Embed Size (px)
Citation preview

Polymerization-Like Co-Assembly of SilverNanoplates and Patchy SpheresBinbin Luo,† John W. Smith,† Zixuan Wu,† Juyeong Kim,†,‡ Zihao Ou,† and Qian Chen*,†,‡,§
†Department of Materials Science and Engineering, ‡Frederick Seitz Materials Research Laboratory, and §Department of Chemistry,University of Illinois at UrbanaChampaign, Urbana, Illinois 61801, United States
*S Supporting Information
ABSTRACT: Highly anisometric nanoparticles have dis-tinctive mechanical, electrical, and thermal properties andare therefore appealing candidates for use as self-assemblybuilding blocks. Here, we demonstrate that ultra-aniso-metric nanoplates, which have a nanoscale thickness but amicrometer-scale edge length, offer many material designcapabilities. In particular, we show that these nanoplates“copolymerize” in a predictable way with patchy spheres(Janus and triblock particles) into one- and two-dimen-sional structures with tunable architectural properties. Wefind that, on the pathway to these structures, nanoplatesassemble into chains following the kinetics of molecular step-growth polymerization. In the same mechanistic framework,patchy spheres control the size distribution and morphology of assembled structures, by behaving as monofunctional chainstoppers or multifunctional branch points during nanoplate polymerization. In addition, both the lattice constant and thestiffness of the nanoplate assemblies can be manipulated after assembly. We see highly anisometric nanoplates as onerepresentative of a broader class of dual length-scale nanoparticles, with the potential to enrich the library of structures andproperties available to the nanoparticle self-assembly toolbox.
KEYWORDS: ultra-anisometric nanoplates, patchy spheres, self-assembly dynamics, colloidal step-growth polymerization,adaptive materials, chain stiffness
Biological systems often employ nanoscale buildingblocks with strictly controlled chemistry or intricatesurface patterning to engineer self-assembled structures
with unique properties and adaptive functional behavior fromthe bottom-up.1−4 However, not all of the tricks up nature’ssleeve hinge on such sophisticated strategies; there aresometimes far simpler ways in which living systems will exploitfeatures of a self-assembly building block to “build in” aparticular set of desired properties. One such feature isanisometry. Consider, for example, the fibrous protein collagen.The staggered arrangement of highly anisometric tropocollagenunits within this protein ultimately allows collagen to undergomultiple modes of tensile deformation and thereby exhibitexceptional elastic energy absorption.5,6 Even though manyindividual tropocollagen units are required to form thisfunctional staggered array, by choosing a highly anisometricbuilding block (i.e., one that is both very thin and very long),collagen can do so without completely compromising bendingflexibility in the final structure. Similarly remarkable propertiesenabled by anisometry can be found in synthetic nanofiberassemblies, as well; in carbon nanotube7−10 and ultrathin metalnanowire10−13 systems, a high degree of anisometry engendersnot just superlative mechanical behavior but also remarkableelectrical and optical properties in assembled structures.
Here, we present material engineering capabilities offered byanisometry in a different but similarly intriguing context: theself-assembly of ultra-anisometric silver nanoplates. Forexample, as is the case for nanoparticle systems in general,these nanoplates have one dimension which is comparable tothe range of interparticle interactions14,15 (for example, a typicalthickness of ∼30 nm). Thus, a balance between van der Waalsattraction and electrostatic repulsion leads to assembly with“loose” packing16 (Figures 1 and S1). Such loose packing allowsone to tune properties like the size and lattice constant ofassembled structureswhich are parameters relevant toplasmonic coupling applications17−20simply by adjustingthe ionic strength of the surrounding solution. At the sametime, these anisometric nanoplates have a micrometer-scaleedge length. This characteristic facilitates imaging the nanoplateself-assembly trajectory not only in situ and starting from thelevel of single nanoparticles but also in a straightforward way:using optical microscopy. Typically, imaging the solution-phaseself-assembly dynamics of nanoparticles which are nanoscopicin all dimensions is not as simple.16,21 We take advantage of the
Received: March 24, 2017Accepted: July 17, 2017Published: July 17, 2017
Artic
lewww.acsnano.org
© XXXX American Chemical Society A DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
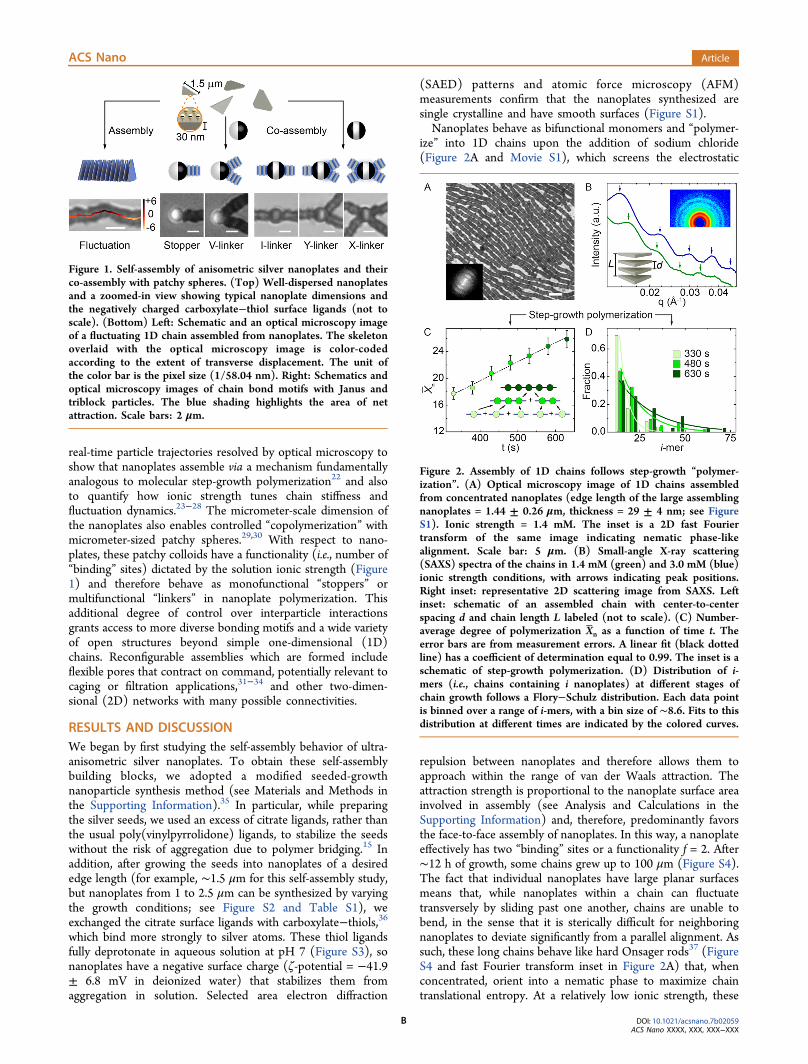
real-time particle trajectories resolved by optical microscopy toshow that nanoplates assemble via a mechanism fundamentallyanalogous to molecular step-growth polymerization22 and alsoto quantify how ionic strength tunes chain stiffness andfluctuation dynamics.23−28 The micrometer-scale dimension ofthe nanoplates also enables controlled “copolymerization” withmicrometer-sized patchy spheres.29,30 With respect to nano-plates, these patchy colloids have a functionality (i.e., number of“binding” sites) dictated by the solution ionic strength (Figure1) and therefore behave as monofunctional “stoppers” ormultifunctional “linkers” in nanoplate polymerization. Thisadditional degree of control over interparticle interactionsgrants access to more diverse bonding motifs and a wide varietyof open structures beyond simple one-dimensional (1D)chains. Reconfigurable assemblies which are formed includeflexible pores that contract on command, potentially relevant tocaging or filtration applications,31−34 and other two-dimen-sional (2D) networks with many possible connectivities.
RESULTS AND DISCUSSIONWe began by first studying the self-assembly behavior of ultra-anisometric silver nanoplates. To obtain these self-assemblybuilding blocks, we adopted a modified seeded-growthnanoparticle synthesis method (see Materials and Methods inthe Supporting Information).35 In particular, while preparingthe silver seeds, we used an excess of citrate ligands, rather thanthe usual poly(vinylpyrrolidone) ligands, to stabilize the seedswithout the risk of aggregation due to polymer bridging.15 Inaddition, after growing the seeds into nanoplates of a desirededge length (for example, ∼1.5 μm for this self-assembly study,but nanoplates from 1 to 2.5 μm can be synthesized by varyingthe growth conditions; see Figure S2 and Table S1), weexchanged the citrate surface ligands with carboxylate−thiols,36which bind more strongly to silver atoms. These thiol ligandsfully deprotonate in aqueous solution at pH 7 (Figure S3), sonanoplates have a negative surface charge (ζ-potential = −41.9± 6.8 mV in deionized water) that stabilizes them fromaggregation in solution. Selected area electron diffraction
(SAED) patterns and atomic force microscopy (AFM)measurements confirm that the nanoplates synthesized aresingle crystalline and have smooth surfaces (Figure S1).Nanoplates behave as bifunctional monomers and “polymer-
ize” into 1D chains upon the addition of sodium chloride(Figure 2A and Movie S1), which screens the electrostatic
repulsion between nanoplates and therefore allows them toapproach within the range of van der Waals attraction. Theattraction strength is proportional to the nanoplate surface areainvolved in assembly (see Analysis and Calculations in theSupporting Information) and, therefore, predominantly favorsthe face-to-face assembly of nanoplates. In this way, a nanoplateeffectively has two “binding” sites or a functionality f = 2. After∼12 h of growth, some chains grew up to 100 μm (Figure S4).The fact that individual nanoplates have large planar surfacesmeans that, while nanoplates within a chain can fluctuatetransversely by sliding past one another, chains are unable tobend, in the sense that it is sterically difficult for neighboringnanoplates to deviate significantly from a parallel alignment. Assuch, these long chains behave like hard Onsager rods37 (FigureS4 and fast Fourier transform inset in Figure 2A) that, whenconcentrated, orient into a nematic phase to maximize chaintranslational entropy. At a relatively low ionic strength, these
Figure 1. Self-assembly of anisometric silver nanoplates and theirco-assembly with patchy spheres. (Top) Well-dispersed nanoplatesand a zoomed-in view showing typical nanoplate dimensions andthe negatively charged carboxylate−thiol surface ligands (not toscale). (Bottom) Left: Schematic and an optical microscopy imageof a fluctuating 1D chain assembled from nanoplates. The skeletonoverlaid with the optical microscopy image is color-codedaccording to the extent of transverse displacement. The unit ofthe color bar is the pixel size (1/58.04 nm). Right: Schematics andoptical microscopy images of chain bond motifs with Janus andtriblock particles. The blue shading highlights the area of netattraction. Scale bars: 2 μm.
Figure 2. Assembly of 1D chains follows step-growth “polymer-ization”. (A) Optical microscopy image of 1D chains assembledfrom concentrated nanoplates (edge length of the large assemblingnanoplates = 1.44 ± 0.26 μm, thickness = 29 ± 4 nm; see FigureS1). Ionic strength = 1.4 mM. The inset is a 2D fast Fouriertransform of the same image indicating nematic phase-likealignment. Scale bar: 5 μm. (B) Small-angle X-ray scattering(SAXS) spectra of the chains in 1.4 mM (green) and 3.0 mM (blue)ionic strength conditions, with arrows indicating peak positions.Right inset: representative 2D scattering image from SAXS. Leftinset: schematic of an assembled chain with center-to-centerspacing d and chain length L labeled (not to scale). (C) Number-average degree of polymerization Xn as a function of time t. Theerror bars are from measurement errors. A linear fit (black dottedline) has a coefficient of determination equal to 0.99. The inset is aschematic of step-growth polymerization. (D) Distribution of i-mers (i.e., chains containing i nanoplates) at different stages ofchain growth follows a Flory−Schulz distribution. Each data pointis binned over a range of i-mers, with a bin size of ∼8.6. Fits to thisdistribution at different times are indicated by the colored curves.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
B

long chains do not attach with each other laterally (for example,see the lateral gap between individual long chains in Figure 2A),indicating a weak interchain attraction in the lateral direction(see Analysis and Calculations in the Supporting Informationfor more details). The face-to-face assembly of nanoplates intochains is also reversible; a decrease in solution ionic strengthleads to the disassembly of chains into individual nanoplates, asshown in Movie S2.The assembly of nanoplates into chains markedly proceeds
by a mechanism characteristic of molecular step-growthpolymerization. As shown in Movie S1 and Figure S5, chainsgrow either by “monomer” addition or by the fusion of existingchains. In addition, we measured the number-average degree ofpolymerization Xn (i.e., ∑nii/∑ni, where ni is the number ofchains containing i nanoplates)38 over the chain growthprocess. Here, we estimated values of i for different chains asi = L/d, where L is the length of the chain measured underoptical microscopy and d is the center-to-center spacingbetween neighboring nanoplates in a chain determined fromSAXS (Figure 2B and Table S2). The measured Xn growslinearly with time t (Figure 2C), which is another qualitativefeature of step-growth polymerization. Similarly, the averagechain length grows linearly with time, which is also consistentwith step-growth polymerization and the fact that the system isa closed system, with a constant number of nanoplates in thefield of view (Figure S5B). We then applied the rate equationfor the externally catalyzed molecular step-growth polymer-ization of bifunctional monomers with identical functionalgroups,38 namely, Xn = 4[M]0kt + 1 (where [M]0 is the initialmolar concentration of silver nanoplates in the field of view), toestimate the nanoplate self-assembly rate constant, k, as 3.2 ×107 M−1 s−1. This rate constant is 3 orders of magnitude largerthan one that has been measured in an inorganic nanocrystalsystem (2.9 × 104 M−1 s−1).38 We attribute this difference tothe fact that the pairwise interaction between nanoplatesthatis, the driving force for self-assemblyis much stronger in thissystem as it scales with the nanoplate surface area. This 1Dchain formation is also observed with nanoplates of differentedge lengths (see, for example, the assembly of nanoplates withan average edge length of 2.17 μm in Movie S3). We observedthe assembly of larger nanoplates at lower ionic strengths (∼0.5mM for 2.17 μm nanoplates, ∼0.8 mM for 1.44 μm nanoplates,etc.), which we attribute to the stronger net attraction betweenlarger nanoplates at similar ionic strengths. For the samereason, we expect that nanoplates would assemble at an evenhigher rate with higher ionic strengths or with larger sizes at thesame ionic strength condition, due to stronger attractions witheach other.We also found that the distribution of chain lengths at
different times follows a Flory−Schulz distribution38 (Figures2D and S6; for more details see Analysis and Calculations in theSupporting Information). Namely, the fraction of chainscontaining i nanoplates (that is, ni/NL, where NL is the totalnumber of chains) is proportional to (1 − p)pi−1. Here, thefitting parameter p is the extent of reaction of nanoplates or theprobability that a nanoplate has “reacted” and become part of achain and is given by p = ([M]0 − [M])/[M]0, where [M] isthe concentration of all species (i.e., chains of any length) at aparticular time. As shown in Figures 2D and S6, as the reactiontime increases, more nanoplates have assembled into longerchains, and the extent of reaction obtained from fitting theFlory−Schulz distribution approaches unity. These two featuresof molecular step-growth polymerizationthat the number-
average degree of polymerization grows linearly with time, andthat the chain length distribution follows a Flory−Schulzdistributionare both ultimately associated with the assump-tion that reactivity is independent of chain length,22 whichfurther suggests that pairwise interactions dominate chaingrowth at this colloidal nanoparticle scale.Seeded growth, although an established synthesis method for
silver nanoplates, can still give rise to particles with some sizedispersity (namely, both the large nanoplates we observedassembling and much smaller nanoplates), as shown in FigureS1. The population of smaller nanoplates in the sample hasedge lengths 22−49% the size of the large nanoplates.However, these smaller nanoplates do not substantially affectthe assembly of large ones because they have a considerablysmaller surface area and, therefore, smaller van der Waalsattraction. In particular, the net attraction between two largenanoplates at 1.4 mM ionic strength was calculated to be about−35 kBT (where kB is the Boltzmann constant and T is thetemperature), favoring step-growth polymerization-like assem-bly. The net attraction between small−small nanoplate andsmall−large nanoplate pairs at the same ionic strengthcondition, based on the average size of the small nanoplates,is only about −4 kBT, which is not strong enough to inducestable assembly into chains.29 Thus, small nanoplates do nottend to assemble with one another or with large nanoplates atrelatively low ionic strength conditions (i.e., below 1.8 mMbased on interaction calculations; see Figure S7D). To confirmthis experimentally, we characterized the assembled chains afterrapid solvent evaporation under vacuum conditions by scanningelectron microscopy (SEM). It is clear that these chains areprimarily composed of large nanoplates of similar size (FigureS8). Meanwhile, unassembled small nanoplates are likely unableto serve as depletion agents that could enhance the self-assembly of large nanoplates due to (i) the insufficient sizeseparation between large and small nanoplates18,39,40 and (ii)the very low number density of small nanoplates in solution.The estimated depletion attraction between two largernanoplates induced by the smaller nanoplates is only about−0.03 kBT (see Analysis and Calculations in the SupportingInformation), which is negligible compared with the total netattraction strength (−35 kBT; see Figure S7B). This is alsoconsistent with the fact that considering only electrostaticrepulsion and van der Waals attraction in assembly very closelypredicts the equilibrium lattice spacings in the assembled chainsmeasured by SAXS (Figure S7C).The length of actively assembling chains can be controlled by
using a colloidal analogue to the molecular polymerization“chain stopper” strategy.32,41 This ability to control chain lengthhas been shown to be an important part of modulating thelongitudinal surface plasmon resonance of chains assembledfrom colloidal nanocrystals17,42,43 and involves the introductionof monofunctional “stoppers” which terminate chain growth. Inthis context, Janus particles with one negatively charged,nanoplate-repelling polystyrene hemisphere and one gold-coated, nanoplate-attracting hemisphere were chosen to serveas colloidal stoppers (Figures 3A and S9). The degree ofpolymerization is directly regulated by the Janus particle−nanoplate concentration ratio; as shown in Figures 3B and S10,as a higher ratio of Janus particles is introduced, chains tend tobe shorter. The fraction of chains capped with one or two Janusparticles on the chain ends is 34% with only 1.2% Janusparticles added to the system and increases to 52% when moreJanus particles are added (3.5%), which further demonstrates
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
C

the effectiveness of the Janus particles as chain stoppers. Morespecifically, we found that the stopper-based control over thed e g r e e o f p o l y m e r i z a t i o n i s m o d e l e d b y
= − + −|Δ |( )X p pe2/ 2(1 ) cc
U k Tn
/s
m
B , where cs is the Janus
particle concentration, cm is the nanoplate concentration, andΔU is the difference between the nanoplate−nanoplate andnanoplate−Janus particle interaction energies at a particularionic strength (for a detailed derivation, see Analysis andCalculations in the Supporting Information). We applied thismodel to fit the data shown in Figure 3B, with e−|ΔU|/kBT as thefitting parameter. The number-average degrees of polymer-ization Xn obtained from fitting this model show goodagreement with experimental results for each concentrationratio condition, as shown in Table S3. The obtained fittingparameter indicates an interaction energy difference of 1.6 kBTto 2.5 kBT for the ionic strength of 1.4 mM used in thisexperiment. Note that this chain stopper strategy is mosteffective only in a certain ionic strength range (regime III inFigures 3A and S11). At relatively low ionic strengths (regimeII in Figure 3A), even though nanoplates start to self-assemble,the attraction between nanoplates and Janus particles issufficiently small (given the curved surface of Janus particlescompared to the flat surface of nanoplates) that they do nottend to assemble stably with one another. Meanwhile, at veryhigh ionic strengths (i.e., regime IV, when electrostaticrepulsions between all particles are greatly screened), Janusparticles self-assemble into clusters, which reduces the numberof Janus particles available to attach to chain ends and arrestchain growth.Fine-tuning the ionic strength also enables direct modulation
of the stiffness of co-assembled chains. Colloidal chain stiffnessis the central control variable in applications of so-calledelectrorheological44−46 and magnetorheological47,48 fluids,where the strength of an applied electric or magnetic field is
used to manipulate chain fluctuations and, consequently, therheological properties of a chain solution. To quantify chainstiffening in this system, from real-time optical microscopymovies, we characterized the transverse fluctuations of chainsfor two ionic strength conditions (1.4 and 3.0 mM; also seeMovie S4). Specifically, we tracked chain “skeletons” for 100frames (about 5.81 s in total) and considered statistics of thetransverse displacement Δh of two points as a function of theirrelative separation |x − x′| along a chain26−28 (Figure 4A−C;
for more details, see Analysis and Calculations in theSupporting Information). At low relative separations, thisrelationship follows a power law of the form ⟨(h(x) −h(x′))2⟩1/2 ∝ |x − x′|α (Figure 4D), where α is referred to asthe “roughness exponent”. At a lower ionic strength (1.4 mM),averaging data from four chains of different chain lengths (seeTable S4) gives α = 0.736 ± 0.018, whereas at a higher ionicstrength (3.0 mM), α = 0.656 ± 0.013. This exponentcharacterizes the steady-state chain roughness, with anexponent of α > 0.5 arising in the case of a biased or directedrandom walk.28 In this context, such a walk is mostly restrictedto the transverse direction, as strong volume exclusion effectsbetween nanoplates impede any substantial chain bending. Weattribute the difference in this exponent between ionic strengthconditions to the more attractive potential between nanoplatesat higher ionic strength, which more strongly disfavors theenthalpy penalties associated with larger transverse misalign-ments. Based on a Boltzmann-type argument, the strength ofinter-nanoplate interactions permits them to slide past oneanother by as much as a few tens of nanometers at both ionic
Figure 3. Control over chain length through co-assembly ofanisometric nanoplates and Janus particles. (A) Schematic (not toscale) showing four ionic strength regimes that generate differentassemblies from a binary mixture of nanoplates and Janus particles.The purple box highlights the “chain stopper” regime. Thefluorescence microscopy image shows a representative tetrahedralcluster formed from Janus particles with a diameter of 2 μm. Forcorresponding optical microscopy images, see Figure S11. (B)Equilibrium i-mer fraction distributions and corresponding Flory−Schulz distribution fits (black curves) at different Janus particle−nanoplate concentration ratios (0, 1.2, and 3.5%). Each data pointis binned over a range of i-mers, with a bin size of ∼53. Ionicstrength = 1.4 mM. The insets are optical microscopy imagesshowing representative structures. Orange arrows indicate Janusparticles attached to chain ends. Scale bars: 3 μm.
Figure 4. Analysis of chain fluctuation. (A) Optical microscopyimage analysis and extraction of h(x). Scale bar: 2 μm. (B)Accumulated height functions for a particular chain over 75 frames(about 4.36 s in total). The height axis has been scaled by ∼110%for clarity between different curves. Pixel size: 1/73.86 nm. (C)Chains stiffen and contract upon an increase in ionic strength.Here, we compare the overall length and the standard deviation ofh(x) at each x over 100 frames (denoted by the shaded area arounda line tracing the temporal average at each point) for a chaincapped with Janus particles on both ends that is initially exposed tolow ionic strength (1.4 mM, top) and later to higher ionic strength(3.0 mM, bottom) (see Movies S4 and S5). (D) Root-mean-squared transverse displacement ⟨(h(x) − h(x′))2⟩1/2 versus relativeseparation |x − x′| at two ionic strength conditions (1.4 and 3.0mM). The inset is a log−log plot of the same data used todetermine the power law scaling.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
D

strengths (see Figure S12 and calculation details in theSupporting Information), but greater degrees of sliding arepossible at a lower ionic strength. The magnitude of the lateralfluctuations we observed under optical microscopy is consistentwith this energetic estimate (Figure S12C). We also calculatedthe persistence length of eight different chains (four for eachionic strength condition). The persistence length is a measureof the flexibility of a chainlike object and can be defined as thelength scale beyond which vectors tangent to the chain contourlose their correlation.24 In each case, the persistence lengthcalculated for a chain exceeds its length by about 1 order ofmagnitude or more (Figure S13 and Table S4; for more details,see Analysis and Calculations in the Supporting Information),which further indicates that the assembled chains are essentially“rigid rods” and unbending. In addition, the persistence lengthalso increases with increasing ionic strength, which is a furtherindication of stiffening.As shown in Figure 4C, chain stiffening is accompanied by
chain contraction. Chains contract because the equilibriumcenter-to-center spacing between nanoplates, d, decreases withincreasing ionic strength (Movie S5). The relative contractionmeasured from the initial and final length of the nanoplateportion of co-assembled chains in optical micrographs (0.70 ±0.08, based on averaging the contraction of three chains; seeTable S5) is consistent both with the d spacing ratiodetermined from SAXS (0.73 ± 0.01) and predictions fromtheoretical calculations16 (0.74; see Figure S7).At higher ionic strengths within regime III, a Janus particle
can accommodate more than one chain on its gold hemisphere,which leads to the formation of 2D, not just 1D, structures. Inparticular, by increasing the ionic strength (while remainingbelow the threshold for Janus particle clustering), one canfurther screen the electrostatic repulsion between nanoplatesand Janus particles and make Janus particles bifunctional, suchthat they connect chains into a “V” shape (Figures 5A and S14).We found that two chains connected by a Janus particle tend tostay in physical contact (Figure 5B), which we hypothesize isdue to either a lateral van der Waals attraction between chainsor an effort to maximize the rotational entropy of the Janusparticle49 (Figure S15). As such, the “bond angle” θ (Figure5B) can be estimated directly from the size of the Janus particle(diameter D) and the width of the chains (W, i.e., the averageedge length of the assembled nanoplates) as θ = 2 tan−1(W/D).If W = 1.15 μm and D = 2 μm, the predicted bond angle is 60°(i.e., the interior angle of an equilateral triangle), which isconsistent with the dominant bond angle observed inexperiments with particles of this size combination (Figures5C and S16A). Movie S6 shows the formation of a triangularring using this combination of particles. The bond anglefluctuates considerably before stabilizing around 60° when thering is closed. As in the contraction of 1D chains, an increase inionic strength leads to the contraction of the triangular ring(Movie S7). In other words, the ring is a breathable pore with atunable pore size. Other assemblies, such as “zigzag” chains andmultimembered rings, were also observed in the binary mixturesystem (Figures 5C and S17 and Movie S8).The library of possible structures is further enriched through
the incorporation of multipatch spheres.30 The triblockparticles used here, for example, have attractive gold patchesat both “north” and “south” poles (Figures 5D and S18). Whenboth of the triblock particle patches have a functionality f = 2,these triblock particles can exhibit I-, V-, Y-, or X-shapedbonding with silver nanoplates (Figure 5D). One can use these
motifs to construct various linear and branched structures(Figure 5E). The broader potential of this strategy lies in themodularity of the size, position, and number of patches on thelinker surface. Moreover, the linker colloids need not bespherical. In fact, this method to assemble many differentarchitectures from only one or two types of “monomers” simplyby varying the linker unit is commonly used in biology50−52 andhas already shown great success in other synthetic systems.53−55
For example, using the patchy spheres implemented here, if theangle between two gold patches is ϕ and the linked chains areof similar length, one would anticipate an m-membered ring toform, with m given by m = 360°/(180° − ϕ) (Figure S19).Going even further, by varying which linkers are present overthe course of nanoplate polymerization, it would be possible toco-assemble an even wider variety of shapes and architectures.
CONCLUSIONSIn summary, we demonstrate that a combination of highlyanisometric silver nanoplates and patchy spheres can be used to“polymerize” a wide variety of adaptive 1D and 2D structures.We show that the ionic strength can be used during assembly tocontrol the functionality of linkers (Janus and triblock particles)and, consequently, the morphologies of structures thatassemble. We also show that ionic strength can be used post-assembly for the dynamic manipulation of structural properties,such as the size, stiffness, and interparticle distance withinassemblies. We hope these results can inspire the bottom-upfabrication of highly adaptive structures with advancedproperties or perhaps serve as the basis for controlled, synthetic
Figure 5. Co-assembly of anisometric nanoplates with patchyspheres. (A) Schematics and representative optical microscopyimages showing the functionality ( f) of the Janus particle changefrom 1 to 2 when the ionic strength is increased from 1.0 to 1.6mM. (B) Schematic showing the bond angle θ. (C) Opticalmicroscopy images of a triangular ring and “zigzag” chain, withcorresponding schematics, assembled from nanoplates with a 1.15± 0.14 μm edge length (large assembling nanoplate population)and Janus particles with a diameter of 2 μm. Ionic strength = 1.6mM. (D) Schematics of a triblock particle with a functionality of 4and the possible bond motifs with nanoplates. (E) Representativeoptical microscopy images and schematics of linear and branchedassemblies from nanoplates and triblock particles. Ionic strength =1.6 mM. Scale bars: 2 μm.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
E

analogues to functional one- and two-dimensional structuresfound in nature.56,57
METHODS AND EXPERIMENTAL DETAILSSynthesis of Anisometric Silver Nanoplates. Silver nanoplates
were synthesized using a modified seeded-growth method.35 Silverseeds were prepared by sequential addition of aqueous solutions ofAgNO3 (25 mL, 0.1 mM), sodium citrate (600 μL, 75 mM), andhydrogen peroxide (30 wt %, 60 μL) to a 125 mL Erlenmeyer flaskstirring at 300 rpm at room temperature. The stirring speed wasincreased to 1050 rpm before rapid injection of aqueous NaBH4 (250μL, 0.1 M). This solution was kept stirring for 5 min after turning blue,then centrifuged at 10 500 rpm for 8 min, and redispersed in a sodiumcitrate solution (10 mL, 0.94 mM). L-Ascorbic acid (0.75 mL, 0.1 M)was added to 20 mL of this diluted seed solution in a 40 mL glass vial.A separate solution was prepared by mixing aqueous AgNO3 (20 mL,1.0 mM), citric acid (0.125 mL, 0.1 M), and sodium citrate (0.1 mL,1.5 mM). This solution was added to the seed solution dropwisethrough a syringe pump at an injection rate of 0.4 mL/min. After 5min (2 mL) of injection, two-thirds of the reaction solution wasremoved, and the remaining amount was used as a source of seeds forthe next growth cycle. The removed products of the first few cycleswere discarded, but samples from cycles 3, 4, and 5 were collected inseparate 8 mL glass vials. The whole growth process was performedunder vigorous shaking with a thermomixer at a shaking speed of 300−1000 rpm.Thiol Modification of Silver Nanoplates. Citrate ligands on as-
synthesized silver nanoplates were exchanged with carboxylate−thiolsby adding 200 μL of a 7.93 mM thiol ligand solution to 2 mL of asilver nanoplate sample solution and allowing the mixture to sitovernight. The final product was washed and stored in 1.5 mL of waterin an 8 mL glass vial.Preparation of Patchy Spheres. Janus particles were prepared by
directional electron beam evaporation of gold onto a monolayer ofcolloidal polystyrene particles on a glass substrate.29 A 2 wt % aqueoussuspension of carboxylate−polystyrene particles was spread on a glassslide and dried into a monolayer. Then, a 2 nm titanium coatingfollowed by a 15 nm gold coating was deposited vertically on themonolayer using a Temescal electron beam evaporation system. Janusparticles were collected by sonication and used for experiments atdifferent concentrations. Triblock particles were prepared following aliterature method.30 A closely packed carboxylate−polystyrene particlemonolayer was deposited onto a silicon wafer and dried in air. Next, 2nm titanium and 25 nm gold coatings were deposited vertically on theparticle monolayer using the same deposition conditions as for Janusparticles. After deposition, particles were lifted from the substrate witha polydimethylsiloxane stamp. The particles inverted on the stampthen underwent a second titanium and gold deposition using the sameconditions as the first deposition. After the second deposition, particleson the stamp were immersed in a gold etching solution for 80 s andthen washed with 200 mL of water three times to collect triblockparticles for later use.Sample Characterization. UV−vis spectra of as-prepared seeds
and SEM images of as-synthesized silver nanoplates were obtained tocharacterize the quality of the samples. The thickness of nanoplateswas measured using both tapping-mode AFM and SEM when thenanoplates were in a standing configuration. The ζ-potential ofcarboxylate−thiol-coated silver nanoplates in water was measuredusing a Malvern Zetasizer Nano. Transmission SAXS spectra werecollected from samples of chains assembled from silver nanoplates for30 min using a home-built setup (Forvis Technologies, Santa Barbara)with a Xenocs GeniX3D Cu Kα ultralow divergence X-ray source(1.54 Å/8 keV) with a divergence of ∼1.3 mrad and a Pilatus 300 K 20Hz hybrid pixel detector (Dectris). FIT2D (software from theEuropean Synchrotron Radiation facility, http://www.esrf.eu/computing/scientific/FIT2D) was used to integrate 2D scatteringplots.Optical Microscopy Imaging and Image Analysis. Bright-field
optical microscopy imaging was performed using a Zeiss inverted
microscope (Axiovert 200) with a 10× air objective (NA = 0.25), a63× air objective (NA = 0.75), with 1.6× post-magnification, and100× oil objectives (NA = 1.45 and 1.30). Optical microscopy movieswere recorded using a complementary metal−oxide−semiconductorcamera (Edmund Optics 5012 M GigE) at a rate of 17.21 to 20 framesper second. Epifluorescence imaging was performed using a Zeissinverted microscope (Observer.Z1) with a 100× oil objective (NA =1.30) and an iXon electron-multiplying charge-coupled device camera.A 532 nm laser line was used to excite fluorescence in Nile redfluorophores contained in the polystyrene particles. Image analysis wasperformed using FIJI/ImageJ.
ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acsnano.7b02059.
Movie S1 (AVI)Movie S2 (AVI)Movie S3 (AVI)Movie S4 (AVI)Movie S5 (AVI)Movie S6 (AVI)Movie S7 (AVI)Movie S8 (AVI)Materials and methods, analysis and calculations, FiguresS1−S22, and Tables S1−S6 (PDF)
AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] Chen: 0000-0002-1968-441XNotesThe authors declare no competing financial interest.
ACKNOWLEDGMENTSWe thank Yingfeng Yang at the University of Illinois for helpfuldiscussions on the “chain stopper” model. This work wassupported by the U.S. Department of Energy, Office of BasicEnergy Sciences, Division of Materials Sciences and Engineer-ing under Award No. DE-FG02-07ER46471, through theFrederick Seitz Materials Research Laboratory at the Universityof Illinois.
REFERENCES(1) McManus, J. J.; Charbonneau, P.; Zaccarelli, E.; Asherie, N. ThePhysics of Protein Self-Assembly. Curr. Opin. Colloid Interface Sci.2016, 22, 73−79.(2) Li, W.; Morin, M.; Gustafsson, E.; Persson, B. A.; Lund, M.;Zackrisson Oskolkova, M. Strings and Stripes Formed by a ProteinSystem Interacting via a Single-Patch Attraction. Soft Matter 2016, 12,9330−9333.(3) Lomakin, A.; Asherie, N.; Benedek, G. B. Aeolotopic Interactionsof Globular Proteins. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9465−9468.(4) Nogales, E.; Whittaker, M.; Milligan, R. A.; Downing, K. H. High-Resolution Model of the Microtubule. Cell 1999, 96, 79−88.(5) Buehler, M. J. Nature Designs Tough Collagen: Explaining theNanostructure of Collagen Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2006,103, 12285−12290.(6) Minary-Jolandan, M.; Yu, M.-F. Nanomechanical Heterogeneityin the Gap and Overlap Regions of Type I Collagen Fibrils withImplications for Bone Heterogeneity. Biomacromolecules 2009, 10,2565−2570.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
F

(7) Bornhoeft, L. R.; Castillo, A. C.; Smalley, P. R.; Kittrell, C.; James,D. K.; Brinson, B. E.; Rybolt, T. R.; Johnson, B. R.; Cherukuri, T. K.;Cherukuri, P. Teslaphoresis of Carbon Nanotubes. ACS Nano 2016,10, 4873−4881.(8) Fan, S.; Chapline, M. G.; Franklin, N. R.; Tombler, T. W.;Cassell, A. M.; Dai, H. Self-Oriented Regular Arrays of CarbonNanotubes and Their Field Emission Properties. Science 1999, 283,512−514.(9) Yu, M.-F.; Lourie, O.; Dyer, M. J.; Moloni, K.; Kelly, T. F.; Ruoff,R. S. Strength and Breaking Mechanism of Multiwalled CarbonNanotubes under Tensile Load. Science 2000, 287, 637−640.(10) Zhang, Y.; Liu, J.; Li, D.; Yan, F.; Wang, X.; Yang, W. Self-Assembly of Ultrathin Gold Nanowires and Single Walled CarbonNanotubes as a Highly Sensitive Substrate for Surface EnhancedRaman Spectroscopy. New J. Chem. 2016, 40, 7286−7289.(11) Chen, Y.; Wang, Y.; Peng, J.; Xu, Q.; Weng, J.; Xu, J. Assemblyof Ultrathin Gold Nanowires: From Polymer Analogue to ColloidalBlock. ACS Nano 2017, 11, 2756−2763.(12) He, Y.; Chen, Y.; Xu, Q.; Xu, J.; Weng, J. Assembly of UltrathinGold Nanowires into Honeycomb Macroporous Pattern Films withHigh Transparency and Conductivity. ACS Appl. Mater. Interfaces2017, 9, 7826−7833.(13) Li, B.; Wen, X.; Li, R.; Wang, Z.; Clem, P. G.; Fan, H. Stress-Induced Phase Transformation and Optical Coupling of SilverNanoparticle Superlattices into Mechanically Stable Nanowires. Nat.Commun. 2014, 5, 4179.(14) Min, Y.; Akbulut, M.; Kristiansen, K.; Golan, Y.; Israelachvili, J.The Role of Interparticle and External Forces in NanoparticleAssembly. Nat. Mater. 2008, 7, 527−538.(15) Bishop, K. J. M.; Wilmer, C. E.; Soh, S.; Grzybowski, B. A.Nanoscale Forces and Their Uses in Self-Assembly. Small 2009, 5,1600−1630.(16) Kim, J.; Jones, M. R.; Ou, Z.; Chen, Q. In Situ ElectronMicroscopy Imaging and Quantitative Structural Modulation ofNanoparticle Superlattices. ACS Nano 2016, 10, 9801−9808.(17) Gao, B.; Rozin, M. J.; Tao, A. R. Plasmonic Nanocomposites:Polymer-Guided Strategies for Assembling Metal Nanoparticles.Nanoscale 2013, 5, 5677−5691.(18) Young, K. L.; Jones, M. R.; Zhang, J.; Macfarlane, R. J.; Esquivel-Sirvent, R.; Nap, R. J.; Wu, J.; Schatz, G. C.; Lee, B.; Mirkin, C. A.Assembly of Reconfigurable One-Dimensional Colloidal Superlatticesdue to a Synergy of Fundamental Nanoscale Forces. Proc. Natl. Acad.Sci. U. S. A. 2012, 109, 2240−2245.(19) Vutukuri, H. R.; Badaire, S.; de Winter, D. A. M.; Imhof, A.; vanBlaaderen, A. Directed Self-Assembly of Micron-Sized Gold Nano-platelets into Oriented Flexible Stacks with Tunable InterplateDistance. Nano Lett. 2015, 15, 5617−5623.(20) Bai, F.; Li, B.; Bian, K.; Haddad, R.; Wu, H.; Wang, Z.; Fan, H.Pressure-Tuned Structure and Property of Optically Active Nano-crystals. Adv. Mater. 2016, 28, 1989−1993.(21) Chen, Q.; Cho, H.; Manthiram, K.; Yoshida, M.; Ye, X.;Alivisatos, A. P. Interaction Potentials of Anisotropic Nanocrystalsfrom the Trajectory Sampling of Particle Motion Using In Situ LiquidPhase Transmission Electron Microscopy. ACS Cent. Sci. 2015, 1, 33−39.(22) Odian, G. Reactivity of Functional Groups. Principles ofPolymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ,2004; pp 40−44.(23) Byrom, J.; Han, P.; Savory, M.; Biswal, S. L. Directing Assemblyof DNA-Coated Colloids with Magnetic Fields to Generate Rigid,Semiflexible, and Flexible Chains. Langmuir 2014, 30, 9045−9052.(24) Li, D.; Banon, S.; Biswal, S. L. Bending Dynamics of DNA-Linked Colloidal Particle Chains. Soft Matter 2010, 6, 4197−4204.(25) Wang, Y.; Hollingsworth, A. D.; Yang, S. K.; Patel, S.; Pine, D. J.;Weck, M. Patchy Particle Self-Assembly viaMetal Coordination. J. Am.Chem. Soc. 2013, 135, 14064−14067.(26) Domínguez-García, P.; Melle, S.; Rubio, M. A. Morphology ofAnisotropic Chains in a Magneto-Rheological Fluid during Aggrega-
tion and Disaggregation Processes. J. Colloid Interface Sci. 2009, 333,221−229.(27) Furst, E. M.; Gast, A. P. Dynamics and Lateral Interactions ofDipolar Chains. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat.Interdiscip. Top. 2000, 62, 6916−6925.(28) Silva, A. S.; Bond, R.; Plouraboue, F.; Wirtz, D. FluctuationDynamics of a Single Magnetic Chain. Phys. Rev. E: Stat. Phys., Plasmas,Fluids, Relat. Interdiscip. Top. 1996, 54, 5502−5510.(29) Chen, Q.; Whitmer, J. K.; Jiang, S.; Bae, S. C.; Luijten, E.;Granick, S. Supracolloidal Reaction Kinetics of Janus Spheres. Science2011, 331, 199−202.(30) Chen, Q.; Diesel, E.; Whitmer, J. K.; Bae, S. C.; Luijten, E.;Granick, S. Triblock Colloids for Directed Self-Assembly. J. Am. Chem.Soc. 2011, 133, 7725−7727.(31) Demortiere, A.; Snezhko, A.; Sapozhnikov, M. V.; Becker, N.;Proslier, T.; Aranson, I. S. Self-Assembled Tunable Networks of StickyColloidal Particles. Nat. Commun. 2014, 5, 3117.(32) Groschel, A. H.; Walther, A.; Lobling, T. I.; Schacher, F. H.;Schmalz, H.; Muller, A. H. E. Guided Hierarchical Co-Assembly ofSoft Patchy Nanoparticles. Nature 2013, 503, 247−251.(33) Kuroki, H.; Islam, C.; Tokarev, I.; Hu, H.; Liu, G.; Minko, S.Tunable Ultrathin Membranes with Nonvolatile Pore Shape Memory.ACS Appl. Mater. Interfaces 2015, 7, 10401−10406.(34) Rybtchinski, B. Adaptive Supramolecular Nanomaterials Basedon Strong Noncovalent Interactions. ACS Nano 2011, 5, 6791−6818.(35) Zhang, Q.; Hu, Y.; Guo, S.; Goebl, J.; Yin, Y. Seeded Growth ofUniform Ag Nanoplates with High Aspect Ratio and Widely TunableSurface Plasmon Bands. Nano Lett. 2010, 10, 5037−5042.(36) Smith, A. M.; Marbella, L. E.; Johnston, K. A.; Hartmann, M. J.;Crawford, S. E.; Kozycz, L. M.; Seferos, D. S.; Millstone, J. E.Quantitative Analysis of Thiolated Ligand Exchange on GoldNanoparticles Monitored by 1H NMR Spectroscopy. Anal. Chem.2015, 87, 2771−2778.(37) Parsons, J. D. Nematic Ordering in a System of Rods. Phys. Rev.A: At., Mol., Opt. Phys. 1979, 19, 1225−1230.(38) Liu, K.; Nie, Z.; Zhao, N.; Li, W.; Rubinstein, M.; Kumacheva,E. Step-Growth Polymerization of Inorganic Nanoparticles. Science2010, 329, 197−200.(39) Israelachvili, J. N. Attractive “Depletion” Forces. Intermolecularand Surface Forces, 3rd ed.; Academic Press: New York, 2011; pp 398−402.(40) Sacanna, S.; Irvine, W. T. M.; Chaikin, P. M.; Pine, D. J. Lockand Key Colloids. Nature 2010, 464, 575−578.(41) Klinkova, A.; Therien-Aubin, H.; Choueiri, R. M.; Rubinstein,M.; Kumacheva, E. Colloidal Analogs of Molecular Chain Stoppers.Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18775−18779.(42) Rosen, D. A.; Tao, A. R. Modeling the Optical Properties ofBowtie Antenna Generated by Self-Assembled Ag Triangular Nano-prisms. ACS Appl. Mater. Interfaces 2014, 6, 4134−4142.(43) Liu, K.; Ahmed, A.; Chung, S.; Sugikawa, K.; Wu, G.; Nie, Z.;Gordon, R.; Kumacheva, E. In Situ Plasmonic Counter for Polymer-ization of Chains of Gold Nanorods in Solution. ACS Nano 2013, 7,5901−5910.(44) Lee, S.; Lee, J.; Hwang, S. H.; Yun, J.; Jang, J. EnhancedElectroresponsive Performance of Double-Shell SiO2/TiO2 HollowNanoparticles. ACS Nano 2015, 9, 4939−4949.(45) Schwarz, G.; Maisch, S.; Ullrich, S.; Wagenhofer, J.; Kurth, D. G.Electrorheological Fluids Based on Metallo-Supramolecular Polyelec-trolyte−Silicate Composites. ACS Appl. Mater. Interfaces 2013, 5,4031−4034.(46) Cheng, Y.; Wu, K.; Liu, F.; Guo, J.; Liu, X.; Xu, G.; Cui, P. FacileApproach to Large-Scale Synthesis of 1D Calcium and TitaniumPrecipitate (CTP) with High Electrorheological Activity. ACS Appl.Mater. Interfaces 2010, 2, 621−625.(47) Carlson, J. D.; Jolly, M. R. MR Fluid, Foam and ElastomerDevices. Mechatronics 2000, 10, 555−569.(48) Jolly, M. R.; Bender, J. W.; Carlson, J. D. Properties andApplications of Commercial Magnetorheological Fluids. J. Intell. Mater.Syst. Struct. 1999, 10, 5−13.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
G

(49) Mao, X.; Chen, Q.; Granick, S. Entropy Favours Open ColloidalLattices. Nat. Mater. 2013, 12, 217−222.(50) Winder, S. J.; Ayscough, K. R. Actin-Binding Proteins. J. Cell Sci.2005, 118, 651−654.(51) Delandre, C.; Amikura, R.; Moore, A. W. MicrotubuleNucleation and Organization in Dendrites. Cell Cycle 2016, 15,1685−1692.(52) Wiese, C.; Zheng, Y. Microtubule Nucleation: γ-Tubulin andBeyond. J. Cell Sci. 2006, 119, 4143−4153.(53) Jorgenson, T. D.; Mohammed, A. M.; Agrawal, D. K.; Schulman,R. Self-Assembly of Hierarchical DNA Nanotube Architectures withWell-Defined Geometries. ACS Nano 2017, 11, 1927−1936.(54) Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. SupramolecularCoordination: Self-Assembly of Finite Two- and Three-DimensionalEnsembles. Chem. Rev. 2011, 111, 6810−6918.(55) Mohammed, A. M.; Velazquez, L.; Chisenhall, A.; Schiffels, D.;Fygenson, D. K.; Schulman, R. Self-Assembly of Precisely DefinedDNA Nanotube Superstructures Using DNA Origami Seeds. Nano-scale 2017, 9, 522−526.(56) Morrow, J. S.; Rimm, D. L.; Kennedy, S. P.; Cianci, C. D.;Sinard, J. H.; Weed, S. A. Of Membrane Stability and Mosaics: TheSpectrin Cytoskeleton. Comprehensive Physiology; John Wiley & Sons,Inc.: New York, 2010; pp 485−540.(57) Albers, S.-V.; Meyer, B. H. The Archaeal Cell Envelope. Nat.Rev. Microbiol. 2011, 9, 414−426.
ACS Nano Article
DOI: 10.1021/acsnano.7b02059ACS Nano XXXX, XXX, XXX−XXX
H





























