Embed Size (px)
Citation preview

© 2000 Macmillan Magazines Ltd
news and views
NATURE CELL BIOLOGY | VOL 2 | FEBRUARY 2000 | cellbio.nature.com E21
Presenilin mutants subvert chaperone function
Mary-Jane Gething
Mutant presenilin proteins, known to promote the development of Alzheimer’s disease through increased generation of Aβ42 peptides, appear to compound this insult by downregulating the signalling pathway that adjusts levels of molecular chaperones in the endoplasmic reticulum in response to stress.
lzheimer’s disease is a progressiveneurodegenerative condition, thehallmark of which is the accumula-
tion in the cerebral cortex of extracellularlesions composed largely of aggregates ofamyloid-β peptide (Aβ; reviewed in ref. 1).Certain mutations in the presenilin-1 andpresenilin-2 proteins cause early-onsetAlzheimer’s disease, in part by increasingthe generation, and secretion into the brain,of Aβ peptides. Now, an exciting paper byKatayama and colleagues2, published inNature Cell Biology, indicates that preseni-lin-1 mutants may in fact stage a two-pronged attack on cells. Not only do theylead to increased production of Aβ pep-tides, but they may also render cells less ableto respond to stress conditions in the endo-plasmic reticulum.
How are the presenilins involved in thegeneration of Aβ peptides? Aβ is generatedfrom the amyloid precursor protein (APP),a transmembrane protein (Fig. 1). A smallproportion of APP is cleaved by a so-calledβ-secretase — just a name until recently,but now revealed to be a new aspartic pro-tease located in the endoplasmic reticulum(ER) and Golgi3. Cleavage of APP by β-secretase produces a soluble ectodomainand a membrane-associated carboxy-termi-nal fragment — the ‘β-stub’. This stub isthen the substrate for cleavage by γ-secre-tase(s) (the identity of which is not yet cer-tain) to form Aβ peptides of 42 and 40amino acids in length (Aβ42 and Aβ40).Germline mutations in presenilins 1 and 2that cause early-onset Alzheimer’s diseaseselectively increase the production andsecretion of the highly amyloidogenic Aβ42peptide. Presenilin-1 (PS1) controls APPprocessing, and either may be a γ-secretase,generating Aβ peptides, or may regulateaccess of the γ-secretase to the β-stub.Inherited and sporadic missense mutationsin the PS1 gene that promote Alzheimer’sdisease cause a dominant gain in function ofPS1 activity, and result in increased γ-secre-tase activity. Although processing of APP togenerate Aβ peptides occurs at variouspoints along the secretory pathway, as wellas at the plasma membrane and in the endo-cytic compartment, it appears that PS1 ena-
bles production of Aβ42 in just pre-Golgicompartments of the secretory pathway,while the less amyloidogenic Aβ40 peptide isgenerated later in the secretory pathway andin the endocytic compartment.
These observations, together with areport4 that overexpression of the ER chaper-one BiP/GRP78 decreases production of Aβpeptides, set the stage for Katayama et al.’sexciting finding2 that missense mutations inPS1 downregulate the ER-to-nucleus (ERN)signalling pathway that adjusts the levels ofmolecular chaperones (protein complexesthat help other proteins to fold correctly) inthe ER in response to need.
In both yeast and mammalian cells, theaccumulation of misfolded polypeptides inthe lumen of the ER (ER stress) results in an‘unfolded-protein response’ (UPR) — thetranscriptional upregulation of genes,located in the nucleus, that encode ERchaperones and folding catalysts (reviewedin ref. 5). The increased production ofchaperones alleviates the increased demandon the existing cellular protein-foldingmachinery. These UPR-controlled genescontain conserved regulatory elements intheir promoters (known as UPREs in yeast,and ERSEs in higher eukaryotes).
The UPR is best understood in the yeastSaccharomyces cerevisiae, where this ERN sig-nalling pathway contains two unique com-ponents — the transmembrane protein Ire1(also known as Ern1) and the transcriptionfactor Hac1, which recognizes UPREelements5. These proteins are not essentialfor vegetative growth but are absolutelyrequired for cell survival under conditionsthat cause ER stress. Ire1 combines in onemolecule a sensor that detects conditions inthe ER lumen, the mechanism for transduc-ing the signal across the ER membrane, andthe means of inducing the synthesis of Hac1.The glycosylated amino-terminal portion ofIre1 is located in the ER lumen, where itapparently senses the decrease in the concen-tration of free BiP that occurs when BiP issequestered into complexes with unfoldedproteins. The C-terminal half of Ire1 carriesan essential protein kinase domain, which isactivated by dimerization of Ire1 and medi-ates trans-autophosphorylation, as well as a
C-terminal domain that functions as anRNA endonuclease (or RNA-cleavingenzyme) following activation by ER stress.This endonuclease activity, together with theRlg1 protein identified previously as a trans-fer-RNA ligase, is required to process theconstitutively synthesized HAC1 messengerRNA precursor to generate an unconven-
A
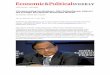
Figure 1 Generation of amyloid-ββββ peptide (Aββββ), the hallmark of Alzheimer’s disease. Amyloid precursor protein (APP) comprises a group of proteins of 695–770 amino acids that are generated by differential mRNA splicing. The residue numbering for the 770-amino-acid form is used here. To generate Aββββ, APP is cleaved twice, first by a ββββ-secretase (ββββ) in the endoplasmic reticulum (ER), and then by a γγγγ-secretase (γγγγ). The latter cleavage event generates Aββββ peptides of either 40 or 42 amino acids; Aββββ42 appears to be generated in pre-Golgi compartments of the secretory pathway, while Aββββ40 is generated in later compartments and in the endocytic pathway. BiP is a molecular chaperone found in the ER, and may promote a conformation of APP that hinders the access of ββββ-secretase. Presenilin-1 mutants characteristic of early-onset Alzheimer’s disease may enhance the production of Aββββ peptides in two ways. They increase γγγγ-secretase activity and, by downregulating chaperone function in the ER2, may increase the susceptibility of APP to cleavage by ββββ-secretase.
BiP
N
C
APP ‘β-stub’ Aβ
β
γ
1
672
770 770
672
711/713
Cytoplasm
Lumen
Amyloidplaques
ER/Golgimembrane
40/42

© 2000 Macmillan Magazines Ltd
news and views
E22 NATURE CELL BIOLOGY | VOL 2 | FEBRUARY 2000 | cellbio.nature.com
tionally spliced mRNA encoding Hac1.The ERN pathway in higher eukaryotes is
less well defined, but two mammalian homo-logues of yeast Ire1 have been identifiedrecently (reviewed in ref. 5). The Ire1α andIre1β proteins are very similar in structure tothe yeast protein, although the sequences oftheir lumenal domains have diverged exten-sively from that of yeast Ire1, and indeedfrom each other. The mammalian Ire1 pro-teins undergo ER-stress-activated trans-autophosphorylation and each retains astress-dependent endonuclease activitycapable of cleaving yeast HAC1 mRNA invivo and in vitro6. However, the endogenoussubstrate(s) of this endonuclease activityremains a mystery because the transcriptionfactor ATF6, which is related to yeast Hac1and recognizes ERSEs in the promoters ofUPR-activated mammalian genes7, is regu-lated by ER-stress-dependent proteolyticcleavage rather than by splicing of itsmRNA8. ATF6 is an ER transmembrane pro-tein, and proteolytic cleavage at or close tothe cytosolic face of the membrane liberatesthe N-terminal cytoplasmic domain, whichcontains the DNA-binding, dimerizationand transactivation domains of the mole-cule. This transcription-factor fragmenttranslocates to the nucleus and activatesERSE-containing target genes8. It remains tobe determined whether the lumenal domainof ATF6 can itself sense the load of unfoldedproteins in the ER, or whether cleavage ofATF6 requires ER-stress-dependent activa-tion of one or both Ire1 proteins.
Katayama et al.2 have now providedintriguing and compelling evidence that theUPR is weakened in Alzheimer’s disease.Expression in neuroblastoma cells of PS1mutants linked to familial Alzheimer’s dis-ease increased the cells’ susceptibility to ERstress and caused a significant decrease in theamount of BiP mRNA, apparently because ofa failure to induce transcriptional activationof UPR-activated genes. Katayama et al. alsoused mice with ‘knocked-in’ gain-of-func-tion PS1 mutants. Primary cultures of neu-rons from embryos of such mice also hadreduced levels of BiP mRNA, while thebrains of patients with sporadic or familialAlzheimer’s disease contained significantlyless BiP and GRP94 (another ER chaperone)than those of age-matched controls. The PS1mutant cells attenuated the UPR at the levelof activation of human Ire1, inhibiting itsautophosphorylation. Ire1 immunoprecipi-tated with PS1, both wild-type and mutant,but only with full-length molecules charac-teristic of the ER, and not with PS1 fragmentsthat localize mainly to the Golgi apparatus.
Curiously, a more recent paper by Niwaet al.6 reports that the UPR is also reduced(about twofold) in cells that lack PS1 (asopposed to cells containing the gain-of-function PS1 mutants used by Katayama etal.2); according to previous studies1, suchPS1-deficient cells lack significant γ-secre-
tase activity. To reconcile the observationsof the two groups, it is necessary to proposethat the PS1 variants used by Katayama etal.2 produce a dominant increase in γ-secre-tase-mediated cleavage of Aβ peptides, buta loss of function in their interaction withIre1. Niwa et al.6 suggest that Ire1α andIre1β are physiological substrates of PS1,which may be responsible for the proteo-lytic cleavages that Niwa et al. (but not, sofar, other researchers) observe in the Ire1proteins. The cleavages apparently generatesoluble cytoplasmic fragments that translo-cate to the nucleus, and, by an as-yet-unknown mechanism, activate transcrip-tion of UPR-regulated genes. Unfortu-nately, Niwa et al.6 did not demonstrate aprecursor–product relationship betweenthe full-length Ire1 proteins and the smallerfragments, and were unable to test whetherthe levels of the fragments were altered inPS1-deficient cells. Furthermore, the frag-ments were present and apparently locatedin the nucleus of at least some cells evenunder unstressed conditions, although ERstress did increase the number of cells thatcould be stained using antibodies raisedagainst peptides corresponding to the C ter-mini of the Ire1 proteins.
So, many intriguing questions remain tobe answered about the mechanism(s) bywhich PS1 mutants promote Alzheimer’sdisease and about the role of PS1 in the UPR.How do the great variety of point mutationsin PS1, scattered along the length of the PS1sequence1, all cause gain-of-function in γ-secretase activity and generation of Aβ42? Isthe detrimental effect of these mutant PS1proteins on Ire1 activation directly linked tothis increase in γ-secretase activity? If this isthe case, why might a lack of PS1 also causedownregulation of the UPR? Or is PS1 a typeof chaperone that positively regulates anundiscovered γ-secretase (analagous to the
activation by SCAP of the SREBP-cleavageprotein9) and independently modulates theactivity of Ire1 proteins? Will the UPR also beaffected in cases of Alzheimer’s disease thatare not associated with mutations in preseni-lin proteins?
One question for which we have possibleanswers concerns the relevance in Alzhe-imer’s disease of the attenuation of the UPRby mutant presenilins. Katayama et al.2 pro-pose that the inability of cells expressingPS1 mutants to induce ER chaperones inresponse to need increases their vulnerabil-ity to various cell-death-inducing stimulithat provoke ER stress.
An alternative, but not mutually exclu-sive, possibility is that an ER chaperone(s)downregulated by mutant PS1 normally sup-presses the formation of amyloidogenic Aβpeptides. How might this work? BiP, which isprominent among the molecular chaperonesregulated by the UPR, recognizes unfoldedpolypeptides in the ER lumen and, by inhib-iting intramolecular or intermolecularaggregation, maintains these polypeptides ina state competent for subsequent folding andoligomerization (reviewed in ref. 10). Likethe majority of newly synthesized polypep-tides that transit through the secretory path-way, APP interacts with BiP in the ER4. Theobserved association between BiP and APP istransient and restricted to the immature,core-glycosylated form of APP that is presentin the ER. Interestingly, the so-called Swed-ish mutant APP, which is linked to Alzhe-imer’s disease and associated with increasedproduction of both Aβ42 and Aβ40, alsointeracts with BiP only transiently and to nogreater an extent than wild-type APP4. Thisindicates that the increased generation of Aβpeptides from the mutant APP results notfrom its misfolding but rather, as previouslysuggested11, from the presence of the pointmutations near the β-cleavage site, which
News and Views contributionsThe News and Views section provides a forum in which new advances in the field of cell biology, as reported in published papers, can be communicated to a wide audience.
Most News and Views pieces are linked to Articles that appear in Nature Cell Biology, but some may focus on papers of exceptional significance that are published elsewhere. Unsolicited contributions will not normally be considered, although prospective authors are welcome to make proposals to the Editorbefore the paper is published.
As a general guideline, News and Views pieces should be about 1,300 words, with one or two display items (figures, boxes and tables). They should make clear the advance (the ‘news’ ) and communicate a sense of excitement, yet provide a critical evaluation of the work in the context of the rest of the field. We encourage personal ‘views’, criticisms and predictions, but authors should not refer to their own work, except in passing.
Detailed guidelines are available on request from [email protected] and on Nature Cell Biology’s Web site (http://cellbio.nature.com).

© 2000 Macmillan Magazines Ltd
news and views
NATURE CELL BIOLOGY | VOL 2 | FEBRUARY 2000 | cellbio.nature.com E23
alter its accessibility to β-secretase.Overexpression of BiP impairs the mat-
uration of APP into the modified formsfound in the Golgi apparatus, and sup-presses the formation of the Aβ40 and Aβ42peptides from both wild-type APP and theSwedish mutant4. Because the levels of bothforms of Aβ peptide are reduced, it is possi-ble that the increased concentration of BiPin the ER lumen promotes a conformationof APP that hinders access of β-secretase.This would decrease the amount of β-stubsavailable for γ-secretase(s), either in the ER(for generation of Aβ42) or in distal com-partments of the secretory pathway (whereAβ40 can be generated).
It seems, therefore, that PS1 mutationsdeliver a ‘double whammy’ to promote thegeneration of Aβ peptides and, conse-quently, Alzheimer’s disease. Not only doPS1 mutants increase γ-secretase activitybut, by downregulating chaperone function
in the ER, they may increase the accessibilityof APP to cleavage by secretase enzymes, aswell as increasing the susceptibility of cellsto ER stress.
This is by no means the first instance inwhich molecular chaperones appear asparticipants in the development of amy-loid diseases, nor the first observation ofamyloid-disease-associated alterations inchaperone levels. A variety of molecularchaperones can modulate the conforma-tional state of yeast and mammalian prionproteins (see, for example, refs 12, 13).Furthermore, the inducibility of someheat-shock-responsive proteins (many ofwhich are molecular chaperones) isaltered in prion-infected cells, which mayalso exhibit an unusual distribution ofHsc73, a cytosolic homologue of BiP14.Katayama et al.’s results2 forewarn those ofus seeking to understand such changesthat cells can use undreamed-of mecha-
nisms to subvert chaperone function. hMary-Jane Gething is in the Department of Biochemistry and Molecular Biology, University of Melbourne, Parkville, Victoria 3052, Australia.e-mail: [email protected]
1. Selkoe, D. J. Trends Cell Biol. 8, 447–453 (1998).
2. Katayama, T. et al. Nature Cell. Biol. 1, 479–485 (1999).
3. Hussain, I. et al. Mol. Cell. Neurosci. 14, 419–427 (1999).
4. Yang, Y., Turner, R. S. & Gaut, J. R. J. Biol. Chem. 273, 25552–
25555 (1998).
5. Kaufman, R. Genes Dev. 13, 1211–1233 (1999).
6. Niwa, M., Sidrauski, C. & Walter, P. Cell 99, 691–702 (1999).
7. Yoshida, H., Haze, K., Yanagi, H., Yura, T. & Mori, K. J. Biol.
Chem. 273, 33741–33749 (1998).
8. Haze, K., Yoshida, H., Yanagi, H., Yura, T. & Mori, K. Mol. Biol.
Cell 10, 3787–3799 (1999).
9. Brown, M. S. & Goldstein, J. L. Cell 89, 331–340 (1997).
10. Gething, M.-J. Semin. Cell Dev. Biol. 10, 465–472 (1999).
11. Selkoe, D. J. Nature 399 (Suppl.), A23–A31 (1999).
12. DebBurman, S. K., Raymond, G. J., Caughey, B. & Lindquist, S.
Proc. Natl Acad. Sci. USA 94, 13938–13943 (1997)
13. Chernoff, Y. O., Newman, G. P., Kumar, J. & Zink, A. D. Mol.
Cell Biol. 19, 8103–8112 (1999).
14. Tatzelt, J., Voellmy, R. & Welch, W. J. Cell. Mol. Neurobiol. 18,
721–729 (1998).
CD95: more than just a death factor?
Anne-Odile Hueber
The CD95 protein delivers crucial signals for lymphocyte death, and may also negatively regulate T-lymphocyte activation by preventing the influx of calcium ions from the cell’s exterior. The block in calcium-ion influx occurs through the activation of acidic sphingomyelinase and the release of ceramide, a metabolite that can also induce cell death.
aintaining a finely tuned, properlyregulated immune response requiresthat the immune system’s watchdogs,
the lymphocytes, are precisely controlled sothat they respond correctly to stimulation byantigens. Perturbation of these controlmechanisms may result in profoundimmune defects, such as lymphoproliferativedisorders, autoimmunity or immunodefi-ciencies. Lepple-Wienhues and colleagues1
now describe a possible link between two dif-ferent ways of controlling circulating T lym-phocytes. They show that CD95, a proteinfound on the surface of T lymphocytes andknown to be involved in inducing the deathof activated T cells, can also limit T-cell acti-vation by interfering with signalling throughtheir antigen receptors.
The pathway by which signalling throughCD95 leads to cell death (apoptosis) has beenstudied extensively. It begins with crosslink-ing of CD95 (also known as APO-1 or Fas) toeither an anti-CD95 antibody or its in vivoligand, CD95L, which may be induciblyexpressed on the same T cell or on a neigh-
bouring one. This crosslinking results in theformation of the death-inducing signallingcomplex (DISC) in the cytoplasm, a complexthat contains CD95, the CD95-associateddeath-domain-containing molecule FADD,and procaspase-8. As a result of associationwith this complex, procaspase-8 is cleaved,generating caspase-8. Caspase-8 is thenreleased and activates a cascade of caspases,the workhorses of the apoptotic machinery— protein-cleaving enzymes that act atcysteine residues within their targets. Thiscascade is activated directly in so-called type-I cells, and indirectly in type-II cells. In thelatter case, caspase-8 cleaves Bid, a pro-apop-totic member of the Bcl-2-protein family,which in turn leads to activation of the apop-togenic function of mitochondria throughthe release of cytochrome c, resulting in thecleavage of other caspases downstream of themitochondria (reviewed in ref. 2).
Until now, the role of CD95 was thoughtto be restricted to inducing cell death, butLepple-Wienhues and colleagues1 provideevidence that this receptor may also limit
M
the proliferation of activated, mature T cellsby downregulating their activation. Bindingof the T-cell antigen receptor (TCR) to itscognate antigen normally induces an influxof calcium ions (Ca2+) into the T cell, andthis influx is needed to activate the T celland allow it to proliferate in response to theantigen, so generating a clone of T cells thatall recognize the same antigen. Lepple-Wienhues et al. report that stimulation ofCD95 inhibits this influx of Ca2+ and that, asa consequence, these cells are unable torespond to stimulation through the TCR.
Increases in the intracellular concentra-tion of ‘free’ (cytosolic) Ca2+ regulate manylymphocyte processes, such as proliferationand acquisition of immune function. In non-excitable cells, such as lymphocytes, increasesresult from activation of phospholipase Cand generation of inositol-1,4,5-trisphos-phate, which, by binding to its receptor onthe endoplasmic reticulum (ER), activatesCa2+-permeable channels on the ER mem-brane and causes the release of stored Ca2+
into the cytoplasm. This release of Ca2+ fromintracellular stores causes an influx of extra-cellular Ca2+ through the opening of Ca2+-release-activated Ca2+ channels (CRACs) inthe plasma membrane. This process is called‘capacitative Ca2+ entry’ or ‘store-operatedCa2+ entry’ (for a review, see ref. 3). Once theCRACs are open, the transmembrane Ca2+
concentration gradient and the electricalpotential across the plasma membrane(membrane potential) provide the drivingforce for Ca2+ entry.
Lepple-Wienhues and colleagues1 findthat stimulation of CD95 does not affect therelease of Ca2+ from intracellular stores, andso presumably must be inhibiting Ca2+
influx into the cytosol by negatively regu-lating CRACs. They then show that the




![Amyloid Precursor Protein (APP) processing in Alzheimer’s ...Presenilin 1 7297 P ELISA, IHC-Fr, IHC-P, WB Presenilin 1 [APS 11] 15456 M ELISA, IF, IHC-P, WB Presenilin 1 [APS 18]](https://img.pdfslide.us/doc/110x75/5f41361f607f43742c6d85e0/amyloid-precursor-protein-app-processing-in-alzheimeras-presenilin-1-7297.jpg)