Embed Size (px)
Citation preview

1
Draft for Comment Purposes (Not for Implementation)
Medical Devices Sector
SAUDI FOOD & DRUG AUTHORITY
Project of GUIDELINES FOR BEST PRACTICES IN MEDICAL
DEVICE MANAGEMENT WITHIN HEALTH FACILITIES
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50

2
51
Contents 52
53
LIST OF ABBREVIATIONS AND ACRONYMS ................................................... 4 54
Chapter 1: Preambles 11 55
1.1 Introduction ....................................................................................................... 11 56
1.2 Health Technology Management Department .................................................. 14 57
2.1.2 Preventable medical errors 15 58
Chapter 2: Planning 17 59
2.1 Introduction 17 60
2.2 Medical Devices Planning Process 19 61
2.3 Checklist 30 62
Chapter 3: Procurement 31 63
3.1 Introduction ....................................................................................................... 31 64
3.2 Procurement Procedures ................................................................................... 33 65
3.3 Operation Costs Consideration ......................................................................... 35 66
3.4 Methods of Acquisition2 ................................................................................... 35 67
3.5 Tender Contents ................................................................................................ 36 68
3.6 Ethical Considerations ...................................................................................... 37 69
3.7 Evaluation Criteria ............................................................................................ 39 70
3.8 Compliance Checking ....................................................................................... 40 71
3.9 Contract with Supplier. ..................................................................................... 40 72
3.10 Payments Scheduling. ....................................................................................... 40 73
3.11 Storage Requirements: ...................................................................................... 40 74
3.12 Tender Award Process and Payment Processing .............................................. 40 75
3.13 Checklist ........................................................................................................... 42 76
Chapter 4: Installation & Commissioning ............................................................... 43 77
4.1 Introduction ....................................................................................................... 43 78
4.2 Delivery............................................................................................................. 44 79
4.3 Medical Device Installation & Commissioning ................................................ 45 80
4.4 Checklist ........................................................................................................... 47 81
Chapter 5: Operations 48 82
5.1 Introduction ....................................................................................................... 48 83
5.2 Operation Activities .......................................................................................... 50 84
5.3 Corrective Maintenance Management .............................................................. 52 85
5.4 Transfer and storage procedures ....................................................................... 53 86

3
5.5 Reusable Medical Devices Reprocessing ......................................................... 54 87
5.6 Computerized Maintenance Management System (CMMS) ............................ 58 88
5.7 Data integrity .................................................................................................... 60 89
5.8 Considerations in selecting a new or replacement CMMS ............................... 63 90
5.9 Reporting adverse events to SFDA ................................................................... 65 91
5.10 Special requirements for specific medical devices ........................................... 69 92
5.11 Special safety requirements .............................................................................. 71 93
5.12 Inventory Accuracy ........................................................................................... 72 94
5.13 Warranty Management...................................................................................... 74 95
5.14 Disaster/Contingency Planning ......................................................................... 75 96
5.15 User Training .................................................................................................... 77 97
5.16 HTM/Clinical Engineering Training programs ................................................ 77 98
5.17 Spare Parts procurement and management ....................................................... 78 99
5.18 On Site Libraries ............................................................................................... 78 100
5.19 Workshop Space and Test Devices ................................................................... 79 101
5.20 Inter Service Coordination ................................................................................ 79 102
5.21 Checklist ........................................................................................................... 80 103
Chapter 6: Disposal & Donation 82 104
6.1 Introduction ....................................................................................................... 82 105
6.2 Checklist: .......................................................................................................... 86 106
Chapter7: Special Topics 87 107
7.1 Introduction ....................................................................................................... 87 108
7.2 Medical device Consumables, Single Use. ....................................................... 90 109
7.3 SFDA requirements with regards to reprocessing of single use devices .......... 91 110
7.4 Reuse of Reusable, Limited Use & Single Patient Use Medical Devices ........ 91 111
7.5 Reuse of Single Use Medical devices ............................................................... 91 112
7.6 Medical Device Implants .................................................................................. 93 113
7.7 Checklist ......................................................................................................... 100 114
8. References 101 115
9. Appendices 102 116
9.1 Planning: ......................................................................................................... 102 117
9.2 Procurement .................................................................................................... 129 118
9.3 Device Inventory Auditing Example .............................................................. 133 119
120
121

4
122
LIST OF ABBREVIATIONS AND ACRONYMS 123
124
AAMI American Association of Medical Instrumentation
AI Artificial Intelligence
BMET Biomedical Engineer/Technician/Technologist
CCTV Closed-Circuit Television
CE mark Conformité Européene (EU certificate of conformity)
CD Construction Documents
CM Corrective Maintenance
CMMS Computerized Maintenance Management System
CPG Clinical Practice Guidelines
CR Computed Radiography
CSSD Central Sterile Supplies Department
CT Computed Tomography
DD Design Development
DHCP Dynamic Host Configuration Protocol
ECG Electrocardiogram
ECRI Emergency Care Research Institute
EHRs Electronic Health Records
EM Equipment Management
EU European Union
FDA Food and Drug Administration
GMDN Global Medical Device Nomenclature
HR Human Resources
HTM Health Technology Management
IDFs The intermediate distribution frames
ICDs Implantable cardioverter-defibrillators
IHE Integrating the Healthcare Enterprise
IR Interventional Radiology
ISO International Organization Standards
IT Information Technology
LDAP Lightweight Directory Access Protocol

5
MDEL Medical Devices Establishment Licensing
MDF Multiple Distribution Frame
MDMA Medical Device Manufacturers Association
MDNR Medical Devices National Registry
MEMDMC Medical Equipment Management Device Management Communication
MRI Magnetic Resonance Imaging
PACU Post Anesthesia Care Unit
PET Positron Emission Tomography
PPM Planned Preventive Maintenance
SAM Service Availability Mapping
SEM Small and Medium Sized Enterprises
SFDA Saudi Food and Drug Authority
SON Statement of Need
SUMD single-use medical device
UMDNS Universal Medical Device Nomenclature System
UPS Uninterruptible Power Supply
WHO World Health Organization
LIST OF DEFINITIONS 125
126
Acceptance
Testing
The initial inspection performed on a piece of medical equipment prior to
it being put into service. When the device first arrives in the health-care
facility, it is checked to ensure it matches the purchase order, it is
functioning as specified, the training for users has been arranged and it is
installed correctly. If a computerized maintenance management system
(CMMS) is available, it is registered into the CMMS.
Medical Device
Accessories
Products intended specifically by its manufacturer to be used together
with a medical device to enable that medical device to achieve its intended
purpose.
Assessment
Process
Tool for determining medical device needs at the healthcare facility and
detecting the gaps in the availability of medical devices by collecting

6
baseline information that can support recommendations for future
planning.
Calibration
Some medical equipment, particularly those with therapeutic energy
output (e.g. defibrillators, electrosurgical units, physical therapy
stimulators, etc.), needs to be calibrated periodically. This means that
energy levels are to be measured and if there is a discrepancy from the
indicated levels, adjustments must be made until the device functions
within specifications. Devices that take measurements (e.g.
electrocardiographs, laboratory equipment, patient scales, pulmonary
function analyzers, etc.) also require periodic calibration to ensure
accuracy compared to known standards.
Certificate of
Need (CON)
Planning tool used to support decision-makers in evaluating investments
of highly specialized and expensive medical equipment, based on
technical, epidemiological, and cost-benefit criteria in order to best
optimize resources.
Clinical
Engineer
A professional who supports and advances patient care by applying
engineering and managerial skills to healthcare technology.
Computerized
Maintenance
Management
Systems
Computer-based software system that is used to automate issues related
to technical support of medical devices and provide support to the
management of the medical device inventory, corrective maintenance,
preventive maintenance, contract management and provides a wide range
of real time data reports on different issues related to the medical device
lifecycle such as downtime, life cycle cost and inventory reports related
to device type, location or selected manufacturers.
Corrective
Maintenance
(CM)
A process used to restore the physical integrity, safety and/or performance
of a device after a failure. Corrective maintenance and unscheduled
maintenance are regarded as equivalent to the term repair. This document
uses these terms interchangeably.

7
Cybersecurity
Body of technologies, processes and practices designed to protect
networks, computers, programs and data from attack, damage or
unauthorized access.
Device
Planning
Core component of hospital planning and design. Some medical devices
require special architectural, electromechanical and infrastructure design
in addition to the unique and specific utility and pre-installation
requirements.
Failure The condition of not meeting intended performance or safety
requirements, and/or a breach of physical integrity. A failure is corrected
by repair and/or calibration.
Global Medical
Devices
Nomenclature
System
(GMDNS)
List of generic names used to identify all medical devices to provide
health authorities, regulators, health care providers, manufacturers and
others with a naming system that can be used to exchange medical device
information and support patient safety.
The GMDNS is used for:
a) Data exchange between manufacturers, regulators and healthcare
authorities
b) Exchange of post-market vigilance information
c) Supporting inventory control in hospitals
d) Purchasing and supply chain management
The GMDNS is recommended by the International Medical Device
Regulators Forum (IMDRF) and is now used by over 70 national medical
device regulators to support their activity. The GMDNS is managed by
the GMDN Agency, a registered charity, which has a Board of Trustees,
which represent regulators and industry.
Healthcare
Needs
Assessment
The identification and definition of prioritized requirements regarding
medical devices.

8
Health
Technology
Management
(HTM)/
Clinical
Engineering
The cost-effective and clinical effective management of all issues related
to the medical device lifecycle. This may consist of a framework of
standard policies and procedures designed to address all different
components and issues related to medical devices. The term health
technology management is used to describe the delivery structure required
to manage the medical devices within healthcare systems.
Inspection Scheduled activities necessary to ensure a piece of medical equipment is
functioning correctly. It includes both performance inspections and safety
inspections. These occur in conjunction with preventive maintenance,
corrective maintenance, or calibration but can also be completed as a
stand-alone activity scheduled at specific intervals.
Inspection and
Preventive
Maintenance
(IPM)
IPM refers to all the scheduled activity necessary to ensure a piece of
medical equipment is functioning correctly and is well maintained. IPM
therefore includes inspection and preventive maintenance (PM). This is
also referred as PPM (Planned Preventive Maintenance).
Medical Device
Any instrument, apparatus, implement, machine, appliance, implant, in
vitro reagent or calibrator, software, material or other similar or related
article:
A. Intended by the manufacturer to be used, alone or in combination,
for human beings for one or more of the specific purpose(s) of: −
Diagnosis, prevention, monitoring, treatment or alleviation of
disease, − Diagnosis, monitoring, treatment, alleviation of or
compensation for an injury or handicap, − Investigation,
replacement, modification, or support of the anatomy or of a
physiological process, − Supporting or sustaining life, − Control
of conception, − Disinfection of medical devices, − Providing
information for medical or diagnostic purposes by means of in
vitro examination of specimens derived from the human body;

9
B. Which does not achieve its primary intended action in or on the
human body by pharmacological, immunological or metabolic
means, but which may be assisted in its intended function by such
means.
Medical Device
Nomenclature
Systems
Classification system to identify the type of medical devices by assigning
a code to the type of device such as CT, MRI and implantable pacemakers.
The classification system is needed for several HTM and regulatory
issues. It must be part of any CMMS coding system to be able to identify
and track certain parameters related to the different types of medical
devices managed by the CMMS.
Performance
Inspections
Activities designed to test the operating status of a medical device. Tests
compare the performance of the device to technical specifications
established by the manufacturer in their maintenance or service manual.
These inspections are not meant to extend the life of the device, but
merely to assess its current condition. Performance inspections are
sometimes referred to as ‘performance assurance inspections.’.
Preventive
Maintenance
(PM)
PM involves maintenance performed to extend the life of the device and
prevent failure. PM is usually scheduled at specific intervals and includes
specific maintenance activities such as lubrication, cleaning (e.g. filters)
or replacing parts that are expected to wear (e.g. bearings) or which have
a finite life (e.g. tubing). The procedures and intervals are usually
established by the manufacturer. In exceptional cases the user may change
the frequency to accommodate local environmental conditions.
Preventive maintenance is sometimes referred to as ‘planned
maintenance’ or ‘scheduled maintenance’. This document uses these
terms interchangeably.
Repair A process used to restore the physical integrity, safety, and/or
performance of a device after a failure. Used interchangeably with
corrective maintenance.
10
Reuse Repeated use or multiple use of any medical device with reprocessing
(cleaning, disinfection or sterilization) between uses.
Safety
Inspections
Safety inspections are performed to ensure the device is electrically and
mechanically safe. These inspections may also include checks for
radiation safety or dangerous gas or chemical pollutants. When these
inspections are done, the results are compared to country or regional
standards as well as to manufacturer’s specifications. The frequency of
safety inspections may be different than planned maintenance and
performance inspections, and are usually based on regulatory
requirements.
Single Patient
Use Medical
Device
Medical devices that can be used for more than one episode of use on one
patient only. The device shall be decontaminated between each use.
Single Use
Medical Device
(SUMD)
Medical device intended for use once, on an individual patient for a single
procedure, and then should be discarded.
Unique Device
Identification
Code (UDI)
Unique device identification code using a globally accepted standard
format which consist of series of numeric or alphanumeric characters and
used as the key access to information stored in a UDI database.
Universal
Medical
Devices
Nomenclature
System
(UMDNS)
Standard, free of charge, monthly updated, international nomenclature
and computer coding system to help better manage classifying medical
device types.
UMDNS facilitates identifying, processing, filing, storing, retrieving,
transferring, and communicating data about medical devices. The
nomenclature is used in applications ranging from hospital inventory and
work-order controls to national agency medical device regulatory systems
and from e-commerce and procurement to medical device databases.
127
128

11
129
Chapter 1: Preambles 130
1.1 Introduction 131
132
133
1.1.1 Objective 134 135
To develop standardized national guideline for best practices in medical device 136
management throughout their life cycle within healthcare facilities. 137
Enhance the efficiency and improve the management of medical devices, their 138
operation and their rational use within healthcare facilities. 139
Minimize the risks associated with the use of medical devices. 140
Ensure the medical devices within the healthcare facility are: 141
- utilized appropriately; 142
- suitable for its intended purpose; 143
- maintained in a safe and reliable condition; 144
- operated in accordance with the manufacturer’s instruction by users and 145
professionals who have obtained and maintained the correct level of 146
knowledge and competency necessary, and disposed of appropriately at the 147
end of its useful life. 148
Periodically monitored for performance, leakage current, isolation, insulation, 149
calibration, adverse events, risks, utilization and cost 150
Perform one of the surveillance tasks that on the official order of SFDA system 151
that mentions the “SFDA shall monitor the obligation of healthcare facilities 152
with international standards of safety performance for medical devices" in 153
accordance with these guidelines for management of health technology /medical 154
devices within healthcare facilities. 155
156
In accordance with these guidelines, the SFDA is working with its partners to build 157
a framework of regulatory procedures to support improving the quality of healthcare 158
in the kingdom. 159
160
161

12
162
1.1.2 Scope 163
164
This guideline will apply to any medical device used as part of the routine care of 165
patients. It will provide procedures and practices that will help ensure that a medical 166
device is managed, operated and maintained as recommended by the manufacturer 167
to ensure that its performance will be maintained throughout its lifetime. The 168
procedures will cover the medical devices life cycle as following: 169
170
1. Planning phase: User requirements, safety, needs assessment, pre-installations 171
requirements; 172
2. Procurement phase: Tender process, technical specifications, life-cycle cost, 173
effective tender evaluation, pre-installation costs; 174
3. Installation phase: Pre-installation, inspection and commissioning, acceptance 175
certificate, connectivity, operating and service manuals; 176
4. Operation phase: Effective lifecycle management of medical technologies; 177
implementation of CMMS, preventive and corrective maintenance, calibration, 178
training users, transport, and storage. 179
5. Disposal phase: Justification for scrapping, safety and hygiene, removal, 180
dismantling and disinfection of medical device. 181
182
1.1.3 Stakeholders within healthcare facility 183 184
Medical device management requires the involvement of staff from many disciplines 185
–technical, clinical, financial, administrative, etc. It is not just the job of managers; 186
it is the responsibility of all members of staff who deal with healthcare technology1. 187
This includes the departments of Biomedical Engineering, information technology, 188
Nursing, Medical, Finance, Projects, General maintenance, Asset management 189
…etc. 190
1 How to organize a system of healthcare technology management

13
191
1.1.4 Background 192 193
Medical devices are for Diagnosis and Treatment. They play a crucial role in patient 194
care and treatment. Once a device is in the market, its safety and performance 195
depends on: 196
Preservation of the integrity of the device through applying proper management 197
procedures in storage, handling, transportation, delivery, installation and 198
testing. 199
Using the devices for the exact intended purpose as per the manufacturer's 200
instruction. 201
Operating them effectively and safely. 202
Device characteristics change with time, therefore, continuous monitoring for 203
safety and performance is essential to ensure that these changes don't have an 204
impact on patient safety. 205
Constant detection, investigation of unexpected incidents and possible adverse 206
events. 207
Proper preventive, corrective Maintenance, calibration and electrical safety tests 208
to preserve its specified performance. 209
Maintain proper documentation for all actions taken on the device including 210
utilization, operation and maintenance cost. 211
Decommission, disposing and replacing unsafe and obsolete items. 212
Ensuring the staff have the right skills to optimally use the medical device. 213
214
Health technology management involves a cycle of activities in the life of 215
equipment, as shown in (Figure 1.1). 216

14
217
Figure 1.1: Medical Device life cycle. 218
219
1.2 Health Technology Management Department 220
221
Medical devices and supplies constitute a good percentage of hospital costs. They 222
play a role in Diagnosing and treating patients in almost every department and ward 223
within a healthcare facility. Managing these sensitive and fast evolving devices 224
requires the availability of trained biomedical engineers and technicians who are 225
capable of identifying and managing risks associated with the technology used 226
within the hospital. Furthermore, most of the medical devices now share information 227
with the hospital network to create and platform for information exchange and 228
artificial intelligence. 229
In principle, no technology can be added or removed without the involvement of 230
Health Technology Management/Clinical Engineering department in the process. 231
232
The recommended organizational structure of the HTM Department (sometimes called 233
Clinical Engineering or Biomedical Department) depends mainly on the size and nature 234
of services within the healthcare facility. The world health organization (WHO) 235
published "HUMAN RESOURCES FOR MEDICAL DEVICES The role of 236
biomedical engineers" as part of Medical Devices technical series to ensure access to 237
quality and safe use of medical devices. The booklet details the responsibilities of the 238
Decommissioning and Disposal
OperationInstalling &
Commissioning
Planning & Technology Assessment
procurement

15
HTM/Clinical Engineering department depending on the hospital size. The following 239
shows some of the responsibilities of this department: 240
1. Planning for new technologies that includes (needs assessments, budgeting, 241
cost analysis, pre-installation requirements …etc. 242
2. Purchasing process: Oversee the procurement cycle, putting specifications, 243
tender processing, technical assessment, contracting, commissioning and 244
acceptance testing. 245
3. Preventive and corrective maintenance of medical devices (in-house) and/or 246
management of service contracts for devices categorized to be maintained by 247
service providers (support the preparation, signing and supervising the 248
implementation of contracts with manufacturers and local suppliers of medical 249
devices, spare parts, chemicals, reagents and consumables). 250
4. Administrative support: Oversee the internal and external administrative 251
communications, human resources and other administrative work related to 252
medical devices. 253
5. OEM & 3rd party Contract Management, negotiations and administration 254
6. Quality control: Monitor the quality of technical support provided to medical 255
devices, ensure safety to department staff, users and patients and monitor the 256
overall performance of the HTM Dept, adverse events reporting, managing 257
recalls on medical devices, incident reporting and investigations 258
7. Medical equipment retirement plan including decommissioning, disposing, 259
and replacing unsafe & obsolete items. In order to ensure safe, accurate, and 260
affordable technology in the hospital and to exclude obsolete, beyond repair 261
and un-economical technology in the hospital. 262
263
2.1.2 Preventable medical errors 264
265
Significant percentage of preventable medical errors are caused by improper use of 266
medical devices. In USA 440,000 patients are dying annually because of preventable 267
medical errors1. The figure below illustrates errors related to medical devices. In 268
another study, 59.5% of drug errors reported to USP from 570 HC facilities are due to 269
implicate use of computers2 . 270
It is the responsibility on clinical engineers when building their HTM system to have 271
amandate to enhance the safety and performance of medical devices by monitoring the 272

16
performance of medical devices, detect any risk related to that devices and take all 273
measures to minimize all adverse events related to medical devices. 274
In most Middle East countries, medical errors including those adverse events related to 275
medical devices are not properly tracked. The role of HTM system and clinical 276
engineers is to be part of any available initiative for tracking medical errors and adverse 277
events related to medical devices and raise a “red flag” to hospital leaders such 278
mechanism is not available. 279
280

17
281
Chapter 2: Planning 282
283
2.1 Introduction 284
285
2.1.1 Objective 286
287
To demonstrate how to conduct the medical devices planning through providing 288
basic tools, references, and examples to do a proper planning and assessment of their 289
current situation and future needs about medical devices. 290
2.1.2 Scope 291
292
This chapter applies to any healthcare facility whose planning for medical devices 293
includes: 294
New construction 295
Renovation 296
Device installation 297
298
to come up with a list of needs of medical devices and supplies from all healthcare 299
providers departments with justifications that based on previous years utilizations, 300
future needs and other objective parameters (product or service, quality, quantity, 301
pre-installation, the condition and performance of current medical devices installed, 302
including: device failures and issues; utilization, performance, maintenance; repair 303
and calibration history). 304
305
2.1.3 Ownership 306 307
The involvement of Committee with minimum members as following: 308
Healthcare provider administration. 309
Finance department. 310
Healthcare technology /Clinical engineering department. 311
End-user department. 312

18
IT department. 313
Facility management department. 314
Supply chain / procurement department. 315
Project management department. 316
317
2.1.4 Background 318 319
Early engagement of all stakeholders in planning the technology needs for a new 320
department or hospital is important to assure having a comprehensive view over the 321
needs to make an educated decision on the proper technologies needed. The 322
following diagram shows the steps involved in technology planning. 323
324
325
326
327
328
329
330
331
332
333
334
335
Figure 2.1: Pre-installation and Planning Processes Work Flow. 336
337
338
339
Needs of New
Medical Device
Healthcare Needs
Assessment
Occupancy
Construction
Design Data Collection and
Needs Assessment
Process
Clinical Program
Planning Process
Schematic
Design
The Required
New Clinical
Services
Device
installation
Commissioning
planning
IT
Infrastructure

19
2.2 Medical Devices Planning Process 340
341
342
Planning can be divided into four major phases: 343
Healthcare needs assessment. 344
Design. 345
Construction. 346
Occupancy. 347
348
2.1.5 Healthcare Needs Assessment 349 350
In this section, the required data collection and analysis steps that are required for 351
specific medical devices needs assessment processes include: 352
- Health service requirements, 353
- Health service availability, 354
- Human resources Information, 355
- Budgeting 356
- Priority setting – local, regional and national 357
358
2.1.5.1 Health Service Requirements 359
360
The following should be considered to understand the overall requirements of the 361
health services in the target area: 362
a. Epidemiological needs (disease priorities). 363
b. Population issues (demography, patient rate). 364
c. Anticipated level of utilization (how many times the devices will be used, is 365
there a demand on advanced or just basic options, impact on quality of care 366
for the patient 367
d. Clinical program guidelines and protocols; national or local 368
recommendations; and internationally recognized standards on diagnosis 369
and treatment of different diseases relevant to this population. 370
371
Example: For a needs assessment for computed tomography (CT) imaging 372
services, it is important to understand the needs for CT of the target population. 373
To determine if more CT scanners are needed, the current region’s number of CT 374

20
scanners per 10,000 population can be compared to the average international and 375
regional standards. The specifications for the CT scanner will also be influenced 376
by the regional disease burden, the clinical scope of the healthcare facility and the 377
availability of qualified human resources. 378
379
2.1.5.2 Health Services Availability 380
381
The following are the main types of information needed to be collected: 382
1) Healthcare provider infrastructure 383
384
- Type, size, and location of facilities, including the number and type of 385
building(s). 386
- Availability and condition of water supply, connections and installation 387
(e.g., Where does the water come from? What is the water quality?). 388
- Electrical power supply, electrical connections and installations (e.g., Is 389
an emergency generator available?). 390
- Waste disposal system (e.g., How is waste handled, segregated, and 391
disposed of?). 392
- Availability of tele-communication infrastructure. 393
- Assessment of electromagnetic compatibility (EMC). 394
395
396
2) Medical Devices 397 398
It is very important to understand and collect all relevant information 399
associated with the current available medical devices. Normally, all 400
needed information should be available in the healthcare facility’s 401
CMMS reporting system and may include the following: 402
- Device type and available quantity. 403
- Manufacturer & model names. 404
- Average lifetime of medical devices identified by installation date. 405
- Location and distribution of medical devices identified by clinical 406
departments, building number, floor level and room number. 407
- Working condition (operational, out of order, awaiting parts, etc.). 408
- Lifecycle costs information. 409

21
- Downtime information. 410
- Availability and consumption rate of spare parts. 411
- Supplier performance assessment. 412
- Any available incident reports. 413
414
2.1.5.3 Human Resource (HR) Information 415
416
Following are some HR related items to consider in the needs assessment process: 417
a. Availability of clinical staff with required qualifications. For example, 418
qualified radiologists and radiographers are needed when considering the 419
need for CT and X-ray systems. 420
b. The availability of outcome based continuing medical education programs 421
(training based on manufacturer requirements) that include on the job 422
training. 423
424
2.1.5.4 Budgeting 425 426
The following financial information are necessary: 427
a. Available budget, project or annual, for medical devices. 428
b. Estimated costs of the medical devices requested by the medical staff. 429
c. Estimated life cycle costs of the needed devices, such as spare parts, 430
maintenance contracts, software licenses, other support costs, consumables, 431
chemicals, training and disinfection. 432
d. The estimated costs for any additional infrastructure. 433
e. Does the device require consumables? Is it an open or closed system? 434
f. The estimated cost of any pre-installation and training requirements. 435
436
2.1.5.5 Priority setting – local, regional and national and final needs 437
assessment recommendations 438 439
After data collection, perform a gap analysis as following: 440
- Interpretation of the collected information. 441
22
- Determine what similar technology is available in the local 442
healthcare providers city (or region) and, where applicable, in 443
the entire country based on the health services demand and 444
overall population health and health delivery organization 445
circumstances of the local area. 446
- Determine the available local, national and international 447
standards. 448
- Consider the possible financial and human resource constraints, 449
as well as prioritized epidemiological requirements. 450
451
The outputs of the gap analysis is a development of a prioritized needs list. (Table 452
2.1) outlines the questions to be asked, the data required to answer those 453
questions, and the tools available to collect and evaluate the data. 454
455
There are several procedures available internationally to identify gaps in medical 456
device availability. For example, a stepwise approach to identify gaps in medical 457
devices (availability matrix and survey methodology) by the World Health 458
Organization provides lists of recommended medical devices for various health 459
screening and treatment processes. 460
461
462
463
464
Questions Data Required Tools Results
1 What do we
want/need in
terms of
health
services?
- Population (target population,
patient’s rate, density of
population).
- Health service provider
availability
- Epidemiological data (disease
priorities…).
- “Certificate of need”
process, see Appendix A
- Clinical practice
guidelines (CPG).
- Survey questionnaires
- Standards of level of
care.
Appropriate
health
services
delivery
2 What do we
have? (Local
conditions/
Limitations).
- Health service availability.
- Lists of available medical
devices.
- Human resources availability.
- Service Availability
Mapping (SAM)
questionnaires?
- Evaluation tools?

23
- infrastructure of medical devices
management ( electrical, water
gases and IT system related to
medical devices
- Inventory management
tool.
- Computerized
maintenance management
system (CMMS).
3 Which
standards/
recommende
d best
practices
exist that
could be
applied or
adapted?
- Standards/ recommendations for
health service delivery coverage.
- Standards/ recommendations for
medical devices
- Standards/ recommendations for
human resources required for
operation/maintenance/
management of medical
equipment.
- (Essential) Medical
device lists; i.e., per
facility type and
department, or per clinical
procedure.
4 Overall Gap (2 – 3): List of
General
Needs
5 What
financial /
human
resources do
we have?
(Constraints).
Budget (capital investment and
operational)
Human resources.
6 Prioritized Needs (4 – 5): Prioritized
List of Needs
Table 2.1: Sample questions to be asked, data required to answer those questions, 465
and the tools available to collect and evaluate the data. 466
467
2.1.6 Design of new departments in hospitals 468
469
Once the healthcare needs assessment is reviewed and approved, preliminary design 470
planning can begin: 471
472
Clinical Program Planning Process 473 474
Design starts with a detailed clinical program planning process. 475
a) Program planning process should describe the following: 476
- Clinical services that will be included in the proposed new or renovated 477
building(s), 478
- Each clinical program’s general requirements (e.g. number of patient, 479
exam and treatment rooms; number of operating rooms; space size needs) 480
within the constraints of the approved healthcare needs assessment (e.g. 481

24
overall building/remodel space size needs and budget estimate for the 482
project). 483
484
Note: Clinical engineering technology assessments are useful at the program 485
planning stage since generic device needs are one of several factors to be decided 486
in the program planning phase (e.g. Do we need a CT scanner in this new 487
building? Do we need basic CT scanner, intermediate, or advanced? What 488
accessories do we need to add to address the clinical needs? Do we have trained 489
staff to run the advanced options? What are the training needs? ). Clinical 490
engineers should get involved in this program planning stage in translating 491
clinical requirements and concerns into technical and facility requirements. At 492
this stage, clinical engineers can provide technology assessments for major 493
clinical systems as well as starting to develop a generic equipment list with 494
generic requirements for required major systems (e.g. Radiology systems, OR 495
rooms and lights) under consideration for each program. This is also the time to 496
discuss in general terms required utilities for the programs and medical devices 497
systems that will need to be installed or remodeled in the new building(s). Of 498
course, even at this early stage, specific national and local regulatory requirements 499
and governmental approvals need to be met and, if necessary, appropriate 500
preliminary local and national regulatory agency approvals obtained. 501
502
b) For the next step in the Program planning process, a “schematic design” plan 503
is drawn describing the program and space requirements for each department, 504
their required support services and “adjacencies” (i.e. what other services 505
need to be nearby). 506
507
Example: Once it is determined, that five operating rooms will be included in a 508
new healthcare facility building then the Post Anesthesia Care Unit (PACU) or 509
Recovery Room needs to be located adjacent and sized accordingly (e.g. 20+ 510
beds). The Central Sterilization Department also needs to be designed in 511
proximity to the operating rooms (ORs), or provisions made for the efficient 512
movement of dirty and clean/sterile instruments back and forth from the ORs to 513
that department. 514

25
515
c) The next step in the design phase is the development of detailed architectural 516
and engineering drawings, often called design development (DD) drawings. 517
The architectural and engineering teams develop detailed drawing sets 518
including the following: 519
- Patient flow 520
- Floor plans. 521
- Interior elevations. 522
- Mechanical. 523
- Electrical. 524
- Plumbing. 525
- Fire prevention. 526
- Radiation Protection 527
- Medical Gasses 528
- Reception area 529
- Nursing station 530
- Changing rooms, utilities, restrooms, storage …etc. 531
- Special power requirements (3 phase, grounding, Uninterrupted power 532
systems, backup generators, isolating service powerlines from medical 533
equipment power lines, Intensive care unit and operating room electrical 534
isolation …etc) 535
- Medical equipment loaded drawings. 536
- Tele- communication, including IT. 537
- Furnishings, finishes and other detailed drawings. 538
539
d) Simultaneously with the architectural design phase, a multidisciplinary team 540
which include clinical engineers and clinical end-users shall develop a 541
medical devices plan and incorporate the medical device plan into the 542
architectural and engineering drawings showing medical devices locations 543
and utility requirements (see: Table 9.1 of utilities). First priority, from an 544
architectural planning standpoint, are the medical device systems that are 545
fixed and therefore, require installation. 546
547

26
e) Technical requirements and selection of major system products based on the 548
program plan, technology assessment reports, standards within the healthcare 549
facility and the community, and the new facility design need to be developed. 550
551
Note: This is an iterative approach with input from the end-users and various 552
support teams, including IT, and clinical engineers. Typically, a multidisciplinary 553
formal drawing set review takes place at critical drawing development thresholds 554
such as 50% completion, and 95% completion. After 100% DD drawing 555
completion, healthcare executive leadership review and approval, funding 556
approval, regulatory review and approval; final schedules and construction 557
documents (CD) can be developed and construction contracts can be bid and let. 558
559
560
2.1.7 Construction 561
562
Once construction starts, clinical engineers need to be available to answer any 563
questions that the design and/or construction teams have regarding device 564
requirements. 565
566
Note: Change orders are expensive and need to be minimized. However, change 567
orders often need to occur to accommodate the selected technologies that may not 568
have been available or known about earlier in the new healthcare facility design 569
process. Often changes in device requirements occur due to changes in technology, 570
particularly when there is a long period of time between the design of the building 571
and construction completion. For example, a plan for installing the magnet for an 572
MRI machine needs to be in place since the magnet comes as one large piece and 573
may require demolishing some walls to be placed in its place. 574
575
2.1.8 Occupancy Planning 576 577
As construction gets well underway, it’s time to finalize occupancy and device 578
installation and commissioning planning. After finalizing the procurement phase 579
(see chapter 3, Procurement), plans and schedules need to be developed for fixed 580
device installation, fixed device moves (de-installation and re-installation), furniture 581

27
installation, and people and mobile medical device moves. Plans also need to be in 582
place so that all medical devices get tested prior to first clinical use. 583
584
The medical devices requiring contractor installation get installed after their 585
respective rooms and utilities are ready for these systems. As construction nears 586
completion, and all building, fire and utility inspections and testing are complete, 587
it’s time to continue medical devices installation and testing (some will call this 588
milestone “Beneficial Occupancy”). 589
590
In addition, with the large amount of medical devices requiring IT connectivity, 591
HTM shall coordinate with IT to install the manufacturer’s recommended IT 592
requirements. After the IT infrastructure is installed and operational, network 593
connected medical device installation and testing can be completed. 594
Clinical end-users will require training on all new medical devices. Clinical 595
engineers, along with clinical educators, both internal and supplier-provided, need 596
to assure that as many clinical staff as possible complete training on all new 597
technology prior to occupancy and first clinical use. 598
599
Note: HTM is responsible for the coordination of all medical device installation and 600
testing. 601
602
2.1.9 Consideration of Emerging Technologies on Medical Devices 603 604
Several emerging technologies will impact future improvements in medical devices. 605
For example, the increasing computerization of medical devices, and their 606
interconnectivity has created a very large impact on medical devices. This 607
integration and convergence of IT and Clinical Engineering is a recurring theme in 608
many emerging technologies. Some of the challenges created by this convergence 609
include the re-focus of Medical Devices resources (funds and staff) toward IT-610
related activities, medical device/IT security-related challenges for networked 611
connected medical devices, and the need for the education of HTM and Clinical 612
Engineering staff in IT technologies. 613
614

28
2.1.9.1 IT Security-related Challenges for Network-connected Medical 615
Devices 616 617
Connected devices and cyber security are rapidly becoming critical topics for 618
regulators as sophisticated IT technology integrates with medical devices. 619
Connected devices’ generally refer to devices wired or wirelessly connected to 620
other machines and those connected to the “Internet of Things” (IoT). 621
MD Specific Considerations: computerized medical devices can be vulnerable to 622
security breaches and potentially impacting the safety & effectiveness of the 623
device. This vulnerability escalates as medical devices are increasingly connected 624
to the internet, hospital networks, and to any other medical device. 625
626
There are numerous sources of cybersecurity risk in medical devices and 627
consideration must be given to all in order to ensure adequate protection which 628
include but are not limited to the following: 629
1. Malware infections of devices that are network connected. 630
2. Unsecured or uncontrolled password distribution. 631
3. Failure to provide adequate software security updates. 632
4. Theft or loss of networked medical device. 633
5. Unauthorized access to hospital network or devices. 634
6. Off-the-shelf software that is not designed to prevent unauthorized access. 635
7. Denial-of-service (DoS) attack renders a device inaccessible to the 636
authorized user. 637
8. Improper disposal of patient data or information. 638
9. Untested or defective software and firmware. 639
10. Misconfigured networks or poor security practices. 640
11. Saudi FDA cyber Security for Medical Devices requirements 641
642
Healthcare facilities need to take special precautions in order to safe-guard their 643
network systems to attempt to reduce/eliminate potential attacks. 644
Ongoing Healthcare Facility Processes Required Regarding Cybersecurity: 645 646
The following may be used to help mitigate cybersecurity threats: 647
648
- Improve cybersecurity awareness. 649

29
- Evaluate shared networks. 650
- Build security requirements into contracts. 651
- Follow cybersecurity hygiene regimes during medical device 652
deployment. 653
- Encourage collaboration and harmonize with network participants. 654
- Address healthcare facility IT process inconsistencies. 655
656
2.1.9.2 Education of HTM Professionals in IT-Related Skills 657 658
All HTM professionals (e.g. Clinical Engineers, BMETs) will need to obtain basic 659
and more advanced training in IT-related topics. HTM professionals are often the 660
only staff that understand a complex network-connected medical device system 661
“end-to-end”. Training will have to occur at all HTM levels. Expertise will need 662
to be developed in all areas of healthcare IT including networks, applications, 663
operating systems, tools and troubleshooting. And most importantly, the HTM 664
professional will have to learn to work closely with their peers in IT, who will 665
continue to be the experts on the infrastructure (e.g. networks and data center), 666
healthcare provider-wide IT services (e.g. LDAP, DHCP) and healthcare provider 667
-wide applications such as the EHR. 668
669
670
671
672
673
674
675
676
677
678
679
680
681
682
683
684
685
686
687
688
689
690
691

30
692
693
694
695
696
697
698
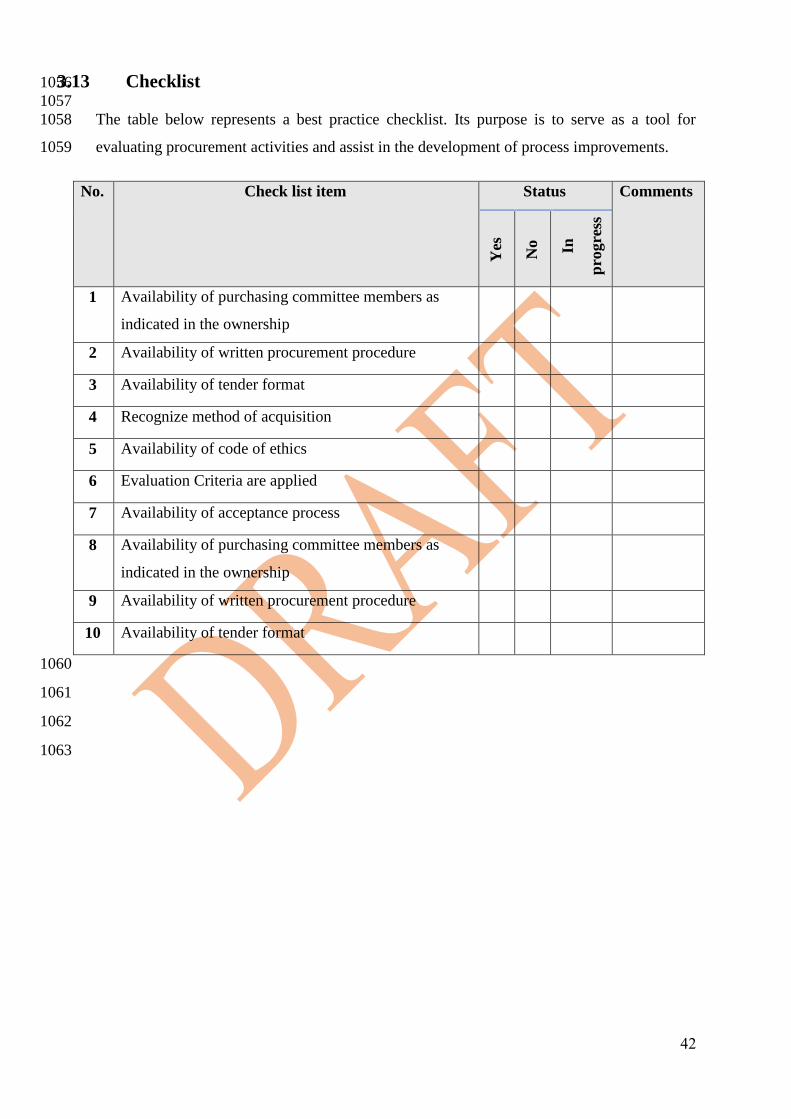
2.3 Checklist 699
700
701
No. Check list item Status Comments
Yes
No
In
pro
gre
ss
1 Availability of the needs assessment process incorporating
a Health Technology Assessment review
2 Involvement of HTM and biomedical staff in the needs
assessment
3 Availability of a medical device planning written
procedure.
4
Availability of Group/ Committee for Medical
Devices/Health Technology Planning with involvement of
all related parties in the ownership.

31
702
Chapter 3: Procurement 703
704
3.1 Introduction 705
706
3.1.1 Objective 707 708
To get satisfactory quality, service and price within a timely delivery schedule (right 709
product or service of the right quality, right price and the right quantity, at the right place 710
and time).5 711
To have effective procurement practice that leads to safe and high quality health care. 712
Other potential benefits of good procurement will include the following: 713
- The most economically advantageous terms for the medical devices acquired – not 714
necessarily the lowest price obtained through tender, but the best deal for the 715
organization’s needs. 716
- Timely delivery. 717
- Satisfactory and well-defined terms of delivery, installation, commissioning, 718
training, payment, warranty and after-sales service. 719
- A greater interest from the suppliers in submitting offers in the future. 720
To acquire a product that is safe, improves quality of care, is cost effective within the 721
context of its application and is manageable (both support and training requirements) 722
within the institution concerned. 723
A product acquisition that fits within the strategic plan and the needs of the organization. 724
A procurement process that follows national regulatory requirements with standardized 725
structure that is transparent, auditable, effective and fair. 726
727
3.1.2 Scope 728 729
This process applies to the healthcare providers who will procure the medical devices and 730
should include the following: 731
Sourcing and solicitation of offers. 732
Evaluation of offers. 733
Review and award of contracts. 734
735

32
Contracting and all phases of contract administration until delivery of the products. 736
Requesting from suppliers that they provide accurate and transparent information in 737
terms of product support and their understanding of a healthcare facility’s needs. 738
739
3.1.3 Ownership 740
741
The mechanism for procuring medical devices should be handled by committee with 742
minimum members of: 743
Purchasing. 744
Legal. 745
Finance. 746
HTM department. 747
End-users. 748
IT. 749
750
3.1.4 Background 751
752
In order to provide safe, effective and high quality patient care, healthcare facilities need to 753
have well-defined medical device acquisition and management programs. Not all devices 754
are created equal, and not all devices are suitable for all healthcare environments. Therefore, 755
it is essential that any new acquisition be thoroughly evaluated to ensure that it is an 756
appropriate choice for a given facility and application. This includes soliciting feedback 757
from a variety of personnel, such as end users and clinical engineers. Multiple devices 758
should be reviewed and compared on predetermined key features listed in technical 759
specifications, which are created by "clinical, technical, and end-user groups" to ensure that 760
the needs of these groups are met (Willson et al.)4 761
The adoption of a value analysis based approach to procurement is also relevant and 762
addresses the need to consider the benefits to quality of care and service delivery using a 763
multidisciplinary, evidence based systematic approach. 764
765
766
767

33
3.2 Procurement Procedures 768
769
770
3.3.1 Pre-procurement Procedures 771
772
Before reaching the point of procurement, as stated in earlier chapters of this document, it 773
is important to use the planning information described in the previous chapter. 774
After that, procurement can move forward and enable a process which includes: 775
- Sourcing and solicitation of offers. 776
- Evaluation of offers. 777
- Review and award of contracts, 778
- Requesting from suppliers to provide accuracy and transparency in terms of product 779
support and understanding of a healthcare facility’s needs. 780
- Contracting and all phases of contract administration until delivery of the product. 781
Ethical and transparency principles need to be adopted in the process, all stakeholders must 782
avoid any perceived conflict of interest. Involved parties must declare any conflict of 783
interest and avoid participating in decisions related to favoring one party over the other. 784
785
Figure 3.1: Purchasing Process. 786
787
788
Tender Requirements: 789 790
Technical & Medical Evaluation Committee
HTM Department
Medical Department
Purchasing Department
Condition of Site/ Works
Planning Phase
Market Testing Pre Supplier
Survey
Supplier List
Invitation to Tender(ITT)
Tender Analyses
Supplier Award
Installation & Commissioning

34
Prepare the tender to obtain the following: 791
- Technical specifications from HTM department. 792
- Clinical application/specifications from the end users department. 793
- Pre-installation requirements from engineering (e.g. architects, physical plant). 794
- Governmental/Legal requirements and tender submission procedure from 795
logistics/procurement. 796
- Regulatory requirements (License for the distributor, Market authorization for 797
the device) 798
- Transparency and conflict of interest policy associated with the tender 799
- Tender evaluation process and important dates. 800
- Process of obtaining further information related to the tender. 801
- Pre-installation requirements request. 802
- Choose your key performance indicators that are going to be used for approving 803
payments (warranty, spare parts, time to respond to service request, required up 804
time, maximum allowed down time, delivery time, installation time, spare parts 805
requirements, length of warranty, training of user, training of biomed in 806
hospital, accessories, availability of an engineer on sight, service and operation 807
manual, preventive maintenance requirements, Electrical safety tests required, 808
Calibration tests requirements …etc) 809
- Any other requirements associated with the medical device (e.g. radiation 810
safety, chemical, medical gases, central supply and sterilization/cleaning 811
requirements). 812
3.3.2 Technical Specification Development of Tender Document, General 813
Considerations: 814
815
- Clearly identify the previously identified clinical needs for the device. 816
- Suppliers need to understand whether you need a product with simple, moderate 817
of advanced options. 818
- Establishing unbiased technical specifications for the technology requested. 819
- It is vital that a Clinical Engineer is involved in the development of the technical 820
specifications to be included in any tender for the medical device under 821
consideration. 822
- The tender evaluation will depend on the specifications, requirements and 823
performance levels, therefore, be very specific and clearly identify your needs. 824

35
- Avoid adding specifications that would exclude certain suppliers over the other 825
unless there is a clearly identified clinical need. 826
- Request device upgrade and update policy 827
- Technology evolving is fast and quick, therefore, healthcare facilities need to have a 828
mechanism to constantly develop and update technical specifications for medical 829
devices in all modalities (Radiology, different laboratory departments, Intensive care, 830
operating rooms, oncology ..etc). Such committees need to include biomedical 831
engineers, clinicians, and technologists. They need to remain up to date with the 832
developments, recalls issued, and assesments done in their areas. 833
834
3.3 Operation Costs Consideration 835
836
The full costs of ownership of the technology includes: 837
- Training (both initial and ongoing) for the operator, clinician and Biomedical engineers. 838
- Software updates. 839
- Maintenance and installation costs, which are effectively identified prior to execution 840
of any agreement. 841
- Running costs (ex: Daily expenses to operate the equipment. This might include the 842
reagent cost, detergent cost, cession cost, etc.) 843
- Preventive maintenance cost if some parts need regular repalcements 844
- Extended warranty cost 845
- Agreement that if at a later date it is necessary to move the device to another location, 846
cost of supplementary reinstallation. 847
- Any medical device that is ‘donated’ or ‘gifted’ to a healthcare facility also requires a 848
thorough life cycle costing analysis prior to acceptance even though there may be no 849
initial capital outlay for the device itself. 850
851
3.4 Methods of Acquisition2 852
853
The suppliers of medical devices to hospitals and healthcare organizations in the Kingdom of 854
Saudi Arabia is for the most part provided by local vendor representatives of the manufacturers. 855
The process of acquiring/selling medical devices follows the International standards under 856
three (3) known methods: 857
36
1) The direct purchase/sale form is used for the medical devices which are owned by the 858
hospitals in general. 859
2) Leased medical devices are owned by the vendors and installed in hospitals according 860
to agreements for the purchase of medical consumables or laboratory reagents as per 861
the consumption by the leased medical devices used/consumed for a certain period of 862
time. 863
3) The third type of acquiring medical devices is through agreements known as 864
“Guaranteed Price Per Reportable Results (GPPRR). In this kind of agreement the 865
device remains the property of the vendor and medical/laboratory consumables are 866
provided by the supplier free of charge while the hospital pays only for reportable test 867
results in compliance with the terms of the agreement. 868
869
3.5 Tender Contents 870
871
A generic template of tender should be designed and subsequently customized to incorporate 872
the specific technology/medical device core requirements. Typical tender items of a generic 873
nature include: 874
Distributor Qualification requirements (licenses, bank guarantees, previous 875
experience, SFDA establishment license, Market authorization for devices …etc) 876
General condition of the contract (sometimes referred to as Terms & Conditions). 877
Contract specification (sometimes referred to as scope of work) which will include the 878
technical specifications of the technology required. 879
Pricing schedule (this enables potential suppliers to present their pricing within a 880
standard layout enabling comparisons to be made during the evaluation process). 881
Proposal evaluation criteria and matrix 882
Tender processing time lines, and 883
Process for seeking further information (all suppliers must have access to the same 884
level of information so that no one has more advantage over the other) 885
Site visiting arrangements process (suppliers need to visit the site, ask clarification 886
questions about the clinical and technical needs, ,etc). 887
Administrative instructions (this will cover matters such as return of tender documents, 888
delivery, application procedure, closure date, mechanism of asking questions …etc.). 889
Announce the tender shall include the following: 890 891

37
- List of medical devices with quantities and specifications, 892
- Performance characteristics like accuracy… etc. 893
- Ability to upgrade medical devices if needed. 894
- Interoperability requirements 895
- Safety requirements. 896
- Regulatory requirements (SFDA establishment license (MDEL) No. for the 897
establishment, SFDA Medical Devices Market Authorizations (MDMA) No. of 898
requested devices...etc.) 899
- Warranty period required. 900
- Extended warranty 901
- Training needs. 902
- Maintenance costs. 903
- Known risks associated with the device(s). 904
- Supplier evaluation criteria. 905
- Pre-purchase questioner. 906
- Compatibility of the operating/environmental conditions of the place where the 907
device will be used. 908
- Manuals (user, operation, instructions for use (IFU), service, spare part list, lists 909
of tool and test equipment required, circuit diagram, planned preventive 910
maintenance manual and checklist as per manufacturer’s requirements). 911
- Decontamination, cleaning, disinfection and sterilization procedures, ensuring 912
the healthcare facility is able to reprocess in line with the manufacturer’s 913
instructions (e.g. by trained technicians). The infection control & CSSD team 914
and should be consulted. 915
- Disposal instructions. 916
917
3.6 Ethical Considerations 918
919
An integral component of any procurement process is a mandatory need to ensure that all 920
ethical considerations are observed. These are as follows: 921
922
• Transparency, confidentiality and fairness 923
• All suppliers should be treated fairly and evenhandedly at all stages of the 924
procurement process. 925

38
• Avoid any perceived conflict of interest between members involved in the 926
tender evaluation and the suppliers. Any such conflict of interest must be 927
declared. (Example: supplier is a relative of one of the decision makers in 928
the tender, one of the tender evaluation committee members have previously 929
benefited from the supplier or may benefit from the current tender, supplier 930
is funding a project running by one of the committee members …etc) 931
• Use of power 932
Buyers should discourage the arbitrary or unfair use of purchasing power or influence. 933
• Corruption 934
Buyers must not tolerate corruption in any form. 935
• Declaring an interest 936
All personal interests should be declared. 937
• Business gifts and hospitality 938
No prospective supplier business gifts or hospitality should be tolerated in the 939
procurement cycle. 940
• Payment terms 941
Payment terms should be objective with clear milestones. Late payments undermine an 942
organization’s credibility. 943
• Supplier relationships and competition 944
Relationships with suppliers, regardless of duration, should be managed professionally. 945
• Encouraging small businesses 946
Buyers should, wherever possible, be aware of opportunities to support the local 947
community and SMEs, whilst maximizing opportunities for global sourcing. 948
• Employment relationship 949
Employees should have legal contracts. 950
• Wages and working hours 951
Living wages are paid. 952
• Treatment of employees 953
No harsh or inhumane treatment is allowed. 954
• Health and safety 955
Working conditions are safe and hygienic. 956
• Discrimination 957
No discrimination is practiced. 958
959

39
3.7 Evaluation Criteria 960
961
Apply the predetermined evaluation criteria for all suppliers after receiving and evaluating the 962
bids by comparing the suppliers' proposals based on the below criteria (supplier competency) 963
per the following: 964
• Service support (Availability of hotline and quick response time, quick delivery of spare 965
parts). 966
• Time to deliver. 967
• Adhering to the regulatory requirements (establishment license, no issued alerts/recalls, 968
MDMA as applicable). 969
• Cost. 970
• Installation cost included or not? 971
• Freight included or not? 972
• Risk of device: 973
- Patient safety – is there a risk of patient harm with this technology? 974
- Risk and clinical effectiveness – will patient outcome be impacted negatively? 975
- Patient experience – will this be better or worse with the provision of this technology? 976
- Risk to health and safety. 977
• Accessories need. 978
• Maintenance costs. 979
• Warranty period. 980
• Cost of consumables. 981
• Lifetime cost. 982
• Meets specification requirements 983
• Meets standards 984
• Review of published hazard and recall data to identify any ongoing technology issues. 985
• Examine the evidence base (documentation, clinical trials data, etc.) for any manufacturer 986
claims cited in the submitted documentation. 987
• Consideration of the use of market trend and price benchmarking systems to identify 988
significant pricing trends and discrepancies. 989
• Supplier Capability: 990
- Staffing structure. 991
- Availability of experienced staff. 992
- Have a system to measure customer satisfaction. 993
- Experience in the industry. 994
40
- Warranty information. 995
- After sale service. 996
• Suppliers history of performance: 997
- Experience in the industry 998
- Previous experience. 999
- References and customers’ recommendations. 1000
• Clinical users evaluation. Evaluate the usability, safety and risk management. Evaluate 1001
that precautions are incorporated into the design to guard against misuse. 1002
• End user license agreements for software products. 1003
• Cybersecurity information (e.g. ability to run anti-virus software). 1004
1005
3.8 Compliance Checking 1006
1007
• Prioritize the above evaluation criteria (i.e. is this requirement specified a ‘high’, 1008
‘medium’ or ‘low’ priority) 1009
• Assign weight for all criteria 1010
• Compare and rank the suppliers responses 1011
1012
3.9 Contract with Supplier. 1013
1014
3.10 Payments Scheduling. 1015
1016
3.11 Storage Requirements: 1017
1018
See tables (9-6) and (9-7) in the appendixes for more details. 1019
1020
3.12 Tender Award Process and Payment Processing 1021
1022
A tender award is often initially provided through a letter confirming supplier status followed 1023
by the issuing of a purchase order document from the institution. This may also be 1024
supplemented for large value items with a contract document. 1025
1026

41
The following shall be considered before tender awarding or payment processing: 1027
• Any contract documentation generated should include the information and terms and 1028
conditions originally specified in the invitation to tender documentation. 1029
• Any advanced payment for a selected technology should be resisted and payment should 1030
be specifically tied to successful acceptance testing, completion of delivery, installation 1031
and all training aspects for the product. On large projects, partial payments based on 1032
objective milestone completions, may be appropriate. 1033
• Any contracts generated should be duplicated for the “Buyer” and “Supplier” and 1034
forwarded to the Supplier for signature in the first instance. Once signed off by both 1035
parties the contract will be in force. 1036
• Unsuccessful suppliers who participated in the tendering process should be entitled to a 1037
debrief including identification of the areas of weakness established during the evaluation 1038
process. 1039
• Any meetings should be held and coordinated by the Procurement Department supported 1040
by Clinical/ biomedical Engineering Department in conjunction with the End-user 1041
Medical Department also involved. 1042
• Any comparisons with other tender responses received should be avoided, as tenders will 1043
have been submitted on a confidential basis. 1044
• Any such meetings should be documented. 1045
1046
1047
1048
1049
1050
1051
1052
1053
1054
1055
42
3.13 Checklist 1056
1057
The table below represents a best practice checklist. Its purpose is to serve as a tool for 1058
evaluating procurement activities and assist in the development of process improvements. 1059
No. Check list item Status Comments
Yes
No
In
pro
gre
ss
1 Availability of purchasing committee members as
indicated in the ownership
2 Availability of written procurement procedure
3 Availability of tender format
4 Recognize method of acquisition
5 Availability of code of ethics
6 Evaluation Criteria are applied
7 Availability of acceptance process
8 Availability of purchasing committee members as
indicated in the ownership
9 Availability of written procurement procedure
10 Availability of tender format
1060
1061
1062
1063

43
1064
1065
Chapter 4: Installation & Commissioning 1066
1067
4.1 Introduction 1068
1069
4.1.1 Objective 1070 1071
• Ensure the medical device supplied matches the specification of the device indicated in 1072
the Supplier’s invitation to tender response. 1073
• Ensure the medical device is in good condition and fully functional after commissioning. 1074
1075
4.1.2 Scope 1076
1077
This process applies to the healthcare provider who will install and commission the medical 1078
devices that will include the following: 1079
1080
• Checking the supplied medical device with related manuals, catalogues, accessories & 1081
official documentation. 1082
• Ensure the medical device is in good condition and fully functional. 1083
• Checking the Calibration, performance and electrical safety tests. 1084
• Getting the related training (both user and technical). 1085
1086
4.1.3 Ownership 1087 1088
The mechanism for installation of medical devices should be handled by committee with 1089
minimum members of: 1090
• Warehouse Department. 1091
• HTM Department. 1092
• End-users Department. 1093
• Assets management. 1094
• Purchase Department. 1095
• Project management department. 1096
1097

44
1098
4.1.4 Background 1099
1100
Pre-installation Requirements, Receipt and Acceptance 1101 1102
- Following completion of the tender process, the Supplier will be responsible for 1103
the delivery of the medical device. 1104
- Prior to delivery and installation, any site preparation work shall be completed. 1105
1106
Note: Pre-installation checks should apply to all medical devices installed into a healthcare 1107
setting, whether they are leased, donated, on trial or purchased. 1108
1109
PurchasingPhysical
Check
Components
Check
Accessories
Check
Disinfection
Requirements
Availability Check
Transport&
Storage
Condition Check
Accompanied
Documents
Check
InstallationConfiguration
Check
Function &
Safety Check
Data Capturing
in Inventory
CMMS
MD label with
Control
Number
Maintenance
Schedule in
CMMS
User and
Technical
Training
Signed
Acceptance
Report
Planning
1110
Figure 4.2: Installation & Commissioning processes. 1111 1112
1113
1114
4.2 Delivery 1115
1116
The healthcare provider shall check for external damage to the package or its 1117
contents. (physical checks). 1118
The medical device delivered matches the original tender acquisition specification. 1119
The healthcare facility should ensure the medical device supplied matches the 1120
specification of the device indicated in the Supplier’s invitation to tender response. 1121
The medical device has been supplied with the appropriate original medical device 1122
manufacturer recommended accessories and consumables. 1123

45
All necessary information and documentation, including instructions for use have 1124
been provided. 1125
Ensure that manufacturers transport and storage requirements are met. Many chemical 1126
reagents lose their effectiveness because they were not transported or stored properly. 1127
4.3 Medical Device Installation & Commissioning 1128
1129
1130
• The device is configured in an appropriate manner. 1131
• Installed according to the original medical devices manufacturer's recommendations and 1132
located where required with oversight of this process by the Clinical Engineering 1133
department. 1134
• Delivery has been achieved with the technology/medical device in good condition and 1135
fully functional. 1136
• Manufacturer’s and regulatory authorities’ Health & Safety requirements during the 1137
installation process are considered and met. 1138
• The availability of disinfection and sterilization contract or competent personnel tools 1139
and spaces. 1140
• Data has been captured and stored on the organizations Computerized Maintenance 1141
Management System (CMMS) for asset tracking purposes. 1142
• Formal training needs according to the manufacturer requirements have been identified 1143
and implemented and data captured for inclusion in training databases for ongoing needs. 1144
• In the case of reusable devices, maintenance requirements have been identified and 1145
scheduled. 1146
• Information about proper storing and transporting. 1147
• Involvement of HTM and End-user departments during the installation and acceptance 1148
process. The entire process should be coordinated by clinical/ biomedical department 1149
which should liaise with other groups as required. 1150
• Report signed from the responsible clinical/biomedical engineer and end user explaining 1151
that the medical device is installed, in good condition and fully functional. 1152
• Confirmation that all documentation has been delivered including local configurations, 1153
as-built drawings, and IT information for connected devices. 1154
• A control number will be assigned and labelled, placed on the unit, along with any other 1155
appropriate labels (e.g. warrant expiration date, loaned device.) 1156
• In case the delivered medical device failed to comply with healthcare facility’s identified 1157
requirements in the tender, the Procurement Department shall be responsible for liaising 1158

46
with the medical device supplier in the event of damaged or inappropriately delivered or 1159
unrepairable medical device. 1160
1161
Note: Medical devices must not be put into use before acceptance check processes have 1162
been performed. 1163
1164
1165
1166
1167
1168
1169
1170
1171
1172
1173
1174
1175
1176
1177
1178
1179
1180
1181
1182
1183
1184
1185
1186
1187
1188
1189
1190
1191
1192
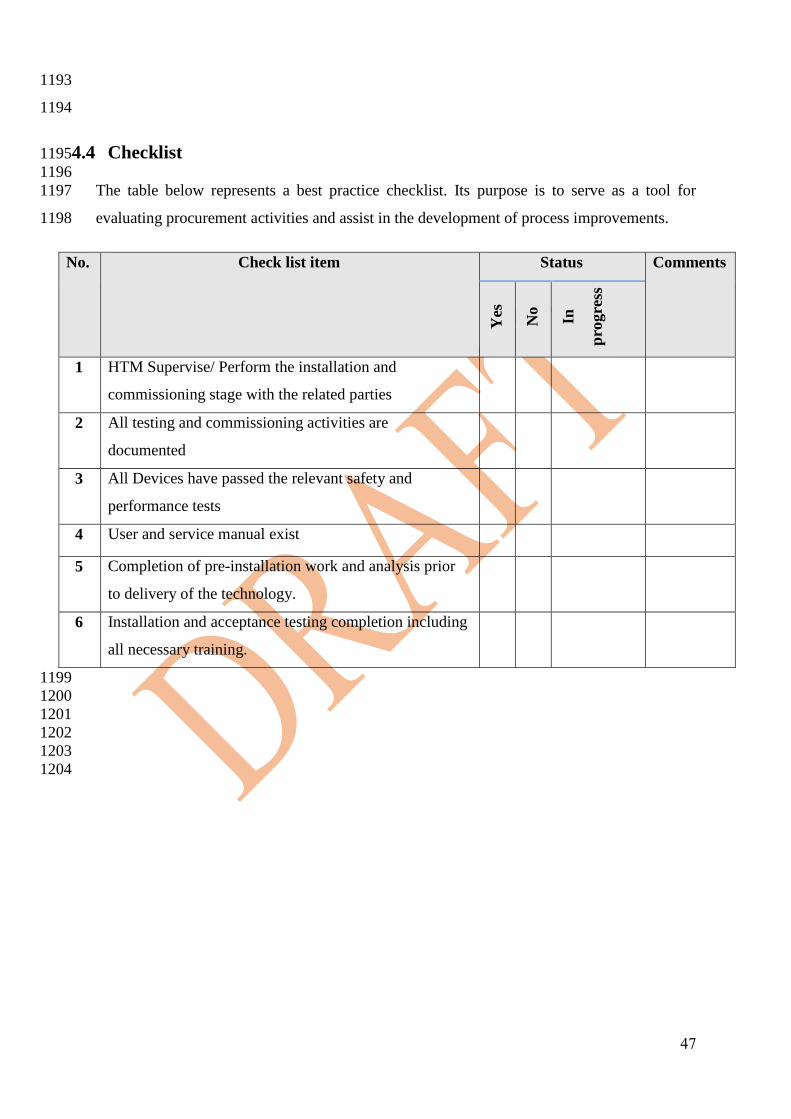
47
1193
1194
4.4 Checklist 1195
1196
The table below represents a best practice checklist. Its purpose is to serve as a tool for 1197
evaluating procurement activities and assist in the development of process improvements. 1198
No. Check list item Status Comments
Yes
No
In
pro
gre
ss
1 HTM Supervise/ Perform the installation and
commissioning stage with the related parties
2 All testing and commissioning activities are
documented
3 All Devices have passed the relevant safety and
performance tests
4 User and service manual exist
5 Completion of pre-installation work and analysis prior
to delivery of the technology.
6 Installation and acceptance testing completion including
all necessary training.
1199
1200
1201
1202
1203
1204

48
1205
Chapter 5: Operations 1206
1207
5.1 Introduction 1208
1209
5.1.1 Objectives: 1210
1211
• Effective maintenance plans of the medical devices in order to increase the lifetime and 1212
decrease the breakdown of medical devices. 1213
• Documentation of medical device history. 1214
• Maintaining the safety and effectiveness of the medical devices. 1215
• Ensuring that the medical device’s users are trained and have the required 1216
skills/knowledge to operate the devices. 1217
• Effective reprocessing of medical devices. 1218
1219
5.1.2 Scope: 1220
1221
This chapter covers all operational activities of medical devices as follows: 1222
• Tracking the medical devices. 1223
• Maintenance of medical devices (i.e. CM, PPM). 1224
• Management of spare parts. 1225
• Calibration. 1226
• Safety inspection. 1227
• Reprocessing. 1228
• Transfer of medical devices. 1229
• Managing the field service notifications (FSNs), recalls and alerts and reporting any 1230
adverse events. 1231
5.1.3 Ownership: 1232
1233
• HTM/Clinical Engineering Department. 1234
• End-user Medical Departments. 1235
• Quality Department. 1236
• CSSD. 1237

49
5.1.4 Background 1238
1239
After the medical devices acceptance phase has been satisfactorily completed, the device can 1240
be used safely and the device is now in the operations phase. 1241
1242 Figure 1 .1: Operations processes. 1243
1244

50
1245
5.2 Operation Activities 1246
1247
The healthcare facility shall do the following: 1248
5.2.1 Create Device History Record 1249 1250
An asset management software (e.g. CMMS) shall be used to manage the medical devices. The 1251
software shall include –but not be limited to- the following information: 1252
1253
Device General Information 1254 1255
Includes: device category, device type (according to GMDN, description, model 1256
name & number, manufacturer, supplier, serial number and asset number, SFDA 1257
MDMA number, warranty period, purchasing costs, device location, assigned 1258
HTM/Clinical Engineering employee and service contract information if applicable, 1259
acceptance date, type of procurement (this could be: lease, reagent rental, deferred 1260
payment, loan, etc.), life expectancy of the medical device and risk class of device, 1261
accessories requirements if any. 1262
1263
Periodic Preventive Maintenance (PPM) 1264 1265
Includes: Frequency of periodic preventive maintenance (PPM), procedure of PPM, 1266
calibration requirements and spare parts used for PPM. 1267
1268
Test equipment required 1269 1270
The characteristics of all measuring or energy delivering devices even the best 1271
designed ones change with time. Meaning if the device is showing a reading of (10), 1272
as the device is used more frequently, the deviation from the actual value increases. 1273
Therefore, special test devices need to be used regularly by a trained biomedical 1274
engineer/technician to verify the level of deviation in the operation of the devices. All 1275
devices having a deviation higher than the acceptable range identified by the 1276
manufacturer must be removed from service and the supplier must re-calibrate them. 1277
Some of the test devices include (patient simulator, infusion pump tester, defibrillator 1278
tester, Xray KV mater, artificial lung, laser power meter, and more. 1279

51
Examples: 1280
o The anesthetic sets up the anesthesia machine for a certain gas and air 1281
percentage, certain gas volume and expects the machine to deliver the 1282
amounts indicated on the machine. The actual delivered parameters by 1283
the machine would change with time, and this requires using an 1284
artificial lung with calibrated sensors to verify that the actual delivered 1285
parameters match the machine settings. If not, the machine should be 1286
removed from service. 1287
o Kilovolt (KV) and milli-ampere-second (mAs) meters need to be 1288
frequently used on Xray machines to ensure that the dose delivered 1289
from an Xray tube matches the machine settings, if not, the machine 1290
needs to be removed from service. 1291
o All devices connected to patients must be checked for leakage current, 1292
insulation resistance, ground resistance and more electrical parameters 1293
to ensure that no excessive currents leading to possible heart failures 1294
enters into the body from that machine. 1295
1296
The supplier needs to identify the processes for: 1297
o Preventive maintenance procedures and frequency 1298
o Calibration verification procedure and frequency 1299
o Test equipment needed for functional tests for the device 1300
o Regular replacement parts needed for the machine 1301
1302
Device History 1303 1304
Includes: 1305
- PPM history: date of each PPM, PPM kits & parts used, who completed the 1306
work, and time spent performing PPM. 1307
- Corrective Maintenance (CM) history: date of failure, failure description, spare 1308
parts used, time duration of maintenance. 1309
- related FSNs/recalls 1310
- The history record of a new device shall be generated when a new device is 1311
ready for clinical operation (installed, tested, and tagged). 1312
1313

52
5.2.2 Plan for PPMs, Spare Parts Inventory and Electrical Safety Tests 1314
1315
Periodic Preventive Maintenances (PPMs) and safety tests are performed to maintain the basic 1316
safety and essential performance of medical devices according to the manufacturer 1317
requirements. In addition, performing PPM will reduce the chance of breakdown of the device 1318
which will increase the device’s utilization and reduce repair cost during the device’s life cycle. 1319
When performing PPM, the following points should be considered: 1320
1321
• The PPM frequency and procedures shall be in line with the manufacturer requirements. 1322
• The PPM frequency is usually indicated by time periods (interval-based), or as per 1323
device’s operation hours (meter-based), or both. 1324
• Only trained personnel shall perform the PPM. 1325
• For some devices, the PPM procedure requires replacing some parts or PPM kits. In this 1326
case, an adequate number of PPM kits shall be kept in stock, and periodically re-stocked, 1327
depending on the number of devices used in the facility and frequency of PPMs. 1328
1329
Note: If the PPM procedure includes using test equipment for performance or calibration 1330
checks, the test equipment shall be tested and calibrated by the manufacturer or a certified 1331
body. 1332
1333
• Before performing PPM, the device shall be clean, decontaminated (if needed), and 1334
working in a good condition. 1335
• After performing PPM, a device’s performance and function tests shall be carried out. 1336
• In all cases, this should include a safety test and safety inspection as part of the PPM. 1337
• The PPM details (date, PPM kits, etc.) shall be recorded in the device’s history in the 1338
CMMS. Data shall be filed for a minimum of five years. 1339
• All PPMs must be signed off by the end-user and clinical engineer/contractor performing 1340
the service. In addition, the device shall be tagged after PPM. The tag shall contain at 1341
least the date of last performed PPM, the due date for the next PPM, and responsible the 1342
engineer or technician. 1343
5.3 Corrective Maintenance Management 1344
1345
1346
If a device has a malfunction, the user shall do the following: 1347
• Inform the HTM/Clinical Engineering department immediately. 1348

53
1349
A work order shall be generated and contain –at least- the following information: 1350
- Device’s description and asset number. 1351
- Location of the device. 1352
- Description of the problem. 1353
- Requestor’s contact details. 1354
1355
• The work order should be assigned to the responsible clinical engineer/technician who 1356
shall have proper training, qualifications, and tools to repair the device. 1357
• After repairing the device, the assigned clinical engineer/technician shall update the work 1358
order by entering details of the used spare parts, time consumed in repairing the device, 1359
and corrective action details. Then close the work order. 1360
• All CM order must be signed off by the end-user and Clinical Engineer/Contractor 1361
performing the service and the device is working as intended by the manufacturer. 1362
• On all electrical medical devices, a test/safety Inspection shall be conducted as part of the 1363
CM. 1364
• Function and calibration tests shall be conducted after installation, any major replacement 1365
and modification. 1366
• If applicable, the device shall be calibrated after the corrective maintenance, as per 1367
manufacturer requirements. 1368
1369
Note: If the CM procedure includes using test equipment for performance or calibration 1370
checks, the test equipment shall be tested and calibrated by the manufacturer or a certified 1371
body. 1372
1373
5.4 Transfer and storage procedures 1374
1375
1376
• When transferring a device to be used in another location or department, a transfer form 1377
shall be filled. The form should contain at least the device description and asset number, 1378
present location, new location, date of transfer. 1379
• A copy of the (transfer form) must be sent to the HTM/Clinical Engineering Department 1380
to update the device’s record in order to keep track of the device for the purpose of the 1381
device’s maintenance, utilization, and future needs assessments. 1382

54
• In case the device need to disassembled and reassembled in different location, qualified 1383
engineer according to the manufacturer recommendations shall conduct this task. 1384
In case the device needs to be stored, it must be stored in accordance with the 1385
manufacturer's storage instructions such as the device’s shelf life, storage room 1386
temperature and humidity requirements ( See: tables (9.6) and (9.7) in the appendixes for 1387
more details). 1388
1389
5.5 Reusable Medical Devices Reprocessing 1390
1391
1392
5.5.1 Steps of the Reprocessing 1393 1394
1395
Pretreatment Disinfection Require
Sterilization?Sterilization
Document
Release or ReprocessEND
Function &
Safety CheckYes
Reprocess
Release
No
1396 Figure 5.2: Reprocessing steps. 1397
1398
The reprocessing process includes several steps and sub-processes. These apply depending on 1399
the risk classification of the medical device, which imply the aimed goal whether it is 1400
disinfection or sterilization. The process of reprocessing includes the following: 1401
1402
1. Pretreatment (to eliminate obvious stains): 1403
a) Collecting. 1404
b) Pre-cleaning. 1405
c) Disassemble. 1406
1407
2. Disinfection: 1408

55
Disinfection can be manual or machine accomplished (e.g. washer-disinfectors 1409
machines according to ISO 15883 standards). The process includes the following sub-1410
processes: 1411
a) Cleaning and rinsing: To eliminate the residual blood, secretion or tissue 1412
before disinfection. 1413
b) Disinfection: To eliminate bacteria, fungus and virus 1414
c) Final rinsing and drying 1415
d) Check for cleanliness and integrity 1416
e) Assembly 1417
f) Visual inspection 1418
3. Functionality check (at the end of disinfection). 1419
4. Sterilization: To eliminate prion pathogens: 1420
a) Packaging (per ISO 11607) 1421
b) Sterilization: (using one of the methods listed below based on 1422
manufacturer requirements): 1423
i. Moist heat sterilization (ISO 17665). 1424
ii. Dry heat sterilization (ISO 20857). 1425
iii. Sterilization through radiation (ISO 11137). 1426
iv. Sterilization through ethylene oxide (ISO 11135). 1427
c) Labeling: 1428
i. Device name and model. 1429
ii. Release date. 1430
iii. Devices expiration date. 1431
iv. Sterility expiration date. 1432
v. Time of use limit and the current time of use. 1433
vi. Assign a lot-number to each reprocessed medical device. 1434
5. Documentation and release or reprocess. 1435
6. Change the lot-number after each validation and any update on the process and maintain 1436
records for 5 years. 1437
7. Transport and store (Disinfected medical devices shall be stored under clean-room 1438
conditions and sterile medical devices shall be stored under the recommended 1439
conditions of the manufacturer of the packaging material. 1440
1441
5.5.2 General Consideration for Reprocessing 1442 1443

56
1. All parts and external and internal components of the medical devices shall be exposed 1444
to the effect of each process. 1445
2. Follow all manufacturer recommendations. 1446
3. Check after each step for any residual stains or degradation of the medical devices. 1447
4. Use desalinated water for rinsing. 1448
5. Maintain checklist and clear comprehendible SOPs for each process. 1449
1450
5.5.3 Risk Classification 1451 1452
Considerations for the risk classification and choosing the reprocessing method: 1453
1. Design, material, technical and functional properties of the device. 1454
2. The risk of the to-be-disinfected medical device can be defined based on: 1455
a) The expected adverse effect as a result of: 1456
i. The usage preceding the disinfection process 1457
ii. The preliminary disinfection process that preceded the main 1458
disinfection. 1459
iii. The transport and the stage of the medical devices. 1460
b) The type of usage following the reprocessing. 1461
3. The amount and typical types of pathogens expected to be on the device’s surfaces after 1462
use and their resistance to the proper disinfection process. 1463
1464
5.5.4 Risk Classes 1465
1466
1. Uncritical: 1467
Medical devices that only come into contact with intact skin. 1468
1469
2. Semi-critical: 1470
Medical devices that come into contact with mucous membranes or diseased skin. 1471
1472
a) Medical devices without reprocessing special requirements (A). 1473
b) Medical devices with more stringent reprocessing requirements (B) if: 1474
i. The judgment of the efficacy of reprocessing is not possible by simply 1475
inspection, 1476

57
ii. The fully avoidance of influential factor on the use or functional safety 1477
of the devices, as a result of reprocessing or transporting of the medical 1478
devices, is not guaranteed, or 1479
iii. The number of uses or the reprocessing-cycle are limited as per 1480
manufacturer's instructions. 1481
1482
3. Critical: 1483
Medical devices for the application of blood, blood products or other sterile drug or 1484
sterile medical devices, and medical devices which, as intended, penetrate the skin or 1485
mucous membrane and are used in contact with blood, or internal tissues or organs, 1486
including wounds 1487
1488
a) Medical devices without reprocessing special requirements (A): 1489
i. The medical device is thermostable (can be sterilized by steam 1490
sterilizer under 134 degrees C). 1491
1492
b) Medical devices with higher reprocessing requirements (B):If the device is 1493
thermostable (can be sterilized by steam sterilizer under 134 degrees C) and: 1494
i. The judgment of the efficacy of reprocessing is not possible by simply 1495
inspection, 1496
ii. The fully avoidance of influential factor on the use or functional safety 1497
of the devices, as a result of reprocessing or transporting of the medical 1498
devices, is not guaranteed, 1499
iii. The number of uses or the reprocessing-cycle are limited as per 1500
manufacturer's instruction, or 1501
1502
c) Medical devices with special high reprocessing requirements (C) if: 1503
i. The medical device is thermolabile (not to be sterilized by steam 1504
steam). 1505
1506
1507
1508
1509
1510
1511

58
5.5.5 Requirements for Each Class 1512
1513
Class Processes Requirements
Uncritical Cleaning
Semi Critical A Disinfection
Optional:
Pre-treatment
Sterilization
Manual process.
Quality Management.
SOPs and validation.
Semi Critical B Pretreatment
Disinfection
Optional:
Sterilization
Automated cleaning and disinfection.
Quality Management.
SOPs and validation.
Critical A Disinfection
Sterilization
Optional:
Pre-treatment
Automated cleaning and disinfection.
Moist Heat Sterilization.
Quality Management.
SOPs and validation.
Critical B Pretreatment
Disinfection
Sterilization
Optional:
Labeling
Evidence of accredited training.
Automated cleaning and disinfection.
Moist Heat Sterilization.
Quality Management.
SOPs and validation.
Critical C Pretreatment
Disinfection
Sterilization
Labeling
Proper Sterilization process
Certification of the Quality Management Process
according to ISO 13485.
Risk Management according to ISO 14971
SOPs and validation.
Table 5.1: Sterilization and Disinfection Risk Classes Requirements. 1514
1515
5.6 Computerized Maintenance Management System (CMMS) 1516
1517
A computerized maintenance management system (CMMS) is a software application that 1518
provides the documentation and management tools used by healthcare technology management 1519
(HTM) programs to collect, store, organize, analyze, and report medical device data for 1520
healthcare facility. 1521
Sophisticated CMMSs support medical devices throughout the entire device lifecycle; from 1522
pre-procurement, acquisition, commissioning, device inventories, corrective maintenance (CM 1523
or unplanned repair) and planned maintenance management, field safety notices/ recalls and 1524
adverse events management, to device “end-of-life” management, replacement analysis and 1525
disposal. 1526
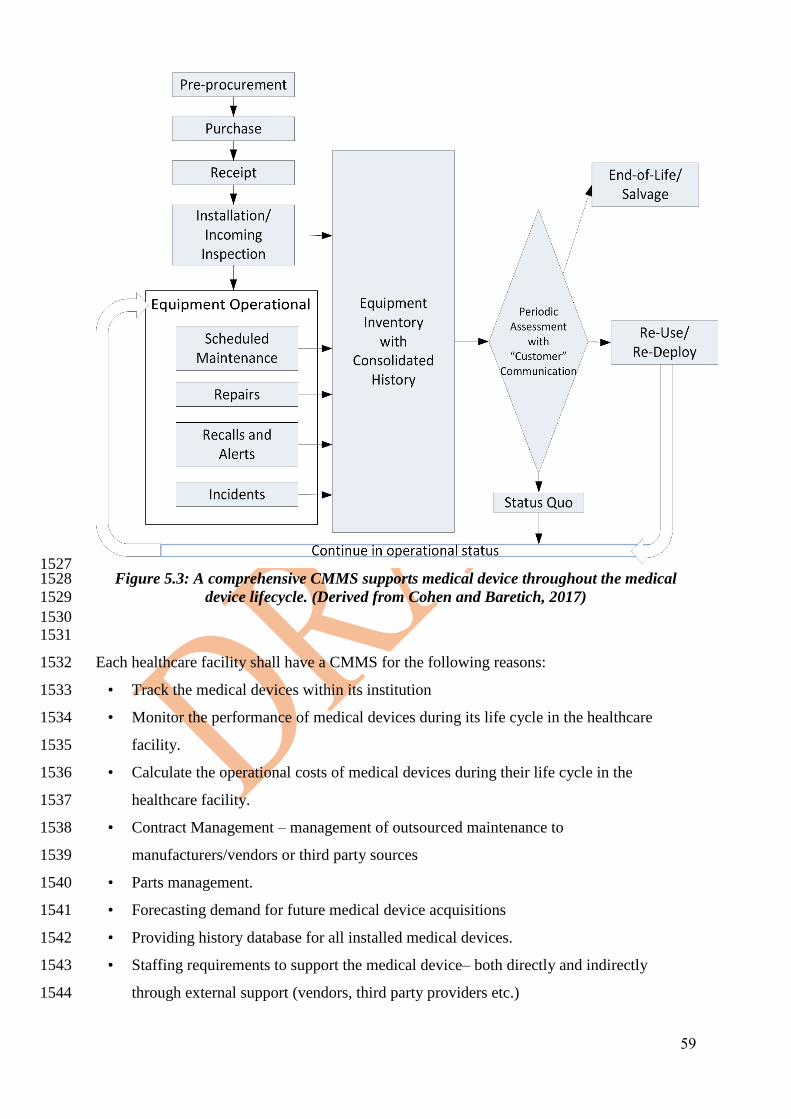
59
1527 Figure 5.3: A comprehensive CMMS supports medical device throughout the medical 1528
device lifecycle. (Derived from Cohen and Baretich, 2017) 1529
1530 1531
Each healthcare facility shall have a CMMS for the following reasons: 1532
• Track the medical devices within its institution 1533
• Monitor the performance of medical devices during its life cycle in the healthcare 1534
facility. 1535
• Calculate the operational costs of medical devices during their life cycle in the 1536
healthcare facility. 1537
• Contract Management – management of outsourced maintenance to 1538
manufacturers/vendors or third party sources 1539
• Parts management. 1540
• Forecasting demand for future medical device acquisitions 1541
• Providing history database for all installed medical devices. 1542
• Staffing requirements to support the medical device– both directly and indirectly 1543
through external support (vendors, third party providers etc.) 1544

60
• Present the responsible engineer for each devices and trained end-users. 1545
• Document reprocessing process. 1546
1547
5.7 Data integrity 1548
1549
Without accurate and complete data, a CMMS is not going to be of much use to the HTM 1550
department. All HTM staff is responsible for assuring that the data they enter and update are 1551
complete and accurate. Incomplete and inaccurate data make it difficult to report useful 1552
information, which can lead to poor decision-making. 1553
1554
Good data quality depends on several factors, including the following: 1555
• Availability of HTM-relevant data sources: Source data needs to be available to the 1556
HTM staff. For example, medical device acquisition cost data, which should be included 1557
on every asset record, require a purchase order or invoice information in order to 1558
accurately capture the device’s price. 1559
• HTM employee accountability: Documentation quality begins at the source, the 1560
employees. CMMS training and documentation expectations and guidelines should be 1561
provided to every HTM employee. Assessment of documentation quality should be 1562
included in employee performance appraisals. 1563
• CMMS data standardization: Operation of a high-data-quality CMMS depends on 1564
multiple levels of data field standards. These include fields defined by the CMMS 1565
supplier, fields defined within the healthcare facility, and fields that use nationally or 1566
internationally published “standardized” definitions. Policies should indicate which data 1567
definitions HTM staff should use. For example, fields that indicate which department 1568
“owns” a medical device can use the organization’s own chart of accounts. Device type 1569
fields can use the Global Medical Device Nomenclature (Global Medical Device 1570
Nomenclature, 2017) or Universal Medical Device Nomenclature System (UMDNS, 1571
2017). HTM supervisory personnel or the CMMS database administrator should be 1572
responsible for managing (adding, deleting, and updating) these and other foundation data 1573
tables (e.g. department, building, medical device type, manufacturer and vendor lists). 1574
• Referential integrity: For the foundational data, both the underlying database structure 1575
and the CMMS application error-checking capability, are important to ensure high data 1576
quality. The database should include appropriate referential integrity in order to make 1577
sure that related fields do not have any “orphan” data. For example, every medical device 1578

61
record should have a manufacturer's reference, and every manufacturer reference should 1579
have a manufacturer name, address, and so on. 1580
• Field definitions: Each CMMS field should have a written definition so every HTM staff 1581
member uses the field the same way. Without field definitions, individual staff may 1582
interpret a single field in different ways. For example, for a network-connected medical 1583
device, what is the definition of the “Port” field on the IT data section of the asset record 1584
in (Figure 5.4)? Is it the physical data port where the Ethernet cable plugs into the wall 1585
or the virtual port used for HL-7 data communication with the electronic medical record?. 1586
Both “ports” are important data, but they serve different purposes. 1587
1588
1589 Figure 5.4: all fields need to be defined. For example, is the “port” field a data port in the 1590
wall or a logical port for HL-7 data or? 1591
1592
• Maintenance activity coding: Where sharing or comparison of maintenance data is 1593
desirable or required, maintenance activities must be consistently and accurately coded. 1594
Dropdown lists and other coding techniques help standardize data, although sometimes 1595
at the expense of detail. Definitions are paramount to make sure “apples to apples” 1596
comparisons can be made. For example, what does the checkbox field “PM-preventable 1597
mean” (Figure 5.5)? Does it only apply to CM workorders, or can it be used to indicate a 1598
change is needed in a PPM procedure? How will the aggregated data from this field be 1599
used? 1600
1601
1602 Figure 5.5: “PM-Preventable” is an example of a field that needs to be defined. When 1603
should technicians check this box? 1604
1605
5.7.1 Error Checking 1606
1607
Certain fields in the CMMS are always required (e.g. Asset Control Number). Other fields may 1608
be required by Healthcare Facility/HTM policy and the CMMS should allow these to be 1609
configurable (e.g. Downtime). 1610
1611

62
The CMMS should provide a robust set of configurable error checks. For example: 1612
• Data types: The CMMS should automatically ensure that each field has the appropriate 1613
data type entered (e.g., string, number, dollar amount, date). 1614
• Range constraints: Most fields have logical ranges, so constraints can be configured to 1615
make sure that data entry is stopped, or a warning issued, if a value is out of the expected 1616
range. For example, a range check for a vendor hourly rate field could be set for a 1617
minimum of $50 USD to a maximum of $999 USD per hour to catch an obviously 1618
erroneous data entry. 1619
• Date checks: Dates (and times) need to have a consistent format throughout the CMMS. 1620
Some dates (e.g., next PPM due date) must be in the future when initially entered. Most 1621
other dates should be current or in the past when entered (e.g., work order completion 1622
date). 1623
• Multiple-field error checking: Multiple-field error checking is more complex and often 1624
requires special CMMS configuration or customization. For example, suppose that 1625
downtime is a required field when an asset is a “critical system,” but optional if it is not. 1626
Multiple-field error checking would be configured so that downtime is always required 1627
on critical system repair work orders. 1628
• HTM business rules: A healthcare provider may have internal requirements that 1629
mandate certain rules in the CMMS. For example, local business rules may authorize 1630
different levels of purchasing authority for parts and services for different levels of HTM 1631
personnel, based on the cost of the order (e.g., a staff BMET is allowed to place parts 1632
orders up to $1,000 USD without additional approval, whereas a BMET supervisor is 1633
allowed to place an order up to $5,000 USD). 1634
In addition to automatic error checking, periodic audits are necessary to maintain data quality. 1635
Some data problems that require data aggregation cannot be immediately caught at data entry. 1636
1637
Example: For a specific BMET, or group of staff, an audit could recognize that an insufficient 1638
number of labor hours have been documented on work orders compared to payroll system-1639
related data (e.g., time card entries). Sampling audits by supervisors can also check for 1640
problems that are difficult to automate, such as verification that work orders are accurate and 1641
documented in sufficient detail. 1642
1643
For a CMMS to be effective it needs to be easy to use. Modern CMMSs allow the use of smart 1644
phones, tablets, and laptops. Pick lists, autocomplete text entry, voice to text, and other modern 1645

63
data entry techniques make them easier to use. Reports can be set up to run at regular intervals 1646
and dashboards can provide near-real-time feedback on ongoing activities (see: section 9.1.3). 1647
1648
5.8 Considerations in selecting a new or replacement CMMS 1649
1650
1651
Suppliers offer a wide variety of CMMS applications at a wide variety of price points. With 1652
such wide availability, it is not recommended that HTM departments try to develop their own 1653
CMMS. Modern CMMSs are complex software applications, that take a lot of time to write 1654
and can be difficult to update and maintain. Most healthcare facilities will already have some 1655
kind of CMMS in place so the rest of this section will focus on CMMS replacement selection 1656
and implementation. If there is no CMMS currently available, and there are no, or very little, 1657
funds available for a CMMS, there are low cost, or even free (with restrictions) CMMSs 1658
available, although they are not typically HTM focused (Hoppe, 2017). 1659
1660
CMMS Software Evaluation and Selection 1661
1662
The evaluation and selection process should be as follows: 1663
1. Assess your current CMMS system for its features that are working well and its 1664
deficiencies. Also, as you assess deficiencies of your current system, take into 1665
consideration that some of those deficiencies might be data related, and poor quality 1666
data are poor quality data regardless of the CMMS features. You don’t want to import 1667
bad data into your new CMMS, so a data clean-up project may also be appropriate. 1668
2. Develop a high-level list of your CMMS needs, including key required and desirable 1669
features, for a new system. 1670
3. Consider the proposed physical location of the new system (cloud-based vs local), 1671
taking into account security considerations, data ownership, hardware, support model, 1672
and cost. 1673
1674
Note: Commercial CMMS suppliers offer vendor-hosted, cloud-based CMMS software that 1675
runs from a web browser and is compatible with a variety of web-enabled devices (e.g. 1676
laptops, tablets and smart phones). 1677
1678
1679
1680

64
4. Invite vendors to provide an on-line demonstration of their software. 1681
1682
Note: To optimize everyone’s time, focus on your requirements list by providing the 1683
suppliers in advance of their demonstrations with a draft high-level list of your requirements 1684
and, if possible, a few scenarios that they can use to show how their products might meet 1685
your requirements. (e.g. demonstrate a COSR report for all imaging systems, show a 1686
dashboard of pending work for today or for this week). From these demonstrations, and the 1687
other information you gather, you can develop a more formal and final requirements 1688
document and pursue your selection process through RFI (Request for Information) and/or 1689
tender/RFP/RFQ (Request for Proposal/Quotation) processes depending on your healthcare 1690
facility’s procurement process requirements. The result should be a written response from 1691
each vendor to each of your requirements and a written pricing proposal. You may also want 1692
to prioritize each specification item (e.g. mandatory, preferred, desirable). 1693
1694
5. Review the supplier’s written responses and see which products meet which 1695
specifications—both “out of the box” and through customization. 1696
1697
Note: Pay particular attention to supplier responses that require customizations: What will 1698
the customizations cost? How long will they take to complete? Contact references and, if 1699
needed, schedule additional demonstrations. 1700
1701
6. Limit the number of suppliers to your top candidates based on pricing and features. 1702
Limit the demonstrations to exactly what you want to see so you can clearly evaluate 1703
their responses. Features can be “scored” by your evaluation team based on how well 1704
they meet your requirements. Pricing comparisons should include both initial pricing 1705
and support and licensing pricing for a period of time (e.g. 5 years, 10 years) so you 1706
can evaluate pricing based on life cycle costs. Then determine which product comes 1707
closest to meeting both feature requirements and budget. 1708
7. Once you decide on a single product, the next step is to negotiate a final price, schedule, 1709
and terms and conditions with the vendor. Get all the vendor promises in writing. Be 1710
clear about the deliverables, responsibilities, expectations, and schedules, particularly 1711
customization and data imports, which sometimes are problematic and can take a long 1712
time. Then start getting ready for the implementation-planning phase. 1713

65
1714
5.9 Reporting adverse events to SFDA 1715
1716
It is a moral responsibility for users, patients and all those using a medical device or accessory 1717
to report adverse events. There are multiple numbers of the same device being used by other 1718
patients in other hospitals and reporting the incident will save others from facing the same 1719
accident (reporting is saving). 1720
Accidents or injuries on patients or users caused by a Medical device or accessory must be 1721
reported to SFDA national center for medical device reporting (NCMDR) to protect patients in 1722
other hospitals from facing the same incident (reporting is saving). The center investigates the 1723
accident and a recall, warning, or field safety notice for the device may result from this 1724
investigation. The released statement will include a corrective actions required to be 1725
implemented on the device to avoid the re-occurrence of the incident in other hospitals. 1726
Corrective actions include but not limited to the following: 1727
• Removing the device from service 1728
• Replacing or adjusting some parts 1729
• Advising nursing on proper method for using the device. 1730
• Regular warning 1731
SFDA requires that manufacturers and distributors track the devices they sold within Saudi. 1732
When a recall is issued, the National center for medical device reporting forces the 1733
manufacturer's representative in Saudi to conduct whatever corrective action needed on all the 1734
similar devices available in Saudi to protect patients and users in other hospitals from facing 1735
the same accident. 1736
It is important to note that recalls on Medical devices and equipment take place within the best 1737
brands, best manufacturers and best devices. Sometimes, a manufacturing problem temporarily 1738
takes place, so all batches of devices manufactured during that design problem need to be 1739
removed from service or fixed. Manufacturers must report such problems to SFDA, in addition, 1740
design problems, false claims, wring instructions for use, faults that have caused or may cause 1741
harm to patients or users must be reported to SFDA so that an investigation and root cause 1742
analysis is done. If the investigation results in an action against the device, SFDA will request 1743
the list of customers who bought the device from the manufacturer and follow up with 1744
manufacture or it's representative to perform the desired action to avoid causing harm to other 1745
patients. 1746
1747

66
5.9.1 How to report an adverse event? 1748
1749
Report adverse events by calling 19999 or by visiting the NCMDR web portal: 1750
(https://ncmdr.sfda.gov.sa/), where the adverse event can be reported and followed up. 1751
What should be reported? 1752
1753
1. An event has occurred 1754 1755
1756
Typical events are: 1757
1758
a) A malfunction or deterioration in the characteristics or performance. 1759
1760
A malfunction or deterioration should be understood as a failure of a device to 1761
perform in accordance with its intended purpose when used in accordance with the 1762
manufacturer's instructions. 1763
The intended purpose means the use for which the device is intended, according to 1764
the data supplied by the manufacturer on the labeling, in the instructions and/or in 1765
promotional materials. 1766
b) An inadequate design or manufacture. 1767
This would include cases where the design or manufacturing of a device is found 1768
deficient. 1769
c) An inaccuracy in the labeling, instructions for use and/or promotional materials. 1770
Inaccuracies include omissions and deficiencies. Omissions do not include the 1771
absence of information that should generally be known by the intended users. 1772
d) A significant public health concern. 1773
This can include an event that is of significant and unexpected nature, such that it 1774
becomes alarming as a potential public health hazard, e.g. human 1775
immunodeficiency virus (HIV) or Creutzfeldt-Jacob Disease (CJD). 1776
e) Use error. 1777
• Use Error Resulting in Death or Serious Injury/ Serious Public Health 1778
Concern. 1779
• Use error related to medical devices, which did result in death or serious 1780
injury or serious public health concern, should be reported to the SFDA. 1781
• Use errors becoming reportable: Use errors become reportable to the SFDA 1782
when you: 1783

67
- Note a change in trend (usually an increase in frequency), or a 1784
change in the pattern of an issue that can potentially lead to death 1785
or serious injury or public health concern. 1786
- Initiate corrective action to prevent death or serious injury or 1787
serious public health concern. 1788
1789
1790
1791
2. The Event Led to One of the Following Outcomes 1792 1793
a) Death of a Patient, User or Other Person 1794
b) Serious Injury of a Patient, User or Other Person. 1795
Serious injury (also known as serious deterioration in state of health) is either: 1796
• Life threatening illness or injury. 1797
• Permanent impairment of a body function or permanent damage to a body 1798
structure. 1799
The term "permanent" means irreversible impairment or damage to a body structure 1800
or function, excluding minor impairment or damage. 1801
Medical intervention is not in itself a serious injury. It is the reason that motivated 1802
the medical intervention that should be used to assess the reportability of an event. 1803
c) No Death or Serious Injury Occurred, but the Event Might Lead to Death or Serious 1804
Injury of a Patient, User or Other Person if the Adverse Event Recurs. 1805
1806
All events do not lead to a death or serious injury. The non-occurrence of such a result 1807
might have been due to circumstances or to the timely intervention of health care 1808
personnel. 1809
The event is considered "adverse" if in the case of recurrence, it could lead to death or 1810
serious injury. 1811
This applies also if the examination of the device or a deficiency in the information 1812
supplied with the device, or any information associated with the device, indicates some 1813
factor which could lead to an event involving death or serious injury. 1814
Include relevant information that might impact the understanding or evaluation of the 1815
adverse event AND that is not included elsewhere in this report. 1816
1817

68
3. The Manufacturer's Device is Associated with the Event 1818 1819
In assessing the link between the device and the event, the manufacturer should take 1820
into account: 1821
a) The opinion, based on available information, from a healthcare professional 1822
b) Information concerning previous, similar events 1823
c) Other information held by the manufacturer 1824
1825
This judgment may be difficult when there are multiple devices and drugs involved. In complex 1826
situations, it should be assumed that all factors, including the device, be associated with the 1827
event. 1828
1829
5.9.2 Dealing with FSNs/Recalls. 1830 1831
Field Safety Correction Action 1832 1833
A field safety corrective action is taken by a manufacturer to reduce the risk of death 1834
or serious deterioration in the state of health associated with the use of a medical 1835
device. Such action should be notified via a field safety notice. 1836
1837
Field Safety Notice (FSN)/Recall 1838 1839
A notification from the SFDA to relevant medical device users in relation to a Field 1840
Safety Corrective Action. 1841
1842
SFDA classifies medical device recalls into three categories, representing the 1843
potential risk to public health: 1844
- Class I High risk. 1845
- Class II Medium risk. 1846
- Class III Low risk. 1847
1848
FSNs/Recalls Weekly Report 1849 1850

69
The NCMDR staff is sending an updated list of recalls on a weekly basis to 1851
healthcare officers. The report contains information such as the manufacturer name, 1852
affected device model and description, and affected lot/batch/serial numbers. 1853
1854
When the health care officer receives the recalls weekly report, he/she shall: 1855
- Ensure that the healthcare facility removes from use any affected 1856
device/product mentioned in the FSNs/Recalls. 1857
1858
1859
1860
If the healthcare facility has any affected device then the officer shall: 1861
- Communicate with the NCMDR Team and Authorized Representative of 1862
the manufacturer to ensure the implementation of the necessary corrective 1863
actions. 1864
- If the FSN requires the user to stop using the device, then the officer shall 1865
take the necessary actions to ensure that the affected device(s) are not in use 1866
until a corrective action is done. 1867
1868
5.10 Special requirements for specific medical devices 1869
1870
Safety of all medical devices for both patients and medical practitioners can be enhanced by 1871
providing user training. However, some especially high risk, medical devices need additional 1872
training to reduce their risk for the following medical devices: 1873
1. Non-implantable active medical devices for: 1874
1875
a. generation and application of electrical energy to directly affect the function of 1876
nerves and / or muscles or the heart activity including defibrillators, 1877
b. intracardiac measurement of electrical quantities or measurement of other 1878
quantities using electrically operated probes or sensors in blood vessels or on 1879
exposed blood vessels, 1880
c. Generation and application of any energy for immediate coagulation, tissue 1881
destruction or destruction of deposits in organs, 1882

70
d. direct introduction of substances and fluids into the bloodstream under potential 1883
pressure build-up, where the substances and fluids can also be processed or 1884
specially treated endogenous substances whose introduction is directly coupled to 1885
a removal function, 1886
e. mechanical ventilation with or without anesthesia, 1887
f. diagnosis using imaging techniques based on the principle of nuclear magnetic 1888
resonance 1889
g. therapy with pressure chambers 1890
h. therapy by means of hypothermia 1891
1892
2. Infant incubators 1893
3. external active components of active implants 1894
1895
The healthcare provider shall not operate or use these high risk? medical devices without 1896
assigning an in-house medical device clinical educator for each type of high risk medical 1897
device. 1898
The Medical Devices Clinical Educator shall be trained by the manufacturer or his 1899
representative on the correct and safe handling, use and operation of the medical device 1900
and their accessories and if the device is subject to be combined and operate with other 1901
devices or within system, the training scope shall be extended to include the whole 1902
combined devices and system. This training shall be based on the IFU and related safety 1903
and maintenance information. This training shall qualify the Medical Devices Clinical 1904
Educator to train the other users of the device and this shall be included in the manufacturer 1905
certification. The healthcare provider shall document this training. 1906
1907
The Medical Devices Clinical Educator’s obligations toward all medical devices assigned 1908
to him are the following: 1909
- Attend the training provided by the manufacturer 1910
- Organize and conduct training for other users of the same medical device from the same 1911
manufacturer in their healthcare provider. 1912
- Document all the training activities that have been executed on the related medical 1913
device and share it with the HTM department. 1914
- Assure the availability of the IFU of this medical device and make it accessible for all 1915
users at all working hours. 1916

71
- Monitor the compliance/ adherence to the requirement of safety and calibration checks 1917
and periodic preventive maintenance. 1918
- Organize on a regular basis discussions to address safety issues regarding the use and 1919
operation of these medical devices with the users, SFDA-Officer, manufacturer or his 1920
authorized representative and the HTM Department and all parties involved in the 1921
operation of these medical devices. Apply corrective action where appropriate. These 1922
discussions shall be documented. 1923
- Update users with and all parties involved in the operation of these medical devices 1924
with all safety issues and recalls issued by the SFDA or the manufacturer. 1925
- Support the SFDA-Officer in case of reporting adverse events, applying SFDA medical 1926
devices recall procedures and manufacturer procedures. 1927
1928
5.11 Special safety requirements 1929
1930
1931
Safety of medical devices for both patients and medical practitioners can be enhanced by 1932
providing proper user training and proper device maintenance. However, some medical devices 1933
needs special safety requirements to eliminate their risk. 1934
1935
5.11.1 Medical Laser Devices 1936
1937
The laser is used in several different applications in healthcare facilities including 1938
dermatology, surgery, and ophthalmology. 1939
When using a laser device, the following precautions should be taken: 1940
• Use of proper protective eyewear for both health practitioner and the patient to protect 1941
the eyes from direct and reflected laser energy. 1942
• Laser warning signs in Arabic and English should be placed on the door of each procedure 1943
room to prevent personnel from entering without wearing protective eyewear. 1944
• Ventilation of airborne contaminants to protect personnel from inhaling noxious fumes. 1945
• Quick operating procedures for using laser equipment shall be posted in each clinic. 1946
• The operator and /or dermatologist shall be licensed by Saudi Commission for Health 1947
Specialties in relevant specialty 1948
• The patient shall sign a consent form that acknowledges procedure hazards and declare 1949
patient's history and physical state, and it should be updated at each visit. 1950

72
• Operators shall be trained on laser emergency incidents, laser safety training and operating 1951
laser equipment. (Training certificate shall be kept on file). 1952
• The eyewear wavelength shall match and cover the device wavelength. 1953
• Eyewear shall be in good condition (Operators shall not use broken eyewear). 1954
• Adequate number of eyewear (at least 3) shall be provided. 1955
• Any reflecting object of the laser beam such as mirrors, metal and any other reflecting 1956
objects shall be covered or removed to avoid inadvertent exposure to direct or scattered 1957
laser radiation. 1958
1959
5.11.2 Ionizing Radiation 1960 1961
Ionizing radiation is a form of energy that acts by removing electrons from atoms and 1962
molecules of materials that include air, water, and living tissue. Ionizing radiation can travel 1963
unseen and pass through these and most other materials. 1964
1965
The main exposure to ionizing radiation is through the use of diagnostic medical exams. 1966
Medical exams that use ionizing radiation include: X-ray, CT scan, PET scan, Fluoroscopy, 1967
and Nuclear Medicine procedures. 1968
1969
The following actions can help reduce the exposure to and risk of harm from diagnostic 1970
ionizing radiation by: 1971
1972
• Checking to see if a test that does not use ionizing radiation can provide similar 1973
information. 1974
• Making certain the least possible amount of radiation needed to obtain a good quality 1975
image is used. 1976
• Using protective lead shielding to prevent exposing other areas of the body to radiation. 1977
• Performing functional tests of quality assurance to ensure that the device is not deviating 1978
from the proper use (KV, mAs, tests, collimator alignment tests, …etc) 1979
1980
5.12 Inventory Accuracy 1981
1982 1983

73
The inventory accuracy shall be maintained so that all devices & spare parts can be tracked 1984
after acquisition by the hospital. All additions, deletions, changes should be made promptly. 1985
The inventory accuracy shall be periodically verified, at least the existence of identification 1986
number, manufacturer and description, with a statistically significant sample each year. 1987
1988
Inventory should include: 1989
1990 • Location of the device. 1991
• Department owning the device. 1992
• Service provider. 1993
• Acceptance date. 1994
• Additional useful information. 1995
• Spare parts. 1996
1997
5.12.1 A Sample Statistical Model for Evaluating Inventory Accuracy 1998 1999
The values chosen below reflect the minimum sample quantities necessary to ensure that the 2000
inventory is at least 85% accurate. A more accurate inventory would require less sampling. The 2001
numbers are derived from the equation: 2002
s = √𝑝(1 − 𝑝)(𝑁 − 𝑛)
𝑛𝑁 2003
2004
where s is the standard error of the sample, p is the mean accuracy of the inventory, n is the 2005
sample size, and N is the population size. 2006
2007
To derive the numbers used here, a mean accuracy for the inventory was assumed to be 90%, 2008
with a standard error of 5%. In this way, the following statement can be made with 95% 2009
confidence: Based upon the sample, if the accuracy of the selected sample is 90% or greater, 2010
then the mean accuracy of the entire population is the same as the accuracy of the sample +/- 2011
5%. This assumes that the selected samples are truly randomly selected. 2012
For example, to find the sample size required, this equation can be rewritten as: 2013
2014
𝑛 =𝑁𝑝(1 − 𝑝)
𝑁𝑠2 + 𝑝(1 − 𝑝) 2015
If the total number of items in the inventory is 100 items, then: 2016

74
𝑛 =(100)(0.9)(1 − 0.9)
(100)(0.5)2 + 0.9(1 − 0.9) 2017
2018
𝑛 = 26.47 2019
This number is then rounded up to the next highest integer. The typical sample sizes used for 2020
accuracy check are: 2021
• < 100 items the audit shall be 27 randomly selected items. 2022
• >100 but < 500 items, the audit shall be 34 randomly selected items. 2023
• 500 but < 1000. The audit shall be at least 35 randomly selected items. 2024
• 1000 the inventory shall consist of 36 randomly selected items. 2025
The accuracy is expected to be > 95%.2 2026
2027
For more details please refer 2013 Association for the Advancement of Medical 2028
Instrumentation ■ ANSI/AAMI EQ56:2013 2029
2030
5.13 Warranty Management 2031
2032 2033
Warranty Management deals with management of all technical services related to medical 2034
devices that are within their warranty period. During this period, the type, quality and 2035
performance of the medical device, as well as the quality of services and technical support 2036
provided by the supplier, is assessed. The responsibility for the device’s technical performance 2037
is primarily with the supplier, and the healthcare facility and HTM program is responsible for 2038
documenting and notifying the supplier of all quality and performance issues so they can get 2039
them rectified free of charge during the pre-agreed-upon warranty period. 2040
2041 2042
• Warranty management procedures shall ensure that all faults of the device occurring 2043
within the contracted warranty period are reported and fixed, and bring the device to its 2044
original working condition. 2045
• The warranty provisions and scope of coverage, are recommended by the HTM/Clinical 2046
Engineering and the device’s clinical department, and based on the device’s purchase 2047
agreement. This would include all warranties provided by manufacturers, local suppliers 2048
and third-party providers. This could include Planned Preventive Maintenance, Corrective 2049
Maintenance, Emergency services, inclusion/exclusion of spare parts, etc. HTM/Clinical 2050
Engineering has to monitor the performance – both in terms of quality of repairs and 2051
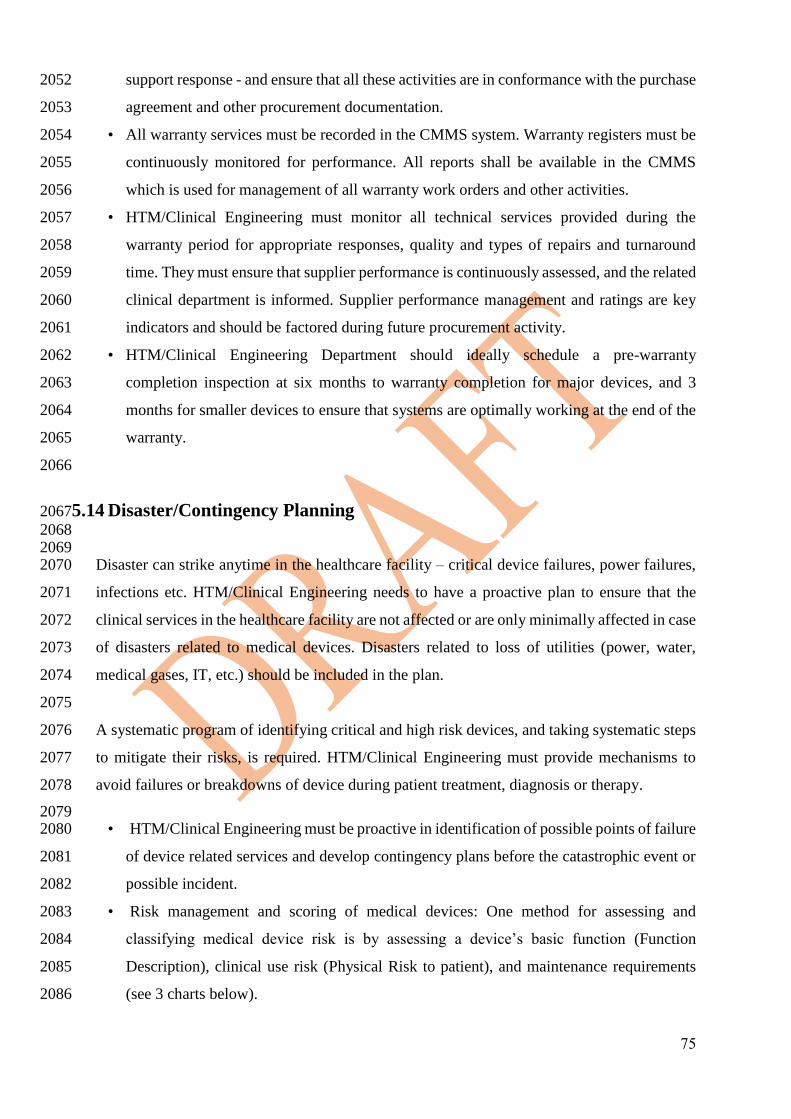
75
support response - and ensure that all these activities are in conformance with the purchase 2052
agreement and other procurement documentation. 2053
• All warranty services must be recorded in the CMMS system. Warranty registers must be 2054
continuously monitored for performance. All reports shall be available in the CMMS 2055
which is used for management of all warranty work orders and other activities. 2056
• HTM/Clinical Engineering must monitor all technical services provided during the 2057
warranty period for appropriate responses, quality and types of repairs and turnaround 2058
time. They must ensure that supplier performance is continuously assessed, and the related 2059
clinical department is informed. Supplier performance management and ratings are key 2060
indicators and should be factored during future procurement activity. 2061
• HTM/Clinical Engineering Department should ideally schedule a pre-warranty 2062
completion inspection at six months to warranty completion for major devices, and 3 2063
months for smaller devices to ensure that systems are optimally working at the end of the 2064
warranty. 2065
2066
5.14 Disaster/Contingency Planning 2067
2068 2069
Disaster can strike anytime in the healthcare facility – critical device failures, power failures, 2070
infections etc. HTM/Clinical Engineering needs to have a proactive plan to ensure that the 2071
clinical services in the healthcare facility are not affected or are only minimally affected in case 2072
of disasters related to medical devices. Disasters related to loss of utilities (power, water, 2073
medical gases, IT, etc.) should be included in the plan. 2074
2075
A systematic program of identifying critical and high risk devices, and taking systematic steps 2076
to mitigate their risks, is required. HTM/Clinical Engineering must provide mechanisms to 2077
avoid failures or breakdowns of device during patient treatment, diagnosis or therapy. 2078
2079 • HTM/Clinical Engineering must be proactive in identification of possible points of failure 2080
of device related services and develop contingency plans before the catastrophic event or 2081
possible incident. 2082
• Risk management and scoring of medical devices: One method for assessing and 2083
classifying medical device risk is by assessing a device’s basic function (Function 2084
Description), clinical use risk (Physical Risk to patient), and maintenance requirements 2085
(see 3 charts below). 2086
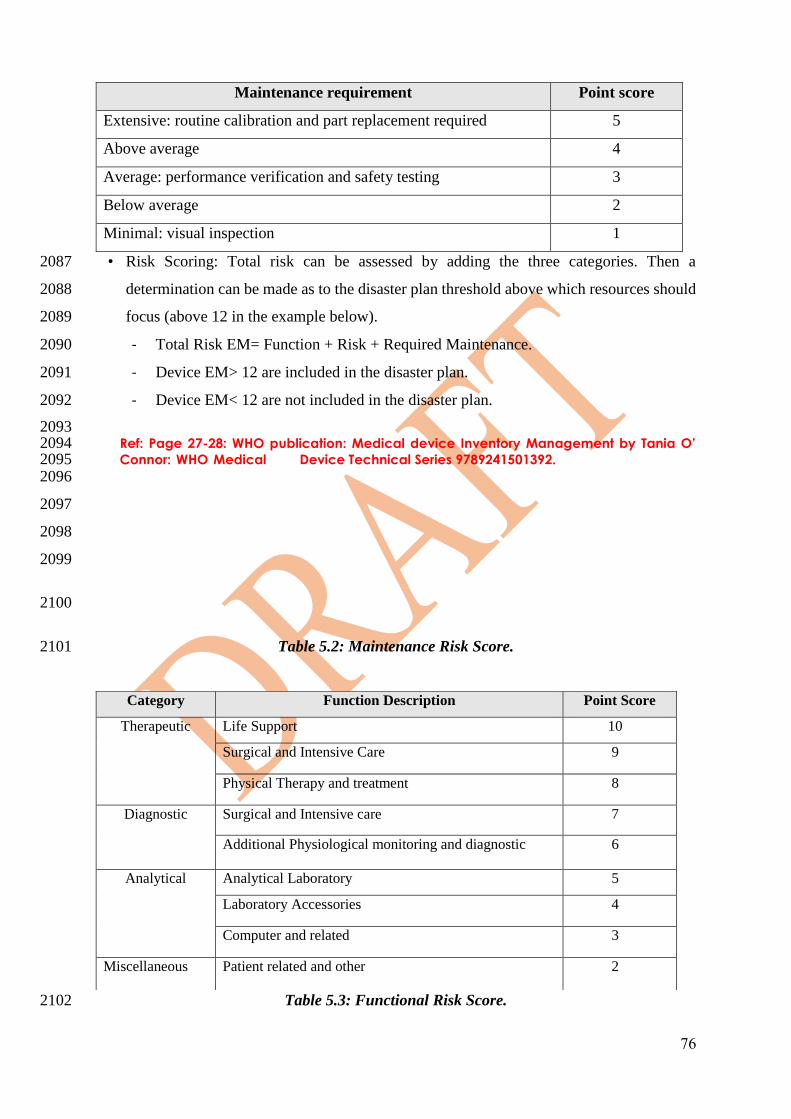
76
• Risk Scoring: Total risk can be assessed by adding the three categories. Then a 2087
determination can be made as to the disaster plan threshold above which resources should 2088
focus (above 12 in the example below). 2089
- Total Risk EM= Function + Risk + Required Maintenance. 2090
- Device EM> 12 are included in the disaster plan. 2091
- Device EM< 12 are not included in the disaster plan. 2092
2093 Ref: Page 27-28: WHO publication: Medical device Inventory Management by Tania O’ 2094 Connor: WHO Medical 2095 Device Technical Series 9789241501392.
2096
2097
2098
2099
2100
Table 5.2: Maintenance Risk Score. 2101
Table 5.3: Functional Risk Score. 2102
Maintenance requirement Point score
Extensive: routine calibration and part replacement required 5
Above average 4
Average: performance verification and safety testing 3
Below average 2
Minimal: visual inspection 1
Category Function Description Point Score
Therapeutic Life Support 10
Surgical and Intensive Care 9
Physical Therapy and treatment 8
Diagnostic Surgical and Intensive care 7
Additional Physiological monitoring and diagnostic 6
Analytical Analytical Laboratory 5
Laboratory Accessories 4
Computer and related 3
Miscellaneous Patient related and other 2

77
2103
2104
Table 5.4: Physical Risk Associated with Clinical Applications. 2105 2106
2107
2108
2109
2110 2111
5.15 User Training 2112
2113 2114 Safe and effective use of medical device requires that users shall receive appropriate training 2115
as per manufacturer requirements. The training should focus on clinical use and the need to 2116
ensure that all features of the technology are used for best possible patient diagnosis and 2117
therapy. 2118
• The key focus of user training programs would be to enable users to operate the device in 2119
a safe and effective manner. An additional objective will be to ensure that users cooperate 2120
in increasing the useful life of the device, while operating the device safely. 2121
• Training will be mandatory when a new device is installed. 2122
• Training may also be required based on failure data or wrongful use of the medical device. 2123
If any special training is required – either in-house or from manufacturers or specialized 2124
agencies- it shall be provided as requested and agreed upon in the initial purchase or 2125
subsequent support agreements for the life of the equipment.. 2126
• HTM/Clinical Engineering will keep records of the training; content and attendees. 2127
2128
5.16 HTM/Clinical Engineering Training programs 2129
2130 2131
The staff in the HTM department shall be trained as follows: 2132
• The required education for his job. 2133
Description of use Risk Point score
Potential patient death 5
Potential patient or operator injury 4
Inappropriate therapy or misdiagnosis 3
Device damage 2
No significant risk 1

78
• The required training from the manufacturer. 2134
2135
5.17 Spare Parts procurement and management 2136
2137 2138
• HTM/Clinical Engineering staff, based on their engineering assessment, shall source 2139
appropriate and genuine spare parts for maintenance based on manufacturer service 2140
manuals and technical bulletins. 2141
• HTM/Clinical Engineering with the assistance of the device supplier should plan for 2142
appropriate inventories of spare parts to minimize downtime. 2143
2144 Note: Spare parts should cover periodic preventive maintenance spares, as well as spares for 2145
breakdowns. 2146
2147
2148
2149
5.18 On Site Libraries 2150
2151 2152
To maintain devices, and to use devices appropriately, users and clinical engineers need access 2153
to operation and service manuals. A systematic method of maintaining service and user 2154
manuals should be developed to ensure that hard or soft copies of these documents are available 2155
on demand. 2156
• HTM/Clinical Engineering must provide technical staff with appropriate manuals and 2157
documented instructions on maintenance and support of all medical devices. 2158
• On site libraries should include manufacturer authorized and authenticated: 2159
- Service and repair manuals. 2160
- Operation manuals. 2161
- Other manuals including software manuals etc. 2162
- These may be provided in hard or soft copies. 2163
- Surveys of all medical device documentation can identify documentation gaps and 2164
requirements. 2165
- All HTM programs should include a plan for a documentation system with safe 2166
storage and easy access for all information. 2167
- This system should be catalogued based on manufacturer and model number for easy 2168
identification and retrieval. 2169

79
- When a new device is commissioned, a copy of the manuals should be provided to 2170
HTM Department. 2171
- It is suggested that all new devices must come with at least two copies of the manuals. 2172
– One for HTM and the other is for the end-users. 2173
2174 2175
5.19 Workshop Space and Test Devices 2176
2177 2178
A well-equipped workshop space is a mandatory requirement for a HTM/Clinical Engineering 2179
Department. The workshop must be equipped with work benches, test equipment, service 2180
toolkits, personal protective equipment, jigs and fixtures necessary to provide competent 2181
technical services. 2182
2183 2184 2185
Workshop requirements: 2186 2187
- The workshops should be of adequate size to accommodate the service teams, 2188
manuals and documentation. 2189
- Test equipment: Appropriate test equipment for analyzing, performance and safety 2190
of medical devices shall be available. 2191
- Ensure that all test equipment are procured and calibrated as per manufacturer 2192
requirements. 2193
- Ensure that the workshop is appropriately equipped with all safety equipment (fire 2194
extinguishers, PPE, etc.). 2195
- Workshop plans, and drawings must be available. 2196
2197 2198
5.20 Inter Service Coordination 2199
2200 2201
Medical devices are dependent for their optimal performance on many support systems such 2202
as air conditioning, medical gases, electrical power, emergency power (e.g. (UPS), IT, and 2203
other utilities. Failure of these services could result in medical device failure resulting in 2204
catastrophes. To prevent this, regular coordination between these facility services and HTM 2205
will help ensure higher uptime for the medical devices. 2206
2207
80
• The HTM/Clinical Engineering Department must coordinate with many other departments 2208
of the healthcare facility, to ensure that quality services are provided to the healthcare 2209
facility. Typically, these include Facilities Management, IT, Quality, Infection Control, 2210
etc. This process addresses the framework of such coordination. 2211
• Close coordination with many support services in the healthcare facility may require a 2212
formal framework. 2213
• Healthcare Administration should facilitate this formal framework. 2214
• A periodic (e.g. monthly) meeting with representatives of all services is suggested. 2215
2216
2217
2218
2219
5.21 Checklist 2220
2221
No. Check list item Status Comments
Yes
No
In
pro
gre
ss
1 Availability of a dedicated HTM/Clinical
Engineering Department with appropriate budgets
and management.
2 Medical device management plan.
3 Availability of CMMS system
4 Availability of PPM, CM, Safety testing and
performance testing policy
5 The Storage area is in compliance with Medical
Devices manufacturer and any regulatory
requirements
6 All medical Devices are calibrated if applicable
7 Availability of Electrical Safety analyser

81
8 Availability of Quality assurance test equipment as
needed
9 Availability of biomedical test equipment as needed
10 All test Equipment are calibrated
11 Availability of SFDA-Officer
12 Availability and applying of written procedure to
manage recalls published by SFDA
13 Availability applying of written procedure to report
adverse events to SFDA
14 Availability of Medical Devices Clinical Educator
2222 2223 2224

82
2225
Chapter 6: Disposal & Donation 2226
2227
6.1 Introduction 2228
2229
2230
6.1.1 Objective 2231 2232
Ensuring safe and cost effective disposal or donation of medical devices. 2233
2234
6.1.2 Scope 2235
2236
This process applies to the healthcare organization and their staff who will dispose or donate 2237
surplus medical devices. 2238
2239
6.1.3 Ownership 2240 2241
• HTM/Clinical Engineering Department. 2242
• End-user Medical Departments. 2243
• Assets management. 2244
• Legal. 2245
• Finance. 2246
• Quality Department. 2247
• CSSD. 2248
• Related Healthcare admin. 2249
2250
6.1.4 Background 2251 2252

83
Decommission and
Disposal
Device may be
recommended for disposal
because of :
- Beyond economic repairs
- New safety requirements
render the device unsafe
- Recent technology
renders the device
redundant
- Hazardous for use
- Recall advice from
regulatory agencies
HTM/ Clinical Engineering
and Facilities Engineering
shall arrange for the device
to be moved into long term
storage facilities pending
safe disposal
Update the device s status record. The
device s history
shall be kept for at
least 5 years.
In the case of
radiation device,
laser systems etc.
governed by
statutory laws and
regulations,
appropriate
authorities must be
informed.
Planning Procurement Installation Operation
2253 2254
Figure 6.1: Decommission and Disposal Process. 2255
2256
6.1.4.1 Donating of Used Medical Devices (Decommissioning and Disposal) 2257
2258
Medical devices may be donated or sold to other healthcare organizations within the Kingdom 2259
of Saudi Arabia or to foreign countries. However, to improve the quality of donations of used 2260
medical devices process, it is suggested to follow the following guidelines: 2261
• Healthcare organizations shall properly plan for the devices that can be donated and 2262
communicate with the anticipated recipients before removal of the devices from service. 2263
• The used medical devices that are proposed to be donated/ or sold shall be removed from 2264
service after conducting the appropriate testing and removal all patients’ data. The 2265
devices shall be cleaned, disinfected from contamination and transported to a warehouse 2266
of suitable environment before delivering them to the recipient. 2267
• The donating organization shall provide maintenance manuals, documents and available 2268
related spare parts to the receiving organization together with any information about 2269
missing parts, if any. 2270
• The recipient shall assure the availability of the necessary consumables or in-vitro 2271
diagnostic devices (IVD), as well as the availability of a suitable location that contains 2272
the basic space, physical installation (alignment, shielding, etc.) and utility-related 2273
services (e.g. such as electricity, drainage, water supply, nitrogen outlets) as appropriate. 2274
• The recipient of the used medical devices shall review their requirements, healthcare 2275
level, patient workload, number of users, their abilities, and their available technical 2276
experience level prior to acceptance of any used medical devices. 2277
2278

84
• Do not sell or donate medical devices to a third party as follows: 2279
- Any used medical devices that have at least 2 years left of their operational life. 2280
- Devices destroyed as junk, dismantled or having parts removed for spares. 2281
2282
• Used medical devices that will be donated or sold to a third party shall: 2283
- Pass mechanical safety tests, calibration and electrical safety tests, including but not 2284
limited to electrical leakage current testing. 2285
- Be accompanied by maintenance and user manuals whenever possible. 2286
- Be accompanied by usage starting date, periodic maintenance record, periodic test 2287
schedules and all reports describing the status of the device during service, if 2288
available. 2289
- Be free from medical any contamination or biological remains, radiological waste, 2290
splashes or any hazardous medical remains. 2291
2292 • All used medical devices that shall be sold or submitted for public tender shall be kept in 2293
a safe storage area (a warehouse with good temperature and humidity control) within or 2294
out of the healthcare organization. Such devices shall not be left in the open air. It is 2295
preferable to wrap these devices for their safety during storage and transport. 2296
• The healthcare organization shall keep a separate record for all disposed of used medical 2297
devices by device. A separate record shall be maintained for identification of the sold 2298
items in addition to the identity of the purchaser. 2299
2300
6.1.4.2 Decommissioning and Disposal of Medical Device 2301 2302
Decommissioning of Medical device is required when a device needs to be removed from 2303
service. In some cases, there are specific protocols for removal of devices (e.g. devices with 2304
radiation sources) or other statutory laws and regulations (e.g. hazardous waste, patient data). 2305
This is a sensitive and hazardous activity, and in specifically identified cases, must only be 2306
done by experienced staff. When decommissioning a device, consider the following points: 2307
2308
• The organization is expected to have a well-defined and written procedure to coordinate 2309
removal and disposal of medical devices that have been decommissioned. 2310
• The reasons for decommissioning must be clearly identified and may include: 2311
- Beyond economic repair. 2312

85
- New safety requirements render the device unsafe. 2313
- Recent technology renders the device redundant. 2314
- Hazardous for use. 2315
- Recall advice from regulatory agencies. 2316
- Re-sale/public auction. 2317
• Disposal of used medical devices shall follow the standards of disposal of medical waste, 2318
including disposal of nuclear and contaminated wastes. 2319
• Disposal of used medical devices shall be carried out within the same healthcare 2320
organization and under the supervision of qualified professionals or licensed 2321
subcontractors who shall destroy the medical devices on behalf of the healthcare 2322
organization. 2323
• The healthcare organization shall keep all the documents pertaining to the destroyed used 2324
medical devices. 2325
• Spare parts may be Dismantled from used medical devices that are not destroyed due to 2326
contamination or fire or non-conformance or recall by the supplier. 2327
• Spare part(s) to be dismantled from the disposed used medical devices shall conform with 2328
the following guidelines: 2329
- Shall not require disinfection before fixing it on another similar medical device. 2330
- Printed circuit boards or any other electronic part shall not be totally or partially 2331
damaged (e.g. due to heat from solder removal process). 2332
- Shall not be contaminated or cracked or damaged. 2333
- Shall not be expired (e.g. a consumable part passed its operational life as calculated 2334
by the number of hours of use). 2335
- An exposed radioactive part. 2336
- A part that directly or indirectly contributes to the clinical measurements and cannot 2337
be calibrated before using it again in another medical device. 2338
- A part in that has been in contact with the patient or touching the patient’s body 2339
directly or through an interface. 2340
• Dismantling shall be conducted only by a trained and licensed biomedical engineering. 2341
• A record of all dismantled used medical devices shall be kept within the respective 2342
healthcare organization. 2343
• HTM/Clinical Engineering and Facilities Engineering shall arrange for the device to be 2344
moved into long term storage facilities pending safe disposal. 2345

86
• The CMMS data for the decommissioned medical device shall be updated and records 2346
shall kept for 5 years or longer. 2347
• In the case of radiation devices, laser systems or other devices governed by statutory 2348
laws and regulations, appropriate authorities must be informed. 2349
2350
6.2 Checklist: 2351
2352
No. Check list item Status Comments
Yes No In
progress
1 Are reasons for decommissioning are identified
and documented
2 Availability and applying of a medical device
written procedure for disposal and donation.
3 Availability of Group/ Committee for Medical
Devices/Health Technology disposal and donation
with involvement of all related parties in the
ownership.
5 The availability and applying written procedures
of dismantled spare part
6 Availability of record of all disposed medical
devices for at least 5 years
7 Devices to be disposed are isolated in storage area
2353

87
Chapter7: Special Topics 2354
7.1 Introduction 2355
2356
7.1.1 Objective: 2357
2358
Define & demonstrate the special requirements for Medical Device Consumables, Single 2359
Use Medical Devices (SUMD), Implants, and Medical Devices Clinical Trials (Clinical 2360
Investigation). 2361
2362
7.1.2 Scope: 2363
2364
This chapter applies to the following products: 2365
2366
- Medical device consumables (reusable). 2367
- Single use medical devices (SUMD). 2368
- Single patient medical devices. 2369
- Implants. 2370
- Medical devices clinical trials (Clinical Investigation). 2371
2372
7.1.3 Background 2373
2374
Consumables and Accessories in medical devices for this chapter are defined as follows6: 2375
2376
1. Accessories are those items which: 2377
2378
• Connect the medical device to the patient (e.g. breathing circuits, ECG leads, 2379
probes/transducers.). 2380
• Assist with the use of the medical device (e.g. internal trays, foot-switches, 2381
computer mouse). 2382
• Adapt its performance (e.g. different sized adaptors, objectives, lenses). 2383
2384
Note: The accessories are often the main link to the patient, the part most handled by 2385
staff flexed, bent, twisted and, sometimes misused. They are subject to a great deal of 2386

88
wear and tear and often are the most vulnerable problem part of a medical device. Even 2387
for medical device last for many years, accessories may need to be replaced regularly, 2388
therefore accessories must be available for the lifetime of the medical device. 2389
2390
2391
2392
2. Consumables are those items which: 2393
2394
• Are used up and need to be replaced frequently (e.g. each case, daily, weekly) during 2395
the operation of the medical device (e.g. X-ray film, disposable electrodes, laboratory 2396
reagents, ultrasound gel, washing powder, copier toner). 2397
• Consumables are needed throughout the lifetime of the medical device. 2398
2399
- Consumables used in conjunction with a primary medical device are in fact 2400
devices in their own right, albeit they may have a secondary function to the 2401
primary device and may have gone through an independent regulatory review 2402
process. 2403
- The availability of a medical device consumable item is significant. It is often 2404
the case that when a device consumable is no longer available for supply and is 2405
specific to the function of a primary medical device, it renders the primary device 2406
unusable. 2407
2408
2409
Medical device consumables may be single use or reusable. There are some advantages 2410
and disadvantages to both types and some of the re-use management aspects are discussed 2411
later in this chapter. 2412
2413
A Primary electro-medical device (e.g. an infusion pump) that uses or relies on the use of 2414
consumables often may use different brands of accessories or consumables which are not 2415
easily interchangeable. There are, however, a number of key principles that apply to the 2416
safe use and application of these components including: 2417
2418
• Functional compatibility with the primary medical device. 2419
• Purchase from an establishment registered and licensed by SFDA. 2420

89
• The product shall be authorized for marketing (e.g. MDMA & MDNR). 2421
• Standardization and rationalization of as much of the consumable stock items as 2422
possible in order to reduce cost and minimize user training requirements. 2423
• Effective stock control – many consumable items will have a specific shelf life and 2424
this requires an effective monitoring and management process to be in place. 2425
• Ability for clinical users to access e-catalogues to inform staff about the range of 2426
consumable products available in stock or to purchase from approved suppliers. 2427
• Appropriate environmental storage facilities. 2428
• The volume of consumables used and the utilization of the primary device will have 2429
a significant impact on a health technology expenditure. 2430
• An effective dissemination of guidelines for the use of medical devices and 2431
consumables. 2432
• Availability of the consumable (when used in conjunction with a primary health 2433
technology) throughout the entire expected lifetime of the primary device. 2434
2435
2436
The availability and usage of medical device consumables and accessories should be of 2437
significant interest to the healthcare facility. A failure of a device consumable will have a 2438
significant impact on the performance of a primary medical device with potential negative 2439
impacts on clinical service delivery and safety. In addition, the expense profile of a primary 2440
device can be significantly altered based on poor management of device consumables. 2441
Methods to consider managing consumable and accessory cost and performing the 2442
following: 2443
• Direct purchase of consumables. 2444
• Guaranteed price per reportable result (GPPRR) (e.g. Suppliers may offer to provide 2445
their devices free of charge against a fixed price for the supply of consumables.) 2446
• Leased medical devices with or without consumables and accessories included in the 2447
lease cost. 2448
2449
2450

90
Note: HTM/Clinical engineers have a significant role to play ensuring that the following 2451
two aspects (single use and re-use) are effectively addressed should a decision to move 2452
towards a different consumable plan be under consideration. 2453
2454
2455
7.2 Medical device Consumables, Single Use. 2456
2457
• Medical device consumable expenditure can be a very significant component of the 2458
whole life cycle cost of a primary medical device. 2459
• There are also consumables that are not always part of the functionality of a primary 2460
electro-medical device such as syringes, needles, etc. 2461
• There is a wide range of consumables in use within healthcare and it would be 2462
impractical to list them all within this document. 2463
• Manufacturers of single use devices indicate which devices are for single use only. 2464
2465
2466
2467 2468
Figure 7.1: Example of manufacturer labelling for Single Use Device 2469 2470
2471
Examples of SUMD include, but are not restricted to, the following: 2472
• Endodontic reamers and files. 2473
• Saliva ejectors. 2474
• Aspirator tips and three-in-one tips. 2475
• Dental burs. 2476
• Matrix bands. 2477
• Single use patient face masks. 2478

91
2479
2480
2481
7.3 SFDA requirements with regards to reprocessing of single use devices 2482
2483
1. Medical devices designed for single use only in surgical procedures performed in the 2484
tissues and internal organs of the human body shall not be reused for any medical 2485
application. 2486
2. Medical devices that are for single use only utilized for the external organs of the 2487
human body can be reprocessed according to the requirements determined by the 2488
SFDA. 2489
3. Sites that reprocess single use medical devices for themselves or any other party will 2490
be treated in accordance with No.7.3.2 above as a new manufacturer of the medical 2491
device and shall be responsible for the safety and efficiency of the product according 2492
to the requirements specified in the SFDA regulations. 2493
4. Healthcare facilities shall comply with the provisions of above requirements when 2494
reprocessing medical devices. 2495
5. The establishment that reprocesses the single use devices will be treated as a new 2496
manufacturer for that reprocessed single use device. 2497
2498
2499
7.4 Reuse of Reusable, Limited Use & Single Patient Use Medical Devices 2500
2501
The reuse of reusable medical devices and single patient use medical devices shall be in 2502
accordance with manufacturer’s guidance. 2503
2504
7.5 Reuse of Single Use Medical devices 2505
2506
Healthcare facilities that use single use devices for patient care have several options: 2507
• Dispose of all single use devices after a single use and purchase a new device. 2508
• Contract with a regulated reprocessing organization and purchase devices from them. 2509

92
• Become a reprocessing organization and comply with standards for reprocessing 2510
single use devices. 2511
• Use only reusable medical devices as labelled by the manufacturer. 2512
2513
The healthcare facility shall follow the SFDA requirements regarding the reuse of 2514
reprocessed single use medical devices. The reprocessed single use medical device will be 2515
treated as a new medical device. 2516
2517
Key good practice actions4 that should be considered by healthcare providers: 2518
2519
a) Be informed about current laws, regulations and guidance that may affect the 2520
healthcare providers’ decisions regarding use of reprocessed single use devices. 2521
b) Establish an interdisciplinary "reuse committee" to consider the issues that should 2522
inform policies, procedures, and protocols. 2523
c) Educate clinical staff about the facility's policy for use of reprocessed single use devices 2524
and provide a supporting rationale. 2525
d) Develop and implement criteria for selecting the reprocessed single use devices the 2526
healthcare provider will use and for evaluating third-party reprocessing organizations. 2527
e) Review provisions in contracts with original equipment manufacturers (of single use 2528
devices) that would prohibit or inhibit third-party reprocessing of their single use 2529
devices. 2530
f) Ensure that the facility's event reporting system captures adverse events related to 2531
reprocessed single use devices and that a procedure is in place for mandatory reporting 2532
of medical-device-related deaths and voluntary reporting of serious adverse events 2533
related to medical devices to the SFDA. 2534
2535
All reprocessing organizations (e.g. hospitals, manufacturers, third-party vendors) are 2536
required to follow the SFDA licensing requirements. 2537
2538
Through a process of clinical and technical testing and evidence the reprocessing 2539
organization will need to identify the maximum number of reprocessing cycles to which a 2540
device can be subjected while maintaining safe device functionality and performance. 2541

93
Once the maximum number of reuse cycles has been reached the product should be 2542
discarded and safely disposed of. 2543
2544
Reprocessing of single use devices generally consists of the following processes: 2545
a) Cleaning. 2546
b) Disinfection (reduces the number of viable microorganisms). 2547
c) Sterilization (kills all microorganisms and is required for devices that likely will 2548
become contaminated with pathogens during use). 2549
d) Functional testing. 2550
e) Relabeling. 2551
f) Repackaging. 2552
2553
Reprocessing may require disassembly and reassembly to facilitate cleaning. Each 2554
reprocessed item must be inspected before reuse and also function-tested and repackaged 2555
for reuse. After every single use of a reprocessed single use device, the device must again 2556
go through the entire reprocessing system. (Vocalic)2. 2557
2558
7.6 Medical Device Implants 2559
2560
Medical device implants range from simple devices, such as intraocular lenses, cosmetic 2561
implants, orthopedic hardware, stents, and joint replacements, to highly complex devices, 2562
such as retinal implants, artificial hearts, implantable cardioverter-defibrillators (ICDs), 2563
and cochlear implants. 2564
2565
Examples of ‘active’ implants can include the following: 2566
• Implantable nerve stimulators. 2567
• Implantable cardiac rhythm management devices (pacemakers, defibrillators, etc.). 2568
• Leads, electrodes, adaptors for the above. 2569
• Cochlear implants. 2570
• Catheters, sensors for active drug administration technologies. 2571

94
2572
A key feature of the management procedures for medical implants concerns the 2573
management of stock and product requirements. There are a number of options to consider 2574
which will help manage the cost of these often expensive devices. Good practice 2575
considerations include the following actions: 2576
• Establish the full range of device implant suppliers and products used by the 2577
healthcare provider. 2578
• Identify the product volumes consumed. 2579
• Consider any unused stock held on the premises and identify reasons, and follow-up 2580
actions, to more efficiently manage implant logistics. 2581
2582
7.6.1 Tracking Medical Device Implants 2583 2584
The healthcare provider has a responsibility to be able to notify its patient population 2585
should a product recall occur. An inability to notify the recipient of an implanted device 2586
that is the subject of a recall is to be avoided. 2587
The following is an example of data that should be collected by healthcare providers to 2588
enable effective implant tracking to take place. 2589
• Any unique identification number, lot number, batch number, model number, or 2590
serial number of the device or other identifier used by the manufacturer to track the 2591
device. 2592
• The name, address, telephone number of the patient in receipt of the device. 2593
• The date the device was provided for use by the patient. 2594
• The name, address, and telephone number of the prescribing clinician. 2595
• The name, address, and telephone number of the clinician regularly following up care 2596
with the patient, if different from the prescribing clinician. 2597
• The date of the patient's death, if applicable. 2598
• At the device explant point: the name, mailing address, and telephone number of the 2599
explanting clinician. 2600
• When applicable, the date the device was permanently retired from use, explanted or 2601
otherwise permanently disposed of. 2602

95
2603
Device tracking good practice areas for consideration for healthcare providers are provided 2604
below: 2605
1. Ensure that the healthcare provider has implemented a device tracking program that 2606
meets current legal and regulatory requirements. 2607
2. Designate a medical device "tracking manager" to oversee the device tracking 2608
process, to act as a liaison with device manufacturers and the SFDA for tracking 2609
purposes, and to ensure that pertinent staff are educated about the facility's tracking 2610
program. 2611
3. Establish a system for collecting and submitting tracking information to 2612
manufacturers and SFDA. Address whether and how patient consent to the release 2613
of his or her identity will be obtained and documented. 2614
4. If a consent process is established, ensure that it includes a mechanism for 2615
documenting refusals. 2616
5. Verify that a patients’ rights to refuse to release identifying information to the 2617
manufacturer for purposes of tracking are being upheld. 2618
6. Address the need to retain device tracking records and determine the length of 2619
retention. 2620
7. Establish a method by which the healthcare provider can monitor changes to any 2621
list of tracked devices and future SFDA regulations and recommendations 2622
regarding tracking and cooperate with tracking procedures implemented by 2623
manufacturers in response to new or revised regulations. 2624
2625
7.6.2 Implanted Medical Devices Registries in Saudi Arabia 2626 2627
In order to monitor the performance of medical device implants, tracking any associated 2628
side effects or complications that may arise in health facilities in Saudi Arabia, the SFDA 2629
has established the (Comprehensive Implanted Medical Device Registry - CIMDR) that 2630
will help facilitate tracing and tracking of these devices and their users, where potential 2631
risks may or could threaten their safety and health. Additionally, this process supports and 2632
encourages medical research conducted by healthcare providers, researchers, innovators, 2633

96
and manufacturers on medical device implants for verification of their effectiveness and 2634
intended use leading to the best healthcare services. It also provides SFDA with a post 2635
market surveillance tool by analyzing the registry data about the safety, effectiveness, and 2636
quality of medical devices implants. 2637
Currently, there are four established registries: Hip joints, knee joints, stents, and ICDs. 2638
SFDA is planning also to include cochlear implants and breast implants registries in 2019. 2639
The registry captured data includes: demographics, diagnosis, patient history, 2640
comorbidities, previous procedures, complaints/symptoms, surgery information, 2641
outcomes, device information, and follow up information. 2642
More information about this national initiative and how health facilities can participate in 2643
the registry, can be found at: http://cimdr.sfda.gov.sa 2644
2645
2646
7.6.3 Reporting Processes - Medical Device Implants 2647
2648
Good practice recommendations for an effective implant reporting process includes the 2649
following: 2650
2651
1. A closed-loop process that, in addition to the distribution of recalls, includes 2652
confirmation that the recall has been received by a responsible party and 2653
documentation of the remediation efforts taken. 2654
2. A written policy is available specifying, for example, to whom incoming recalls 2655
should be sent, how recalls should be processed, and how the response to those 2656
recalls should be documented. 2657
3. A manufacturer or other organization that issues a recall might direct the recall to 2658
a specific department, to an individual physician, or to others. This data needs to 2659
be captured within a centralized recalls management process. 2660
4. All parties will need to be educated about the process for forwarding recalls to the 2661
correct individual or department. 2662
2663

97
Recalled implanted devices require careful clinical management and follow-up. The 2664
primary responsibility for the clinical management of recalled implants usually rests with 2665
the patient’s clinician and/or referring clinician. When a recall affects an implanted device, 2666
the healthcare provider should notify both clinicians so that they may provide appropriate 2667
follow-up care to patients. 2668
2669
Clinicians whose names are included on the device registration may be notified of the 2670
product recalls directly by the manufacturer. The clinician concerned should report the 2671
receipt of this recall so that this information can be captured in the healthcare providers’ 2672
central medical device recall /patient safety reporting system. 2673
2674
2675
2676
7.6.4 Explanted Medical Devices 2677
2678
Implanted medical devices may be removed for numerous reasons: 2679
• Device may have reached the end of its useful life; or 2680
• Product defect or malfunction may necessitate the explant. 2681
2682
In the case of a recall involving an implant, when the manufacturer notifies the implant 2683
clinician or hospital, an incident report should be filed within the healthcare providers own 2684
health technology recalls or patient safety incident reporting management system. Such 2685
recalls should also be reported to the SFDA for regulatory purposes. Where an implant is 2686
deemed to be a patient safety risk by the implant clinician this matter should be reported to 2687
the SFDA and the original device manufacturer (not the distributor). 2688
2689
It is also recommended that prior patient consent for post-mortem device evaluation and 2690
explantation be documented and, in the absence of such consent, seek approval be obtained 2691
for explantation from family members. Some medical device companies post contact 2692
information and information about where and how to return the devices on their web sites. 2693
2694
2695
98
7.6.5 Medical Devices Clinical Trials (Clinical Investigation) 2696
2697
7.6.5.1 Objectives 2698
2699
To ensure the compliance of medical devices clinical trials to the Saudi Food and 2700
Drug Authority (SFDA) requirements. 2701
2702
7.6.5.2 Scope 2703
2704
Any party who wishes to conduct medical devices clinical trials in KSA. 2705
2706
7.6.5.3 Background 2707
2708
A clinical investigation is defined as a “systematic investigation in one or more 2709
human subjects, undertaken to assess the safety or performance of a medical 2710
device”. 2711
2712
Note: “Clinical trial” and “clinical study” are synonyms for “clinical 2713
investigation”. The objective of a clinical investigation is to assess the safety and 2714
performance/efficacy of the device in question and evaluate whether the device is 2715
suitable for the purpose(s) and the population(s) for which it is intended (ISO 2716
14155:2011). Clinical investigations must take into account scientific principles 2717
underlying the collection of clinical data along with accepted ethical standards 2718
surrounding the use of human subjects. The clinical investigation objectives and 2719
design should be documented in the clinical investigation plan. 2720
2721
2722
7.6.5.4 Ownership 2723
2724
• Institutional Review Board (IRB). 2725
• Principal investigator. 2726
2727

99
7.6.5.5 Requirements 2728
2729
• Shall comply with the Law of Ethics of Research on Living Creatures by National 2730
Committee of Bioethics (NCBE). 2731
• Shall request “No Objection Letter” issued by SFDA before starting the study 2732
recruitments. 2733
• Should be in accordance with: 2734
- Declaration of Helsinki. 2735
- ISO 14155 (or any equivalent standard GCP). 2736
2737
2738
2739
2740
2741
2742
2743
2744
2745
2746
2747
2748
2749
2750
2751
2752
2753
2754
2755
2756
2757
2758
2759
2760
2761
2762
2763
2764
2765
2766
2767
2768
2769

100
7.7 Checklist 2770
2771
The table below represents a best practice checklist. Its purpose is to serve as a tool for 2772
evaluating medical device consumables, single use devices and implant activities and assist 2773
in the development of process improvements. 2774
2775
No Checklist item Status Comments
Yes
No
In
pro
gre
ss
1 Identify the stock of medical device consumables and
implants needed.
2 Provision of an effective stock control system for medical
consumables and implants.
3 Implement a medical device implant tracking process.
4 Availability of Registration Documents by SFDA for any
Medical Device intended to be used for Clinical
Investigation
2776 2777
2778
2779
2780
2781
2782

101
2783
8. References 2784
2785
2786
2787
2788
2789
2790
2791
2792
2793
2794
2795
2796
2797
2798
2799
2800
2801
2802
2803
2804
2805
2806
2807
2808
2809
2810
2811
2812
2813
2814
2815
2816
2817
2818
2819
2820
2821
2822
2823 2824 2825

102
2826
9. Appendices 2827
2828
9.1 Planning: 2829
2830
- Appendix 9.1.2 Includes facility planning examples for Operating rooms, 2831
intensive care units and Imaging. 2832
- Appendix 9.1.3 Includes sample emerging technology descriptions for 2833
multiple modality systems, 3-D printing in healthcare; integrating the 2834
Healthcare Enterprise (IHE) profiles for device integration; medical device 2835
surveillance monitoring; genomics; and a few examples of the use of 2836
artificial intelligence (AI) and big data in healthcare and HTM. 2837
2838
Table 9.1: Typical healthcare facility Utilities for Medical Devices 7
Electrical power - Emergency electrical power
Fire detection and prevention
Elevators (move planning relevant)
Domestic water - Water for dialysis and other special water treatment systems
Heating, ventilation and air conditioning (HVAC)
Hot water
Building temperature control
Cooling & Heating
Humidification
Sanitary sewer
Storm sewer
Medical gases and vacuum
Compressed medical air
Oxygen
Vacuum
Other piped specialty gases (CO2, NO, N2)
Steam
For heating and air conditioning
For sterilizers
Natural gas
For steam
For electricity if co-generation plant
IT *
Wiring infrastructure
Wireless infrastructure
Data closets (MDF, IDFs)
Data centers
Other, non-IT-based communication technologies requiring separate wiring
103
Nurse call
Telephone
2839
9.1.1 Appendix A: Certificate of Need Process 2840 2841
A certificate of need is a review and approval process, typically from a government 2842
agency, to proceed with a major project (e.g. building, large cost equipment purchase) 2843
taking into account the overall needs of the community being served. Many countries 2844
do not have this process in place but it may be something decision makers may want 2845
to consider. For those countries who have or will implement this process, it is 2846
important to take into consideration the following when applying for a certificate of 2847
need. 2848
2849
General Data 2850
- Place. 2851
- Catchment area. 2852
- Epidemiology information. 2853
- Mortality/morbidity. 2854
- Applicant’s data. 2855
2856
Description of need 2857
- Service characteristics. 2858
- Clinical procedures required. 2859
- Number of referred patients to another site. 2860
- Other available equipment in the area. 2861
Proposal 2862
- Medical equipment. 2863
- Staff. 2864
- Infrastructure. 2865
Resources needed 2866

104
- Investment. 2867
- Operational costs. 2868
- Sources of financing. 2869
2870
9.1.2 Planning Examples: 2871 2872
9.1.2.1 Operating Rooms: 2873
Operating Rooms (ORs) provide a safe and fully controlled environment for 2874
patients undergoing diagnostic and surgical procedures under anesthesia and pre-2875
operative care including post procedure recovery. Medical devices are the 2876
backbone of all types of ORs, ranging from the “same day”, short stay OR up to 2877
the most advanced open heart and neurosurgery ORs. Effective lifecycle 2878
management of medical devices ranging from the basic single use devices and 2879
surgical tools, up to the most advanced robotic surgery and imaging equipment 2880
plays a key role in clinical effectiveness and patient’s surgical outcomes. 2881
2882
9.1.2.2 Medical Devices Storage and Special Areas: 2883
From a planning perspective, it is an essential and major requirement to include 2884
sufficient OR storeroom space for medical devices and equipment. Depending on 2885
the type of OR, storage requirements of at least 10 m2 per OR should be provided. 2886
It is important to ensure the following when designing the OR storage and medical 2887
equipment special areas: 2888
a) Storerooms are best designed in an elongated rectangular to allow easy access 2889
to all items. 2890
b) The design of the Operating Unit should allow for ease of access to the storage 2891
areas for delivery of operating room consumables. Controlled access from an 2892
external corridor is highly desirable. 2893
c) Mobile equipment bays need to be provided for equipment such as portable X-2894
ray equipment, stretchers, trolleys, warming devices and mobile equipment. 2895
d) Equipment bays are best designed as elongated rectangular shapes and may be 2896
combined for space efficiency with an area for testing operating room 2897

105
equipment. This room may be co-located with a general storeroom, or a 2898
dedicated room for testing equipment may be necessary. Direct corridor access 2899
to this room is recommended, with controlled access to the remainder of the 2900
ORs. 2901
2902
9.1.2.3 Planning and Design Considerations: 2903
Several factors must be considered when designing operating rooms including 2904
expected utilization, specialties cases expected, and layout of the operating rooms. 2905
The following must be considered: 2906
a) The number of operating rooms and recovery beds and sizes of the support 2907
areas shall be based on the expected surgical workload 2908
b) The surgical suites corridor layout shall be located and arranged to prevent 2909
non-surgery-related traffic through the suite. This is important for infection 2910
control purposes. 2911
c) The operating room suite shall be designed with a sterile core to have no cross 2912
traffic of clean supplies and soiled/decontaminated supplies and areas. Flow 2913
of clean and soiled/decontaminated supplies and equipment to the OR suites 2914
themselves shall be designed to not compromise universal precautions and 2915
aseptic techniques. 2916
d) The surgical suite shall be divided into three distinct areas; unrestricted, semi-2917
restricted, and restricted. 2918
e) Consideration should be given to the effects of building vibration, as building 2919
vibration could interfere with the accuracy of patient position, testing and 2920
delicate surgeries (e.g. ophthalmic procedures). 2921
f) Patient corridors should be a minimum of 8’-0” wide, to accommodate 2922
wheelchairs, equipment, and gurneys. 2923
g) Operating rooms shall have the following: 2924
1. Provision shall be made for a closed-circuit television (CCTV) system. 2925
2. Communication needs are as follows: patient data computer outlets, 2926
nurse call/intercom system, telephone with speakerphone capability. 2927

106
3. All floors of the ORs to be homogeneous with a coved floor base 2928
extending no less than 6” above the finish floor for floor cleaning 2929
purposes. No floor drains are permitted. 2930
2931
9.1.2.4 OR Space Requirements: 2932
OR space requirements vary depending on the standards implemented and the 2933
operating rooms’ layout implemented (e.g. single or double corridor). It is always 2934
the role of hospital planners to consider local needs and national standards for 2935
recommended spaces. 2936
It is important to understand the medical equipment quantities and requirements 2937
in the operating rooms in order to plan for the utility and space requirements. 2938
Radiology equipment is also used in the OR such as the RF C-arms and mobile x-2939
ray units and radiation protection precautions and shielding must be considered. 2940
2941
No. OR space Recommended space (m2)
1 Operating Room, General 41
2 Operating Room, Special Purpose 65
3 Operating Room, Neurosurgery 42
4 Equipment Room, Neurosurgery 17
5 Operating Room, Cardiac 65
6 Pump Room, Cardiac 17
7 Cystoscopy Room 42
8 Cystoscopy Instrument Prep / Storage Room 19
Table 9.2: Recommended Sizes for OR Spaces (Sample)4 2942
2943
Following is a sample list of medical equipment and devices typically found in 2944
operating rooms: 2945
1. Operating table. 2946
2. Operating lights (ceiling mounted and mobile). 2947
3. Surgical and Neurosurgical microscopes. 2948

107
4. Robotics surgery (Orthopedic, neurosurgery, etc.). 2949
5. Patient monitoring systems. 2950
6. Anesthesia and patient ventilation equipment. 2951
7. Electrosurgical units. 2952
8. Surgical sets. 2953
9. Medical gases (O2, Nitrous, Vacuum, Air 4 Bar & 11 Bar). 2954
2955
Modern ORs are complex. Surgical robotics, other minimally invasive surgical 2956
instrumentation, new and larger imaging devices, and more and more other 2957
equipment are being added to ORs all the time. Therefore, new ORs are being 2958
planned larger and larger. In addition, in busy ORs, efficiency and throughput are 2959
critical. All of these factors need to be taken into consideration in all facets and 2960
phasing of the planning processes for new and remodeled operating rooms. 2961
2962
9.1.2.5 Cardiac Operating Rooms: 2963
The cardiac OR requires some special nonstandard spaces such as a perfusion 2964
room. The perfusion room is for the preparation of perfusion equipment, and for 2965
set-up for cardiac procedures. The perfusion room must be near the cardiac OR 2966
and adjacent to a perfusion storage area with the following main requirements: 2967
a) Heavy duty shelving for storage of perfusion fluids and equipment. 2968
b) Computer workstation for a perfusion technician including power and data 2969
outlets. 2970
c) Bench, sink and cupboard unit for servicing of the perfusion machine. 2971
2972
9.1.2.6 Imaging and Radiology Rooms: 2973
Imaging departments are one of the most strategic departments in any hospital 2974
because of both the diagnostic imaging role in clinical outcomes and the high cost 2975
of imaging equipment. The major imaging rooms in a typical diagnostic imaging 2976
department may include the following: 2977

108
a) Chest x-ray rooms: A specific or specialized radiography room used for routine 2978
chest X-rays and those radiographic procedures that can or should be 2979
performed in an upright position. 2980
b) Computed Radiography (CR): CR uses a special plate technology, scanning 2981
and computer processing, to produce a digital image of a patient’s organ or 2982
body part. This digital image can then be output to a digital reading station, 2983
sent to PACS, and/or printed to a dry processor. 2984
c) Computed Tomography (CT): CT employs ionizing radiation to produce axial 2985
(cross sectional) body section images. Data obtained by X-ray transmission 2986
through the patient is computer analyzed to produce these images. This series 2987
of sectional, planar images can be manipulated to produce different planar 2988
views of the areas of interest and eliminate overlying structures such as bone. 2989
Manipulations of data allow for the selective view of either dense tissues such 2990
as bones or diffuse tissues such as the heart, brain, or lung. CT is used for both 2991
head and body imaging and is applicable to diagnosis, biopsy, and therapy 2992
planning. 2993
d) General radiology rooms: Images of the skull, chest, abdomen, spine, and 2994
extremities are produced by the basic radiographic process. 2995
e) Magnetic Resonance Imaging (MRI): This technique utilizes magnetic and 2996
radio frequency fields to produce computer calculated images of human 2997
anatomy (body tissue) and can also provide body chemistry analysis. While 2998
immersed in a magnetic field, the portion of the body to be scanned is exposed 2999
to energy in the radio frequency range. The effects of this exposure on atomic 3000
nuclei position are read by the computerized system and converted into 3001
images. MRI reflects tissue density and body chemistry. 3002
f) Interventional Radiology (IR): This clinical subspecialty uses fluoroscopy, CT 3003
and ultrasound to guide percutaneous (through the skin) procedures such as 3004
performing biopsies, draining fluids, inserting catheters, or dilating or stenting 3005
narrowed ducts or vessels. IR procedures are complex, requiring a team of 3006
doctors and technicians. As such, they are often performed in the surgical suite, 3007
and scheduled in advance as they require special preparation. IR / Special 3008

109
Procedure rooms can be categorized as angiography rooms and/or 3009
vascular/neuro-vascular rooms. 3010
g) Ultrasound: High frequency sound waves are utilized to determine the 3011
location, size and shape of internal organs based on the differential rates of 3012
reflection of different density tissues. In addition, images can be observed in 3013
real time to reveal motion, and ultrasound systems add coloration to indicate 3014
arterial and venous blood flow. Cyst aspiration and fluid removal are also 3015
procedures done with ultrasound imaging assistance. 3016
3017
One important factor to be considered by hospital planners during the planning of 3018
a radiology department is the radiation protection for patients, radiologists, 3019
radiographers and other hospital staff. This is can be done by providing the 3020
required shielding on the walls, doors and even windows to make sure that leaked 3021
radiation is within allowed limits according to the local national standards. 3022
Space requirements for imaging equipment vary between different international 3023
standards. The location of the imaging equipment within the radiology department 3024
is essential. The following diagram illustrates an example radiology department. 3025
3026
3027
3028
3029
3030
3031
3032
3033
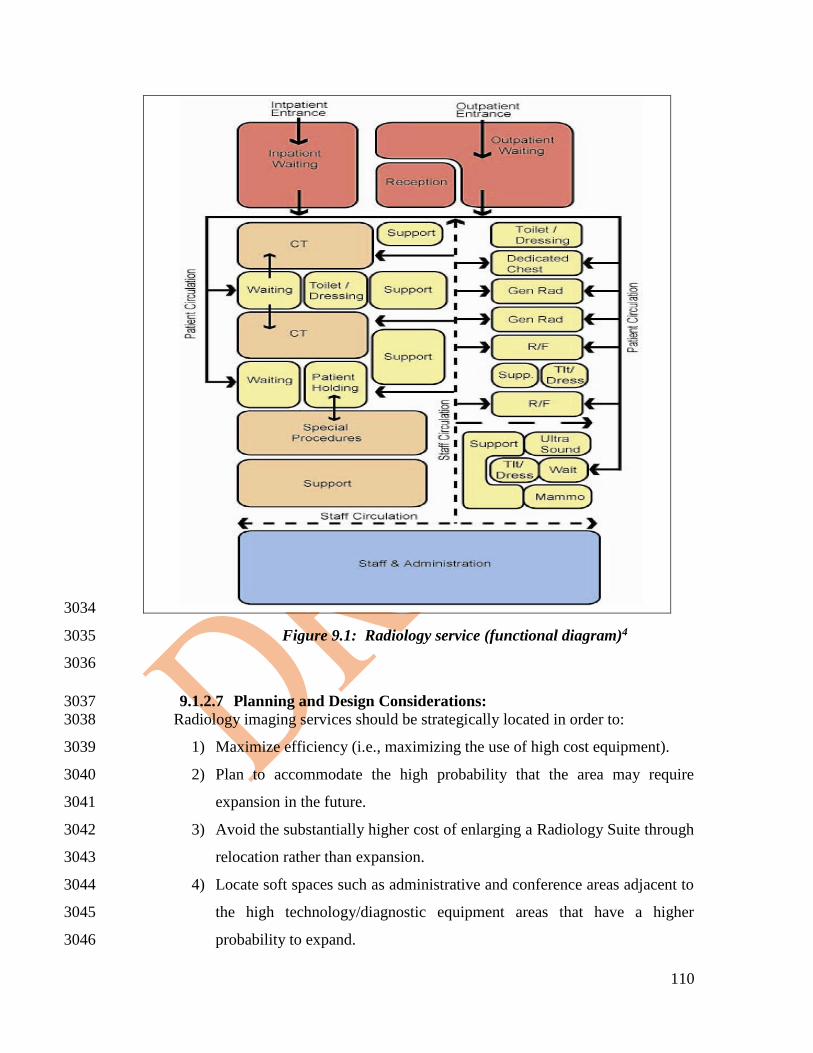
110
3034
Figure 9.1: Radiology service (functional diagram)4 3035
3036
9.1.2.7 Planning and Design Considerations: 3037
Radiology imaging services should be strategically located in order to: 3038
1) Maximize efficiency (i.e., maximizing the use of high cost equipment). 3039
2) Plan to accommodate the high probability that the area may require 3040
expansion in the future. 3041
3) Avoid the substantially higher cost of enlarging a Radiology Suite through 3042
relocation rather than expansion. 3043
4) Locate soft spaces such as administrative and conference areas adjacent to 3044
the high technology/diagnostic equipment areas that have a higher 3045
probability to expand. 3046
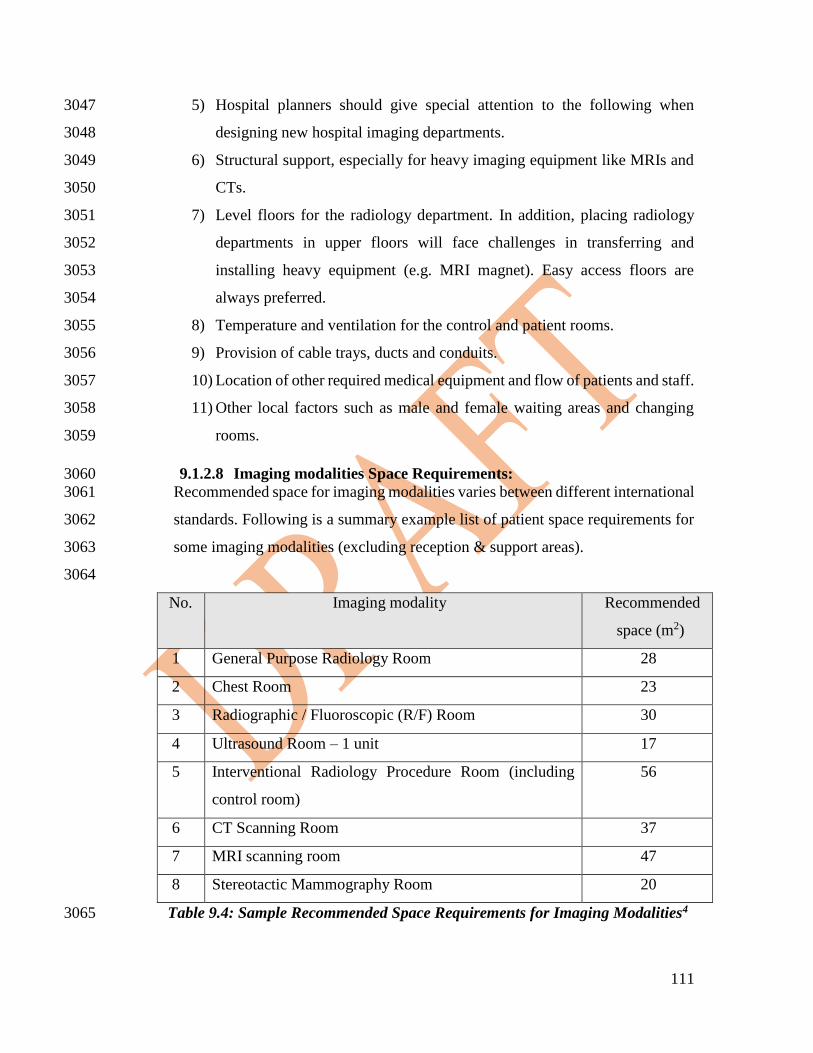
111
5) Hospital planners should give special attention to the following when 3047
designing new hospital imaging departments. 3048
6) Structural support, especially for heavy imaging equipment like MRIs and 3049
CTs. 3050
7) Level floors for the radiology department. In addition, placing radiology 3051
departments in upper floors will face challenges in transferring and 3052
installing heavy equipment (e.g. MRI magnet). Easy access floors are 3053
always preferred. 3054
8) Temperature and ventilation for the control and patient rooms. 3055
9) Provision of cable trays, ducts and conduits. 3056
10) Location of other required medical equipment and flow of patients and staff. 3057
11) Other local factors such as male and female waiting areas and changing 3058
rooms. 3059
9.1.2.8 Imaging modalities Space Requirements: 3060
Recommended space for imaging modalities varies between different international 3061
standards. Following is a summary example list of patient space requirements for 3062
some imaging modalities (excluding reception & support areas). 3063
3064
No. Imaging modality Recommended
space (m2)
1 General Purpose Radiology Room 28
2 Chest Room 23
3 Radiographic / Fluoroscopic (R/F) Room 30
4 Ultrasound Room – 1 unit 17
5 Interventional Radiology Procedure Room (including
control room)
56
6 CT Scanning Room 37
7 MRI scanning room 47
8 Stereotactic Mammography Room 20
Table 9.4: Sample Recommended Space Requirements for Imaging Modalities4 3065

112
9.1.2.9 Imaging Room Layout: 3066
Clinical engineers must always refer to the latest manufacturer’s recommendation 3067
for an imaging modality’s layout to make sure the layout accommodates all 3068
possible configurations. The following figure demonstrates a sample MRI 3069
scanning room layout. 3070
3071
Figure 9.2: MRI Scanning Room Layout4. 3072
113
In summary, new imaging systems and departments are high cost, complex and 3073
strategic investments where good, prior planning is paramount to a successful 3074
long-term efficient operation. HTM involvement in imaging equipment planning 3075
and support is required in order to optimize this important healthcare diagnostic 3076
and treatment tool. 3077
3078
9.1.2.10 Intensive Care Units: 3079
There are several types of intensive care units depending on patient type and age 3080
such as Neonatal ICU, Pediatric ICU, Adult ICU and Coronary Care Units (CCU) 3081
in cardiac centers. In this section, discussion will be kept generic to critical care. 3082
The intensive care unit provides a concentration of clinical expertise, technology 3083
and therapeutic resources, which are coordinating care for the critically ill patient. 3084
3085
Typical list of medical equipment needed for all types of ICU units 3086
include: 3087
- Patient monitoring system. 3088
- Patient ventilator. 3089
- Infusion pump. 3090
- Bedhead unit. 3091
- ICU electrical bed. 3092
- Suction unit. 3093
3094
9.1.2.11 Planning and Design Considerations: 3095
The level of intensive care available should support the role of the hospital. The 3096
role of an ICU will vary, depending on staffing, facilities and support services as 3097
well as the type and number of patients it needs to manage. 3098
3099
9.1.2.12 Main Functional Areas: 3100
a) Intensive Care Units normally consist of the following main functional areas: 3101
b) Family/visitor waiting areas. 3102
c) Patient treatment areas including patient beds and treatment rooms. 3103

114
d) Support areas including clean and dirty utility rooms, storeroom, linen room, 3104
housekeeping/cleaner's room. 3105
e) Administrative / office areas. 3106
f) Staff amenities areas. 3107
3108
The following figure illustrates an example of the ICU functional relation and 3109
layout diagrams: 3110
3111
Figure 9.3: Intensive Care Unit Functional Relationship Diagram3 3112
3113

115
9.1.2.13 Medical Equipment Support: 3114
Depending on the location of the main HTM unit (or biomedical department) and 3115
its distance from the ICU, it must be noted that fastest response is required to 3116
support medical devices in the ICU units. A 24 hours on-call emergency 3117
biomedical support service is recommended. It is common practice to have a full-3118
time biomedical engineer or technician covering OR and ICU during working 3119
hours and on-call emergency service after working hours. The competency list of 3120
biomedical staff covering ICU and OR must include several technical and clinical 3121
skills that must be continuously updated according to an effective outcome-based 3122
competency development program. The training of such biomedical support staff 3123
must be part of the technical specification and tender requirements for OR and 3124
ICU medical devices. 3125
3126
9.1.2.14 Laboratory Support: 3127
All ICU units must have available 24-hr clinical laboratory services. When this 3128
service cannot be provided by the central hospital laboratory, a satellite laboratory 3129
within or immediately adjacent to, the ICU must serve this function. Satellite 3130
facilities must be able to provide minimum chemistry and hematology testing, 3131
including arterial blood gas analysis. It is common practice to have a hematology 3132
analyzer within the ICU unit operated by the ICU nursing staff that are trained by 3133
laboratory staff. 3134
3135
9.1.2.15 Storage Areas: 3136
A dedicated storage area, that is out of traffic paths but conveniently located for 3137
easy access by staff, is required to store mobile equipment such as 3138
cardiopulmonary resuscitation trolleys and mobile x-ray machines, Storage space 3139
considerations should be given to the increasing amount of equipment used in 3140
these units. 3141
3142

116
9.1.2.16 Design and Space Planning: 3143
The recommended space requirements for intensive care units varies between 3144
different international standards, yet it is very common to recommend enough 3145
spaces to allow patient treatment and staff access. 3146
3147
9.1.2.17 Patient Treatment Areas: 3148
Patient bed layout is very important in the ICU. It must provide continuous direct 3149
or indirect visualization for clinical staff. The preferred design is to allow a direct 3150
line of vision between the patient and the central staff station. In ICUs with a 3151
modular design, patients should be visible from their respective nursing 3152
substations. Sliding glass doors and glass partitions facilitate this arrangement and 3153
increase access to the room in emergency situations. 3154
3155
9.1.2.18 Bedside Monitoring: 3156
Bedside patient monitoring systems should be located to permit easy access and 3157
viewing, and should not interfere with the visualization of, or access to, the patient. 3158
The bedside nurse, and/or monitor technician, must be able to observe the 3159
monitored status of each patient at a glance. This goal can be achieved either by a 3160
central monitoring station, or by bedside monitors that permit the observation of 3161
more than one patient simultaneously. Neither of these methods is intended to 3162
replace bedside observation. 3163
Weight-bearing surfaces that support the monitoring equipment should be sturdy 3164
enough to withstand high levels of strain over time. It should be assumed that 3165
monitoring equipment will increase in volume and connectivity over time. 3166
Therefore, space, electrical and IT facilities should be designed accordingly3. 3167
3168
9.1.2.19 Environmental Conditions (Acoustics): 3169
Signals from patient call systems, alarms from monitoring equipment, and 3170
telephones add to auditory sensory overload in critical care units for both patients 3171
and staff. Without reducing their importance or sense of urgency, such signals 3172
should be modulated to a level that will alert staff members yet be rendered less 3173

117
intrusive. For these reasons, floor coverings that absorb sound should be used while 3174
keeping infection control, maintenance, and equipment movement needs under 3175
consideration. Walls and ceilings should be constructed of materials with high 3176
sound absorption capabilities. Ceiling soffits and baffles help reduce echoed 3177
sounds. Doorways should be offset, rather than being placed in symmetrically 3178
opposed positions, to reduce sound transmission. Counters, partitions, and glass 3179
doors are also effective in reducing noise levels3. 3180
3181
9.1.2.20 ICU Space Requirements: 3182
Spaces recommended for the whole ICU and the different sections within the ICU 3183
vary widely between different international standards. Where an open plan 3184
arrangement is provided, bed spaces shall be arranged so that there is a clearance 3185
of at least 1200 mm from the side of the bed to the nearest fixed obstruction 3186
(including bed screens) or wall. At the head of the bed, at least 900 mm clearance 3187
shall be allowed between the bed and any fixed obstruction or wall. 3188
3189
When an open plan arrangement is provided, a circulation space of 2200 mm 3190
minimum clear width shall be provided beyond the dedicated cubicle space4. 3191
Separate cubicles and single patient bedrooms including isolation rooms, shall 3192
have minimum dimensions of 3900 mm x 3900 mm. Following are the main types 3193
of isolation rooms: 3194
Neutral or standard room air pressure, for example standard air conditioning, also 3195
known as Class S. 3196
Positive room air pressure where an immune-compromised patient is protected 3197
from airborne transmission of any infection, Class P. 3198
Negative room air pressure, where others are protected from any airborne 3199
transmission from a patient who may be an infection risk, Class N. 3200
Negative room air pressure with additional barriers, including an Anteroom, also 3201
known as Class Q for quarantine isolation. 3202
3203

118
No. ICU Recommended space (m2)
1 Patient Room, Intensive Care 28
2 Patient Room, Airborne Infection
Isolation
28
3 Nursing station 17
4 Medication Room 10
5 Storage, ICU Equipment 17
6 Workroom, Nurse 11
7 Utility Room, Clean / soiled 11
Table 9.5: Sample recommended spaces for ICU. 3204
3205
3206
9.1.3 Appendix: Emerging Technology Examples: 3207
3208
3209
9.1.3.1 Emerging Technologies Introduction 3210
For new and emerging technologies, it is difficult to predict what will become a 3211
clinical success in the near-future and nearly impossible to predict what will be 3212
commercially successful five to ten years from now. It is also impossible to include 3213
a comprehensive list of emerging technologies in a few pages. Therefore, this 3214
section reviews a few, new and emerging technologies chosen for their potential 3215
impact on healthcare providers and HTM. Included are the following: Multiple 3216
modality systems; 3D printing in healthcare; integrating the Healthcare Enterprise 3217
(IHE) profiles for device integration; medical device surveillance monitoring; 3218
genomics; and a few examples of the use of artificial intelligence (AI) and big data 3219
in healthcare and HTM. 3220
3221
9.1.3.2 Multiple Modality Imaging Systems 3222
Multi-modality imaging such as combinations of CT, MRI, fMRI (functional MRI), 3223
ultrasound, PET, and SPECT imaging combine two of these modalities into one 3224
system with a common gantry. These systems typically use one modality focused 3225
on structure (anatomy) and one focused on function (physiology). Since the patient 3226

119
is placed on a common gantry, problems with patient registration to the correct 3227
reproducible imaging system position are minimized compared to using two 3228
different physical systems and localizing the patient to anatomic landmarks. 3229
3230
Example: in PET/CT, a widely-used technology, the CT scanner is used to image 3231
the brain or other structures, while the PET subsystem provides information about 3232
the function (physiology) of the area in the image. Positron emission tomography 3233
(PET) uses small amounts of radioactive pharmaceuticals called radiotracers, a 3234
special camera detector that detects the radiotracers, and a computer to develop 3235
human-readable images used to evaluate organ and tissue functions. PET scans 3236
measure important body functions, such as blood flow, oxygen use, and glucose 3237
metabolism to help doctors evaluate how well organs and tissues are functioning. 3238
PET is used for brain imaging of patients with seizure disorders, cancer diagnosis 3239
and tumor metastasis determination, heart muscle evaluation and other functions. 3240
Currently, almost all PET scans are performed on instruments that are combined 3241
PET and CT scanners. The combined PET/CT scans provide images that pinpoint 3242
the anatomic location of abnormal physiologic activity. The combined scans have 3243
been shown to provide more accurate diagnoses than the two scans performed 3244
separately. (Griffeth, 2005). 3245
3246
Similar types of multi-modality systems are under development and early 3247
implementation for MR/PET and CT/MR. MR/PET systems have been developed 3248
to stage cancer progression and for the diagnosis and treatment planning for other 3249
diseases. MR imaging has major strengths compared with CT, including superior 3250
soft-tissue contrast resolution, and functional imaging capability through 3251
specialized techniques such as MR spectroscopy. Furthermore, the lack of ionizing 3252
radiation from MR scanners, compared to CT, is highly appealing, particularly for 3253
pediatric and pregnant patients. (Savage, 2013). 3254
3255
In the research stage are systems that combine CT and MR. MR provides high 3256
contrast images and allows doctors to measure functional and even molecular 3257

120
changes. CT provides greater structural detail. Together, they allow doctors to get 3258
a superior picture of processes in action, such as imaging certain types of plaques 3259
on artery walls that are particularly unstable and prone to causing heart attacks or 3260
strokes. A combination of structural, functional, and molecular information is 3261
needed to tell just how dangerous the plaque may be. There are many challenges in 3262
building combined MR/CT systems, including the moving metal of rotating CT 3263
gantries causing interference with MR’s magnetic fields. 3264
3265
Also available are combined CT/radiation therapy linear accelerators and several 3266
combinations of ultrasound scanners with the various other imaging modalities. 3267
These multi-modality systems are all very expensive and complex. Repair and 3268
maintenance of them are primarily the responsibility of the OEM. However, where 3269
HTM staff is appropriately trained, and already supporting the base modality (e.g. 3270
CT scanners), they can be trained to provide some service support for the additional 3271
modality (e.g. PET/CT). In some locations, shared agreements are offered where 3272
HTM shares responsibilities with the OEM and the Healthcare Provider, receives a 3273
discounted service contract. 3274
3275
9.1.3.3 3-D Printing in Healthcare 3276
3-D printers have been used for several years for rapid prototyping of simple and 3277
complex parts in the research and development environment. Parts are made from 3278
various thermoplastic and sintered metal materials. This additive manufacturing 3279
technology has slowly made its way into healthcare and is emerging as a tool for 3280
manufacturing implant parts and custom one-time-use designs, sometimes 3281
originating from patient CT or MRI scans. Applications include custom prosthetics 3282
(e.g. artificial hands); custom assistive devices (e.g. canes, crutches); parts for, and 3283
complete, artificial ankles, knees and other orthopedic implants; casts for fracture 3284
immobilization; and crowns, bridges and other dental products. Another application 3285
is 3-D modeling of anatomy and disease (e.g. tumors) to assist surgeons in complex 3286
surgical planning or construction of complex models of body parts (e.g. heart) for 3287
teaching purposes in lieu of using processed cadavers. (Crawford, 2017). 3288

121
3289
For HTM, 3-D printers have been used to make simple custom parts, such as covers 3290
for protruding connectors prone to damage (Clark, 2017). 3-D printed parts can also 3291
be used for making simple replacement parts for obsolete equipment where the 3292
parts are no longer sold stretching the operational lifespan of these devices. 3293
Regulatory guidance on 3-D printed repair parts is still in its infancy, however, 3294
manufacturer approval should be obtained whenever possible (FDA, 2017). 3295
3296
9.1.3.4 Integrating the Healthcare Enterprise and Emerging Connectivity 3297
Standards 3298
Integrating the Healthcare Enterprise (IHE) is an initiative by healthcare 3299
professionals and the medical device and healthcare IT companies and communities 3300
to improve the way medical devices and computer systems in healthcare share 3301
information. IHE promotes the coordinated use of established standards to address 3302
specific clinical needs in support of patient care. IHE’s Patient Care Devices group 3303
(IHE-PCD) develops medical device integration profiles for a variety of 3304
connectivity “use cases” based on existing standards such as HL-7, DICOM and 3305
IEEE 11073. As more medical device manufacturers develop products that meet 3306
these IHE profiles, and more Healthcare Providers implement them, it will become 3307
much easier to implement the integration of medical devices into the electronic 3308
medical record and other healthcare IT systems. The intent of these IHE PCD 3309
profiles is to give a uniform methodology for representing common data to facilitate 3310
interoperability of systems between different vendors. IHE PCD currently includes 3311
14 different profiles in use or under development ranging from alarm management 3312
to waveform communication (IHE PCD Wiki, 2017). 3313
3314
The following example describes how the implementation of the IHE PCD DEC 3315
profile may help Healthcare Providers and HTM in their device integration projects 3316
now and in the near future. 3317
The Device Enterprise Communication (DEC) profile provides vendor 3318
independent, consistent communication of PCD data to the Electronic Medical 3319

122
Record (EMR) and other clinical repositories. Examples of patient care devices in 3320
this profile include: ICU physiological monitors, vital signs monitors, point of care 3321
blood analyzers, infusion pumps, point of care glucometers, anesthesia systems, 3322
ventilators, and dialysis systems. For example, DEC allows patient monitors to 3323
automatically send vital signs data to the electronic medical record reducing staff 3324
time spent manually entering data and significantly reducing transcription errors. 3325
The data is time stamped with consistent enterprise time. This profile also provides 3326
an option to address the binding of patient identification information (e.g. patient 3327
name, medical record number) with the data from the device (IHE PCD Wiki, 3328
2017). 3329
3330
Another emerging connectivity standards-related technology is FHIR. In order to 3331
improve device integration processes, researchers and the HL-7 organization have 3332
developed a new connectivity standard called Fast Healthcare Interoperability 3333
Resources (FHIR, 2017). Currently (2017) FHIR is published as a “Standard for 3334
Trial Use”. FHIR is a more granular way to exchange data without the rigid 3335
workflow of traditional HL-7. FHIR has a higher emphasis on conformance and 3336
reference implementations than prior versions of HL-7. It has drivers for mobile 3337
healthcare applications and the cloud, medical device integration, and incorporates 3338
more flexible, yet well organized, custom workflow than the HL-7 version 2.x, 3339
currently in widespread use. As FHIR matures beyond the trial use, it is likely that 3340
new applications, and new IHE PCD profiles, will make use of FHIR. 3341
3342
For HTM, IHE and/or FHIR will simplify the integration of medical devices 3343
making support easier. As more and more devices are connected, standards-based 3344
connectivity makes implementing, monitoring and troubleshooting problems 3345
easier. HL-7 private tags and other proprietary constructs are significantly reduced. 3346
New project development time is shortened, problems are decreased, and problem 3347
resolution is quicker. 3348
3349

123
9.1.3.5 Device Surveillance Monitoring 3350
Medical device surveillance monitoring provides self-test data and other device 3351
status information such as battery status to the HTM department automatically from 3352
networked devices. As more and more medical equipment is networked, devices 3353
don't only have the capability to send out clinical data to the EMR, but to also send 3354
device self-test results and other device-status-related information. On some 3355
products, service-related surveillance monitoring is currently available using 3356
proprietary applications. (Zoll, 2017). 3357
3358
In order for medical devices to provide the above described self-reporting, 3359
surveillance features in a standards-based methodology, the IHE PCD group has 3360
drafted a surveillance monitoring profile using HL7-based messaging called the 3361
Medical Equipment Management Device Management Communication 3362
(MEMDMC) (IHE PCD, 2017). MEMDMC covers device identification, 3363
configuration and condition messages (e.g., self-test passed/failed, battery low) and 3364
status (e.g., on/off/standby, battery power/line power). Once a device is already 3365
connected to the network, and delivering clinical data to the EMR, with MEMDMC 3366
that device can also connect to the CMMS or some other HTM-focused application 3367
to send device management and maintenance-related information allowing 3368
problems to be seen prior to any downtime or other clinical impact. IHE PCD’s 3369
MEMDMC profiles has been developed to encode in a standardized manner some 3370
of these maintenance management data. Devices that use this profile will be able to 3371
send to HTM automatically self-tests, battery levels, utilization information (e.g. 3372
hours meter), network parameters, date-sensitive connected consumable (e.g., 3373
defibrillator pads) information that they are nearing their expiration date, and many 3374
other parameters. 3375
3376
9.1.3.6 Artificial Intelligence and Big Data 3377
Clinical decision support systems have been available for several years. For 3378
example, these systems use pattern recognition and other image analysis techniques 3379
to identify potential tumors and other abnormal structures within normal structures, 3380

124
and then display them to a radiologist or pathologist for confirmation and final 3381
diagnosis. These systems also help radiologists and pathologists screen large 3382
numbers of routine scans quicker (e.g. screening mammography). Artificial 3383
intelligence (AI)-based decision making is the next step after computer-aided 3384
decision support. AI can help detect abnormalities on medical images and supervise 3385
diagnostic processes (e.g. what test to do next?). 3386
3387
Technology giants, such as IBM, Google, and Microsoft are building artificial 3388
intelligence solutions to design personalized treatments for some cancers. For 3389
example, instead of spending the time to read through large amounts of research 3390
material themselves, physicians can use IBM’s Watson, the company’s advanced 3391
artificial intelligence platform, to read through dozens of research papers to find 3392
information relevant to a specific patient’s case. Watson has been used in oncology, 3393
radiology and genomics (IBM-Medical Sieve, 2017, IBM-Watson, 2016). 3394
3395
9.1.3.7 Big Data: 3396
Using data analytics electronic health records (EHRs) and other large healthcare 3397
data sets, can be analyzed to help improve the quality and coordination of care, 3398
reduce costs and avoid unnecessary use of resources. Clinical registries, typically 3399
medical specialty or disease focused, provide repositories for large amounts of data 3400
for a specific disease or medical specialty. For example, in 2016, the American 3401
Academy of Dermatology introduced a clinical registry called DataDerm 3402
(DataDerm, 2016). This database contains data on millions of patients from 3403
thousands of dermatologists throughout the US. As more data is collected these can 3404
be used for data analyses, patterns and clinically-relevant trends in diagnosis and 3405
treatment (e.g. which medications work best for a specific skin problem). 3406
3407
Big data can be used in real-time to alert clinicians that a patient’s condition is 3408
trending downward, prior to any obvious symptoms. Within EMRs, and as separate 3409
products, applications are emerging for “early warning” clinical alerts based on 3410
algorithms developed from big data. These applications use multiple parameters, 3411

125
typically vital signs trends plus lab values, and in some cases real-time analysis of 3412
respiratory and cardiac waveforms, with complex computer algorithms and 3413
statistics to predict the risk that an already ill patient will become significantly more 3414
ill very soon. These systems can currently identify that there is a high-risk that the 3415
patient is currently developing serious clinical problems, including early signs of 3416
sepsis, potential for significant bleeding problems, likely respiratory failure leading 3417
to the need for intubation, and some cardiac problems (Moorman, 2017). 3418
3419
For example, if the early warning system shows signs that there is a high risk that 3420
sepsis may be occurring a blood culture can be obtained earlier than typical, and if 3421
positive, early treatment with special antibiotics to help prevent further morbidity 3422
or mortality. Early treatment of sepsis is one key to survival from sepsis. From the 3423
HTM department standpoint, the interface of physiological monitors to the EMR 3424
for vital signs data collection, and waveform analysis in the future, becomes critical 3425
for the proper function of these early warning systems. 3426
3427
9.1.3.8 HTM and Big Data: 3428
Reliability centered maintenance (RCM) and related maintenance strategies have 3429
been used in the airline industry for many years. In the past few years there have 3430
been attempts to use similar data-driven methodologies in HTM to help optimize 3431
planned maintenance. Related efforts are currently underway, from groups under 3432
the auspices of AAMI, to develop standardized data definitions for a variety of 3433
maintenance–related data, and collect data using these standard definitions, with 3434
the ultimate goal to be able to share that data amongst HTM, HTM regulators and 3435
accrediting bodies, and the medical device industry. (Ridgway, 2017). Whether the 3436
HTM field will ever have a widely-shared maintenance data repository remains to 3437
be seen. If this effort ever comes to fruition, there will be an opportunity to use data 3438
analytics to analyze the data and further optimize HTM’s scheduled maintenance 3439
programs. 3440
3441

126
9.1.3.9 Genomic Testing: 3442
The Human Genome Project (HGP) was an international, collaborative research 3443
program whose goal was the complete mapping and understanding of all the genes 3444
of the human being. It was completed in 2003 revealing that there are about 20,500 3445
human genes. Researchers deciphered the human genome in three major ways: The 3446
order or sequence of the bases in our genome’s DNA. The development of maps 3447
which show the locations of genes for major sections of the human chromosomes. 3448
And the production of linkage maps which show how inherited traits, such as those 3449
of genetic diseases, are tracked over generations (Human Genome Project, 2016). 3450
DNA sequence information is important to scientists investigating the functions of 3451
genes. DNA sequencing is a laboratory technique used to determine the exact 3452
sequence of the DNA bases, adenine (A), cytosine (C), guanine (G), and thymine 3453
(T) in a DNA molecule. The DNA base sequence carries the information a cell 3454
needs to assemble protein and RNA molecules. Increasing the speed and reducing 3455
the cost of DNA sequencing technology continues and is resulting in the increased 3456
availability of such testing. 3457
3458
In the clinical arena, genetic screening tests are used for the possible presence of 3459
genetic diseases, or mutant forms of genes associated with increased risk of 3460
developing genetic disorders. Types of mutations include base substitutions (one 3461
base substituted for another); base deletions (one or more bases missing in a DNA 3462
strand), and base additions (one or more bases added to the DNA strand). 3463
3464
From a technology perspective, in order for a potential DNA sample to be analyzed, 3465
it needs to be amplified (copied), fragmented and then analyzed. One common 3466
methodology for amplification is PCR (polymerase chain reaction). This method 3467
involves using short DNA sequences called primers to select the portion of the 3468
genome to be copied. The temperature of the sample is repeatedly raised and 3469
lowered to help a DNA replication enzyme copy the target DNA sequence. The 3470
technique can produce a billion copies of the target sequence in just a few hours. 3471

127
Various other techniques using bacteria and yeast are used to first fragment and 3472
then grow specific segments of the DNA strands for further analysis using 3473
electrophoresis and other analysis techniques. Different sequencers use different 3474
analytic techniques and offer varying speed and accuracy. Choosing a DNA 3475
sequencer and method will typically depend on the clinical and research objectives 3476
and available budget. 3477
3478
Clinical examples of DNA analysis include: Screening for well-known genetic 3479
diseases such as cystic fibrosis, finding mutations that cause intellectual 3480
disabilities; locating certain mutations that cause one form of colorectal cancer that 3481
is treatable with aspirin; tracking donor tissue DNA to track transplant rejection 3482
sooner than when symptoms appear; and tracking tumor DNA to discover cancer 3483
spread sooner. Medications for various cancer and cardiovascular diseases have 3484
been developed that are specific for diseases confirmed by DNA testing. 3485
The increasing use of comprehensive genomic profiling in clinical practice for the 3486
genomic characterization of tumors is having an increasingly important impact on 3487
patients with cancer, empowering the rational use of targeted therapies and driving 3488
smarter, faster drug development. New sequencers and analyzers, and large 3489
databases of DNA test results, are bringing personalized cancer treatment to the 3490
marketplace (Mesko, 2017). 3491
3492
One challenge for the widespread use of genetic testing in clinical practice is that 3493
current electronic medical records (EMRs) store genetic test results only in free text 3494
reports. As the amount of genetic data continues to rapidly grow, EMRs will need 3495
to become capable of allowing physicians to effectively process this new type of 3496
information. Genetic results differ from traditional laboratory tests because of their 3497
persistent nature, broad scope, and complex interpretation. While current EMRs 3498
present laboratory results in a structured and predictable format, genetic data are 3499
often stored as free text and presented as cumbersome, highly detailed reports. 3500
Building EMRs that can adapt to the genetic information needs of the clinician is a 3501
challenge that is yet to be solved. 3502

128
Technology support for sequencers, and the other complex equipment used for 3503
genetic testing, is typically provided by the OEM. Biomedical engineering support 3504
has made some progress with a few third-party and in-house providers supporting 3505
clinical and research systems. As more and more systems and companies develop 3506
products, and the products become less expensive, increased technical training and 3507
support opportunities should become available. 3508
3509
9.1.3.10 Emerging Technology Final Comments: 3510
Of course, there are many more emerging technologies. Medical devices are getting 3511
smaller and smaller, with some integrated into cell phones (e.g. cell phone-based 3512
ultrasound machines). Whether in a patient room, in an MRI gantry, or in surgery, 3513
more and more companies and hospitals are providing entertainment and relaxation 3514
audio and video experiences for the patient. For example, MRI exams are very 3515
noisy and claustrophobic, and can take one hour or longer. The patient needs to 3516
remain very still during the MRI exam. Providing, soothing, relaxing, or distracting 3517
audio and video entertainment, preferably as chosen by the patient, can improve the 3518
patient experience, and reduce exam re-takes. Similar systems are being offered for 3519
operating rooms, and procedure areas. (GE Healthcare, 2017). 3520
3521
There are many other new technologies on the horizon including: Vocal analytics 3522
that use voice patterns to detect various diseases and robotic devices that not only 3523
scan for, and automatically identify suspect skin lesions, but can treat them using 3524
robot-guided laser irradiation (Mesko, 2017). 3525
3526
The above discussion is just a small sample of emerging technologies. Modern 3527
technology is rapidly changing and improving. It remains to be seen which 3528
technologies will have widespread positive impact on population health, healthcare 3529
providers, the practice of medicine, and HTM. 3530
3531
3532

129
9.2 Procurement 3533
3534
Table (9.6): SFDA Storage requirements for medical devices. 3535
Storage Area 1 Storage areas should: Be designed or adapted for the storage of medical devices. Be clean. Be equipped with thermometers/hygrometers. The
thermometers/hygrometers should be placed to allow effective monitoring where temperatures/humidity are most likely to fluctuate or rise.
Have surfaces and shelves, if applicable, made of or covered by an impermeable material to enable proper and safe cleaning.
Be suitably spaced to allow cleaning and inspection. Include a physically separate area for keeping damaged,
defective, expired, counterfeit or recalled medical devices. This area should be clearly labeled and controlled to prevent the use of these devices until a final decision is taken on their fate.
Be adequately lit. Be adequately ventilated. Have an emergency plan set up and used in case of an electricity
shutdown (power outage), if applicable.
Traceability in the Storage
Area
2 In the case of a recall/field safety notice by the SFDA or the manufacturer, the healthcare providers should be able to trace a product in the storage area by its lot/batch/serial number.
3 The expiration dates of medical devices in the storage area should be monitored through periodic inventories to avoid unintended dispatch of expired medical devices.
Transportation
4 Vehicles used to transport medical devices should be properly designed and equipped to ensure protection from different environmental and weather conditions in which it operates.

130
5 Medical devices should be transported in such a manner that does NOT allow exceeding of appropriate temperature and relative humidity conditions which could negatively affect their integrity and quality.
6 Medical devices should be transported and carried carefully in a manner that corresponds to the special transport precautions and requirements.
7 Any case of spoilage or breakage should be reported.
8 The transporting vehicle or containers should be adequately suitable for the intended purpose and cleaned.
Manufacturer’s Instructions/
Requirements
9 Medical devices should be stored, handled and transported under conditions specified by the manufacturer’s instructions/requirements to prevent deterioration. These conditions might be related to one or more of the following:
o Temperature (all the medical devices should be kept during storage and/or transportation at temperature ranges specified by the manufacturer).
o Moister and humidity. o Exposure to light. o The direction the package should face. o The maximum number of packages stacked above each other. o Other specific instructions/requirements.
Note 1: If the packaging labelling do not include information about the required storage and transportation conditions of a medical device, healthcare providers should obtain such information from the manufacturer and/or its authorized representative located within the KSA. Note 2: If the manufacturer does not specify the temperature values or not define the storage conditions on the packaging labelling, see Annex 1 to determine these values and the set of storage definitions. Note 3:

131
If the manufacturer requires medical devices to be stored or transported under certain conditions (e.g. temperature and humidity), healthcare providers should monitor and periodically record these conditions.
Sterile Medical Devices
10 In addition to manufacturer-specific instructions, medical devices that are dispatched in a sterile state, should be stored, handled and transported in a manner that protects their packaging from:
o exposure to moisture. o direct sunlight. o Damage. o dirt and unclean environment.
Note: Sterile medical devices should be considered unsterile if packaging loses its integrity.
Staff
11 Staff involved in the storage, handling and/or transport of medical devices should:
o have proper knowledge about these activities, and o be able to deal with those devices that have different storage and
transportation requirements, if applicable.
Written Procedures
12 Healthcare providers should have a written procedure that describes the practices taken to ensure medical devices are stored, handled and/or transported based on the manufacturer’s instructions/requirements. The written procedure ideally should be part of a quality management system.
3536
3537
3538
3539
3540
3541
3542
3543
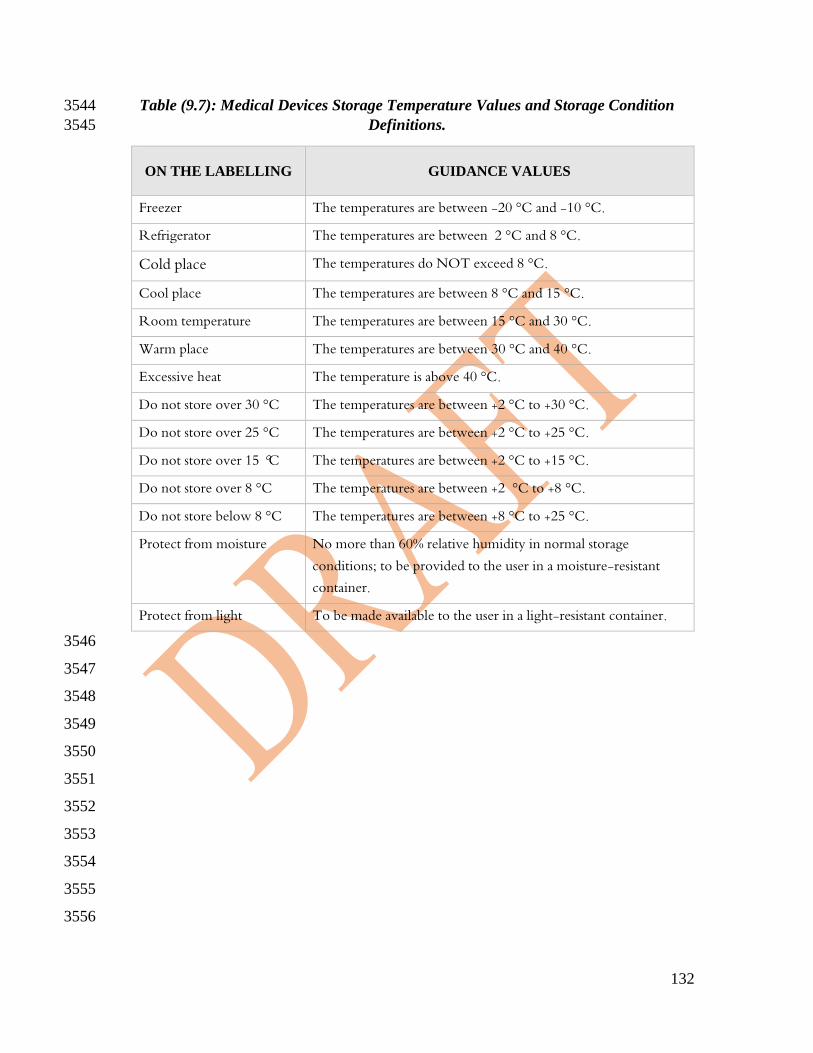
132
Table (9.7): Medical Devices Storage Temperature Values and Storage Condition 3544
Definitions. 3545
ON THE LABELLING GUIDANCE VALUES
Freezer The temperatures are between -20 °C and -10 °C.
Refrigerator The temperatures are between 2 °C and 8 °C.
Cold place The temperatures do NOT exceed 8 °C.
Cool place The temperatures are between 8 °C and 15 °C.
Room temperature The temperatures are between 15 °C and 30 °C.
Warm place The temperatures are between 30 °C and 40 °C.
Excessive heat The temperature is above 40 °C.
Do not store over 30 °C The temperatures are between +2 °C to +30 °C.
Do not store over 25 °C The temperatures are between +2 °C to +25 °C.
Do not store over 15 °C The temperatures are between +2 °C to +15 °C.
Do not store over 8 °C The temperatures are between +2 °C to +8 °C.
Do not store below 8 °C The temperatures are between +8 °C to +25 °C.
Protect from moisture No more than 60% relative humidity in normal storage conditions; to be provided to the user in a moisture-resistant container.
Protect from light To be made available to the user in a light-resistant container. 3546
3547
3548
3549
3550
3551
3552
3553
3554
3555
3556

133
9.3 Device Inventory Auditing Example 3557
3558
Equipment inventories can be audited using sampling techniques of which there are many 3559
techniques. A random sample is taken based on the inventory size (population) and 3560
accuracy expected. If the results do not meet the accuracy expected, then a complete audit 3561
is required or depending on the method selected, a second random sample taken if those 3562
results are not met, then a complete inventory audit must be completed. 3563
3564
Sampling
Procedure
ANSI/ASQC
Z1.4
Product Population
Range
Step 1
Sample
Size
Step 1
Accept
Step 1
Reject
Move to step
IF
unacceptable
is:
Step 2
Sample
Size
Step 2
Accept
Step 2
Reject
Single 151 280 32 3 4
Double 151 280 20 1 4 2 to 3 20 4 5
Single 280 500 50 5 6
Double 280 500 32 2 5 3 to 4 32 6 7
Single 501 1,200 80 7 8
Double 501 1,200 50 3 7 4 to 6 50 8 9
Single 1,201 3,200 125 10 11
Double 1,201 3,200 80 5 9 6 to 8 80 12 13
Single 3,201 10,000 200 14 15
Double 3,201 10,000 125 7 11 8 to 10 125 18 19
Single 10,001 35,000 315 21 22
Double 10,001 35,000 200 11 16 12 to 15 200 26 27
Table 9.8: Attribute sampling for inventory audit. Reference: ANSI/ASQC Z1.4. 3565 See: https://www.sqconline.com/sampling-attributes-plan. 3566
3567
3568
For example, in the chart above, (Table 9.8), if there is a population of 5,000 devices, based 3569
on a +- 4% accuracy requirement, two options are provided for an inventory audit. 3570
Option 1: Single sampling: 3571
a) 5,000 devices in device population 3572
b) 4% quality limit (see references for other quality limits) 3573
c) From chart Figure 5.x: 3574

134
c.1.1 200 device identification numbers randomly are selected 3575
from CMMS or other source. Note the sample has to be 3576
randomly selected or this process does not work! 3577
c.1.2 If the identification numbers match (serial numbers, asset 3578
id numbers etc.) the device passed audit. Otherwise it is 3579
counted as unacceptable. 3580
c.1.3 If 14 or fewer of the 200 devices audited are acceptable, 3581
the process is finished and the audit is complete. 3582
c.1.4 If 15 or more are unacceptable the audit is unacceptable 3583
and all devices would have to be inventoried. 3584
3585
Option 2: Double sampling: 3586
a) 5,000 devices in device population 3587
b) 4% quality limit (see references for other quality limits) 3588
c) From chart Figure 5.x: 3589
c.1.1 125 device identification numbers are randomly selected 3590
from CMMS or other source. Note the sample has to be 3591
randomly selected or this process does not work! 3592
c.1.2 If the identification numbers match (serial numbers, asset 3593
id numbers etc) the device passed audit. Otherwise it is 3594
counted as unacceptable. 3595
c.1.3 If 7 or fewer of the 125 devices audited are acceptable, 3596
the process is finished and the audit is complete. 3597
c.1.4 If 11 or more audited devices are unacceptable the audit 3598
is unacceptable and all devices would have to be 3599
inventoried. 3600
c.1.5 If between 8 an 10 device id audits were unacceptable 3601
then randomly select a second sample of 125 devices and: 3602
c.1.5.1.1 If a total of 18 or fewer of the 250 devices 3603
audited are acceptable, the process is finished 3604
and the audit is complete. 3605

135
c.1.5.1.2 If 19 or more of the 250 audited devices are 3606
unacceptable the audit is unacceptable and all 3607
devices would have to be inventoried. 3608
3609