Embed Size (px)
Citation preview

Journal of Controlled Release 150 (2011) 63–69
Contents lists available at ScienceDirect
Journal of Controlled Release
j ourna l homepage: www.e lsev ie r.com/ locate / jconre l
PEGylated TNF-related apoptosis-inducing ligand (TRAIL)-loaded sustained releasePLGA microspheres for enhanced stability and antitumor activity
Tae Hyung Kim a,1, Hai Hua Jiang a,1, Chan Woong Park a, Yu Seok Youn b, Seulki Lee c,Xiaoyuan Chen c, Kang Choon Lee a,⁎a College of Pharmacy, SungKyunKwan University, 300 Chonchon-dong, Jangan-ku, Suwon City 440-746, Republic of Koreab College of Pharmacy, Pusan National University, Busan 609-735, Republic of Koreac Laboratory for Molecular Imaging and Nanomedicine, NIBIB, NIH, Bethesda, MD 20892, USA
⁎ Corresponding author. Drug Targeting Laboratory, ColleUniversity, 300 Chonchon-dong, Jangan-ku, Suwon 440-746290 7724.
E-mail address: [email protected] (K.C. Lee).1 Both authors contributed equally to this work.
0168-3659/$ – see front matter © 2010 Elsevier B.V. Aldoi:10.1016/j.jconrel.2010.10.037
a b s t r a c t
a r t i c l e i n f oArticle history:Received 11 August 2010Accepted 31 October 2010Available online 6 November 2010
Keywords:TRAILPEGylationPLGA microspheresStabilitySustain releaseAntitumor
The purpose of this workwas to develop an effective PEGylated TNF-related apoptosis-inducing ligand (PEG-TRAIL)delivery system for antitumor therapy based on local injection to tumor sites that has a sustained effect withoutprotein aggregation or an initial release burst. The authors designed poly (lactic-co-glycolic) acid (PLGA)microspheres that deliver PEG-TRAIL locally and continuously at tumor sites with sustained biological activity andcompared its performance with that of TRAIL microspheres. TRAIL or PEG-TRAIL was microencapsulated into PLGAmicrospheres using a double-emulsion solvent extraction method. Prepared TRAIL and PEG-TRAIL microspheresshowed entirely spherical, smooth surfaces. However, PEG-TRAIL microspheres exhibited a 2.07-fold higherencapsulation efficiency than TRAIL microspheres, and exhibited a tri-phasic in vitro release profile with a lowerinitial burst (15.8%) than TRAILmicrospheres (42.7%). Furthermore, released PEG-TRAIL showed a continued abilityto induce apoptosis over 14 days. In vivo pharmacokinetic studies also demonstrated that PEG-TRAILmicrospheres had a sustained release profile (18 days), and that the steady-state concentration of PEG-TRAIL inrat plasma was reached at day 3 andmaintained until day 15; its steady-state concentration in rat plasma changedfrom 1444.3±338.4 to 2697.7±419.7 pg/ml. However, TRAIL microspheres were released out within 2 days afteradministration. Finally, in vivo antitumor tests revealed that tumor growths were significantly more inhibited by asingle dose of PEG-TRAILmicrospheres than TRAILmicrosphereswhen delivered at 300 μg of TRAIL/mouse. Tumorstaken frommouse treatedwith PEG-TRAIL microspheres showed 78.3% tumor suppression at 24 days, and this was3.02-fold higher than that observed for TRAILmicrospheres (25.9% tumor inhibition). Furthermore, these improvedpharmaceutical characteristics of PEG-TRAIL microspheres resulted in superior therapeutic effects withoutdetectable side effects. These findings strongly suggest that PEG-TRAIL microspheres offer a new therapeuticstrategy for the treatment of cancers.
ge of Pharmacy, SungKyunKwan, Republic of Korea. Fax:+82 31
l rights reserved.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
TNF-related apoptosis-inducing ligand (TRAIL), which is expressedas a type 2 transmembrane protein, is considered an optimalcandidate for cancer therapy due to its tumor cell specificity andnegligible normal cell cytotoxicity [1–5]. This selective and potentactivity of TRAIL is due to its binding affinity to death receptors 4(DR4) and 5 (DR5), which are overexpressed in cancer cells [6,7].TRAIL to DR4 or DR5 binding leads to the recruitment of an adaptormolecule Fas-associated death domain (FADD), which then signalstumor cell death via caspase-dependent apoptotic pathways [8,9].
Although TRAIL has a potent apoptotic effect on tumor cells, TRAIL hasa short biological half-life and is rapidly cleared from the circulationafter systemic administration, and thus, may require frequentadministration to preserve therapeutic levels in blood [10–12].Therefore, a long-term delivery system for TRAIL administration isrequired.
To address these issues, we developed a chemical derivativeof trimeric active TRAIL using PEG, a water-soluble, biocompatiblepolymer [13]. N-terminal-specific PEGylated TRAIL (PEG-TRAIL)showed enhanced stability/solubility, pharmacokinetic characteris-tics, and antitumor efficacy without any hepatotoxic side effects [13].However, systemic administration may achieve insufficient concen-tration in the tumor bed, and therefore, a long-term delivery systemthat can localize the agent and enable its controlled release isrequired.
Poly (lactic-co-glycolic acid) (PLGA)-based systems are mostwidely to achieve a sustained protein delivery [14–18]. The sustained

64 T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
protein delivery systems have a number of important clinicaladvantages, which include reduced dosage frequency and improvedefficacy without toxicity. However, some problems associated withmicrosphere fabrication and agent release system, such as exposure ofthe protein to an aqueous/organic interface, freeze-drying, andphysical instability in an acidic microenvironment, are questionable[19–23]. Furthermore, it has been established that protein entrap-ment within PLGA microspheres often results in protein degradationor inactivation [23].
In recent years, a number of papers have been published on thesuccessful incorporation of proteins into PLGA microspheres, withrespect to protein drug stability, loading, and encapsulation efficiency.For example, PEGylated proteins, such as, lysozyme, insulin, andepidermal growth factor have been encapsulated within PLGAmicrospheres to minimize nonspecific adsorption or aggregation atthe oil/water interface and onto the PLGA surface [24–26]. Further-more, PEGylated proteins have been shown to have better releaseprofiles, encapsulation efficiencies, and better preserved biologicalactivities than their native proteins.
In previous studies, we demonstrated that PEG-TRAIL exhibitedtypical cytotoxic and apoptotic effects in HCT116 cells with minimalactivity loss. In addition, PEG-TRAIL was found to have a betterphysiological stability and a better pharmacokinetic profile thanTRAIL. This leads to the expectation that the microencapsulation ofPEG-TRAIL within PLGA microspheres would result in a significantincrease in the therapeutic potency of the agent due to the presence ofa protective stealth of PEG chains around the TRAIL molecules.
In this study, TRAIL or PEG-TRAIL microspheres were fabricatedusing the double-emulsion solvent extraction method. The micro-spheres were then characterized and studied in vitro. The biologicalactivities and the pharmacokinetics of released TRAIL and PEG-TRAILwere also investigated. Finally, the tumoricidal activities of TRAIL andof PEG-TRAIL microspheres were assessed in HCT 116 tumor bearingBALB/c athymic mice.
2. Materials and methods
2.1. Materials and animals
PEG-TRAIL was prepared as previously described [13]. Briefly,trimeric TRAIL was constructed by incorporating trimer-formingzipper sequences, and then N-terminal-specific PEGylation (PEGMw: 5000) was performed. Although slight activity loss occurredafter PEGylation, PEG-TRAIL successfully enhanced stability andpharmacokinetics. Furthermore, in vivo antitumor tests revealedthat PEG-TRAIL treatment improved therapeutic potentials as com-pared with TRAIL in tumor xenograft animal models. PLGA (50:50)with free carboxyl end groups (Resomer RG504H) was supplied byBoehringer Ingelheim (Ingelheim, Germany). Polyvinyl alcohol (Mw30,000–70,000) was purchased from Sigma (St. Louis, MO). All otherchemicals were of analytical grade and were used as obtained. Celllines were obtained from the Korean Cell Line Bank (Seoul). Animalswere obtained from the Hanlim Experimental Animal Laboratory(Seoul), and were cared for according to the National Institutes ofHealth (NIH) guidelines concerning the care and use of laboratoryanimals (NIH publication 80-23, revised 1996).
2.2. Preparations of TRAIL and PEG-TRAIL microspheres
TRAIL andPEG-TRAILmicrosphereswere preparedusing thedouble-emulsion solvent extraction method with slight modification [27].Briefly, 200 μl of protein solution (2.5 mg/ml) was emulsified in 3 ml ofdichloromethane containing PLGA (15%, w/v) by homogenization(Powergen 700, Fischer Scientific, Germany) for 15 s. The resultingw/o emulsion was gradually injected using a glass syringe with a 22-Gneedle into a 50-ml PVA solution to produce a w/o/w emulsion. The
resultant w/o/w emulsion was then stirred for 4 h to allow thedichloromethane to evaporate. After 4 h of extensive stirring, thehardened microspheres were centrifuged, washed three times, andlyophilized. Empty microspheres were prepared in the same waywithout TRAIL or PEG-TRAIL.
Microsphere morphologies were observed using a field emissionscanningelectronmicroscope (FE-SEM; LEOSUPRA55GENESIS 2000) atan accelerating voltage of 15,000 V. Average particle sizes weredetermined using a laser diffraction particle size analyzer (Mastersizer,Malvern Instruments, USA).
2.3. TRAIL and PEG-TRAIL distributions in microspheres
To observe the extents of protein distributions in microspheres,purified TRAIL or PEG-TRAIL was labeled using a fluoresceinisothiocyanate (FITC)-labeling kit (American Qualex Antibodies, SanClemente, CA). FITC-labeled TRAIL (TRAILFITC) or PEG-TRAIL (PEG-TRAILFITC)-loaded PLGA microspheres were then produced as de-scribed previously. After removing free FITC by washing three timeswith normal saline buffers, fluorescence image of microspheres wasobtained using confocal laser scanning microscopy (CLSM, Carl ZeissMeta LSM510, Germany).
2.4. Protein encapsulation efficiencies
Theamountof protein loaded inmicrosphereswasdeterminedusinga“two-step”extractionmethod [28].About20 mgofmicrosphereswasfirstdissolved in 2 ml of acetonitrile. After centrifugation, the supernatant-containing polymer was discarded and the pellet was dissolved in 0.1 MNaOH/5% sodium dodecyl sulfate solution (NaOH/SDS) at 37 °C for 24 h.After extraction, the encapsulated protein amount was determined usingamicro-BCA protein assay kit (Pierce Chemical Co., Rockford, IL). Sampleswere assayed in triplicate.
2.5. In vitro release studies
In vitro protein release profiles frommicrospheres were determinedas follows. Approximately 20 mg of microspheres was suspended in2 ml phosphate-buffered saline (0.1 M PBS, pH 7.4) containing 0.02%(w/v) Tween 80, and 0.02% (w/v) sodium azide. Sample tubes wereplaced in an air-bath incubator maintained at 37 °C and rotated at30 rpm. At designated times, media were collected by centrifugationand replenished with fresh PBS buffer. Amounts of released proteinwere analyzed using a micro-BCA protein assay.
2.6. Degradation studies
Preweighed microspheres (5 mg) were placed in individual tubescontaining 0.5 ml of PBS buffer. The tubeswere then placed in an air-bathincubator maintained at 37 °C and rotated at 30 rpm. At predeterminedtimes, themicrosphereswere collected by centrifugation and then rinsedwith distilled water to remove residual buffer. The microspheres werethen vacuum-dried overnight for further experiments. SEM was used toinvestigate the surface morphologies of microspheres.
2.7. Flow cytometry analysis
To determine the apoptotic activities of released protein on HCT116 cells, apoptosis and necrosis levels were determined using Annexin-V-FLUOS staining kits (Roche, Applied Science, Mannheim, Germany), aspreviously described [13]. Released TRAIL and PEG-TRAIL concentrationsat 1, 3, 5 and 14 days were equivalent to 100 ng/ml, and their apoptoticactivities were compared with free TRAIL or PEG-TRAIL (100 ng/ml) byusing a FACS Calibur (BD Bioscience Mountain View, CA, USA).
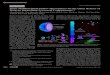
Fig. 1. Images of PLGA microspheres prepared by the w/o/w double-emulsion solventextraction method. Scanning electron micrographs of (A) TRAIL and (B) PEG-TRAILmicrospheres. CLSM images of (C) TRAILFITC and (D) PEG-TRAILFITC microspheres.
65T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
2.8. In vivo pharmacokinetics
The pharmacokinetic characteristics of TRAIL and PEG-TRAILmicrospheres were investigated in male Sprague–Dawley rats(200 g body weight) using a subcutaneous injection into the back ofeach animal (100 μg/rat). At predetermined intervals, blood samplesobtained via a caudal vein were collected in heparinized tubes. Plasmasamples were obtained by centrifugation and stored at −70 °C untilrequired for assay. The concentrations of TRAIL or PEG-TRAIL in ratplasma were measured using commercial TRAIL ELISA kits (BioSourceInternational, Inc, Camarillo, CA).
2.9. In vivo antitumor activity
BALB/c nu/nu male mice (6–7 weeks old) were used as a tumorxenograft model. Tumors were established by inoculating HCT 116cells (4×106 cells/mice, 50 μl/injection) s.c. into a dorsal flank of eachmouse. Five days after inoculation, when tumors reached 50–75 mm3,animals were randomly divided into five treatment groups of 7animals each. The first group of mice was received a single s.c.injection of empty PLGA microspheres as a control. The second andthird groups received an s.c. injection of free TRAIL or PEG-TRAIL at50 μg/mouse/every 3 days. The fourth and fifth groups of mice
Fig. 2. In vitro release kinetics of TRAIL or PEG-TRAIL from PLGAmicrospheres. Means±SDs (n=3).
received a single s.c. injection of TRAIL or PEG-TRAIL microspheres(300 μg/mouse). Tumor diameters were measured using a digitalcaliper and tumor volumes (mm3) were calculated using the formula:tumor volume=length×width2×0.5.
Liver toxicities and in vivo tumor cell apoptosis were alsoinvestigated in HCT116 tumor-bearing BALB/c athymic mice. At24 days after administration, liver and tumor tissues of all groupswere recovered from euthanized animals. Sections (5 μm) were thencut from 10% neutral buffered formalin-fixed and paraffin-embeddedtissue blocks. Apoptotic cell death levels in liver and tumor tissueswere assessed using TdT-mediated dUTP nick end labeling (TUNEL)assays (in situ cell death detection kits, Roche, Applied Science,Mannheim, Germany).
2.10. Statistical analysis
Data are expressed as means±SDs, and the Student's t-test wasused throughout.
3. Results and discussion
3.1. Preparation and characterization of TRAIL or PEG-TRAILmicrospheres
The external surface morphologies of TRAIL and PEG-TRAILmicrospheres prepared by the double-emulsion solvent extractionmethod were investigated by scanning electron microscopy. TRAILand PEG-TRAIL microspheres exhibited a spherical shape with asmooth, uniform surface morphology (Fig. 1A and B). Microspheresizes were measured using a coulter counter. The average diametersof lyophilized TRAIL and PEG-TRAIL microspheres were 14.4±1.06and 12.9±1.98 μm, respectively (Supplementary Fig. S2). Thisinvestigation revealed no apparent difference between the micro-spheres in terms of diameter or surface morphology.
The encapsulation efficiencies of TRAIL and PEG-TRAIL micro-spheres were 40.40±0.8 and 83.97±1.6%, respectively. PEG-TRAILmicrospheres exhibited a 2.07-fold greater encapsulation efficiency,
Fig. 3. Degradation of TRAIL (left) and of PEG-TRAIL (right)-loaded PLGA microspheresafter immersion in release medium. (A, D): initial, (B, E): 1 day, (C, F): 7 days.
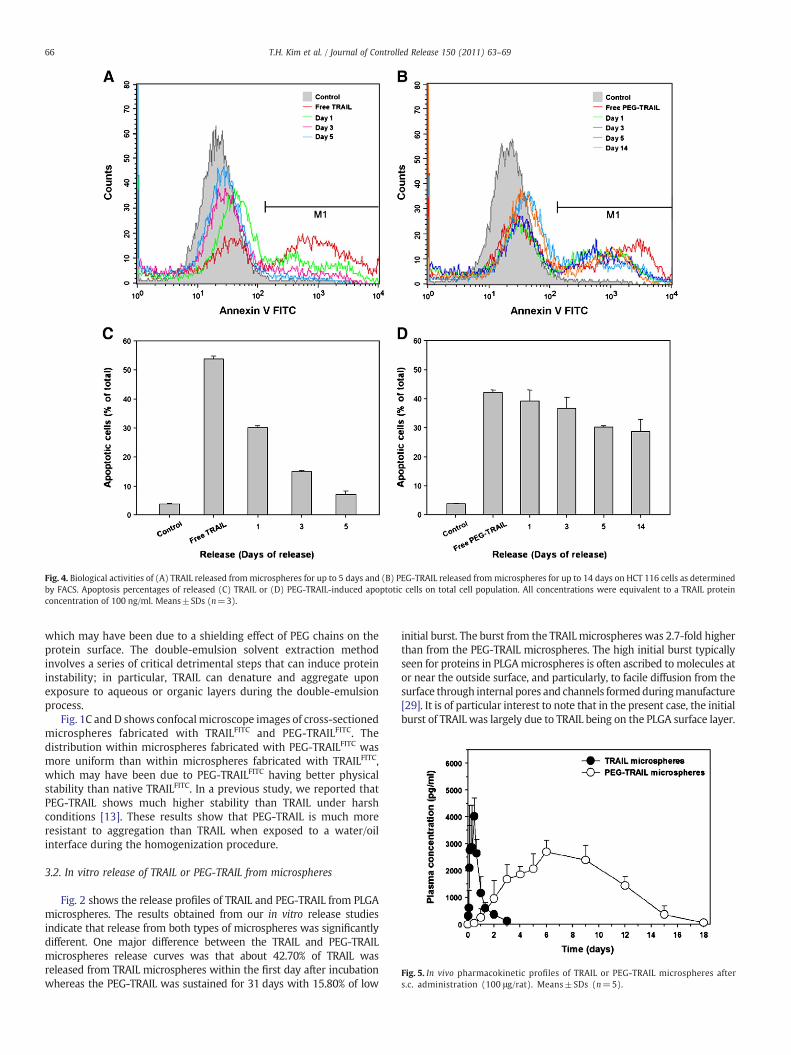
Fig. 4. Biological activities of (A) TRAIL released frommicrospheres for up to 5 days and (B) PEG-TRAIL released frommicrospheres for up to 14 days on HCT 116 cells as determinedby FACS. Apoptosis percentages of released (C) TRAIL or (D) PEG-TRAIL-induced apoptotic cells on total cell population. All concentrations were equivalent to a TRAIL proteinconcentration of 100 ng/ml. Means±SDs (n=3).
Fig. 5. In vivo pharmacokinetic profiles of TRAIL or PEG-TRAIL microspheres afters.c. administration (100 μg/rat). Means±SDs (n=5).
66 T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
which may have been due to a shielding effect of PEG chains on theprotein surface. The double-emulsion solvent extraction methodinvolves a series of critical detrimental steps that can induce proteininstability; in particular, TRAIL can denature and aggregate uponexposure to aqueous or organic layers during the double-emulsionprocess.
Fig. 1C and D shows confocalmicroscope images of cross-sectionedmicrospheres fabricated with TRAILFITC and PEG-TRAILFITC. Thedistribution within microspheres fabricated with PEG-TRAILFITC wasmore uniform than within microspheres fabricated with TRAILFITC,which may have been due to PEG-TRAILFITC having better physicalstability than native TRAILFITC. In a previous study, we reported thatPEG-TRAIL shows much higher stability than TRAIL under harshconditions [13]. These results show that PEG-TRAIL is much moreresistant to aggregation than TRAIL when exposed to a water/oilinterface during the homogenization procedure.
3.2. In vitro release of TRAIL or PEG-TRAIL from microspheres
Fig. 2 shows the release profiles of TRAIL and PEG-TRAIL from PLGAmicrospheres. The results obtained from our in vitro release studiesindicate that release from both types of microspheres was significantlydifferent. One major difference between the TRAIL and PEG-TRAILmicrospheres release curves was that about 42.70% of TRAIL wasreleased from TRAIL microspheres within the first day after incubationwhereas the PEG-TRAIL was sustained for 31 days with 15.80% of low
initial burst. The burst from the TRAIL microspheres was 2.7-fold higherthan from the PEG-TRAIL microspheres. The high initial burst typicallyseen for proteins in PLGAmicrospheres is often ascribed to molecules ator near the outside surface, and particularly, to facile diffusion from thesurface through internal pores and channels formedduringmanufacture[29]. It is of particular interest to note that in the present case, the initialburst of TRAIL was largely due to TRAIL being on the PLGA surface layer.
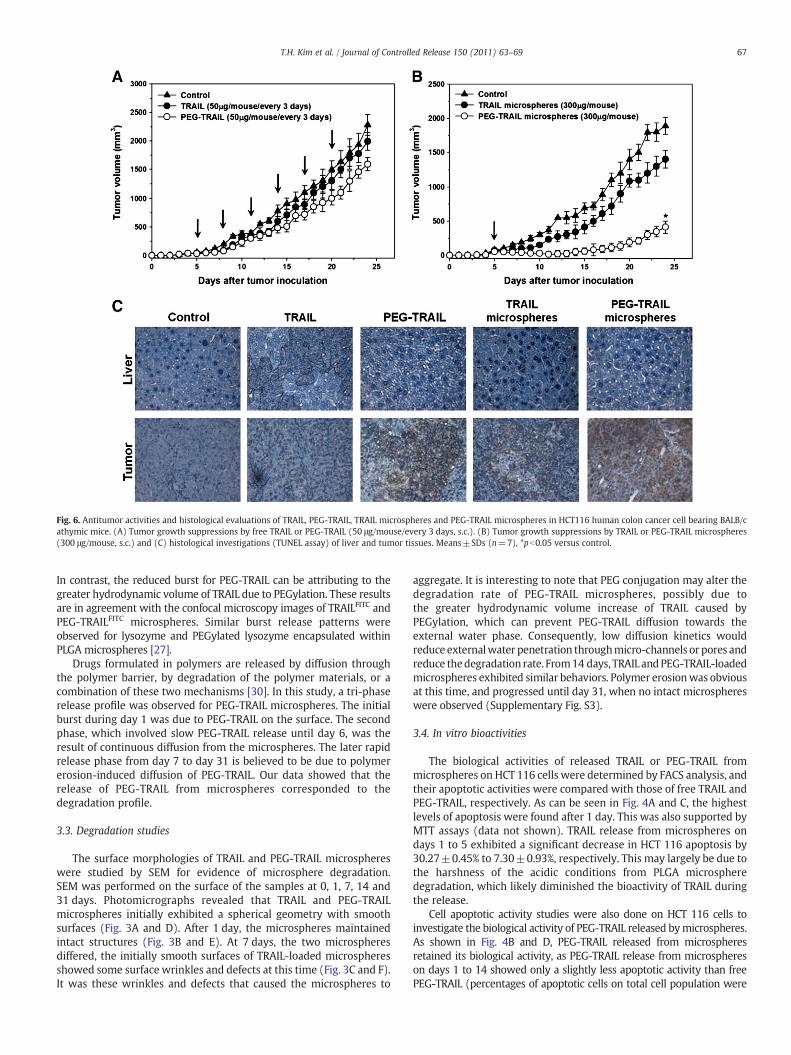
Fig. 6. Antitumor activities and histological evaluations of TRAIL, PEG-TRAIL, TRAIL microspheres and PEG-TRAIL microspheres in HCT116 human colon cancer cell bearing BALB/cathymic mice. (A) Tumor growth suppressions by free TRAIL or PEG-TRAIL (50 μg/mouse/every 3 days, s.c.). (B) Tumor growth suppressions by TRAIL or PEG-TRAIL microspheres(300 μg/mouse, s.c.) and (C) histological investigations (TUNEL assay) of liver and tumor tissues. Means±SDs (n=7), *pb0.05 versus control.
67T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
In contrast, the reduced burst for PEG-TRAIL can be attributing to thegreater hydrodynamic volume of TRAIL due to PEGylation. These resultsare in agreement with the confocal microscopy images of TRAILFITC andPEG-TRAILFITC microspheres. Similar burst release patterns wereobserved for lysozyme and PEGylated lysozyme encapsulated withinPLGA microspheres [27].
Drugs formulated in polymers are released by diffusion throughthe polymer barrier, by degradation of the polymer materials, or acombination of these two mechanisms [30]. In this study, a tri-phaserelease profile was observed for PEG-TRAIL microspheres. The initialburst during day 1 was due to PEG-TRAIL on the surface. The secondphase, which involved slow PEG-TRAIL release until day 6, was theresult of continuous diffusion from the microspheres. The later rapidrelease phase from day 7 to day 31 is believed to be due to polymererosion-induced diffusion of PEG-TRAIL. Our data showed that therelease of PEG-TRAIL from microspheres corresponded to thedegradation profile.
3.3. Degradation studies
The surface morphologies of TRAIL and PEG-TRAIL microsphereswere studied by SEM for evidence of microsphere degradation.SEM was performed on the surface of the samples at 0, 1, 7, 14 and31 days. Photomicrographs revealed that TRAIL and PEG-TRAILmicrospheres initially exhibited a spherical geometry with smoothsurfaces (Fig. 3A and D). After 1 day, the microspheres maintainedintact structures (Fig. 3B and E). At 7 days, the two microspheresdiffered, the initially smooth surfaces of TRAIL-loaded microspheresshowed some surface wrinkles and defects at this time (Fig. 3C and F).It was these wrinkles and defects that caused the microspheres to
aggregate. It is interesting to note that PEG conjugation may alter thedegradation rate of PEG-TRAIL microspheres, possibly due tothe greater hydrodynamic volume increase of TRAIL caused byPEGylation, which can prevent PEG-TRAIL diffusion towards theexternal water phase. Consequently, low diffusion kinetics wouldreduce externalwater penetration throughmicro-channels or pores andreduce thedegradation rate. From14 days, TRAIL andPEG-TRAIL-loadedmicrospheres exhibited similar behaviors. Polymer erosionwas obviousat this time, and progressed until day 31, when no intact microsphereswere observed (Supplementary Fig. S3).
3.4. In vitro bioactivities
The biological activities of released TRAIL or PEG-TRAIL frommicrospheres on HCT 116 cells were determined by FACS analysis, andtheir apoptotic activities were compared with those of free TRAIL andPEG-TRAIL, respectively. As can be seen in Fig. 4A and C, the highestlevels of apoptosis were found after 1 day. This was also supported byMTT assays (data not shown). TRAIL release from microspheres ondays 1 to 5 exhibited a significant decrease in HCT 116 apoptosis by30.27±0.45% to 7.30±0.93%, respectively. This may largely be due tothe harshness of the acidic conditions from PLGA microspheredegradation, which likely diminished the bioactivity of TRAIL duringthe release.
Cell apoptotic activity studies were also done on HCT 116 cells toinvestigate the biological activity of PEG-TRAIL released bymicrospheres.As shown in Fig. 4B and D, PEG-TRAIL released from microspheresretained its biological activity, as PEG-TRAIL release from microsphereson days 1 to 14 showed only a slightly less apoptotic activity than freePEG-TRAIL (percentages of apoptotic cells on total cell population were

68 T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
42.23±0.86% for free PEG-TRAIL and 39.16±3.73% to 28.73±4.21% ondays 1 and 14, respectively, for released PEG-TRAIL). Taken together,these results show that the biological activity of released TRAIL waspreserved for up to 14 days. A control sample taken from emptymicrospheres had no effect on cell viability (data not shown).
3.5. In vivo pharmacokinetics
Thepharmacokinetics of TRAIL and of PEG-TRAILmicrosphereswasanalyzed using TRAIL ELISA kits. The pharmacokinetic profiles of TRAILor PEG-TRAIL microspheres are shown in Fig. 5. Our resultsdemonstrated that PEG-TRAIL microspheres caused sustained release(18 days), whereby the steady-state concentration was reached atday 3 and maintained until day 15. The steady-state concentrations ofPEG-TRAIL in rat plasma changed between1444.3±338.4 and 2697.7±419.7 pg/ml. However, TRAIL microspheres were released out of TRAILwithin the 2 days after injection. This high initial burst of TRAILmicrospheres was largely due to TRAIL on the surfaces of thePLGA microparticles. This result presented experimental evidence thatPEG-TRAILmicrospheres showed sustained release in vivo and that theyhave potential as a long-lasting delivery system.
3.6. In vivo antitumor activity
The antitumor effect of free TRAIL or PEG-TRAIL was investigated inHCT116 tumor bearing mice. Five days after inoculation, micewere treated with free TRAIL (50 μg/mouse, s.c.) or free PEG-TRAIL(50 μg/mouse, s.c.) every 3 days, and tumor volumes were continuouslymonitored. The tumor growth rate was highest in control group; TRAILand PEG-TRAILwere found to have slight antitumor effects. Tumors takenfrom the animals treatedwith free TRAIL or PEG-TRAIL showed 12.6% and30.1% tumor suppression at 24 days, respectively (Fig. 6A).
In microsphere cases, TRAIL or PEG-TRAIL microspheres loadedwith equivalent amounts of protein (300 μg/mouse) were injectedinto tumor bearing nude mice, and tumor growths were examined.Fig. 6B shows that tumor growthwas significantly more inhibited by asingle dose of PEG-TRAIL microspheres than a single dose of TRAILmicrospheres. Tumors taken from the animals treatedwith PEG-TRAILmicrospheres showed 78.3% tumor suppression (pb0.05) at 24 daysversus controls, and this suppression was 3.02-fold higher than thatobserved for TRAIL microspheres (25.9% tumor inhibition). This lowertumor inhibition of TRAIL microspheres is probably due to the harshconditions of the microencapsulation process. In particular, the acidicpH condition probably causedmicrosphere degradation and extensivedamage to TRAIL, and thus, interfered with the binding of TRAIL todeath receptors on HCT116 tumor cells. TUNEL assays of liver tissuesshowed that free TRAIL induced apoptosis whereas other groups didnot, and TUNEL assays of tumor tissues from TRAIL, PEG-TRAIL, TRAILmicrosphere groups showed slightly induced tumor cell destruction.However, PEG-TRAIL microspheres did not have any noticeablehepatotoxic effect (Fig. 6C), and that tumor tissues from PEG-TRAILmicrosphere-treated mice showed substantial tumor cell apoptosis(Fig. 6C). The body weight change during in vivo antitumor activitytest also indicated that the absence of serious toxic-side effects byTRAIL, PEG-TRAIL, TRAIL microsphere and PEG-TRAIL microspheretreatments (Supplementary Fig. S1). In the present study, we foundthat a single injection of microspheres loaded with only PEG-TRAIL(300 μg/mouse) showed preserved antitumor activity for 14–15 days.Accordingly, we suggest that this systemmay overcome the problemsassociatedwith high doses and frequent administrations of PEG-TRAIL.
4. Conclusions
In this study, PEG-TRAIL microspheres were found to have highencapsulation efficiencies and tri-phases in vitro release profile with alower initial burst than TRAIL microspheres. Furthermore, PEG-TRAIL
microspheres preserved the biological activity of released PEG-TRAIL for14 days. In addition, our in vivo pharmacokinetic studies demonstrate asustained release profile from PEG-TRAIL microspheres for15 days. These improved pharmaceutical characteristics of PEG-TRAILmicrospheres resulted in superior therapeutic effects in an animal colontumormodelwithout frequent administration or detectable side effects.These findings strongly suggest that the PEG-TRAIL microspherestrategy has therapeutic potential for the treatment of cancer.
Acknowledgement
This study was supported by the Ministry of Education, Scienceand Technology of Korea [Grant 2009K001604].
Appendix A. Supplementary data
Supplementary data to this article can be found online atdoi:10.1016/j.jconrel.2010.10.037.
References
[1] T.S. Griffith, D.H. Lynch, TRAIL: a molecule with multiple receptors and controlmechanisms, Curr. Opin. Immunol. 10 (1998) 559–563.
[2] A. Ashkenazi, V.M. Dixit, Apoptosis control by death and decoy receptors, Curr.Opin. Cell Biol. 11 (1999) 255–260.
[3] A. Ashkenazi, R.C. Pai, S. Fong, S. Leung, D.A. Lawrence, S.A. Marsters, C. Blackie, L.Chang, A.E. McMurtrey, A. Hebert, L. DeForge, I.L. Koumenis, D. Lewis, L. Harris, J.Bussiere, H. Koeppen, Z. Shahrokh, R.H. Schwall, Safety and antitumor activity ofrecombinant soluble Apo2 ligand, J. Clin. Invest. 104 (1999) 155–162.
[4] L.E. French, J. Tschopp, The TRAIL to selective tumor death, Nat. Med. 5 (1999)146–147.
[5] A. Almasan, A. Ashkenazi, Apo2L/TRAIL: apoptosis signaling, biology, andpotential for cancer therapy, cytokine, Growth Factor Rev. 14 (2003) 337–348.
[6] S.G. Hymowitz, H.W. Christinger, G. Fuh, M. Ultsch, M. O'Connell, R.F. Kelley, A.Ashkenazi, A.M. de Vos, Triggering cell death: the crystal structure of Apo2L/TRAILin a complex with death receptor 5, Mol. Cell 4 (1999) 563–571.
[7] H.N. LeBlanc, A. Ashkenazi, Apo2L/TRAIL and its death and decoy receptors, CellDeath Differ. 10 (2003) 66–75.
[8] G.M. Cohen, Caspases: the executioners of apoptosis, Biochem. J. 326 (1997) 1–16.[9] R.W. Johnstone, A.J. Frew, M.J. Smyth, The TRAIL apoptotic pathway in cancer
onset, progression and therapy, Nat. Rev. Cancer 8 (2008) 782–798.[10] H. Xiang, C.B. Nguyen, S.K. Kelley, N. Dybdal, E. Escandón, Tissue distribution,
stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-relatedapoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearingnude mice, Drug Metab. Dispos. 32 (2004) 1230–1238.
[11] M. Jo, T.H. Kim, D.W. Seol, J.E. Esplen, K. Dorko, T.R. Billiar, S.C. Strom, Apoptosisinduced in normal human hepatocytes by tumor necrosis factor-relatedapoptosis-inducing ligand, Nat. Med. 6 (2000) 564–567.
[12] S.K. Kelley, L.A. Harris, D. Xie, L. Deforge, K. Totpal, J. Bussiere, J.A. Fox, Preclinicalstudies to predict the disposition of Apo2L/tumor necrosis factor-relatedapoptosis-inducing ligand in humans: characterization of in vivo efficacy,pharmacokinetics, and safety, J. Pharmacol. Exp. Ther. 299 (2001) 31–38.
[13] S.Y. Chae, T.H. Kim, K. Park, C.H. Jin, S. Son, S. Lee, Y.S. Youn, K. Kim, D.G. Jo, I.C.Kwon, X. Chen, K.C. Lee, Improved antitumor activity and tumor targeting of NH(2)-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducingligand, Mol. Cancer Ther. 9 (2010) 1719–1729.
[14] M.J. Alonso, R.K. Gupta, C. Min, G.R. Siber, R. Langer, Biodegradable microspheresas controlled-release tetanus toxoid delivery systems, Vaccine 12 (1994)299–306.
[15] J. Mullerad, S. Cohen, D. Benharroch, R.N. Apte, Local delivery of IL-1 alphapolymeric microspheres for the immunotherapy of an experimental fibrosarcoma,Cancer Invest. 21 (2003) 720–728.
[16] J. Mullerad, S. Cohen, E. Voronov, R.N. Apte, Macrophage activation for theproduction of immunostimulatory cytokines by delivering interleukin 1 viabiodegradable microspheres, Cytokine 12 (2000) 1683–1690.
[17] A. Sanchez, R.K. Gupta,M.J. Alonso, G.R. Siber, R. Langer, Pulsed controlled-releasedsystem for potential use in vaccine delivery, J. Pharm. Sci. 85 (1996) 547–552.
[18] O. Benny, M. Duvshani-Eshet, T. Cargioli, L. Bello, A. Bikfalvi, R.S. Carroll, M.Machluf, Continuous delivery of endogenous inhibitors from poly(lactic-co-glycolic acid) polymeric microspheres inhibits glioma tumor growth, Clin. CancerRes. 11 (2005) 768–776.
[19] O.L. Johnson, J.L. Cleland, H.J. Lee, M. Charnis, E. Duenas, W. Jaworowicz, D.Shepard, A. Shahzamani, A.J. Jones, S.D. Putney, A month-long effect from a singleinjection of microencapsulated human growth hormone, Nat. Med. 2 (1996)795–799.
[20] S.D. Putney, P.A. Burke, Improving protein therapeutics with sustained-releaseformulations, Nat. Biotechnol. 16 (1998) 153–157.
[21] K. Fu, D.W. Pack, A.M. Klibanov, R. Langer, Visual evidence of acidic environmentwithin degrading poly(lactic-co-glycolic acid) (PLGA) microspheres, Pharm. Res.17 (2000) 100–106.

69T.H. Kim et al. / Journal of Controlled Release 150 (2011) 63–69
[22] G. Zhu, S.R. Mallery, S.P. Schwendeman, Stabilization of proteins encapsulated ininjectable poly (lactide-co-glycolide), Nat. Biotechnol. 18 (2000) 52–57.
[23] X. Li, Y. Zhang, R. Yan, W. Jia, M. Yuan, X. Deng, Z. Huang, Influence of processparameters on the protein stability encapsulated in poly-DL-lactide-poly(ethyleneglycol) microspheres, J. Control. Release 68 (2000) 41–52.
[24] M. Diwan, T.G. Park, Pegylation enhances protein stability during encapsulation inPLGA microspheres, J. Control. Release 73 (2001) 233–244.
[25] K.D. Hinds, K.M. Campbell, K.M. Holland, D.H. Lewis, C.A. Piché, P.G. Schmidt,PEGylated insulin in PLGA microparticles. In vivo and in vitro analysis, J. Control.Release 104 (2005) 447–460.
[26] T.H. Kim, H. Lee, T.G. Park, PEGylated recombinant human epidermal growthfactor (rhEGF) for sustained release from biodegradable PLGA microspheres,Biomaterials 23 (2002) 2311–2317.
[27] S. Cohen, T. Yoshioka, M. Lucarelli, L.H. Hwang, R. Langer, Controlled deliverysystems for proteins based on poly(lactic/glycolic acid) microspheres, Pharm. Res.8 (1991) 713–720.
[28] M. Diwan, H. Dawar, G.P. Talwar, Induction of early and bioeffective antibodyresponse in rodents with the luteinizing hormone-releasing hormone vaccinegiven as a single dose in biodegradable microspheres along with alum, Prostate 35(1998) 279–284.
[29] Y. Yeo, K. Park, Control of encapsulation efficiency and initial burst in polymericmicroparticle systems, Arch. Pharm. Res. 27 (2004) 1–12.
[30] X.S. Wu, Preparation, characterization, and drug delivery applications of microspheresbased on biodegradable lactic/glycolic acid polymers, in: D.L. Wise, D.J. Trantolo, D.E.Altobelli, M.J. Yaszemski, J.D. Gresser, E.R. Schwartz (Eds.), Encyclopedic Handbook ofBiomaterials and Bioengineering, Part A: Materials, vol. II, Marcel Dekker, New York,1995, pp. 1151–1200.