among children <5 (Figure 4). These newborns mainly die from prematurity and low birth weight (31%), neonatal infections (25%), birth asphyxia (lack of oxygen at birth) and birth trauma (23%). A non-negligible proportion of newborns die from congenital anomalies (6.7%), other non-infectious perinatal causes (5.7%), tetanus (3.4%) and diarrhoeal diseases (2.6%). Figure 5 shows the causes of neonatal deaths
21
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION
Figure 1Changes in infant mortality rates between 1990 and 2007
Data from reference (8).
0
20
40
60
80
100
120
Africa
Infa
nt d
eath
s per
100
0 liv
e bi
rths
EasternMediterranean
South-East Asia
WesternPaci�c
Americas Europe World
1990 2000 2007
Figure 2 Changes in neonatal mortality rates between 1995 and 2004
Adapted from references (9, 10).
0
10
20
30
40
50
Africa
Neo
nata
l dea
ths p
er 1
000
live
birt
hs
EasternMediterranean
South-East Asia
WesternPaci�c
Americas Europe World
1995 2000 2004
22
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
Figure 3Child mortality rates by cause and region, 2004
Africa
Eastern Mediterranean
South-East Asia
Europe
Western Pacic
Americas
High income
0 10 20 30 40
Deaths per 1000 children aged 0–4 years
Perinatal conditions
Diarrhoeal diseases
Respiratory diseases
Malaria
Other
Redrawn from reference (11).
Figure 4Causes of death in neonates and children <5 years of age
Adapted from reference (11).
Neonatal deaths37%
Acute respiratory infections
(postneonatal)17%
Diarrhoeal diseases
(postneonatal)16%
Malaria 7%
Measles 4%HIV/AIDS 2%
Other infectious and parasitic
diseases 9%
Noncommunicable diseases (postneonatal)
4%Injuries (postneonatal)
4%
Diarrhoeal diseases 2.6%Other 3.0%Neonatal tetanus 3.4%Other non-infectious perinatal causes 5.7%Congenital anomalies 6.7%
Birth asphyxia and birth trauma 23%
Neonatal infections 25%
Prematurity and low birth weight 31%
Deaths among children <5 Neonatal deaths
23
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION
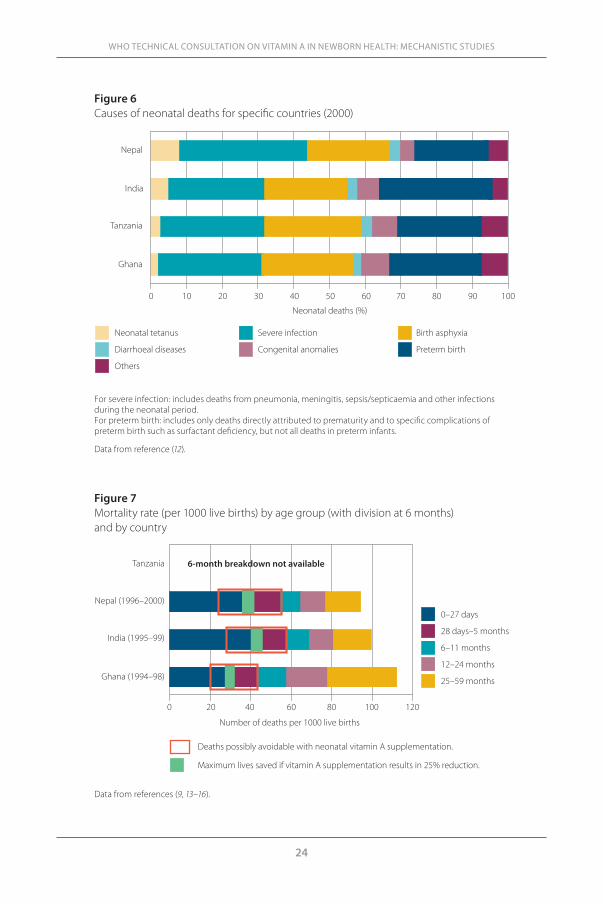
by WHO regions. In all regions, >50% of deaths occur due to conditions not preventable by vitamin A supplementation and in the first week of life (e.g. prematurity/low birth weight and birth asphyxia/trauma). Neonatal vitamin A supplementation may have an impact only on infections and diarrhoea, which represent ~30% of neonatal deaths in each region.
A similar pattern is observed in Ghana, Tanzania and India (Figure 6), where WHO-commissioned RCTs are currently investigating the impact on neonatal vitamin A supplementation on infant mortality. Such an interven-tion may improve infant survival by reducing the frequency and/or severity of infections and diarrhoea in the neonatal period. The biological mechanism underlying this potential benefit is unclear but is presumed to include improv-ing immune and mucosal barrier functions. We have attempted to define the proportion of all deaths (0–6 months) that could be potentially prevented by supplemental vitamin A at birth in each of these countries.
Neonatal vitamin A supplementation could potentially be beneficial for neonates and also for infants up to 6 months of age (assuming that children >6 months are covered by current vitamin A supplementation policy). If neo-natal vitamin A supplementation could reduce by 25% the third of neonatal deaths related to infection and diarrhoea and 25% of the other deaths up to
Data from reference (11).
Figure 5 Causes of neonatal deaths by WHO regions
0 10 20 30 40 50 60 70 80 90 100
Africa
Americas
South-East Asia
Europe
Eastern Mediterranean
Western Paci�c
Neonatal deaths (%)
Preterm birth and low birth weight Neonatal tetanus Neonatal infection
Diarrhoeal diseases Birth asphyxia and birth trauma Congenital anomalies
Others Potentially amenable to vitamin A supplementation
24
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
Data from references (9, 13–16).
Figure 7Mortality rate (per 1000 live births) by age group (with division at 6 months) and by country
0 20 40 60 80 100 120
Ghana (1994–98)
India (1995–99)
Nepal (1996–2000)
Tanzania 6-month breakdown not available
Deaths possibly avoidable with neonatal vitamin A supplementation.
Maximum lives saved if vitamin A supplementation results in 25% reduction.
Number of deaths per 1000 live births
0–27 days
28 days–5 months
6–11 months
12–24 months
25–59 months
Figure 6 Causes of neonatal deaths for specific countries (2000)
For severe infection: includes deaths from pneumonia, meningitis, sepsis/septicaemia and other infections during the neonatal period. For preterm birth: includes only deaths directly attributed to prematurity and to specific complications of preterm birth such as surfactant deficiency, but not all deaths in preterm infants.
Data from reference (12).
Ghana
Tanzania
India
Nepal
0 10 20 30 40 50 60 70 80 90 100
Neonatal deaths (%)
Neonatal tetanus Severe infection Birth asphyxia
Diarrhoeal diseases Congenital anomalies Preterm birth
Others

25
6 months of age (Figure 7), this would amount to 3–6 lives saved per 1000 live births. Although these figures may seem small, in South-East Asia and Afri-ca this intervention could actually save the lives of 0.2–0.4 million children. Considering the low cost of vitamin A capsules, it is worth testing neonatal vitamin A supplementation as an ancillary measure alongside other interven-tions, including improved skilled care in pregnancy, at birth and postnatally for a substantive improvement of infant health and survival.
Key messages
n In Africa and South-East Asia 45–60 infants/1000 die within the first 6 months of life.
n As <5 and infant mortality rates decline, neonatal deaths assume a higher proportion of the total mortality due to slower declines in neonatal deaths.
n About two-thirds of neonatal deaths result from causes that are not pre-ventable/treatable by neonatal vitamin A supplementation (e.g. congenital anomalies, prematurity, asphyxiation).
n If neonatal vitamin A supplementation could reduce the remaining one-third of neonatal deaths and all other deaths up to 6 months of age by 25%, this would amount to 3–6 lives saved per 1000 live births. (Note: This is the maximally optimistic estimate assuming that deaths after 6 months of age are covered by the current vitamin A supplementation policy.)
n This could amount to 0.2–0.4 million children’s lives saved in South-East Asia and Africa.
n The large proportion of perinatal deaths already “predestined” by pregnancy- related and obstetric problems limit the potential efficacy of postnatal inter-ventions and also require urgent attention.
PRESENTATIONS MADE AT THE TECHNICAL CONSULTATION

26
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
References1. Gogia S, Sachdev HS. Neonatal vitamin A supplementation for prevention of
mortality and morbidity in infancy: systematic review of randomised controlled trials. British Medical Journal, 2009, 228:b919.
2. Shenai JP et al. Clinical trial of vitamin A supplementation in infants susceptible to bronchopulmonary dysplasia. Journal of Pediatrics, 1987, 111: 269–277.
3. Tyson JE et al. Vitamin A supplementation for extremely-low-birth-weight in-fants. National Institute of Child Health and Human Development Neonatal Research Network. New England Journal of Medicine, 1999, 340:1962–1968.
4. Kawaguchi R et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science, 2007, 315:820–825.
5. Wu L, Ross AC. Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT, and CYP26B1 expression in neonatal lung. Journal of Lipid Research, 2010, 51:378–387.
6. Bhutta ZA et al. What works? Interventions for maternal and child undernutri-tion and survival. Lancet, 2008, 371:417–440.
7. WHO statistical information system. (http://www.who.int/whosis/en/, accessed 24 November 2010).
8. World health statistics 2009. Geneva, World Health Organization, 2009. (http://www.who.int/whosis/whostat/2009/en/index.html, accessed 4 October 2011).
9. The World Health Report 2005: make every mother and child count. Geneva, World Health Organization, 2005. (http://www.who.int/whr/2005/en/index.html, accessed 4 October 2011).
10. Ahman E, Zupan J. Neonatal and perinatal mortality: country, regional and glob-al estimates, 2004. Geneva, World Health Organization, 2007. (http://whqlibdoc.who.int/publications/2007/9789241596145_eng.pdf, accessed 4 October 2011).)
11. The global burden of disease: 2004 update. Geneva, World Health Organiza-tion, 2008. (http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf, accessed 4 October 2011).
12. Mortality country fact sheet 2006: Ghana, India, Nepal. Geneva, World Health Organization, 2006. (http://www.who.int/whosis/mort/profiles/en/index.html, accessed 4 October 2011).
13. Ministry of Health and Population [Nepal], New ERA and Macro International. Nepal Demographic and Health Survey 2006. Kathmandu, Ministry of Health and Population, New ERA, and ORC Macro, 2007.
14. International Institute for Population Sciences (IIPS) and Macro International. National Family Health Survey 2005/2006. Mumbai, IIPS and ORC Macro, 2007.
15. Ghana Statistical Service (GSS), Ghana Health Service (GHS), and Macro Inter-national. Ghana Demographic and Health Survey 2008. Accra, GSS, GHS, and ORC Macro, 2009.
16. National Bureau of Statistics (NBS) [Tanzania] and ORC Macro. Tanzania De-mographic and Health Survey 2004–05. Dar es Salaam, NBS and ORC Macro, 2005.

27
annex 1List of participants


29
ANNEX 1. LIST OF PARTICIPANTS
A. Experts
Dr María Nieves García-Casal (rapporteur)
Centro de Medicina Experimental, Laboratorio de Fisiopatología
Instituto Venezolano de Investigaciones Científicas
Caracas, Bolivarian Republic of Venezuela
Ms Alison GreigMicronutrient Initiative (MI)Ottawa, Canada
Dr Alain LabriqueDepartments of International Health
and EpidemiologyProgram in Global Disease
Epidemiology and ControlBloomberg School of Public HealthJohns Hopkins UniversityBaltimore, United States of America
Dr Samuel NewtonKintampo Health Research CentreKintampo, Ghana
Dr Ellen G. Piwoz Integrated Health Solutions
DevelopmentGlobal Health ProgramBill & Melinda Gates FoundationSeattle, United States of America
Professor Andrew M. PrenticeMRC International Nutrition GroupNutrition and Public Health
Intervention Research UnitLondon School of Hygiene and Tropical
MedicineLondon, England
Dr A. Catharine Ross (by phone)Department of Nutritional SciencesPennsylvania State UniversityUniversity Park, United States of
America
Dr Mathilde SavyMRC International Nutrition GroupNutrition and Public Health
Intervention UnitLondon School of Hygiene and Tropical
MedicineLondon, England
Dr Andrew SerazinHuman BiologyBill & Melinda Gates FoundationSeattle, United States of America
Dr Jayant P. ShenaiDivision of NeonatologyVanderbilt University School of
MedicineVanderbilt Children’s HospitalNashville, United States of America
Dr Charles B. StephensenUSDA Western Human Nutrition
Research CenterUniversity of California – Davis Davis, United States of America
Dr Sherry TanumihardjoDepartment of Nutritional SciencesUniversity of Wisconsin – MadisonMadison, United States of America
Professor David I. ThurnhamSchool of Biomedical SciencesUniversity of UlsterColeraine, England
Mr Arnold TimmerUNICEF Nutrition SectionNew York, United States of America
Dr Keith P. West JrDepartment of International HealthCenter of Human NutritionJohns Hopkins Bloomberg School of
Public HealthBaltimore, United States of America

30
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
B. WHO Secretariat
Dr Rajiv BahlMedical OfficerNewborn and Child Health and
Development UnitDepartment of Child and Adolescent
Health and Development
Dr Francesco BrancaDirectorDepartment of Nutrition for Health and
Development
Ms Emily CerconeIntern (rapporteur)Micronutrients UnitDepartment of Nutrition for Health and
Development
Ms Sueko MatsumuraInternMicronutrients UnitDepartment of Nutrition for Health and
Development
Dr Juan Pablo Peña-RosasCoordinatorMicronutrients UnitDepartment of Nutrition for Health and
Development
Dr Luz Maria de RegilEpidemiologistMicronutrients UnitDepartment of Nutrition for Health and
Development
Dr Lisa M. RogersTechnical Officer Micronutrients UnitDepartment of Nutrition for Health and
Development

annex 2Background papers


33
a2.1Vitamin A supplementation in newborns: a conceptual framework for biological plausibilityJayant P. Shenai
Department of Neonatology, Vanderbilt Children’s Hospital, Nashville, Tennessee, United States of America
Corresponding author: Jayant P. Shenai; [email protected]
Abstractn Vitamin A is a fat-soluble micronutrient essential for optimal growth. It promotes orderly growth and differentiation of epithelial cells, and its defi-ciency affects various organ systems, including the lung. Vitamin A is trans-ferred from the mother to the fetus, particularly in late gestation, in most animal species, with the plasma vitamin A concentration in the fetus appear-ing to be maintained within a normal range despite variations in the maternal vitamin A status and intake. Preterm infants are susceptible to lung injury from insults such as hyaline membrane disease, trauma from mechanical ven-tilation, oxygen toxicity and airway infection. Bronchopulmonary dysplasia (BPD), a common cause of chronic lung disease, is a major source of mortality and morbidity among these infants. Preterm infants with BPD often manifest clinical, biochemical and histopathological evidence of vitamin A deficiency. In addition to the prevention of prematurity as the primary perinatal health-care goal, strategies for reducing the occurrence of BPD among preterm sur-vivors are clearly warranted, and optimal vitamin A nutrition is one such strategy. Vitamin A supplementation from early postnatal life in these infants not only improves their vitamin A status, but also ameliorates the lung dis-ease, as evidenced by a decreased incidence of BPD and associated morbidity. Although clinical trials to date have significantly informed our understand-ing of the role of vitamin A in BPD among preterm survivors, much remains to be studied.
ANNEX 2: BACKGROUND PAPERS

34
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
IntroductionVitamin A is a fat-soluble micronutrient recognized since 1912 as a con-stituent of diet essential for growth (1, 2). Vitamin A compounds (retinoids) occur in three natural forms: retinol, retinaldehyde (retinal) and retinoic acid. Retinol (vitamin A alcohol) is present in the form of retinyl esters in foods of animal origin, and is also formed in vivo from its precursor β-carotene, which is present in foods of plant origin. Retinyl esters are derived from esterifica-tion of retinol and constitute the main storage forms of vitamin A; retinyl esters in animal and human tissues include principally retinyl palmitate and stearate, and also oleate and linoleate. Retinaldehyde is derived from revers-ible oxidation of retinol and, in combination with various lipoproteins, forms the visual pigment of the retina (3). The photoisomerization of retinaldehyde is necessary for vision (4). Retinoic acid is derived from irreversible oxidation of retinaldehyde in the tissues (5) and is the active metabolite of vitamin A in functions related to growth and differentiation (6).
Vitamin A is transported in plasma as retinol bound to a specific carrier protein, retinol-binding protein (RBP). The human RBP polypeptide chain has ~180 amino acid residues, a molecular weight (MW) of ~21 kD and a sin-gle binding site for one molecule of retinol (7, 8). The RBP is synthesized in the liver and secreted into plasma as the retinol–RBP complex (9), where it interacts with another protein, transthyretin, to circulate as RBP–transthyre-tin complex (9). Transthyretin is a stable, symmetrical tetramer composed of four identical subunits with a MW of ~55 kD (10). Vitamin A is distributed to the target tissues in the form of retinol–RBP complex bound to transthyre-tin, and its cellular uptake depends on a specific plasma membrane receptor that recognizes RBP (11, 12). The mechanisms by which vitamin A circulates within the cell and influences nuclear metabolism, genomic expression, and growth and differentiation of the target tissue remain under investigation.
Vitamin A promotes orderly growth and differentiation of epithelial cells, and its deficiency affects various organ systems, including the lung (13). The histopathological changes in the respiratory system generally precede other consequences of vitamin A deficiency involving the genitourinary system, eye and skin (13). This review focuses on the perinatal aspects of vitamin A metabolism and effects of vitamin A deficiency in relation to lung injury in preterm infants, and provides a rationale for vitamin A supplementation as a means to reduce infant mortality and morbidity associated with chronic lung disease.
Acquisition of vitamin A by the fetusVitamin A is transferred from the mother to the fetus, particularly in late ges-tation, in most animal species (14–20) and RBP synthesized by the fetal liver

35
may help extract vitamin A from the placental circulation. In early gestation, maternal retinol–RBP complex appears to be the predominant source of fetal vitamin A. Other possible sources are swallowed amniotic fluid (21, 22) and maternal lipoproteins, which contain retinyl esters (18).
Human transplacental transfer of vitamin A has been studied by examin-ing paired samples of maternal blood and fetal or umbilical cord blood at vari-ous gestational ages (23–27). The ratio of maternal to fetal concentration of plasma vitamin A in healthy human pregnancies is ~2:1. When maternal vita-min A status is marginal/deficient, fetal plasma vitamin A concentration is often maintained within normal limits and may even exceed maternal plasma vitamin A concentration (23, 28, 29). Conversely, following maternal vitamin A supplementation, the cord blood vitamin A concentration remains similar to that in unsupplemented controls (23, 30, 31). Thus, fetal plasma vitamin A concentration appears to be maintained despite variations in the mater-nal vitamin A status and intake. The regulatory mechanisms underpinning this homeostasis remain unclear, nor is it known whether such mechanisms can compensate successfully for extreme maternal vitamin A deprivation or excess.
Vitamin A status at birthPlasma vitamin A indexes
Plasma vitamin A concentration is the most common biochemical marker of vitamin A status. In healthy human adults, it ranges from 20 to 80 µg/dL (32). In children, including infants, concentrations <20 µg/dL are indicative of vita-min A deficiency (33). Most preterm infants are born with deficient concentra-tions (34, 35). Plasma concentration of RBP is another biochemical marker of vitamin A status. In healthy human adults, this averages 4.6 ± 1.0 mg/dL (36). Concentrations in infants and children are ~60% of adult values (25), and those <2.5 mg/dL are indicative of vitamin A deficiency (37). Again, most preterm infants are born with deficient plasma RBP concentrations (35, 38).
Another useful biochemical marker of vitamin A status is the plasma RBP response to vitamin A administration. This functional measure of vitamin A status is based on the varied rates of secretion of RBP from the liver into the plasma, which depends on the availability of vitamin A (39, 40). In vita-min A-deficient animals, RBP secretion is blocked, resulting in high liver and low plasma concentrations of RBP. When these animals are given vita-min A, hepatic RBP is mobilized, leading to a rapid decrease in the liver and a concomitant increase in the plasma RBP concentration (39). In vitamin A-sufficient animals, RBP does not accumulate in the liver, and minimal or no change in plasma RBP concentration is observed after vitamin A adminis-tration (39). The per cent increase in plasma RBP concentration (∆-RBP) from
ANNEX 2: BACKGROUND PAPERS

36
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
its baseline value in response to vitamin A administration, therefore, is useful for determining the vitamin A status. A high ∆-RBP value (>8%) is indicative of vitamin A deficiency (41). In preterm neonates, the plasma RBP response to vitamin A administration, characterized by high ∆-RBP values, provides fur-ther evidence that these infants are vitamin A deficient at birth (41), probably because of deprivation of transplacental vitamin A supply that results from their delivery at an early gestational age.
Tissue vitamin A storage
Around 90% of the total body reserve of vitamin A is normally stored in the liver in most animal species (42). The liver vitamin A concentration (100–300 µg/g in healthy human adults (43)) is low during infancy relative to later childhood (44, 45). Liver vitamin A concentration <40 µg/g is indicative of low vitamin A reserve and <20 µg/g is considered deficient (44–46). Most preterm infants are born with low liver vitamin A stores (45, 47–50), and in the absence of an adequate intake of vitamin A, these infants are at further risk of vitamin A deficiency.
Other organs, including the developing lung, are also capable of storing vitamin A (51–53). Significant vitamin A storage occurs in the fetal lung dur-ing the latter third of prenatal life in rats (52, 53). Depletion of these stores begins before birth and continues into the early postnatal period, suggesting that the developing lung may depend on these local stores of vitamin A during the period of active growth and differentiation associated with rapid alveo-larization of the lung. Thus prematurely born animals may be susceptible to the adverse pulmonary effects of vitamin A deficiency. The fetal lung stores of vitamin A can be augmented by prenatal maternal administration of supple-mental vitamin A (54) and depleted by prenatal interventions such as mater-nal glucocorticosteroid treatment (55). Whether the developing fetal lung of humans is capable of vitamin A uptake, esterification and storage remains undetermined. Moreover, whether administration of vitamin A to human mothers with imminent preterm deliveries can augment the lung vitamin A stores of their newborn infants remains unexplored.
Vitamin A deficiencyVitamin A deficiency results in a progressive sequence of changes in the epi-thelial lining of the pulmonary conducting airways (Figure A2.1.1) (13, 56, 57). In proximal airways, in which basal cells exist as stem cells for replace-ment of lining epithelial cells after their attrition from natural causes or injury or disease, basal cell proliferation is stimulated. As these normally discontin-uous basal cells become confluent, the differentiated ciliated columnar cells and non-ciliated secretory cells are displaced from the basement membrane

37
ANNEX 2: BACKGROUND PAPERS
Figure A2.1.1Progressive changes in epithelial lining of pulmonary conducting airways in relation to vitamin A deficiency. (A) Normal ciliated columnar and non-ciliated secretory cells in vitamin A sufficiency. (B) Basal cell hyperplasia as initiating event in vitamin A deficiency. (C) Necrotizing tracheobronchitis in early stages of vitamin A deficiency. (D) Squamous metaplasia in advanced stages of vitamin A deficiency. (E) Keratinized squamous metaplasia in advanced stages of vitamin A deficiency.
Adapted from reference (13).© Wolbach SB, Howe PR, 1925. Originally published in Journal of Experimental Medicine. doi: 10.1084/jem.42.6.753.

38
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
and lose their blood supply and become necrotic. The resultant histopatholog-ical change – necrotizing tracheobronchitis – is characteristic of early vitamin A deficiency. In more advanced vitamin A deficiency states, the basal cells continue to proliferate, lose their phenotypic future and develop as layers of stratified squames that become keratinized. The resultant histopathological change is described as squamous metaplasia, whose pathophysiological con-sequences include: the loss of normal secretions of goblet cells and of other secretory cells, normal water homeostasis across tracheobronchial epithelium and mucociliary transport with resultant predisposition to airway infection; and narrowing of lumina and loss of distensibility of airways with resultant increase in airway resistance and work of breathing. The histopathological changes and the associated pathophysiological consequences are reversible with restoration of normal vitamin A status (58, 59).
Preterm infants are susceptible to lung injury from such insults as hyaline membrane disease, trauma from mechanical ventilation, oxygen toxicity and airway infection (60), and the development of the chronic lung disease called bronchopulmonary dysplasia (BPD) (60). The histopathological changes of BPD – now described as “classical” BPD − consist of necrotizing tracheobron-chitis in early stages and squamous metaplasia in advanced stages (Figure A2.1.2) of the disease, which are remarkably similar to those seen in vitamin A deficiency (61, 62). Preterm infants who have BPD often manifest clinical, biochemical and histopathological evidence of vitamin A deficiency (63–65). Vitamin A supplementation from early postnatal life can improve their vita-min A status and also ameliorate the lung disease, as evidenced by a decreased incidence of BPD and of the associated morbidity (66, 67).
Recent advances in neonatal care, including the use of surfactant, continu-ous positive airway pressure, efficient strategies of mechanical ventilation and enhanced nutrition have altered the epidemiology of BPD (68). In contrast to the classical BPD, seen in preterm infants with significant initial lung disease and need for high oxygen and ventilator support, the “new” BPD is emerging as a disease of the extremely preterm infant, often without much preceding lung illness or need for intensive support (68). Whereas airway injury – char-acterized by epithelial metaplasia, smooth muscle hypertrophy and parenchy-mal fibrosis – has been the hallmark of classical BPD, the new BPD features a striking decrease in alveolar septation and microvascular development (69). Normally, the gas-exchange region of the immature lung is composed of large saccules (70), which undergo subdivision by an outgrowth of septae from their walls into smaller, more numerous structures termed alveoli. This septation is critical for increasing the alveolar number and surface area for gas exchange. In humans, the alveolar septation occurs in late gestation and the first few years after birth (71, 72). Several postnatal factors can inhibit this process, such as oxygen exposure, stretch injury from assisted ventilation, activation
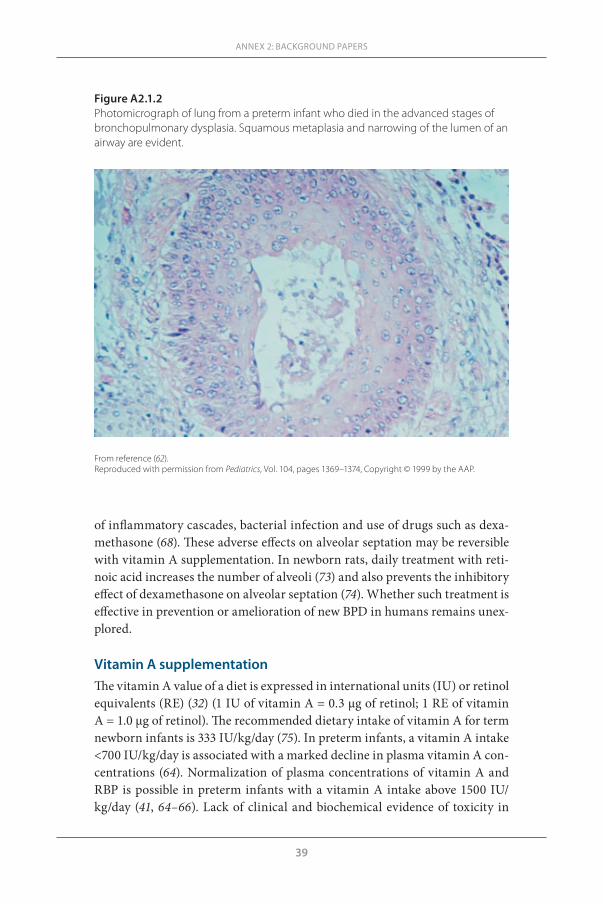
39
of inflammatory cascades, bacterial infection and use of drugs such as dexa-methasone (68). These adverse effects on alveolar septation may be reversible with vitamin A supplementation. In newborn rats, daily treatment with reti-noic acid increases the number of alveoli (73) and also prevents the inhibitory effect of dexamethasone on alveolar septation (74). Whether such treatment is effective in prevention or amelioration of new BPD in humans remains unex-plored.
Vitamin A supplementationThe vitamin A value of a diet is expressed in international units (IU) or retinol equivalents (RE) (32) (1 IU of vitamin A = 0.3 µg of retinol; 1 RE of vitamin A = 1.0 µg of retinol). The recommended dietary intake of vitamin A for term newborn infants is 333 IU/kg/day (75). In preterm infants, a vitamin A intake <700 IU/kg/day is associated with a marked decline in plasma vitamin A con-centrations (64). Normalization of plasma concentrations of vitamin A and RBP is possible in preterm infants with a vitamin A intake above 1500 IU/kg/day (41, 64–66). Lack of clinical and biochemical evidence of toxicity in
ANNEX 2: BACKGROUND PAPERS
Figure A2.1.2Photomicrograph of lung from a preterm infant who died in the advanced stages of bronchopulmonary dysplasia. Squamous metaplasia and narrowing of the lumen of an airway are evident.
From reference (62).Reproduced with permission from Pediatrics, Vol. 104, pages 1369–1374, Copyright © 1999 by the AAP.

40
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
studies of infant vitamin A supplementation and maintenance of their plasma vitamin A concentrations below 80 µg/dL suggest that a total vitamin A intake in the range of 1500–2800 IU/kg/day is safe for preterm infants (41, 65, 66, 76).
The potential role of vitamin A in prevention or amelioration of lung dis-ease has led to development of local guidelines for vitamin A supplementa-tion in preterm infants at risk for BPD (62). According to these guidelines, preterm infants meeting the following criteria are eligible for vitamin A sup-plementation: birth weight <1250 g, estimated gestational age at birth <31 weeks, appropriate growth for gestational age, need for mechanical ventila-tion at 24 hours’ postnatal age and no major congenital anomalies. In addi-tion to standard vitamin A intake from parenteral and enteral sources, these infants are given supplemental vitamin A (retinyl palmitate) in two phases. Phase I entails an intramuscular injection of 2000 IU/kg/dose on postnatal day 1 and on alternate days thereafter until establishment of full enteral feed-ing, that is, when energy intake by enteral route exceeds 75% of total ener-gy intake. Phase II entails an orogastric administration of 4000 IU/kg/dose on the day of full feeds and daily thereafter until discharge. Each infant is monitored for optimal vitamin A status by serial measurements of plasma concentrations of vitamin A (desired range 30–60 µg/dL) and RBP (desired concentration >2.5 mg/dL). To avoid toxicity, each infant is also monitored by careful physical examination, periodic blood chemistry profiles and haema-tological indexes. Brain ultrasonography to detect intracranial haemorrhage and non-invasive assessment of intracranial pressure, such as with fontanelle tonometry, are also useful. The goal of the guidelines, monitoring and careful assessment for potential toxicity, is to promote optimal vitamin A nutrition in vulnerable preterm infants at risk for lung disease and associated morbidity.
Vitamin A toxicityHypervitaminosis A may result from acute intoxication or from chronic inges-tion of vitamin A. A single large dose of vitamin A in excess of 100 000 IU may cause acute intoxication in infants (32). The clinical manifestations of acute hypervitaminosis A include increased intracranial pressure. Prolonged inges-tion of vitamin A in excess of 25 000 IU/day may cause chronic intoxication in infants (32, 77, 78). The clinical manifestations of chronic hypervitaminosis A include increased intracranial pressure, bone and joint pains, and mucocu-taneous lesions; hepatomegaly and hepatic injury, hypercalcaemia and hae-matological abnormalities are sometimes seen (79). Radiographic findings of chronic hypervitaminosis A in infants <6 months include widened metaphy-ses, especially of the distal ulna, and radiolucent zones in the radius proximal to the metaphysis (80). Cortical hyperostosis of multiple long bones and soft tissue changes are seen typically in infants >6 months (80). In hypervitamino-

41
ANNEX 2: BACKGROUND PAPERS
sis A, the plasma vitamin A concentration is generally in excess of 100 µg/dL, whereas the plasma RBP concentration usually remains normal (81).
Hypervitaminosis A is reversible with restriction of vitamin A intake. Infusion of 2-hydroxypropyl-β-cyclodextrin as a means of solubilizing reti-noids and enhancing their urinary excretion has been used occasionally as adjunct therapy (82).
ConclusionsMost preterm infants are born with low plasma concentrations of vitamin A and RBP, and with limited liver tissue reserves of vitamin A. These infants are probably vitamin A deficient at birth because of deprivation of the transpla-cental supply that results from their delivery at an early gestational age. BPD, a common cause of chronic lung disease, is a major source of mortality and morbidity among preterm infants. Besides prevention of prematurity as the primary perinatal health-care goal, clear strategies for reducing the occur-rence of BPD among preterm survivors are clearly warranted. These strategies involve the modulation of a delicate balance between injury and healing of the immature lung. Optimal vitamin A nutrition is one such strategy to comple-ment other approaches.
Preterm infants with BPD often manifest clinical, biochemical and histo-pathological evidence of vitamin A deficiency. Vitamin A supplementation from early postnatal life in these infants not only improves their vitamin A status, but also ameliorates the lung disease, as evidenced by a decreased inci-dence of BPD and of the associated morbidity. Although significant strides have been made towards our understanding of the role of vitamin A in pre-venting BPD among preterm survivors, much remains to be studied, includ-ing: precise requirement of vitamin A in these infants, and the optimal mode and duration of its administration; clinical applications of diagnostic meas-ures that can evaluate molecular mechanisms of vitamin A action in the developing lung; the therapeutic role of vitamin A metabolites, including reti-noic acid, in promoting lung healing; and the role of vitamin A in the patho-genesis of other diseases of preterm infants, such as necrotizing enterocolitis and retinopathy of prematurity. Finally, further study of vitamin A supple-mentation in all vulnerable neonates will be important to promote optimal vitamin A nutrition on a global basis.

42
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
References1. Hopkins FG. Feeding experiments illustrating the importance of accessory fac-
tors in normal dietaries. Journal of Physiology, 1912, 44:425–460.2. McCollum EV, Davis M. The influence of certain vegetable fats on growth. Jour-
nal of Biological Chemistry, 1915, 2:179.3. Hubbard R. The molecular weight of rhodopsin and the nature of the rhodopsin-
digitonin complex. Journal of General Physiology, 1954, 37:381–399.4. Wald G. Molecular basis of visual excitations. Science, 1968, 162:230.5. Emerick RJ, Zile M, DeLuca HF. Formation of retinoic acid from retinol in the
rat. Biochemical Journal, 1967, 102:606–611.6. Blaner WS, Olson JA. Retinol and retinoic acid metabolism. In: Sporn MB, Rob-
erts AB, Goodman DS, eds. The retinoids: biology, chemistry, and medicine, 2nd ed. New York, NY, Raven Press, 1994:229.
7. Kanai M, Raz A, Goodman DS. Retinol-binding protein: the transport protein for vitamin A in human plasma. Journal of Clinical Investigation, 1968, 47:2025–2044.
8. Raz A, Shiratori T, Goodman DS. Studies on the protein-protein and protein-ligand interactions involved in retinol transport in plasma. Journal of Biological Chemistry, 1970, 245:1903–1912.
9. Soprano DR, Blaner WS. Plasma retinol-binding protein. In: Sporn MB, Roberts AB, Goodman DS, eds. The retinoids: biology, chemistry, and medicine, 2nd ed. New York, NY, Raven Press, 1994:257.
10. Kanda Y et al. The amino acid sequence of human plasma prealbumin. Journal of Biological Chemistry, 1974, 249:6796–6805.
11. Rask L, Peterson PA. In vitro uptake of vitamin A from the retinol-binding plas-ma protein to mucosal epithelial cells from the monkey’s small intestine. Journal of Biological Chemistry, 1976, 251:6360–6366.
12. Bok D, Heller J. Transport of retinol from the blood to the retina: an autoradio-graphic study of the pigment epithelial cell surface receptor for plasma retinol-binding protein. Experimental Eye Research, 1976, 22:395–402.
13. Wolbach SB, Howe PR. Tissue changes following deprivation of fat-soluble vita-min A. Journal of Experimental Medicine, 1925, 42:753–777.
14. Moore T. Vitamin A transfer from mother to offspring in mice and rats. Interna-tional Journal for Vitamin and Nutrition Research, 1971, 41:301–306.
15. Branstetter RF et al. Vitamin A transfer from cows to calves. International Jour-nal for Vitamin and Nutrition Research, 1973, 43:142–146.
16. Mitchell GE Jr, Rattray PV, Hutton JB. Vitamin A alcohol and vitamin A palmi-tate transfer from ewes to lambs. International Journal for Vitamin and Nutrition Research, 1975, 45:299–304.
17. Takahashi YI, Smith JE, Goodman DS. Vitamin A and retinol-binding protein metabolism during fetal development in the rat. American Journal of Physiology, 1977, 233:E263–E272.
18. Ismadi SD, Olson JA. Dynamics of the fetal distribution and transfer of vitamin A between rat fetuses and their mother. International Journal for Vitamin and Nutrition Research, 1982, 52:112–119.

43
19. Donoghue S et al. Placental transport of retinol in sheep. Journal of Nutrition, 1982, 112:2197–2203.
20. Vahlquist A, Nilsson S. Vitamin A transfer to the fetus and to the amniotic fluid in rhesus monkey (Macaca mulatta). Annals of Nutrition and Metabolism, 1974, 28:321–333.
21. Wallingford JC, Milunsky A, Underwood BA. Vitamin A and retinol-binding protein in amniotic fluid. American Journal of Clinical Nutrition, 1983, 38:377–381.
22. Sklan D et al. Retinol transport proteins and concentrations in human amniotic fluid, placenta, and fetal and maternal sera. British Journal of Nutrition, 1985, 54:577–583.
23. Lund CJ, Kimble MS. Plasma vitamin A and carotene of the newborn infant with consideration of fetal-maternal relationships. American Journal of Obstet-rics and Gynecology, 1973, 46:207.
24. Baker H et al. Vitamin profile of 174 mothers and newborns at parturition. American Journal of Clinical Nutrition, 1975, 28:59–65.
25. Vahlquist A et al. The concentrations of retinol-binding protein, prealbumin, and transferrin in sera of newly delivered mothers and children of various ages. Scandinavian Journal of Laboratory and Clinical Investigation, 1975, 35:569–575.
26. Baker H et al. Vitamin levels in low-birth-weight newborn infants and their mothers. American Journal of Clinical Nutrition, 1977, 129:521–524.
27. Jansson L, Nilsson B. Serum retinol and retinol-binding protein in mothers and infants at delivery. Biology of the Neonate, 1983, 43:269–271.
28. Venketachalam PS, Belavady B, Gopalan C. Studies on vitamin A nutritional status of mothers and infants in poor communities in India. Journal of Pediat-rics, 1962, 61:262–268.
29. Butte NF, Calloway DH. Proteins, vitamin A, carotene, folacin, ferritin and zinc in Navajo maternal and cord blood. Biology of the Neonate, 1982, 41:273–278.
30. Lewis JM et al. Supplements of vitamin A and of carotene during pregnancy: their effect on the levels of vitamin A and carotene in the blood of mother and of newborn infant. American Journal of Diseases of Children, 1947, 73:143–150.
31. Barnes AC. The placental metabolism of vitamin A. American Journal of Obstet-rics and Gynecology, 1951, 61:368–372.
32. Underwood BA. Vitamin A in human nutrition. In: Sporn MB, Roberts AB, Goodman DS, eds. The retinoids: biology, chemistry, and medicine, 2nd ed. New York, NY, Raven Press, 1994:211.
33. Pitt GAJ. The assessment of vitamin A status. Proceedings of the Nutrition Soci-ety, 1981, 40:173–178.
34. Brandt RB et al. Serum vitamin A in premature and term neonates. Journal of Pediatrics, 1978, 92:101–104.
35. Shenai JP et al. Plasma vitamin A and retinol-binding protein in premature and term neonates. Journal of Pediatrics, 1981, 99:302–305.
36. Smith FR, Goodman DS. The effects of diseases of liver, thyroid, and kidneys on the transport of vitamin A in human plasma. Journal of Clinical Investigation, 1971, 50:2426–2436.
ANNEX 2: BACKGROUND PAPERS

44
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
37. Vahlquist A et al. Plasma vitamin A transport and visual dark adaptation in dis-eases of the intestine and liver. Scandinavian Journal of Laboratory and Clinical Investigation, 1978, 38:301–308.
38. Bhatia J, Ziegler EE. Retinol-binding protein and prealbumin in cord blood of term and preterm infants. Early Human Development, 1983, 8:129–133.
39. Muto Y et al. Regulation of retinol-binding protein metabolism by vitamin A status in the rat. Journal of Biological Chemistry, 1972, 247:2542–2450.
40. Smith JE et al. The effects of chylomicron vitamin A on the metabolism of reti-nol-binding protein in the rat. Journal of Biological Chemistry, 1973, 248:1544–1549.
41. Shenai JP et al. Plasma retinol-binding protein response to vitamin A adminis-tration in infants susceptible to bronchopulmonary dysplasia. Journal of Pediat-rics, 1990, 116:607–614.
42. Goodman DS, Blaner WS. Biosynthesis, absorption, and hepatic metabolism of retinol. In: Sporn MB, Roberts AB, Goodman DS, eds. The retinoids: biology, chemistry, and medicine, volume 2. New York, NY, Academic Press, 1984:1.
43. Pearson WN. Blood and urinary vitamin levels as potential indices of body stores. American Journal of Clinical Nutrition, 1967, 20:514–527.
44. Huque T. A survey of human liver reserves of retinol in London. British Journal of Nutrition, 1982, 47:165–172.
45. Olson JA, Gunning DB, Tilton RA. Liver concentrations of vitamin A and ca-rotenoids, as a function of age and other parameters, of American children who died of various causes. American Journal of Clinical Nutrition, 1984, 39:903–910.
46. Olson JA. Evaluation of vitamin A status in children. World Review of Nutrition and Dietetics, 1978, 31:130–134.
47. Iyengar L, Apte SV. Nutrient stores in human foetal livers. British Journal of Nu-trition, 1972, 27:313–317.
48. Olson JA. Liver vitamin A reserves of neonates, preschool children and adults dying of various causes in Salvador, Brazil. Archivos Latinoamericanos de Nutri-ción, 1979, 29:521–545.
49. Montreewasuwat N, Olson JA. Serum and liver concentrations of vitamin A in Thai fetuses as a function of gestational age. American Journal of Clinical Nutri-tion, 1979, 32:601–606.
50. Shenai JP, Chytil F, Stahlman MT. Liver vitamin A reserves of very low birth weight neonates. Pediatric Research, 1985, 19:892–893.
51. Goodman DS, Huang HS, Shiratori T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. Journal of Lipid Research, 1965, 6:390–396.
52. Zachman RD, Kakkad B, Chytil F. Perinatal rat lung retinol (vitamin A) and retinyl palmitate. Pediatric Research, 1984, 18:1297–1299.
53. Shenai JP, Chytil F. Vitamin A storage in lungs during perinatal development in the rat. Biology of the Neonate, 1990, 57:126–132.
54. Shenai JP, Chytil F. Effect of maternal vitamin-A administration on fetal lung vitamin-A stores in the perinatal rat. Biology of the Neonate, 1990, 58:318–325.
55. Shenai JP, Chytil F. Effect of maternal dexamethasone treatment on fetal lung vitamin A stores in the perinatal rat. International Journal for Vitamin and Nu-trition Research, 1994, 64:93–97.

45
56. Wong YC, Buck RC. An electron microscopic study of metaplasia of the rat tra-cheal epithelium in vitamin A deficiency. Laboratory Investigation, 1971, 24:55–66.
57. McDowell EM, Keenan KP, Huang M. Effects of vitamin A-deprivation on ham-ster tracheal epithelium: a quantitative morphologic study. Virchows Archiv. B, Cell Pathology Including Molecular Pathology, 1984, 45:197–219.
58. Wolbach SB, Howe PR. Epithelial repair in recovery from vitamin A deficiency. Journal of Experimental Medicine, 1933, 57:511–526.
59. McDowell EM, Keenan KP, Huang M. Restoration of mucociliary tracheal epi-thelium following deprivation of vitamin A: a quantitative morphologic study. Virchows Archiv. B, Cell Pathology Including Molecular Pathology, 1984, 45:221–240.
60. Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. New Eng-land Journal of Medicine, 1967, 276:357–368.
61. Stahlman MT, Gray ME. Lung injury and repair in the neonate. In: Brigham KL, Stahlman MT, eds. Respiratory distress syndromes: molecules to man. Nashville, TN, Vanderbilt University Press, 1990:3.
62. Shenai JP. Vitamin A supplementation in very low birth weight neonates: ration-ale and evidence. Pediatrics, 1999, 104:1369–1374.
63. Hustead VA et al. Relationship of vitamin A (retinol) status to lung disease in the preterm infant. Journal of Pediatrics, 1984, 105:610–615.
64. Shenai JP, Chytil F, Stahlman MT. Vitamin A status of neonates with bron-chopulmonary dysplasia. Pediatric Research, 1985, 19:185–188.
65. Shenai JP et al. Sequential evaluation of plasma retinol-binding protein response to vitamin A administration in very-low-birth-weight neonates. Biochemical and Molecular Medicine, 1995, 54:67–74.
66. Shenai JP et al. Clinical trial of vitamin A supplementation in infants susceptible to bronchopulmonary dysplasia. Journal of Pediatrics, 1987, 111:269–277.
67. Tyson JE et al. Vitamin A supplementation for extremely-low-birth-weight in-fants. New England Journal of Medicine, 1999, 340:1962–1968.
68. Jobe AH. The new BPD: an arrest of lung development. Pediatric Research, 1999, 46:641–643.
69. Coalson JJ. Pathology of new bronchopulmonary dysplasia. Seminars in Neona-tology, 2003, 8:73–81.
70. Massaro GD, Massaro D. Formation of pulmonary alveoli and gas-exchange surface area: quantitation and regulation. Annual Review of Physiology, 1996, 58:73–92.
71. Langston C et al. Human lung growth in late gestation and in the neonate. Amer-ican Review of Respiratory Disease, 1984, 129:607–613.
72. Zeltner TB, Burri PH. The postnatal growth of the human lung II. Morphology. Respiration Physiology, 1987, 67:269–282.
73. Massaro GD, Massaro D. Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. American Journal of Physiology, 1996, 270:305–310.
ANNEX 2: BACKGROUND PAPERS

46
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
74. Whitney D et al. Gene expression of cellular retinoid-binding proteins: modu-lation by retinoic acid and dexamethasone in postnatal rat lung. Pediatric Re-search, 1999, 45:2–7.
75. FAO/WHO. Requirement of vitamin A, thiamine, riboflavin and niacin: report of a joint FAO/WHO Expert Group. Geneva, World Health Organization, 1967.
76. Wang SG et al. Assessment of vitamin A supplementation in very-low-birth-weight neonates. Pediatric Research, 2002, 51:313A.
77. Mahoney CP et al. Chronic vitamin A intoxication in infants fed chicken liver. Pediatrics, 1980, 65:893–897.
78. Farris WA, Erdman JW Jr. Protracted hypervitaminosis A following long-term, low-level intake. Journal of the American Medical Association, 1982, 247:1317.
79. Goodman DS. Vitamin A and retinoids in health and disease. New England Journal of Medicine, 1984, 310:1023–1031.
80. Persson B, Tunell R, Ekengren K. Chronic vitamin A intoxication during the first half year of life: description of 5 cases. Acta Paediatrica Scandinavica, 1965, 54:49–60.
81. Smith FR, Goodman DS. Vitamin A transport in human vitamin A toxicity. New England Journal of Medicine, 1976, 294:805–808.
82. Carpenter TO et al. Severe hypervitaminosis A in siblings: evidence of variable tolerance to retinol intake. Journal of Pediatrics, 1987, 111:507–512.

47
ANNEX 2: BACKGROUND PAPERS
a2.2Metabolism of vitamin A in early lifeA. Catharine Ross
Department of Nutritional Sciences, Pennsylvania State University, University Park, Pennsylvania, United States of America
Corresponding author: A. Catharine Ross; [email protected]
Abstractn Neonates are born with low hepatic reserves of vitamin A. Even in infants born in affluent communities, plasma retinol levels are low as compared with parturient women or other control groups. Thus neonates resemble the vitamin A-marginal adult, but whether this resemblance holds true at the level of cellular and organ systems retinol metabolism is not known. The neo-nate’s organ systems are immature at birth, but again, whether this imma-turity affects neonatal vitamin A metabolism is mostly speculative. At birth energy metabolism changes abruptly from glucose to fat as the primary fuel, as human milk provides nearly 50% of calories from fat. The adaptations of the neonate to a high-fat diet may also be beneficial for the absorption of fat-soluble vitamin A. A few animal studies have directly tested the response of the neonatal liver and lungs to a large dose of vitamin A, similar in amount to a 50 000 IU dose of vitamin A in human neonates, and have observed signifi-cantly increased tissue vitamin A concentrations in the postprandial period. Little is known about the rate of production of the retinol transport protein, retinol-binding protein (RBP), in neonates and it remains unclear why low plasma retinol and RBP levels are found in this age group, even with adequate diet. Renal immaturity may contribute to increased urinary retinol excretion. Overall, there are many gaps in present knowledge, as well as many research opportunities, concerning vitamin A absorption, transport, storage and utili-zation in the neonatal period.

48
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
IntroductionIt has been known for decades that at birth vitamin A levels are low in ani-mals and humans. Even neonates born to well-nourished mothers have low hepatic vitamin A reserves (1–5). Thus, the early months of life, concomitant with breastfeeding, are considered a critical period for building up vitamin A stores to a level that will be sufficient to prevent vitamin A deficiency post-weaning. This is especially important if the child’s future diet is likely to be low in vitamin A. Infants ingest vitamin A from breast milk until the time of weaning, and some from formulas that contain vitamin A in amounts similar to those present in breast milk. Infants also may obtain vitamin A from com-plementary foods, which generally are introduced around the age of 6 months, and, additionally, from paediatric vitamin-mineral supplements intended for healthy infants without significant risk of nutritional deficiencies, or in sup-plements intended to prevent deficiencies in at-risk populations. The World Health Organization (WHO)/United Nations Children’s Fund (UNICEF) nutritional strategy aims to ensure that young children living in areas with inadequate intake of vitamin A receive the vitamin through a combination of breastfeeding, dietary improvement, food fortification and supplementa-tion (6). Currently, vitamin A supplements used for the purpose of increasing the child’s reserves and preventing vitamin A deficiency onset are adminis-tered at 4–6 month intervals in a single bolus dose determined by the child’s age (e.g. 50 000–200 000 IU (= 15–60 mg retinol)).
This review begins with an overview of vitamin A metabolism and its status in the neonate, followed by an organ systems-oriented discussion of vitamin A metabolism, proceeding through in several steps: (1) intestinal digestion, absorption and initial transport of newly absorbed vitamin A; (2) hepatic uptake, storage and mobilization; (3) metabolism of retinol by target tissues, including uptake and metabolism; and (4) excretion. At each step, considera-tion is given to the maturational state of the tissues in neonates and whether this is likely to affect vitamin A metabolism. Although vitamin A metabolism has been studied for now nearly a century (7), the current state of knowledge of vitamin A transport, metabolism and cellular functions is still incom-plete for the adult-age animal in which it has been most studied, and in adult humans. In neonatal-age animals and humans this knowledge is even more fragmentary; indeed, for many aspects, it is necessary to extrapolate from knowledge in adult animals and humans. High-quality trial evidence from studies in children and neonatal animal models is deemed to be crucial (8) but owing to the limited information on neonates, this review also includes infor-mation from studies conducted in vitamin A-deficient animals, based on the rationale that adult animals with marginal or deficient vitamin A status have low levels of vitamin A compounds (retinoids) in tissues, resembling the situ-

49
ation in neonates. However, it is not assured that results from such studies are necessarily relevant for understanding the vitamin A metabolism in neonates, and therefore these data must be considered only as a way to possibly predict, or generate hypotheses about, how vitamin A is metabolized in neonates. Epi-thelial tissues are particularly sensitive to vitamin A status in the marginal to deficient range. Rapidly turning over tissues such as the gut epithelium and haematopoietic system are also altered in states of vitamin A deficiency. Tis-sue maturation, such as occurs in the small intestine and lungs in the postna-tal period, is also dependent on adequate dietary vitamin A (9, 10).
Overview of vitamin A metabolism: pathways, regulatory factors and physiology Figure A2.2.1 shows a typical but simplified schematic of vitamin A metabo-lism (excluding reactions that are specific to the retina) (11). Most tissues and cell types are capable of several of these reactions, but some cells may not be capable of the entire pathway. Many other apparently minor reactions and by-products have also been identified. The parent vitamin A molecule, retinol, is transformed through esterification to retinyl ester, a stable form of vitamin A that is highly efficient for storage. Quantitatively, retinyl ester is the major form of vitamin A and is found in its most concentrated form in the retinal pigment epithelial (RPE) cells, and in the hepatic stellate cells (HSCs or Ito cells). It is also present in lower concentrations in other tissues. Retinyl ester is also the major form in which in chylomicrons (CMs) and CM remnants transport vitamin A from the intestine to the systemic circulation. However, neither the retinol nor the retinyl ester molecule has any known biological activity. Oxidative activation of retinol is essential for bioactivity. Retinol is oxidized, first, to retinal, which is required by rod and cone cells for vision, in a reversible reaction. Thereafter, retinal is irreversibly oxidized to the car-boxylic acid metabolite of vitamin A, retinoic acid (RA), the form essential for cell differentiation and normal pattern of gene expression. The irreversibility of this oxidation reaction acts as a one-way gate (Figure A2.2.1), so that RA is separated from the vitamin A storage system consisting of the esterifying enzyme lecithin:retinol acyltransferase (LRAT) and a variety of retinyl ester hydrolases (REHs), as well as from the cleavage enzymes that convert carot-enoids to retinal and the oxidoreductases that reduce retinal to retinol (11). This enables the organism to regulate the metabolism of dietary forms of vitamin A, i.e. retinol and carotenoids, separately from the formation of the hormonal form of the vitamin, RA. Nonetheless, RA does play a role in how the body handles dietary vitamin A (see below), because RA is a regulator of the LRAT system, which esterifies retinol in liver, lung and possibly other tissues (12). Both retinol and RA are oxidized on their β-ionone ring,
ANNEX 2: BACKGROUND PAPERS

50
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
generally at carbon 4, to form polar metabolites which then can be fur-ther conjugated to form water-soluble metabolites (retinyl- and retinoyl-β-glucuronides). The latter forms are found transiently in plasma after high doses of vitamin A (13) and are usual end-products of retinoid metabolism excreted in bile and faeces (14, 15).
Table A2.2.1 lists the major proteins required for vitamin A metabolism, which fall into three functional categories: nuclear retinoid receptors, cellular retinoid-binding proteins and enzymes catalyzing the metabolic transforma-tion of retinoids. Nuclear retinoid receptors of the retinoic acid receptor (RAR) family, comprising RARα, β and γ, are members of the superfamily of nuclear hormone receptors, as are their dimerization partners, retinoid X receptors (RXRα, β and γ) and other structurally related receptors that bind glucocorti-coids, thyroid hormone, vitamin D, fatty acids and sterol metabolites. Nuclear retinoid receptors are involved in retinoid metabolism through their role in regulating several transporters, enzymes and binding proteins that are central to the metabolism of retinoids (16). During several steps in retinol metabolism, specific forms of cellular retinoid-binding proteins function as chaperones for retinol, RA and for retinal in the retina (17, 18). Nearly all these proteins dis-criminate in binding the all-trans isomers of these compounds while failing
Figure A2.2.1 Vitamin A metabolism from the biochemical perspective. Several steps are physio-logically reversible, allowing for extensive interconversion of retinol with retinyl ester and retinal. Some oxidative steps are irreversible and, because retinoic acid (RA) cannot be converted to retinol, this hormonal form of the vitamin is essentially metabolically isolated from retinol. The concentration of RA is controlled by the rates of aldehyde dehydrogenases that produce RA and various cytochrome P450 enzymes that degrade it.
Oxidative activation reactions
Carotenoids
RE Retinol Retinal RA polar storage metabolites
Retinyl-β- Retinoyl, 4-oxy- glucuronide retinoyl-β-glucuronides
Oxidative inactivation reactions
Reproduced with permission by Lippincott Williams & Wilkins (11).
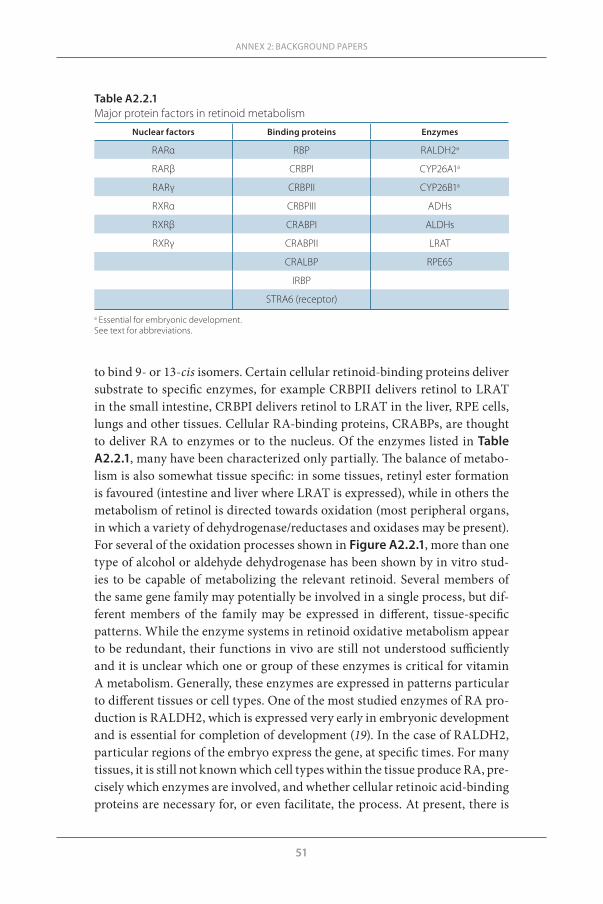
51
to bind 9- or 13-cis isomers. Certain cellular retinoid-binding proteins deliver substrate to specific enzymes, for example CRBPII delivers retinol to LRAT in the small intestine, CRBPI delivers retinol to LRAT in the liver, RPE cells, lungs and other tissues. Cellular RA-binding proteins, CRABPs, are thought to deliver RA to enzymes or to the nucleus. Of the enzymes listed in Table A2.2.1, many have been characterized only partially. The balance of metabo-lism is also somewhat tissue specific: in some tissues, retinyl ester formation is favoured (intestine and liver where LRAT is expressed), while in others the metabolism of retinol is directed towards oxidation (most peripheral organs, in which a variety of dehydrogenase/reductases and oxidases may be present). For several of the oxidation processes shown in Figure A2.2.1, more than one type of alcohol or aldehyde dehydrogenase has been shown by in vitro stud-ies to be capable of metabolizing the relevant retinoid. Several members of the same gene family may potentially be involved in a single process, but dif-ferent members of the family may be expressed in different, tissue-specific patterns. While the enzyme systems in retinoid oxidative metabolism appear to be redundant, their functions in vivo are still not understood sufficiently and it is unclear which one or group of these enzymes is critical for vitamin A metabolism. Generally, these enzymes are expressed in patterns particular to different tissues or cell types. One of the most studied enzymes of RA pro-duction is RALDH2, which is expressed very early in embryonic development and is essential for completion of development (19). In the case of RALDH2, particular regions of the embryo express the gene, at specific times. For many tissues, it is still not known which cell types within the tissue produce RA, pre-cisely which enzymes are involved, and whether cellular retinoic acid-binding proteins are necessary for, or even facilitate, the process. At present, there is
ANNEX 2: BACKGROUND PAPERS
Table A2.2.1Major protein factors in retinoid metabolism
Nuclear factors Binding proteins Enzymes
RARα RBP RALDH2a
RARβ CRBPI CYP26A1a
RARγ CRBPII CYP26B1a
RXRα CRBPIII ADHs
RXRβ CRABPI ALDHs
RXRγ CRABPII LRAT
CRALBP RPE65
IRBP
STRA6 (receptor)
a Essential for embryonic development.See text for abbreviations.

52
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
no definitive description of the levels of nuclear retinoid receptors, retinoid-binding proteins and enzymes expressed in neonatal tissues. However, genes for each of the six known nuclear retinoid receptors, cellular retinoid-binding proteins, and most of the enzymes are expressed during embryonic develop-ment, in temporally and spatially regulated patterns (20, 21). Thus it can be inferred that the neonate is born with the capability of metabolizing retinoids and responding to them.
Figure A2.2.2 summarizes the physiological metabolism of vitamin A at the levels of the intestine, liver and target organ levels. Vitamin A absorp-tion, storage, transport and elimination depend on adequate functioning of the pancreas, intestine, liver and kidneys, as well as metabolism of retinol to retinal and RA in appropriate target organs. RA is essential for normal cell proliferation and well-regulated cell differentiation in tissues throughout the body, but epithelial tissues and cells that turn over rapidly (e.g. in the haema-topoietic system), are particularly sensitive to inadequate amounts of RA. As RA itself is metabolized rapidly, usually within hours, a constant resupply is necessary to maintain tissue levels. It can reasonably be hypothesized that organs such as the intestinal mucosa, respiratory system and immune system that continue to undergo significant maturation in the postnatal period will
Figure A2.2.2 Physiology of retinol metabolism. Pathways of retinol metabolism that are discussed in this review include: (1) luminal digestion; (2) intestinal retinyl ester formation; (3) intravascular CM processing; (4) hepatic uptake; (5) hepatic re-esterification and storage; (6) hydrolysis of retinyl ester and secretion of holo-RBP; (7) uptake of retinol from RBP in peripheral tissues; (8) metabolism of retinol and formation of RA in target tissues; (9) nuclear actions of RA affecting tissue maturation, integrity and metabolism; (10) recycling of retinol and return of RA to liver for further metabolism and elimination. Not illustrated: filtration and recovery of RBP and retinol in the kidneys. See text for abbreviations.
Reproduced with permission by Lippincott Williams & Wilkins (11).
INTESTINE LIVER TARGET ORGANS
ChyloR E ,ß-C
B rushborderhydro-lases
micellizationand uptake
variouslipases
Dietary R E
micellization
Retinol
PL
R E
CRB P -II& LRAT
c hylo.remnant
(LPL)
blood
lymph
R E Retinol
R Estorage R E
RBP4+
CYP26hydroxylation,glucuronidation
plasma
TTR R B P4
recycling
R A
NuclearRAR , RXR
oxymetabolites
R A CRABPStra6
Retinol CRBP
R E
CRB P -I& LRAT
R EH
R EH
ß-c aroteneR EH
cleavageRetinol
biliaryexcretion
1
2
3
4
5 6 7
8
9
10

53
have a higher requirement for RA, and thus for vitamin A, compared with less rapidly turning over tissues. Conversely, the proper metabolism of vitamin A is likely to depend on adequate organ maturation.
Vitamin A status at birthPlasma retinol in the neonate
Comparisons of levels of retinol and its transport proteins, RBP and tran-sthyretin (TTR), in cord blood plasma or newborn plasma with maternal plasma have consistently shown lower levels in neonates, even in industri-alized countries where vitamin A deficiency is unlikely. In a Swedish study, newborn plasma retinol levels averaged 1.0 µM as compared with 1.7 µM in parturient mothers, and 2.1 µM in non-pregnant controls (22). In an Indian study, mean values were lower for both mothers and infants, and still lower in cord blood than maternal plasma (12.5 vs 22.7 µg/dL, or 0.44 vs 0.79 µM) (23). Moreover, levels are even lower in preterm (<37 weeks’ gestation) than in full-term neonates. In a study of pregnant women in India who had received a pro-vitamin A supplement in the form of red palm oil, plasma retinol lev-els at parturition were higher in supplemented mothers and their infants as compared with control mothers and their infants, but lower in infants (mean 0.73 µM) compared with mothers (1.20 µM) in the supplemented group (24). Even in countries not known for vitamin A deficiency, a substantial propor-tion of infants are born with plasma retinol concentrations <0.7 µM, a value considered indicative of vitamin A deficiency in older children. But in neo-nates this value is not so unusual and its use as an indicator of deficiency needs further validation. In a study of Swedish children from birth to 16 years of age, the transport proteins for retinol, RBP and TTR were also found to be lower at birth (about 50% of concentrations in non-pregnant women) (25). Levels then increased rapidly over the first 6 months to a maximum of 31 µg/mL but then declined to a mean level of about 26 µg/mL, which was maintained until the age of puberty when adult values were gradually attained. Concentrations of TTR showed a similar pattern of development (25).
Additionally, RA is present in plasma at low concentrations, bound to albu-min. Neonatal plasma RA concentrations were also lower than those of par-turient mothers (22). Plasma is known to also contain low concentrations of several oxidized retinoids including cis and di-cis isomers of RA, oxidized forms of RA, and conjugated water-soluble forms of retinol and RA (13, 26, 27).
Liver retinol in the neonate
The neonatal liver has little vitamin A and thus the accumulation of retinyl ester in the HSCs has barely begun at birth. However, HSCs are more abun-dant in neonatal liver (28, 29), suggesting that a lack of this type of storage
ANNEX 2: BACKGROUND PAPERS

54
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
cell is not an explanation. In a small US study of liver autopsy specimens from infants and children (0–10 years), 5/7 infants <1 month of age had liver total retinol concentrations in the range of 10 µg retinol/g (35 nmol/g) (30). Similar values were found (9 and 15 µg retinol/g (31 and 52 nmol/g liver)) in the liver of neonatal rats born to mothers fed a vitamin A-restricted and a high vitamin A diet, respectively (5). These data imply that even in well-nourished popula-tions, infants begin life with low hepatic vitamin A stores. The liver concen-tration of vitamin A, a “gold standard” for assessment of vitamin A status, is often considered deficient, in adults, if it is <20 µg/g liver (70 nmol/g liver), but whether this value is meaningful in neonates is as yet unproven. Moreover, in adult vitamin A-deficient rats, only approximately 30% of total body retinol is present in liver. For neonates, the vitamin A content in other tissues is largely unknown. An exception is in some studies of the lungs where the ontogeny of vitamin A contents has been studied (see later). Several animal studies show that hepatic vitamin A levels at birth are not increased appreciably by supple-mentation during pregnancy (2, 3, 5). Therefore, it appears that low vitamin A at birth is physiologically normal. However, studies are needed on multiple tissues to better understand the newborn’s vitamin A status and its physi-ological significance.
Milk as a source of vitamin A
The rapid growth of the infant in the postnatal period necessarily results in a dilution of existing vitamin A concentrations as tissues expand, unless new vitamin A is taken in by the infant from the mother’s milk, formula or sup-plements to supply the expanding body tissues and allow for maintenance or accrual of tissue vitamin A stores (31). The amount of vitamin A that neonates receive from colostrum and milk depends significantly on the mother’s vitamin A nutritional status. Studies of lactating animals illustrate that maternal diet is a strong determinant of the concentration of vitamin A in milk (5, 32, 33). Poor protein and calorie status were also shown to reduce milk output, and thus vitamin A secretion in milk and accrual by the nursling pups (34). Vita-min A in breast milk changes rapidly in concentration with changes in mater-nal dietary vitamin A, as best observed in a cross-over study in lactating rats that were switched in mid-lactation from a high to a low vitamin A diet, or vice versa. Within 2 days, the breast milk vitamin A concentration in rats switched to the high vitamin A diet “matched” and did not differ from that of rats fed on the high vitamin A diet continuously, while in contrast the milk of rats switched to the low vitamin A diet nearly matched the milk of rats con-tinuously fed on the low vitamin A diet (33). Breast milk retinol peaked within 24 hours and declined by 48 hours after administration of high-dose vitamin A supplements to lactating sows (35). Thus, overall, breast milk vitamin A

55
levels reflect the mother’s recent diet or supplementation more than it does her long-term stores as indicated by liver vitamin A concentration.
The role of specific organs in vitamin A metabolismIntestinal metabolismVitamin A absorption and chylomicron formationNumerous studies in adult animals and a limited number of human studies show that retinyl ester must be digested to retinol in the lumen of the intes-tine prior to absorption. Digestion depends on adequate pancreatic function and adequate lipid intake for micellization of the retinol and other products of lipid digestion. Retinol then undergoes intestinal uptake, re-esterification within enterocytes, followed by incorporation of these newly formed retinyl ester molecules into the lipid core of the nascent CMs that transport dietary fat (36, 37) (Figure A2.2.2). After assembly of CMs within the endoplasmic reticulum of the enterocyte, the CM particle is secreted through the Golgi apparatus and released into the intestinal lymphatic system within the lamina propria, and transported through the mesenteric and thoracic lymph ducts to the venous blood stream.
Vitamin A status and intestinal adaptation A series of studies on intestinal adaptation in adult animals, which were designed to understand the compensatory adaptation of the gut following bowel resection, have provided evidence that vitamin A, and specifically its metabolite RA, are important for the processes through which adaptation occurs, including an increase in crypt cell proliferation and villus height, leading to increased area of the absorptive mucosal surface. Vitamin A defi-ciency in rats inhibited the adaptation response after partial small bowel resection (38) by reducing crypt cell proliferation, which was increased by resection, by increasing apoptosis within crypt cells, and by reducing by 44% the rate of migration of enterocytes from crypt to villi tip, which is necessary for normal intestinal mucosal renewal. The adaptive increase in villus height and crypt depth after resection was not seen in vitamin A-deficient rats (38). Rats that were partially replenished with vitamin A also had a reduced rate of crypt cell proliferation 48 hours after resection. Retinoic acid, administered either intravenously to rats studied 6 hours later, or given as an implanted pel-let for 14 days, resulted in significant trophic effects in bowel-resected rats (9). The 14-day treatment with RA also had trophic effects in sham-resected rats, indicating that a tropic effect of RA occurs even on the normal small bowel. RA inhibited apoptosis and stimulated crypt cell proliferation and enterocyte migration post-resection (9). The investigators suggested that the proadaptive effects of RA might be mediated via changes in the extracellular matrix for-
ANNEX 2: BACKGROUND PAPERS

56
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
mation, transcription factors and various signalling pathways. Other studies in pregnant rats provide evidence that the administration of 10 mg retinyl acetate/day from gestational day 7 to 15 increased the levels of alkaline phos-phatase in 15-day fetal rat small intestine, indicating an effect of maternal retinoid on intestinal maturation before birth (39). The level of CRBPII, an abundant protein in adult enterocytes (40), which is expressed prenatally and throughout the suckling period (41), was also increased adaptively in adult rats with a shortened jejunum (42). Overall, several lines of evidence indicate that vitamin A, through its active metabolite RA, has a trophic effect on gut maturation pre- and postnatally.
Luminal fat and retinyl ester hydrolysisAt birth, the infant’s major fuel source changes from glucose to fat (43). The pancreas and intestine must abruptly adapt to the digestion and absorption of new fuels in milk, namely lactose and a high concentration of triglycerides. The pancreas is functionally immature at birth. Enteral feeding as compared with total parenteral nutrition (TPN) has a trophic effect on the gastrointes-tinal tract in both preterm and term neonates. For piglets in both neonatal age groups, the weight of the pancreas increased 30–75% and amylase activity increased 0.5- to 13-fold after birth, more so in enteral-fed than in TPN-fed piglets (44). Thus, adequate nutrition and stimulation of the gastrointestinal tract are important for the postnatal development of the pancreas. Neonates are deficient in pancreatic co-lipase-dependent triglyceride lipase (also known as bile salt-stimulated lipase, BSSL) as compared with older infants and adults. Nevertheless, fat absorption is well developed at birth and is commensurate with the high fat intake of the infant. BSSL is also expressed in the mammary gland of some species during lactation, and secreted into the milk (45). This enzyme and other lipases, such as carboxyl ester lipase (CEL), in milk may explain the ability of neonates to digest dietary fat (46).
More than 95% of breast milk vitamin A is in the form of retinyl ester, which is present as medium and long-chain fatty acid esters (47). Most vita-min A supplements also contain retinyl ester, except in a single form such as retinyl palmitate. The digestion of retinyl ester and the presence in the lumen of products of fat digestion are both necessary for retinol micelliza-tion, which must occur prior to absorption of the digested lipid products (48). In adults, dietary retinyl ester is hydrolysed in the intestine by the pancreatic enzyme pancreatic triglyceride lipase and an intestinal brush border enzyme, phospholipase B (48). However, BSSL in human milk can digest milk retinyl ester (49), and BSSL and pancreatic lipase-related protein 2 (PLRP2) both have broad substrate specificity (45). It therefore seems likely that BSSL and/or CEL activity, or some combination of lipases from the pancreas, intestine and/or breast milk will be sufficient for the digestion of retinyl ester by the neonate,

57
since fat which is present in much larger amounts is digested. Thus digestion and micelle formation probably are not likely to be limiting in the absorption of vitamin A contained in milk. If vitamin A is administered to breastfeeding neonates who are actively absorbing fat, retinyl ester will likely be digested and absorbed along with the milk fat.
Intestinal absorption phaseThe neonatal intestine is structurally and functionally immature (50). Chil-dren with lower serum retinol concentrations, and thus presumably poorer vitamin A nutritional status, have been shown to more likely have impaired intestinal integrity, as assessed by a lactulose/mannitol test commonly used to assess intestinal barrier function (51). Research on the ontogeny of expres-sion of proteins needed for normal vitamin A metabolism has shown that important components of the “basic machinery” for intestinal retinyl ester formation, including CRBPII and LRAT, are already expressed in the small intestine, in which CBRPII and LRAT activities were detectable by gestational days 18–19 in the rat (41). Levels of these proteins rose after birth and reached high levels in the middle of the suckling period, and then declined after wean-ing to adult levels. Thus, it appears that the capacity for intestinal absorption of vitamin A is well developed in the neonate.
Secretion of CMs Each CM particle contains a constant amount of the principal apolipoprotein (apo)-B-48, without which it cannot be formed, as a congenital deficiency in apo-B has been shown to severely impair fat absorption, including the absorp-tion of fat-soluble vitamins (52). The amount of apo-B in lymph does not change significantly during active fat absorption, however, other apoproteins may be involved in the adaptation to a high fat diet. To study fat absorption in neonates, Black and co-workers developed the 2-day old neonatal pig as a model, to which they administered a high-fat intraduodenal lipid infusion versus a low fat infusion for 24 hours (53). In these studies, the jejunal expres-sion of apo-A-IV mRNA increased sevenfold, then decreased as the piglets were weaned from a high-fat breast milk diet. Black suggested that apo-A-IV plays a special role in facilitating intestinal lipid absorption in the suckling newborn by increasing the amount of lipid transported per CM particle (53).
The vitamin A content of the CM particle is highly variable, ranging from essentially none if the meal being absorbed contains no vitamin A to several retinyl ester molecules after a vitamin A-containing meal, to a rather large number of retinyl ester molecules per CM after ingestion of a high dose of vitamin A. The ratio of vitamin A to triglycerides (TGs) contained in CM col-lected from the mesenteric lymph duct of adult rats increased nearly linearly with the amount of vitamin A infused into the duodenum, which was varied
ANNEX 2: BACKGROUND PAPERS

58
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
over an eightfold range (54). Overall, a variety of physiological data support the idea that the CM transport system provides a flexible, expandable means for absorbing dietary fat, and, simultaneously, for absorbing vitamin A.
Retinoic acid formation in the postprandial periodWithin the intestine, a small but possibly physiologically important portion of the newly absorbed vitamin A is oxidized to form RA, which then is trans-ported from the intestine via the portal blood system, bound to serum albu-min. RA concentration in portal blood increased after the intestinal infusion of β-carotene in ferrets (55) and after consumption of liver (high in vitamin A) or vitamin A supplementation in pigs (56). The portal and central veins of the pig contains RA generated from supplemental vitamin A in the intestine, which was released into the systemic circulation, and well as glucuronides of RA and retinol itself (56). Because portal blood flows directly to the liver, the RA produced in the small intestine may be rapidly taken up and metabo-lized therein (57) while, in contrast, RA that is not extracted into the liver may pass through (56) and reach the peripheral organs. Quantitatively, the proportion of vitamin A oxidized to RA in enterocytes is uncertain. The RA formed within the enterocyte may be important for maintaining a normal differentiation status, as shown for exogenous RA (above), while the RA that enters the circulation could be important for uptake by distal tissues and for the maintenance of their normal state of differentiation. RA that has been taken up from plasma by the liver, as has been shown in adult rats (57), may serve as a signal of the body’s vitamin A status and may be a major regulator of retinol homeostasis (12). However, nearly all the above information has been obtained in adult-age animals and data specifically from neonates are lacking.
Intravascular chylomicron metabolism
CM vitamin A, in the form of intestinally synthesized retinyl ester, can be detected in postprandial plasma after the consumption of high amounts of vitamin A. However, due to the rapid clearance of CMs from plasma within minutes after entry, neither CMs nor the retinyl ester component of the CM is detected in fasting plasma, except when CM metabolism is significantly impaired.
CMs are processed lipolytically in peripheral tissues to form CM remnants, which are then taken up, mainly but not exclusively, by the liver. Lipoprotein-lipase (LPL) which is highly expressed in skeletal and cardiac muscle and adipose tissue, and in other tissues, lipolyses the TG component of the CM and thus provides fatty acids for energy metabolism or for storage. The LPL pro-tein is bound to the capillary endothelium by glycosaminoglycans and, there-fore, circulating CM are metabolized by LPL while still in the blood stream; however, they seem to disappear from plasma due to being temporarily bound

59
to LPL on the endothelial surface (54). The activity of LPL, and therefore the uptake of CM lipids into LPL-containing tissues, is sensitive to changes in nutritional status, such as fasting and feeding, and to hormonal status (58). A proportion of the CM retinyl ester is transferred into LPL-containing tissues during CM binding (54, 59). After LPL has acted on the CM, the lipid-depleted CM remnant, still containing most of the newly absorbed retinyl ester that was present in the original CM, is taken up by CM receptors which are pre-dominant on hepatic parenchymal cells. Whereas uptake of CM retinyl ester by liver accounts for most of the clearance of CM vitamin A, 30–40% of CM retinyl ester is taken up by extrahepatic tissues, which is likely to be mediated in part by LPL (see above). The rapid postprandial metabolism of vitamin A, including lipolysis and tissue uptake, generally reaches completion within 2–6 hours after its ingestion.
CM retinyl ester also undergoes exchange with other plasma lipoproteins, including the low- (LDL) and high-density (HDL) plasma lipoproteins. The exchange of retinyl ester from CMs to lipoproteins is most likely of greatest quantitative importance when the CM retinyl ester content is high, as will occur after vitamin A supplementation. Because lipoproteins are taken up by lipoprotein receptors that have a different tissue distribution compared with LPL, uptake with lipoproteins provides another route for small quantities of the newly absorbed retinyl ester to reach certain types of cells (60). LDL recep-tors are widely distributed and HDL receptors are found predominantly on steroidogenic tissues. Scavenger-type receptors also take up lipoproteins and may be involved in the uptake of vitamin A carried by lipoproteins.
Hepatic metabolismLiver maturity at birth The liver does not obtain full maturity until 2 years after birth (61). As soon as the umbilical supply is interrupted, hepatic functions such as transamina-tion, glutamyl transferase, synthesis of coagulation factors, bile production and transport are increased (61). Preterm infants are at special risk of hepat-ic decompensation because their immaturity results in a delay in achieving normal detoxifying and synthetic function. Hypoxia and conditions such as sepsis are also serious causes of liver dysfunction in neonates (61). Postna-tal development includes enlargement of the hepatocytes and expansion and multiplication of the liver lobules (62), and pronounced changes in sinusoidal structure (63). Hepatocellular maturation is characterized by a progressive transition from an architecture in which hepatocyte plates are at least two cells thick, to the adult pattern in which liver cell plates are predominantly one cell in thickness (63). The effect of these structural alterations on the uptake, metabolism and storage of vitamin A is not known. Vitamin A status in utero could potentially be a factor in postnatal maturation, as RA has been shown to
ANNEX 2: BACKGROUND PAPERS

60
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
induce maturation characterized as an increase in albumin production and a decrease in α-fetoprotein production in fetal rat liver cells (64). RBP is detect-able in liver of the fetal rat around gestational day 16 (4).
In the adult liver, vitamin A is stored as retinyl ester primarily in lipid droplets within perisinusoidal HSCs (see below). Stellate-appearing cells with staining characteristic of HSCs have been detected in neonatal mouse liver (29), and are abundant in human liver <1 month of age but then decrease by 6 months (28). Thus, although the liver is still undergoing structural remodelling in the neonatal period, the hepatic cells types involved in vitamin A metabolism in the adult liver are already in place.
Chylomicron uptake and initial processingAfter the initial hepatic uptake of CM retinyl ester via CM receptors, the retinyl ester molecules are rapidly hydrolysed. The first report of the liver of adult rats injected with tritium-labelled vitamin A-containing CMs found an increase in unesterified 3H-retinol, followed by an increase in esterified 3H-retinol, accompanied by a change in the composition of the retinyl ester. Compared with the composition of CM retinyl ester, which somewhat resem-bled the fatty acids in the diet, the hepatic retinyl ester was mostly retinyl pal-mitate and retinyl stearate, reflecting the post-uptake metabolism of retinol in the liver (65).
The next steps in the physiological processing of vitamin A are highly dependent on vitamin A status. Blomhoff et al. (66) first showed that the fate of CM vitamin A once taken up by the liver differs in vitamin A-adequate (control) and vitamin A-deficient rats. In the control rats, CM vitamin A first appeared in the hepatic parenchymal cell fraction and then most was trans-ferred over the next 240 minutes into the HSC fraction. At the end of the 4-hour experiment, ~65% of the injected dose of CM vitamin A remained in the liver. In contrast, in vitamin A-deficient liver, transfer of the vitamin A to the HSC fraction did not occur, and yet the parenchymal cell fraction lost vitamin A. By the end of the experiment, <35% of the injected dose of CM vitamin A remained in the liver in the vitamin A-deficient group. These results complement those showing that the uptake of CM vitamin A into the liver of the vitamin A-deficient rat results in a very rapid secretion into plasma of retinol bound to RBP (67, 68) (see below).
In the vitamin A-adequate state, HSCs are known to be the principal site of retinyl ester storage, with >50% of total body vitamin A in liver and >90% of the liver vitamin A in the form of retinyl ester located in these cells. As noted early, the neonatal liver contains HSCs that appear similar morphologically to those in adult liver, but, since liver total vitamin A is very low in neonates, most HSCs must therefore contain very little vitamin A. It would be expect-ed that the retinyl ester content of the HSCs would increase after vitamin A

61
supplementation. In a 12-hour study in 8-day postnatal rats, we observed an increment in liver vitamin A that was equivalent to ~22% of the retinyl pal-mitate in the vitamin A supplement;1 this number thus provides a minimal estimate of absorption and hepatic storage since some of the vitamin A is likely to have been stored elsewhere and some of may have been metabolized. Because retinyl ester has the physical property of viscous oil, it is a highly efficient storage form. HSC retinyl ester is dissolved along with triacylglyc-erols, cholesteryl esters and other lipids in lipid droplets (69), which appear as multiple, clustered lipid droplets on electron microscopy. The proportion of retinyl ester, compared with other lipids, within lipid droplets and the size of the lipid droplets increases after consumption of higher levels of vitamin A (69, 70). In vitamin A-adequate or vitamin A-supplemented conditions, the vitamin A-containing HSCs themselves can be visualized by light micros-copy (71). Conversely, during and after vitamin A depletion, the amount of retinyl ester in HSCs falls and eventually disappears. Whereas vitamin A depletion is a slow process, vitamin A repletion can increase the vitamin A content of HSCs rapidly, within the postprandial period following intake of a large dose of vitamin A.
Retinol esterification and hydrolysisEsterification and hydrolysis work oppositely, esterification to accrue any excess retinol as retinyl ester and hydrolysis to release retinol as it is needed. The balance of these processes is thought to determine whether retinol storage is increased or decreased. The physiological mechanism for the formation of retinyl ester in the liver HSC involves, first, binding of retinol to CRBPI, which may occur immediately upon uptake, and then the transfer of retinol bound to CRBPI to the enzyme LRAT, which transfers fatty acid from membrane-associated lecithin and generates retinyl ester. Hepatic LRAT is localized pri-marily in HSCs (72). LRAT activity is sensitive to vitamin A status: in rats fed a vitamin A-deficient diet, the decline in LRAT activity followed the decline in plasma retinol nearly exactly (73). LRAT mRNA levels were also low in vitamin A-deficient animals (74, 75). The low expression of LRAT mRNA and enzymatic activity were completely reversed by 24 hours following a single oral dose of retinol (73), and even more rapidly, by 4 hours, after adminis-tration of a small dose of RA (76, 77). Thus, RA is the principal metabolite
ANNEX 2: BACKGROUND PAPERS
1 This calculation is based on a dose of 6 mg retinol/kg given once according to daily body weight (mean weight ~15.0 g on postnatal day 7), which in sum equalled ~314 nmol of retinol, and on an observed increase, 6 hours after dosing, in liver vitamin A of 127 (197 – 70) nmol/g with a mean liver weight of 0.55 g, equalling 70 nmol per liver. Thus, the increment in total retinol stored in the liver of vitamin A-supplemented rats was ~70 nmol retinol. Therefore, the minimal uptake and retention in the liver of the vitamin A-supplemented neonates during the 6-hour postprandial period was about 22% of the administered dose.

62
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
of retinol that functions as a positive feedback regulator of LRAT expression in the liver, as has recently been confirmed by molecular studies (78). Since LRAT activity in the intestine is not regulated in this manner and does not fall during vitamin A deficiency, the intestine is maintained ready to absorb and esterify vitamin A whenever it becomes available (73). The ready state of intestinal LRAT is important for the response to vitamin A supplementation. Conversely, in mice lacking LRAT, vitamin A absorption is low (79). Overall, the liver-specific regulation of LRAT by the animal’s vitamin A status and the effect of RA repletion appear to be beneficial adaptations, which may limit retinol esterification and storage in HSCs as vitamin A deficiency develops, and thereby preserve retinol for oxidative metabolism and RA formation (see Figure A2.2.1 and (12)). Oppositely, when RA is in abundance the upregula-tion of LRAT maintains or increases the capacity to store retinol as retinyl ester.
Indirect evidence for absorption and studies from clinical trials in neonatesIn a randomized, double-blind, placebo-controlled South African hospital trial (designed to determine whether a 25 000 IU enteral dose of vitamin A, given to low-birth-weight (LBW) neonates by nasogastric tube between 36 and 60 hours after delivery and repeated on days 4 and 8, would be absorbed and well tolerated), the mean serum retinol concentration was significantly higher in the vitamin A-treated group (~46 ± 17 µg/dL vs 13 ± 6.5 µg/dL) (80). Toxic effects – vomiting, drowsiness, irritability or bulging of the fontanelles – were not observed. The investigators concluded that high-dose enteral vitamin A is well absorbed in LBW neonates. Additional indirect evidence can be found in a study of Kenyan infants in which an improved relative dose response (RDR) test was used as an indicator of adequate vitamin A storage. The RDR test was reported to be improved in those infants who had received a supplement of 100 000 IU of vitamin A, versus placebo, at week 14 (81); however, the differ-ence in the proportion of infants showing improvement was quite small (69% with deficient scores in the vitamin A group vs 78 % in the placebo group).
Retinyl ester mobilization, RBP synthesis and releaseThe mobilization of hepatic retinyl ester occurs on an “as needed” basis. HSC retinyl ester is gradually hydrolysed when net vitamin A intake is below the vitamin A needs of the individual. The molecular signals for mobilization of vitamin A from HSCs are still not well understood, but the presence of apo-RBP in plasma and/or low levels of circulating retinol or RA are possible signals to the liver that more retinol is needed by peripheral tissues. Essential-ly all the HSC retinyl ester can be hydrolysed and mobilized. Thus, negligible amounts of vitamin A remain in the liver in the depleted state. REH activity in the liver of rats did not differ between vitamin A-deficient and vitamin

63
A-adequate animals (72), suggesting that REH activity is not an important point of regulation of retinoid homeostasis.
Liver hepatocytes are the major source of RBP. Although the concentration of plasma RBP is relatively low compared with that of many other plasma pro-teins, the rate of synthesis of RBP protein is high, because of the rapid turno-ver of RBP, which has a plasma half-life of ~11–12 hours in adult men (82), and the intrahepatic degradation of a portion of the newly synthesized RBP, such that not all is secreted. Adequate RBP synthesis is dependent on an adequate supply of amino acids and energy (calories) (83, 84). The process of RBP secre-tion from the liver is unusual in that it is dependent on the binding of reti-nol to the newly synthesized apo-RBP protein. After RBP is synthesized in the endoplasmic reticulum, the apoprotein moves to the Golgi apparatus and then “waits” for a molecule of retinol to become associated with it, before it is released as holo-RBP. Little is known about the control of this process. How-ever, if the concentration of hepatic retinol falls into the inadequate range, RBP accumulates in the liver. The administration of vitamin A to vitamin A-deficient rats, either orally or directly as an injection of CM vitamin A or as free retinol resulted in a very rapid secretion of holo-RBP from liver into plasma (67, 68). These observations provided the biological rationale for the RDR test subsequently used in human clinical and field studies (85, 86). Pulse labelling studies using radiolabelled amino acids have shown that a portion of the newly synthesized RBP in hepatocytes is degraded intracellularly because of protein misfolding or when more RBP is synthesized than is required (84). In any case, the liver synthesizes more RBP than is secreted.
The predominant form of vitamin A in fasting plasma is unesterified reti-nol bound to RBP. Retinol is also the form in which vitamin A is most often measured as an indicator of vitamin A status. However, plasma retinol is not in itself functional and, moreover, its level may not signify the vitamin A sta-tus of tissues. The rate of RBP synthesis can also affect plasma retinol levels. Plasma RBP levels are sensitive to nutritional status with respect to protein, calories and other micronutrients, such as zinc, which affect secretion. RBP synthesis in the liver is reduced in states of inflammation, even when liver vitamin A is not depleted (87). Table A2.2.2 lists several conditions with reduced serum retinol and RBP levels, either in children or in relevant ani-mal models of human nutrition or disease conditions. Because information is lacking on the rate of neonatal hepatic RBP synthesis, it is not known whether RBP accumulates in the neonatal liver as it does in the liver of older animals with similar low hepatic vitamin A stores.
Retinoid oxidation and elimination in bileAnother physiological function of the liver is the elimination of retinol and its metabolites. Studies using isotopically labelled vitamin A have shown that
ANNEX 2: BACKGROUND PAPERS
64
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
most of the vitamin A or its metabolic products produced by oxidation and/or conjugation are eliminated in the faeces (15). These results together with the detection of numerous products of vitamin A metabolism in bile imply that the liver plays a major role in eliminating vitamin A through processes of oxidation and conjugation. As noted earlier, conjugation reactions are slow to develop in the liver of neonates, as is well known for bilirubin conjugation (61). However, it is not known whether this also affects the conjugation of retinoid metabolites. Since similar UDP-glucuronidyl transferases are involved, this is a possibility to be considered.
In adult animals, dynamical models have predicted that the fractional cata-bolic rate for retinol and the mass of retinol disposed per day does not increase in proportion to the intake of vitamin A or vitamin A storage (98), which may
Table A2.2.2Examples of conditions associated with low plasma retinol or retinol transport proteins
Condition Effect on vitamin AInfants Prematurity Lower plasma retinol, RBP and TTR compared with full-
term infants (88–90)
Steroid therapy Plasma retinol and RBP increased transiently by treatment; possible mobilization of retinol from liver stores (91)
Children >6 months
Measles infection Low plasma retinol and low RBP to TTR ratio regardless of retinol supplementation during acute phase response to measles (92)
Malarial infection Proportional decreases in retinol, RBP and TTR during inflammation (93)
Reduced gut barrier function
Inverse correlation of lactulose/mannitol test of gut permeability with serum retinol and RBP (51)
Acute illness (accidental kerosene poisoning)
Low plasma retinol; low RBP and TTR; infection-associated reduction in retinol transport associated with acute phase response (94)
Animal studies
Inflammation Reduced plasma retinol, RBP and TTR in rat model of endotoxin-induced inflammation during control feeding or marginal vitamin A deficiency; increased liver retinyl esters and reduced retinol; slow clearance of plasma retinyl esters (92)
Diabetes Low serum retinol, low hepatic RBP mRNA in BB rat model of spontaneous diabetes; reduced mobilization of liver retinol on RBP; possible role of low zinc (95)
Iron deficiency Low plasma retinol and increased liver retinyl esters in rats fed low-iron diets; impaired mobilization of retinol from storage in liver, reduced absorption, slowed utilization (96, 97)
See text for abbreviations.

65
ANNEX 2: BACKGROUND PAPERS
explain in part the propensity for vitamin A accumulation over time in tissues when intake exceeds requirements. Overall, it appears that retinoid oxidation is well controlled, but does not completely keep up with the intake of vitamin A when intake is high.
Peripheral tissue vitamin A metabolism Retinol in peripheral tissues A number of tissues can be considered “peripheral” in the sense that vitamin A reaches them mainly in the form of holo-RBP, after the initial processing of newly absorbed dietary vitamin A in the liver. In most tissues, the most abundant form of vitamin A is retinyl ester, often contained in stellate-like cells within peripheral organs, but at concentrations well below those present in the liver. However, the peripheral tissue pools of vitamin A do add up, as indicated by a tracer kinetic study conducted in rats with marginal vitamin A status in which 44% of the whole-body vitamin A was estimated by modelling to be present in extrahepatic tissues (99). According to studies using retinol tracer kinetics and mathematical modelling, vitamin A in certain kinetically defined compartments turns over rapidly, while vitamin A also resides in a slowly turning over compartment that exchanges less readily or barely at all with plasma. This slowly turning over pool represents in part hepatic and in part peripheral tissue stores of retinyl ester (74, 100). The uptake of vitamin A uptake into peripheral tissues does not necessarily lead to catabolism. Indeed, retinol can emerge again as evidenced by the recycling of retinol between plas-ma, liver and peripheral tissues that has been observed by modelling plasma retinol kinetics in adult rats and humans (74). No similar studies have been reported in neonates.
Uptake of retinol from RBPThe mechanism of uptake of retinol from plasma into cells has remained speculative for many years. There is now greater clarity on this process for some tissues because a receptor for RBP, STRA6, has been identified (101). STRA6 is abundant in the RPE cells, where it is located on the apical surface, proximal to the blood vessels. STRA6 is also expressed in the lungs and pla-centa. The STRA6 protein forms a membrane-spanning channel that facili-tates the uptake of retinol from holo-RBP-TTR into the cells, where it is likely that CRBP picks it up. In model cells engineered to express both STRA6 and LRAT, the total uptake of retinol, originally bound to RBP-TTR, is higher. Thus, STRA6 and CRBP may work in concert to facilitate uptake, with LRAT facilitating the storage of retinol in cells that express all three proteins. The removal of retinol by LRAT may allow for continued uptake of retinol from RBP (101). The RBP protein itself apparently does not enter the cell.
The transport of retinol by RBP is not only important for providing retinol

66
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
to target tissues, but also for the “reverse retinol transport” of retinol from peripheral tissues back to the liver in the process known as retinol recy-cling (74). New information suggests that RBP (referred to as RBP4) is also an adipokine, and this aspect has focused attention on extrahepatic RBP, such as is found in several adipose tissue beds (102, 103). The RBP produced in periph-eral tissues could be important for recycling retinol back to plasma.
Other mechanisms of vitamin A uptake in peripheral tissuesDespite new knowledge about STRA6, the ways that vitamin A may enter tissues is still not completely understood, and number of pathways appear to be possible based on studies in various models. The liver either does not express STRA6 or at only low levels, yet retinol enters liver from plasma and exits again many times before undergoing irreversible catabolism, as predicted by mathematical modelling of kinetic data (104, 105). As noted earlier, peripheral tissues that express LPLs may derive vitamin A directly from CMs in the post-prandial period, when the CMs are bound to the LPLs during lipo lysis (60). Plasma also contains low concentrations of RA, generally 5–20 nM (22), and RA and its oxidation products may enter cells. Although RA cannot be reduced to form retinal and retinol, it is capable of acting directly in many vitamin A-dependent processes. Most of the oxidized metabolites of RA may be catabolites with little if any biological activity, but in some cells oxidized metabolites of RA have exhibited regulatory activity (106). Thus, more needs to be learned about the mechanisms of retinoid uptake by cells and tissues and how they may differ with age and nutritional status.
LungsIn humans, the lungs undergo alveolar septation from birth to ~2 years, whereas in mice and rats septation the period from postnatal day 4–14 is criti-cal, and septation is complete by postnatal day 28, a week after the typical age of weaning. In studies in mice, the expression of RAR during development has been shown to be important for pulmonary development and repair pro-cesses (107, 108), and treatment with RA has been shown to induce septation in the lungs of neonatal rats and mice (109). In the rat, significant retinyl ester accumulation in the fetal lungs begins in the third trimester, but the stored retinyl ester becomes quickly depleted during late gestation and early post-natal life (110). Thus, low vitamin A status in preterm infants (111–114) could be a contributing factor to poor lung maturation and increased susceptibility to respiratory diseases (115, 116). Additional research is needed in this area.
In a rat model, vitamin A, mostly as retinyl ester, accumulated in late gesta-tion, declines just before birth and then stays relatively low during the suckling period, while liver retinyl ester accumulates during this period (100). When postnatal day 5–8 rats were supplemented directly with vitamin A at dose

67
resembling 50 000 IU in human neonates, lung retinyl ester increased more than fivefold, and increased even more dramatically if one-tenth the amount of RA was added to the vitamin A dose (117, 118). Thus, the lungs are clearly capable of storing more vitamin A if it is provided and they are stimulated to do so by RA. A further molecular analysis showed that LRAT was increased 6 and 12 hours after vitamin A treatment, and that STRA6 and LRAT, as well as CYP26B1, an oxidative enzyme, were increased in the lungs after neonates were supplemented with vitamin A enriched with RA (118).
Renal metabolism RBP filtration and reuptakeThe combination of RBP, a 21 kDa protein with its co-transport protein TTR results in a complex of ~75 kDa. The greater size of the complex reduces the rate of glomerular filtration and loss of RBP in urine, and therefore increases the plasma half-life of retinol and RBP. Nonetheless, the plasma half-life of holo-RBP bound to TTR is still short, ~11–12 hours in adult humans, com-pared with other plasma proteins such as albumin (half-life 20 days). Apo-RBP, which does not bind effectively to TTR, is rapidly filtered in the glomerulus and, consequently, the half-life of apo-RBP is very short, ~4 hours (82).
Studies on the mechanism of renal handling of RBP have identified a re uptake process that recovers a substantial portion of filtered RBP. The pro-tein megalin, a multiligand receptor which resides on the surface of renal tubular cells, has been shown to bind RBP and several other nutrient trans-port proteins including transferrin. Megalin, in cooperation with another renal tubule protein, cubulin, is involved in the endocytic reuptake and thus in the physiological conservation of retinol and RBP. Christensen et al. (119) found that mice lacking megalin lose much more RBP and retinol in urine. Megalin also functions in the reuptake of TTR (120).
In a recent study on urinary RBP and retinol excretion in LBW and full-term infants, both groups excreted retinol and RBP in urine but the rate of excretion was significantly higher in the LBW group (121). Renal function is low at birth, increasing over the first few months of life (122). Whether the rate of glomerular filtration of RBP-TTR is higher, reuptake is less efficient, or both, in neonates and especially in LBW neonates is not yet known. This area is also important for further research because factors that increase retinol excretion would be expected to increase the requirement for vitamin A.
Summary Based on our current understanding of vitamin A metabolism in general and, in some cases, on direct studies in neonates, it can be anticipated that vitamin A given as an oral supplement will undergo rapid metabolism in the postpran-
ANNEX 2: BACKGROUND PAPERS

68
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
dial period, not only in the intestine but also in liver and some extrahepatic organs. The efficient absorption of vitamin A will depend on a well-developed capacity for fat digestion and absorption. Direct evidence from studies in ani-mal models and indirect evidence from clinical studies suggest that absorp-tion is generally efficient in neonates. Nonetheless, infants having problems with fat digestion or absorption, or having no dietary fat to absorb along with the dose of vitamin A, would be expected to absorb the vitamin A supplement much less efficiently. In animals, LRAT activity is high in the small intestine and it is not reduced during vitamin A deficiency. From these data it can be deduced that the intestine stands ready to absorb vitamin A when the vitamin is administered along with an appropriate amount of fat (12). Based on stud-ies in animals that were vitamin A deficient or marginal in status, it can be anticipated that CM remnant vitamin A uptake into the liver occurs at a nor-mal rate regardless of vitamin A status (66); however, the transfer of retinol to HSCs and its use for retinyl ester formation should be expected to be low in vitamin A deficiency. Nevertheless, healthy neonatal rats having low retinyl ester reserves but nursed by vitamin A-adequate mothers and without signs of deficiency are able to absorb and rapidly store in the liver more than 20% of a bolus oral dose of vitamin A, equivalent to 50 000 IU in neonates (117, 123). Therefore, the transfer of retinol to HSCs might occur efficiently in neonates even though pre-existing liver vitamin A stores are low. It may also be antici-pated that a portion of the newly absorbed retinol will appear in plasma as holo-RBP.
Although the RDR test may be able to “report” on liver vitamin A adequa-cy, we do not know enough about the basic regulation of neonatal hepatic RBP synthesis and secretion to know whether the RDR test can be interpreted in the same way for neonates as for older children. In animals, low vitamin A status is associated with low LRAT activity in the liver and lungs, but LRAT expression in the lungs is responsive to vitamin A supplementation (118). Thus, treatment with vitamin A may have an immediate effect on tissue reti-nol metabolism and retinyl ester concentrations in the postprandial period. At the same time that retinyl ester is formed, retinoid oxidation may also be increased, as suggested by the increased expression of CYP26 genes in liver and lungs after vitamin A and RA administration (118). Over the longer term, vitamin A supplementation in the neonatal period alone may not be enough to prevent low tissue vitamin A levels from gradually developing, as observed in rats supplemented with vitamin A at neonatal age and then fed a vitamin A-deficient diet after weaning, in whom normal plasma but low lung vitamin A levels were found (124). Full-term and especially LBW infants may also be prone to urinary loss of retinol, RBP and TTR, which could increase their need for vitamin A (121). Overall, the state of knowledge of vitamin A metab-

69
olism in the neonate is very fragmentary. Making logical deductions from the situation of adult animals and humans with low vitamin A status is helpful in predicting what may happen in neonates, but direct studies are needed, par-ticularly with respect to the cellular and molecular processes by which vita-min A is stored in liver, secreted as holo-RBP, filtered and reabsorbed in the kidneys, and used for RA production in the rapidly growing infant.
AcknowledgementSupport from National Institutes of Health grants DK41479, HD66892 and CA90214 contributed to the author’s research cited in this review.
References1. Dann WJ. The transmission of vitamin A from parents to young in mammals.
Biochemical Journal, 1932, 26:1072–1080.2. Dann WJ. The transmission of vitamin A from parents to young in mammals:
Effect of the liver reserves of the mother on the transmission of vitamin A to the foetal and suckling rat. Biochemical Journal, 1934, 8:2141–2146.
3. Moore T. Vitamin A transfer from mother to offspring in mice and rats. Interna-tional Journal for Vitamin and Nutrition Research, 1971, 41:301–306.
4. Takahashi YI, Smith JE, Goodman DS. Vitamin A and retinol-binding protein metabolism during fetal development in the rat. American Journal of Physiology, 1977, 233:E263–E272.
5. Davila ME et al. Vitamin A during lactation: Relationship of maternal diet to milk vitamin A content and to the vitamin A status of lactating rats and their pups. Journal of Nutrition, 1985, 115:1033–1041.
6. Vitamin A supplementation. Geneva, World Health Organization. (http://www.who.int/vaccines/en/vitamina.shtml, accessed December 2009).
7. McCollum EV, Davis M. The necessity of certain lipins in the diet during growth. Journal of Biological Chemistry, 1913,15:167–175.
8. Klassen TP et al. Children are not just small adults: the urgent need for high-quality trial evidence in children. Public Library of Science Medicine, 2008, 5:e173.
9. Wang JL et al. Retinoic acid stimulates early cellular proliferation in the adapt-ing remnant rat small intestine after partial resection. Journal of Nutrition, 1997, 127:1297–1303.
10. Baybutt RC, Hu L, Molteni A. Vitamin A deficiency injures lung and liver pa-renchyma and impairs function of rat type II pneumocytes. Journal of Nutrition, 2000, 130:1159–1165.
11. Ross AC. Vitamin A and carotenoids. In: Shils ME et al., eds. Modern nutri-tion in health and disease. Baltimore, MD, Lippincott Williams and Wilkins, 2006:351–375.
12. Ross AC, Zolfaghari R. Regulation of hepatic retinol metabolism: perspectives from studies on vitamin A status. Journal of Nutrition, 2004, 134:269S–275S.
ANNEX 2: BACKGROUND PAPERS

70
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
13. Penniston KL, Tanumihardjo SA. Elevated serum concentrations of β-glucuro-nide metabolites and 4-oxoretinol following treatment with vitamin A in lactat-ing sows: a model for evaluating supplementation of lactating women. American Journal of Clinical Nutrition, 2005, 81:851–858.
14. Zachman RD, Dunagin PE Jr, Olson JA. Formation and enterohepatic circula-tion of metabolites of retinol and retinoic acid in bile duct-cannulated rats. Jour-nal of Lipid Research, 1966, 7:3–9.
15. Varma RN, Beaton GH. Quantitative aspects of the urinary and fecal excretion of radioactive metabolites of vitamin A in the rat. Canadian Journal of Physiol-ogy and Pharmacology, 1972, 50:1026–1037.
16. Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene, 2004, 328:1–16.
17. Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochemical Journal, 2000, 348:481–495.
18. Ross AC. Cellular metabolism and activation of retinoids: roles of the cellular retinoid-binding proteins. Journal of the Federation of American Societies for Ex-perimental Biology, 1993, 7:317–327.
19. Niederreither K et al. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nature Genetics, 1999, 21:444–448.
20. Sonneveld E, van der Saag PT. Metabolism of retinoic acid: implications for de-velopment and cancer. International Journal for Vitamin and Nutrition Research, 1998, 68:404–410.
21. Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signal-ing pathway during mouse embryogenesis. Annual Review of Pharmacology and Toxicology, 2006, 46:451–480.
22. Berggren Söderlund M, Fex GA, Nilsson-Ehle P. Concentrations of retinoids in early pregnancy and in newborns and their mothers. American Journal of Clini-cal Nutrition, 2005, 81:633–636.
23. Agarwal K et al. Factors affecting serum vitamin A levels in matched maternal-cord pairs. Indian Journal of Pediatrics, 2008, 75:443–446.
24. Radhika MS et al. Red palm oil supplementation: a feasible diet-based approach to improve the vitamin A status of pregnant women and their infants. Food and Nutrition Bulletin, 2003, 24:208–217.
25. Vahlquist A et al. The concentrations of retinol-binding protein, prealbumin, and transferrin in the sera of newly delivered mothers and children of vari-ous ages. Scandinavian Journal of Clinical and Laboratory Investigation, 1975, 35:569–575.
26. Arnhold T et al. Identification of 9-cis-retinoic acid, 9,13-di-cis-retinoic acid, and 14-hydroxy-4,14-retro-retinol in human plasma after liver consumption. Life Sciences, 1996, 59:PL169–PL177.
27. Horst RL et al. 9,13-Di-cis-retinoic acid is the major circulating geometric iso-mer of retinoic acid in the periparturient period. Archives of Biochemistry and Biophysics, 1995, 322:235–239.

71
28. Sugitani T et al. Comparison human infantile develpmental hepatic stellate cell changes with liver disesase by immunohistochemical study. Gastroenterology, 1998, 114:A1348.
29. Nitou M, Ishikawa K, Shiojiri N. Immunohistochemical analysis of development of desmin-positive hepatic stellate cells in mouse liver. Journal of Anatomy, 2000, 197:635–646.
30. Dahro M, Gunning D, Olson JA. Variations in liver concentrations of iron and vitamin A as a function of age in young American children dying of the sudden infant death syndrome as well as of other causes. International Journal for Vita-min and Nutrition Research, 1983, 53:13–18.
31. Stoltzfus RJ, Humphrey JH. Vitamin A and the nursing mother-infant dyad: evi-dence for intervention. Advances in Experimental Medicine and Biology, 2002, 503:39–47.
32. Green MH et al. Increased rat mammary tissue vitamin A associated with in-creased vitamin A intake during lactation is maintained after lactation. Journal of Nutrition, 2001, 131:1544–1547.
33. Akohoue SA, Green JB, Green MH. Dietary vitamin A has both chronic and acute effects on vitamin A indices in lactating rats and their offspring. Journal of Nutrition, 2006, 136:128–132.
34. Pasatiempo AMG, Ross AC. Effects of food or nutrient restriction on milk vita-min A transfer and neonatal vitamin A stores in the rat. British Journal of Nutri-tion, 1990, 63:351–362.
35. Penniston KL, Valentine AR, Tanumihardjo SA. A theoretical increase in in-fants’ hepatic vitamin A is realized using a supplemented lactating sow model. Journal of Nutrition, 2003, 133:1139–1142.
36. Goodman DS et al. The intestinal absorption and metabolism of vitamin A and β-carotene in man. Journal of Clinical Investigation, 1966, 45:1615–1623.
37. Huang HS, Goodman DS. Vitamin A and carotenoids. I. Intestinal absorption and metabolism of 14C-labeled vitamin A alcohol and β-carotene in the rat. Journal of Biological Chemistry, 1965, 240:2839–2844.
38. Swartz-Basile DA et al. Vitamin A deficiency inhibits intestinal adaptation by modulating apoptosis, proliferation, and enterocyte migration. American Jour-nal of Physiology. Gastrointestinal and Liver Physiology, 2003, 285:G414–G432.
39. Nikawa T et al. Vitamin A up-regulates expression of bone-type alkaline phos-phatase in rat small intestinal crypt cell line and fetal rat small intestine. Journal of Nutrition, 1998, 128:1869–1877.
40. Crow JA, Ong DE. Cell-specific immunohistochemical localization of a cellular retinol-binding protein (type two) in the small intestine of rat. Proceedings of the National Academy of Sciences of the United States of America, 1985, 82:4707–4711.
41. Ong DE et al. Ontogeny of two vitamin A-metabolizing enzymes and two reti-nol-binding proteins present in the small intestine of the rat. Journal of Lipid Research, 1991, 32:1521–1527.
ANNEX 2: BACKGROUND PAPERS

72
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
42. Takase S, Goda T, Shinohara H. Adaptive changes of intestinal cellular retinol-binding protein, type II following jejunum-bypass operation in the rat. Biochim-ica et Biophysica Acta, 1993, 1156:223–231.
43. Herrera E, Amusquivar E. Lipid metabolism in the fetus and the newborn. Dia-betes/Metabolism Research and Reviews, 2000, 16:202–210.
44. Sangild PT et al. Preterm birth affects the intestinal response to parenteral and enteral nutrition in newborn pigs. Journal of Nutrition, 2002, 132:2673–2681.
45. Li X et al. Bile salt-stimulated lipase and pancreatic lipase-related protein 2 are the dominating lipases in neonatal fat digestion in mice and rats. Pediatric Re-search, 2007, 62:537–541.
46. Miller R, Lowe ME. Carboxyl ester lipase from either mother’s milk or the pan-creas is required for efficient dietary triglyceride digestion in suckling mice. Journal of Nutrition, 2008, 138:927–930.
47. Ross AC, Davila ME, Cleary MP. Fatty acids and retinyl esters of rat milk: effects of diet and duration of lactation. Journal of Nutrition, 1985, 115:1488–1497.
48. Harrison EH. Mechanisms of digestion and absorption of dietary vitamin A. Annual Review of Nutrition, 2005, 25:87–103.
49. Fredrikzon B et al. Bile salt-stimulated lipase in human milk: evidence of activ-ity in vivo and of a role in the digestion of milk retinol esters. Pediatric Research, 1978, 12:1048–1052.
50. Lebenthal A, Lebenthal E. The ontogeny of the small intestinal epithelium. Jour-nal of Parenteral and Enteral Nutrition, 1999, 33 (Suppl. 5):S3–S6.
51. Quadro L et al. Retinol and retinol-binding protein: gut integrity and cirulating immunoglobulins. Journal of Infectious Diseases, 2000, 182:S97–S102.
52. Kayden HJ, Traber MG. Clinical, nutritional and biochemical consequences of apolipoprotein B deficiency. Advances in Experimental Medicine and Biology, 1986, 201:67–81.
53. Black DD. Development and physiological regulation of intestinal lipid absorp-tion. I. Development of intestinal lipid absorption: cellular events in chylomi-cron assembly and secretion. American Journal of Physiology. Gastrointestinal and Liver Physiology, 2007, 293:G519–G524.
54. Ross AC, Pasatiempo AM, Green MH. Chylomicron margination, lipolysis, and vitamin A uptake in the lactating rat mammary gland: implications for milk retinoid content. Experimental Biology and Medicine (Maywood, NJ), 2004, 229:46–55.
55. Wang X-D et al. Intestinal perfusion of β-carotene in the ferret raises retinoic acid level in portal blood. Biochimica et Biophysica Acta, 1993, 1167:159–164.
56. Arnhold T et al. Porcine intestinal metabolism of excess vitamin A differs fol-lowing vitamin A supplementation and liver consumption. Journal of Nutrition, 2002, 132:197–203.
57. Kurlandsky SB et al. Plasma delivery of retinoic acid to tissues in the rat. Journal of Biological Chemistry, 1995, 270:17850–17857.
58. Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regula-tion, and role in disease. Journal of Molecular Medicine, 2002, 80:753–769.

73
59. van Bennekum AM et al. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. Journal of Lipid Research, 1999, 40:565–574.
60. Blomhoff R, Skrede B, Norum KR. Uptake of chylomicron remnant retinyl ester via the low density lipoprotein receptor: Implications for the role of vitamin A as a possible preventive for some forms of cancer. Journal of Internal Medicine, 1990, 228:207–210.
61. Beath SV. Hepatic function and physiology in the newborn. Seminars in Neona-tology, 2003, 8:337–346.
62. Papp V et al. Architectural changes during regenerative and ontogenic liver growth in the rat. Liver Transplantation, 2009, 15:177–183.
63. Alexander B, Guzail MA, Foster CS. Morphological changes during hepatocel-lular maturity in neonatal rats. Anatomical Record, 1997, 248:104–109.
64. Chou JY, Ito F. Role of retinoic acid in maturation of fetal liver cells in vitro. Biochemical and Biophysical Research Communications, 1984, 118:165–175.
65. Goodman DS, Huang HS, Shiratori T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. Journal of Lipid Research, 1965, 6:390–396.
66. Blomhoff R et al. In vivo uptake of chylomicron [3H]retinyl ester by rat liver: evi-dence for retinol transfer from parenchymal to nonparenchymal cells. Proceed-ings of the National Academy of Sciences of the United States of America, 1982, 79:7326–7330.
67. Muto Y et al. Regulation of retinol-binding protein metabolism by vitamin A status in the rat. Journal of Biological Chemistry, 1972, 247:2542–2550.
68. Smith JE, Goodman DS. Retinol-binding protein and the regulation of vitamin A transport. Federation Proceedings, 1979, 38:2504–2509.
69. Hendriks HFJ, Brouwer A, Knook DL. The role of hepatic fat-storing (stellate) cells in retinoid metabolism. Hepatology, 1987, 7:1368–1371.
70. Blaner WS et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochimica et Biophysica Acta, 2009, 1791:467–473.
71. Wake K. Development of vitamin A-rich lipid droplets in multivesicular bodies of rat liver stellate cells. Journal of Cell Biology, 1974, 63:683–691.
72. Matsuura T et al. Lecthin: retinol acyltransferase and retinyl ester hydrolase ac-tivities are differentially regulated by retinoids and have distinct distributions between hepatocyte and nonparenchymal cell fractions of rat liver. Journal of Nutrition, 1997, 127:218–224.
73. Randolph RK, Ross AC. Vitamin A status regulates hepatic lecithin: retinol acyltransferase activity in rats. Journal of Biological Chemistry, 1991, 266:16453–16457.
74. Cifelli CJ, Green JB, Green MH. Use of model-based compartmental analysis to study vitamin A kinetics and metabolism. In: Litwak G, ed. Vitamins and hor-mones. San Diego, CA, Academic Press, 2007:161–195.
75. Zolfaghari R, Ross AC. Lecithin: retinol acyltransferase from mouse and rat liv-er: cDNA cloning and liver-specific regulation by dietary vitamin A and retinoic acid. Journal of Lipid Research, 2000, 41:2024–2034.
ANNEX 2: BACKGROUND PAPERS

74
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
76. Matsuura T, Ross AC. Regulation of hepatic lecithin: retinol acyltransferase ac-tivity by retinoic acid. Archives of Biochemistry and Biophysics, 1993, 301:221–227.
77. Shimada T et al. Regulation of hepatic lecithin: retinol acyltransferase activity by retinoic acid receptor-selective retinoids. Archives of Biochemistry and Biophys-ics, 1997, 344:220–227.
78. Zolfaghari R, Ross AC. An essential set of basic DNA response elements is re-quired for receptor-dependent transcription of the lecithin:retinol acyltrans-ferase (Lrat) gene. Archives of Biochemistry and Biophysics, 2009, 489:1–9.
79. O’Byrne SM et al. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). Journal of Biological Chemistry, 2005, 280:35647–35657.
80. Coutsoudis A et al. Absorption of high-dose enteral vitamin A in low-birth-weight neonates. South African Medical Journal, 1996, 86:1337–1339.
81. Ayah RA et al. The effects of maternal and infant vitamin A supplementation on vitamin A status: a randomised trial in Kenya. British Journal of Nutrition, 2007, 98:422–430.
82. Goodman DS. Plasma retinol-binding protein. In: Sporn MB, Roberts AB, Good-man DS, eds. The retinoids, volume 2. Orlando, FL, Academic Press, 1984:42–88.
83. Smith JE, Muto Y, Goodman DS. Tissue distribution and subcellular localiza-tion of retinol-binding protein in normal and vitamin A-deficient rats. Journal of Lipid Research, 1975, 16:318–323.
84. Kaji EH, Lodish HF. Unfolding of newly made retinol-binding protein by dithiothreitol. Sensitivity to retinoids. Journal of Biological Chemistry, 1993, 268:22188–22194.
85. Underwood BA, Loerch JD, Lewis KC. Effects of dietary vitamin A deficiency, retinoic acid and protein quantity and quality on serially obtained plasma and liver levels of vitamin A in rats. Journal of Nutrition, 1979, 109:796–806.
86. Underwood BA. Methods for assessment of vitamin A status. Journal of Nutri-tion, 1990, 120:1459–1463.
87. Rosales FJ et al. Effects of acute inflammation on plasma retinol, retinol-binding protein, and its mRNA in the liver and kidneys of vitamin A-sufficient rats. Jour-nal of Lipid Research, 1996, 37:962–971.
88. Mupanemunda RH et al. Postnatal changes in serum retinol status in very low birth weight infants. Early Human Development, 1994, 38:45–54.
89. Sasanow SR et al. Effect of gestational age upon prealbumin and retinol binding protein in preterm and term infants. Journal of Pediatric Gastroenterology and Nutrition, 1986, 5:111–115.
90. Bhatia J, Ziegler EE. Retinol-binding protein and prealbumin in cord blood of term and preterm infants. Early Human Development, 1983, 8:129–133.
91. Shenai JP, Mellen BG, Chytil F. Vitamin A status and postnatal dexamethasone treatment in bronchopulmonary dysplasia. Pediatrics, 2000, 106:547–553.
92. Rosales FJ, Ross AC. Acute inflammation induces hyporetinemia and modifies the plasma and tissue response to vitamin A supplementation in marginally vitamin A-deficient rats. Journal of Nutrition, 1998, 128:960–966.

75
93. Rosales FJ et al. Relation of serum retinol to acute phase proteins and malarial morbidity in Papua New Guinea children. American Journal of Clinical Nutri-tion, 2000, 71:1582–1588.
94. Filteau SM et al. Use of the retinol-binding protein: transthyretin ratio for as-sessment of vitamin A status during the acute-phase response. British Journal of Nutrition, 2000, 83:513–520.
95. Lu Jl et al. The metabolic availability of vitamin A is decreased at the onset of diabetes in BB rats. Journal of Nutrition, 2000, 130:1958–1962.
96. Rosales FJ et al. Iron deficiency in young rats alters the distribution of vitamin A between plasma and liver and between hepatic retinol and retinyl esters. Journal of Nutrition, 1999, 129:1223–1228.
97. Jang JT et al. Kinetic analysis shows that iron deficiency decreases liver vitamin A mobilization in rats. Journal of Nutrition, 2000, 30:1291–1296.
98. Green MH, Green JB, Lewis KC. Variation in retinol utilization rate with vita-min A status in the rat. Journal of Nutrition, 1987, 117:694–703.
99. Lewis KC et al. Retinol metabolism in rats with low vitamin A status: A com-partmental model. Journal of Lipid Research, 1990, 31:1535–1548.
100. Von Reinersdorff D, Green MH, Green JB. Development of a compartmental model describing the dynamics of vitamin A metabolism in men. Advances in Experimental Medicine and Biology, 1998, 445:207–223.
101. Kawaguchi R et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science, 2007, 315:820–825.
102. Tsutsumi C et al. Retinoids and retinoid-binding protein expression in rat adi-pocytes. Journal of Biological Chemistry,1992, 267:1805–1810.
103. Kloting N et al. Serum retinol-binding protein is more highly expressed in vis-ceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metabolism, 2007, 6:79–87.
104. Green MH, Green JB. Vitamin A intake and status influence retinol balance, utilization and dynamics in rats. Journal of Nutrition, 1994, 124:2477–2485.
105. Green MH et al. Vitamin A metabolism in rat liver: a kinetic model. American Journal of Physiology, 1993, 264:G509–G521.
106. Sonneveld E et al. Retinoic acid hydroxylase (CYP26) is a key enzyme in neu-ronal differentiation of embryonal carcinoma cells. Developmental Biology, 1999, 213:390–404.
107. McGowan SE. Contributions of retinoids to the generation and repair of the pul-monary alveolus. Chest, 2002, 121:206S–208S.
108. Massaro GD, Massaro D, Chambon P. Retinoic acid receptor-alpha regulates pulmonary alveolus formation in mice after, but not during, perinatal period. American Journal of Physiology. Lung Cellular and Molecular Physiology, 2003, 284:L431–L433.
109. Massaro D, Massaro GD. Pre- and postnatal lung development, maturation, and plasticity – invited review: Pulmonary alveoli: formation, the “call for oxygen”, and other regulators. American Journal of Physiology. Lung Cellular and Molecu-lar Physiology, 2002, 282:L345–L358.
ANNEX 2: BACKGROUND PAPERS

76
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
110. Shenai JP, Chytil F. Vitamin A storage in lungs during perinatal development in the rat. Biology of the Neonate, 1990, 57:126–132.
111. Shenai JP et al. Plasma vitamin A and retinol-binding protein in premature and term neonates. Journal of Pediatrics, 1981, 99:302–305.
112. Shenai JP et al. Vitamin A supplementation and bronchopulmonary dysplasia – revisited. Journal of Pediatrics, 1992, 121:399–401.
113. Spears K, Cheney C, Zerzan J. Low plasma retinol concentrations increase the risk of developing bronchopulmonary dysplasia and long-term respiratory dis-ability in very-low-birth-weight infants. American Journal of Clinical Nutrition, 2004, 80:1589–1594.
114. Mactier H, Weaver LT. Vitamin A and preterm infants: what we know, what we don’t know, and what we need to know. Archives of Disease in Childhood. Fetal and Neonatal Edition, 2005, 90:F103–F108.
115. Zachman RD. Role of vitamin A in lung development. Journal of Nutrition, 1995, 125:1634S–1638S.
116. Chan V, Cheeseman P, Gamsu HR. Vitamin A status in preterm and term in-fants at birth. Journal of Perinatal Medicine, 1993, 21:59–62.
117. Ross AC et al. Vitamin A combined with retinoic acid increases retinol uptake and lung retinyl ester formation in a synergistic manner in neonatal rats. Journal of Lipid Research, 2006, 47:1844–1851.
118. Wu L, Ross AC. Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT and CYP26B1 expression in neonatal lung. Journal of Lipid Research, 2010, 51:378–387.
119. Christensen EI et al. Evidence for an essential role of megalin in transepithelial transport of retinol. Journal of the American Society of Nephrology, 1999, 10:685–695.
120. Sousa MM et al. Evidence for the role of megalin in renal uptake of transthyre-tin. Journal of Biological Chemistry, 2000, 275:38176–38181.
121. Nagl B et al. Urinary vitamin A excretion in very low birth weight infants. Pedi-atric Nephrology, 2009, 24:61–66.
122. Maples HD, James LP, Stowe CD. Special pharmacokinetic and pharmacody-namic considerations in children. In: Burton ME et al., eds. Applied pharmacoki-netics and pharmacodynamics: principles of therapeutic drug, 4th ed. Baltimore, MD, Lippincott Williams and Wilkins, 2006:213–230.
123. Wu L. Investigation on the mechanism of vitamin A uptake, accumulation and metabolism in the lungs of the neonatal rat [thesis]. University Park, PA, Pennsyl-vania State University, 2008.
124. Ross AC, Li N-q. Lung retinyl ester is low in young adult rats fed a vitamin A-deficient diet after weaning, despite neonatal vitamin A supplementation and maintenance of normal plasma retinol. Journal of Nutrition, 2007, 137:2213–2218.

77
a2.3Vitamin A deficiency and the neonatal immune system: possible effects of vitamin A supplementation at birthKimberly Livingston, Charles. B Stephensen
USDA Western Human Nutrition Research Center, University of California, Davis, California, United States of America
Corresponding author: Charles B. Stephensen; [email protected]
Abstractn The immune system in early infancy must deal with the dual challenge of exposure to pathogenic microorganisms and initial colonization with com-mensal microflora. These challenges require the development of protective as well as regulatory responses. Vitamin A plays a role in the development of both protective (“proinflammatory”) and regulatory (“anti-inflammatory”) responses by the innate and adaptive immune systems. Since vitamin A defi-ciency is common in pregnant women, newborn infants are at high risk of vitamin A deficiency. Vitamin A deficiency is known to impair important aspects of both protective and regulatory immunity. Thus the provision of vitamin A supplements at birth may prevent adverse consequences of vitamin A deficiency on immune function during the neonatal period, although inter-vention trials conducted to date do not offer conclusive results with regard to an impact on infant mortality. This review examines possible immunologi-cal mechanisms by which vitamin A supplementation at birth might increase infant survival.
ANNEX 2: BACKGROUND PAPERS

78
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
IntroductionVitamin A deficiency is common in many developing countries. Worldwide, 190 million infants and young children have low serum retinol concentrations indicative of inadequate vitamin A stores (<0.70 µmol/L) with the majority living in developing countries in South Asia and Africa (1). Vitamin A defi-ciency increases the severity of common infections of childhood (2), presuma-bly because of impaired resistance to, or recovery from, infection (3); however, vitamin A supplementation does not necessarily decrease morbidity (4). Vitamin A supplementation between 6 months and 5 years in those at risk of vitamin A deficiency can decrease overall mortality (5–8). While the spe-cific biological mechanisms underlying this effect in humans have not been clearly defined, vitamin A supplementation in deficient individuals probably restores impaired immune function, thus improving recovery from infections and decreasing mortality. Another important mechanism may be improved maturation of other organ systems such as the lung (9).
All infants are born with low vitamin A stores (10), and thus are at risk of deficiency if intake is not adequate during the neonatal period. Infants born to vitamin A-deficient mothers have even lower serum retinol (11) and may have functional vitamin A deficiency at birth, with the risk further increased by the lower vitamin A concentrations in the breast milk (12). Infections can also increase the risk as they decrease the efficiency of absorption, alter retinol metabolism and, if severe, cause direct loss of vitamin A in the urine (3, 12).
Although vitamin A supplementation at >6 months of age decreases mor-tality, between 1 and 6 months of age this effect has not been shown (8, 13, 14). Studies in Indonesia (15), India (16) and Bangladesh (17) have demonstrated lower mortality when infants were supplemented soon after birth, but other studies (Nepal and Zimbabwe) have found no beneficial effect on mortal-ity (14, 18). In a pooled analysis of two independent trials conducted at the same site in Guinea-Bissau (one in infants with normal and one in infants with low birth weight), Benn and colleagues (19, 20) demonstrated no overall effect on mortality. But they found a significant interaction between vitamin A supplementation and sex, and suggested that mortality was lower in boys and higher in girls having vitamin A supplements; the higher mortality may be associated with the concomitant administration of the diphtheria-tetanus-pertussis vaccine. Further data from this group suggest that the effect of vita-min A supplementation may be modified by vaccination status (21, 22). These observations were not derived from an a priori hypothesis postulating a sex difference in response to vitamin A supplementation in early infancy, and as a mechanism to explain these observations is not immediately apparent, such interactions should be evaluated in future studies.
The above-mentioned data suggest that vitamin A supplementation at birth

79
may decrease infant mortality under certain circumstances. The disparate findings at different study sites, particularly the hypothetic higher mortality in Guinea-Bissau among girls, make it important to consider the mechanisms underlying the benefits and risks of vitamin A supplementation at birth. This review considers how such supplementation may alter the neonatal immune response to infection. The effects of vitamin A supplementation on the immune system at birth may differ from its effects later in infancy because of fundamental differences between the immune systems of neonates and older infants. While the immune system of neonates comprises the same cells and tissues as it does later in life, the responses of the immune system to pathogens are more “moderate”, or less “inflammatory”, in neonates, as discussed below. Such moderate responses could impair clearance of pathogenic organisms, but this shortcoming may be less important for neonates than it would be for older infants because neonates are protected by maternally derived immunity, including from breast milk. The major immunological challenge that comes with colonization of the gut and other body surfaces with commensal flora at birth may also produce excessive immune activation that would be damaging to the host if the immune system responded as it would later in life.
Vitamin A and innate immunityEpithelium
The first lines of defence against pathogens for any animal are its epithelial tissues. Changes in the epithelium due to vitamin A deficiency were first docu-mented in 1925 (23) as patches of squamous metaplasia in the respiratory tract of vitamin A-deficient rats. This phenomenon is not limited to the respiratory tract and is seen at mucosal epithelium throughout the body. Mucus-producing goblet cells are also lost as a result of vitamin A deficiency and can be restored rapidly after treatment with the vitamin A metabolite retinoic acid (24).
Preterm infants, who have lower plasma retinol than term infants (25), have immature lungs, which leaves them susceptible to bronchopulmonary dys-plasia (BPD) (reviewed in (26)). BPD is a chronic lung disease characterized by inflammation and scarring and is frequently accompanied by squamous metaplasia (27). When infants with BPD are treated with an intramuscular injection of vitamin A, morbidity due to BPD diminishes (28).
Squamous metaplasia and decreased mucus production can decrease pro-tection of epithelial surfaces from bacterial pathogens. These changes may increase adherence of some pathogens, as can occur in the respiratory tract (3) and urinary bladder (29), and can increase tissue damage by viral infection in the gut (30, 31). These epithelial changes may increase the risk of invasive diseases, which could be life-threatening. Thus treatment with vitamin A at birth may decrease the risk of invasive disease and mortality.
ANNEX 2: BACKGROUND PAPERS

80
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
Granulocytes
Neutrophils are a part of the initial response of the innate immune system to invasive infection that will result when microbial pathogens cross a mucosal barrier. Although blood neutrophil levels are higher at birth than in adults and older infants (reviewed in (32) and (33)), neonatal neutrophils are less effective in dealing with pathogens than are neutrophils later in life, having lower adherence to activated endothelium (34), lower chemotaxis towards sites of inflammation, less effective bacterial killing, and lower content of anti-bacterial molecules in cytoplasmic granules (35).
Neutrophils develop in the bone marrow and require the activity of reti-noic acid, mediated by retinoic acid receptor (RAR)α (36). Although vita-min A is required for normal development, its deficiency does not result in neutropenia. In rats, a paradoxical increase in vitamin A levels occurs in the bone marrow as a result of vitamin A deficiency (37), perhaps suggesting an adaptive response to maintain normal development of neutrophils and other cells of the immune system. There is, however, a morphological change in neutrophils in these vitamin A-deficient rats, with the majority being hyper-segmented (37). Neutrophils in deficient animals have impaired chemotaxis, adhesion and bacterial killing (38), as is also true of neonatal neutrophils. When cord blood cells are cultured with retinoic acid there is a shift towards neutrophils and away from eosinophils and basophils (39), which is consistent with the role of RARα in mediating neutrophil development in vivo. However, when mature neutrophils are cultured with retinoic acid, the oxidative burst is inhibited (40, 41) but under what situations such an exposure to retinoic acid might occur during an in vivo response to infection is not clear.
Based on the above, it is possible that vitamin A deficiency at birth could blunt this neutrophilia and thus decrease the number of neutrophils that would be available to respond to infection. In addition, if such neutrophils have a further functional impairment, in addition to the normal neonatal hyporesponsiveness, clearance of bacterial pathogens could be impaired. This potential defect in neutrophil function resulting from vitamin A deficiency could have a synergistic effect with the impaired mucosal barrier function also seen in vitamin A deficiency in that an increased risk of invasive bacterial infection in combination with impaired neutrophil function could further increase the risk of a more severe infection and perhaps death.
Natural killer cells
Natural killer (NK) cells are cytotoxic lymphocytes that play an important role in the early protection against viral infections (42). The NK activity in neonates is low (43) but reaches adult levels as early as 5 months (44). Vitamin A-deficient animals have lower numbers of NK cells with decreased cytotox-icity (45). Treatment with retinoic acid restores the NK cell population and

81
increases cytotoxic activity (46), which has also been demonstrated in neona-tal mice (47).
NK cells play an important role in protecting against viral and other infec-tion (48), thus their lower activity in neonates would seem to compromise an important aspect of innate immunity. Perhaps this decreased activity is related to a lesser need during this period, given the protection provided by maternally acquired immunity, and the possibility that adult-level responses might cause excessive tissue damage in response to a viral infection early in life. However, a further decrease in NK activity resulting from vitamin A defi-ciency during the neonatal period could significantly impair the initial host response to a viral infection. Thus supplementation with vitamin A at birth might restore NK numbers and activity to “normal” levels, improving resist-ance to viral infections.
Monocytes/macrophages
Monocytes develop in the bone marrow and travel through the bloodstream to tissue sites, where they differentiate into macrophages. Macrophages are normally found in tissues and their numbers increase during most infec-tions, particularly infection by viruses and other intracellular pathogens (49). Their preferred method of killing is through phagocytosis, as is seen with neutrophils. Macrophages also secrete many cytokines that promote inflam-mation by attracting other immune cells. Although the number of neonatal monocytes in peripheral blood does not differ from adults, there are signifi-cant “defects” in their function. The phagocytic capacity of neonatal mono-cytes is low, with neonatal cells ingesting fewer bacteria than adult cells (50). More over, the production of cytokines, including tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-12, following exposure to bacteria or bacterial products is lower in neonatal than adult monocytes. This differ-ence seems to be due to a lower level of cell-surface expression of receptors for microbial products that normally trigger cytokine gene expression. In par-ticular, expression of toll-like receptors (TLR) is lower in neonatal than adult cells (32, 51, 52). There is also a decreased response of macrophages to inter-feron (IFN)-γ in neonates (53). IFN-γ activates macrophages to augment bac-terial killing (54) and a decreased responsiveness on the part of macrophages could decrease clearance of intracellular bacteria as well as other intracellular pathogens, perhaps including viruses such as human immunodeficiency virus (HIV) and measles (32).
The number of macrophages found in secondary lymphoid tissues of vita-min A-deficient mice can be higher than in control animals (55) and similarly, when mice are treated with retinoic acid, the number of monocytes in blood may decrease (56). While the number of macrophages may be high in vitamin A deficiency, their ability to ingest bacteria is impaired (3, 57), and this ability
ANNEX 2: BACKGROUND PAPERSANNEX 2: BACKGROUND PAPERS

82
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
can be enhanced with vitamin A supplementation (3). Vitamin A deficiency in mice has also been shown to cause higher IL-12 production by macrophages, which may result in a T helper (Th)1 bias, further resulting in IFN-γ produc-tion and further activation of macrophages (3).
As with NK cells, vitamin A deficiency seems to reproduce some of the same “defects” in pathogen clearance by macrophages that are seen in this cell type in neonates. (Higher IL-12 production by vitamin A-deficient mac-rophages runs counter to this trend.) Since both cell types are involved in responses to viral infections, it is possible that vitamin A supplementation at birth could enhance macrophage function and improve this component of innate immunity, thus improving neonatal responses to some infections.
Vitamin A and adaptive immunityAntigen-presenting cells
Antigen-presenting cells (APCs), such as dendritic cells (DCs), are found in peripheral tissues. When they encounter microbial products during an infec-tion or immunization they are activated to take up and process antigen, pro-duce specific cytokines (which may vary depending on the type of microbe encountered) and other signalling molecules (e.g. retinoic acid in some cases) and migrate to the draining lymph node. Activation will increase the expression of major histocompatibility complex (MHC) and co-stimulatory molecules on DCs to enhance presentation of antigen and activation of anti-gen-specific T-cells. The pattern of cytokines produced by the activated APCs will help to regulate T-cell differentiation (e.g. IL-12 will promote Th1 cell development) (reviewed in (58)). Although DCs from cord blood have similar surface markers as DCs of older infants, cord blood DCs have lower expres-sion of MHC II and co-stimulatory molecules upon stimulation (59, 60), which leads to decreased activation of T-cells. Cord blood DCs also produce less IL-12 (60) and when these cells are cultured with naive T-cells, less IFN-γ is produced (59). The latter observation demonstrates decreased development of Th1 cells by neonates. This phenomenon can also be viewed as a bias toward Th2 development. One reason for decreased responsiveness of neonatal APCs to microbial challenge appears to be decreased expression of TLR, as is seen for neonatal monocytes (52). Despite the impairment of neonatal DC func-tion, differentiated neonatal APCs are able to efficiently present antigen to T-cells and stimulate proliferation (61) if the inflammatory signal accompany-ing the antigen is of sufficient strength (62). As a result, “live” vaccines (e.g. oral polio vaccine) may better stimulate immune responses early in infancy than would killed vaccines using the relatively weak adjuvant alum (63).
Vitamin A has diverse effects on APC development and function. In vita-min A-deficient medium, myeloid progenitor cells preferentially develop into granulocytes, however, when these cells are cultured with retinoic acid

83
myeloid DC development is stimulated (64). Moreover, monocytes cultured with retinoic acid and granulocyte macrophage-colony stimulating factor (GM-CSF) differentiate to myeloid DCs in culture (65) and also promote DC maturation, including enhanced MHC II expression and antigen presentation to T-cells when accompanied by an appropriate inflammatory signal (66). However, it has also been reported that all-trans retinoic acid inhibits cord blood DC maturation, decreasing T-cell activation and altering cytokine pro-duction to favour development of a Th2 phenotype (67, 68). Vitamin A may also increase presentation of lipid antigens by APCs via increased surface expression of CD1d and increased expression of matrix metalloproteinase 9, which could enhance APC migration from peripheral tissues to lymph nodes where APCs interact with T-cells (68).
While results vary, some of the preceding studies indicate that the activ-ity of retinoic acid on APCs ex vivo reproduces, to a degree, the normally immature state of neonatal DCs. Vitamin A deficiency in neonates may thus have the opposite effect and promote a bias towards, rather than away from, Th1 development. Treatment with vitamin A supplements could thus restore a more “normal” level of activity to neonatal APCs and decrease such a Th1 bias. Such treatment with vitamin A should not inhibit responses that would ordinarily develop in the neonate in response to infection or a robust vaccine because when sufficient inflammatory stimulus is present, retinoic acid helps promote, rather than inhibiting, DC maturation (66).
Thymic function
The thymus is an important organ of the immune system where T-cell devel-opment occurs. The rate of production of T-cells by the thymus peaks within the first few months of life (69). The retinoic receptors RXRα and β and RARα and γ are expressed in the thymus of young children, and the mRNA expres-sion changes with age (both RARα and RXRα decrease and RARγ increas-es) (70). When RXRα was knocked out of the thymus in mice, the development of CD4+ T-cells was disrupted (71). Moreover, CD4+ T-cell development is enhanced when thymic explants are treated with retinoic acid (70) and thymic stromal cells produce compounds with RAR-agonist activity (72) (pre-sumably retinoic acid itself) that could support the normal development of thymocytes.
These data indicate that vitamin A deficiency impairs thymic develop-ment. Such impairment could result in long-term loss of T-cell diversity and thus potentially diminish the efficacy of the adaptive immune response to infections later in life. Conversely, vitamin A supplementation at birth may improve thymic development and thus prevent a decrease in the diversity and effectiveness of the T-cell response to infections and, as a result, increase sur-vival.
ANNEX 2: BACKGROUND PAPERS

84
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
T-cells
T-cells play an important role in both cell-mediated and humoral immunity. Helper T-cells (Th) can be divided into multiple subtypes based on their pat-tern of cytokine production and function. Th1 and Th2 cells were the first subsets identified (73). Th1 cells produce IFN-γ, which helps promote a cell-mediated response to intracellular pathogens that can involve cytotoxic CD8 T-cells as well as macrophages activated by IFN-γ. Th2 cells produce IL-4, IL-5 and IL-13, which help promote B-cell responses and development of antibody-mediated immunity (74) as well as promoting eosinophil development (75).
Neonatal T-cells, in general, are less responsive to stimulation via the T-cell receptor (TCR) than adult T-cells (76) and also have lower levels of co-stimu-latory receptors (77). Combined with the lower stimulatory capacity of neona-tal APCs, this lower responsiveness of neonatal T-cells further diminishes the basal T-cell mediated response in neonates and also appears to leave these cells with an innate bias toward Th2 rather than Th1 development (78). This bias can be overcome, however, with appropriate stimulation (78, 79) (see above).
While studies in neonates are lacking, considerable data from older ani-mals indicate that vitamin A deficiency can produce a Th1 bias (3), while reti-noic acid itself can act directly on T-cells to promote Th2 development (80). Thus a vitamin A-deficient newborn may have an immune system biased more toward Th1 development (both because of effects on DCs, discussed above, and because of direct effects on T-cells) than would ordinarily be the case. Vitamin A supplementation at birth could thus act to restore the normal Th2 bias of the neonatal period. This Th2 bias may be beneficial to the infant because a less inflammatory response to the frequent encounters with novel microbial antigens (all such antigens would be novel for the newborn) may protect the neonate from potentially deleterious effects of inflammation. Since the neonate should be protected efficiently by maternal immunity this appar-ent hyporesponsiveness of T-cells during this period appears not to be del-eterious. It is worth noting, however, that high-level vitamin A treatments in mice can produce pathogenic Th2-mediated inflammation in an experimen-tal model of asthma (81), thus the possibility of unexpected outcomes should always be kept in mind.
In addition to its effect on Th1/Th2 balance, retinoic acid also modulates development of inducible T regulatory (iTreg) cell and Th17 cells (82–85) in the gut. Th17 cells reside in the mucosa of the gut and protect the host from microbial pathogens by producing cytokines (IL-17A, IL-17F) that stimulate the production of antimicrobial peptides by epithelial cells (86) and attract neutrophils, which help kill microbes that cross the epithelial barrier (87). The main function of the iTregs is to control the damage that excessive effec-tor T-cells cause to the host (86). Transforming growth factor (TGF)-β is the main factor that determines if a T-cell will differentiate to a Treg or Th17 cell.

85
However, when retinoic acid is present in conjunction with TGF-β, the Treg cell lineage is favoured over the Th17 compared with TGB-β alone (84). The in vivo story may not be this simple, however, since vitamin A deficiency in mice increases another type of IL-10-producing regulatory T-cell, called Tr1 (88), and can also increase Th17 cell development (89). Thus it is not clear which way the Treg/Th17 balance might tip in vitamin A-deficient humans. Treg cells down-regulate inflammatory responses by producing IL-10 and TGF-β, which can decrease T-cell proliferation and thus modulate an inflammatory T-cell response (90). These regulatory mechanisms may control the responses which might otherwise develop against commensal microorganisms in the gut.
An important focus of Treg activity in the neonate (as well as adult) is the intestine, due to the colonization of the gut with commensal microflora that begins at birth. Within 4 days of birth the percentage of circulating Tregs nearly doubles and stays higher until 18 months of age (91). Retinoic acid is a key regulator of mucosal immunity in the gut, including Treg development. Intestinal CD103+ DCs have the ability to metabolize retinol to retinoic acid, an activity not found in most DCs from other anatomical sites (92, 93). This activity helps to promote Treg differentiation after exposure to orally admin-istered antigen (85). Moreover, retinoic acid also promotes migration of adap-tive Tregs to the gut by upregulating α4β7 and CCR9 expression (83). Other lymphocytes which develop in the gut, including other T-cells as well as mem-ory B-cells and IgA-producing plasma cells, also express α4β7 and CCR9 as a result of exposure to retinoic acid produced by gut-derived DCs (94). Thus, in addition to promoting tolerance, retinoic acid seems to be important for targeting protective responses to the gut, such as IgA-mediated responses that develop in response to enteric pathogens and oral vaccines (95).
These data indicate that vitamin A deficiency at birth could alter the balance of Treg/Th17 development in the gut and also impair protective, Th2-mediated, responses that develop in the gut in response to pathogen expo-sure. For this reason vitamin A supplementation at birth could restore the normal pattern of Treg/Th17 and Th2 development in the gut. Such treatment might thus facilitate the normal adaptation of the neonatal immune system to colonization of the gut with commensal flora, prevent “damaging” inflamma-tion in the short term and promote protective Th2 responses to enteric patho-gens that would have both short- and long-term benefits for the infant.
B-cells
Naive B-cells develop into antibody-producing plasma cells and memory B-cells following appropriate exposure to antigen. Thus B-cells are responsible for development of the humoral immune response. Some antibody respons-es develop without T-cell help (e.g. polyvalent antigens), and these are not
ANNEX 2: BACKGROUND PAPERS

86
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
impaired by vitamin A deficiency (96) but many humoral responses require such help. These responses often rely on Th2-derived cytokines to induce B-cell proliferation and isotype switching (97). Although neonates have a bias towards a Th2 response, the actual antibody response of neonates is lower than an adult response. Co-stimulatory receptor expression on neonatal B-cells is also lower, which decreases their response to T-cell dependent antigens (98). Additionally, serum antibody responses to T-independent responses are lower in neonates, leading to depressed responses to polysaccharide vaccines (99). In spite of the lower neonatal antibody response, the development of memory B-cells does not differ from adults (98). The serum antibody response to vac-cination in neonates is also shorter in duration because of an adequate plas-ma-cell pool in the bone marrow (100), which occurs because neonatal bone marrow stromal cells cannot promote survival of the plasma cells (101).
Vitamin A deficiency impairs Th2-dependent antibody responses. Both IgE and IgG1 responses are diminished during vitamin A deficiency because the activated B-cells fail to proliferate efficiently (3). Retinoic acid treatment also increased the synthesis of IgM from cord blood B-cells (102). Vitamin A not only influences the production of serum antibody, it is also enhances TGF-β-mediated development of IgA-producing plasma cells in the gut (103, 104). Based on these data, neonatal vitamin A supplementation would be expected to enhance serum and, perhaps more importantly, secretory IgA responses. This latter effect, particularly in combination with the postulated effects of vitamin A on Th17/Treg balance in the gut, could directly affect resistance to enteric pathogens and host adaptation to commensal gut flora thus improving gut health and, perhaps, survival.
ConclusionsThis brief review has identified several mechanisms by which vitamin A supplementation at birth could enhance immune function compromised by vitamin A deficiency. In particular, development of an appropriate balance of Th17, Treg and secretory IgA activity in the gut would seem to be uniquely sensitive to vitamin A status. Establishing this balance in the neonate may also be critical, given the rapid changes that must occur in the neonatal immune system as it responds to the unique challenge provided by colonization of the gut with commensal flora (105). Thus supplementation in the neonatal period may affect infant health to a degree not seen a few weeks later. This neonatal effect may differ qualitatively than the protective effects conferred by vitamin A supplementation after 6 months of age. These mechanisms remain specula-tive but could be evaluated in human studies, and such data would be useful in helping to interpret results of community intervention trials that, by their nature, do not provide such mechanistic information.

87
ANNEX 2: BACKGROUND PAPERS
References1. Global prevalence of vitamin A deficiency in populations at risk 1995–2005. WHO
Global Database on Vitamin A Deficiency. Geneva, World Health Organization, 2009.
2. Sommer A et al. Increased mortality in children with mild vitamin A deficiency. Lancet, 1983, 2:585–588.
3. Stephensen CB. Vitamin A, infection, and immune function. Annual Review of Nutrition, 2001, 21:167–192.
4. Grotto I et al. Vitamin A supplementation and childhood morbidity from di-arrhea and respiratory infections: a meta-analysis. Journal of Pediatrics, 2003, 142:297–304.
5. Sommer A et al. Impact of vitamin A supplementation on childhood mortality. A randomised controlled community trial. Lancet, 1986, 1:1169–1173.
6. West KP Jr et al. Efficacy of vitamin A in reducing preschool child mortality in Nepal. Lancet, 1991, 338:67–71.
7. Rahmathullah L et al. Reduced mortality among children in southern India re-ceiving a small weekly dose of vitamin A. New England Journal of Medicine, 1990, 323:929–935.
8. Daulaire NM et al. Childhood mortality after a high dose of vitamin A in a high risk population. British Medical Journal (Clinical Research Ed), 1992, 304:207–210.
9. Biesalski HK, Nohr D. Importance of vitamin-A for lung function and develop-ment. Molecular Aspects of Medicine, 2003, 24:431–440.
10. Olson JA, Gunning DB, Tilton RA. Liver concentrations of vitamin A and ca-rotenoids, as a function of age and other parameters, of American children who died of various causes. American Journal of Clinical Nutrition, 1984, 39:903–910.
11. Shirali GS, Oelberg DG, Mehta KP. Maternal-neonatal serum vitamin A concen-trations. Journal of Pediatric Gastroenterology and Nutrition, 1989, 9:62–66.
12. Miller M et al. Why do children become vitamin A deficient? Journal of Nutri-tion, 2002, 132 (Suppl. 9):2867S–2880S.
13. WHO/CHD Immunisation-Linked Vitamin A Supplementation Study Group. Randomised trial to assess benefits and safety of vitamin A supplementation linked to immunisation in early infancy. Lancet, 1998, 352:1257–1263.
14. West KP Jr et al. Mortality of infants <6 mo of age supplemented with vitamin A: a randomized, double-masked trial in Nepal. American Journal of Clinical Nutrition, 1995, 62:143–148.
15. Humphrey JH et al. Impact of neonatal vitamin A supplementation on infant morbidity and mortality. Journal of Pediatrics, 1996, 128:489–496.
16. Rahmathullah L et al. Impact of supplementing newborn infants with vitamin A on early infant mortality: community based randomised trial in southern India. British Medical Journal (Clinical Research Ed), 2003, 327:254.
17. Klemm RD et al. Newborn vitamin A supplementation reduced infant mortality in rural Bangladesh. Pediatrics, 2008, 122:e242–250.
18. Malaba LC et al. Effect of postpartum maternal or neonatal vitamin A supple-mentation on infant mortality among infants born to HIV-negative mothers in Zimbabwe. American Journal of Clinical Nutrition, 2005, 81:454–460.

88
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
19. Benn CS et al. Effect of 50,000 IU vitamin A given with BCG vaccine on mor-tality in infants in Guinea-Bissau: randomised placebo controlled trial. British Medical Journal (Clinical Research Ed), 2008, 336:1416–1420.
20. Benn CS et al. Vitamin A supplementation and BCG vaccination at birth in low birthweight neonates: two by two factorial randomised controlled trial. British Medical Journal (Clinical Research Ed), 2010, 340:c1101.
21. Benn CS et al. The effect of high-dose vitamin A supplementation administered with BCG vaccine at birth may be modified by subsequent DTP vaccination. Vaccine, 2009, 27:2891–2898.
22. Benn CS et al. Does vitamin A supplementation interact with routine vaccina-tions? An analysis of the Ghana Vitamin A Supplementation Trial. American Journal of Clinical Nutrition, 2009, 90:629–639.
23. Wolbach SB, Howe PR. Tissue changes following deprivation of fat-soluble A vitamin. Journal of Experimental Medicine, 1925, 42:753–777.
24. Rojanapo W, Lamb AJ, Olson JA. The prevalence, metabolism and migration of goblet cells in rat intestine following the induction of rapid, synchronous vita-min A deficiency. Journal of Nutrition, 1980, 110:178–188.
25. Shenai JP et al. Plasma vitamin A and retinol-binding protein in premature and term neonates. Journal of Pediatrics, 1981, 99:302–305.
26. Darlow BA, Graham PJ. Vitamin A supplementation for preventing morbidity and mortality in very low birthweight infants. Cochrane Database of Systematic Reviews, 2000, 2:CD000501.
27. Shenai JP, Chytil F, Stahlman MT. Vitamin A status of neonates with bron-chopulmonary dysplasia. Pediatric Research, 1985, 19:185–188.
28. Shenai JP et al. Clinical trial of vitamin A supplementation in infants susceptible to bronchopulmonary dysplasia. Journal of Pediatrics, 1987, 111:269–277.
29. Liang FX et al. Cellular basis of urothelial squamous metaplasia. Journal of Cell Biology, 2005, 171:835–844.
30. Reifen R, Mor A, Nyska A. Vitamin A deficiency aggravates rotavirus infection in CD-1 mice through extensive involvement of the gut. International Journal for Vitamin and Nutrition Research, 2004, 74:355–361.
31. Ahmed F, Jones DB, Jackson AA. Effect of vitamin A deficiency on the immune response to epizootic diarrhoea of infant mice (EDIM) rotavirus infection in mice. British Journal of Nutrition, 1991, 65:475–485.
32. Johnston RB Jr. Function and cell biology of neutrophils and mononuclear phagocytes in the newborn infant. Vaccine, 1998, 16:1363–1368.
33. Koenig JM, Yoder MC. Neonatal neutrophils: the good, the bad, and the ugly. Clinics in Perinatology, 2004, 31:39–51.
34. Anderson DC et al. Diminished lectin-, epidermal growth factor-, complement binding domain-cell adhesion molecule-1 on neonatal neutrophils underlies their impaired CD18-independent adhesion to endothelial cells in vitro. Journal of Immunology, 1991, 146:3372–3379.
35. Bektas S, Goetze B, Speer CP. Decreased adherence, chemotaxis and phagocytic activities of neutrophils from preterm neonates. Acta Paediatrica Scandinavica, 1990, 79:1031–1038.

89
36. Robertson KA et al. Multiple members of the retinoic acid receptor family are capable of mediating the granulocytic differentiation of HL-60 cells. Molecular and Cellular Biology, 1992, 12:3743–3749.
37. Twining SS et al. Retinol is sequestered in the bone marrow of vitamin A-defi-cient rats. Journal of Nutrition, 1996, 126:1618–1626.
38. Twining SS et al. Vitamin A deficiency alters rat neutrophil function. Journal of Nutrition, 1997, 127:558–565.
39. Upham JW et al. Retinoic acid modulates IL-5 receptor expression and selective-ly inhibits eosinophil-basophil differentiation of hemopoietic progenitor cells. Journal of Allergy Clinical Immunology, 2002, 109:307–313.
40. Wolfson M et al. Inhibitory effect of retinoic acid on the respiratory burst of adult and cord blood neutrophils and macrophages: potential implication to bronchopulmonary dysplasia. Clinical and Experimental Immunology, 1988, 72:505–509.
41. Higuchi H, Nagahata H. Effects of vitamins A and E on superoxide production and intracellular signaling of neutrophils in Holstein calves. Canadian Journal of Veterinary Research, 2000, 64:69–75.
42. Yokoyama WM, Altfeld M, Hsu KC. Natural killer cells: tolerance to self and in-nate immunity to viral infection and malignancy. Biology of Blood and Marrow Transplantation, 2010, 16 (Suppl. 1):S97–S105.
43. Gaddy J, Risdon G, Broxmeyer HE. Cord blood natural killer cells are function-ally and phenotypically immature but readily respond to interleukin-2 and in-terleukin-12. Journal of Interferon and Cytokine Research, 1995, 15:527–536.
44. Yabuhara A, Kawal H, Komiyama A. Development of natural killer cytotoxicity during childhood: marked increases in number of natural killer cells with ad-equate cytotoxic abilities during infancy to early childhood. Pediatric Research, 1990, 28:316–321.
45. Zhao Z, Murasko DM, Ross AC. The role of vitamin A in natural killer cell cyto-toxicity, number and activation in the rat. Nature Immunology, 1994, 13:29–41.
46. Zhao Z, Ross AC. Retinoic acid repletion restores the number of leukocytes and their subsets and stimulates natural cytotoxicity in vitamin A-deficient rats. Journal of Nutrition, 1995, 125:2064–2073.
47. Ma Y, Ross AC. The anti-tetanus immune response of neonatal mice is aug-mented by retinoic acid combined with polyriboinosinic:polyribocytidylic acid. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102:13556–13561.
48. Lodoen MB, Lanier LL. Natural killer cells as an initial defense against patho-gens. Current Opinion in Immunology, 2006, 18:391–398.
49. Naito M. Macrophage heterogeneity in development and differentiation. Ar-chives of Histology and Cytology, 1993, 56:331–351.
50. Strunk T et al. Differential maturation of the innate immune response in human fetuses. Pediatric Research, 2004, 56:219–226.
51. Velilla PA, Rugeles MT, Chougnet CA. Defective antigen-presenting cell func-tion in human neonates. Clinical Immunology, 2006, 121:251–259.
ANNEX 2: BACKGROUND PAPERS

90
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
52. Sadeghi K et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. Journal of Infectious Dis-eases, 2007, 195:296–302.
53. Marodi L et al. Cytokine receptor signalling in neonatal macrophages: defective STAT-1 phosphorylation in response to stimulation with IFN-gamma. Clinical and Experimental Immunology, 2001, 126:456–460.
54. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and toll-like recep-tor signaling during macrophage activation by opposing feedforward and feed-back inhibition mechanisms. Immunological Reviews, 2008, 226:41–56.
55. Smith SM, Levy NS, Hayes CE. Impaired immunity in vitamin A-deficient mice. Journal of Nutrition, 1987, 117:857–865.
56. Miller SC, Kearney SL. Effect of in vivo administration of all trans-retinoic acid on the hemopoietic cell populations of the spleen and bone marrow: profound strain differences between A/J and C57BL/6J mice. Laboratory Animal Science, 1998, 48:74–80.
57. Wiedermann U et al. Vitamin A deficiency predisposes to Staphylococcus au-reus infection. Infection and Immunity, 1996, 64:209–214.
58. Sozzani S. Dendritic cell trafficking: More than just chemokines. Cytokine and Growth Factor Reviews, 2005, 16:581–592.
59. Naderi N et al. Cord blood dendritic cells prevent the differentiation of naive T-helper cells towards Th1 irrespective of their subtype. Clinical and Experimental Medicine, 2009, 9:29–36.
60. Langrish CL et al. Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clinical and Experimental Immunology, 2002, 128:118–123.
61. Matthews NC, Power UF, Reen DJ. Neonatal human autologous dendritic cells pulsed with recombinant protein antigen prime the generation of non-polarized CD4 T-cell effectors. International Immunology, 2007, 19:703–712.
62. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nature Reviews. Immunology, 2004, 4:553–564.
63. Wilson CB, Kollmann TR. Induction of antigen-specific immunity in human neonates and infants. Nestlé Nutrition Workshop Series. Paediatric Programme, 2008, 61:183–195.
64. Hengesbach LM, Hoag KA. Physiological concentrations of retinoic acid favor myeloid dendritic cell development over granulocyte development in cultures of bone marrow cells from mice. Journal of Nutrition, 2004, 134:2653–2659.
65. Mohty M et al. All-trans retinoic acid skews monocyte differentiation into in-terleukin-12-secreting dendritic-like cells. British Journal of Haematology, 2003, 122:829–836.
66. Geissmann F et al. Retinoids regulate survival and antigen presentation by im-mature dendritic cells. Journal of Experimental Medicine, 2003, 198:623–634.
67. Tao Y, Yang Y, Wang W. Effect of all-trans-retinoic acid on the differentiation, maturation and functions of dendritic cells derived from cord blood monocytes. FEMS Immunology and Medical Microbiology, 2006, 47:444–450.
68. Jin CJ et al. All-trans retinoic acid inhibits the differentiation, maturation, and function of human monocyte-derived dendritic cells. Leukemia Research, 2010, 34:513–520.

91
69. Bains I et al. Quantifying the development of the peripheral naive CD4+ T-cell pool in humans. Blood, 2009, 113:5480–5487.
70. Zhou X, Wang W, Yang Y. The expression of retinoic acid receptors in thymus of young children and the effect of all-transretinoic acid on the development of T cells in thymus. Journal of Clinical Immunology, 2008, 28:85–91.
71. Stephensen CB, Borowsky AD, Lloyd KC. Disruption of Rxra gene in thymo-cytes and T lymphocytes modestly alters lymphocyte frequencies, proliferation, survival and T helper type 1/type 2 balance. Immunology, 2007, 121:484–498.
72. Kiss I et al. Retinoid receptor-activating ligands are produced within the mouse thymus during postnatal development. European Journal of Immunology, 2008, 38:147–155.
73. Mosmann TR, Cherwinski H, Bond MW. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. Journal of Immunology, 1986, 136:2348–2357.
74. Liew FY. TH1 and TH2 cells: a historical perspective. Nature Reviews. Immunol-ogy, 2002, 2:55–60.
75. Sanderson CJ. Interleukin-5, eosinophils, and disease. Blood, 1992, 79:3101–3109.76. Adkins B. T-cell function in newborn mice and humans. Immunology Today,
1999, 20:330–335.77. Delespesse G et al. Maturation of human neonatal CD4+ and CD8+ T lympho-
cytes into Th1/Th2 effectors. Vaccine, 1998, 16:1415–1419.78. Adkins B. Development of neonatal Th1/Th2 function. International Reviews of
Immunology, 2000, 19:157–171.79. Yu HR et al. Different antigens trigger different Th1/Th2 reactions in neonatal
mononuclear cells (MNCs) relating to T-bet/GATA-3 expression. Journal of Leu-kocyte Biology, 2003, 74:952–958.
80. Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nature Reviews. Immunology, 2008, 8:685–698.
81. Schuster GU, Kenyon NJ, Stephensen CB. Vitamin A deficiency decreases and high dietary vitamin A increases disease severity in the mouse model of asthma. Journal of Immunology, 2008, 180:1834–1842.
82. Ziegler SF, Buckner JH. FOXP3 and the regulation of Treg/Th17 differentiation. Microbes and Infection, 2009, 11:594–598.
83. Benson MJ et al. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. Jour-nal of Experimental Medicine, 2007, 204:1765–1774.
84. Mucida D et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science, 2007, 317:256–260.
85. Sun CM et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. Journal of Experimental Medi-cine, 2007, 204:1775–1785.
86. Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nature Reviews. Immunology, 2009, 9:883–889.
87. Liu JZ, Pezeshki M, Raffatellu M. Th17 cytokines and host-pathogen interactions at the mucosa: dichotomies of help and harm. Cytokine, 2009, 48:156–160.
ANNEX 2: BACKGROUND PAPERS

92
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
88. Maynard CL et al. Contrasting roles for all-trans retinoic acid in TGF-beta-mediated induction of Foxp3 and Il10 genes in developing regulatory T cells. Journal of Experimental Medicine, 2009, 206:343–357.
89. Cha HR et al. Downregulation of Th17 cells in the small intestine by disrup-tion of gut flora in the absence of retinoic acid. Journal of Immunology, 2010, 184:6799–6806.
90. Levings MK, Roncarolo MG. T-regulatory 1 cells: a novel subset of CD4 T cells with immunoregulatory properties. Journal of Allergy and Clinical Immunology, 2000, 106:S109–S112.
91. Grindebacke H et al. Dynamic development of homing receptor expression and memory cell differentiation of infant CD4+CD25high regulatory T cells. Journal of Immunology, 2009, 183:4360–4370.
92. Iwata M et al. Retinoic acid imprints gut-homing specificity on T cells. Immu-nity, 2004, 21:527–538.
93. Manicassamy S, Pulendran B. Retinoic acid-dependent regulation of immune responses by dendritic cells and macrophages. Seminars in Immunology, 2009, 21:22–27.
94. Duriancik DM, Lackey DE, Hoag KA. Vitamin A as a regulator of antigen pre-senting cells. Journal of Nutrition, 2010, 140:1395–1399.
95. Mora JR et al. Generation of gut-homing IgA-secreting B cells by intestinal den-dritic cells. Science, 2006, 314:1157–1160.
96. Ross AC. Vitamin A status: relationship to immunity and the antibody response. Proceedings of the Society for Experimental Biology and Medicine, 1992, 200:303–320.
97. Poudrier J, Owens T. Th1 and Th2 help for B cells: differential capacity for in-duction of autonomous responsiveness to IL-2. International Immunology, 1995, 7:1021–1027.
98. Siegrist CA, Aspinall R. B-cell responses to vaccination at the extremes of age. Nature Reviews. Immunology, 2009, 9:185–194.
99. Siegrist CA. The challenges of vaccine responses in early life: selected examples. Journal of Comparative Pathology, 2007, 137 (Suppl. 1):S4–S9.
100. Pihlgren M et al. Delayed and deficient establishment of the long-term bone marrow plasma cell pool during early life. European Journal of Immunology, 2001, 31:939–946.
101. Pihlgren M et al. Reduced ability of neonatal and early-life bone marrow stromal cells to support plasmablast survival. Journal of Immunology, 2006, 176:165–172.
102. Wang W, Napoli JL, Ballow M. The effects of retinol on in vitro immunoglobulin synthesis by cord blood and adult peripheral blood mononuclear cells. Clinical and Experimental Immunology, 1993, 92:164–168.
103. Suzuki K et al. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity, 2010, 33:71–83.
104. Watanabe K et al. Requirement for Runx proteins in IgA class switching acting downstream of TGF-beta 1 and retinoic acid signaling. Journal of Immunology, 2010, 184:2785–2792.
105. Conroy ME, Shi HN, Walker WA. The long-term health effects of neonatal micro-bial flora. Current Opinion in Allergy and Clinical Immunology, 2009, 9:197–201.

93
ANNEX 2: BACKGROUND PAPERS
a2.4Mammalian models for understanding mechanisms of retinol and retinoid actionsSherry A. Tanumihardjo
University of Wisconsin-Madison, Department of Nutritional Sciences, Madison, Wisconsin, United States of America
Corresponding author: Sherry A. Tanumihardjo; [email protected]
Abstractn Animal models cannot completely replace humans when attempting to understand the intricacies of vitamin A and carotenoid metabolism. Stark differences exist among species, particularly concerning carotenoid uptake and storage. Nonetheless, when appropriately applied, mammalian models can shed light on understanding the metabolism and mechanisms of vitamin A action for extrapolation to the human condition. Vitamin A has multiple functions in the human body. The specificity of some of its functions is quite fascinating. For example, the flipping of the retinal molecule from the cis to trans configuration allows us to see at night. Without vitamin A, mammals cannot grow, reproduce or fight off disease. Therefore, vitamin A is truly an essential nutrient. This review includes snapshots of how mammals have been used in the past to elucidate vitamin A metabolism, binding protein function and provitamin A activity.

94
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
IntroductionMany models have been used to discern the mechanisms of retinol and reti-noid action. Although liver was known to cure night blindness in ancient times, vitamin A was not discovered until 1913 AD, using a rat model (1). By feeding rats a purified diet and supplementing with butter fat, egg yolk or olive oil, McCollum and Davis discovered a lipid factor that was necessary to maintain growth but supplying purified protein, carbohydrate, fat and salt was not enough for animals to maintain growth. They concluded that “certain accessory articles in certain food-stuffs” found in lipid extracts were essential for normal growth (1).
Since then, vitamin A has not only been found to be essential for growth but also for vision, cellular differentiation, immune function and reproduc-tion. The functions of retinoids in embryonal development (2) and repro-duction (3) have been reviewed. Vitamin A deficiency and excess lead to embryonic maldevelopment and therefore balance is important during preg-nancy. Deficiency can result in fetal death or congenital abnormalities that involve the heart, eye, and respiratory, urogenital and circulatory systems (4). Retinoid-based therapies are used in a number of clinical applications includ-ing skin disorders. Excess vitamin A can also lead to fetal death and defects including exencephaly, cleft lip, cleft palate and eye defects (2).
Other models have also been developed and used as surrogates for the human condition to discover the multiple needs for vitamin A by the host. Human population-based studies have limitations, such as inability to control bias and confounders, invasive procedures, interruption of routine, difficult logistics and limited funding (5). Furthermore, in human field studies, blood is usually the only tissue that can be studied, but this does not necessarily reflect what is happening at the tissue level (5). This review focuses on mam-malian models that are commonly used for studies involving mechanisms of action and biological activity of vitamin A (Table A2.4.1).
RodentsMiceKnockout modelsMice (Mus) are a very common experimental model, because they easily grow in captivity. Genetically engineered mice have greatly improved our under-standing of many biological mechanisms including those of vitamin A. The crucial roles of the retinoic acid receptor and the retinoid X receptor have been studied using both knockouts and targeted mutagenesis in mice (6) models generated for human disease states.
The role of retinol-binding protein (RBP) is now better understood after the development of knockout mouse models (7, 8). Mice lacking RBP do not main-
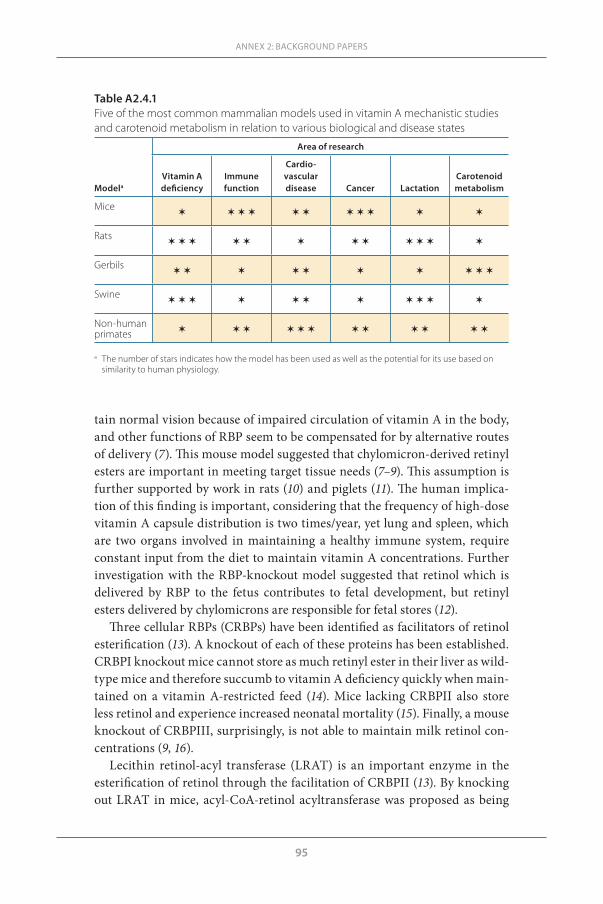
95
tain normal vision because of impaired circulation of vitamin A in the body, and other functions of RBP seem to be compensated for by alternative routes of delivery (7). This mouse model suggested that chylomicron-derived retinyl esters are important in meeting target tissue needs (7–9). This assumption is further supported by work in rats (10) and piglets (11). The human implica-tion of this finding is important, considering that the frequency of high-dose vitamin A capsule distribution is two times/year, yet lung and spleen, which are two organs involved in maintaining a healthy immune system, require constant input from the diet to maintain vitamin A concentrations. Further investigation with the RBP-knockout model suggested that retinol which is delivered by RBP to the fetus contributes to fetal development, but retinyl esters delivered by chylomicrons are responsible for fetal stores (12).
Three cellular RBPs (CRBPs) have been identified as facilitators of retinol esterification (13). A knockout of each of these proteins has been established. CRBPI knockout mice cannot store as much retinyl ester in their liver as wild-type mice and therefore succumb to vitamin A deficiency quickly when main-tained on a vitamin A-restricted feed (14). Mice lacking CRBPII also store less retinol and experience increased neonatal mortality (15). Finally, a mouse knockout of CRBPIII, surprisingly, is not able to maintain milk retinol con-centrations (9, 16).
Lecithin retinol-acyl transferase (LRAT) is an important enzyme in the esterification of retinol through the facilitation of CRBPII (13). By knocking out LRAT in mice, acyl-CoA-retinol acyltransferase was proposed as being
ANNEX 2: BACKGROUND PAPERS
Table A2.4.1Five of the most common mammalian models used in vitamin A mechanistic studies and carotenoid metabolism in relation to various biological and disease states
Modela
Area of research
Vitamin A deficiency
Immune function
Cardio-vascular disease Cancer Lactation
Carotenoid metabolism
Mice✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶
Rats✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶
Gerbils✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶
Swine ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶
Non-human primates ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶ ✶
a The number of stars indicates how the model has been used as well as the potential for its use based on
similarity to human physiology.

96
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
involved in retinyl ester formation in the liver, mammary gland and adipose tissue (17). In mice lacking hormone-sensitive lipase, retinyl ester concentra-tions increase in white adipose tissue while other metabolites decrease, i.e. retinol, retinaldehyde and all-trans retinoic acid (18), demonstrating that this lipase is an important retinyl ester hydrolase to provide retinoids for adipose tissue differentiation.
Once provitamin A carotenoids are ingested, they may be cleaved in the intestinal epithelium by 15, 15′ carotenoid mono-oxygenase (CMO1) (19). This process results in the formation of two molecules of retinal (vitamin A) from β-carotene whereas α-carotene or β-cryptoxanthin cleavage results in one molecule of retinal and one molecule of α-retinal or 3-hydroxyretinal, respectively. CMO1 is expressed in the intestinal epithelium, liver, lung and skin epidermis (20). CMO1 knockout C57Bl/6J mice were developed origi-nally by Wyss and colleagues (21) and have been used by several other inves-tigators (22, 23).
ImmunitySeveral important studies concerning the relationship between vitamin A and immunity have recently been done in mice (24–27). Vitamin A is critical to support T helper cell development through immune response initiation (28). The importance of vitamin A for the immune system has been reviewed (29).
Host defences within the gastrointestinal tract exclude bacteria and other substances. There is a complex interaction between bacteria and local immune responses and non-immunological processes. This barrier is an effective protector of the intestinal mucosa from invading pathogens (30). The gut-associated lymphoid tissue (GALT) is a component of the mucosa-associated lymphoid tissue. Approximately 50–60% of the body’s total immunity under-lies the mucosal surfaces of the upper and lower respiratory tract, small intes-tine and colon, and accounts for 70–80% of antibody production (primarily IgA) by the body (31). GALT consists of organized lymphoid aggregates rep-resented by the Peyer’s patches, the appendix, mesenteric lymph nodes and solitary lymph nodes. The non-organized lymphoid aggregates consist of intraepithelial lymphocytes and lamina propria lymphocytes (32). Gut- associated dendritic cells in GALT induce gut-homing receptors on B-cells via a mechanism that depends on retinoic acid (26, 33, 34).
Immunological communication exists between the gastrointestinal tract and mucosal surfaces and the postulated system is called the com-mon mucosal immune hypothesis. In the system, antigens are processed in the Peyer’s patches and delivered to antigen-presenting cells. Naive T- and B-lymphocytes are sensitized to antigens taken up by the Peyer’s patches. After sensitization, cells migrate to the mesenteric lymph nodes and to the thoracic duct and are distributed by the bloodstream to various effector sites

97
including the GALT, respiratory tract, mammary glands and the genitouri-nary tract. The type and route of nutrition significantly impact the system including histology, cytokine levels, IgA levels, and intestinal and respira-tory immunity (31). For example, total parenteral nutrition decreases GALT by reducing the Peyer’s patches, lamina propria, and intraepithelial space of T- and/or B-cells, and lowers the CD4+/CD8+ ratio within the lamina pro-pria (35). These changes in the GALT are correlated with decreased respirato-ry IgA levels (36). Furthermore, decreased GALT and IgA levels are associated with reduced ability of mice to clear respiratory infections and the influenza virus (31). Vitamin A, besides its importance in maintaining mucosal tissue, may augment an immune response to infection by modulating an immune response from the GALT. Due to the communication between the GALT and other mucosal surfaces, vitamin A may directly affect the immune response.
Disease statesMouse models are also used in cancer-related studies to evaluate DNA dam-age and repair, point mutations, and numerical and structural chromosomal alteration (37, 38). Some mouse models spontaneously form tumours or induce melanoma, which responds to retinoic acid (39, 40). Animal models are essen-tial in understanding the metabolism and breakdown of potential anticarci-nogenic compounds (41). Experimental models can predict safety and efficacy of potential chemopreventive agents through understanding mechanisms of action (38). The use of carotenoids and vitamin A in the application of a vari-ety of cancer models has been reviewed (38).
SAMP1/YP mice, used as a model for Crohn’s disease, develop spontane-ous chronic intestinal lesions in the ileum (42) and this has been used as an inflammation response model to low and high vitamin A therapies (43). Reti-noid receptor null mice have been developed to model a number of diseases and human conditions including prostate cancer, retinal dysfunction, obesity prevention and dermatitis (reviewed in (6)).
RatsReproduction and embryonic developmentRats (Rattus) are an easy model to work with for vitamin A depletion and deficiency studies because they are born with low stores, similar to human infants. Rats have been used extensively in studies related to reproduction and embryonic development (reviewed in (3)). Timed dosing studies with retinol or retinoic acid have determined that the need for vitamin A is greater during late rather than early gestation (44, 45).
ANNEX 2: BACKGROUND PAPERS

98
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
Enzymatic activityRats have been used to determine important enzyme functions and regulation, such as lecithin:retinol acyltransferase (46). In rodent studies, the diet can be easily manipulated to the point of deficiency that is not ethical in human studies to look at effects of vitamin A levels or other nutrients, such as fat, on metabolism (47). The role of diet on enzyme activity, e.g. lecithin:retinol acyl-transferase and P450 CYP26, has been studied in rats (reviewed in (48)). Diet manipulation is key in determining the essentiality of vitamin A in a variety of life processes.
Assessment methodologiesRats have successfully been used to model vitamin A assessment methods and whole body vitamin A tracers. The original studies that led to the development of the relative dose response (RDR) (49) and modified relative dose response (MRDR) were performed in rats (50–52). In regard to tracer studies, rats have been administered tritiated vitamin A (53) and vitamin A2 (3,4-didehydro-retinol) (47) and the metabolism has been traced for days. Multiple blood samples can be obtained from a single rat but the volume needs to be moni-tored to be compliant with local ethical committee standards. For example, 250 µL was taken multiple times per rat over 54 days in the tritiated retinol study investigating the effect of inflammation on hepatic mobilization (53). However, in the vitamin A2 tracer study, 1 mL blood was collected serially no more than three times from each rat so that <10% of each rat’s total blood vol-ume was removed within 1 week (47). Although the number of blood samples that can be taken from a rat is limited due to the animal’s body size, unlike the swine model (below), the number of rats can be adjusted to cover the time span of interest because the model is relatively inexpensive to maintain.
LactationRats have been used to study vitamin A metabolism during lactation. A series of studies have shown that chylomicrons contribute significant vitamin A to milk concentrations (54, 55). Lipoprotein lipase activity increases during lactation in mammary glands, allowing chylomicron retinyl ester incorpora-tion into milk before reaching the liver as part of chylomicron remnants. The effects of maternal diet on milk concentrations and pup outcome can be easily measured in rodent studies because feed needs are much less than with other models (56). However, there are some volume limitations, which may cause researchers to seek other models.
Lung developmentRats have been used as a model for lung development. Orally administer-ing retinoic acid with vitamin A assists the uptake of vitamin A into lung

99
tissues (57). This finding led to mechanistic studies that showed that LRAT, the retinol-binding protein receptor called STRA6 (stimulated by retinoic acid gene) and the cytochrome gene of CYP26B1 were enhanced after retinoic acid co-supplementation (58).
Carotenoid metabolismRats do not absorb carotenoids intact until they are fed pharmacological lev-els (>0.02%), which is equivalent to a 70 kg person eating about 163 typical carrots each day (59). Thus, extrapolation of provitamin A carotenoid efficacy and metabolism findings in the rat needs to be done carefully. Lycopene, a non-provitamin A carotenoid implicated in the prevention of various can-cers and related compounds, has been much studied in two strains of rats, i.e. Copenhagen (60) and Fisher 344 (61); rats were chosen because they have a prostate that is histologically similar to humans (61, 62).
GerbilsPlant sources of provitamin A carotenoids are important for health world-wide (63). Although underestimated as a good source of vitamin A in some studies, increased consumption can supply vitamin A as well as other poten-tially beneficial phytochemicals. Models that are useful for carotenoid metab-olism are few and have been reviewed (59). Mice, rats and swine are “white”-fat animals, and, therefore, preferentially cleave provitamin A carotenoids to vitamin A as opposed to absorbing and storing them intact in tissues such as the adipose tissue. One model that has been used extensively to evaluate bioef-ficacy of provitamin A carotenoids is the Mongolian gerbil (Meriones unguic-ulatus). In addition to the original studies performed with β-carotene (64, 65), gerbils have been used to evaluate the bioefficacy of α-carotene (66) and β-cryptoxanthin (67, 68).
The advantages of using gerbils are that they are relatively inexpensive as an animal model and true bioefficacy can be determined because the entire liver is available for analysis. Because the average size of an adult animal is 70 g, the amount of feed needed for test foods is not as intimidating (6 g/day) as what would be needed to feed other models, such as preruminant calves (e.g. 0.7–0.9 kg dry weight/day (69)), and non-human primates (e.g. 250 g/day for rhesus macaques (70)).
Although the model has been validated for studying the most common pro-vitamin A carotenoids in the human diet, gerbils lack the utility for studying absorption and metabolism of the dihydroxy xanthophylls (67, 71). However, gerbils do absorb, store and distribute lycopene from dietary sources (72).
ANNEX 2: BACKGROUND PAPERS

100
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
Domesticated speciesSwine
Swine (Sus domesticus) have been used extensively in nutrition research. Although there is a misconception that pig studies are expensive, the mar-ket price for young piglets (US$40–50) is less than the average cost for some specialty rats, e.g. Zucker rats (US$40 for lean and US$160 for obese). Pigs and humans have similar gastrointestinal anatomy, morphology and physiol-ogy. In fact, the relative sizes of sections of human and pig gastrointestinal tracts are similar, including gastric cell types and villi (73–75). Moreover, the nutrient requirements of pigs are similar to or greater than humans. The first study on vitamin A deficiency as a cause of congenital malformations was per-formed in pigs (76). Thus, the pig makes a good model for looking at vitamin A metabolism, storage and expression in milk.
The vitamin A content of adult pig liver is very close to that in humans, with a reported range of 0.48–1.3 µmol/g liver. Because fat is not readily trans-ferred through the placenta, including fat-soluble vitamins, liver reserves of vitamin A are low in newborn pigs. Pigs weaned onto a purified diet providing 1–1.6 times the National Research Council’s micronutrient requirements for a month, have low vitamin A stores with a mean range of 0.06–0.09 µmol/g liver. Young piglets used in development of a retinyl ester hydrolase assay had low vitamin A stores with a range of 0.0–0.11 µmol/g liver (77), compared with vitamin A deficiency in humans, which is defined as vitamin A stores <0.07 µmol/g liver.
Newborn pigs obtain their vitamin A from the milk of lactating sows and are thus are a good choice for studying the vitamin A needs of humans. Piglets are a good model for infants due to the similarities in physiology, anatomy (73, 78, 79), including form and function of the gastrointestinal tract, and diges-tive physiology (73–75). Adaptations of the MRDR test to infants was tested and validated in the piglet model (80). This response test was then applied after graded doses to young piglets as a model for infant supplementation practices (81). An extension to these studies on vitamin A in essential organs used 3,4-didehydroretinol as a tracer for chylomicron delivery and concluded that chylomicron delivery to lung and spleen is important in maintaining con-centrations (11). Differences in vitamin A metabolism have been found after ingestion of liver (82) and supplements by pigs (82, 83). When applied as a model for the lactating mother–nursing infant dyad, higher doses of vitamin A (equivalent to about 400 000 IU vitamin A to a human) resulted in a higher detoxification response than a lower dose (equivalent to about 200 000 IU vitamin A to a human) (83). Several vitamin A metabolites can be followed in the serum after a high dose of vitamin A (Figure A2.4.1). The water-soluble glucuronides allow the vitamin to be excreted in the urine.

101
Vitamin A concentration in milk in lactating sows can be easily measured. Extrapolations of milk concentrations obtained in sows to liver reserves of a nursing infant revealed that a higher dose of vitamin A may not increase liver reserves any higher than a lower dose (84); a point confirmed in a subsequent study where piglets were killed at two separate time points after dosing the mothers with two different levels of vitamin A (85). Another application of the lactating sow model has been the correlation of serum ratios of 3, 4-di -dehydroretinol to retinol to that in milk (86).
Although swine have been used extensively to study vitamin A metabolism and storage after administration of large doses, they are white-fat animals. Therefore, they predominantly cleave provitamin A carotenoids to vitamin A and have limited utility in evaluating the efficacy of food sources.
Preruminant calves
Preruminant calves have been used for studies involving carotenoid metabo-lism (59), as their β-carotene metabolism is similar to that in human infants after meal feeding (87). The calves absorb other carotenoids such as lutein,
ANNEX 2: BACKGROUND PAPERS
Figure A2.4.1 Formation of important vitamin A metabolites involved in the storage and detoxification of the vitamin. Dietary forms include the provitamin A carotenoids (β-carotene illustrated) and retinyl esters (retinyl palmitate illustrated). Both can lead to retinol. Retinoic acid, formed from the oxidation of retinal, is a necessary growth factor but in high amounts it is toxic. Therefore, retinoic acid can be catabolized to 4-oxo-retinoic acid or the water soluble glucuronide for excretion.
Retinol Retinal
All trans-Retinyl palmitate
O
OHOHOH
HOOCO
Retinyl β-glucuronide
O
OHOHOH
HOOCO
O
OH
O
Retinoic acid
O (CH2)14CH3
O
CH2OHCHO
β-Carotene
OH
O
O 4-oxo-Retinoic acid
Retinoyl β-glucuronide
β-caroteneAll trans-retinyl palmitate
Retinal Retinol
Retinoic acid Retinyl β-glucuronide
4-oxo-retinoic acid
Retinoyl β-glucuronide

102
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
lycopene and α-carotene (88). Furthermore, they utilize β-carotene for vita-min A (89). Thus, when large sample volumes are needed, the preruminant calf is a viable model for some applications.
Non-human primates
Genetic similarity to humans makes non-human primates an informative model for biomedical research. Great apes are genetically most similar to humans, however, they are rarely used as a model for vitamin A metabolism and carotenoid bioavailability (90, 91).
In 2001, it was discovered that rhesus monkeys held in captivity had hypervitaminosis A (92), and subsequently they have been used as a model for hypervitaminosis A. This is a dietary maintenance confounder instead of an experimentally induced state of vitamin A nutriture (70). Several pub-lished studies have exploited this model. Analyses of necropsy samples col-lected to determine tissue vitamin A concentration in rhesus and marmoset monkeys (93) have shown that, unlike humans, marmosets have a tremen-dous ability to store vitamin A in the kidney. Hypervitaminosis A was also documented in wild vervet monkeys held in captivity and fed on standard lab chow for only 2 years (94). Aborted fetuses from Old World monkeys on these lab diets had elevated levels of retinyl esters (95). Serum retinol levels were normal in all monkeys analysed; retinyl esters were elevated in the rhesus and marmoset monkeys (84), but not in the vervet monkeys (94).
Using this condition to understand the effects of hypervitaminosis A on stable isotope methodology, vitamin A labelled with 13C was administered to rhesus monkeys from the same colony in which the original observation of hypervitaminosis was made (96). Stable isotope methodology qualitatively predicted hypervitaminosis A at the individual level and quantitatively pre-dicted vitamin A liver concentration at the group level. Mathematical model-ling of these data revealed that the exchangeable pool of vitamin A is much less than the estimated body pool of vitamin A (96). Thus, during a hypervita-minotic state, some pools of vitamin A are not accessible and are most likely “walled off” in an effort to protect the body from the deleterious effects of the condition. A subsequent study of the individual 13C enrichments of hepatic retinyl esters after dosage with labelled vitamin A revealed that the 13C-retinol was readily taken up and stored in the liver as esters even in the presence of these extreme liver vitamin A concentrations (97). The retinyl ester stores in hepatic stellate cells act not only as a long-term reservoir for times of inad-equate intake, but also as a detoxification mechanism for this vitamin.

103
ANNEX 2: BACKGROUND PAPERS
ConclusionsIn the application of animal models to the human condition, it is important to carefully consider the limitations of each model and to interpret the results with these caveats in mind. Nonetheless, animal models allow researchers to evaluate more tissues than can be sampled from humans, especially when considering infants and young children. Vitamin A is essential to all mamma-lian physiology, but depending on the desired outcome and application, the species needs to be considered before a model is chosen. Of course, resources are often a limiting factor, but when properly applied, use of small mammals such as mice, rats and gerbils can yield essential insights into the mechanisms of action of vitamin A.
References1. McCollum EV, Davis M. The necessity of certain lipins in the diet during growth.
Journal of Biological Chemistry, 1913, 15:167–175.2. Ross SA et al. Retinoids in embryonal development. Physiological Reviews, 2000,
80:1021–1054.3. Clagett-Dame M, DeLuca HF. The role of vitamin A in mammalian reproduc-
tion and embryonic development. Annual Review of Nutrition, 2002, 22:347–381.4. Zile MH. Vitamin A and embryonic development: on overview. Journal of Nutri-
tion, 1998, 128:455S–458S.5. Keusch G. Interpretation of animal and human intervention trials. Nutrition Re-
views, 1995, 53:S47–S51.6. Metzger D, Chambon P. Contribution of targeted conditional somatic mutagene-
sis to deciphering retinoid X receptor functions and to generating mouse models of human disease. Handbook of Experimental Pharmacology, 2007, 178:511–524.
7. Quadro L et al. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO Journal, 1999, 18:4633–4644.
8. Quadro L et al. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mo-lecular Aspects of Medicine, 2003, 24:421–430.
9. Vogel S et al. Characterization of a new member of the fatty acid-binding pro-tein family that binds all-trans retinol. Journal of Biological Chemistry, 2001, 276:1353–1360.
10. Ross AC, Li N. Lung retinyl ester is low in young adult rats fed a vitamin A-defi-cient diet after weaning, despite neonatal vitamin A supplementation and main-tenance of normal plasma retinol. Journal of Nutrition, 2007, 137:2213–2218.
11. Sun T, Surles RL, Tanumihardjo SA. Vitamin A concentrations in piglet extra-hepatic tissues respond differently ten days after vitamin A treatment. Journal of Nutrition, 2008, 138:1101–1106.
12. Quadro L et al. Pathways of vitamin A delivery to the embryo: Insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology, 2005, 146:4479–4490.

104
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
13. Moise AR et al. Delivery of retinoid-based therapies to target tissues. Biochemis-try, 2007, 46:4449–4458.
14. Ghyselinck NB et al. Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO Journal, 1999, 18:4903–4914.
15. Xueping E et al. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. Journal of Biological Chemistry, 2002, 277:36617–36623.
16. Piantedosi R et al. Cellular retinol-binding protein type III is needed for reti-noid incorporation into milk. Journal of Biological Chemistry, 2005, 280:24286–24292.
17. O’Byrne SM et al. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). Journal of Biological Chemistry, 2005, 280:35647–35657.
18. Strom K et al. Hormone sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL. FASEB Journal, 2009, 23:2307–2316.
19. Leuenberger MG, Engeloch-Jarret C, Woggon WD. The reaction mechanism of the enzyme-catalyzed central cleavage of β-carotene to retinal. Angewandte Che-mie (International edition in English), 2001, 40:2613–2617.
20. Wyss A et al. Expression pattern and localization of β, β-carotene 15, 15′-dioxy-genase in different tissues. Biochemical Journal, 2001, 354:521–529.
21. Hessel S et al. CMO1 deficiency abolishes vitamin A production from β-carotene and alters lipid metabolism in mice. Journal of Biological Chemistry, 2007, 282:33553–33561.
22. Fierce Y et al. In vitro and in vivo characterization of retinoid synthesis from β-carotene. Archives of Biochemistry and Biophysics, 2008, 472:126–138.
23. Lindshield BL et al. Lycopene biodistribution is altered in 15,15′-carotenoid mo-nooxygenase knockout mice. Journal of Nutrition, 2008, 138:2367–2371.
24. Johansson-Lindbom B, Agace WW. Vitamin A helps gut T cells find their way in the dark. Nature Medicine, 2004, 10:1300–1301.
25. Iwata M et al. Retinoic acid imprints gut-homing specificity on T cells. Immu-nity, 2004, 21:527–538.
26. Mora JR et al. Generation of gut-homing IgA-secreting B cells by intestinal den-dritic cells. Science, 2006, 314;1157–1160.
27. Mucida D et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science, 2007, 317:256–260.
28. Carman JA, Smith SA, Hayes CE. Characterization of a helper T lymphocyte defect in vitamin A-deficient mice. Journal of Immunology, 1989, 142:388–393.
29. Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nature Reviews. Immunology, 2008, 8:685–698.
30. Langkamp-Henken B, Glezer JA, Kudsk KA. Immunologic structure and func-tion of the gastrointestinal tract. Nutrition in Clinical Practice, 1992, 7:100–108.
31. Kudsk K. Current aspects of mucosal immunology and its influence by nutrition. American Journal of Surgery, 2002, 183: 390–398.
32. Croitoru K, Beinenestock J. Characteristics and functions of mucosa-associated lymphoid tissue. In: Ogra P et al., eds. Handbook of mucosal immunology. San Diego, CA, Academic Press, 1994:141–149.

105
33. Uematsu S et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nature Immunology, 2008, 9:769–776.
34. Mora JR, von Andrian UH. Role of retinoic acid in the imprinting of gut-homing IgA-secreting cells. Seminars in Immunology, 2009, 21:28–35.
35. Li J et al. Effects of parenteral and enteral nutrition on gut-associated lymphoid tissue. Journal of Trauma Injury, Infection and Critical Care, 1995, 39:44–52.
36. King BK, Kudsk KA. A temporal study of TPN-induced changes in gut-associat-ed lymphoid tissue and mucosal immunity. Archives of Surgery, 1997, 132:1303–1309.
37. Leenders MWH, Nijkamp MW, Rinkes INMB. Mouse models in liver cancer research: A review of current literature. World Journal of Gastroenterology, 2008, 14:6915–6923.
38. De Flora S, Bagnasco M, Vainio H. Modulation of genotoxic and related effects by carotenoids and vitamin A in experimental models: Mechanistic issues. Mu-tagenesis, 1999, 14:153–172.
39. Murakoshi M et al. Potent preventive action of α-carotene against carcinogen-esis: Spontaneous liver carcinogenesis and promoting stage of lung and skin carcinogenesis in mice are suppressed more effectively by α-carotene than by β-carotene. Cancer Research, 1992, 52:6583–6587.
40. Niles RM. Vitamin A (retinoids) regulation of mouse melanoma growth and dif-ferentiation. Journal of Nutrition, 2003, 133:282S–286S.
41. Russell RM. The enigma of β-carotene in carcinogenesis: What can be learned from animal studies. Journal of Nutrition, 2004, 134:262S–268S.
42. Kang SG et al. Identification of a chemokine network that recruits FoxP3 regula-tory T cells into chronically inflamed intestine. Gastroenterology, 2007, 132:966–981.
43. Kang SG et al. High and low vitamin A therapies induce distinct Fox P3+ T-cell subsets and effectively control intestinal inflammation. Gastroenterology, 2009, 137:1391–1402.
44. Wellik D, DeLuca HF. Retinol in addition to retinoic acid is required for success-ful gestation in vitamin A-deficient rats. Biology of Reproduction, 1995, 53:1392–1397.
45. White JC, Highland M, Clagett-Dame M. Abnormal development of the sinuat-rial venous valve and posterior hindbrain may contribute to late fetal resorption of vitamin A-deficient rat embryos. Teratology, 2000, 62:374–384.
46. Zolfaghari R, Ross AC. An essential set of basic DNA response elements is re-quired for receptor-dependent transcription of the lecithin:retinol acyltrans-ferase (Lrat) gene. Archives of Biochemistry and Biophysics, 2009, 489:1–9.
47. Escaron AL, Green MH, Tanumihardjo SA. Plasma turnover of 3,4-didehydro-retinol (vitamin A2) increases in vitamin A-deficient rats fed low versus high dietary fat. Journal of Lipid Research, 2009, 50:694–703.
48. Ross AC. Retinoid production and catabolism: Role of diet in regulating retinol esterification and retinol acid oxidation. Journal of Nutrition, 2003, 133:291S–296S.
ANNEX 2: BACKGROUND PAPERS

106
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
49. Loerch JD, Underwood BA, Lewis KC. Response of plasma levels of vitamin A to a dose of vitamin A as an indicator of hepatic vitamin A reserves in rats. Journal of Nutrition, 1979, 109:778–786.
50. Tanumihardjo SA, Barua AB, Olson JA. Use of 3, 4-didehydroretinol to assess vi-tamin A status in rats. International Journal for Vitamin and Nutrition Research, 1987, 57:127–132.
51. Tanumihardjo SA et al. Use of the modified relative dose response (MRDR) as-say in rats and its application to humans for the measurement of vitamin A sta-tus. European Journal of Clinical Nutrition, 1990, 44:219–224.
52. Tanumihardjo SA, Olson JA. A modified relative dose-response assay employing 3, 4-didehydroretinol (vitamin A2) in rats. Journal of Nutrition, 1988, 118:598–603.
53. Gieng SH et al. Model-based compartmental analysis indicates a reduced mo-bilization of hepatic vitamin A during inflammation in rats. Journal of Lipid Research, 2007, 48: 904–913.
54. Green MH et al. Vitamin A intake affects the contribution of chylomicrons vs. retinol-binding protein to milk vitamin A in lactating rats. Journal of Nutrition, 2001, 131:1279–1282.
55. Ross AC, Pasatiempo AM, Green MH. Chylomicron margination, lipolysis, and vitamin A uptake in the lactating rat mammary gland: Implications for milk retinoid content. Experimental Biology and Medicine, 2004, 229:46–55.
56. Davila ME et al. Vitamin A during lactation: Relationship of maternal diet to milk vitamin A content and to the vitamin A status of lactating rats and their pups. Journal of Nutrition, 1985, 115:1033–1041.
57. Ross AC et al. Vitamin A combined with retinoic acid increases retinol uptake and lung retinyl ester formation in a synergistic manner in neonatal rats. Journal of Lipid Research, 2006, 47:1844–1851.
58. Wu L, Ross AC. Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT and CYP26B1 expression in neonatal lung. Journal of Lipid Research, 2010, 51:378–387.
59. Lee CM et al. Review of animal models in carotenoid research. Journal of Nutri-tion, 1999, 129:2271–2277.
60. Liu AG et al. Feeding tomato and broccoli powders enriched with bioactives im-proves bioactivity markers in rats. Journal of Agricultural and Food Chemistry, 2009, 57:7304–7310.
61. Campbell JK et al. Phytoene, phytofluene, and lycopene from tomato powder differentially accumulate in tissues of male Fisher 344 rats. Nutrition Research, 2007, 27:794–801.
62. Boslan MC. Use of animal models in defining efficacy of chemoprevention agents against prostate cancer. European Urology, 1999, 35:459–463.
63. Tanumihardjo SA. Food-based approaches for ensuring adequate vitamin A nu-trition. Comprehensive Reviews in Food Science and Food Safety, 2008, 7:373–781.
64. Pollack J et al. Mongolian gerbils (Meriones unguiculatus) absorb β-carotene intact from a test meal. Journal of Nutrition, 1994, 124:869–873.
65. Deming DM et al. Amount of dietary fat and type of soluble fiber independently modulate postabsorptive conversion of β-carotene to vitamin A in Mongolian gerbils. Journal of Nutrition, 2000, 130:2789–2796.

107
66. Tanumihardjo SA, Howe JA. Twice the amount of α-carotene isolated from car-rots is as effective as β-carotene in maintaining the vitamin A status of Mongo-lian gerbils. Journal of Nutrition, 2005, 135:2622–2626.
67. Davis CR et al. The xanthophyll composition of biofortified maize (Zea mays sp.) does not influence the bioefficacy of provitamin A carotenoids in Mongolian gerbils (Meriones unguiculatus). Journal of Agricultural and Food Chemistry, 2008, 56:6745–6750.
68. Davis CR et al. β-Cryptoxanthin from supplements or carotenoid enhanced maize maintains liver vitamin A in Mongolian gerbils (Meriones unguiculatus) better than or equal to β-carotene supplements. British Journal of Nutrition, 2008, 100:786–793.
69. Jenkins KJ, Hidiroglou M. Tolerance of the preruminant calf for excess manga-nese or zinc in milk replacer. Journal of Dairy Science, 1991, 74:1047–1053.
70. Penniston KL, Tanumihardjo SA. Vitamin A intake of captive rhesus monkeys exceeds National Research Council Recommendations. American Journal of Pri-matology, 2006, 68:1114–1119.
71. Molldrem KL, Tanumihardjo SA. Lutein supplements are not bioavailable in the Mongolian gerbil while consuming a diet with or without cranberries. Interna-tional Journal for Vitamin and Nutrition Research, 2004, 74:153–160.
72. Mills JP, Simon PW, Tanumihardjo SA. β-Carotene from red carrot maintains vitamin A status but lycopene bioavailability is lower relative to tomato paste in Mongolian gerbils. Journal of Nutrition, 2007, 137:1395–1400.
73. Tumbleson M. Swine in biomedical research, volumes I–III. New York, NY, Ple-num Press, 1986.
74. Miller ER, Ullrey DE. The pig as a model for human nutrition. Annual Review of Nutrition, 1987, 7:361–382.
75. Leddin DJ. Characterization of small intestinal water, sodium, and potassium transport and morphology in the pig. Canadian Journal of Physiology and Phar-macology, 1992, 70: 113–115.
76. Hale F. The relation of vitamin A to anophthalmos in pigs. American Journal of Ophthalmology, 1935, 18:1087–1093.
77. Cooper DA, Olson JA. Properties of liver retinyl ester hydrolase in young pigs. Biochimica and Biophysica Acta, 1986, 884:251–258.
78. Book SA, Bustad LK. The fetal and neonatal pig in biomedical research. Journal of Animal Science, 1974, 38:997–1002.
79. Mount LE. The climatic physiology of the pig. London, Edward Arnold, 1968.80. Valentine AR, Tanumihardjo SA. Adjustments to the modified relative dose re-
sponse (MRDR) test for assessment of vitamin A status minimize the blood vol-ume used in piglets. Journal of Nutrition, 2004, 134:1186–1192.
81. Surles RL et al. One-time graded doses of vitamin A to weanling piglets enhance hepatic retinol but do not always prevent a deficient vitamin A status. American Journal of Clinical Nutrition, 2007, 86:1045–1053.
82. Arnhold T et al. Porcine intestinal metabolism of excess vitamin A differs fol-lowing vitamin A supplementation and liver consumption. Journal of Nutrition, 2002, 132:197–203.
ANNEX 2: BACKGROUND PAPERS

108
WHO TECHNICAL CONSULTATION ON VITAMIN A IN NEWBORN HEALTH: MECHANISTIC STUDIES
83. Penniston KL, Tanumihardjo SA. Elevated serum concentrations of β-glucuro-nide metabolites and 4-oxoretinol following treatment with vitamin A in lactat-ing sows: a model for evaluating supplementation of lactating women. American Journal of Clinical Nutrition, 2005, 81: 851–858.
84. Penniston KL, Thayer JC, Tanumihardjo SA. Serum vitamin A esters are high in captive rhesus (Macaca mulatta) and marmoset (Callithrix jacchus) monkeys. Journal of Nutrition, 2003, 133:4202–4206.
85. Valentine AR, Tanumihardjo SA. One-time vitamin A supplementation of lactating sows enhances hepatic retinol of offspring independent of dose size. American Journal of Clinical Nutrition, 2005, 81:427–433.
86. Surles RL, Li J, Tanumihardjo SA. The modified-relative-dose-response values in serum and milk are positively correlated over time in lactating sows with ad-equate vitamin A status. Journal of Nutrition, 2006, 136:939–945.
87. Poor CL et al. Evaluation of the preruminant calf as a model for the study of hu-man carotenoid metabolism. Journal of Nutrition, 1992, 122:262–268.
88. Bierer TL, Merchen NR, Erdman Jr JW. Comparative absorption and transport of five common carotenoids in preruminant calves. Journal of Nutrition, 1995, 125:1569–1577.
89. Hoppe PP et al. Dietary β-carotene elevated plasma steady-state and tissue con-centration of β-carotene and enhances vitamin A balance in preruminant calves. Journal of Nutrition, 1996, 126:202–208.
90. Gagneux P et al. Proteomic comparison of human and great ape blood plasma reveals conserved glycosylation and differences in thyroid hormone metabolism. American Journal of Physical Anthropology, 2001, 115:99–109.
91. García AL et al. Great apes show highly selective plasma carotenoids and have physiologically high plasma retinyl esters compared to humans. American Jour-nal of Physical Anthropology, 2006, 131:236–242.
92. Penniston KL, Tanumihardjo SA. Subtoxic hepatic vitamin A concentrations in captive rhesus monkeys (Macaca mulatta). Journal of Nutrition, 2001, 131:2904–2909.
93. Mills JP, Penniston KL, Tanumihardjo SA. Extra-hepatic vitamin A concentra-tions in captive Rhesus (Macaca mulatta) and Marmoset (Callithrix jacchus) monkeys fed excess vitamin A. International Journal for Vitamin and Nutrition Research, 2005, 75:126–132.
94. Mills JP, Tanumihardjo SA. Vitamin A toxicity is biochemically evident in wild-caught African green vervet monkeys (Chlorocebus aethiops) after two years in captivity. Comparative Medicine, 2006, 56:423–427.
95. Mills JP, Terasawa E, Tanumihardjo SA. Excessive preformed vitamin A intake by mothers amplifies early fetal liver retinyl ester storage in captive Old World monkeys. Comparative Medicine, 2007, 57:505–511.
96. Escaron AL et al. Mathematical modeling of serum 13C-retinol in captive rhe-sus monkeys provides new insights on hypervitaminosis A. Journal of Nutrition, 2009, 139:2000–2006.
97. Escaron AL, Tanumihardjo SA. Orally ingested 13C2-retinol is incorporated into hepatic retinyl esters in a nonhuman primate (Macaca mulatta) model of hyper-vitaminosis A. Comparative Medicine, 2010, 60:71–76.


For more information, please contact:
Department of Nutrition for Health and DevelopmentWorld Health OrganizationAvenue Appia 20, CH-1211 Geneva 27, SwitzerlandFax: +41 22 791 4156E-mail: [email protected]/nutrition
ISBN 978 92 4 150316 7



![Vitamin KBK.ppt [Read-Only]ocw.usu.ac.id/.../bbc115_slide_vitamins.pdf · Vitamin A : k t i EiE xcessive intake Retinol intake > 1000 μg/d Ƶbone fx risk Retinol intake 3000 μg/d](https://img.pdfslide.us/doc/110x75/5ff48784cb598568bd64b641/vitamin-kbkppt-read-onlyocwusuacidbbc115slide-vitamin-a-k-t-i.jpg)








![Vitamin A-deficiency, fish eating status and the ... · PDF file10.06.2011 · Vitamin A Retinol [Vitamin A1] Marine Fishes Adult Amphibia Reptiles Aves *1 Mammals Freshwater Fishes*2](https://img.pdfslide.us/doc/110x75/5a79b8d17f8b9a99188b50b3/vitamin-a-deficiency-fish-eating-status-and-the-a-retinol-vitamin-a1-marine.jpg)