Embed Size (px)
Citation preview

doi: 10.1136/hrt.2008.156299 2010 96: 552-559Heart
Holger M Nef, Helge Möllmann, Christian Hamm, et al. pulmonary hypertensionUpdated classification and management of
http://heart.bmj.com/content/96/7/552.full.htmlUpdated information and services can be found at:
These include:
MaterialSupplemental http://heart.bmj.com/content/suppl/2010/03/29/96.7.552.DC1.html
References http://heart.bmj.com/content/96/7/552.full.html#ref-list-1
This article cites 23 articles, 19 of which can be accessed free at:
serviceEmail alerting
box at the top right corner of the online article.Receive free email alerts when new articles cite this article. Sign up in the
Topic collections
(4771 articles)Epidemiology � (18785 articles)Clinical diagnostic tests �
(11845 articles)Hypertension � (1651 articles)Echocardiography �
(22640 articles)Drugs: cardiovascular system � (1 articles)Primary pulmonary hypertension �
Articles on similar topics can be found in the following collections
Notes
http://heart.bmj.com/cgi/reprintformTo order reprints of this article go to:
http://heart.bmj.com/subscriptions go to: HeartTo subscribe to
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

PULMONARY HYPERTENSION
Updated classification andmanagement of pulmonaryhypertensionHolger M Nef,1,2 Helge Mollmann,1 Christian Hamm,1
Friedrich Grimminger,1,2 Hossein-Ardeschir Ghofrani1,2
Pulmonary arterial hypertension (PAH) is a severedisease characterised by a progressive increase ofpulmonary pressure and resistance leading to rightheart failure. PAH is commonly diagnosed at a latestage of the disease and is associated with progres-sive clinical deterioration and premature death.According to the most recent consensus conference,pulmonary hypertension (PH) is categorised intofive main groups: group 1, PAH; group 2, PH asso-ciated with left sided heart diseases; group 3, PHassociated with lung disease and/or hypoxaemia;group4, PHdue to chronic thrombotic and/or embolicdisease; and group 5, miscellaneousdsummarising avariety of rare and not well characterised disorderscharacterised by non-specific signs and symptoms.1
Screening for PH is usually made by transthoracicechocardiography on the basis of the velocity of theregurgitant tricuspid jet, and final confirmation isdone by right heart catheterisation.2
Without treatment, the prognosis for patientswith significant PH is poor. The reported medianlife expectancy of idiopathic PAH is 2.8 years fromthe diagnosisw1; however, recent meta-analysis oftrials in the field of PAH have provided indicationsof a beneficial influence of PAH treatments onsurvival.3 Current PAH specific medications,including prostanoids, endothelin receptor antago-nists (ERA), and phosphodiesterase 5 inhibitors(PDE5i), have sought to address the pulmonaryvascular endothelial dysfunction and vaso-constriction associated with this condition. Severalnovel drugs that have already produced encouragingresults in animal models aim to target more directlythe structural vascular changes (remodelling) andare currently under clinical investigation (eg, solubleguanylyl cyclase activators/stimulators, tyrosinekinase inhibitors, 5-HT2B receptor antagonists).This article reviews the clinical classification,
which has been recently updated. Moreover, estab-lished approaches to evaluate PH and the currentlyrecommended treatments for PAH are summarised.
DEFINITION OF PHThe haemodynamic definition of PH has not beenevidence based. Under normal circumstances,resting mean pulmonary artery pressure (mPAP)was shown to be 14.063.3 mm Hg, which wasindependent of gender and ethnicity.4
Thus, during the 4th World Symposium on PHheld in Dana Point 2008, new thresholds for mPAPwere introduced. An mPAP <21 mm Hg wasdefined as normal, from 21e25 mm Hg was cate-gorised as borderline, and anmPAP>25 mm Hgwasdesignated as manifest PH. Correspondingly, echo-cardiographic systolic tricuspid regurgitant velocitythresholds<2.5 m/s is defined as normal, 2.5e2.8 m/sas borderline, and >2.8 m/s is highly indicative for amanifest PH (table 1).w2 mPAP during exercise isdependent on exercise level and age. During mildexercise, PAP was 19.464.8 mm Hg in subjects<50 years compared to 29.468.4 mmHg in subjects>50 years. While the mPAP at rest is virtually inde-pendent of age and rarely exceeds 20 mm Hg, exer-cise mPAP is age related and frequently exceeds30 mm Hg, especially in elderly individuals, whichmakes it difficult to define normal mPAP valuesduring exercise.4 Therefore, exercise values of mPAPwere not included in the new definition of PH.1
UPDATED CLASSIFICATION OF PHThe World Health Organization has hosted confer-ences on PH introducing a classification scheme in1973 and proposing the Evian classification in1988.w3 This classification was based on similarpathophysiological mechanism, clinical presentationand therapeutic options, and wasmodified further inVenice in 2003, where the term idiopathic pulmo-nary arterial hypertension (IPAH) replaced the termprimary pulmonary hypertension.w4 The otherprominent change made at the Venice meeting wasto move pulmonary veno-occlusive disease (PVOD)and pulmonary capillary haemangiomatosis (PCH)from separate categories into a single subcategory ofPAH. In 2008 the 4th World Symposium on PHprovided the opportunity to slightly modify theprevious clinical classification (figure 1).1
PAH (group 1)Idiopathic and heritable PAHDue to the fact that PH occurs in different clinicalconditions, IPAH corresponds to sporadic diseasewithout family history of PAH and typical riskfactors. If PH is detected in a familial context,mutations of the bone morphogenetic proteinreceptor type 2 (BMPR2) gene can be documented inapproximately 70% of cases.5 However, given thefact that in 30% of families with PAH no BMPR2
< Supplementary referencesare published online only athttp://heart.bmj.com/content/vol96/issue71Kerckhoff Heart and ThoraxCenter, Departments ofCardiology(Franz-Groedel-Institute) andPneumology, Bad Nauheim,Germany2University Hospital Giessen andMarburg GmbH, Klinikstrasse36, Giessen, Germany
Correspondence toDr med. Holger M Nef, KerckhoffHeart and Thorax Center,Benekestr. 2-8, D-61231 BadNauheim, Germany;[email protected]
552 Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

mutation has been identified, it was decided toabandon the term ‘familial PAH’ in the new classi-fication and to replace it with the term ‘heritablePAH’. Heritable forms of PAH include germlinemutations (BMPR2, activin-receptor like kinase 1,endoglin) and familial cases without identifiedgermline mutations.1 However, this new categorydoes not imply a recommendation for general genetictesting of patients with IPAH or in familial cases ofPAH. Instead, genetic testing should be performed aspart of an extensive programme that includes geneticcounselling and discussion of risk, benefits, andlimitations of such testing.6
Drug and toxin induced PAHPAH is also a rare side effect of certain weightreducing agents, such as fenfluramine and dexfen-fluramine. However, the incidence of drug inducedPAH is decreasing as these agents are no longeravailable. Recently, a class effect of selective sero-tonin reuptake inhibitors was claimed after use inpregnant women. In a caseecontrol study, after20 weeks of gestation newborns showed anincreased risk for developing PAH.Additionally, the occurrence of PAH after meth-
amphetamine abuse (inhaled, smoked, or intra-venous), which has already caused a significantincrease of drug related new PAH cases in the southwest of the USA, could be documented.IPAH and many forms of the formerly called
secondary PAH share histopathological common-alities, natural history and response to treatment,and on this basis these conditions are considered asbeing ‘associated pulmonary arterial hypertension’.These forms include the following entities.
PAH associated with connective tissue diseasePAH is a well recognised complication of connec-tive tissue diseases such as systemic sclerosis andsystemic lupus erythematosus (SLE). It can, butmust not necessarily, occur in conjunction withdifferent degrees of interstitial lung diseases. Theprevalence of PAH in patients with systemic scle-rosis has been reported to be up to 16%. Patientswith PAH associated with systemic sclerosis have aparticularly poor prognosis compared to those withsystemic sclerosis without PAH.6 w5
PAH associated with congenital heart diseaseCongenital heart disease is relatively common,affecting around 1% of the population. Within thispopulation 15% eventually develop PAH.7 Asdetermined by the level of pulmonary vascularresistance, the most severe form of PAH is Eisen-
menger ’s syndrome, which is associated with thereversal of an initial left to right shunt causingcyanosis and limited exercise capacity.w6 During theDana Point meeting, the pathologic and pathophy-siologic classification of congenital heart diseasewith systemic-to-pulmonary shunts were redefinedto provide more detailed description of eachcondition.1
PAH associated with HIV infectionPAH is a rare (estimated prevalence in patients withHIV is 0.5%) but relatively well documentedcomplication of HIV infection.8 With the advent ofhighly active antiretroviral therapy and notablyimproved survival, PAH and other non-infectiousmanifestations of HIV infection are increasinglyresponsible for HIV associated morbidity and poorprognosis. In patients with HIV, the HIV-1 envelopeglycoprotein GP120 may stimulate the productionof endothelin by macrophages.9 HIVassociated PAHshows a clinical picture similar to IPAH and seemsto be independent of the degree of immunosup-pression. The prevalence of HIVassociated PAHwasevaluated more recently and showed a stable prev-alence of 0.46%.
Portopulmonary hypertensionPortal hypertension might lead to development ofPAH which is known as portopulmonary hyper-tension (POPH). Prospective studies have shownthat 2e6% of patients with portal hypertensionhave PH. Risk factors for development of POPH areidentified as female sex and autoimmune hepatitis.The prognosis is related to the presence and severityof cirrhosis and to cardiac function.10 w7
PAH associated with schistosomiasisSince 2008 PAH associated with schistosomiasishas been added to group 1. In the previous classi-fication this form of PH had been categorised ingroup 4. It was thought that embolic obstruction ofpulmonary arteries by schistosoma eggs wasresponsible for the development of PAH. However,more recent publications point to similarities ofPAH associated with schistosomiasis and IPAHshowing the same histological findingsdforexample, plexiform lesions. From the latest data it isassumed that more than 200 million people areinfected with any of the three species of schisto-soma and that 4e8% of patients develop PH inassociation with hepatosplenic disease.11 12
Chronic haemolytic anaemiaThere is growing evidence that chronic hereditaryhaemolytic anaemias, including sickle cell disease(SCD), thalassaemia, hereditary spherocytosis,stomacytosis, and microangiopathic haemolyticanaemia, are today associated with PAH. It is esti-mated that PAH occurs most frequently in patientswith SCD. However, the mechanism leading to PAHin patients with SCD is not fully elucidated. Chronichaemolysis might be followed by high rates of nitricoxide (NO) consumption, thereby not activatingsmooth muscle guanosine monophosphate, a potent
Table 1 New thresholds of pulmonary hypertension determined at the 4th WorldSymposium held in Dana Point 2008
Definition of pulmonary hypertension
Invasive(mean pulmonary artery pressure)
Normal <21 mm Hg
Borderline 21e25 mm Hg
Manifest >25 mm Hg
Non-invasive(systolic tricuspid regurgitant velocitythreshold)
Normal <2.5 m/s
Borderline 2.5e2.8 m/s
Manifest >2.8 m/s
Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299 553
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

vasodilator/antiproliferative mediator.13 However,there is some discussion regarding the relevance ofmoderate elevations of pulmonary artery (PA) pres-sures in view of the hyperdynamic circulation ofpatients particularly suffering from SCD, wherepulmonary vascular resistance typically is in thenormal range; therefore, a proposed benefit fromtreatment with PAH specific drugs is yet to beshown.The most recent classification for PH placed
PVOD and PCH with PH in a dedicated subgroupof group 1 (so called group 1). In PVOD, histo-logical changes similar to those found in the smallpulmonary arteries (ie, intimal fibrosis and medialhypertrophy) of IPAH patients are present inaddition in small post-capillary venules. Regardingdiagnosis, untreated PVOD is often difficult todistinguish clinically from IPAH. Indications forPVOD can be derived from high resolutioncomputed tomography (CT) scans of the lungs,showing interlobular septal thickening, groundglass opacities, and enlarged mediastinal lymphnodes. PVOD/PCH remains a difficult disorder totreat, as often initiation of PAH specific treatments,particularly intravenous prostanoids, can result inpulmonary oedema formation with severe aggra-vation of hypoxaemia. Thus, early listing for lungtransplantation remains the only option formanagement of PVOD patients.
PH owing to left heart disease (group 2)All forms of PH owing to left heart disease areincluded in group 2. Either left ventricular diseaseor valvular disease may lead to an increased leftventricular end-diastolic pressure resulting in highpressure in the pulmonary artery. In most affectedpatients the pulmonary vascular resistance isnormal (<250 dyn$s$cm�5) and the trans-pulmonary gradient is <12 mm Hg. The most
recent classification acknowledges the increasingnumber of patients with left sided heart dysfunc-tion with preserved systolic function. Thus thisdistinct aetiology has been added to left heartsystolic dysfunction and left heart valvular disease.1
Currently no drug is approved for the treatment ofthis subcategory of PH, and adequate treatment ofthe underlying cardiac disease remains the recom-mended standard.
PH owing to lung disease and/or hypoxia (group 3)Group3 includes PHassociatedwith lungdisease and/or hypoxaemia. This includes chronic obstructivepulmonary disease (COPD) and interstitial lungdiseases, as well as PH associatedwith prolonged stayin high altitudes (‘chronic mountain sickness’). Theprevalence of PH in this group remains largelyunknown; however, recent data indicate for COPDassociated PH an incidence of approximately 25%with manifest PH (defined by mPAP>25 mm Hg)and 1e2%with severe PH (ie, mPAP >40 mmHg).14
Currently numerous trials are investigating thetherapeutic benefit of treating PH, both in patientswith interstitial lung disease as well as COPD.
Chronic thromboembolic pulmonary hypertension(group 4)In the new classification, chronic thromboembolicpulmonary hypertension (CTEPH) remains ingroup 4. The former Venice classification dividedCTEPH into two subgroupsdproximal and distalCTEPH. However, since there is no consensusamong experts on how to distinguish betweenproximal and distal CTEPH, only a single categoryof CTEPH is proposed in the new definition ofgroup 4. Recent prospective data have suggestedthat the incidence of CTEPH after initial pulmonaryembolism may be higher than previously estimated.The incidence of symptomatic CTEPH within2 years of a first time pulmonary embolism was3.8%. In addition to the residual clot in the proximalpulmonary arteries, another major problem inCTEPH is the development of varying degrees ofsmall vessel arteriopathy. These small vesselchanges can contribute to the development andprogression of PH in CTEPH. With few differences,the general pathology of these distal vessels inCTEPH appears to mimic that seen in PAH.15 w8
PH with unclear aetiologies (group 5)Group 5 comprises several forms of PH for whichthe aetiology is unknown or multifactorial. Chronicmyeloproliferative disorders including poly-cythaemia vera, essential thrombocythaemia, andchronic myeloid leukaemia are listed; systemicdisorders leading to PH (sarcoidosis, Langerhan’s cellhistiocytosis, lymphangioleiomyomatosis, neuro-fibromatosis), and metabolic disorders (type Iglycogen storage disease, Gaucher disease, hyper-thyroidism, hypothyroidism) are also summarised inthis group. Lastly, some rare cases of PH have beenobserved in a number of miscellaneous conditions(tumour, end stage renal failure, mediastinalfibrosis).1
Figure 1 Updated clinical classification of pulmonary hypertension according tothe proposals of the 4th World Symposium on Pulmonary Hypertension held inDana Point 2008.
554 Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

DIAGNOSTICS OF PHClinical presentationThe diagnostic workup of PH requires a series ofinvestigations to confirm the diagnosis and toclarify the clinical class and subtype of PH.However, PAH is difficult to diagnose. The clinicalcardinal symptom of PH is dyspnoea (either onexertion or even at rest) and early fatigue; as thedisease advances, patients can develop chest paindue to right ventricular dysfunction, palpitations,oedema, and ascites, and can experience syncope.w1
A high level of suspicion is needed to establish thediagnosis. A typical delay of 2e3 years betweenonset of symptoms and making a diagnosis is stillfrequently observed.
ElectrocardiogramIn PH the electrocardiogram (ECG) may demon-strate signs of right ventricular hypertrophy, such asright axis deviation, T inversion in the anterior leads,persistent S wave, and right bundle branch block
(figure 2). However, the ECG’s sensitivity (55e73%)and specificity (70%) are inadequate to be an effec-tive screening tool for the detection of PAH. Somefeatures of the ECG in patients with IPAH mayhave prognostic value: a P wave amplitude in lead IIof $0.25 mV has shown to predict a 2.8-timeshigher risk of death over a 6 year follow-up period.7
Chest x-rayThe chest x-ray appears to be inferior to the ECG indetecting PH. Nevertheless, peripheral lung vascularmarkings, and hilar pulmonary artery prominencewith a hilar-to-thoracic ratio >44%, might bespecific for the diagnosis of PH (figure 3). Even it isnot sensitive, it may still uncover underlying lungdisease. Furthermore, changes in heart diameter mayreflect beneficial (reduction in diameter) or detri-mental (increased diameter) courses of the disease,particularly in response to medical treatments.
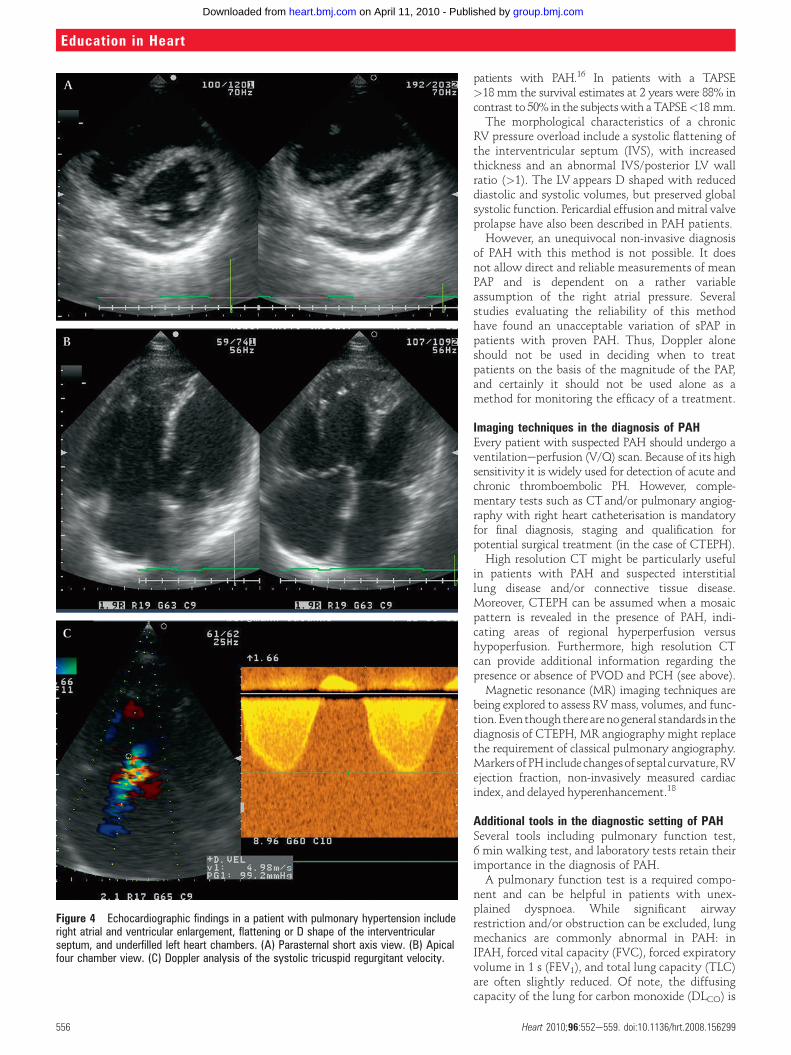
EchocardiographyIf PH is suspected based on the history, symptoms,and physical examination, transthoracic echo-cardiography should be performed (figure 4). Itprovides a quantitative assessment on pulmonaryartery pressure (PAP). After Dana Point 2008 newthresholds of the velocity of the tricuspid regur-gitation (TR) jet were defined as described above.Utilising the modified Bernoulli equation (4v2) andadding the right atrial pressure, the systolic pulmo-nary artery pressure (sPAP) can be estimated non-invasively. sPAP >40 mm Hg generally warrantsfurther evaluation in patients with unexplaineddyspnoea.w9 Doppler echocardiography can alsoaccurately estimate the right ventricular outflowtract (RVOT) acceleration time, which correlateslinearly with sPAP. An acceleration time <100 msreflects an abnormal PAP. Moreover, the peak earlydiastolic and the end-diastolic velocities of pulmo-nary regurgitation flow correlate with the mean PAPand pulmonary artery end-diastolic pressure.Echocardiography not only provides Doppler
analysis but also helps to exclude valvular, primarymyocardial and congenital causes of elevated rightsided pressure. Furthermore, morphological aspectsof the right ventricle (RV) can be assessed. Most ofthe patients present with enlarged right sidedchambers, RV hypertrophy, and reduced global RVsystolic function due to chronic overload byincreased pulmonary resistances. A reduced RVsystolic function can be measured using either theTei index or tricuspid annulus plane systolicexcursion (TAPSE).16 The Tei index is defined as theisovolumic contraction time (ICT) and isovolumicrelaxation time (IRT) divided by the ejection time(ET). The normal value of the index is 0.2860.04.An increased RV Tei index is associated with leftventricle (LV) abnormalities or increased PAP andcorrelates with a worse outcome. When the sPAP>40 mm Hg is combined with an RV Tei index>0.36, the accuracy of echocardiography forpredicting PAH might increase.17
Anatomic M mode is used to measure TAPSE andhas also been reported to predict survival rates in
Figure 2 Electrocardiogram demonstrating the changes of right ventricular hyper-trophydfor example, incomplete right bundle branch block, inversion of the T wave inanterior leads, persistent S wave in V6.
Figure 3 Chest x-ray showing decreased peripheral lung vascular markings, and hilarpulmonary artery prominence with a hilar-to-thoracic ratio>44%, which is specific but notsensitive for the diagnosis of pulmonary hypertension.
Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299 555
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from
patients with PAH.16 In patients with a TAPSE>18 mm the survival estimates at 2 years were 88% incontrast to 50% in the subjectswith a TAPSE<18 mm.The morphological characteristics of a chronic
RV pressure overload include a systolic flattening ofthe interventricular septum (IVS), with increasedthickness and an abnormal IVS/posterior LV wallratio (>1). The LV appears D shaped with reduceddiastolic and systolic volumes, but preserved globalsystolic function. Pericardial effusion andmitral valveprolapse have also been described in PAH patients.However, an unequivocal non-invasive diagnosis
of PAH with this method is not possible. It doesnot allow direct and reliable measurements of meanPAP and is dependent on a rather variableassumption of the right atrial pressure. Severalstudies evaluating the reliability of this methodhave found an unacceptable variation of sPAP inpatients with proven PAH. Thus, Doppler aloneshould not be used in deciding when to treatpatients on the basis of the magnitude of the PAP,and certainly it should not be used alone as amethod for monitoring the efficacy of a treatment.
Imaging techniques in the diagnosis of PAHEvery patient with suspected PAH should undergo aventilationeperfusion (V/Q) scan. Because of its highsensitivity it is widely used for detection of acute andchronic thromboembolic PH. However, comple-mentary tests such as CTand/or pulmonary angiog-raphy with right heart catheterisation is mandatoryfor final diagnosis, staging and qualification forpotential surgical treatment (in the case of CTEPH).High resolution CT might be particularly useful
in patients with PAH and suspected interstitiallung disease and/or connective tissue disease.Moreover, CTEPH can be assumed when a mosaicpattern is revealed in the presence of PAH, indi-cating areas of regional hyperperfusion versushypoperfusion. Furthermore, high resolution CTcan provide additional information regarding thepresence or absence of PVOD and PCH (see above).Magnetic resonance (MR) imaging techniques are
being explored to assess RVmass, volumes, and func-tion.Eventhoughtherearenogeneral standards in thediagnosis of CTEPH, MR angiography might replacethe requirement of classical pulmonary angiography.Markers ofPHinclude changesof septal curvature,RVejection fraction, non-invasively measured cardiacindex, and delayed hyperenhancement.18
Additional tools in the diagnostic setting of PAHSeveral tools including pulmonary function test,6 min walking test, and laboratory tests retain theirimportance in the diagnosis of PAH.A pulmonary function test is a required compo-
nent and can be helpful in patients with unex-plained dyspnoea. While significant airwayrestriction and/or obstruction can be excluded, lungmechanics are commonly abnormal in PAH: inIPAH, forced vital capacity (FVC), forced expiratoryvolume in 1 s (FEV1), and total lung capacity (TLC)are often slightly reduced. Of note, the diffusingcapacity of the lung for carbon monoxide (DLCO) is
Figure 4 Echocardiographic findings in a patient with pulmonary hypertension includeright atrial and ventricular enlargement, flattening or D shape of the interventricularseptum, and underfilled left heart chambers. (A) Parasternal short axis view. (B) Apicalfour chamber view. (C) Doppler analysis of the systolic tricuspid regurgitant velocity.
556 Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

also reduced in proportion to the PAH severity19;this is not somuch due to thickening of the diffusionbarrier (such as in interstitial lung diseases) but morebecause of an overall reduction of gas exchangesurface area (due to vascular pruning in PAH).The 6 min walking test is sensitive to restrictions
in cardiac and pulmonary performance, therebypredicting morbidity and mortality in patients withPAH. However, this test is affected by many factorsincluding age, body height, and general fitness.Although commonly used as the primary efficacyend point in clinical trials in PAH, in clinical practiceit should be considered complementary to cardio-pulmonary exercise testing and other parameters forthe judgement of the individual clinical course ofthe patients.Laboratory tests should include antinuclear anti-
bodies (ANA) and other markers of autoimmunediseases, screening for HIV and viral hepatitis, andmarkers of coagulation disorders (eg, protein S andC, lupus anticoagulants, von Willebrand factor).Moreover, the cardiac biomarker brain natriureticpeptide (BNP, NT-proBNP) is a valuable tool in thedetermination of RV overload and for controllingtreatment effects and prognosis prediction.
Right heart catheterisation and vasoreactivitytestingThe inability to assess reliably the transpulmonaryblood flow and pulmonary venous pressure repre-sents the most relevant limitations of echo-cardiography in the context of PH assessment.Thus, right heart catheterisation remains the goldstandard for diagnosis and allows concomitantvasoreactivity testing.18
Haemodynamics determine prognosis in PAH,but more importantly the measurement of thepulmonary capillary wedge pressure (PCWP) at restand after application of vasodilators is essential forthe differential diagnosis of PH. Moreover, aninvaluable component is the transpulmonarygradient (mPAPePCWP), which is significantlyelevated in patients with PAH, but usually not inthose with PH due to left heart disease.According to the new guidelines the diagnosis of
PH requires an elevated mPAP >25 mm Hg, and avascular resistance of more than 3 Wood units, inthe presence of a normal PCWP (<15 mm Hg). Anelevated peripheral vascular resistance (PVR) repre-sents a robust parameter reflecting an increasedtranspulmonary gradient and decreased cardiacoutput. It is only elevated if the vascular obstruc-tion occurs mainly inside the precapillary pulmo-nary resistance vessels.A limited number of patients with IPAH may
benefit from calcium channel blockers (CCBs).Therefore, the current recommendation for thetreatment of PAH proposes that the acute responseof the pulmonary circulation to a short actingpulmonary vasodilator should be used as the basisfor selecting patients for high dose CCB treatment.According to a recently published analysis, a posi-tive vasoreactive response was defined as a drop inmPAP of >10 mm Hg, a reduction below an abso-lute value of 40 mm Hg, and concomitant normal-isation of cardiac output during administration of ashort acting vasodilator such as inhaled NO.20
However, the vast majority of PAH patients (>95%)are non-responders (according to the abovementioned criteria), in whom the use of CCBsshould be generally discouraged due to potentialdetrimental side effects such as negative inotropyand chronotropy.There are some discrepancies and difficulties in
the interpretation of the PCWP after administrationof vasodilators. In patients with congestive heartfailure and PH, the inhalation of NO results in areduction of PVR without changes in PAP andcardiac output as a consequence of increased PCWP.In contrast, after application of sildenafil a signifi-cant reduction of PAP and an increase of cardiacindex could be observed whereas the PVR remainsunaffected due to reduced PCWP.2
UPDATED TREATMENT STRATEGIES FOR PHAt the 4th World Symposium on PH in Dana Point,several important changes in the treatmentstrategy for patients with PAH were discussed.There are currently approved treatments only forthose conditions in group I (PAH); however, aconsensus statement was also achieved regardingthe treatment of the other PH entities (figure 5).21
Non-pharmacological treatmentPhysical activity, as applied during controlledrehabilitation programmes, may have a positiveimpact on exercise capacity and quality of life ofPAH patients.w10 However, potentially hazardous
Figure 5 Treatment algorithm of pulmonary arterial hypertension (PAH) following therecommendation of the 4th World Symposium on PAH held in Dana Point 2008. APAH,diseases associated with pulmonary arterial hypertension; CCB, calcium channelblockers; ERA, endothelin receptor antagonist; FC, functional class; HLuTx, heart and lungtransplantation; IPAH, idiopathic pulmonary arterial hypertension; IV, intravenous; LuTx,lung transplantation; NYHA, New York Heart Association; PDE5I, phosphodiesterase-5-inhibitors; SC, subcutaneous; AeC, level of evidence.
Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299 557
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

symptoms such as severe dyspnoea, syncope, andchest pain should be clearly avoided.
Pharmacological treatmentIn PAH patients, a specific treatment with ERA orPDE5i is now recommended for patients in func-tional class II. This is partly based on the inter-pretation of the results from the EARLY trial,which investigated the effects of treatment withthe ERA bosentan in PAH patients in functionalclass II. After 6 months PVR was reduced signifi-cantly in bosentan treated patients (compared toplacebo), whereas the 6 min walk distance wasunchanged. Moreover, there was a significant delayin time to clinical worsening in the bosentan treatedgroup.In the ARIES-1 and 2 trials ambrisentan
improved exercise capacity after 12 weeks in PAHpatients both in functional class II and III, anobservation also seen for sildenafil in the SUPER-1trial. In Europe, bosentan, ambrisentan and silde-nafil are approved for the treatment of patients infunctional class II.The treatment recommendations suggest either
PDE5i, ERA or inhaled iloprost with the highestdegree of evidence for patients in functional classIII. Parenteral prostanoids (eg, intravenousepoprostenol, intravenous iloprost, intravenous orsubcutaneous treprostinil) should be primarilygiven to PAH patients in functional class IV.Combination therapy (ERA and/or PDE5i and/or
prostanoids) is proposed if the clinical response tomonotherapy is not adequate. An adequateresponse includes maintenance of/improvementtowards functional class I and II, no signs of rightheart failure (eg, normal cardiac output, absence ofsyncope or oedema), and a 6-min walking distance>400 m.For other forms of PH such as PH due to left
heart disease or chronic lung disease, it remains
valid that the underlying disease should be treatedas efficiently as possible. In patients with chroniclung disease even a mild PH (mPAP 20e30 mm Hg)might be detrimental due to worsening of thephysical activity and gas exchange, which arepredictors of a worse prognosis. In the absence ofcontrolled clinical trial data, a general recom-mendation for administration of PAH specific treat-ments does not exist here. However, a targeted PAHtreatment may be beneficial in selected patients withan ‘out of proportion PH’, but these treatmentsshould exclusively be initiated in experienced centres.Pulmonary endarterectomy (PEA) remains the
treatment of choice for CTEPH. If patients aretechnically inoperable and/or if comorbiditiespreclude surgical treatment, specific PAH treatmentmay be considered, but these patients should atpresent be included in clinical trials.Since there is currently no cure for PH/PAH,
further development and progress in medicaltreatment are highly desirable. A number ofpromising novel compounds are currently underinvestigation. These include soluble guanylatecyclase (sGC) stimulators/activators, tyrosinekinase inhibitors, and serotonin antagonists.
sGC stimulatorsRecently, classes of drugs have been developed thatcan activate sGC independently of NO. The sGCstimulators (eg, BAY63-2521) can stimulate thesGC directly and enhance the sensitivity of thereduced enzyme to low levels of NO. In patientswith IPAH a significant elevation of sGC expressioncould be documented and in several animal modelsof PAH a positive effect of BAY63-2125 (riociguat)could be observed.22 In a phase II trial the effects ofriociguat on pulmonary haemodynamics could bedemonstrated.23
Tyrosine kinase inhibitorsGiven the PAH induced morphological changes ofthe vascular wall (‘vascular remodelling’) the diseasehas been characterised as chronic proliferativedisease. These morphological changes are partic-ularly induced by peptidergic growth factors such asplatelet derived growth factor (PDGF) that elicittheir signals via activation of receptor tyrosinekinases. Hence, there is both experimental andclinical evidence for a therapeutic efficacy of thetyrosine kinase inhibitors, such as imatinib, whichprovides the basis for ‘reverse remodeling’ strategiesand indeed represent a promising novel approach forthe treatment of PAH.24 25
Serotonin receptor antagonistsSeveral observations suggest a role for serotonin (5-HT) in the pathogenesis of PAH. In particular, anincreased 5-HT2B receptor expression has beenevaluated. Terguride is a partial dopamine-D2receptor antagonist, an a-2 receptor antagonist, anda 5-HT2A and 5-HT2B receptor antagonist, whichshowed antiproliferative, antithrombotic, and anti-fibrotic effects. In monocrotaline induced PAH,terguride results in a dose dependent reduction of
You can get CPD/CME credits for Education in Heart
Education in Heart articles are accredited by both the UK Royal College ofPhysicians (London) and the European Board for Accreditation in Cardiologydyou need to answer the accompanying multiple choice questions (MCQs). Toaccess the questions, click on BMJ Learning: Take this module on BMJLearning from the content box at the top right and bottom left of the onlinearticle. For more information please go to: http://heart.bmj.com/misc/education.dtl< RCP credits: Log your activity in your CPD diary online (http://www.
rcplondon.ac.uk/members/CPDdiary/index.asp)dpass mark is 80%.< EBAC credits: Print out and retain the BMJ Learning certificate once you have
completed the MCQsdpass mark is 60%. EBAC/ EACCME Credits can now beconverted to AMA PRA Category 1 CME Credits and are recognised by allNational Accreditation Authorities in Europe (http://www.ebac-cme.org/newsite/?hit¼men02).
< Please note: The MCQs are hosted on BMJ Learningdthe best availablelearning website for medical professionals from the BMJ Group. If prompted,subscribers must sign into Heart with their journal’s username and password.All users must also complete a one-time registration on BMJ Learning andsubsequently log in (with a BMJ Learning username and password) on everyvisit.
558 Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from

PAP and right ventricular hypertrophy. In a phase IItrial (TERPAH) the effect of terguride in PAHpatients is currently under investigation.
Acknowledgements The authors are thankful for the supportgiven by the Excellence Cluster Cardio-Pulmonary System (ECCPS,Coordinator Professor W Seeger, University of Giessen Lung Center,Klinikstrasse 36, D-35392 Giessen).
Competing interests In compliance with EBAC/EACCME guidelines,all authors participating in Education in Heart have disclosed potentialconflicts of interest that might cause a bias in the article. HA Ghofranihas received honoraria and research funds from Actelion, BayerSchering, ErgoNex Pharma, GlaxoSmithKline, Novartis, and Pfizer.F Grimminger has received honoraria and research funds fromActelion, Bayer Schering, Novartis, and Pfizer. C Hamm receivedspeaker fees from Actelion and GlaxoSmith Kline. H Moellmann and HNef received speakers fee from Actelion.
Provenance and peer review Not commissioned; not externallypeer reviewed.
REFERENCES1. Simonneau G, Robbins I, Beghetti M, et al. Updated clinical
classification of pulmonary hypertension. J Am Coll Cardiol2009;54:43e54.
< Updated classification of pulmonary hypertension after the4th World Symposium held in Dana Point 2008.
2. Ghofrani HA, Wilkins MW, Rich S. Uncertainties in the diagnosisand treatment of pulmonary arterial hypertension. Circulation2008;118:1195e201.
< Important paper dealing with distinctive features in thediagnosis of pulmonary hypertension.
3. Galie N, Manes A, Negro L, et al. A meta-analysis of randomizedcontrolled trials in pulmonary arterial hypertension. Eur Heart J2009;30:394e403.
< Epidemiologic registry indicating the improved outcome ofPAH patients on optimised treatment.
4. Kovacs G, Berghold A, Scheidl S, et al. Pulmonary arterial pressureduring rest and exercise in healthy subjects. A systematic review.Eur Respir J 2009 34:888e94.
5. Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency ofBMPR2 exonic deletions/duplications in familial pulmonary arterialhypertension. Am J Respir Crit Care Med 2006;174:590e8.
6. McGoon M, Gutterman D, Steen V, et al. Screening, earlydetection, and diagnosis of pulmonary arterial hypertension: ACCPevidence-based clinical practice guidelines. Chest2004;126:14Se34S.
7. Barst RJ (ed). Pulmonary arterial hypertension: diagnosis andevidence-based treatment. John Wiley & Sons, 2008.
8. Sitbon O, Lascoux-Combe C, Delfraissy JF, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviraltherapy era. Am J Respir Crit Care Med 2008;177:108e13.
9. Sitbon O, Beghetti M, Petit J, et al. Bosentan for the treatment ofpulmonary arterial hypertension associated with congenital heartdefects. Eur J Clin Invest 2006;36 (Suppl 3):25e31.
10. Herve P, Le Pavec J, Sztrymf B, et al. Pulmonary vascularabnormalities in cirrhosis. Best Pract Res Clin Gastroenterol2007;21:141e59.
11. Lapa M, Dias B, Jardim C, et al. Cardiopulmonary manifestations ofhepatosplenic schistosomiasis. Circulation 2009;119:1518e23.
12. Butrous G, Ghofrani HA, Grimminger F. Pulmonary vasculardisease in the developing world. Circulation 2008;118:1758e66.
13. Gladwin MT, Kato GJ. Cardiopulmonary complications of sicklecell disease: role of nitric oxide and hemolytic anemia. HematologyAm Soc Hematol Educ Program 2005:51e7.
14. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension inCOPD. Eur Respir J 2008;32:1371e85.
15. Hoeper MM,Mayer E, Simonneau G, et al. Chronic thromboembolicpulmonary hypertension. Circulation 2006;113:2011e20.
< Excellent review on the state-of-the-art management andtreatment of patients with chronic thromboembolicpulmonary hypertension.
16. Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annulardisplacement predicts survival in pulmonary hypertension. Am JRespir Crit Care Med 2006;174:1034e41.
< Important paper on TAPSE indicating its predictive value inPAH patients.
17. Vonk MC, Sander MH, van den Hoogen FH, et al. Right ventricleTei-index: a tool to increase the accuracy of non-invasive detectionof pulmonary arterial hypertension in connective tissue diseases.Eur J Echocardiogr 2007;8:317e21.
18. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009expert consensus document on pulmonary hypertension a report ofthe American College of Cardiology Foundation Task Force onExpert Consensus Documents and the American Heart Associationdeveloped in collaboration with the American College of ChestPhysicians; American Thoracic Society, Inc.; and the PulmonaryHypertension Association. J Am Coll Cardiol 2009;53:1573e619.
< Expert consensus on the optimal treatment of pulmonaryhypertension.
19. Sun XG, Hansen JE, Oudiz RJ, et al. Pulmonary function in primarypulmonary hypertension. J Am Coll Cardiol 2003;41:1028e35.
20. Sitbon O, Humbert M, Jais X, et al. Long-term response to calciumchannel blockers in idiopathic pulmonary arterial hypertension.Circulation 2005;111:3105e11.
21. Hoeper MM, Barbera JA, Channick RN, et al. Diagnosis,assessment, and treatment of non-pulmonary arterialhypertension pulmonary hypertension. J Am Coll Cardiol 2009;54:S85e96.
< Nice review on pulmonary hypertension associated with leftheart disease or chronic lung disease.
22. Schermuly RT, Stasch JP, Pullamsetti SS, et al. Expression andfunction of soluble guanylate cyclase in pulmonary arterialhypertension. Eur Respir J 2008; 32:881e91.
23. Grimminger F, Weimann G, Frey R, et al. First acutehaemodynamic study of soluble guanylate cyclase stimulatorriociguat in pulmonary hypertension. Eur Respir J 2009;33:785e92.
24. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatmentof pulmonary arterial hypertension. N Engl J Med2005;353:1412e13.
25. Schermuly RT, Dony E, Ghofrani HA, et al. Reversal ofexperimental pulmonary hypertension by PDGF inhibition. J ClinInvest 2005;115:2811e21.
Heart 2010;96:552e559. doi:10.1136/hrt.2008.156299 559
Education in Heart
group.bmj.com on April 11, 2010 - Published by heart.bmj.comDownloaded from




























