Embed Size (px)
Citation preview

Polymer International 43 (1997) 210È216
Preparation, Characterization andBiodegradation Characteristics of
Poly(adipic -lactide)anhydride-co-D,L
K. J. Zhu* & Yuan Lei
Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, PeopleÏs Republic of China
(Received 13 November 1996 ; accepted 19 December 1996)
Abstract : Poly(adipic anhydride-co-D,L-lactide) (P(AA-LA)) has been synthesizedby ring-opening polymerization of adipic anhydride (AA) and D,L-lactide (LA)using stannous octoate as catalyst in bulk and in solution. The copolymers werecharacterized by IR, nuclear magnetic resonance, gel permeation chromato-graphy and di†erential scanning calorimetry. The physical properties can betailored by varying the copolymer compositions, and showed low glass transitiontemperature, melting temperature and good solubility in common solvents. Invitro tests showed that after rapid weight loss in the Ðrst day, a constant degra-dation rate was observed. The release proÐles of model drugs, bovine serumalbumin and norethindrone over 16 days followed closely that of the degradationof copolymers containing higher amounts of AA (AA[ 64 mol%), suggestingthat the release mechanism was controlled predominantly by surface erosion.However, a large deviation from the close correlation of polymer degradationand drug release was observed for copolymers containing lower amounts of AA(\30 mol%). These materials may be useful in protein delivery systems.
Polym. Int. 43, 210È216 (1997)No. of Figures : 10. No. of Tables 3. No. of Refs : 14
Key words : Poly(adipic anhydride-co-D,L-lactide), surface erosion copolymers,drug controlled release
INTRODUCTION
Controlled release matrices composed of biodegradablepolymers such as poly(D,L-lactide), poly(glycolide) andtheir copolymers generally degrade in a homogeneousmanner.1,2 This leads to a progressive loosening of thematrix from the surface and inner layers, which causeschange in both permeability and mechanical strength ofthe devices. It would be desirable for a matrix to bedegraded heterogeneously from the surface to innerlayers. Such erosion would lead to zero-order drugrelease.3 Poly(orthoester)4 and poly(anhydride) havebeen reported as bioerodible materials ; the latter especi-ally has been extensively studied by Langer et al.5h7Recently we studied the preparation of surface biode-gradable polymers with hydrophobic characteristics in asearch for new matrices for controlled release of peptide
* To whom all correspondence should be addressed.
and protein drugs. We assumed that these kinds ofmatrices might facilitate the stability of peptide andprotein in the release process, which is a major problemin the area of peptide and protein controlled release.8For this purpose, we prepared a group of novel copoly-mers which exhibited surface biodegradable character-istics. In this communication we report somepreliminary results on the preparation and degradationcharacteristics of poly(adipic anhydride-co-D,L-lactide)(P(AA-LA)).
EXPERIMENTAL
Synthesis of monomers
D,L-Lactide (LA) was synthesized according to the liter-ature.9 Crude products were recrystallized three timesfrom ethyl acetate, giving material with a melting point
2101997 SCI. Polymer International 0959-8103/97/$17.50 Printed in Great Britain(

Characterization and biodegradation of P(AA-L A) 211
of 125È126¡C. Adipic anhydride (AA) was synthesizedas described by Hill.10 Adipic acid (50 g) and aceticanhydride (40 ml) were reÑuxed together for 4 h. Theacetic acid formed in the reaction and the excess aceticanhydride were removed by distillation in a vacuum.The distillate was collected between 105 and 120¡Cunder vacuum (0É1 mmHg). The solid phase of the distil-late was separated by centrifuging, and the liquid wasredistilled. Colourless AA was obtained (boiling point98È100¡C at 0É1 mmHg, yield c. 40%), and sealed in adried glass tube for use.
Polymerization
For polymerization in bulk, well dried LA and AA wereplaced into a polymerization tube under nitrogen atmo-sphere, and stannous octoate (0É2 mol%, dissolved inpetroleum ether) was added. The tube was connected toa vacuum system and heated at 60¡C for 30 min toremove solvent, the temperature was then raised to130¡C and the tube was sealed. The polymerization wascarried out at 130¡C for 48 h.
For polymerization in solution, well dried monomers,toluene and catalyst were placed into a polymerizationtube and sealed under reduced pressure, then heated at90¡C for 20 h. The product mixtures were precipitatedfrom n-hexane.
The crude products obtained from both bulk andsolution polymerization were extracted with a solventmixture of ethyl acetate and n-hexane (2 : 1 v/v) toremove possible LA and PLA. Then the products werefurther extracted with a solvent mixture of methylenechloride and ethyl acetate (1 : 5É5 v/v) to remove pos-sible AA and PAA. The characteristics of the copoly-mers are given in Table 1.
Characterization
The copolymers were characterized by IR, nuclear mag-netic resonance (NMR), gel permeation chromato-graphy (GPC) and di†erential scanning calorimetry.The Fourier transform infrared (FTIR) spectra wereobtained from a Nicolet FTIR spectrometer model
5DXC. Samples were either Ðlm-cast in chloroform onKBr plates or pressed into KBr pellets. The NMRspectra were recorded on a JEOL 90Q NMR spectro-meter at room temperature in with tetra-CDCl3methylsilane as the internal standard. The molecularweight was determined by GPC (Waters-208 with poly-styrene standards, k-Styragel columns of 103, 104 and
chloroform as solvent, Ñow rate 1É5 mlmin~1). A105 Ó,Perkin Elmer DSC-7 was used for the determination oftransition temperature. The sample size was about10 mg and the heating rate was 10¡Cmin~1. Scanningelectron micrographs were obtained on a JSM-T20instrument. The specimens were sputter-coated withgoldÈpalladium for visualization in the scanning elec-tron microscope.
Degradation and drug release in vitro
Drug-incorporated matrices were formulated as follows.The micronized copolymers were sieved into a particlesize range of 90È150 km and mixed with the same sizerange of model drugs. The mixture was pressed into cir-cular discs (diameter 15 mm, thickness 2 mm) under apressure of 500 kg cm~2 at 60¡C. To study the degrada-tion behaviour of the copolymers, the same procedureswere conducted as for sample preparation, except fordrug mixing.
Degradation of the copolymers in vitro was measuredby immersion of samples in phosphate-bu†ered saline(PBS; pH 7É4) at 37¡C; samples were recovered period-ically, and weight loss and molecular weight were deter-mined. The drug release kinetics were followed bymeasuring the UV absorbance at 240 nm for nor-ethindrone (NET) and at 280 nm for bovine serumalbumin (BSA).11 The degradation and release experi-ments were done in triplicate.
RESULTS AND DISCUSSION
Polymerization
The polymerization data are partially shown in Tables2 and 3. It was found that the copolymerization of AA
TABLE 1. Characteristics of PAA and P(AA-LA) copolymersa
Sample code Composition M1n
M1w
M1w/M1
nT
gT
m(AA/LA by mole) (Ã104) (Ã104) (¡C) (¡C)
PAA 100/0 1·18 2·59 2·2 – 71
P(AA-LA) É82 82/18 1·31 2·75 2·1 – 58
P(AA-LA)-64 64/36 1·36 2·99 2·2 – 57
P(AA-LA)-45 45/55 1·42 3·26 2·3 36 55
P(AA-LA)-30 30/70 1·55 3·25 2·1 42 –
P(AA-LA)-15 15/85 1·61 3·86 2·4 49 –
PLA 0/100 1·66 3·65 2·2 56 –
a The compositions were determined by 1H NMR; molecular weights were measured by
GPC using chloroform as solvent at 25¡C; and values were obtained by DSC.Tg
Tm
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997
212 K. J. Zhu, Y . L ei
TABLE 2. Partial data of bulk copolymerization of adipic anhydride (AA) and lactide
(LA)a
AA/LA Catalyst Polym. Polym. Yield M1w
M1w/M1
n(by mole) conc. (Ã10É3, mol%) temp. (¡C) time (h) (%) (Ã104)
0·2 2·0 120 30 82 3·2 2·2
0·3 3·5 120 30 80 3·3 2·3
0·5 3·5 120 30 76 3·0 2·0
1·0 3·5 120 30 73 2·7 2·1
1·5 3·5 120 30 69 2·2 2·3
0·5 3·5 110 48 85 3·6 2·4
0·5 3·5 130 30 75 2·9 2·0
0·5 3·5 140 30 65 2·5 2·2
0·5 12·0 120 30 78 2·8 2·3
0·5 3·5 120 48 82 2·6 2·2
was used as catalyst ; was measured by GPC using chloroform as solvent at 25¡C.a Sn(oct)2
M1w
and LA can be carried out in both bulk and solution,but the molecular weight of the resulting polymers washigher in bulk than in solution. Several catalysts weree†ective for AA and LA reaction ; however, AlEt3ÈH2Oand Al(o-iPr) were more efficient (Table 3). If toxicity3of catalyst was of concern, was preferred forSn(oct)2use. The concentration of catalysts in the range 2 to12 ] 10~3mol% had no signiÐcant e†ect on the yieldand molecular weight of the copolymers. A reactiontemperature in the range 110 to 130¡C for bulk and 70to 90¡C for solution polymerization was preferred.When the temperature was over 140¡C in bulk poly-merization, the product became dark and the molecularweight decreased. A non-polar solvent (such as toluene)was more efficient than a polar solvent (such asdimethylsulphoxide) in solution polymerization. It wasalso noticed that the AA fraction in the copolymer washigher than that in the feed, indicating that AA wasmore reactive than LA. The reactivity ratios were calcu-
lated by the FinemanÈRoss method12 as r1(AA)\ 3É46and r2(LA)\ 0É46.
Characterization
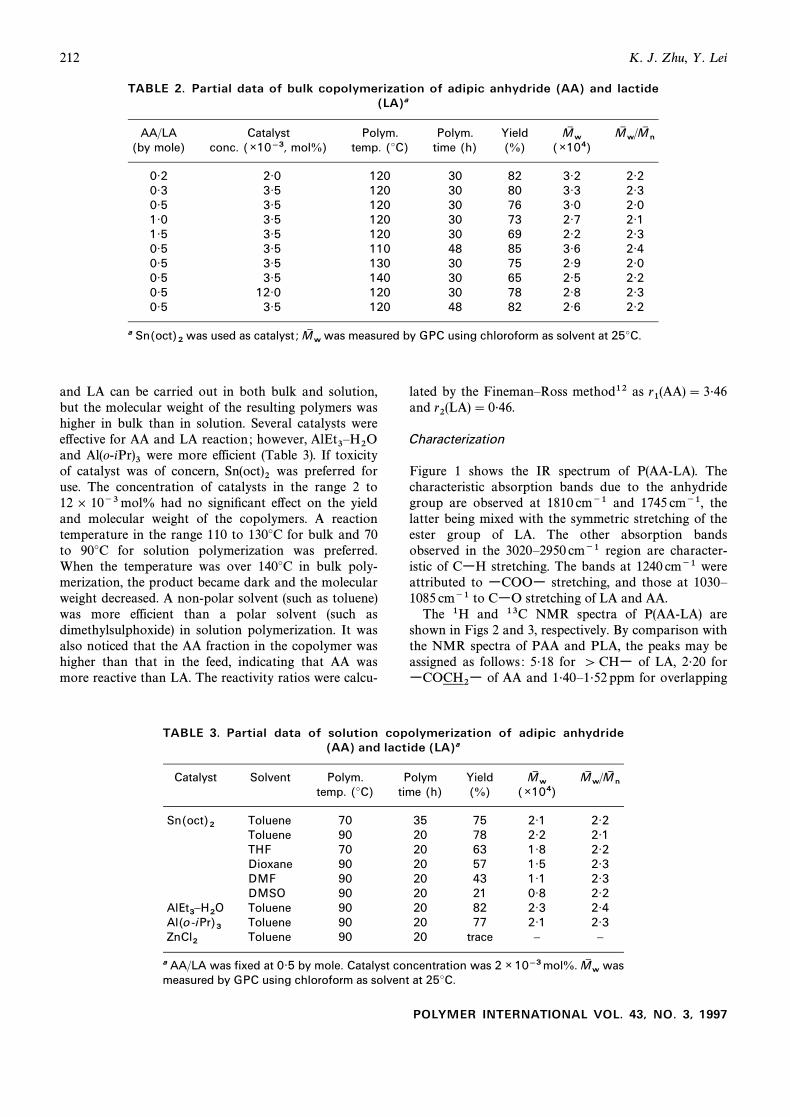
Figure 1 shows the IR spectrum of P(AA-LA). Thecharacteristic absorption bands due to the anhydridegroup are observed at 1810 cm~1 and 1745 cm~1, thelatter being mixed with the symmetric stretching of theester group of LA. The other absorption bandsobserved in the 3020È2950 cm~1 region are character-istic of CwH stretching. The bands at 1240 cm~1 wereattributed to wCOOw stretching, and those at 1030È1085 cm~1 to CwO stretching of LA and AA.
The 1H and 13C NMR spectra of P(AA-LA) areshown in Figs 2 and 3, respectively. By comparison withthe NMR spectra of PAA and PLA, the peaks may beassigned as follows : 5É18 for [ CHw of LA, 2É20 for
of AA and 1É40È1É52 ppm for overlappingwCOCH2w
TABLE 3. Partial data of solution copolymerization of adipic anhydride
(AA) and lactide (LA)a
Catalyst Solvent Polym. Polym Yield M1w
M1w/M1
ntemp. (¡C) time (h) (%) (Ã104)
Sn(oct)2
Toluene 70 35 75 2·1 2·2
Toluene 90 20 78 2·2 2·1
THF 70 20 63 1·8 2·2
Dioxane 90 20 57 1·5 2·3
DMF 90 20 43 1·1 2·3
DMSO 90 20 21 0·8 2·2
AlEt3–H
2O Toluene 90 20 82 2·3 2·4
Al(o-iPr)3
Toluene 90 20 77 2·1 2·3
ZnCl2
Toluene 90 20 trace – –
a AA/LA was fixed at 0·5 by mole. Catalyst concentration was 2 Ã10É3 mol%. wasM1w
measured by GPC using chloroform as solvent at 25¡C.
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997
Characterization and biodegradation of P(AA-L A) 213
Fig. 1. Infrared spectrum of poly(adipic anhydride-co-D,L-lactide) (KBr).
Fig. 2. 1H NMR spectrum of poly(adipic anhydride-co-D,L-lactide) in (a) 1É40È1É52 ppm of LA,CDCl3 . (CH3
of AA) ; (b) 2É20 ppm of AA) ; (c)COCH2CH2 (COCH25É18 ppm (CH of LA).
Fig. 3. 13C NMR spectrum of poly(adipic anhydride-co-D,L-lactide) in (a) 16É3 ppm of LA) ; (b) 24É2 ppmCDCl3 . (CH3
of AA) ; (c) 33É5 ppm of AA) ; (d)(COCH2CH2 (COCH268É8 ppm (CH of LA) ; (e) 168É8 ppm (CO of LA) ; (f) 173É9 ppm
(CO of AA).
Fig. 4. Molecular weight changes of PAA and P(AA-LA)versus degradation time in 0É1 M pH 7É4 phosphate bu†er at37¡C. of PAA, P(AA-LA)-82, P(AA-LA)-64 and P(AA-M1 n0LA)-15 were 1É18 ] 104, 1É31 ] 104, 1É36 ] 104 and
1É61 ] 104, respectively.
of of LA and of AA. In the 13CCH3 wCOCH2CH2wNMR spectrum the bands at 16É3, 68É8 and 168É8 ppmare due to wCH and wCOw of LA, respec-wCH3 ,tively ; and the bands at 24É2, 33É5 and 173É9 ppm corre-spond to and wCOwwCOCH2CH2w, wCOCH2wof AA, respectively.
The DSC data for P(AA-LA) are given in Table 1.The morphology of P(AA-LA) is related to the composi-tions of the copolymers. If the AA content in thecopolymers was less than 30 mol%, there was only oneglass transition temperature (amorphous state).However, the copolymers containing more than45 mol% AA exhibited melting peaks (semicrystallinestate). The values of and for P(AA-LA) were lowerTg Tm
Fig. 5. Degradation proÐles of poly(adipic anhydride-co-D,L-lactide) in 0É1 M pH 7É4 phosphate bu†er at 37¡C.
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997
214 K. J. Zhu, Y . L ei
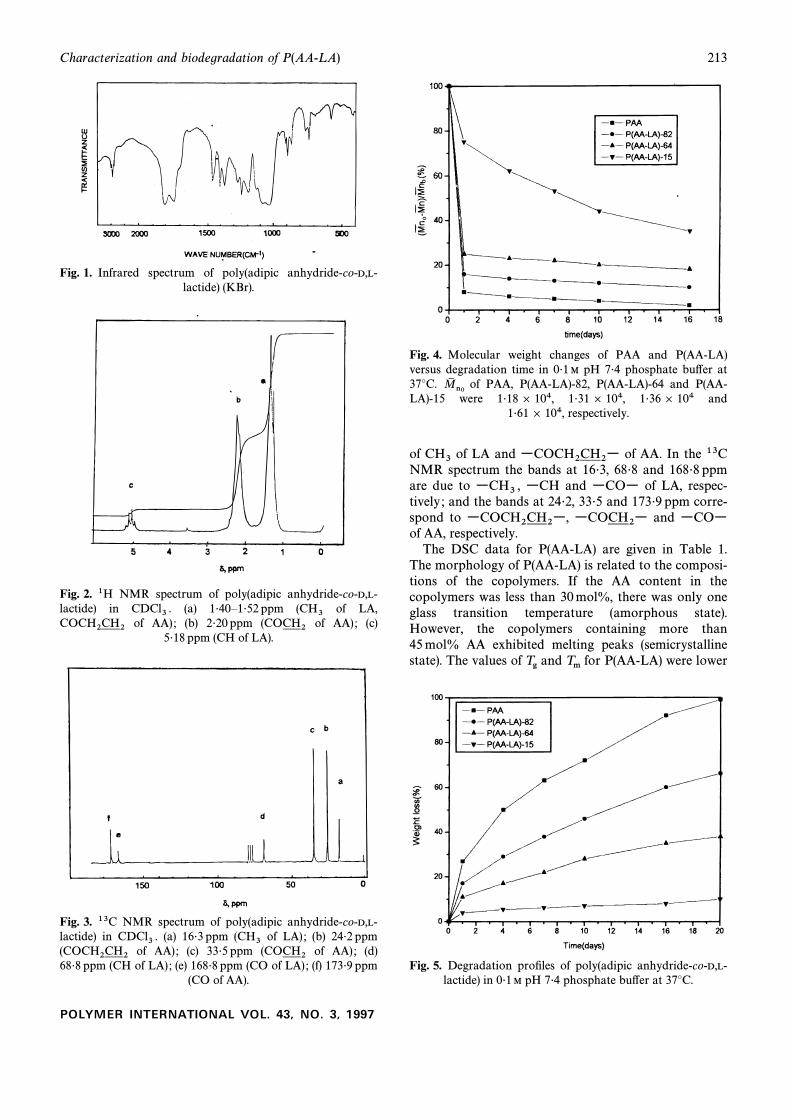
Fig. 6. Scanning electron micrographs of surface morphology of P(AA-LA)-82, after (a) 0 day, (b) 3 days, (c) 10 days and (d) 16days in 0É1 M pH 7É4 phosphate bu†er at 37¡C.
than those of the homopolymers. This may be advanta-geous for the formulation of unstable drugs, such asproteins, under mild conditions.
In vitro degradation
The copolymers su†ered hydrolysis and dissolution insaline solution ; this process can be followed by determi-nation of the molecular weight and weight loss of thematrix. Figures 4 and 5 show the molecular weightchanges and weight loss of PAA and P(AA-LA) in thetest period, respectively. It can be seen that the molecu-lar weight of PAA, P(AA-LA)-82 and P(AA-LA)-64decreased rapidly during the Ðrst day (more than 70%),while P(AA-LA)-15 only dropped by 15%, then allslowed down. On the other hand, the matrix of PAAwas completely degraded in 20 days, but for P(AA-LA)-
15 only 10% mass was lost in the same period. Theweight loss of PAA and P(AA-LA) copolymers showeda near-constant rate of degradation after a rapid loss inthe Ðrst day, which was caused by dissolution of lowmolecular weight fractions. One contributing factor tothis degradation behaviour may be the chemical struc-ture of the copolymer, containing anhydride and estergroups in the backbone. It is known that anhydridegroups are more susceptible to hydrolysis in water thanester groups. However, upon hydrolysis of the endgroups, wCOOH groups were formed. This local acidicenvironment decreases the anhydride degradation13 butaccelerates ester degradation.14 It may lead to constantdegradation of the copolymers in the later stages.
The external shape of the disc samples remainedunchanged even after 16 days, although the sample dis-integrated after removal from the vial unless handled
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997
Characterization and biodegradation of P(AA-L A) 215
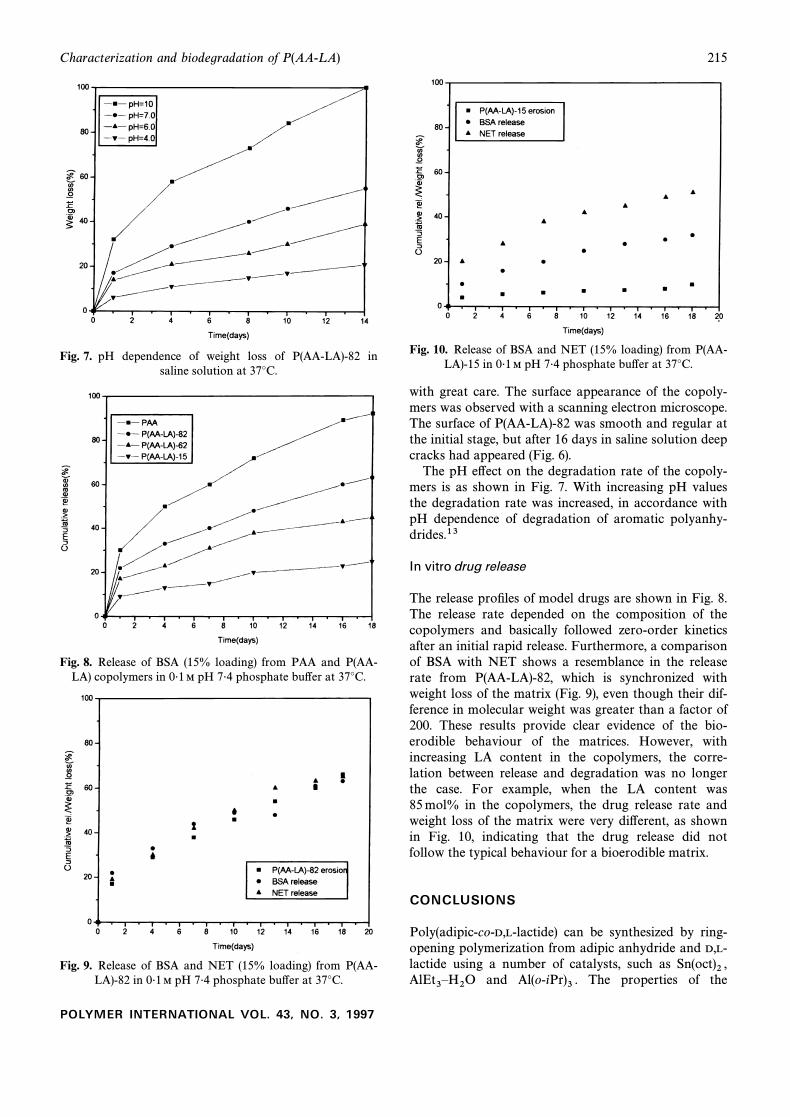
Fig. 7. pH dependence of weight loss of P(AA-LA)-82 insaline solution at 37¡C.
Fig. 8. Release of BSA (15% loading) from PAA and P(AA-LA) copolymers in 0É1 M pH 7É4 phosphate bu†er at 37¡C.
Fig. 9. Release of BSA and NET (15% loading) from P(AA-LA)-82 in 0É1 M pH 7É4 phosphate bu†er at 37¡C.
Fig. 10. Release of BSA and NET (15% loading) from P(AA-LA)-15 in 0É1 M pH 7É4 phosphate bu†er at 37¡C.
with great care. The surface appearance of the copoly-mers was observed with a scanning electron microscope.The surface of P(AA-LA)-82 was smooth and regular atthe initial stage, but after 16 days in saline solution deepcracks had appeared (Fig. 6).
The pH e†ect on the degradation rate of the copoly-mers is as shown in Fig. 7. With increasing pH valuesthe degradation rate was increased, in accordance withpH dependence of degradation of aromatic polyanhy-drides.13
In vitro drug release
The release proÐles of model drugs are shown in Fig. 8.The release rate depended on the composition of thecopolymers and basically followed zero-order kineticsafter an initial rapid release. Furthermore, a comparisonof BSA with NET shows a resemblance in the releaserate from P(AA-LA)-82, which is synchronized withweight loss of the matrix (Fig. 9), even though their dif-ference in molecular weight was greater than a factor of200. These results provide clear evidence of the bio-erodible behaviour of the matrices. However, withincreasing LA content in the copolymers, the corre-lation between release and degradation was no longerthe case. For example, when the LA content was85 mol% in the copolymers, the drug release rate andweight loss of the matrix were very di†erent, as shownin Fig. 10, indicating that the drug release did notfollow the typical behaviour for a bioerodible matrix.
CONCLUSIONS
Poly(adipic-co-D,L-lactide) can be synthesized by ring-opening polymerization from adipic anhydride and D,L-lactide using a number of catalysts, such as Sn(oct)2 ,
and Al(o-iPr) The properties of theAlEt3ÈH2O 3 .
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997

216 K. J. Zhu, Y . L ei
copolymers can be tailored by adjusting the composi-tion. Degradation and drug release tests showed thattypical surface erosion behaviour of the matrices andzero-order kinetics for drug release were achieved withhigher AA contents of the copolymers. The materialswith lower and may be advantageous for formu-T g T mlation of peptide and protein drugs.
ACKNOWLEDGEMENT
We are indebted to the National Natural Science Foun-dation of China for their support of this work.
REFERENCES
1 Heller, J. & Baker, R. W., in Controlled Release of BioactiveMaterials, ed. R. W. Baker. Academic Press, New York, 1980, pp.17.
2 Pitt, C. G., Gratzl, M. M., Kummel, G. L., Surles, J. & Schindler,A., Biomaterials, 2 (1981) 215.
3 Hopfenberg, H. B., Pap. Meet-Am. Chem. Soc., Div. Org. Coat.Plast. Chem. 1976, 36 229È234.
4 Heller, J., Biomaterials, 11 (1990) 659.5 Tamada, J. & Langer, R., J. Biomater. Sci. Polym. Edn, 3 (1992)
315.6 Domb, A. & Langer, R., Macromol. Chem. Macromol. Symp., 19
(1988) 189.7 Yasuhiko, T. & Langer, R., Pharm. Res., 10 (1993) 391.8 Pitt, C. G., Int. J. Pharm., 59 (1990) 173.9 Gilding, D. K. & Reed, A. M., Polymer, 20 (1979) 1459.
10 Hill, J. W., J. Am. Chem. Soc., 52 (1930) 4110.11 Crotts, G. & Park, T. G., J. Contr. Rel., 35 (1995) 91.12 Fineman, M. & Ross, S. D., J. Polym. Sci., 5 (1950) 259.13 Leong, K. W., Brott, B. C. & Langer, R., J. Biomed. Mater. Res.,
19 (1985). 941.14 Hu†man, K. R. & Casey, D., J. Polym. Sci., Polym. Chem. Edn., 23
(1985) 1939.
POLYMER INTERNATIONAL VOL. 43, NO. 3, 1997





























