Embed Size (px)
Citation preview

PNEUMOTRIESTE 2011 PNEUMOTRIESTE 2011
Carlo AlberaCarlo Albera
Università di Torino Università di Torino
Facoltà di Medicina e Chirurgia San Luigi GonzagaFacoltà di Medicina e Chirurgia San Luigi Gonzaga
Dipartimento di Scienze Cliniche e Biologiche Dipartimento di Scienze Cliniche e Biologiche
Ambulatorio Interstiziopatie Polmonari / Malattie RareAmbulatorio Interstiziopatie Polmonari / Malattie Rare
Pulmonary hypertension and diffuse parenchymal diseases: therapeutic options

Pulmonary hypertension and diffuse parenchymal diseases: therapeutic options
To date no approved drugs

Thank you for your attention

Clinical Classification of Pulmonary Hypertension(Dana Point 2008)
3. PH due to lung dis/hypoxiaemia3.1 COPD3.2 ILD3.3 Other pulmonary diseases with mixed
restrictive and obstructive pattern3.4 Sleep-disordered breathing3.5 Alveolar hypoventilation disorders3.6 Chronic exposure to high altitude3.7 Developmental abnormabilities
4. Chronic thromboembolic PH
2. PH due to left heart disease2.1 Systolic dysfunction2.2 Diastolic dysfunction2.3 Valvular diseases
1’. PVOD and/or Pulm capill Haemang.
1. Pulmonary Arterial Hypertension1.1 Idiopatic1.2 Heritable
1.2.1 BMPR21.2.2 ALK1, endoglin (+ / - hereditary haemorrhaigc
teleangectasia)
1.2.3 Unknown1.3 Drugs and toxins induced1.4 Associated with (APAH)
1.4.1 Connective Tissue Diseases1.4.2 HIV infection1.4.3 Portal hypertension1.4.4 Congenital heart disease1.4.5 Schistosomiasis1.4.6 Chronic haemolityc anaemia
1.5 Persistent PH of the newborn
5. PH with unclear and/or multifact mechs5.1 Haematological dis: myieloproliferative,
splenectomy5.2 Systemic diseases: sarcoidosis, pulmonary
Langerhans cell histiocytosis, lymphangioleiomyomatosis
5.3 Metabolic dis: glycogen storage disease. Gaucher disease, thyroid disorders
5.4 Others: tumoral obstruction, fibrosing mediastinitis, CRF on dialysis

DPLD of knownDPLD of knowncause orcause or
associationassociation
Respiratory bronchiolitisRespiratory bronchiolitisinterstitial lung diseaseinterstitial lung disease
CryptogenicCryptogenicorganising pneumoniaorganising pneumonia
IP other thanIP other thanidiopathicidiopathic
pulmonary fibrosispulmonary fibrosis
Diffuse Parenchymal Lung DiseaseDiffuse Parenchymal Lung Disease
ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonia AJRCCM 2002; 165: 277-304Pneumonia AJRCCM 2002; 165: 277-304

Pulmonary hypertension and ILDs
What from guidelines ?

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Treatment of selected complications and comorbid conditions
There is an increasing awareness of complications and comorbid conditions frequently associated with IPF.
These include acute exacerbation of IPF, pulmonary hypertension, gastroesophageal reflux disease, obesity, emphysema, and obstructive sleep apnea.
It is unknown if treating these comorbidities influences clinical outcomes.

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Question: Should pulmonary hypertension be treated in patients with IPF ? (1)
Recommendation:
Pulmonary hypertension should not be treated in the majority of patients with IPF, but treatment may be a reasonable choice in a minority (weak recommendation, very low-quality evidence).
Values:
This recommendation places a high value on cost and the potential for drug-related morbidity, and a low value on very low-quality data suggesting a possible benefit in selected patients.

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Question: Should pulmonary hypertension be treated in patients with IPF ? (2)
Remarks:
In patients with moderate to severe pulmonary hypertension documented by right heart catheterization (i.e., mPAP ≥ 35 mm Hg), in line with the interpretation of a weak recommendation, a trial of vasomodulatory therapy may be indicated.
The committee recognizes the need for clinical trials of vasomodulatory therapies in this patient population.
(Vote: 8 for use, 14 against use, 1 abstention, 8 absent.)

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Monitoring for Complications and Comorbidities:
Comorbidities including pulmonary hypertension, pulmonary embolism, lung cancer, and coronary artery disease are known to occur in IPF.
While the development of these comorbidities may influence survival, the role of routine screening to identify such complications in patients with IPF is unknown. (???)
Thus, a recommendation for routine screening cannot be made

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Monitoring for Complications and Comorbidities:
pulmonary hypertension (1)
In patients demonstrating progressive disease, the identification of PH may impact consideration for lung transplantation in eligible patients, and evaluation is indicated.
Echocardiography is inaccurate in estimating pulmonary hemodynamics in patients with fibrotic lung disease and should not be relied upon to assess the presence and severity of pulmonary hypertension.

ATS/ERS/JRS/ALAT EBM GUIDELINES Am J Respir Crit Care Med 183, 788–824, 2011
Monitoring for Complications and Comorbidities:
pulmonary hypertension (2)
BNP levels have been shown to correlate with the presence of moderate to severe PH, but have not been validated as a screening tool
At the present time, right heart catheterization is required to confirm the presence of PH.

European Heart Journal (2009) 30, 2493–2537

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
In COPD the presence of PH is associated withshorter survival and frequent episodes of exacerbation.
In interstitial lung diseases PH is a poor prognostic factor and PAP is the most important predictor of mortality.

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Diagnosis
echocardiography is the best screening tool for the assessment of PH.
its diagnostic value in advanced respiratory diseases is lower than in PAH
Echocardiographic estimation of PAPs l ILDs may be inaccurate.

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Diagnosis
The specificity of systolic PAP in detecting PH is low
The negative predictive value is acceptable.
Indications for echocardiography for the screening of PH in COPD and ILDs are:
(i) exclusion of significant PH (ii) evaluation of concomitant left heart disease; (iii)selection of patients for RHC

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Diagnosis
A definite diagnosis of PH relies on measurements at RHC.
The indication for RHC in advanced lung disease are:
(i)proper diagnosis of PH incandidates for surgical treatments (transplantation, lung volume reduction); (ii)suspected ‘out of proportion’ PH potentially amenable to be enrolled in RCT with specific PAH drug therapy;(iii) frequent episodes of RV failure;(iv) inconclusive echocardiographic study in cases with a high level of suspicion

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Therapy
Currently there is no specific therapy for PH associated with COPD or interstitial lung diseases.
Long-term O2 administration has been shown partially toreduce the progression of PH in COPD.
Nevertheless, with this treatment PAP rarely returns to normal values and the structural abnormalities of pulmonary vessels remain unaltered.
In interstitial lung diseases, the role of long-term O2 therapy in PH progression is less clear.

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Therapy
Treatment with conventional vasodilators is not recommended because they may impair gas exchange due to:
•the inhibition of hypoxic pulmonary vasoconstriction •lack of efficacy after long-term use.
Published experience with specific PAH drug therapy is scarce and consists of the assessment of acute effects and uncontrolled studies in small series.

European Heart Journal (2009) 30, 2493–2537
PH due to lung diseases and/or hypoxaemia (group 3)
Therapy
The treatment of choice for patients with COPD or interstitial lung diseases and associated PH who are hypoxaemic is long-term O2 therapy.
Patients with ‘out of proportion’ PH due to lung diseases (dyspnoea insufficiently explained by lung mechanical disturbances and mPAP >40-45mmHg at rest) should be referred to expert centres and enrolled in clinical trials targeting PAH-specific drug therapy.
The use of targeted PAH therapy in patients with COPD or interstitial lung diseases and mean PAP <40 mmHg is currently discouraged because there are no systematic data regarding its safety or efficacy.

a) Class of recommendation. b) Level of evidence.
Recommendations for PH due to lung diseases
Statement Class (a)
Level (b)
TTE is recommended as screening tool for the assessement of PH due to lung diseases
I C
RHC is recommended for a definite diagnosis of PH due to lung diseases
I C
The optimal treatment of the underlying lung disease including long-term O2 therapy in patients with chronic hypoxaemia is recommended in patients with PH due to lung diseases
I C
Patients with ‘out of proportion’ PH due to lung diseases should be enrolled in RCTs targeting PAH-specific drugs
IIa C
The use of PAH-specific drug therapy is not recommended in patients with PH due to lung diseases
III C
European Heart Journal (2009) 30, 2493–2537

Take home message from guidelines
Actually there are no EBM-supported therapeutic options for the treatment of PH associated to ILDs
The therapy of ILDs is probably the best way to prevent/delay the development of associated PH
Effective antifibrotic drug(s) may offer in the next future some new chances
The treatment of ILDs-associated PH represent a still unmetmedical need
ILDs- associated PH if “Out proportion” may be considered andmanaged in a different way

Some additional data on ILDs and PH

PATOLOGIA N° Casi totali N° Centri Centro e N° Casi
iPAH 39 8 San Luigi 16Molinette 156 altri Centri 8
Sarcoidosi cronicaed extrapolmonare
242 16 San Luigi 133Molinette 2714 altri centri 82
IPF 210 10 San Luigi 142Molinette 428 altri centri 26
Report 2010 Registro Malattie Rare Regione Piemonte/Val d’Aosta
www.malattierarepiemonte.it

Nathan SD Int J Clin Pract.20081742-1241
Dana Point Group V
Dana Point Group III

IP other thanIP other thanidiopathicidiopathic
pulmonary fibrosispulmonary fibrosis
Diffuse Parenchymal Lung DiseaseDiffuse Parenchymal Lung Disease
ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonia AJRCCM 2002; 165: 277-304Pneumonia AJRCCM 2002; 165: 277-304
SARCOIDOSIS

Pulmonary Hypertension and Pulmonary Hypertension and SarcoidosisSarcoidosis
5. PH with unclear and/or multifact mechs
5.1 Haematological dis: myieloproliferative, splenectomy
5.2 Systemic diseases: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis
5.3 Metabolic dis: glycogen storage disease. Gaucher disease, thyroid disorders
5.4Others: tumoral obstruction, fibrosing mediastinitis, CRF on dialysis
Dana Point group 5

Handa,T et al. CHEST 2006; 129:1246–1252
Prospective, observational study Prospective, observational study
246 consecutive sarcoidosis patients246 consecutive sarcoidosis patients
PH defined as estimated sPAP PH defined as estimated sPAP > > 40 mm Hg 40 mm Hg
212 patients evaluated for sPAP212 patients evaluated for sPAP
12 patients12 patients (5.7%) had PH(5.7%) had PH..
Decreased lung volume increases the risk of PH developing Decreased lung volume increases the risk of PH developing in patients with sarcoidosis. in patients with sarcoidosis.

Arthritis Research & Therapy 2007, 9(Suppl 2):S8

Arthritis Research & Therapy 2007, 9(Suppl 2):S8
Endothelin-1 levels in BAL fluidEndothelin-1 / albumin levels in
bronchoalveolar lavage fluid (BAL) fluid
in sarcoid patients and healthy controls.

Arthritis Research & Therapy 2007, 9(Suppl 2):S8
Suspected if:
•unexplained dyspnoea in sarcoidosis•worsening dyspnoea in steroid-refractory sarcoidosis
Pathogenetic mechanism of PH in sarcoidosis appears to be multifactorial:
•direct compression of the pulmonary arteries•fibrotic destruction of the lung vasculature •hypoxia and a pulmonary vasculopathy •vasculitis•veno-occlusive PH (ECHO vs RHC !!!)

Pulmonary Hypertension and Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis Idiopathic Pulmonary Fibrosis
(IPF) (IPF)
3.PH due to lung dis/hypoxemia
3.1 COPD3.2 ILD3.3 Other pulmonary diseases with mixed
restrictive and obstructive pattern3.4 Sleep-disordered breathing3.5 Alveolar hypoventilation disorders3.6 Chronic exposure to high altitude3.7 Developmental abnormabilities
Dana Point group 3

Pulmonary Hypertension in IPFPulmonary Hypertension in IPF
•PathogenesisPathogenesis
•Prevalence, detection, diagnosisPrevalence, detection, diagnosis
•Course and significanceCourse and significance
•TherapyTherapy
Pulmonary Hypertension in IPF
•PathogenesisPathogenesis
•Prevalence, detection, diagnosisPrevalence, detection, diagnosis
•Course and significanceCourse and significance
•TherapyTherapy

Pathogenesis of PAHPathogenesis of PAH
Genetic predisposition(mutations BMPR-2, ALK-
1, 5HT polymorphism, NOS)
Genetic predisposition(mutations BMPR-2, ALK-
1, 5HT polymorphism, NOS)
Risk factors andassociated conditions
(e.g. HIV, anorexigens)
Risk factors andassociated conditions
(e.g. HIV, anorexigens)
Matrix, platelets, inflammatory cells
Matrix, platelets, inflammatory cells
Endothelial cells
Endothelial cells
Smooth muscle cells
Smooth muscle cells
PULMONARY VASCULAR INJURYPULMONARY VASCULAR INJURYPULMONARY VASCULAR INJURYPULMONARY VASCULAR INJURY
Vasoconstriction – proliferation
inflammation – thrombosis
Vasoconstriction – proliferation
inflammation – thrombosis
Pulmonary vascular diseasePulmonary vascular diseaseClinical syndrome of PAHClinical syndrome of PAH
Pulmonary vascular diseasePulmonary vascular diseaseClinical syndrome of PAHClinical syndrome of PAH
1. Adapted from Gaine S JAMA 2000; 284: 3160–3168
BMPR-2: bone morphogenetic protein receptor type 2; ALK-1: activin receptor-like kinase 1; 5HT: 5-hydroxytryptamine or seratonin; NOS: nitric oxide synthase
IPF- associated PHHypoxiaHypoxia
Local milieu (cytokines,chemokines, growth factors,etc) Local milieu (cytokines,chemokines, growth factors,etc) → → fibrosisfibrosis
Loss of capillary and/or angiogenesis, in situ Loss of capillary and/or angiogenesis, in situ thrombosisthrombosis

Patel N et al CHEST 2007; 132:998–1006
(n 376)
(n 376)
Pulmonary artery branch in an area of dense fibrosis.
Medial thickening and significant intimal proliferation.
Pulmonary artery/arteriole in a lobule marked medial thickening of relatively nonfibrotic lung tissue.
bronchiole-vascular bundle in an area medial + intimal thickening +elastic tissue of relatively nonfibrotic lung tissue duplication.
PH in IPF shares pathologic features of hypoxia-inducedPH in IPF shares pathologic features of hypoxia-induced PH but also demonstrates marked intimal changes, which PH but also demonstrates marked intimal changes, which
likely reflect a local and systemic cytokine effect.likely reflect a local and systemic cytokine effect.

Pulmonary Hypertension in IPFPulmonary Hypertension in IPF
•PathogenesisPathogenesis
•Prevalence, detection, diagnosisPrevalence, detection, diagnosis
•Course and significanceCourse and significance
•TherapyTherapy

Author, year # Pts Disease Diagn PHCut off
PH Prevalence
Hassan2005
Retrosp’94-’96
88 UIPATS/biopsy(first visit)
TTEPAPs >
35 mmHg
84 % 31% (# 27) > 50 mmHgMedian surv 0.7 y
Vs Median surv 4 y
Gagermeier 2005
Retrosp
117 UIP biopsy
TTERVsP >
30 mmHg
40 % 17%(# 20) > 45 mmHg
Lettieri2006
Retrosp’98-’04
79 UIPATS+biopsy
RHCPAPm>
25 mmHg
31 % 29% vs 6% 1y mortality
Nathan 2007
Retrosp‘97-’05
118 UIP73% SLB
RHCPAPm>
25 mmHg
40.7 %
Swanson2008
Prospec‘04-’05
92 UIPATS+biopsy
TTERVsP >
40 mmHg
53 % 40 / 53 underwent RHCIn 75% RHC confirmed
ECHO12% nn ; 13% Venous PH

Diagnosis of P(A)H: a four-stage Diagnosis of P(A)H: a four-stage processprocess
Galiè N et al Eur Heart J 2004; 25: 2243–2278
1. Suspicion of pulmonary hypertension
2. Detection of pulmonary hypertension
3. Pulmonary hypertension class identification
4. Pulmonary arterial hypertension evaluation
•Symptoms and physical examination
•Screening procedures
•Incidental findings
•Electrocardiogram
•Chest radiograph
•Transthoracic Dopplerechocardiography
•Pulmonary function tests and arterial blood gases
•Ventilation/perfusion lung scan
•High-resolution CT
•Spiral CT
•Pulmonary angiography
•Type (blood tests and immunology, HIV test, abdominal ultrasound)
•Exercise capacity (6MWD, peak oxygen consumption)
•Haemodynamics (RHC)Nt-pro BNP
Angio-RMN
Pulmonary Hypertension in IPFPulmonary Hypertension in IPF
•PathogenesisPathogenesis
•Prevalence, detection, diagnosisPrevalence, detection, diagnosis
•Course and significanceCourse and significance
•TherapyTherapy

Koh et al. Br J Rheumatol 1996;35:989-993Koh et al. Br J Rheumatol 1996;35:989-993
PAH associata a sclerodermia: PAH associata a sclerodermia: curve di sopravvivenzacurve di sopravvivenza
Anni dalla diagnosi di PAHAnni dalla diagnosi di PAH
PAH associata a sclerodermia (n = 17)PAH associata a sclerodermia (n = 17)
Coinvolgimento Coinvolgimento polmonare (polmonare (senzasenza PAH) PAH)
n = 73) n = 73)
Nessun coinvolgimento degli Nessun coinvolgimento degli organi interni (es. cuore) organi interni (es. cuore) (n = 138) (n = 138)
00
2020
4040
6060
8080
100100
00 22 66 88 1010 121244
% d
i so
pra
vviv
enza
% d
i so
pra
vviv
enza

Hamada K et al CHEST 2007; 131:650–656Hamada K et al CHEST 2007; 131:650–656
5-year survival rates of patients with IPF 5-year survival rates of patients with IPF grouped by mPAP grouped by mPAP
mPAP <17 mm HgmPAP <17 mm Hg
mPAP mPAP ≥≥ 17 mm Hg17 mm Hg

Song JW et al Respiratory Medicine (2009) 103, 180 186
Comparison of the survival curves between the patients with sPAP ≥40 mmHg and those with sPAP < 40 mmHg.
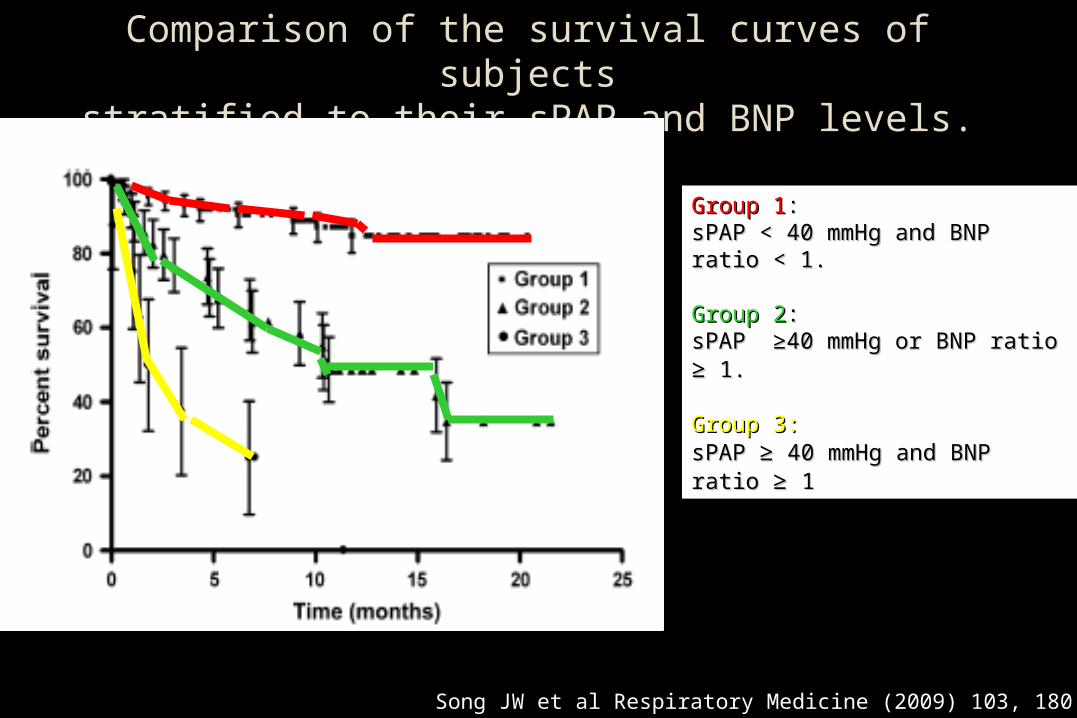
Song JW et al Respiratory Medicine (2009) 103, 180 186
Comparison of the survival curves of subjectsstratified to their sPAP and BNP levels.
Group 1Group 1::sPAP < 40 mmHg and BNP ratio sPAP < 40 mmHg and BNP ratio < 1. < 1.
Group 2Group 2: : sPAP ≥40 mmHg or BNP ratio sPAP ≥40 mmHg or BNP ratio ≥≥ 1. 1.
Group 3:Group 3: sPAP sPAP ≥≥ 40 mmHg and BNP ratio 40 mmHg and BNP ratio ≥≥ 1 1

CHEST 2007; 132:998–1006
Reanalysis of 376 patients listed for lung transplantation in the UNOS registry from 2004 to 2005 who underwent catheterization.
28% had PH with PCWP ≤ 15 mm Hg (median mPAP, 31 mm Hg)

Patel N et al CHEST 2007; 132:998–1006
(n 376)PH is present in 20% to 40% of patients with IPFwho are evaluated for lung transplantation.
DlCO% and other indicators of gas diffusion abnormality are predictive of PH in IPF more than other measures of lung function.

110 IPF patients wer evaluated.
The prevalence of emphysema in the IPF cohort was 28%
Pulmonary arterial hypertension (PAH) was evaluated with TTEand defined by an eSPAP > 45 mmHg
All IPF patients with emphysema showed PAHAll IPF patients with emphysema showed PAH
IPF with emphysema was highly associated with severe PAH(eSPAP: 82.3 ± 20.2 mmHg versus 56.7± 15.3 mmHg, p < 0.0001).

Chest; Prepublished online February 18, 2009;

Rossato Silva D et al J Bras Pneumol. 2008;34(10):779-786

eSPAP (mmHg) 56.7 ― 15.3eSPAP (mmHg) 56.7 ― 15.3
eSPAP (mmHg) 82.3 ―20.2eSPAP (mmHg) 82.3 ―20.2
Chest; Prepublished online February 18, 2009;

Based on analogy to primary pulmonary hypertension, more specific interventions aiming at the restoration of endothelial vasoconstrictor-dilator imbalance could be undertaken.
Randomized controlled trials of oral therapy endothelin receptor blockers and phosphodiesterase 5 inhibitors are being considered. (Naeije, Proc Am Thorac Soc, 2005)
Advanced PH in the presence of underlying lung disease of less severity raises the possibility of a primary vasculopathy that is at least partly independent from the underlying parenchymal process. (Girgis, Clin Chest Med, 2007)
““Out of proportion” PH in IPF Out of proportion” PH in IPF

Pulmonary Hypertension in IPFPulmonary Hypertension in IPF
•PathogenesisPathogenesis
•Prevalence, detection,diagnosisPrevalence, detection,diagnosis
•Course and significanceCourse and significance
•TherapyTherapy

Treatment of ph in the context of ILD : A still unmet need
The primary treatment approach is directed to control the underlying ILD
Treatment recommendations for patients with PH haveonly been established for PAH patients, which refers to GroupI patients of the Dana Point classification
No clear evidence of that the same approach may be useful in all cases of ILD-associated PH
Screening for PH in each patient with ILD is mandatory
RCS are needed
Do we have any active drug for IPF ?
Off label drugs:careful and limited use !!
ECHO and RHC to be included in the diagnostic workout and follow-up in IPF ?
RCS : in progress; study design: end-points !!

CHEST 2006; 130:182–189CHEST 2006; 130:182–189

CHEST 2006; 130:182–189
Immunosuppressive Therapy inImmunosuppressive Therapy inConnective Tissue Diseases-Associated PAHConnective Tissue Diseases-Associated PAH
No patients with SSC associated PAH did respond
PAH associated with SLE or MCTD might respond to a treatment combining glucocorticosteroids and cyclophosphamide.

57

Work in progress

ARTEMIS-PH - Study of Ambrisentan in Subjects With Pulmonary Hypertension Associated With
Idiopathic Pulmonary Fibrosis (Phase III)
This study will compare the efficacy and safety of ambrisentan to placebo in subjects with pulmonary hypertension associated with idiopathic pulmonary fibrosis.X

Double Blind, Randomized Trial of Bosentan for Sarcoidosis Associated Pulmonary Hypertension (BOSAPAH) (Phase II-III)
Patients with advanced sarcoidosis often develop pulmonary hypertension.
The purpose of this study is to determine if bosentan (Tracleer) will help sarcoidosis associated pulmonary hypertension.

Pulmonary Arterial Hypertension Secondary to Idiopathic Pulmonary Fibrosis And Treatment With Sildenafil (Phase IV)
•Pulmonary Arterial Hypertension (PAH) in the setting of Idiopathic Pulmonary Fibrosis(IPF)is a risk factor for morbidity and mortality in the peri-lung transplant(LT) setting.
•Currently there is no significant data to support the use of pulmonary vasodilators for PAH in the setting of interstitial lung disease such as IPF.
•The majority of IPF patients have PAH either at rest or during exercise.
•The study hypothesis is that sildenafil may improve morbidity and mortality in the peri-LT setting in both IPF cohorts with either resting or exercise PAH.

80 patients enrolled in the study.
Primary outcome :
•proportion of patients with an increase in the 6-minute walk distance of 20% or more.
Key secondary outcomes:
•changes in oxygenation•degree of dyspnea•quality of life
The difference in the primary outcome was not significant
A controlled trial of sildenafil in advanced IPF
Zisman DA, et al N Engl J Med. 2010 12;363(7):620-8.

There were small but significant differences in key secondary endpoints:
arterial oxygenationDlCOdegree of dyspneaquality of life
favoring the sildenafil group.
Serious adverse events were similar in the two study groups.
This study did not show a benefit for sildenafil for the primary outcome.
The presence of some positive secondary outcomes creates clinical equipoise for further research.
Zisman DA, et al N Engl J Med. 2010 12;363(7):620-8.

Treprostinil Therapy For Patients With Interstitial Lung Disease And Severe Pulmonary Arterial Hypertension (Phase III)
Using Either Intravenous (IV) or Subcutaneous (SQ) Treprostinil to Treat Pulmonary Hypertension Related to Underlying Interstitial Lung Disease
The hypothesis is that IV or SQ Treprostinil can improve 6 minute walk distance, hemodynamics and quality of life in patients with interstitial lung disease and severe secondary pulmonary arterial hypertension.

Prognostic significance of PH in IPF could be considerd as a background to introduce PH evaulation in diagnosis /follow-up of patients with IPF
Summary and conclusions (1)
The treatment of PH in IPF is mainly the treatment of parenchimal disease in order to prevent PH development
Both screening and diagnosing for PH patients with IPF should be included among procedures used to manage selected ILDs in dailypracticeMany measures of ILDs outcome (DLCO, PAO2, 6 min wt, QOL) usually employed to evaluate disease porgression (parenchimal) in IPF patients are influenced not only by parenchimal changes, but also by both the presence and the level of associated PH

When PH develops lesson learned from previous experiences in treating PAH must be taken in account (in PAH associated with PSS no drug approved for patients with PAH associated to UIP-like parenchymal disease)
Summary and conclusions (2)
Early detection, diagnosis of PH in IPF probably will play a not negligible role in our daily practice for the clinical management of patients with UIP
Treatment of PH associated to ILDs is a still unmet need and even in presence of negative results of RC trials further studies are recruiting patients





























