Embed Size (px)
Citation preview

SHORT COMMUNICATION
Proteome Analysis Using SelectiveIncorporation of Isotopically LabeledAmino Acids
Timothy D. Veenstra, Suzana Martinovic, Gordon A. Anderson,Ljiljana Pasa-Tolic, and Richard D. SmithEnvironmental and Molecular Sciences Laboratory, Pacific Northwest National Laboratories, Richland,Washington, USA
A method is described for identifying intact proteins from genomic databases using acombination of accurate molecular mass measurements and partial amino acid content. Aninitial demonstration was conducted for proteins isolated from Escherichia coli (E. coli) using amultiple auxotrophic strain of K12. Proteins extracted from the organism grown in naturalisotopic abundance minimal medium and also minimal medium containing isotopicallylabeled leucine (Leu-D10), were mixed and analyzed by capillary isoelectric focusing (CIEF)coupled with Fourier transform ion cyclotron resonance mass spectrometry (FTICR). Theincorporation of the isotopically labeled Leu residue has no effect on the CIEF separation of theprotein, therefore both versions of the protein are observed within the same FTICR spectrum.The difference in the molecular mass of the natural isotopic abundance and Leu-D10isotopically labeled proteins is used to determine the number of Leu residues present in thatparticular protein. Knowledge of the molecular mass and number of Leu residues present canbe used to unambiguously identify the intact protein. Preliminary results show the efficacy ofthis method for unambiguously identifying proteins isolated from E. coli. (J Am Soc MassSpectrom 2000, 11, 78–82) © 2000 American Society for Mass Spectrometry
Proteomics is rapidly becoming an important toolto study the global events that occur within a cellunder a specific set of conditions. There is a need
to develop methods that allow proteomic studies to beperformed quickly and with high sensitivity. The pres-ently predominant strategy of 2-D PAGE separationfollowed by in-gel tryptic digestion and tandem massspectrometry (MS) identification of the resultant pep-tides is too slow and insensitive to meet future demandsfor high throughput proteomics of complex organisms.A strategy based on one-dimensional isoelectric sepa-ration followed by matrix-assisted laser desorption/ionization coupled with time-of-flight (MALDI-TOF)mass spectrometry, has shown the ability to measurethe molecular masses of hundreds of proteins within asingle experiment [1,2]. A two-dimensional liquid chro-matographic system has been developed which usessize-exclusion liquid chromatography followed by re-versed-phase liquid chromatography to fractionate pro-teins extracted from E. coli cells and analyze the intactproteins using MALDI-TOF or electrospray ionization(ESI)/MS [3]. Efforts in our laboratory have included
the separation of cell lysate proteins via CIEF coupledwith analysis of the intact proteins by Fourier transformion cyclotron resonance mass spectrometry (FTICR)[4–6]. Although these approaches have the potential forrapid and sensitive proteome analysis, they are gener-ally incapable of proteome-wide unambiguous proteinidentification based on molecular mass alone, even withthe mass measurement accuracy afforded using FTICR(due to issues such as unexpected modifications). It isobvious that more information besides the molecularmass of the intact protein is generally required forunambiguous protein identification.
We describe here a strategy combining molecularmass measurement and partial amino acid content tounambiguously identify intact proteins isolated from E.coli K12 strain CP78 [7]. The organism is grown inminimal medium and minimal medium supplementedwith isotopically labeled leucine containing 10 deute-rium atoms (Leu-D10). The presence of the isotopicallylabeled Leu in the medium causes the cell to transcribeproteins with Leu-D10 in place of natural isotopic abun-dance Leu. The mass difference between the proteinsproduced in cells grown in the different types of mediais used to determine the number of Leu residuespresent for detected proteins. Knowledge of the mea-sured molecular mass and the number of Leu greatly
Address correspondence to Dr. Richard D. Smith, Environmental andMolecular Sciences Laboratory, P.O. Box 999, MSIN: K8-98, Richland, WA99352. E-mail: [email protected]
© 2000 American Society for Mass Spectrometry. Published by Elsevier Science Inc. Received August 27, 19991044-0305/00/$20.00 Revised September 22, 1999PII S1044-0305(99)00120-8 Accepted September 24, 1999

constrains possible protein assignments and generallyallows the unambiguous identification of the proteinsbeing observed, and extensions of this approach shouldbe broadly applicable for high-throughput protein iden-tification. Additionally, the mass difference between thepredicted and the experimental accurate mass measure-ment provides insights into the nature of the differ-ences, which can generally be attributed to specifictypes of post-translational modifications. Although se-lective labeling of specific amino acid residues has beenused in nuclear magnetic resonance spectroscopy tostudy purified proteins [8–10], this represents, to thebest of our knowledge, the first report of its applicationin the field of proteomics for the identification of intactproteins isolated from E. coli.
Experimental
Materials
Deuterated leucine (Leu-D10, 97.9%) was purchasedfrom Cambridge Isotopes Laboratories (Andover, MA).
Sample Preparation
The E. coli K12 strain CP78 (lueB2, argH2, thr2, his2) [7]was used in this study. The cells were grown in naturalisotopic abundance M9 minimal medium [11] contain-ing Leu and M9 medium containing Leu-D10. The cellswere grown at 37 °C with shaking (225 rpm), andharvested by centrifugation at 10,000 3 g for 30 s. Afterresuspending the cells in 200 mL of PBS buffer, theywere lysed by mechanical agitation at 4600 rpm for 60 sin the presence of 0.1 mm diameter zirconium/silicabeads (Biospec Products, Bartlesville, OK) using a Mini-Beadbeater (Biospec Products). The cell lysate wasrecovered and centrifuged at 10,000 3 g for 5 min toremove any cell debris. The protein extract was pro-cessed using a dual microdialysis system [12] and finalprotein concentration was determined using the Brad-ford assay [13].
Capillary Isoelectric Focusing Separation
The protein extract was separated by CIEF using 50 cm(50 mm i.d. and 192 mm o.d.) fused silica capillarycoated internally with linear polyacrylamide [14]. Thecapillary was filled with a sample solution containing;0.25 mg/mL of protein isolated from E. coli and 0.5%Pharmalyte 3-10 (Amersham Pharmacia Biotech, Pisca-taway, NJ). CIEF focusing was done at a constantvoltage (;13 kV) for 15 min using a high-voltage powersupply (Glassman High-Voltage, Whitehouse Station,NJ). Phosphoric acid (20 mM) and sodium hydroxide(20 mM) were used as the anolyte and catholyte,respectively. The CIEF separation was coupled to a 7tesla Fourier transform ion cyclotron resonance massspectrometer (FTICR). The IEF capillary was interfacedutilizing a coaxial liquid sheath flow configuration [5,
15]. A sheath liquid (50% CH3OH/49% H2O/1%CH3COOH, pH 2.6) was delivered at a flow rate of 2mL/min using a Harvard Apparatus 22 syringe pump(South Natick, MA). Gravity mobilization was appliedconcurrently with cathodic mobilization by raising theinlet reservoir 5 cm above the level of the electrosprayneedle.
Mass Spectrometry and Data Analysis
ESI-FTICR mass spectra were acquired using 7 teslaFTICR mass spectrometer [16] equipped with an Odys-sey data system (Finnigan FTMS, Madison, WI). Ionswere transferred from the ESI interface to the trap usingan rf quadrupole for collisional focusing at 200 mtorr,followed by two sets of rf-only quadrupoles in thehigher vacuum region of the mass spectrometer. Posi-tive ion mass spectra were acquired using a standardexperimental sequence for ion injection and accumula-tion, pump down, excitation, and detection. The totalspectrum acquisition time was about 4 s. Ion accumu-lation was accomplished by injecting 1025 torr of N2
into the trap via a piezoelectric pulse valve (Lasertech-niques, Albuquerque, NM). Background pressure in theICR trap was maintained at 1029 torr using a customcryopumping assembly [16]. The analysis of the CIEF-FTICR data and the interpretation of the results wereaccomplished using ICR-2LS software developed at ourlaboratory to assist protein identification and databasesearching [5, 6].
Results and Discussion
The present work provides an approach that augmentsaccurate molecular mass information with additionalinformation on the number of one or more specificamino acid residues to assist protein identification. Toobtain this additional information, a multiple auxotro-phic E. coli K12 strain, CP78, was grown in naturalisotope abundance minimal medium and also in mini-mal medium to which Leu-D10 had been added. Afterextraction and processing, cytosolic proteins weremixed and analyzed by on-line CIEF-FTICR [5]. Theisotopically labeled Leu-D10 residue has 10 nonex-changeable deuterium atoms substituted for hydrogen,creating a theoretical mass difference of 10 Da betweenthe unlabeled (Leu) and isotopically labeled residue(Leu-D10). In many cases we can then use the measuredmolecular mass and partial amino acid composition torapidly and unambiguously identify intact E. coli pro-teins. It has been our experience that only a smallpercentage of observed intact proteins can be unambig-uously identified solely by their measured molecularmasses [5].
As shown in Figure 1, the natural isotopic abundanceand Leu-D10 isotopically labeled versions of the proteinbehave identically during CIEF separation and areobserved within the same FTICR spectrum. In Figure1A, both protein versions display identical charge state
79J Am Soc Mass Spectrom 2000, 11, 78–82 PROTEOME ANALYSIS WITH LABELED AMINO ACIDS
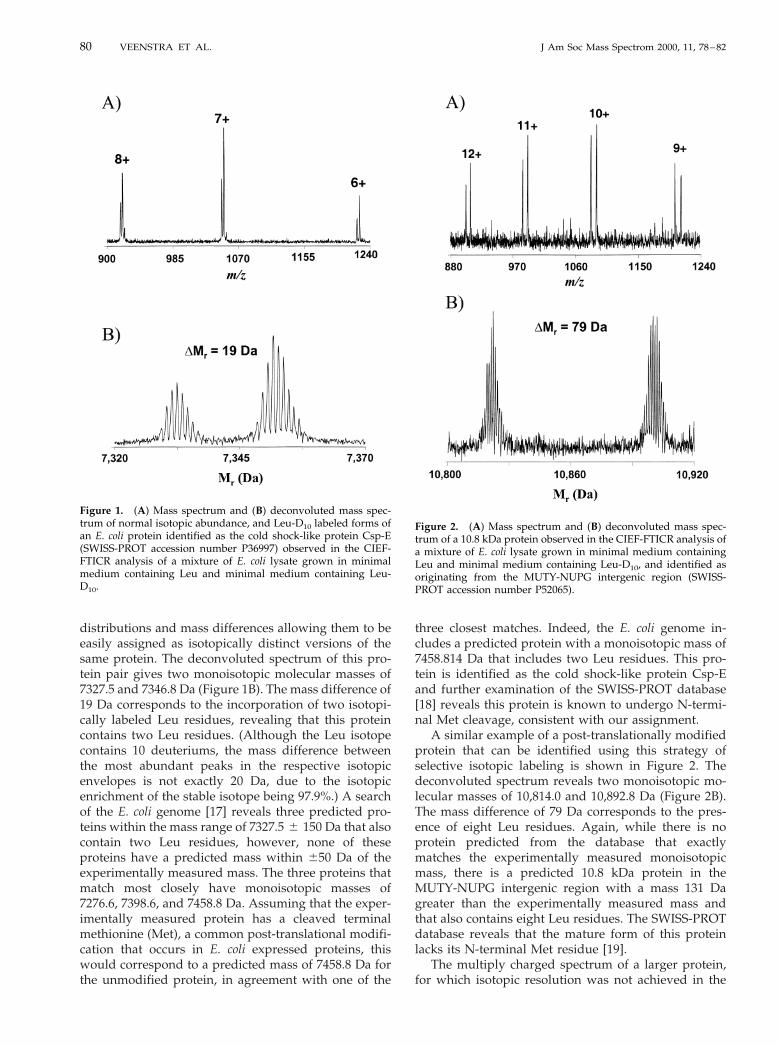
distributions and mass differences allowing them to beeasily assigned as isotopically distinct versions of thesame protein. The deconvoluted spectrum of this pro-tein pair gives two monoisotopic molecular masses of7327.5 and 7346.8 Da (Figure 1B). The mass difference of19 Da corresponds to the incorporation of two isotopi-cally labeled Leu residues, revealing that this proteincontains two Leu residues. (Although the Leu isotopecontains 10 deuteriums, the mass difference betweenthe most abundant peaks in the respective isotopicenvelopes is not exactly 20 Da, due to the isotopicenrichment of the stable isotope being 97.9%.) A searchof the E. coli genome [17] reveals three predicted pro-teins within the mass range of 7327.5 6 150 Da that alsocontain two Leu residues, however, none of theseproteins have a predicted mass within 650 Da of theexperimentally measured mass. The three proteins thatmatch most closely have monoisotopic masses of7276.6, 7398.6, and 7458.8 Da. Assuming that the exper-imentally measured protein has a cleaved terminalmethionine (Met), a common post-translational modifi-cation that occurs in E. coli expressed proteins, thiswould correspond to a predicted mass of 7458.8 Da forthe unmodified protein, in agreement with one of the
three closest matches. Indeed, the E. coli genome in-cludes a predicted protein with a monoisotopic mass of7458.814 Da that includes two Leu residues. This pro-tein is identified as the cold shock-like protein Csp-Eand further examination of the SWISS-PROT database[18] reveals this protein is known to undergo N-termi-nal Met cleavage, consistent with our assignment.
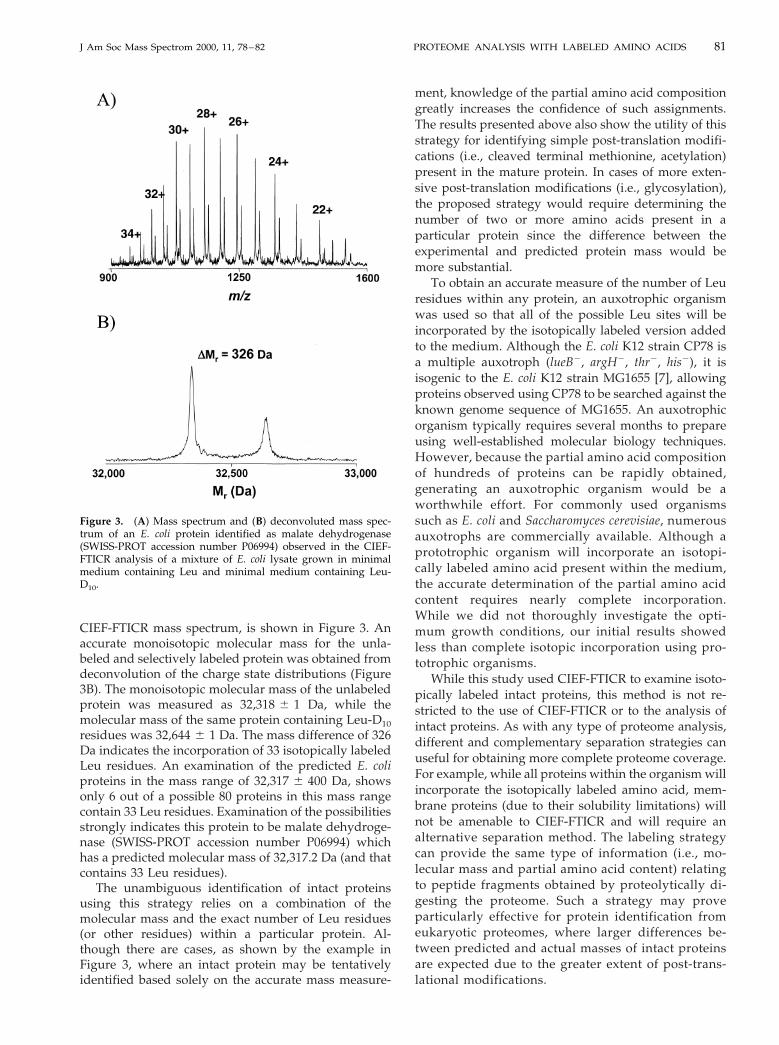
A similar example of a post-translationally modifiedprotein that can be identified using this strategy ofselective isotopic labeling is shown in Figure 2. Thedeconvoluted spectrum reveals two monoisotopic mo-lecular masses of 10,814.0 and 10,892.8 Da (Figure 2B).The mass difference of 79 Da corresponds to the pres-ence of eight Leu residues. Again, while there is noprotein predicted from the database that exactlymatches the experimentally measured monoisotopicmass, there is a predicted 10.8 kDa protein in theMUTY-NUPG intergenic region with a mass 131 Dagreater than the experimentally measured mass andthat also contains eight Leu residues. The SWISS-PROTdatabase reveals that the mature form of this proteinlacks its N-terminal Met residue [19].
The multiply charged spectrum of a larger protein,for which isotopic resolution was not achieved in the
Figure 1. (A) Mass spectrum and (B) deconvoluted mass spec-trum of normal isotopic abundance, and Leu-D10 labeled forms ofan E. coli protein identified as the cold shock-like protein Csp-E(SWISS-PROT accession number P36997) observed in the CIEF-FTICR analysis of a mixture of E. coli lysate grown in minimalmedium containing Leu and minimal medium containing Leu-D10.
Figure 2. (A) Mass spectrum and (B) deconvoluted mass spec-trum of a 10.8 kDa protein observed in the CIEF-FTICR analysis ofa mixture of E. coli lysate grown in minimal medium containingLeu and minimal medium containing Leu-D10, and identified asoriginating from the MUTY-NUPG intergenic region (SWISS-PROT accession number P52065).
80 VEENSTRA ET AL. J Am Soc Mass Spectrom 2000, 11, 78–82
CIEF-FTICR mass spectrum, is shown in Figure 3. Anaccurate monoisotopic molecular mass for the unla-beled and selectively labeled protein was obtained fromdeconvolution of the charge state distributions (Figure3B). The monoisotopic molecular mass of the unlabeledprotein was measured as 32,318 6 1 Da, while themolecular mass of the same protein containing Leu-D10
residues was 32,644 6 1 Da. The mass difference of 326Da indicates the incorporation of 33 isotopically labeledLeu residues. An examination of the predicted E. coliproteins in the mass range of 32,317 6 400 Da, showsonly 6 out of a possible 80 proteins in this mass rangecontain 33 Leu residues. Examination of the possibilitiesstrongly indicates this protein to be malate dehydroge-nase (SWISS-PROT accession number P06994) whichhas a predicted molecular mass of 32,317.2 Da (and thatcontains 33 Leu residues).
The unambiguous identification of intact proteinsusing this strategy relies on a combination of themolecular mass and the exact number of Leu residues(or other residues) within a particular protein. Al-though there are cases, as shown by the example inFigure 3, where an intact protein may be tentativelyidentified based solely on the accurate mass measure-
ment, knowledge of the partial amino acid compositiongreatly increases the confidence of such assignments.The results presented above also show the utility of thisstrategy for identifying simple post-translation modifi-cations (i.e., cleaved terminal methionine, acetylation)present in the mature protein. In cases of more exten-sive post-translation modifications (i.e., glycosylation),the proposed strategy would require determining thenumber of two or more amino acids present in aparticular protein since the difference between theexperimental and predicted protein mass would bemore substantial.
To obtain an accurate measure of the number of Leuresidues within any protein, an auxotrophic organismwas used so that all of the possible Leu sites will beincorporated by the isotopically labeled version addedto the medium. Although the E. coli K12 strain CP78 isa multiple auxotroph (lueB2, argH2, thr2, his2), it isisogenic to the E. coli K12 strain MG1655 [7], allowingproteins observed using CP78 to be searched against theknown genome sequence of MG1655. An auxotrophicorganism typically requires several months to prepareusing well-established molecular biology techniques.However, because the partial amino acid compositionof hundreds of proteins can be rapidly obtained,generating an auxotrophic organism would be aworthwhile effort. For commonly used organismssuch as E. coli and Saccharomyces cerevisiae, numerousauxotrophs are commercially available. Although aprototrophic organism will incorporate an isotopi-cally labeled amino acid present within the medium,the accurate determination of the partial amino acidcontent requires nearly complete incorporation.While we did not thoroughly investigate the opti-mum growth conditions, our initial results showedless than complete isotopic incorporation using pro-totrophic organisms.
While this study used CIEF-FTICR to examine isoto-pically labeled intact proteins, this method is not re-stricted to the use of CIEF-FTICR or to the analysis ofintact proteins. As with any type of proteome analysis,different and complementary separation strategies canuseful for obtaining more complete proteome coverage.For example, while all proteins within the organism willincorporate the isotopically labeled amino acid, mem-brane proteins (due to their solubility limitations) willnot be amenable to CIEF-FTICR and will require analternative separation method. The labeling strategycan provide the same type of information (i.e., mo-lecular mass and partial amino acid content) relatingto peptide fragments obtained by proteolytically di-gesting the proteome. Such a strategy may proveparticularly effective for protein identification fromeukaryotic proteomes, where larger differences be-tween predicted and actual masses of intact proteinsare expected due to the greater extent of post-trans-lational modifications.
Figure 3. (A) Mass spectrum and (B) deconvoluted mass spec-trum of an E. coli protein identified as malate dehydrogenase(SWISS-PROT accession number P06994) observed in the CIEF-FTICR analysis of a mixture of E. coli lysate grown in minimalmedium containing Leu and minimal medium containing Leu-D10.
81J Am Soc Mass Spectrom 2000, 11, 78–82 PROTEOME ANALYSIS WITH LABELED AMINO ACIDS

Conclusions
The ability to unambiguously identify intact proteins ina single experiment, through a combination of molecu-lar mass measurement and determination of partialamino acid compositions, offers an attractive strategyfor high-throughput proteomic studies. The strategypresented requires minimal sample handling and pro-cessing and is therefore applicable to such analyses. Theapproach can obviously be extended to the labeling ofadditional amino acid residues and the analysis ofproteolytic products. From a single CIEF-FTICR exper-iment we have been able to measure the mass anddetermine the number of Leu residues for hundreds ofdifferent proteins. Efforts are currently underway inour laboratory to identify these proteins and use thisstrategy for the proteome-wide identification of intactproteins isolated from E. coli as well as Saccharomycescerevisiae. We anticipate this basic approach will be animportant complement to the set of tools available forrapid protein identification [5].
AcknowledgmentsThe authors would like to express their appreciation to Dr. B. E.Wright (Division of Biological Sciences, University of Montana)for the generous gift of the E. coli K12 strain CP78. This researchwas supported by the U.S. Department of Energy Office ofBiological and Environmental Research and internal Pacific North-west National Laboratory (PNNL) Directed Research and Develop-ment. PNNL is operated by Battelle Memorial Institute for the U.S.Department of Energy under contract DEAC06-76RLO 1830.
References1. Chong, B. E.; Wall, D. B.; Lubman, D. M.; Flynn, S. J. Rapid
Commun. Mass Spectrom. 1997, 11, 1900–1908.2. Loo, J. A.; Brown, J.; Critchley, G.; Mitchell, C.; Andrews, P. C.;
Ogorzalek Loo, R. R. Electrophoresis 1999, 20, 743–748.
3. Opiteck, G. J.; Ramirez, S. M.; Jorgenson, J. W.; Moseley, M. A.,III. Anal. Biochem. 1998, 258, 349–361.
4. Yang, L.; Lee, C. S.; Hofstadler, S. A.; Pasa-Tolic, L.; Smith,R. D. Anal. Chem. 1998, 70, 3235–3241.
5. Jensen, P. K.; Pasa-Tolic, L.; Anderson, G. A.; Horner, J. A.;Lipton, M. S.; Bruce, J. E.; Smith, R. D. Anal. Chem. 1999, 71,2076–2084.
6. Pasa-Tolic, L.; Jensen, P. K.; Anderson, G. A.; Lipton, M. S.;Peden, K. K.; Martinovic, S.; Tolic, N.; Bruce, J. E.; Smith, R. D.J. Am. Chem. Soc. 1999, 121, 7949–7950.
7. Wright, B. E.; Longacre, A.; Reimers, J. M. Proc. Natl. Acad. Sci.U.S.A. 1999, 96, 5089–5094.
8. Xia, B.; Pikus, J. D.; Xia, W.; McClay, K.; Steffan, R. J.; Chae,Y. K.; Westler, W. M.; Markley, J. L.; Fox, B. G. Biochemistry1999, 38, 727–739.
9. McIntosh, L. P.; Griffey, R. H.; Muchmore, D. C.; Nielson,C. P.; Redfield, A. G.; Dahlquist, F. W. Proc. Natl. Acad. Sci.U.S.A. 1987, 84, 1244–1248.
10. Griffey, R. H.; Redfield, A. G.; Loomis, R. E.; Dahlquist, F. W.Biochemistry 1985, 24, 817–822.
11. Sambrook, J.; Fritsch, E. F.; Maniatis, T. Molecular cloning: alaboratory manual, 2nd ed.; Cold Spring Harbor Laboratory:New York, 1989.
12. Liu, C. L.; Hofstadler, S. A.; Bresson, J. A.; Udseth, H. R.;Tsukuda, T.; Smith, R. D.; Snyder, A. P. Anal. Chem. 1998, 70,1797–1801.
13. Bradford, M. M. Anal. Biochem. 1976, 72, 248–254.14. Kilar, F.; Hjerten, S. Electrophoresis 1989, 10, 23–29.15. Smith, R. D.; Wahl, J. H.; Goodlett, D. R.; Hofstadler, S. A.
Anal. Chem. 1993, 65, A574–A584.16. Winger, B. E.; Hofstadler, S. A.; Bruce, J. E.; Udseth, H. R.;
Smith, R. D. J. Am. Soc. Mass Spectrom. 1993, 4, 566–577.17. Blattner, F. R.; Plunkett, G., 3rd; Bloch, C. A.; Perna, N. T.;
Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J. D.; Rode,C. K.; Mayhew, G. F.; Gregor, J.; Davis, N. W.; Kirkpatrick,H. A.; Goeden, M. A.; Rose, D. J.; Mau, B.; Shao, Y. Science1997, 277, 1453–1474.
18. Bairoch, A.; Apweiler, R. Nucleic Acids Res. 1999, 27, 49–54.19. Link, A. J.; Robison, K.; Church, G. M. Electrophoresis 1997, 18,
1259–1313.
82 VEENSTRA ET AL. J Am Soc Mass Spectrom 2000, 11, 78–82





























