Embed Size (px)
Citation preview

PAX6 aniridia and interhemispheric brain anomalies
Hana Abouzeid12 Mohamed A Youssef24 Nihal ElShakankiri5 Philippe Hauser3 Francis L Munier12
Daniel F Schorderet26
1Jules-Gonin Eye Hospital University of Lausanne Switzerland 2IRO ndash Institute for Research in Ophthalmology Sion Switzerland3Department of Radiology CHUV University of Lausanne Switzerland 4Department of Paediatrics Genetics Unit University ofAlexandria Egypt 5Department of Ophthalmology Paediatric Unit University of Alexandria Egypt 6EPFL - Ecole polytechniquefeacutedeacuterale de Lausanne Lausanne Switzerland
Purpose To report the clinical and genetic study of patients with autosomal dominant aniridiaMethods We studied ten patients with aniridia from three families of Egyptian origin All patients underwent fullophthalmologic general and neurological examination and blood drawing Cerebral magnetic resonance imaging wasperformed in the index case of each family Genomic DNA was prepared from venous leukocytes and direct sequencingof all the exons and intronndashexon junctions of the Paired Box gene 6 (PAX6) was performed after PCR amplificationPhenotype description including ophthalmic and cerebral anomalies mutation detection in PAX6 and phenotype-genotypecorrelation was acquiredResults Common features observed in the three families included absence of iris tissue corneal pannus with differentdegrees of severity and foveal hypoplasia with severely reduced visual acuity In Families 2 and 3 additional findingssuch as lens dislocation lens opacities or polar cataract and glaucoma were observed We identified two novel (c170-174delTGGGC [pL57fs17] and c475delC [pR159fs47]) and one known (c718CgtT [pR240X]) PAX6 mutationsin the affected members of the three families Systemic and neurological examination was normal in all ten affectedpatients Cerebral magnetic resonance imaging showed absence of the pineal gland in all three index patients Severehypoplasia of the brain anterior commissure was associated with the pL57fs17 mutation absence of the posteriorcommissure with pR159fs47 and optic chiasma atrophy and almost complete agenesis of the corpus callosum withpR240XConclusions We identified two novel PAX6 mutations in families with severe aniridia In addition to common phenotypeof aniridia and despite normal neurological examination absence of the pineal gland and interhemispheric brain anomalieswere observed in all three index patients The heterogeneity of PAX6 mutations and brain anomalies are highlighted Thisreport emphasizes the association between aniridia and brain anomalies with or without functional impact such asneurodevelopment delay or auditory dysfunction
In the majority of cases aniridia is a panophthalmopathythat is characterized by the absence or hypoplasia of the irisand is associated with other ocular anomalies such as cataractfoveal hypoplasia corneal opacity (pannus) colobomaanterior chamber angle (with secondary glaucoma) or opticnerve malformations mainly hypoplasia (OMIM 106210)Mostly inherited in an autosomal dominant mode aniridia canhave variable expressivity and about one-third of the patientsare sporadic cases Mutations in the Paired Box gene 6(PAX6) located on chromosome 11p13 [1] have been shownto cause aniridia [2] PAX6 encodes a transcription factoressential for the development of the structures and axes of theeye [3] Knocking out PAX6 from the mouse genome resultsin the absence of the eye the so-called Small eye (Sey) mouse[45]
Correspondence to Francis L Munier MD Jules-Gonin EyeHospital 15 ave de France CH-1004 Lausanne Switzerland Phone +41 21 626 8580 FAX +41 21 626 8544 emailfrancismunierfa2ch
Furthermore PAX6 plays a major role in braindevelopment [67] where it is expressed in the telencephalonthe diencephalon the caudal part of the rhombencephalon themyelencephalon and the spinal cord [38] The early death ofa baby with a compound heterozygous PAX6 mutation hasbeen reported and the brain of this child displayed major brainand cytoarchitectonic abnormalities [9] Brain imaging(magnetic resonance imaging [MRI]) performed in patientsheterozygous for PAX6 mutations often reveals variousmalformations as well Absence of the brain anterior orposterior commissure absence of the pineal gland and apresent but reduced in size corpus callosum have all beenreported [10-14] These anomalies involve the braininterhemispheric fibers and can thus affect the auditoryfunctions that depend on these fibers [10]
Genotypendashphenotype correlations have shown thatmutations causing premature termination codons areassociated with aniridia and missense mutations are related tonon-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects [15]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gtReceived 19 July 2009 | Accepted 12 October 2009 | Published 17 October 2009
copy 2009 Molecular Vision
2074
The aim of this study was to analyze three families ofEgyptian origin to describe their clinical phenotypeincluding brain imaging and to report the results of theirmolecular screening We present two novel and one knownnonsense mutations associated with brain anomalies
METHODSThis study was approved by the Ethics Committee of theFaculty of Medicine of the University of Alexandria Egyptand was conducted in accordance to the tenets of theDeclaration of Helsinki Written informed consent wasobtained from each participant or parent Ten patients withaniridia belonging to three families of Egyptian origin wereincluded in this study as well as six first-degree relatives(Figure 1) The three families were from the Governorate of
Alexandria in northwestern Egypt All subjects underwentfull ophthalmic general and neurological examinationrespectively at the Departments of OphthalmologyPediatrics and Neurology of the University of AlexandriaEgypt Special attention was paid to assessing the presence ofassociated anomalies such as Wilmsrsquo tumor urogenitalanomalies or mental retardation in all subjects Weperformed a cerebral MRI in each index patient (Patient III-1of Family 1 IV-3 of Family 2 and I-1 of Family 3 Table 1)MRIs were reviewed by two different radiologists MRIacquisition techniques included conventional T1- and T2-weighted multisection images (5-mm slice) on a GE SignaHDx 15Tesla MRI (General Electric Company FairfieldCT)
Figure 1 Pedigrees and mutationsequences of the three Egyptian familieswith autosomal dominant aniridia Maleand female subjects are represented bysquares and circles respectively andaffected family members have darkenedsymbols
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2075
TAB
LE 1
OC
ULA
R A
ND
CER
EBR
AL
ASS
ESSM
ENT
OF T
HE
TEN
PATI
ENTS
WIT
H A
NIR
IDIA
Patie
ntnu
mbe
r
PAX
6 M
utat
ion
Bra
in M
RI f
indi
ngs
A
ge a
t ex
amin
atio
nC
orne
al p
annu
sG
lauc
oma
Len
s sta
tus
Exo
n
D
NA
Prot
ein
Fam
ily 1
II-7
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
46y
01
015
yes
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l aph
akia
III-
17
c68
2-68
6del
TGG
GC
Q
57fx
17ab
sent
pin
eal g
land
an
terio
r com
mis
sure
seve
re h
ypop
lasi
a
19y
005
00
5no
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l ant
erio
r pol
arca
tara
ct w
ith p
erip
hera
lle
ns o
paci
ties
supe
rior
disl
ocat
ion
III-
27
c68
2-68
6del
TGG
GC
Q
57fx
17N
A17
y0
050
05
yes
nops
eudo
phak
icII
I-4
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
11y
005
00
5m
ildno
fain
t cor
tical
opa
citie
ssu
perio
r dis
loca
tion
Fam
ily 2
III-
58
g30
5862
27de
lC
R15
9fx4
7N
A36
y0
10
2ye
sno
bila
tera
l ant
erio
r pol
arca
tara
ct f
aint
cor
tical
opac
ities
III-
68
g30
5862
27de
lC
R15
9fx4
7N
A44
yH
ML
Pse
vere
yes
unco
ntro
lled
supe
rior d
islo
catio
n
IV-1
8g
3058
6227
delC
R
159f
x47
NA
10y
01
01
nono
bila
tera
l aph
akia
IV-3
8g
3058
6227
delC
R
159f
x47
abse
nt p
inea
l gla
nd
abse
nt p
oste
rior
com
mis
sure
17y
01
01
yes
nobi
late
ral p
oste
rior p
olar
cata
ract
sup
erio
rdi
sloc
atio
nFa
mily
3 I-1
9g
3057
9567
CgtT
R24
0Xab
sent
pin
eal g
land
opt
icch
iasm
a at
roph
y a
lmos
tco
mpl
ete
agen
esis
of t
heco
rpus
cal
losu
m
35y
01
01
mild
nono
rmal
II-1
9g
3057
9567
CgtT
R24
0XN
A3m
NA
nono
norm
al
The
aste
risk
indi
cate
d a
nove
l mut
atio
n R
E ri
ght e
ye L
E le
ft ey
e N
A n
ot a
vaila
ble
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2076
Vis
ual A
cuity
R
EL
E
Molecular analyses DNA from patients was extractedfrom peripheral leucocytes as previously described [16]Direct bidirectional resequencing of all PCR-amplifiedcoding exons and adjacent junctions was performed with theABI Dye Terminator version 1 (Applied Biosystems FosterCity CA) in a final reaction volume of 10 μl andelectrophoresed on a 3130XL ABI genetic analyzer (AppliedBiosystems) Sequences were compared to the referencesequence NM_0002803 using Chromas version 223(Technelysium Tewantin Australia) The adenine of theATG translation start site was set to 1 Primers and annealingtemperature for PAX6 exons are listed in Table 2 In shortamplification was performed in a thermal cycler (GeneAmp9700 Applied Biosystems) in a total volume of 30 μl EachPCR contained 100 ng genomic DNA 09 nl of each primerand 15 μl master mix 2times (Qiagen HombrechtikonSwitzerland) with or without betaine PCR reactions wereperformed as follows an initial denaturation step was carriedout for 10 min followed by 35 cycles of 1 min at 92 degC 1 minat the specific annealing temperature (Table 2) and 1 min at72 degC A final extension cycle at 72 degC for 10 min wasperformed Identified mutations were evaluated in 96 ethnic-matched controls by denaturing high-performance liquidchromatography (DHPLC) on a WAVE system(Transgenomics Crewe UK) Buffer A contained 01 Mtriethylammonium acetate (TEAA Transgenomics) Buffer Bcontained 01 M TEAA and 25 acetonitrile HPLC grade(Sigma-Aldrich Co St Louis MO) The flow rate was set at15 mlmin and the Buffer B gradient was increased by 5per min for 2 min The optimum temperature was determinedby the Wavemaker software (Transgenomic) for each DNAfragment and a time shift was applied as needed (Table 2)Initial Buffer B concentrations and temperatures for eachfragment are listed in Table 2
RESULTSClinical findings Patients clinical features and mutationsdescription are detailed in Table 1 All ten affected patientsfrom the three families had bilateral aniridia with almostcomplete absence of iris tissue at its base and bilateral fovealhypoplasia Pendular nystagmus consecutive to early severelyreduced visual acuity was observed in all affected subjectsexcept patient III-6 of Family 2 (Figure 1) who presented aleft exotropia Corneal vascularization of different degreeswas observed in the three families (Figure 2) Lensabnormalities and glaucoma were present in Families 1 and 2only In Family 1 the two affected subjects with glaucomahad controlled pressure after bilateral trabeculectomywhereas patient III-6 of Family 2 had uncontrolled highpressure Systemic and neurological examinations werenormal in all ten affected subjects In particular no kidney orurogenital anomalies on ultrasonography no mentalretardation no cerebellar abnormal signs and no olfactory orhearing difficulties were observed or reported
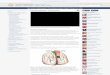
Cerebral MRI of Patients III-1 of Family 1 showed asevere hypoplasia of the anterior commissure of the brain andan absent pineal gland the posterior commissure of the brainwas normal (Figure 3A) In Patient IV-3 of Family 2 a totalabsence of both the pineal gland and the posterior commissureof the brain was observed but the anterior commissure wasnormal (Figure 3C) Patient I-1 of Family 3 harbored analmost complete agenesis of the corpus callosum with a smallamount of remnant tissue localized at the virtual connectionbetween the genu and the body of the corpus callosum (Figure4A) In the same patient Probst bundles were identified(Figure 4C) Concomitant ectasia of the ventricles and atriumhypoplasia of the optic chiasm (Figure 4C) and total absenceof the pineal gland were observed as well (Figure 4C) Bothanterior and posterior commissures of the brain were normal
Molecular findings In Family 1 a c170-174delTGGGCmutation was identified in exon 7 generating a frameshift andan early termination 17 codons downstream (pL57fs17) Themutation was present at the heterozygous state in the fouraffected members of Family 1 and absent in the two unaffectedsubjects (Figure 1) Family 2 also exhibited an unreporteddeletion The deletion of c475delC in exon 8 generates aframeshift and a new stop codon 47 amino acids downstream(pR159fs47) This mutation was present in the three affectedsubjects and absent in the two unaffected individuals (Figure1) The two mutations were not detected in 96 ethnicallymatched healthy individuals and have not been previouslyreported according to the Human PAX6 Mutation Database[17] In Family 3 the previously described c718CgtT(pR240X) nonsense mutation in exon 9 [21819] wasdetected in the two affected members and not in the unaffectedmother (Figure 1)
DISCUSSIONWe report three PAX6 mutations segregating in three familieswith aniridia originating from the northwestern part of EgyptTwo of the three mutations are novel frameshifting deletionsand one is a previously described nonsense mutation To ourknowledge this is the first report of aniridia mutations fromthis part of the world It is interesting to note that the pathologyof the PAX6-related genome is far from being fully determinedwhen one considers that only one of the identified mutantswas already known From a clinical point of view the tenpatients of this series harbored common but extensive featuresof ocular-isolated aniridia associated with the wide spectrumof already described MRI brain malformations These includeabsence of the pineal gland and hypogenesis of the corpuscallosum and of the anterior or posterior commissure Theinterpretation of the observed MRI malformations is limitedby the lack of a full assessment of the neurologicaldevelopment refined examinations such aselectroencephalogram neurocognitive tests or sleep studywere not available Nevertheless the patients did not harbor
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2077
TAB
LE 2
LIS
T O
F IN
ITIA
L B
UFF
ER B
CO
NC
ENTR
ATI
ON
S AN
D T
EMPE
RA
TUR
ES FO
R E
AC
H D
NA
FRA
GM
ENT
PCR
WA
VE
Exo
nSe
nse
prim
er (5
rsquo-3rsquo
)A
ntis
ense
pri
mer
(5rsquo-
3rsquo)
Ann
ealin
g te
mpe
ratu
re degC
Tem
pera
ture
degC
star
t B1
TGTT
GC
GG
AG
TGA
TTA
GTG
GTC
CTG
GG
AA
GG
AG
AC
AG
AG
A60
+be
tain
608
579
2A
CA
CA
CTT
GA
GC
CA
TCA
CC
AC
TCC
TGC
GTG
GA
AA
CTT
CT
60 +
beta
in59
360
63
GTG
GG
TGTA
ATG
CTG
GG
AC
TC
CC
AA
TCTG
TTTC
CC
CTA
CA
60 +
beta
in56
597
4C
CC
CA
AG
AG
GTT
GA
GTG
GA
TG
TCG
CG
AG
TCC
CTG
TGTC
60 +
beta
in61
457
15
TGA
GG
ATG
CA
TTG
TGG
TTG
TG
TGG
AA
GG
AG
AG
GG
GA
AA
GT
60 +
beta
in59
560
86
TTC
AG
GC
AG
TGTT
TAA
GA
AA
AG
TTA
CTC
AC
AC
ATC
CG
TTG
GA
CA
5554
358
77
TGC
AG
ATG
CA
AA
AG
TCC
AA
GC
TCTG
TTC
CC
CC
AG
GTA
CA
A60
+be
tain
574
618
TTTC
CA
CG
GTG
TATC
TGC
AA
AA
GC
CC
TGA
GA
GG
AA
ATG
GT
60 +
beta
in59
659
99
AC
CTT
GG
GA
ATG
TTTT
GG
TGC
AC
TGA
AA
AG
ATG
CC
CA
GA
GA
60 +
beta
in56
760
310
AG
GTG
GG
AA
CC
AG
TTTG
ATG
CA
TGG
CA
GC
AG
AG
CA
TTTA
G60
+be
tain
575
5811
TTC
AG
TCTG
CTA
AA
TGC
TCTG
CTG
TGA
GG
GC
TGTG
TCTG
TTC
60 +
beta
in59
6012
AC
CA
CA
CC
GG
GTA
ATT
TGA
AC
TCTC
AA
GG
GTG
CA
GA
CA
CA
60 +
beta
in58
758
413
TAG
CTC
GA
GG
CC
CA
ATC
TTA
TAA
AC
AC
GC
CC
TCC
CA
TAA
G60
+be
tain
583
6014
TTTC
TGA
AG
GTG
CTA
CTT
TTA
TTTG
AA
GTC
CA
TTC
CTT
CC
CC
AG
T55
535
602
15A
AA
CTT
AA
GTG
TTTT
GA
AG
TTG
TTC
AC
CC
CA
GA
TTG
AA
AA
TGC
CA
GT
60 +
beta
in54
360
5
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2078
Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

The aim of this study was to analyze three families ofEgyptian origin to describe their clinical phenotypeincluding brain imaging and to report the results of theirmolecular screening We present two novel and one knownnonsense mutations associated with brain anomalies
METHODSThis study was approved by the Ethics Committee of theFaculty of Medicine of the University of Alexandria Egyptand was conducted in accordance to the tenets of theDeclaration of Helsinki Written informed consent wasobtained from each participant or parent Ten patients withaniridia belonging to three families of Egyptian origin wereincluded in this study as well as six first-degree relatives(Figure 1) The three families were from the Governorate of
Alexandria in northwestern Egypt All subjects underwentfull ophthalmic general and neurological examinationrespectively at the Departments of OphthalmologyPediatrics and Neurology of the University of AlexandriaEgypt Special attention was paid to assessing the presence ofassociated anomalies such as Wilmsrsquo tumor urogenitalanomalies or mental retardation in all subjects Weperformed a cerebral MRI in each index patient (Patient III-1of Family 1 IV-3 of Family 2 and I-1 of Family 3 Table 1)MRIs were reviewed by two different radiologists MRIacquisition techniques included conventional T1- and T2-weighted multisection images (5-mm slice) on a GE SignaHDx 15Tesla MRI (General Electric Company FairfieldCT)
Figure 1 Pedigrees and mutationsequences of the three Egyptian familieswith autosomal dominant aniridia Maleand female subjects are represented bysquares and circles respectively andaffected family members have darkenedsymbols
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2075
TAB
LE 1
OC
ULA
R A
ND
CER
EBR
AL
ASS
ESSM
ENT
OF T
HE
TEN
PATI
ENTS
WIT
H A
NIR
IDIA
Patie
ntnu
mbe
r
PAX
6 M
utat
ion
Bra
in M
RI f
indi
ngs
A
ge a
t ex
amin
atio
nC
orne
al p
annu
sG
lauc
oma
Len
s sta
tus
Exo
n
D
NA
Prot
ein
Fam
ily 1
II-7
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
46y
01
015
yes
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l aph
akia
III-
17
c68
2-68
6del
TGG
GC
Q
57fx
17ab
sent
pin
eal g
land
an
terio
r com
mis
sure
seve
re h
ypop
lasi
a
19y
005
00
5no
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l ant
erio
r pol
arca
tara
ct w
ith p
erip
hera
lle
ns o
paci
ties
supe
rior
disl
ocat
ion
III-
27
c68
2-68
6del
TGG
GC
Q
57fx
17N
A17
y0
050
05
yes
nops
eudo
phak
icII
I-4
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
11y
005
00
5m
ildno
fain
t cor
tical
opa
citie
ssu
perio
r dis
loca
tion
Fam
ily 2
III-
58
g30
5862
27de
lC
R15
9fx4
7N
A36
y0
10
2ye
sno
bila
tera
l ant
erio
r pol
arca
tara
ct f
aint
cor
tical
opac
ities
III-
68
g30
5862
27de
lC
R15
9fx4
7N
A44
yH
ML
Pse
vere
yes
unco
ntro
lled
supe
rior d
islo
catio
n
IV-1
8g
3058
6227
delC
R
159f
x47
NA
10y
01
01
nono
bila
tera
l aph
akia
IV-3
8g
3058
6227
delC
R
159f
x47
abse
nt p
inea
l gla
nd
abse
nt p
oste
rior
com
mis
sure
17y
01
01
yes
nobi
late
ral p
oste
rior p
olar
cata
ract
sup
erio
rdi
sloc
atio
nFa
mily
3 I-1
9g
3057
9567
CgtT
R24
0Xab
sent
pin
eal g
land
opt
icch
iasm
a at
roph
y a
lmos
tco
mpl
ete
agen
esis
of t
heco
rpus
cal
losu
m
35y
01
01
mild
nono
rmal
II-1
9g
3057
9567
CgtT
R24
0XN
A3m
NA
nono
norm
al
The
aste
risk
indi
cate
d a
nove
l mut
atio
n R
E ri
ght e
ye L
E le
ft ey
e N
A n
ot a
vaila
ble
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2076
Vis
ual A
cuity
R
EL
E
Molecular analyses DNA from patients was extractedfrom peripheral leucocytes as previously described [16]Direct bidirectional resequencing of all PCR-amplifiedcoding exons and adjacent junctions was performed with theABI Dye Terminator version 1 (Applied Biosystems FosterCity CA) in a final reaction volume of 10 μl andelectrophoresed on a 3130XL ABI genetic analyzer (AppliedBiosystems) Sequences were compared to the referencesequence NM_0002803 using Chromas version 223(Technelysium Tewantin Australia) The adenine of theATG translation start site was set to 1 Primers and annealingtemperature for PAX6 exons are listed in Table 2 In shortamplification was performed in a thermal cycler (GeneAmp9700 Applied Biosystems) in a total volume of 30 μl EachPCR contained 100 ng genomic DNA 09 nl of each primerand 15 μl master mix 2times (Qiagen HombrechtikonSwitzerland) with or without betaine PCR reactions wereperformed as follows an initial denaturation step was carriedout for 10 min followed by 35 cycles of 1 min at 92 degC 1 minat the specific annealing temperature (Table 2) and 1 min at72 degC A final extension cycle at 72 degC for 10 min wasperformed Identified mutations were evaluated in 96 ethnic-matched controls by denaturing high-performance liquidchromatography (DHPLC) on a WAVE system(Transgenomics Crewe UK) Buffer A contained 01 Mtriethylammonium acetate (TEAA Transgenomics) Buffer Bcontained 01 M TEAA and 25 acetonitrile HPLC grade(Sigma-Aldrich Co St Louis MO) The flow rate was set at15 mlmin and the Buffer B gradient was increased by 5per min for 2 min The optimum temperature was determinedby the Wavemaker software (Transgenomic) for each DNAfragment and a time shift was applied as needed (Table 2)Initial Buffer B concentrations and temperatures for eachfragment are listed in Table 2
RESULTSClinical findings Patients clinical features and mutationsdescription are detailed in Table 1 All ten affected patientsfrom the three families had bilateral aniridia with almostcomplete absence of iris tissue at its base and bilateral fovealhypoplasia Pendular nystagmus consecutive to early severelyreduced visual acuity was observed in all affected subjectsexcept patient III-6 of Family 2 (Figure 1) who presented aleft exotropia Corneal vascularization of different degreeswas observed in the three families (Figure 2) Lensabnormalities and glaucoma were present in Families 1 and 2only In Family 1 the two affected subjects with glaucomahad controlled pressure after bilateral trabeculectomywhereas patient III-6 of Family 2 had uncontrolled highpressure Systemic and neurological examinations werenormal in all ten affected subjects In particular no kidney orurogenital anomalies on ultrasonography no mentalretardation no cerebellar abnormal signs and no olfactory orhearing difficulties were observed or reported
Cerebral MRI of Patients III-1 of Family 1 showed asevere hypoplasia of the anterior commissure of the brain andan absent pineal gland the posterior commissure of the brainwas normal (Figure 3A) In Patient IV-3 of Family 2 a totalabsence of both the pineal gland and the posterior commissureof the brain was observed but the anterior commissure wasnormal (Figure 3C) Patient I-1 of Family 3 harbored analmost complete agenesis of the corpus callosum with a smallamount of remnant tissue localized at the virtual connectionbetween the genu and the body of the corpus callosum (Figure4A) In the same patient Probst bundles were identified(Figure 4C) Concomitant ectasia of the ventricles and atriumhypoplasia of the optic chiasm (Figure 4C) and total absenceof the pineal gland were observed as well (Figure 4C) Bothanterior and posterior commissures of the brain were normal
Molecular findings In Family 1 a c170-174delTGGGCmutation was identified in exon 7 generating a frameshift andan early termination 17 codons downstream (pL57fs17) Themutation was present at the heterozygous state in the fouraffected members of Family 1 and absent in the two unaffectedsubjects (Figure 1) Family 2 also exhibited an unreporteddeletion The deletion of c475delC in exon 8 generates aframeshift and a new stop codon 47 amino acids downstream(pR159fs47) This mutation was present in the three affectedsubjects and absent in the two unaffected individuals (Figure1) The two mutations were not detected in 96 ethnicallymatched healthy individuals and have not been previouslyreported according to the Human PAX6 Mutation Database[17] In Family 3 the previously described c718CgtT(pR240X) nonsense mutation in exon 9 [21819] wasdetected in the two affected members and not in the unaffectedmother (Figure 1)
DISCUSSIONWe report three PAX6 mutations segregating in three familieswith aniridia originating from the northwestern part of EgyptTwo of the three mutations are novel frameshifting deletionsand one is a previously described nonsense mutation To ourknowledge this is the first report of aniridia mutations fromthis part of the world It is interesting to note that the pathologyof the PAX6-related genome is far from being fully determinedwhen one considers that only one of the identified mutantswas already known From a clinical point of view the tenpatients of this series harbored common but extensive featuresof ocular-isolated aniridia associated with the wide spectrumof already described MRI brain malformations These includeabsence of the pineal gland and hypogenesis of the corpuscallosum and of the anterior or posterior commissure Theinterpretation of the observed MRI malformations is limitedby the lack of a full assessment of the neurologicaldevelopment refined examinations such aselectroencephalogram neurocognitive tests or sleep studywere not available Nevertheless the patients did not harbor
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2077
TAB
LE 2
LIS
T O
F IN
ITIA
L B
UFF
ER B
CO
NC
ENTR
ATI
ON
S AN
D T
EMPE
RA
TUR
ES FO
R E
AC
H D
NA
FRA
GM
ENT
PCR
WA
VE
Exo
nSe
nse
prim
er (5
rsquo-3rsquo
)A
ntis
ense
pri
mer
(5rsquo-
3rsquo)
Ann
ealin
g te
mpe
ratu
re degC
Tem
pera
ture
degC
star
t B1
TGTT
GC
GG
AG
TGA
TTA
GTG
GTC
CTG
GG
AA
GG
AG
AC
AG
AG
A60
+be
tain
608
579
2A
CA
CA
CTT
GA
GC
CA
TCA
CC
AC
TCC
TGC
GTG
GA
AA
CTT
CT
60 +
beta
in59
360
63
GTG
GG
TGTA
ATG
CTG
GG
AC
TC
CC
AA
TCTG
TTTC
CC
CTA
CA
60 +
beta
in56
597
4C
CC
CA
AG
AG
GTT
GA
GTG
GA
TG
TCG
CG
AG
TCC
CTG
TGTC
60 +
beta
in61
457
15
TGA
GG
ATG
CA
TTG
TGG
TTG
TG
TGG
AA
GG
AG
AG
GG
GA
AA
GT
60 +
beta
in59
560
86
TTC
AG
GC
AG
TGTT
TAA
GA
AA
AG
TTA
CTC
AC
AC
ATC
CG
TTG
GA
CA
5554
358
77
TGC
AG
ATG
CA
AA
AG
TCC
AA
GC
TCTG
TTC
CC
CC
AG
GTA
CA
A60
+be
tain
574
618
TTTC
CA
CG
GTG
TATC
TGC
AA
AA
GC
CC
TGA
GA
GG
AA
ATG
GT
60 +
beta
in59
659
99
AC
CTT
GG
GA
ATG
TTTT
GG
TGC
AC
TGA
AA
AG
ATG
CC
CA
GA
GA
60 +
beta
in56
760
310
AG
GTG
GG
AA
CC
AG
TTTG
ATG
CA
TGG
CA
GC
AG
AG
CA
TTTA
G60
+be
tain
575
5811
TTC
AG
TCTG
CTA
AA
TGC
TCTG
CTG
TGA
GG
GC
TGTG
TCTG
TTC
60 +
beta
in59
6012
AC
CA
CA
CC
GG
GTA
ATT
TGA
AC
TCTC
AA
GG
GTG
CA
GA
CA
CA
60 +
beta
in58
758
413
TAG
CTC
GA
GG
CC
CA
ATC
TTA
TAA
AC
AC
GC
CC
TCC
CA
TAA
G60
+be
tain
583
6014
TTTC
TGA
AG
GTG
CTA
CTT
TTA
TTTG
AA
GTC
CA
TTC
CTT
CC
CC
AG
T55
535
602
15A
AA
CTT
AA
GTG
TTTT
GA
AG
TTG
TTC
AC
CC
CA
GA
TTG
AA
AA
TGC
CA
GT
60 +
beta
in54
360
5
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2078
Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

TAB
LE 1
OC
ULA
R A
ND
CER
EBR
AL
ASS
ESSM
ENT
OF T
HE
TEN
PATI
ENTS
WIT
H A
NIR
IDIA
Patie
ntnu
mbe
r
PAX
6 M
utat
ion
Bra
in M
RI f
indi
ngs
A
ge a
t ex
amin
atio
nC
orne
al p
annu
sG
lauc
oma
Len
s sta
tus
Exo
n
D
NA
Prot
ein
Fam
ily 1
II-7
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
46y
01
015
yes
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l aph
akia
III-
17
c68
2-68
6del
TGG
GC
Q
57fx
17ab
sent
pin
eal g
land
an
terio
r com
mis
sure
seve
re h
ypop
lasi
a
19y
005
00
5no
yes
bila
tera
ltra
becu
lect
om
y
bila
tera
l ant
erio
r pol
arca
tara
ct w
ith p
erip
hera
lle
ns o
paci
ties
supe
rior
disl
ocat
ion
III-
27
c68
2-68
6del
TGG
GC
Q
57fx
17N
A17
y0
050
05
yes
nops
eudo
phak
icII
I-4
7c
682-
686d
elTG
GG
C
Q57
fx17
NA
11y
005
00
5m
ildno
fain
t cor
tical
opa
citie
ssu
perio
r dis
loca
tion
Fam
ily 2
III-
58
g30
5862
27de
lC
R15
9fx4
7N
A36
y0
10
2ye
sno
bila
tera
l ant
erio
r pol
arca
tara
ct f
aint
cor
tical
opac
ities
III-
68
g30
5862
27de
lC
R15
9fx4
7N
A44
yH
ML
Pse
vere
yes
unco
ntro
lled
supe
rior d
islo
catio
n
IV-1
8g
3058
6227
delC
R
159f
x47
NA
10y
01
01
nono
bila
tera
l aph
akia
IV-3
8g
3058
6227
delC
R
159f
x47
abse
nt p
inea
l gla
nd
abse
nt p
oste
rior
com
mis
sure
17y
01
01
yes
nobi
late
ral p
oste
rior p
olar
cata
ract
sup
erio
rdi
sloc
atio
nFa
mily
3 I-1
9g
3057
9567
CgtT
R24
0Xab
sent
pin
eal g
land
opt
icch
iasm
a at
roph
y a
lmos
tco
mpl
ete
agen
esis
of t
heco
rpus
cal
losu
m
35y
01
01
mild
nono
rmal
II-1
9g
3057
9567
CgtT
R24
0XN
A3m
NA
nono
norm
al
The
aste
risk
indi
cate
d a
nove
l mut
atio
n R
E ri
ght e
ye L
E le
ft ey
e N
A n
ot a
vaila
ble
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2076
Vis
ual A
cuity
R
EL
E
Molecular analyses DNA from patients was extractedfrom peripheral leucocytes as previously described [16]Direct bidirectional resequencing of all PCR-amplifiedcoding exons and adjacent junctions was performed with theABI Dye Terminator version 1 (Applied Biosystems FosterCity CA) in a final reaction volume of 10 μl andelectrophoresed on a 3130XL ABI genetic analyzer (AppliedBiosystems) Sequences were compared to the referencesequence NM_0002803 using Chromas version 223(Technelysium Tewantin Australia) The adenine of theATG translation start site was set to 1 Primers and annealingtemperature for PAX6 exons are listed in Table 2 In shortamplification was performed in a thermal cycler (GeneAmp9700 Applied Biosystems) in a total volume of 30 μl EachPCR contained 100 ng genomic DNA 09 nl of each primerand 15 μl master mix 2times (Qiagen HombrechtikonSwitzerland) with or without betaine PCR reactions wereperformed as follows an initial denaturation step was carriedout for 10 min followed by 35 cycles of 1 min at 92 degC 1 minat the specific annealing temperature (Table 2) and 1 min at72 degC A final extension cycle at 72 degC for 10 min wasperformed Identified mutations were evaluated in 96 ethnic-matched controls by denaturing high-performance liquidchromatography (DHPLC) on a WAVE system(Transgenomics Crewe UK) Buffer A contained 01 Mtriethylammonium acetate (TEAA Transgenomics) Buffer Bcontained 01 M TEAA and 25 acetonitrile HPLC grade(Sigma-Aldrich Co St Louis MO) The flow rate was set at15 mlmin and the Buffer B gradient was increased by 5per min for 2 min The optimum temperature was determinedby the Wavemaker software (Transgenomic) for each DNAfragment and a time shift was applied as needed (Table 2)Initial Buffer B concentrations and temperatures for eachfragment are listed in Table 2
RESULTSClinical findings Patients clinical features and mutationsdescription are detailed in Table 1 All ten affected patientsfrom the three families had bilateral aniridia with almostcomplete absence of iris tissue at its base and bilateral fovealhypoplasia Pendular nystagmus consecutive to early severelyreduced visual acuity was observed in all affected subjectsexcept patient III-6 of Family 2 (Figure 1) who presented aleft exotropia Corneal vascularization of different degreeswas observed in the three families (Figure 2) Lensabnormalities and glaucoma were present in Families 1 and 2only In Family 1 the two affected subjects with glaucomahad controlled pressure after bilateral trabeculectomywhereas patient III-6 of Family 2 had uncontrolled highpressure Systemic and neurological examinations werenormal in all ten affected subjects In particular no kidney orurogenital anomalies on ultrasonography no mentalretardation no cerebellar abnormal signs and no olfactory orhearing difficulties were observed or reported
Cerebral MRI of Patients III-1 of Family 1 showed asevere hypoplasia of the anterior commissure of the brain andan absent pineal gland the posterior commissure of the brainwas normal (Figure 3A) In Patient IV-3 of Family 2 a totalabsence of both the pineal gland and the posterior commissureof the brain was observed but the anterior commissure wasnormal (Figure 3C) Patient I-1 of Family 3 harbored analmost complete agenesis of the corpus callosum with a smallamount of remnant tissue localized at the virtual connectionbetween the genu and the body of the corpus callosum (Figure4A) In the same patient Probst bundles were identified(Figure 4C) Concomitant ectasia of the ventricles and atriumhypoplasia of the optic chiasm (Figure 4C) and total absenceof the pineal gland were observed as well (Figure 4C) Bothanterior and posterior commissures of the brain were normal
Molecular findings In Family 1 a c170-174delTGGGCmutation was identified in exon 7 generating a frameshift andan early termination 17 codons downstream (pL57fs17) Themutation was present at the heterozygous state in the fouraffected members of Family 1 and absent in the two unaffectedsubjects (Figure 1) Family 2 also exhibited an unreporteddeletion The deletion of c475delC in exon 8 generates aframeshift and a new stop codon 47 amino acids downstream(pR159fs47) This mutation was present in the three affectedsubjects and absent in the two unaffected individuals (Figure1) The two mutations were not detected in 96 ethnicallymatched healthy individuals and have not been previouslyreported according to the Human PAX6 Mutation Database[17] In Family 3 the previously described c718CgtT(pR240X) nonsense mutation in exon 9 [21819] wasdetected in the two affected members and not in the unaffectedmother (Figure 1)
DISCUSSIONWe report three PAX6 mutations segregating in three familieswith aniridia originating from the northwestern part of EgyptTwo of the three mutations are novel frameshifting deletionsand one is a previously described nonsense mutation To ourknowledge this is the first report of aniridia mutations fromthis part of the world It is interesting to note that the pathologyof the PAX6-related genome is far from being fully determinedwhen one considers that only one of the identified mutantswas already known From a clinical point of view the tenpatients of this series harbored common but extensive featuresof ocular-isolated aniridia associated with the wide spectrumof already described MRI brain malformations These includeabsence of the pineal gland and hypogenesis of the corpuscallosum and of the anterior or posterior commissure Theinterpretation of the observed MRI malformations is limitedby the lack of a full assessment of the neurologicaldevelopment refined examinations such aselectroencephalogram neurocognitive tests or sleep studywere not available Nevertheless the patients did not harbor
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2077
TAB
LE 2
LIS
T O
F IN
ITIA
L B
UFF
ER B
CO
NC
ENTR
ATI
ON
S AN
D T
EMPE
RA
TUR
ES FO
R E
AC
H D
NA
FRA
GM
ENT
PCR
WA
VE
Exo
nSe
nse
prim
er (5
rsquo-3rsquo
)A
ntis
ense
pri
mer
(5rsquo-
3rsquo)
Ann
ealin
g te
mpe
ratu
re degC
Tem
pera
ture
degC
star
t B1
TGTT
GC
GG
AG
TGA
TTA
GTG
GTC
CTG
GG
AA
GG
AG
AC
AG
AG
A60
+be
tain
608
579
2A
CA
CA
CTT
GA
GC
CA
TCA
CC
AC
TCC
TGC
GTG
GA
AA
CTT
CT
60 +
beta
in59
360
63
GTG
GG
TGTA
ATG
CTG
GG
AC
TC
CC
AA
TCTG
TTTC
CC
CTA
CA
60 +
beta
in56
597
4C
CC
CA
AG
AG
GTT
GA
GTG
GA
TG
TCG
CG
AG
TCC
CTG
TGTC
60 +
beta
in61
457
15
TGA
GG
ATG
CA
TTG
TGG
TTG
TG
TGG
AA
GG
AG
AG
GG
GA
AA
GT
60 +
beta
in59
560
86
TTC
AG
GC
AG
TGTT
TAA
GA
AA
AG
TTA
CTC
AC
AC
ATC
CG
TTG
GA
CA
5554
358
77
TGC
AG
ATG
CA
AA
AG
TCC
AA
GC
TCTG
TTC
CC
CC
AG
GTA
CA
A60
+be
tain
574
618
TTTC
CA
CG
GTG
TATC
TGC
AA
AA
GC
CC
TGA
GA
GG
AA
ATG
GT
60 +
beta
in59
659
99
AC
CTT
GG
GA
ATG
TTTT
GG
TGC
AC
TGA
AA
AG
ATG
CC
CA
GA
GA
60 +
beta
in56
760
310
AG
GTG
GG
AA
CC
AG
TTTG
ATG
CA
TGG
CA
GC
AG
AG
CA
TTTA
G60
+be
tain
575
5811
TTC
AG
TCTG
CTA
AA
TGC
TCTG
CTG
TGA
GG
GC
TGTG
TCTG
TTC
60 +
beta
in59
6012
AC
CA
CA
CC
GG
GTA
ATT
TGA
AC
TCTC
AA
GG
GTG
CA
GA
CA
CA
60 +
beta
in58
758
413
TAG
CTC
GA
GG
CC
CA
ATC
TTA
TAA
AC
AC
GC
CC
TCC
CA
TAA
G60
+be
tain
583
6014
TTTC
TGA
AG
GTG
CTA
CTT
TTA
TTTG
AA
GTC
CA
TTC
CTT
CC
CC
AG
T55
535
602
15A
AA
CTT
AA
GTG
TTTT
GA
AG
TTG
TTC
AC
CC
CA
GA
TTG
AA
AA
TGC
CA
GT
60 +
beta
in54
360
5
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2078
Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

Molecular analyses DNA from patients was extractedfrom peripheral leucocytes as previously described [16]Direct bidirectional resequencing of all PCR-amplifiedcoding exons and adjacent junctions was performed with theABI Dye Terminator version 1 (Applied Biosystems FosterCity CA) in a final reaction volume of 10 μl andelectrophoresed on a 3130XL ABI genetic analyzer (AppliedBiosystems) Sequences were compared to the referencesequence NM_0002803 using Chromas version 223(Technelysium Tewantin Australia) The adenine of theATG translation start site was set to 1 Primers and annealingtemperature for PAX6 exons are listed in Table 2 In shortamplification was performed in a thermal cycler (GeneAmp9700 Applied Biosystems) in a total volume of 30 μl EachPCR contained 100 ng genomic DNA 09 nl of each primerand 15 μl master mix 2times (Qiagen HombrechtikonSwitzerland) with or without betaine PCR reactions wereperformed as follows an initial denaturation step was carriedout for 10 min followed by 35 cycles of 1 min at 92 degC 1 minat the specific annealing temperature (Table 2) and 1 min at72 degC A final extension cycle at 72 degC for 10 min wasperformed Identified mutations were evaluated in 96 ethnic-matched controls by denaturing high-performance liquidchromatography (DHPLC) on a WAVE system(Transgenomics Crewe UK) Buffer A contained 01 Mtriethylammonium acetate (TEAA Transgenomics) Buffer Bcontained 01 M TEAA and 25 acetonitrile HPLC grade(Sigma-Aldrich Co St Louis MO) The flow rate was set at15 mlmin and the Buffer B gradient was increased by 5per min for 2 min The optimum temperature was determinedby the Wavemaker software (Transgenomic) for each DNAfragment and a time shift was applied as needed (Table 2)Initial Buffer B concentrations and temperatures for eachfragment are listed in Table 2
RESULTSClinical findings Patients clinical features and mutationsdescription are detailed in Table 1 All ten affected patientsfrom the three families had bilateral aniridia with almostcomplete absence of iris tissue at its base and bilateral fovealhypoplasia Pendular nystagmus consecutive to early severelyreduced visual acuity was observed in all affected subjectsexcept patient III-6 of Family 2 (Figure 1) who presented aleft exotropia Corneal vascularization of different degreeswas observed in the three families (Figure 2) Lensabnormalities and glaucoma were present in Families 1 and 2only In Family 1 the two affected subjects with glaucomahad controlled pressure after bilateral trabeculectomywhereas patient III-6 of Family 2 had uncontrolled highpressure Systemic and neurological examinations werenormal in all ten affected subjects In particular no kidney orurogenital anomalies on ultrasonography no mentalretardation no cerebellar abnormal signs and no olfactory orhearing difficulties were observed or reported
Cerebral MRI of Patients III-1 of Family 1 showed asevere hypoplasia of the anterior commissure of the brain andan absent pineal gland the posterior commissure of the brainwas normal (Figure 3A) In Patient IV-3 of Family 2 a totalabsence of both the pineal gland and the posterior commissureof the brain was observed but the anterior commissure wasnormal (Figure 3C) Patient I-1 of Family 3 harbored analmost complete agenesis of the corpus callosum with a smallamount of remnant tissue localized at the virtual connectionbetween the genu and the body of the corpus callosum (Figure4A) In the same patient Probst bundles were identified(Figure 4C) Concomitant ectasia of the ventricles and atriumhypoplasia of the optic chiasm (Figure 4C) and total absenceof the pineal gland were observed as well (Figure 4C) Bothanterior and posterior commissures of the brain were normal
Molecular findings In Family 1 a c170-174delTGGGCmutation was identified in exon 7 generating a frameshift andan early termination 17 codons downstream (pL57fs17) Themutation was present at the heterozygous state in the fouraffected members of Family 1 and absent in the two unaffectedsubjects (Figure 1) Family 2 also exhibited an unreporteddeletion The deletion of c475delC in exon 8 generates aframeshift and a new stop codon 47 amino acids downstream(pR159fs47) This mutation was present in the three affectedsubjects and absent in the two unaffected individuals (Figure1) The two mutations were not detected in 96 ethnicallymatched healthy individuals and have not been previouslyreported according to the Human PAX6 Mutation Database[17] In Family 3 the previously described c718CgtT(pR240X) nonsense mutation in exon 9 [21819] wasdetected in the two affected members and not in the unaffectedmother (Figure 1)
DISCUSSIONWe report three PAX6 mutations segregating in three familieswith aniridia originating from the northwestern part of EgyptTwo of the three mutations are novel frameshifting deletionsand one is a previously described nonsense mutation To ourknowledge this is the first report of aniridia mutations fromthis part of the world It is interesting to note that the pathologyof the PAX6-related genome is far from being fully determinedwhen one considers that only one of the identified mutantswas already known From a clinical point of view the tenpatients of this series harbored common but extensive featuresof ocular-isolated aniridia associated with the wide spectrumof already described MRI brain malformations These includeabsence of the pineal gland and hypogenesis of the corpuscallosum and of the anterior or posterior commissure Theinterpretation of the observed MRI malformations is limitedby the lack of a full assessment of the neurologicaldevelopment refined examinations such aselectroencephalogram neurocognitive tests or sleep studywere not available Nevertheless the patients did not harbor
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2077
TAB
LE 2
LIS
T O
F IN
ITIA
L B
UFF
ER B
CO
NC
ENTR
ATI
ON
S AN
D T
EMPE
RA
TUR
ES FO
R E
AC
H D
NA
FRA
GM
ENT
PCR
WA
VE
Exo
nSe
nse
prim
er (5
rsquo-3rsquo
)A
ntis
ense
pri
mer
(5rsquo-
3rsquo)
Ann
ealin
g te
mpe
ratu
re degC
Tem
pera
ture
degC
star
t B1
TGTT
GC
GG
AG
TGA
TTA
GTG
GTC
CTG
GG
AA
GG
AG
AC
AG
AG
A60
+be
tain
608
579
2A
CA
CA
CTT
GA
GC
CA
TCA
CC
AC
TCC
TGC
GTG
GA
AA
CTT
CT
60 +
beta
in59
360
63
GTG
GG
TGTA
ATG
CTG
GG
AC
TC
CC
AA
TCTG
TTTC
CC
CTA
CA
60 +
beta
in56
597
4C
CC
CA
AG
AG
GTT
GA
GTG
GA
TG
TCG
CG
AG
TCC
CTG
TGTC
60 +
beta
in61
457
15
TGA
GG
ATG
CA
TTG
TGG
TTG
TG
TGG
AA
GG
AG
AG
GG
GA
AA
GT
60 +
beta
in59
560
86
TTC
AG
GC
AG
TGTT
TAA
GA
AA
AG
TTA
CTC
AC
AC
ATC
CG
TTG
GA
CA
5554
358
77
TGC
AG
ATG
CA
AA
AG
TCC
AA
GC
TCTG
TTC
CC
CC
AG
GTA
CA
A60
+be
tain
574
618
TTTC
CA
CG
GTG
TATC
TGC
AA
AA
GC
CC
TGA
GA
GG
AA
ATG
GT
60 +
beta
in59
659
99
AC
CTT
GG
GA
ATG
TTTT
GG
TGC
AC
TGA
AA
AG
ATG
CC
CA
GA
GA
60 +
beta
in56
760
310
AG
GTG
GG
AA
CC
AG
TTTG
ATG
CA
TGG
CA
GC
AG
AG
CA
TTTA
G60
+be
tain
575
5811
TTC
AG
TCTG
CTA
AA
TGC
TCTG
CTG
TGA
GG
GC
TGTG
TCTG
TTC
60 +
beta
in59
6012
AC
CA
CA
CC
GG
GTA
ATT
TGA
AC
TCTC
AA
GG
GTG
CA
GA
CA
CA
60 +
beta
in58
758
413
TAG
CTC
GA
GG
CC
CA
ATC
TTA
TAA
AC
AC
GC
CC
TCC
CA
TAA
G60
+be
tain
583
6014
TTTC
TGA
AG
GTG
CTA
CTT
TTA
TTTG
AA
GTC
CA
TTC
CTT
CC
CC
AG
T55
535
602
15A
AA
CTT
AA
GTG
TTTT
GA
AG
TTG
TTC
AC
CC
CA
GA
TTG
AA
AA
TGC
CA
GT
60 +
beta
in54
360
5
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2078
Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

TAB
LE 2
LIS
T O
F IN
ITIA
L B
UFF
ER B
CO
NC
ENTR
ATI
ON
S AN
D T
EMPE
RA
TUR
ES FO
R E
AC
H D
NA
FRA
GM
ENT
PCR
WA
VE
Exo
nSe
nse
prim
er (5
rsquo-3rsquo
)A
ntis
ense
pri
mer
(5rsquo-
3rsquo)
Ann
ealin
g te
mpe
ratu
re degC
Tem
pera
ture
degC
star
t B1
TGTT
GC
GG
AG
TGA
TTA
GTG
GTC
CTG
GG
AA
GG
AG
AC
AG
AG
A60
+be
tain
608
579
2A
CA
CA
CTT
GA
GC
CA
TCA
CC
AC
TCC
TGC
GTG
GA
AA
CTT
CT
60 +
beta
in59
360
63
GTG
GG
TGTA
ATG
CTG
GG
AC
TC
CC
AA
TCTG
TTTC
CC
CTA
CA
60 +
beta
in56
597
4C
CC
CA
AG
AG
GTT
GA
GTG
GA
TG
TCG
CG
AG
TCC
CTG
TGTC
60 +
beta
in61
457
15
TGA
GG
ATG
CA
TTG
TGG
TTG
TG
TGG
AA
GG
AG
AG
GG
GA
AA
GT
60 +
beta
in59
560
86
TTC
AG
GC
AG
TGTT
TAA
GA
AA
AG
TTA
CTC
AC
AC
ATC
CG
TTG
GA
CA
5554
358
77
TGC
AG
ATG
CA
AA
AG
TCC
AA
GC
TCTG
TTC
CC
CC
AG
GTA
CA
A60
+be
tain
574
618
TTTC
CA
CG
GTG
TATC
TGC
AA
AA
GC
CC
TGA
GA
GG
AA
ATG
GT
60 +
beta
in59
659
99
AC
CTT
GG
GA
ATG
TTTT
GG
TGC
AC
TGA
AA
AG
ATG
CC
CA
GA
GA
60 +
beta
in56
760
310
AG
GTG
GG
AA
CC
AG
TTTG
ATG
CA
TGG
CA
GC
AG
AG
CA
TTTA
G60
+be
tain
575
5811
TTC
AG
TCTG
CTA
AA
TGC
TCTG
CTG
TGA
GG
GC
TGTG
TCTG
TTC
60 +
beta
in59
6012
AC
CA
CA
CC
GG
GTA
ATT
TGA
AC
TCTC
AA
GG
GTG
CA
GA
CA
CA
60 +
beta
in58
758
413
TAG
CTC
GA
GG
CC
CA
ATC
TTA
TAA
AC
AC
GC
CC
TCC
CA
TAA
G60
+be
tain
583
6014
TTTC
TGA
AG
GTG
CTA
CTT
TTA
TTTG
AA
GTC
CA
TTC
CTT
CC
CC
AG
T55
535
602
15A
AA
CTT
AA
GTG
TTTT
GA
AG
TTG
TTC
AC
CC
CA
GA
TTG
AA
AA
TGC
CA
GT
60 +
beta
in54
360
5
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2078
Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

Figure 2 Slit-lamp photographs A Right eye of patient II-7 from Family 1 Note the significant heavy corneal vascularization sparing thenasal area The iris base is very thin almost invisible a typical feature of aniridia The patient was aphakic since cataract surgery performedin childhood Best-corrected visual acuity was 01 B Right eye patient III-4 from Family 1 showing heavy corneal vascularization (pannus)and superior dislocation of the lens Almost no iris residual tissue is visible as typically seen in aniridia Note as well the small anterior polarcataract and the associated faint peripheral cortical opacities C and D Right and left eye of patient IV-3 from Family 2 Note the bilateralposterior polar cataract shaped like the petals of a flower and the superior lens dislocation E and F Fundus photographs Right and left eyeof patient IV-1 from Family 2 Foveal hypoplasia is observed with macular pigment epithelium alterations
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2079
major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

major neurodevelopmental delay that could be detectedclinically by a senior geneticist and pediatrician
In a large genotypendashphenotype correlation of the PAX6Mutation Database Tzoulaki et al [15] established thatmutations introducing a premature termination codon (PTC)were predominantly associated with aniridia whereas non-aniridia phenotypes such as isolated foveal hypoplasiamicrophthalmia and optic nerve defects were predominantlycaused by missense mutations These authors showed that thesecond most frequent type of PAX6 mutations wasframeshifting insertions or deletions and missense mutationswere the third In the present series of patients with typicalaniridia the three identified mutations caused a PTC and twoof them were frameshifting deletions thus confirming thefrequent association of PTC and of frameshifting mutationswith aniridia [15]
The pR240X mutation segregating in Family 3 haspreviously been described [21819] with more than 20independent records in the PAX6 Mutation Database [17]This mutation is a CgtT transition that occurs on a CpGdinucleotide a structure known for its high mutability [20]This CpG in exon 9 located in the homeobox coding regionis a mutation hotspot since CpG dinucleotides of the last thirdof PAX6 tend to be methylated and thus more inclined toundergo spontaneous deamination of cytosine resulting inCgtT transition [21] A sense-strand deamination of CpG in aCGA codon creates a termination codon CTA It has beenproposed that nonsense-mediated decay (NMD) themechanism responsible for the elimination of mRNAs thatcontain premature termination codons before the last exon
[22] is highly involved in aniridia since the majority ofaniridia PAX6 mutations introduce premature terminationcodons [15] Thus NMD impedes truncated proteinformation and loss-of-function of one allele is responsible forthe development of aniridia through haploinsufficiency Thismechanism may explain how different mutations and differentputative truncated proteins can induce similar phenotypesThe Family 1 and 2 unreported mutations pL57fs17 andpR159fs47 are both likely to result in haploinsufficiencythrough NMD
Hypoplasia of the anterior commissure [101214] of theposterior commissure [10] and of the corpus callosum[10-1214] have been reported in heterozygous carriers ofPAX6 mutations Absence or hypoplasia of the anteriorcommissure is present in up to one-third of PAX6 mutationcarriers [1013] Abnormal anterior MRI anomalies can beassociated with subtle neurological deficits such as olfactorydifficulties [14] hearing difficulties [10] and deficits inexecutive and social cognition [11] or with aniridia only[12] We did observe anomalies of these three brain structuresin our three imaged patients (Table 1) Although no brainanomalies can yet be directly related to any specific PAX6mutation we report the third observation of MRI anomaliesassociated with the pR240X mutation [1014] In contrastwith both Sisodiya et al [14] and Bamiou et al [10] whoreported a hypoplastic anterior commissure but a normalcorpus callosum we observed in Patient I-1 of Family 3 analmost complete agenesis of the corpus callosum with anormal anterior commissure (Table 1Figure 4C)Interestingly we observed Probst bundles in this patient
Figure 3 Axial cerebral T2-weighted magnetic resonance images A Patient III-1 from Family 1 Dashed arrow severe hypoplasia of theanterior commissure Arrow head normal posterior commissure Lower arrow absence of the pineal gland B Normal magnetic resonanceimaging (MRI) images Dashed arrow normal anterior commissure Arrow head normal posterior commissure Lower arrow normal pinealgland C Patient IV-3 from Family 2 Dashed arrow normal anterior commissure Arrow head absent posterior commissure Lower arrowabsent pineal gland
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2080
(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

(Figure 4C) which highlights the severity of the corpuscallosum hypogenesis in this case Indeed Probst bundlesrepresent fiber tracts that grow caudally along the medialsurface of the ipsilateral cerebral hemisphere that would havecrossed the midline in the case of normal corpus callosumdevelopment [23] their presence is common in patients withcorpus callosum hypogenesis and twice as frequent in patientswith corpus callosum agenesis [24]
We hypothesize that a specific mutation can cause brainanomalies but that the brain anomaly can be expresseddifferently Being a transcription factor PAX6 interacts withseveral brain developmental genes and transcription factorssuch as the Homeobox gene expressed in ES cells the Hesx1gene [3] whose interaction with PAX6 could be altered by thepresence of a mutant protein causing corpus callosum andbrain commissure hypogenesis It has recently been
demonstrated in mouse that the transcription factors Emptyspiracles homeobox 2 Emx2 and Pax6 are essential forcortical regionalization at the beginning of neuronogenesis[25] From this perspective one can hypothesize thatabnormal interaction between mutated PAX6 protein and anormal EMX2 protein could be responsible for the presenceof the interhemispheric brain anomalies Moreover thepossibility of digenism is not excluded with the presence ofan undetected EMX2 mutation added to the PAX6 one to resultin corpus callosum and commissure dysgenesis Last theexistence of at least two promoters is described in the literatureto mediate Pax6 expression in different tissues [2627] Thusone promoter mediates expression in the brain anddifferential PAX6 transcription through alternate promoterusage could be involved in neural development [28]
Figure 4 Coronal cerebral T2-weightedmagnetic resonance images A PatientI-1 from Family 3 Dashed arrow showssevere hypogenesis of the corpuscallosum with small amount of remnanttissue localized at the virtual connectionbetween the genu and the body of thecorpus callosum arrow head shows theatrophic optic chiasm B Normal MRIimages Dashed arrow shows normalcorpus callosum and arrow head showsnormal optic chiasm C Patient I-1 fromFamily 3 Dashed arrow shows lateralcallosal bundles of Probst which arehemispheric connection fibers that didnot cross the midline and that are seenin callosal dysgenesis Superomedialmargins of the lateral ventricles areindented by the Probst bundles Arrowhead shows remnants of the corpuscallosum Lower arrow shows normalposterior commissure D Normal MRIimage with dashed arrow pointingnormal corpus callosum
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2081
The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

The auditory fibers travel through the interhemisphericpathways and it has been demonstrated that children withPAX6 mutations and abnormalities of the interhemisphericpathways on MRI harbor reduced auditory capacities even inthe presence of normal audiograms [10] We did not observeany major clinical auditory deficits in any of the ten studiedpatients although the three patients with available MRIsshowed abnormal interhemispheric pathways However wecannot conclude on the auditory status since we did notperform any specific tests including the study of auditory-evoked potentials
Mitchell et al [13] published an MRI study of 24 aniridiapatients with PAX6 mutations and found absence of the pinealgland in 1324 patients (54) These authors concluded thatthis observation may be common in aniridia patients Indeedwe have previously reported absence of the pineal gland inaniridia patients [29] as we do in the three patients of thepresent series in whom we performed MRI As previouslymentioned sleep study was not performed and thus we couldnot assess the functional consequences of the absence ofpineal glands Mice homozygous for mutations in the Pax6gene harbor a wide variety of neurodevelopmentalabnormalities including absence of the corpus callosum andpineal gland [3031]
PAX6 affects the development and function of the centralnervous system and the eye as well as the pancreas and thehypothalamopituitary axis through the hypothalamus [3]which shares with the retina a common embryologic originmdashthe neural plate Unfortunately we were not able to performelectroretinography or endocrine testing to study the effect ofthe PAX6 mutations on the retina and hypothalamopituitaryaxis
Finally WAGR syndrome (OMIM 194072) whichincludes Wilmsrsquo tumor aniridia genitourinary anomaliesand mental retardation is caused by either microscopic orsubmicroscopic deletion of chromosome 11p13-p12 in aregion containing both the Wilms tumor 1 WT1 gene and thePAX6 genes A contiguous syndrome the WAGRO syndrome(OMIM 612469) includes the features of WAGR with obesityand is caused by a similar deletion that includes the Brain-derived neurotrophic factor BDNF gene as well Bothsyndromes were clinically excluded by normal kidney andurogenital ultrasonography performed on our patients
In summary we describe three mutations found in threefamilies from northwestern Egypt adding two novelmutations to the existing spectrum of PAX6 mutations Twoof the three mutations are frameshifting small deletions andone is a previously described nonsense mutation While theten familial aniridia patients of this series harbored typicalfeatures of ocular-isolated aniridia we observed a widespectrum of MRI brain malformations including absence ofthe pineal gland hypogenesis of the corpus callosum withProbst bundles and hypoplasia of the anterior or posterior
commissure in patients not harboring major neurologicaldevelopment anomalies Correlation between phenotype andgenotype of PAX6 mutations is still in its infancy in regard tobrain malformations The wide spectrum of PAX6-relatedanomalies and the fact that aniridia is frequently caused byPAX6 mutations should prompt physicians facing aniridia toperform an examination of the central nervous system (at leastwith an MRI) an ultrasonography of the kidney and of theurinary pathways a study of the kidney functions and mostimportantly a complete assessment of the pituitary hormonesand hypothalamic-releasing hormones
ACKNOWLEDGMENTSThis study was supported by grant 320030_127558 form theSwiss National Science Foundation (to Drs Schorderet andMunier)
REFERENCES1 Mannens M Bleeker-Wagemakers EM Bliek J Hoovers J
Mandjes I van Tol S Frants RR Heyting C Westerveld ASlater RM Autosomal dominant aniridia linked to thechromosome 11p13 markers catalase and D11S151 in a largeDutch family Cytogenet Cell Genet 1989 5232-6 [PMID2575483]
2 Glaser T Walton DS Maas RL Genomic structureevolutionary conservation and aniridia mutations in thehuman PAX6 gene Nat Genet 1992 2232-9 [PMID1345175]
3 Haubst N Favor J Goumltz M The Role of Pax6 in the NervousSystem during Development and in Adulthood MasterControl Regulator or Modular Function In Gerald ThielEditor Transcription Factors in the Nervous SystemWeinheim Wiley-VCH Verlag GmbH amp Co 2006 p 23-51
4 Hill RE Favor J Hogan BL Ton CC Saunders GF HansonIM Prosser J Jordan T Hastie ND van Heyningen V Mousesmall eye results from mutations in a paired-like homeobox-containing gene Nature 1991 354522-5 [PMID 1684639]
5 Quiring R Walldorf U Kloter U Gehring WJ Homology ofthe eyeless gene of Drosophila to the Small eye gene in miceand Aniridia in humans Science 1994 265785-9 [PMID7914031]
6 Stoykova A Gruss P Roles of Pax-genes in developing andadult brain as suggested by expression patterns J Neurosci1994 141395-412 [PMID 8126546]
7 Jones L Loacutepez-Bendito G Gruss P Stoykova A Molnaacuter ZPax6 is required for the normal development of the forebrainaxonal connections Development 2002 1295041-52[PMID 12397112]
8 Norman MG McGillivray BC Kalousek DK Hill A PoskittKJ Congenital Malformations of the Brain New YorkOxford University Press 1995
9 Glaser T Jepeal L Edwards JG Young SR Favor J Maas RLPAX6 gene dosage effect in a family with congenitalcataracts aniridia anophthalmia and central nervous systemdefects Nat Genet 1994 7463-71 [PMID 7951315]
10 Bamiou DE Free SL Sisodiya SM Chong WK Musiek FWilliamson KA van Heyningen V Moore AT Gadian DLuxon LM Auditory interhemispheric transfer deficits
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
2082
hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083

hearing difficulties and brain magnetic resonance imagingabnormalities in children with congenital aniridia due toPAX6 mutations Arch Pediatr Adolesc Med 2007161463-9 [PMID 17485622]
11 Ellison-Wright Z Heyman I Frampton I Rubia K Chitnis XEllison-Wright I Williams SC Suckling J Simmons ABullmore E Heterozygous PAX6 mutation adult brainstructure and fronto-striato-thalamic function in a humanfamily Eur J Neurosci 2004 191505-12 [PMID 15066147]
12 Free SL Mitchell TN Williamson KA Churchill AJ ShorvonSD Moore AT van Heyningen V Sisodiya SM QuantitativeMR image analysis in subjects with defects in the PAX6 geneNeuroimage 2003 202281-90 [PMID 14683729]
13 Mitchell TN Free SL Williamson KA Stevens JM ChurchillAJ Hanson IM Shorvon SD Moore AT van Heyningen VSisodiya SM Polymicrogyria and absence of pineal gland dueto PAX6 mutation Ann Neurol 2003 53658-63 [PMID12731001]
14 Sisodiya SM Free SL Williamson KA Mitchell TN Willis CStevens JM Kendall BE Shorvon SD Hanson IM MooreAT van Heyningen V PAX6 haploinsufficiency causescerebral malformation and olfactory dysfunction in humansNat Genet 2001 28214-6 [PMID 11431688]
15 Tzoulaki I White IM Hanson IM PAX6 mutations genotype-phenotype correlations BMC Genet 2005 627 [PMID15918896]
16 Abouzeid H Munier FL Thonney F Schorderet DF Ten novelRB1 gene mutations in patients with retinoblastoma Mol Vis2007 131740-5 [PMID 17960112]
17 Human PAX 6 Mutation Database [Internet] Edinburgh MRCHuman Genetics Unit 2007-[cited 2009 Feb 10] Availablefrom httplsdbhgumrcacukhomephpselect_db=PAX6
18 Redeker EJ de Visser AS Bergen AA Mannens MMMultiplex ligation-dependent probe amplification (MLPA)enhances the molecular diagnosis of aniridia and relateddisorders Mol Vis 2008 14836-40 [PMID 18483559]
19 Robinson DO Howarth RJ Williamson KA van Heyningen VBeal SJ Crolla JA Genetic analysis of chromosome 11p13and the PAX6 gene in a series of 125 cases referred withaniridia Am J Med Genet A 2008 146A558-69 [PMID18241071]
20 Cooper DN Krawczak M Cytosine methylation and the fate ofCpG dinucleotides in vertebrate genomes Hum Genet 198983181-8 [PMID 2777259]
21 Nachman MW Crowell SL Estimate of the mutation rate pernucleotide in humans Genetics 2000 156297-304 [PMID10978293]
22 Byers PH J Clin Invest 2002 1093-6Killing the messengernew insights into nonsense-mediated mRNA decay [PMID11781342]
23 Probst FP Congenital defects of the corpus callosumMorphology and encephalographic appearances Acta RadiolSuppl 1973 3311-152 [PMID 4202700]
24 Hetts SW Sherr EH Chao S Gobuty S Barkovich AJAnomalies of the corpus callosum an MR analysis of thephenotypic spectrum of associated malformations AJR AmJ Roentgenol 2006 1871343-8 [PMID 17056927]
25 Muzio L DiBenedetto B Stoykova A Boncinelli E Gruss PMallamaci A Emx2 and Pax6 control regionalization of thepre-neuronogenic cortical primordium Cereb Cortex 200212129-39 [PMID 11739261]
26 Plaza S Saule S Dozier C High conservation of cis-regulatoryelements between quail and human for the Pax-6 gene DevGenes Evol 1999 209165-73 [PMID 10079359]
27 Kammandel B Chowdhury K Stoykova A Aparicio S BrennerS Gruss P Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity Dev Biol 199920579-97 [PMID 9882499]
28 Okladnova O Syagailo YV Moumlssner R Riederer P Lesch KPRegulation of PAX-6 gene transcription alternate promoterusage in human brain Brain Res Mol Brain Res 199860177-92 [PMID 9757029]
29 Dansault A David G Schwartz C Jaliffa C Vieira V de laHoussaye G Bigot K Catin F Tattu L Chopin C Halimi PRoche O Van Regemorter N Munier F Schorderet D DufierJL Marsac C Ricquier D Menasche M Penfornis A AbitbolM Three new PAX6 mutations including one causing anunusual ophthalmic phenotype associated withneurodevelopmental abnormalities Mol Vis 200713511-23 [PMID 17417613]
30 Schmahl W Knoedlseder M Favor J Davidson D Defects ofneuronal migration and the pathogenesis of corticalmalformations are associated with Small eye (Sey) in themouse a point mutation at the Pax-6-locus Acta Neuropathol1993 86126-35 [PMID 8213068]
31 Estivill-Torrus G Vitalis T Fernandez-Llebrez P Price DJ Thetranscription factor Pax6 is required for development of thediencephalic dorsal midline secretory radial glia that form thesubcommissural organ Mech Dev 2001 109215-24 [PMID11731235]
Molecular Vision 2009 152074-2083 lthttpwwwmolvisorgmolvisv15a223gt copy 2009 Molecular Vision
The print version of this article was created on 14 October 2009 This reflects all typographical corrections and errata to thearticle through that date Details of any changes may be found in the online version of the article
2083