Embed Size (px)
Citation preview

ASSOCIATION MAPPING
Statistica: L’unica scienza che permette a esperti diversi di utilizzare informazioni uguali per trarre conclusioni diverse.
Evan Esar

Analisi QTL
1. Disporre di popolazioni idonee– Da specifico incrocio– Individui ‘diversi’
2. Misurare il carattere (fenotipo)3. Descrivere il genotipo4. Trovare una associazione statistica tra marcatori e fenotipo
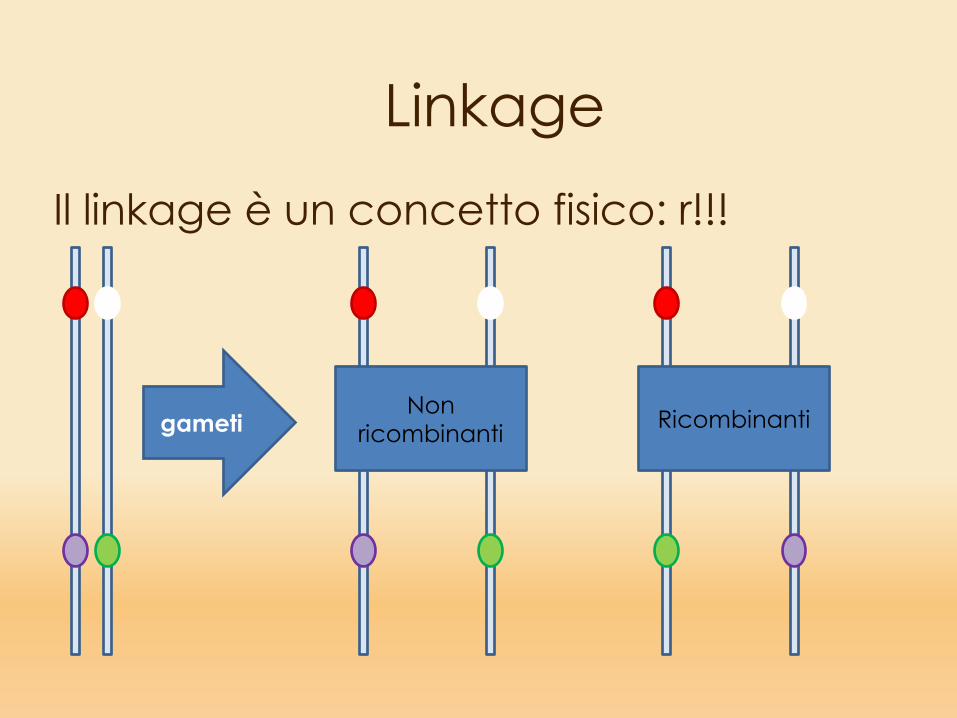
LinkageIl linkage è un concetto fisico: r!!!
gametiNon
ricombinanti Ricombinanti

Popolazioni di mappa(RILs, DH, F2…..)
1. Pochi fenotipi2. Richiedono molto tempo3. Hanno «solo» 2 alleli per locus4. «pochi» eventi possibili di ricombinazione5. Spesso basso potere di risoluzione
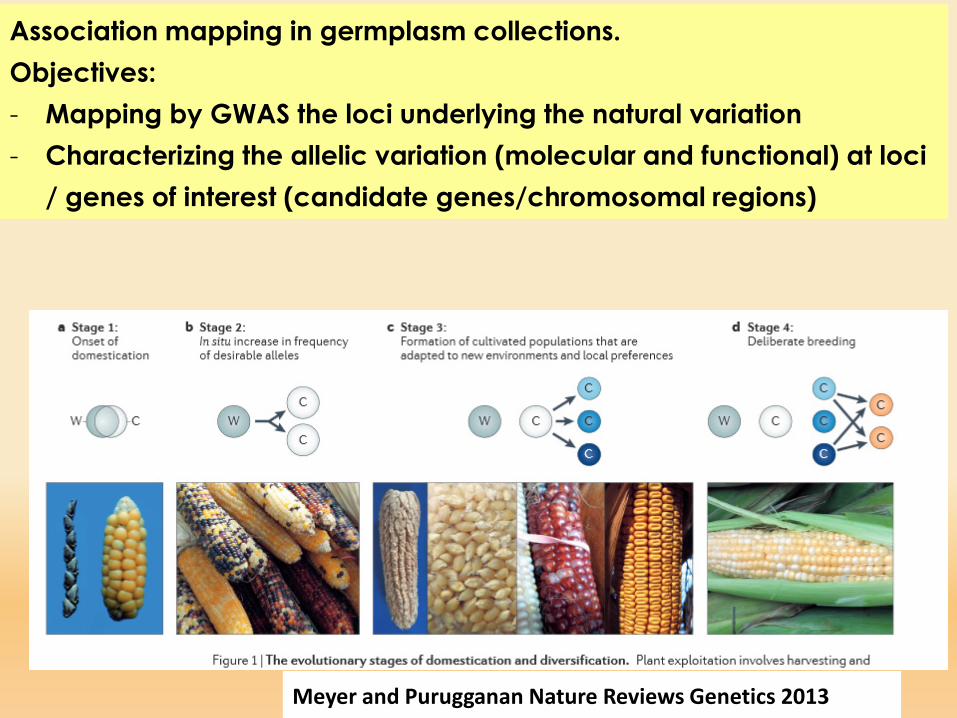
Association mapping in germplasm collections.Objectives:- Mapping by GWAS the loci underlying the natural variation- Characterizing the allelic variation (molecular and functional) at loci
/ genes of interest (candidate genes/chromosomal regions)
Meyer and Purugganan Nature Reviews Genetics 2013
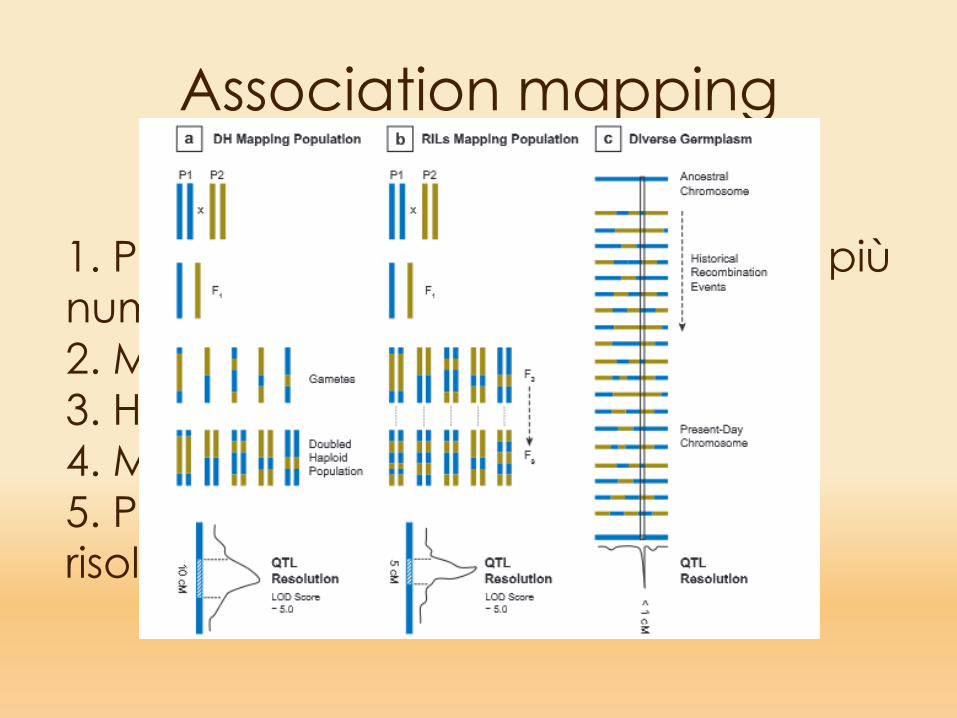
Association mapping
1. Popolazioni potenzialmente molto più numerose2. Materiale ‘già pronto’3. Hanno molti alleli per locus4. Molti eventi di ricombinazione5. Potenzialmente alto potere di risoluzione

Mapping of genetic factors controlling Mendelian and quantitative traits:2) Mapping methods:
a) Linkage analysis in plants
maps genes using gametic phase disequilibrium created in a single- or multiple-generation cross
"ad Hock" Segregating populations are created, maintained, genotyped and phenotypedPopulations: F2 individuals, BC individuals, F3 progenies, RI lines, intermated lines,
multiparental populations
b) Association mapping
Relies on segregating variation in natural populations.multiparental populations with complex pedigreescrop germplasm collections with various degrees of relatedness between accessionsnatural populations
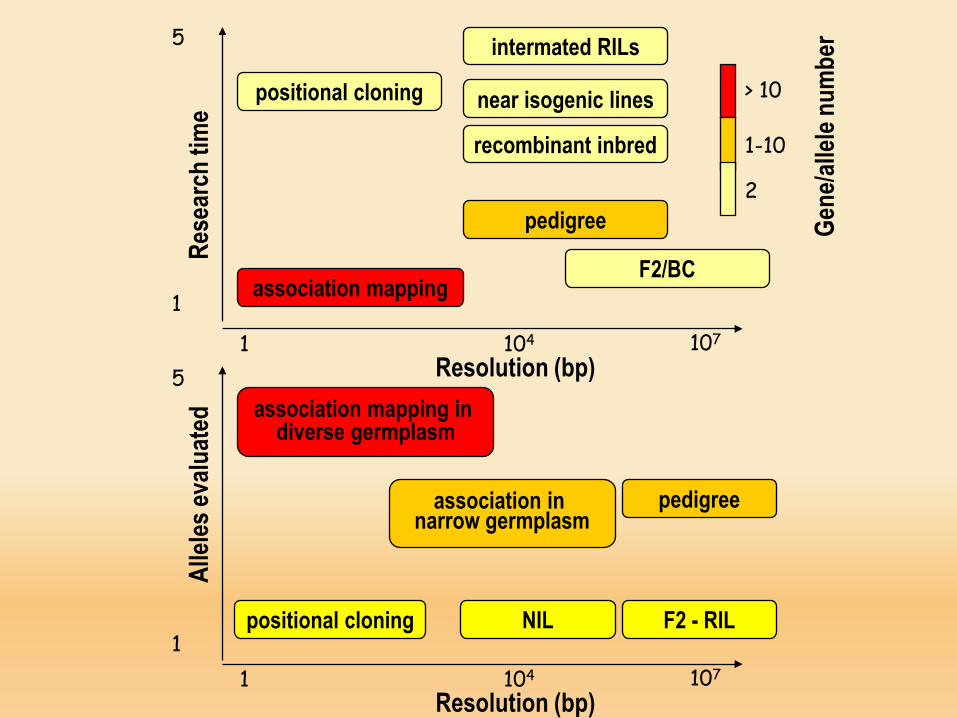
positional cloning
association mapping
pedigree
F2/BC
recombinant inbred
near isogenic lines
intermated RILs
Gene
/allel
e num
ber
2
1-10
> 10
1
5
Rese
arch
tim
e
1 104 107
Resolution (bp)association mapping in
diverse germplasm
association in narrow germplasm
pedigree
F2 - RILNILpositional cloning
1 104 107
Resolution (bp)
5
1
Allel
es ev
aluat
ed

LINKAGE DISEQUILIBRIUM
~~~
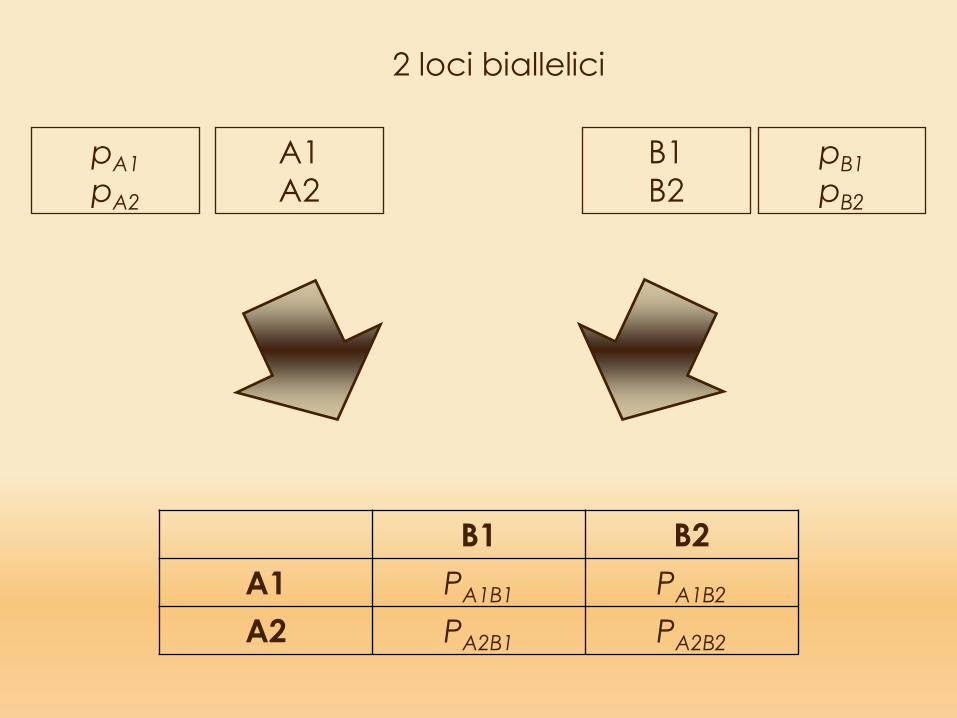
2 loci biallelici
A1A2
B1B2
pA1pA2
pB1pB2
B1 B2A1 PA1B1 PA1B2
A2 PA2B1 PA2B2
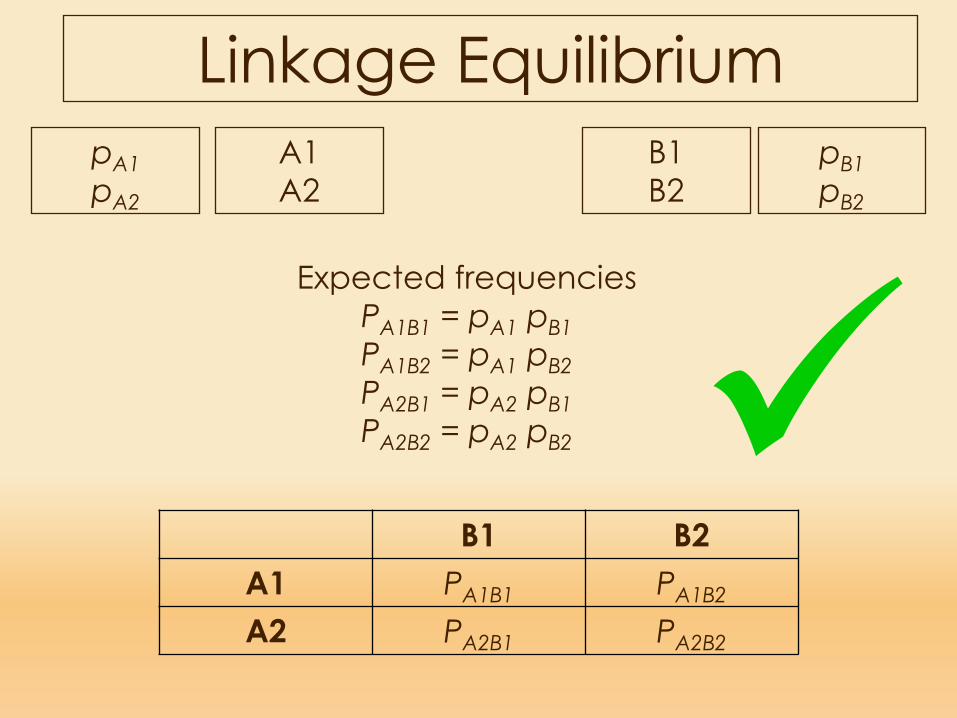
Expected frequenciesPA1B1 = pA1 pB1PA1B2 = pA1 pB2PA2B1 = pA2 pB1PA2B2 = pA2 pB2
Linkage Equilibrium A1A2
B1B2
pA1pA2
pB1pB2
B1 B2A1 PA1B1 PA1B2
A2 PA2B1 PA2B2
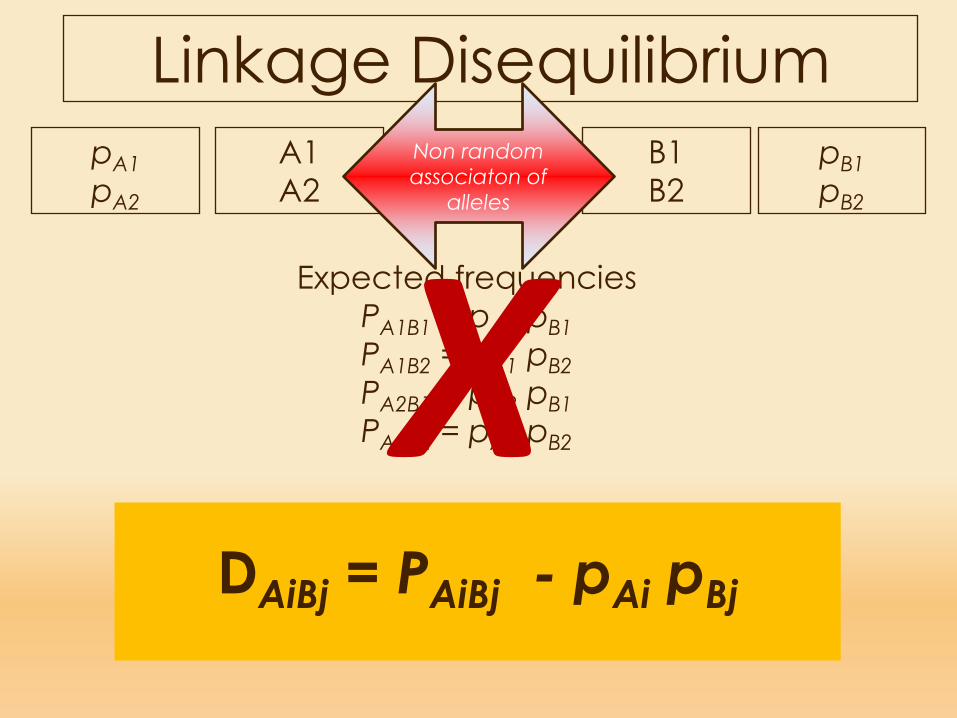
Expected frequenciesPA1B1 = pA1 pB1PA1B2 = pA1 pB2PA2B1 = pA2 pB1PA2B2 = pA2 pB2X
Linkage DisequilibriumA1A2
B1B2
pA1pA2
pB1pB2
Non random associaton of
alleles
B1 B2A1 PA1B1 PA1B2A2 PA2B1 PA2B2
DAiBj = PAiBj - pAi pBj
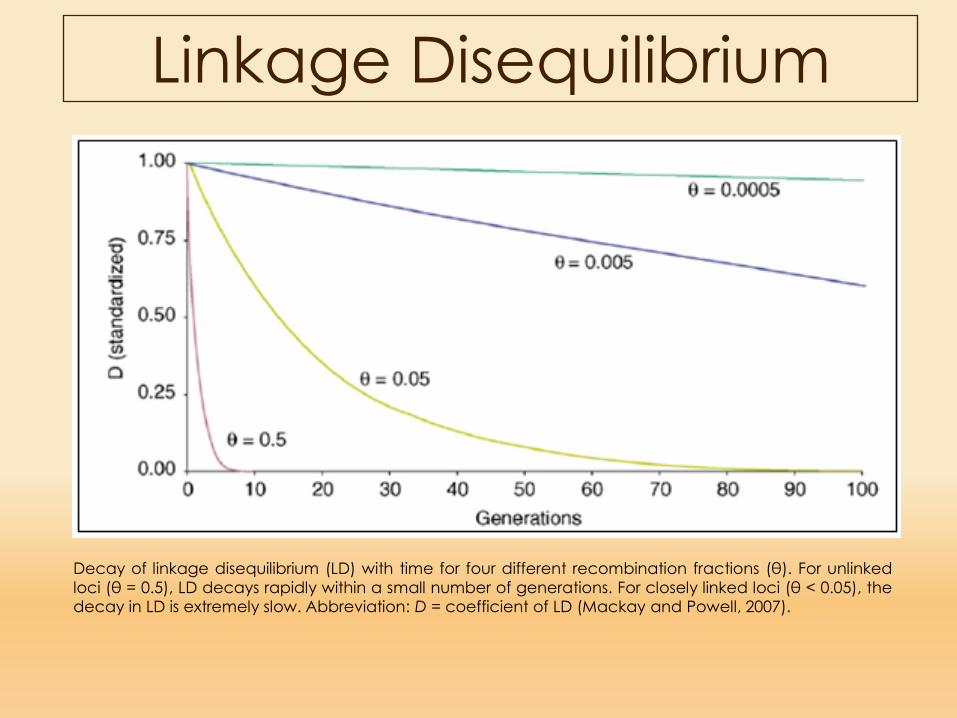
Linkage Disequilibrium
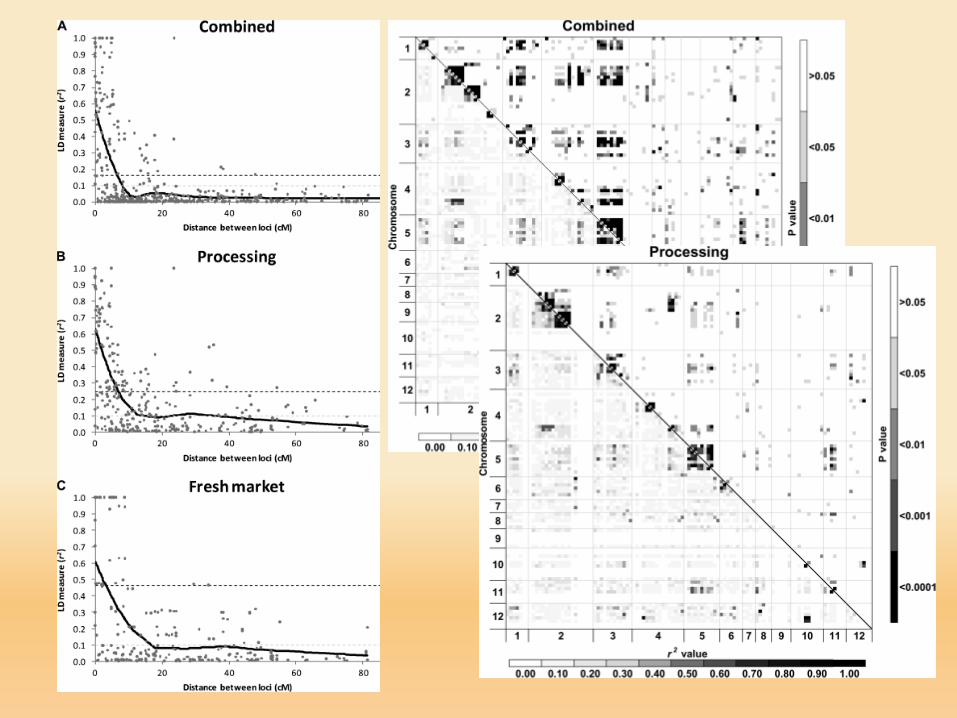
Decay of linkage disequilibrium (LD) with time for four different recombination fractions (θ). For unlinkedloci (θ = 0.5), LD decays rapidly within a small number of generations. For closely linked loci (θ < 0.05), thedecay in LD is extremely slow. Abbreviation: D = coefficient of LD (Mackay and Powell, 2007).

Linkage DisequilibriumDAiBj = PAiBj - pAi pBj
Dmin = -min (pA1pB2, pA2pB1)
D’ = |D|/DminD < 0
Dmax = min (pA1pB1, pA2pB2)
D’ = D/DmaxD > 0
D dipende dalla frequenza allelica caratteristica ‘rischiosa’!!!!D normalizzato D’

Linkage Disequilibrium
r2 = DAiBj2 / (pA1 pA2 pB1 pB2)
Quadrato del coefficiente di correlazione tra due marcatori
Riflette il potere statistico di calcolare LD
Basso r2 campione GRANDE (N) per trovare LD tra imarcatori

D = (0.43 – 0.70 * 0.45) = 0.115Dmax = min (0.70 * 0.55, 0.30 * 0.45) = 0.135D’ = 0.115 / 0.135 = 0.8581r2 = (0.115)2 / 0.70 * 0.30 * 0.55 * 0.45 = 0.2545
0.385 0.135
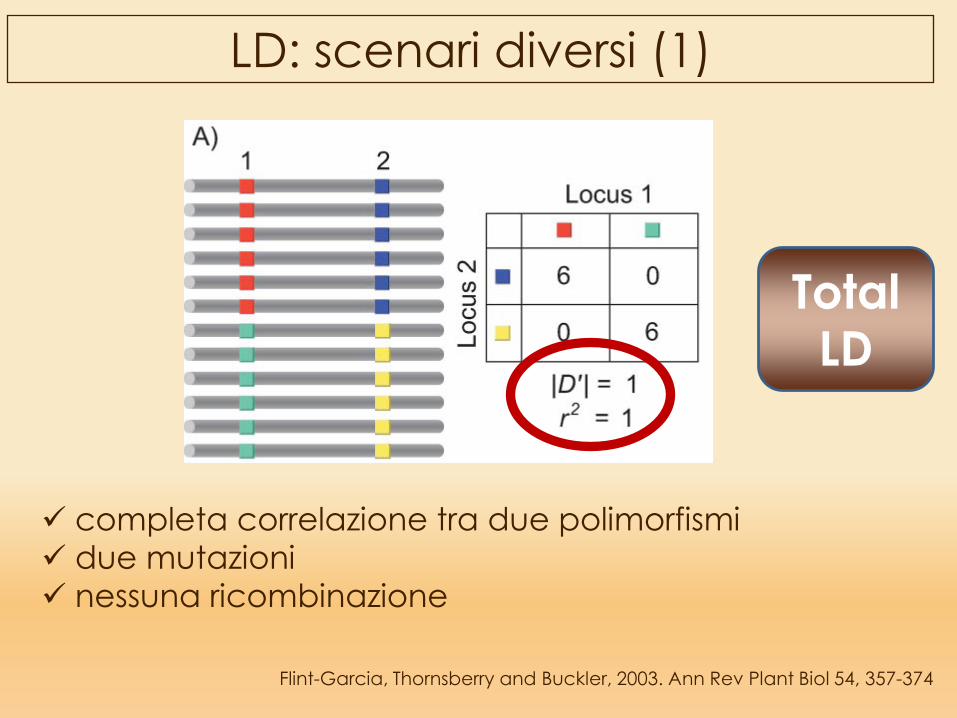
Flint-Garcia, Thornsberry and Buckler, 2003. Ann Rev Plant Biol 54, 357-374
LD: scenari diversi (1)
completa correlazione tra due polimorfismi due mutazioni nessuna ricombinazione
Total LD
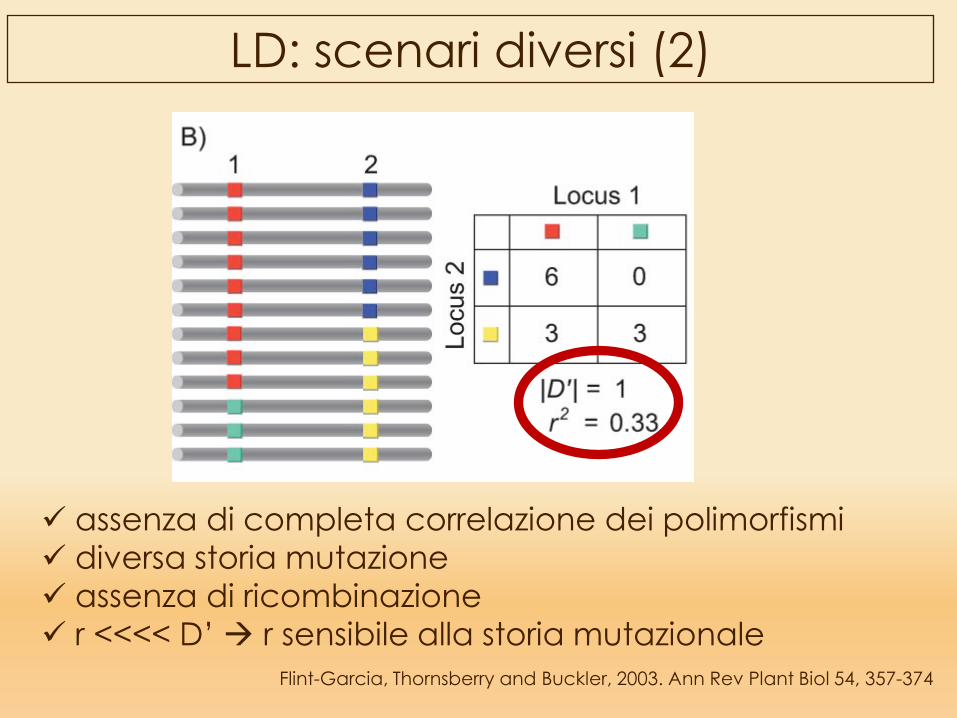
Flint-Garcia, Thornsberry and Buckler, 2003. Ann Rev Plant Biol 54, 357-374
LD: scenari diversi (2)
assenza di completa correlazione dei polimorfismi diversa storia mutazione assenza di ricombinazione r <<<< D’ r sensibile alla storia mutazionale
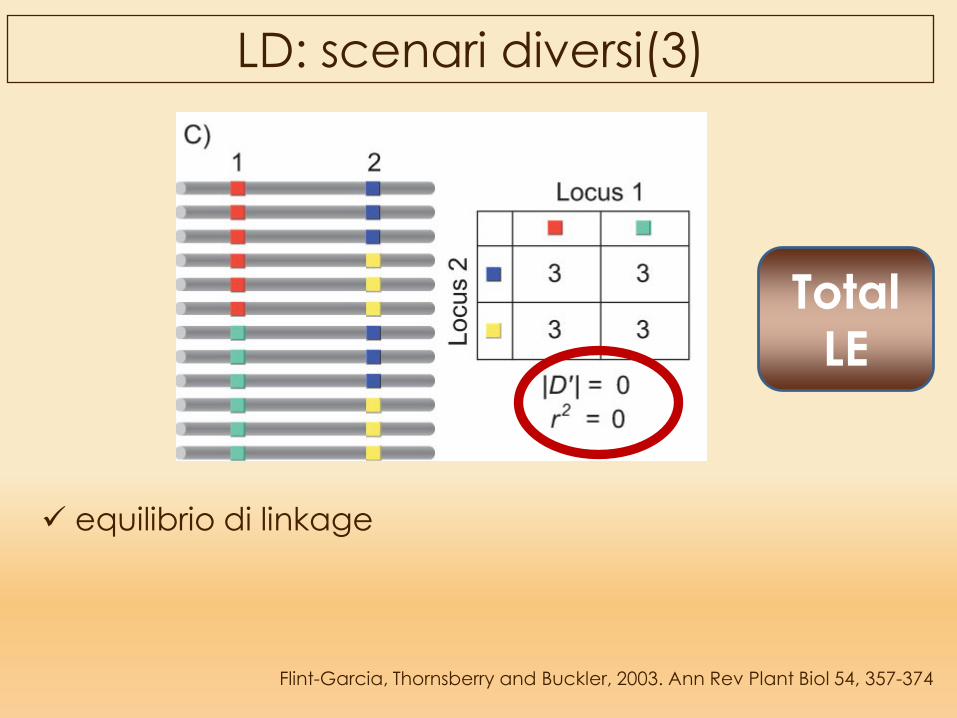
Flint-Garcia, Thornsberry and Buckler, 2003. Ann Rev Plant Biol 54, 357-374
LD: scenari diversi(3)
equilibrio di linkage
Total LE
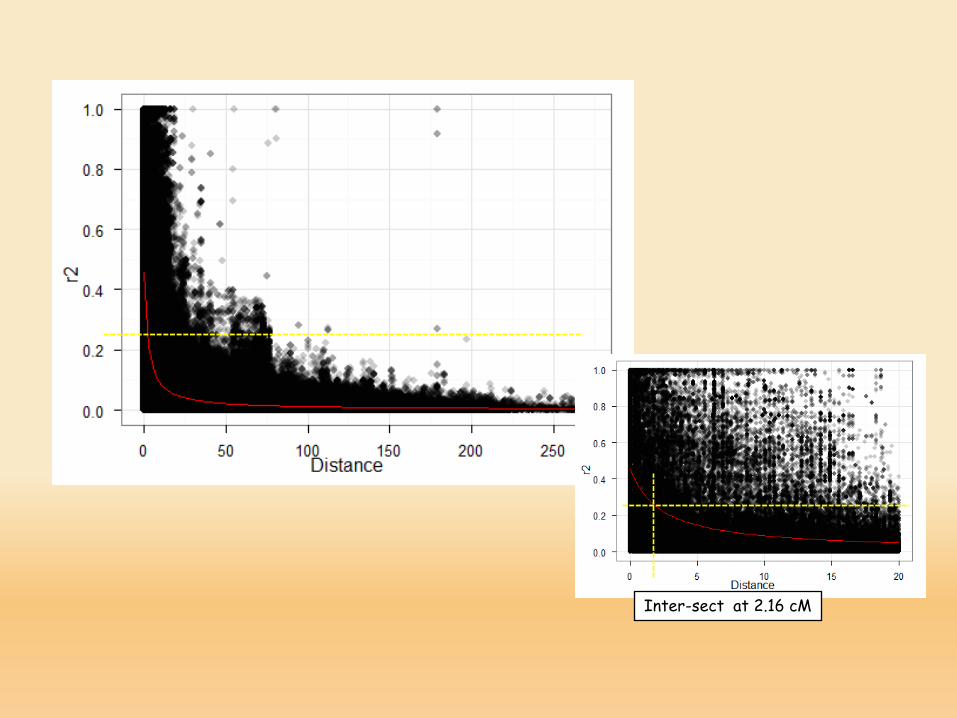
Inter-sect at 2.16 cM
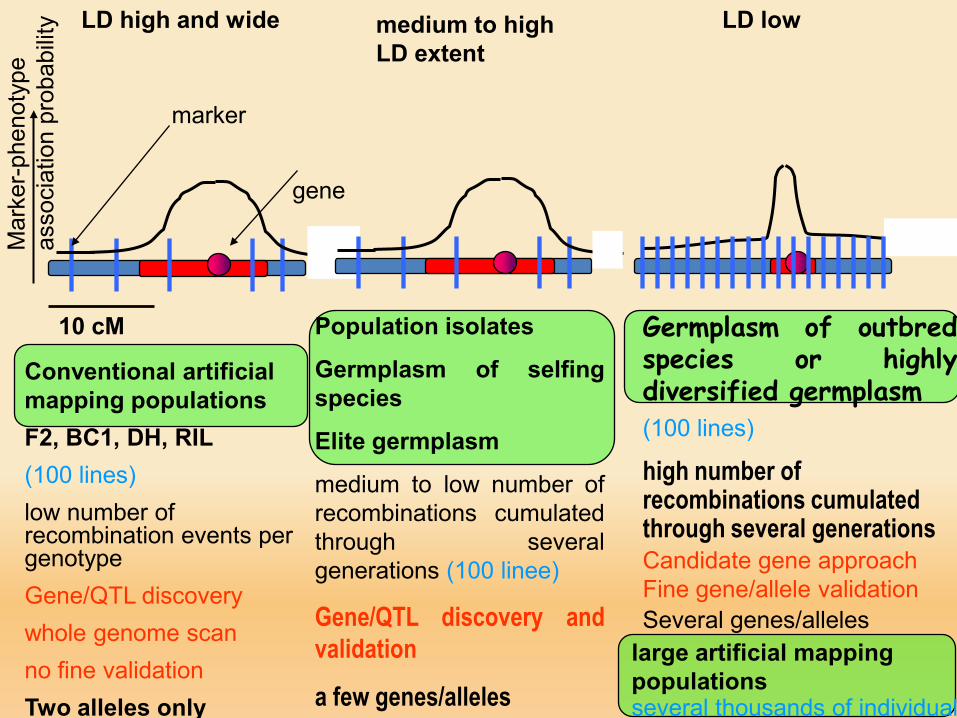
LD high and wide
Conventional artificial mapping populationsF2, BC1, DH, RIL(100 lines)low number of recombination events per genotypeGene/QTL discovery whole genome scanno fine validationTwo alleles only
large artificial mapping populationsseveral thousands of individual
gene
Population isolates
Germplasm of selfingspecies
Elite germplasm
medium to low number ofrecombinations cumulatedthrough severalgenerations (100 linee)
Gene/QTL discovery andvalidation
a few genes/alleles
LD lowmedium to high LD extent
10 cM
marker
Mar
ker-p
heno
type
as
soci
atio
n pr
obab
ility
Germplasm of outbredspecies or highlydiversified germplasm(100 lines)
high number of recombinations cumulated through several generationsCandidate gene approachFine gene/allele validationSeveral genes/alleles

Species Mating system, germplasm and LD range (average distance where r2 = 0.10)Human Outcrossing- Nigerian: 5 kb- European: 80 kbCattle Outcrossing: 10 cMDrosophila Outcrossing: <1 kbMaize Outcrossing- Diverse maize: 1 kb or less- Diverse inbred lines: 1.5 kb- Elite (cultivated) lines: > 100 kbArabidopsis Selfing: 250 kbSugarcane Outcrossing/vegetative: 10 cM propagation
Wheat and Barley selfing- Diverse barley: less than 1 cM - Elite (cultivated) germplasm up to 1-20 cM up to 60 cM
Haplotype blocks: extensive regions showing no recombinations and inherited from a know "foundation genotype"


Phenotyping
Dati raccolti con un adeguato numero di replicheentro ambienti, tra ambienti e anni diversi
Disegni sperimentali adeguati (e.g., α-lattice)
Metodi statistici di analisi appropriati
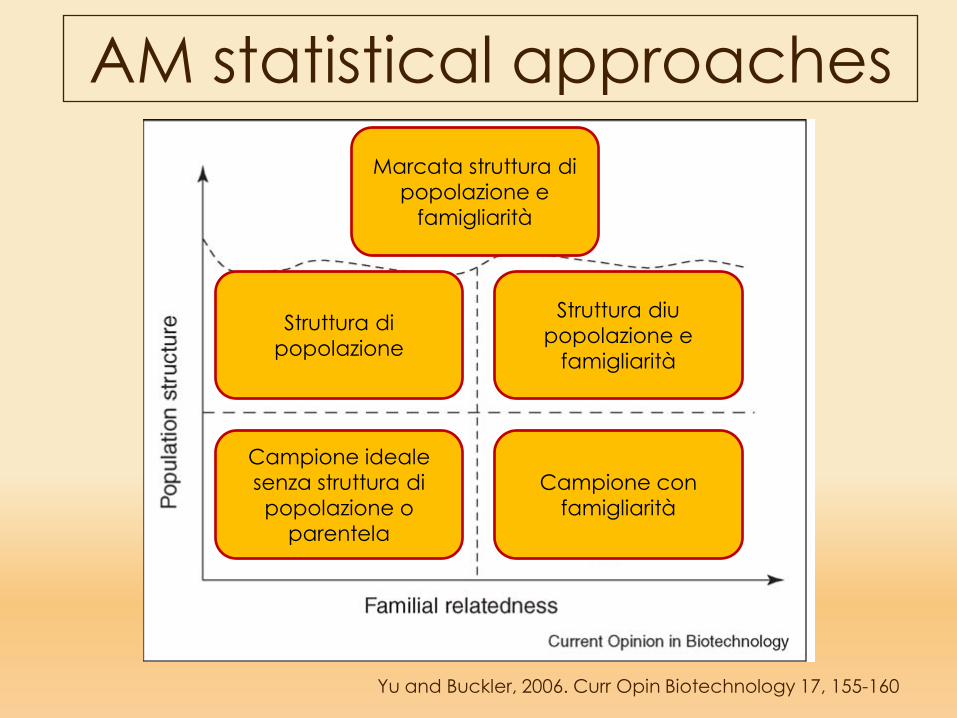
AM statistical approaches
Yu and Buckler, 2006. Curr Opin Biotechnology 17, 155-160
Campione ideale senza struttura di popolazione o
parentela
Campione con famigliarità
Struttura di popolazione
Marcata struttura di popolazione e
famigliarità
Struttura diu popolazione e
famigliarità
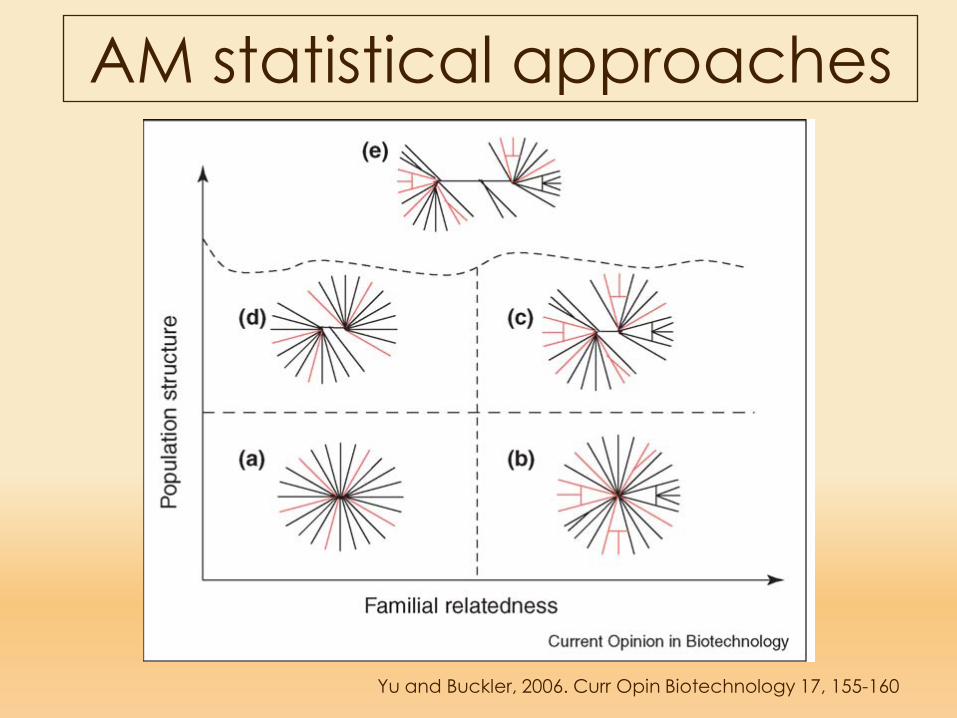
AM statistical approaches
Yu and Buckler, 2006. Curr Opin Biotechnology 17, 155-160

When population subdivision is present, it ispossible to find statistical associations between a disease phenotype and arbitrary markers that have no physical linkage to causative loci
Such associations occur because population subdivision (or any other form of nonrandom mating) permits marker-allele frequencies to vary among segments of the population, as the result of genetic drift or founder effects (Slatkin 1991).
Pop structure causes: - natural selection (e.g. main adaptation traits: vernalization and photoperiod sensitivity)- breeder's selection (based on ideotypes, particularly in inbred crops, founders are successfull cultivars)
Linkage disequilibrium and population structure
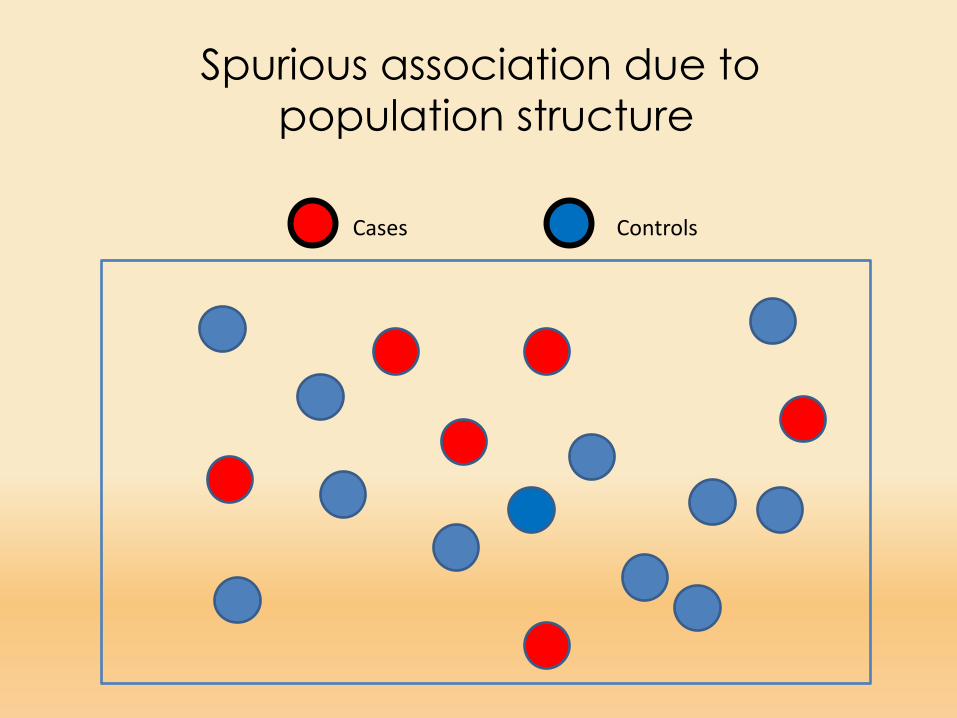
Cases Controls
Spurious association due to population structure
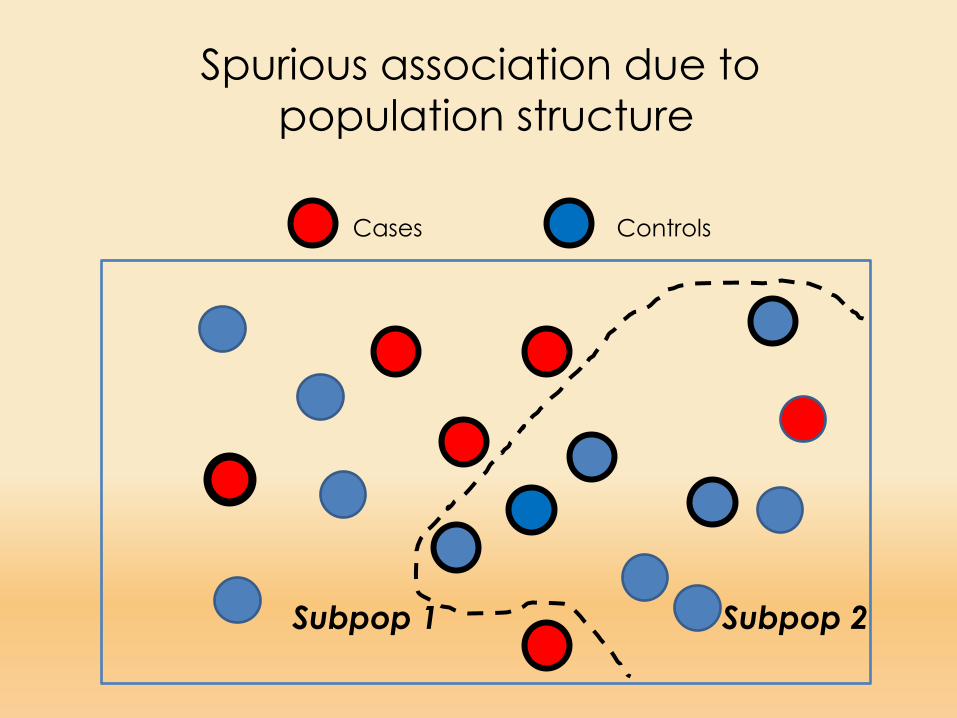
Cases Controls
Subpop 1 Subpop 2
Spurious association due to population structure
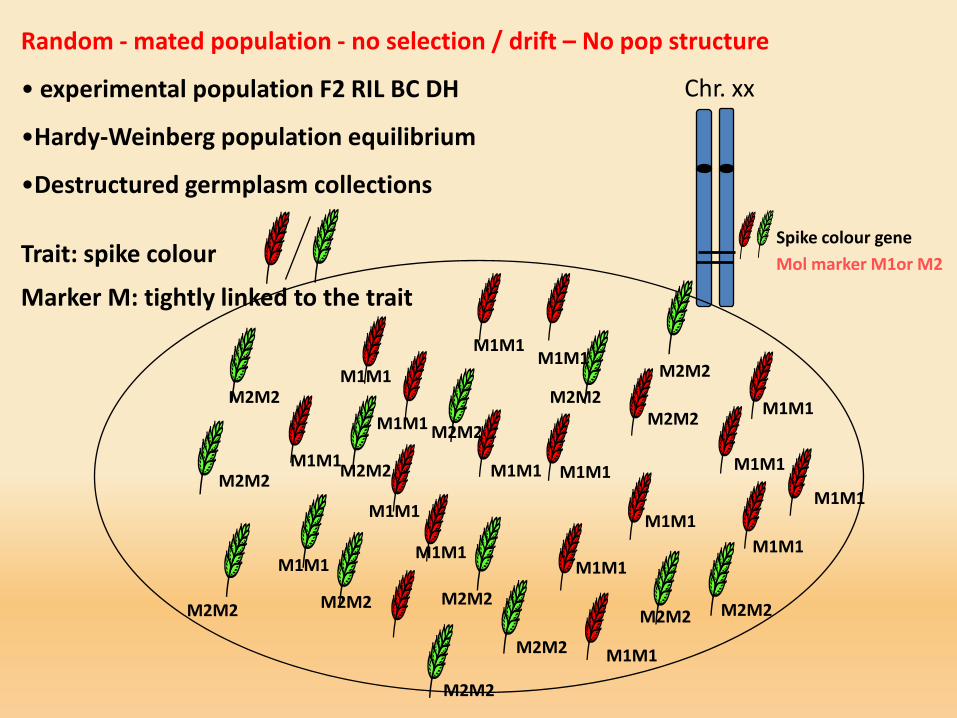
Chr. xx
Random - mated population - no selection / drift – No pop structure
• experimental population F2 RIL BC DH
•Hardy-Weinberg population equilibrium
•Destructured germplasm collections
Mol marker M1or M2
M1M1
M2M2
M2M2
M2M2
M2M2M2M2M2M2
M1M1
M1M1M1M1
M1M1
M2M2 M1M1
M1M1
M1M1
M1M1
M1M1
M1M1M1M1
M1M1
M1M1
M1M1
M1M1
M1M1
M2M2
M2M2
M2M2
M2M2
M2M2M2M2
M2M2
Trait: spike colour
Marker M: tightly linked to the trait
Spike colour gene
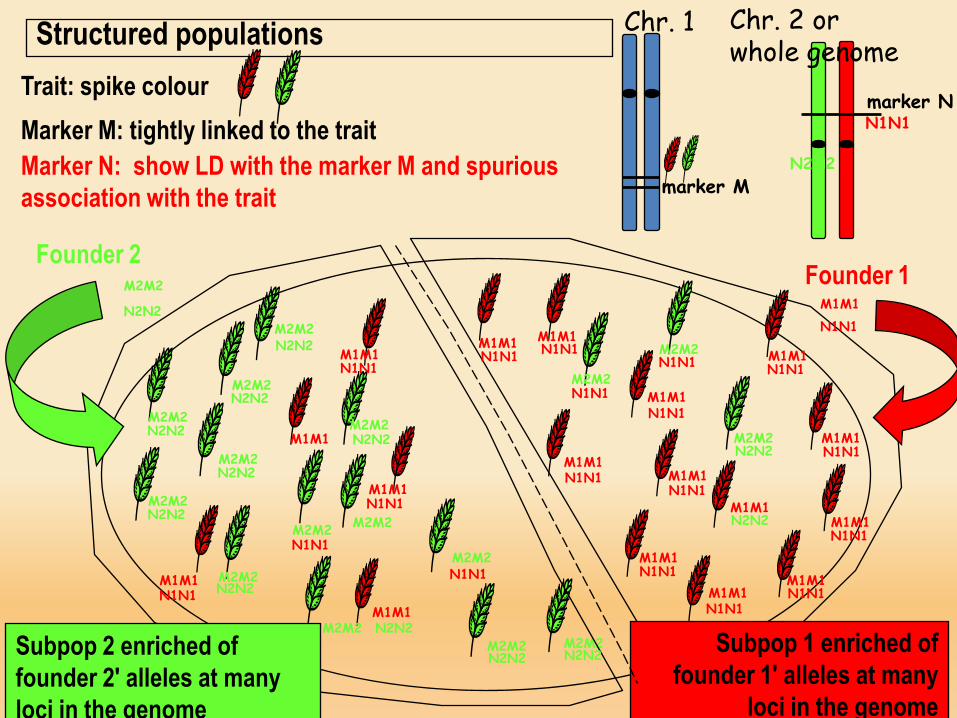
Chr. 1Structured populations
marker M
M2M2M2M2
M2M2
M2M2
M2M2
M2M2
M1M1
M1M1
M1M1
M1M1
M1M1
M1M1M1M1
M1M1
M1M1
M1M1
M1M1
M1M1
M1M1
M1M1
M1M1
M2M2
M2M2
M2M2
M2M2
M2M2
M2M2
M2M2
M1M1
M1M1M2M2
M2M2
Founder 2
marker N
Founder 1
M2M2
N2N2
N1N1
N1N1
N1N1
N1N1
N1N1
N1N1
N1N1N1N1
N2N2
N2N2
N1N1
N1N1
N1N1 N1N1
N1N1
N1N1
N2N2
N2N2
N2N2
N2N2
N2N2
N2N2
N2N2
N1N1
N2N2
N2N2
N2N2
N2N2
N1N1
N1N1N1N1
M2M2M1M1
Subpop 2 enriched of founder 2' alleles at many loci in the genome
Subpop 1 enriched of founder 1' alleles at many
loci in the genome
Trait: spike colourMarker M: tightly linked to the traitMarker N: show LD with the marker M and spurious association with the trait
N1N1
N1N1
Chr. 2 or whole genome
known or unknown population history
Kinship (coancestry, relatedness)
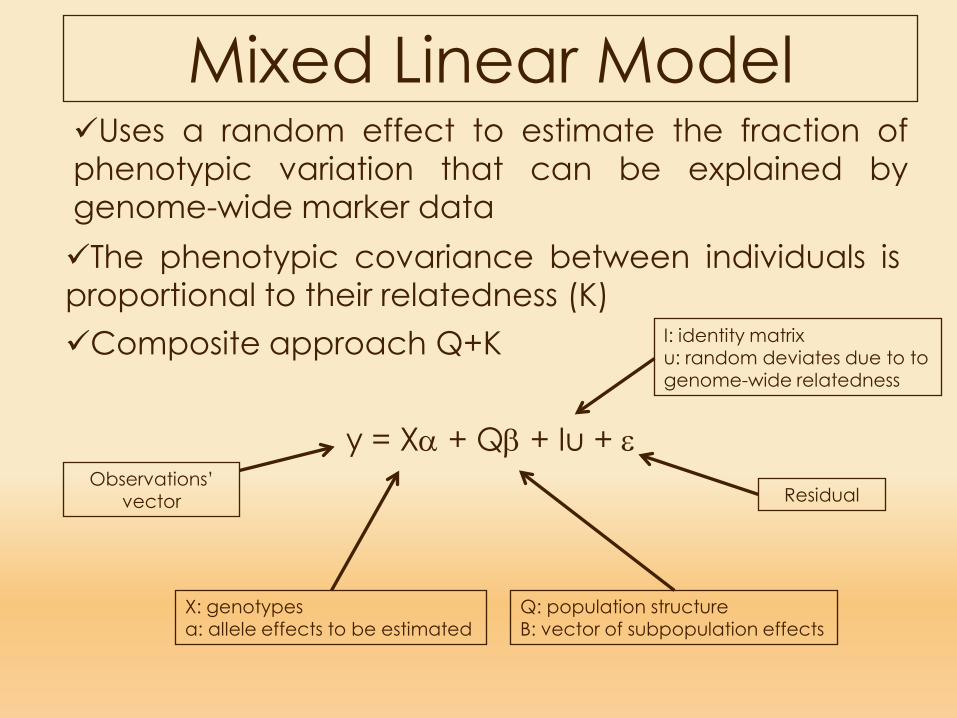
Mixed Linear Model
The phenotypic covariance between individuals isproportional to their relatedness (K)Composite approach Q+K
y = Xα + Qβ + Iu + εObservations’
vector
X: genotypesa: allele effects to be estimated
Q: population structureB: vector of subpopulation effects
Residual
I: identity matrixu: random deviates due to to genome-wide relatedness
Uses a random effect to estimate the fraction ofphenotypic variation that can be explained bygenome-wide marker data
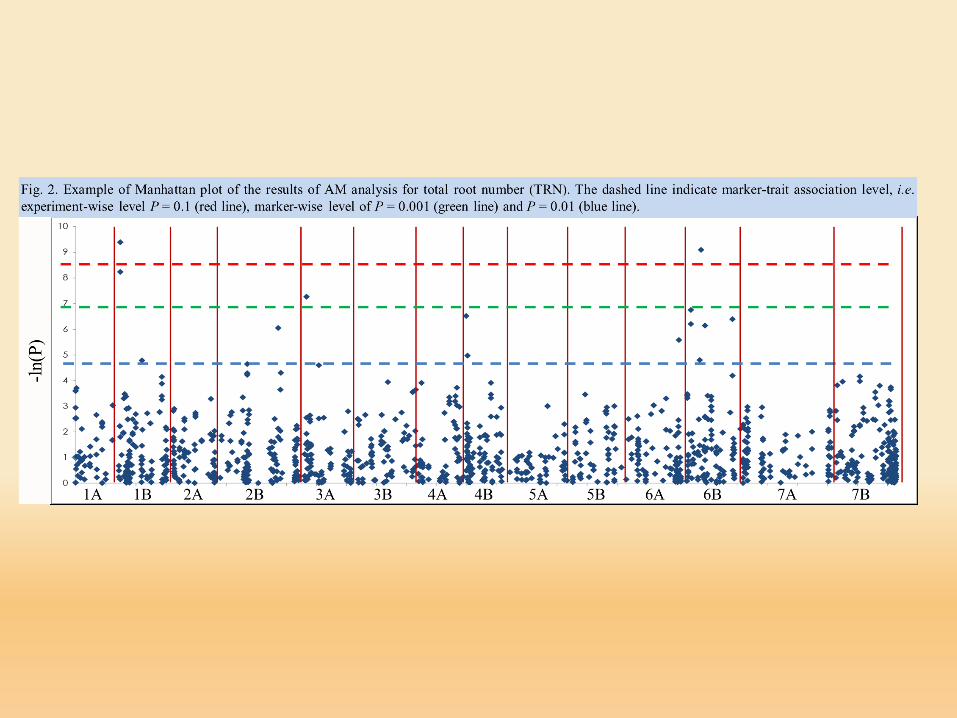
www.maizegenetics.net/tassel
Trait Analysis by aSSociation, Evolution and Linkage





























