Embed Size (px)
Citation preview

The Pennsylvania State University
The Graduate School
The Huck Institutes of the Life Sciences
REGULATION OF GENE TRANSCRIPTION BY THE ARYL HYDROCARBON
RECEPTOR –NEW TARGETS AND MECHANISMS OF REGULATION
A Dissertation in
Integrative Biosciences
by
Rushang Dilipkumar Patel
© 2008 Rushang Dilipkumar Patel
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
August 2008

The dissertation of Rushang Dilipkumar Patel was reviewed and approved* by the following:
Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Dissertation Advisor Chair of Committee
Curtis Omiecinski Professor of Veterinary Science H. Thomas and Dorothy Willits Hallowell Chair
Jeffrey M. Peters Associate Professor of Environmental Toxicology
Robert Mitchell Professor Emeritus of Biology
Naomi S. Altman Associate Professor of Statistics
Peter Hudson Willaman Professor of Biology Director, Huck Institutes of the Life Sciences
*Signatures are on file in the Graduate School

iii
ABSTRACT
Adaptation in response to changes in internal as well as external environment is
imperative to sustenance of life. Modulation of gene expression is a critical component of
this adaptive response and is mediated by activation of various transcription factors.
Individual signaling pathways have been well characterized for many transcription factor
systems. Aryl hydrocarbon receptor (AHR) is a transcription factor that is activated by a
variety of structurally diverse ligands, including the environmental contaminant dioxin,
the cigarette smoke constituent benzo[a]pyrene and the therapeutically prescribed drug
omeprazole. Prior to activation, AHR is primarily located in a cytoplasmic complex with
chaperone and co-chaperone proteins. Ligand-binding is believed to initiate a
conformational change that leads to nuclear translocation, dissociation from the
chaperones and heterodimerization with AHR-nuclear translocator (ARNT). AHR-ARNT
heterodimer recognizes and binds to a consensus DNA sequence (TNGCGTG),
commonly referred to as a dioxin response element (DRE), to drive transcription of target
genes. Phase I and II xenobiotic metabolism enzymes have been the well-characterized
targets of AHR-mediated transactivation. This sequence of coordinate events has been
described as the classical pathway of AHR activity. Different lines of evidence suggest
that AHR serves physiologically relevant functions, though the details have not been
elucidated. The goal of this research project was to identify previously uncharacterized
targets of AHR-mediated gene regulation and to investigate the hypothesis that AHR
functions through mechanisms that are independent of DNA-binding. The advances in
performing genome-wide transcriptional profiling at the time of commencement of this

iv
project, encouraged the use of DNA-microarray technology for identifying new target
genes. Epiregulin, a potent mitogen belonging to the epidermal growth factor family, was
discovered to be regulated by AHR in immortalized murine hepatocytes. The fact that a
number of AHR ligands have been associated with carcinogenesis signifies that the
induction of growth factors like epiregulin might be a potential mechanism for AHR-
mediated tumor enhancement. The next phase of this project led to the identification of
the constitutive androstane receptor (CAR), another receptor involved in drug
metabolism, as an in vivo target of AHR activation. This association between AHR-CAR
highlights the possibility of crosstalk between AHR and other pathways. Exposure to
divergent stimuli leads to simultaneous activation of multiple signaling pathways. This
suggests that it is essential to study the networking of various pathways to be able to
predict the biological outcomes. The third phase of this project focuses on the ability of
AHR to modulate the inflammatory pathway and on the involved mechanism. AHR
activation can repress the acute-phase response (APR) gene expression, implicated in
disorders like septic shock and Alzheimer’s, partly by antagonizing NF-κB mediated
gene regulation through a non-classical mechanism not involving DRE. Serum amyloid
family members, C-reactive protein and haptoglobin were found to be repressed by AHR,
signifying that AHR regulates multiple members of the APR. Thus, this research has led
to the identification of multiple AHR-regulated genes. It also presents a model to study
AHR-mediated gene repression, an aspect that has therapeutic potential.

v
TABLE OF CONTENTS
LIST OF FIGURES .....................................................................................................vii
LIST OF TABLES....................................................................................................... ix
ACKNOWLEDGEMENTS.........................................................................................x
Chapter 1 INTRODUCTION......................................................................................1
1.1 History and Characterization .........................................................................2 1.1.1 Discovery of AHR:...............................................................................2 1.1.2 AHR structure:......................................................................................3 1.1.3 AHR alleles and polymorphisms:.........................................................6
1.2 AHR activation: ..............................................................................................8 1.2.1 Exogenous ligands:...............................................................................8 1.2.2 Endogenous ligands:.............................................................................9 1.2.3 Ligand-independent AHR activation:...................................................11
1.3 AHR pathway: ................................................................................................11 1.4 AHR mouse models:.......................................................................................16
1.4.1 AHR-null mouse models: .....................................................................16 1.4.2 Other transgenic AHR mouse models: .................................................18 1.4.3 Biosensor mouse models based on AHR: ............................................21
1.5 AHR Regulated Genes:...................................................................................21 1.5.1 Phase I and Phase II enzymes:..............................................................23 1.5.2 Other AHR regulated genes: ................................................................25
1.6 Potential physiological roles of AHR:............................................................31 1.6.1 Reproduction: .......................................................................................31 1.6.2 Cardiovascular:.....................................................................................32 1.6.3 Development: .......................................................................................33 1.6.4 Endocrinal homeostasis: .......................................................................34
1.7 Interaction of AHR with other signaling pathways: .......................................35 1.7.1 AHR and estrogen signaling:................................................................36 1.7.2 AHR and inflammatory signaling: .......................................................38
1.8 Overview and significance of research:..........................................................46
Chapter 2 THE ARYL HYDROCARBON RECEPTOR DIRECTLY REGULATES EXPRESSION OF THE POTENT MITOGEN EPIREGULIN...54
2.1 Abstract:..........................................................................................................55 2.2 Introduction.....................................................................................................56 2.3 Materials and methods....................................................................................59 2.4 Results.............................................................................................................63 2.5 Discussion.......................................................................................................72

vi
Chapter 3 AHR ACTIVATION REGULATES CONSTITUTIVE ANDROSTANE RECEPTOR (CAR) LEVELS IN MURINE AND HUMAN LIVER...................................................................................................................78
3.1 Abstract...........................................................................................................79 3.2 Introduction.....................................................................................................80 3.3 Materials and Methods ...................................................................................82 3.4 Results: ...........................................................................................................86 3.5 DISCUSSION.................................................................................................105
Chapter 4 AHR REPRESSES CYTOKINE MEDIATED ACUTE PHASE RESPONSE BY A DNA-INDEPENDENT MECHANISM................................110
4.1 Abstract...........................................................................................................111 4.2 Introduction.....................................................................................................113 4.3 Materials and Methods: ..................................................................................116 4.4 Results: ...........................................................................................................120 4.5 Discussion:......................................................................................................150
Chapter 5 CONCLUSIONS AND FUTURE DIRECTIONS.....................................157
Bibliography ................................................................................................................168

vii
LIST OF FIGURES
Figure 1.1: Modular domain architecture of AHR ..................................................4
Figure 1.2: Classical AHR pathway. ........................................................................15
Figure 1.3: NF-κB pathway and the possible levels at which AHR can exert repression.............................................................................................................42
Figure 1.4: Alternate models proposed for AHR-mediated activation of gene transcription. .......................................................................................................49
Figure 1.5: Alternate models proposed for AHR-mediated repression of gene transcription. .......................................................................................................50
Figure 2.1: TCDD increases Epiregulin mRNA. .....................................................64
Figure 2.2: Epiregulin promoter occupancy by the ligand-activated AhR. .........66
Figure 2.3: AhR binds DRE in rat Epiregulin promoter. ......................................69
Figure 2.4: Epiregulin and TCDD increase primary mouse keratinocyte proliferation in a dose-dependent manner. ......................................................71
Figure 2.5: The DRE is absent in the human epiregulin promoter. ......................77
Figure 3.1: CAR mRNA levels increase in response to the AhR-ligand BNF. .....94
Figure 3.2: CAR up-regulation is AhR-dependent. .................................................97
Figure 3.3: Temporal and spatial patterns of CAR expression. .............................99
Figure 3.4: AhR-dependent CAR up-regulation leads to increased CAR-mediated transcriptional activity. .....................................................................102
Figure 3.5: CAR induction in response to AhR ligands in primary human hepatocyte culture...............................................................................................104
Figure 4.1: Functional dissociation of the properties of AHR involved in Saa3 repression.............................................................................................................122
Figure 4.2: AHR functional mutants.........................................................................125
Figure 4.3: AHR represses Saa3 induction by various cytokines. .........................127

viii
Figure 4.4: Dose-response and ligand-specificity analysis of AHR mediated repression of Saa3. ..............................................................................................129
Figure 4.5: AHR-mediated Saa3 repression is due to direct transcriptional inhibition..............................................................................................................131
Figure 4.6: AHR activation represses other Saa-family member gene expression. ...........................................................................................................133
Figure 4.7: AHR represses Saa induction by physiologically attainable cytokine concentrations......................................................................................136
Figure 4.8: ChIP assay to determine the effect of AHR activation on Saa1, Saa2 and Saa3 promoters...................................................................................138
Figure 4.9: Effect of HDAC inhibition on Saa expression. ....................................140
Figure 4.10: AHR activation induces SOCS genes...................................................142
Figure 4.11: AHR activation represses other APR genes as well. .........................144
Figure 4.12: AHR-mediated repression in human cells..........................................146
Figure 4.13: Saa repression in human cells is AHR-dependent..............................148
Figure 4.14: AHR-mediated NF-κB suppression is gene-specific...........................149
Figure 5.1: Schematic for a screen to identify ‘Selective AHR Modulators – SARM’. ................................................................................................................166

ix
LIST OF TABLES
Table 1.1: Modular domain architecture of AHR. ..................................................5
Table 1.2: Allelic variation in murine AHR. ............................................................7
Table 1.3: Genes regulated by the classical AHR pathway.....................................30
Table 3.1: Sequence information for primers used in qPCR.................................85
Table 3.2: BNF-mediated differentially regulated genes, sorted by Biological Process (BP)/Molecular Function (MF)............................................................88
Table 4.1: List of genes regulated by A78D-AHR and WT-AHR. .........................121

x
ACKNOWLEDGEMENTS
I would like to take this opportunity to thank everyone who helped me in my
research. Dr. Gary Perdew, my thesis advisor, has been a great source of encouragement
and advice. His optimism and patience have been invaluable in my learning. Dr. Jeffrey
Peters has been kind enough to share advice and lab equipment for animal work and Dr.
Dae Joon Kim, a former member of the Peters lab, did the primary keratinocyte
experiments in Chapter 2. Dr. Curtis Omiecinski procured primary human hepatocytes
used for experiments in Chapter 3. Dr. Naomi Altman provided valuable suggestion for
microarray data analysis. I would also like to thank Dr. Brett Hollingshead for animal
treatments in Chapter 3. Dr. Iain Murray helped in the isolation of primary mouse
hepatocytes used in Chapter 4 and for his overall help when I joined the lab. Dr. Ann
Kusnadi performed dose-response experiment in Chapter 4. I would also like to
acknowledge DNA-microarray facility and the animal care facility. Finally, I am grateful
for the love and support of my parents, my wife and one-year old daughter, Roma!

xi
‘Maatru Devo Bhaava’
- ancient Hindu teaching
‘Maatru devo bhaava’ appears at the beginning of the Vedaas – the four books of the
foundation of Hindu religion. It means ‘Always hold mother as God’. I dedicate this
thesis to my mother who devoted her life to nurture mine.

Chapter 1
INTRODUCTION

2
1.1 History and Characterization
1.1.1 Discovery of AHR:
Even before the existence of aryl hydrocarbon receptor (AHR) was conceived,
researchers detected aryl hydrocarbon hydroxylase (initially B[a]P hydroxylase) activity
in mammalian cell cultures that was inducible in response to aromatic hydrocarbons (1-
3). The observation that some mouse strains (C57BL/6) are responsive to the inducer of
this hydroxylase activity while others (DBA/2) are not, and that ‘responsiveness’ is
inherited in a dominant fashion, further indicated the involvement of a receptor protein in
this mechanism (4, 5). Subsequent studies that examined the characteristics of induction
of aryl hydrocarbon hydroxylase substantiated the notion of aryl hydrocarbon receptor. In
a landmark report, Poland et. al. determined various [3H]TCDD-binding properties, such
as dissociation constant, of the hepatic cytosol from C57BL/6 mice and provided
convincing evidence that TCDD-binding species is a receptor (6). Examination of 23
other dioxins tested for their binding-properties to hepatic cytosol, revealed a correlation
with their ability to induce hydroxylase activity. Later on, the synthesis of a photoaffinity
ligand, 2-azido-3-iodo-7,8-dibromadibenzo-p-dioxin (7), facilitated the purification of
AHR and generation of antibodies to the receptor (8-10). These initial observations and
tools laid the basis for further research on AHR.

3
1.1.2 AHR structure:
The field of AHR has always drawn inspiration from the advancements in steroid
receptor biology, especially the glucocorticoid receptor (GR). Though AHR behaves
biochemically in a manner similar to GR, the architectural layout of AHR’s functional
domains differs significantly from that of GR. Nuclear receptors have an amino-terminal
activation function (AF-1) domain, a central DNA-binding domain (DBD) and a carboxy-
terminal ligand-binding domain (LBD) which also encompasses the activation function
(AF-2) domain. The LBD also serves dimerization, coregulator recruitment and nuclear
localization functions (reviewed multiple times, including (11). AHR, on the other hand,
is a basic-helix-loop-helix (bHLH) PAS (Period (Per), Aryl Hydrocarbon Receptor
Nuclear Translocator (ARNT), Single Minded (Sim)) protein. The bHLH family of
transcriptional regulators are involved in critical cellular processes (12). In general, the
basic region imparts DNA binding ability while the HLH region serves as a dimerization
domain along with a second one located in the PAS region. The PAS domain is a 250-
300 amino acid region comprising of two degenerate 50 amino acids subdomains (PAS A
and PAS B). Like the LBD of the steroid receptors, the PAS domain mediates a number
of functions – dimerization with other bHLH/PAS proteins, association with chaperones
in the cytoplasm and ligand binding (reviewed in (13)). A systematic deletion analysis of
AHR and ARNT proteins has helped identify the role of various amino-terminal domains
in the AHR signaling pathway (14, 15).

4
While the amino-terminal domains of AHR are responsible for generic functions
like ligand and DNA binding, the carboxy-terminal domains impart a control over the
transcriptional activation by mediating interactions with coregulator proteins. The initial
report involved generation of chimeric proteins with the DNA-binding domain of yeast
protein Gal4 and various deletion mutants of AHR and ARNT (16). Results from this
study also demonstrated that the amino-terminal domains of AHR and ARNT were
devoid of transactivation potential. A schematic representation of the distribution of AHR
domains is presented in Figure 1.1 , and Table 1.1 summarizes the location and functions
of various AHR domains.
Figure 1.1: Modular domain architecture of AHR
AHR is composed of many domains. From the amino terminal to the carboxy terminal,these are the basic domain, helix-loop-helix (HLH) domain, Per-ARNT-Sim (PAS) A and B domains,

5
Table 1.1: Modular domain architecture of AHR.
Motif / Domain Amino acid span Functions
Basic 12 – 39 DNA binding Helix-loop-helix 40 – 80 Dimerization, HSP90 binding Pas A 116 – 179 Dimerization Pas B 269 – 336 Dimerization, HSP90 binding, ligand binding (PAC) PAS – associated C-terminal domain 342 – 380 It is proposed to contribute to the PAS domain
fold Acidic subdomain 491 – 593 Gln-rich subdomain 594 – 648 PST subdomain 648 – 805
Part of the transactivation domain (C-terminal)
NLS 12 – 38 Nuclear localization signal NES 62 – 72 Nuclear export signal

6
1.1.3 AHR alleles and polymorphisms:
AHR demonstrates inter- as well as intra-species variation. In mice, there are four
allelic variations in inbred strains that differ significantly in their biochemical properties
and transactivation potential. As described, this allelic variation, in fact, helped in
identification of AHR. The differences in the four mouse alleles are summarized in
Table 1.2. In addition, the Han/Wistar (Kuopio) strain of rats is significantly more
resistant to the toxic effects of TCDD as compared to other rat strains. A point mutation
that leads to alternate splice variant of AHR is believed to be responsible for this
variation (17). Polymorphisms in human AHR have also been documented. Arg554Lys,
Pro517Ser and Val570Iso are the only polymorphisms supported by phenotypic effects
(18). Even these polymorphisms do not affect CYP1A1 induction individually. Paired
polymorphisms at the 554 and 570 amino acids can more effectively inhibit CYP1A1
induction in vitro (19). Since all of these mutations are in the transactivation domain, it is
likely that they alter the cohort of coregulator proteins recruited by AHR for gene
regulation, leading to phenotypic variation. Since the physiological roles of AHR have
not been conclusively established yet, it is difficult to assess the phenotypic association of
polymorphisms outside the realm of xenobiotic metabolism.

7
Table 1.2: Allelic variation in murine AHR.
Allele Allele Definition
Ahrb-1 • high affinity, relatively heat stable, 95 kDa receptor. • ten nucleotide differences between the coding sequences of the
DBA/2J and C57BL/6J receptors. Five of the ten differences would cause amino acid changes.
• one of these, a C to T transition in exon 11 would change the arginine codon in the DBA/2J allele to a termination codon.
• C57, C58 and MA/My Ahrb-2 • high affinity, heat labile, 104 kDa receptor containing 848 amino
acids. • BALB/cBy, A and C3H
Ahrb-3 • high affinity, 105 kDa receptor with slightly more heat stability. • M. spretus, M. caroli and MOLF/Ei
Ahrd • 104 kDa receptor that is stabilized by molybdate. • affinity for ligand 10-100 fold lower than that of the receptor
produced by the C57BL/6J allele. • DBA, AKR and 129

8
1.2 AHR activation:
AHR has always been described as a ligand-activated transcription factor.
Majority of research has been conducted by activating AHR with exogenous ligands,
though there is an active ongoing search for an endogenous ligand as well. Non-ligand
based AHR activation has also been reported. Important findings from the literature
related to AHR activation are discussed here.
1.2.1 Exogenous ligands:
A number of aromatic hydrocarbons are capable of binding to AHR. Most of
these can be classified as either polycyclic aromatic hydrocarbons (PAH) or halogenated
aromatic hydrocarbons (HAH) (20). The structure of the ligand binding domain of AHR
has not been established by x-ray crystallography studies. However, structure-activity
relationship and binding affinity studies suggest that the ligand pocket can accommodate
a compound that is planar, 12-14 Å long, 12 Å wide and 5 Å deep (21, 22). Generally,
halogenated compounds have a higher affinity for the receptor. PAH, such as
benzo[a]pyrene (B[a]P) and 3-methyl cholanthrene (3-MC), and HAH, such as
polychlorinated dibenzo-p-dioxins (PCDD, including TCDD), biphenyls and
dibenzofurans, are generated as a result of industrial processes like paper bleaching,
waste incineration, and combustion, as well as cigarette smoking, automobile exhaust and
charbroiled foods (23, 24). Polychlorinated biphenyls are also found in insulation

9
materials, adhesives and flame retardants. Thus, compounds capable of activating AHR
are ubiquitous and impossible to avoid.
1.2.2 Endogenous ligands:
In the absence of a proven physiologically relevant endogenous ligand, AHR is
still classified as an orphan receptor. There are a number of candidates (e.g. bilirubin)
that can claim to be the endogenous activator of AHR, but, most of them have
significantly lower affinities for the receptor as compared to TCDD (reviewed in (25,
26)). Consequently, a very high plasma concentration would be required for each of these
candidates to activate the receptor. However, there are three important caveats that
challenge the search for an endogenous ligand. First, though it would be highly
improbable to achieve the required plasma concentration physiologically, it is possible
that the local availability of an endogenous ligand might be adequate to activate the
receptor. Second, the suitability of a ligand is measured by its ability to activate the
prototypical AHR target gene Cyp1a1. Cyp1a1 does not have an explained role in many
of the pathophysiological processes attributed to AHR. An endogenous ligand cannot be
expected to activate AHR with an intention to upregulate Cyp1a1 to the same extent as
exogenous environmental pollutants. Therefore, to identify an endogenous AHR ligand, it
is essential to explicitly describe the role of AHR in cellular processes including the site
of action and a measurable endpoint that is unrelated to xenobiotic metabolism. Finally,
in many cases the proposed ligand may be unstable, making it difficult to differentiate
whether the parent compound or a metabolite binds the receptor.

10
Endogenous AHR ligands include food derivatives, though not ‘truly’
endogenous, as well as de novo compounds synthesized in the body. Indole 3-carbinol
(I3C) and its acid condensation products indolo-(3,2,-b)-carbazole (ICZ) and
diindolylmethane (DIM) (27-29) as well as curcumin (30), carotinoids and flavanoids,
such as quercetin (31), are all derived from diet and have been shown to activate AHR.
Indigo and indirubin are tryptophan metabolites, previously used for dyeing fabrics.
These indigoids have been isolated from human urine and can activate the mammalian
receptor in cultured cells (32, 33). Tryptamine (TA) and indole acetic acid (IAA) (34) and
6-formylindolo[3,2-b]carbazole (FICZ, a UV irradiation photoproduct (35, 36)) are
derived from tryptophan and have been shown to bind AHR and drive DRE-mediated
transcription. Arachidonic acid metabolites such as Lipoxin A4 (37) and prostaglandin G2
(38) as well as heme metabolites like bilirubin and biliverdin (39, 40) have also
demonstrated AHR activation capabilities. Interestingly, AHR mediated induction of
UDP-glucuronyl transferase can detoxify bilirubin and thus serve as a feedback
mechanism. Equilenin, an estrogenic molecule present in the widely prescribed hormone
replacement therapy, Premarin, can also activate AHR. Equilenin induced CYP1A1
activity is independent of the ligand-affinity of different AHR alleles (41). Thus,
candidate endogenous ligands are generated from a variety of sources under diverse
scenarios.

11
1.2.3 Ligand-independent AHR activation:
AHR can also be activated in the absence of exogenous ligand, probably through
post-translational modification. When rat epithelial cells (42), murine hepatoma cells (43)
and human keratinocytes (44) were cultured in suspension, enhanced CYP1A1
transcription was detected. However, this response could also be attributed to the release
or synthesis of a ligand endogenously. Even when adhered to tissue culture containers,
cell confluence, and thus cell-cell contact, has been shown to modulate AHR activity; a
higher DRE-driven reporter activity was noted when cells were sparsely seeded (45). An
increase in cAMP can also activate AHR in Hepa1c1c7 cells, leading to its nuclear
translocation and induction of target genes (46).
1.3 AHR pathway:
AHR signaling pathway can be broadly divided into three phases:
• the ‘resting’ cytoplasmic AHR complex,
• nuclear translocation upon ligand activation and exchange of partner proteins and
• DNA-binding and transcriptional activation of target genes.
Chemical crosslinking studies in Hepa1c1c17 cell fractions have demonstrated
that in the absence of a ligand, AHR exists as a heteromeric, predominantly as a
tetrameric, complex in cytosol (47). Prior to this study, it had already been established
that 90-kDa heat shock protein (HSP90; current gene symbol: HSP90AA1) could

12
associate with cytosolic AHR (48, 49). Later on, it was realized that in fact two molecules
of HSP90 are present in the AHR complex (50). In vitro translation of AHR deletion
mutants using reticulocyte lysate revealed that amino acid sequences 1-166 and 289-347
of AHR mediate HSP90 association (51). A number of studies have examined the role of
HSP90 in the AHR-complex. It is believed that the most important functions of HSP90
are to maintain the receptor in a ligand-responsive state and stabilize it from proteolytic
turnover (52, 53).
Hepatitis B virus X-associated protein 2 (XAP2, also referred to as ARA9 or AIP)
was later identified to be the fourth member of the tetrameric complex (54, 55). Further
studies using cos-1 and Hepa1c1c7 cells revealed that XAP2 can independently associate
with AHR as well as HSP90 through three tetracotripeptide sites and that the AHR and
XAP2 binding sites on HSP90 do not overlap (56, 57). Elucidation of the role of XAP2 in
AHR complexes has led to conflicting results obtained from cell culture and mouse
models. Based on cell culture experiments, it seems that XAP2 plays an important role in
determining the subcellular localization of AHR (57, 58), while studies with a
hepatocyte-specific XAP2 gain-of-function/overexpression transgenic mouse model did
not reveal an alteration in localization or activity of AHR (59). Since XAP2 knock-out
mice are embryonic lethal (60), tissue-specific XAP2 knock-out mice are required to
definitively address this issue. HSP90 associated cytoplasmic molecular chaperone
machinery is common to a number of other receptors. An effort to confirm whether
observations from progesterone receptor could be extrapolated to AHR, revealed that p23
is also a member of AHR cytoplasmic complex (61). Though p23 has not been

13
extensively studied in the context of AHR-complex, it is believed that it facilitates ligand-
responsiveness and transformation of AHR to a DNA-binding state (62, 63).
Ligand-binding is believed to initiate a series of ill-defined conformational
changes in AHR that promote nuclear translocation, dissociation from the cytosolic
complex and heterodimerization with aryl hydrocarbon nuclear translocator (ARNT).
This is perhaps one of the least characterized aspects of AHR pathway. It is not clear
when and how the AHR dissipates its cytoplasmic complex, though it has been
demonstrated with photo-affinity ligands that in Hepa1c1c7 cells, AHR-HSP90 complex
can be isolated from the nuclear fraction (64). However, XAP2 has not been isolated
from nuclear forms of AHR (57). Furthermore, a bipartite nuclear localization sequence
(NLS) and a nuclear export sequence (NES) have been identified in AHR (65).
Microinjection of AHR fragments fused to glutathione S-transferase (GST)-green
fluorescent protein (GFP), nuclear export inhibitor – Leptomycin B and co-
immunoprecipitation identified chromosome region maintenance 1 (CRM-1) to be a
facilitator of AHR export (66). Similarly, an association between importer protein β-
importin and the AHR-complex has been show to be involved in nuclear import (67).
After nuclear localization, AHR dimerizes with another bHLH-PAS protein,
ARNT (68). Even before the identification of ARNT, it was known that the nuclear form
of AHR exists as a heterodimer (47). ARNT serves as a common dimerization partner to
other bHLH-Pas proteins and can even function as a homodimer (69). It is primarily
nuclear and its function is not dependent on ligand-activation (reviewed in (70)). AHR-

14
ARNT heterodimer recognizes cognate response element (known as dioxin response
element (DRE) or xenobiotic response element (XRE)) sequences in promoters/enhancers
and recruits a number of coactivators to induce transcription of target genes. The binding
sequence for AHR-ARNT was identified initially by an 82-bp Cyp1a1 enhancer driven
reporter assays (71), and then narrowed down to the core DRE (5`-TNGCGTG-3`) by
electrophoretic mobility shift assays using synthetic oligonucleotides (72). Based on the
information from other bHLH proteins, it is known that ARNT binds the GTG half-site
while AHR recognizes TNGC half-site. However, this core sequence is not sufficient to
form a functional enhancer and flanking nucleotides must contribute to the DRE (72).
However, a position weight matrix has not been identified for these flanking nucleotides.
A simplified schematic of AHR pathway is presented in Figure 1.2 .
Thus, extensive research focusing on AHR activation has highlighted the details
of various steps involved in AHR signaling, though there remain certain aspects requiring
further experiments. Many of these ambiguous aspects might not have a single face, but
could be context-specific instead; for example, different ligands might transform the
receptor in diverse ways which would, in turn, dictate differential coregulator recruitment
and gene regulation. It is also noteworthy that the human and mouse AHR differ
significantly (73-75), possibly resulting in the development of alternate viewpoints with
respect to the potential adverse effects of AHR activation. In addition to the ‘classic’
pathway described above, new developments have focused on alternate modes of AHR
activity, such as receptor cross-talk (discussed later).
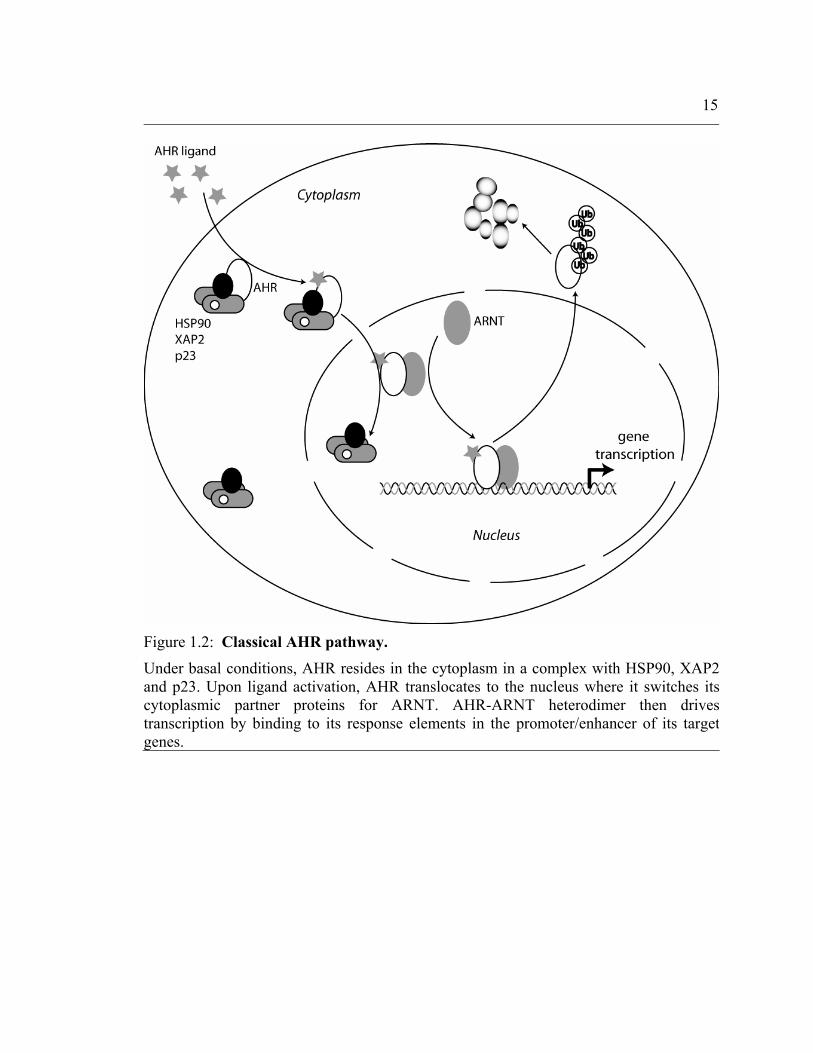
15
Figure 1.2: Classical AHR pathway.
Under basal conditions, AHR resides in the cytoplasm in a complex with HSP90, XAP2and p23. Upon ligand activation, AHR translocates to the nucleus where it switches its cytoplasmic partner proteins for ARNT. AHR-ARNT heterodimer then drives transcription by binding to its response elements in the promoter/enhancer of its targetgenes.

16
1.4 AHR mouse models:
Most of the available mammalian AHR biology information has resulted from
experiments using the murine system. Over the last few years, a number of mouse models
have been developed to explore the role of AHR. Both, loss-of-function as well as gain-
of-function models have provided insight into the physiological relevance of AHR.
1.4.1 AHR-null mouse models:
Three AHR knock-out mouse models have been reported, each generated with a
slightly different strategy. The first AHR knock-out mouse was developed in Dr.
Gonzalez’s lab. Exon 1 (subsequently referred to as Δ1/Δ1) of the Ahr gene in J1
embryonic stem cells was partially replaced by a neomycin (neo) cassette by homologous
recombination using an engineered fragment from 129SvJ genomic library. Screened
clones were injected into C57BL/6 embryos to generate chimeric mice which were then
back-crossed to the C57BL/6N background (76). The second AHR knock-out mouse was
generated in Dr. Bradfield’s lab by replacing exon 2 (subsequently referred to as Δ2/Δ2)
with a neo cassette in R1 embryonic stem cells. The final animals were derived on a
C57BL/6J background (77). In a third AHR knock-out model, generated in Dr. Fujii-
Kuriyama’s lab, a lacZ gene with a poly-A tail was fused downstream of exon 1 in CCE
embryonic stem cells and the male chimeras were mated with C57BL/6J females to
generate heterozygotes (Ahr+/-). These animals were then interbred (78).Δ1/Δ1 and Δ2/Δ2

17
AHR knock-out models have been more widely utilized as compared to the third model,
probably due to the fact that the mixed genetic background of the third model makes it
difficult to compare the results with established mouse lines.
The main phenotypic features observed in Δ1/Δ1 model include a 40-50%
neonatal mortality and hypocellularity of spleen and peripheral lymph nodes, but not in
the thymus, during the first few weeks of life (76). However, the mice that did survive
were fertile. Subsequent studies also revealed fibrotic cardiomyopathy, vascular
abnormalities and fibrosis in uterus and liver, polyp formation in stomach, enhanced
susceptibility to Helicobacter infection, almost 50% rectal prolapse rate and skin changes
resembling psoriasis (79). Δ2/Δ2 mice did not exhibit most of these phenotypic features,
but did suffer from persistent embryonic vascular structures (most notably a patent ductus
venosus) (80), microscopic fatty changes in the liver, fibrosis of the portal regions in the
liver, spleenomegaly with increase in mononuclear cell fraction (77). Unlike Δ1/Δ1 mice,
Δ2/Δ2 mice demonstrate reduced fertility, probably correlating with decrease ovarian
germ cells (81) and reduced pre-antral and antral follicles (82). However, all three AHR
knock-out models do share growth retardation and a smaller liver size as compared to
wild-type mice. Also, all three models are resistant to teratogenic and toxic effects of
TCDD (78, 83). The reason for differences in the observed phenotypes is not evident,
though genetic variation and environmental factors have been proposed (84). An AHR
and NRF2 double knock-out mouse has also been generated to study the interaction of
these two receptors in regulation of phase II xenobiotic metabolism enzymes (85).

18
1.4.2 Other transgenic AHR mouse models:
In addition to the knock-out approach, several other models have employed
different AHR manipulations in an attempt to delineate the AHR’s contribution to
cellular processes. Poellinger and co-workers used a previously engineered constitutively
active AHR (CA-AHR) mutant (deletion of amino acids 288-421 involving the ligand
binding domain) (86) and subcloned it to express under the mouse Ig heavy chain intron
enhancer and a modified simian virus 40 promoter. Transgenic mice were created by
pronuclear injection into fertilized C57BL/6 × CBA eggs and backcrossed for two
generations to C57BL/6 strain (87). The CA-AHR was expressed in liver, lung and
stomach, in addition to the expected expression in thymus and spleen. These mice
demonstrated cystic tumors in the glandular portion of the stomach and exhibited
histological resemblance to hamartomas and intestinal metaplasia. Males suffered a
higher frequency as well as severity of tumors. Another noticeable trait was a significant
reduction in longevity, with most mice not surviving beyond 12 months (87). Later, these
mice were crossed with C3H/He mice and the offspring were treated with N-
nitrosodiethylamine (single intraperitoneal injection at six weeks). After thirty-five
weeks, the CA-AHR mice demonstrated a significantly higher burden of liver tumors. A
comparison of gene expression profile revealed down-regulation of heat shock proteins in
CA-AHR mice liver, though a definite link was not established with the observed
phenotype (88).

19
Another model involved the study of an AHR mutant incapable of nuclear
translocation and DRE-binding. As discussed, the AHR possesses a bipartite NLS
sequence within the basic region that overlaps the DRE-binding domain. Arg, His and
Arg were changed to Ala, Gly and Ser at the 37-39th positions by PCR in a 15 kb region
from the 129SvJ genome that was homologous to the AHR sequence surrounding its
second exon. The mutated fragment was introduced in GS1 embryonic stem cells for
homologous recombination. Targeted clones were injected into C57BL/6 blastocysts and
subsequently backcrossed to Ahrd allele bearing C57BL/6 mice. The resulting Ahrnls/nls
mice behaved exactly like the Ahr-/- mice with respect to TCDD toxicity and
teratogenicity as well as AHR-DRE mediated gene expression. However, Ahrnls/nls mice
had normal fertility, unlike the Ahr-/- mice (89).
Bradfield and co-workers described vascular malformations (ductus venosus) in
Ahr-/- mice (80). In an effort to determine whether AHR activation during embryogenesis
could rescue this vascular aberration, they generated AHR- and ARNT-hypomorphic
mice (90, 91). A 15 kb region homologous to the Ahr locus was modified to introduce a
neomycin cassette and flank exon 2 as well as the neo cassette with loxp sites.
Ahrfxneo/fxneo (hypomorphic AHR) mice were generated using a strategy similar to that of
Ahrnls mice. AHR protein expression as well as its transcriptional activity in these
hypomorphic mice was approximately 10-15% of the wild-type. Vascular anomalies in
the Ahrfxneo/fxneo mice mimicked those of Ahr-/- mice, but were completely prevented by
treating pregnant mice with dioxin. This result clearly establishes a role for AHR in
embryonic vascular development.

20
Persistent ductus venosus in mice lacking a functional AHR could be due either to
AHR function in the hepatocytes or in endothelial cells. Bradfield and co-workers
employed Cre-lox technology and derived conditional AHR knock-out mice to determine
the answer. First, the Ahrfxneo/fxneo mice were crossed to a transgenic line that expresses
Cre under the EIIa promoter to remove the neo cassette. Next, the Ahrfxneo/+CreEIIa mice
were backcrossed to C57BL/6 strain and then, again crossed to the hepatocyte specific
CreAlb mice or the endothelial cell specific CreTek mice. Finally, the animals were
backcrossed again to the C57BL/6 strain. Liver angiography demonstrated that ductus
venosus was only observed in the endothelial cell-specific AHR knock-out mice
(Ahrfx/fxCreTek) and not in the hepatocyte-specific AHR knock-out mice (Ahrfx/fxCreAlb).
However, other AHR expression in hepatocytes was found to mediate other aspects of
TCDD toxicity (92).
Since the mouse and human AHR differ in their properties, including ligand-
binding and the C-terminal domain, attempt has been made to generate a mouse model
that expresses the human AHR (hAHR). Human Ahr cDNA expressed under the control
of 129SvJ mouse Ahr promoter, was introduced in E14 embryonic stem cells for
homologous recombination. Ultimately, the hAHR knock-in mice were backcrossed to
C57BL/6 strain. The authors concluded that hAHR knock-in mice are resistant to the
transcriptional and teratogenic effects of TCDD (93). However, the report does not
demonstrate AHR protein expression.

21
1.4.3 Biosensor mouse models based on AHR:
Gene transcription is often used as the end-point for determining the suitability of
a compound as an AHR ligand. However, in vivo experiments require sacrificing the
animal and thus limit the range of possible experiments. A mouse model with an easily
measurable marker under AHR regulation can serve to detect and study potential ligands.
To this end, a transgenic mouse on C57BL/6 background, with a DRE-driven secreted
alkaline phosphatase (SEAP) has been generated. Upon oral administration of AHR
ligands, only TCDD elicited a sustained (>40 days) serum SEAP activity. The mice also
demonstrated reliable increase in SEAP upon exposure to cigarette smoke (94).
1.5 AHR Regulated Genes:
Gene regulation is the most well characterized function of AHR. In fact, until
recently, the majority of efforts in the field of AHR biology have been focused on
understanding the details of how the ligand-activated AHR enhances transcription of its
target genes. The classical AHR-gene battery comprises of xenobiotic metabolizing
enzymes. AHR has been shown to regulate the expression of enzymes involved in both
Phase I as well as Phase II metabolism reactions. These enzymes serve to modify
chemical groups on xenobiotics with an intention to render them more polar and thus,
easy to excrete. Regulation of these enzymes signifies the role of AHR in metabolic
adaptation. A third group of proteins that are involved in the efflux of PhaseI/II substrates
have been ‘unofficially’ labeled as Phase III. AHR has also been found to regulate some

22
members of this group. Unlike many other transcription factors, the cognate sequence
recognized by AHR – the dioxin response element (DRE) – is relatively well defined.
This, and the use of AHR-specific ligand TCDD, provides a relatively simple model to
define whether a gene is a direct AHR target or not.
This section is devoted to AHR-mediated gene regulation. After a brief overview
of the established AHR target genes, the xenobiotic metabolizing enzymes are presented.
A significant number of other genes have been described to be directly regulated by
AHR. Most of these have been identified from high-throughput screens. These novel
target genes are unrelated to xenobiotic metabolism and are expressed in a variety of
tissues, suggesting a broader role for AHR in cellular processes. Unfortunately, in many
cases, following the initial characterization, these target genes have not been further
researched. In addition to the directly regulated genes described here, AHR has been
found to interact with other signaling pathways, such as the estrogen receptor and NF-κB,
and influence the expression of genes regulated through those pathways. Such ‘co-
regulated’ genes have been discussed in the respective sections. Since the mechanism of
AHR-mediated gene repression has not been established, down-regulated target genes
have been discussed in detail. Genes whose regulation most likely involves the
prototypical AHR-DRE pathway are summarized in a tabular format.

23
1.5.1 Phase I and Phase II enzymes:
Cyp1a1: Cytochrome P450 family 1 subfamily A member 1 (Cyp1a1) is a
monooxygenase localized to endoplasmic reticulum. It is the prototypical AHR target
gene and induction of this gene has been extensively utilized as an end-point to study
various aspects of AHR activation. In rodents as well as humans, well-characterized
DREs have been described in the promoter/enhancer regions of the Cyp1a1 gene. Unlike
Cyp1b1, Cyp1a1 expression is almost exclusively regulated by AHR and this provides a
simple model to assess AHR transcriptional activity. Cyp1a1 is expressed primarily in the
liver and lung tissues and is also believed to function in the intestine (95). Cyp1a1-null
mice demonstrated significantly higher sensitivity to oral benzo[a]pyrene than Cyp1a2-
null or Cyp1b1-null animals. Due to its involvement in detoxification of polycyclic
aromatic hydrocarbons, it is expected that Cyp1a1 imparts a protection against chemical-
induced carcinogenesis. A specific African-American Cyp1a1 polymorphism has been
associated with adenocarcinoma of the lungs in individuals who smoke (96). A second
polymorphism has also been associated with breast cancer risk in African-American
women (97).
Cyp1a2: Cyp1a2 expression is primarily restricted to the liver and of all the Cyp1
family members, Cyp1a2 has been associated with the metabolism of common drugs
more than others. Drugs like omeprazole, phenytoin and rifampin induce Cyp1a2 activity,
while others like quinolone antibiotics and fluvoxamine (antidepressant) inhibit its
activity. Cyp1a2 metabolizes many anti-psychotic and anti-depressant drugs, theophylline

24
(broncho-dilator) and warfarin (anti-coagulant). Since some of these drugs have a narrow
therapeutic index, co-administration of a Cyp1a2 inhibitor can elevate the plasma levels
of these drugs and unexpectedly result in adverse reactions. Cyp1a2 also metabolizes
caffeine, an ingredient commonly found in hot beverages.
In humans, Cyp1a1 and Cyp1a2 are located on the same chromosome within 25
kb of each other. Functional DREs have been identified individually for both genes.
Interestingly, an attempt to reassess the regulation of these genes revealed that the DRE
cluster closer to Cyp1a1 can function bidirectionally to regulate Cyp1a2 as well (98).
Cyp1b1: Cyp1b1 is expressed in extra-hepatic tissues in humans as well as
rodents. Cyp1b1 can also be regulated by signals other than AHR activation. Primary
congenital glaucoma has been linked to Cyp1b1 polymorphisms (99), suggesting that in
addition to phase I metabolism, Cyp1b1 is also involved in developmental processes.
Cyp2s1: Dioxin induces Cyp2s1 in mice through three upstream DREs.
Chromatin immunoprecipitation (ChIP) assays demonstrated presence of AHR-ARNT
heteromer at Cyp2s1 promoter. Interestingly, another ARNT based heteromer, hypoxia
inducible factor-1 (HIF-1)-ARNT, can also upregulate Cyp2s1 by binding the same
regulatory promoter region.
Cyp2a5: Cyp2a5 is expressed in liver, kidney and various tissues of the
respiratory tract. Using TCDD in primary hepatocytes and 3-MC in vivo, Arpiainen and

25
co-workers reported induction of Cyp2a5 upon AHR activation (100). Induction of
Cyp2a5 varied with the ligand-affinity of AHR in C57BL6 and DBA/2 mice. The
increase in functional activity of Cyp2a5 was assessed by a coumarin 7-hydroxylation
assay. Reporter assays with a 3 kb Cyp2a5 promoter construct identified a functional
DRE. However, mutation of this DRE did not completely abolish reporter activity.
Subsequently, the same group published another report claiming that Cyp2a5
transcription can be controlled by the binding of ARNT homodimers to an E-box site in
the promoter. Unlike other AHR-ARNT regulated genes, ARNT transactivation domain
was required for Cyp2a5 transcription (69). CYP2A13 is the human homologue of
murine Cyp2a5.
AHR also regulates various Phase II enzymes such as NAD(P)H menadione
oxido-reductase 1 (NQO1), glutathione S-transferase A2 (GSTA2), UDP
glycosyltransferase 1 family, polypeptide A1 (UGT1A1), UGT1A6 and aldehyde
dehydrogenase 3 family, member A1 (ALDH3A1), as reviewed in (101). This clearly
establishes a pivotal role for AHR in regulating xenobiotic metabolism.
1.5.2 Other AHR regulated genes:
NRF2: NF-E2 p45-related factor (NRF2) is a transcription factor that is activated
by electrophilic compounds, binds antioxidant response elements (ARE) and regulates
phase II metabolism enzymes. The gene battery of NRF2 and AHR overlap and probably
this led investigators to hypothesize that these two transcription factors might

26
functionally interact. TCDD treatment induced NRF2 protein as well as Nrf2 mRNA in
Hepa1c1c7 cells. This induction was lost upon AHR-knockdown by siRNA oligos. ChIP
assays demonstrated recruitment of AHR to three imperfect DRE sequences found in the
putative Nrf2 regulatory region (102). Conversely, another report demonstrated that
NRF2 can regulate constitutive expression of AHR. Pharmacological activation of NRF2
induced Ahr, Cyp1a1 and Cyp1b1 mRNA in mouse embryonic fibroblasts. NRF2 binds
an ARE located 230 bp upstream of Ahr transcription start site (103). Both these studies
were performed using murine models and further studies are required to verify this effect
in humans. It has also been suggested that AHR-mediated CYP1A1 upregulation can lead
to an increase in electrophiles in the cell, which in turn can activate NRF2 (104).
EGR1: Early growth response 1 (EGR1) was identified as a differentially
regulated gene in by toxicogenomic approaches in human lung epithelial cells (105).
Subsequently, it was proposed that TCDD prolongs the half-life of EGR1 mRNA rather
than direct transcriptional induction. However, this conclusion was loosely based on the
inability of TCDD to drive expression from reporter plasmids (106).
c-myc: Two different reports have been published on the effect of AHR on c-myc
expression in human mammary epithelial cell-lines. The first report demonstrated that
RELA (p65) subunit of NF-κB and AHR positively interact to induce c-myc RNA (107).
AHR and RELA physically interact with each other and increase reporter activity from a
c-myc promoter construct in a dose-dependent manner by forming a novel transcriptional
complex that binds the κB element in the c-myc promoter. Most experiments required

27
over-expression of AHR and RELA, and an increase in c-myc protein was not
demonstrated. The second report from the same laboratory claimed that AHR
constitutively represses c-myc transcription in the same cell-lines (108). Five DRE-like
and one DRE are present in the 3.2 kb promoter of human c-myc gene. Reporter assays
using wild-type and mutated constructs revealed that at least two response elements are
functional. Inhibition of constitutive AHR activity by transient expression of AHR-
repressor (AHRR) led to an increase in c-myc RNA and protein. These results provide
contrasting roles of AHR at the same promoter and should be explored further in primary
cells.
MHC Q1b: Major histocompatibility complex Q1b is a non-classical class I MHC
whose function is not well defined. MHC Q1b was identified by differential display as the
sole TCDD-responsive gene that was down-regulated in Hepa1c1c7 cells. A relatively
low dose TCDD treatment (100 pM) repressed MHC Q1b RNA by sixty percent after 16
h. AHR dependence was verified by transfecting Hepa1c1c7 cells with a dominant
negative AHR mutant (R39A) that is capable of heterodimerization, but incapable of
binding DNA. Interestingly, when cells were treated with the translational inhibitor
actinomycin D, MHC Q1b RNA levels remained unchanged even at 12 h (109). Although
microRNA concept was not prevalent at that time, these results strongly suggest AHR
mediated microRNA upregulation as a potential mechanism for MHC Q1b down-
regulation.

28
T-cadherin: T-cadherin is an atypical member of the cadherin family of adhesion
molecules that is abundantly expressed in heart and vascular tissues. A DRE-like element
is present in the 5` untranslated region (UTR) of rat, mouse and human T-cadherin genes.
However, it was not determined if this DRE was functional in TCDD-mediated down-
regulation. Vascular smooth muscle cells (VSMC) obtained from Wistar Kyoto rat aortas
showed a decrease in T-cadherin RNA after 20 h of AHR activation. Notably, the cells
had to be treated with high doses of AHR ligands (75-100 nM TCDD or 10-30 µM
B[a]P). AHR antagonism by α-naphthoflavone pre-treatment abolished T-cadherin down-
regulation. However, cycloheximide and actinomycin D treatment had no effect on
TCDD-mediated T-cadherin regulation (110).
Dystrophin Dp71: Dystrophin is a 427 kDa cytoskeletal protein, the dysfunction
of which leads to Duchenne muscular dystrophy, an X-linked inherited disorder. The
Dp71 isoform is expressed in multiple tissues and bears four DRE-like motifs in its
promoter. AHR activation by 50 µM β-naphthoflavone (BNF) for 24 h repressed Dp71
protein by forty percent in Hepa1c1c7 cells. This effect was also observed at 24 h in mice
injected with 100 mg/kg BNF (111).
Spp1: Secreted phosphoprotein 1 (commonly known as osteopontin) expression
was found to be negatively regulated in the tissues of transgenic mice expressing a
constitutively active AHR mutant. These mice have an increased incidence of gastric
tumors. Interestingly, immunohistochemistry revealed that the suppression of osteopontin

29
was confined to the corpus portion of stomach and this correlated with tumor occurrence
(112).
In addition to the genes discussed above, a number of other genes have been
described to be induced by AHR. These target genes are enlisted in Table 1.3 along with
the species, cell-type and significant experimental details.

30
Table 1.3: Genes regulated by the classical AHR pathway. Gene Function Species /
Cell-type Experiments to demonstrate AHR dependence
Ref.
Cycloxygenase-2 (a.k.a. Prostaglandin G/H synthase 2
catalyzes the conversion of arachidonic acid products to prostaglandin
Rat insulinoma, human breast cancer cells, Human lung fibroblasts, murine lymphatic tissue
Reporter assays, chemical AHR antagonists
(113-116)
Small inducible cytokine A1 (CCL1)
chemotactic for monocytes but not for neutrophils
Primary human macrophages, Murine lungs
siRNA, chemical AHR antagonists, ChIP assay
(117)
Suppressor of cytokine signaling 2 (SOCS2)
part of a classical negative feedback system that regulates cytokine signal
Murine B-cell lymphoma cells (CH12.LX)
AHR-deficient B cells
(118)
Paraoxonase1 (PON-1) Protective role in organophosphate poisoning and cardiovascular diseases
Human hepatoma cell-line (Huh7), Balb/c mice liver
siRNA, Quercetin activated significantly better than TCDD
(119)
DNA polymerase kappa (POLK)
DNA polymerase specifically involved in DNA repair
Murine testes No induction in AHR knock-out mice testes
(120)
Scinderin (a.k.a Adversin)
Ca(2+)-dependent actin filament-severing protein that is presumed to have a regulatory function in exocytosis
Immature thymocytes in murine thymic cortex
TCDD responsiveness correlates with ligand affinity of different mouse strains
(121, 122)
N-myristoyltransferase 2
Adds a myristoyl group to the N-terminal glycine residue
Rat hepatoma cells (5L), Murine liver
AHR-deficient BP8 cells
(123)
Gulonolactone oxidase enzyme for ascorbic acid biosynthesis
Murine liver Induction varied with mouse strains correlating with ligand affinity of AHR
(124)
Slug Transcriptional repressor Human keratinocytes cell-line (HaCaT)
ChIP assays, siRNA
(125)
Hairy and Enhancer of Split homolog-1 (HES-1)
Transcriptional repressor of genes that require a bHLH protein for their transcription
Human mammary carcinoma cells (T47D)
(126)
Plasminogen activator inhibitor-1 (PAI-1)
Mouse hepatoma cell-line
AHR-deficient hepatoma cells, AHR antagonists
(127)
Breast cancer resistance protein (BCRP)
may function as a major control point in the regulation of fibrinolysis
Human intestinal cell line (Caco-2)
AHR antagonists (128)

31
1.6 Potential physiological roles of AHR:
Though AHR is best known for its regulation of enzymes involved in xenobiotic
metabolism, several arguments can be made for the physiological importance of the
receptor. AHR orthologues can be traced across the animal kingdom to worms and flies.
This argues against xenobiotic metabolism as being the sole reason for AHR’s existence,
as the evolutionary pressures could not have been the same during pre-historic times and
throughout animal taxa. In the last couple of decades, interest in identifying the ‘true’
physiological function of AHR has significantly increased amongst the researchers in this
field. Development of a variety of mouse models and the availability of high-throughput
genomic and proteomic technologies has certainly inspired this quest. Though a
physiological function has not been precisely outlined for AHR, various lines of
evidence, as discussed below, that indicate a cellular role for AHR are emerging. Most of
these potential physiological roles have been inferred from the phenotypes observed in
AHR knock-out mice and, ironically, from TCDD toxicity studies. Certain aspects of this
topic are elaborated further in the next section.
1.6.1 Reproduction:
As discussed, AHR knock-out mice demonstrate various levels of impairment in
reproduction. AHR-knock out males apparently posses the same potency as wild-type
males, though studies are required to confirm this observation. In sexually mature mice
(8-11 week old), AHR is most abundantly expressed in the oocytes (129). Studies have
revealed a significant decrease in the efficiency of follicle maturation. This inefficiency is

32
due to inadequate levels of estrogen synthesis in the ovary and not due to alterations in
the pattern or levels of gonadotrophins (130, 131). Administration of exogenous estrogen
partially rescued this phenotype. Ovaries from AHR knock-out mice weigh significantly
less as compared to their wild-type counterparts, in proportion to total body weight. Also,
the estrous cycle of knock-out mice was found to be dysregulated. Further exploration
identified cooperative regulation of Cyp19 gene by AHR and Ad4BP/SF-1 through DRE-
binding (130). Cyp19 enzyme is essential for estradiol synthesis during folliculogenesis.
AHR has also been shown to participate at other stages in successful reproduction,
including dynamic tissue changes during blastocyst implantation (132) and development
of lactation structures in the mammary gland (133).
1.6.2 Cardiovascular:
AHR knock-out mice demonstrate a decrease in liver weight and this is most
likely due to the persistence of a fetal vascular shunt, ductus venosus, resulting in an
inadequate blood supply. These effects are observed in the absence of exogenous AHR
activation, indicating that AHR might function to ensure proper vascular development. In
addition to the ductus venosus anomaly, AHR knock-out mice also demonstrate vascular
abnormalities in the kidney and the eye (persistent embryonic hyaloid artery) (80).
Though these effects are labeled as ‘vascular’, they might simply be the result of AHR’s
involvement in cell proliferation and apoptosis, the implications of which would
obviously be most noticeable in the development of blood vessels.

33
Adult AHR knock-out mice also demonstrate other effects on the cardiovascular
system such as cardiac hypertrophy and hypertension (79, 134, 135). However, the
anatomical cardiac lesions did not demonstrate the expected changes in molecular
biomarkers (135). Elevated arterial pressures in AHR knock-out mice were accompanied
by increased levels of two potent vasoactive peptides – angiotensin II and endothelin-1.
Treatment of these mice with captopril, a commonly used antihypertensive, improved
some aspects of cardiac functions (136).
1.6.3 Development:
Experiments performed in C.elegans and D. melanogster indicate that AHR and
ARNT orthologues (AHR-1 and AHA-1 in C. elegans, and Spineless and Tango in D.
melanogaster) perform important functions in developmental processes such as neuronal
differentiation, appendage development and regulation of photoreceptor mosaic
(reviewed in (137)). Even in mammalian systems, AHR contributes to anatomic
development and cell differentiation. Increased incidence of hydronephrosis, tortuous
ureters and a reduction in kidney size was noticed in TCDD treated wild-type mice, but
not in AHR knock-out mice (83). Wilms’ tumor suppressor gene (Wt1) regulates
mesenchymal-to-epithelial transition during nephrogenesis. AHR activation leads to an
abnormal shift in the relative expression of different splice variants of Wt1, which in turn
perturbs gene expression during renal cell differentiation (138). AHR activation in
undifferentiated neuronal cells induces formation of specialized axon-like structures and
alters molecular milieu to resemble catecholinergic neuron-like properties (139). A

34
previous study also showed that β-naphthoflavone exposure inactivated STAT3 mediated
transcription, resulting in inhibition of astrocytic differentiation of C6 glioma cell-line
(140). However, this study did not conclusively demonstrate the requirement of AHR in
this process.
1.6.4 Endocrinal homeostasis:
Owing to the fact that AHR can be activated by a variety of structurally diverse
compounds, any influence that AHR activation might have on other hormonal signaling
pathways would be interesting. Consequently, a number of groups have investigated the
effects of PAH and HAH on disruption of other hormonal systems, especially the
estrogen, thyroid and retinoic acid pathway. In many cases, the absolute requirement of
AHR has not been assessed, making it difficult to isolate AHR-independent effects of
aromatic hydrocarbons from the AHR-mediated ones. However, TCDD exposure of wild-
type and AHR knock-out animals demonstrated that AHR was essential for TCDD-
mediated reduction in thyroid hormone levels (141). In the same study, TCDD-mediated
reduction in liver retinoid content was also found to be AHR dependent.
Besides the preceding overview of potential physiological AHR functions, a
number of studies have examined the effect of constitutive as well as induced activity of
AHR on cell proliferation (reviewed in (142)). However, the evidence is controversial
and the only conclusion that can be drawn at this time is that AHR’s influence on cell
growth is context-specific. Also, an increasing amount of evidence implicating the

35
influence of AHR on immune system is accumulating. AHR seems to cross-talk with
inflammatory signaling pathways and, as a result, this aspect of AHR biology is
discussed in the subsequent section.
1.7 Interaction of AHR with other signaling pathways:
Nuclear receptors and other transcription factors are known to regulate the
expression of their target genes though direct association with their cognate response
elements in response to activation signals. The discovery and characterization of these
relatively straight forward signaling pathways has served to identify the molecular
phenomena for many biological processes. However, it would be naïve to expect these
independent signaling cascades to illuminate the details of complex homeostatic
physiology. Since organisms are simultaneously exposed to a variety of stimuli, an
interaction, or cross-talk, between different signaling systems is inevitable. In fact, this
interaction is more likely to dictate biological outcomes, rather than activation of
individual transcription factors. The advent of high capacity molecular biology tools has
inspired investigators to probe the effects of cross-talk between different receptors.
AHR also interacts with other transcription factors. Perhaps the most studied
interactions of AHR are with the estrogenic and inflammatory signaling, which are
described in further detail below. In addition, AHR communicates with transforming
growth factor-beta (TGF-β) driven developmental processes. All three isoforms of TGF-β

36
are altered either by the constitutive presence of AHR or via its ligand-dependent
activation (143-145). This alteration is unlikely to be mediated via a DRE-dependent
mechanism, as the TGF-β promoters lack a canonical DRE. Though the mechanistic
aspects of this cross-talk have not been resolved as yet, it is possible that AHR mediated
plasminogen activator inhibitor-2 (PAI-2) induction (146) can lower TGF-β levels. It has
also been proposed that strong exogenous activation (TCDD exposure) or an absolute
deficiency of AHR can lead to increased all-trans-retinoic acid levels, and thus enhanced
retinoic acid receptor/ 9-cis-retinoic acid receptor (RAR/RXR) signaling (147, 148). In
fact, feeding a vitamin A deficient diet to AHR knock-out mice attenuated liver fibrosis
and elevated TGF-β levels (149), suggesting that accumulation of retinoids in AHR-
deficient mouse livers plays a role in the observed abnormalities. Decreased levels of
Cyp2C39, a potential AHR target gene, in AHR knock-out mice might partially explain
this accumulation of retinoids (150). Further experiments are required to fully elucidate
the details of this cross-talk.
1.7.1 AHR and estrogen signaling:
Interaction of AHR and estrogen receptor (ER) has been an interesting aspect of
AHR function. In a cell-type specific manner, AHR has been shown to modulate ER-
mediated gene induction. Cathepsin D in MCF-7 (human mammary tumor) cells, pS2 in
MCF-7, HeLa (human cervical cancer cells) and Hepa1c1c7 (mouse liver tumor) cells
(151) and heat shock protein 27 in MCF-7 cells (152) are some examples of ER target
genes whose induction by 17β-estradiol is inhibited by TCDD. The concept of inhibitory

37
DRE (iDRE) has been favored by some research groups to explain the inhibitory effect of
AHR (153). Additional proposed mechanisms of repression include metabolism of
estrogen (17β-estradiol) by cytochrome enzymes induced via AHR (154) and
proteosomal degradation of ER by AHR (155, 156). Based on the most recent report,
AHR seems to perform a non-genotropic task by acting as an E3 ubiquitin ligase in a
cullin 4B complex (156) to facilitate degradation of specific target proteins such as ER.
AHR-ER interaction studies are further complicated by the fact that many AHR ligands
themselves possess estrogenic properties, most recently revisited by Abdelrahim et.
al.(157). Thus, multiple mechanisms might be responsible for AHR mediated ER
repression.
Interestingly, in addition to interaction of the two pathways upon activation with
respective ligands, AHR activation can even recruit unliganded ER, along with the
coactivators p300, to estrogen response elements (ERE) to drive gene transcription (158).
In the same report, it was demonstrated that AHR activation also correlates with
estrogenic effects of AHR activation in ovarectomized mouse uteri, such as increased
uterine weight and enhanced proliferation of glandular epithelium. Along the same line,
another report described that ER activation leads to recruitment of unliganded AHR to
ER target gene promoters, specifically breast cancer 1, early onset isoform 1 (BRCA-1).
However, upon providing AHR ligand, the activating complex transforms to a inhibitory
complex (159). Moreover, ARNT – the heterodimerization partner of AHR, can
independently function as a coactivator for estrogen-activated ER (160) by associating
via its C-terminal domain. C-terminal domain of ARNT is dispensable for the functioning

38
of AHR-ARNT heteromer. However, the requirement of AHR in this process was not
investigated adequately. Even, an isoform of ARNT, ARNT-2, was able to coactivate ER.
Though the effect of AHR activation on ER-driven transcription has been highly focused
on, it is also known that in response to estrogen treatment ER can transrepress AHR
target gene expression (161). ER mediates its repressive effects by physically associating
with the AHR-ARNT complex at Cyp1a1/Cyp1b1 promoters, as demonstrated by
successive ChIP assays. Similar to the AHR-ER transcriptional interference, cross-talk
has also been demonstrated between dioxin and testosterone signaling pathways in
prostate cancer cells (162) and in vivo (163).
1.7.2 AHR and inflammatory signaling:
Due to its immense clinical significance, inflammation and immunity-related
processes have been widely examined for interaction with numerous surface and soluble
receptors. An especially favorite topic has been the effect of glucocorticoids on
inflammatory pathways, resulting in identification of therapeutic avenues to regulate
‘over-expression’ of inflammation. Other nuclear receptors, such as ER, progesterone
receptor (PR), androgen receptor (AR) and peroxisome proliferator-activated receptor-
alpha and -gamma (PPARα and PPARγ) are also known to influence NF-κB driven
transcription (164, 165). Inflammatory signaling involves activation of multiple
interconnected pathways with several possible substitutions at different steps in these
pathways, resulting in an extremely intricate and yet unresolved network of molecular

39
switches. In general, the signaling can be initiated by a wide variety of
cytokine/chemokine molecules or exogenous entities like lipopolysaccharide (LPS).
These molecules bind their respective receptors leading to a coordinated and sequential
recruitment/post-translational modification of adaptor proteins. These changes activate
one or more of many transcription factors involved in inflammation – NF-κB, C/EBP,
STAT, AP-1 or IRFs. These transcription factors translocate to the nucleus and bind their
response elements in the promoters of their target genes. Two critical components that
have drawn wide attention lately are cofactor exchange (166) (swapping of repressors for
activators) and modification of histone code (167). Different nuclear receptors utilize
separate methods of cross-talk with inflammatory signaling and this makes it difficult to
delineate the precise mechanisms.
A bilateral transcriptional interference also exists between AHR and inflammation
associated transcription factors. Inflammatory cytokines are capable of altering
expression of cytochrome P450 genes (168, 169). Specifically, IL-1β, IL-6 and TNF-α
have been shown to downregulate Cyp1a1 expression (170, 171). RELA subunit of NF-
κB can physically bind to AHR and can also deacetylate histones at the Cyp1a1 promoter
(172, 173). Conversely, with the help of a ‘triple cytokine receptor-null’ mouse model, it
has also been proposed that IL1-like cytokines may in fact contribute to certain aspects of
dioxin toxicity (174). It is interesting to note that investigations have focused
significantly more on communication of nuclear receptors with NF-κB as compared to
that with CEBP, STAT or AP-1 pathways. Owing to the relative simplicity of AHR
signaling and the lack of a defined physiological function for AHR, the ‘NF-κB affecting

40
AHR’-arm of the crosstalk is relatively simple and perhaps of less clinical relevance than
its counterpart.
The effect of AHR activation on inflammatory signaling has been of greater
interest and has been more widely explored. AHR activation perturbs inflammatory
proceedings at the molecular as well as systemic levels. Figure 1.3 is a simplified
schematic of NF-κB pathway demonstrating the steps at which AHR can possibly act to
influence inflammatory signaling. A number of studies have revealed that AHR
activation can alter cytokine levels and the downstream signaling. B[a]P, an AHR ligand
found in cigarette smoke, can induce IL-8 in primary human macrophages in an AHR
dependent fashion through a DRE found in IL-8 promoter (175). The same group has also
demonstrated the chemokine CCL1 to be an AHR target gene (117). CCL1, along with
other cytokines – B-cell activating factor of TNF family (BAFF) and B-lymphocyte
chemoattractant (BLC), has also been claimed to be an AHR target in another study
(176). TCDD exposure of murine fetal thymus cultures leads to increased IL-1β, IL-6 and
TNF-α, though the requirement of AHR for this response remains to be firmly established
(177). In addition to direct induction of cytokine transcription, AHR activation can also
increase IL-1β expression by prolonging IL-1β mRNA half-life, though the precise
mechanism has not been evaluated (178). Recently, IL-22 was also found to be
upregulated upon AHR activation (179).Contrary to the AHR-mediated induction of
cytokine expression observed in the above studies, AHR has also been reported to repress
cytokine levels under certain circumstances. TCDD pre-treatment attenuated IL-6
induction by LPS in bone marrow stromal cells (180). In fact, AHR mediated inhibition

41
of IL-6 expression could present therapeutic opportunities (181). Wild-type mice (AHR
present) generate less IL-12 and IL-10 than AHR knock-out mice, upon challenging with
Listeria monocytogenes (182). Hence, AHR exerts a complex effect on cytokine
expression that is highly cell-type and context-specific. Further studies are required to
better understand the consequence of AHR activation on different cytokines. Finally,
AHR can also regulate the expression of inflammatory mediators other than cytokines,
such as arachidonic acid derivatives – prostaglandin E2 – and the enzymes involved in
arachidonic acid metabolism, for example cyclooxygenase-2 (113, 183). Thus, regulating
cytokine levels might be a critical part of AHR-inflammation cross-talk.

42
Figure 1.3: NF-κB pathway and the possible levels at which AHR can exertrepression. Under basal conditions, NF-κB resides in the cytoplasm in association with IκB-containing inhibitor complex. Inflammatory molecules (A) bind to their receptors (B) andthis initiates a cascade of protein recruitment and post-translational modifications (C) which result in degradation of IκB and release of NF-κB (D). NF-κB itself undergoes acetylation (Ac) and phosphorylation (P) (E) to assemble a coactivator complex at thepromoter to drive transcription of its target genes (F). This is facilitated by acetylation ofhistones (gray circles on the DNA) at the promoter (G). Disruption of any of these steps(A-G) by AHR could lead to repression.

43
In addition to altering cytokine levels, AHR can operate at various steps in the
intracellular part of the inflammatory network. Altering absolute protein levels or the
subcellular localization (cytoplasmic vs. nuclear) of inflammatory transcription factors
would be an obvious way to effectively regulate inflammation. TCDD can induce
prostaglandin endoperoxide H synthase-2 in rat hepatocytes by activating AHR and by
increasing C/EBP levels (184, 185). The effect of TCDD on AP-1 family members has
been shown to be cell-type specific – induction in liver derived cells (186), but repression
in immunological cells (187). In DC2.4, a dendritic cell-line, TCDD exposure resulted in
decreased levels of transcriptionally active RELA/p50 dimers, while transcriptionally
repressive p50 homodimers were unaffected (188). In a macrophage cell-line, AHR
activation induces interferon gamma responsive factor-3 (IRF-3) through an AHR/RELB
complex bound to NF-κB element in the IRF-3 promoter (176). AHR may have a
protective effect on RELB, as AHR deficient mice demonstrate rapid degradation of
RELB resulting in a enhanced inflammatory response (189). Though regulation of NF-κB
and other transcription factor levels can provide a simple explanation, it is unlikely to be
the major component because of the gene-specificity of AHR-mediated suppression. The
selectivity of transcriptional repression can be more rationally explained by a model
wherein one receptor alters post-translational modification of the other receptor and/or
the chromatin remodeling at the specific gene promoter. One of the very few studies
analyzing such effects demonstrated that B[a]P mediated activation of MAP kinase
ERK1/2 in primary human macrophages was responsible for induction of TNF-α (190).
Similarly, coactivators p300/CBP and steroid receptor coactivator-1 were found to
attenuate NF-κB mediated repression of AHR driven reporter activity (191). The same

44
study also showed that TNF-α can inhibit AHR-ligand induced acetylation of histone H4
at the Cyp1a1 promoter thereby exerting repression of AHR regulated genes. AHR and
NF-κB/cytokines may also cooperate with each other through physical and functional
interaction, as in the induction of c-myc expression in breast cancer cells (107) and
induction of cyclin A in rat liver progenitor cell model (192). These reports again
emphasize that the AHR-inflammatory pathways interaction is highly context-specific.
The multitude of effector cells – T cells, B cells, macrophages, dendritic cells,
granulocytes, etc. and their subtypes – make the immune system highly versatile. A
balance between different cell types is critical during development as well as under
pathological circumstances. At the systemic level, AHR can alter immune/inflammatory
outcomes by perturbing this balance in immune cell-types, affecting immune-related
disease processes. In humans and mice, AHR activation increases T(H)17 T cells, which
might be responsible for elevated severity of autoimmune encephalomyelitis in wild-type
mice as compared to AHR knock-out mice (179). AHR activation by TCDD elevates
macrophage infiltration in numerous organs including liver, lung and heart, through
induction of monocyte chemoattractant protein-1 (MCP-1) and keratinocyte
chemoattractant (193). AHR-dependent induction of MCP-1 in murine (ApoE knock-out
mice) and human (primary endothelial cells) models has also been linked to rapid
progression of atherosclerosis (194). Two studies examining the role of AHR in
artificially induced infections in mice revealed that the constitutive presence (182) as well
as ligand activation of AHR (195) reduced pathogenic burden and improved survival.
When a transgenic mouse model expressing the constitutively active form of AHR in

45
keratinocytes was examined, severe skin lesions resembling allergic dermatitis developed
(196). However, at this time, it is not clear whether the effects described above are simply
a consequence of AHR-mediated alteration in cytokine levels, or are due to other
additional effects of AHR on inflammation-responsive gene transcription.
Contrary to the inflammation-supportive roles of AHR described in the previous
paragraph, some studies support an anti-inflammatory function for AHR. Prolonged AHR
activation by TCDD exposure made mice more susceptible to bacterial infection (197-
199). However, this might be due to a reduction in AHR levels by continuous TCDD
treatment. In a recent study, AHR was implicated as the receptor for a novel low
molecular weight compound capable of attenuating allergic lung inflammation (181).
Also, endotoxin exposure through inhalation causes a greater accumulation of neutrophils
as well as inflammatory cytokines in AHR knock-out mice lungs as compared to wild-
types (189). AHR may exert its anti-inflammatory effect in a different ways, one of
which could be by up-regulating expression of anti-inflammatory gene products, such as
suppressor of cytokine signaling 2 (Socs2) (118). Finally, long-term TCDD exposure in a
tumor allograft C57BL/6 mice model, led to diminished activation of CD4+ as well as
CD8+ T-cells. This effectively resulted in decreased production of type 1 cytokines and
the related antibody isotypes (200). Thus, based on the current evidence, it is very
difficult to draw a conclusion regarding the role of AHR in immune
processes/inflammation.

46
In conclusion, AHR can functionally interact with multiple signaling pathways.
This exponentially increases the scope of AHR activity and presents exciting scopes for
further research.
1.8 Overview and significance of research:
The aryl-hydrocarbon receptor (AhR) is a ligand activated transcription factor
classically associated with regulation of a battery of xenobiotic metabolizing enzymes.
The list of AhR ligands encompasses halogenated aromatic hydrocarbons (HAH) and
polycyclic aromatic hydrocarbons (PAH) as well as non-classical ligands like omeprazole
and thiabendazole. Many of these compounds are persistent and widespread
environmental contaminants of considerable concern due to their potential for adverse
health effects. There is also a growing body of evidence supporting the concept of
endogenous ligand(s) for the AhR, even though no single molecule has been identified so
far. Phylogenetics can provide vital information regarding the properties and functions of
proteins. AHR homologues are found across the animal kingdom, including mammals,
fish, avians and even invertebrates. In the invertebrates, AHR lacks an ability to bind
exogenous ligands; an observation that strongly emphasizes a role for AHR in
development and homeostasis (201).
As described in the previous sections, besides its established role in xenobiotic
metabolism, there is an increasing body of evidence implicating AhR in a myriad of

47
pathophysiological processes involving the immune system, liver and the cardiovascular
system as well as in cell cycle control. AhR activation in response to xenobiotic exposure
leads to a variety of adverse effects, including tumor promotion. A transgenic mouse
model expressing a constitutively active AhR demonstrated increased occurrence of
hepatic and gastric tumors (87, 88). The fact that benzo[a]pyrene, a known AhR ligand, is
unable to exhibit its carcinogenic effects in the skin of AhR null mice, provides further
evidence supporting a role for AhR in tumorigenesis (202). Conversely, studies using
mice deficient in AhR have provided clues to its critical role in growth and development.
AhR null mice exhibit increased hepatic fibrosis, decreased liver weight, abnormalities of
the immune system in the form of a reduced number of peripheral lymphocytes (76, 77),
decreased fertility, vascular abnormalities in heart, liver and uterus, delayed wound
healing, impaired skin homeostasis and an overall slower growth (79). These studies have
also shown that AhR might have a role in the formation of primordial follicles and in
regulating the number of antral follicles, thus affecting development of the mouse ovary
(81). Also, the incidence of cardiac hypertrophy in AhR null mice suggests a role for
AhR in normal cardiovascular development and angiogenesis (136). However, there is no
known association between most of these effects and the established role of AHR in
xenobiotic metabolism and it has not been clearly defined how AhR mediates these
effects.
Evolutionary conservation, the possibility of an endogenous ligand, implication in
developmental and pathological processes and above else, inability to explain these
phenomena, solely on the basis of DRE-driven metabolism genes, warrants the necessity

48
to search for AHR regulated genes in different ontological groups as well as alternate
models of receptor function. Sequence analysis of the human and the mouse genomes
revealed that only 48 of the 133 mouse-rat orthologous genes with a DRE between -1500
to +1500 had a human ortholog (203). Many of these putative AhR targets did not show
any change in mRNA levels as determined by microarray and real time PCR analysis. To
explain these observations a number of possibilities have been proposed, which are
partially supported by preliminary evidence. Examples of certain DRE-independent
models of AHR-regulated transcription are presented in Figure 1.4 (activation models)
and in Figure 1.5 (repression models).

49
Figure 1.4: Alternate models proposed for AHR-mediated activation of gene transcription.
(A) Classical DRE-dependent mechanism. AHR-ARNT heterodimer directly binds o the DRE to drive gene transcription.
(B) AHR-ARNT may tether to another protein to bind DNA. This could be a proteinthat cannot drive transcription on its own.
(C) In response to its activation signal, another transcription factor can bind to itsresponse element and induce its own target genes. However, ligand-activated AHR-ARNT may recruit the nonactivated form of this transcription factor to drive gene expression.
The legend key also refers to the next figure.

50
Figure 1.5: Alternate models proposed for AHR-mediated repression of gene transcription.
(A) ARNT has multiple heterodimerization partners. If the ARNT pool is limiting,activation of AHR could sequester ARNT away from other ARNT-based transcriptional complexes (left to right).
(B) DRE can overlap with other response elements. Upon ligand-activation, AHR-ARNT heterodimer would bind to DRE and inhibit DNA-binding of the other transcription factor.
(C) ‘Co-factor exchange’ model. Transcription factors recruit coactivators uponactivation (left). AHR can associate with this complex and switch coactivators with corepressors, thereby converting it to a repressive complex.
Refer to Figure 1.4 for legend key.

51
Of all the views presented, non-genotropic mode of AHR activity is the most
favored. Recently it was demonstrated that AHR could drive the expression of a reporter
by binding to a novel ‘response element’ in the enhancer region of the Cyp1A2 gene
(204). Using a DNA-binding mutant (replacing Arg39 by Ile – a mutation different from
the one proposed here), it was proved that AHR can function in the absence of its ability
to bind the DNA. Further, electrophoretic mobility gel shift assays (EMSA) revealed that
the AHR-ARNT heterodimer binds to Cyp1A2 enhancer through another factor
constitutively present in nuclear extracts from HepG2 cells. All the experiments in this
report employed the cell-culture system, the focus was on the enhancer of a single gene,
an AHR null background was not used and the bacterially expressed AHR-ARNT used
for EMSA formed aggregates. Despite the pitfalls, this report was the first description of
AHR-ARNT heterodimer modulating gene transcription through protein-protein
interactions at a gene promoter.
The concept of functional dissociation of response element dependent and
independent activities has been previously demonstrated in other receptors. Reichardt et.
al. (205) generated DNA-binding mutant glucocorticoid receptor (GR) expressing mice
by introducing a previously characterized mutation (Ala458Thr) in GR’s D-loop. This
mutant GR failed to transactivate target genes through the glucocorticoid response
element (GRE). However, even in the absence of DNA-binding, it retained
transrepression by interacting with transcription factors AP-1 and NFκB. Using these
mice, it was possible to segregate DNA-binding-dependent and -independent functions of
GR. Later, in a separate report, Bladh et. al. (206) were able to further isolate NFκB

52
repression from AP-1 repression by using a different GR mutant. Similarly, Valentine et.
al. (207) were able to discriminate between transactivation (DNA-binding-dependent)
and transrepression (DNA-binding-independent) properties of the estrogen receptor (ER)
by incorporating mutations in the receptor’s ligand-binding domain. These studies
exemplify the possibility of a functional role for transcription factors that extends beyond
DNA-binding.
To summarize, the following facts inspired this research project:
• the known battery of AHR regulated xenobiotic metabolism enzymes is unlikely
to explain many of the pathophysiological effects associated with AHR
• not all genes altered upon AHR activation possess a consensus DRE within a
reasonable region of their putative promoter
• microarray experiments reveal that many genes are repressed by activation of
AHR.
• AHR can regulate gene expression in the absence of direct DNA binding
• functional dissociation of nuclear receptor domains can help segregate their
functions, as seen in the case of GR and ER
Overall hypothesis:
AHR modulates expression of genes involved in varied cellular processes
through the classical DRE-dependent mechanism as well as mechanism(s)
independent of direct DNA binding.

53
This series of studies was designed to expand the battery of known AHR
regulated genes, and to characterize the ability of AHR to function in a DNA-independent
manner. The experiments described in Chapter 2 deal with identification of a growth
factor, epiregulin, as a classic AHR target gene. Chapter 3 describes another important
transcription factor, constitutive androstane receptor (CAR), as an AHR target gene.
However, a consensus DRE does not exist in putative murine CAR promoter. Though the
precise mechanism remains to be elucidated, CAR is unlikely to be a classical AHR
target gene. Chapter 4 provides evidence for functional dissociation of AHR domains. A
DNA-binding mutant of AHR is found to repress NF-κB mediated gene transcription to
the same extent as the wild-type AHR. Thus, this research has identified novel AHR
regulated genes as well as contributed to expanding the scope of AHR function.

Chapter 2
THE ARYL HYDROCARBON RECEPTOR DIRECTLY REGULATES EXPRESSION OF THE POTENT MITOGEN EPIREGULIN

55
2.1 Abstract:
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is known to cause a large number of
adverse effects, mediated largely by its binding to the aryl-hydrocarbon receptor (AhR)
and subsequent modulation of gene expression. It is thought that AhR mediates these
effects through the untimely and disproportionate expression of specific genes. However,
the exact mechanism, or the genes involved, through which TCDD leads to these effects
is still unknown. This study reports the discovery of a novel target gene, epiregulin,
which is regulated by TCDD-activated AhR. Epiregulin is a growth regulator which
belongs to the epidermal growth factor (EGF) family. Using real time quantitative PCR
(qPCR), it was established that TCDD upregulates epiregulin gene expression. The
promoter region of epiregulin has a dioxin responsive element (DRE) 56 nucleotides
upstream of the transcription start site, along with three potential Sp1 binding sites.
Chromatin immunoprecipitation (ChIP) assays with an anti-AhR antibody showed
promoter occupancy upon TCDD treatment. Luciferase reporter assays using a vector
harboring the first 125 base pairs of the epiregulin rat promoter revealed an increase in
signal on TCDD treatment, which was lost upon mutation of the DRE. Epiregulin and
TCDD treatment mediated a dose-dependent increase in primary mouse keratinocyte
growth. These results demonstrate that AhR directly increases epiregulin expression,
which could play an important role in TCDD mediated tumor promotion observed in
rodent models.

56
2.2 Introduction
Aside from its role in xenobiotic metabolism, there is an increasing body of
evidence implicating AhR in a diverse range of patho-physiological processes involving
the immune system, liver and the cardiovascular system (208) as well as in cell cycle
control (209). AhR activation in response to xenobiotic exposure leads to a variety of
adverse effects, including tumor promotion. Studies using mice deficient in AhR have
provided clues to its role in growth and development. AhR null mice exhibit increased
hepatic fibrosis, decreased liver weight, abnormalities of the immune system in the form
of a reduced number of peripheral lymphocytes (76, 77), decreased fertility, vascular
abnormalities in heart, liver and uterus, delayed wound healing, impaired skin
homeostasis and an overall slower growth (79). In addition, studies using null mice have
also shown that AhR might have a role in the formation of primordial follicles and in
regulating the number of antral follicles, thus affecting development of the mouse ovary
(81). Also, the incidence of cardiac hypertrophy in AhR null mice suggests a role for
AhR in normal cardiovascular development and angiogenesis (136). However, it has not
been clearly defined how AhR mediates these functions. Differential regulation of genes,
other than those involved in xenobiotic metabolism, could possibly account for these
AhR mediated effects. Alternate mechanisms have also been suggested in the recent
years to explain the different functions of AhR. For example, it has been shown to
interact with retinoblastoma (Rb) (210) and nuclear factor κ-B (211) transcription factors
and modulate cell proliferation. Co-expression of AhR and BRG-1 restored Rb-

57
sensitivity to a tumor cell line, C33A, which could otherwise progress through the cell
cycle even in the presence of active Rb protein (212).
A number of exogenous and endogenous ligands are capable of activating the
AhR (213). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is known to be the most potent
exogenous AhR ligand. While there is evidence suggesting that TCDD exerts almost all
its toxic effects through the AhR, the exact mechanisms underlying these effects are not
clear. TCDD has been shown to be more effective as a tumor promoter than as a tumor
initiator, due to the fact that TCDD is essentially not metabolized (reviewed in (214)).
TCDD has been shown to promote skin tumor formation in hairless mice (215) as well as
promote liver tumor formation (reviewed in (216)). The importance of AhR in tumor
promotion is also highlighted by the results obtained in a recent study using transgenic
mice expressing a constitutively active AhR where a significant increase in
hepatocarcinogenesis was noted compared to wild-type mice (88). In addition, studies
using constitutively active AhR have demonstrated that the receptor can induce tumors in
the stomach (87). The fact that benzo[a]pyrene, a known AhR ligand, is unable to exhibit
its carcinogenic effects in the skin of AhR null mice, provides further evidence
supporting a role for AhR in tumorigenesis (202).
Epidermal growth factor (EGF) and other members of its family are potent
peptide growth factors and are involved in a plethora of physiological and pathological
processes through their signaling properties (217). Mutations and/or over-expression of
EGF-family members cause cells to acquire an oncogenic phenotype. Epiregulin is a

58
relatively newly identified member of the EGF family, isolated from the conditioned
medium of NIH/3T3/clone 7 cells (218). It is secreted as a 46-amino acid single chain
polypeptide with contrasting growth regulatory properties, inhibiting the growth of
several epithelial cell lines while promoting growth of other cell types, like vascular
smooth muscle cells (211), hepatocytes (218, 219) and keratinocytes (220). Epiregulin is
expressed during development as well as later on in the genitourinary (221, 222) tract,
gastrointestinal tract, vascular smooth muscle cells and skin. In the latter two tissues, it
has been shown to function in a paracrine as well as autocrine fashion (223).
This study demonstrates, for the first time, that ligand activated AhR binds to its
cognate element in the epiregulin promoter and mutation of this binding element results
in loss of AhR-driven reporter activity. AhR increases epiregulin transcription in cultured
primary mouse keratinocytes, immortalized hepatocytes and mouse hepatoma derived
cell line. It is also shown that epiregulin is capable of significantly enhancing the
proliferation of cultured primary mouse keratinocytes. This could possibly account for
one of the mechanisms by which TCDD exerts its tumor promotional effects in rodent
skin.

59
2.3 Materials and methods
Cell culture
Primary mouse keratinocytes were obtained from 2-day old neonatal C57BL/6
mice and cultured using a previously described method (224). NIH/3T3 cells and
Hepa1c1c7 were grown in α-minimal essential medium (α-MEM) supplemented with 10
% FBS (HyClone Laboratories, Logan, UT), 100 IU/ml penicillin, and 0.1 mg/ml
streptomycin (Sigma) at 37 °C in 5% CO2 atmosphere. Hepatocytes from 2-week old
C57BL/6N mice were infected with SV40 virus to prepare temperature sensitive SV40
immortalized mouse hepatocytes in our laboratory (225) using a previously described
protocol with modifications (226, 227). Three cell lines of immortalized hepatocytes
were established. Cells from line 2 were used for experiments. These cells were
maintained in 4% FBS and 0.1 μM dexamethasone at 34°C.
Real Time Quantitative PCR (qPCR)
Real time qPCR was performed on the DNA Engine Opticon (MJ Research, Inc.)
using DyNAmo Hot Start SYBR Green qPCR kit purchased from MJ Research, Inc.
cDNA synthesis was carried out using High Capacity cDNA Archive Kit from Applied
Biosystems. The reverse transcription reactions were set up according to the
manufacturer’s instructions. cDNA synthesized from 50 ng of total RNA was used per
qPCR reaction. Epiregulin mRNA was detected using 5`-
TGGGTCTTGACGCTGCTTTGTCTA-3` and 5`-

60
AAGCAGTAGCCGTCCATGTCAGAA-3` primers. Epiregulin promoter in ChIP assays
was detected using 5`-TTCCTGAGAGGGAGGATGACAT-3` and 5`-
CCCACCAAGTCGCTGTGACT-3` primers. Thermal cycling conditions were setup
according to the manufacturer’s protocol.
Plasmids
The following plasmids were used for reporter assays : pEpi125 – derived by
inserting rat epiregulin promoter region -125/+12 (genomic contig: NW_047424.1) in
pGL3-Basic luciferase vector, pmutABC – pEpi125 with all three GT and CT boxes
mutated (228), pmutARNT – pEpi125 with ARNT half-site of DRE mutated and
pmutAhR – pEpi125 with AhR half-site of DRE mutated. pEpi125 and pmutABC were a
kind gift of Dr. Kaoru Miyamoto (Fukui Medical University, Japan). pmutARNT and
pmutAhR were modified forms of pEpi125, generated by site-directed mutagenesis using
the QuikChange mutagenesis kit (Stratagene). Forward and reverse primer pairs used to
mutate the ARNT and AhR half-sites, respectively were (5`-
GTAAGTCCTCGCTGGCCTAAGCACC-3` and 5`-
GGTGCTTAGGCCAGCGAGGACTTAC-3`) and (5`-
GTAAGTCCTCGAGTGCCTAAGCACC-3` and 5`-
GGTGCTTAGGCACTCGAGGACTTAC-3`).
Transient transfections and luciferase reporter assays
These experiments were carried out using NIH/3T3 cells, obtained from the
American Type Culture Collection (ATCC). Transfections were performed using

61
Lipofectamine Plus (Invitrogen). Cells at 70% confluence in 6-well plates were washed
with PBS and 1.5 ml Optimem (Life Technologies) containing 5 μl of Lipofectamine, 1
μl of Plus and 1.5 μg DNA were added per well. The DNA was comprised of 500 ng of a
luciferase reporter vector harboring wild-type or mutated versions of the rat epiregulin
promoter, 100 ng of pDJM/β-gal to control for transfection efficiency and empty
expression vector. After 4 h cells were washed with PBS and α-MEM containing 10%
FBS and antibiotics was added. Cells were allowed to grow overnight and treated with 10
nM TCDD or DMSO the next day for 7 h and lysed with 1X cell lysis buffer (25 mM
Tris buffer pH 7.8, 2 mM DTT, 2 mM EDTA, 10% glycerol and 1% Triton X-100).
Luciferase activity from cell lysates was measured using a Turner TD-20e luminometer
(Turrner Designs, Sunnyvale, CA) and values were normalized to β-gal values.
Chromatin Immunoprecipitation assay and PCR
Chromatin Immunoprecipitation (ChIP) assays were performed as described by
Spencer et. al. (229) with some modifications. Briefly, 80% confluent cells were treated
with 10 nM TCDD or DMSO for 100 min and crosslinked for 8 min at room temperature
with 0.33% formaldehyde (SIGMA, F-8775). Cells were harvested and lysed with 750
μl/flask lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8). Lysates were
sonicated with Branson Sonifier 250 using 40% duty cycle and output set at 4 for 8 cycles
of 12 pulses each. Protein-DNA complexes were immunoprecipitated from 200 μl lysate
using either 6 μl rabbit polyclonal anti-AhR antibody (Biomol, SA-210), control IgG or
no antibody and 50 μl Goat Anti-Rabbit IgG-Agarose resin (SIGMA, A1027). Resin was

62
washed once each with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20
mM Tris-HCl pH 8 and 150 mM NaCl), 1X RIPA buffer (0.1% SDS, 0.1% sodium
deoxycholate, 1% Triton X-100, 1 mM EDTA, 0.5 mM EGTA, 140 mM NaCl, 10 mM
Tris-HCl pH 8) and 1 X MENG buffer (25 mM MOPS, 2 mM EDTA, 0.02% sodium
azide, 10% glycerol pH 7.5) in that order. Immunoprecipitated protein-DNA complexes
were incubated in 300 μl digesting buffer (50 mM Tris-HCl pH 8, 1 mM EDTA, 100 mM
NaCl, 0.5% SDS, 100 μg/ml Proteinase K). DNA was isolated by phenol-chloroform
extraction and concentrated using ethanol precipitation. PCR was performed using
following primers: Epiregulin (5`- TTCCTGAGAGGGAGGATGACAT-3` and 5`-
CCCACCAAGTCGCTGTGACT-3`; located 107 bases upstream and 75 bases
downstream relative to TATA box), Cyp1A1 (5`-GCCGAGCATCGCACGCAAACC-3`
and 5`-GGATCCACGCGAGACAGCAGG-3`; located 1168 and 784 bases upstream
relative to TATA box) and GAPDH (5`-CATGGCCTTCCGTGTTCCTA-3` and 5`-
GCGGCACGTCAGATCCA-3`). 10% DMSO was added to the reactions.
Keratinocyte proliferation studies
Equivalent number of keratinocytes were seeded in 12 well plates and cultured in
low calcium medium (0.05 mM) containing 8% chelexed FBS for 24 h before treatment.
To test the effect of epiregulin or TCDD on keratinocyte growth, cells were cultured in
low calcium medium containing epiregulin (R&D Systems, 1068-EP), at a concentration
from 1 – 20 ng/ml, or TCDD at a concentration from 0.1 – 1.0 nM, continuously with
medium change after 24 and 72 h. After 24, 72, or 120 h incubation, cell number was
measured using a Z1 coulter® particle counter (Beckman Counter, Inc., Hialeah, FL).

63
2.4 Results
TCDD treatment upregulates Epiregulin transcription
The principal mode of action of AhR is through binding its cognate cis-regulatory
element, the DRE, in the promoter region of its target genes. AhR heterodimerizes with
ARNT and then recruits coregulators to enhance transcription (reviewed in (230)).
Despite the fact that AhR is implicated in a diverse range of physiological processes, as
discussed in the introduction, few genes outside the gamut of xenobiotic metabolism have
been reported to be directly regulated by the AhR pathway. In an attempt to identify
novel target genes, whose expression is under the regulation of AhR, microarray
experimentsi were performed using SV-40 immortalized mouse hepatocytes. These
experiments revealed epiregulin as one of the target genes whose mRNA levels increased
on TCDD treatment. Microarray results were confirmed with real-time qPCR
(Figure 2.1). Effect of TCDD on epiregulin mRNA levels was also tested in Hepa1c1c7,
a mouse hepatoma cell line. Epiregulin was upregulated by 2.6 fold, 90 min after TCDD
exposure. As TCDD exerts significant adverse effects on skin, including tumor
promotion, upregulation of epiregulin in response to TCDD was assessed in primary
keratinocytes. Primary mouse keratinocytes were treated with 10 nM TCDD for 90 min
and change in the level of epiregulin mRNA was compared by real time qPCR. TCDD
increased epiregulin mRNA expression by 3.3 fold (Figure 2.1) in primary mouse
keratinocytes also. Thus AhR activation resulted in epiregulin upregulation in primary
cells as well as immortalized cell lines.

64
Figure 2.1: TCDD increases Epiregulin mRNA.
Primary mouse keratinocytes, SV-40 immortalized hepatocytes and Hepa1c1c7 cells were treated with 10 nM TCDD and total RNA was extracted at 90 min. Real time qPCRwas performed using cDNA synthesized from 50 ng RNA. Experiments were performedfour times with primary keratinocytes and twice each with immortalized hepatocytes and Hepa1c1c7 cells. qPCR was performed in duplicate for each biological replicate. Datafrom representative experiments are presented as fold increase in relative fluorescenceunits upon TCDD treatment compared to untreated samples. Data is normalized toGAPDH mRNA levels within each cell type. * p<0.05 as determined by Student’s t-test.

65
AhR binds the DRE in the epiregulin promoter
In accordance with the currently accepted theory, AhR must bind its response
element in the promoter of a gene to directly regulate its expression. A search for binding
sites for AhR-ARNT heterodimer by sequence analysis identified a consensus DRE
(TCGCGTG) 56 nucleotides upstream of the transcription start site in the epiregulin
promoter in mouse and rat genomic DNA. To assess if AhR binds the DRE in the
epiregulin promoter sequence, ChIP assays were performed with SV40 virus
immortalized mouse hepatocytes treated with TCDD. DNA fragments isolated from ChIP
assays were analyzed using PCR (Figure 2.2 A). CYP1A1 was used as a positive control.
GAPDH and immunoprecipitations with no antibody or with control IgG were used as
negative controls. CYP1A1 showed a strong signal in the TCDD treated sample,
compared to a carrier solvent treated sample, while there was no difference in the case of
GAPDH (data not shown). Immunoprecipitations with control IgG or no antibody
showed minimal background signals. While the epiregulin signal was more intense in
TCDD samples compared to carrier solvent treated samples, the difference was less than
that of CYP1A1. This difference could be explained by the fact that epiregulin has only
one DRE in its promoter while the CYP1A1 promoter has multiple DREs, and that AhR
upregulates CYP1A1 expression by a greater magnitude than epiregulin. These results
were confirmed by realtime qPCR (Figure 2.2 B) performed on DNA isolated from ChIP
assays. The relative signal for epiregulin in TCDD treated samples was 1.7 fold greater
than carrier solvent treated samples. Combined, these results demonstrate an increase in
epiregulin promoter occupancy by AhR in response to TCDD treatment.

66
Figure 2.2: Epiregulin promoter occupancy by the ligand-activated AhR.
(A) SV-40 immortalized mouse hepatocytes were treated with TCDD or carrier solvent(DMSO), crosslinked with formaldehyde, lysed and sonicated. Protein-DNA complexes were immunoprecipitated with anti-AhR antibody, control IgG or no antibody as outlinedin materials and methods. The crosslinked DNA was resolved by heating, PCR amplifiedusing appropriate primers and visualized on agarose gel. Immunoprecipitations usinganti-AhR antibody were done in duplicate. Experiment was repeated three times andsimilar results were obtained. (B) Real time qPCR revealed an increase in AhR binding to the DRE in epiregulinpromoter. Reverse crosslinked DNA from ChIP assay was analyzed by real time qPCR using primers surrounding the DRE in Epiregulin promoter. Data is represented asaverage relative fluorescence units in TCDD and carrier solvent treated samples acrosstwo ChIP assays, each measured in duplicate. (* p-value = 0.014 on Student’s t-test) AhR IP = immunoprecipitation using anti-AhR antibody; IgG IP = control IgG antibody; NoAb IP = no antibody; RFU = relative fluorescence units; Input = 10% of lysate used forimmunoprecipitation with different antibodies.

67
AhR driven promoter activity is dependent on the DRE
To further confirm the role of the DRE in modulating epiregulin gene expression,
a luciferase reporter assay was performed using a construct containing the first 125 bases
of the rat epiregulin promoter cloned into pGL3-Basic vector (pEpi125). There is a single
nucleotide difference between the first 125 bases of the mouse (genomic contig:
NT_039308.3) and the rat (genomic contig: NW_047424.1) epiregulin promoters, which
is not a part of the DRE or the Sp1 binding sites. This construct has been previously used
by Sekiguchi et. al to study transcriptional regulation of the epiregulin gene in the rat
ovary (222). They showed by deletion and mutation analyses that the region
encompassing 125 bp upstream of the transcription start site was essential for controlling
transcription of the epiregulin gene. According to their results, Sp1/Sp3 proteins bind to
one or more of the two CT boxes and one GT box within this 125 bp region and are
involved in regulating epiregulin gene expression.
The relative contributions of the AhR and Sp1/Sp3 were studied using plasmids
derived from pEpi125 by mutating their binding sites, the DRE and the CT and GT
boxes, respectively (Figure 2.3 A). Luciferase reporter plasmid pEpi125 was transiently
transfected in NIH/3T3 cells, while pDJM/β-gal vector was cotransfected to control for
transfection efficiency. Reporter assays were also carried out with pmutABC plasmid,
derived from pEpi125 by mutating both the CT boxes as well as the GT box, pmutARNT
plasmid, derived from pEpi125 by mutating the ARNT half-site of DRE and pmutAhR
plasmid, derived from pEpi125 by mutating the AhR half-site of DRE (Figure 2.3 C).

68
There was a two-fold increase (2.3 fold) in luciferase reporter activity observed upon
TCDD treatment (Figure 2.3 B) with the pEpi125 transfections. Surprisingly, a similar
increase in luciferase activity (2.2 fold) was observed with the pmutABC transfections
compared to vehicle treated control samples. There was no increase in luciferase activity
on TCDD treatment when the AhR and ARNT binding half-sites were mutated in
pmutARNT and pmutAhR, respectively (~1.2 fold in pmutARNT and pmutAhR each).
Interestingly, the reporter gene activity in pmutABC transfections was 3.2 times lower
compared to pEpi125 transfections, both in TCDD and carrier solvent treated samples.
This decrease in reporter activity was not observed when the DRE was mutated
(pmutAhR and pmutARNT transfections). These results, along with ChIP assays,
demonstrate that the activated AhR binds to the DRE within the epiregulin promoter and
is responsible for the observed increase in transcriptional activity.

69
Figure 2.3: AhR binds DRE in rat Epiregulin promoter.
(A) NIH/3T3 cells were transfected in 6-well dishes with pEpi125, pmutABC, pmutARNT or pmutAhR plasmids using Lipofectamine Plus protocol as described inmaterials and methods. Cells were subjected to no treatment, carrier solvent (DMSO) or10 nM TCDD. Transcriptional activity was assessed by measuring luciferase assaysignals. Data is shown as mean and standard deviation of triplicate transfections.Experiment was repeated four times with similar results. Statistical analysis is performedusing Student’s t-test; * p<0.001 in both cases (significant); # p>0.05 in both cases (insignificant). (B)Graphic representation of the fold-increase in luciferase assay data from panel A. (C)Schematic diagram of first 125 bases of rat epiregulin promoter present within theplasmid pEpi125. Transcription start site is indicated by an arrow. DRE is shown as two half-sites (AhR and ARNT) represented by ovals. Box A and C are the two CT boxes andBox B is the GT box. In pmutABC, pmutARNT and pmutAhR, mutation of respectiveelements are shown as blacked out boxes/ovals.

70
Effect of Epiregulin and TCDD on Mouse Keratinocyte Proliferation
To characterize the functional role of increased epiregulin expression on cell
growth, the effect of epiregulin treatment was determined using primary mouse
keratinocytes. It has previously been shown that epiregulin stimulates proliferation of
cultured human keratinocytes in a dose-dependent manner and acts as an autocrine
growth factor (220). Recombinant human epiregulin at a concentration of 1 ng/ml
resulted in a three-fold increase in cell growth. In the present study, primary mouse
keratinocytes showed a dose-dependent increase in cell number when treated with
recombinant mouse epiregulin, even in the presence of other growth factors found in fetal
bovine serum (Figure 2.4 A). Epiregulin stimulated a statistically significant proliferation
of primary mouse keratinocytes across a dose range of 1-20 ng/ml at 24, 72 and 120 h of
treatment, as determined by ANOVA (p<0.05). Significant increase in proliferation was
observed at all three-time points with the higher doses (10 and 20 ng/ml), whereas 1
ng/ml epiregulin induced proliferation only at 72 and 120 h.

71
Figure 2.4: Epiregulin and TCDD increase primary mouse keratinocyte proliferation in a dose-dependent manner.
105 cells were seeded per well in 12-well plates. After 24 h, fresh medium containing either recombinant mouse epiregulin (A) or TCDD (B) was added at indicated doses. Cells were counted 24, 72 and 120 h after adding epiregulin or TCDD. Data is presented as the mean and standard error of cells measured in triplicate wells. Data presented waschecked for statistical significance by ANOVA and Tukey HSD test (see text). Observedincrease in proliferation was significant for both epiregulin and TCDD (p<0.05).

72
Similar cell proliferation studies were performed using TCDD as a stimulant
(Figure 2.4 B). Primary mouse keratinocytes were treated with 0.1 nM, 0.5 nM and 1 nM
TCDD, or vehicle solvent, and cells counted at 24, 72 and 120 h after addition of TCDD.
TCDD-induced cell proliferation was statistically significant, as determined by ANOVA
(p<0.05). Individually, all three doses induced statistically significant increase in cell
proliferation when compared to vehicle treated cells. However, a statistically significant
difference was not observed amongst the different doses, as determined by Tukey’s HSD
test. When the effects of different doses were compared at individual time-points, an
increase in the number of cells was observed at all three time-points with 0.1 nM and 0.5
nM TCDD treatments. Primary mouse keratinocytes treated with 1 nM TCDD showed a
slight reduction in cell numbers at the 120 h time point. This could be due to increasing
toxicity owing to prolonged TCDD exposure at this latest time point.
2.5 Discussion
TCDD causes a wide range of toxic effects including tumor promotion, but the
exact mechanisms responsible for these effects are not known. However, most of the
efforts investigating genes regulated by AhR have been focused on enzymes involved in
xenobiotic metabolism. Though there are indications for involvement of AhR in a
number of cellular processes (231), there is no well described role for the AhR in normal
cellular function or tumor promotion that has been delineated at the gene expression

73
level. In this report we have examined the ability of the AhR to regulate expression of the
potent mitogen epiregulin.
Skin and liver have been observed to be two of the most common sites of tumor
development in TCDD tumor promotion studies. AhR-dependent upregulation of genes
involved in growth promotion could be an important mechanism for TCDD mediated
carcinogenesis. One of the functional classes of genes that could mediate these effects
would be growth factors. The ability of TCDD to increase epiregulin mRNA in cell types
known to exhibit TCDD-mediated toxicity, as revealed by the microarrays and real time
qPCR experiments in hepatocytes and keratinocytes, support the hypothesis that TCDD
exposure could stimulate the growth potential of precancerous cells through the enhanced
expression of potent mitogens like epiregulin. Further, results from the ChIP assay and
luciferase reporter assay, reveal that an increase in epiregulin mRNA levels is brought
about by ligand-activated AhR binding to the DRE in the epiregulin promoter and thus,
epiregulin is directly regulated by the AhR in response to TCDD. Earlier studies have
demonstrated that two of the three Sp1 binding elements, in the epiregulin promoter, are
actively involved in regulating epiregulin mRNA levels. A cooperative interaction
between AhR-ARNT and Sp1 in the CYP1A1 gene promoter has been reported
previously (232). According to their results, binding of either AhR-ARNT or Sp1 to its
element in the CYP1A1 promoter facilitated binding of the other factor and increased
reporter activity in a synergistic fashion. The DRE and GC/CT boxes are found in close
proximity in a number of other AhR regulated gene promoters like UDP-glucuronosyl
transferase, aldehyde dehydrogenase-3 and quinine reductase. An interaction between the

74
AhR/ARNT complex and Sp1 has been shown to exist even in the absence of exogenous
ligand, for the Cathepsin D gene promoter (233). Since the epiregulin promoter has both
the AhR and Sp1 binding elements, it was interesting to determine whether this kind of
cooperative interaction existed at the epiregulin promoter. Notably, as revealed in the
reporter assays, even when all three Sp1 binding sites were mutated within the epiregulin
promoter (pmutABC transfections), the fold-increase in luciferase activity on TCDD
induction was similar to that observed in the wild-type promoter, 2.2 and 2.3 fold
respectively. However, the overall amount of luciferase signal decreased by
approximately two thirds in pmutABC transfections. This shows that Sp1 is not necessary
for AhR-mediated induction of epiregulin gene expression, but enhances the overall
transcription rate of the epiregulin promoter. After demonstrating that TCDD activated
AhR increases epiregulin transcription, the next logical step would be to show a
corresponding increase in epiregulin secreted in conditioned media. However attempts to
detect epiregulin using Western blot were unsuccessful (data not shown).
Increased epiregulin mRNA expression has been observed in pancreatic cancer
tissue samples (234) as well as in prostate and pancreatic cell lines (234, 235). It has been
proposed that over-expression of epiregulin is an important factor in growth regulation of
malignant fibrous histiocytoma (236), colon cancer cells (237) and various gynecological
malignancies (238). These data, along with the fact that ligands and receptors of the EGF-
family have been implicated in a number of tumors, suggest that epiregulin might be an
important factor in mediating TCDD dependent skin tumor promotion in mice. To show
that modulation of epiregulin levels has a functionally significant role in primary mouse

75
keratinocytes, cells were treated with increasing concentrations of epiregulin. Epiregulin
treatment caused an increase in proliferation of primary mouse keratinocytes as observed
at three time points (24, 72 and 120 h), even at a concentration as low as 1 ng/ml. It is
worth noting that this increase occurred in the presence of 8% fetal bovine serum.
Different concentrations of TCDD also increased cell proliferation, albeit at a lower rate
than epiregulin. Statistical analysis revealed this effect of TCDD to be significant, even
though apparently modest. Although it is not possible to rule out the contribution of other
growth factors, it is possible that this effect is due, at least in part, to an increase in
epiregulin level.
Results from these studies also suggest that caution should be taken when
extrapolating results across different species. The International Agency for Research on
Cancer (IARC) updated its classification of TCDD as a group 1 carcinogen in 1997 from
its previous evaluation that classified it as a group 2B (possible) human carcinogen (239).
This upgrade has been debated in the literature (240, 241). However, it is clear from these
discussions that not all data obtained from animal studies can be applied directly to
humans. The toxic effects of TCDD appear to be less pronounced in humans than in
rodents. A number of factors could be responsible for this, including differences in AhR,
its cytosolic or nuclear binding partners, its affinity for ligands, endogenous ligands,
transcriptional coregulators or the battery of genes whose transcription is modulated by
AhR. If a gene involved in a cellular process is regulated differently in humans and mice,
it could be an important reason for possible differences. We compared the first 125 bases
of epiregulin promoter sequence from human, mouse and rat and found that the DRE is

76
absent in humans (Figure 2.5) . Thus, TCDD-activated AhR may not regulate expression
of epiregulin in humans. However, further studies in human tissue samples or cell lines
will be needed to clarify this issue. In conclusion, we have shown for the first time that
epiregulin is a direct target gene for the AhR pathway in rodents. Considering the
mitogenic potential of epiregulin and the possibility that it might not be regulated through
the AhR pathway in humans, these results point to a potential inter-species difference in
regulation of a gene that could participate in TCDD-mediated tumor promotion.

77
Figure 2.5: The DRE is absent in the human epiregulin promoter.
First 125 bases of epiregulin promoter, in mouse, rat and human, were aligned.Nucleotides representing Box A, B and C and DRE are enclosed. The DRE sequence is conserved in rodents but lost in human epiregulin promoter. Box A and C are conservedin all three species while Box B is lost in human promoter.

78
Chapter 3
AHR ACTIVATION REGULATES CONSTITUTIVE ANDROSTANE RECEPTOR (CAR) LEVELS IN MURINE AND HUMAN LIVER.

79
3.1 Abstract
The aryl-hydrocarbon receptor (AhR) is a bHLH/PAS (basic helix-loop-helix/Per-
Arnt-Sim) transcription factor that can be activated by exogenous as well as endogenous
ligands. AhR is traditionally associated with xenobiotic metabolism. In an attempt to
identify novel target genes, C57BL/6J mice were treated with β-naphthoflavone (BNF), a
known AhR ligand, and genome-wide expression analysis studies were performed using
high-density microarrays. Constitutive androstane receptor (CAR) was found to be one of
the differentially regulated genes. Real-time quantitative polymerase chain reaction
(qPCR) verified the increase in CAR mRNA level. BNF treatment did not increase CAR
mRNA in AhR-null mice. Time-course studies in mice revealed that the regulation of
CAR mRNA mimicked that of Cyp1A1, a known AhR-target gene. In order to
demonstrate that the increase in CAR mRNA translates to an increase in functional CAR
protein, mice were sequentially treated with BNF (6 hours) followed by the selective
CAR agonist, TCPOBOP (3 hours). qPCR revealed an increase in the mRNA level of
Cyp2b10, previously known to be regulated by CAR. This also suggests that CAR protein
is present in limiting amounts with respect to its transactivation ability. Finally, CAR was
also upregulated in primary human hepatocytes in response to AhR activation by TCDD
and benzo[a]pyrene. In conclusion, this study identifies a novel mode of up-regulating
CAR and potentially expands the role of AhR in drug metabolism. This is the first study
demonstrating in vivo upregulation of CAR through chemical exposure.

80
3.2 Introduction
Though a number of genes have been characterized to be regulated by AhR in a
DRE-dependent manner, alternative modes of receptor function as well as novel target
genes must be identified to adequately explain the wide spectrum of patho-physiologic
effects associated with AhR. An emerging aspect of transcription factor biology is the
ability of various factors to interact with members of different signaling pathways.
Recent reports focusing on receptor cross-talk have highlighted the ability of AhR to
influence the activity of other proteins involved in gene regulation, including NFkB
(173), estrogen receptor (ER) (158, 161, 242) and TGF-β1 (243).
The constitutive androstane receptor (CAR, also known as Nr1i3) is a member of
the nuclear receptor family. It is found in the cytoplasm in a complex with heat shock
protein 90 (HSP90) and CAR cytoplasmic retention protein (CCRP) (244). A unique
feature of CAR is that it can be activated by two distinct mechanisms. Ligands like 1,4-
bis [2-(3,5,-dichloropyridyloxy) benzene (TCPOBOP) can directly bind to CAR and
activate the receptor (245). Alternately, CAR activity can be induced indirectly by a
phenobarbital-responsive protein phosphatase-2A dependent signaling cascade (246).
Activated CAR undergoes nuclear translocation, heterodimerizes with 9-cis retinoic acid
receptor (RXR) to bind its response element and drives the transcription of its target
genes. Using cell-culture models, it has been demonstrated that activation of the
glucocorticoid receptor can upregulate transcriptional activity at the CAR promoter (247)

81
and that IL-1β mediated NF-κB activation inhibits this upregulation by interfering with
chromatin remodeling (248).
In the current study, we have identified CAR to be an AhR target gene.
Previously, it has been shown that both AhR and CAR play a significant role in response
to exogenous stimuli as well as patho-physiologic events in the liver. Both the receptors
induce numerous xenobiotic metabolism enzymes. AhR is also known to affect vascular
development as demonstrated by persistent ductus venosus and microvasculature
abnormalities in the liver of AhR-null mice (80). Recently it has been demonstrated that
TCDD exposure severely impairs the regenerative ability of partially excised mouse
livers (249), an effect most likely mediated through AhR. On the other hand, TCDD is
also known to be a potent tumor promoter in the mouse liver (250). CAR is similarly
involved in a number of physiologic processes in the liver including bilirubin metabolism
and hepatocyte proliferation as discussed below. As shown in this study, AhR-activation
increases CAR mRNA in liver as well as extra-hepatic tissues and follows a temporal
pattern similar to Cyp1A1, a known AhR target gene. This increase in CAR mRNA
correlates with an increase in the transcriptional activity of CAR. Since a broad range of
compounds can activate AhR, an AhR-mediated increase in CAR activity could
potentially lead to unexpected effects on drug metabolism. Considering the importance of
AhR and CAR in liver biology, knowledge of the interaction between the two receptors
will be useful in interpreting the observations made in relation to these receptors.

82
3.3 Materials and Methods
Mice and the treatments:
Adult (10 ± 2 weeks) male wild-type C57BL/6J mice were purchased from the
Jackson laboratory. AhR knock-out (AhR-KO) mice in a C7BL/6J background were a
kind gift of Dr. Bradfield (McArdle Laboratory for Cancer Research, University of
Wisconsin-Madison Medical School). All mice were maintained in a temperature and
light-controlled facility and had free access to water and diet. All experiments were
performed in compliance with the standards for animal use and care set by the
Pennsylvania State University’s animal research program. Mice were injected
intraperitoneally (I.P.) with BNF dissolved in corn oil (CO) or corn oil alone (control).
The volume of injection was adjusted in proportion to the body weight. Mice were
sacrificed by carbon-dioxide inhalation and liver-tissue samples were collected. Tissues
were frozen immediately in liquid nitrogen and stored at -80°C.
Microarray experiments:
Liver samples were homogenized in TRI-Reagent® with Ultra Turrax T25 basic
disperser from IKA® Works, Inc. (Wilmington, NC). RNA was isolated from tissues
using TRI Reagent® (Sigma-Aldrich Co.) and was further purified with RNeasy® kits
(Qiagen Inc.) according to the manufacturer’s protocol with minor modifications. The
quality of RNA was analyzed on 1% agarose-formaldehyde gel and with Agilent 2100
bioanalyzer and RNA-6000 Nano Chip kit (Agilent Technologies, Inc.). GeneChip®
One-Cycle Target Labeling and Control Reagent package (Affymetrix, CA) was used to

83
label 8.0 μg total RNA for each microarray. The GeneChip® Hybridization, Wash, and
Stain kit (Affymetrix, CA) was used for processing the microarrays. Liver gene
expression profiles were compared between BNF (10 mg/kg) treated versus vehicle-
control mice using GeneChip® Mouse Genome 430 2.0 arrays. The arrays were scanned
with GeneChip® Scanner 3000 at the microarray core facility of the Huck Institutes of
Life Sciences, Pennsylvania State University.
Microarray data analysis:
Background adjustment, normalization and summarization were performed on the
raw data files (.CAB files) using RMAexpress (0.3 Release). Summarized data was
further analyzed by Significance Analysis of Microarrays (SAM, Version 2.23A). The
data was input in the log scale (base 2) and the default settings were accepted as the
choice of analysis parameters i.e. T-statistic, 100 permutations, 10 neighbors for K-
nearest neighbors imputer. A delta-value of 0.35 and 2-fold change was used as the
threshold for significant differential expression. 97 probe-sets, corresponding to 80
distinct genes/ESTs, were found to be significantly changed with a false discovery rate of
5.7 percent using the settings detailed above.
Post-processing of the significantly altered genes was performed using the
DAVID Bioinformatic Resources 2006 online tool. Genes were clustered using the Gene
Functional Classification Tool using the lowest classification stringency settings. To
reduce redundancy, the list of terms associated with the clustered genes was restricted to
biological processes and/or molecular function (according to Gene Ontology

84
classification) with a p-value < 0.05, as computed by DAVID. A subset of genes was
selected to confirm differential expression by real-time quantitative PCR.
Real-time quantitative PCR (qPCR):
Total RNA isolated from mice livers, as mentioned above, was reverse
transcribed using the High Capacity cDNA Archive® kit (Applied Biosystems) according
to the manufacturer’s protocol. cDNA made from 25 ng of RNA was used for each qPCR
reactions. qPCR was performed on DNA Engine Opticon® system using DyNAmo™
SYBR® Green qPCR Kit purchased from New England Biolabs, Inc. Sequence
information for the qPCR primers is provided in Table 3.1.

85
Table 3.1: Sequence information for primers used in qPCR.
Gene Primer Set (FP=forward primer; RP=reverse primer)
CAR FP: 5`-GGAGCGGCTGTGGAAATATTGCAT-3`
RP: 5`-TCCATCTTGTAGCAAAGAGGCCCA-3`
Cyp1a1 FP: 5`-CTCTTCCCTGGATGCCTTCAA-3`
RP: 5`-GGATGTGGCCCTTCTCAAATG-3`
PXR FP: 5`-TTCATGTGGAGCCAAAGAAACGGC-3`
RP: 5`-TCCTGGAATGTGGGAACCTTTCCT-3`
Cyp2b10 FP: 5`-TTCTGCGCATGGAGAAGGAGAAGT-3`
RP: 5`-TGAGCATGAGCAGGAAGCCATAGT-3`
GAPDH FP: 5`-CATGGCCTTCCGTGTTCCTA-3`
RP: 5`-GCGGCACGTCAGATCCA-3`
CAR (human) FP: 5`-AGTGCTTAGATGCTGGCATGAGGA-3`
RP: 5`-TGCTCCTTACTCAGTTGCACAGGT-3`
Rpl13a (human) FP: 5`-CCTGGAGGAGAAGAGGAAAGAGA-3`
RP: 5`-GAGGACCTCTGTGTATTTGTCAA-3`

86
Primary human hepatocyte culture:
Primary human hepatocytes were obtained from the Liver Tissue Procurement
and Distribution System (LTPDS) at the University of Pittsburgh, through NIH Contract
#NO1-DK-9-2310, and generously provided by Dr. Stephan C. Strom. Donor organs not
designated for transplantation were used to isolate hepatocytes according to a three step
collagenase perfusion protocol (251). Preparations enriched for hepatocytes were
received plated in collagen-coated T25 flasks. Upon arrival, the media was changed to
William’s Media E supplemented with 1% penicillin/streptomycin, 10 mM HEPES, 20
μM glutamine, 25 nM dexamethasone, 10 nM insulin, 30 mM linoleic acid, 1 mg/ml
BSA, 5ng/ml selenious acid, 5 μg/ml transferring, as described previously (252). All cells
were maintained at 37°C under 5% CO2. All culturing materials were purchased from
Invitrogen (Carlsbad, CA), unless otherwise noted.
3.4 Results:
AhR activation alters the expression of various genes in mouse liver:
In an effort to identify novel AhR target genes, we performed genome-wide
expression profiling studies using high-density microarrays (Affymetrix). 10-week old
male wild-type C57BL/6J mice were treated with 10 mg/kg BNF in corn oil by I.P.
injections, for 5 h, following which, RNA was isolated from their livers. The RNA was
processed and hybridized to GeneChip® Mouse Genome 430 2.0 arrays. After pre-
processing and analyzing the data as described in the methods, significantly altered genes
were classified on the basis of gene ontology (biological process (BP) and/or molecular

87
function (MF)) to identify patterns of biological importance. The ‘Gene Functional
Classification’ module, implemented in the online version of DAVID, clusters genes on
the basis of the similarity of different ontology terms associated with the genes. The list
of terms for each cluster comprised of overlapping and redundant entries and thus had to
be manually limited to include those with high statistical significance of association and
to prevent repetition of parent/child terms. The results are presented in Table 3.2 . The
table also includes whether a dioxin response element (DRE) is present in the putative
regulatory region (-5000 to +1000 bases relative to transcription start site) of the
respective genes. To validate the microarray results, qPCR was performed on fourteen
genes that included four previously known AhR targets and the results were similar to the
changes observed on the microarrays. The induction/repression observed by qPCR is
given as a fold-change in parenthesis. The alteration of genes belonging to diverse
functional categories further supports the role of AhR in a variety of cellular processes.

88
Genes induced by BNF treatment
Affy
Id
Fold
Cha
nge
Gen
e N
ame
BP/
MF
Term
s (p
<0.0
5)
Pres
ence
of D
RE
I
qPC
R c
onfir
mat
ion
Gene Group 1 Enrichment Score: 2.24
1 1425477_x_at, 1451721_a_at, 1450648_s_at
2.8 histocompatibility 2, class ii antigen a, beta 1
antigen presentation, exogenous antigen via MHC class II
2 1435290_x_at, 1452431_s_at, 1438858_x_at
2.6 histocompatibility 2, class ii antigen a, alpha
positive regulation of T cell differentiation
3 1425519_a_at 2.4 cd74 antigen (invariant polypeptide of major histocompatibility complex, class ii antigen-associated)
MHC class II receptor activity +
4 1417025_at 2.3 histocompatibility 2, class ii antigen e beta
antigen processing, exogenous antigen via MHC class II
+
5 1422527_at 2.2 histocompatibility 2, class ii, locus dma immune response +
Gene Group 2 Enrichment Score: 1.98
1 1449009_at 5.7 t-cell specific gtpase
hydrolase activity, acting on acid anhydrides
2 1420549_at 3.9 guanylate nucleotide binding protein 1 nucleoside-triphosphatase activity
+
3 1419518_at 2.7 tubulin, alpha 8 purine nucleotide binding
4 1425351_at, 1426875_s_at, 1451680_at
2.6 neoplastic progression 3 GTPase activity
5 1417141_at, 1458589_at 2.5 interferon gamma induced gtpase
Table 3.2: BNF-mediated differentially regulated genes, sorted by Biological Process(BP)/Molecular Function (MF)

89
6 1419748_at, 1438431_at 2.4 Atp-binding cassette, sub-family d (ald),
member 2 7 1418392_a_at 2.3 guanylate nucleotide binding protein 4 + 8 1423597_at 2.2 atpase, aminophospholipid transporter
(aplt), class i, type 8a, member 1 + 9 1425156_at 2.2 riken cdna 9830147j24 gene 10 1417101_at 2.1 heat shock protein 2 + 11 1443870_at 2.0 Atp-binding cassette, sub-family c
(cftr/mrp), member 4 Gene Group 3 Enrichment Score: 1.95
1 1422217_a_at 266.6
cytochrome p450, family 1, subfamily a, polypeptide 1
monooxygenase activity + +
2 1423627_at 3.4 nad(p)h dehydrogenase, quinone 1
generation of precursor metabolites and energy
+ +
3
1422904_at, 1422905_s_at, 1435459_at, 1453435_a_at
2.7 flavin containing monooxygenase 2 electron transport +
4 1450715_at 2.6 cytochrome p450, family 1, subfamily a, polypeptide 2
oxygen and reactive oxygen species metabolism
+ +
5 1454930_at 2.4 leucine rich repeat containing 35 FAD binding 6 1449525_at 2.3 flavin containing monooxygenase 3 NADP binding + 7 1447411_at 2.3 udp-glucose dehydrogenase heme binding + 8 1449565_at 2.2 cytochrome p450, family 2, subfamily g,
polypeptide 1
9 1416612_at 2.0 cytochrome p450, family 1, subfamily b, polypeptide 1 + +
Gene Group 4 Enrichment Score: 0.94
1 1419647_a_at 3.1 immediate early response 3 establishment of localization +
2 1419748_at, 1438431_at 2.4 Atp-binding cassette, sub-family d (ald),
member 2 transport
3 1421346_a_at 2.1 solute carrier family 6 (neurotransmitter transporter, taurine), member 6 +
Gene Group 5 Enrichment Score: 0.65
1 1419647_a_at 3.1 immediate early response 3 transmembrane receptor activity +
2 1417625_s_at 2.9 chemokine orphan receptor 1 MHC class II receptor activity +
3 1425477_x_at, 1451721_a_at, 1450648_s_at
2.8 histocompatibility 2, class ii antigen a, beta 1
cell surface receptor linked signal transduction
4 1448147_at 2.7 tumor necrosis factor receptor superfamily, member 19
defense response + +
5 1417025_at 2.3 histocompatibility 2, class ii antigen e beta
G-protein coupled +

90
receptor activity
6 1446850_at 2.1 phosphatidic acid phosphatase type 2b + 7 1417894_at 2.1 g protein-coupled receptor 97 Gene Group 6 Enrichment Score: 0.13
1 1421818_at, 1450381_a_at 3.3 b-cell leukemia/lymphoma 6
ligand-dependent nuclear receptor activity
+
2 1451814_a_at 3.3 Hiv-1 tat interactive protein 2, homolog (human)
regulation of transcription, DNA-dependent
+
3 1455267_at 3.2 estrogen-related receptor gamma
regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism
+
4 1416543_at, 1457117_at 2.3 nuclear factor, erythroid derived 2, like 2
regulation of cellular metabolism
5 1425392_a_at 2.3 nuclear receptor subfamily 1, group i, member 3 +
6 1429177_x_at 2.1 Sry-box containing gene 17 + Gene Group 7 Enrichment Score: 0.11
1 1418191_at 3.8 ubiquitin specific peptidase 18
ubiquitin-dependent protein catabolism
+ +
2 1436532_at 3.6 doublecortin and cam kinase-like 3 purine nucleotide binding
+
3 1451453_at 3.1 death-associated kinase 2 cellular protein metabolism + +
4 1422962_a_at 2.9 proteosome (prosome, macropain) subunit, beta type 8 (large multifunctional peptidase 7)
cellular macromolecule metabolism
+
5 1419518_at 2.7 tubulin, alpha 8 endopeptidase activity +
6 1417801_a_at 2.7 protein tyrosine phosphatase, receptor-type, f interacting protein, binding protein 2
lipid transport +
7 1428484_at 2.5 oxysterol binding protein-like 3 +
8 1450696_at 2.2 proteosome (prosome, macropain) subunit, beta type 9 (large multifunctional peptidase 2)
+
9 1417101_at 2.1 heat shock protein 2 + 10 1424518_at 2.0 riken cdna 2310016f22 gene + 11 1424518_at 2.0 cdna sequence bc020489 +

91
Genes that could not be classified
Affy
Id
Fold
Cha
nge
Gen
e N
ame
Pres
ence
of
DR
E I
qPC
R
conf
irmat
ion
1 1427912_at 5.6 carbonyl reductase 3 + 2 1452913_at 3.7 purkinje cell protein 4-like 1 3 1451486_at 3.7 riken cdna 1200006f02 gene + 4 1456319_at 3.5 Est x83313
5 1424167_a_at, 1430780_a_at
3.1 phosphomannomutase 1
6 1428670_at 2.7 riken cdna 2610305j24 gene 7 1443138_at 2.6 sulfotransferase family 5a, member 1 + 8 1423410_at 2.5 meiosis expressed gene 1 + 9 1424296_at,
1455959_s_at 2.5 glutamate-cysteine ligase, catalytic subunit 10 1428120_at 2.4 f-box and wd-40 domain protein 9 + 11 1426936_at 2.4 hypothetical loc433593 12 1433685_a_at
, 1455654_at 2.3 riken cdna 6430706d22 gene 13 1417185_at 2.3 lymphocyte antigen 6 complex, locus a + 14 1438855_x_at 2.3 tumor necrosis factor, alpha-induced protein
2 + 15 1450699_at 2.3 selenium binding protein 1 + 16 1424594_at 2.3 lectin, galactose binding, soluble 7 + 17 1431240_at 2.3 c-type lectin domain family 2, member h + 18 1433710_at 2.2 hypothetical protein mgc36552 + 19 1431806_at 2.2 riken cdna 4931408d14 gene 20 1436576_at 2.2 riken cdna a630077b13 gene + 21 1418727_at 2.2 nucleoporin 155 + 22 1454890_at 2.2 Angiomotin + + 23 1418346_at 2.2 insulin-like 6 + + 24 1419149_at 2.0 serine (or cysteine) peptidase inhibitor, clade
e, member 1 + 25 1426215_at 2.0 dopa decarboxylase 26 1431648_at 2.0 riken cdna 4930528f23 gene 27 1438294_at 2.0 ataxin 1 +

92
Genes repressed by BNF treatment
Affy
Id
Fold
Cha
nge
Gen
e N
ame
Pres
ence
of
DR
E I
qPC
R
conf
irmat
ion
1
1444296_a_at, 1444297_at, 1448092_x_at
3.5 serine (or cysteine) peptidase inhibitor, clade a, member 4, pseudogene 1 +
2 1433898_at 2.3 solute carrier family 25, member 30 +
3 1448724_at 2.3 cytokine inducible sh2-containing protein + +
4 1437073_x_at 2.2 expressed sequence av025504
5 1418288_at 2.1 lipin 1 +
6 1452426_x_at 2.1 hypothetical protein
7 1436186_at 2.0 e2f transcription factor 8

93
AhR activation increases CAR mRNA in mouse liver:
CAR was one of the genes observed to be upregulated in the livers of BNF treated
mice as compared to the vehicle (corn oil) alone. The results obtained from the
microarray experiments were verified by real-time quantitative PCR (qPCR) and are
presented in Figure 3.1. qPCR data revealed 2.1-fold increase in CAR mRNA on BNF
treatment which correlated well with microarray results. The currently accepted model of
AhR-dependent transcription is based on the AhR-ARNT heterodimer binding to a
consensus DRE in the regulatory region of a target gene. However, sequence analysis of
the putative mouse CAR promoter (-5000 to +1000 bases relative to transcription start
site) did not reveal any sequences matching the DRE (TNGCGTG). This observation
generated further interest in examining the role of AhR in regulating CAR expression and
whether the observed increase in CAR mRNA had a functional significance.
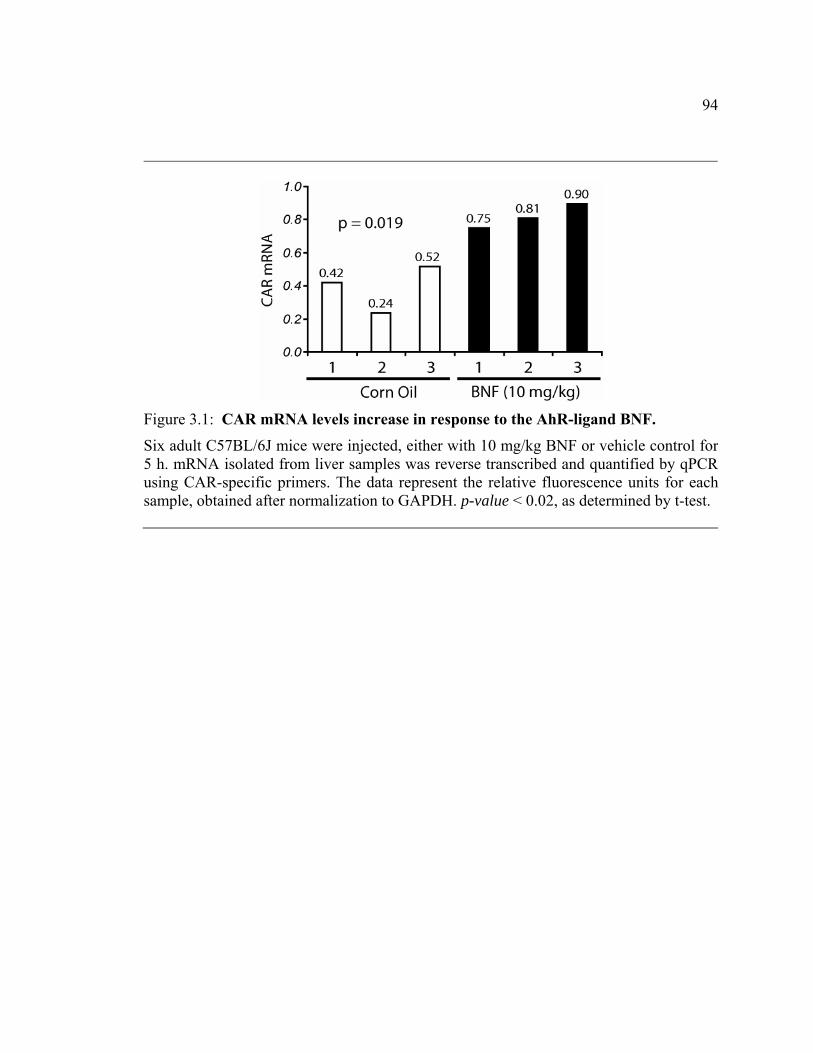
94
Figure 3.1: CAR mRNA levels increase in response to the AhR-ligand BNF.
Six adult C57BL/6J mice were injected, either with 10 mg/kg BNF or vehicle control for5 h. mRNA isolated from liver samples was reverse transcribed and quantified by qPCRusing CAR-specific primers. The data represent the relative fluorescence units for eachsample, obtained after normalization to GAPDH. p-value < 0.02, as determined by t-test.

95
Presence of AhR is essential for BNF-mediated increase in CAR mRNA:
BNF is an established AhR ligand and to the best of our knowledge, there are no
reports confirming the ability of BNF to directly activate any transcription factor other
than AhR. However, the possibility of a BNF-mediated effect that is independent of AhR
has to be considered. BNF exposure is known to cause oxidative stress, which can
possibly activate the nuclear factor erythroid 2 related factor 2 (Nrf2). Nrf2 is a
transcription factor known to regulate genes involved with protection against cellular
stress, such as Nqo1 (253). Additionally, Nqo1 is upregulated directly by AhR. Since the
AhR knock-out (AhR-KO) mice are devoid of AhR transcriptional activity, any increase
in Nqo1 mRNA in AhR-KO mice can be attributed to oxidant stress.
To exclude the contribution of such an effect in the observed upregulation of
CAR mRNA, age-matched AhR-KO (77) and wild-type mice were injected with 50
mg/kg BNF or vehicle alone for 5 h. As compared to the previous experiment, mice were
treated with a higher dose of BNF to definitively exclude such an effect. Liver CAR
mRNA levels did not demonstrate a significant difference between BNF and vehicle
treated AhR-KO mice, as determined by qPCR (Figure 3.2 B). Also, Nqo1 mRNA levels
were the same between control and BNF treated AhR-KO mice (data not shown). This
suggests that with the experimental parameters used in this study, the observed increase
in CAR mRNA is not a result of BNF generated oxidant stress. Wild-type mice
demonstrated an increase in CAR mRNA on BNF treatment similar to that in the
microarray experiments (Figure 3.2 B). Cyp1A1 mRNA levels were determined as a
control (Figure 3.2 A) for AhR activity. This finding, along with the absence of a

96
consensus DRE in the CAR promoter, makes it necessary to examine the mode by which
AhR influences CAR mRNA levels. Although it is possible to distinguish a secondary
effect from direct transcription with the use of a protein synthesis inhibitor like
cycloheximide, the inability to significantly induce transcription of CAR in established
cell-lines has precluded such experiments.

97
Figure 3.2: CAR up-regulation is AhR-dependent.
AhR knock-out (AhR KO) mice, or wild-type (WT) mice, were injected with 50 mg/kg BNF or vehicle alone. Liver mRNA, collected after 5 h, was reverse transcribed andquantified by qPCR using (A) Cyp1A1– and (B) CAR–specific primers. Normalized relative fluorescence values obtained with Cyp1A1, or CAR, primers are presented for individual mice. Statistical analysis was performed using t-test.

98
Temporal and spatial patterns of CAR upregulation mimics Cyp1A1:
As CAR plays a significant role in regulating many drug metabolism enzymes,
most of which are distinct from the known AhR-target genes, an increase in CAR levels
in response to AhR activation can significantly expand the role of AhR in controlling
xenobiotic metabolism. Time course experiments were performed to determine whether
AhR-mediated increase in CAR mRNA was sustained over a longer period. Adult mice
were injected 50 mg/kg BNF or vehicle alone and sacrificed at 6, 12 and 24 h. Maximal
induction (3-fold) of CAR was observed 6 h after BNF exposure and was sustained till 24
h (Figure 3.3 A). Induction of Cyp1A1, a known AhR target gene, demonstrated a similar
temporal pattern, although the level of induction was greater than that of CAR (data not
shown). The decline in CAR and Cyp1A1 mRNA at the later time-points is most likely
due to a decrease in BNF levels, and subsequent loss of AhR activity, as a result of
metabolism.

99
Figure 3.3: Temporal and spatial patterns of CAR expression.
(A) CAR mRNA levels were quantified by qPCR on liver samples from C57BL/6J micetreated with 50 mg/kg BNF or vehicle for the indicated times. Data are represented asmean (n=4) and standard deviation; *p-value<0.05 as determined by t-test. (B) qPCR quantification of CAR mRNA levels in kidney obtained from mice treated for6 h in the above time-course experiment. *p-value<0.05 as determined by t-test.

100
Increase in CAR levels in extra-hepatic tissues could significantly complement
the hepatic clearance of xenobiotics. The ability of AhR to upregulate CAR mRNA in
kidney and small intestine, both of which play a significant role in drug metabolism, was
determined by qPCR. A statistically significant increase in CAR mRNA levels was
observed in kidney after 6 h of 50mg/kg BNF treatment (Figure 3.3 B). CAR
upregulation (2.2-fold) was also noted in small intestine (terminal ileum); however,
increased inter-sample variability prevented statistical verification of the results (data not
shown).
Increase in CAR mRNA results in increased CAR transcriptional activity:
We wanted to confirm that an increase in CAR mRNA would lead to a
corresponding increase in the functional capacity of CAR, as determined by changes in
CAR-mediated transcription of its target genes. Cyp2b10 mRNA levels were chosen as a
marker of CAR’s transcriptional activity. Previously, it has been reported that activation
of CAR by TCPOBOP, a mouse-CAR-specific ligand, leads to an increase in hepatic
Cyp2b10 levels in mice (254). Adult mice were treated with either BNF or vehicle
control, to upregulate CAR levels. Six hours later, the mice were treated with TCPOBOP
to activate CAR protein, or with vehicle control. Three hours after TCPOBOP treatment,
liver Cyp2b10 mRNA levels were measured by qPCR. Mice treated with BNF-
TCPOBOP demonstrated 2.4-fold increase in hepatic Cyp2b10 mRNA compared to
vehicle-TCPOBOP (Figure 3.4 B). These results demonstrate that the observed increase
in CAR mRNA translates to a similar increase in CAR activity. Interestingly, even in the
absence of subsequent TCPOBOP treatment, an increase (2-fold) in Cyp2b10 mRNA was

101
observed in BNF treated mice compared to vehicle (Figure 3.4 B). However, the absolute
levels of Cyp2b10 mRNA were significantly lower without TCPOBOP exposure. Based
on these results, it is reasonable to assume that the AhR-dependent increase in CAR
mRNA leads to a functionally significant change in CAR activity. Although a Western
blot would serve to directly confirm an increase in CAR protein, the lack of a quality
commercially available antibody to murine-CAR has prevented the demonstration of such
an increase in protein level.

102
Figure 3.4: AhR-dependent CAR up-regulation leads to increased CAR-mediated transcriptional activity. Adult C57BL/6J mice were treated with an AhR ligand (50 mg/kg BNF for 6 h), to up-regulate CAR levels, or vehicle control. Subsequently, the mice were treated with a CAR ligand (3 mg/kg TCPOBOP for 3 h), or vehicle control, to activate CAR protein. CAR(A), Cyp2b10 (B) and PXR (C) mRNA levels were quantified in liver samples by qPCR.BNF/TCPOBOP treatment is indicated by a ‘+’ beneath the bars. Absence of a ‘+’ indicated vehicle treatment. Data are represented as mean (n=4) and standard deviationfor each group. Statistical analysis was performed by t-test.

103
Pregnane-X-receptor (PXR), another transcription factor involved in regulating
xenobiotic metabolism enzymes, is also capable of inducing Cyp2b10 mRNA (242).
Alterations in PXR levels were monitored by qPCR to determine if it contributed to the
observed increase in Cyp2b10. PXR mRNA levels were found to be similar across all
treatments (Figure 3.4 C), confirming that the increase in Cyp2b10 mRNA was most
likely due to the AhR-mediated increase in CAR levels.
CAR induction in primary human hepatocytes with different AhR ligands:
CAR mRNA levels were determined after treating primary human hepatocytes
with TCDD and benzo[a]pyrene (BaP). 10 nM TCDD treatment for 24 h and 1 μM BaP
for 12 h resulted in a statistically significant increase in CAR mRNA as compared to
control ( Figure 3.5 ). Increase in CAR mRNA with BaP treatment for 24 h was less than
that observed at 12 h, possibly due to faster metabolism of BaP as compared to TCDD.
Although there is only a modest increase in CAR mRNA levels, it should be noted that
this increase is in presence of 25 nM dexamethasone in the media. As discussed
previously, dexamethasone is known to induce CAR level (247). Increase in CAR mRNA
was further confirmed in primary human hepatocytes obtained from a second individual,
as shown in Figure 3.5 .

104
Figure 3.5: CAR induction in response to AhR ligands in primary human hepatocyte culture. Primary human hepatocytes were cultured in 6-well plates and treated with 10 nM TCDD or 1 μM BaP for the indicated time periods. RNA was isolated and analyzed by qPCR.Bars represent mean and standard deviation of triplicate treatments for the firstindividual. * indicates statistical significance at p<0.05, as determined by t-test. The same experiment was repeated in duplicate for the second individual.

105
3.5 DISCUSSION
Transcription factors play a significant role in coordinating the responses of a cell
to various external stimuli. Traditionally, these factors are thought to function by binding
to defined consensus DNA sequences and driving the transcription of a certain array of
target genes. However, a number of receptors have also been found to function in an
alternate manner by influencing the functional capacity of other receptors. This
significantly expands the range of effects attributed to the individual receptors. At the
same time it also makes it challenging to interpret the results of genome-wide studies. As
observed in the results of microarray experiments performed as a part of this study, AhR-
activation leads to alteration in the expression of a variety of genes. The number of genes,
whose expression was repressed by AhR activation, is small as compared to the number
of up-regulated genes (7 down-regulated genes versus 73 up-regulated genes). A probable
explanation for this discrepancy is the short time period of AhR activation used here.
Although 5 h is enough to upregulate the expression of direct as well as, in some cases,
indirect target genes, it might not allow adequate time to notice a decline in the levels of
down regulated genes.
AhR has been demonstrated to extensively participate in cross-talk with a number
of other receptors. It can directly interact with the NF-κB (173), the retinoblastoma (RB)
protein (210), Sp1 transcription factor (232) and the estrogen receptor. Interaction of the
AhR with each of these proteins has been shown to modulate signal transduction by these

106
proteins. In the case of AhR-ER, crosstalk has been postulated to arise from multiple
mechanisms including binding to inhibitory DREs, ER ligand depletion due to increased
metabolism by AhR induced enzymes, AhR-mediated proteasome-dependent ER
degradation and inaccessibility to their respective response element because of protein-
protein interaction (158, 161, 242). In this report, the ability of AhR to influence CAR-
mediated signal transduction exemplifies regulation of receptor quantities as an additional
mechanism of AhR-crosstalk.
Like AhR, CAR has been implicated in regulating the expression of a number of
xenobiotic metabolism enzymes. CAR and PXR play a significant role in controlling the
levels of Cyp2B and Cyp3A family of enzymes. These enzymes are believed to influence
the metabolism of a number of currently available drugs (255). An alteration in the
functional capacity of CAR, or of PXR, would affect the levels of these enzymes and
potentially generate unanticipated changes in the pharmacokinetic properties of their
target drugs. The observations recorded in this study demonstrate that in vivo activation
of AhR alters Cyp2b10 levels by mediating an increase in CAR, without affecting the
expression of PXR. This can have significant practical implications as AhR can be
activated in response to a wide variety of compounds that are ubiquitous and are often
encountered by humans. Common examples of AhR ligands include benzo[a]pyrene (a
constituent of cigarette smoke), TCDD (generated by combustion of organic matter),
flavones (present in common food items), indole 3-carbinol (found in cruciferous
vegetables), tryptophan metabolites as well as drugs like omeprazole (reviewed in (26)).
As shown in Figure 3.5, AhR-mediated CAR induction occurs in human cells as well,

107
and therefore is not limited to only the murine model. We also demonstrate that a variety
of AhR ligands, as exemplified by the use of TCDD and BaP, can lead to increased CAR
levels. Thus, varying degrees of exposure to AhR ligands can result in significant inter-
individual differences in CAR activity and consequent disparity in xenobiotic metabolism
potential.
The physiological role of CAR extends beyond that of regulating xenobiotic
metabolism. Because of its ability to control the expression of genes involved in hepatic
uptake and clearance of bilirubin, CAR has been implicated in stress response during
periods of hyperbilirubinemia (256). Also, bilirubin and biliverdin can function as AhR
ligands (39, 40). It is possible that AhR-dependent increase in CAR levels is one of the
mechanisms by which bilirubin indirectly increases CAR activity to protect the body
from the adverse effects of elevated bilirubin levels. Recent reports have also associated
CAR activity with thyroid hormone homeostasis during states of restricted calorie intake
(250) as well as with hepatocyte proliferation (257) and tumor formation (258). An AhR-
mediated increase in CAR activity may potentially alter the outcome of these states as
well.
The activity of many transcription factors is regulated by their tissue-specific
expression. Constitutive expression of CAR is principally observed in liver and small
intestine, and most studies related to CAR have explored its functions primarily in a
hepatic context. In contrast, AhR is expressed in a wide variety of tissues and can
potentially induce CAR levels in these tissues, as demonstrated by an increase in renal

108
CAR expression reported here. Even in hepatic tissue, where maximal constitutive CAR
levels are observed, it is possible to increase CAR activity by increasing its levels.
Figure 3.4 clearly demonstrates that an increase in CAR (approximately 2-fold) resulted
in a proportionate increase in its transcriptional activity as demonstrated by a 2-fold
increase in Cyp2b10 mRNA. Collectively, these results suggest that in extra-hepatic
tissues, where the constitutive level of CAR is extremely low or absent, AhR-dependent
induction of CAR expression can significantly enhance its functions and might reveal
additional roles for this receptor. Additional experiments, such as profiling genome-wide
expression changes in extra-hepatic tissues as well as effects of long-term exposure to
AhR-ligands in wild-type and CAR knock-out mice, can provide further information.
In conclusion, these results demonstrate the ability of activated AhR to increase
CAR activity, which can alter drug metabolism. Inhalation of cigarette smoke has been
known to markedly increase Cyp1A1 levels in murine livers, thus indicating AhR
activation (259). Similar circumstances can lead to unexpected changes in CAR-
dependent drug metabolism. Thus, it may be important to be aware of the effects of AhR
activation while determining the dosage of certain drugs in different patient populations.
Additionally, the direct transcription models of receptor function cannot explain all the
observations from large-scale gene expression studies. There is clearly a need to explore
different mechanisms by which transcription factors can influence gene expression. Our
results demonstrate a link between AhR and CAR, and although further studies are
required to determine the exact molecular mechanism, the information presented here

109
will be helpful in interpreting experimental results related to AhR and CAR, particularly
in liver.

110
Chapter 4
AHR REPRESSES CYTOKINE MEDIATED ACUTE PHASE RESPONSE BY A DNA-INDEPENDENT MECHANISM.

111
4.1 Abstract
In the recent years, there has been considerable focus on identifying novel
mechanisms for transcription factor activity. DNA-binding independent effects have been
identified for receptors such as GR and ER. An endpoint for analyzing these effects has
been the ability to influence the activity of other transcription factors. We investigated the
ability of AhR to function in the absence of direct DNA-binding. A previously
characterized DNA-binding mutant (A78D) form of AhR, along with the wild-type (WT)
form of AhR, was transiently expressed in Simian virus 40 immortalized AhR-null mouse
hepatocytes. Changes in gene expression were analyzed using Affymetrix microarrays.
Serum amyloid A3 (Saa3) was one of the genes whose expression was repressed 2-fold
with the DNA-binding mutant (A78D) form as well as the WT from of AhR. Saa3 is an
acute-phase protein and is significantly induced by pro-inflammatory cytokines,
particularly interleukin (IL)-6 and IL-1β. Microarray results were verified with real-time
PCR in an independent experiment, which also included two additional mutants of AhR –
a heterodimerization and a nuclear localization mutant – neither of which were able to
repress Saa3 expression. Subsequent experiments, conducted in Hepa1c1c7 cells,
demonstrated that AhR activation with different ligands can suppress IL-mediated Saa3
mRNA expression. Moreover, the fact that TCDD failed to repress Saa3 expression in
primary AhR-null hepatocytes demonstrated that the effect is AhR-dependent. Chromatin
immunoprecipitation assays revealed that AhR activation diminishes the acetylation of
histones as well as recruitment of the p65 component of NFκB at the Saa3 promoter in

112
response to IL-6 and IL-1β. Other acute-phase response genes, notably Saa1, Saa2 and C-
reactive protein, were also observed to be significantly repressed on AhR activation.
Finally, AHR-mediated inflammatory inhibition was verified in human liver cells. Our
results provide evidence for a DNA-binding independent mode of AhR activity that may
be critical in influencing inflammatory outcomes. This study also demonstrates that AhR
can modulate the acute-phase response, thus establishing a role for AhR in inflammatory
signaling.

113
4.2 Introduction
All organisms are programmed to react rapidly to environmental as well as
intrinsic challenges by inducing physiological alterations that favor survival. The acute
phase response (APR) is a set of such alterations that enable an organism to restore
homeostasis in response to insults, such as infection, inflammation, stress and even
neoplasm. Acute phase changes can be broadly classified into two groups – first,
neuroendocrine and behavioral changes such as fever, somnolence, anorexia and
lethargy, and second, an alteration in the expression of certain proteins, known as acute
phase proteins (APP) (reviewed in (260)). Examples of APP include members of the
complement and coagulation cascades, antiproteases, transport proteins and, the most
famous of all, C-reactive protein (CRP) and serum amyloid A (SAA). Liver serves as the
primary site of synthesis and secretion of acute-phase proteins. Collectively, an increase
in the expression of these proteins serves to facilitate the immune and metabolic
responses necessary to restore homeostasis. Induction of most acute phase proteins is
mediated by cytokine signaling (261) through the activation of NF-κB, CEBP and Stat
pathways.
NF-κB is a transcription factor that functions as a principal regulator of
inflammatory and immune responses. It exists as a dimer formed by various
combinations of seven proteins translated from five NF-κB/REL genes (reviewed in
(262)). The majority of gene regulatory effects mediated by NF-κB are attributed to the

114
heterodimer composed of the p50 and RELA (p65) subunits. Under basal conditions, the
RELA/p50 complex is sequestered in the cytoplasm by its association with a member of
the IκB family of inhibitor proteins, IκBα (263, 264). Cytokine stimulation leads to
phosphorylation, ubiquitylation and subsequent degradation of IκB by the proteasome
complex. This releases NF-κB which then translocates to the nucleus and binds cognate
κB elements to drive transcription of its target genes. Phosphorylation (265-269) and
acetylation (270, 271) act as nuclear molecular switches to modulate the transcriptional
potential of NF-κB (272).
A number of activated nuclear receptors can interact with NF-κB signaling to alter
biological outcomes (164, 165). Recently, there has been evidence suggesting that aryl
hydrocarbon receptor (AHR) also influences NF-κB activity (173). Traditionally, AHR
activation has been associated with induction of enzymes involved in xenobiotic
metabolism. However, there is a growing body of evidence implicating a role for AHR in
a wide array of cellular processes (137, 142) and DRE-dependent induction of target
genes is unlikely to be able to explain all the patho-physiological effects attributed to
AHR.
Receptor cross-talk with other transcription factors has been an exciting emerging
aspect of nuclear receptor biology. Understanding the networking of various signaling
pathways can elucidate hitherto unexplained physiological phenomena. Here, we present
evidence demonstrating the ability of the aryl-hydrocarbon receptor (AHR) to repress
nuclear factor-kappa B (NF-κB) mediated induction of an important aspect of

115
inflammatory reaction – the acute phase response (APR). Cytokine mediated induction of
an APR gene, serum amyloid A (Saa), can be significantly repressed by activated AHR in
mouse and human cell lines as well as primary mouse hepatocytes. The observed
repression is due to a direct effect on Saa transcription and may involve NF-κB
regulation at multiple levels. Interestingly, DNA-binding is not essential for this new
AHR function. A previously described AHR mutant (273) incapable of binding to its
cognate response element (A78D-AHR) repressed Saa transcription with the same
efficiency as the wild-type AHR. Since DNA affinity studies have revealed that AHR
binds only to a unique cognate DNA sequence, we believe that A78D-AHR can be
described as a DNA-binding mutant. AHR-mediated repression is not limited to Saa, but
extends to an array of other APR genes as well. This is the first report demonstrating the
ability of AHR to repress a facet of inflammatory response in a non-traditional manner.

116
4.3 Materials and Methods:
Reagents:
Cycloheximide, Actinomycin D and TSA were purchased from Sigma Co. TCDD
was a kind gift from Dr. Stephen Safe, Texas A & M University. Anti-RELA antibody
(sc-372) was purchased from Santa Cruz Biotechnology Inc. and anti-acetyl-histone H4
(Lys5) antibody was purchased from Upstate. Recombinant interleukins were purchased
from PeproTech Inc. Hepa1c1c7 and Huh7 cells were obtained from American Type
Culture Collection. Primers for PCR reactions were purchased from Integrated DNA
Technology.
Cell Culture:
Hepa1c1c7 and Huh7 established cell-lines were cultured in α-minimum essential
medium under 5% CO2 at 37°C. Primary bone marrow (BM) cells were isolated from
lower limb bones of 8-12 weeks old C57BL/6 mice. Cells were flushed out and were
dispersed by gentle manipulation with a 1 ml pipette. BM cells were cultured overnight in
Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 8% fetal bovine
serum (FBS) and penicillin/streptomycin. Non-adherent cells were centrifuged and plated
in DMEM supplemented with 10 ng/ml granulocyte-monocyte colony stimulating factor
(GM-CSF) and 2 mM glutamine. Half the volume of medium was replaced everyday for
4 days prior to treatment.

117
RNA isolation and Real-time PCR:
RNA was isolated from cells with TRI® reagent (Sigma-Aldrich Co.) and reverse
transcribed with High Capacity cDNA Archive kit (Applied Biosystems ). Quantitative
real-time PCR was performed on iQ systems (BioRad) using iQ SYBR Green master mix
(BioRad), according to the manufacturer’s protocol. Expression values of genes of
interest were normalized to that of the housekeeping gene ribosomal protein L13a
(RpL13a).
Chromatin Immunoprecipitation assay:
Chromatin immunoprecipitation assays (ChIP) were performed as described
previously in Chapter 2. Briefly, cells cultured in 150 mm culture dishes were crosslinked
with 1% formaldehyde for 8 min at 37°C and sonicated to generate 500 - 700 bp
fragments. The sonicated lysate was diluted to 2 A260 units and 1 ml of this lysate was
subjected to immunoprecipitation with the respective antibodies and protein sepharose A
resin. Enrichment of promoter fragments was determined by PCR/real-time PCR. Primer
sequences are:
Saa1ChIP-F: AGAGCGACACACACACACTGTCTT Saa1ChIP-R: AGGTGAGAGGAGGCAGGCATTTAT Saa2ChIP-F: TACTACACCCCAGAAGATTGCCAC Saa2ChIP-R: AGGTGAGAGGAGGCAGGCATTTAT Saa3ChIP-F: GCGCAATCTGGGGAAAGAAGATGT Saa3ChIP-R: TGAGTGGCTTCTGTCCTTTGCTGA

118
siRNA:
AHR knock-down was attained with siRNA oligos purchased from Dharmacon
RNAi Technologies, Thermo Fisher Scientific Inc. Approximately 60% confluent Huh7
cells were transfected with 120 nM scrambled or anti-AHR oligos using 6 μl Dharmafect-
1 transfection reagent. Culture medium was changed after 24 h and the cells were
allowed to recover for an additional 12 h before treatment.
ELISA:
Huh7 cells were treated with TCDD and interleukins for 10 h or 24 h under
serum-free conditions. 100 μl of culture media was analyzed for SAA protein levels using
Human SAA ELISA kit purchased from Anogen, Yes Biotech Laboratories Ltd.,
according to the manufacturer’s protocol.
Microarrays:
RNA was isolated from 106 sorted cells using TRI® reagent and cleaned with
RNeasy® columns. RNA integrity was confirmed by Bioanalyzer (Applied Biosystems).
Samples were then hybridized to Affy mouse 2.0A genome chips. Labeling, hybridization
and washing was performed at the microarray core facility, the Pennsylvania State
University. Data was processed and significantly altered genes were identified using
GeneChip Operating Software (GCOS). Genes that were declared as increased or as
decreased in wild-type AHR (WT-AHR) and DNA-binding mutant AHR (A78D-AHR)
transfected cells, as compared to control transfections were enlisted.

119
Animal care:
C57BL6/J mice were maintained at the Penn State University’s animal care
facility. Animals had ad libitum access to regular chow and water, and were maintained
on a 12 h light cycle. Animals were euthanized by CO2 exposure. Guidelines for animal
care provided by the Pennsylvania State University’s animal care program were adhered
to. Conditional AHR knock-out mice (Ahrfx/fxCreAlb) were kindly provided by Dr.
Christopher Bradfield.
Statistics:
Results were analyzed by t-test and deemed significant at p<0.05, unlessotherwise
noted.

120
4.4 Results:
Identification of DNA-binding independent effects of AHR
The established mechanism of AHR function involves binding of the AHR-ARNT
heterodimer to DNA with a consensus DRE sequence. As discussed, this mechanism is
unlikely to be able to explain most of the effects observed with AHR activation or those
observed in the AHR knock-out mouse. In an effort to determine the possibility of a
DNA-binding independent manner of AHR function, simian virus 40 (SV40)
immortalized AHR null mouse hepatocytes (274) were transfected with either the wild-
type (WT) AHR or DNA-binding mutant (A78D) AHR expressing plasmid or the empty
vector. The A78D-AHR mutant has previously been shown to bind ligand, translocate to
the nucleus and heterodimerize with ARNT, yet it fails to bind DNA at its response
element. Green fluorescent protein (GFP) expressing plasmid was cotransfected along
with AHR in a ratio of 1:3. Transfected cells were sorted for GFP expression and RNA
and protein were isolated from the sorted cells. AHR expression was confirmed by
Western blot (Figure 4.1 A) and the RNA was used for microarray experiments. Data
analysis was performed to identify the subset of genes altered in WT-AHR as well as
A78D-AHR transfected cells but not in the control (Table 4.1). Saa3 was found to be
repressed by the wild-type and DNA-binding mutant AHR, and was selected for further
analysis. Conversely, Cyp1a1 was upregulated only by WT-AHR, and not by A78D-
AHR.

121
Gen
e Ti
tle
Gen
e Sy
mbo
l
Rat
io
A78
D v
s C
ontr
ol
Rat
io
WT
vs
Con
trol
aryl-hydrocarbon receptor Ahr 2.6 2.6 trimethyllysine hydroxylase, epsilon Tmlhe 2.1 2.7 LIM domain containing preferred translocation partner in lipoma Lpp 1.7 2.2 acidic (leucine-rich) nuclear phosphoprotein 32 family, member A Anp32a 1.7 1.8 denticleless homolog (Drosophila) Dtl 1.6 1.6 DNA segment, Chr 9, ERATO Doi 306, expressed D9Ertd306e 1.5 1.6 PREDICTED: LIM domain only 7 [Mus musculus], mRNA sequence
Lmo7 1.5 1.7
germ cell-less homolog (Drosophila) Gcl 1.4 1.6 zinc finger protein 207 Zfp207 1.4 1.6 WD repeat domain 26 Wdr26 1.4 1.6 protein tyrosine phosphatase, receptor type, J Ptprj 1.4 1.7 RIKEN cDNA 3300001H21 gene 3300001H2
1Rik 0.7 0.6
zinc finger protein 179 Zfp179 0.7 0.6 Ceruloplasmin Cp 0.7 0.5 procollagen, type VI, alpha 1 Col6a1 0.7 0.6 matrilin 2 Matn2 0.7 0.6 interferon, alpha-inducible protein 27 Ifi27 0.7 0.5 vanin 3 Vnn3 0.7 0.6 Transferring Trf 0.7 0.6 procollagen, type VI, alpha 2 Col6a2 0.7 0.6 procollagen, type VI, alpha 2 Col6a2 0.7 0.6 myxovirus (influenza virus) resistance 1 Mx1 0.7 0.6 complement component factor h Cfh 0.7 0.6 complement component 1, s subcomponent C1s 0.7 0.5 slit homolog 3 (Drosophila) Slit3 0.7 0.6 Kruppel-like factor 10 Klf10 0.7 0.6 lipocalin 2 Lcn2 0.6 0.6 lipopolysaccharide binding protein Lbp 0.6 0.5 serine (or cysteine) peptidase inhibitor, clade A, member 3M Serpina3m 0.6 0.6 proteoglycan 4 (megakaryocyte stimulating factor, articular superficial zone protein)
Prg4 0.6 0.5
complement component 3 C3 0.6 0.5 FBJ osteosarcoma oncogene Fos 0.6 0.4 serine (or cysteine) peptidase inhibitor, clade A, member 3N Serpina3n 0.5 0.5 STEAP family member 4 Steap4 0.5 0.4 serine (or cysteine) peptidase inhibitor, clade A, member 3G Serpina3g 0.5 0.6 serum amyloid A 3 Saa3 0.4 0.4
Table 4.1: List of genes regulated by A78D-AHR and WT-AHR.

122
Control
WTA78
D H1Δ K14
A0
25
50
75
100
* *
Saa3
mR
NA
Control
WTA78
D H1Δ K14
A0
100
200
300
400
500
600
Cyp
1A1
mR
NA
A
B
C
D
Figure 4.1: Functional dissociation of the properties of AHR involved in Saa3repression. (A) Western blot analysis of WT-AHR and A78D-AHR protein expression in SV40 immortalized AHR-null mouse hepatocytes transfected with a combination of GFP andWT-AHR/A78D-AHR/control vector in a ratio of 1:3, using Lipo2000 transfectionreagent. 106 cells were sorted for GFP expression. (B) Schematic representation of murine WT-AHR domains and the deletion/mutation (arrowheads) for each AHR mutant. (C) and (D) Real-time PCR on RNA isolated from SV40 immortalized AHR-null hepatocytes transfected with WT-AHR or various AHR mutants for 24 h. Data representsmean and standard deviation of triplicate measurements obtained from two independentexperiments. WT, wild-type; A78D, DNA-binding mutant; ΔH1, heterodimerization mutant; K14A, nuclear localization mutant.

123
Generation and characterization of AHR-heterodimerization mutant
It is necessary to examine whether heterodimerization of AHR and ARNT is
essential for the DNA-independent effects of AHR. An AHR-heterodimerization mutant
(ΔH1-AHR) was made by deleting amino acids 43 – 51, which encompasses the helix-1
of the helix-loop-helix domain. Experiments involving characterization of ΔH1-AHR
mutant were performed by a colleague and will not be described here.
Saa3 repression requires heterodimerization and nuclear translocation
The conventional AHR pathway requires nuclear translocation and
heterodimerization with ARNT. A previously described AHR mutant (K14A-AHR)
incapable of translocating to the nucleus (67, 74) and the heterodimerization mutant
(ΔH1-AHR) described above, were expressed in SV40-immortalized AHR knock-out
cells along with A78D-AHR and ΔH1-AHR 4.2 . Saa3 expression was found to be
repressed by the WT-AHR and A78D-AHR, but not by K14A-AHR and ΔH1-AHR
(Figure 4.1 C). Thus, DNA-binding independent gene regulation by AHR appears to
require nuclear translocation and heterodimerization. As expected, Cyp1a1 – a gene
known to be upregulated through the conventional activated AHR pathway, was induced
in the cells transfected with WT-AHR, but not with A78D-AHR or ΔH1-AHR
(Figure 4.1 D). K14A-AHR minimally induced Cyp1a1; however, this may be due
overexpression that may occur in transient transfection experiments leading to a certain
degree of diffusion of the receptor into the nucleus. It should be noted that activation of

124
AHR by TCDD did not alter the extent of Saa3 repression in SV40-immortalized AHR
null hepatocytes (data not shown).

125
Figure 4.2: AHR functional mutants.
Simplified AHR pathway is presented. Critical steps are marked by arrowheads –chaperone binding, ligand-binding, nuclear translocation, AHR-ARNT heterodimerization and DNA-binding at the DRE. Different AHR mutants (A78D, ΔH1 and K14A) result in a progressive loss of functionalities, as depicted by a reducingnumber of arrowheads.

126
Saa3 repression under different experimental conditions:
AHR-mediated Saa3 repression observed in immortalized AHR null cells was
confirmed in other model systems to ensure that the observed effect was not an artifact of
the SV40 driven immortalization. Saa3 transcription was induced in Hepa1c1c7 cells, a
mouse hepatoma derived cell-line, by treatment with different pro-inflammatory
cytokines – interleukin-1β (IL1B), interleukin-6 (IL6), tumor necrosis factor-α (TNFA)
or a combination of IL1B and IL6 ( Figure 4.3 A and B). Activated AHR repressed the
induction of Saa3 by approximately fifty percent for each cytokine treatment.

127
Control
TCDDIL-6
TCDD + IL-6
+ IL-6
βIL-1
+ IL-6
β
TCDD + IL-1
0
5
1040
60
80
100
Saa3
mR
NA
*
*
Control
TCDD βIL-1 β
TCDD + IL-1 α
TNF α
TCDD + TNF
0
25
50
75
100
*
*
Saa3
mR
NA
A
B
Figure 4.3: AHR represses Saa3 induction by various cytokines.
Hepa1c1c7 cells were treated with 10 nM TCDD or vehicle control. After 30 min, cells were treated with 2 ng/ml IL1B, IL6, TNFA or a combination of IL1B and IL6 for anadditional 6 h. Data represents mean and standard deviation of triplicate measurements.

128
Repression of Saa3 induction by AHR ligands provides an interesting therapeutic
avenue. However, it is essential to confirm that Saa3 repression is not limited to higher
doses of an AHR ligand. To this end, repression of cytokine-induced Saa3 was studied
with decreasing doses of TCDD in Hepa1c1c7 cells. TCDD was able to effectively
repress Saa3 even at the lowest dose tested (200 pM) (Figure 4.4 A). Benzo[a]pyrene
(B[a]P), beta-naphthoflavone (β-NF), alpha-naphthoflavone (α-NF) and M50354 are
examples of established AHR ligands. M50354 is a recently described AHR agonist
compound capable of attenuating atopic allergic responses (275, 276). Hepa1c1c7 cells
were treated with different AHR ligands (26) to determine if Saa3 repression was a
TCDD-specific effect. The established AHR ligands, were all able to repress Saa3
induction (Figure 4.4 C).

129
Control M)
μ
NF (2
α
M)μ
NF (2
β
M)μ
B[a]P (2
M)μ
M5035
4 (2
TCDD (1 nM)
024
6
8
1012
14
Saa3
mR
NA
** * * *
Control M)
μ
NF (2
α
M)μ
NF (2
β
M)μ
B[a]P (2
M)μ
M5035
4 (2
TCDD (1 nM)
0
10
20
30
Cyp
1a1
mR
NA
-0.2 0.2 0.6 1.0 1.4 1.80123
TCDDTCDD + IL
8 10
10
20
30
40
50
TCDD (nM)
Saa3
mR
NA
(fold
cha
nge)
-0.2 0.2 0.6 1.0 1.4 1.80
10
20
30
40
50
60 TCDD
8 10TCDD (nM)
Cyp
1a1
mR
NA
(fold
cha
nge)
C
D
A
B
Figure 4.4: Dose-response and ligand-specificity analysis of AHR mediated repression of Saa3. (A and B) Analysis of TCDD dose-response of AHR-mediated Saa3 repression.Hepa1c1c7 cells treated with increasing doses of TCDD (0.2 nM to 10 nM) for 30 minprior to interleukin (IL1B + IL6, 2ng/ml each) treatment. (A) Closed triangles represent repression of IL-induced Saa3 mRNA by various doses of TCDD, as determined by real-time PCR. Closed squares represent uninduced Saa3 mRNA levels, as a control. (B) TCDD-driven Cyp1a1 mRNA induction, as measured by real-time PCR. (C and D) Various classes of AHR ligands can suppress Saa3. Hep1c1c7 cells were treated with different AHR ligands at the described doses for 30 minprior to interleukin (IL1B + IL6, 2ng/ml each) treatment. Saa3 mRNA (C) and Cyp1a1mRNA (D) were measured by real-time PCR. Data represents the mean and standarddeviation of triplicate measurements.

130
AhR mediated Saa3 repression is a direct transcriptional effect
In order to ascertain that AHR directly effects the transcription of Saa3,
Hepa1c1c7 cells were pretreated with cycloheximide, a translation inhibitor. Though
cycloheximide treatment elevated the constitutive level of Saa3 expression, it did not
alter its repression by AHR activation (Figure 4.5 A, B and C). This indicates that AHR-
mediated Saa3 repression is a direct effect and not secondary to changes in the expression
of another protein. Gene repression can be mediated by a decrease in transcription rate or
by alteration of mRNA stability. AHR could also possibly upregulate the expression of a
microRNA which could regulate Saa3 mRNA levels. After challenging with TCDD and
interleukins, Hepa1c1c7 cells were treated with Actinomycin D, a transcription inhibitor,
and followed to 4 hours for changes in Saa3 mRNA level. Decay rate of Saa3 mRNA
was not altered by activated AHR and Saa3 mRNA appeared to be quite stable
(Figure 4.5 D).

131
0 1 2 3 40
500
1000
15002000
2500
30003500
DMSOTCDD
Time (h)
Saa3
mR
NA
Control
TCDD βIL-1 β
TCDD + IL-1
Control
TCDD βIL-1 β
TCDD + IL-1
0100200300400500600700800900
No CHXCHX
*
*Saa3
mR
NA
Control
TCDDIL-6
TCDD + IL-6
Control
TCDDIL-6
TCDD + IL-6
0
50
100
150
200
No CHXCHX
*
*
Saa3
mR
NA
Control
TCDD αTNF α
TCDD + TNF
Control
TCDD αTNF α
TCDD + TNF
0250500
7501000
125015001750
No CHXCHX
*
*Sa
a3 m
RN
AA B
C D
Figure 4.5: AHR-mediated Saa3 repression is due to direct transcriptionalinhibition. Real-time PCR on RNA from Hepa1c1c7 cells treated first with (black bars), or without(open bars), the translational inhibitor – cycloheximide (10 µg/ml) for 30 min, then with TCDD (10 nM) for 30 min and finally with one of the cytokines – TNFα (A), IL1β (B) or IL6 (C) for 6 h. Data represent the mean and standard deviation of triplicatemeasurements. (D) Real-time PCR to measure the effect of activated AHR on Saa3 mRNA decay rate. Hepa1c1c7 cells were treated with vehicle control or TCDD (10 nM) for 30 min and then with interleukins (2 ng/ ml each of IL1β and IL6) for 3 h to induce Saa3. Then, the cells were treated with the translational inhibitor, Actinomycin D and RNA samples collectedat 30 min, 1 h, 2 h and 4 h. Data represent the mean and standard deviation of triplicate measurements.

132
AhR activation represses other Saa-family member genes
All members of the SAA family are upregulated simultaneously in an acute phase
response. Hence, we examined the effect of AHR activation on the expression of Saa1
and Saa2 in Hepa1c1c7 cells (Figure 4.6 A and B). Cytokine-mediated induction of both
Saa1 and Saa2 was repressed by AHR activation by 75 and 85 percent respectively. This
is significant as Saa1 and Saa2 are the major hepatic serum amyloid isoforms.
Interestingly, Saa1 and Saa2 did not appear to be repressed in the previous microarray
results from WT-AHR or A78D-AHR transfected SV40-immortalized mouse
hepatocytes, the reason for which is not clear.

133
Control
TCDD IL
TCDD + IL
0
25
50
75
100 *
Saa1
mR
NA
Control
TCDD IL
TCDD + IL
0
50
100
150
200
250 *
Saa1
mR
NA
Control
TCDD IL
TCDD + IL
0
30
60
90
120
150
Saa1
mR
NA
Control
TCDD IL
TCDD + IL
0
30
60
90
120
150 *
Saa2
mR
NA
Control
TCDD IL
TCDD + IL
0
50
100
150
200
250 *
Saa2
mR
NA
Control
TCDD IL
TCDD + IL
0
30
60
90
120
150
Saa2
mR
NA
A
C
E
B
D
F
Figure 4.6: AHR activation represses other Saa-family member gene expression.
(A and B) Real-time PCR on RNA from TCDD (10 nM, 30 min) followed by interleukin (2 ng/ ml each of IL1B and IL6) treated Hepa1c1c7 cells. Effect of AHR activation oninduction of other SAA family members, Saa1 (A) and Saa2 (B), was determined. Data represents the mean and standard deviation of triplicate measurements. Experiment was repeated thrice with similar results. (C and D) Real-time PCR on RNA from primary mouse hepatocytes treated with TCDD(10 nM for 30 min) followed by interleukin (2 ng/ ml each of IL1B and IL6) for 24 h.Prior to treatment, cells were transferred to α-MEM with 1 mg/ml bovine serum albumin for 24 h. Repression of Saa1 (C) and Saa2 (D) mRNA was measured. (E and F) Real-time PCR measurement of Saa1 (E) and Saa2 (F) mRNA, as described above, in AHR-deficient primary mouse hepatocytes obtained from Ahrfx/fxCreAlb mice (liver-specific AHR knock-out).

134
Saa repression is AhR dependent
AHR-deficient or AHR-expressing primary hepatocytes were isolated from
Ahrfx/fxCreAlb (hepatocyte-specific AHR knock-out) (92) or wild-type C57BL6/J mice
respectively. Saa transcription was induced in these cells by interleukins. TCDD was able
to restrict the induction of Saa1 and Saa2 in AHR-expressing (Figure 4.6 C and D), but
not in AHR-deficient hepatocytes (Figure 4.6 E and F). This, along with the observation
that AHR transfection in AHR knock-out cells is required for suppressing Saa3
(Figure 4.1 C), clearly establishes that AHR is essential for Saa repression.
Saa repression can occur under physiological conditions
Different cytokines can have counter-regulatory effects on various aspects of an
inflammatory response. It is possible that IL1β and IL6 mediated Saa induction might not
truly simulate the response obtained with a combination of cytokines, as expected in an
inflammatory response. To confirm the ability of AHR to repress Saa induction under
such circumstances, primary bone-marrow cells were isolated from C57BL6 mice and
were cultured to promote differentiation into macrophages. Following a three-day LPS
challenge, the conditioned culture medium was collected off of the macrophages and
used to treat Hepa1c1c7 cells. To differentiate the effect of secreted cytokines from those
of LPS, LPS-containing culture medium was incubated in the absence of macrophages
and used as a control. AHR-activation was able to repress the induction of Saa1, Saa2
and Saa3 in response to the macrophage-conditioned-media (Figure 4.7 A, B and C). This
demonstrates AHR’s ability to repress Saa induction under physiologically attainable

135
concentration/combination of cytokines. In contrast to Saa1 and Saa2, macrophage-
conditioned media was unable to induce Saa3 to a significantly higher level as compared
to LPS alone.

136
Control
TCDDMCM
TCDD + MCM
LPS
TCDD + LPS
0
100
200
300
400
500
600 *Sa
a1 m
RN
A
Control
TCDDMCM
TCDD + MCM
LPS
TCDD + LPS
0
25
50
75 *
Saa2
mR
NA
Control
TCDDMCM
TCDD + MCM
LPS
TCDD + LPS0
10
20
30
40
50
60
70 *
Saa3
mR
NA
A B
C
Figure 4.7: AHR represses Saa induction by physiologically attainable cytokineconcentrations. Primary murine bone marrow cells were cultured to promote differentiation intomacrophages, as outlined in the text. After a 3 d LPS challenge, the conditioned mediawas collected from macrophage containing plates (MCM – macrophage conditioned media) and used to treat Hepa1c1c7 cells for 6 h following TCDD (10 nM, 30 min) pre-treatment. ‘LPS’ refers to LPS-spiked media that was maintained under similar cultureconditions in the absence of any cells, and thus was devoid of any cytokines secreted bymacrophages. Saa1 (A), Saa2 (B) and Saa3 (C) mRNA levels were determined by real-time PCR. Data represent the mean and standard deviation of triplicate measurements.

137
Mechanistic insight into AHR mediated Saa repression
The fact that AHR directly represses Saa3 and that the K14A-AHR (nuclear
localization) mutant failed to repress Saa3 induction, indicate that AHR likely effects the
formation of transcription complex at the Saa promoters. Chromatin immunoprecipitation
(ChIP) assays in Hepa1c1c7 cells demonstrate that activated AHR reduced the presence
of RELA (p65) subunit of NF-κB at the Saa3 and Saa2 promoters in response to
interleukin treatment (Figure 4.8 A, B and C). AHR has previously been shown to
physically interact with RELA (172) and this might contribute to preventing RELA
recruitment to Saa promoters in response to interleukins.

138
Figure 4.8: ChIP assay to determine the effect of AHR activation on Saa1, Saa2 and Saa3 promoters. Hepa1c1c7 cells were treated with TCDD (10 nM for 30 min) prior to interleukins (2ng/ml each of IL1B and IL6 for 20 min). Immunoprecipitation was performed withantibodies for RELA and acetylated histones (K5). Changes at the Saa3 promoter were assessed by regular PCR (A), while changes at Saa1 and Saa2 promoters were analyzed by real-time PCR (B and C). Data represents one of three independent experiments.

139
Chromatin immunoprecipitation with an acetylated-histone 4 antibody
demonstrated that AHR activation also reduced histone acetylation at Saa1, Saa2 and
Saa3 promoters (Figure 4.8 A, B and C). These results lead us to explore the contribution
of histone deacetylases (HDAC) in AHR-mediated Saa repression. If AHR activation
increases HDAC activity at the Saa promoters to repress transcription, then trichostatin A
(TSA), an HDAC inhibitor, treatment should reverse the repression by blocking HDAC
activity (Figure 4.9 A). To this end, Hepa1c1c7 cells were treated with TSA before
activating AHR and NF-κB, and Saa1 and Saa3 mRNA levels were monitored by real-
time PCR (Figure 4.9 B and C). HDAC inhibition by TSA treatment increased the basal
rate of Saa1 transcription by two-fold. Interestingly, HDAC inhibition increased
interleukin-mediated Saa1 induction by only five-fold in the absence of AHR activation,
but, by ten-fold in the presence of AHR activation. Saa3 transcription demonstrated
similar changes. This clearly suggests that AHR-mediated Saa repression involves an
increase in HDAC activity. HDAC activity has previously been implicated in regulating
NFKB transcriptional activity (270, 271, 277). However, the results obtained here cannot
be precisely attributed to HDAC activity at the Saa promoter because, acetylation /
deacetylation have been known to regulate NFKB pathway at multiple levels (278).

140
Figure 4.9: Effect of HDAC inhibition on Saa expression.
(A) Schematic representation of the possible consequence of HDAC inhibition bytrichostatin A (TSA). Hepa1c1c7 cells were treated with TSA (30 min) prior to TCDD(10 nM for 30 min) and interleukin treatment (2 ng/ml each of IL1B and IL6 for 6 h). Consequence of TSA treatment on changes in Saa1 (B) and Saa3 (C) mRNA were determined by real-time PCR. Data is represented as fold-change caused by TSA as compared to the expression in the absence of TSA, under each treatment condition. Threemeasurements were averaged before obtaining fold-values.

141
Suppressors of cytokine signaling (SOCS) proteins exert an anti-inflammatory
effect through inhibition of the JAK-STAT pathway (recently reviewed in (279)) and
might be an additional mechanism for AHR-mediated Saa repression. In fact, SOCS2 has
been shown to be an AHR target-gene (118). The mRNA levels of SOCS1, SOCS2 and
SOCS3 in response to interleukin treatment in the presence or absence of AHR activation
was tested. In Hepa1c1c7 cells both TCDD and interleukins were able to induce SOCS2
and SOCS3 (Figure 4.10 A and C). Treatment with a combination of TCDD and
interleukins had an additive effect on the induction of these genes. However, in primary
mouse hepatocytes, only SOCS3 was inducible (four-fold) with interleukin treatment.
TCDD alone had no effect on SOCS3 expression in primary mouse hepatocytes, but, in
combination with interleukins, an eight-fold induction of SOCS3 was observed
(Figure 4.10 D). SOCS1 was not altered in either model.

142
DMSOTCDD IL
TCDD + IL
0
300
600
900 *SO
CS2
mR
NA
Control
TCDD IL
TCDD + IL
0
30
60
90
120
Socs
2 m
RN
A
DMSOTCDD IL
TCDD + IL
0
300
600
900
1200 **
SOC
S3 m
RN
A
Control
TCDD IL
TCDD + IL
0
20
40
60
80 *So
cs3
mR
NA
A B
C D
Figure 4.10: AHR activation induces SOCS genes.
Hepa1c1c7 cells (A and C) were treated with TCDD (10 nM for 30 min) prior tointerleukins (2 ng/ml of IL1B and IL6 for 6 h). Primary mouse hepatocytes (B and D)were similarly treated, but for 24 h with interleukins. Socs2 (A and B) and Socs3 (C and D) mRNA levels were measured by real-time PCR. Data represents mean and standard deviation of triplicate measurements.

143
AHR-mediated suppression extends to other APR genes as well
After confirming the repression of Saa1 and Saa2 in primary mouse hepatocytes,
expression of other acute phase response genes was also examined by real-time PCR
(Figure 4.11 A-I). AHR activation was able to repress induction of many of the acute
phase genes including C-reactive protein (CRP), LPS-binding protein (LBP),
haptoglobin, alpha-2-macroglobulin and alpha-1-acid glycoprotein-1. This suggests that
AHR represses the acute-phase probably through a central transcriptional regulatory
mechanism common to most acute-phase genes.

144
Control
TCDD IL
TCDD + IL
0
50
100
150Sa
a1 m
RN
A
Control
TCDD IL
TCDD + IL
0
100
200
300 *
Saa2
mR
NA
Control
TCDD IL
TCDD + IL
0
30
60
90
120
Saa3
mR
NA
Control
TCDD IL
TCDD + IL
0
75
150
225
300 *
Crp
mR
NA
Control
TCDD IL
TCDD + IL
0
75
150
225
300 *Lb
p m
RN
A
Control
TCDD IL
TCDD + IL
0
5
10
15
20 *
A2m
mR
NA
Control
TCDD IL
TCDD + IL
0
50
100
150
Orm
-1 m
RN
A
Control
TCDD IL
TCDD + IL
025
5075
100
125150175
Apc
s m
RN
A
Control
TCDD IL
TCDD + IL
0
2550
75100
125150
175 *H
p m
RN
A
A B C
D E F
G H I
Figure 4.11: AHR activation represses other APR genes as well.
Real-time PCR on RNA from primary mouse hepatocytes treated with TCDD (10 nM, 30min) followed by interleukin (2 ng/ ml each of IL1B and IL6) for 24 h. Repression ofdifferent APR genes was assayed. CRP, C-reactive protein; LBP, LPS-binding protein; Orm-1, acid-1 glycoprotein; A2m, alpha-2-macroglobulin; Hp, haptoglobin; Apcs, serum amyloid (P component).

145
Saa repression in human hepatocyte-derived cells
Another important question to address is whether human cells would elicit a
similar response. The human AHR was capable of repressing SAA induction similar to the
mouse AHR. SAA3 is not expressed in human liver (280), and SAA1 and SAA2 have a
very high sequence similarity which did not allow designing a unique primer-set for
detecting SAA2. Hence, SAA1 mRNA levels were monitored to assess the effect of AHR
activation on acute phase response. Huh7 cells, a human hepatocarcinoma derived cell-
line, were treated with vehicle or TCDD to activate the AHR and then with human IL1B
and IL6. Activation of AHR repressed SAA1 mRNA induction by seventy-five percent
(Figure 4.12 A). Changes in the level of secreted SAA protein were determined by
ELISA and were found to mimic changes in mRNA (Figure 4.12 B). Since it is not
possible to differentiate between different SAA family members by ELISA, this
repression of SAA reflects the changes in the levels of all secreted SAA family members.

146
Control
TCDD IL
TCDD + IL
0
25
50
75
100
125 *
SAA
1 m
RN
A
Ctrl
TCDD
IL (10 h
)
TCDD+IL (1
0h)
IL (24 h
)
TCDD+IL (2
4h)
0
10
20
30
40
50
60 *
SAA
con
cent
ratio
n(n
g/m
l)
A
B
Figure 4.12: AHR-mediated repression in human cells.
(A) Real-time on RNA from Huh7 cells treated with TCDD (10 nM, 30 min) followed byrecombinant human interleukins (2 ng/ml each of IL1B and IL6) for 6 h. Human SAA1mRNA abundance was assayed. (B) ELISA to quantify SAA protein secreted by Huh7 cells, treated for 10 h or 24 h withTCDD and interleukins. Just prior to treatment, cells were transferred to serum-free media.

147
Although TCDD exerts its effects almost exclusively through the AHR, we
wanted to confirm that the observed TCDD-mediated Saa1 repression in human cells is
indeed AHR dependent. AHR was knocked-down in Huh7 cells using AHR siRNA
oligos. As expected, loss of AHR resulted in a loss of SAA1 repression with TCDD
treatment (Figure 4.13 A and C). AHR knock-down was verified by the loss of its
transcriptional activity, as demonstrated by a loss of CYP1A1 mRNA induction
(Figure 4.13 B). Interestingly, the loss of AHR resulted in an enhanced induction of SAA1
with interleukin treatment (Figure 4.13 A). In order to confirm that this was not an off-
target effect of the AHR siRNA oligo sequence, a second anti-AHR siRNA oligo was
transfected into Huh7 cells by electroporation. AHR knock-down by this second AHR
siRNA also resulted in enhanced SAA1 induction by interleukins. This suggests that AHR
might function to constitutively suppress the level of SAA1 transcription, and possibly
suppress the expression of acute phase response in general. However, AHR-mediated
repression of inflammatory genes is not a universal phenomenon. Two known NF-κB
regulated genes, interleukin-8 (IL-8) and NF-kappa-B inhibitor alpha (NFKBIA,
commonly known as IκBα) were found to be induced by interleukins, but remained
unaffected by co-treatment with TCDD ( Figure 4.14 A and B).

148
Con
trol
TCD
D IL
TCD
D +
IL
Con
trol
TCD
D IL
TCD
D +
IL
0
100
200
300
400
500
600
700
800
Scramble anti-AhR
*
SAA
1 m
RN
A
IL T+IL IL T+IL0
50
100
150
scrambled anti-AhR
*
SAA
1 m
RN
A
Con
trol
TCD
D IL
TCD
D +
IL
Con
trol
TCD
D IL
TCD
D +
IL
0
5000
10000
15000
20000
25000
30000
Scramble anti-AhR
CYP
1A1
mR
NA
A
B
C
Figure 4.13: Saa repression in human cells is AHR-dependent.
(A and B) siRNA-driven AHR knock-down in Huh7 cells. 36 h after siRNA transfection,cells were treated with TCDD and interleukins, as in (Figure 4.12 A). SAA1 mRNA (A) and CYP1A1 (B) mRNA levels were determined by real-time PCR. (C) An alternate representation of data from (A). SAA1 induction, upon interleukin exposure of scrambled and anti-AHR siRNA transfected cells, was scaled to 100 units.

149
Control
TCDD IL
TCDD+ IL
0
50
100
150
200
250IL
-8 m
RN
A
Control
TCDD IL
TCDD+ IL
0
30
60
90
120
NFK
BIA
A B
Figure 4.14: AHR-mediated NF-κB suppression is gene-specific.
Huh7 cells were treated with TCDD (10 nM for 30 min) followed by interleukins (2 ng/ml of IL1B and IL6 each for 6 h). mRNA changes were measured for IL8 andNFKBIA (IκBα) by real-time PCR. Data represents mean and standard deviation oftriplicate measurements.

150
4.5 Discussion:
The immune system is perhaps the most dynamic ‘organ’ system in our body. It
plays a key role in our survival, empowering us to fight off infections and other foreign
particles. Though the significance of immunity and inflammation in our lives is
indisputable, the regulation of such a versatile and potent system is of equal importance.
Dysregulated inflammatory/immune response underlies many diseases such as asthma,
lupus and rheumatoid arthritis. Acute phase response dominates the initial reaction to
perceived insults and commences a series of biochemical and neuroendocrine changes
that facilitate mounting an inflammatory/immune response. Acute phase proteins serve
various tasks in this process, for example, CRP binds to phosphocholine on microbial
surfaces to promote recognition, fibrinogen and haptoglobin promote wound healing and
complement factors promote coagulation that may help ward off infection (reviewed in
(260)). However, persistent activation of APR has its own perils (281). Elevated CRP is
associated with increased cardiovascular risk and has been proposed to be a better
prognosticator of atherosclerosis and related events than lipid levels (282, 283). SAA is
an apolipoprotein for high-density lipoproteins (HDL) and influences cholesterol
metabolism in favor of inflammation. Conversely, constant elevation of SAA, and even
alpha-2-macroglobulin, leads to extracellular amyloid plaques that interfere with organ
function and underlie the pathology of diseases such as Alzheimer’s and prion diseases
(284). Thus, therapeutic approaches to regulate APP production are being actively

151
researched. Our results demonstrate that AHR activation has the potential to repress APP
expression.
At the transcriptional level, the induction of SAA, and most other APPs, is
regulated by NF-κB, CCAAT-enhancer-binding protein β (C/EBP-β or NFIL6) and signal
transducer and activator of transcription-3 (STAT3) (285-287). The potential of these
pro-inflammatory transcription factors has been shown to be repressed by many activated
NRs. The inhibitory effects of glucocorticoid receptor (GR), estrogen receptor (ER) and
peroxisome proliferators-activated receptor family (PPAR) on NF-κB induced gene
transcription has received wide attention (11, 164). NF-κB signaling allows multiple
levels of regulation which have been utilized by NRs to interact with this pathway.
Cytokines engage distinct receptors on cell surface to commence inflammatory signaling.
The intracellular portions of these cytokine-receptors then recruit and activate a family of
adaptor proteins through various post-translational modifications. Eventually, signaling
from different cytokine receptors converges on phosphorylation-dependent activation of
the IKK complex (IκB-kinase complex), which in turn releases NF-κB. Activated AHR
can possibly inhibit any of these cell-surface receptors or the immediate downstream
cytoplasmic signaling to repress NF-κB activity. However, in the context of APR gene
regulation, AHR effectively repressed Saa3 mRNA when induced separately by IL1β,
IL6 and TNFα. Also, the K14A-AHR, nuclear localization mutant, was unable to repress
Saa3 mRNA induction. This demonstrates that AHR-mediated acute-phase gene
suppression is not due to an effect on upstream cytokine signaling, but is primarily a
nuclear phenomenon.

152
Following the degradation of IκB, NF-κB translocates to the nucleus and binds κB
cognate response elements found in the promoter/enhancer region of its target genes.
Activation of AHR diminished cytokine-induced association of RELA subunit of NF-κB
with its response elements in Saa2 and Saa3 promoters, as shown by ChIP assay. Other
groups have previously demonstrated the ability of AHR to physically interact with the
RELA subunit of NF-κB by immunoprecipitation-Western blot (IP-WB) experiments
(172). While a direct physical interaction between the two proteins can certainly explain
the reduction in RELA recruitment to Saa promoters, it cannot be the sole mechanism for
AHR-NF-κB cross-talk. The rationale for this argument is derived from the fact that AHR
activation is unable to universally repress NF-κB driven gene expression (Figure 4.14).
Also, we did not observe a significant reduction in RELA or p50 protein levels upon
AHR activation (data not shown). Based on recent studies, it is evident that degradation
of IκBα is not sufficient to induce transcriptional activity of NF-κB. Phosphorylation and
acetylation of NF-κB at multiple sites is essential for optimizing DNA-binding and
transactivation of target genes (267, 271, 288). A ChIP assay with an anti-acetylated
histone 4 (anti-AcH4) antibody revealed that cytokine-responsive histone acetylation at
Saa promoters is reduced upon activation of AHR. This observation encouraged us to
determine the effect that HDAC inhibition would have on Saa regulation. Though
Trichostatin A (TSA) pre-treatment enhanced basal as well as induced levels of Saa
mRNA, the derepression effect was significantly higher (10-fold increase in TCDD + IL
samples as opposed to, 5-fold increase in IL samples) in AHR activated samples. This
strongly suggests that AHR-mediated NF-κB repression also involves an alteration of the

153
‘histone code’. It is also possible that like GR (289), AHR recruits HDAC to the activated
NF-κB complex, resulting in decreased acetylation of critical lysine residues (218, 221
and 310) on RELA. Extensive protein interaction studies and ChIP assay would be
needed to explore this hypothesis. Though universal mechanisms like direct physical
interaction between AHR and NF-κB may play a role, commonly used experimental
techniques like electrophorectic mobility shift assays and reporter-based experiments
should be interpreted cautiously as these do not simulate promoter-specific chromatin
dynamics.
NF-κB binding to its response elements establishes an activated transcriptional
complex and drives target gene expression, including that of IκBα (290). Increased IκBα
can terminate transcription by retrieving active NF-κB complexes from the nucleus back
to the cytoplasm (291), as has been shown for some nuclear receptor (NR)-NF-κB
interactions (292). Another possible feedback mechanism is a receptor-dependent
reduction in the cytokines, such as AHR-mediated reduction in TNFA and IL6 (177,
180). However, these mechanisms are unlikely to contribute to APR repression because
inhibiting protein translation by cycloheximide pre-treatment did not affect AHR’s ability
to suppress NF-κB. Even in the absence of protein synthesis, a decrease in mRNA levels
can be due to altered mRNA stability. When Hepa1c1c7 cells were treated with
Actinomycin D, a transcriptional inhibitor, no difference in Saa3 mRNA decay rate was
observed between TCDD treated and control samples. These experiments clearly
demonstrate that AHR represses APR gene induction by a direct effect on transcription.

154
AHR can be activated by a variety of ligands that are ubiquitously present in the
environment. TCDD is perhaps the most potent inducer of AHR transcriptional activity
and is believed to exert its toxicity through activating AHR. The lack of Saa repression
upon TCDD treatment in AHR-deficient primary hepatocytes isolated from Ahrfx/fxCreAlb
mice proves that AHR is essential even for TCDD-mediated repression. All AHR ligands
tested were able to repress Saa mRNA, indicating that this is not a ligand-specific effect.
From the inflammatory viewpoint, an important aspect that is often overlooked is the
complexity of the cytokine ‘network’. Signal amplification and feedback loops are
integral to cytokine signaling and thus, the effects observed with a single cytokine at a
defined dose might be counter-regulated by another cytokine. To this end, a
physiologically feasible mix of inflammatory mediators was obtained by challenging
bone-marrow derived primary mouse macrophages. AHR-activation was able to repress
Saa induction by this mix of inflammatory mediators, suggesting that AHR-mediated
APR regulation could occur under inflammatory situations. A larger repertoire of tools
and models are available to study murine AHR biology as compared to human AHR.
However, the murine and human AHR have considerable differences, including nucleo-
cytoplasmic shuttling and transactivation domain sequence (73, 74), and hence
extrapolating observations from murine-models to humans requires caution. We verified
the ability of AHR to repress SAA induction in human liver derived cell-line. AHR
activation repressed SAA1 induction by more than seventy percent. This repression was
also observed in levels of secreted SAA protein. AHR knock-down by siRNA confirmed
AHR-dependency of TCDD-mediated SAA1 repression in human cells.

155
Ligand-activated receptors have multiple domains that impart different
functionalities, such as DNA-binding, ligand-binding, dimerization, nuclear trafficking
signals and co-regulator recruitment. However, depending on the manner of activation
and the physiological context, the functionalities of soluble receptors can be dissociated
from their biological roles, as in the case of GR (205) and ER (207). Here, we
demonstrate for the first time that DNA-binding is not essential for AHR-mediated
repressive effects on NF-κB transactivation, while heterodimerization with ARNT and
nuclear translocation are required. This report is also the first attempt at thoroughly
characterizing a heterodimerization mutant of AHR. Based on Figure 4.1 B and C, it is
clear that heterodimerization with ARNT is essential for AHR-mediated repression of
acute-phase genes. Besides xenobiotic metabolism enzymes, this is also the first report
identifying a functional role for AHR in an entire biological process, and not just
individual gene regulation.
In conclusion, the data presented in this report demonstrate the interaction of
AHR and NF-κB signaling pathways to regulate multiple APR gene expression, an
important aspect of the inflammatory reaction. This identifies a novel physiological
function performed by the AHR in murine as well as human systems. AHR-mediated
transcriptional repression is not conducted in the classical DRE-dependent fashion, but
most likely involves multiple mechanisms. Altered post-translational modifications of
NF-κB proteins and histone code upon AHR activation seem to play an important part.
Finally, in order to therapeutically utilize the ability of AHR to function as a repressor of
acute phase response, and possibly other inflammatory phenomena, it is necessary to

156
identify/design ‘selective ligands’. Selective AHR modulators (SARM), like the selective
ER modulators (SERM – e.g. Tamoxifen), would induce the beneficial effects of AHR
without eliciting its potentially harmful effects. This concept has been described further
as future directions in the next chapter.

157
Chapter 5
CONCLUSIONS AND FUTURE DIRECTIONS

158
AHR is a unique receptor – it is the only ligand inducible bHLH-PAS domain
transcription factor, differs from steroid receptors in terms of modular domain
architecture and can respond to structurally diverse chemicals that are so ubiquitous that
it is impossible to escape them. In fact, excessive exposure to some of these ligands has
been implicated in adverse health effects, including carcinogenesis. Though a
considerable research effort has been invested in understanding AHR biology, and an
appreciable amount of information has indeed been accumulated, two key issues that
have not been satisfactorily answered are the identification of an endogenous ligand and a
clear understanding of the physiological role of AHR. Both of these pieces of information
can be instrumental in unveiling the therapeutic potential of manipulating AHR activity
to attain desired biological effects. AHR knock-out mice are not embryonic lethal,
however they exhibit remarkable phenotypic features including reproductive inferiority,
delayed growth and heightened susceptibility to certain infections.
At the time of commencement of my research, non-xenobiotic gene regulation
comprised a minimal amount of knowledge on AHR biology. With the simultaneously
growth of high-capacity genome and proteome analysis technology, there have been
multiple reports identifying new AHR target genes in divergent scenarios. This has
certainly expanded the range of implications of AHR activation, but, ‘we are not there
yet.’ My research involved identifying the cellular processes influenced by AHR
activation that are unrelated to xenobiotic metabolism. The approach involved applying

159
DNA microarray technology to identify previously unknown AHR regulated genes. Since
the classical DRE-driven gene regulation is unlikely to explain all the pathophysiological
phenomena associated with AHR, an attempt was also made to identify alternate means
of AHR function. As a result, two novel target genes and a DRE-independent mode of
AHR activity have been characterized.
Epiregulin is an epidermal growth factor family member and functions to promote
cell proliferation. As a group, growth factors and receptors belonging to the epidermal
growth factor family have been implicated in numerous cancers. In fact, inhibitors of
epidermal growth factor receptor are in clinical use as anti-tumor agents (for example
(293, 294)). Epiregulin has also been associated with numerous tumors (295, 296) and
immune disorders such as dermatitis (220, 297). Interestingly, carcinogenesis is also one
of the adverse effects of AHR ligands (240, 298) and AHR-mediated epiregulin
upregulation might contribute to tumor development. This hypothesis should be tested by
tumor studies involving treatment of epiregulin knock-out mice (299) with AHR ligands.
After the discovery of epiregulin as a direct AHR target gene, another epidermal growth
factor family member – amphiregulin – has also been described to be regulated by AHR
(300). Thus, one mechanism of AHR-mediated alteration of cell growth and proliferation
could be related to its effect on growth factor expression.
Chapter 3 describes the impact of AHR activation on the expression and the
activity of constitutive androstane receptor (CAR). AHR-mediated CAR induction differs
significantly as compared to the regulation of epiregulin. First, CAR upregulation was

160
detected in vivo in the liver of C57BL/6 mice. Second, β-naphthoflavone (BNF) was
utilized to activate AHR, as opposed to TCDD. Though there is no definite evidence to
establish a ligand-specific AHR transactivation profile, it should be noted that there are
important differences in biochemical properties of these two ligands. Third, genomic
sequence analysis did not reveal a DRE within a reasonable putative CAR promoter.
Though, enhancer regions for gene regulation have been described several kilobases
upstream of transcription start sites, and such a DRE-containing regulatory region might
exist for CAR, it is not very common. It would have been exciting to elaborate the
mechanism by which AHR regulates CAR expression in the absence of a consensus
DRE. However, the lack of induction of CAR expression in cell-culture system
discouraged attempts to delineate mechanistic details. Whether upregulation of CAR is a
DRE-independent mechanism of gene upregulation can be tested by using transgenic
mice expressing the DNA-binding mutant (A78D) form of AHR instead of the wildtype
AHR.
Though the particulars of inducible CAR expression might remain pending until a
suitable cell-culture system is established, the observation that AHR activation can
upregulate CAR activity might be of clinical relevance. CAR is an important
transcription factor associated with metabolism of many pharmaceutical compounds via
its ability to upregulate cytochrome P450 enzymes. Thus, AHR-mediated CAR
upregulation could have clinically relevant effect on drug metabolism. The likelihood of
this possibility is further enhanced when we consider that it is possible to achieve an
increase in the AHR-mediated transcription in the lungs of smokers. ‘Personalized

161
medicine’ is an attractive concept and can be implemented at various levels in healthcare.
Though we may not be close enough to anticipate fabricating individualized drug
molecules, efforts have certainly been initiated to individualize the dosing of
medications. Genetic variability in the population renders some of us as ‘fast’, ‘normal’
or ‘slow’ metabolizers of certain drugs. Administration the same dose of a drug to
various patients can result either in an overdose and undesired adverse effects in ‘slow’
metabolizers, or inadequate treatment in ‘fast’ metabolizers. Affymetrix, a pioneer in
DNA-microarray technology, and Roche pharmaceuticals have launched a microarray-
based diagnostic platform to detect variations in CYP2D6 and CYP2C19, two clinically
relevant drug metabolizing enzymes. Healthcare facilities in Europe are utilizing this
technology to predict patient phenotype and regulate drug dosage between individuals. In
addition to genetic variation, environmental determinants, such as smoking or dietary
habits, can also potentially determine individual drug requirements. Thus, AHR-mediated
CAR induction can have prospective therapeutic implications that need to be investigated
further.
The term ‘AHR activation’ typically conveys the classic DRE-dependent
transactivation pathway. Research on AHR has almost exclusively focused on its
transactivation potential, baring a few reports on AHR-mediated ER inhibition. After
describing activation of two previously uncharacterized target genes in Chapter 2 and 3,
efforts were focused on addressing the hypothesis that AHR mediates DRE-independent
regulation of transcriptional responses. The ability of AHR to repress the acute phase
response without binding a DRE, clearly establishes a non-classical mechanism of AHR

162
activity. This is also the first evidence (outside of the classical xenobiotic metabolism
regulation) where the receptor has been shown to regulate an entire biological process, as
opposed to controlling the expression of individual genes. The use of different AHR
mutants and primary as well as established cell-culture systems permitted a thorough
characterization of AHR-mediated suppression of cytokine signaling, while raising new
questions which will require additional research. The following avenues would be a fair
extension of the research presented in Chapter 4.
The mechanism for AHR-mediated NF-κB repression needs to be elucidated
further. ‘Cofactor exchange’ is a concept that has gained considerable popularity.
Normally, transcription factors associate with coactivators to induce transcription of
positively regulated genes, and with corepressors to suppress the transcription of
negatively regulated genes. According to the ‘cofactor exchange’ model, activation of
one transcription factor can influence the activity of another by exchanging coactivators
with corepressors, and vice versa. Specific examples of this model are provided by
glucocorticoid receptor (GR) and peroxisome-proliferator-activated receptor-γ (PPAR-γ)
mediated repression of inflammatory response genes (reviewed in (11)). Some
inflammatory gene promoters bear an inhibitory nuclear-receptor co-repressor (NCoR)-
based complex that is lost in response to cytokine signaling. Upon ligand activation
PPAR-γ can be sumoylated, which in turn leads to its association with NCoR. This
stabilizes the inhibitory complex and prevents the switch to a coactivator complex at the
promoters of a subset of NF-κB regulated genes, resulting in continued repression of
transcription (301). Genes that normally do not have NCoR-based repressive complexes

163
are not affected by PPAR-γ activation. Certain genes require NF-κB/IRF3 (interferon-
regulatory factor 3) association to form activator complexes that would drive
transcription in response to inflammatory signaling. Activated GR can disrupt
RELA/IRF3 association and avert formation of an activator complex thereby repressing
the transcription of these genes. AHR-mediated inflammatory repression can possibly
involve similar mechanics. Extensive chromatin immunoprecipitation studies will by
necessary to study the dynamics of transcriptional complexes on the promoters of AHR-
sensitive inflammatory genes. Another factor that dictates the composition of
transactivation complexes is the cis element (the DNA sequence of the
promoter/enhancer, including the ‘flexible’ bases within the core response elements)
(302). This allosteric effect of DNA has been documented for GR (303) and NF-κB
(304). To this end, microarray studies can be performed to identify AHR-sensitive and
AHR-resistant inflammatory genes. A bioinformatics approach to promoter analysis
might provide valuable information that would help explain the selectivity of AHR-
mediated repression. Furthermore, the results from Chapter 4 have established a role of
histone deacetylases (HDAC) in mediating AHR-dependent repression of acute phase
genes. Whether HDAC activity affects only chromatin remodeling or does it also alter
acetylation of NF-κB, remains to be answered.
A number of disease states result from dysregulated immune responses. For AHR-
mediated inflammatory repression to be advantageous, it is essential to determine the
effect of AHR activation in in vivo models. A number of mouse models with phenotypic
resemblance to human immune-related diseases have been described. For example,

164
DNase II-null/interferon type I receptor (IFNIR)-null mice and mice with an induced
deletion of the DNase II gene develop arthritis of multiple joints resembling human
rheumatoid arthritis (305). This correlates with the upregulation of a subset of cytokines,
especially TNF-α, in the affected joints and high serum levels of rheumatoid factor.
Systemic lupus erythematosus (SLE) is another autoimmune disorder characterized by a
chronic, remitting, relapsing, inflammatory, and often febrile multisystemic disorder of
connective tissue. Dnase1-deficient mice show the classic symptoms of SLE, namely the
presence of anti-nuclear antibodies, immune complex deposition in glomeruli, and
consequent glomerulonephritis (306). This correlates with reduced Dnase1 activity in the
serum from SLE patients compared to normal subjects (307). Patients with rheumatoid
arthritis and other chronic inflammatory diseases develop amyloidosis, and it also can be
induced in mice by increasing SAA concentrations through injection of silver nitrate or
casein. 2 or 3 weeks after the inflammatory stimulus, systemic AA deposits develop in
mice, identical to those with amyloidosis. Administration of protein extracted from AA
amyloid-laden mouse spleen or liver further shortens this lag phase (308). It would be
interesting to note whether AHR activation by chronic administration of AHR ligands
would have any protective effects in these kinds of disease models.
Every coin has two faces. It would be nice to have only the winning face.
Activation of AHR also has two faces – activation of xenobiotic metabolism enzymes
which bears the potential to generate harmful metabolites, and repression of
inflammatory response which can be exploited therapeutically. However, in order to take
advantage of the positive effects of AHR activation without suffering the consequences

165
of xenobiotic metabolism enzyme upregulation, it is necessary to identify selective
ligands. In nuclear receptors, agonist (defined with reference to a positive transcriptional
effect) binding induces a conformational change, particularly in the form of a physical
shift of the AF2 domain. This event spawns the formation of surfaces with affinity for the
LXXLL motifs of transcriptional coactivators. Moreover, creation of these surfaces
hinders the association of corepressors due to the structurally incompatible nature of their
interaction motifs. Thus, it is possible to choose a ligand that induces a conformational
change favoring association of corepressors rather than coactivators. Such a preferential
modulator of transcription factor activity can be of a therapeutic value. Tamoxifen and
raloxifene are examples of function-specific ER ligands that allow repressive effects of
ER without inducing its transactivation potential (309). Similarly, transcriptional effects
of ER and androgen receptor (AR) can be dissociated from their anti-apoptotic effects
with the use of selective synthetic ligands (310). Identification of acute phase genes as a
candidate for AHR-mediated repression has now made it possible to screen for selective
AHR ligands. Based on the experiments optimized for Chapter 4, a high throughput
screen can be established for analyzing libraries of compounds in search for the selective
ligand. A schematic representation of the screen is presented in Figure 5.1.

166
Figure 5.1: Schematic for a screen to identify ‘Selective AHR Modulators – SARM’.
Huh7 cells can be treated with interleukins (IL) and a library of compounds (X). SAAexpression in conditioned media can be assessed by ELISA. Compounds capable ofrepressing SAA (top graph) would then be used to treat AHR-deficient Huh7 cells along with IL. Compounds that cannot repress SAA in the absence of AHR (bottom graph) aremost likely to be AHR activators. This subset of compounds should then be used toassess their ability to induce classical AHR target genes (e.g. CYP1A1) by real-time PCR (qPCR). Compounds that cannot induce CYP1A1 would be potential SARMs. C=control.

167
Regulation of inflammatory responses by AHR is a significant contribution to the
field of AHR-biology. It has widened the scope of AHR research and will spur many
follow-up studies. However, it is prudent to continue the quest for involvement of AHR
in other biological processes. Microarray studies can be initiated to analyze the
interaction of AHR with other signaling pathways; for example, a comparison of gene
regulation in response to dexamethasone treatment in the presence or absence of AHR
ligands may generate a list of GR-regulated genes which are sensitive to AHR activation.
An in vivo mouse model would be a more physiologically relevant system to assay cross-
talk properties of AHR. A liver-specific transgenic model expressing A78D-AHR in
AHR knock-out mice has been generated in our lab. However, inferior fertility of AHR
knock-out mice has made it difficult to obtain enough age- and sex-matched animals to
perform meaningful studies. As a result, we are now attempting to generate A78D-AHR
expressing mice on a liver-specific AHR knock-out (Ahrfx/fxCreAlb) model, which breed
normally. This mouse model will also be valuable in re-assessing the hepatotoxic effects
of TCDD and determining the relative contributions of genotropic versus non-genotropic
effects of AHR activation. Primary hepatocytes from these mice can also be utilized to
search for other genes regulated by AHR in a DRE-independent fashion.

Bibliography
1. Nebert DW, Gelboin HV. Substrate-inducible microsomal aryl hydroxylase in mammalian cell culture. I. Assay and properties of induced enzyme. J Biol Chem 1968;243:6242-6249. 2. Nebert DW, Gelboin HV. Substrate-inducible microsomal aryl hydroxylase in mammalian cell culture. II. Cellular responses during enzyme induction. J Biol Chem 1968;243:6250-6261. 3. Poland AP, Glover E, Robinson JR, Nebert DW. Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically "nonresponsive" to other aromatic hydrocarbons. J Biol Chem 1974;249:5599-5606. 4. Nebert DW, Gielen JE. Genetic regulation of aryl hydrocarbon hydroxylase induction in the mouse. Fed Proc 1972;31:1315-1325. 5. Nebert DW, Goujon FM, Gielen JE. Aryl hydrocarbon hydroxylase induction by polycyclic hydrocarbons: simple autosomal dominant trait in the mouse. Nat New Biol 1972;236:107-110. 6. Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 1976;251:4936-4946. 7. Poland A, Glover E, Ebetino H, Kende A. Photoaffinity labelling of the Ah receptor. Food Chem Toxicol 1986;24:781-787. 8. Perdew GH, Poland A. Purification of the Ah receptor from C57BL/6J mouse liver. J Biol Chem 1988;263:9848-9852. 9. Bradfield CA, Glover E, Poland A. Purification and N-terminal amino acid sequence of the Ah receptor from the C57BL/6J mouse. Mol Pharmacol 1991;39:13-19. 10. Poland A, Glover E, Bradfield CA. Characterization of polyclonal antibodies to the Ah receptor prepared by immunization with a synthetic peptide hapten. Mol Pharmacol 1991;39:20-26. 11. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol 2006;6:44-55. 12. Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 2000;20:429-440. 13. Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol 2000;40:519-561. 14. Reisz-Porszasz S, Probst MR, Fukunaga BN, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT). Mol Cell Biol 1994;14:6075-6086.

169
15. Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem 1995;270:29270-29278. 16. Jain S, Dolwick KM, Schmidt JV, Bradfield CA. Potent transactivation domains of the Ah receptor and the Ah receptor nuclear translocator map to their carboxyl termini. J Biol Chem 1994;269:31518-31524. 17. Pohjanvirta R, Wong JM, Li W, Harper PA, Tuomisto J, Okey AB. Point mutation in intron sequence causes altered carboxyl-terminal structure in the aryl hydrocarbon receptor of the most 2,3,7,8-tetrachlorodibenzo-p-dioxin-resistant rat strain. Mol Pharmacol 1998;54:86-93. 18. Harper PA, Wong JY, Lam MS, Okey AB. Polymorphisms in the human AH receptor. Chem Biol Interact 2002;141:161-187. 19. Wong JM, Okey AB, Harper PA. Human aryl hydrocarbon receptor polymorphisms that result in loss of CYP1A1 induction. Biochem Biophys Res Commun 2001;288:990-996. 20. Poland A, Glover E. Chlorinated biphenyl induction of aryl hydrocarbon hydroxylase activity: a study of the structure-activity relationship. Mol Pharmacol 1977;13:924-938. 21. McKinney JD, Singh P. Structure-activity relationships in halogenated biphenyls: unifying hypothesis for structural specificity. Chem Biol Interact 1981;33:271-283. 22. Waller CL, McKinney JD. Three-dimensional quantitative structure-activity relationships of dioxins and dioxin-like compounds: model validation and Ah receptor characterization. Chem Res Toxicol 1995;8:847-858. 23. Tiernan TO, Taylor ML, Garrett JH, VanNess GF, Solch JG, Wagel DJ, Ferguson GL, et al. Sources and fate of polychlorinated dibenzodioxins, dibenzofurans and related compounds in human environments. Environ Health Perspect 1985;59:145-158. 24. Lawther PJ, Waller RE. Coal fires, industrial emissions and motor vehicles as sources of environmental carcinogens. IARC Sci Publ 1976:27-40. 25. Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol 2008;21:102-116. 26. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 2003;43:309-334. 27. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A 1991;88:9543-9547. 28. Perdew GH, Babbs CF. Production of Ah receptor ligands in rat fecal suspensions containing tryptophan or indole-3-carbinol. Nutr Cancer 1991;16:209-218. 29. Jellinck PH, Forkert PG, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem Pharmacol 1993;45:1129-1136. 30. Ciolino HP, Daschner PJ, Wang TT, Yeh GC. Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem Pharmacol 1998;56:197-206.

170
31. Ciolino HP, Daschner PJ, Yeh GC. Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem J 1999;340 ( Pt 3):715-722. 32. Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, Kitagawa H, Miller CA, 3rd, et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem 2001;276:31475-31478. 33. Peter Guengerich F, Martin MV, McCormick WA, Nguyen LP, Glover E, Bradfield CA. Aryl hydrocarbon receptor response to indigoids in vitro and in vivo. Arch Biochem Biophys 2004;423:309-316. 34. Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, Denison MS. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998;37:11508-11515. 35. Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, Grafstrom AK. Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J Biol Chem 1987;262:15422-15427. 36. Fritsche E, Schafer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hubenthal U, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A 2007;104:8851-8856. 37. Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry 1999;38:7594-7600. 38. Seidel SD, Winters GM, Rogers WJ, Ziccardi MH, Li V, Keser B, Denison MS. Activation of the Ah receptor signaling pathway by prostaglandins. J Biochem Mol Toxicol 2001;15:187-196. 39. Phelan D, Winter GM, Rogers WJ, Lam JC, Denison MS. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch Biochem Biophys 1998;357:155-163. 40. Sinal CJ, Bend JR. Aryl hydrocarbon receptor-dependent induction of cyp1a1 by bilirubin in mouse hepatoma hepa 1c1c7 cells. Mol Pharmacol 1997;52:590-599. 41. Jinno A, Maruyama Y, Ishizuka M, Kazusaka A, Nakamura A, Fujita S. Induction of cytochrome P450-1A by the equine estrogen equilenin, a new endogenous aryl hydrocarbon receptor ligand. J Steroid Biochem Mol Biol 2006;98:48-55. 42. Monk SA, Denison MS, Rice RH. Transient expression of CYP1A1 in rat epithelial cells cultured in suspension. Arch Biochem Biophys 2001;393:154-162. 43. Sadek CM, Allen-Hoffmann BL. Suspension-mediated induction of Hepa 1c1c7 Cyp1a-1 expression is dependent on the Ah receptor signal transduction pathway. J Biol Chem 1994;269:31505-31509. 44. Sadek CM, Allen-Hoffmann BL. Cytochrome P450IA1 is rapidly induced in normal human keratinocytes in the absence of xenobiotics. J Biol Chem 1994;269:16067-16074. 45. Cho YC, Zheng W, Jefcoate CR. Disruption of cell-cell contact maximally but transiently activates AhR-mediated transcription in 10T1/2 fibroblasts. Toxicol Appl Pharmacol 2004;199:220-238.

171
46. Oesch-Bartlomowicz B, Huelster A, Wiss O, Antoniou-Lipfert P, Dietrich C, Arand M, Weiss C, et al. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: divergent signaling pathways. Proc Natl Acad Sci U S A 2005;102:9218-9223. 47. Perdew GH. Chemical cross-linking of the cytosolic and nuclear forms of the Ah receptor in hepatoma cell line 1c1c7. Biochem Biophys Res Commun 1992;182:55-62. 48. Perdew GH. Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem 1988;263:13802-13805. 49. Denis M, Cuthill S, Wikstrom AC, Poellinger L, Gustafsson JA. Association of the dioxin receptor with the Mr 90,000 heat shock protein: a structural kinship with the glucocorticoid receptor. Biochem Biophys Res Commun 1988;155:801-807. 50. Chen HS, Perdew GH. Subunit composition of the heteromeric cytosolic aryl hydrocarbon receptor complex. J Biol Chem 1994;269:27554-27558. 51. Perdew GH, Bradfield CA. Mapping the 90 kDa heat shock protein binding region of the Ah receptor. Biochem Mol Biol Int 1996;39:589-593. 52. Coumailleau P, Poellinger L, Gustafsson JA, Whitelaw ML. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J Biol Chem 1995;270:25291-25300. 53. Pongratz I, Mason GG, Poellinger L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J Biol Chem 1992;267:13728-13734. 54. Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol Cell Biol 1998;18:978-988. 55. Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J Biol Chem 1997;272:11452-11456. 56. Carver LA, LaPres JJ, Jain S, Dunham EE, Bradfield CA. Characterization of the Ah receptor-associated protein, ARA9. J Biol Chem 1998;273:33580-33587. 57. Meyer BK, Perdew GH. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry 1999;38:8907-8917. 58. Meyer BK, Petrulis JR, Perdew GH. Aryl hydrocarbon (Ah) receptor levels are selectively modulated by hsp90-associated immunophilin homolog XAP2. Cell Stress Chaperones 2000;5:243-254. 59. Hollingshead BD, Patel RD, Perdew GH. Endogenous hepatic expression of the hepatitis B virus X-associated protein 2 is adequate for maximal association with aryl hydrocarbon receptor-90-kDa heat shock protein complexes. Mol Pharmacol 2006;70:2096-2107. 60. Lin BC, Sullivan R, Lee Y, Moran S, Glover E, Bradfield CA. Deletion of the aryl hydrocarbon receptor-associated protein 9 leads to cardiac malformation and embryonic lethality. J Biol Chem 2007;282:35924-35932. 61. Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen

172
receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones 1996;1:237-250. 62. Shetty PV, Bhagwat BY, Chan WK. P23 enhances the formation of the aryl hydrocarbon receptor-DNA complex. Biochem Pharmacol 2003;65:941-948. 63. Kazlauskas A, Poellinger L, Pongratz I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J Biol Chem 1999;274:13519-13524. 64. Perdew GH. Comparison of the nuclear and cytosolic forms of the Ah receptor from Hepa 1c1c7 cells: charge heterogeneity and ATP binding properties. Arch Biochem Biophys 1991;291:284-290. 65. Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem 1998;273:2895-2904. 66. Ikuta T, Tachibana T, Watanabe J, Yoshida M, Yoneda Y, Kawajiri K. Nucleocytoplasmic shuttling of the aryl hydrocarbon receptor. J Biochem 2000;127:503-509. 67. Petrulis JR, Kusnadi A, Ramadoss P, Hollingshead B, Perdew GH. The hsp90 Co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J Biol Chem 2003;278:2677-2685. 68. Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992;256:1193-1195. 69. Arpiainen S, Lamsa V, Pelkonen O, Yim SH, Gonzalez FJ, Hakkola J. Aryl hydrocarbon receptor nuclear translocator and upstream stimulatory factor regulate Cytochrome P450 2a5 transcription through a common E-box site. J Mol Biol 2007;369:640-652. 70. Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol 2004;36:189-204. 71. Denison MS, Fisher JM, Whitlock JP, Jr. Inducible, receptor-dependent protein-DNA interactions at a dioxin-responsive transcriptional enhancer. Proc Natl Acad Sci U S A 1988;85:2528-2532. 72. Denison MS, Fisher JM, Whitlock JP, Jr. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J Biol Chem 1988;263:17221-17224. 73. Ramadoss P, Perdew GH. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol Pharmacol 2004;66:129-136. 74. Ramadoss P, Perdew GH. The transactivation domain of the Ah receptor is a key determinant of cellular localization and ligand-independent nucleocytoplasmic shuttling properties. Biochemistry 2005;44:11148-11159. 75. Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 (XAP2) in human versus mouse Ah receptor complexes. Biochemistry 2004;43:700-709.

173
76. Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995;268:722-726. 77. Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 1996;93:6731-6736. 78. Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 1997;2:645-654. 79. Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol 1997;34:605-614. 80. Lahvis GP, Lindell SL, Thomas RS, McCuskey RS, Murphy C, Glover E, Bentz M, et al. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc Natl Acad Sci U S A 2000;97:10442-10447. 81. Benedict JC, Lin TM, Loeffler IK, Peterson RE, Flaws JA. Physiological role of the aryl hydrocarbon receptor in mouse ovary development. Toxicol Sci 2000;56:382-388. 82. Benedict JC, Miller KP, Lin TM, Greenfeld C, Babus JK, Peterson RE, Flaws JA. Aryl hydrocarbon receptor regulates growth, but not atresia, of mouse preantral and antral follicles. Biol Reprod 2003;68:1511-1517. 83. Peters JM, Narotsky MG, Elizondo G, Fernandez-Salguero PM, Gonzalez FJ, Abbott BD. Amelioration of TCDD-induced teratogenesis in aryl hydrocarbon receptor (AhR)-null mice. Toxicol Sci 1999;47:86-92. 84. Lahvis GP, Bradfield CA. Ahr null alleles: distinctive or different? Biochem Pharmacol 1998;56:781-787. 85. Noda S, Harada N, Hida A, Fujii-Kuriyama Y, Motohashi H, Yamamoto M. Gene expression of detoxifying enzymes in AhR and Nrf2 compound null mutant mouse. Biochem Biophys Res Commun 2003;303:105-111. 86. McGuire J, Okamoto K, Whitelaw ML, Tanaka H, Poellinger L. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: delineation of overlapping repression and ligand binding functions within the PAS domain. J Biol Chem 2001;276:41841-41849. 87. Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A 2002;99:9990-9995. 88. Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, Schwarz M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res 2004;64:4707-4710. 89. Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem 2003;278:17767-17774. 90. Walisser JA, Bunger MK, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A 2004;101:16677-16682.

174
91. Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. J Biol Chem 2004;279:16326-16331. 92. Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci U S A 2005;102:17858-17863. 93. Moriguchi T, Motohashi H, Hosoya T, Nakajima O, Takahashi S, Ohsako S, Aoki Y, et al. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc Natl Acad Sci U S A 2003;100:5652-5657. 94. Kasai A, Hiramatsu N, Hayakawa K, Yao J, Kitamura M. Direct, continuous monitoring of air pollution by transgenic sensor mice responsive to halogenated and polycyclic aromatic hydrocarbons. Environ Health Perspect 2008;116:349-354. 95. Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, et al. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol 2006;69:1103-1114. 96. Taioli E, Crofts F, Trachman J, Demopoulos R, Toniolo P, Garte SJ. A specific African-American CYP1A1 polymorphism is associated with adenocarcinoma of the lung. Cancer Res 1995;55:472-473. 97. Taioli E, Trachman J, Chen X, Toniolo P, Garte SJ. A CYP1A1 restriction fragment length polymorphism is associated with breast cancer in African-American women. Cancer Res 1995;55:3757-3758. 98. Ueda R, Iketaki H, Nagata K, Kimura S, Gonzalez FJ, Kusano K, Yoshimura T, et al. A common regulatory region functions bidirectionally in transcriptional activation of the human CYP1A1 and CYP1A2 genes. Mol Pharmacol 2006;69:1924-1930. 99. Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet 1997;6:641-647. 100. Arpiainen S, Raffalli-Mathieu F, Lang MA, Pelkonen O, Hakkola J. Regulation of the Cyp2a5 gene involves an aryl hydrocarbon receptor-dependent pathway. Mol Pharmacol 2005;67:1325-1333. 101. Kohle C, Bock KW. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem Pharmacol 2007;73:1853-1862. 102. Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem 2005;280:20340-20348. 103. Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, et al. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 2007;27:7188-7197. 104. Marchand A, Barouki R, Garlatti M. Regulation of NAD(P)H:quinone oxidoreductase 1 gene expression by CYP1A1 activity. Mol Pharmacol 2004;65:1029-1037.

175
105. Martinez JM, Afshari CA, Bushel PR, Masuda A, Takahashi T, Walker NJ. Differential toxicogenomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin in malignant and nonmalignant human airway epithelial cells. Toxicol Sci 2002;69:409-423. 106. Martinez JM, Baek SJ, Mays DM, Tithof PK, Eling TE, Walker NJ. EGR1 is a novel target for AhR agonists in human lung epithelial cells. Toxicol Sci 2004;82:429-435. 107. Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000;19:5498-5506. 108. Yang X, Liu D, Murray TJ, Mitchell GC, Hesterman EV, Karchner SI, Merson RR, et al. The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene 2005;24:7869-7881. 109. Dong L, Ma Q, Whitlock JP, Jr. Down-regulation of major histocompatibility complex Q1b gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem 1997;272:29614-29619. 110. Niermann T, Schmutz S, Erne P, Resink T. Aryl hydrocarbon receptor ligands repress T-cadherin expression in vascular smooth muscle cells. Biochem Biophys Res Commun 2003;300:943-949. 111. Bermudez de Leon M, Gomez P, Elizondo G, Zatarain-Palacios R, Garcia-Sierra F, Cisneros B. Beta-naphthoflavone represses dystrophin Dp71 expression in hepatic cells. Biochim Biophys Acta 2006;1759:152-158. 112. Kuznetsov NV, Andersson P, Gradin K, Stein P, Dieckmann A, Pettersson S, Hanberg A, et al. The dioxin/aryl hydrocarbon receptor mediates downregulation of osteopontin gene expression in a mouse model of gastric tumourigenesis. Oncogene 2005;24:3216-3222. 113. Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2005;289:L391-399. 114. Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, Tuscano J, et al. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol 2007;171:1538-1548. 115. Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis 2005;22:149-156. 116. Yang F, Bleich D. Transcriptional regulation of cyclooxygenase-2 gene in pancreatic beta-cells. J Biol Chem 2004;279:35403-35411. 117. N'Diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, Monteiro P, et al. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem 2006;281:19906-19915. 118. Boverhof DR, Tam E, Harney AS, Crawford RB, Kaminski NE, Zacharewski TR. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces suppressor of cytokine signaling 2 in murine B cells. Mol Pharmacol 2004;66:1662-1670.

176
119. Gouedard C, Barouki R, Morel Y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol Cell Biol 2004;24:5209-5222. 120. Ogi T, Mimura J, Hikida M, Fujimoto H, Fujii-Kuriyama Y, Ohmori H. Expression of human and mouse genes encoding polkappa: testis-specific developmental regulation and AhR-dependent inducible transcription. Genes Cells 2001;6:943-953. 121. Svensson C, Silverstone AE, Lai ZW, Lundberg K. Dioxin-induced adseverin expression in the mouse thymus is strictly regulated and dependent on the aryl hydrocarbon receptor. Biochem Biophys Res Commun 2002;291:1194-1200. 122. Svensson C, Lundberg K. Immune-specific up-regulation of adseverin gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol 2001;60:135-142. 123. Kolluri SK, Balduf C, Hofmann M, Gottlicher M. Novel target genes of the Ah (dioxin) receptor: transcriptional induction of N-myristoyltransferase 2. Cancer Res 2001;61:8534-8539. 124. Braun L, Kardon T, El Koulali K, Csala M, Mandl J, Banhegyi G. Different induction of gulonolactone oxidase in aromatic hydrocarbon-responsive or -unresponsive mouse strains. FEBS Lett 1999;463:345-349. 125. Ikuta T, Kawajiri K. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res 2006;312:3585-3594. 126. Thomsen JS, Kietz S, Strom A, Gustafsson JA. HES-1, a novel target gene for the aryl hydrocarbon receptor. Mol Pharmacol 2004;65:165-171. 127. Son DS, Rozman KK. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces plasminogen activator inhibitor-1 through an aryl hydrocarbon receptor-mediated pathway in mouse hepatoma cell lines. Arch Toxicol 2002;76:404-413. 128. Ebert B, Seidel A, Lampen A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005;26:1754-1763. 129. Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A 2002;99:4465-4470. 130. Baba T, Mimura J, Nakamura N, Harada N, Yamamoto M, Morohashi K, Fujii-Kuriyama Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol Cell Biol 2005;25:10040-10051. 131. Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, Flaws JA. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol Reprod 2007;76:1062-1070. 132. Kitajima M, Khan KN, Fujishita A, Masuzaki H, Koji T, Ishimaru T. Expression of the arylhydrocarbon receptor in the peri-implantation period of the mouse uterus and the impact of dioxin on mouse implantation. Arch Histol Cytol 2004;67:465-474. 133. Hushka LJ, Williams JS, Greenlee WF. Characterization of 2,3,7,8-tetrachlorodibenzofuran-dependent suppression and AH receptor pathway gene expression in the developing mouse mammary gland. Toxicol Appl Pharmacol 1998;152:200-210.

177
134. Thackaberry EA, Gabaldon DM, Walker MK, Smith SM. Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1alpha in the absence of cardiac hypoxia. Cardiovasc Toxicol 2002;2:263-274. 135. Vasquez A, Atallah-Yunes N, Smith FC, You X, Chase SE, Silverstone AE, Vikstrom KL. A role for the aryl hydrocarbon receptor in cardiac physiology and function as demonstrated by AhR knockout mice. Cardiovasc Toxicol 2003;3:153-163. 136. Lund AK, Goens MB, Kanagy NL, Walker MK. Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol Appl Pharmacol 2003;193:177-187. 137. McMillan BJ, Bradfield CA. The aryl hydrocarbon receptor sans xenobiotics: endogenous function in genetic model systems. Mol Pharmacol 2007;72:487-498. 138. Ramos KS, Hadi Falahatpisheh M, Nanez A, He Q. Modulation of biological regulatory networks during nephrogenesis. Drug Metab Rev 2006;38:677-683. 139. Akahoshi E, Yoshimura S, Ishihara-Sugano M. Over-expression of AhR (aryl hydrocarbon receptor) induces neural differentiation of Neuro2a cells: neurotoxicology study. Environ Health 2006;5:24. 140. Takanaga H, Kunimoto M, Adachi T, Tohyama C, Aoki Y. Inhibitory effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cAMP-induced differentiation of rat C6 glial cell line. J Neurosci Res 2001;64:402-409. 141. Nishimura N, Yonemoto J, Miyabara Y, Fujii-Kuriyama Y, Tohyama C. Altered thyroxin and retinoid metabolic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin in aryl hydrocarbon receptor-null mice. Arch Toxicol 2005;79:260-267. 142. Barouki R, Coumoul X, Fernandez-Salguero PM. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett 2007;581:3608-3615. 143. Zaher H, Fernandez-Salguero PM, Letterio J, Sheikh MS, Fornace AJ, Jr., Roberts AB, Gonzalez FJ. The involvement of aryl hydrocarbon receptor in the activation of transforming growth factor-beta and apoptosis. Mol Pharmacol 1998;54:313-321. 144. Gaido KW, Maness SC, Leonard LS, Greenlee WF. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-dependent regulation of transforming growth factors-alpha and -beta 2 expression in a human keratinocyte cell line involves both transcriptional and post-transcriptional control. J Biol Chem 1992;267:24591-24595. 145. Guo J, Sartor M, Karyala S, Medvedovic M, Kann S, Puga A, Ryan P, et al. Expression of genes in the TGF-beta signaling pathway is significantly deregulated in smooth muscle cells from aorta of aryl hydrocarbon receptor knockout mice. Toxicol Appl Pharmacol 2004;194:79-89. 146. Sutter TR, Guzman K, Dold KM, Greenlee WF. Targets for dioxin: genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science 1991;254:415-418. 147. Andreola F, Fernandez-Salguero PM, Chiantore MV, Petkovich MP, Gonzalez FJ, De Luca LM. Aryl hydrocarbon receptor knockout mice (AHR-/-) exhibit liver retinoid accumulation and reduced retinoic acid metabolism. Cancer Res 1997;57:2835-2838. 148. Schmidt CK, Hoegberg P, Fletcher N, Nilsson CB, Trossvik C, Hakansson H, Nau H. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the endogenous metabolism of all-trans-retinoic acid in the rat. Arch Toxicol 2003;77:371-383.

178
149. Andreola F, Calvisi DF, Elizondo G, Jakowlew SB, Mariano J, Gonzalez FJ, De Luca LM. Reversal of liver fibrosis in aryl hydrocarbon receptor null mice by dietary vitamin A depletion. Hepatology 2004;39:157-166. 150. Andreola F, Hayhurst GP, Luo G, Ferguson SS, Gonzalez FJ, Goldstein JA, De Luca LM. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J Biol Chem 2004;279:3434-3438. 151. Zacharewski TR, Bondy KL, McDonell P, Wu ZF. Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on 17 beta-estradiol-induced pS2 expression. Cancer Res 1994;54:2707-2713. 152. Porter W, Wang F, Duan R, Qin C, Castro-Rivera E, Kim K, Safe S. Transcriptional activation of heat shock protein 27 gene expression by 17beta-estradiol and modulation by antiestrogens and aryl hydrocarbon receptor agonists. J Mol Endocrinol 2001;26:31-42. 153. Safe S, Wormke M, Samudio I. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J Mammary Gland Biol Neoplasia 2000;5:295-306. 154. Spink DC, Lincoln DW, 2nd, Dickerman HW, Gierthy JF. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes an extensive alteration of 17 beta-estradiol metabolism in MCF-7 breast tumor cells. Proc Natl Acad Sci U S A 1990;87:6917-6921. 155. Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, Safe S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol Cell Biol 2003;23:1843-1855. 156. Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007;446:562-566. 157. Abdelrahim M, Ariazi E, Kim K, Khan S, Barhoumi R, Burghardt R, Liu S, et al. 3-Methylcholanthrene and other aryl hydrocarbon receptor agonists directly activate estrogen receptor alpha. Cancer Res 2006;66:2459-2467. 158. Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003;423:545-550. 159. Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, Romagnolo DF. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res 2006;66:2224-2232. 160. Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc Natl Acad Sci U S A 2003;100:6517-6522. 161. Beischlag TV, Perdew GH. ER alpha-AHR-ARNT protein-protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription. J Biol Chem 2005;280:21607-21611. 162. Jana NR, Sarkar S, Ishizuka M, Yonemoto J, Tohyama C, Sone H. Cross-talk between 2,3,7,8-tetrachlorodibenzo-p-dioxin and testosterone signal transduction pathways in LNCaP prostate cancer cells. Biochem Biophys Res Commun 1999;256:462-468.

179
163. Lin TM, Ko K, Moore RW, Simanainen U, Oberley TD, Peterson RE. Effects of aryl hydrocarbon receptor null mutation and in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on prostate and seminal vesicle development in C57BL/6 mice. Toxicol Sci 2002;68:479-487. 164. De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 2006;25:6868-6886. 165. Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab 2006;17:321-327. 166. Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 2006;20:1405-1428. 167. Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol 2005;6:439-445. 168. Morgan ET. Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev 1997;29:1129-1188. 169. Stoilov I, Krueger W, Mankowski D, Guernsey L, Kaur A, Glynn J, Thrall RS. The cytochromes P450 (CYP) response to allergic inflammation of the lung. Arch Biochem Biophys 2006;456:30-38. 170. Gharavi N, El-Kadi AO. Down-regulation of aryl hydrocarbon receptor-regulated genes by tumor necrosis factor-alpha and lipopolysaccharide in murine hepatoma Hepa 1c1c7 cells. J Pharm Sci 2005;94:493-506. 171. Paton TE, Renton KW. Cytokine-mediated down-regulation of CYP1A1 in Hepa1 cells. Biochem Pharmacol 1998;55:1791-1796. 172. Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem 1999;274:510-515. 173. Tian Y, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions: mechanisms and physiological implications. Chem Biol Interact 2002;141:97-115. 174. Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol 2005;67:1393-1398. 175. Podechard N, Lecureur V, Le Ferrec E, Guenon I, Sparfel L, Gilot D, Gordon JR, et al. Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation. Toxicol Lett 2008;177:130-137. 176. Vogel CF, Sciullo E, Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun 2007;363:722-726. 177. Lai ZW, Hundeiker C, Gleichmann E, Esser C. Cytokine gene expression during ontogeny in murine thymus on activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol 1997;52:30-37. 178. Henley DV, Bellone CJ, Williams DA, Ruh TS, Ruh MF. Aryl hydrocarbon receptor-mediated posttranscriptional regulation of IL-1beta. Arch Biochem Biophys 2004;422:42-51.

180
179. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links T(H)17-cell-mediated autoimmunity to environmental toxins. Nature 2008. 180. Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health 2003;2:16. 181. Lawrence BP, Denison MS, Novak H, Vorderstrasse BA, Harrer N, Neruda W, Reichel C, et al. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low molecular weight compound. Blood 2008. 182. Shi LZ, Faith NG, Nakayama Y, Suresh M, Steinberg H, Czuprynski CJ. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J Immunol 2007;179:6952-6962. 183. Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer 2007;59:248-257. 184. Vogel C, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, Abel J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis 2000;21:2267-2274. 185. Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem 2004;279:8886-8894. 186. Hoffer A, Chang CY, Puga A. Dioxin induces transcription of fos and jun genes by Ah receptor-dependent and -independent pathways. Toxicol Appl Pharmacol 1996;141:238-247. 187. Suh J, Jeon YJ, Kim HM, Kang JS, Kaminski NE, Yang KH. Aryl hydrocarbon receptor-dependent inhibition of AP-1 activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in activated B cells. Toxicol Appl Pharmacol 2002;181:116-123. 188. Ruby CE, Leid M, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-alpha and anti-CD40-induced activation of NF-kappaB/Rel in dendritic cells: p50 homodimer activation is not affected. Mol Pharmacol 2002;62:722-728. 189. Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol 2007;170:855-864. 190. Lecureur V, Ferrec EL, N'Diaye M, Vee ML, Gardyn C, Gilot D, Fardel O. ERK-dependent induction of TNFalpha expression by the environmental contaminant benzo(a)pyrene in primary human macrophages. FEBS Lett 2005;579:1904-1910. 191. Ke S, Rabson AB, Germino JF, Gallo MA, Tian Y. Mechanism of suppression of cytochrome P-450 1A1 expression by tumor necrosis factor-alpha and lipopolysaccharide. J Biol Chem 2001;276:39638-39644. 192. Umannova L, Zatloukalova J, Machala M, Krcmar P, Majkova Z, Hennig B, Kozubik A, et al. Tumor necrosis factor-alpha modulates effects of aryl hydrocarbon receptor ligands on cell proliferation and expression of cytochrome P450 enzymes in rat liver "stem-like" cells. Toxicol Sci 2007;99:79-89.

181
193. Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys 2007;461:169-175. 194. Knaapen AM, Curfs DM, Pachen DM, Gottschalk RW, de Winther MP, Daemen MJ, Van Schooten FJ. The environmental carcinogen benzo[a]pyrene induces expression of monocyte-chemoattractant protein-1 in vascular tissue: a possible role in atherogenesis. Mutat Res 2007;621:31-41. 195. Vorderstrasse BA, Lawrence BP. Protection against lethal challenge with Streptococcus pneumoniae is conferred by aryl hydrocarbon receptor activation but is not associated with an enhanced inflammatory response. Infect Immun 2006;74:5679-5686. 196. Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J, Hosoya T, et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol 2005;25:9360-9368. 197. Hinsdill RD, Couch DL, Speirs RS. Immunosuppression in mice induced by dioxin (TCDD) in feed. J Environ Pathol Toxicol 1980;4:401-425. 198. Luster MI, Boorman GA, Dean JH, Harris MW, Luebke RW, Padarathsingh ML, Moore JA. Examination of bone marrow, immunologic parameters and host susceptibility following pre- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Int J Immunopharmacol 1980;2:301-310. 199. Sugita-Konishi Y, Kobayashi K, Naito H, Miura K, Suzuki Y. Effect of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on the susceptibility to Listeria infection. Biosci Biotechnol Biochem 2003;67:89-93. 200. Kerkvliet NI, Baecher-Steppan L, Shepherd DM, Oughton JA, Vorderstrasse BA, DeKrey GK. Inhibition of TC-1 cytokine production, effector cytotoxic T lymphocyte development and alloantibody production by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol 1996;157:2310-2319. 201. Hahn ME. Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact 2002;141:131-160. 202. Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 2000;97:779-782. 203. Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res 2004;32:4512-4523. 204. Sogawa K, Numayama-Tsuruta K, Takahashi T, Matsushita N, Miura C, Nikawa J, Gotoh O, et al. A novel induction mechanism of the rat CYP1A2 gene mediated by Ah receptor-Arnt heterodimer. Biochem Biophys Res Commun 2004;318:746-755. 205. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998;93:531-541. 206. Bladh LG, Liden J, Dahlman-Wright K, Reimers M, Nilsson S, Okret S. Identification of endogenous glucocorticoid repressed genes differentially regulated by a glucocorticoid receptor mutant able to separate between nuclear factor-kappaB and activator protein-1 repression. Mol Pharmacol 2005;67:815-826.

182
207. Valentine JE, Kalkhoven E, White R, Hoare S, Parker MG. Mutations in the estrogen receptor ligand binding domain discriminate between hormone-dependent transactivation and transrepression. J Biol Chem 2000;275:25322-25329. 208. Heid SE, Walker MK, Swanson HI. Correlation of cardiotoxicity mediated by halogenated aromatic hydrocarbons to aryl hydrocarbon receptor activation. Toxicol Sci 2001;61:187-196. 209. Elizondo G, Fernandez-Salguero P, Sheikh MS, Kim GY, Fornace AJ, Lee KS, Gonzalez FJ. Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol 2000;57:1056-1063. 210. Ge NL, Elferink CJ. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J Biol Chem 1998;273:22708-22713. 211. Koo BH, Kim DS. Factor Xa induces mitogenesis of vascular smooth muscle cells via autocrine production of epiregulin. J Biol Chem 2003;278:52578-52586. 212. Puga A, Marlowe J, Barnes S, Chang CY, Maier A, Tan Z, Kerzee JK, et al. Role of the aryl hydrocarbon receptor in cell cycle regulation. Toxicology 2002;181-182:171-177. 213. Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact 2002;141:3-24. 214. Dunson DB, Haseman JK, van Birgelen AP, Stasiewicz S, Tennant RW. Statistical analysis of skin tumor data from Tg.AC mouse bioassays. Toxicol Sci 2000;55:293-302. 215. Poland A, Palen D, Glover E. Tumour promotion by TCDD in skin of HRS/J hairless mice. Nature 1982;300:271-273. 216. Dragan YP, Schrenk D. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Addit Contam 2000;17:289-302. 217. Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res 2003;284:2-13. 218. Toyoda H, Komurasaki T, Uchida D, Takayama Y, Isobe T, Okuyama T, Hanada K. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem 1995;270:7495-7500. 219. Komurasaki T, Toyoda H, Uchida D, Nemoto N. Mechanism of growth promoting activity of epiregulin in primary cultures of rat hepatocytes. Growth Factors 2002;20:61-69. 220. Shirakata Y, Komurasaki T, Toyoda H, Hanakawa Y, Yamasaki K, Tokumaru S, Sayama K, et al. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J Biol Chem 2000;275:5748-5753. 221. Sekiguchi T, Mizutani T, Yamada K, Kajitani T, Yazawa T, Yoshino M, Miyamoto K. Expression of epiregulin and amphiregulin in the rat ovary. J Mol Endocrinol 2004;33:281-291. 222. Sekiguchi T, Mizutani T, Yamada K, Yazawa T, Kawata H, Yoshino M, Kajitani T, et al. Transcriptional regulation of the epiregulin gene in the rat ovary. Endocrinology 2002;143:4718-4729.

183
223. Takahashi M, Hayashi K, Yoshida K, Ohkawa Y, Komurasaki T, Kitabatake A, Ogawa A, et al. Epiregulin as a major autocrine/paracrine factor released from ERK- and p38MAPK-activated vascular smooth muscle cells. Circulation 2003;108:2524-2529. 224. Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, Yuspa SH. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol 1995;254:3-20. 225. Murray IA, Reen, R.K., Leathery, N., Ramadoss, P., Bonati, L., Gonzalez, F.J., Peters, J.M., and Perdew G.H. Evidence that ligand binding is a key determinant of Ah receptor-mediated transcriptional activity. Arch. Biochem. Biophys. (In Press) 2005. 226. Chou JY. Establishment of rat fetal liver lines and characterization of their metabolic and hormonal properties: use of temperature-sensitive SV40 virus. Methods Enzymol 1985;109:385-396. 227. Lorenzen A, Kennedy SW, Bastien LJ, Hahn ME. Halogenated aromatic hydrocarbon-mediated porphyrin accumulation and induction of cytochrome P4501A in chicken embryo hepatocytes. Biochem Pharmacol 1997;53:373-384. 228. Sato K, Nakamura T, Mizuguchi M, Miura K, Tada M, Aizawa T, Gomi T, et al. Solution structure of epiregulin and the effect of its C-terminal domain for receptor binding affinity. FEBS Lett 2003;553:232-238. 229. Spencer VA, Sun JM, Li L, Davie JR. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods 2003;31:67-75. 230. Swanson HI, Whitelaw ML, Petrulis JR, Perdew GH. Use of [125I]4'-iodoflavone as a tool to characterize ligand-dependent differences in Ah receptor behavior. J Biochem Mol Toxicol 2002;16:298-310. 231. Carlson DB, Perdew GH. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol 2002;16:317-325. 232. Kobayashi A, Sogawa K, Fujii-Kuriyama Y. Cooperative interaction between AhR.Arnt and Sp1 for the drug-inducible expression of CYP1A1 gene. J Biol Chem 1996;271:12310-12316. 233. Shelly M, Pinkas-Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, Alimandi M, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem 1998;273:10496-10505. 234. Zhu Z, Kleeff J, Friess H, Wang L, Zimmermann A, Yarden Y, Buchler MW, et al. Epiregulin is Up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem Biophys Res Commun 2000;273:1019-1024. 235. Torring N, Jorgensen PE, Sorensen BS, Nexo E. Increased expression of heparin binding EGF (HB-EGF), amphiregulin, TGF alpha and epiregulin in androgen-independent prostate cancer cell lines. Anticancer Res 2000;20:91-95. 236. Yamamoto T, Akisue T, Marui T, Nakatani T, Kawamoto T, Hitora T, Nagira K, et al. Expression of betacellulin, heparin-binding epidermal growth factor and epiregulin in human malignant fibrous histiocytoma. Anticancer Res 2004;24:2007-2010. 237. Baba I, Shirasawa S, Iwamoto R, Okumura K, Tsunoda T, Nishioka M, Fukuyama K, et al. Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res 2000;60:6886-6889.

184
238. Freimann S, Ben-Ami I, Hirsh L, Dantes A, Halperin R, Amsterdam A. Drug development for ovarian hyper-stimulation and anti-cancer treatment: blocking of gonadotropin signaling for epiregulin and amphiregulin biosynthesis. Biochem Pharmacol 2004;68:989-996. 239. Fiorito F, Pagnini U, De Martino L, Montagnaro S, Ciarcia R, Florio S, Pacilio M, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases Bovine Herpesvirus type-1 (BHV-1) replication in Madin-Darby Bovine Kidney (MDBK) cells in vitro. J Cell Biochem 2008;103:221-233. 240. Cole P, Trichopoulos D, Pastides H, Starr T, Mandel JS. Dioxin and cancer: a critical review. Regul Toxicol Pharmacol 2003;38:378-388. 241. Steenland K, Bertazzi P, Baccarelli A, Kogevinas M. Dioxin revisited: developments since the 1997 IARC classification of dioxin as a human carcinogen. Environ Health Perspect 2004;112:1265-1268. 242. Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, Blumberg B, Guzelian PS, et al. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev 2000;14:3014-3023. 243. Puga A, Tomlinson CR, Xia Y. Ah receptor signals cross-talk with multiple developmental pathways. Biochem Pharmacol 2005;69:199-207. 244. Kobayashi K, Sueyoshi T, Inoue K, Moore R, Negishi M. Cytoplasmic accumulation of the nuclear receptor CAR by a tetratricopeptide repeat protein in HepG2 cells. Mol Pharmacol 2003;64:1069-1075. 245. Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol 2000;20:2951-2958. 246. Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol 1999;19:6318-6322. 247. Pascussi JM, Busson-Le Coniat M, Maurel P, Vilarem MJ. Transcriptional analysis of the orphan nuclear receptor constitutive androstane receptor (NR1I3) gene promoter: identification of a distal glucocorticoid response element. Mol Endocrinol 2003;17:42-55. 248. Assenat E, Gerbal-Chaloin S, Larrey D, Saric J, Fabre JM, Maurel P, Vilarem MJ, et al. Interleukin 1beta inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology 2004;40:951-960. 249. Mitchell KA, Lockhart CA, Huang G, Elferink CJ. Sustained aryl hydrocarbon receptor activity attenuates liver regeneration. Mol Pharmacol 2006;70:163-170. 250. Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem 2004;279:19832-19838. 251. Strom SC, Pisarov LA, Dorko K, Thompson MT, Schuetz JD, Schuetz EG. Use of human hepatocytes to study P450 gene induction. Methods Enzymol 1996;272:388-401. 252. Sidhu JS, Liu F, Omiecinski CJ. Phenobarbital responsiveness as a uniquely sensitive indicator of hepatocyte differentiation status: requirement of dexamethasone and extracellular matrix in establishing the functional integrity of cultured primary rat hepatocytes. Exp Cell Res 2004;292:252-264.

185
253. Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A 1996;93:14960-14965. 254. Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000;407:920-923. 255. Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol 1999;39:1-17. 256. Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. Induction of bilirubin clearance by the constitutive androstane receptor (CAR). Proc Natl Acad Sci U S A 2003;100:4156-4161. 257. Ledda-Columbano GM, Pibiri M, Concas D, Molotzu F, Simbula G, Cossu C, Columbano A. Sex difference in the proliferative response of mouse hepatocytes to treatment with the CAR ligand, TCPOBOP. Carcinogenesis 2003;24:1059-1065. 258. Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, Moore DD. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol 2005;19:1646-1653. 259. Koide A, Fuwa K, Furukawa F, Hirose M, Nishikawa A, Mori Y. Effect of cigarette smoke on the mutagenic activation of environmental carcinogens by rodent liver. Mutat Res 1999;428:165-176. 260. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 1999;340:448-454. 261. Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis 1999;19:141-155. 262. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell 2002;109 Suppl:S81-96. 263. Baldwin AS, Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649-683. 264. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004;4:499-511. 265. Mosialos G, Gilmore TD. v-Rel and c-Rel are differentially affected by mutations at a consensus protein kinase recognition sequence. Oncogene 1993;8:721-730. 266. Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-[kappa]B is regulated by the I[kappa]B-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 1997;89:413-424. 267. Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by [zeta]PKC in NF-[kappa]B transcriptional activation. EMBO J. 2003;22:3910-3918. 268. Wang D, Baldwin AS. Activation of nuclear factor-[kappa]B-dependent transcription by tumor necrosis factor-[alpha] is mediated through phosphorylation of RelA/p65 on serine 529. J. Biol. Chem. 1998;273:29411-29416. 269. Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. I[kappa]B kinases phosphorylate NF-[kappa]B p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999;274:30353-30356.

186
270. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-[kappa]B action regulated by reversible acetylation. Science 2001;293:1653-1657. 271. Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-[kappa]B. EMBO J. 2002;21:6539-6548. 272. Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol 2004;5:392-401. 273. Levine SL, Petrulis JR, Dubil A, Perdew GH. A tetratricopeptide repeat half-site in the aryl hydrocarbon receptor is important for DNA binding and trans-activation potential. Mol Pharmacol 2000;58:1517-1524. 274. Murray IA, Reen RK, Leathery N, Ramadoss P, Bonati L, Gonzalez FJ, Peters JM, et al. Evidence that ligand binding is a key determinant of Ah receptor-mediated transcriptional activity. Arch Biochem Biophys 2005;442:59-71. 275. Morales JL, Krzeminski J, Amin S, Perdew GH. Characterization of the antiallergic drugs 3-[2-(2-phenylethyl) benzoimidazole-4-yl]-3-hydroxypropanoic acid and ethyl 3-hydroxy-3-[2-(2-phenylethyl)benzoimidazol-4-yl]propanoate as full aryl hydrocarbon receptor agonists. Chem Res Toxicol 2008;21:472-482. 276. Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H, Yamamoto M, et al. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J Immunol 2005;175:7348-7356. 277. Kiernan R. Post-activation turn-off of NF-[kappa]B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003;278:2758-2766. 278. Quivy V, Van Lint C. Regulation at multiple levels of NF-kappaB-mediated transactivation by protein acetylation. Biochem Pharmacol 2004;68:1221-1229. 279. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007;7:454-465. 280. Kluve-Beckerman B, Drumm ML, Benson MD. Nonexpression of the human serum amyloid A three (SAA3) gene. DNA Cell Biol 1991;10:651-661. 281. Bone RC. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med 1996;24:163-172. 282. Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GD, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004;350:1387-1397. 283. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000;342:836-843. 284. Veerhuis R, Boshuizen RS, Familian A. Amyloid associated proteins in Alzheimer's and prion disease. Curr Drug Targets CNS Neurol Disord 2005;4:235-248. 285. Ray A, Ray BK. Serum amyloid A gene expression level in liver in response to different inflammatory agents is dependent upon the nature of activated transcription factors. DNA Cell Biol 1997;16:1-7. 286. Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, Yoshizaki K. Essential role of STAT3 in cytokine-driven NF-kappaB-mediated serum amyloid A gene expression. Genes Cells 2005;10:1051-1063.

187
287. Shimizu H, Yamamoto K. NF-kappa B and C/EBP transcription factor families synergistically function in mouse serum amyloid A gene expression induced by inflammatory cytokines. Gene 1994;149:305-310. 288. Vermeulen L, De Wilde G, Damme PV, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-[kappa]B p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003;22:1313-1324. 289. Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1[beta]-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000;20:6891-6903. 290. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-[kappa]B controls expression of inhibitor I[kappa]B[alpha]: evidence for an inducible autoregulatory pathway. Science 1993;259:1912-1915. 291. Arenzana-Seisdedos F. Inducible nuclear expression of newly synthesized I[kappa]B[alpha] negatively regulates DNA-binding and transcriptional activities of NF-[kappa]B. Mol. Cell. Biol. 1995;15:2689-2696. 292. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 2003;24:488-522. 293. Galizia G, Lieto E, De Vita F, Orditura M, Castellano P, Troiani T, Imperatore V, et al. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene 2007;26:3654-3660. 294. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169-181. 295. Nishimura T, Andoh A, Inatomi O, Shioya M, Yagi Y, Tsujikawa T, Fujiyama Y. Amphiregulin and epiregulin expression in neoplastic and inflammatory lesions in the colon. Oncol Rep 2008;19:105-110. 296. Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230-3237. 297. Shirakata Y, Kishimoto J, Tokumaru S, Yamasaki K, Hanakawa Y, Tohyama M, Sayama K, et al. Epiregulin, a member of the EGF family, is over-expressed in psoriatic epidermis. J Dermatol Sci 2007;45:69-72. 298. Schwarz M, Appel KE. Carcinogenic risks of dioxin: mechanistic considerations. Regul Toxicol Pharmacol 2005;43:19-34. 299. Shirasawa S, Sugiyama S, Baba I, Inokuchi J, Sekine S, Ogino K, Kawamura Y, et al. Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune-related responses of keratinocyte and macrophage. Proc Natl Acad Sci U S A 2004;101:13921-13926. 300. Choi SS, Miller MA, Harper PA. In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin induces amphiregulin gene expression in the developing mouse ureter. Toxicol Sci 2006;94:163-174.

188
301. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-[gamma]. Nature 2005;437:759-763. 302. Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature 1998;392:885-888. 303. Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, et al. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci U S A 2003;100:13845-13850. 304. Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell 2004;118:453-464. 305. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2006;443:998-1002. 306. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 2000;25:177-181. 307. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 2001;28:313-314. 308. Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A 2002;99:6979-6984. 309. Brzozowski AM. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753-758. 310. Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 2001;104:719-730.

VITA
Rushang Dilipkumar Patel Education 07/2002 – 08/2008 Ph.D., Integrative Biosciences - Molecular Medicine Option The Pennsylvania State University, University Park, PA, USA 11/1996 - 03/2002 MBBS (Bachelor of Medicine and Bachelor of Surgery) M.S.University, Baroda Medical College, India Membership and Honorary/Professional Societies Society of Toxicology, U.S.A. Gujarat Medical Council, India. Publications Patel RD, Hollingshead BD, Omiecinski CJ, Perdew GH.. Aryl-hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver.. Hepatology. 2007, Jul; 46(1):209-218. Patel RD, Kim DJ, Peters JM, Perdew GH.. The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin.. Toxicological Sciences. 2006, Jan; 89(1):75-82. Hollingshead BD, Patel RD, Perdew GH.. Endogenous hepatic expression of the hepatitis B virus X-associated protein 2 is adequate for maximal association with aryl hydrocarbon receptor-90-kDa heat shock protein complexes.. Molecular Pharmacology. 2006, Sep; 70(6):2096-2107. Chiaro CR, Patel RD, Marcus CB, Perdew GH. Evidence for an Ah receptor-mediated cytochrome P450 auto-regulatory pathway.. Molecular Pharmacology. 2007, Nov;72(5):1369-79. Other Awards/Accomplishments 2006 Research Excellence Award - Society of Toxicology, U.S.A. 2006 2nd prize, Annual Graduate Student Research exhibit, Penn State University. 2005 Research Grant - College of Agricultural Sciences, Penn State University. 2002 – 2004 Life Sciences Consortium Fellowship, Penn State University. 1996 – 1998 Recipient of Baroda Educational Trust scholarship for two consecutive years in Baroda Medical College for securing 2nd (1996-1997) and 1st (1997-1998) ranks respectively. 1994 – 1995 4th rank in Senior- and 5th rank in Junior-Mathematics Olympiad (state level) Gujarat, India.


![WELCOME [bmb.natsci.msu.edu] · 2018. 3. 13. · Poster: 30 Mentor(s): John LaPres (Biochemistry) The Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that](https://img.pdfslide.us/doc/110x75/600f8c864b2b00243c56cf13/welcome-bmb-2018-3-13-poster-30-mentors-john-lapres-biochemistry.jpg)


























