Embed Size (px)
Citation preview

Kidney International, Vol. 66 (2004), pp. 1918–1925
ION CHANNELS – MEMBRANE TRANSPORT – INTEGRATIVE PHYSIOLOGY
Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2and the serum- and glucocorticoid-dependent kinases
HAMDY M. EMBARK, CHRISTOPH BOHMER, MONICA PALMADA, JEYAGANESH RAJAMANICKAM,AMANDA W. WYATT, SABINE WALLISCH, GIOVAMBATTISTA CAPASSO, PETRA WALDEGGER,HANNSJORG W. SEYBERTH, SIEGFRIED WALDEGGER, and FLORIAN LANG
Department of Physiology I, University of Tubingen, Germany; Department of Nephrology, University of Napoli, Napoli, Italy; andDepartment of Pediatrics, University of Marburg, Germany
Regulation of ClC-Ka/barttin by the ubiquitin ligase Nedd4-2and the serum- and glucocorticoid-dependent kinases.
Background. ClC-Ka and ClC-Kb, chloride channels partici-pating in renal tubular Cl− transport, require the coexpressionof barttin to become functional. Mutations of the barttin genelead to the Bartter’s syndrome variant BSND, characterizedby congenital deafness and severe renal salt wasting. Barttinbears a proline-tyrosine motif, a target structure for the ubiq-uitin ligase Nedd4-2, which mediates the clearance of channelproteins from the cell membrane. Nedd4-2 is, in turn, a targetof the serum- and glucocorticoid-inducible kinase SGK1, whichphosphorylates and, thus, inactivates the ubiquitin ligase. ClC-Ka also possesses a SGK1 consensus site in its sequence. Wehypothesized that ClC-Ka/barttin is stimulated by SGK1, anddown-regulated by Nedd4-2, an effect that may be reversed bySGK1 and its isoforms, SGK2 or SGK3.
Methods. To test this hypothesis, ClC-Ka/barttin was het-erologously expressed in Xenopus oocytes with or without theadditional expression of Nedd4-2, SGK1, SGK2, SGK3, consti-tutively active S422DSGK1, or inactive K127NSGK1.
Results. Expression of ClC-Ka/barttin induced a slightly in-wardly rectifying current that was significantly decreased uponcoexpression of Nedd4-2, but not the catalytically inactive mu-tant C938SNedd4-2. The coexpression of S422DSGK1, SGK1, orSGK3, but not SGK2 or K127NSGK1 significantly stimulated thecurrent. Moreover, S422DSGK1, SGK1, and SGK3 also phospho-rylated Nedd4-2 and thereby inhibited Nedd4-2 binding to itstarget. The down-regulation of ClC-Ka/barttin by Nedd4-2 wasabolished by elimination of the PY motif in barttin.
Conclusion. ClC-Ka/barttin channels are regulated by SGK1and SGK3, which may thus participate in the regulation of trans-port in kidney and inner ear.
Both ClC-Ka and ClC-Kb, members of the ClC fam-ily of chloride channels [1], are expressed in the kidney
Key words: Bartter, deafness, ubiquitination, surface expression, ionchannel.
Received for publication February 20, 2003and in revised form September 26, 2003, and April 27, 2004Accepted for publication May 18, 2004
C© 2004 by the International Society of Nephrology
[2]. Functional expression of ClC-Ka and ClC-Kb chan-nels requires the coexpression of barttin, an essential bsubunit of the channels [3, 4]. The functional significanceof ClC-Kb and barttin is illustrated by monogenic dis-orders. Defects of the ClC-Kb gene CLCNKB underlyclassic Bartter’s syndrome, which is characterized by re-nal salt wasting [5]. Mutations in the barttin gene BSNDcause an even more pronounced salt-losing phenotypeaccompanied by sensorineural deafness [6].
The ubiquitin ligase Nedd4-2 can regulate several chan-nels, such as the epithelial sodium channel ENaC [7–9]via interaction with proline-tyrosine (PY) motifs presenton the target protein. Barttin carries a PY motif [3],and accordingly, replacement of the tyrosine in the PYmotif by alanine enhances the currents induced by ClC-Kb/barttin [3]. The ubiquitin ligase Nedd4-2 is phospho-rylated, and its interaction with ENaC is thus inhibitedby the serum- and glucocorticoid-dependent kinase 1(SGK1) [7, 8]. This effect provides a mechanistic explana-tion for the strong stimulatory effect of SGK1 on ENaC-induced sodium currents [10–17].
Two further isoforms, SGK2 and SGK3 [18], are sim-ilarly able to stimulate ENaC activity [19]. All threeisoforms belong to the family of Ser/Thr kinases, andrecognize the same consensus sequence in their targets(Arg-Xaa-Arg-Xaa-Xaa-Ser/Thr). ClC-Ka channels con-tain this sequence (Arg-Val-Arg-Thr-Thr-187Thr), and arethus, putative targets of the kinases. SGK1-3 are activatedby IGF1, insulin, and oxidative stress through a signalingcascade involving phosphatidylinositol 3 kinase (PI3 ki-nase) and phosphoinositide-dependent kinase PDK1 [18,20, 21]. The activation of SGK1 by PDK1 involves phos-phorylation of the serine at position 422; replacement ofthis serine by aspartate (S422DSGK1) yields a constitu-tively active kinase [20]. The transcription of SGK1, butnot SGK2 and SGK3 [22], has been shown to be sensitiveto cell volume [2, 23, 24] and a variety of hormones [22],including glucocorticoids [25–27] and mineralocorticoids[12, 14, 15, 28].
1918
Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases 1919
The present study has been performed to test whetherthe Cl− channels induced by coexpression of ClC-Ka andbarttin are sensitive to regulation by the ubiquitin ligaseNedd4-2 and the SGK isoforms SGK1, SGK2, and SGK3.
METHODS
Expression in Xenopus laevis oocytes and voltage-clampanalysis
cRNA encoding wild-type human ClC-Ka [4], wild-type human barttin [4], and PY-deficient barttin(Y98Abarttin), wild-type Xenopus Nedd4-2 [7], and cat-alytically inactive Nedd4-2 (C938SNedd4-2), wild-typehuman SGK1 [24], constitutively active (S422DSGK1) orinactive (K127NSGK1) human SGK1 [20], wild-type hu-man SGK2 [18], or wild-type human SGK3 [18] weresynthesized in vitro as previously described [29]. Dissec-tion of Xenopus laevis ovaries, collection, and handling ofthe oocytes have been described in detail elsewhere [29].Oocytes were injected with 5 ng of ClC-Ka, 5 ng of barttin,15 ng of Nedd4-2, 7.5 ng of SGK1, S422DSGK1, SGK2, orSGK3 cRNA. Control oocytes were injected with H2O.Electrophysiologic experiments were performed at roomtemperature 2 to 3 days after injection of the respectivecRNAs. The currents were determined in two-electrodevoltage-clamp experiments utilizing a pulse protocol of800 ms pulses from −150 mV to +50 mV. The inter-mediate holding-voltage was −60 mV. Steady-state cur-rent at the end of each voltage step was taken for dataanalysis. The data were filtered at 10 Hz, and recordedwith MacLab digital to analog converter and softwarefor data acquisition and analysis (ADInstruments, Cas-tle Hill, Australia). The control bath solution (ND96)contained 96 mmol/L NaCl, 2 mmol/L KCl, 1.8 mmol/LCaCl2, 1 mmol/L MgCl2, and 5 mmol/L HEPES, pH 7.4.The final solutions were titrated to the pH indicated usingHCl or NaOH. The flow rate of the superfusion was 20mL/min, and a complete exchange of the bath solutionwas reached within about 10 seconds.
Detection of Nedd4-2 phosphorylation
For determination of Nedd4-2 phosphorylation, 30cells of each group were homogenized in lysis buffer con-taining 50 mmol/L Tris (pH 7.5), 0.5 mmol/L EDTA (pH8.0), 0.5 mmol/L EGTA, 100 mmol/L NaCl, 1% TritonX-100, 100 lmol/L sodium orthovanadate, and proteininhibitor cocktail (Roche, Mannheim, Germany) at therecommended concentration. Proteins were separatedon a 7% polyacrylamide gel and transferred to nitrocel-lulose membranes. After blocking with 5% nonfat drymilk in PBS/0.15% Tween 20 for 1 hour at room tem-perature, blots were incubated at 4◦C overnight with arabbit anti-phosphoserine328 Nedd4-2 antibody (Pineda,Berlin, Germany, diluted 1:250 in PBS/0.15% Tween
H2O A
1.5
0.0
−1.5
µA
−150 −100 −50 0 50
H2OCIC-Ka + barttin
B
V, mV
Bro
mid
e
Nitr
ate
Iodi
de
CIC-Ka + barttin
500 ms 1 µA
1.0
0.8
0.6
0.4
0.2
0.0
Px/
Pcr
C
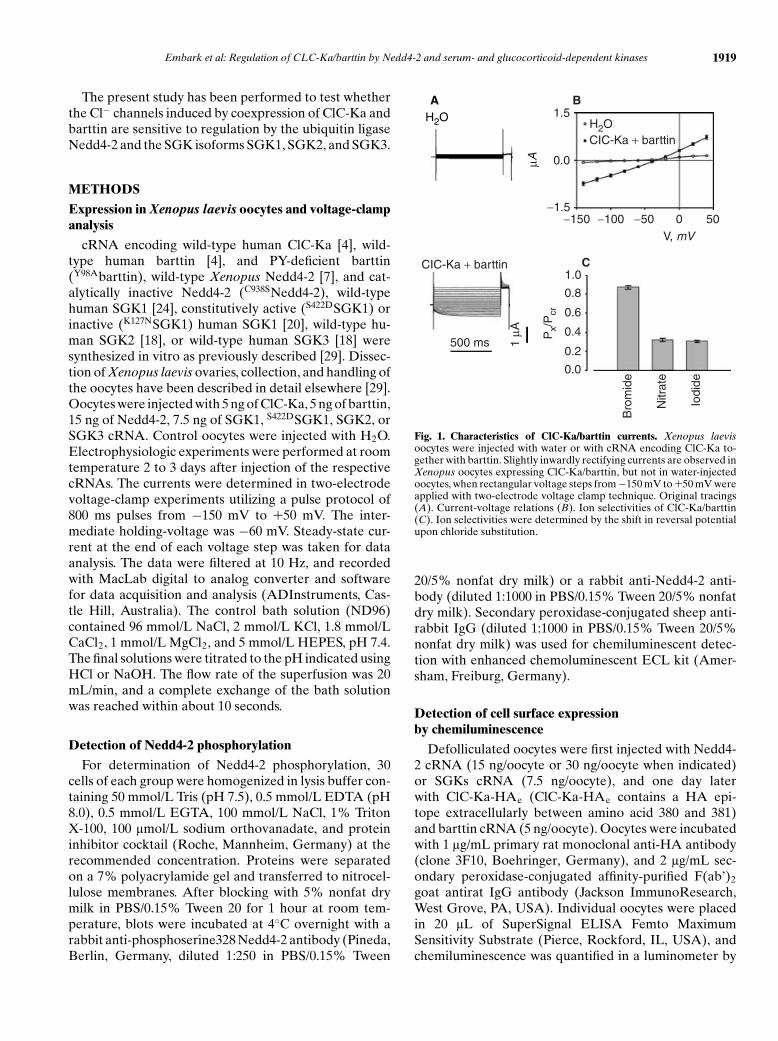
Fig. 1. Characteristics of ClC-Ka/barttin currents. Xenopus laevisoocytes were injected with water or with cRNA encoding ClC-Ka to-gether with barttin. Slightly inwardly rectifying currents are observed inXenopus oocytes expressing ClC-Ka/barttin, but not in water-injectedoocytes, when rectangular voltage steps from −150 mV to +50 mV wereapplied with two-electrode voltage clamp technique. Original tracings(A). Current-voltage relations (B). Ion selectivities of ClC-Ka/barttin(C). Ion selectivities were determined by the shift in reversal potentialupon chloride substitution.
20/5% nonfat dry milk) or a rabbit anti-Nedd4-2 anti-body (diluted 1:1000 in PBS/0.15% Tween 20/5% nonfatdry milk). Secondary peroxidase-conjugated sheep anti-rabbit IgG (diluted 1:1000 in PBS/0.15% Tween 20/5%nonfat dry milk) was used for chemiluminescent detec-tion with enhanced chemoluminescent ECL kit (Amer-sham, Freiburg, Germany).
Detection of cell surface expressionby chemiluminescence
Defolliculated oocytes were first injected with Nedd4-2 cRNA (15 ng/oocyte or 30 ng/oocyte when indicated)or SGKs cRNA (7.5 ng/oocyte), and one day laterwith ClC-Ka-HAe (ClC-Ka-HAe contains a HA epi-tope extracellularly between amino acid 380 and 381)and barttin cRNA (5 ng/oocyte). Oocytes were incubatedwith 1 lg/mL primary rat monoclonal anti-HA antibody(clone 3F10, Boehringer, Germany), and 2 lg/mL sec-ondary peroxidase-conjugated affinity-purified F(ab’)2
goat antirat IgG antibody (Jackson ImmunoResearch,West Grove, PA, USA). Individual oocytes were placedin 20 lL of SuperSignal ELISA Femto MaximumSensitivity Substrate (Pierce, Rockford, IL, USA), andchemiluminescence was quantified in a luminometer by
1920 Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases
1.5
0.0
−1.5−150 −100 −50 0 50
V, mV
A
NoninjectedCIC-Ka + barttinCIC-Ka + barttin + Nedd4-2CIC-Ka + barttin + SGK1CIC-Ka + barttin + SGK1 + Nedd4-2
µA
1.5
0.0
−1.5−150 −100 −50 0 50
V, mV
B
NoninjectedCIC-Ka + barttinCIC-Ka + barttin + Nedd4-2CIC-Ka + barttin + C938SNedd4-2
µA
16
14
12
10
86
4
2
0
Slo
pe c
ondu
ctan
ce, µ
S
Non
inje
cted
CIC
-Ka
+ba
rttin
CIC
-Ka
+ ba
rttin
+N
edd4
-2
CIC
-Ka
+ba
rttin
+C
938S
Ned
d4-2
*
D16
14
12
10
8
6
4
2
0
Slo
pe c
ondu
ctan
ce, µ
S
Non
inje
cted
CIC
-Ka
+ ba
rttin
CIC
-Ka
+ ba
rttin
+ N
edd4
-2
CIC
-Ka
+ ba
rttin
+ S
GK
1
CIC
-Ka
+ ba
rttin
+ S
GK
1 +
Ned
d4-2
*
* *
C
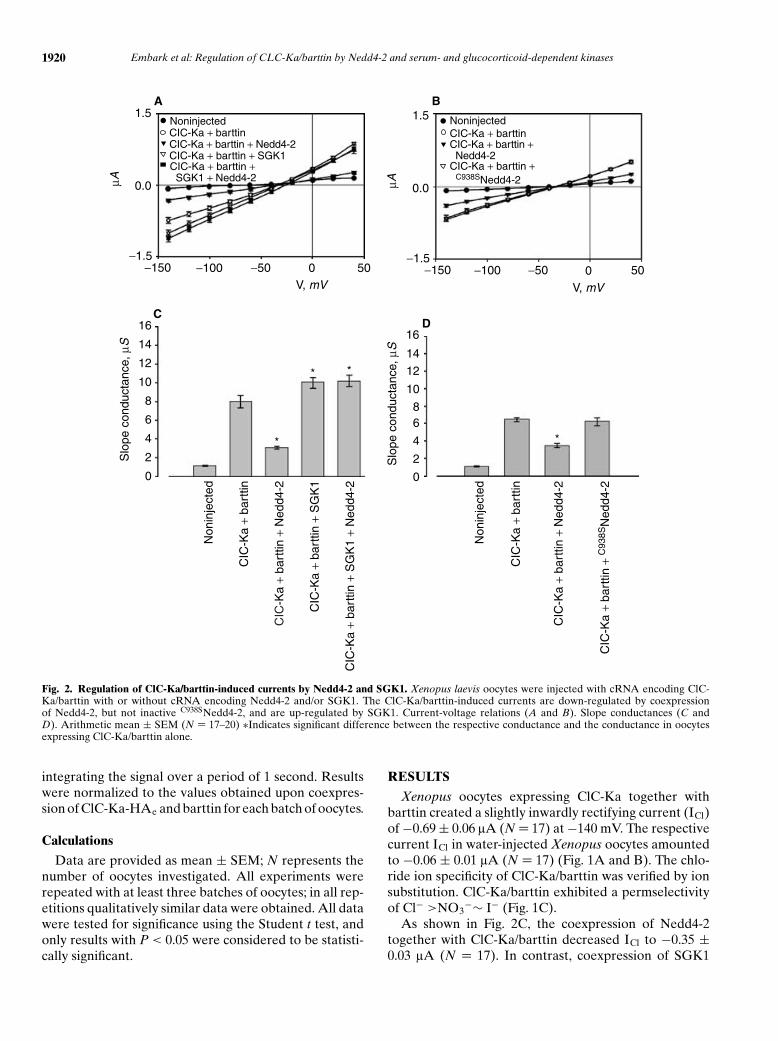
Fig. 2. Regulation of ClC-Ka/barttin-induced currents by Nedd4-2 and SGK1. Xenopus laevis oocytes were injected with cRNA encoding ClC-Ka/barttin with or without cRNA encoding Nedd4-2 and/or SGK1. The ClC-Ka/barttin-induced currents are down-regulated by coexpressionof Nedd4-2, but not inactive C938SNedd4-2, and are up-regulated by SGK1. Current-voltage relations (A and B). Slope conductances (C andD). Arithmetic mean ± SEM (N = 17–20) ∗Indicates significant difference between the respective conductance and the conductance in oocytesexpressing ClC-Ka/barttin alone.
integrating the signal over a period of 1 second. Resultswere normalized to the values obtained upon coexpres-sion of ClC-Ka-HAe and barttin for each batch of oocytes.
Calculations
Data are provided as mean ± SEM; N represents thenumber of oocytes investigated. All experiments wererepeated with at least three batches of oocytes; in all rep-etitions qualitatively similar data were obtained. All datawere tested for significance using the Student t test, andonly results with P < 0.05 were considered to be statisti-cally significant.
RESULTS
Xenopus oocytes expressing ClC-Ka together withbarttin created a slightly inwardly rectifying current (ICl)of −0.69 ± 0.06 lA (N = 17) at −140 mV. The respectivecurrent ICl in water-injected Xenopus oocytes amountedto −0.06 ± 0.01 lA (N = 17) (Fig. 1A and B). The chlo-ride ion specificity of ClC-Ka/barttin was verified by ionsubstitution. ClC-Ka/barttin exhibited a permselectivityof Cl− >NO3
−∼ I− (Fig. 1C).As shown in Fig. 2C, the coexpression of Nedd4-2
together with ClC-Ka/barttin decreased ICl to −0.35 ±0.03 lA (N = 17). In contrast, coexpression of SGK1
Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases 1921
1.5
0.0
−1.5−150 −100 −50 0 50
V, mV
A
CIC-Ka + barttinCIC-Ka + barttin + Nedd4-2CIC-Ka + barttin + K127NSGK1
CIC-Ka + barttin + S422DSGK1
CIC-Ka + barttin + K127NSGK1 + Nedd4-2
CIC-Ka + barttin + S422DSGK1 + Nedd4-2
µA
Slo
pe c
ondu
ctan
ce, µ
S
CIC
-Ka
+ ba
rttin
CIC
-Ka
+ ba
rttin
+ N
edd4
-2
CIC
-Ka
+ ba
rttin
+ K
127N
SG
K1
CIC
-Ka
+ ba
rttin
+ K
127N
SG
K1
+ N
edd4
-2
CIC
-Ka
+ ba
rttin
+ S
422D
SG
K1
+ N
edd4
-2
CIC
-Ka
+ ba
rttin
+ S
422D
SG
K1
**
**
16
14
12
10
8
6
4
2
0
B
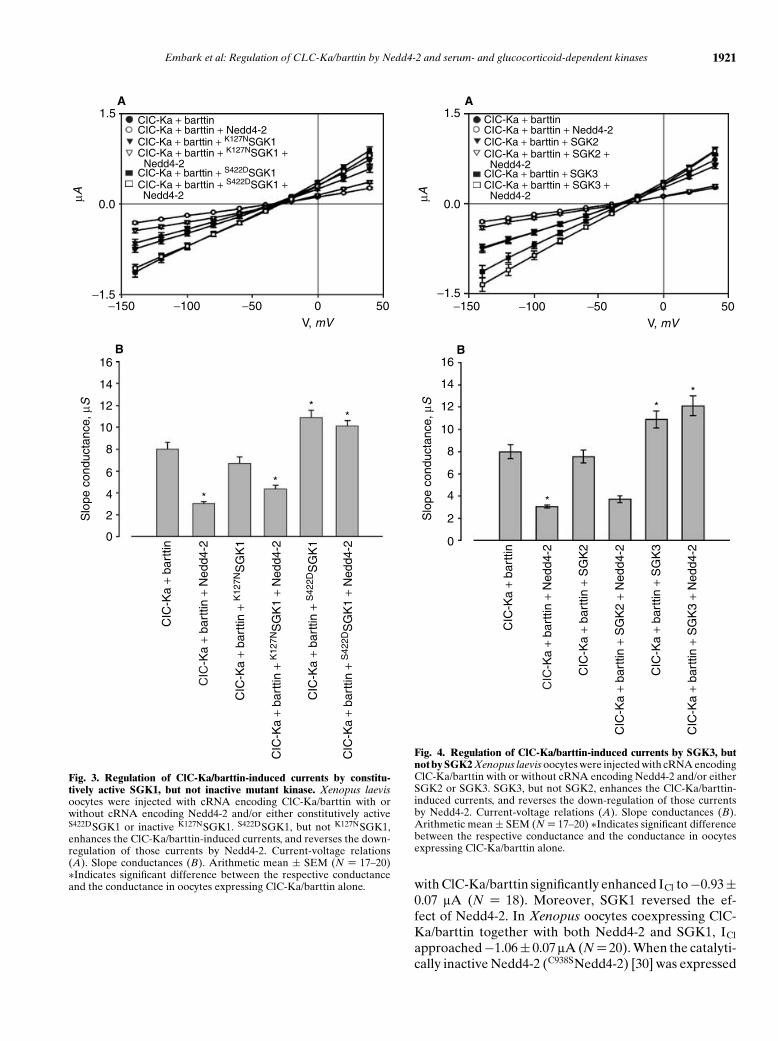
Fig. 3. Regulation of ClC-Ka/barttin-induced currents by constitu-tively active SGK1, but not inactive mutant kinase. Xenopus laevisoocytes were injected with cRNA encoding ClC-Ka/barttin with orwithout cRNA encoding Nedd4-2 and/or either constitutively activeS422DSGK1 or inactive K127NSGK1. S422DSGK1, but not K127NSGK1,enhances the ClC-Ka/barttin-induced currents, and reverses the down-regulation of those currents by Nedd4-2. Current-voltage relations(A). Slope conductances (B). Arithmetic mean ± SEM (N = 17–20)∗Indicates significant difference between the respective conductanceand the conductance in oocytes expressing ClC-Ka/barttin alone.
1.5
0.0
−1.5−150 −100 −50 0 50
V, mV
A
CIC-Ka + barttinCIC-Ka + barttin + Nedd4-2CIC-Ka + barttin + SGK2
CIC-Ka + barttin + SGK3
CIC-Ka + barttin + SGK2 + Nedd4-2
CIC-Ka + barttin + SGK3 + Nedd4-2 µ
A
16
14
12
10
8
6
4
2
0
Slo
pe c
ondu
ctan
ce, µ
S
CIC
-Ka
+ ba
rttin
CIC
-Ka
+ ba
rttin
+ N
edd4
-2
CIC
-Ka
+ ba
rttin
+ S
GK
2
CIC
-Ka
+ ba
rttin
+ S
GK
2 +
Ned
d4-2
CIC
-Ka
+ ba
rttin
+ S
GK
3 +
Ned
d4-2
CIC
-Ka
+ ba
rttin
+ S
GK
3
*
**
B
Fig. 4. Regulation of ClC-Ka/barttin-induced currents by SGK3, butnot by SGK2 Xenopus laevis oocytes were injected with cRNA encodingClC-Ka/barttin with or without cRNA encoding Nedd4-2 and/or eitherSGK2 or SGK3. SGK3, but not SGK2, enhances the ClC-Ka/barttin-induced currents, and reverses the down-regulation of those currentsby Nedd4-2. Current-voltage relations (A). Slope conductances (B).Arithmetic mean ± SEM (N = 17–20) ∗Indicates significant differencebetween the respective conductance and the conductance in oocytesexpressing ClC-Ka/barttin alone.
with ClC-Ka/barttin significantly enhanced ICl to −0.93 ±0.07 lA (N = 18). Moreover, SGK1 reversed the ef-fect of Nedd4-2. In Xenopus oocytes coexpressing ClC-Ka/barttin together with both Nedd4-2 and SGK1, ICl
approached −1.06 ± 0.07 lA (N = 20). When the catalyti-cally inactive Nedd4-2 (C938SNedd4-2) [30] was expressed
1922 Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases
B
0
1
2
3
4
5
6
7
8
9
10
Rat
io o
f int
ensi
ty, p
-N42
/N42
Ned
d4-2
Ned
d4-2
+ S
GK
1
Ned
d4-2
+ S
GK
2
Ned
d4-2
+ S
GK
3
*
*
A
α-pN42
α-N42
Ned
d4-2
Ned
d4-2
+ S
GK
1
Ned
d4-2
+ S
GK
2
Ned
d4-2
+ S
GK
3
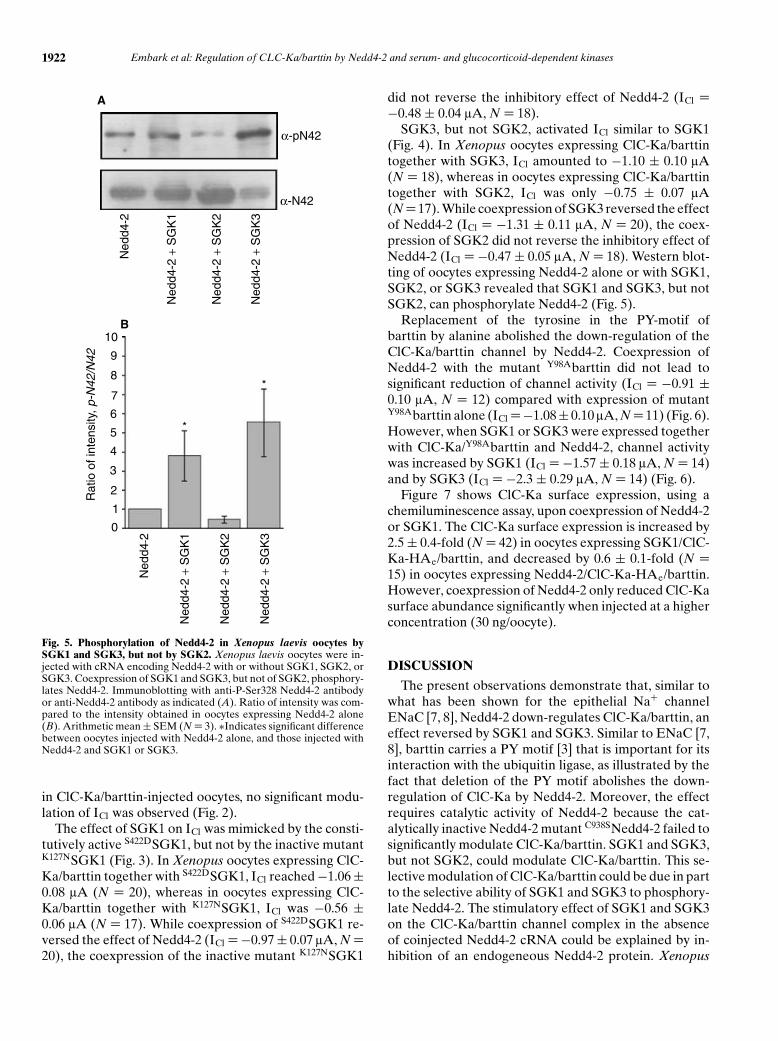
Fig. 5. Phosphorylation of Nedd4-2 in Xenopus laevis oocytes bySGK1 and SGK3, but not by SGK2. Xenopus laevis oocytes were in-jected with cRNA encoding Nedd4-2 with or without SGK1, SGK2, orSGK3. Coexpression of SGK1 and SGK3, but not of SGK2, phosphory-lates Nedd4-2. Immunoblotting with anti-P-Ser328 Nedd4-2 antibodyor anti-Nedd4-2 antibody as indicated (A). Ratio of intensity was com-pared to the intensity obtained in oocytes expressing Nedd4-2 alone(B). Arithmetic mean ± SEM (N = 3). ∗Indicates significant differencebetween oocytes injected with Nedd4-2 alone, and those injected withNedd4-2 and SGK1 or SGK3.
in ClC-Ka/barttin-injected oocytes, no significant modu-lation of ICl was observed (Fig. 2).
The effect of SGK1 on ICl was mimicked by the consti-tutively active S422DSGK1, but not by the inactive mutantK127NSGK1 (Fig. 3). In Xenopus oocytes expressing ClC-Ka/barttin together with S422DSGK1, ICl reached −1.06 ±0.08 lA (N = 20), whereas in oocytes expressing ClC-Ka/barttin together with K127NSGK1, ICl was −0.56 ±0.06 lA (N = 17). While coexpression of S422DSGK1 re-versed the effect of Nedd4-2 (ICl = −0.97 ± 0.07 lA, N =20), the coexpression of the inactive mutant K127NSGK1
did not reverse the inhibitory effect of Nedd4-2 (ICl =−0.48 ± 0.04 lA, N = 18).
SGK3, but not SGK2, activated ICl similar to SGK1(Fig. 4). In Xenopus oocytes expressing ClC-Ka/barttintogether with SGK3, ICl amounted to −1.10 ± 0.10 lA(N = 18), whereas in oocytes expressing ClC-Ka/barttintogether with SGK2, ICl was only −0.75 ± 0.07 lA(N = 17). While coexpression of SGK3 reversed the effectof Nedd4-2 (ICl = −1.31 ± 0.11 lA, N = 20), the coex-pression of SGK2 did not reverse the inhibitory effect ofNedd4-2 (ICl = −0.47 ± 0.05 lA, N = 18). Western blot-ting of oocytes expressing Nedd4-2 alone or with SGK1,SGK2, or SGK3 revealed that SGK1 and SGK3, but notSGK2, can phosphorylate Nedd4-2 (Fig. 5).
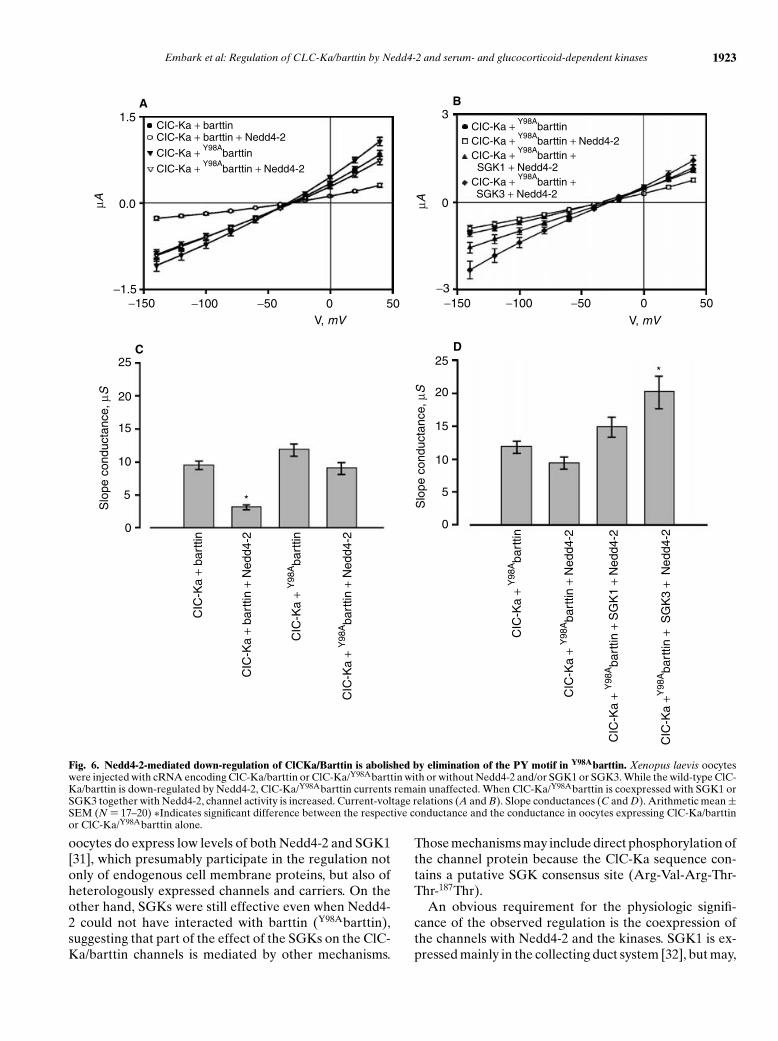
Replacement of the tyrosine in the PY-motif ofbarttin by alanine abolished the down-regulation of theClC-Ka/barttin channel by Nedd4-2. Coexpression ofNedd4-2 with the mutant Y98Abarttin did not lead tosignificant reduction of channel activity (ICl = −0.91 ±0.10 lA, N = 12) compared with expression of mutantY98Abarttin alone (ICl =−1.08 ± 0.10 lA, N = 11) (Fig. 6).However, when SGK1 or SGK3 were expressed togetherwith ClC-Ka/Y98Abarttin and Nedd4-2, channel activitywas increased by SGK1 (ICl = −1.57 ± 0.18 lA, N = 14)and by SGK3 (ICl = −2.3 ± 0.29 lA, N = 14) (Fig. 6).
Figure 7 shows ClC-Ka surface expression, using achemiluminescence assay, upon coexpression of Nedd4-2or SGK1. The ClC-Ka surface expression is increased by2.5 ± 0.4-fold (N = 42) in oocytes expressing SGK1/ClC-Ka-HAe/barttin, and decreased by 0.6 ± 0.1-fold (N =15) in oocytes expressing Nedd4-2/ClC-Ka-HAe/barttin.However, coexpression of Nedd4-2 only reduced ClC-Kasurface abundance significantly when injected at a higherconcentration (30 ng/oocyte).
DISCUSSION
The present observations demonstrate that, similar towhat has been shown for the epithelial Na+ channelENaC [7, 8], Nedd4-2 down-regulates ClC-Ka/barttin, aneffect reversed by SGK1 and SGK3. Similar to ENaC [7,8], barttin carries a PY motif [3] that is important for itsinteraction with the ubiquitin ligase, as illustrated by thefact that deletion of the PY motif abolishes the down-regulation of ClC-Ka by Nedd4-2. Moreover, the effectrequires catalytic activity of Nedd4-2 because the cat-alytically inactive Nedd4-2 mutant C938SNedd4-2 failed tosignificantly modulate ClC-Ka/barttin. SGK1 and SGK3,but not SGK2, could modulate ClC-Ka/barttin. This se-lective modulation of ClC-Ka/barttin could be due in partto the selective ability of SGK1 and SGK3 to phosphory-late Nedd4-2. The stimulatory effect of SGK1 and SGK3on the ClC-Ka/barttin channel complex in the absenceof coinjected Nedd4-2 cRNA could be explained by in-hibition of an endogeneous Nedd4-2 protein. Xenopus
Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases 1923
3
0
−3−150 −100 −50 0 50
V, mV
B
µA
25
20
15
10
5
0
Slo
pe c
ondu
ctan
ce, µ
S
CIC
-Ka
+ ba
rttin
CIC
-Ka
+ ba
rttin
+ N
edd4
-2
CIC
-Ka
+ Y
98Aba
rttin
CIC
-Ka
+ Y
98Aba
rttin
+ N
edd4
-2
*
C25
20
15
10
5
0
Slo
pe c
ondu
ctan
ce, µ
S
CIC
-Ka
+Y
98Aba
rttin
CIC
-Ka
+ Y
98Aba
rttin
+N
edd4
-2
CIC
-Ka
+ Y
98Aba
rttin
+S
GK
1 +
Ned
d4-2
CIC
-Ka
+Y98
Aba
rttin
+ S
GK
3 +
Ned
d4-2
*
D
1.5
0.0
−1.5−150 −100 −50 0 50
V, mV
A µ
A
CIC-Ka + barttinCIC-Ka + barttin + Nedd4-2
CIC-Ka + Y98Abarttin + Nedd4-2
CIC-Ka + Y98Abarttin
CIC-Ka + Y98Abarttin
CIC-Ka + Y98Abarttin + Nedd4-2
CIC-Ka + Y98Abarttin +
SGK1 + Nedd4-2CIC-Ka + Y98A
barttin + SGK3 + Nedd4-2
Fig. 6. Nedd4-2-mediated down-regulation of ClCKa/Barttin is abolished by elimination of the PY motif in Y98Abarttin. Xenopus laevis oocyteswere injected with cRNA encoding ClC-Ka/barttin or ClC-Ka/Y98Abarttin with or without Nedd4-2 and/or SGK1 or SGK3. While the wild-type ClC-Ka/barttin is down-regulated by Nedd4-2, ClC-Ka/Y98Abarttin currents remain unaffected. When ClC-Ka/Y98Abarttin is coexpressed with SGK1 orSGK3 together with Nedd4-2, channel activity is increased. Current-voltage relations (A and B). Slope conductances (C and D). Arithmetic mean ±SEM (N = 17–20) ∗Indicates significant difference between the respective conductance and the conductance in oocytes expressing ClC-Ka/barttinor ClC-Ka/Y98Abarttin alone.
oocytes do express low levels of both Nedd4-2 and SGK1[31], which presumably participate in the regulation notonly of endogenous cell membrane proteins, but also ofheterologously expressed channels and carriers. On theother hand, SGKs were still effective even when Nedd4-2 could not have interacted with barttin (Y98Abarttin),suggesting that part of the effect of the SGKs on the ClC-Ka/barttin channels is mediated by other mechanisms.
Those mechanisms may include direct phosphorylation ofthe channel protein because the ClC-Ka sequence con-tains a putative SGK consensus site (Arg-Val-Arg-Thr-Thr-187Thr).
An obvious requirement for the physiologic signifi-cance of the observed regulation is the coexpression ofthe channels with Nedd4-2 and the kinases. SGK1 is ex-pressed mainly in the collecting duct system [32], but may,

1924 Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases
3.0
2.5
2.0
1.5
1.0
0.5
0.0
Cel
l sur
face
exp
ress
ion,
norm
aliz
ed
H2O
CIC
-Ka-
HA
+ b
artti
n +
SG
K1
(S42
2D)
CIC
-Ka-
HA
+ b
artti
n
CIC
-Ka-
HA
+ b
artti
n +
Ned
d4-2
(15
ng)
CIC
-Ka-
HA
+ b
artti
n +
Ned
d4-2
(30
ng)
*
*
Fig. 7. Regulation of ClC-Ka surface expression by SGK1 and Nedd4-2. Surface expression of extracellularly HA-tagged ClC-Ka (ClC-Ka-Hae) was assessed by chemiluminescence in oocytes expressing ClC-Ka-HAe/barttin alone, and in oocytes coexpressing ClC-Ka-HA/barttintogether with S422DSGK1 or Nedd4-2. Arithmetic mean ± SEM (N =15–44). ∗Indicates significant difference between oocytes injected withClC-Ka-HA/barttin alone and those injected with ClC-Ka-HA/barttintogether with S422DSGK1 or Nedd4-2. Results are normalized to thevalues obtained upon coexpression of ClC-Ka-HAe/barttin for eachbatch of oocytes.
under pathophysiologic conditions, be expressed in fur-ther nephron segments, such as the thick ascending limb[13]. Barttin is expressed thoughout the distal nephron(i.e., medullary and cortical thick ascending limb, dis-tal convoluted tubule, connecting tubule, cortical andmedullary collecting duct) [4]. ClC-K1 and ClC-K2, therat homologs of human ClC-Ka and ClC-Kb, are similarlylocalized in all distal nephron segments [4].
Because SGK1 is under transcriptional control of glu-cocorticoids [25, 26] and mineralocorticoids [12, 14, 15,28], and is activated by insulin and IGF1 [20], the presentmechanism may well participate in the regulation of re-nal tubular Cl− transport by those hormones. The func-tional significance of the regulation by SGK1 is reflectedin the SGK1 knockout mouse (SGK1−/−). NaCl excre-tion in this mouse is normal under salt repletion, andis decreased during salt-deficient diet [33]. However,the adaptation following salt-free diet is insufficient [i.e.,the Na+ and Cl− excretion remains significantly higherin the SGK1−/− mouse as compared to its wild-typelittermate (SGK1+/+)]. As a result, in contrast to theSGK1+/+ mouse, the SGK1−/− mouse suffers from
weight loss, a decline in blood pressure, and decreasedrenal glomerular filtration rate at low salt diet [33]. Thesechanges occur despite a larger increase of plasma aldos-terone concentration in the SGK1−/− mouse comparedto the SGK1+/+ mouse [33]. The renal NaCl loss is atleast partially due to insufficient up-regulation of ENaCin the cell membrane, but according to the results of thepresent study may be paralleled by defective regulationof Cl− channels.
SGK1 has been shown to also act on the surfaceexpression and/or function of several other transport pro-teins, such as the nonselective cation channel, voltage-sensitive K+ channels (Kv1.2–5), the slow-activating K+
channel KCNE1/KCNQ1, the inward rectifying K+ chan-nel ROMK1, CFTR, the cardiac Na+ channel SCN5A,Na+, K+-ATPase, the Na+/H+ exchanger NHE3, the an-ionic amino acid transporter EAAT1, and the system Ntransporter SN1 [16, 22, 31, 34–42]. These results suggestthat SGK1 may have a similarly broad function in trans-port regulation, as has been documented for the proteinkinase A (PKA). The effects of SGK1 are partially sharedby SGK2 and SGK3, even though the affinities to the tar-get proteins are not identical, and marked differences dooccur between the efficacy of the three kinases depend-ing on the regulated channel and additional proteins in-volved.
CONCLUSION
The present observations disclose that ClC-Ka/barttinchannels are targets for the ubiquitin ligase Nedd4-2 andthe serum- and glucocorticoid-inducible kinase isoformsSGK1 and SGK3.
ACKNOWLEDGMENTS
The authors acknowledge the technical assistance of B. Noll, and themeticulous preparation of the manuscript by T. Loch and L. Subasic.This study was supported by the Deutsche Forschungsgemeinschaft, Nr.La 315/4–4, La 315/5–1 and Wa 1088/3–1, the Bundesministerium furBildung, Wissenschaft, Forschung und Technologie (Center for Interdis-ciplinary Clinical Research) 01 KS 9602, and the Kempkes Foundationof the University of Marburg.
Reprint requests to Prof. Dr. Florian Lang, Physiologisches Institut,Universitat Tubingen, Gmelinstr. 5, D-72076 Tubingen, Germany.E-mail: [email protected]
REFERENCES
1. JENTSCH TJ, FRIEDRICH T, SCHRIEVER A, YAMADA H: The CLC chlo-ride channel family. Pflugers Arch 437:783–795, 1999
2. WALDEGGER S, GABRYSCH S, BARTH P, et al: h-SGK serine-threonineprotein kinase as transcriptional target of p38/MAP kinase pathwayin HepG2 human hepatoma cells. Cell Physiol Biochem 10:203–208,2000
3. ESTEVEZ R, BOETTGER T, STEIN V, et al: Barttin is a Cl- channelbeta-subunit crucial for renal Cl- reabsorption and inner ear K+secretion. Nature 414:558–561, 2001
4. WALDEGGER S, JECK N, BARTH P, et al: Barttin increases surface ex-pression and changes current properties of ClC-K channels. PflugersArch 444:411–418, 2002

Embark et al: Regulation of CLC-Ka/barttin by Nedd4-2 and serum- and glucocorticoid-dependent kinases 1925
5. SIMON DB, BINDRA RS, MANSFIELD TA, et al: Mutations in the chlo-ride channel gene, CLCNKB, cause Bartter’s syndrome type III.Nat Genet 17:171–178, 1997
6. BIRKENHAGER R, OTTO E, SCHURMANN MJ, et al: Mutation of BSNDcauses Bartter syndrome with sensorineural deafness and kidneyfailure. Nat Genet 29:310–314, 2001
7. DEBONNEVILLE C, FLORES SY, KAMYNINA E, et al: Phosphorylationof Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surfaceexpression. EMBO J 20:7052–7059, 2001
8. SNYDER PM, OLSON DR, THOMAS BC: Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the ep-ithelial Na+ channel. J Biol Chem 277:5–8, 2002
9. VERREY F, LOFFING J, ZECEVIC M, et al: SGK1: Aldosterone-inducedrelay of Na+ transport regulation in distal kidney nephron cells. CellPhysiol Biochem 13:21–28, 2003
10. ALVAREZ DLR, ZHANG P, NARAY-FEJES-TOTH A, et al: The serumand glucocorticoid kinase sgk increases the abundance of epithelialsodium channels in the plasma membrane of Xenopus oocytes. JBiol Chem 274:37834–37839, 1999
11. BOHMER C, WAGNER CA, BECK S, et al: The shrinkage-activatedNa(+) conductance of rat hepatocytes and its possible correlationto rENaC. Cell Physiol Biochem 10:187–194, 2000
12. CHEN SY, BHARGAVA A, MASTROBERARDINO L, et al: Epithelialsodium channel regulated by aldosterone-induced protein sgk. ProcNatl Acad Sci USA 96:2514–2519, 1999
13. LANG F, KLINGEL K, WAGNER CA, et al: Deranged transcrip-tional regulation of cell-volume-sensitive kinase hSGK in diabeticnephropathy. Proc Natl Acad Sci USA 97:8157–8162, 2000
14. NARAY-FEJES-TOTH A, CANESSA C, CLEAVELAND ES, et al: SGK is analdosterone-induced kinase in the renal collecting duct. Effects onepithelial na+ channels. J Biol Chem 274:16973–16978, 1999
15. SHIGAEV A, ASHER C, LATTER H, et al: Regulation of sgk by aldos-terone and its effects on the epithelial Na(+) channel. Am J PhysiolRenal Physiol 278:F613–F619, 2000
16. WAGNER CA, OTT M, KLINGEL K, et al: Effects of the ser-ine/threonine kinase SGK1 on the epithelial Na(+) channel (ENaC)and CFTR: Implications for cystic fibrosis. Cell Physiol Biochem11:209–218, 2001
17. PEARCE D: SGK1 regulation of epithelial sodium transport. CellPhysiol Biochem 13:13–20, 2003
18. KOBAYASHI T, DEAK M, MORRICE N, COHEN P: Characterization ofthe structure and regulation of two novel isoforms of serum- andglucocorticoid-induced protein kinase. Biochem J 344:189–197, 1999
19. FRIEDRICH B, WARNTGES S, KLINGEL K, et al: Up-regulation of thehuman serum and glucocorticoid-dependent kinase 1 in glomeru-lonephritis. Kidney Blood Press Res 25:303–307, 2002
20. KOBAYASHI T, COHEN P: Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinosi-tide 3-kinase is mediated by 3-phosphoinositide-dependent proteinkinase-1 (PDK1) and PDK2. Biochem J 339:319–328, 1999
21. PARK J, LEONG ML, BUSE P, et al: Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulatedsignaling pathway. EMBO J 18:3024–3033, 1999
22. LANG F, COHEN P: Regulation and physiological roles of serum-and glucocorticoid-induced protein kinase isoforms. Sci STKE2001:RE17, 2001
23. BELL LM, LEONG ML, KIM B, et al: Hyperosmotic stress stimulatespromoter activity and regulates cellular utilization of the serum-and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J Biol Chem 275:25262–25272, 2000
24. WALDEGGER S, BARTH P, RABER G, LANG F: Cloning and character-ization of a putative human serine/threonine protein kinase tran-scriptionally modified during anisotonic and isotonic alterations ofcell volume. Proc Natl Acad Sci USA 94:4440–4445, 1997
25. BRENNAN FE, FULLER PJ: Rapid upregulation of serum andglucocorticoid-regulated kinase (sgk) gene expression by corticos-teroids in vivo. Mol Cell Endocrinol 166:129–136, 2000
26. WEBSTER MK, GOYA L, GE Y, et al: Characterization of sgk, a novelmember of the serine/threonine protein kinase gene family whichis transcriptionally induced by glucocorticoids and serum. Mol CellBiol 13:2031–2040, 1993
27. FIRESTONE GL, GIAMPAOLO JR, O’KEEFFE BA: Stimulus-dependentregulation of serum and glucocorticoid inducible protein kinase(SGK) transcription, subcellular localization and enzymatic activity.Cell Physiol Biochem 13:1–12, 2003
28. COWLING RT, BIRNBOIM HC: Expression of serum- andglucocorticoid-regulated kinase (sgk) mRNA is up-regulatedby GM-CSF and other proinflammatory mediators in humangranulocytes. J Leukoc Biol 67:240–248, 2000
29. WAGNER CA, FRIEDRICH B, SETIAWAN I, et al: The use of Xeno-pus laevis oocytes for the functional characterization of heterolo-gously expressed membrane proteins. Cell Physiol Biochem 10:1–12,2000
30. ABRIEL H, LOFFING J, REBHUN JF, et al: Defective regulation of theepithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest103:667–673, 1999
31. BOHMER C, WILHELM V, HENKE G, et al: Serum and glucocorticoidsensitive kinases in the regulation of the cardiac Na+ channel. Car-diovasc Res 57:1079–1084, 2003
32. LOFFING J, ZECEVIC M, FERAILLE E, et al: Aldosterone induces rapidapical translocation of ENaC in early portion of renal collectingsystem: Possible role of SGK. Am J Physiol Renal Physiol 280:F675–F682, 2001
33. WULFF P, VALLON V, HUANG DY, et al: Impaired renal Na(+) re-tention in the sgk1-knockout mouse. J Clin Invest 110:1263–1268,2002
34. EMBARK HM, BOHMER C, VALLON V, et al: Regulation of KCNE1-dependent K(+) current by the serum and glucocorticoid-induciblekinase (SGK) isoforms. Pflugers Arch 445:601–606, 2003
35. GAMPER N, FILLON S, FENG Y, et al: K+ channel activation by all threeisoforms of serum- and glucocorticoid-dependent protein kinaseSGK. Pflugers Arch 445:60–66, 2002
36. SETIAWAN I, HENKE G, FENG Y, et al: Stimulation of Xenopus oocyteNa(+),K(+)ATPase by the serum and glucocorticoid-dependent ki-nase sgk1. Pflugers Arch 444:426–431, 2002
37. WAGNER CA, BROER A, ALBERS A, et al: The heterodimeric aminoacid transporter 4F2hc/LAT1 is associated in Xenopus oocyteswith a non-selective cation channel that is regulated by the ser-ine/threonine kinase sgk-1. J Physiol 526 Pt 1:35–46, 2000
38. YOO D, KIM BY, CAMPO C, et al: Cell surface expression of theROMK (Kir 1.1) channel is regulated by the aldosterone-inducedkinase, SGK-1, and protein kinase A. J Biol Chem 278:23066–23075,2003
39. YUN CC, PALMADA M, EMBARK HM, et al: The serum andglucocorticoid-inducible kinase SGK1 and the Na+/H+ exchangeregulating factor NHERF2 synergize to stimulate the renal outermedullary K+ channel ROMK1. J Am Soc Nephrol 13:2823–2830,2002
40. YUN CC, CHEN Y, LANG F: Glucocorticoid activation of Na(+)/H(+)exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. JBiol Chem 277:7676–7683, 2002
41. YUN CC: Concerted roles of SGK1 and the Na+/H+ exchanger reg-ulatory factor 2 (NHERF2) in regulation of NHE3. Cell PhysiolBiochem 13:29–40, 2003
42. LANG F, HENKE G, EMBARK HM, et al: Regulation of channels by theserum and glucocorticoid-inducible kinase—Implications for trans-port, excitability and cell proliferation. Cell Physiol Biochem 13:41–50, 2003





![Role of the ubiquitin system in regulating ion transportdoc.rero.ch/record/310765/files/424_2010_Article_893.pdf · ClC5 Endocytosis WWP2, Nedd4-2 [84, 176] ClC2 Endocytosis Nedd4-2](https://img.pdfslide.us/doc/110x75/5f23cac04037bc42af3360ce/role-of-the-ubiquitin-system-in-regulating-ion-clc5-endocytosis-wwp2-nedd4-2-84.jpg)




![Glucocorticoid-induced Cell Death Requires …...[CANCER RESEARCH 59, 1378–1385, March 15, 1999] Glucocorticoid-induced Cell Death Requires Autoinduction of Glucocorticoid Receptor](https://img.pdfslide.us/doc/110x75/5e5646d0314f24389e233453/glucocorticoid-induced-cell-death-requires-cancer-research-59-1378a1385.jpg)


















