Embed Size (px)
Citation preview

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2727272727

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2626262626
ContentsPage
1. Introduction : 1
2. Overview of the current regulatory framework for recombinant pharma sector : 3
3. Guidelines for safety in biotechnology : 6
4. Analysis of the current regulatory framework : 7
5. Recommendations : 95.1 Recommended Procedure for Regulation of Recombinant Pharma Products Derived
from Living Modified Organisms (LMOs) : 95.2 The step-wise regulatory procedures /protocols for five scenarios : 145.3 Functions of Regulatory Committees/ Competent Authorities under Rules 1989 of EPA
and Drugs & Cosmetics Act, 1940 and Rules, 1945 : 205.4 The time lines for decisions : 215.5 Documents to be submitted by the applicant to the regulatory authorities for
obtaining clearances : 215.6 Recommendations on other Linked issues : 215.7 Standing Technical Advisory Committee on Biotechnology Regulation : 23
6 Proposed independent institutional mechanism-National Biotechnology RegulatoryAuthority/Commission. : 24
7 AnnexuresI. Minutes of the Consultative Meeting : 25II. Constitution of the Task Force on Recombinant Pharma Sector : 29III. Minutes of the First Meeting of the Task Force : 35IV. Summary of views/recommendations received from Experts : 42V. Minutes of the Second Meeting of the Task Force : 53
VI. Constitution of Drafting Committee : 60VII. Minutes of the Third Meeting of the Task Force : 62VIII. Minutes of the Fourth Meeting of the Task Force : 67IX. Minutes of the Fifth Meeting of the Task Force : 71X. Proforma for submission of information to the IBSC : 75XI. Proforma for submission of information to the RCGM : 79XII. Proforma for submission of information to the GEAC : 82XIII. Proposed structure of National Biotechnology Regulatory Authority/Commission : 88

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
11111
1.1 In view of the rapid advancements in the area of Biotechnology andits potential benefits in the healthcare sector, various stakeholdershave voiced the need for a transparent and effective regulatory systemfor recombinant pharma sector at various fora.
1.2 It was in this context that a Consultative Meeting was convenedunder the Chairmanship of Secretary, Ministry of Environment andForests (MoEF) on 22nd March 2004, involving pharma industry,industry associations and officials of other concerned administrativeministries. The Minutes of the meeting are placed at Annexure ‘I’. Inaccordance with the decision taken in the Consultative meeting theMoEF constituted a Task Force on Recombinant Pharma Sector underDr. R.A. Mashelkar, DG, CSIR and Secretary Ministry of Science &Technology vide OM No 12/7/2004-CS dated 20.4.2004 (Annexure‘II’) with the following terms of reference:i) To recommend a transparent and streamlined regulatory
mechanism and process for the use of living Modified Organisms(LMOs) in the pharmaceutical industry during the various stagesof R & D, testing, manufacture and use.
ii) To recommend regulatory process and mechanisms for import ofLMOs in the pharma sector.
iii) In developing the regulatory mechanism/process, the Task Forcemay take into account the generic norms, for re-engineering ofregulatory process set forth by the Govindarajan Committee.
1.3 The first meeting of the Task Force was held on 12th May 2004 whereinSecretary (E&F) made a presentation on the Current RegulatoryFramework and addressed some of the perceived problems and stepsinitiated by the MoEF to address issues related to delay andtransparency. The members of the Task Force also expressed theirviews on the major issues that need to be addressed by the Task Forcewhile recommending measures to streamline the regulatory process.Views expressed by the members are summarized in the Minutes ofthe meeting at Annexure ‘III’. Consequent to the Task Force meetingthe Chairman invited views of various experts on the proposedreforms. These have been summarized at Annexure ‘IV’.
1.4 The second meeting of the Task Force was held on 15th June 2004wherein Secretary DBT presented a Framework for Modification ofExisting Regulatory System and the Institutional Structure for the
IntroductionIntroductionIntroductionIntroductionIntroduction
1

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
22222
proposed National Biotechnology Regulatory Authority/Commission.The minutes of the second meeting of the Task Force along with theviews of the members of the Task Force in the context of therecommendation made by Secretary DBT are placed at Annexure ‘V’.During the meeting it was decided to constitute a Drafting Committeeto prepare a ‘Framework for Streamlining the Current RegulatoryProcess’ reflecting the consensus achieved by the group for furtherconsideration of the Task Force. In accordance the Ministry ofEnvironment & Forests has constituted a Drafting Committee underthe Chairmanship of Secretary DBT vide OM No 12/7/2004-CS dated22nd June 2004 (Annexure ‘VI’), comprising:
i. Dr. M.K. Bhan Secretary DBT Chairman
ii. Mr. D.D. Verma, Joint Secretary, MOEF Member
iii. Dr. Amit Ghosh, Director, IMTECH Member
iv. Dr. R. Warrier, Addl. Director, MOEF Member
1.5 The terms of reference of this Committee are as follows:
a) Review the current regulatory process under the “Rules forManufacture, Use, Import, Export and Storage of HazardousMicroorganisms/Genetically Engineered Organisms or Cells,1989” notified under the Environment (Protection) Act, 1986in terms of regulatory objective, decision rule and informationrequirement of each regulatory body during the various stages ofR&D, testing, manufacture, import and use.
b) Propose amendments in the regulatory mechanism and processfor the use of recombinant organisms in the pharmaceuticalindustry during the various stages of R&D, testing, manufacture,import and use with a view to introduce transparency andeliminate duplicity of approvals.
c) Revised formats for submission of information to variousregulatory agencies.
1.6 The Chairman of the Drafting Committee and Secretary, DBT co-opted two more expert members as follows:
1. Dr. K.K. Tripathi, Advisor DBT
2. Dr. T.V. Ramaniah, Director DBT
1.7 The draft report prepared by the Drafting Committee andrecommendations made therein was deliberated extensively in themeetings held on 3rd September 2004 and 3rd December 2004 and13th June 2005 respectively. The minutes of the meetings are placed atAnnexure VII, VIII and IX.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
33333
OverOverOverOverOverview of the Curview of the Curview of the Curview of the Curview of the Current Rrent Rrent Rrent Rrent RegulatoregulatoregulatoregulatoregulatoryyyyyFFFFFramework for Rramework for Rramework for Rramework for Rramework for Recombinant Pharecombinant Pharecombinant Pharecombinant Pharecombinant Pharma Sectorma Sectorma Sectorma Sectorma Sector
2.1 The Ministry of Environment & Forests has notified the Rules for theManufacture, Use, Import, Export and Storage of HazardousMicroorganisms/Genetically Engineered Organisms or Cells 1989 (Rules1989) under the Environment (Protection) Act, 1986. The recombinantpharma products are regulated under these Rules from the research andproduct development stage up to its release into the environment.
2.2 ‘Rules 1989’ also define the regulatory authorities responsible foraccording various approvals. Presently there are three regulatoryauthorities for the recombinant DNA (rDNA) research, productdevelopment and commercialization in the recombinant pharmasector. Brief description and responsibilities of these regulatoryauthorities are as follows:
(i) Institutional Biosafety Committee (IBSC)It is mandatory that the institutions intending to carry outresearch activities involving genetic manipulation of organismsconstitute the IBSC. The Rules mandate the inclusion of nomineeof DBT in the constitution of the IBSC. The IBSC is the nodalpoint for interaction within the institution for implementationof the rDNA Biosafety Guidelines. The activities of IBSC includetraining of personnel on biosafety and instituting healthmonitoring programme for laboratory personnel.
(ii) Review Committee on Genetic Manipulation (RCGM)The RCGM is serviced by the Department of Biotechnology. Itsmandate is to monitor the safety related aspects in respect ofongoing research projects and activities involving geneticallyengineered organisms/hazardous microorganisms. The RCGMis also responsible to bring out manuals or guidelines specifyingthe procedure for regulatory process with respect to activitiesinvolving genetically modified organisms in research, utilizationand their application including in industry with a view to ensureenvironmental safety. All on-going projects involving rDNAtechnology and controlled field experiments are reviewed byRCGM to ensure that adequate precautions and containmentconditions are followed as per the guidelines. It is also empoweredunder Rules 1989 to lay down procedures restricting orprohibiting production, sale, import and use of geneticallyengineered organisms or cells as per the Schedule of Rules 1989.
2

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
44444
(iii) Genetic Engineering Approval Committee (GEAC):
The Genetic Engineering Approval Committee (GEAC)serviced by the Ministry of Environment and Forests isresponsible for approval of activities involving large-scale useof genetically modified/hazardous microorganisms and productsthereof in research and industrial production from theenvironment angle. The GEAC is also responsible for approvalof proposals relating to release of genetically modified/hazardousmicroorganisms and products into the environment includingexperimental field trials.
2.3 The State Biotechnology Coordination Committee (SBCC) andDistrict Level Committee (DLC) under Rules 1989 have theresponsibility for post-release monitoring of genetically modifiedorganisms/hazardous microorganisms and products thereof.
2.4 The Drugs and Cosmetics Act 1940 and the Rules 1945 also regulatethe recombinant pharma products, as amended from time to time.The Authority to regulate the recombinant pharma products is theDrugs Controller General of India (DCGI) and the State DrugsController. The Recombinant Drugs Advisory Committee (RDAC)constituted by Ministry of Health and Family Welfare supports the DCGI.
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
55555
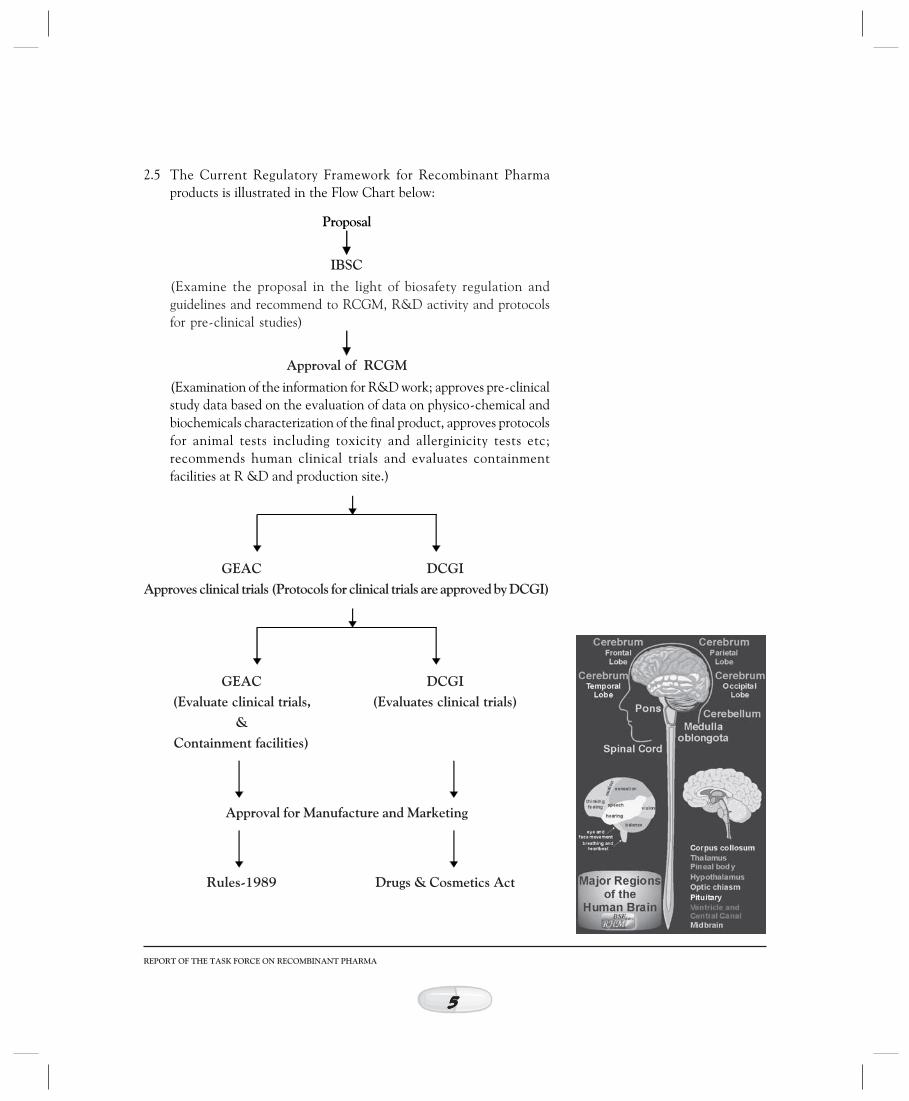
2.5 The Current Regulatory Framework for Recombinant Pharmaproducts is illustrated in the Flow Chart below:
Proposal
IBSC
(Examine the proposal in the light of biosafety regulation andguidelines and recommend to RCGM, R&D activity and protocolsfor pre-clinical studies)
Approval of RCGM
(Examination of the information for R&D work; approves pre-clinicalstudy data based on the evaluation of data on physico-chemical andbiochemicals characterization of the final product, approves protocolsfor animal tests including toxicity and allerginicity tests etc;recommends human clinical trials and evaluates containmentfacilities at R &D and production site.)
GEAC DCGIApproves clinical trials (Protocols for clinical trials are approved by DCGI)
GEAC DCGI(Evaluate clinical trials, (Evaluates clinical trials)
&Containment facilities)
Approval for Manufacture and Marketing
Rules-1989 Drugs & Cosmetics Act

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
66666
3.1 The Department of Biotechnology has evolved various guidelines,which have been adopted by RCGM and GEAC for the regulation ofrecombinant pharma products in the country. These guidelinesinclude:
a) Recombinant DNA Safety Guidelines, 1990: These guidelinesinclude procedure for large-scale production and deliberaterelease of GMOs and products thereof into the environmentand the shipment and import of GMOs for laboratory research.The guidelines may be viewed at http://www.envfor.nic.in/divisions/csurv/geac/biosafety.html
b) Guidelines for generating pre-clinical and clinical data for r-DNA based vaccines, diagnostics and other biologicals, 1999.The guidelines may be viewed at http://www.envfor.nic.in/divisions/csurv/geac/biosafety.html
3.2 The current practice of according approval for phase III clinical trials,both by the GEAC and the DCGI is based on the procedure outlinedin the 1999 guidelines.
Guidelines for Safety inGuidelines for Safety inGuidelines for Safety inGuidelines for Safety inGuidelines for Safety inBiotechnologyBiotechnologyBiotechnologyBiotechnologyBiotechnology
3

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
77777
4
4.1 An analysis of the current regulatory framework was carried out inthe context of regulatory objectives of various Authorities, views ofthe members of the Task Force, other experts, industry associationsas well as the developments in the international scenario. Theproblems identified by the Task Force in implementation ofrecombinant pharma projects/research activities in the country are:
Multiple approval system has sometime led to cumbersome andlengthy approval process.
Regulatory objectives of the different agencies in the regulatorychain are sometimes overlapping leading to duplication in theapproval process.
Information sought tends to be sometimes duplicated and inpiecemeal.
Opportunity to an applicant to present proposal to the regulatoris not a common practice.
Frequencies of meetings of regulatory authorities are irregularand inadequate.
Documents/information required by the regulatory authoritiesfor review of the proposal is not clearly articulated.
The sequential approval procedure under Rules 1989 of EPA andDrugs & Cosmetics Act and Rules are ambiguous.
Environmental impacts of the GMOs/LMOs per se and theproducts thereof per se have not been clearly identified in pharmasector.
Regulatory Committees lack expertise in inter-disciplinary areasand professional support staff.
The risk assessment and management of LMOs /GMOs, biosafetyand risk categories as per WHO or other international standards/guidelines are not fully adhered to.
Imports of GMOs/LMOs as well as the non-GM microorganismsfor R&D work has been made cumbersome with the new PlantQuarantine Order, issued by the Ministry of Agriculture,hampering the R&D, quality control and manufacturingactivities of the pharma sector.
Analysis of the CurAnalysis of the CurAnalysis of the CurAnalysis of the CurAnalysis of the CurrentrentrentrentrentRRRRRegulatoregulatoregulatoregulatoregulatory Fy Fy Fy Fy Frameworkrameworkrameworkrameworkramework

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
88888
There is no effective mechanism for evaluation of Post Marketingsurveillance data of recombinant pharma products and feedback onproduct efficacy and environmental safety.
4.2 Several views were expressed by stakeholders in favour of anindependent National Biotechnology Regulatory Authority/Commission for providing a professionally managed single windowmechanism for giving various clearances including biosafety issues.It was recognized that evolving such an institutional mechanismwould involve a lengthy evolutionary process requiring extensiveconsultations among various stakeholders. However, the feasibilityof setting up such an institutional mechanism should be explored.The Task Force therefore, addressed the issue of streamlining theexisting regulatory procedures for immediate redressal. The Task Forceenumerated the following points for streamlining of the existingregulatory system.
a) to clearly define the roles of each regulatory Committee or Competentauthority and enumerate decision making criteria under Rules 1989of EPA to remove ambiguity in the existing regulatory process
b) to clearly enumerate the stepwise procedure involved in the biosafetyregulations of LMOs and the products thereof for indigenousdevelopment as well as imported products.
d) to evolve a documentation system to be submitted by the applicantto the regulatory Committees/ Competent authorities for obtainingclearances.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
99999
Analyzing the above issues and relevance of the suggestions made by thestakeholders regarding streamlining of the current regulatory frameworkand procedures outlined in the DBT guidelines for implementation ofRules 1989 of EPA, the Task Force came to the conclusion that (i) there isno overlapping of regulatory objectives up to the evaluation of pre-clinicalstudy data generated by the applicant on the recombinant pharmaceuticalproducts. (ii) The regulatory process can be expedited if the regulatoryobjectives of GEAC and DCGI are clearly defined. The Task Forcerecommends that the regulatory objective of GEAC should be confined toregulation of proposals which involve the large scale use of LMOs fromenvironmental angle. Evaluation of the product safety, efficacy, clinicaltrials and market authorization is the mandate of the DCGI; (iii) thecurrent regulatory procedure under ‘Rules 1989" of EPA is applicable toall recombinant pharma product(s) irrespective of the risk group of LMOs.Therefore the current regulatory procedure and protocols for productdevelopment, clinical trials and market authorization should be elaboratedto address various scenarios in the development and marketing ofrecombinant pharma products. The specific recommendations of the TaskForce on the regulatory mechanism are stated below:
5.1 Recommended Procedure for Regulation of Recombinant PharmaProducts Derived from Living Modified Organisms (LMOs).
5.1.1 As mentioned earlier, the Task Force recommends that the currentregulatory procedure and protocols for product development andclinical trials should be elaborated to include various scenarios inthe development and release of recombinant pharma products.Taking into consideration the regulatory objective of GEAC, which,is confined to regulation of LMOs, two protocols are recommended(i) products derived from LMOs but the end product is not a LMOand (ii) Product derived from LMO where the end product is aLMO. The product where the end product is a LMO has thepotential for propagating/replicating in the environment andtherefore needs a higher level of regulation as compared to productsderived from LMOs where the end product is not a LMO. Furtherthe magnitude and probability of environmental risk depends onthe extent of use of LMOs within the R&D and production units.In case of imports this risk is not there especially in cases oftherapeutic proteins in finished form. Taking into considerationvarious aspects, the Task Force recommends that the regulatoryprocedure needs to be rationalized for the following five scenarios:
RRRRRecommendationsecommendationsecommendationsecommendationsecommendations
5

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1010101010
a. Indigenous product development, manufacture and marketingof pharmaceutical products derived from LMOs but the endproduct is not a LMO.
b. Indigenous product development, manufacture and marketingof pharmaceutical products where the end product is a LMO
c. Import and marketing of LMOs as Drugs/Pharmaceuticals infinished formulations
d. Import and marketing of LMOs as Drugs/Pharmaceuticals inbulk for making finished formulation
e. Import and marketing of products derived from LMOs as Drugs/Pharmaceuticals in bulk and/or finished formulations wherethe end product is not a LMO.
5.1.2 The Task Force recommends the following procedure for each ofthe scenario:
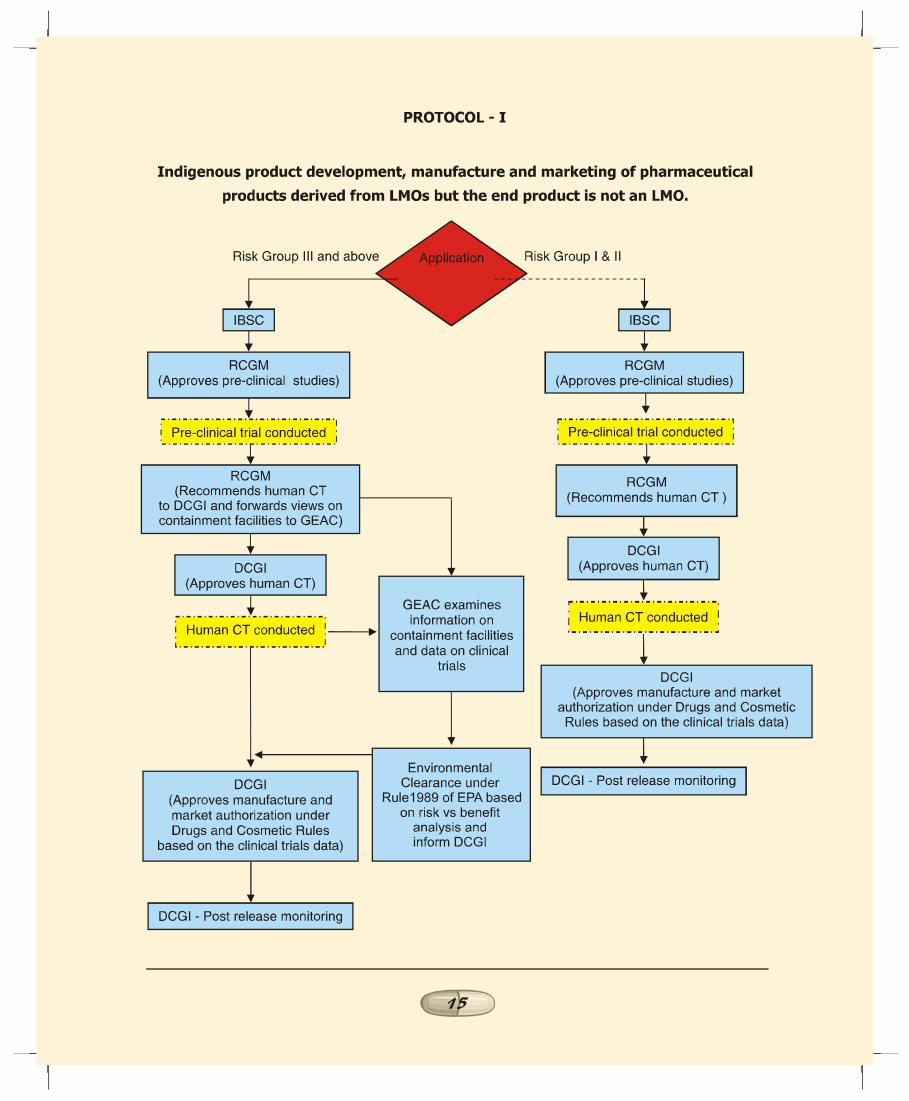
a. Indigenous Product Development, Manufacture andMarketing of Pharmaceutical Products Derived from LMOsbut the End Product is not a LMO (Protocol I):
1. As mentioned earlier there is no overlapping of regulatoryobjectives, up to the evaluation of pre-clinical study datagenerated by the applicant on the recombinantpharmaceutical products derived from LMOs. Accordingly,RCGM to continue the practice of examining the protocolsand pre-clinical study data submitted by the applicant andforward their recommendation to DCGI for taking a viewon the human clinical trials.
2. Since the end products do not contain any LMO, the issueof environmental release during the clinical trials does notarise. Therefore no approval from GEAC is required forconducting human clinical trials. As per the Drugs andCosmetic Act and Rules, it is the mandate of DCGI toexamine the pre-clinical and human clinical trial data forassessing the efficacy and safety of the product. ThereforeDCGI to accord marketing approval for such products.
3. As per the current regulatory process, approval of GEAC isrequired for activities beyond 20 litres fermentationcapacity even for research activities. The Task Force is ofthe view that the nature of organisms and extent of riskinvolved should be the criteria for assessing the safety ofthe product. The Task Force endorses the suggestion thatthis category where the end product is not a LMO may befurther divided into two parts namely (i) organisms fallingunder Risk Group I & II and (ii) Risk Group III and above.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1111111111
4. Approval of GEAC is not required for products producedwith the use of organisms falling under Risk Group I & II atany stage of product development, trials, manufacture andmarketing. In this case, RCGM, based on the report ofIBSC to evaluate the adequacy of the containment facilitiesinstalled by the applicant for handling LMOs during R&Dstage and in the production units. The GEAC to forwardtheir views on the safety of the product from environmentalangle to the DCGI.
5. However, for proposals involving use of LMOs of RiskGroup III & above, approval of GEAC would be necessaryprior to manufacture and marketing of the product. Theinformation in this regard to be submitted to GEAC wouldinclude process as well as product specifications, details ofcontainment facilities and human clinical trials data. Whileevaluation of the environmental risk, GEAC to take intoconsideration, the recommendation of RCGM on thecontainment facility, purity profile and pre-clinical studydata as well as results of human clinical trials. GEAC toinform DCGI about it’s decision.
6. DCGI in consultation with RDAC to approve the protocolsand conduct of human clinical trials. The clinical trial datato be submitted to the DCGI by the applicant. DCGI toexamine the human clinical trials data and to take intoconsideration the recommendation of GEAC (whereapplicable) prior to according market authorization forcommercial release of the product as per the Drugs andCosmetics Act & Rules. DCGI to endorse its decision tothe GEAC for record.
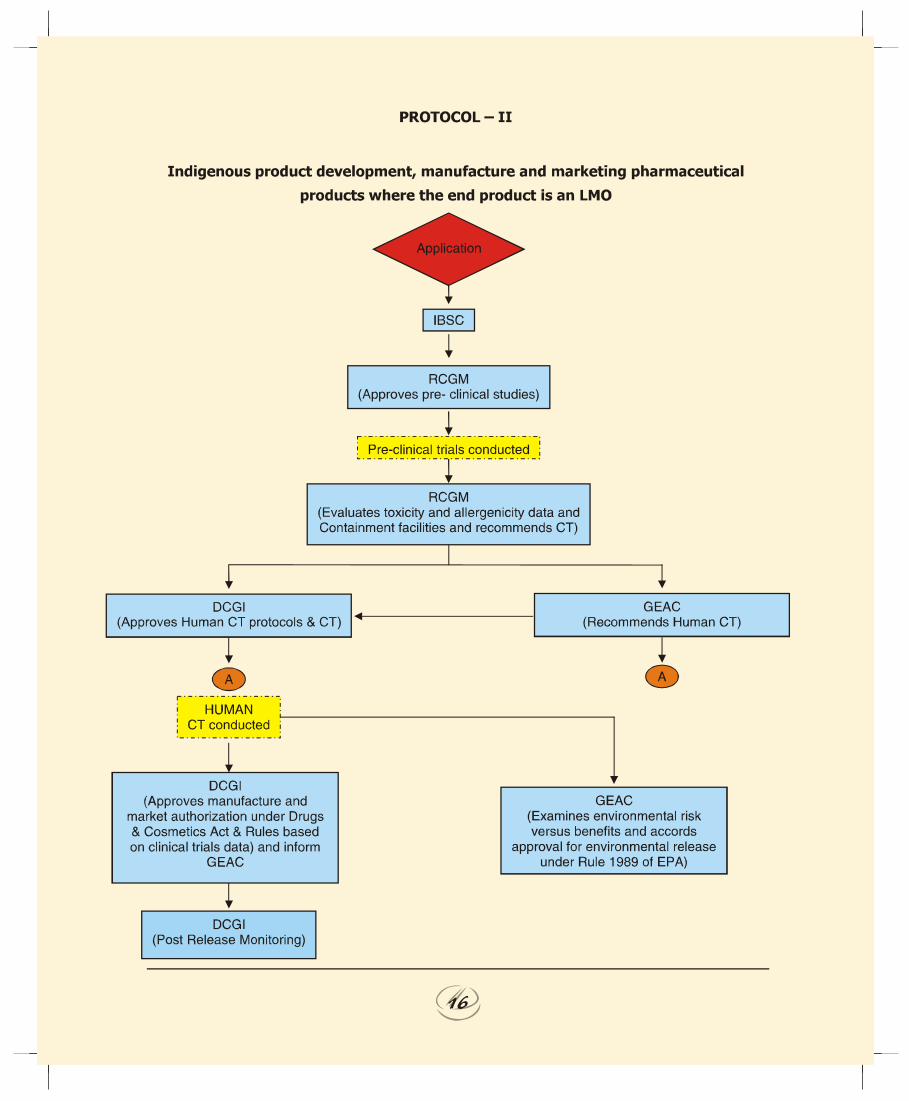
b. Indigenous Product Development, Manufacture andMarketing of Pharmaceutical Products where the EndProduct is a LMO ( Protocol II).1. RCGM to continue the practice of examining the protocols
and pre-clinical data submitted by the applicant(s) andforward their recommendation to DCGI and GEAC fortaking a view on the human clinical trials. RCGM to alsoforward their views to the GEAC on the adequacy ofcontainment facilities installed by the applicant forhandling of LMOs during the R&D and production stage.
2. Since the end product is a LMO, the probability of risk dueto accidental release is higher and therefore GEAC will beresponsible to evaluate the environmental impact causedby handling and large-scale use/release of LMOs.Accordingly GEAC to approve phase III human clinical

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1212121212
trials. The GEAC to forward their views on the safety ofthe product for conduct of human clinical trials fromenvironmental angle to the DCGI.
3. The GEAC to also ensure that the containment facilityand infrastructure available with the applicant is adequateto handle and safe use of LMOs at all stages of productdevelopment and manufacture. The GEAC to approveenvironmental release based on the environmental risksvs. benefits analysis which takes into consideration therecommendation of RCGM and results of the clinical trials.
4. DCGI to examine the data on the toxicity, allergenicityand QC tests and recommendation of RCGM through theRDAC and approve the protocols and conduct of humanclinical trials under Drugs & Cosmetics Act and Rules.The DCGI also to take into consideration the views ofGEAC on the safety of the product for conduct of humanclinical trials from environmental angle. DCGI to beresponsible for evaluation of product efficacy and safetyprior to market authorization.
5. Prior to granting approval for commercial release of LMOsas drugs, the DCGI should ensure that necessary clearanceby GEAC under Rules 1989 of EPA have been obtained bythe applicant. The information on approval for marketauthorization should be intimated to the GEAC. DCGI tobe responsible for post market surveillance.
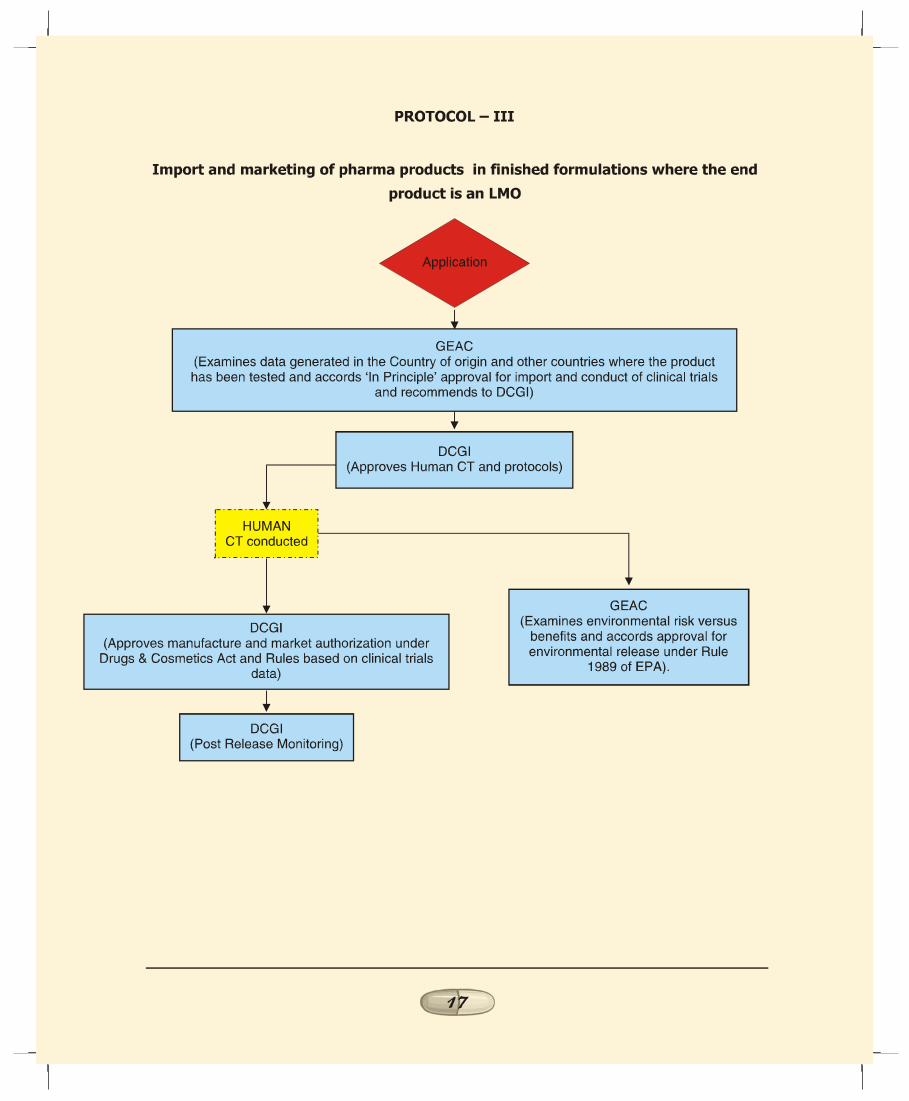
c. Import and marketing of recombinant pharma productsin finished formulation where the end product is a LMO(Protocol III):1. In case of import of LMOs per se as products, in the absence
of any product development within the country theprobability of environmental risk would be less than thatof indigenously manufactured products involving the useof LMOs.
2. Since this scenario pertains to import of LMOs, the onlyactivity envisaged within the country prior to issue ofmarket authorization is the conduct of human clinical trialsand therefore the procedure outlined in Protocol II fromclinical trial stage would be applicable in this case.
3. Accordingly, GEAC to evaluate the environmental impactcaused by handling and large-scale use of LMOs, based onthe data available from the country of origin and othercountries where the product has been tested. Based on theassessment, GEAC to accord ‘in principle’ approval forimport and recommend to DCGI the safety of the product

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1313131313
from environmental angle for conduct of human clinicaltrials. Based on the results of the clinical trials GEAC toaccord approval for marketing of the product in the Countryfrom environmental angle.
4. DCGI to examine the data on the toxicity, allergenicityand QC tests and recommendation of GEAC through theRDAC and approve the protocols and conduct of humanclinical trials under Drugs & Cosmetics Act and Rules.DCGI to be responsible for evaluation of product efficacyand safety prior to market authorization as well as postmarket surveillance.
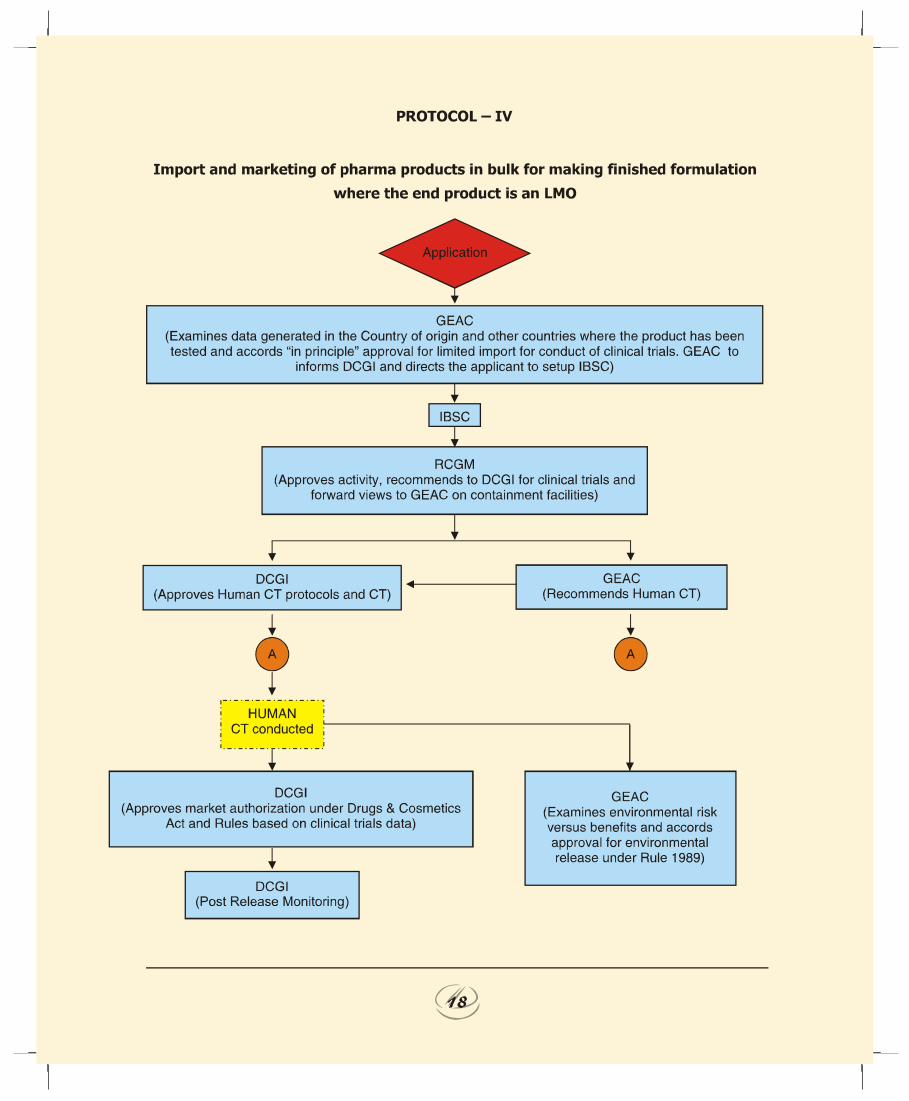
d. Import and marketing of recombinant pharma products inbulk for making finished formulation where the end productis a LMO (Protocol IV):1. In case of import of LMOs per se as bulk, the probability of
environmental risk would be higher as compared to importof finished formulation in view of the fact that the activitywould involve partial manufacturing beside storage of LMOsin bulk.
2. GEAC to evaluate the environmental impact caused byhandling and large-scale use of LMOs, based on the dataavailable from the country of origin and other countrieswhere the product has been tested and accord approval forimport. Based on the assessment GEAC to accord ‘inprinciple’ approval for import and also to recommend toDCGI the safety of the product from environmental anglefor conduct of phase-III human clinical trials.
3. Subsequent to approval of GEAC for import, the proposalinvolves setting up of processing facilities for makingfinished formulations. Therefore, constitution of the IBSCis mandatory.
4. This case being similar to Protocol II, the Task Forcerecommends that the procedure outlined for obtaining theapproval of RGCM, GEAC and DCGI for setting up facilitiesfor making finished formulation, conduct of human clinicaltrials, environmental release and market authorizationprior to marketing the product within the country wouldbe applicable in this case.
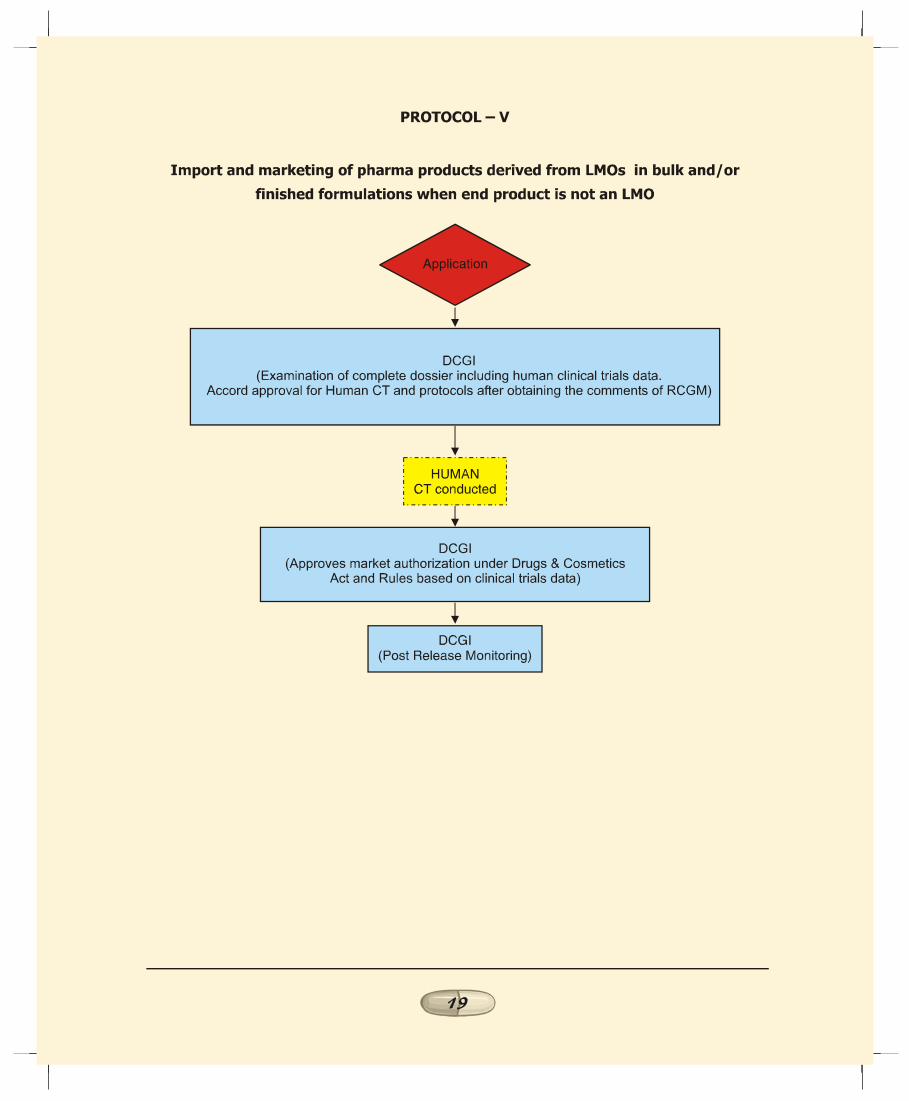
e. Import and marketing of recombinant pharma productsin bulk/finished form where the end product is not a LMO(Protocol V):1. This case involves import of products derived from LMOs
where the end product is not a LMO. There would no

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1414141414
manufacturing facilities in case of finished formulation andin case of bulk imports, only partial processing facilities.This scenario in terms of environmental risk falls underthe least risk category since there is no use of LMOs withinthe country. Therefore approval under Rules 1989 of EPAis not applicable in this case.
2. In such cases, DCGI to evaluate the data generated in othercountries where the product has been tested and approveprotocols for human clinical trials if any. The Task Forcerecommends that the views of RCGM may be obtained onthe process, purity profile, pre-clinical and clinical trialsdata and product specifications prior to approving humanclinical trials or marketing of the product. Alternatively,DCGI may empower RCGM to review the proposal in thefirst instance prior to referring the matter to DCGI by theapplicant.
5.1.3 Common Recommendations applicable for all five scenarios:1. Approval for import of recombinant organisms for the purpose
of research and development is within the purview of RCGM.2. The regulatory agency to authorize human clinical trials would
be the DCGI in all cases. However, approval of GEAC isnecessary prior to conduct of Phase-III clinical trials whereapplicable as outlined in Protocol II, III and IV.
3. In situations where approval of both DCGI & GEAC ismandatory, clearances from these regulatory agencies need notbe interlinked and both agencies can process their caseconcurrently.
5.2 The step-wise regulatory procedures for each of the fivescenarios discussed above is illustrated in the flow charts asProtocols I to V.
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1515151515
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1616161616
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1717171717
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1818181818
REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
1919191919

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2020202020
5.3 Functions of the Regulatory Committees/ Competent Authoritiesunder Rules 1989 of EPA and Drugs & Cosmetics Act, 1940and Rules 1945.
a. IBSC to examine proposals involving r-DNA work; to ensureadherence of Recombinant DNA Safety Guidelines of 1990including preparation of emergency plans; examine protocolsfor pre-clinical studies and inspection of containment facilitiesat R&D and production units. Throughout the productdevelopment, preclinical and clinical trials and manufacturing,the IBSC would act as a nodal point for interaction withstatutory bodies.
As per the Recombinant DNA Safety guidelines of 1990, IBSCcan approve experiments utilizing the organisms and geneticelements from Risk Group-I and II under intimation to RCGM.This practice would continue. For experimental use of organismsfalling in Risk Group III & above, IBSC would make itsrecommendation to RCGM. IBSC would also examine theprotocols for toxicity/allergenicity studies as per national andinternational guidelines and make their recommendations toRCGM. IBSC to recommend to RCGM import/ exchange ofGMOs/LMOs, vectors, gene constructs, plasmids, etc., forresearch purposes.
b. RCGM to approve experiments utilizing the organisms andgenetic elements of all Risk Group organisms. This is as per theRecombinant DNA Safety guidelines of 1990. RCGM toapprove protocols for preclinical studies for all products. RCGMto submit its recommendation on the preclinical studies/datadirectly to the DCGI. For the products from Risk group III &above organisms, RCGM to examine the information oncontainment facilities at the R&D and production sites as wellas the results of the pre-clinical studies and submit theirrecommendations both to the GEAC and DCGI. Approval forimport of recombinant organisms for the purpose of research isthe mandate of RCGM.
c. GEAC to confine its regulatory role in terms of productapproval if the proposals involve use of LMOs falling in RiskGroup III & above as well as use of LMOs in open environmentspecifically when the end products are living modified organisms(LMOs) per se. In this context GEAC to approve activitiesinvolving large scale use of LMOs in industrial production andapplication; authorize large-scale production and release ofLMOs into the environment; adopt procedures restricting orprohibiting production, sale, import and use of LMOs for

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2121212121
applications under EPA; authorize agencies or persons to havepowers to initiate punitive actions under EPA against defaulters.
d. DCGI to examine preclinical studies data on animal toxicity& allergenicity and QC data and the protocol for human clinicaltrials, recommend conduct of human clinical trial and approveproduction of trial batches. Prior to market authorization DCGIto examine human clinical trial report, test reports from alaboratory designated by DCGI. The responsibility of post marketsurveillance is the mandate of DCGI. For products and processesinvolving Risk Group III & above organisms, DCGI will makeavailable its decision on Phase-III clinical trials to the GEAC.
5.4 The time lines for decisions:
5.4.1 A consensus on the following timelines emerged for variousapprovals by the regulatory Committees / Competent authorities.
· RCGM approval for pre-clinical animal studies: 45 days· RDAC approval for Human Clinical Trials protocol: 45 days· RDAC (DCGI) examination of clinical trial data
and response: 90 days· Simultaneous DCGI & GEAC decisions: 45 days
5.5 Documentation to be submitted by the applicant to the regulatoryauthorities for obtaining clearances.
5.5.1 The Task Force observed that formats for preparing documentsto be submitted by the applicant to the IBSC, RCGM and GEAChave been evolved and is adequate for taking a view on theproposals. The proforma under reference are attached asAnnexure X, XI and XII.
5.5.2 The Task Force recommends that a format for submission of data tothe DCGI by the applicant be evolved by the DCGI at the earliest.
5.6 Recommendations on other Linked issues:
1. The definition of LMOs will include only those organismsmodified by r-DNA techniques through human interventions.MoEF in this regard would issue necessary amendments to Rules1989 of EPA.
2. Since the responsibility of according market authorization forrecombinant drugs is being entrusted to DCGI, there is an urgentneed for strengthening the Committees under the Drugs &Cosmetics Act and Rules.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2222222222
3. The expertise in the various regulatory agencies under Rules1989 of EPA should be further strengthened.
4. There is a need for creation of an independent inspection facilityto audit the manufacturing and containment facilities set upby the applicants involved in the production of recombinantdrugs. This would also ensure acceptability of the Indian r-DNApharmaceutical products in the global market. Since there isno single agency with adequate field level support system tocarry out an independent inspection, the Task Forcerecommends that the Government may set up a separate agencyfor this purpose.
5. The products emanating from mono-clonals derived fromrDNA technology in the form of therapeutic proteins/drugswould attract the provisions of Rule 1989 of EPA, and can betreated under Protocol I as Risk Category I & II.
6. Enzymes /industrial products from GMOs would attract theprovisions of Rule 1989 of EPA. In such cases, RCGM may beauthorized to approve such proposals under intimation toGEAC.
7. If there is a change in the host organism or expression construct,fresh permission will be required to be sought from RCGM forthe change by providing adequate data on bio-equivalence. Ifthe data is found to be inadequate then RCGM may prescribelimited pre-clinical and/or clinical studies to be conducted toestablish bio-equivalence. This would also be applicable tofinished imported products intended for marketing.
8. No imported recombinant pharma product should be allowedto be introduced in the Indian market without adequateevaluation of clinical trial data or clinical evaluation in theCountry. The Task Force recommends that the efficacy andsafety of the imported product should be evaluated for its efficacyon the Indian population before issue of market authorization.
9. For import of GMO / LMO for research/contract manufacturingor similar service, where the product (which is not an LMO) isto be exported out of India, a procedure should be laid down sothat the companies can explore opportunities for this businesswhile the safety aspect is also adequately addressed. A suggestedprocedure is: IBSC to examine proposal and recommend toRCGM; RCGM to approve if within Risk Group I and II. Iforganism is of Risk Group III or above, GEAC permission willbe required. DCG(I) need not play any role.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2323232323
10. On the issue of seeking approvals of PPA/DCGI/GEAC underRules 1989 of EPA and PQO by Customs Authorities on theimports of microorganisms, GMOs/ LMOs for R&D purpose itis suggested that the earlier practice of permitting the importwith the approval of RCGM should continue and PPA/DCGIto issue instructions to Custom Authorities to clear theconsignment based on RCGM approval.
11. Regarding the constraints faced by the industry for import ofnon-GMOs, PPA may issue instruction to Customs Authoritiesto clear the consignment based on the declaration of theimporter/exporter on certification of the nature of the non-GM organisms etc.
5.7 Standing Technical Advisory Committee on BiotechnologyRegulation
5.7.1 Since several modifications have been made in the existingregulatory mechanism for recombinant pharma; during itsimplementation, several anomalies may become apparent. To addressthese issues, the Task Force recommends, constitution of a StandingTechnical Advisory Committee on Biotechnology Regulation toredress and look into various regulatory aspects and make issue-based recommendations on case-by-case basis. Prior to any deviationfrom the proposed regulatory mechanism, which when comes invogue, the views of this Committee should be obtained in the firstinstance.
5.7.2 The terms of reference of the proposed Standing Technical AdvisoryCommittee should be to address the issues emanating from theoverlapping/ conflicting Rules in various Acts applicable that areregulating biotechnology activities at R&D, import, export, trials,release, etc. and also to frame guidelines from time to time tofacilitate the growth of biotechnology in the country. Since theissues involved are highly technical and complex, the Task Forcerecommends that the Standing Technical Advisory Committeeshould comprise of an expert body instead of an inter-ministerialbody. An eminent Scientist should head the Standing TechnicalAdvisory Committee and the members should include ChairmanGEAC, Chairman RCGM, Member-Secretary GEAC, Member-Secretary RCGM, DCGI and Experts on Immunobiologicals,Biogenerics, Plant Breeding, Molecular Biology, EnvironmentalSciences and other relevant areas.
5.7.3 The Committee can be administratively supported by any of theconcerned agencies.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2424242424
6.1 The group believes that the creation of a professionally managedsingle authority would send a strong signal to the internationalcommunity and promote trade and investment as well as ensuretimely and effective regulation. While it is desirable to establish anindependent professionally competent authority, if possible, forproviding single window approvals, the Group also recognized thefact unless the existing relevant statutory requirements under EPA/Seeds Act/ DCGI are harmonized; setting up a NBRA may lead to“one more window clearance instead of a single window clearance”.It is therefore recommended that such harmonization is an essentialprerequisite for establishing the national biotechnology regulatoryauthority.
6.2 On the issue of the model for NBRA, the Group recommends thatone of the models for National Biotechnology Regulatory Authority/Commission (NBRA), similar to the FDA system was proposedby Secretary DBT in the second meeting of the Task Force heldon 15th June 2004 may be considered by the Government. Theproposed model recommends that the NBRA would comprise offour wings namely: a) Agricultural products / Transgenic Crops b)Pharmaceutical/ Drugs and Industrial Products c) Transgenic Foods/Feed and d) Transgenic Animals/ aquaculture. Professionals who havebeen well trained in regulatory affairs would manage the four wingsof the authority. This will facilitate more interactive regulatoryprocess. A Vice Chairman would head the four wings of the Secretariat.The recommendation of the Secretariat would be forwarded to ApexCommittee with Statutory Powers. The members of the ApexCommittee would comprise of representatives from all stakeholdersMinistries/ Departments. The Apex Committee would report to theChairman. The proposed model for NBRA is given at Annexure- XII.
6.3 Alternate models of how a National Biotechnology RegulatoryAuthority can be created also needs to be examined.
6.4 In view of the complexities, the Task Force recommends that aninter-ministerial group be established to examine the model proposedby Secretary DBT among various others administrative Departments/Ministries, for functioning of the proposed authority and makespecific proposals with respect to the implementation including thebudgetary requirements.
Proposed Independent InstitutionalProposed Independent InstitutionalProposed Independent InstitutionalProposed Independent InstitutionalProposed Independent InstitutionalMechanism-National BiotechnologyMechanism-National BiotechnologyMechanism-National BiotechnologyMechanism-National BiotechnologyMechanism-National BiotechnologyRRRRRegulatoregulatoregulatoregulatoregulatory Ay Ay Ay Ay Authority/Commissionuthority/Commissionuthority/Commissionuthority/Commissionuthority/Commission
6

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2525252525
PrefaceIndian biotechnology industry is on the growth path. Its turnover has already crossed a billion dollarmark, but what is more heartening is its growth rate of around 40%. Biopharmaceuticals will be a majorgrowth sector in India, where the Indian competitive advantage is increasingly evident. An appropriatepolicy and regulatory framework can be beneficial to the biotech industry as well as the Indian societythat directly benefits from the biopharmaceutical therapeutics.
We need to ensure a high level of safety and efficacy of the biopharmaceutical products. This impliesthat we need to have rigorous drug quality control systems and enforcement mechanisms in place. Themultiple regulatory systems in India have been a cause of concern for some time for the industry. On theother hand, public concerns about the clinical trials and environmental safety need also to be given aserious attention.
The present Task Force was formed due to a proactive initiative taken by the Ministry of Environmentand Forest to address these issues. The specific mandate of the Task Force was to review the currentframework for recombinant pharma (r-pharma) and make suggestions to streamline it to the best extentpossible. This initiative could not have been more timely.
The review and recommendations of the Task Force are based on a consultative approach involving alarge number of stakeholders spanning diverse interests. We are truly grateful to all of them for theirvaluable inputs.
The Task Force has recommended regulatory procedures (protocols) for the use of Living ModifiedOrganisms (LMO) in the manufacture / import of pharma products. The recommendations made in thisreport should hopefully help in the removal of the overlapping regulatory functions between the variousregulatory agencies.
I was indeed privileged to have been asked to chair a Committee having as its members outstandingprofessionals as well as senior representatives of key Ministries. I particularly appreciate the fact that allthe members rose beyond their fields of specialization as well as narrow territories of their departmentsand took a view that was holistic and truly in national interest.
We are indebted to Hon’ble Shri A. Raja, Union minister for Environment and Forests for his interest andto Dr. Prodipto Ghosh, Secretary, Ministry of Environment and Forests for his guidance and creativeparticipation in all our meetings.
My special thanks are due to Dr. M.K. Bhan, Secretary, Department of Biotechnology for guiding theDrafting Committee throughout the preparation of the report. I congratulate the members of the DraftingCommittee for their excellent contribution.
Finally, my sincere thanks go to Shri D. D. Verma, Joint Secretary, MoEF and Member Secretary of theTask Force and Dr. Ranjini Warrier, Additional Director, MoEF for their commitment, dedication anduntiring efforts.
The Task Force Members are confident that the implementation of the task force would serve the purposeof making Indian r-DNA pharmaceutical product companies globally competitive as well as addressingthe issues of environmental safety concerns of the society.
DR R A MASHELKARAUGUST 2005

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2525252525
Minutes of the Consultative meeting convened in the Ministry of Environment & Forests with representatives from the Industry Associations and other Central Departments on 22nd March 2004 to discuss the regulatory reforms in the recombinant Pharma & Food sectors.
1.0 A meeting was held on 22.3.2004 under the Chairpersonship of Dr. Prodipto Ghosh Secretary (E&F) to discuss the regulatory reforms in the recombinant Pharma & Food sectors. This meeting was attended by Mrs. Veena Chhotray, Additional Secretary & Chairperson GEAC, Shri D.D. Verma, Joint Secretary, Dr. R. Warrier, Additional Director and Member Secretary GEAC, Dr. M. Hota, Joint Director, representatives of DCGI, DBT, M/o Food Processing Industries, FICCI, CII and ABLE and other individual companies. List of the participants is annexed.
2.0 The Secretary (E&F) welcomed the participants and gave a brief background on the purpose of this meeting. Making a reference to the streamlining exercise currently being done in the Agriculture sector for transgenic crops by the Task Force under Prof. M.S. Swaminathan, he emphasized that the purpose of this meeting is to initiate a similar exercise to streamline the regulatory process in the recombinant Pharma and Food sector. He also pointed out that the current regulatory framework for GMOs needs to be harmonized with the commitments under the Cartagena Biosafety Protocol which has come into force on 11th September 2003. He then requested Dr. R. Warrier, Additional Director, MoEF to make a presentation on the objectives of the meeting, analysis of the regulatory framework for transgenic crops carried out by MoEF and the proposed draft recommendations of the Task Force under Prof. Swaminathan vis-à-vis the current regulatory framework. Dr. Warrier also briefed the participants on the requirements under the Biosafety Protocol with specific reference to the Advanced Information Agreement (AIA), Decision procedure and Identification and Documentation required for transboundary movement of LMOs for the purpose of contained use, intentional release and Food/Feed and processing (FFP).
3.0 Before inviting the industry association to present their views, Secretary (E&F) further clarified that the basic concerns of MoEF pertains to Living Modified Organisms (LMOs) which can replicate in the environment during various stages of handling and processing of LMOs for production of recombinant products even though the end product itself may not be a LMO for this purpose, if need be necessary steps for amendment in the 1989 Rules would be taken. He also placed before the participants a “Road Map” for their consideration. He informed the participants that the present meeting is more of a brain storming session to be followed by constitution of a Task Force for suggesting measures to streamline the current regulatory process. The meeting will also decide on the terms of reference of the Task Force and its composition.
4.0 Dr. Kiran Shaw Majumdar, representing the Industry Associations informed that investment in the biotechnology sector is low because it has been categorized as a high risks sector. It was stated that delay in getting approvals from various regulatory agencies was the main reason for this sector to be listed as a high- risk category. Referring to the earlier initiatives for streamlining the regulatory process she informed that a dedicated conference in Agro Biotech sector was held in 2001 where several international regulators and representative of DBT, DCGI and GEAC were all present. She presented briefly the reforms suggested by CII as an outcome of the above conference for consideration of the Ministry. As per the proposed reforms RCGM would approve pre-clinical trials and RDAC under DCGI would approve the Phase-III clinical trials. The applicant would approach GEAC only after Phase-III clinical trials. It was clarified by Dr. Shaw
Annexure-IAnnexure-IAnnexure-IAnnexure-IAnnexure-I

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2626262626
that upto Phase-III clinical trials there would be only pilot scale operation and the scale up of operation would be only at the manufacturing stage. Therefore, approval of GEAC is not required for Phase-III human clinical trials. Secretary (E&F) was of the view that if the end product is not a LMO, GEAC would not like to be concerned with regulation of that product. However, the process involving LMOs needs to be regulated to safeguard unintentional release. The mandate of GEAC is to assess whether the environmental risk is within manageable limit and the accrued benefits arising from the product would be more than the environmental risks. Therefore, at what stage the GEAC approval is required needs to be decided taking into consideration the associated risk factors in case of unintentional release. Secretary (E&F) requested the industry representatives to clarify on how to generate the data to take the above decision.
5.0 The following points were also raised by representatives of other associations.
1. Some of the industry representatives and DBT officials were of the view that IBSC should form part of the regulatory framework and a representative of DBT should be on the IBSC to ensure that adequate caution and safety is ensured while developing a recombinant technology.
2. An inventory of Micro-organism is well documented and classified under risk category I, II and III. Since risk category-I is known not to have any adverse impact and meets the 95% requirement of the Pharma Industry, micro-organism under risk category-I may be exempted from the present regulation. However, risk category-II and III could be subjected to stringent regulation.
3. There is an overlap in the requirements under the Plant protection varietal Act of November 2003 and the GEAC mandate. Under the Plant Protection Varietal Act approval for import of Biological material (both LMO and non LMO) is mandatory. Since approval from various regulatory agencies for import of Biological materials take more than 6 months, the R&D activities in the country has slow down. It was suggested that the current regulatory framework should be harmonized to avoid duplication in the approvals granted should be avoided.
4. While amending the 1989 rules under EPA it was suggested that the role and responsibility of various regulatory agencies should be clearly demarcated, timelines for taking a decisions defined and protocols for carrying out various studies should form part of the notification so that inter-ministerial coordination is reduced and requisite information is submitted to the GEAC along with the application.
5. There is a need for close inter-ministerial coordination between GEAC for environmental release and MOA for commercial release. Secretary E&F clarified that GEAC mandate is only to assess the environmental risk vs. economic benefits and make its recommendation to the MOA but would not be involved in the commercial release of the crops.
6. A representative of the industry association should be nominated on the GEAC.
7. The proposals submitted for consideration of the GEAC are referred to the Experts who take a very long time to submit their comments. It was suggested that individual proponents be given an opportunity to make a presentation before the GEAC and clarify the concerned issues. Chairperson GEAC clarified that such a practice is being encouraged in the GEAC.
The representatives of various industry associations thanked the Secretary (E&F) for initiating the present exercise to streamline the necessary desired changes in the existing regulatory mechanism. While offering full support they requested the Govt. to ensure coordinated regulatory mechanism by different agencies of the Govt.
After detailed deliberations, it was decided that an informal Task Force under MOEF comprising of representatives from DBT, DCGI, FICCI, CII and ABLE and two experts would be set up for suggesting measures to streamline the

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2727272727
regulatory process for the recombinant Pharma sector. The terms of reference of the committee would be to determine the regulatory objective of each regulating agencies, decision rule, information/documents required and optimum timeline for according approval as well as measures to harmonize the domestic regulatory framework with the international obligation under Cartagena Protocol. The Task Force will submit its report by 31st May 2004. It was also decided that reforms in the regulatory process for the Food sector would be taken up subsequently.
Some of the industry representatives requested the Ministry that until the revised regulatory mechanism is put in place, as an interim measure the GEAC may agree with the revised guidelines recommended by DBT. Chairperson GEAC clarified that we are bound by the 1989 Rules notified under EPA, 1986 and therefore, until the revised system is formally notified, the current practice of approval of GEAC for Phase-III clinical trials would be mandatory.
The meeting ended with a vote of thanks to the Chair.
List of the Participants who attended the Consultative meeting with the major industry associations in the Pharma & Food sectors on 22.3.2004 in
the Ministry of Environment & Forests, New Delhi
S.No. Name & Designation
1. Dr. Prodipto Ghosh, Secretary (E&F)
2. Mrs. Veena Chhotray, Add. Secretary MoEF
3. Shri D.D. Verma, Joint Secretary MoEF
4. Dr. Sandeep Khanna, Joint Secretary, M/o of Consumer Affairs & Food (PPD)
5. Mr. Ashwini Kumar, DCGI
6. Dr. Vasantha Muthuswamy, Sr. Deputy Director General, ICMR, Delhi
7. Dr. T.V. Ramaniah, Director DBT
8. Mr. Juggi Lal, Joint Commissioner (S&R), M/o of Consumer Affairs
9. Dr. K.K. Tripathi, Advisor DBT
10. Dr. R. Warrier, Additional Director & Member Secretary GEAC, MoEF
11. Dr. M. Hota, Joint Director, MoEF
12. Ms. Madhu Gupta, Research Assistant, MoEF
13. Ms. Kiran Majumdar, CMD, Biocon
14. Ms. Sandhya Tiwari, Director, CII
15. Shri Kumar S., President, R&D Biocon
16. Mr. Rajesh Jain, Jt. Mg. Director, Panacea Biotech Ltd.
17. Mr. S.K. Bahl, Director, Serum Institute of India Ltd.
List continued on next page

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2828282828
18. Mr. Sunil K. Tadepalli, General Manager (Biotechnology), Ranbaxy Laboratories Ltd.
19. Mr. A. Sundara Rajan, Asst. Vice President, Cadilla Healthcare Ltd.
20. Col. L.J.S. Gill, Vice President, Business Development, Wockahardt Ltd.
21. Mr. S. Sethi, Resident Manager, Wockhardt ltd.
22. Mr.K.I. Varaprasad Reddy, MD, Shanta Biotechnics
23. Mr. N.S. Katoch, GM Corporate Affairs, Cadbury India Ltd.
24. Mr. Bhagirath Chaudhary, Forum Leader, FICCI
25. Mr. Sahl Singhal, FICCI
26. Mr. Ashwin Shroff, FICCI
27. Mr. S. Ganesh, FICCI
28. Mr.T. Eshwarprasad, Head, Bharat Biotech
29. Mr. Kaushik Samanta, Asst. Manager, Dr. Reddy Laboratory, (Safety Health &
Environment),Hyderabad
30. Brig. B.S. Butalia (Retd.), Cargill India Pvt. Ltd.
31. Dr. Partha R. Dasgupta, CII
32. Rashi Bahadur, CII
33. Rakesh Bamzai, Vice President, Biocon Ltd. Bangalore
34. Mr. P. Swan, FICCI
**********
List of the Participants who attended the Consultative meeting with the major industry associations in the Pharma & Food sectors on 22.3.2004 in
the Ministry of Environment & Forests, New Delhi (continued from last page)

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
2929292929
Annexure-IIAnnexure-IIAnnexure-IIAnnexure-IIAnnexure-II
F. No. 12/7/2004 - CS Government of India
Ministry of Environment & Forests (CS Division)
Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi
E-mail: [email protected] Telefax: 2436 3964, 2436 1613
Dated 20th April 2004
OFFICE MEMORANDUM
Subject: Task Force on Recombinant Pharma Sector.
1. In accordance with the decision taken in the Consultative Meeting under the Chairmanship of Secretary (E&F) on 30th January 2004, it has been decided to set up a Task Force on recombinant Pharma Sector with a view to streamline the regulatory process under the “Rules for Manufacture, Use, Import Export and Storage of hazardous micro organisms / Genetically Engineered Organisms or Cells, 1989” notified under the Environment (Protection) Act, 1986.
2. The composition of the Task Force shall be as follows:
i. Dr. R. A. Mashelkar Chairman
ii. Secretary (E&F) Member
iii. Secretary DBT Member
iv. Secretary MOH Member
v. Director General, ICMR Member
vi. Director General, DCGI Member
vii. Dr. Amit Ghosh, Co-Chairman GEAC Expert Member
viii. Dr. C.M. Gupta, Chairman RCGM Expert Member
ix. Nominee of ABLE Member
x. Nominee of CII Member
xi. Nominee of FICCI Member
xii. Shri Desh Deepak Verma, JS, MoEF Member Secretary

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3030303030
3. The Task Force shall have the following terms of reference:
i) To recommend a transparent streamlined regulatory mechanism and process for the use of LMO’s in the Pharmaceutical Industry during the various stages of R & D, testing, manufacture and use.
ii) To recommend regulatory process and mechanism for import of LMOs in the Pharma sector.
In developing the regulatory mechanism/process the Task Force may take into account the generic norms the re-engineering of regulatory process set forth by the Govindrajan Committee.
4. The Task Force shall submit its report by 31st May 2004.
5. The Chairman may also Co-opt persons with required expertise as and when required.
6. The expenditure on TA/DA for the chairman and other non official members of the Task Force and other contingency expenditure will be met from the Biodiversity Conservation Scheme.
(R. Warrier)
Additional Director
Dr. R.A. Mashelkar, Director General CSIR & Secretary Govt. of India, Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi – 110 001 Fax No. 2371 0618
Secretary Ministry of Environment & Forests, Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi – 110 003
Secretary Department of Biotechnology Block No. 2, 7th floor CGO Complex, Lodi Road New Delhi-110 003
Secretary Ministry of Health & Family Welfare, Nirman Bhawan, New Delhi.
Director General Indian Council of Medical Research Ansari Nagar, P.B. No. 4508 New Delhi - 110 029

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3131313131
Director General
Drug Controller General of India
Directorate General of Health services,
Nirman Bhawan,
New Delhi – 110 011
Dr. Amit Ghosh, Director IMTECH Sector 39-A, Chandigarh – 160 036
Dr. C.M. Gupta Director, Central Drug Research Institute, Chattar Manzil Palace, Post Box No. 173, Lucknow – 226 001 (UP)
President Association of Biotechnology Lead Enterprises(ABLE) 2ND Floor No.13, 4th Block 10, Main Road, Koromangale Banglore-34
Secretary General Confederation of Indian Industries, India Habitat Centre, 4th Floor, Core 4 A, Lodi Road, New Delhi-110003
Secretary General Federation of Indian Chambers Tansen Marg, New Delhi – 110 001
Shri Desh Deepak Verma Joint Secretary Ministry of Environment & Forests, Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi – 110 003

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3232323232
F. No. 12/7/2004 - CS Government of India
Ministry of Environment & Forests (CS Division)
Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi
E-mail: [email protected] Telefax: 2436 3964, 2436 1613
Dated 11th May 2004
OFFICE MEMORANDUM
Subject: Task Force on Recombinant Pharma Sector.
In continuation to this Ministry’s notification dated 30th April 2004, this is to inform that the Composition of the Task Force has been amended to include Ms. Veena Chhotray, Additional Secretary & Chairperson GEAC and representative of Indian Pharmaceutical Alliance (IPA) as member of the Task Force.
(Dr. Ranjini Warrier) Additional Director
Dr. R.A. Mashelkar, Director General CSIR & Secretary Govt. of India, Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi – 110 001 Fax No. 2371 0618
Secretary Ministry of Environment & Forests, Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi – 110 003
Secretary Department of Biotechnology Block No. 2, 7th floor CGO Complex, Lodi Road New Delhi-110 003
Secretary Ministry of Health & Family Welfare, Nirman Bhawan, New Delhi.
ºÉiªÉ¨Éä´É VɪÉiÉä

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3333333333
Ms. Veena Chhotray Additional Secretary & Chairperson GEAC Representative of IPA Ministry of Environment & Forests CGO Complex, Lodhi Road, New Delhi – 110 003
Director General Indian Council of Medical Research Ansari Nagar, P.B. No. 4508 New Delhi - 110 029
Director General Drug Controller General of India Directorate General of Health services, Nirman Bhawan, New Delhi - 110 011
Dr. Amit Ghosh, Director IMTECH Sector 39-A, Chandigarh – 160 036
Dr. C.M. Gupta Director, Central Drug Research Institute, Chattar Manzil Palace, Post Box No. 173, Lucknow – 226 001 (UP)
Ms. Kiran Mazumdar Shaw President Association of Biotechnology Lead Enterprises(ABLE) 2ND Floor No.13, 4th Block 10, Main Road, Koromangale Banglore-34
Secretary General Confederation of Indian Industries, India Habitat Centre, 4th Floor, Core 4 A, Lodi Road, New Delhi-110003
Secretary General Federation of Indian Chambers Tansen Marg, New Delhi – 110 001

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3434343434
President Representative of Indian Pharmaceutical Alliance (IPA) Vision Consulting Group 201, Darvesh Chambers, 743, P.D. Hinduja Road, Khar, Mumbai – 400 052
Shri Desh Deepak Verma Joint Secretary Ministry of Environment & Forests, Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi – 110 003
******

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3535353535
Minutes of the first meeting of the Task Force on Recombinant Pharma Sector held on 12th May 2004.
The first meeting of the Task Force on Recombinant Pharma Sector was held on 12th May 2004 under the Chairmanship of Dr R. A. Mashelkar, Director General CSIR at Department of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi. List of participants is annexed.
At the outset, the Chairman welcomed the members and thanked Secretary (E&F) for taking the initiative of addressing a long-standing issue with respect to regulatory approval processes relating to recombinant pharma products. He congratulated MoEF, DBT & MOH for their constant efforts in trying to streamline the regulatory process. However, he mentioned, there was scope for considerable improvement. Briefly touching upon the terms of reference of the Task Force he requested the members to work as far as possible, towards the ultimate aim of ensuring a ‘single window clearance’, which is symbolic of a ‘hassle free’ process. He said that it was important to focus on how time lines could be reduced without compromising safety and efficacy evaluation. He also urged the members that in the ensuing meetings of Task Force, efforts should be to look for solutions rather than mere reflection of problems.
He initiated the meeting by inviting Dr. Prodipto Ghosh, Secretary (E&F) to make a presentation on the current regulatory process. To begin with Dr. Ghosh clarified that the Ministry of Environment & Forests was essentially concerned only with LMOs or their products. By the same logic, products containing GMOs, which had no possibility of replication in the environment, was not really the concern of Environment Ministry. In his presentation Secretary (E&F) addressed some of the perceived problems and the steps already initiated to address the issues related to delay and transparency.
Secretary (E&F) informed the Task Force that certain steps had already been taken by the GEAC in a pro- active manner:
1. The GEAC has eliminated the unpredictability of its meetings by meeting on the 2nd Wednesday of every month. This has been made effective since January 2004.
2. GEAC has addressed the transparency issue by posting all the decisions taken at GEAC meetings on the website of MoE&F.
3. GEAC has started inviting Industry to be present at GEAC meetings to clarify any issues that may be raised at GEAC meetings pertaining to applications in order to eliminate delays.
4. A Task Force for Agri-Biotech products under the Chairmanship of Prof. M.S. Swaminathan has already submitted a recommendation for Regulatory reforms in this sector.
5. The MOEF has initiated the Mashelkar Task Force for Recombinant Therapeutics and will implement the recommendations of this Task Force after it is finalized.
Other key steps taken by the MOEF and GEAC are the following:
1. GEAC will confine its regulatory role in terms of product approvals only if the products are Living Modified Organisms (LMOs).
2. GEAC will exclude itself from product approvals pertaining to products emanating from LMOs but not LMOs themselves. Viz. Recombinant Therapeutics.
Annexure-IIIAnnexure-IIIAnnexure-IIIAnnexure-IIIAnnexure-III

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3636363636
3. GEAC will be responsible to evaluate the environmental impact caused by LMOs, assess environmental risks and estimate the benefits that are likely to accrue to society.
4. GEAC will approve products only if the environmental impact realizes a net benefit to society. This is more relevant to Agri-Biotech sector.
After a brief discussion on the issues raised by Secretary (E&F), the Chairman invited each of the members to present their preliminary views on the major issues needing redressal in the regulatory process in recombinant pharma products. The views expressed by various members are summarized below:
Dr. N.K. Ganguly, DG, ICMR:
Referring to the report of the Expert Committee on “A Comprehensive Examination of Drug Regulatory Issues, including the Problem of Spurious Drugs” constituted by Ministry of Health under the Chairmanship of Dr. R.A. Mashelkar, he informed the Task Force that the Committee has recommended complete re-vamping of the Drug Regulatory system including the setting up a National Drug Authority. He also informed the Committee that land had been acquired for setting up a Federal Drug Authority Building. Besides, a Testing Lab at NIB costing Rs. 360 Crores has been set up. Once the testing lab was made functional the time lag for testing would decrease from 3 month to 1.5 months. He further stated that schedule Y of the Drugs & Cosmetics Act is being revised as per DBT guidelines. As recommended by DBT, the MOH has also constituted a special recombinant Drug Advisory Committee that includes a representative from DBT.
Dr. Ganguly, particularly emphasized the urgent need for streamlining the approval process as per 1989 Rules notified under EPA 1986, in view of the international time frame and competitiveness. The following suggestions were made:
1. Single window approach to streamline the delays due to duplication in the appraisal process.
2. The review process should follow fixed schedules and timelines.
3. Any product that does not replicate in the environment should not be considered by the GEAC. Since the end product of recombinant drugs basically contains functional proteins the matter can be dealt under the Drugs Control Regulations.
Dr. M.K. Bhan, Secretary, DBT:
Dr. Bhan advocated for an independent competent professional biosafety regulatory authority. He was of the view that the professional competence was a critical matter to anticipate any biosafety situation in advance because of the different products involved in the recombinant pharma sector. He also felt that the current practice of getting the proposals reviewed by out side experts needed reconsideration because most of them are extremely busy persons and were hard pressed for time to give their valuable time in providing comments. Instead, an ideal situation would be an institutional framework with in-house competent experts from multidisciplinary fields dealing with different aspects simultaneously and finalizing the decisions concurrently. Such an institution could work under a single regulatory authority that could oversee the bio-safety concerns right from the stage of planning and research to product development. Dr. Bhan said that the independent regulatory body could be more like the FDA in USA that could emphasize on playing a facilitating role in the approval process without diluting the biosafety regulatory aspects.
Dr. Bhan also felt that Industry did not have comprehensive documentation to reflect a professional level of product development evaluation. He suggested that it be made mandatory to encourage industries to have a “Product Development Plan” . The product development plan is usually discussed with the regulatory authorities early in the development phase and this practice is followed globally.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3737373737
Prof. Mashelkar said that this would be an ideal model but it would require an evolutionary process to get there as it might require time and a change in mindset. However, he requested Dr. Bhan to present a framework for such a model at the next meeting.
Smt. Mahima Datla, Representative of FICCI:
1. There should be a single central regulatory body such as Biotechnology Regulatory Authority (BRA) that looks into the regulatory related issues in drugs, vaccines, biologicals and food. It should be supported by independent back-end expert bodies on drugs, biologicals and food.
2. On the risk assessment & management of microorganism, bio-safety and risk categories of microorganisms as per the World Health Organization (WHO) or other international body standards and guidelines should be accepted.
3. All biologicals, genetically modified or not may fall into the bio-safety classifications as defined in WHO and FDA.
4. Clinical trials and basic R& D should not come under the purview of the GEAC
5. Recombinant pharma products that are derived from microorganism/genetically modified organism need not to fall within the ambit of rules 1989 as they are by and large a functional protein.
6. All drugs whether recombinant or not should be treated under Indian Pharmacopiea.
Dr. Kiran Mazumdar, Nominee of ABLE:
Referring to the analysis of the regulatory process carried out by the Task Force on Agriculture Biotechnology she suggested a similar model may be adopted in the case of recombinant pharma products. Expressing the concerns of the industries due to delay in the regulatory process she made the following suggestions:
1. The duplications in the review process should be resolved.
2. The roles of different regulatory authorities should be defined.
3. RCGM, which is a body of experts, should examine and approve the pre-clinical trials.
4. The safety and efficacy of the products could be evaluated by DCGI. However, DCGI should have sufficient experts to understand the protocol, evaluate the clinical trials data and accord approval.
5. Since GEAC is concerned only with the environmental risks, approval of GEAC may be obtained right in the initial stage of freezing the manufacturing process.
6. Documents/information required for review of the proposal should be clearly indicated.
Smt. Shaw also stated that there was no need to differentiate between r-DNA Drugs and conventional Drugs with respect to Pre-Clinical and Clinical Trials. The safety and efficacy parameters for both types of drugs was the same. The issue is about whether Recombinant Drugs can be measured in terms of “Bio-equivalance” or whether they need to undergo Phase III clinical trials. She welcomed Dr. Prodipto Ghosh’s proposal to limit GEAC’s role to LMOs. She suggested that since LMOs would only be present in the initial stages of the process, that GEAC limit their role to ensuring that the LMO is indeed “inactivated” before release into the environment. Further, that once a facility has been approved for a class of Recombinant Drugs, like USFDA, there need not be separate inspections for each subsequent product but more in terms of annual inspections.
She mentioned that the companies, which are manufacturing pharma products, have to undergo a lengthy multiple regulatory process even at the stage of import of microbes for research and development.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3838383838
Finally she also highlighted what Industry perceived as an unfair practice to permit Imported Recombinant Drugs to be marketed without Clinical Trials whilst Indian Manufacturers were subjected to an inordinately long regulatory approval process which was disincentive to Indigenous Biotech companies and instead encouraged a Biotech Drug sector based on trading imported drugs
Mr. Ashwini Kumar, DCGI:
Referring to the initiatives taken by DCGI to adopt the recommendation of the Expert Committee on “A Comprehensive Examination of Drug Regulatory Issues, including the Problem of Spurious Drugs” he suggested the following measures:
1. Single window clearance for pharma products including recombinant drugs through DCGI.
2. The 1989 Rules under EPA needs to be revisited in the context of the decision taken by MoEF to regulate only LMOs.
3. Environmental impact/ risk of therapeutic proteins does not exist, therefore the safety aspects concerning containment facility could be looked after by IBSC/RCGM.
Representatives of CII:
1. The recombinant therapeutic drugs should be regulated by DCGI in the same lines as that of chemicals and biological drugs.
2. On the issue of bio-safety aspects, concerning manufacturing units, the DBT guidelines should be followed. This aspect should be monitored by IBSC/RCGM.
3. GEAC should deal with the case only when the final product contains LMOs.
4. The regulatory bodies need to be strengthened with requisite expertise.
5. The regulatory objective and decision rules of the regulatory agencies need to be defined.
Dr. C.M. Gupta, Director, CDRI & Chairman RCGM:
1. The toxicity data submitted by the proponent may be evaluated either by RCGM or Recombinant Drug Advisory Committee.
2. There is no clear guideline on schedule Y of the Drugs and Cosmetics Act on Biogenerics and animal modules for generating toxicity data.
3. Approval of GEAC for conducting Phase-III clinical trials is not required.
4. In the case of mixed vaccines (formulation containing approved drugs) there is a need to generate toxicity data.
Mrs. Rita Teaotia, JS, MOH:
1. The responsibilities of DCGI and GEAC need to be defined.
2. Approval of GEAC for conducting Phase-III clinical trials is not required.
3. Restructuring of existing laws to avoid overlapping, ensure fixed timelines and transparency in the decision- making.
Dr. Amit Ghosh, Director, IMTECH & Co-Chairman, GEAC:
1. Lack of requisite documentation by the applicant is often the cause of delay in decision-making.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
3939393939
2. There is a lot of duplication in the regulatory process involving DCGI and GEAC.
3. Approval of GEAC for Phase-III clinical trials is not required.
4. Decision on import of recombinant products can be based on the product safety & efficacy data generated in the clinical trials and approvals obtained in the country of origin.
In response to an enquiry on the issue of field trials as per the Rules, 1989 which inter- alia states that the GEAC shall also be responsible for approval of proposals relating to release of Genetically Modified Organisms and products in to the environment including experimental field trials, Dr. Bhan clarified that the experimental field trials of r-DNA pharma products are that of individually randomized clinical trials done in a population. The term field trials does not include the clinical trials conducted in the contained environment of hospitals.
Representative of Indian Pharmaceutical Alliance:
1. The regulatory agencies for approval of the products have been satisfactory. The system has worked well but needs fine-tuning to address issues related to duplication of efforts and delays. The proposal of Secretary (E&F) can be applied to recombinant vaccines and biologicals as well.
2. The aim of the regulatory agencies is to minimize the risk associated with the trials of the product on humans. The sequential review during the product development is therefore very crucial.
3. Although phase III clinical trials of the above products are carried out by the clinicians in the hospitals under controlled conditions, yet the population being tested and administered the drugs are not in-patient in the hospital wards but go back to their places of residence and interact with the population at large. The role of GEAC is very important in the process of regulatory approval of recombinant biologics meant for human use because of above reason.
4. Evaluation of the process and quality of the recombinant product at each sequential step of approval is important. In terms of product quality and efficacy the review should ensure its similarity to the product approved earlier.
5. Better coordination between RCGM, DCGI and GEAC while review of the product.
6. More experts should be co-opted in the Committees for product review.
7. The time frames for approvals by each agency should be predefined with their documentary requirements and ability to track the approval of the product.
8. Installed capacities of the products of different companies should be made available on the web site of DBT to help develop different products without any crowding on the few to avoid the well known story of penicillin industry in the country.
Shri D.D. Verma, JS, MoEF:
1. Since some of the regulatory agencies are also involved in the promotion of biotechnology, evaluation of environmental risk by an impartial body such as GEAC is necessary. In response, Secretary DBT clarified that the Department of Biotechnology is committed to ensure that the bio-safety aspects are adequately addressed in the promotion of biotechnology.
2. In reference to the point raised by the nominee of ABLE, it is clarified that the difference in the regulatory process of imported products and indigenous products is because of different levels of environmental risks involved in the manufacturing process and in the import process. Since the manufacturing process involves the risk of release of LMOs into the environment during the manufacturing, appropriate safety measures need to be taken.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4040404040
Mrs. Veena Chhotray, AS, MoEF:
1. The regulatory objectives of various regulatory agencies need to be defined for better coordination.
2. Prescribed procedures and guidelines should be followed until formalized through necessary amendments in the Act/Rules.
3. While there is often an emphasis in expediting the decision-making process for promotion of biotechnology, equal care should be taken to ensure that the bio-safety concerns are not compromised.
Dr. Prodipto Ghosh, Secretary (E&F):
In response to various issues raised by members, following points were clarified:
1. The issue of single window clearance was examined by the Govindrajan Committee wherein it was concluded that perspective and regulatory objectives of various agencies being different, single window clearance may not be always possible. Further, it would require radical change in the statutory provisions.
2. Referring to the role of IBSC in the regulating system, he stated that there should be a clear demarcation in the regulatory and promotional roles played by various agencies and the promotional roles should be separated out from the regulatory provisions.
3. Referring to the observations made by Dr. Shaw regarding disparity in the regulatory process for import and manufacture it was clarified that GEAC would be involved in the regulation if any process or activities involve the use of LMOs. Therefore if the import of recombinant pharma products does not contain an LMO in the end product approval of GEAC may not be required. However, if product development and manufacturing of recombinant products involve use of LMOs it would require the approval of GEAC. Extending the scope of mandate of the regulating agencies is a broader policy issue and need separate consideration.
4. Evaluation of the multifaceted environmental risk involved in the handling and use of LMO is the mandate of GEAC. However, the GEAC may make use of the information available with DCGI/RCGM or accepted international practices in the decision making process.
Concluding remarks of Chairman:
Appreciating the frank and positive approach of the participants, the Chairman stated that he already saw signs of solutions emerging and arriving at a consensus might not be a difficult proposition. He further emphasized that there is a need to take upon the challenge and evolve an expert, professionalized and sound regulatory mechanism, which is, in par with international competitiveness of Indian Pharmaceutical Industry.
Referring to the terms of reference of the Task Force, he requested Secretary (E&F) to extend the tenure of the Committee upto 30th June 2004. Before concluding the meeting he informed the members that the formation of the Task Force had generated a lot of enthusiasm and he had been received several phone calls and e-mails from a wide segment of persons. Emphasizing the need for a wide consultative process, the Chairman suggested that he would write to various stakeholders (50 numbers) to seek their views on the proposed reforms and invite some of them to present their views in the next meeting of the Task Force. He also requested the members to suggest the names of persons to be consulted in this regard. He also invited Secretary DBT to make a presentation in the next meeting.
The meeting ended with a vote of thanks to the Chair.
*******

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4141414141
List of the Participants who attended the 1st meeting of the Task Force on Pharma Sector held on 12th May, 2004 under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR in Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi.
S. No. Name of the participants
1. Dr. R.A. Mashelkar, Chairman
2. Dr. Prodipto Ghosh, Secretary (E&F)
3. Dr. M.K. Bhan, Secretary, DBT
4. Mrs. Veena Chhotray, AS, MoEF & Chairperson GEAC
5. Dr. N.K. Ganguly, DG, ICMR
6. Shri Ashwini Kumar, DG, DCGI
7. Shri D.D. Verma, JS, MoEF
8. Ms. Rita Teaotia, JS, MOH
9. Dr. Amit Ghosh, Director, IMTECH & Co-Chairman GEAC
10. Dr. C.M. Gupta, Director, CDRI
11. Ms. Kiran Mazumdar Shaw, Representative of ABLE
12. Shri K.K. Tripathi, Advisor, DBT
13. Dr. T.V. Ramaniah, Scientist-F, DBT
14. Dr. R. Warrier, Additional Director, MoEF
15. Ms. Mahima Datta, FICCI
16. Shri Bhagirath Chaudhary, FICCI
17. Shri D.B. Patankar, CII
18. Ms. Sandhya Tewari, Director, CII
19. Shri M.K. Sahib, Wockhardt
20. Col. J.S. Gill, Vice President, Wockhardt

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4242424242
TASK FORCE ON R-PHARMA UNDER Dr. R.A. MASHELKAR, DG, CSIR
Summary of Views/ Recommendations received from Experts for Streamlining the Current Regulatory Framework on Recombinant Pharma.
—————————————————————————————————————————————
1. Shri J.V.R. Prasada Rao, Secretary, Ministry of Health:
1. The Rules framed by the Ministry of Environment and Forests in 1989 have led to the creation of ambiguities, overlapping of functions and consequent delays in licensing of products for market. These Rules have led to excessive environment concern about recombinant pharma and products which are basically therapeutic proteins like Insulin, Erythropoietin, Streptokinase, growth hormones, antigens etc. which are not associated with any environmental hazards. Separate clearance by a body like GEAC under the Ministry of Environment for products such as these, is not scientific.
2. Moreover, over time, the GEAC has also taken up the role of sanctioning various phases of clinical trials and scrutinizing of efficacy data etc. that is clearly the role of the office of DCGI. For this purpose, the GEAC itself does not have a technical support structure and depends upon the comments of the office of DCGI. Such dual processes and procedures have created a regulatory situation unprecedented anywhere in the world.
3. The only environment issues involved in the production of recombinant pharma products are confined to the level of manufacturing operations and seek the corresponding containment facilities. Such manufacturing facilities are required to seek environmental approval prior to production. Further, these manufacturing facilities are audited by the regulatory officials along with the expert support to examine the status of compliance with GMP, as well as the containment facilities which form an integral part of GMP.
4. In view of the above, it would be appropriate to amend the 1989 Rules so that the regulatory evaluation and approval of recombinant pharma products rests only with the office of the DCGI as is the practice in other countries. The RCGM under DBT may support the office of the DCGI in the initial evaluation of recombinant pharma products in respect of their molecular characteristics, amino acid sequence, purity, specification and safety.
2. Dr. Mrs. Manju Sharma, Former Secretary DBT:
1. Based on extensive consultation DBT had prepared a “Note” incorporating suggestions of experts, industries and others concerned with biotechnology regulatory methods. The recommendations made in the note may be considered by the Task Force.
2. One issue that needs to be looked into urgently is whether after RCGM has looked at the proposal, it has to go to GEAC or only DCGI for conducting Phase-I, II and III trials.
3. GEAC should consider the proposal only for commercialization.
3. Prof. G. Padmanaban, Distinguished Biotechnologist & Honorary Professor, Dept. of Biochemistry:
1. This subject is better handled by the Department of Biotechnology rather than the Ministry of Environment.
Annexure-IVAnnexure-IVAnnexure-IVAnnexure-IVAnnexure-IV

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4343434343
2. The present role of IBSC for R&D purposes should continue. IBSC needs to be strengthened.
3. IBSC can also clear requests for import of GMOs for R&D purposes in academic institutions.
4. IBSC clearance reports are not being regularly sent to DBT and this has to be insisted upon.
5. RCGM needs to be split into two, one dealing with pharma and medical uses and the other Agricultural uses.
6. RCGM (Pharma) could regulate testing and trials as well as import and export of GMOs for medical applications. The trial data can be evaluated and approved by RCGM instead of GEAC.
7. Inputs of RCGM are provided to DCGI which is the final Authority for giving approval.
8. The linkage between RCGM and DCGI has to be an efficient one to avoid delays.
9. Permission for import of GMOs by R&D labs or industry should be based on the provision of adequate information. Though the applicant may not like to reveal the entire DNA sequence information which are proprietary, the industry should declare the names of the gene(s) of commercial interest and tools to identify the same, besides declaration regarding the nature of gene, organism etc.
10. The RCGM (pharma) committee should meet 4 times a year and clear applications for the industry. But, all the necessary clarifications etc. should be obtained by the secretariat before the meeting, enabling a clear decision at the first meeting itself. The Chairman and members of the committee should be contacted as soon as the request is received and discussions carried out and clarifications obtained through email before the meeting.
11. All decisions taken should be posted on a website.
4. Mr. P.M. Bhargava:
1. A single window clearance for all drugs derived from recombinant DNA technology under the auspicious of an Independent Committee that would discharge all the functions of RCGM, GEAC and DCGI. The Committee could operate under a National Biotechnology Commission.
2. The requirements that a company applying for approval must satisfy in each case, must be clearly spelt out. They should be precise, capable of being followed, assessable and in the public domain. Three sets of such requirements would need to be laid out for the following three stages of the approval process:
R&D work on genetically engineered drugs: this would require registration of the R&D laboratory.
Clinical trials after the usual, mandatory preclinical work have been done. This would include proof-of-concept trials. If the product is claimed to be identical with a product already in the market, irrevocable proof of such identity must be provided; if such proof is provided, only abbreviated clinical trials may be required.
Commercial release.
3. The time schedule for approval in each case must be specified with a clear statement that if the approval is not received within a predefined time, the company assume that the approval has been granted.
4. The basis for granting approval must be available in public domain to ensure transparency and demonstrate professionalism in the National interest.
5. The Committee should comprise of members having high level of professional competence, personal integrity and honesty and high public credibility. All members should be appointed by name and not ex- officio. It should have members from scientists, industrialist, legalist and NGOs.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4444444444
6. A mechanism for reliable and objective monitoring system must be put in place and the results of the monitoring must be put in public domain. Responsible NGOs should participate in such monitoring system.
7. In case of dispute the applicant can approach the Biotechnology Commission, which must have the power to take a final decisions in such cases.
5. Dr. Krishna M. Ella, CMD, Bharat Biotech International Ltd.:
Based on the US model, India could evolve a system with three wings:
1. Genetic Engineering Approval Committee (GEAC).
Like the EPA, the GEAC could not only oversee the testing of GM organisms but also oversee exports, containment, hygiene & Environmental Protection.
2. Indian Council of Agricultural Research (ICAR).
Like USDA, ICAR could oversee Agriculture Biotechnology and Animal Biotechnology. A nominee from GEAC could monitor the environmental impact and ICMR nominee could evaluate safety for human consumption.
3. Drugs Controller General of India (DCGI) & Biotech Controller General of India (BCGI).
The role of DCGI should be redefined to constitute Biotech Controller General of India (BCGI). The present body (DCGI) could assume the role of regulatory body for drugs, chemicals, intermediate and bulk systems.
BCGI (proposed body) could act as a regulatory body for Biotech, Biologicals, Diagnostics, Veterinary, Animal Biotechnology.
4. DBT should assume the role of a facilitator and must monitor only academic and research activity. As a facilitator DBT cannot be a regulator.
6. Dr. K.S.N. Prasad, Head R&D, Shantha Biotechnics Pvt. Ltd.
1. Independent competent authority as proposed by Secretary DBT needs to be considered even if it amounts to radical change from the present regulatory process.
2. IBSC, RCGM and GEAC were set up with a view to oversee safe handling and use of DNA derived products and not envisaged as a regulator.
3. Total revamp of DCGI is required for its functioning effectively without the involvement of IBSC, RCGM and GEAC.
4. Currently GEAC is involved for operation beyond 20 l fermentation volumes. GEAC should be involved only for large-scale operation beyond 100 l fermentation volumes.
5. The regulatory oversight of biologics should be de-linked from regulation of experimental work involving genetic manipulation. The role of IBSC and RCGM should be limited to overseeing experimental work involving genetic manipulation only.
6. The role of GEAC should be limited to controlling the release of genetically modified organisms and their products, into the environment wherever such concerns exist. For example, field studies on transgenic plants, use of genetically modified microbes for insect control, etc. Clinical studies on recombinant biologics should be out of the purview of GEAC.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4545454545
7. The responsibility for the approval of all biologics through various stages of development starting from preclinical safety and efficacy studies to the final approval and issue of manufacturing license should be entrusted to DCGI.
8. An expert Committee should be constituted to assist and advise DCGI in the regulation of biologics (recombinant as well as non-recombinant) at various stages of development (at present an expert committee is involved only in the final stage of approval after clinical trials). The committee should have a core team and special invitees with expertise in the relevant areas depending on the product under consideration.
9. Financial resources available to Drugs Control Organisation should be augmented immediately. Having a small cess on the drugs sold in the market can generate the necessary revenues.
10. Administrative and other measures should be initiated to ensure the honesty and integrity of Drugs Control Organization.
11. Measures should be initiated to protect confidentially of all the documents submitted to the regulatory authorities.
12. Laboratories for testing biologics should be made functional without delay.
13. Guidance documents for industry and formats for various submissions should be prepared in consultation with foreign experts and made available on the websites.
14. Mechanism should be created to facilitate constant interaction between regulators and industry.
15. A long-term plan should be chalked out to overhaul the Drugs Control Department so that the standards of regulatory oversight are improved gradually, eventually reaching the highest international standards. For chalking out such a program, it may be necessary to appoint a small committee, preferably involving regulatory experts from US and Europe to study the system and prepare a long-term plan at state and central level.
7. Dr. Amit Ghosh, Director IMTech:
1. If the product (drug) is licensed in its country of origin and is being marketed there, it should be treated at par with any other drug not produced through genetic engineering (GE) route. It’s import should be regulated as per the rules existing for the drugs not derived through the GE route.
2. For carrying out clinical trials, approval from DCGI should be adequate.
3. If the product to be imported is to be “used” in animals, rules followed by the Department of Animal Husbandry should be followed.
4. Exceptions to the above include:
Live recombinant vaccines
Import of an LMO as the starting material for manufacturing a “product”.
5. RCGM should continue to provide guidelines, approve and monitor ongoing research, give permission for import of LMOs for research purposes (if, however, the company intends to use the imported LMO eventually for manufacturing, GEAC must be informed), recommend the level of containment and monitor its compliance. It should also continue to approve pre-clinical trials.
6. DCGI will approve Phase-III trials.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4646464646
7. Before the company initiates the manufacturing, it should submit a detailed protocol (together with a copy of the RCGM approval) of the process to GEAC, elaborating especially the containment facility it proposes to put in place to prevent the release of LMOs to the environment. Proposal should also state what would be the contingency plan in case there is an accident.
8. Dr. S.K. Mahajan, Member GEAC:
1. Better coordination between the regulatory agencies to avoid the recent controversies and complaints for non-transparency.
2. The 1989 Rules notified under EPA 1986 cover LMOs as well as any recombinant substances derived from these. However, the terms of reference of TFRP refer to only LMOs. This is probably because the Cartagena Protocol is mostly concerned with LMOs. However, this dichotomy can cause some confusion in the approval process and lead to complaints of non-transparency. The new procedure must give clear instructions on approval of GEOs and recombinant substances derived from them.
3. All genetically engineered organisms (both LMO and GMO) should be regulated since they may contain hazardous impurities like DNA/ RNA/ Toxic proteins. The levels of the proteins often vary from batch to batch or manufacture to manufacture. These may not be seen in short term toxicological trials but their potential risks cannot be ignored.
4. In respect of drugs imported from other countries especially those having relatively less stringent regulatory system should be permitted with great caution.
5. Accredited laboratories in the countries where the quality of recombinant drugs can be independently checked should be set up.
6. While simplification and unambiguous enunciation of the regulatory procedures is highly desirable, our procedures should not excessively favour import and marketing and make local development unattractive. This can happen if the requirements for data on quality assurance and human trials (which must be submitted by local manufacturers) are frequently waived in the case of import on grounds such as certificates of questionable credibility. The original data in support of the imported drugs must be carefully scrutinized and wherever felt necessary, human trials should be conducted within the country.
7. While legitimate interests of drug companies (importers as well as local manufacturers) should guide the development of expeditious, non-cumbersome and transparent regulatory procedures, the interests of consumers in respect of safety and efficacy of the drugs must take precedence over all other considerations. We cannot, therefore, dilute the essential regulatory requirements for the locally developed or imported drugs merely because of pressures generated by influential lobbyists.
8. The autonomous body proposed for facilitating single window clearance should not be under the control of one of the promotional Ministries (DBT, ICAR, MOA, CSIR, DSIR). In fact while reorganizing the regulatory setup, RCGM should be moved out of DBT or its regulatory responsibilities should be curtailed.
9. On the issue of scientist vs. IAS to head the GEAC, there are advantage and disadvantage in both systems. The administration of Scientific Departments has been benefited from the presence of both scientists as well as IAS. It is the attitude, competence and integrity of individuals heading an organization (rather than scientists vs. IAS), discussion on this matter is unwarranted.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4747474747
9. Dr. S. R. Nair, MD, Biotech Consortium India Ltd. (BCIL):
1. A single window agency should receive and process applications rather than multiple agencies such as DBT, MoEF or the Drug controller General of India etc. as at present.
2. With the rapid developments in biotech pharma sector, the guidelines should be frequently modified and simplified in the light of the experience in the application and use of these products.
3. Duplication of generation of data should be avoided.
4. Modification and guidelines should be available on website.
5. Data on performance and side effects of approved products should be closely monitored and compared with similar products globally as well as in the country.
6. Processing of application should be done online.
7. While taking a decision on the import of LMOs, the experience of countries already using them should be given emphasis provided that the regulatory process in those countries is sound and well established.
8. Pre-cautionary principle should be limited only to any specific and special effects relevant to India.
10. Mr. S.C. Singhal, VP, Dr. Reddy’s laboratories:
1. The present approval process is quite cumbersome, multicentric, difficult and time consuming.
2. Interdepartmental discipline and understanding needs more clarity.
3. A single point regulatory appraisal and approval system is the need of today to expedite the approvals. All requirements for the permission should be available at one place and process must be explicitly clear stating requirements at every step.
4. We need to have recognized labs where the biologically derived products can be tested for physio- chemical and biological properties, which is acceptable to any regulatory agency in the world.
5. The requirements of clinical trials are negotiable today from product to product. There has to be more clarity and specificity so that work is performed accordingly.
6. With the invention of process analytical technology, the size of clinical trials should be kept meaningful to ascertain safety and efficacy of therapeutics.
7. Industry should be encouraged to reach the regulations with facts without the fear of delays.
8. For each processing step timeliness must be fixed to bring more efficiency into the process.
9. While the idea of separating recombinant therapeutic from LMOs is very good, the approval of therapeutic protein should be done by DCGI and should be treated like other drugs when it comes to clinical trials.
10. With the rise in requirement of clinical trials in our country more emphasis should be given to approve agencies engaged in clinical trials and should be issued recognition by the government.
11. It will be a good idea to insist on clinical trials in India for imported recombinant drugs.
11. Dr. V. Prakash, Director, Central Food Technological Research Institute, Mysore:
1. Use of LMOs in Pharmaceutical industry should be regulated during the various stages of R&D, testing, manufacture, import, export and use.
2. There is a need to put in place a “National Biosafety Board” which provide regulations for:
Safe Biotechnology Research.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4848484848
Safe Handling of LMOs and products
3. To ensure good manufacturing practices for specific pharmaceutical product as laid down by International Regulatory bodies such as WHO (http://www.who.int/medicines/organization/qsm/activities/ qualityassurance/gmp/gmpthree_bio.html).
4. Cartagena Biosafety Protocol (CPB) on LMOs does not apply to transboundary movement of LMOs, which are pharmaceuticals for humans. Therefore a regulatory process and mechanism for import of LMOs intended for pharmaceutical use could be established based on the guidelines of Cartagena Protocol on Biosafety. The regulatory process and mechanism for import could include:
Setting up of a Biosafety Clearing House for LMOs used in Pharma sector
Risk Assessment of LMOs
Risk Management of LMOs.
12. Dr. Seyed E. Hasnain, Director, Centre for DNA Fingerprinting and Diagnostics:
1. Any product which is made using identical DNA clone or GMP conditions, then an Autonomous agency may be given the responsibility to create a signature (genetic, chemical etc.) of the product and match the same with an already existing product in the market. If such matching is present then permission should be automatically given to the new products without asking them to go through the tedious processes essentially tantamounting to reinventing the wheel.
2. There should be one regulatory authority (chaired by an eminent scientist) which should have the mandate to receive the application, review and accord approval. This body will comprise of representatives from MOH, DBT, CSIR, DSIR, MOEF and DCGI etc.
3. The authority should meet twice a month to consider all applications received.
13. Dr. Lalji Singh, Director, CCMB:
1. Under 1989 Rules the proposals that are referred to GEAC include import as well as manufacture of r- Pharma (drugs, therapeutics vaccines). Detailed and exhaustive information regarding clinical efficacy of the product need not be provided for assessing the suitability of the product from an environmental angle. Other agencies of the government are assessing clinical efficacy.
2. Similarly, detailed information about the procedures for production, including floor diagrams of production facilities and procedures for effluent treatment, are not necessary for proposals related to imports of finished products. This information along with details about the containment facilities is required only for products being manufactured in India,
14. Dr. Samir Bhattacharya, Director, Indian Institute of Chemical Biology:
1. The product coming out from the genetically modified organisms requires to be examined for its similarity with the naturally available product already used for intended purpose and known to be non-toxic.
2. While genetically modified organisms should not be treated equally with natural organism, it is important to note that sometimes the product of GMOs represent a higher quantity of the product produced from natural organism. Such products from GMOs should have less regulatory barriers for research and marketing.
3. In order to encourage the Pharma industry to import and proper use of LMOs, the regulatory mechanism should have enough flexibility. However, it should not leave any porosity to risk biosafety and be prone to bioterrorism.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
4949494949
15. Dr. Cyrus S. Poonawalla, Chairman, Serum Institute of India Ltd. & Dr. P. Pushpangadan, Director, National Botanical Research Institute:
1. A statutory and autonomous National Biotechnology Regulatory Authority (NBRA) with two separate wings – one for Agriculture and Food Biotechnology and the other for Medical and Pharmaceutical Biotechnology as recommended by the Task Force under Prof. M.S. Swaminathan may be considered.
2. Composition and function of the Pharmaceutical and Regulatory wings needs to be finalized through a multi-stakeholder consultation process.
3. The existing regulatory committees such as RCGM, GEAC etc. should be strengthened to include expert on biosafety, biotechnology, bioethics, bioinformatics, environment and ecology.
4. Developing new biosafety guidelines for the use of LMOs and rDNA molecules in pharmaceutical industries in the line of the NIH Guidelines for Research involving the recombinant DNA molecules.
5. Development of skilled human resources in undertaking risk assessment and risk analysis related to LMOs. Some of the key information required in risk assessment include:
Organisms, organization and people involved
DNA donor and the receiving organisms
Conditions of release and the target environment
Interactions between transgenic and the environment
Monitoring, waste disposal treatment and control
6. Access to information on DNA technology and decisions taken by the regulatory agencies.
7. The relevant article of Cartagena Biosafety Protocol should be integrated into the new regulatory mechanisms and guidelines.
16. Mr. Paramvir Singh Ahuja, Director, Institute of Himalayan Bioresource Technology:
1. A single window approach rather than having different regulatory wings looking at different issues.
2. Duplication needs to be avoided between the agencies assigned to look at acquisition, production system, storage system and disposal system.
3. Biotechnology sector is highly competitive and is ridden with IPR interest; hence critical minimum disclosure is desired before the regulatory agencies.
17. Prof. P. Reddanna, University of Hyderabad:
1. Academic institutes and industrial R&D units have been severely effected by:
Most of the Airports are not equipped to handle perishable items due to lack of proper storage and handling facilities and mechanism for immediate clearance.
Elaborate procedures and importing / exporting materials.
2. The Task Force may look into these aspects and suggest appropriate measures to Govt. for implementation.
18. Dr. N.H. Wadia, President, Moving Academy of Medicine and Biomedicine:
1. Many recommendations of our regulatory bodies are vague resulting in arbitrary decisions creating bottlenecks for R&D so crucial for industrial growth. There is a need to make them more defined so as to bring transparency.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5050505050
2. Another suggestion is that we must introduce and implement the concept of a single window clearance so that files are not shuffled from desk to desk without arriving at a decision. This would cut down the red tape drastically.
19. Mr. D.S. Sankholkar, Head, Corporate Regulatory Affairs (Technical), Hindustan Lever Ltd.:
1. The 1989 Rules and the specific Protocols for research and scale up of biotechnological processes using LMO or other substrates, involving the Institutional Bio-Safety Committee (IBSC) provide a reasonable regulatory framework and should function well with the following improvements:
Harmonization of formats for Risk Assessment and Authorization to import of LMOs.
Mandatory standards for acceptance testing/ quarantine procedures; transportation, storage, handling contingency planning, disposal etc of LMOs.
Certified panel of experts who can act on behalf of DBT and be nominated on IBSC for expeditiously processing applications.
2. There appears to be duality of regulating authorities between GEAC and DCGI office, when an LMO is under consideration for clinical or field trials or marketing as a drug or a pharmaceutical or for new crop introduction.
3. It would always be good to have a single window authority for clearance procedures.
4. Clear-cut guidelines on the data requirements for biotechnology-based products do not exist as of date under “Schedule Y” of Drugs and Cosmetics Act and the same would need to be developed and introduced.
5. Any regulation that is likely to be made now should take into consideration its future adaptation for areas of usage like cosmeceuticals as well as nutritional products.
20. Mr. K. Dharmalingam, Sr. Professor & Head, Department of Genetic Engineering, Madurai Kamraj University:
1. IMTCC to be nodal agency to import and supply all material needed for the investigators.
2. A group of laboratories with expertise with specific organisms must be co-opted to act as referral labs for identification and confirmation of microbial strains. These labs could form a consortium under IMTCC.
3. A positive action would be to make it mandatory that every strain developed is to be deposited in the IMTCC as per their guidelines before the PhD thesis or the paper is accepted/ submitted for publication. Sooner or later we will end up paying heavily even for classical strains developed.
4. Clear GMP regulations are non-existent in India. It is high time to come up with our own clear-cut practical GMP stipulations. These must take into account the existing US and European norms and converting them to suit India.
5. While R&D lab architecture could be flexible, laboratory organization for manufacture under GMP condition could be defined, perhaps more than one modular organization could be drawn up with all details, including ducting, plastering etc.
6. The major focus should be on the requirements of startup companies and not organized sectors who could afford the imported designs & equipment. Certification of indigenous manufacturers of fermentors etc., also should be streamlined with specifications, therefore new equipment manufacturers will have competitive edge.
21. Mr. K.P. Gopinathan, Hon. Professor/ CSIR Emeritus Scientist, Department of Microbiology & Cell Biology:
1. The terms of reference of the Task Force item 1, may be modified from “for the use of LMOs in the Pharmaceuticals….” To read as “for the development and use of LMO….”.
2. It is important to have a central inventory, for the imported as well as indigenously developed LMOs, and every stakeholder should deposit the information at this inventory.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5151515151
3. The regulatory body however, insists on getting the full DNA sequences, rather than the overall modified gene information, and this creates problems for getting clearances for import of the recombinant constructs or LMOs even from their collaborators or principals abroad. This procedure could be simplified.
4. Besides, the LMOs, even the storage, handling, maintenance and disposal of pathogenic organisms (bacteria, viruses, pathogenic organism etc), whether genetically manipulated or not should be included under similar set of rules.
5. “Proper Disposal methods” for LMOs and pathogenic materials should form an important component of the guidelines.
6. Labeling must be made mandatory. The existing methodologies for evaluation and application of the end precuts (eg., vaccines/ pharmaceuticals) can be continued.
7. Issues related to “Stem Cell Research” (Banking, Applications and fundamental studies) be also included in the purview of the recommendation of this Task Force.
22. Prof. Arun Balakrishnan, Director, Anna University:
1. The concept of a single window administration, wherein the scientists, technocrats and business administrators, legal etc can be placed.
2. The R&D activities of various institutions and University of the state in the various areas of biotechnology should be tapped and converted to commercial ventures.
3. In order to effectively administer Biotechnology towards business development, it will be essential to develop a single body that will regulate the biotechnology reforms and needs for the development of such industry.
4. The interfacing between administrators, scientists and business developers is a key to the success of business development that can be accomplished.
5. The availability of Dept. of Biotechnology and Business development as indicated in the flow sheet should be an autonomous body, functioning and advising the Chief Minister of the State.
23. Dr. Somesh Sharma, Chief Scientific Officer, Nicholas Piramal India Ltd.:
Regulations governing the Biological Research Centres (BRCs) should include:
1. The BRCs must put in place procedures to manage the health and safety of all who may be put at risk by its activities. This requires a suitable and sufficient assessment of the risks to health and safety to which any person whether employed by them or not, may be exposed to through their work. These assessments must be reviewed regularly when changes in procedures or regulations demand, and must be recorded. The distribution of microorganisms to other outside the workplace extends these duties to protect others.
2. The BRCs must ensure that all strains are assigned to appropriate risk/ hazard groups. Unless proven otherwise, all unknown biological agents must be considered hazardous. Hazard information must be recorded and made available to recipients of the material.
3. The BRCs must do their best to ensure that non-indigenous pathogens are not distributed unless the recipient has a current license.
4. The BRCs must ensure that information on ownership of IP is passed to third parties /recipients of the organism.
5. The BRCs must ensure transparency retaining the link between country of origin and end user of genetic resources. Biological materials must be received and supplied within the spirit of the Convention on

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5252525252
Biological Diversity (CBD) ensuring material transfer agreements are in place. A BRC must maintain contact and follow recommendations of its national CBD Contact Point and National Focal Point.
6. The BRCs must issue an appropriate Safety Data Sheet with every culture consignment.
7. The BRCs must ensure that staff responsible for distribution of cultures have a current IATA Shippers training certificate and ensure organisms are packed and shipped in accordance with IATA requirements, if applicable. Non- infections microorganisms may be sent by (air) mail, according to the UPU requirements.
8. The BRCs must have procedures to check the validity of customers that wish to receive dangerous organisms and if in doubt must be required by local law to REFUSE to supply.
24. Prof. D. Balasubramaniam, Director of Research, L.V. Prasad Eye Institute:
1. The regulations that have been formulated by the USDA-APHIS seem comprehensive, in relation to the production, transport and consumption of LMOs/ GMOs. These, along with the role played by the FDA seem applicable in a general manner to us as well. The one aspect of worry we would soon have to face is about gene delivery/ gene therapy. Here too, the guidelines that are being formulated and revised by Inder Verma and his group might be worth considering, along with views of ICMR/ Health Ministry/ DBT.
25 Prof. Dipankar Chatterji, Indian Institute of Science:
1. Storage of hazardous microorganisms should be in one place in the country (P4-facility) in the line of small poxvirus now being kept in USA/ Russia.
2. Manufacture, construction of plasmid based systems, import and export of hazardous microorganism should be carried out through a centrally operated scientific agency (like CSIR/ICMR) where a project should be submitted first by the PI, evaluated like CSIR projects, presented in front of a committee before a decision is reached by the committee. The guidelines for such an endeavor should be clear to the committee members and they can meet 3-4 times a year. In the case of extremely dangerous organisms, a site visit to the PI’s laboratory is necessary before permission is granted.
3. Every patent, publication should clearly state in which facility such work had been carried and who financed them.
****

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5353535353
Minutes of the 2nd meeting of the Task Force on recombinant Pharma Sector held on 15th June 2004.
The 2nd meeting of the Task Force on recombinant Pharma Sector was held on 15th June 2004 under the Chairmanship of Dr R. A. Mashelkar, Director General CSIR at Department of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi. List of participants is annexed.
1.0 Opening Remarks of the Chairman At the outset, the Chairman welcomed the members and informed the Committee that subsequent to the first meeting of the Task Force he has written to 57 persons including Experts, Industry, Policy makers, experts and others. He briefed the Committee on the various response received from 27 persons. He also stated that the response from the media and the experts have been very encouraging.
2.0 Confirmation of Minutes The minutes were confirmed subject to the following amendments.
The name of “Dr. Amit Ghosh” in the sentence “In response to an enquiry from Dr. Ghosh, on the issue of field trials as per the Rules, 1989 ————————” at page 6 para 4 stands deleted.
3.0 Presentation by Secretary DBT The Chairman initiated the meeting by inviting Dr. M.K. Bhan, Secretary DBT to make a presentation on the Current Regulatory Framework, Proposed Modifications and Institutional Structure for the Biosafety Regulatory Authority. Dr. Bhan informed the Committee that his presentation covers two aspects. The first part relating to the current regulatory mechanism and proposed modification has been developed with a view to simplify the process and shorten the timelines for approvals. The second part relates to the National Biotechnology Regulatory Authority/ Commission, which is in consonance with the recommendation of Prof. M.S. Swaminathan Task Force for Agriculture and Food Product. Dr. Bhan in his presentation addressed the following issues:
1. General approval procedures for recombinant pharma products and informed the Committee that there is no over lapping of regulatory objectives up to the pre-clinical evaluation of the data generated by the company. He suggested that the need for human clinical trials both by DCGI and GEAC is an issue that needs to be addressed by the Committee.
2. He recommended the following modification to the current regulatory process:
RCGM to continue the practice of examining the pre-clinical data submitted by the company and forward their recommendation to DCGI for necessary action and GEAC for information.
RDAC/DCGI approves the protocol and recommends for conducting human clinical trials; DCGI Examines the Human Clinical Trials data and recommends to GEAC directly.
GEAC to approve environmental/ commercial release.
Annexure-Annexure-Annexure-Annexure-Annexure-VVVVV

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5454545454
3. Referring to the views expressed by Secretary E&F in the first meeting of the Task Force that MoEF is concerned only with LMOs, he suggested that MoEF may like to consider recombinant pharma proposals where the end product is an LMO.
4. He also recommended the following timelines for approvals:
RCGM approval for pre-clinical animal studies: 45 days RDAC approval for Human Clinical Trials protocol: 45 days RDA (DCGI) examination of trial data and approval: Case specific Simultaneous DCGI & GEAC approvals: 45 days
5. On the issue of imports of GMOs/ LMOs for R&D purpose he informed the Committee that Customs Authorities are seeking approvals of PPA/DCGI/GEAC under Rules 1989 of EPA and PQO w.e.f. 1.1.2004. He suggested that the earlier practice of permitting the import with the approval of RCGM should continue and requested PPA/DCGI to issue instructions to Custom Authorities to clear the consignment based on RCGM approval.
6. On the issue of import of transgenic material for research, he suggested that no change in the present procedure of GMOs/LMOs is required. The procedure is in accordance with the Allocation of Business of DBT, EPA and NBPGR. However, PQO needs clarification for Biocontrol Agents imports.
7. Regarding the constraints faced by the industry for import of non-GMOs, he suggested that PPA/ DCGI to issue instruction to Customs Authorities to clear the consignment based on the declaration of the importer and exporter on certification of the nature of the organism etc. DCGI or PA (only one agency) to issue instructions in this regard.
8. He further suggested that the ultimate goal should be to establish an independent professionally competent authority for providing single window approvals. Referring to the FDA approval system in USA, he suggested a similar institutional framework for the National Biotechnology Regulatory Authority/ Commission. He recommended that the NBRA would comprise of four wings namely: a) Agricultural products / Transgenic Crops b) Pharmaceutical/ Drugs and Industrial Products c) Transgenic Foods/Feed and d) Transgenic Animals/ aquaculture. The four wing Secretariat would be headed by a Vice Chairman. The recommendation of the Secretariat would be forwarded to Apex Committee with Statutory Powers. The members of the Apex Committee would comprise of representatives from all stakeholders Ministries/ Departments. The Apex Committee would report to the Chairman. The presentation also included details of the Secretariat and areas of relevant expertise as well as functions of the Apex Committee.
4.0 Discussions on the proposal suggested by Secretary DBT
After a brief discussion, the Chairman invited each of the members to present their views on the recommendation made by Secretary DBT in his presentation. The views expressed by various members are summarized below:
Secretary (E&F):
1. On the issue of whether GEAC will exclude itself from product approvals pertaining to products emanating from LMOs but not LMOs themselves. viz. Recombinant Therapeutics, it was clarified that

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5555555555
GEAC will confine its regulatory role in terms of product approvals only if the products are Living Modified Organisms (LMOs). However, if product development and manufacturing of recombinant products involve use of LMOs it would require the approval of GEAC. Similarly import of recombinant pharma products, which does not contain an LMO in the end product approval of GEAC, may not be required.
2. In the case of manufacturing units, GEAC will be responsible to evaluate the environmental impact caused by handling and use of LMOs, assess environmental risks within certain pre-determined limit and estimate the benefits that are likely to accrue to society. The GEAC accords approval only if the net benefits >> environmental/ risks. It was clarified by Dr. Bhan that in pharma/ biological products this information is available only after human clinical trials are completed.
3. On the issue of environmental risks during handling and use of LMOs, it was stated by DCGI that prescribed WHO guidelines for containment facilities and safety of operation are currently in practice. Dr. Bhan suggested that this concern could be addressed by a mechanism for certification of facilities. Secretary E&F clarified that while mechanisms for safe handling and use of LMOs are in place, GEAC would still be concern with any accidental release and associated risks. Further the expected risk should take into account the probability of accidental release as well as potential damages likely to occur.
4. On the timelines suggested by Secretary DBT, Secretary E&F informed the Committee that the Ministry has brought out a document on good practices in environmental regulations and this has been implemented in the Ministry.
Mr. Ashwini Kumar, DCGI:
1. Environmental impact/ risk of therapeutic proteins does not exist; therefore the safety aspects concerning containment facility could be looked after by IBSC/RCGM.
2. Information on societal benefits of a specific pharma product is available in literature.
3. While referring the proposals to experts what exactly needs to be evaluated should be defined. Dr. Bhan suggested that this issue can be addressed by bringing out relevant manuals.
Dr. Samir Sangitrao ABLE representative:
1. There should be a central testing facility for biopharmaceutical products on the lines of Central Drug Laboratory, which is available for testing the pharmaceutical products. This will ensure that quality products come to Indian market.
2. Regulations should also address the issue of Contract Research and Contract Manufacturing, which are emerging as big commercial opportunities. The regulations should be designed so as to enable industry to capture such business speedily, while keeping in place reasonable measures to ensure compliance to biosafety. For example, in this case, only plant approval by the relevant body may be required, and no product data may be required.
3. In the case of import of Drug substance and Drug product, DCG (I) should ensure GMP is maintained and check all the documents so that Indian counter parts are not at a disadvantage and Clinical trials should be mandatory for imported products.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5656565656
4. After the committee has finalized the drug approval system, they should prepare a drug approval manual, which should contain the flow sheet of the system, all the relevant forms, copies of Rules, Act and Law governing these regulations. There should be also a single regulatory help desk to assist industry and provide updated information as and when required.
5. To avoid confusion, only one body {DCG (I) as it is a drug} should communicate the changes in the regulatory system if done.
Dr. Amit Ghosh, Director, IMTECH & Co-Chairman, GEAC:
1. There is no need to distinguished between GMO and LMOs in pharma products. Secretary E&F, however clarified that the distinction is relevant to GEAC functioning, which involves assessment of environmental risks. LMOs could replicate and cause damage to environment whereas GMOs may not. Further, GMOs may not require the same level of environmental risk assessment, as in the case of LMOs, as it cannot propagate in the environment.
2. The research and development as well as Phase-I II and III are carried out in contained conditions and therefore these activities do not pose any environmental risks. However, what needs to be examined is the adequacy of the emergency plan in case of accidental release.
3. Decision on import of recombinant products can be based on the product safety & efficacy data generated in the clinical trials and approvals obtained in the country of origin.
Dr. Vasantha Muthuswamy, ICMR:
1. Environmental impact/ risk of therapeutic proteins does not exist; therefore therapeutic proteins can be approved by DCGI.
Dr. Krishna M. Ella, CMD, Bharat Biotech International Ltd.:
1. Based on the US model, India could evolve a system with three wings. He suggested that:
Like the EPA, the GEAC could not only oversee the testing of GM organisms but also oversee exports, containment, hygiene & Environmental Protection.
Like USDA, ICAR could oversee Agriculture Biotechnology and Animal Biotechnology. A nominee from GEAC could monitor the environmental impact and ICMR nominee could evaluate safety for human consumption.
Drugs Controller General of India (DCGI) could oversee the product purity, safety and efficacy.
2. He mentioned that while therapeutic vaccines under go rigorous scrutiny, more virulent vaccines like the small pox do not require approval of RCGM/GEAC because it is a non-recombinant vaccine.
3. The composition of the Committee needs to be reviewed and strengthened.
4. On the issue of an independent single Authority, he supported the proposal of Secretary DBT. He stated that this is an excellent mechanism provided the industry does not have to approach the linked Ministries. Expressing apprehension that NBRA may be lead to “one more window clearance instead of a single window clearance”, he suggested that strengthening of the current regulatory mechanism may be sufficient to address the concern of the Industry.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5757575757
Dr. D.B. Patankar CII representative:
1. Dr. Patankar made the following suggestions during the presentation of Dr. Bhan’s proposals:
He supported Dr. Bhan’s proposals that only DCGI along with RDAC should have a role in approving clinical trial protocols as well as clinical trial results. Neither GEAC nor RCGM nor IBSC should have any role in it. He also supported the proposed approval time lines.
There is zero risk to environment from the release of the recombinant protein itself in almost all cases of therapeutic proteins. The only potential risk is during the manufacturing process.
Manufacturing plant approval from biosafety angle can be made by RCGM. During discussion on this topic, Dr. Bhan also pointed out the possibility that for risk group 1 category of host-vector systems, which bears low individual risk and low community risk as per the classification, even IBSC itself may be allowed to inspect the plant from biosafety point of view. Dr. Patankar supported this viewpoint.
GEAC role can be limited to where the products themselves contain LMO or where the risk group of the organism is higher, presenting a higher potential risk to environment.
Dr. Patankar suggested that during review of clinical trial protocol or results by DCGI, one of the members of the RDAC be kept a floating member to be taken based on the particular therapy area of the drug in question (e.g. oncologist for cancer drug, etc.) while the other members can be fixed. This will help in better review of the protocol or data, since currently there are fixed members who may not be able to review all products equally thoroughly.
2. In response to a question from Dr. Mashelkar at the end of the presentation, Dr. Patankar said that industry would be quite happy if the proposals as presented by Dr. Bhan for the simplification of procedures, which included elimination of duplication of work by different committees, defining the roles of each committee to their areas of expertise, and also introduction of specific timelines for approvals by different bodies at every stage, etc., were implemented.
3. In response to Secretary (E&F)’s comments that the expected risk should take into account probability of accidental release to environment as well as potential for damage in the case of such accidental release, Dr. Patankar mentioned that the potential damage in case of accidental release is already built into the system of assigning risk groups as per biosafety guidelines. He also pointed out that most recombinant proteins for pharmaceutical use are made in lowest risk group host-vector systems.
4. Once a product is approved, if the company develops a new formulation of the same product, where there is no change in the biosafety aspect, manufacturing process or any other aspect of the bulk molecule, the new product should go directly to DCGI and should not go again to IBSC, RCGM or GEAC.
5. Industry is willing to work in a manner that can simplify the work of the regulatory bodies so that they can perform their task to ensure quality products reach the market, without compromising safety to people or environment. As an example, Dr. Patankar cited that applications could be submitted in separate volumes for pre-clinical data, clinical data, etc. to facilitate sending the relevant portion to the relevant experts, etc.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5858585858
Dr. C.M. Gupta, Director, CDRI & Chairman RCGM: 1. In the case of mixed vaccines (formulation containing approved drugs) there is a need to generate
toxicity data.
2. The current regulatory mechanism/ procedures needs to be simplified and timelines for approval by various regulators, as suggested by Dr. Bhan may be accepted.
3. The single window mechanism if worked out would be excellent. However, the Authority should not be under any Ministry.
Representative of Indian Pharmaceutical Alliance: 1. The industry is not looking for less regulation but robust regulation that stands scrutiny.
2. The information required by various agencies needs to be pre-defined. Mechanism for scrutiny of pre- submission data would be useful.
3. Drug safety is essentially matter of human safety. This aspect needs to be looked into DCGI.
4. Considering the present infrastructure, the proposed timelines may not be feasible.
5.0 Concluding remarks of Chairman: Prof. Mashelkar said that the institutional framework for National Biosafety Regulatory Authority as proposed by Dr. Bhan would be an ideal model. Referring to the recommendation of the Expert Committee on “A Comprehensive Examination of Drug Regulatory Issues including the Problem of Spurious Drugs”, he explained that experience has been that the Govt. is wary about any new institutional structure. He explained that the concept of a single window clearance would require time, resources as well as change in mindset. However, this should not prevent the Task Force from making this recommendation.
He suggested constitution of a Drafting Committee to prepare the draft recommendation for consideration of the Task Force based on the discussions held in the two Task Force meetings and recommendations received from experts. He also suggested that in the next meeting he would like invite about 6-8 persons for presenting their views on the regulatory reforms. To have focus inputs, Secretary (E&F) suggested that, the Task Force hold further consultations after preparation of the draft report.
The representative of ABLE suggested that the Drafting committee to be instituted by Dr. Mashelkar, to finalize the recommendations should include an industry nominee. Also, they should visit at least one company to get first hand information of the various systems like contained facility, biosafety, etc. The drafting committee should also go through all the forms and applications in detail so that the information required to be submitted is relevant and appropriate to the area of scrutiny of the respective regulatory body.
The meeting concluded with the following decisions: a) A Drafting Committee to be constituted by the Chairman to prepare a Zero Draft which will form the
basis for further discussion in the next meeting of the Task Force.
b) Few persons would be invited to present their views on the Zero Draft.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
5959595959
6.0 The meeting ended with a vote of thanks to the Chair.
List of the Participants who attended the 2nd meeting of the Task Force on Pharma Sector held on 15th June, 2004 under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR in Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi.
S. No. Name of the participants
1. Dr. R.A. Mashelkar, Chairman 2. Dr. Prodipto Ghosh, Secretary (E&F) 3. Dr. M.K. Bhan, Secretary, DBT 4. Mrs. Veena Chhotray, AS, MoEF & Chairperson GEAC 5. Shri Ashwini Kumar, DG, DCGI 6. Shri D.D. Verma, JS, MoEF 7. Dr. Amit Ghosh, Director, IMTECH & Co-Chairman GEAC 8. Dr. C.M. Gupta, Director, CDRI 9. Dr. Vasantha Muthuswamy, ICMR
10. Dr. Samir Sangitrao, Representative of ABLE/ Intas Pharmaceutical 11. Shri K.K. Tripathi, Advisor, DBT 12. Dr. T.V. Ramaniah, Scientist-F, DBT 13. Dr. R. Warrier, Additional Director, MoEF 14. Shri Bhagirath Chaudhary, FICCI 15. Dr. Krishna Ella, FICCI 16. Shri D.B. Patankar, CII 17. Shri N.S. Yadav, Representative of IPA (Dr. Reddy) 18. Shri Raghu Cidambi, Representative of IPA (Bharat Biotech) 19. Col. L.J.S. Gill, Representative of IPA (Wockhardt)

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6060606060
F. No. 12/7/2004 - CS Government of India
Ministry of Environment & Forests (CS Division)
Paryavaran Bhawan, CGO Complex, Lodhi Road, New Delhi
E-mail: [email protected] Telefax: 2436 3964, 2436 1613
Dated 22nd June 2004
OFFICE MEMORANDUM
Subject: Drafting Committee for preparation of Draft Report on the recommendations of the Task Force on Recombinant Pharma Sector.
1. In accordance with the decision taken in the 2nd Meeting of the Task Force under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR on 15th June 2004, it has been decided to set up a drafting Committee for preparation of the Draft Report on the recommendations of the Task Force on Recombinant Pharma Sector.
2. The composition of the Task Force shall be as follows: i. Dr. M. K. Bhan, Secretary DBT Chairman ii. Mr. D. D Verma, Joint Secretary MoEF Member iii. Dr. Amit Ghosh, Director IMTECH Member iv Dr. R. Warrier Additional Director, MoEF Member
3. The Drafting Committee shall address the following issues:
a) Review the current regulatory process under the “Rules for Manufacture, Use, Import, Export and Storage of hazardous micro organisms / Genetically Engineered Organisms or Cells, 1989” notified under the Environment (Protection) Act, 1986 in terms of the regulatory objective, decision rule and information requirement of each regulatory body during the various stages of R & D, testing, manufacture, import and use.
b) Propose amendments in the regulatory mechanism and process for the use of recombinant organisms in the Pharmaceutical Industry during the various stages of R & D, testing, manufacture, import and use with a view to introduce transparency and eliminate duplicity of approvals.
c) Revised formats for submission of information to various regulatory agencies.
Annexure-Annexure-Annexure-Annexure-Annexure-VIVIVIVIVI

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6161616161
4. In developing the regulatory mechanism/process the Drafting Committee Task Force shall take into account the views expressed by various members of the Task Force and other experts as well guidelines and best practices adopted by other countries.
5. Dr. O.P. Agarwal, Head (RDPD) will provide inputs on behalf of CSIR/Dr. Amit Ghosh (who is out of Delhi) as and when needed. Dr. Rajendra Prasad, Scientific Secretary to DG, CSIR will provide an appropriate link with the Chairman for the Drafting Committee.
6. The Drafting Committee shall submit its report by 30th June 2004.
7. The Chairman may also co-opt persons with required expertise as and when required.
8. The expenditure on TA/DA for non-official members of the Drafting Committee and other contingency expenditure will be met from the Biodiversity Conservation Scheme.
(R. Warrier) Additional Director
To, The Chairman & Members of the Drafting Committee Copy to: The Chairman & Members of the Task Force

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6262626262
Annexure-Annexure-Annexure-Annexure-Annexure-VIIVIIVIIVIIVII
Minutes of the third meeting of the Task Force on recombinant Pharma Sector held on 3rd September 2004.
1.0 The third meeting of the Task Force on recombinant Pharma Sector was held on 3rd September 2004 under the Chairmanship of Dr R. A. Mashelkar, Director General CSIR at Department of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi. List of participants is annexed.
2.0 At the outset, the Chairman welcomed the members and thanked Secretary (DBT) for his initiative in drafting the ‘Draft Report on r-Pharma’. He congratulated the members of the Drafting Committee for an excellent draft report, which had optimally addressed the concerns of the industry and regulators.
3.0 He initiated the meeting by inviting Mr. D.D. Verma, JS, MoEF to make a presentation on the salient features of the Draft Report on r-Pharma. To begin with Mr. D.D. Verma, JS, MoEF briefed the Committee on the approach adopted by the Committee while drafting the recommendations and accordingly, two sets of recommendations have been made. The first set of recommendations would address streamlining of the current regulatory process and the second set would address the long-term objective of setting up a National Biotechnology Regulatory Authority/ Commission. The presentation made by JS (MoEF) covered the recommendations of the Drafting Committee on the following aspects:
A. Streamlining of the Current Regulatory Process.
1. Regulatory objective and functions of various regulatory agencies.
2. Protocols for five scenarios.
− Development/ production/ release of r-drugs derived from the use of LMOs through R&D within the country.
− Development/ production/ release of r-drugs where end product is LMO through R&D within the country.
− Import and marketing of LMOs as drugs in finished formulation. − Import and marketing of LMOs as drugs in bulk for making finished formulation. − Import and marketing of drugs derived from LMOs in bulk and or finished formulation.
3. Timelines
4. Inter- Ministerial Standing Committee.
B. National Biotechnology Regulatory Authority/ Commission.
4. After the presentation by JS (MoEF), Dr. R. Warrier, Additional Director, MoEF, introduced the recommendations of the Drafting Committee on the five protocols and the rational behind it. The Chairman invited the Members to give their comments on the recommendations made by the Drafting Committee. Views of the Members on the respective protocols are summarized below:

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6363636363
Dr. Kiran Mazumdar, Nominee of ABLE made the following suggestions:
a) GEAC should take note/approve containment facilities for the use of LMOs for a scale greater than 20 l only for commercial scale operations.
b) GEAC should not be involved in approving containment facilities for the use of LMOs if the activity pertains to R&D irrespective of the scale of the facility. IBSC should have this approval function upto R&D phase. The above system will enable R&D programs to continue in an uninterrupted manner. GEAC approvals will introduce an additional time of 30-45 days for R&D scale-up. IBSC, being closely involved with the R&D program will eliminate this likely delay.
c) GEAC should approve containment facilities for the use of LMOs for commercial scale production in a facility specific rather than a product specific manner. She further explained that if a Company qualifies with respect to one product then it should be assumed that if the same facility is used for other products, the qualification should hold good. It is a common practice for companies to use the same plant for multiple products. Even USFDA provides such facility approvals.
d) To a query from Dr. Bhan, Secretary, DBT, it was clarified that the kind of rigorous inspection conducted by USFDA is not being practices in India and therefore, environmental implications can be addressed only by creating stringent inspection facilities.
She also briefed the Task Force on the recommendations received from Dr. Varaprasad Reddy, M/s Shantha Biotechnics. The following suggestions made by Dr. Reddy were noted:
a) The definition of LMOs should exclude organisms modified by non-recombinant methods.
b) Initial R&D work often requires growing LMOs at more than 20 l scale. RCGM should, therefore, have authority to approve experimental work upto 100 l scale. GEAC should be involved only if the culture volumes exceed 100 l.
c) Approval of clinical trials wherein LMO itself is used as a drug is best done by DCGI supported by a committee consisting of such experts as well as nominees of Ministry of Environment.
d) Import of LMOs and recombinant nucleic acids may be approved by an appropriate authority in DBT or by RCGM directly.
e) Pre clinical data on all indigenously developed products derived from/ based on LMOs are evaluated by RCGM, whereas for all such imported products (except when the LMOs are imported as bulk and formulated), the evaluation of pre clinical and clinical data is done by DCGI only. There is no rationale for this discrimination.
Dr. Prodipto Ghosh, Secretary, MoEF:
a) On the definition of LMOs, he clarified that LMOs will include only those organisms modified by recombinant methods. MoEF in this regard would issue necessary amendments.
b) On the issue of GEAC according approval for containment facilities beyond 20 l fermentation capacity, he was of the view that the GEAC’s role is not specifically limited to certification. GEAC would still be concerned with any accidental release and associated risks. Further the expected risk should take into

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6464646464
account the probability of accidental release as well as potential damages likely to occur. The regulatory objective of GEAC is to evaluate the net societal benefits vs. risks where an environmental risk is less than the predetermined environmental risks and therefore, the information required to make that decisions should be made available to the GEAC. This would include process and product specifics.
c) The probability of occurrence of an accidental release of an LMO during the R&D stage being low, the GEAC may not be necessarily involved. However, for all operations involving the use of an LMO at a commercial level would mandate the approval of GEAC.
Dr. Amit Ghosh, Director, IMTech and Co-Chairman GEAC:
a) The consequences of an impact/ environmental risks arising from the use of LMOs for a given fermentation capacity would be the same irrespective of the fact that the LMO is used for research purpose or for commercial production. Therefore approval of GEAC should be linked to the risk involved rather than the nature of the activity.
Dr. K.K. Tripathi, Advisor, DBT:
a) Regarding the issue of GEAC approving containment facilities for the use of LMOs for commercial scale production in a facility specific rather than a product specific manner, he informed that the USFDA is recommending that the facility for a product should be dedicated keeping in view the GMP/ GLP.
Mrs. Veena Chhotray, Additional Secretary, MoEF & Chairperson GEAC:
a) The scope of 1989 Rules would need to be redefined in several important aspects eg. Definition of LMOs, empowering DCGI under 1989 Rules or evoking exemption clause etc.
b) The role of IBSC in the protocols needs to be amended to be in consonance with the existing DBT guidelines.
c) The responsibility of DCGI could also include Post Market Surveillance.
d) The Timelines suggested in the draft report may not be realistic due to various operational issues.
e) Since the responsibility of according market authorization for recombinant drugs is being entrusted to DCGI, the need for strengthening the Committees under the Drugs & Cosmetics Rules should be emphasized.
5. After detailed deliberations the Protocols were adopted subject to the following amendments:
Protocol - I Development/ production/ release of r-drugs derived from the use of LMOs through R&D within the country.
a) For risk category I & II, the RCGM will approve the containment facilities and pre clinical data and make its recommendations to DCGI. The stipulation of clearance for containment facilities > 20 l is not considered necessary.
b) For risk category III & IV the GEAC clearance is mandatory. This would include the process as well as product approval. The GEAC would take a final view on the proposal only after receipt of the

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6565656565
recommendations from RCGM on the containment facilities and from DCGI on the Phase-III clinical trials data.
c) Before the final approval by DCGI for commercial release, environmental clearance from GEAC should be made mandatory. *
d) The role of DCGI would include Post Market Surveillance. *
Protocol II Development/ production/ release of r-drugs where end product is LMO through R&D within the country.
The members of the Task Force adopted the Protocol recommended by the Drafting Committee.
Protocol III Import and marketing of LMOs as drugs in finished formulation. The members of the Task Force adopted the Protocol recommended by the Drafting Committee.
Protocol IV Import and marketing of LMOs as drugs in bulk for making finished formulation. The members of the Task Force adopted the Protocol recommended by the Drafting Committee.
Protocol V Import and marketing of drugs derived from LMOs in bulk and or finished formulation. The members of the Task Force adopted the Protocol recommended by the Drafting Committee.
6. Other General Recommendations:
1. The definition of LMOs will include only those organisms modified by recombinant methods. MoEF in this regard would issue necessary amendments.
2. On the recommendations of the Drafting Committee regarding the constitution of an ‘Inter- Ministerial Standing Committee’ to address various anomalies in the interpretation of the recommendations/ guidelines/ rules/ acts etc, it was agreed that this Committee would be serviced by DBT.
3. The Members adopted the timelines recommended by the Drafting Committee for according approvals.
4. It was agreed that there is a need to set up an Inter Ministerial group to examine the feasibility of setting up a National Biotechnology Regulatory Authority/ Commission and makes specific recommendation with respect to its implication and budgetary requirements.
5. Specific recommendation for strengthening the various regulatory agencies should be included in the report of the Task Force.
6. There is a need for creation of an independent inspection facility to audit the manufacturing and containment facilities set up by the companies involved in the production of recombinant drugs. This would also ensure acceptability of the Indian pharmaceutical companies in the global market.
7. Concluding remarks of Chairman: Appreciating the frank and positive discussions in the meeting, the Chairman suggested the following action points for completing the work assigned to the Task Force by 30th September 2004.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6666666666
a. The Drafting Committee would suitably amend the draft report to incorporate the adopted recommendations and circulate it to the members of the Task Force for their acceptance within a week time.
b. The amended draft to be put on the MoEF website for inviting comments from the various stakeholders and public.
c. An advertisement would be put up in the National Newspaper informing the public about the availability of the draft report on MoEF website for their comments within a stipulated time.
d. The Task Force would be reconvened after due consultation process has been completed, during the last week of September for final acceptance of the Draft Report.
Dr. Bhan, Secretary DBT pointed out the absence of the representatives from Ministry of Health, DCGI and ICMR. Considering the importance of their participation in the streamlining exercise, he suggested that the Chairman may like to take up the matter with Secretary Ministry of Health and DG, ICMR/ DCGI to ensure their participation in the next Task Force meeting. The Chairman agreed with the suggestion.
8. Vote of Thanks The meeting ended with a vote of thanks to the Chair.
List of the Participants who attended the 3rd meeting of the Task Force on Pharma Sector held on 3rd September 2004 under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR in Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi.
S. No. Name of the participants
1. Dr. R.A. Mashelkar, Chairman
2. Dr. Prodipto Ghosh, Secretary (E&F)
3. Dr. M.K. Bhan, Secretary, DBT
4. Mrs. Veena Chhotray, AS, MoEF & Chairperson GEAC
5. Shri D.D. Verma, JS, MoEF
6. Dr. Amit Ghosh, Director, IMTECH & Co-Chairman GEAC
7. Ms. Kiran Mazumdar Shaw, Representative of ABLE
8. Shri K.K. Tripathi, Advisor, DBT
9. Dr. T.V. Ramaniah, Scientist-F, DBT
10. Dr. R. Warrier, Additional Director, MoEF
11. Dr. Rajendra Prasad, Scientific Sec. To DG CSIR
12. Ms. Sandhya Tewari, Director, CII

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6767676767
Minutes of the fourth meeting of the Task Force on recombinant Pharma Sector held on 3rd December 2004.
1.0 The fourth meeting of the Task Force on recombinant Pharma Sector was held on 3rd December 2004 under the Chairmanship of Dr R. A. Mashelkar, Director General CSIR at CSIR Headquarters, Anusandhan Bhawan, 2, Rafi Marg, New Delhi. List of participants is annexed.
2.0 At the outset, the Chairman welcomed the members and informed the Committee that this meeting has been specifically convened with a view to discuss and finalize the revised draft report. He initiated the meeting by inviting Mr. D.D. Verma, JS, MoEF to make a presentation on the salient features of the revised Draft Report on r-Pharma.
3.0 To begin with Mr. D.D. Verma, JS, MoEF briefed the Committee on the discussions and recommendations accepted in the third meeting of the task force held on 3.9.2004. He informed the Committee that in the previous meeting of the Task Force the general recommendations with respect to the regulatory objectives and functions of the regulatory bodies, time lines and constitution of an inter-ministerial standing committee were accepted by the members of the Task Force. However, extensive discussions were held on the substantive issues related to the five protocols and accordingly the protocols have been suitably amended. It was further explained that in the meeting held on 3.9.2004, substantive changes were suggested only with respect to protocol I and in the remaining protocols there was a general consensus and only minor changes have been made in protocol II, III, IV & V. He, therefore, proposed that only the following five protocols be taken up in this meeting for getting the views of the members of the task force.
− Development/ production/ release of r-drugs derived from the use of LMOs through R&D within the country.
− Development/ production/ release of r-drugs where end product is LMO through R&D within the country.
− Import and marketing of LMOs as drugs in finished formulation.
− Import and marketing of LMOs as drugs in bulk for making finished formulation.
− Import and marketing of drugs derived from LMOs in bulk and or finished formulation.
4.0 After the brief introduction by JS (MoEF), Dr. R. Warrier, Additional Director, MoEF, introduced the revised protocols and the rationale behind each of them. The Chairman invited the Members to give their comments on the recommendations made by the Drafting Committee. Discussion on the respective protocols are summarized below:
Protocol I Development/ production/ release of r-drugs derived from the use of LMOs through R&D conducted within the country.
Dr Warrier explained that protocol I pertains to use of LMOs in the manufacture and production of recombinant drugs within the country but the end products per se do not contain LMOs. This protocol has been divided into parts based on the risk factor and level of containment required and accordingly separate step wise procedure has been developed for the use of Risk Group I & II category and Risk Group III and above. In the case of Risk Group
Annexure-Annexure-Annexure-Annexure-Annexure-VIIIVIIIVIIIVIIIVIII

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6868686868
I & II, the role of GEAC has been eliminated. However, for Risk Group III & IV, environmental clearance from the GEAC would be mandatory. During the deliberations, various issues regarding the role of GEAC for > 20 L fermentation capacity, authorization levels for phase-I & II clinical trials and the stage at which the GEAC clearance would be required for Risk Group III & IV were discussed. The following clarifications were submitted to the Committee:
a) The new proposal eliminates the current practice of obtaining the approval of GEAC for >20 L fermentation even for commercial manufacturing.
b) The new proposal also eliminates the role of GEAC for Risk Group I & II microorganism in Protocol I.
c) The regulatory agency to authorize Phase I & II clinical trials would be the DCGI, for the products from the Risk Group I and II LMOs.
d) Since the end product does not contain LMOs in Protocol I, approval of GEAC is not required for Phase –III clinical trials irrespective of the Risk category.
e) However, for Risk Group III & above where environmental clearance from GEAC is required, the GEAC would take into account the results of Phase –III clinical trials and recommendation of RCGM on the containment facility while evaluating the environment risk vs. benefit.
With the above clarifications, the Protocol I was accepted. It was agreed that detailed stepwise procedure for the protocol would be prepared wherein the above issues would be made explicit in the final report.
Protocol II Development/ production/ release of r-drugs where end product is LMO per se through R&D conducted within the country.
It was explained that Protocol II pertains to the manufacture and production of recombinant drugs within the country where the end product per se is an LMO and therefore would require the approval of GEAC prior to its release for Phase III clinical trials and market authorization under the 1989 Rules.
To a query from DG ICMR on the need for approval of GEAC prior to conduct of Phase–III clinical trials, it was clarified that the regulatory objective / mandate of DCGI and GEAC being different, the applicant will have to comply with both the statutory requirement under Drugs and Cosmetic Rules as well as 1989 Rules under EPA. While the DCGI is responsible for assessing the safety and efficacy of the product, the GEAC is responsible for assessing the environmental impact due to large-scale use/release of LMOs in the environment. Since the phase III clinical trials are conducted in a wider population for assessing the safety and efficacy of the product, these trials do not fall under the category of limited contained trials and therefore the probability of environmental release is more in such case. Further the GEAC would also like to ensure that the adequacy of the containment facility and infrastructure available with the company is adequate to handle the safe use of LMOs at the initial stage itself. However, it was agreed that the DCGI and GEAC clearances need not be interlinked and both the regulatory agencies can process the case concurrently. It was also clarified that the regulatory agency for authorizing Phase I & II clinical trials would be the DCGI.
It was suggested by some members that the flow chart for protocol II with respect to approval of DCGI and GEAC for Phase III clinical trials may be suitably reworded to reflect the above discussions. The role of DCGI and GEAC may also be made explicit in the protocol to avoid any ambiguity. With the above modification, the members of the Task Force adopted the Protocol II. *

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
6969696969
Protocol III Import and marketing of LMOs as drugs in finished formulation.
The members of the Task Force adopted the Protocol III.
Protocol IV Import and marketing of LMOs as drugs in bulk for making finished formulation.
The members of the Task Force adopted the Protocol IV.
Protocol V Import and marketing of drugs derived from LMOs in bulk and or finished formulation.
The members of the Task Force adopted the Protocol V.
6. Other General Recommendations:
1. The products emanating from mono-clonals prepared involving rDNA technology in the form of therapeutic proteins/drugs would attract the provisions of Rule 1989, and can be treated under Protocol I.
2. Enzymes /industrial products from GMOs would attract the provisions of Rule 1989. In such cases, RCGM may be authorized to approve such proposals under intimation to GEAC.
3. Once RCGM permission is sought for preclinical studies or bio-containment facilities, no further change in the host organism or expression construct should be allowed. If there is a change in the host organism or expression construct, fresh permission from RCGM should be sought for conducting preclinical studies, as well as for bio-containment facilities. A new clinical study should also be required if the host organism or expression construct is changed.
4. For import of GMO / LMO for research/contract manufacturing or similar service, where the product (which is not an LMO) is to be exported out of India, a procedure should be laid down so that the companies can explore opportunities for this business while the safety aspect is also adequately addressed. A suggested procedure is: IBSC to examine proposal and recommend to RCGM; RCGM to approve if within Risk Group I and II. If organism is of Risk Group III or above, GEAC permission will be required. DCG(I) need not play any role.
5. Detailed explanatory note on the rationale behind each recommendation would be provided in the textural part of the final report.
7. Concluding remarks of Chairman: Appreciating the frank and positive discussions in the meeting, the Chairman suggested the following action points:
a. The Drafting Committee would suitably amend the draft report to incorporate the adopted recommendations and the amended draft final report would be put on the MoEF website for inviting comments from the various stakeholders and public by end of December 2004.
b. The Task Force would be reconvened after due consultation process has been completed, during the last week of January 2005 for final acceptance of the Draft Report.
8. Vote of Thanks The meeting ended with a vote of thanks to the Chair.
*******

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7070707070
List of the Participants who attended the 4th meeting of the Task Force on Pharma Sector held on 3rd December 2004 under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR in Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi.
S. No. Name of the participants
1. Dr. R.A. Mashelkar, DG CSIR & Chairman
2. Dr. Prodipto Ghosh, Secretary (E&F)
3. Dr. M.K. Bhan, Secretary, DBT
4. Mr. Suresh Chandra, Special Secretary, MoEF & Chairperson GEAC
5. Dr. N. K. Ganguly, DG ICMR
6. Shri D.D. Verma, JS, MoEF
7. Ms. Kiran Mazumdar Shaw, Biocon & Representative of ABLE
8. Shri K.K. Tripathi, Advisor, DBT
9. Dr. T.V. Ramaniah, Scientist-F, DBT
10. Dr. R. Warrier, Additional Director, MoEF
11. Mr. A. B. Ramtake,
12. Ms. Sandhya Tewari, Director, CII
13. Shri D. B. Patankar, Intas Pharmaceuticals & representative of CII
14. Shri R. K. Bhatia, FICCI
15. Shri Vishal Gandhi, FICCI
16. Dr. Raghu Cidambi, Dr. Reddy’s Laboratories & representative of IPA
*******

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7171717171
Minutes of the fifth meeting of the Task Force on recombinant Pharma Sector held on 15th June 2005.
1.0 The fifth meeting of the Task Force on recombinant Pharma Sector was held on 15th June 2005 under the Chairmanship of Dr R. A. Mashelkar, Director General CSIR at CSIR Headquarters, Anusandhan Bhawan, 2, Rafi Marg, New Delhi. List of participants is annexed.
2.0 At the outset, the Chairman welcomed the Members. He informed the Committee that subsequent to the meeting held on 3rd December 2004, the draft final report was placed on MoEF website on 9th March 2005 upto 15th April 2005 for inviting comments from various stakeholders and comments have been received only from CII and DCGI. He congratulated members of the Task Force and Drafting Committee for an excellent report, which had optimally addressed the concerns of the industry and regulators. He further informed that this is the beginning of a long-term initiative and the matter does not end with the acceptance of the report.
3.0 The Chairman initiated the meeting by inviting Shri Desh Deepak Verma, Joint Secretary, MoEF, to make a presentation on the salient features of the revised Draft Final Report on r-Pharma. To begin with Joint Secretary, MoEF briefed the Committee on the discussions and recommendations accepted in the fourth meeting of the task force held on 3.12.2004. This was followed by a presentation by Dr Warrier. In her presentation she explained the structure of the report and changes incorporated therein. She further introduced the final recommendations of the Task Force and the rationale behind each of them.
4.0 The Chairman, opening the floor for discussion on the draft final report, requested Dr Warrier to take up the comments from CII and DCGI for consideration of the Committee. Details of the deliberations on various issues are summarized below:
A. Structure of the report: Dr Warrier informed the Committee that for further clarity section 5.1 on the role of the regulatory agencies has been shifted and introduced after section 5.3 relating to the step wise procedure/protocols for the five categories. Some of the Members suggested certain editorial changes, which was noted for appropriate incorporation in the report.
B. Regulatory objective and functions of various regulatory agencies. The Committee noted the comments received from CII wherein it was stated that the role of RDAC in the DCGI office is not addressed. Therefore, all the functions of RDAC should be included under section 5.1 (d). The committee accepted the above suggestion.
C. Step wise Procedure/Protocols for five categories
Protocol I Development/ production/ release of r-drugs derived from the use of LMOs through R&D conducted within the country.
Representative of DCGI pointed out that it was agreed by the Task Force and MoEF that GEAC will confine its regulatory role in terms of product approval only if the end products are LMOs. However, in
Annexure-IXAnnexure-IXAnnexure-IXAnnexure-IXAnnexure-IX

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7272727272
para 5.2.2, a (5) at page 14, it has been mentioned that proposal involving use of LMOs under risk Group III & above, requires approval of GEAC. Since Protocol I pertains to products where end products are not LMOs, DCGI recommends that approval of GEAC may not be necessary in this case.
It was clarified that in the 2nd meeting of the Task Force held on 15th June 2004, various issues regarding the role of GEAC for > 20 L fermentation capacity, authorization levels for phase-I & II clinical trials and the need for GEAC clearance for Risk Group III & IV were discussed. While it was agreed that there is no rational for regulating > 20 L fermentor capacity, some Members were of the view that consequences of an impact/ environmental risks arising from the use of LMOs for a given fermentation capacity would be the same irrespective of the fact that the LMO is used for research purpose or for commercial production. Therefore approval of GEAC should be linked to the risk involved rather than the nature of the activity. Accordingly it was agreed that proposal involving use of LMOs under risk Group III & above, requires approval of GEAC.
The Committee further noted that Protocol I in which category maximum number of proposals coming before the GEAC fall, have been streamlined to the extent that:
The new proposal eliminates the current practice of obtaining the approval of GEAC for >20 L fermentation even for commercial manufacturing.
The new proposal also eliminates the role of GEAC for Risk Group I & II microorganism in Protocol I.
The regulatory agency to authorize Phase I & II clinical trials would be the DCGI, for the products from the Risk Group I and II LMOs.
Since the end product does not contain LMOs in Protocol I, approval of GEAC is not required for Phase –III clinical trials irrespective of the Risk category.
With the above clarifications, the Task Force adopted Protocol I without any amendment.
Protocol II Development/ production/ release of r-drugs where end product is LMO per se through R&D conducted within the country.
The members of the Task Force adopted Protocol II.
Protocol III Import and marketing of LMOs as drugs in finished formulation.
The members of the Task Force adopted Protocol III.
Protocol IV Import and marketing of LMOs as drugs in bulk for making finished formulation.
The members of the Task Force adopted Protocol IV.
Protocol V Import and marketing of drugs derived from LMOs in bulk and or finished formulation.
The representatives of CII and DCGI, pointed out that in para 5.2.2, e, 2 (pg.17), it has been recommended that views of RCGM may be obtained on the process, impurity profile, pre-clinical and clinical trial data prior to approving human clinical trials. They were of the view that this recommendation would create an additional window at DBT for clearing the proposal as against the objectives of rationalizing the regulatory approval. It was suggested that the above functions may be confined to RDAC.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7373737373
It was pointed out that DCGI in their comments (no 5) have specifically requested for clubbing functions of RCGM with those of RDAC to strengthen the functioning of RDAC. The Chairman, referring to a letter from Secretary, Ministry of Health to Secretary DBT requesting for support of RCGM in the evaluation of r- DNA products by DCGI, stated that the matter may be reviewed after RDAC has been adequately strengthened.
After detailed deliberations, the Task Force adopted Protocol V without any amendment.
D Timelines for approvals. To a suggestion from CII that specific timelines for RDAC approval should be defined it was agreed that a uniform time frame cannot be stipulated for all products. However, Members were of the view that the matter cannot be left open-ended and specific timelines need to be stipulated. It was agreed by the representative of DCGI that a time frame of 90 days could be considered.
In response to a suggestion from Secretary E& F it was agreed that the title of section 5.4 be amended to ‘Time lines for Decision” instead of ‘Time lines for Approvals’.
On the issue of timelines for GEAC decision, Joint Secretary explained that the GEAC has adopted the ‘Guidelines for Good Practices in Environmental Regulations” formulated by MoEF. These have been put into practise since April 2004 and have been found to be very effective. On behalf of the GEAC/ MoEF, he assured the Committee that timelines adopted by GEAC would be compliant with the “Good Practices in Environmental Regulations”. The Committee appreciated the initiative taken by MoEF in this regard.
E. Documentation to be submitted by the applicant to the regulatory authorities for obtaining clearances. The Task Force observed that documents to be submitted by the applicant to the IBSC, RCGM and GEAC under the ‘Rules 1989’ have been evolved and is adequate for taking a view on the proposals. However, there is a need for evolving a format for submission of documents for r-DNA products to DCGI. The Committee advised DCGI to take necessary action at the earliest.
F Recommendations on other Linked issues: With respect to recommendation no 4 under section 5.6 regarding the need for creating an independent inspection facility to audit the manufacturing and containment facilities through out the project life- cycle, views were expressed that an agency/organization be identified and recommended by the Task Force for implementing the above recommendation. After detailed deliberations, it was concluded that as of date there is no single agency with adequate field level support system to carry out an independent inspection. It was therefore decided to recommend that a separate agency may be set up by the Government for this purpose.
G. Inter-ministerial Standing Committee on Biotechnology Regulation. In response to a suggestion from MoEF, it was agreed that the Standing Committee recommended by the Task Force at section 5.7 should comprise of an expert body instead of an inter-ministerial body, as the issues involved are highly technical and complex. Accordingly it was agreed that the title of section 5.7 be amended as ‘Standing Technical Advisory Committee on Biotechnology Regulation’.
5.0 After detailed deliberations, the draft final report of the Task Force and recommendations made therein were accepted with the above-discussed amendments.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7474747474
6.0 Concluding remarks of Chairman: Appreciating the positive outcome of the meeting, the Chairman desired to incorporate the adopted recommendations and submit the final report within two weeks after suitable editorial amendments. In his closing remarks, the Chairman thanked Secretary E & F for setting up the Task Force and for the support given by MoEF to complete the work assigned to the Committee. He thanked Secretary DBT and congratulated members of the Drafting Committee for the excellent report. Thanking the Members of the Task Force he stated that their co-operation in resolving controversial issues has significantly contributed in finalizing the report and making his task easier. Secretary E&F on behalf of MoEF thanked the Chairman for his stewardship and valuable guidance in finalizing the report. He thanked the members of the Task Force and assured the Committee that MoEF will take all measures to implement the recommendations of the Task Force. Dr K K Tripathi, Advisor DBT, on behalf of Secretary DBT and Chairman of the Drafting Committee thanked the Chairman and Secretary E & F for this initiative and interest taken in finalizing the report. Members of the industry association thanking the Chairman and Secretary E& F and stated that they were happy with the outcome of this initiative and measures recommended by the Task Force for streamlining the current regulatory framework on r-Pharma.
7.0 The meeting ended with a vote of thanks to the Chair.
*******
List of the Participants who attended the 5th meeting of the Task Force on Pharma Sector held on 13th June 2005 under the Chairmanship of Dr. R.A. Mashelkar, DG, CSIR in Dept. of Scientific & Industrial Research, Anusandhan Bhawan, 2, Rafi Marg, New Delhi.
S. No. Name of the participants
1. Dr. R.A. Mashelkar, DG CSIR & Chairman 2. Dr. Prodipto Ghosh, Secretary (E&F) 3. Shri D.D. Verma, JS, MoEF 4. Dr Amit Ghosh, IMTech 5. Dr Vasantha Muthuswamy, ICMR 6. Shri K.K. Tripathi, Advisor DBT 7. Dr. T.V. Ramaniah, Director DBT 8. Mr. A. B. Ramtake, DCGI 9. Shri D. B. Patankar, representative of CII
10. Ms. Jaya Goyal, CII 11. Mr. C. Omprakash, representative of ABLE 12. Shri R. K. Bhatia, FICCI 13. Shri Vishal Gandhi, FICCI 14. Dr. R. Warrier, Additional Director, MoEF

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7575757575
A. PROFORMA FOR SUBMITTING APPLICATION TO IBSC FOR IMPORT/ EXCHANGE OF GMOS/ LMOS AND PRODUCTS THEREOF FOR RESEARCH AND DEVELOPMENT PURPOSES
1. Name of the Applicant Designation (a) Address (Registered Office)
Telephone No.
Telex No.
Fax No.
(b) Address (Research Station)
Telephone No.
Telex No.
Fax No.
2. Application for (to indicate the purpose) :
3. Description of the GMOs/ LMOs/ product thereof (in scientific terms in narrative manner providing information on (a) Morphology; (b) Number of copies of the genes incorporated; (c) Status of approval in country of origin)
4. Source of GMOs/ LMOs Products thereof:
Name of the Agency
Contact person’s
Address
Telephone No.
Telex No.
Fax No.
5. Quantity of GMOs/ LMOs/ products thereof to be Imported/ exchanged:
Annexure-Annexure-Annexure-Annexure-Annexure-XXXXX

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7676767676
6. Details on:
(a) Source of nucleic acid(s) :
(b) Nucleic acid sequence (Please enclose the nucleic acid
sequence map of the target gene) :
(c) Vector(s) (Please enclose the map of the vector):
(d) Sequence of the genes incorporated/ to be incorporated into the host organism.
(e) Host(s) that carrying the vector(s)/ target gene(s) :
(f) Manipulative procedures in outline :
7. Objectives of the proposal:
8. Summary of the proposed work plant utilizing GMOs/ LMOs/ products thereof:(This should indicate schematic lab work, green house whenever applicable)
9. Mode of shipment :
10. Information on containment facilities installed at R&D and production premises (whichever is applicable)
11. Standard Operating Procedures (SOP) on decontamination, disposal mechanisms & risk management (in brief) :
12. Any other relevant points(s)
13. Declaration :
I declare that the information provided in the above format is correct and accurate to the best of my knowledge. The “Biosafety Guidelines” brought out by the Department of Biotechnology, Ministry of Science & Technology, Govt. of India will be and is being strictly followed. The imported/ exchanged material will be utilized for the said purpose only. In case any untoward incident occurs, the Chairman of the IBSC and the Member- Secretary of the RCGM will be informed immediately.Place:Date: Signature of the Applicant
14. Recommendations:The proposal set out above has been considered by the “Institutional Biosafety Committee” in its meeting held on __________________ and is forwarded and recommended to RCGM for further necessary action.Place:Date: Signature of the Chairman, IBSC(Note: Please submit 20 copies of the application to the Department of Biotechnology for placing the same in the meeting of RCGM)

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7777777777
B. PROFORMA FOR SUBMITTING APPLICATION TO IBSC FOR IMPORT/ EXCHANGE OF GMOS/ LMOS AND PRODUCTS THEREOF FOR RESEARCH AND DEVELOPMENT PURPOSES
1. Name of the Applicant
Designation
(a) Address (Registered Office)
Telephone No.
Telex No.
Fax No.
(b) Address (Research Station)
Telephone No.
Telex No.
Fax No.
2. Application for (to indicate the purpose) :
3. Description of the GMOs/ LMOs/ product thereof (in scientific terms in narrative manner providing information on (a) Morphology; (b) Number of copies of the genes incorporated; (c) Status of approval in country of origin)
4. Source of GMOs/ LMOs Products thereof:
Name of the Agency
Contact person’s
Address
Telephone No.
Telex No.
Fax No.
5. Quantity of GMOs/ LMOs/ products thereof to be
Imported/ exchanged:

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7878787878
6. Details on:
(g) Source of nucleic acid(s) :
(h) Nucleic acid sequence (Please enclose the nucleic acid sequence map of the target gene)
(i) Vector(s) (Please enclose the map of the vector):
(j) Sequence of the genes incorporated/ to be incorporated into the host organism.
(k) Host(s) that carrying the vector(s)/ target gene(s) :
(l) Manipulative procedures in outline :
7. Objectives of the proposal:
8. Summary of the proposed work plant utilizing GMOs/ LMOs/ products thereof:(This should indicate schematic lab work, green house whenever applicable)
9. Mode of shipment :
10. Information on containment facilities installed at R&D and production premises (whichever is applicable)
11. Standard Operating Procedures (SOP) on decontamination, disposal mechanisms & risk management (in brief) :
12. Any other relevant points(s)
13. Declaration :
I declare that the information provided in the above format is correct and accurate to the best of my knowledge. The “Biosafety Guidelines” brought out by the Department of Biotechnology, Ministry of Science & Technology, Govt. of India will be and is being strictly followed. The imported/ exchanged material will be utilized for the said purpose only. In case any untoward incident occurs, the Chairman of the IBSC and the Member- Secretary of the RCGM will be informed immediately.
Place:
Date: Signature of the Applicant
14. Recommendations:The proposal set out above has been considered by the “Institutional Biosafety Committee” in its meeting held on __________________ and is forwarded and recommended to RCGM for further necessary action.Place:Date: Signature of the Chairman, IBSC(Note: Please submit 20 copies of the application to the Department of Biotechnology for placing the same in the meeting of RCGM)

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
7979797979
PROFORMA FOR SUBMITTING APPLICATION TO IBSC TO CARRY OUT RESEARCH & DEVELOPMENT WORK ON GMOs/ LMOs/ r-DNA PRODUCTS
Application for clearance by IBSC/ approval of RCGM to carry out research involving genetic engineering activity for the development of r-DNA products.
1. Name of the Applicant
Designation
(a) Address (Registered Office)
Telephone No.
Telex No.
Fax No.
(b) Address (Research Station)
Telephone No.
Telex No.
Fax No.
2. Basic information on application:
2.1 Purpose
2.2 New Yes No
2.3 Ongoing Project Yes No
If yes, No. & Date(s) of previous Permit(s) issued :
2.4 If yes, briefly state the purpose for which permission(s) granted.
2.5 Category of experiments as per the Guidelines of DBT
3. Objectives of the proposal
4. Description of the GMOs employed in the research proposal (in scientific terms; for new application only)
4.1 Description of GMOs
4.2 Description of the target gene(s)
4.3 Number of copies of the genes incorporated
4.4 Description of the target gene product(s)
Annexure-Annexure-Annexure-Annexure-Annexure-XIXIXIXIXI

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8080808080
5. Details on:
5.1 Source of nucleic acid(s):
5.2 Nucleic acid sequence (Please enclose the nucleic acid sequence map of the target gene):
5.3 Vector(s) (Please enclose the map of the vector gene):
5.4 Host(s) that carrying the vector(s)/ target gene(s):
5.5 Manipulative procedures:
5.6 Anticipated functions of Product(s)
6. Summary of the proposed work plan utilizing GMOs (please check it from the following areas and provide the details of work plan).
6.1 Basic transformation and laboratory work to assess the expression of the target gene
6.2 Standardization of fermentation procedures below 20 lt. capacity (if applicable)
7. Assessment of toxicity and allergenicity of the product (if yes, please provide the following information)
i) Production / fermentation procedures adopted
ii) Purification procedures adopted; state briefly the processing chemicals used in the purification steps.
iii) Physico-chemical characterization of the product; please provide limits of residues with there characterization/ identification.
iv) Biochemical/immunological characterization of the product
v) Information on Five batches production data
vi) Toxicity and Allergenicity protocols and the address of the lab/ Institute where these studies are proposed to be conducted.
vii) Institutional Animal Ethics Committee’s Approval.
viii) Acceptability criteria of the bulk and the formulated material wherever ready for animal experiments.
8. Site/ Location of the research work:
9. Proposed containment facility (Please indicate the level of containment proposed and attach IBSC inspection report):
10. Standard operating procedures (SOPs) for decontamination and disposal mechanisms
11. Risk management measures practiced (Emergency plan)
12. Any other relevant information
13. Declaration:
I declare that the information provided in the above format is correct and accurate to the best of my knowledge. The “Safety Guidelines” brought out by the Department of Biotechnology, Ministry of Science & Technology,

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8181818181
Govt. of India will be and is being strictly followed. In case any untoward incident occurs, the Chairman of the IBSC and the Member-Secretary of the RCGM will be informed immediately.
Place:
Date: Signature of the Applicant
14. Recommendations:
The proposal set out above has been considered by the “Institutional Biosafety Committee” in its meeting held on ___________ and is forwarded to RCGM for further necessary action.
Place:
Date: Signature of the Chairman, IBSC
(Note: Please submits 20 copies of the application to the Department of Biotechnology for placing the same in the meeting of RCGM)

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8282828282
FORM – I
APPLICATION FOR ENVIRONMENTAL APPROVAL OF CLINICAL, VETERINARY AND FOOD PRODUCTS DERIVED FROM GENETICALLY MODIFIED ORGANISMS / HAZARDOUS
MICRO-ORGANISMS.
*****
Part A
(a) Not all the points included will apply to every case. It is to be expected, therefore, that individual applicant will address only the particular parameters that are appropriate to individual situations. In each case where it is not technically possible or it does not appear necessary to give the information, the reasons shall be stated.
(b) The details required in response to each parameter are also likely to vary according to the nature and scale of the proposed release.
(c) The description of the methods used or the reference to the standardized or internationally recognized methods shall also be mentioned in the form together with the name of the body or bodies responsible for carrying out the studies.
(d) A 1-2 page summary of the proposal shall be appended with application.
Part B
1. Name of the Applicant
2. Name of organization/firm
3. Approval required
(i) Manufacture
(ii) Import
(iii) Marketing
4. Quantity per year of the product to be manufactured/imported/marketed.
Part C
1. Name of the product:
(i) Human medicine
(ii) Veterinary medicine
Annexure-Annexure-Annexure-Annexure-Annexure-XIIXIIXIIXIIXII

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8383838383
(iii) Food product
(iv) Others (please specify)
2. Intended use (Pathological, Metabolic and Immune Response)
3. Safety Concern:
(i) Donor (heterologous Nucleic acid segments from sources)
(ii) Vector (DNA) molecules to which heterologous nucleic acid segments are joined for transfer to hosts)
(iii) Hosts (living cells or organisms into which rDNA molecules are introduced)
4. Production method (the production method should give the details of the cell lines used, the information on the production of the recombinant proteins within or outside the cells, the concentration of the products in units per litre of the fermented broth as well as the concentration in physical weights. The description should also include in brief the methods applied for reducing the genomic particles, proteins and living contaminants including viruses, bacteria, fungi, parasites. Etc.)
(a) Characterisation of the system of production used
(i) Details of the expression system:
Description of the host cell line
Identification of the genera and the species.
The risks involved in handling the cell line.
The classification of the cell line according to the Government of India’s Biosafety guidelines or any other accepted Recombinant cell line usage guideline.
The method(s) of maintenance and growth of the cell line.
The nature and hazards of using substrates, inducing agents, etc.
(ii) Characteristics of the target gene and vector
The full description of the source of the target gene.
The composition of the vector used indicating the promoter sequence as well as the regulatory mechanisms utilized in the expression cassette.
Schematic diagrams of the expression cassette to describe fully the marker genes used.
The restriction sites related to specific endo nucleases, and the cell lines used for shuttling and amplification of the expression cassette.
The method of constructing the target gene along with all the sequences added or deleted.
The extent of target gene amplification into the host genome.
The target gene should also contain, along with the nucleotide sequence, the description of the amino acids below the codons.
(iii) Approaches adopted for expression of the gene.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8484848484
The description of the transcripted messenger RNA with its analysis of sequence and identification procedure.
The translation information indicating whether the protein product is Chimaric, whether the expression is found as inclusion bodies or as intracellular protein or whether the protein is secreted out.
The extent of the target protein produced as the percentage of the total cell dry mass as well as its percentage compared to the total proteins of the cell.
The quantity of the target protein produced after the cell growth per litre of the fermented broth.
The full sequence of the recombinant gene alongwith the promoters the marker genes and the terminator se sequence.
(b) Description of the production process
(i) Production set up
The handling of the stock cell lines.
The evaluation and uses of the cell lines in pre-fermentor processes.
The preparation of the seed vessel.
The main production fermentation conditions need to be described in brief.
(ii) Growth Kinetics
Graphical plots of the versus substrate usage, optical density change, biomass formation, protein products formation, inducing agents used and their effects on the target protein formation.
The effects of change of standard parameters like pH dissolved oxygen, temperature etc. in the main production fermentor.
(iii) Fermentation parameters
The pH, temperature, aeration, and rpm of the shaft.
Volume of seed to the volume of main fermentor.
Main substrates used inducing agents used if any.
The fermentation time and the concentration of the target protein in the fermented broth.
(c) In process Control Methods
All the analytical methods used for this in-process control must be described to ensure the regulatory authorities that the chances of entry of unwanted products have been minimized.
(d) Description of the raw and processing materials used
Description of all the raw materials and processing materials used starting from the stock cells handling to the finished dosage form must be in a tabular form.
Grades of materials used and their usual sourcing.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8585858585
(e) Description of the plant and Machinery used
List of all the equipment alongwith the indicative capacity.
Names of the manufacturers.
List of main production equipment and the quality control equipment.
(f) Description of the building and facilities created for the manufacture
The manufacturing area.
The flow of the process/operation starting from the stock culture area to the finishing area.
The plot plan of various floors used in the manufacture and processing.
The building diagram in the plan and/or elevation in the outline indicating the cleanliness maintained in different sections such as class 10,000, class 1000 and class 100 areas etc.
Description of the air handling system which is very important.
A separate write-up indicating the movement of operating personnel in the area.
5. Approaches for the extraction and purification of the product. The purification methods should particularly indicate the modes of elimination of viruses, bacteria, fungi, parasites, etc., if the cell lines are mammalian origin. It should also include methods for the removal of genomic particles, and proteins irrespective of cell lines/hosts used for production, removal of adjuvants, reagents chemicals including vectors, donor and recipient organisms foreign DNA and adventitious materials associated with production methods.
The methods of handling the cells.
The methods of isolating the cell soup containing the target protein the methods of enzyme treatment, if any.
The methods of concentration, precipitation (if applied), reconstitution, salt separation ( if applicable), absorption and desorption methods, chromatographic methods used if any, ultracentrifugation methods if used.
Sterilization and final dosage formulation.
Processes to obtain the product in the finished dosage form.
6. Quality Control and Quality Assurance
(a) Bulk material:
Test relating to the identification of the stock cells.
The plasmid construct retention in the cells during cell growth.
The media used.
The procedures adopted for testing and the percentage retention of the target plasmid.
Description of the contamination test of the stock culture to assess and monitor the microbial contamination including the media used and the procedures adopted.
The criteria of acceptance of contamination free culture.
Characterisation of the bio-active molecule produced by monitoring physical & chemical properties of the molecules by the following or more authentic method should be provided.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8686868686
o Microscopic examination including light, phase contract and electron microscopic studies.
o UV Spectroscopic analysis to show the absorption spectra of the product and comparison of this with authentic material.
o Density gradient centrifugation to show single peak.
o Pattern in High performance liquid chromatography to indicate how many peaks or if only one peak is noticed in the produced and purified bulk.
o Iso-electric focussing analysis to show the pl values.
o Western Blot and SDS-PAGE to map the peptide/target protein.
o Existence of special bonds like disulfide bonds by breaking the protein and subjecting to SDS-PAGE mapping.
o Immunodiffusion Test to find out the absence of contaminants or the extent of the presence of contaminants.
o Amino acid composition of the purified protein and its comparison with the authentic material.
o N-Terminal Amino Acid Sequence analysis and its comparison with the gene construct used.
o Determination of the biological activity in the animal model.
o Determination of Contaminants per milligram or any convenient unit of the manufacturing bulk purified protein is also to be carried out to indicate the extent of contaminant nucleic acid stretches, proteins, carbohydrates, lipids, detergents, salts and other processing chemicals used in the purification. The impacts of the presence of these contaminants are also to be indicated with authentic references if any to ensure that the risks associated with their presence are minimal. The limits of contaminants and the acceptance criteria need to be quantified for each contaminant.
(b) Formulated Material:
The ingredients incorporated subsequently in the manufacture. The specification of the final product.
The Quality Control Department would have to certify that the final product has the results within the specifications prescribed and accepted by the Regulatory Authorities.
The documentation should therefore indicate the following specifically:
(i) Product presentation.
(ii) Physical appearance.
(iii) Product inserts, literature and label claim
(iv) Volume/Quantity per pack/dose.
(v) Potency of the product.
(vi) Particulate matter limits for liquid.
(vii) Preservative usage percentage.
(viii) PH
(ix) Other extraneous materials used their extent such as contents of DNA, RNA, Carbohydrates, Lipids, processing materials like detergents, salts, etc.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8787878787
(x) Protein content in the product.
(xi) Sterility status
(xii) Toxicity information
(xiii) Pyrogen status.
7. Possible Hazards to environment from release of GMO/nucleic acid/micro-organisms or products.
8. Clinical field trials done in India/abroad (whether permission of Institutional Ethics Committee obtained)
9. Stability and Shelf Life of product.
10. Method of disposal of vials of syringes/wastes matter.
11. Regulatory status in India/abroad (enclosed certificates like free sale certificate/GMP certificate/certificates granted by Health & Environment Authorities of the country of origins signed by applicant. In case the certificate is issued by the concerned authority of country of origin, the certificate should be endorsed/ authenticated by Indian Embassy/High Commission/Consulate in that country.
Every certificate shall be accompanied by other statutory information like manufacturing batch, no. date of manufacture, date of analysis, date of release of the certificate, signatory to the certificate etc.
The final formulated product needs to be certified for acceptance by the manufacturer.
Applicant’s signature with seal.

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8888888888
A Model for Single Window Biotechnology Regulatory Authority/Commission
This is in consonance with the recommendations of Prof. M.S. Swaminathan Task Force for Agriculture and Food Products as Indian Biotechnology Regulatory Authority (IBRA)
CHAIRMAN
Annexure-Annexure-Annexure-Annexure-Annexure-XIIIXIIIXIIIXIIIXIII
Apex Committee with Statutory Powers, consisting of all stakeholder Ministries/Departments
Vice-Chairman Secretariat
Agriculture Products/ Transgenic Crops
Pharmaceutical/Drugs and Industrial Products
Transgenicfoods/feed TransgenicAnimals/ aquaculture
The Secretariat will have Professionally competent and Experienced Technical Officers in relevant areas of specialization

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
8989898989
Details of the Secretariat and their expertise
Agriculture Products/ Transgenic Crops
Molecular biologist
Plant biologist
Plant Geneticist
Plant breeder
Agronomist
Plant Ecologist
Environment biologist
Agriculture economist
Environment biologist
Pharmaceutical/
Drugs and Industrial Products
Molecular biologist
Pharmacologist
Toxicologist
Epidemiologist
Microbiologist
Biochemist
Analytical Chemist
Physicist
Environment
biologist
Transgenic foods/ feed
Molecular biologist
Nutritionist
Pharmacologist
Toxicologist
Epidemiologist
Microbiologist
Biochemist
Analytical Chemist
Physicist
Environment
biologist
Transgenic Animals/ aquaculture
Molecular biologist
Animal Breeder
Poultry Breeder
Nutrition Expert
Animal Pathologist
Fishery Expert
Pharmacologist
Toxicologist
Epidemiologist
Microbiologist
Biochemist
Analytical Chemist
Physicist
Environment biologist

REPORT OF THE TASK FORCE ON RECOMBINANT PHARMA
9090909090
Functioning of the National Biotechnology Authority/Commission
Proposal
Secretariat
Scrutiny and Processing by Relevant Division of the Secretariat
Examination by the specific sub-committee
Submission to the Apex Committee
Decision making process for commercialization
Licensing/Release/Notification etc. by the DCG(I), concerned Ministry etc.
Functions of the Apex Committee
Single window clearance with on the spot disposal of proposals Policy Framework To generate comprehensive documentation protocols for industry for comprehensive product development evaluation Approval of rDNA products Capacity Building Risk Assessment Risk Management Facilitation of application processing Monitoring of rDNA Research in the Country for Societal benefits and to avoid DUAL-USE of the Biotechnology through Institutional Biosafety Committees

MINISTRY OF ENVIRONMENT & FORESTSGOVERNMENT OF INDIA
AUG 2005
Report of the Task Forceon
Recombinant Pharma
Ministry of Environment & ForestsGovernment of India
Paryavaran Bhavan, CGO Complex, Lodhi RoadNew Delhi - 110 003. (INDIA)
Telephone:+91-11-2436 1896, 2436 0721E-mail: [email protected]
website: http://envfor.nic.in
http://www.envfor.nic.in/divisions/csurv/geac/geac_home.html





























