Embed Size (px)
Citation preview

PRESENTED BY:P.SHARATH CHANDRA M.PHARM(SECOND YEAR)DEPT. OF PHARMACEUTICSKLE UNIVERSITY’S COLLEGE OF PHARMACY,BELGAUM
VALIDATION-DESIGN, DEVELOPMENT OF PROCESS VALIDATION, METHODS FOR PHARMACEUTICAL OPERATIONS INVOLVED IN THE PRODUCTION OF PHARMACEUTICAL DOSAGE FORMS.

The concept of Validation was first proposed by two Food and Drug Administration (FDA) officials, Ted Byers and Bud Loftus, in the mid 1970’s in order to improve the quality of Pharmaceuticals.
USFDA Definition for Process validation: Process validation is establishing documented
evidence which provides a high degree of assurance that a specific process (such as the manufacture of pharmaceutical dosage forms) will consistently produce a product meeting its predetermined specifications and quality characteristics.
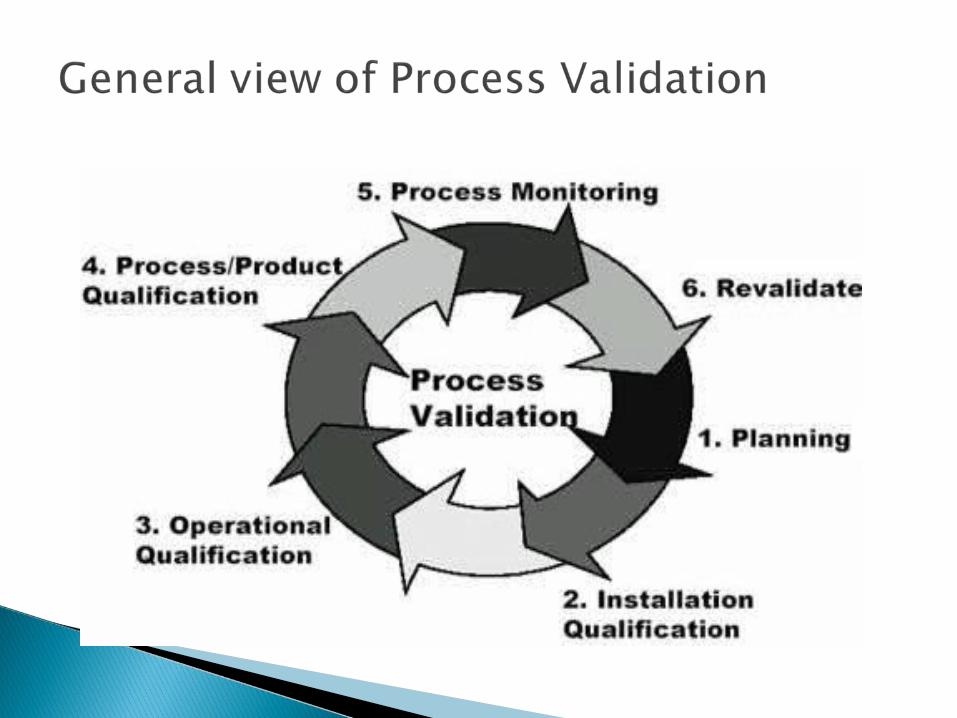

It would not be feasible to use the equipments without knowing whether it will produce the product we wanted or not.
The pharmaceutical industry uses expensive materials, sophisticated facilities & equipments and highly qualified personnel.
The efficient use of these resources is necessary for the continued success of the industry. The cost of product failures, rejects, reworks, and recalls, complaints are the significant parts of the total production cost.
Detailed study and control of the manufacturing process- validation is necessary if failure to be reduced and productivity improved.

Department /Designation Responsibility
Manager Production Responsible for manufacturing of batches and review of protocol and report.
Manager QC Responsible for analysis of samples collected.
Executive QC Responsible for samples collection and submission to QC.
Manager Maintenance Providing utilities and engineering Support.
Executive Production Responsible for preparation of protocol and manufacturing of validation batches.
Manager QA Responsible for protocol authorization and preparation of summary report.
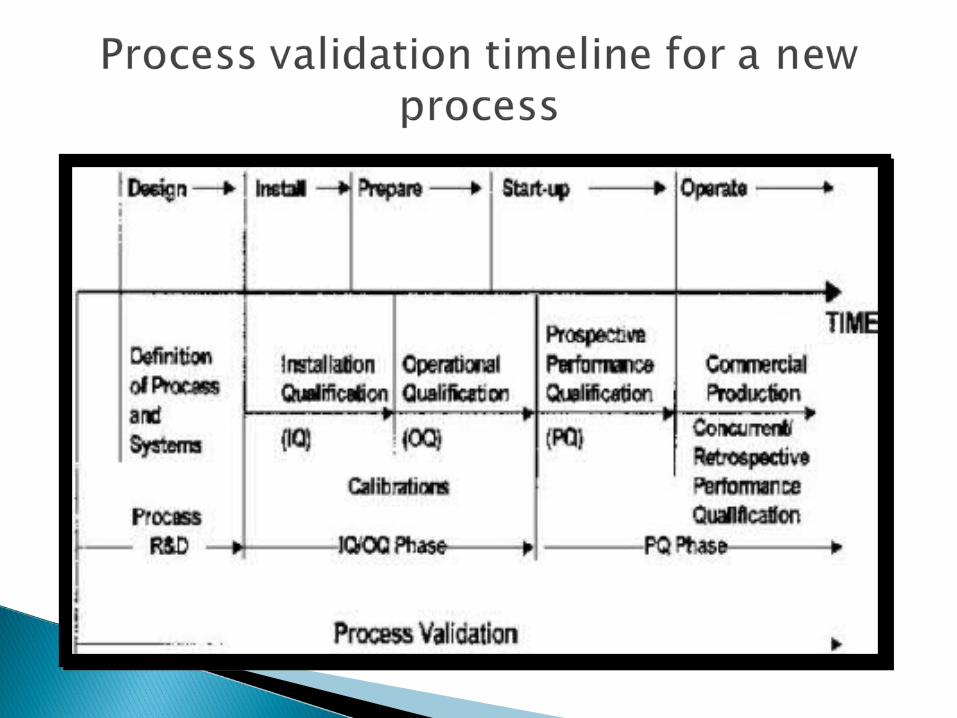
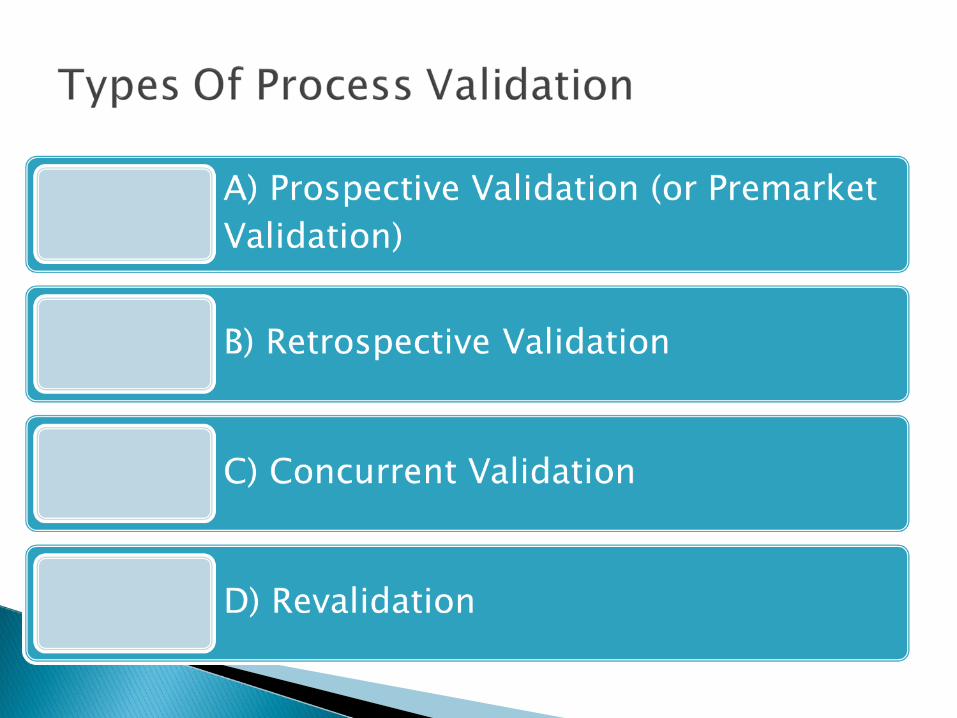
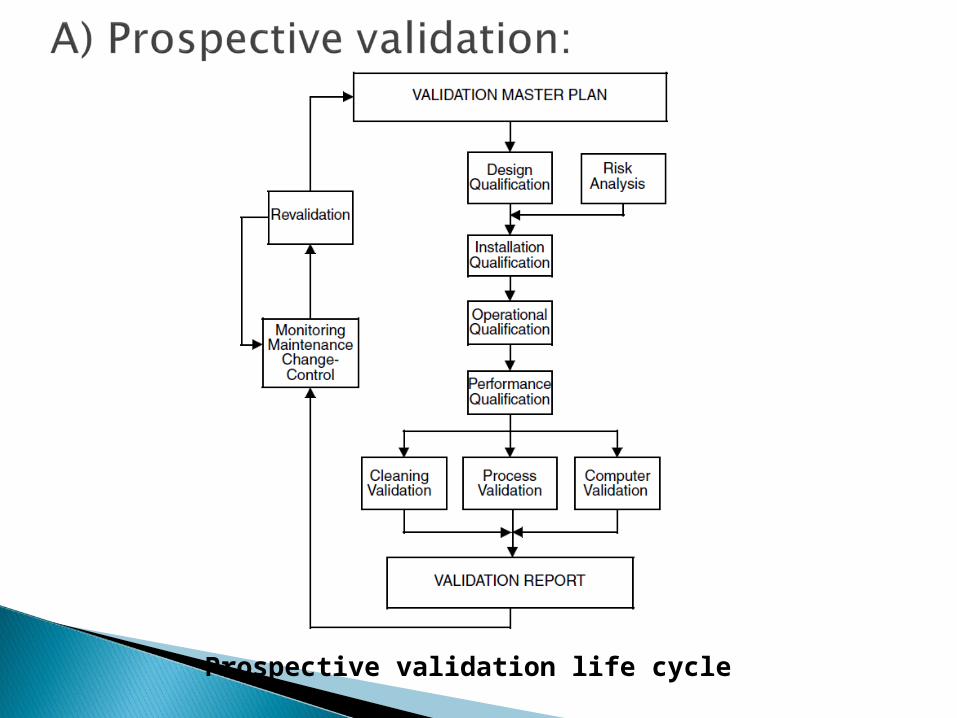
Prospective validation life cycle
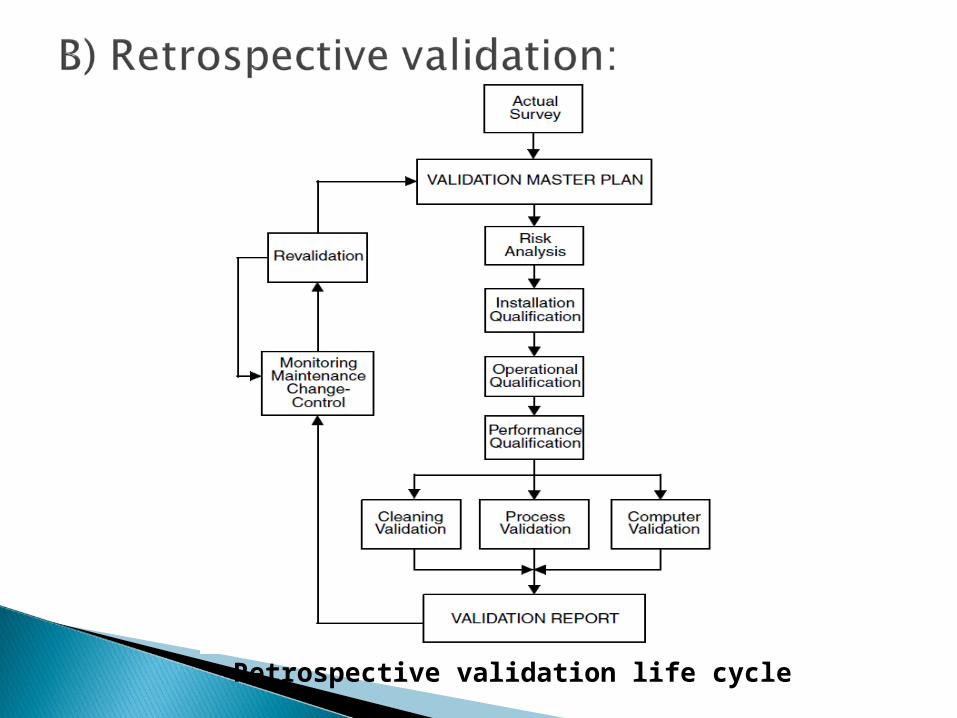
Retrospective validation life cycle
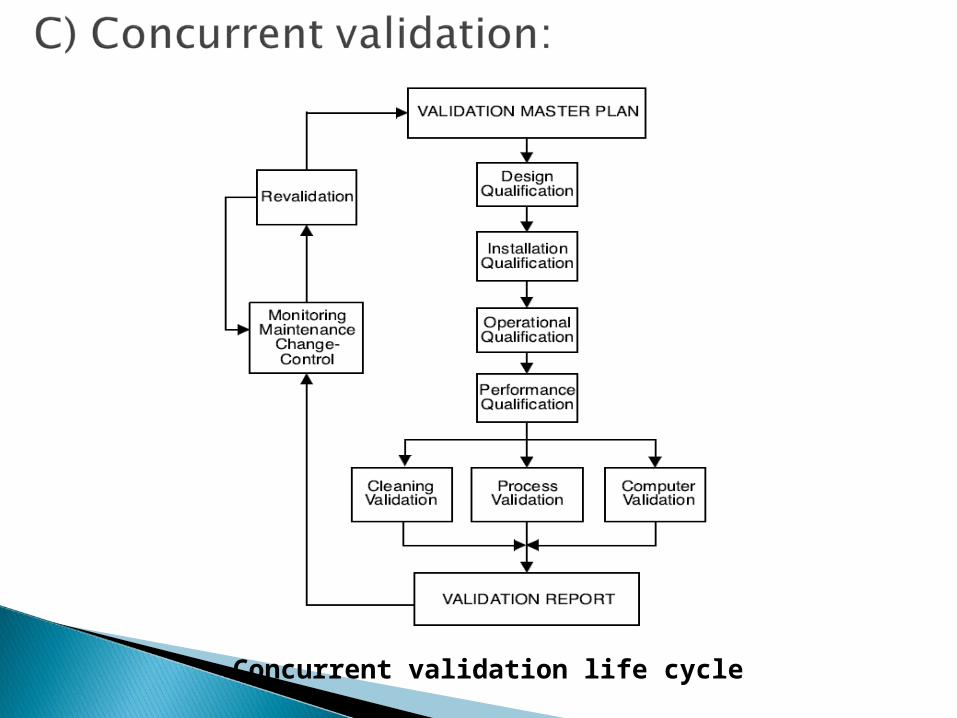
Concurrent validation life cycle

Revalidation means repeating the original validation effort or any part of it, and includes investigative review of existing performance data.
This approach is essential to maintain the validated status of the plant, equipment, manufacturing processes and computer systems.
Change Control: Written procedures should be in place to describe the
actions to be taken if a change is proposed to a product component, process equipment, process environment, processing site.
All changes must be formally requested, documented and accepted by the validation team.

A written plan stating how validation will be conducted,
including test parameters, product characteristics,
production and packaging equipment, and decision
points on what constitutes acceptable test results.
Validation Master Plan A comprehensive document describing the applicable
validation requirements for a given facility, and providing a plan for meeting those requirements.
The VMP provides a “road map” for validation, to establish a sequence of events followed by facilities audits and inspections.

1. Title 2. Objective & Scope 3. Responsibility 4. Protocol Approval 5. Validation Team 6. Product Composition 7. Process Flow Chart 8. Manufacturing Process9. Review of Equipments / Utilities 10. Review of Raw Materials and Packing Materials 11. Review of Analytical and Batch Manufacturing Records 12. Review of Batch Quantities for Validation (Raw Materials) 13. Review of Batch Quantities for Validation (Packing Materials) 14. HSE Requirements 15. Review of Process Parameters 16. Validation Procedure 17. Sampling Location 18. Documentation 19. Acceptance Criteria 20. Summary 21. Conclusion

Introduction: Validation policy, Scope, Location and Schedule
Organizational structure: Personnel responsibilities Plant/ process /product description: Rational for
inclusions or exclusions and extent of validation. Specific process considerations that are critical and those
requiring extra attention. List of products/ processes/ systems to be validated,
summarized in a matrix format, validation approach. Re-validation activities, actual status and future planning. Key acceptance criteria Documentation format Reference to the required SOP’s Time plans of each validation project and sub-project.

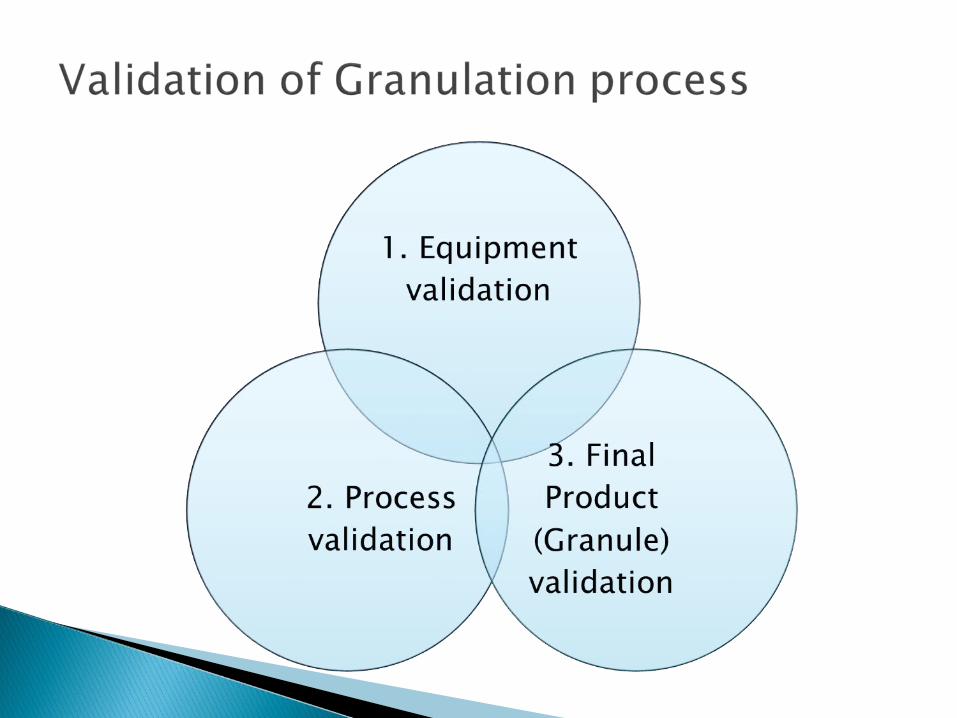

Various equipment utilized in granulation process are 1) Mixer/Granulation 2) Dryer 3) Blender 4) Mills 5) Sieves
In an ideal situation the equipment used to manufacture dosage forms would be selected based upon such factors asformulation,safety requirements,handling or production efficiencies and commercial demands.

Equipment validation steps
Mixer/Blender Dryer Mills
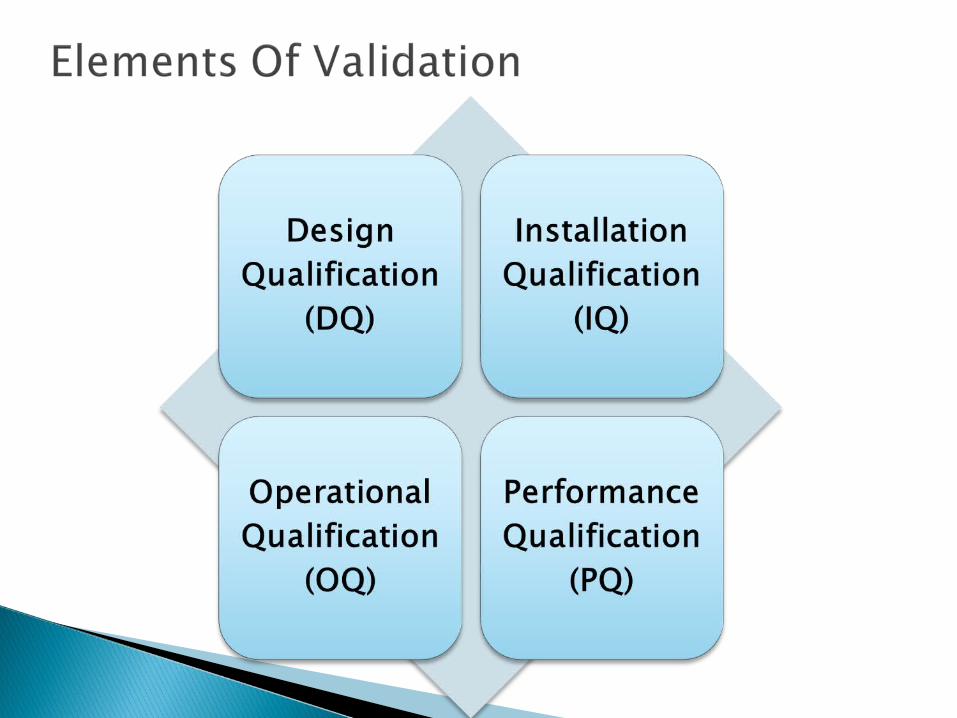
1)Design qualification
1) Check extra paddle, choppers provided 2) Verify paddle is mounted to shaft properly. 3) Granulation end point detection system. 4) Options to introduce the granulating fluid (e.g. dump, meter, spray) 5) Verify automated charging of discharging system.
1) Cabinet with heater.
2) Check position of heaters.
3) Check fans provided.
4) Exhausting system.
5) Verify air handle.
6) Verify inlet or outlet system.
7) Explosion relief duct to avoid explosion.
1) Extra hammers, stationery knives are provided. 2) Verify the location and size of screen in mills. 3) Feeding and discharging system.

3)Operational Qualification
1) Perform calibration requirements. 2) Operate the equipment at low, medium and high-speed rate. 3) Verify all that switches and push buttons are functioning properly. 4) Establish procedure for operation, maintenance, and calibration. 5) Training program for staff.
1) Run three batches of each product and analyze for -Active ingredient uniformity -Moisture content. -Particle size distribution -Tap density. Based on these data fixing a drying end point of process. (E.g. moisture content)
1) Run the mills at high, low, medium speed and determine -Particle size distribution -Range of units From this data set the time and speed of mills.

4)Performance Qualification
Carry out operations for different samples and each product shall meet its predetermined characteristics.
Carry out operations for different samples and determine drying temperature and time and characteristics of product.
Determine milling time and speed for different products. - Particle size distribution of each sample at different time and speed.

Validating a granulation process involves identification of critical parameters, which must be controlled to ensure the consistent production of granulation.
Various operations carried out in granulation process are:
1) Mixing /Blending 2) Wet Granulation 3) Drying 4) Milling

I) Mixing/Blending: Factors in creating uniform blending: -Mixing or blending technique -Mixing/ Blending speed -Mixing time -Drug uniformity -Excipients uniformity. -Equipment capacity or load.
II) Wet granulation: Parameters considered in wet granulation process are I) Binder addition II) Binder concentration III) Amount of binder solution or granulating solvent IV) Binder solution or granulating solvent addition rate V) Mixing time VI) Granulation end point

To break up the lumps and aggregates and enhance drying of granules
Factors to be considered Equipment size and capacity Screen size Milling speed Feed rate
IV) Drying:
Type of drying technique (Tray, Fluid, and Microwave) required for formulation needs to be determined and justified depending on drug formulation properties and equipment availability.

Parameters to be considered in drying 1) Inlet and outlet temperature: Inlet temperature is critical to drying efficiency of
granules and should be set high enough to maximize drying without affecting chemical or physical stability of granules.
2) Air flow: Sufficient to ensure removal of moisture from wet
granules 3) Moisture uniformity: 4) Equipment capability or capacity: Larger load will require more moisture to be
removed on drying and will affect the drying time.


Factors to consider during compression are: Tooling Compression speed Compression/ejection force Tablet compression variables
Fill volume
Pre- and compression force
Turntable speed
Dwell time
Granule size and feed
Ejection force, lubrication

Key areas to consider for tablet coating: Tablet properties Equipment type (Conventional or perforated pan and
fluid bed coaters) Coater load Pan speed Spray guns Application/spray rate Tablet flow: Inlet/outlet temperature and airflow Coating solution (The concentration and viscosity of
the coating solution) Coating weight (A minimum and maximum coating
weight) Residual solvent level

Guideline on General Principles of Process Validation http://www.fda.gov/cder/guidance/pv.htm
Guidance for Industry: For the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products. CDER CVM November 1994. www.fda.gov/CDER/GUIDANCE/cmc2.pdf
Working Party on Control of Medicines and Inspections Final Version of Annex 15 to the EU Guide to Good
Manufacturing Practice Title: Qualification and validation http://pharmacos.eudra.org/F2/eudralex/vol-4/pdfs-en/
v4an15.pdf ICH Q7a Section 12 on validation http://www.fda.gov/cder/meeting/ICH_Q7A/index.htm A WHO guide to good manufacturing practice (GMP)
requirements. Part 2: Validation Chaloner-Larsson, G., Anderson, R., and Egan, A. 1997. World
Health Organization, Geneva.

Process validation is a requirement of cGMP regulation for finished pharmaceutical products.
It is a key element in assuring that the quality goals are met.
Successfully validating a process may reduce the dependence upon intensive inprocess and finished product testing.
Validation of a new or existing product involves the efforts of scientists at various stages of the product development life cycle.
Scientificinformation obtained during the preformulation stage can form the basis for a well-designed and comprehensive validation program.

1)Nash RA., Wachter A.H., Pharmaceutical Process Validation, 3 rd Edition, Marcel Dekker publication, P.g, 173-182.
2) Lachman L., Lieberman H.A., Kania J.L., The Theory and Practice of Industrial Pharmacy, 3 rdedition, Varghese Publishing House, P.g.No. 318-320.
3) N. K. Jain & S. N. Sharma, A Text Book of Professional Pharmacy, VallabhPrakashan, P.g.no. -295-297
4) Haider S.I., Validation Standard Operating Procedures St. Lucie Press Publishers, P.g. No. 345-353.






























