Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Mar. 2010, p. 1653–1660 Vol. 76, No. 50099-2240/10/$12.00 doi:10.1128/AEM.02254-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Probing the Molecular Determinant for the Catalytic Efficiency ofL-Arabinose Isomerase from Bacillus licheniformis�†
Ponnandy Prabhu,1 Marimuthu Jeya,2 and Jung-Kul Lee2,3*Department of Bioscience and Biotechnology,1 Department of Chemical Engineering,2 and Institute of Biomedical Science and
Technology,3 Konkuk University, 1 Hwayang-Dong, Gwangjin-Gu, Seoul 143-701, South Korea
Received 18 September 2009/Accepted 26 December 2009
Bacillus licheniformis L-arabinose isomerase (L-AI) is distinguished from other L-AIs by its high degree ofsubstrate specificity for L-arabinose and its high turnover rate. A systematic strategy that included a sequencealignment-based first screening of residues and a homology model-based second screening, followed bysite-directed mutagenesis to alter individual screened residues, was used to study the molecular determinantsfor the catalytic efficiency of B. licheniformis L-AI. One conserved amino acid, Y333, in the substrate bindingpocket of the wild-type B. licheniformis L-AI was identified as an important residue affecting the catalyticefficiency of B. licheniformis L-AI. Further insights into the function of residue Y333 were obtained by replacingit with other aromatic, nonpolar hydrophobic amino acids or polar amino acids. Replacing Y333 with thearomatic amino acid Phe did not alter catalytic efficiency toward L-arabinose. In contrast, the activities ofmutants containing a hydrophobic amino acid (Ala, Val, or Leu) at position 333 decreased as the size of thehydrophobic side chain of the amino acid decreased. However, mutants containing hydrophilic and chargedamino acids, such as Asp, Glu, and Lys, showed almost no activity with L-arabinose. These data and amolecular dynamics simulation suggest that Y333 is involved in the catalytic efficiency of B. licheniformis L-AI.
L-Arabinose isomerase (L-AI) is an enzyme that mediates invivo isomerization between L-arabinose and L-ribulose as wellas in vitro isomerization of D-galactose and D-tagatose (20).L-Ribulose (L-erythro-pentulose) is a rare and expensive keto-pentose sugar (1) that can be used as a precursor for theproduction of other rare sugars of high market value, such asL-ribose. Despite being a common metabolic intermediate indifferent organisms, L-ribulose is scarce in nature. The marketfor rare and unnatural sugars has been growing, especially inthe sweetener and pharmaceutical industries. For example,several modified nucleosides derived from L-sugars have beenshown to act as potent antiviral agents and are also useful inantigen therapy. Derivatives of rare sugars have also been usedas agents against hepatitis B virus and human immunodefi-ciency virus (2, 22).
For these reasons, interest in the enzymology of rare sugarshas also been increasing. Various forms of L-AI from a varietyof organisms have been characterized, and some have shownpotential for industrial use. Several highly thermotolerant en-zyme forms from Thermotoga maritima (12), Thermotoga nea-politana (10), Bacillus stearothermophilus (18), Thermoanaero-bacter mathranii (9), and Lactobacillus plantarum (5) have beencharacterized previously. All of these reported L-AIs tend tohave broad specificity, although a few L-AIs with high degreesof substrate specificity for L-arabinose have also been docu-mented.
The enzyme properties of L-AIs have been examined by
engineering several forms by error-prone PCR and site-di-rected mutagenesis. Galactose conversion was reportedly en-hanced 20% following site-directed introduction of a doublemutation (C450S-N475K) into L-AI (16). Error-prone PCRmanipulation of L-AI from Geobacillus stearothermophilus re-sulted in a shift in temperature specificity from 60 to 65°C andincreased isomerization activity (11). All of these previouslyreported mutational studies have been aimed at improvingenzymatic properties for industrial application. However, eventhough the three-dimensional (3D) structure of Escherichiacoli L-AI has been determined previously (15), few new struc-tural studies have been performed to decipher the reactionmechanism of this enzyme. Rhimi et al. (19) have reported animportant role for D308, F329, E351, and H446 in catalysis, asindicated by findings from site-directed mutagenesis. Nonethe-less, detailed analysis of the important molecular determinantscontrolling the catalytic activities of the L-AIs is still lacking.
Previously, we have reported the cloning and characteriza-tion of a novel L-AI from Bacillus licheniformis (17). Thisenzyme can be distinguished from other L-AIs by its wide pHrange, high degree of substrate specificity for L-arabinose, andextremely high turnover rate. In the present paper, we reportthe identification of an important amino acid residue respon-sible for the catalytic efficiency of L-AIs, as determined by asystematic screening process composed of sequence alignmentand molecular dynamics (MD) simulation, followed by site-directed mutagenesis. Using the crystal structure of E. coli L-AIas a template, we have built a 3D model of B. licheniformisL-AI. Analysis of the 3D model of B. licheniformis L-AI dockedwith L-arabinose, followed by a systematic screening process,showed that Y333 interacted with the substrate, suggesting thatthis residue in B. licheniformis L-AI may be essential for catal-ysis. We further characterized the role of Y333 in B. licheni-formis L-AI binding of and catalytic efficiency for L-arabinose.
* Corresponding author. Mailing address: Department of Chem-ical Engineering, Konkuk University, 1 Hwayang-dong, Gwangjin-gu,Seoul 143-701, South Korea. Phone: 82-2-450-3505. Fax: 82-2-458-3504. E-mail: [email protected].
† Supplemental material for this article may be found at http://aem.asm.org/.
� Published ahead of print on 4 January 2010.
1653
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

MATERIALS AND METHODS
Materials, bacterial strains, and culture conditions. Reagents for PCR andEx-Taq DNA polymerase were purchased from Promega (Madison, WI). Re-striction enzymes were obtained from New England Biolabs (MA). The pQE-80L expression vector, a plasmid isolation kit, and an Ni-nitrilotriacetic acidSuperflow column for purification were from Qiagen (Hilden, Germany). Oli-gonucleotide primers were obtained from Bioneer (Daejeon, South Korea).Electrophoresis reagents were from Bio-Rad, and all chemicals for assays werefrom Sigma-Aldrich (St. Louis, MO). A plasmid containing the wild-type B.licheniformis L-AI gene (17) was used for the production of wild-type B. licheni-formis L-AI protein. The araA gene from E. coli W3110 was amplified by PCRusing two oligonucleotide primers, 5�-CCGGAATTCATGACGATTTTTGATAATTATG-3� (an EcoRI restriction site is underlined) and 5�-ATTACTCGAGGCGACGAAACCCGTAATAC-3� (an XhoI restriction site is underlined). ThearaA gene released from the pGEM-T Easy vector (Promega) was ligated withthe pET-28a vector to give pET-araA, in which araA is under the control of theT7 promoter. The cloned gene was confirmed to be free of point mutations byDNA sequencing. E. coli BL21(DE3) was transformed with the recombinantplasmid for protein expression. E. coli strains harboring wild-type and mutated B.licheniformis L-AI genes for protein expression were grown in Luria-Bertani (LB)medium supplemented with ampicillin (100 �g/ml) at 37°C. E. coli strains har-boring wild-type and mutated genes for E. coli L-AI were grown in LB mediumsupplemented with kanamycin (50 �g/ml). Isopropyl-�-D-thiogalactopyranoside(IPTG) was then added to the culture medium at a final concentration of 0.5mM, and incubation continued with shaking at 37°C.
Site-directed mutagenesis in B. licheniformis L-AI. Site-directed mutagenesiswas carried out by using the QuikChange site-directed mutagenesis kit fromStratagene (La Jolla, CA). The recombinant plasmid pQE-araA (17) containingthe wild-type L-AI gene was used as the DNA template. The plasmids containingthe correct mutant genes were then used to transform E. coli BL21(DE3), andcolonies selected by ampicillin resistance were used for protein expression.
Purification and protein quantification. Wild-type and mutant enzymes werepurified by the same procedure. Cell pellets were suspended in 20 mM sodiumphosphate buffer (pH 7.5). The cell suspension was incubated on ice for 30 minin the presence of 1 mg/ml lysozyme. Cell disruption was carried out by sonica-tion at 4°C for 5 min, and the lysate was centrifuged at 14,000 � g for 20 min at4°C to remove the cell debris. The resulting crude extract was retained forpurification. The cell extract was applied to an Ni-nitrilotriacetic acid Superflowcolumn (3.4 by 13.5 cm; Qiagen) previously equilibrated with a binding buffer (50mM NaH2PO4, 300 mM NaCl, pH 8.0). Unbound proteins were washed out fromthe column with a washing buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mMimidazole, pH 8.0). Then the protein was eluted from the column with an elutionbuffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). Enzymefractions were analyzed by sodium dodecyl sulfate–12% polyacrylamide gel elec-trophoresis (SDS–12% PAGE) and visualized by staining with Coomassie blueR250. Protein concentrations were determined by the Bradford method usingbovine serum albumin as a standard protein (3).
Enzyme assay and determination of kinetic parameters. L-AI activity wasmeasured by determination of the amount of L-ribulose formed. Under standardconditions, the reaction mixture contained 1 mM MnCl2, �15 �g of enzyme, 250mM L-arabinose (the substrate), and 20 mM phosphate buffer (pH 7.5) to bringthe final volume to 100 �l. The reaction mixture was incubated at 50°C for 5 minand then cooled on ice to stop the reaction. The L-ribulose generated wasevaluated by the cysteine-carbazole-sulfuric acid method, and the absorbance at560 nm was measured (6). Kinetic parameters for B. licheniformis L-AI weredetermined by using a mixture of 20 mM phosphate buffer (pH 7.5), 1 mM Mn2�,and 1 to 1,200 mM substrate (L-arabinose). One unit of L-AI activity was definedas the amount of enzyme catalyzing the formation of 1 �mol ketosugar per minunder the above-specified conditions.
Homology modeling. The 3D homology models of the wild-type and all mutantproteins were generated using the Build Homology Models module in theMODELER application of Discovery Studio 2.1 (DS 2.1; Accelrys Software Inc.,San Diego, CA). The crystal structure of E. coli L-AI (Protein Data Bankaccession code 2ajtA) was used as a template. Comparative modeling was per-formed to generate the most probable structure of the query protein by align-ment with template sequences, simultaneously satisfying spatial restraints andlocal molecular geometry. The fitness of the model sequences in the present 3Denvironment was evaluated by the Profile-3D Score/Verify Protein tool inMODELER as implemented in DS 2.1. A discrete optimized protein energy(DOPE) score in MODELER was also calculated to determine the quality ofprotein structures. The root mean square deviation (RMSD) between the modelsand the template was calculated by superimposing the models onto the template
crystal structure. The evaluated 3D model was used for docking and postdockinganalyses. Hydrogen atoms were first added to the 3D models, and then the addedhydrogen atoms were minimized for stable energy conformation and relaxationof the conformation from close contacts. Different substrate molecules weredocked into the substrate binding pockets (SBP) of B. licheniformis L-AI and amutant model by using C-DOCKER, an MD simulation-annealing-based algo-rithm module from DS 2.1 (23). Different poses were then created using randomrigid-body rotation followed by simulated annealing. Before docking, the struc-tures of the protein, the substrate, and the corresponding complexes were sub-jected to energy minimization using a CHARMm (4) force field as implementedin DS 2.1. A full potential final minimization was then used to refine the substrate(ligand) poses. The substrate orientation which gave the lowest interaction en-ergy was chosen for another round of docking. Based on the C-DOCKERenergy, the docked conformation of the substrate was retrieved for postdockinganalysis.
Analytical methods and protein database search. Optical spectra were re-corded with a Cary 100 Bio UV-Vis spectrophotometer (Varian, Palo Alto, CA).Circular dichroism (CD) experiments were performed with a Jasco J-815 spec-trophotometer at 20°C. Each spectrum was recorded in the 190- to 300-nmregion at the rate of 100 nm/min. CD spectra were corrected with respect to thebaseline, and the measured ellipticity for each sample was expressed in millide-grees. The amino acid sequences deduced from the araA gene sequences of B.licheniformis were compared with those of related enzymes from other sources byusing the BLAST network at the National Center for Biotechnology Informa-tion. The multiple-sequence alignment was performed with the ClustalW pro-gram.
RESULTS
Sequence alignment. To locate the conserved residues inB. licheniformis L-AI, the amino acid sequence from B.licheniformis was aligned with other L-AI sequences fromBacillus halodurans, Lactobacillus lactis, Lactobacillus plan-tarum, Thermotoga maritima, E. coli, Klebsiella pneumoniae,Thermoanaerobacter mathranii, Thermotoga neapolitana, Bacil-lus stearothermophilus, Geobacillus stearothermophilus, Ther-mus sp., Geobacillus thermodenitrificans, and Bacillus subtilis.Multiple-sequence alignment of L-AIs from these organismsrevealed 60 different amino acids, including the 4 active siteresidues (E306, E331, H348, and H447 in B. licheniformisL-AI), that were totally conserved (100% identical) throughoutthe sequences (see Fig. S1 in the supplemental material).
Homology modeling. B. licheniformis L-AI had a level ofsequence identity to E. coli L-AI of 50%. By taking advantageof the X-ray crystal structure of E. coli L-AI (Protein DataBank entry 2ajtA) and molecular modeling, a homology modelof B. licheniformis L-AI was constructed (Fig. 1A). The gener-ated model was then validated by Ramachandran plots (14). Inthe B. licheniformis L-AI model, 97.8% of residues were lo-cated within the allowed regions, with 91.8% of the residues inthe favorable region and 6% of the residues in the remainingallowed region. Only 2.2% of the residues were located in theoutlier regions of the Ramachandran plot. The Profile-3Dscore for the model was 175, versus the maximum expectedscore of 212. The constructed model was also evaluated bysuperimposing it onto the template crystal structure, and theRMSD between the model and the template was 0.51 Å basedon C-� atoms.
The substrate L-arabinose was docked into the homologymodel using DS 2.1 software. A total of 34 amino acid residues,including the 4 active site residues, were found within 5 Å ofthe SBP (see Fig. S2 in the supplemental material). Putativeactive site residues of E. coli L-AI had previously been pro-posed to be E306, E333, H350, and H450 based on the crystal
1654 PRABHU ET AL. APPL. ENVIRON. MICROBIOL.
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG. 1. (A) Complete model of L-AI from B. licheniformis, with the four catalytic residues depicted in stick form. (B) Superimposition of theproposed catalytic residues of E. coli L-AI (brown) and B. licheniformis L-AI (violet), showing the similar arrangements and orientations of activesite residues.
1655
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

structure of E. coli L-fucose isomerase (21). Upon superimpo-sition, residues E306, E331, H348, and H447 of B. licheniformisL-AI corresponded to the proposed putative catalytic residues(E306, E333, H350, and H450) of E. coli L-AI (Fig. 1B).
In order to confirm these findings, site-directed mutagenesisto change each of these four residues to Ala was performed.The E306A, E331A, H348A, and H447A mutant proteinshad no measurable isomerase activity, supporting a role for thecorresponding mutated residues as active site residues re-quired for isomerization (15). Upon confirming the role of the4 putative catalytic residues, we turned our focus to 12 otherconserved residues within 5 Å of the SBP of B. licheniformisL-AI. The roles of 5 of these 12 conserved residues, F279,D308, F329, E351, and H446, have been described previously(19). The roles of the remaining seven residues (M185, T276,Y333, L345, M349, I370, and W439) were therefore investi-gated by further site-directed mutagenesis (Fig. 2).
Alanine substitution for selected residues. To probe thefunctional roles of the selected conserved residues, all selected
residues were individually mutated to Ala. The recombinantenzymes carrying an M349A, W439A, T276A, L345A, I370A,M185A, or Y333A mutation were expressed and purified (Fig.3). When the activities of the mutants with L-arabinose wereanalyzed and compared with that of wild-type B. licheniformisL-AI, only the substitution at Y333 was found to cause anysignificant change in L-AI activity (data not shown). The spe-cific activity of the Y333A mutant was determined to be 3�mol/min/mg of protein for L-arabinose, which correspondedto 2.8% of that of the wild-type enzyme (105 �mol/min/mg ofprotein). This pronounced loss of activity indicated that Y333in the SBP near the substrate significantly modulated the cat-alytic efficiency for the substrate. Thus, this residue was con-sidered to be a crucial determinant of the catalytic efficiency ofB. licheniformis L-AI. The role of position 333 in catalyticefficiency was therefore further investigated by thorough site-directed mutagenesis.
Site-directed mutagenesis at position 333. Tyr in position333 was replaced with nonpolar aromatic, nonpolar hydropho-
FIG. 2. Conserved residues (pink) around the catalytic amino acids (colored according to elemental properties) selected for site-directedmutagenesis analysis.
1656 PRABHU ET AL. APPL. ENVIRON. MICROBIOL.
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

bic, and polar/charged residues by site-directed mutagenesis.All mutants were expressed at a level similar to the wild type(17). All purified mutant proteins exhibited similar CD spectra,with ellipticity minima of comparable amplitudes in the 220- to230-nm range (Fig. 4). This observation is a good indicationthat all enzymes were properly folded. When Y333 was re-placed with polar, charged amino acids (yielding mutationsY333K, Y333E, and Y333D), the mutant enzymes showed noactivity toward L-arabinose. When Y333 was replaced withnonpolar, aliphatic, and hydrophobic amino acids (generatingY333I, Y333V, and Y333A mutants), the activity with L-arabi-nose was significantly decreased. The specific activities ofY333I, Y333V, and Y333A mutants were 28, 18, and 3 �mol/min/mg of protein, respectively, which were 27, 17, and 2.8% ofwild-type B. licheniformis L-AI activity, respectively.
To further investigate the role of the aromatic ring of Y333in the active site, the residue was replaced with Phe or Trp.Neither of these replacements altered the activity significantly,as the activities of Y333F and Y333W mutants were 91 and 76
�mol/min/mg of protein, respectively, compared to that of thewild type of 105 �mol/min/mg of protein. This finding suggeststhat the aromatic ring of Tyr, Phe, or Trp is likely to beinvolved in binding the pyranosyl ring of L-arabinose. Site-directed mutagenesis in E. coli L-AI was performed to mutateY335, which is the residue corresponding to B. licheniformisL-AI Y333, to Ala. The Y335A mutant exhibited significantlydecreased L-AI activity toward L-arabinose (less than 2% ofwild-type E. coli L-AI activity), indicating that the residue atposition 335 is critical for the activity of E. coli L-AI.
Kinetic analyses of wild-type and Tyr333 mutant enzymes.The kinetic parameters determined for purified wild-typeand mutant B. licheniformis L-AI enzymes acting on L-arabi-nose are shown in Table 1. Among the mutants containing anonpolar aliphatic residue, the decrease in catalytic effi-ciency (the kcat/Km ratio) was correlated with the decrease inthe size of the amino acid side chain. Comparisons of thekinetic parameters for Y333 mutants to those for the wild typesuggested that the maximum turnover rate and catalytic effi-ciency were maintained by aromatic amino acids at position333 (Table 1).
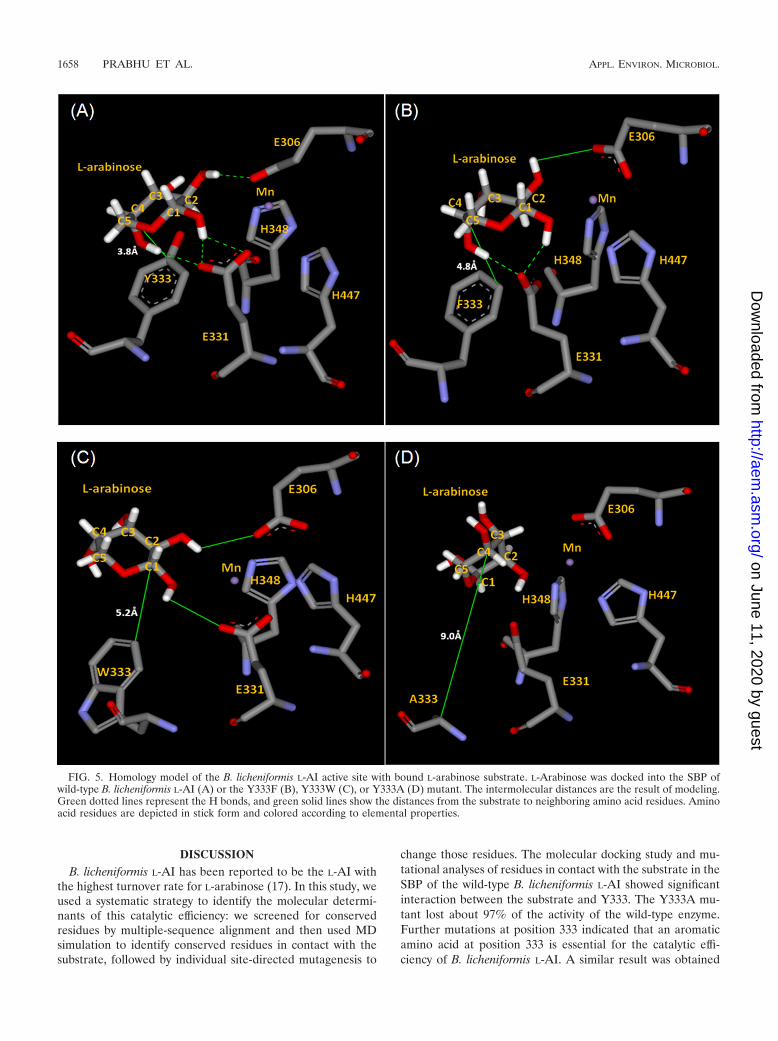
Changes in �(�G) were determined based on kinetic param-eters for the six generated mutant enzymes (Table 1). We theninvestigated the relationship between �(�G) and the SBP of B.licheniformis L-AI by analyzing the active site structures of themutant B. licheniformis L-AI models. In order to investigate theinteraction between the substrate and each amino acid residuethat had been mutated in the B. licheniformis L-AI models, thedistance between each residue and L-arabinose was calculatedfrom the predicted model using MD simulation (Fig. 5). Whencompared to the wild-type B. licheniformis L-AI, the Y333I andY333V mutants showed significant decreases in catalytic effi-ciency (to 4.0 and 2.8 min1 mM1, respectively) and in-creased �(�G) values (5.76 and 6.71 kJ mol1, respectively),which were probably the result of the increased distance be-tween the residue and the substrate. However, the Y333F (Fig.5B) and Y333W (Fig. 5C) mutant enzymes showed no signif-icant changes in catalytic efficiency.
FIG. 3. SDS-PAGE analysis of wild-type B. licheniformis L-AI(WT) and mutants (identified by the relevant mutation) selected foralanine scanning, with the molecular masses given in kilodaltons. LaneM contains the protein markers, and lanes Y333A, I370A, M185A,T276A, L345A, W439A, and M349A correspond to the purified mu-tant B. licheniformis L-AI enzymes with molecular masses of �53 kDa.
FIG. 4. CD spectra of wild-type and mutant B. licheniformis L-AIs.The CD spectra of wild-type (solid line), Y333F (dashed line), Y333W(dotted line), and Y333A (dashed-dotted line) enzymes were recordedat 20°C on a J-815 spectrophotometer (Jasco Corp., Tokyo, Japan).Each spectrum was recorded in the range from 190 to 300 nm by usinga cuvette with an optical path length of 0.5 cm. , ellipticity; mdeg,millidegrees.
TABLE 1. Kinetic parameters determined for the B. licheniformisL-AI wild type and Y333 mutantsa
Enzymeb
Vmax(�mol/min/
mg ofprotein)
Km(mM) kcat
c (min1)
kcat/Kmratio
(min1
mM1)
�(�G)d
(kJ mol1)
Wild type 235 � 5 369 � 7 12,450 � 265 34 � 1.36 0Y333F
mutant213 � 9 492 � 17 11,290 � 476 23 � 1.76 1.05
Y333Wmutant
168.5 � 15 620 � 55 8,930 � 792 14.4 � 2.5 2.30
Y333Imutant
56.9 � 1.5 750 � 22 3,016 � 79 4 � 0.22 5.76
Y333Vmutant
44 � 2 816 � 35 2,332 � 106 2.8 � 0.24 6.71
Y333Amutant
NDe ND ND ND ND
a The Vmax, Km, and kcat values presented are means � standard deviations.b Y333D, Y333E, and Y333K mutants do not show activity with L-arabinose.c The kcat values were calculated by considering the enzyme to be a monomeric
form. Values are means � standard deviations of results from three experiments.d �(�G) � RT � ln (kcat/Km)mut/(kcat/Km)wt�, where (kcat/Km)mut and (kcat/
Km)wt are the kcat/Km ratios for the mutant and the wild type, respectively, Ris the ideal gas constant, and T is the temperature.
e ND, not determined due to the very low activity of the enzyme.
VOL. 76, 2010 CATALYTIC EFFICIENCY OF L-ARABINOSE ISOMERASE 1657
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
DISCUSSIONB. licheniformis L-AI has been reported to be the L-AI with
the highest turnover rate for L-arabinose (17). In this study, weused a systematic strategy to identify the molecular determi-nants of this catalytic efficiency: we screened for conservedresidues by multiple-sequence alignment and then used MDsimulation to identify conserved residues in contact with thesubstrate, followed by individual site-directed mutagenesis to
change those residues. The molecular docking study and mu-tational analyses of residues in contact with the substrate in theSBP of the wild-type B. licheniformis L-AI showed significantinteraction between the substrate and Y333. The Y333A mu-tant lost about 97% of the activity of the wild-type enzyme.Further mutations at position 333 indicated that an aromaticamino acid at position 333 is essential for the catalytic effi-ciency of B. licheniformis L-AI. A similar result was obtained
FIG. 5. Homology model of the B. licheniformis L-AI active site with bound L-arabinose substrate. L-Arabinose was docked into the SBP ofwild-type B. licheniformis L-AI (A) or the Y333F (B), Y333W (C), or Y333A (D) mutant. The intermolecular distances are the result of modeling.Green dotted lines represent the H bonds, and green solid lines show the distances from the substrate to neighboring amino acid residues. Aminoacid residues are depicted in stick form and colored according to elemental properties.
1658 PRABHU ET AL. APPL. ENVIRON. MICROBIOL.
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

with E. coli L-AI, chosen as another model L-AI enzyme. Theseresults suggest that this position can be considered a crucialdeterminant for the catalytic efficiency of all L-AIs.
In the crystal structures of E. coli L-fucose isomerase and E.coli L-AI, C-1 and C-2 of the substrate have been shown totransfer protons via an enediol intermediate (15, 21). Isomer-ization between L-arabinose and L-ribulose is depicted in Fig.6. During the aldose-ketose interconversion, two hydrogen at-oms are transferred via an enediol intermediate. The protontransfer is facilitated by two residues, Glu306 and Glu331,corresponding to the mechanism suggested for E. coli L-AI andE. coli L-fucose isomerase. Thus, the binding of C-1 and C-2 ofthe substrate to the catalytic residues gains importance forcompletion of the isomerization reaction. When L-arabinosewas docked into the active site pocket of wild-type B. licheni-formis L-AI, hydrogen bonding between C-1, C-2, and C-4 ofthe substrate and the E331, E306, and E331 residues, respec-tively, occurred, with bond lengths of 2.28, 2.07, and 2.3 Å,respectively (Fig. 5A). In the Y333A mutant, there can be noH bonding because of the increased distance between C-1 andC-2 of the substrate and the active site residues E331 and E306(Fig. 5D). This increased distance in the Y333A mutant ade-quately explained the reduced specific activity of the Y333Amutant for the substrate. When Tyr was replaced with Phe, thekcat was retained at its maximum, as the H bonding betweenC-1, C-2, and C-4 of the substrate and the active site E331,E306, and E331 residues could be retained with the lengths of2.1, 3.07, and 2.06 Å, respectively (Fig. 5B).
The turnover rates for the mutants containing nonpolaraliphatic residues were as follows: rate for the Y333I mutant �rate for the Y333V mutant � rate for the Y333A mutant. Thispattern correlated with the decreasing size of the side chainand its decreasing hydrophobicity (Table 1). When Y333 wasreplaced with a charged polar amino acid such as Arg, Asp, orGlu, the mutants exhibited no L-AI activity because thecharged amino acid disrupted the essential hydrophobic inter-action with the substrate. These results demonstrate that po-sition 333 requires an aromatic amino acid, which probablyfunctions to position the arabinopyranosyl ring so that theimportant H bond interaction with the active site residues ispossible. Indeed, analysis of the 3D models of the variousmutants showed that a slight modification of the position of
L-arabinose in the active site occurred, which induced a changein the distance between E306/E331 and the substrate. Thisresulted in less efficient isomerization of L-arabinose by themutant B. licheniformis L-AIs than by wild-type B. licheniformisL-AI.
Previously reported X-ray crystal structures of rabbit phos-phoglucose isomerase (13) and mouse phosphoglucose isomer-ase (7) with cyclic forms of the substrate that mimic the enediolintermediate led to proposed mechanisms for the ring-opening(or ring-closing) and isomerization steps in the multistep cat-alytic mechanism. Similar aldose-ketose isomerization mecha-nisms for yihS-encoded proteins from E. coli and Salmonellaenterica (8) have been described previously. Strongly conservedaromatic amino acids (Trp51, Tyr111, Trp316, Phe329, andTrp375) in YihS protein were found to interact with the sub-strate (8). In the present study, our postdocking analysis sug-gests that the aromatic side chain of Y333 is involved in asimilar stabilizing interaction of the protein-ligand complex(Fig. 5). Indeed, the �(�G) value for the wild-type B. licheni-formis L-AI was much lower than the values determined for themutant B. licheniformis L-AIs (Table 1). This was due probablyto the stabilization of the protein-ligand complex by the inter-action between the residue Y333 and L-arabinose. The func-tion of the aromatic group of Y333 (or Trp or Phe) would bethe constraint of reaction intermediates (chain forms) afterring opening to facilitate the following ring closure.
ACKNOWLEDGMENTS
This work was supported by the 21C Frontier Microbial Genomicsand Applications Center Program, Ministry of Education, Science andTechnology, Republic of Korea. It was also supported by a grant (code2008A0080126) from the Agricultural Research and DevelopmentPromotion Center.
REFERENCES
1. Ahmed, Z. 15 August 2001. Production of natural and rare pentoses usingmicroorganisms and their enzymes. Electron. J. Biotechnol. doi:10.4067/S0717-34582001000200008.
2. Beach, J. W., L. S. Jeong, A. J. Alves, D. Pohl, H. O. Kim, C. N. Chang, S. L.Doong, R. F. Schinazi, Y. C. Cheng, and C. K. Chu. 1992. Synthesis ofenantiomerically pure (2�R,5�S)-()-1-(2-hydroxymethyloxathiolan-5-yl)cy-tosine as a potent antiviral agent against hepatitis B virus (HBV) and humanimmunodeficiency virus (HIV). J. Org. Chem. 57:2217–2219.
3. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72:248–254.
4. Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan,and M. Karplus. 1983. CHARMm: a program for macromolecular energy,minimization, and dynamics calculations. J. Comput. Chem. 4:187–217.
5. Chouayekh, H., W. Bejar, M. Rhimi, K. Jelleli, M. Mseddi, and S. Bejar.2007. Characterization of an L-arabinose isomerase from the Lactobacillusplantarum NC8 strain showing pronounced stability at acidic pH. FEMSMicrobiol. Lett. 277:260–267.
6. Dische, Z., and E. Borenfreund. 1951. A new spectrophotometric method forthe detection and determination of keto sugars and trioses. J. Biol. Chem.192:583–587.
7. Graham Solomons, J. T., E. M. Zimmerly, S. Burns, N. Krishnamurthy,M. K. Swan, S. Krings, H. Muirhead, J. Chirgwin, and C. Davies. 2004. Thecrystal structure of mouse phosphoglucose isomerase at 1.6A resolution andits complex with glucose 6-phosphate reveals the catalytic mechanism ofsugar ring opening. J. Mol. Biol. 342:847–860.
8. Itoh, T., B. Mikami, W. Hashimoto, and K. Murata. 2008. Crystal structureof YihS in complex with D-mannose: structural annotation of Escherichia coliand Salmonella enterica yihS-encoded proteins to an aldose-ketose isomer-ase. J. Mol. Biol. 377:1443–1459.
9. Jorgensen, F., O. C. Hansen, and P. Stougaard. 2004. Enzymatic conversionof D-galactose to D-tagatose: heterologous expression and characterisation ofa thermostable L-arabinose isomerase from Thermoanaerobacter mathranii.Appl. Microbiol. Biotechnol. 64:816–822.
10. Kim, B. C., Y. H. Lee, H. S. Lee, D. W. Lee, E. A. Choe, and Y. R. Pyun. 2002.
FIG. 6. Scheme of the reaction catalyzed by B. licheniformis L-AI.The possible reaction mechanism is suggested based on the consider-ation of E. coli L-fucose isomerase and E. coli L-AI. The isomerizationreaction comprises the transfer of two hydrogen atoms (indicated bywhite arrows). The two bases Glu306 and Glu331 transfer the twoprotons via an enediol intermediate.
VOL. 76, 2010 CATALYTIC EFFICIENCY OF L-ARABINOSE ISOMERASE 1659
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

Cloning, expression and characterization of L-arabinose isomerase fromThermotoga neapolitana: bioconversion of D-galactose to D-tagatose using theenzyme. FEMS Microbiol. Lett. 212:121–126.
11. Kim, H. J., J. H. Kim, H. J. Oh, and D. K. Oh. 2006. Characterization of amutated Geobacillus stearothermophilus L-arabinose isomerase that increasesthe production rate of D-tagatose. J. Appl. Microbiol. 101:213–221.
12. Lee, D. W., H. J. Jang, E. A. Choe, B. C. Kim, S. J. Lee, S. B. Kim, Y. H.Hong, and Y. R. Pyun. 2004. Characterization of a thermostable L-arabinose(D-galactose) isomerase from the hyperthermophilic eubacterium Thermo-toga maritima. Appl. Environ. Microbiol. 70:1397–1404.
13. Lee, J. H., and C. J. Jeffery. 2005. The crystal structure of rabbit phospho-glucose isomerase complexed with D-sorbitol-6-phosphate, an analog of theopen chain form of D-glucose-6-phosphate. Protein Sci. 14:727–734.
14. Lovell, S. C., I. W. Davis, W. B. Arendall III, P. I. de Bakker, J. M. Word,M. G. Prisant, J. S. Richardson, and D. C. Richardson. 2003. Structurevalidation by C� geometry: �,� and C� deviation. Proteins 50:437–450.
15. Manjasetty, B. A., and M. R. Chance. 2006. Crystal structure of Escherichiacoli L-arabinose isomerase (ECAI), the putative target of biological tagatoseproduction. J. Mol. Biol. 360:297–309.
16. Oh, H. J., H. J. Kim, and D. K. Oh. 2006. Increase in D-tagatose productionrate by site-directed mutagenesis of L-arabinose isomerase from Geobacillusthermodenitrificans. Biotechnol. Lett. 28:145–149.
17. Prabhu, P., M. K. Tiwari, M. Jeya, P. Gunasekaran, I. W. Kim, and J. K. Lee.
2008. Cloning and characterization of a novel L-arabinose isomerase fromBacillus licheniformis. Appl. Microbiol. Biotechnol. 81:283–290.
18. Rhimi, M., and S. Bejar. 2006. Cloning, purification and biochemical char-acterization of metallic-ions independent and thermoactive L-arabinoseisomerase from the Bacillus stearothermophilus US100 strain. Biochim. Bio-phys. Acta 1760:191–199.
19. Rhimi, M., M. Juy, N. Aghajari, R. Haser, and S. Bejar. 2007. Probing theessential catalytic residues and substrate affinity in the thermoactive Bacillusstearothermophilus US100 L-arabinose isomerase by site-directed mutagene-sis. J. Bacteriol. 189:3556–3563.
20. Roh, H. J., P. Kim, Y. C. Park, and J. H. Choi. 2000. Bioconversion ofD-galactose into D-tagatose by expression of L-arabinose isomerase. Biotech-nol. Appl. Biochem. 31(Pt. 1):1–4.
21. Seemann, J. E., and G. E. Schulz. 1997. Structure and mechanism of L-fucoseisomerase from Escherichia coli. J. Mol. Biol. 273:256–268.
22. Ma, T., S. B. Pai, Y. L. Zhu, J. S. Lin, K. Shanmuganathan, J. Du, C. Wang,H. Kim, M. G. Newton, Y. C. Cheng, and C. K. Chu. 1996. Structure-activityrelationships of 1-(2-deoxy-2-fluro-�-L-arabino-furanosyl)pyrimidine nucleo-sides as anti-hepatitis B virus agents. J. Med. Chem. 39:2835–2843.
23. Wu, G., D. H. Robertson, C. L. Brooks III, and M. Vieth. 2003. Detailedanalysis of grid-based molecular docking: a case study of CDOCKER—aCHARMm-based MD docking algorithm. J. Comput. Chem. 24:1549–1562.
1660 PRABHU ET AL. APPL. ENVIRON. MICROBIOL.
on June 11, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from





























