Embed Size (px)
Citation preview

APPLIED ANI) ENVIRONMENTAL MICROBIOLOGY, June 1994, p. 2113-21190099-224()/94/$04.0(( +Copyright ©) 1994, American Society for Microbiology
Phylogenetic Analysis of the Hyperthermophilic Pink FilamentCommunity in Octopus Spring, Yellowstone National Park
ANNA-LOUISE REYSENBACH, GENE S. WICKHAM, AND NORMAN R. PACE*
Department of Biology and Institute for Molecular and Cellular Biology,Indiana University, Bloomington, Indiana 47405
Received 7 February 1994/Accepted 13 March 1994
The phylogenetic diversity of a well-known pink filament community associated with the 84 to 88°C outflowfrom Octopus Spring, Yellowstone National Park, was examined. Three phylogenetic types ("phylotypes"),designated EM 3, EM 17, and EM 19, were identified by cloning and sequencing the small subunit rRNA genes
(16S rDNA) obtained by PCR amplification of mixed-population DNA. All three phylotypes diverge deeplywithin the phylogenetic domain Bacteria sensu Woese (C. R. Woese, 0. Kandler, and M. L. Wheelis, Proc. Natl.Acad. Sci. USA 87:4576-4579, 1990). No members of the Archaea or Eucarya were detected. EM 3 comprises a
unique lineage within the Thermotogales group, and EM 17 and EM 19 are affiliated with the Aquificales. A totalof 35 clones were examined, of which the majority (26 clones) were of a single sequence type (EM 17) closelyrelated to Aquifex pyrophilus. In situ hybridization with clone-specific probes attributes the majority sequence,
EM 17, to the pink filaments.
The pioneering studies by Brock and his colleagues (forexample, see references 5 through 8) of Yellowstone NationalPark hot springs established that organisms grow at near-
boiling temperatures. Particularly noteworthy examples ofsuch organisms, because of their large accumulations, were thepink filaments from the outflow of Octopus Spring that Brocket al. described (5-8). Although artificial substrates such as
glass slides or cotton string inserted into the outflow were
rapidly colonized by the filaments, attempts to culture theorganisms were unsuccessful (6).
Recently, the use of molecular phylogenetic techniques hasalleviated, to some extent, our reliance on culture enrichmenttechniques for the analysis of microbial diversity (25, 33). Themolecular phylogenetic approach uses the analysis of phyloge-netically informative molecules such as small subunit rRNA(or 16S-like rRNA) to characterize organisms. The 16S-likerRNAs (or their genes) can bc isolated from naturally occur-
ring communities directly or by cloning, and their sequences
can be determined. Comparison of the sequences with a database of aligned rRNA sequences of characterized organismsidentifies the unknown sequences within a phylogenetic frame-work. Some properties of otherwise unknown organisms may
be inferred from rRNA sequences, on the basis of the knownproperties of their relatives. In addition, sequence informationprovides for the design of specific oligonucleotide probes (1,12), for in situ identification, and for estimates of abundanceand dynamics. Many novel, yet uncultivated, organisms havebeen identified by these techniques (for example, see refer-ences 2, 3, and 1 1).
In this study we used a molecular phylogenetic approach tocharacterize the pink filament community at the outflow ofOctopus Spring. The 16S-like rRNA genes of the mixed-population DNA were amplified by PCR, cloned, sorted, andsequenced. Fluorescent oligonucleotide probes specific for theobtained 16S rDNAs were used to determine the sequencecorresponding to the pink filaments. We show that the pinkfilaments are most closely related among cultured organisms to
* Corresponding author. Phone: (812) 855-6154. Fax: (812) 855-6705.
the hydrogen-oxidizing bacterium Aquifex pyrophilus (9, 19)and its close relative Hydrogenobacter thermophillus (9, 22).
MATERIALS AND METHODS
Sample collection. Pink filaments attached to rock surfaceswere collected from the hot water outflow from OctopusSpring, Yellowstone National Park, by using sterile forceps.Filaments were observed in the outflow only at temperaturesbetween 84 and 88°C. The filaments were rinsed briefly infilter-sterilized Octopus Spring water and frozen immediatelyon dry ice. In the laboratory, samples were stored at -70°C.Additional samples were fixed upon collection in 2.5% (vol/vol) glutaraldehyde in phosphate-buffered saline (PBS) (PBScontains [per liter] 8 g of NaCl, 0.2 g of KCl, 1.44 g ofNa2HPO4, and 0.24 g of KH2PO4 [pH 7.4]) for scanningelectron microscopy. Samples for fluorescence microscopywere washed in PBS and then placed in 4% paraformaldehydein PBS for 6 h, rinsed in PBS, and stored in 50% ethanol inPBS at 4°C until the samples reached the laboratory, wherethey were stored at -200C.
Microscopy. Paraformaldehyde-fixed samples were stainedwith 4',6'-diamidino-2-phenylindole (DAPI) for 5 min (26)and viewed with a Zeiss Axioskop photomicroscope. Forscanning electron microscopy, glutaraldehyde-fixed sampleswere passed through an ethanol dehydration series (10 to100%), critical point dried, mounted on scanning electronmicroscope stubs, and sputter coated with gold-palladium. Thesamples were viewed with a Cambridge 5250 MK2 scanningelectron microscope.
Extraction of DNA and amplification and cloning of 16SrRNA genes. Approximately 100 mg (wet weight) of pinkfilaments was thawed, and cells were disrupted and DNA was
isolated essentially according to the procedure described byBollet et al. (4). The filaments were resuspended in 150 ,ud ofTE (10 mM Tris-HCl, pH 8; 1 mM EDTA) containing 3.3%sodium dodecyl sulfate and incubated for 30 min at 65°C. Thelysate was centrifuged, the supernatant was removed (no DNAwas recovered from this fraction), and the pellet was treated in
a microwave (900 W) twice for 1 min each time and resus-
pended in TE. DNA was extracted three times with equal
2113
Vol. 60, No. 6
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
2114 REYSENBACH ET AL.
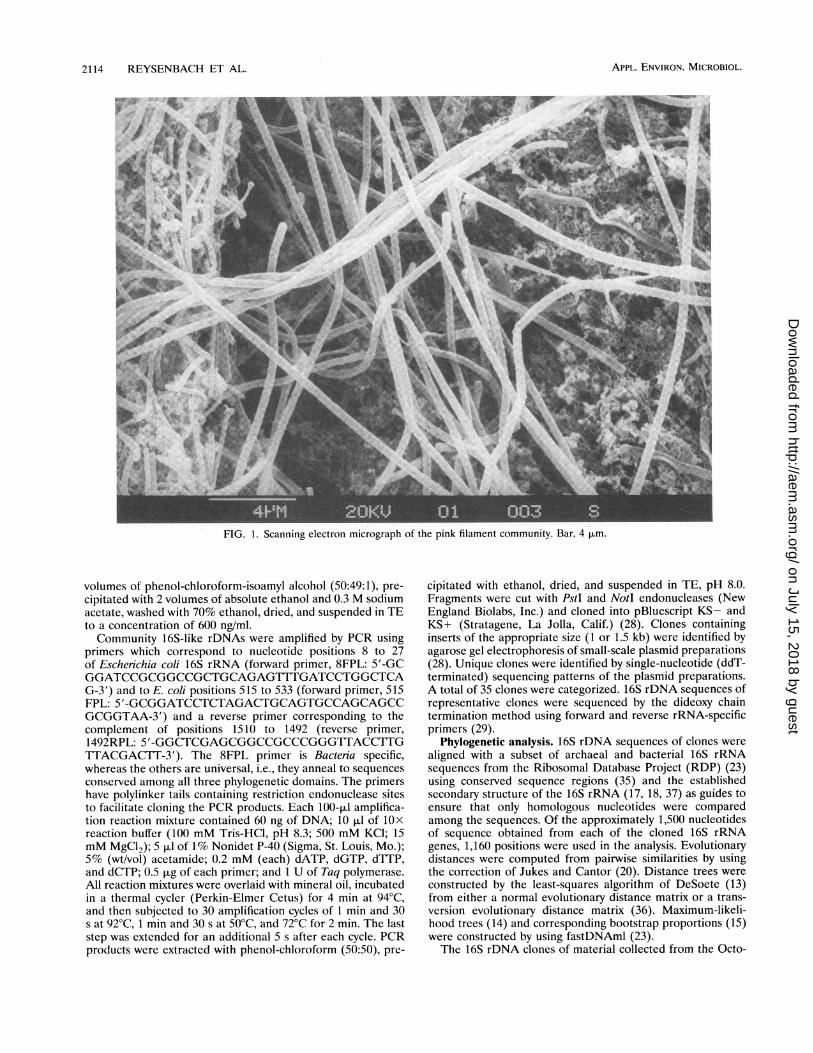
FIG. 1. Scanning electron micrograph of the pink filament community. Bar, 4 pLm.
volumes of phenol-chloroform-isoamyl alcohol (50:49:1), pre-cipitated with 2 volumes of absolute ethanol and 0.3 M sodiumacetate, washed with 70% ethanol, dried, and suspended in TEto a concentration of 600 ng/ml.Community 16S-like rDNAs were amplified by PCR using
primers which correspond to nucleotide positions 8 to 27of Escherichia coli 16S rRNA (forward primer, 8FPL: 5'-GCGGATCCGCGGCCGCTGCAGAGTlTTGATCCTGGCTCAG-3') and to E. coli positions 515 to 533 (forward primer, 515FPL: 5'-GCGGATCCTCTAGACTGCAGTGCCAGCAGCCGCGGTAA-3') and a reverse primer corresponding to thecomplement of positions 1510 to 1492 (reverse primer,1492RPL: 5'-GGCTCGAGCGGCCGCCCGGGTTACCTTGTTACGACTT-3'). The 8FPL primer is Bacteria specific,whereas the others are universal, i.e., they anneal to sequencesconserved among all three phylogenetic domains. The primershave polylinker tails containing restriction endonuclease sitesto facilitate cloning the PCR products. Each 100-,ul amplifica-tion reaction mixture contained 60 ng of DNA; 10 ,ul of 1Oxreaction buffer (100 mM Tris-HCl, pH 8.3; 500 mM KCl; 15mM MgCl2); 5 pAl of 1% Nonidet P-40 (Sigma, St. Louis, Mo.);5% (wt/vol) acetamide; 0.2 mM (each) dATP, dGTP, dTTP,and dCTP; 0.5 ,ug of each primer; and 1 U of Taq polymerase.All reaction mixtures were overlaid with mineral oil, incubatedin a thermal cycler (Perkin-Elmer Cetus) for 4 min at 94°C,and then subjected to 30 amplification cycles of 1 min and 30s at 92°C, 1 min and 30 s at 50°C, and 72°C for 2 min. The laststep was extended for an additional 5 s after each cycle. PCRproducts were extracted with phenol-chloroform (50:50), pre-
cipitated with ethanol, dried, and suspended in TE, pH 8.0.Fragments were cut with PstI and NotI endonucleases (NewEngland Biolabs, Inc.) and cloned into pBluescript KS- andKS+ (Stratagene, La Jolla, Calif.) (28). Clones containinginserts of the appropriate size (1 or 1.5 kb) were identified byagarose gel electrophoresis of small-scale plasmid preparations(28). Unique clones were identified by single-nucleotide (ddT-terminated) sequencing patterns of the plasmid preparations.A total of 35 clones were categorized. 16S rDNA sequences ofrepresentative clones were sequenced by the dideoxy chaintermination method using forward and reverse rRNA-specificprimers (29).
Phylogenetic analysis. 16S rDNA sequences of clones werealigned with a subset of archaeal and bacterial 16S rRNAsequences from the Ribosomal Database Project (RDP) (23)using conserved sequence regions (35) and the establishedsecondary structure of the 16S rRNA (17, 18, 37) as guides toensure that only homologous nucleotides were comparedamong the sequences. Of the approximately 1,500 nucleotidesof sequence obtained from each of the cloned 16S rRNAgenes, 1,160 positions were used in the analysis. Evolutionarydistances were computed from pairwise similarities by usingthe correction of Jukes and Cantor (20). Distance trees wereconstructed by the least-squares algorithm of DeSoete (13)from either a normal evolutionary distance matrix or a trans-version evolutionary distance matrix (36). Maximum-likeli-hood trees (14) and corresponding bootstrap proportions (15)were constructed by using fastDNAml (23).The 16S rDNA clones of material collected from the Octo-
APPL. ENVIRON. MICROBIOL.
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

MOLECULAR PHYLOGENY OF THE HYPERTHERMOPHILIC COMMUNITY 2115
Escherichia coli
Clostridium butyricum
Synechococcus PCC 6301
0.05
B BACTERIA
ARCHAEA&
EUCARYA
FIG. 2. Phylogenetic analysis of rDNAs obtained from the pinkfilament community. (A) Phylogenetic tree determined by maximum-likelihood analysis of complete 16S rRNA sequences. Boldface typeindicates organisms derived from the pink filament community atOctopus Spring, Yellowstone National Park. The scale bar represents0.05 fixed mutation per nucleotide position. Numbers at nodes repre-sent bootstrap values for that node (based on 100 bootstrap resam-
plings). (B) Diagrammatic phylogenetic tree based on this and previ-ous (35) analyses showing the relative positions of the Thermotogalesand Aquificales in the context of the known bacterial kingdoms (sensuWoese et al. [38]).
pus Spring outflow were checked for the presence of chimericsequences (21, 24, 30) using the CHECK_CHIMERA programof the RDP (23). No chimeric sequences were detected by thismethod. Moreover, no idiosyncrasies in long-range secondarystructures were evident; mispairings might be expected ifdifferent portions of a sequence were derived from differentorganisms.
Oligonucleotide synthesis and labeling. Hybridizationprobes complementary to unique regions of the 16S rRNAsequences were designed. The following oligonucleotideprobes were synthesized on an Applied Biosystems automatedDNA synthesizer: clone-specific probes complementary to the1474-to-1447 region of the 16S rRNA, EM 17 (5'-GTCCCCTGCCTCCCTTGC-3'), EM 19 (5'-CACACATTCGTCCCT1T17-3'), EM 3 (5'-TGCCTTCACCCCACCTTC-3'); a bacte-rial probe, EUB (5'-ACCGCTTGTGCGGGCCC-3'); and a
universal probe (5'-GWATTACCGCGGCKGCTG-3', where"W" is A or T and "K" is G or T) (12). An rRNA-like probe(5'-GTGCCAGCMGCCGCGG-3', where "M" is A or C) thatbinds to the complement of the 16S rRNA served as a controlfor nonspecific binding. The sequences of the clone-specificprobes were checked with the RDP collection of sequences(23) to ensure that there would be no cross-hybridization withany other small subunit rRNA sequences of known organisms-.An amino group (Aminolink II; Applied Biosystems) wasadded to the 5' ends of oligonucleotide probes during synthesisand then coupled with fluorescein isothiocyanate (FITC; Mo-lecular Probes, Inc., Eugene, Oreg.) as follows (12). A reactionmixture (250 p.l) consisting of 100 ,ug of 5'-aminoethyl oligo-nucleotide, 200 mM carbonate buffer (pH 9.0), and 40 pul ofFITC (10 mg/ml) was incubated at room temperature over-night in the dark. The unincorporated dye was separated fromthe oligonucleotide on a Sephadex G-25 column equilibratedwith 10 mM Tris-HCl (pH 7.5). The oligonucleotide fractionswere collected in a microtiter plate, pooled, lyophilized, andresuspended in 50 p.l of sterile water. Labeled oligonucleotidewas separated from unlabeled oligonucleotide on a 20%nondenaturing polyacrylamide gel. The labeled oligonucleo-tide was excised from the gel, eluted overnight in sterile,double-distilled water, lyophilized, and resuspended in sterile,double-distilled water to a final concentration of 50 ,ug/ml.
In situ hybridization. Ethanol-fixed samples were smearedonto gelatin-coated glass slides (16) and air dried. Control cellsof Bacillus megaterium and E. coli were grown overnight inLuria broth, pelleted, washed, and then resuspended in PBS toa final concentration of about 2 x 107/ml. A 30-,ul portion ofthis suspension was smeared onto the gelatin-coated slides andair dried. In situ hybridizations were done as described byDeLong et al. (12) and viewed directly with a Nikon Micro-phot-FXA photomicroscope.
Nucleotide sequence accession numbers. The GenBank ac-cession numbers for the EM 3, EM 17, and EM 19 sequencesare (respectively) U05660, U05661, and U05662.
RESULTS
Microscopy. Pink filaments were harvested (see Materialsand Methods) from the outflow of Octopus Spring (84 to88°C). Fluorescence microscopy of DAPI-stained preparationsindicated that cells were generally intact and that the pinkfilaments dominated the community, although small numbersof cocci and short rods were also present. Scanning electronmicroscopy of the filaments (Fig. 1) confirmed the fluorescencemicroscopy observations. The filaments are clearly segmented(Fig. 1).
Cloning and sequence analysis of 16S rRNA genes. DNAwas isolated from pink filaments (see Materials and Methods)and 16S rRNA genes were amplified (27) and cloned. Clonelibraries were screened for unique types by single-nucleotide(ddT) sequencing patterns of the plasmid preparations, and arepresentative of each type was sequenced. No archaeal 16SrRNA genes were obtained from the clone library derived byPCR from the mixed-population DNA by using universalprimers (515FPL and 1492RPL), which sample only the 3'-proximal two-thirds of the gene but likely would detect rDNAof any type of organism. Moreover, attempts to recoverarchaeal sequences by using Archaea-specific primers wereunsuccessful. Subsequent amplifications were carried out withthe Bacteria-specific primer 8FPL, in conjunction with theuniversal 1492RPL primer, to amplify essentially the entire 16SrRNA gene. In both amplifications the same three types ofclones, named EM 3, EM 17, and EM 19, were obtained. The
VOL. 60, 1994
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
2116 REYSENBACH ET AL.
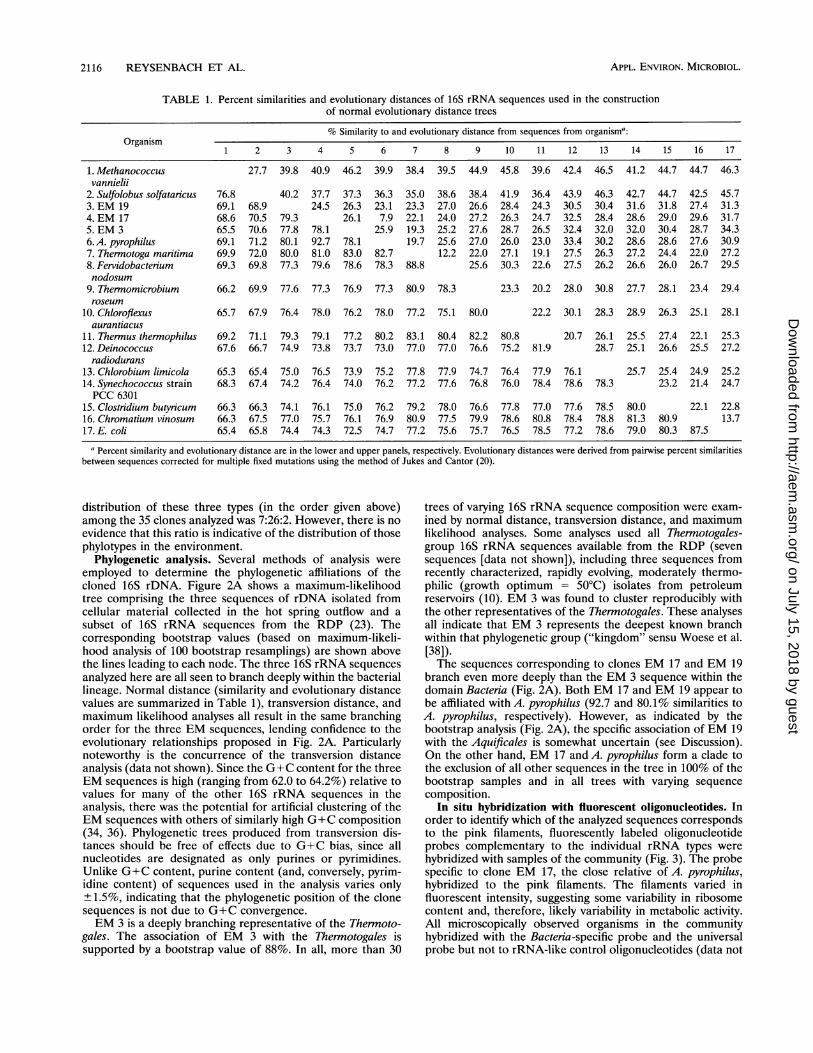
TABLE 1. Percent similarities and evolutionary distances of 16S rRNA sequences used in the constructionof normal evolutionary distance trees
% Similarity to and evolutionary distance from sequences from organism':Organism
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
1. Methanococcus 27.7 39.8 40.9 46.2 39.9 38.4 39.5 44.9 45.8 39.6 42.4 46.5 41.2 44.7 44.7 46.3vannielii
2. Sulfolobus solfataricus 76.8 40.2 37.7 37.3 36.3 35.0 38.6 38.4 41.9 36.4 43.9 46.3 42.7 44.7 42.5 45.73. EM 19 69.1 68.9 24.5 26.3 23.1 23.3 27.0 26.6 28.4 24.3 30.5 30.4 31.6 31.8 27.4 31.34. EM 17 68.6 70.5 79.3 26.1 7.9 22.1 24.0 27.2 26.3 24.7 32.5 28.4 28.6 29.0 29.6 31.75. EM 3 65.5 70.6 77.8 78.1 25.9 19.3 25.2 27.6 28.7 26.5 32.4 32.0 32.0 30.4 28.7 34.36.A. pyrophilus 69.1 71.2 80.1 92.7 78.1 19.7 25.6 27.0 26.0 23.0 33.4 30.2 28.6 28.6 27.6 30.97. Therinotoga maritima 69.9 72.0 80.0 81.0 83.0 82.7 12.2 22.0 27.1 19.1 27.5 26.3 27.2 24.4 22.0 27.28. Fervidobacterium 69.3 69.8 77.3 79.6 78.6 78.3 88.8 25.6 30.3 22.6 27.5 26.2 26.6 26.0 26.7 29.5nodosum
9. Thermomicrobium 66.2 69.9 77.6 77.3 76.9 77.3 80.9 78.3 23.3 20.2 28.0 30.8 27.7 28.1 23.4 29.4roseum
10. Chlorofiexus 65.7 67.9 76.4 78.0 76.2 78.0 77.2 75.1 80.0 22.2 30.1 28.3 28.9 26.3 25.1 28.1aurantiacus
11. Thennus thernophilus 69.2 71.1 79.3 79.1 77.2 80.2 83.1 80.4 82.2 80.8 20.7 26.1 25.5 27.4 22.1 25.312. Deinococcus 67.6 66.7 74.9 73.8 73.7 73.0 77.0 77.0 76.6 75.2 81.9 28.7 25.1 26.6 25.5 27.2radiodurans
13. Chlorobium limicola 65.3 65.4 75.0 76.5 73.9 75.2 77.8 77.9 74.7 76.4 77.9 76.1 25.7 25.4 24.9 25.214. Synechococcus strain 68.3 67.4 74.2 76.4 74.0 76.2 77.2 77.6 76.8 76.0 78.4 78.6 78.3 23.2 21.4 24.7PCC 6301
15. Clostridium butyricum 66.3 66.3 74.1 76.1 75.0 76.2 79.2 78.0 76.6 77.8 77.0 77.6 78.5 80.0 22.1 22.816. Chromatium vinosum 66.3 67.5 77.0 75.7 76.1 76.9 80.9 77.5 79.9 78.6 80.8 78.4 78.8 81.3 80.9 13.717. E. coli 65.4 65.8 74.4 74.3 72.5 74.7 77.2 75.6 75.7 76.5 78.5 77.2 78.6 79.0 80.3 87.5
a Percent similarity and evolutionary distance are in the lower and upper panels, respectively. Evolutionary distances were derived from pairwise percent similaritiesbetween sequences corrected for multiple fixed mutations using the method of Jukes and Cantor (20).
distribution of these three types (in the order given above)among the 35 clones analyzed was 7:26:2. However, there is noevidence that this ratio is indicative of the distribution of thosephylotypes in the environment.
Phylogenetic analysis. Several methods of analysis wereemployed to determine the phylogenetic affiliations of thecloned 16S rDNA. Figure 2A shows a maximum-likelihoodtree comprising the three sequences of rDNA isolated fromcellular material collected in the hot spring outflow and asubset of 16S rRNA sequences from the RDP (23). Thecorresponding bootstrap values (based on maximum-likeli-hood analysis of 100 bootstrap resamplings) are shown abovethe lines leading to each node. The three 16S rRNA sequencesanalyzed here are all seen to branch deeply within the bacteriallineage. Normal distance (similarity and evolutionary distancevalues are summarized in Table 1), transversion distance, andmaximum likelihood analyses all result in the same branchingorder for the three EM sequences, lending confidence to theevolutionary relationships proposed in Fig. 2A. Particularlynoteworthy is the concurrence of the transversion distanceanalysis (data not shown). Since the G+C content for the threeEM sequences is high (ranging from 62.0 to 64.2%) relative tovalues for many of the other 16S rRNA sequences in theanalysis, there was the potential for artificial clustering of theEM sequences with others of similarly high G+C composition(34, 36). Phylogenetic trees produced from transversion dis-tances should be free of effects due to G+C bias, since allnucleotides are designated as only purines or pyrimidines.Unlike G+C content, purine content (and, conversely, pyrim-idine content) of sequences used in the analysis varies only+1.5%, indicating that the phylogenetic position of the clonesequences is not due to G+C convergence.EM 3 is a deeply branching representative of the Thermoto-
gales. The association of EM 3 with the Thermotogales issupported by a bootstrap value of 88%. In all, more than 30
trees of varying 16S rRNA sequence composition were exam-ined by normal distance, transversion distance, and maximumlikelihood analyses. Some analyses used all Thernotogales-group 16S rRNA sequences available from the RDP (sevensequences [data not shown]), including three sequences fromrecently characterized, rapidly evolving, moderately thermo-philic (growth optimum = 50°C) isolates from petroleumreservoirs (10). EM 3 was found to cluster reproducibly withthe other representatives of the Thermotogales. These analysesall indicate that EM 3 represents the deepest known branchwithin that phylogenetic group ("kingdom" sensu Woese et al.[38]).The sequences corresponding to clones EM 17 and EM 19
branch even more deeply than the EM 3 sequence within thedomain Bacteria (Fig. 2A). Both EM 17 and EM 19 appear tobe affiliated with A. pyrophilus (92.7 and 80.1% similarities toA. pyrophilus, respectively). However, as indicated by thebootstrap analysis (Fig. 2A), the specific association of EM 19with the Aquificales is somewhat uncertain (see Discussion).On the other hand, EM 17 and A. pyrophilus form a clade tothe exclusion of all other sequences in the tree in 100% of thebootstrap samples and in all trees with varying sequencecomposition.
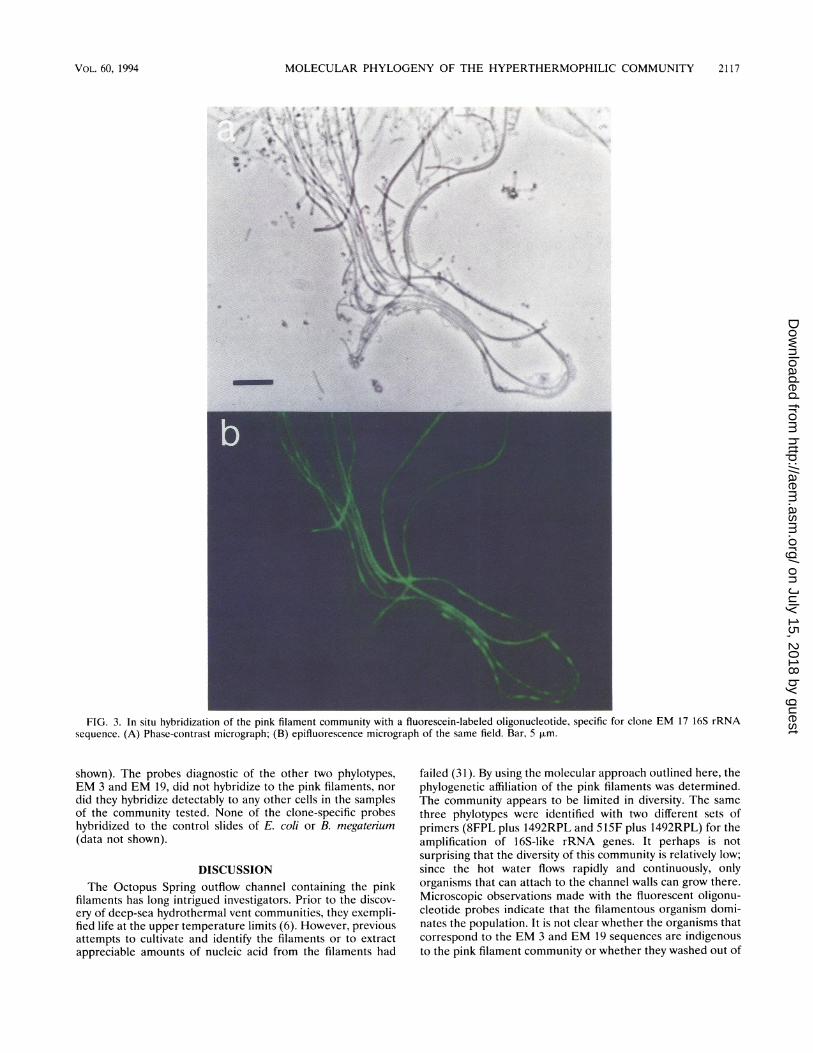
In situ hybridization with fluorescent oligonucleotides. Inorder to identify which of the analyzed sequences correspondsto the pink filaments, fluorescently labeled oligonucleotideprobes complementary to the individual rRNA types werehybridized with samples of the community (Fig. 3). The probespecific to clone EM 17, the close relative of A. pyrophilus,hybridized to the pink filaments. The filaments varied influorescent intensity, suggesting some variability in ribosomecontent and, therefore, likely variability in metabolic activity.All microscopically observed organisms in the communityhybridized with the Bacteria-specific probe and the universalprobe but not to rRNA-like control oligonucleotides (data not
APPL. ENvIRON. MICROBIOL.
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
MOLECULAR PHYLOGENY OF THE HYPERTHERMOPHILIC COMMUNITY 2117
FIG. 3. In situ hybridization of the pink filament community with a fluorescein-labeled oligonucleotide, specific for clone EM 17 16S rRNAsequence. (A) Phase-contrast micrograph; (B) epifluorescence micrograph of the same field. Bar, 5 ,um.
shown). The probes diagnostic of the other two phylotypes,EM 3 and EM 19, did not hybridize to the pink filaments, nordid they hybridize detectably to any other cells in the samplesof the community tested. None of the clone-specific probeshybridized to the control slides of E. coli or B. megaterium(data not shown).
DISCUSSIONThe Octopus Spring outflow channel containing the pink
filaments has long intrigued investigators. Prior to the discov-ery of deep-sea hydrothermal vent communities, they exempli-fied life at the upper temperature limits (6). However, previousattempts to cultivate and identify the filaments or to extractappreciable amounts of nucleic acid from the filaments had
failed (31). By using the molecular approach outlined here, thephylogenetic affiliation of the pink filaments was determined.The community appears to be limited in diversity. The samethree phylotypes were identified with two different sets ofprimers (8FPL plus 1492RPL and 515F plus 1492RPL) for theamplification of 16S-like rRNA genes. It perhaps is notsurprising that the diversity of this community is relatively low;since the hot water flows rapidly and continuously, onlyorganisms that can attach to the channel walls can grow there.Microscopic observations made with the fluorescent oligonu-cleotide probes indicate that the filamentous organism domi-nates the population. It is not clear whether the organisms thatcorrespond to the EM 3 and EM 19 sequences are indigenousto the pink filament community or whether they washed out of
VOL. 60, 1994
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

2118 REYSENBACH ET AL.
20qoAAG
CG
I cucG UG GC U
C AUCg9 UA
A 200
AAG G G G
G
A
A 200
A
g g g g C g UU
GC C C C G U
Thermotoga maritima
G C C C UUU
C G G GUA
EM 17
G
C
CC CuG G G C UC
G A A C C C G U
Aquifexpyrophilus
A
A
G
U
G
220
200
A G G G
U C U
GGAC
C
c U
CLG
Escherichia cofi
FIG. 4. Putative secondary structure of the 195-to-220 region (E.coli numbering) of several 16S rRNAs (9, 17).
the Octopus Spring source pool and were entrapped in thepink filament mass. Indeed, one of the clone sequences (EM 3)has also been obtained from the 92°C source pool (34a).
All three phylotypes identified branch deeply within thephylogenetic domain Bacteria, grouping together with otherknown thermophilic genera. It was surprising that no repre-sentatives of the Archaea were detected, since most cultivatedhyperthermophiles (organisms that grow optimally above80°C) are members of the Archaea (32).The pink filaments, represented by clone EM 17, are clearly
related to A. pyrophilus. Comparison of the EM 17 and A.pyrophilus sequences with a partial 16S rRNA sequence (-400nucleotides [9]) of H. thermophilus TK-H revealed 95.9 and92.8% similarities, respectively, indicating that EM 17 is moreclosely related to H. thermophilus than toA. pyrophilus. EM 17shares a secondary structural feature with A. pyrophilus thatmay be a defining signature of this particular lineage (Fig. 4)(9). The helix at positions 198 to 219 in the 16S rRNAsecondary structure (E. coli numbering) of both sequences ischaracterized by a CUC bulge 5 nucleotides 5' from thecapping loop and a single nucleotide bulge (an A in A.pyrophilus and a U in EM 17) 5 nucleotides 3' from the cappingloop. This motif is found elsewhere only in the 8 group ofproteobacteria (35).The phylogenetic affiliations of EM 3 and EM 17 with
specific bacterial lineages are clear; however, the placement ofEM 19 is not definitive. Although the EM 19 sequence clusterswith those of EM 17 and A. pyrophilus in most analyses (65%of the bootstrap samplings), it is not closely related to either ofthose sequences or other known ones, and deep branchings inphylogenetic trees are intrinsically less reliable than peripheralones. The placement of EM 19 varied slightly among treesconstructed by using different sets of 16S rRNA sequences. Inmore than 90% of all trees of varying composition examined,
EM 19 clusters (albeit distantly) with the Aquificales. However,in a small minority of trees, EM 19 emerged as a distinctbranch either intermediate between the Thermotogales and theAquificales or as the deepest branch in the Bacteria. Establish-ing the correct branching order of EM 19 may depend on thedetermination of 16S rRNA sequences from additional organ-isms which diverge deeply within the bacterial domain and ontheir inclusion in tree analyses.
Figure 2B summarizes the phylogenetic positions of theAquificales and Thermotogales in the context of other knownbacterial kingdoms. Burggraf et al. (9) reported a bootstrapvalue of 98% for the identification of the Thermotogales andthe Aquificales as the two most deeply diverging groups in thebacterial domain. The bootstrap analysis performed on themaximum-likelihood analysis shown in Fig. 2A supports thatbranching at a confidence level of 93%. Although Burggraf etal. (9) found that A. pyrophilus emerged as the deepestbacterial lineage in 62% of the bootstrap samples, our boot-strap analysis supports this topology at a level of only 35%.Bootstrap values supporting the Thernotogales and Aquificalesas sister groups or the Thermotogales alone as the deepestgroup are 42 and 8%, respectively. Furthermore, multiplemaximum-likelihood analyses of the same data set (differingonly in the order of addition of taxa to the tree) resulted in twotypes of trees, one in which the Aquificales are the deepestbranch and one in which the Aquificales and the Thermotogalesemerge as sister groups. The log likelihoods for the two treeswere essentially the same. Therefore, it is not possible with theavailable data to resolve unequivocally whether the Aquificalesare the deepest known lineage within the Bactenia or whetherthey share that distinction with the Thennotogales.
In situ hybridization using the clone-specific probes clearlyattributes clone EM 17 to the pink filaments. However, wewere unable to detect the community members correspondingto clones EM 3 and EM 19 with the probes used. Althoughsome morphotypes different from the pink filaments weredetected microscopically, these did not bind the EM 3- andEM 19-specific probes. They did, however bind universal andbacterial probes. It is possible that the unidentified morpho-types correspond to the EM 3 and EM 19 sequences but thatthe binding of the specific probes was inefficient and so notdetected. It is also possible that DNA corresponding to theunidentified morphotypes was not recovered or amplified.The pink filamentous organism groups phylogenetically with
other hydrogen-oxidizing bacteria, A. pyrophilus and H. ther-mophilus. This correlation between phylogeny and physiologysuggests that the pink filaments, too, may be hydrogen oxidiz-ers. If so, it would help explain the previous lack of success atcultivating them by using organic compound-based media. Onthe basis of the phylogenetic results, attempts to cultivate thepink filaments as hydrogen oxidizers are under way (24a).Thermophilic, hydrogen-oxidizing organisms appear to play asignificant biological role in both terrestrial and marine ther-mal springs (22), but little is known about their diversity anddistribution. The rRNA sequences and the identification of thepink filamentous organism by in situ hybridization will allowrecognition of this group in other environments.
ACKNOWLEDGMENTS
This work was supported in part by a contract from the Departmentof Energy to N.R.P.We give many thanks to Jim Brown, Fred Rainey, Esther Angert,
Susan Barns, and Rocco Mancinelli for helpful discussions. We thankCarl Woese for providing the A. pyrophilus 16S rRNA sequence andRobert Huber and Siegfried Burggraf for providing us with theunpublished partial sequence of H. thermophilus TK-H. We thank
APPL. ENVIRON. MICROBIOL.
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

MOLECULAR PHYLOGENY OF THE HYPERTHERMOPHILIC COMMUNITY 2119
Bernadette Pace for supplying Taq polymerase. The microscopy wouldnot have been accomplished without the help of Susan Strome andMimi Zolan. We are indebted to the staff at the Research Division ofYellowstone National Park, who have made our forays into the Parkextremely easy. Special thanks go to Douglas Adams and his ElectricMonk that enabled us to see everything in a shade of pink.The first two authors contributed equally to the work.
REFERENCES1. Amann, R., W. Ludwig, and K. Schleifer. 1992. Identification and
in situ detection of individual bacterial cells. FEMS Microbiol.Lett. 100:45-50.
2. Angert, E. R., K. D. Clements, and N. R. Pace. 1993. The largestbacterium. Nature (London) 362:239-241.
3. Barns, S. M., R. E. Fundyga, M. W. Jefferies, and N. R. Pace. 1994.Remarkable archaeal diversity detected in a Yellowstone NationalPark hot spring environment. Proc. Natl. Acad. Sci. USA 91:1609-1613.
4. Bollet, C., M. J. Gevaudan, X. de Lamallerie, C. Zandotti, and P.de Micco. 1991. A simple method for the isolation of chromosomalDNA from Gram positive or acid-fast bacteria. Nucleic Acids Res.19:1955.
5. Bott, T. L., and T. D. Brock. 1969. Bacterial growth rates above90°C in Yellowstone Hot Springs. Science 164:1411-1412.
6. Brock, T. D. 1967. Life at high temperatures. Science 158:1012-1019.
7. Brock, T. D., M. L. Brock, T. L. Bott, and M. R. Edwards. 1971.Microbial life at 90 C: the sulfur bacteria of Boulder Spring. J.Bacteriol. 107:303-314.
8. Brock, T. D., and G. K. Darland. 1970. Limits of microbialexistence: temperature and pH. Science 169:1316-1318.
9. Burggraf, S., G. J. Olsen, K. 0. Stetter, and C. R. Woese. 1992. Aphylogenetic analysis of Aquifex pyrophilus. Syst. Appl. Microbiol.15:352-356.
10. Davey, M. E., W. A. Wood, R. Key, K. Nakamura, and D. A. Stahl.1993. Isolation of three species of Geotoga and Petrotoga: twonew genera representing a new lineage in the bacterial line ofdescent distantly related to the 'Thermnotogales." Syst. Appl.Microbiol. 16:191-200.
11. DeLong, E. F. 1992. Archaea in coastal marine environments.Proc. Natl. Acad. Sci. USA 89:5685-5689.
12. DeLong, E. F., G. S. Wickham, and N. R. Pace. 1989. Phylogeneticstains: ribosomal RNA-based probes for the identification ofsingle cells. Science 243:1360-1363.
13. DeSoete, G. 1983. A least squares algorithm for fitting additivetrees to proximity data. Psychometrika 48:621-626.
14. Felsenstein, J. 1981. Evolutionary trees form DNA sequences: amaximum likelihood approach. J. Mol. Evol. 17:368-376.
15. Felsenstein, J. 1985. Confidence limits on phylogenies: an ap-proach using the bootstrap. Evolution 39:783-791.
16. Giovannoni, S. J., E. F. DeLong, G. J. Olsen, and N. R. Pace. 1988.Phylogenetic group-specific oligodeoxynucleotide probes for iden-tification of single microbial cells. J. Bacteriol. 170:720-726.
17. Gutell, R. R. 1993. Collection of small subunit (16S- and 16S-like)ribosomal RNA structures. Nucleic Acids Res. 21:3051-3t)54.
18. Gutell, R. R., and C. R. Woese. 1990. Higher order structuralelements in ribosomal RNAs: pseudo-knots and the use of non-canonical pairs. Proc. Natl. Acad. Sci. USA 87:663-667.
19. Huber, R., T. Whilharm, D. Huber, A. Trincone, S. Burggraf, H.Konig, R. Rachel, I. Rockinger, H. Fricke, and K. 0. Stetter. 1992.Aquifex pyrophilus gen. nov., sp. nov., represents a novel group ofmarine hyperthermophilic hydrogen-oxidizing bacteria. Syst. Appl.Microbiol. 15:340-351.
20. Jukes, T. H., and C. R. Cantor. 1969. Evolution of proteinmolecules, p. 21-132. in H. N. Munro (ed.), Mammalian proteinmetabolism. Academic Press, New York.
21. Koczynski, E. D., M. M. Bateson, and D. M. Ward. 1994. Recog-nition of chimeric small-subunit ribosomal DNAs composed ofgenes from uncultivated microorganisms. Appl. Environ. Micro-biol. 60:746-748.
22. Kristjansson, J. K., A. Ingason, and G. A. Alfredsson. 1985.Isolation of thermophilic obligately autotrophic hydrogen-oxidiz-ing bacteria, similar to Hydrogenobacter thermophilius, from Icelan-dic hot springs. Arch. Microbiol. 140:321-325.
23. Larsen, N., G. J. Olsen, B. L. Maidak, M. J. McCaughey, R.Overbeek, T. J. Macke, T. L. Marsh, and C. R. Woese. 1993. TheRibosomal Database Project. Nucleic Acids Res. 21:3021-3023.
24. Liesack, W., H. Weyland, and E. Stackebrandt. 1991. Potentialrisks of gene amplification by PCR as determined by 16S rDNAanalysis of a mixed-culture of strict barophilic bacteria. Microb.Ecol. 21:191-198.
24a.Mancinelli, R. Personal communication.25. Pace, N. R., D. A. Stahl, D. J. Lane, and G. J. Olsen. 1986. The
analysis of naturally microbial populations by ribosomal RNAsequences. Adv. Microb. Ecol. 9:1-55.
26. Porter, K. G., and Y. S. Feig. 1980. The use of DAPI for identifyingand counting aquatic microflora. Limnol. Oceanogr. 25:943-948.
27. Reysenbach, A.-L., L. J. Giver, G. S. Wickham, and N. R. Pace.1992. Differential amplification of rRNA genes by polymerasechain reaction. Appl. Environ. Microbiol. 58:3417-3418.
28. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecularcloning: a laboratory manual, 2nd ed. Cold Spring Harbor Labo-ratory, Cold Spring Harbor, N.Y.
29. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencingwith chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA78:5463-5467.
30. Shuldiner, A. R., A. Nirula, and J. Roth. 1989. Hybrid DNAartifact from PCR of closely related target sequences. NucleicAcids Res. 17:440)9.
31. Stahl, D. A., D. J. Lane, G. J. Olsen, and N. R. Pace. 1985.Characterization of a Yellowstone hot spring microbial commu-nity by 5S rRNA sequences. Appl. Environ. Microbiol. 49:1379-1384.
32. Stetter, K. O., G. Fiala, G. Huber, R. Huber, and A. Segerer. 1990.Hyperthermophilic microorganisms. FEMS Microbiol. Rev. 75:117-124.
33. Ward, D. M., M. M. Bateson, R. Weller, and A. L. Ruff-Roberts.1992. Ribosomal RNA analysis in microorganisms as they occur innature. Adv. Microb. Ecol. 12:219-286.
34. Weisburg, W. G., S. J. Giovannoni, and C. R. Woese. 1989. TheDeinococcus-Themnus phylum and the effect of rRNA compositionon phylogenetic tree construction. Syst. AppI. Microbiol. 11:128-134.
34a.Wickham, G. S., and N. R. Pace. Unpublished data.35. Woese, C. R. 1987. Bacterial evolution. Microbiol. Rev. 51:221-
271.36. Woese, C. R., L. Achenbach, P. Rouviere, and L. Mandelco. 1991.
Archaeal phylogeny: reexamination of the phylogenetic position ofArchaeoglobus fulgidus in light of certain composition-inducedartifacts. Syst. Appl. Microbiol. 14:364-371.
37. Woese, C. R., R. R. Gutell, R. Gupta, and H. F. Noller. 1983.Detailed analysis of the higher-order structure of 16S-like ribo-somal ribonucleic acids. Microbiol. Rev. 47:621-669.
38. Woese, C. R., 0. Kandler, and M. L. Wheelis. 1990. Towards anatural system of organisms: proposal for the domains Archaea,Bacteria, and Eucarya. Proc. NatI. Acad. Sci. USA 87:4576-4579.
VOL. 60, 1994
on July 15, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from





























