Embed Size (px)
Citation preview

ARTICLE IN PRESS
Pathophysiology and Treatment ofHypertrophic Cardiomyopathy
Mark V. Sherrid
All patients with hypertrophic cardiomyopathy(HCM) should have five aspects of care addressed.An attempt should be made to detect the presenceor absence of risk factors for sudden arrhythmicdeath. If the patient appears to be at high risk,discussion of the benefits and risks of ICD areindicated, and many such patients will be im-planted. Symptoms are appraised and treated.Bacterial endocarditis prophylaxis is recommen-ded. Patients are advised to avoid athletic compe-tition and extremes of physical exertion. Firstdegree family members should be screened withechocardiography and ECG.n 2006 Elsevier Inc. All rights reserved.
Hypertrophic cardiomyopathy (HCM) is
viewed as a complex and challengingcardiac disease. Its pathophysiology, diagnosis
and treatment span the gamut of cardiologic
disciplines: In pathophysiology one must con-
sider left ventricular outflow obstruction, mitral
regurgitation, ischemia, atrial fibrillation, sud-
den death, diastolic dysfunction, molecular
biology and genetics. Diagnostic testing with
echocardiography, nuclear scintigraphy, stresstesting, catheterization, 24 hour ECG, and MRI
may be applied. Treatment may involve the
implanted defibrillator, pharmacologic agents,
surgery, transcoronary intervention, or pacing.
But, when these lists are examined, one recog-
nizes that this is same exact spectrum encoun-
tered in more common cardiac diseases. The
Progress in Cardiovascular Diseases, Vol. 0, No. 0 (August), 20
From the Hypertrophic Cardiomyopathy Program andEchocardiography Laboratory, Department of Medicine,
Division of Cardiology, St. Luke’s-Roosevelt Hospital
Center, College of Physicians and Surgeons, ColumbiaUniversity, New York, NY.
Address reprint requests to Mark V. Sherrid, MD, 1000
10th Avenue 3B-30, New York City, NY 10019.
E-mail: [email protected]/$ - see front matter
n 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.pcad.2006.08.001
challenge in HCM is learning the disease-specificpathophysiology and treatment indications.
Genetics, Pathology, Diagnosis
The inherited nature of HCM was noted as early
as the modern description of the disease.1
Hypertrophic cardiomyopathy is inherited as an
autosomal dominant trait; roughly half of
patients have another family member with
HCM. Unexplained hypertrophy occurs in
1:500 in the general population, making it the
most common inherited cardiac disorder. Sarco-
meric mutations of 10 genes that code for
myofilaments or their supporting proteins havebeen identified as a cause of HCM.2-5 In a cohort
of referred, unrelated patients with HCM, rough-
ly 40% of patients with HCM were found to have
sarcomeric mutations. In the remaining 60%,
none of the known genotype abnormalities were
found.5 Younger age at diagnosis, marked wall
thickness, and a family history of HCM increase
the frequency that a patient will be gene positive.Echocardiographic appearance also appears to
predict a high likelihood of sarcomeric-protein
mutation HCM; a reversed septal curvature
causing a crescent-shaped LV cavity predicts
gene-positive patients as compared with those
with localized subaortic bulge and preserved
septal curvature.6,7 The most common mutations
found are in the b-myosin heavy chain and inmyosin-binding protein C. Although the bulk of
genetically determined HCM occurs on 8 genes,
many hundreds of HCM-causing mutations are
dispersed over the many loci of these genes. All
of these genes may cause different phenotypes
and have different prognoses. Even among
families with the same mutation on a particular
loci individuals vary with respect to phenotypeand prognosis. This has markedly delayed
genotype-phenotype correlation. The pathophys-
iologic linkage between mutations and hypertro-
phy appears to be mediated by mutation-induced
functional abnormalities.8
06: pp 1-29 1
ARTICLE IN PRESS
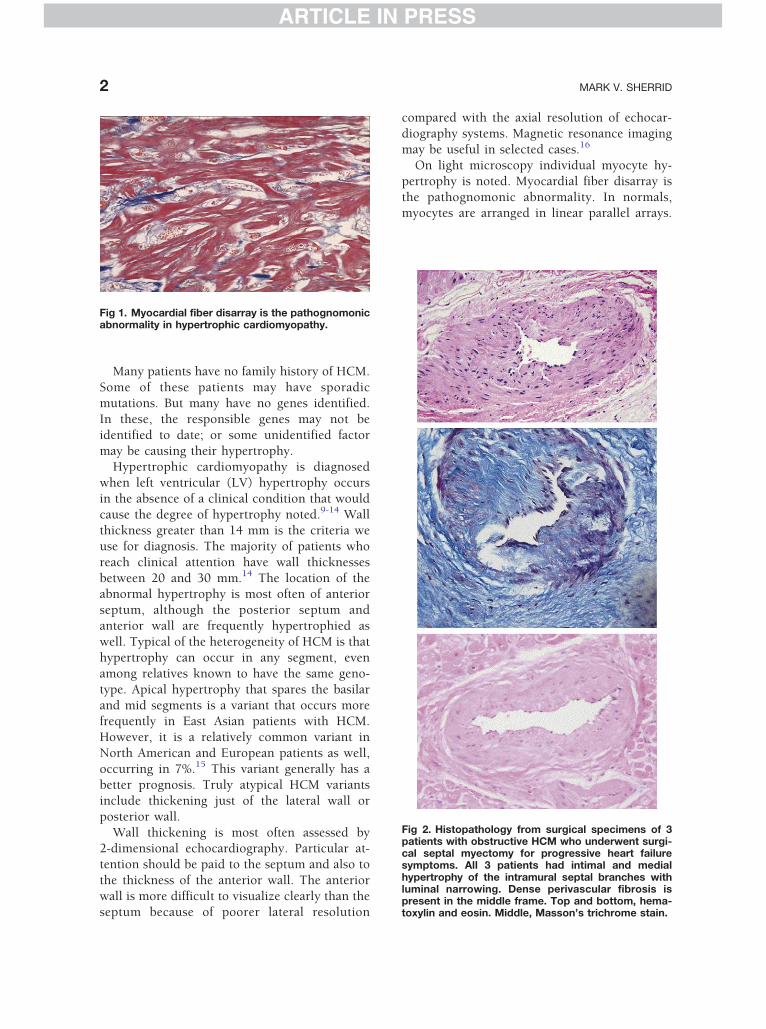
Fig 1. Myocardial fiber disarray is the pathognomonicabnormality in hypertrophic cardiomyopathy.
Fig 2. Histopathology from surgical specimens of 3patients with obstructive HCM who underwent surgi-cal septal myectomy for progressive heart failuresymptoms. All 3 patients had intimal and medialhypertrophy of the intramural septal branches withluminal narrowing. Dense perivascular fibrosis ispresent in the middle frame. Top and bottom, hema-toxylin and eosin. Middle, Masson’s trichrome stain.
MARK V. SHERRID2
ARTICLE IN PRESS
Many patients have no family history of HCM.
Some of these patients may have sporadic
mutations. But many have no genes identified.
In these, the responsible genes may not be
identified to date; or some unidentified factor
may be causing their hypertrophy.Hypertrophic cardiomyopathy is diagnosed
when left ventricular (LV) hypertrophy occurs
in the absence of a clinical condition that would
cause the degree of hypertrophy noted.9-14 Wall
thickness greater than 14 mm is the criteria we
use for diagnosis. The majority of patients who
reach clinical attention have wall thicknesses
between 20 and 30 mm.14 The location of theabnormal hypertrophy is most often of anterior
septum, although the posterior septum and
anterior wall are frequently hypertrophied as
well. Typical of the heterogeneity of HCM is that
hypertrophy can occur in any segment, even
among relatives known to have the same geno-
type. Apical hypertrophy that spares the basilar
and mid segments is a variant that occurs morefrequently in East Asian patients with HCM.
However, it is a relatively common variant in
North American and European patients as well,
occurring in 7%.15 This variant generally has a
better prognosis. Truly atypical HCM variants
include thickening just of the lateral wall or
posterior wall.
Wall thickening is most often assessed by2-dimensional echocardiography. Particular at-
tention should be paid to the septum and also to
the thickness of the anterior wall. The anterior
wall is more difficult to visualize clearly than the
septum because of poorer lateral resolution
compared with the axial resolution of echocar-
diography systems. Magnetic resonance imaging
may be useful in selected cases.16
On light microscopy individual myocyte hy-
pertrophy is noted. Myocardial fiber disarray is
the pathognomonic abnormality. In normals,myocytes are arranged in linear parallel arrays.
ARTICLE IN PRESS
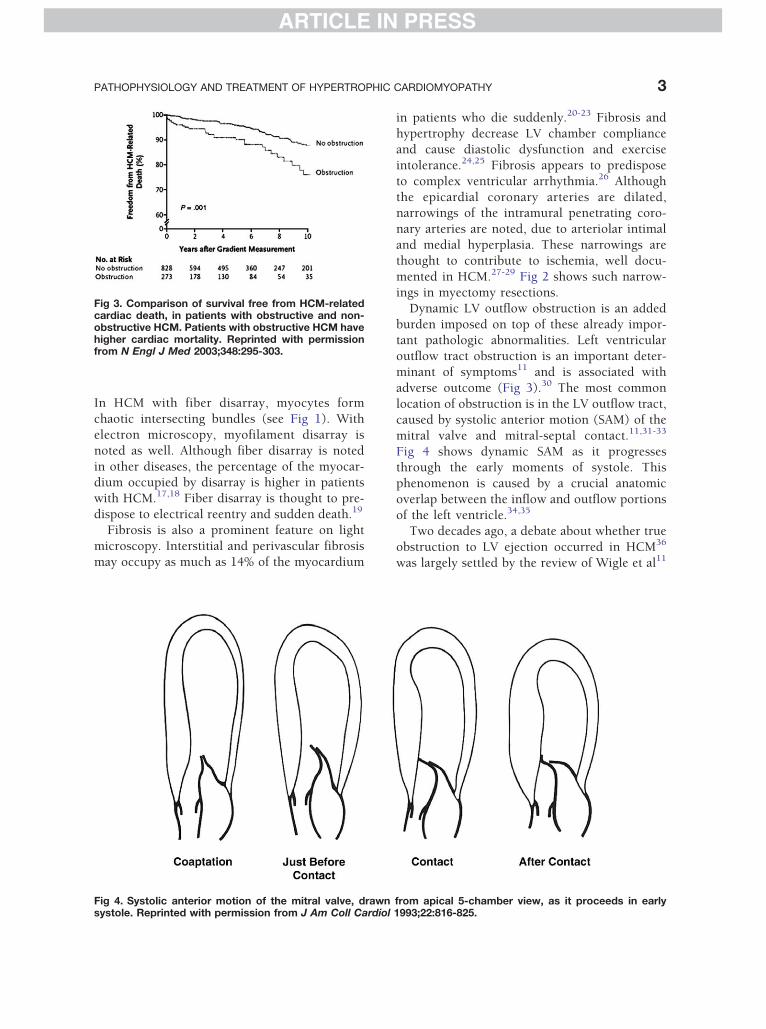
Fig 3. Comparison of survival free from HCM-relatedcardiac death, in patients with obstructive and non-obstructive HCM. Patients with obstructive HCM havehigher cardiac mortality. Reprinted with permissionfrom N Engl J Med 2003;348:295-303.
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 3
ARTICLE IN PRESS
In HCM with fiber disarray, myocytes form
chaotic intersecting bundles (see Fig 1). With
electron microscopy, myofilament disarray is
noted as well. Although fiber disarray is noted
in other diseases, the percentage of the myocar-dium occupied by disarray is higher in patients
with HCM.17,18 Fiber disarray is thought to pre-
dispose to electrical reentry and sudden death.19
Fibrosis is also a prominent feature on light
microscopy. Interstitial and perivascular fibrosis
may occupy as much as 14% of the myocardium
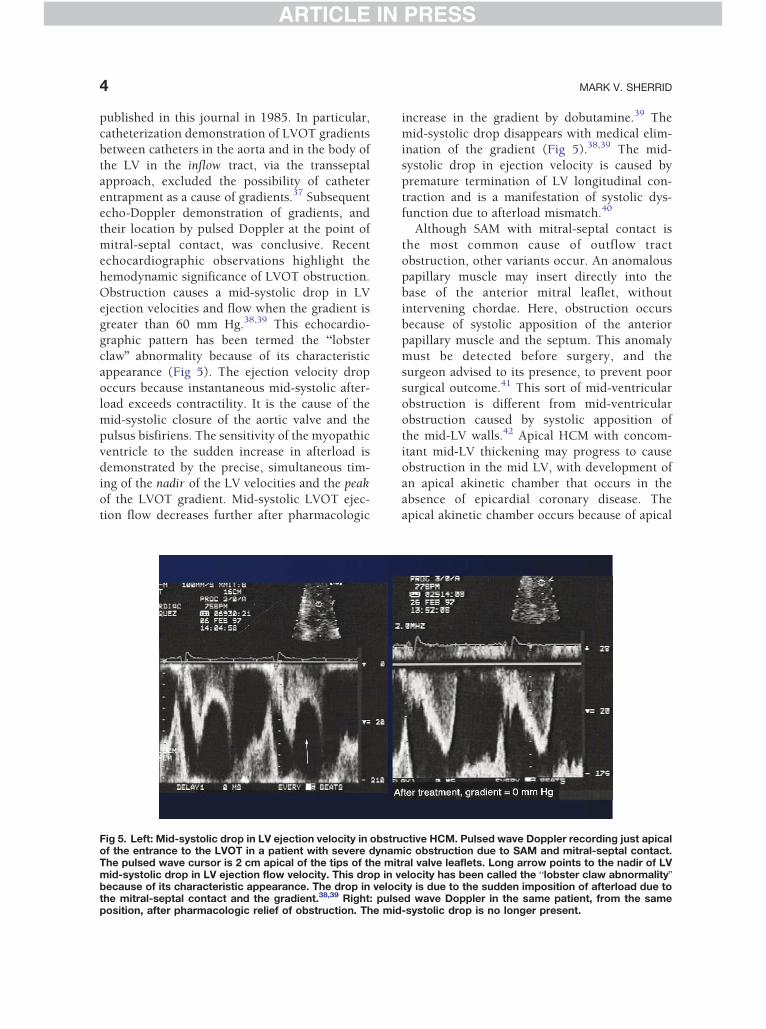
Fig 4. Systolic anterior motion of the mitral valve, drawnsystole. Reprinted with permission from J Am Coll Cardiol
in patients who die suddenly.20-23 Fibrosis and
hypertrophy decrease LV chamber compliance
and cause diastolic dysfunction and exercise
intolerance.24,25 Fibrosis appears to predispose
to complex ventricular arrhythmia.26 Although
the epicardial coronary arteries are dilated,narrowings of the intramural penetrating coro-
nary arteries are noted, due to arteriolar intimal
and medial hyperplasia. These narrowings are
thought to contribute to ischemia, well docu-
mented in HCM.27-29 Fig 2 shows such narrow-
ings in myectomy resections.
Dynamic LV outflow obstruction is an added
burden imposed on top of these already impor-tant pathologic abnormalities. Left ventricular
outflow tract obstruction is an important deter-
minant of symptoms11 and is associated with
adverse outcome (Fig 3).30 The most common
location of obstruction is in the LV outflow tract,
caused by systolic anterior motion (SAM) of the
mitral valve and mitral-septal contact.11,31-33
Fig 4 shows dynamic SAM as it progressesthrough the early moments of systole. This
phenomenon is caused by a crucial anatomic
overlap between the inflow and outflow portions
of the left ventricle.34,35
Two decades ago, a debate about whether true
obstruction to LV ejection occurred in HCM36
was largely settled by the review of Wigle et al11
from apical 5-chamber view, as it proceeds in early1993;22:816-825.
ARTICLE IN PRESS
MARK V. SHERRID4
ARTICLE IN PRESS
published in this journal in 1985. In particular,
catheterization demonstration of LVOT gradients
between catheters in the aorta and in the body of
the LV in the inflow tract, via the transseptal
approach, excluded the possibility of catheter
entrapment as a cause of gradients.37 Subsequentecho-Doppler demonstration of gradients, and
their location by pulsed Doppler at the point of
mitral-septal contact, was conclusive. Recent
echocardiographic observations highlight the
hemodynamic significance of LVOT obstruction.
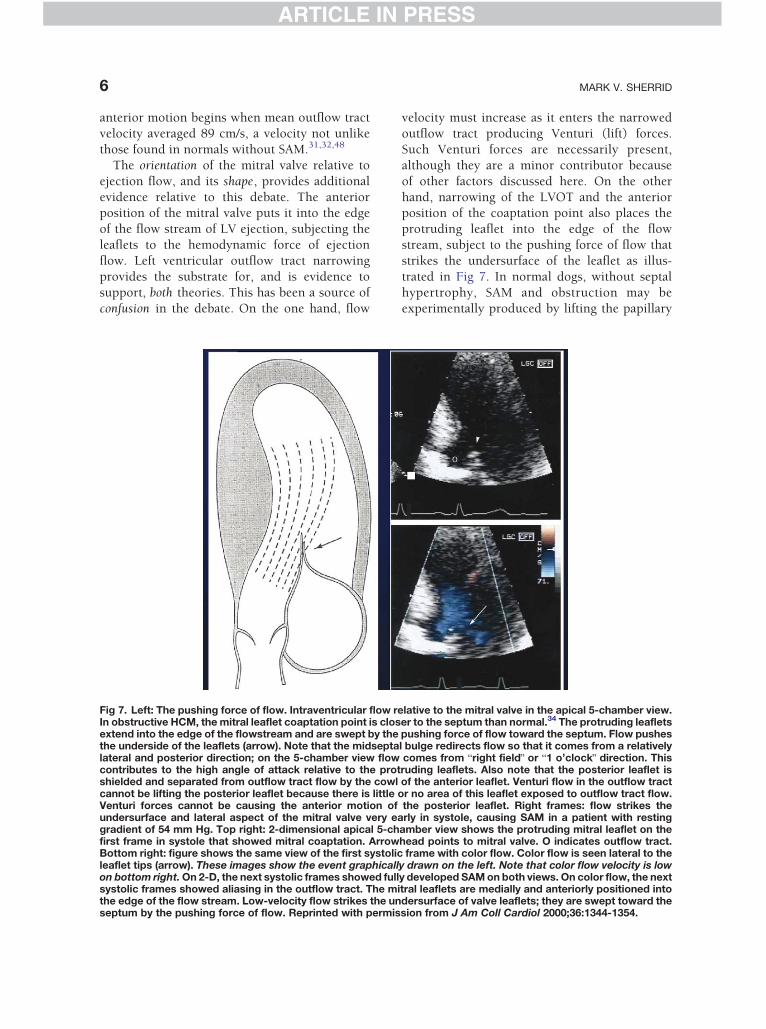
Obstruction causes a mid-systolic drop in LV
ejection velocities and flow when the gradient is
greater than 60 mm Hg.38,39 This echocardio-graphic pattern has been termed the blobster
clawQ abnormality because of its characteristic
appearance (Fig 5). The ejection velocity drop
occurs because instantaneous mid-systolic after-
load exceeds contractility. It is the cause of the
mid-systolic closure of the aortic valve and the
pulsus bisfiriens. The sensitivity of the myopathic
ventricle to the sudden increase in afterload isdemonstrated by the precise, simultaneous tim-
ing of the nadir of the LV velocities and the peak
of the LVOT gradient. Mid-systolic LVOT ejec-
tion flow decreases further after pharmacologic
Fig 5. Left: Mid-systolic drop in LV ejection velocity in obstruof the entrance to the LVOT in a patient with severe dynamThe pulsed wave cursor is 2 cm apical of the tips of the mitmid-systolic drop in LV ejection flow velocity. This drop in vbecause of its characteristic appearance. The drop in velocthe mitral-septal contact and the gradient.38,39 Right: pulsposition, after pharmacologic relief of obstruction. The mid
increase in the gradient by dobutamine.39 The
mid-systolic drop disappears with medical elim-
ination of the gradient (Fig 5).38,39 The mid-
systolic drop in ejection velocity is caused by
premature termination of LV longitudinal con-
traction and is a manifestation of systolic dys-function due to afterload mismatch.40
Although SAM with mitral-septal contact is
the most common cause of outflow tract
obstruction, other variants occur. An anomalous
papillary muscle may insert directly into the
base of the anterior mitral leaflet, without
intervening chordae. Here, obstruction occurs
because of systolic apposition of the anteriorpapillary muscle and the septum. This anomaly
must be detected before surgery, and the
surgeon advised to its presence, to prevent poor
surgical outcome.41 This sort of mid-ventricular
obstruction is different from mid-ventricular
obstruction caused by systolic apposition of
the mid-LV walls.42 Apical HCM with concom-
itant mid-LV thickening may progress to causeobstruction in the mid LV, with development of
an apical akinetic chamber that occurs in the
absence of epicardial coronary disease. The
apical akinetic chamber occurs because of apical
ctive HCM. Pulsed wave Doppler recording just apicalic obstruction due to SAM and mitral-septal contact.ral valve leaflets. Long arrow points to the nadir of LVelocity has been called the blobster claw abnormalityQity is due to the sudden imposition of afterload due toed wave Doppler in the same patient, from the same-systolic drop is no longer present.

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 5
ARTICLE IN PRESS
blood trapping, high apical chamber pressures,
and supply-demand mismatch at the apex.
Severe symptoms may accompany this develop-
ment; the akinetic chamber may harbor thrombi
and also become a source of monomorphic
ventricular tachycardia.43
Left ventricular outflow tract obstruction is
quantified by measuring the pressure drop, the
gradient across the narrowing. This is most
commonly done with continuous wave Doppler
echocardiography.44 Pulsed Doppler correlation
with the 2-dimensional echocardiogram allows
determination of the site of obstruction, which
must be ascertained in every patient, especially ifintervention is contemplated. Resting obstruc-
tion is considered present when a resting
gradient of 30 mm Hg is present. Changing
preload and afterload may provoke a gradient by
increasing the overlap between the inflow and
outflow portions of the LV. Patients who have no
resting gradient but who have gradients greater
than 30 mm Hg after maneuvers have latentobstruction, and patients with mild obstruction
that rises above 30 mm Hg after maneuvers have
provocable obstruction. Typically, Valsalva, ex-
ercise, and standing may be used to provoke
obstruction and, on occasion, exercise in the
post-prandial state.45 As the main reason for
provoking gradient is to correlate patient symp-
toms with obstruction and to provide a target fortherapy, one should only use physiologic maneu-
vers, such as standing or exercise or Valsalva.
Dobutamine and amyl nitrite are not physiologic
stimuli and should not be used to provoke
gradient. Also, dobutamine may provoke gra-
dients in normals. Clinically, we perform tread-
mill stress exercise echocardiography on all
patients with HCM who are able to exercise.An exception would be for patients with resting
gradients of more than 150 mm Hg where little
available information will be gained.
Cardiac catherization may also demonstrate
the severity and location of gradients in
obstructed patients.11 During the procedure,
gradients may be provoked by Valsalva and the
introduction of premature ventricular beats.
Fig 6. The debate between the Venturi (pulling)mechanism and the flow drag (pushing) mechanismof systolic anterior motion of the mitral valve. Reprin-ted with permission from J Am Coll Cardiol 2000;36:1344-1354.
Pathophysiology of SAM
Understanding the hemodynamic mechanism of
SAM is crucial to developing successful treat-
ment strategies. There is agreement about the
anatomic features that expose the mitral valve to
the hydrodynamic effect of flow and thus
predispose to SAM. These are the septal bulge,
large mitral leaflets that are anteriorly positioned
in the LV cavity because of anterior displacementof the papillary muscles, and residual portions of
the leaflets that extend past the coaptation point
and protrude into the outflow tract.11,41,46-49
Initial reports advanced the hypothesis that
SAM might be caused by a Venturi mechanism, a
local underpressure in the LVOT caused by
narrowing of the outflow tract and rapid early
ejection. An alternate theory is that the mitralvalve is swept into the septum by the pushing
force of flow, referred to as the drag force.31,32,48
Contrasting points favoring the Venturi mecha-
nism of SAM vs the flow drag mechanism are
shown in Fig 6. Data pertinent to the debate
about the cause of SAM focuses on the geometry
of the LV relative to the mitral valve, the velocity
of the flow in the LVOT, and the shape of themitral valve. These are admittedly not the sort of
data cardiologists are usually called on to
evaluate, but a brief review may be illuminating.
Systolic anterior motion begins at a time of low
Doppler velocities in the LV. This is not compat-
ible with the Venturi mechanism, which posits a
high-velocity ejection flow pulling the protruding
mitral leaflet toward the septum.32 Systolic
ARTICLE IN PRESS
MARK V. SHERRID6
ARTICLE IN PRESS
anterior motion begins when mean outflow tract
velocity averaged 89 cm/s, a velocity not unlike
those found in normals without SAM.31,32,48
The orientation of the mitral valve relative to
ejection flow, and its shape, provides additional
evidence relative to this debate. The anteriorposition of the mitral valve puts it into the edge
of the flow stream of LV ejection, subjecting the
leaflets to the hemodynamic force of ejection
flow. Left ventricular outflow tract narrowing
provides the substrate for, and is evidence to
support, both theories. This has been a source of
confusion in the debate. On the one hand, flow
Fig 7. Left: The pushing force of flow. Intraventricular flow reIn obstructive HCM, the mitral leaflet coaptation point is closextend into the edge of the flowstream and are swept by thethe underside of the leaflets (arrow). Note that the midseptalateral and posterior direction; on the 5-chamber view flowcontributes to the high angle of attack relative to the protshielded and separated from outflow tract flow by the cowlcannot be lifting the posterior leaflet because there is little oVenturi forces cannot be causing the anterior motion ofundersurface and lateral aspect of the mitral valve very eagradient of 54 mm Hg. Top right: 2-dimensional apical 5-chfirst frame in systole that showed mitral coaptation. ArrowBottom right: figure shows the same view of the first systolicleaflet tips (arrow). These images show the event graphicallon bottom right. On 2-D, the next systolic frames showed fullsystolic frames showed aliasing in the outflow tract. The mithe edge of the flow stream. Low-velocity flow strikes the unseptum by the pushing force of flow. Reprinted with permis
velocity must increase as it enters the narrowed
outflow tract producing Venturi (lift) forces.
Such Venturi forces are necessarily present,
although they are a minor contributor because
of other factors discussed here. On the other
hand, narrowing of the LVOT and the anteriorposition of the coaptation point also places the
protruding leaflet into the edge of the flow
stream, subject to the pushing force of flow that
strikes the undersurface of the leaflet as illus-
trated in Fig 7. In normal dogs, without septal
hypertrophy, SAM and obstruction may be
experimentally produced by lifting the papillary
lative to the mitral valve in the apical 5-chamber view.er to the septum than normal.34 The protruding leafletspushing force of flow toward the septum. Flow pushesl bulge redirects flow so that it comes from a relativelycomes from bright fieldQ or b1 o’clockQ direction. Thisruding leaflets. Also note that the posterior leaflet isof the anterior leaflet. Venturi flow in the outflow tractr no area of this leaflet exposed to outflow tract flow.the posterior leaflet. Right frames: flow strikes therly in systole, causing SAM in a patient with resting
amber view shows the protruding mitral leaflet on thehead points to mitral valve. O indicates outflow tract.
frame with color flow. Color flow is seen lateral to they drawn on the left. Note that color flow velocity is lowy developed SAM on both views. On color flow, the nexttral leaflets are medially and anteriorly positioned intodersurface of valve leaflets; they are swept toward thesion from J Am Coll Cardiol 2000;36:1344-1354.

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 7
ARTICLE IN PRESS
muscles anteriorly with ligatures exposing the
valve to drag forces.50 Similarly, SAM and
obstruction may occur as a complication of
mitral annuloplasty for prolapse when mitral
coaptation is displaced anteriorly by the ring.
Consequently, annuloplasty techniques havebeen developed to ensure that postoperative
mitral coaptation is posterior in the LV, explic-
itly out of the way of the ejection stream and
drag forces.
Orientation
In patients with obstructive HCM there is a highangle of attack of the Doppler ejection flow stream
onto the mitral valve leaflets. This orientation
precludes significant Venturi effects and impli-
cates drag.31 In the apical 5-chamber view, local
flow direction comes from an angle lateral to the
protruding leaflet. The mean angle at time of
mitral coaptation was lateral by 218; mean angle
just before septal contact increased to 458. Dragincreases as systole progresses. As the mitral valve
is pushed toward the septum, the angle of attack
relative to flow increases. An analogy is a partly
opened door in a drafty corridor: the draft catches
the door and sets it in motion; as it presents a
greater surface to the wind the forces on it
increase, until it is slammed shut. These events
are shown graphically in Figs 4 and 8-10.A major contributor to the high positive angle
of attack of flow onto the mitral valve is the mid-
septal bulge which is the rule in patients with
high resting gradients, occurring in 92% of
patients with resting obstruction.11 This occurs
because the midseptal bulge forces the outflow to
sweep from a relatively posterior and lateral
direction in the LV, as shown in Fig 6. Whenviewed in the echocardiographic apical 5-cham-
ber view, flow comes from bright fieldQ or
b1 o’clockQ direction and strikes the undersurface
of the valve from a posterior and lateral direction
and with a high angle.31 In contrast, subaortic
basilar septal thickening that just narrows the
outflow tract is uncommon in patients with
resting gradients, found in just 12%.11
Shape of the Mitral Valve
The mitral valve resembles other biologic struc-
tures with high drag coefficient. The valve has a
sharp anterior edge with no streamlining, and
there is a concavity under the cowl of the
protruding leaflet.47,48,51,52 The mitral valve in
obstructive HCM displays increased drag coeffi-
cient with increasing velocity of flow due to
increased contractility, an adverse feature similarto other examples in nature.52 Vogel and others
have extensively studied and quantified such
shape reconfiguration with increase in velocity
(ibid, pp 113-126).
Other evidence indicating the drag mechanism
stems from posterior leaflet SAM. In almost all
patients with SAM and obstruction, the posterior
leaflet moves anteriorly as well, underneath theanterior leaflet.53,54 But the posterior leaflet is
separated from the flow in the LVOT by the cowl
of the anterior leaflet as shown in Fig 7. Venturi
forces in the LVOT cannot be lifting the
posterior leaflet because there is little or no area
of the posterior leaflet that is exposed to LVOT
flow. In light of this and the previously men-
tioned geometric observations, it is concludedthat the posterior leaflet is pushed anteriorly.
This mechanism is shown in Fig 7. By Occam’s
razor, is it likely that the anterior and posterior
leaflets, which share a coaptation plane, have
different causes for SAM? It is more reasonable
that the anterior motion of the anterior leaflet is
caused by the same force that triggers the
abnormal posterior leaflet motion: both arecaused by flow drag.
Chordal slack plays a permissive role and is
necessary for SAM to occur. Without chordal
slack no SAM would occur because the leaflets
would be tethered. Systolic anterior motion is
anteriorly directed mitral valve prolapse.31 In both
conditions, the mitral valve is often large and is
pushed by flow from its normal position, withmitral regurgitation as a result.
Figs 8 and 9 show the sequence of events in
late diastole and early systole in a patient with
severe SAM just before mitral-septal contact.
Low-velocity flow is seen behind the mitral
valve, shown as dark blue color flow. No high-
velocity flow is seen in the LVOT until the mitral
valve is actually touching the septum and agradient has developed. It is the low-velocity
flow behind the valve that pushes it into the
septum, well before any high-velocity flow
develops in the outflow tract. Fig 10 shows a
similar example.
ARTICLE IN PRESS
MARK V. SHERRID8
ARTICLE IN PRESS
Therapeutic Implications of Drag asthe Cause of SAM
Recently, new methods to relieve obstruction
have been developed: revised surgical techni-
ques, alcohol septal ablation (ASA), and dual-
chamber pacing. Interventions are not always
successful and the reason for heterogeneity in
response is not clear. Understanding the central
role of flow drag in the pathogenesis of SAM may
prevent treatment failures.55,56
Fig 11 (second panel) shows how inadequate
myectomy resection focused on just the sub-
valvular septum, targeted to widen the outflow
tract and to reduce Venturi forces, may result in
persistent SAM and obstruction. A limited
myectomy misses the impact of the mid-ven-
tricular septal bulge which redirects LV flow so
that it comes from a relatively posterolateraldirection. This sort of resection results in
persistent SAM and either outflow obstruction
or mitral regurgitation because flow still must
course around the remaining septal hypertro-
phy, and it still catches the mitral valve and
pushes it into the septum, and causes mitral
regurgitation. To alleviate this sort of residual
SAM, Messmer57 and Schoendube et al58 havepopularized the extended myectomy, diagram-
matically shown in Fig 11 (third panel). More
extensive resection redirects flow away from the
mitral valve precluding drag-induced SAM. A
large decrease in the angle of attack of flow
relative to the mitral valve has been shown after
successful myectomy; flow is made more parallel
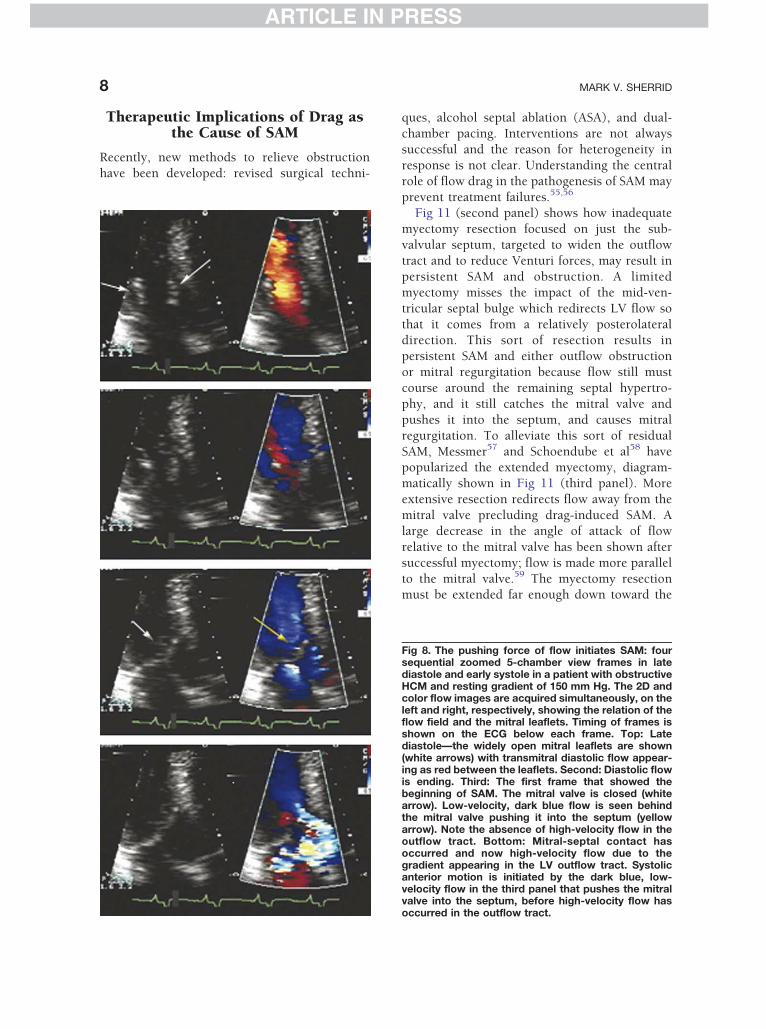
to the mitral valve.59 The myectomy resectionmust be extended far enough down toward the
Fig 8. The pushing force of flow initiates SAM: foursequential zoomed 5-chamber view frames in latediastole and early systole in a patient with obstructiveHCM and resting gradient of 150 mm Hg. The 2D andcolor flow images are acquired simultaneously, on theleft and right, respectively, showing the relation of theflow field and the mitral leaflets. Timing of frames isshown on the ECG below each frame. Top: Latediastole—the widely open mitral leaflets are shown(white arrows) with transmitral diastolic flow appear-ing as red between the leaflets. Second: Diastolic flowis ending. Third: The first frame that showed thebeginning of SAM. The mitral valve is closed (whitearrow). Low-velocity, dark blue flow is seen behindthe mitral valve pushing it into the septum (yellowarrow). Note the absence of high-velocity flow in theoutflow tract. Bottom: Mitral-septal contact hasoccurred and now high-velocity flow due to thegradient appearing in the LV outflow tract. Systolicanterior motion is initiated by the dark blue, low-velocity flow in the third panel that pushes the mitralvalve into the septum, before high-velocity flow hasoccurred in the outflow tract.
ARTICLE IN PRESS
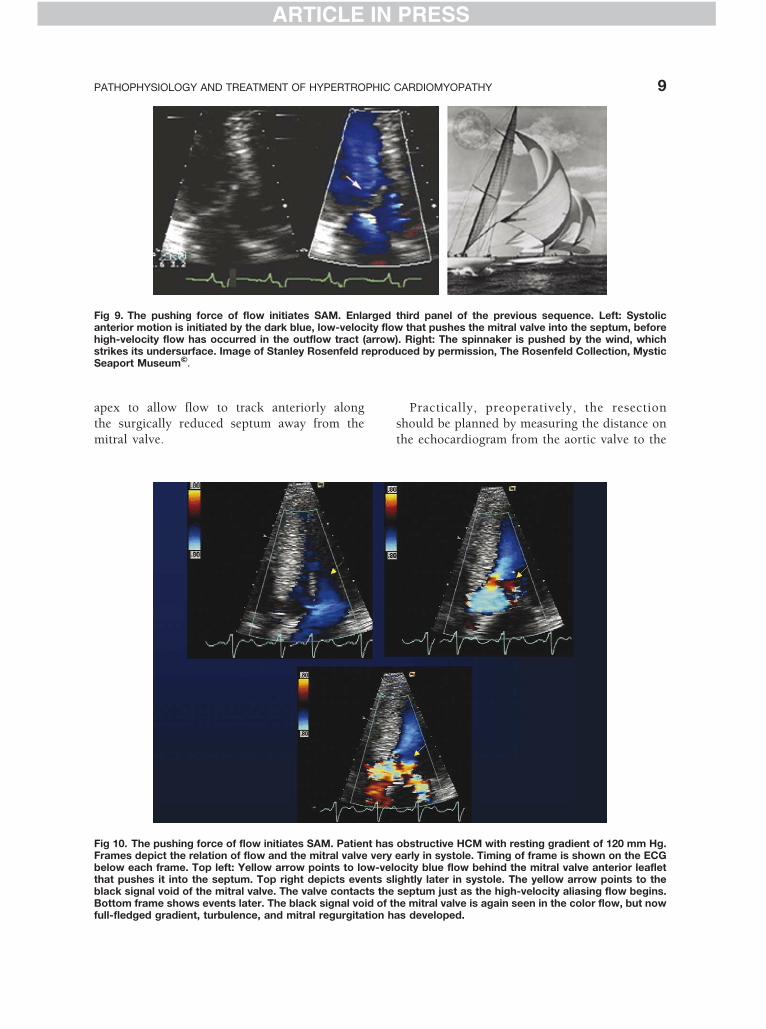
Fig 9. The pushing force of flow initiates SAM. Enlarged third panel of the previous sequence. Left: Systolicanterior motion is initiated by the dark blue, low-velocity flow that pushes the mitral valve into the septum, beforehigh-velocity flow has occurred in the outflow tract (arrow). Right: The spinnaker is pushed by the wind, whichstrikes its undersurface. Image of Stanley Rosenfeld reproduced by permission, The Rosenfeld Collection, MysticSeaport Museum'.
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 9
ARTICLE IN PRESS
apex to allow flow to track anteriorly along
the surgically reduced septum away from the
mitral valve.
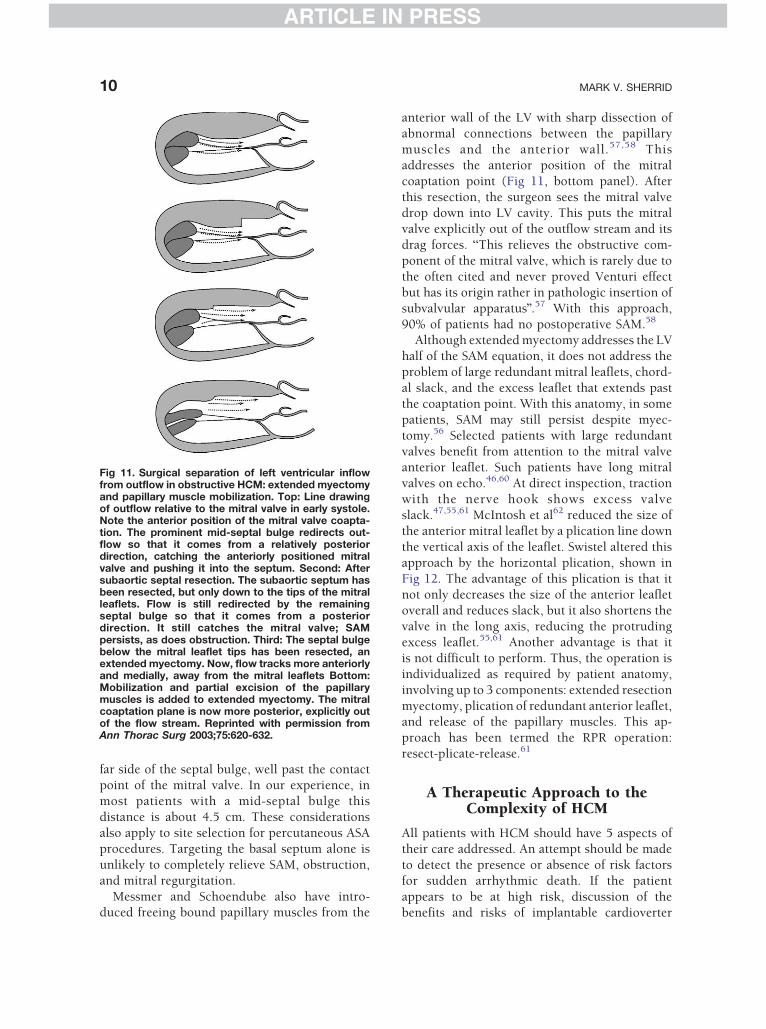
Fig 10. The pushing force of flow initiates SAM. Patient hasFrames depict the relation of flow and the mitral valve verybelow each frame. Top left: Yellow arrow points to low-velthat pushes it into the septum. Top right depicts events slblack signal void of the mitral valve. The valve contacts theBottom frame shows events later. The black signal void of tfull-fledged gradient, turbulence, and mitral regurgitation h
Practically, preoperatively, the resection
should be planned by measuring the distance on
the echocardiogram from the aortic valve to the
obstructive HCM with resting gradient of 120 mm Hg.early in systole. Timing of frame is shown on the ECGocity blue flow behind the mitral valve anterior leafletightly later in systole. The yellow arrow points to theseptum just as the high-velocity aliasing flow begins.
he mitral valve is again seen in the color flow, but nowas developed.
ARTICLE IN PRESS
Fig 11. Surgical separation of left ventricular inflowfrom outflow in obstructive HCM: extended myectomyand papillary muscle mobilization. Top: Line drawingof outflow relative to the mitral valve in early systole.Note the anterior position of the mitral valve coapta-tion. The prominent mid-septal bulge redirects out-flow so that it comes from a relatively posteriordirection, catching the anteriorly positioned mitralvalve and pushing it into the septum. Second: Aftersubaortic septal resection. The subaortic septum hasbeen resected, but only down to the tips of the mitralleaflets. Flow is still redirected by the remainingseptal bulge so that it comes from a posteriordirection. It still catches the mitral valve; SAMpersists, as does obstruction. Third: The septal bulgebelow the mitral leaflet tips has been resected, anextended myectomy. Now, flow tracks more anteriorlyand medially, away from the mitral leaflets Bottom:Mobilization and partial excision of the papillarymuscles is added to extended myectomy. The mitralcoaptation plane is now more posterior, explicitly outof the flow stream. Reprinted with permission fromAnn Thorac Surg 2003;75:620-632.
MARK V. SHERRID10
ARTICLE IN PRESS
far side of the septal bulge, well past the contactpoint of the mitral valve. In our experience, in
most patients with a mid-septal bulge this
distance is about 4.5 cm. These considerations
also apply to site selection for percutaneous ASA
procedures. Targeting the basal septum alone is
unlikely to completely relieve SAM, obstruction,
and mitral regurgitation.
Messmer and Schoendube also have intro-duced freeing bound papillary muscles from the
anterior wall of the LV with sharp dissection of
abnormal connections between the papillary
muscles and the anterior wall.57,58 This
addresses the anterior position of the mitral
coaptation point (Fig 11, bottom panel). After
this resection, the surgeon sees the mitral valvedrop down into LV cavity. This puts the mitral
valve explicitly out of the outflow stream and its
drag forces. bThis relieves the obstructive com-
ponent of the mitral valve, which is rarely due to
the often cited and never proved Venturi effect
but has its origin rather in pathologic insertion of
subvalvular apparatusQ.57 With this approach,
90% of patients had no postoperative SAM.58
Although extended myectomy addresses the LV
half of the SAM equation, it does not address the
problem of large redundant mitral leaflets, chord-
al slack, and the excess leaflet that extends past
the coaptation point. With this anatomy, in some
patients, SAM may still persist despite myec-
tomy.56 Selected patients with large redundant
valves benefit from attention to the mitral valveanterior leaflet. Such patients have long mitral
valves on echo.46,60 At direct inspection, traction
with the nerve hook shows excess valve
slack.47,55,61 McIntosh et al62 reduced the size of
the anterior mitral leaflet by a plication line down
the vertical axis of the leaflet. Swistel altered this
approach by the horizontal plication, shown in
Fig 12. The advantage of this plication is that itnot only decreases the size of the anterior leaflet
overall and reduces slack, but it also shortens the
valve in the long axis, reducing the protruding
excess leaflet.55,61 Another advantage is that it
is not difficult to perform. Thus, the operation is
individualized as required by patient anatomy,
involving up to 3 components: extended resection
myectomy, plication of redundant anterior leaflet,and release of the papillary muscles. This ap-
proach has been termed the RPR operation:
resect-plicate-release.61
A Therapeutic Approach to theComplexity of HCM
All patients with HCM should have 5 aspects oftheir care addressed. An attempt should be made
to detect the presence or absence of risk factors
for sudden arrhythmic death. If the patient
appears to be at high risk, discussion of the
benefits and risks of implantable cardioverter

ARTICLE IN PRESS
Fig 12. Horizontal plication of the anterior mitralleaflet to reduce leaflet length and leaflet/chordalslack. Plication is performed by placing three to fourfine mattress sutures of 5-0 polypropylene in ahorizontal rather than longitudinal orientationthrough the fibrotic area of the leaflet. The width ofthe mattress sutures is dictated by the degree ofredundancy of the leaflet and mobility whenassessed by the nerve hook. This modification moredirectly reduces leaflet-chordal slack and excesslength than a suture line in the longitudinal orienta-tion. Reprinted with permission from Ann ThoracSurg 2003;75:620-632.
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 11
ARTICLE IN PRESS
defibrillator (ICD) is indicated, and many such
patients will be implanted. Symptoms of dys-
pnea, angina, syncope, and fatigue are appraised
and treated. Bacterial endocarditis prophylaxis isrecommended.63 Patients are advised to avoid
athletic competition and extremes of physical
exertion. First-degree family members should be
screened with echocardiography and ECG.
As a routine, screening for hyperlipidemia
should be performed and there should be
aggressive treatment for hyperlipidemia as the
combination of HCM and coronary diseasecauses excess mortality above that seen in
HCM alone.64
Risk Factors for Susceptibility toSudden Cardiac Death
Sudden cardiac death (SCD) was a prominent
feature in the modern description of HCM1 and isits most dreaded complication.9,65-72 An initially
reported incidence of up to 4% per year was
overestimated in the early HCM literature because
of referred-patient selection bias. Reports had
come from selected centers where the sickest
patients had been referred.73-75 Community-
based, more recent series have shown a yearly
HCM-related mortality of 1.5% per year.73,76-80
Ability to predict which patients with HCM
will experience sudden death has long been aclinical goal. The need for risk stratification has
become even more focused since the advent of
SCD prevention with the ICD for both primary
and secondary prevention. The benefit of ICD
implantation in high-risk patients is sudden
death prevention with appropriate shock rates
of 4.5% per year for primary prevention and 11%
per year for secondary prevention.67
In patients who have experienced SCD or
sustained ventricular tachycardia, the judgment
to implant an ICD for secondary prevention is
straightforward because of subsequent high an-
nual rates of recurrent malignant arrhythmia.67,81
Because patients with HCM may present at
young age, and since the risk period for sudden
arrhythmic death may be long and cumulative,decision making about primary prevention may
be difficult. For primary prevention, risk factors
that are observed to stratify risk for SCD in
HCM include massive wall thickening (N30
mm),65,66 unexplained syncope,66,71 family his-
tory of SCD in a first-degree family member—
the relative dying at age less than 40 years,82
ventricular tachycardia—3 or more beats on 24-or 48-hour ECG monitoring,83 inadequate
rise—or frank drop—in blood pressure with
exercise in patients younger than 40 years,66,84-
86 and resting obstruction gradient of 30 mm
Hg or more.30 Certain risk factors, that is,
nonsustained ventricular tachycardia, are con-
sidered to have limited weight, when they occur
in isolation.87 Ventricular tachycardia, occur-ring without syncope, in patients older than
30 years is not a risk factor for SCD, whereas
it is a predictor in young patients younger than
30 years.83
The problem with risk stratification is that
each risk factor has relatively low positive
predictive value for SCD.66,88 Absence of any
risk factors offers the patient and clinician somemeasure of assurance that the risk of SCD is
low.66 The presence of 1 risk factor is very
common in HCM, whereas sudden death is
uncommon. Risk factors may coexist in the same
patient, and when they do, individual risk for

ARTICLE IN PRESS
Fig 13. Risk of mortality stratified by increasingnumber of risk factors. For each 5-mm increase inmaximum LV wall thickness, the chance of dyingincreases. But even more striking increases in mor-tality depend on the number of additional risk factorspresent. Reprinted with permission from Curr ProblCardiol 2004;29:239-291, as compiled from the data ofElliott et al.66
MARK V. SHERRID12
ARTICLE IN PRESS
death increases (Fig 13).66 At present, most
clinicians would agree that the presence of 2 risk
factors would be enough to consider implanta-
tion of an ICD and would individually tailor
therapy depending on age and patient circum-
stances. For example, the low incidence of SCD
after myectomy would make the necessity of ICD
implantation debatable for patients undergoingsurgery. Implantation of an ICD in patients with
1 risk factor would depend on physician judg-
ment and patient choice. There should be
discussion with the patient of the benefits and
risks of the ICD, and the pros and cons of
implantation, and the reason for the physician’s
considered recommendation.
To date, genotype analysis has not yet beenfruitful in predicting high risk. Initially, families
with thin myofilament disease, tropinin, and atropomyosin mutations were thought to be at
higher risk. These mutations are uncommon,
occurring in less than 5% of patients. However,
recent data indicate that they have no definite
prognostic characteristics.88-91 Because HCM
may be caused by any of the many mutationson each individual gene, a myriad number of
disease-causing mutations have been discovered.
Thus, there is yet limited prognostic information
collected on individual mutations (although this
work is ongoing, see http://www.cardiogenomics.
org). In addition, there are modifier genes that
may accentuate or attenuate the individual
prognostic effect of particular mutations.92,93
Risks of ICD Implantation andICD Management in HCM
Risks of ICD implantation may be thought of,
and communicated to patients, as 4 bI’s,Q im-plantation risk, infection, inappropriate shock,and never using the device —insurance risk (youbuy the policy but don’t die). Implantation risksinclude perforation of the great vessels, lung, andcardiac chambers, and the infrequent need forsurgery to correct perforation. The risk ofimplantation is not just restricted to the initialprocedure, but extends a trail of risk into thepatient’s future as they require generator replace-ments roughly every 5 to 7 years and may requirelead removal because of fracture or infection.Lead infection is a dreaded complication becauseit requires removal of infected leads that may befixed in situ at either the right ventricle, rightatrium, or the great veins. Removal of leads hasdeveloped into a specialty of its own, because ofthe need for specialized laser technology and hasa greater than 1% potential for major complica-tions, including death.94 Inappropriate shockoccurs frequently in patients with HCM. Youngpatients may overexert, even if this is proscribed,and sinus tachycardia may be the incitingarrhythmia, especially if a dose of b-blocker isforgotten. Atrial fibrillation is a frequent compli-cation of HCM, and this arrhythmia rivals sinustachycardia as a cause for inappropriate shock.Because the trigger for device intervention is raterelated, most young patients receiving an ICDshould be maintained on b-blockade.
Young patients with HCM should understandthat having an ICD is analogous to purchasing aterm insurance policy. Although there is a 4%per year chance of appropriate shock andprevention of cardiac death, it is also possiblethat malignant arrhythmia will never occur intheir particular case. They will never need deviceintervention. This group will grow old andcontract another lethal illness, die from it, andnever need the device, despite decades of itssurveillance. After discussing the risks andbenefits of ICD implantation, most patients willdecide to have implantation done.
Families are encountered who have had mul-
tiple sudden deaths.82 In these families, it would

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 13
ARTICLE IN PRESS
seem prudent to consider ICD implantation for all
first-degree relatives who are diagnosed with
overt HCM. Because of modifier genes and
incomplete penetrance, it is not clear at this time
whether relatives with very mild thickening
(V14 mm), or genotype-positive family memberswho are completely phenotype-negative with no
wall thickening, should be implanted.
Recommendation toAvoid Competition
Athletes who die suddenly on the playing field are
most often found to have structural heart disease.At autopsy, HCM is the most common structural
heart disease found.95 Because of these observa-
tions, it is recommended that patients with HCM
should avoid competition and extremes of exer-
tion. This recommendation should be discussed
with each patient.96 Moreover, explain also that
exercise would not in any case be expected to
improve the patient’s cardiac condition, whichmight be a patient-held misconception. This
recommendation may be barely relevant in
severely symptomatic patients or in the elderly
who limit themselves. However, in the young, or
in asymptomatic middle-aged patients, this rec-
ommendation may have profound effect. Athlet-
ics and sport occupy a central role in many
patients’ lifestyles. In some, athletic prowess andsuccess have intoxicating appeal. In some ath-
letes, the diagnosis of HCM crushes ambition for
fame and fortune.97 The guidelines allow recrea-
tional sports activity and specifically allow exer-
cise to maintain muscular tone. There is inevitable
ambiguity in the intensity of activity allowed. We
try to clarify by pointing out that, in competition,
athletes will often push beyond limiting symp-toms to win and this is to be specifically avoided,
whether in formal competition or in pickup
games. We also recommend avoiding activities
where syncope would have disastrous effect such
as scuba diving or surfing. We recommend that
patients not lift more than 40 lb.
In a family with sudden death, there is the
appropriate concern that individuals who aregenotype positive might be at risk for SCD if
they compete in sports, even if they have no
clinical signs of HCM. There are no data to
guide recommendations here. However, with
current knowledge, such genotype-positive, phe-
notype-negative patients might be guided to
avoid competition.
Symptoms
Symptoms of dyspnea and exercise intoleranceare related to LV diastolic dysfunction and also to
LV outflow tract obstruction when it is present.
Reduced exercise tolerance correlates with an
inability to increase stroke volume as assessed by
cardiopulmonary stress testing.98 In nonob-
structed patients, inability to increase stroke
volume is due to decreased chamber compliance.
When outflow gradients exceed 60 mm Hg, amid-systolic drop in LV ejection velocities and
volumetric flow has been shown, which may
contribute to inability to increase stroke vol-
ume.38,39 Moreover, further decrement in flow
occurs after pharmacologic increase in gradient
with dobutamine.39 In addition, dynamic ob-
struction is almost invariably associated with
mitral regurgitation, a byproduct of SAM.11,99
Grade of mitral regurgitation correlates with
posterior leaflet length; it is particularly severe
when the posterior leaflet is not long enough to
cover the extent of displacement of the anterior
leaflet, as it is pushed into the septum.100
Chest discomfort of an anginal nature occurs
frequently in patients with HCM with and
without obstruction. There is ample evidence ofischemia: pacing-induced myocardial lactate pro-
duction and reversible stress-induced scinti-
graphic perfusion defects are the most widely
studied manifestations.101 In addition, there is
evidence from multiple sources of inadequate
vasodilator reserve.102,103 This has pointed to
arteriolar narrowing and microvascular dysfunc-
tion as the most likely cause for ischemia. Theepicardial coronary arteries are dilated in HCM
and overall coronary flow is increased, to provide
the hypertrophied myocardium. In contrast, the
arterioles show intimal and medial hyperplasia,
resulting in narrowing of these vessels.27 Because
of dilatation of epicardial vessels, coronary flow
velocity is normal, whereas velocities in the
arterioles are double that of the epicardium andalso twice that found in normals or hyper-
tensives.28 These data lend credence to impor-
tance of arteriolar narrowings as a physiologically
significant cause of ischemia. Ischemia may
predict adverse outcome.101 Left ventricular

ARTICLE IN PRESS
MARK V. SHERRID14
ARTICLE IN PRESS
outflow tract obstruction exacerbates ischemia,
by increasing LV work, and simultaneously
decreasing aortic diastolic, and thus LV perfusion
pressure.104 Surgical relief of obstruction
decreases pacing-induced lactate production.
Syncope is the most multifactorial of HCMsymptoms.105 In any given patient, the clinical
circumstances of syncope must be considered,
although in many cases ambiguity about etiology
prevails. Syncope that occurs without any
circumstantial cause must be considered due to
ventricular arrhythmia until proven otherwise.
Sudden inappropriate vasodilatation due to
autonomic dysfunction in the absence of ar-rhythmia also occurs.106 Postexercise syncope
may be due to arrhythmia, obstruction, or a
paradoxical fall in blood pressure.107 Inappro-
priate vasodilatation after exercise occurs in 25%
of patients.84,107 Typical neurally mediated
syncope occurs in HCM. Circumstances that
may suggest this etiology are associated gastro-
intestinal symptoms.General fatigue is a common nonspecific
complaint. When fatigue occurs, it is often
difficult to distinguish between HCM-related
fatigue and that induced by b-blockade.
Fig 14. A schematic summary of the pharmacologic therapFutura.111
Decrease in dose, or elimination of b-blockade,
may allow differentiation.
Watchful Waiting in Asymptomaticand Mildly Symptomatic HCM
The prognosis in large community-based pop-
ulations of patients with HCM is generally
good.73,77,78 Indeed, survival to old age is
common with diagnosis of HCM.108 These
observations must be considered in the approach
to the patient with no or only mild symptoms,
New York Heart Association (NYHA) class I or
II, who are not deemed to be at high risk forsudden death. In such patients, as no medical,
surgical, or interventional therapy has been
shown in randomized trials to improve mortality
or prevent disease progression (such trials have
not been done in HCM), the approach of
watchful waiting is often appropriate. There is
no urgency to begin pharmacologic therapy in
asymptomatic patients. In mildly symptomaticobstructed patients, after pharmacologic therapy
is begun, there is no urgency to progress rapidly
to myectomy or alcohol ablation. Such patients
may be treated expectantly, moving deliberately
y of HCM. Reprinted with permission from Blackwell-
ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 15
ARTICLE IN PRESS
to more aggressive therapies only when symp-
toms progress.
Pharmacologic Treatment ofSymptoms
Nonobstructive HCM
In symptomatic nonobstructive HCM (Fig 14),
symptoms are due to diastolic dysfunction,
impaired relaxation early in diastole, and de-
creased chamber compliance in late diastole. The
pathology is small LV volumes, hypertrophy,
fiber disarray, and fibrosis. There are few agents
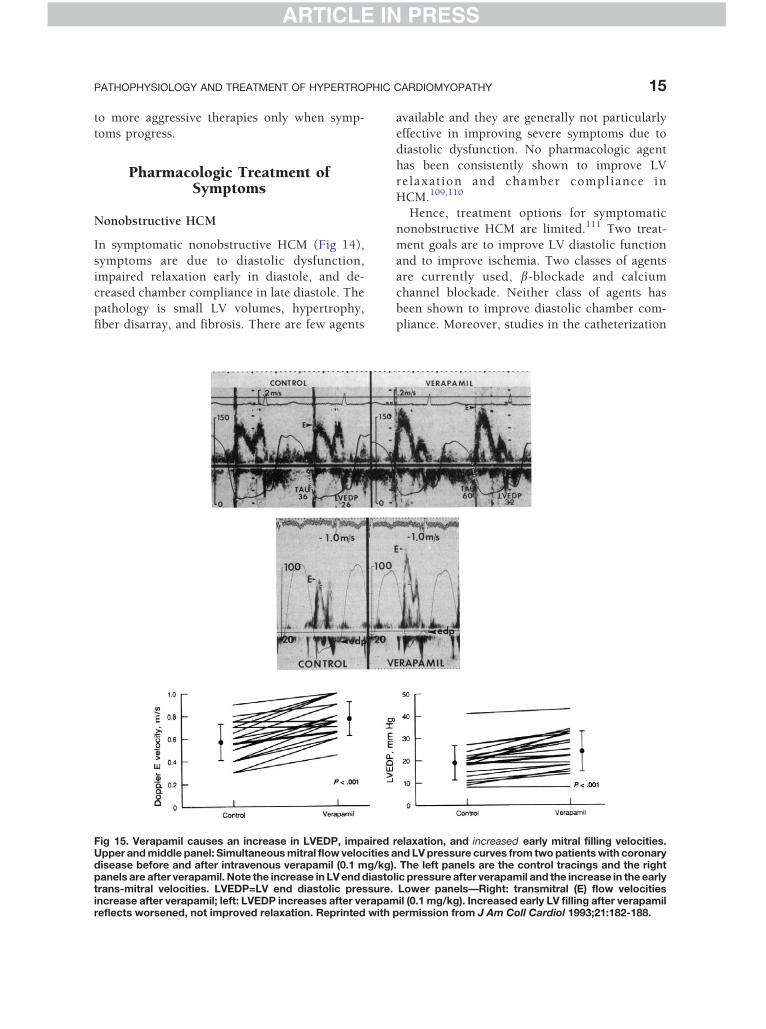
Fig 15. Verapamil causes an increase in LVEDP, impairedUpper and middle panel: Simultaneous mitral flow velocities adisease before and after intravenous verapamil (0.1 mg/kg)panels are after verapamil. Note the increase in LV end diastotrans-mitral velocities. LVEDP=LV end diastolic pressure.increase after verapamil; left: LVEDP increases after verapamreflects worsened, not improved relaxation. Reprinted with
available and they are generally not particularly
effective in improving severe symptoms due to
diastolic dysfunction. No pharmacologic agent
has been consistently shown to improve LV
relaxation and chamber compliance in
HCM.109,110
Hence, treatment options for symptomatic
nonobstructive HCM are limited.111 Two treat-
ment goals are to improve LV diastolic function
and to improve ischemia. Two classes of agents
are currently used, b-blockade and calcium
channel blockade. Neither class of agents has
been shown to improve diastolic chamber com-
pliance. Moreover, studies in the catheterization
relaxation, and increased early mitral filling velocities.nd LV pressure curves from two patients with coronary
. The left panels are the control tracings and the rightlic pressure after verapamil and the increase in the earlyLower panels—Right: transmitral (E) flow velocitiesil (0.1 mg/kg). Increased early LV filling after verapamil
permission from J Am Coll Cardiol 1993;21:182-188.

ARTICLE IN PRESS
MARK V. SHERRID16
ARTICLE IN PRESS
laboratory have shown that neither intravenous
b-blockade nor verapamil improved early dia-
stolic relaxation in the hypertrophic left ventri-
cle.109,110 The data about the effect of verapamil
on early diastolic relaxation are controversial.
One source of confusion concerns data indicatingan increase in early diastolic peak filling rate as
assessed on serial radionuclide ventriculogra-
phy.112-114 This had initially been interpreted as
an improvement in diastolic function (ie, fast
filling is better) until the work of Nishimura
et al.115 They simultaneously measured LV filling
with high-fidelity catheters and Doppler echocar-
diography, before and after verapamil IV, inpatients with coronary disease115 (Fig 15). In this
revealing study, LV diastolic pressures rose after
verapamil, s increased, indicating impaired re-
laxation, but early transmitral echo Doppler dia-
stolic velocities increased. With current knowledge
of diastology, it is now understood that verapamil
actually caused worsening, restrictive LV diastolic
dysfunction, increasing early velocities because ofincreased left atrial pressure. This paper showed
that in a coronary population that verapamil was
not lusitropic, and that the faster early filling
velocities reported in nuclear studies, may actu-
ally be detecting worsened diastolic function.
Verapamil’s positive contribution in the patho-
physiology of nonobstructive HCM appears to be
relief of ischemia. Verapamil improves myocardialperfusion as assessed by stress radionuclide perfu-
sion imaging116 and may thus improve symptoms.
b-Blockade, and, to a lesser degree, verapamil,
may cause chronotropic incompetence in
HCM.117 As diastolic dysfunction may limit the
exercise-induced increase in stroke volume,
patients with HCM often rely on increased heart
rate to increase cardiac output. In such patients,pharmacologic limitation of heart rate rise may
impair exercise capacity.
Whereas disopyramide has been shown to
improve diastolic function in obstructed
patients, by decreasing gradient and systolic
load,118,119 it has not been shown to improve
diastolic function in nonobstructed patients and
should be avoided in this group, pendingfurther investigation.
For the unusual patient with fluid retention,
diuretics may be helpful by relieving dyspnea and
uncommon edema. Overdiuresis should be
avoided as patients with HCM are often preload
dependent for adequate cardiac output. If patients
initially present with edema, another diagnosis
should be sought as this is very unusual. Amyloid
may be suspected in this clinical situation,
especially if the ECG QRS voltage is low.
In animal models of HCM, aldosterone antag-onism has been shown to improve or prevent
fibrosis and hypertrophy.120 Similarly, statin
therapy has been shown to prevent phenotype
in genotype-positive animals.121 As a new desig-
nation, these pharmacologic agents may be
termed fibrotardive. Clinical trials would seem
appropriate for these new approaches as there is
currently no good pharmacologic treatment foradvanced symptoms in nonobstructive HCM.
Obstructive HCM
Pharmacologic therapy of symptoms in obstruc-
tive HCM is succesful in two-thirds of patients
(Fig 14). Negatively inotropic drugs improve
dynamic LV outflow obstruction by decreasingejection acceleration122 (see Figs 16 and 17).
Decreasing ejection acceleration decreases flow
velocities early in systole, decreasing early drag
forces on the mitral valve, delaying mitral-septal
contact, and reducing gradient. Delaying the
early trigger of SAM may allow reassertion of
chordal tension by papillary muscle shortening
to provide countertraction to prevent SAM, evencompletely.
b-Blockade is the initial treatment for symp-
tomatic obstructed patients.111,123 b-Blockers
decrease the sympathetic-mediated rise in gradi-
ent with exercise and improve symptoms. How-
ever, b-blockade is not expected to reduce
resting gradient124 and less than half of patients
have sustained improvement in symptoms.For patients with refractory symptoms and
gradients after b-blockade, there is regional
variation in the choice of the next drug trial. In
many centers, the next trial selected is substitu-
tion of verapamil for b-blocker.111,125,126
In other centers, disopyramide is added to
b-blockade.12,111,119,127-130 Verapamil, a potent
calcium channel blocker, has both negativeinotropic properties but also is a vasodilator. It
has been shown to decrease gradient and
improve symptoms.131 In a limited number of
patients, exercise tolerance has been shown to
increase as well.125,126,132
ARTICLE IN PRESS
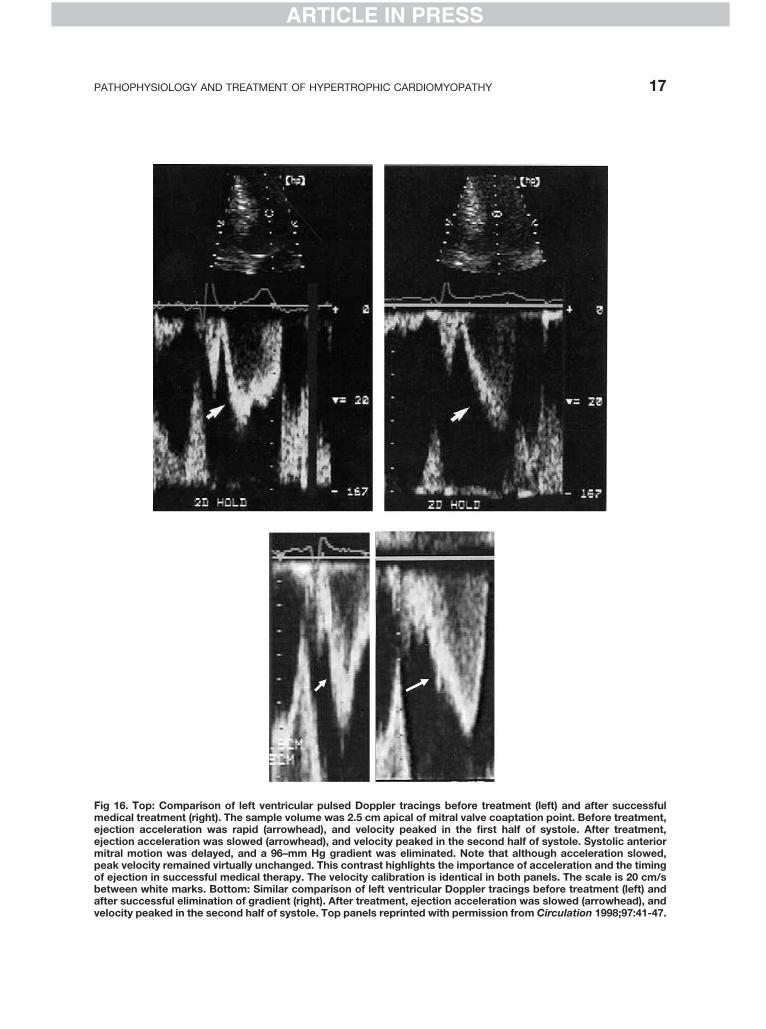
Fig 16. Top: Comparison of left ventricular pulsed Doppler tracings before treatment (left) and after successfulmedical treatment (right). The sample volume was 2.5 cm apical of mitral valve coaptation point. Before treatment,ejection acceleration was rapid (arrowhead), and velocity peaked in the first half of systole. After treatment,ejection acceleration was slowed (arrowhead), and velocity peaked in the second half of systole. Systolic anteriormitral motion was delayed, and a 96–mm Hg gradient was eliminated. Note that although acceleration slowed,peak velocity remained virtually unchanged. This contrast highlights the importance of acceleration and the timingof ejection in successful medical therapy. The velocity calibration is identical in both panels. The scale is 20 cm/sbetween white marks. Bottom: Similar comparison of left ventricular Doppler tracings before treatment (left) andafter successful elimination of gradient (right). After treatment, ejection acceleration was slowed (arrowhead), andvelocity peaked in the second half of systole. Top panels reprinted with permission from Circulation 1998;97:41-47.
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 17
ARTICLE IN PRESS
ARTICLE IN PRESS
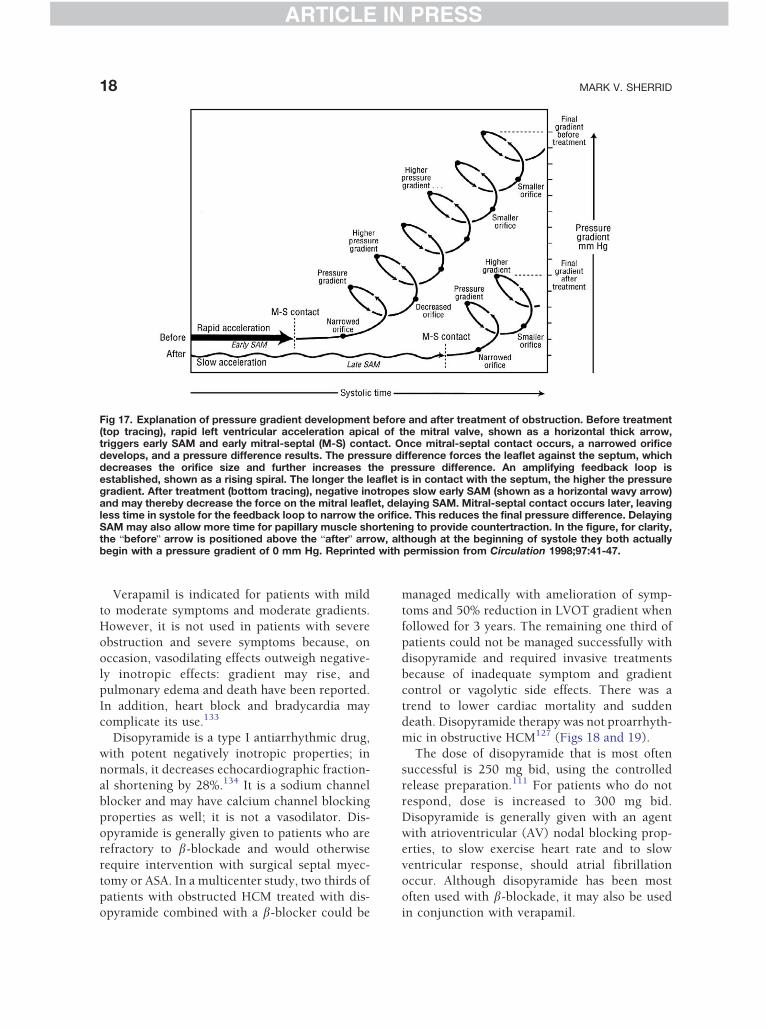
Fig 17. Explanation of pressure gradient development before and after treatment of obstruction. Before treatment(top tracing), rapid left ventricular acceleration apical of the mitral valve, shown as a horizontal thick arrow,triggers early SAM and early mitral-septal (M-S) contact. Once mitral-septal contact occurs, a narrowed orificedevelops, and a pressure difference results. The pressure difference forces the leaflet against the septum, whichdecreases the orifice size and further increases the pressure difference. An amplifying feedback loop isestablished, shown as a rising spiral. The longer the leaflet is in contact with the septum, the higher the pressuregradient. After treatment (bottom tracing), negative inotropes slow early SAM (shown as a horizontal wavy arrow)and may thereby decrease the force on the mitral leaflet, delaying SAM. Mitral-septal contact occurs later, leavingless time in systole for the feedback loop to narrow the orifice. This reduces the final pressure difference. DelayingSAM may also allow more time for papillary muscle shortening to provide countertraction. In the figure, for clarity,the bbeforeQ arrow is positioned above the bafterQ arrow, although at the beginning of systole they both actuallybegin with a pressure gradient of 0 mm Hg. Reprinted with permission from Circulation 1998;97:41-47.
MARK V. SHERRID18
ARTICLE IN PRESS
Verapamil is indicated for patients with mild
to moderate symptoms and moderate gradients.
However, it is not used in patients with severe
obstruction and severe symptoms because, on
occasion, vasodilating effects outweigh negative-
ly inotropic effects: gradient may rise, and
pulmonary edema and death have been reported.
In addition, heart block and bradycardia maycomplicate its use.133
Disopyramide is a type I antiarrhythmic drug,
with potent negatively inotropic properties; in
normals, it decreases echocardiographic fraction-
al shortening by 28%.134 It is a sodium channel
blocker and may have calcium channel blocking
properties as well; it is not a vasodilator. Dis-
opyramide is generally given to patients who arerefractory to b-blockade and would otherwise
require intervention with surgical septal myec-
tomy or ASA. In a multicenter study, two thirds of
patients with obstructed HCM treated with dis-
opyramide combined with a b-blocker could be
managed medically with amelioration of symp-
toms and 50% reduction in LVOT gradient when
followed for 3 years. The remaining one third of
patients could not be managed successfully with
disopyramide and required invasive treatments
because of inadequate symptom and gradient
control or vagolytic side effects. There was a
trend to lower cardiac mortality and suddendeath. Disopyramide therapy was not proarrhyth-
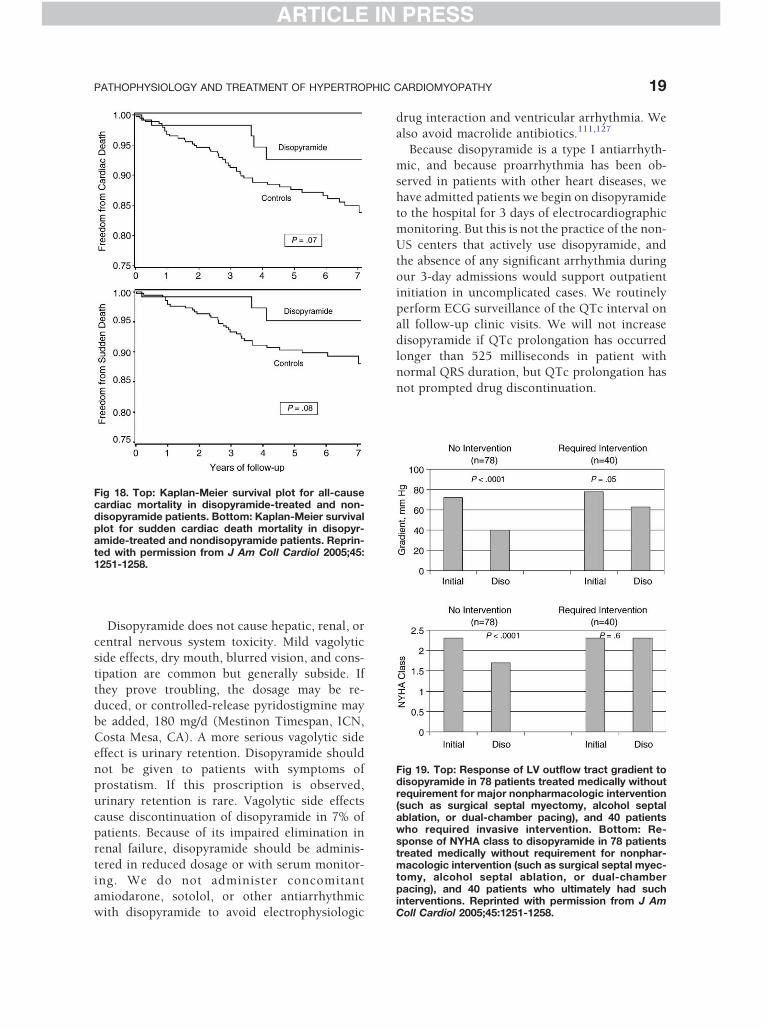
mic in obstructive HCM127 (Figs 18 and 19).
The dose of disopyramide that is most often
successful is 250 mg bid, using the controlled
release preparation.111 For patients who do not
respond, dose is increased to 300 mg bid.
Disopyramide is generally given with an agent
with atrioventricular (AV) nodal blocking prop-erties, to slow exercise heart rate and to slow
ventricular response, should atrial fibrillation
occur. Although disopyramide has been most
often used with b-blockade, it may also be used
in conjunction with verapamil.
ARTICLE IN PRESS
Fig 18. Top: Kaplan-Meier survival plot for all-causecardiac mortality in disopyramide-treated and non-disopyramide patients. Bottom: Kaplan-Meier survivalplot for sudden cardiac death mortality in disopyr-amide-treated and nondisopyramide patients. Reprin-ted with permission from J Am Coll Cardiol 2005;45:1251-1258.
Fig 19. Top: Response of LV outflow tract gradient todisopyramide in 78 patients treated medically withoutrequirement for major nonpharmacologic intervention(such as surgical septal myectomy, alcohol septalablation, or dual-chamber pacing), and 40 patientswho required invasive intervention. Bottom: Re-sponse of NYHA class to disopyramide in 78 patientstreated medically without requirement for nonphar-macologic intervention (such as surgical septal myec-tomy, alcohol septal ablation, or dual-chamberpacing), and 40 patients who ultimately had suchinterventions. Reprinted with permission from J AmColl Cardiol 2005;45:1251-1258.
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 19
ARTICLE IN PRESS
Disopyramide does not cause hepatic, renal, or
central nervous system toxicity. Mild vagolytic
side effects, dry mouth, blurred vision, and cons-
tipation are common but generally subside. If
they prove troubling, the dosage may be re-duced, or controlled-release pyridostigmine may
be added, 180 mg/d (Mestinon Timespan, ICN,
Costa Mesa, CA). A more serious vagolytic side
effect is urinary retention. Disopyramide should
not be given to patients with symptoms of
prostatism. If this proscription is observed,
urinary retention is rare. Vagolytic side effects
cause discontinuation of disopyramide in 7% ofpatients. Because of its impaired elimination in
renal failure, disopyramide should be adminis-
tered in reduced dosage or with serum monitor-
ing. We do not administer concomitant
amiodarone, sotolol, or other antiarrhythmic
with disopyramide to avoid electrophysiologic
drug interaction and ventricular arrhythmia. We
also avoid macrolide antibiotics.111,127
Because disopyramide is a type I antiarrhyth-
mic, and because proarrhythmia has been ob-
served in patients with other heart diseases, we
have admitted patients we begin on disopyramideto the hospital for 3 days of electrocardiographic
monitoring. But this is not the practice of the non-
US centers that actively use disopyramide, and
the absence of any significant arrhythmia during
our 3-day admissions would support outpatient
initiation in uncomplicated cases. We routinely
perform ECG surveillance of the QTc interval on
all follow-up clinic visits. We will not increasedisopyramide if QTc prolongation has occurred
longer than 525 milliseconds in patient with
normal QRS duration, but QTc prolongation has
not prompted drug discontinuation.

ARTICLE IN PRESS
MARK V. SHERRID20
ARTICLE IN PRESS
Atrial fibrillation occurs frequently in patients
with HCM in 25% to 30% of older patients, both
with and without obstruction.135-138 Left atrial
dilatation is the most frequent substrate. The
advent of atrial fibrillation is often marked by a
deterioration of symptoms and is a frequent causeof hospitalization. There is a dramatic increase in
embolic potential and stroke after the develop-
ment of atrial fibrillation.136 This pertains both to
patients with paroxysmal atrial fibrillation and
also to the young. Such patients also experience
higher mortality. Patients with atrial fibrillation
should be anticoagulated with warfarin.136 Amio-
darone is an effective antiarrhythmic for preven-tion of recurrence, but because of its long-term
toxicity it is not a good solution for younger
patients. Such patients may be controlled with
sotolol or dofetilide. In selected patients, radio-
frequency ablation of the orifices of the pulmo-
nary veins prevents recurrence.139
Surgical Septal Myectomy
Myectomy is the treatment of choice for patients
who fail medical therapy.55,61,140-148 Candidates
for myectomy have persistent disabling symp-
toms and gradients of greater than 50 mm Hg at
rest or after physiologic provocation. Myectomy
has been successfully performed for 30 years,
and in experienced centers it can be performedwith low surgical mortality, 1%, and is uniformly
successful in reducing both gradient and symp-
toms. Postoperative survival has been excellent
with series reporting annual cardiac mortality of
1% per year.9,13,141,142,145,146 Medications for
obstruction may be reduced or stopped postop-
eratively. Gradient reduction after surgery is
greater than that observed after ASA.149 Inpatients with paroxysmal atrial fibrillation, a
modified intraoperative maze procedure may be
done concomitantly with myectomy in an at-
tempt to prevent postoperative fibrillation. The
opportunity to address atrial fibrillation directly
(and the mitral valve) is a benefit of surgery over
alcohol ablation. Intraoperative transesophageal
echocardiography both before the resection andafter rewarming and weaning from bypass
is essential.60,150,151 Imaging is done before
removing the canulas. If resting or provocable
obstruction persists, or if there is more than mild
mitral regurgitation, the patient must be placed
back on bypass and further resection/repair must
be attempted; if this is not possible, then mitral
valve replacement is rarely required.
Krajcer et al152 introduced the idea that as
obstruction is most often caused by SAM of the
mitral valve, routine mitral valve replacementmight be a logical and successful way to relieve
obstruction. However, this approach has the
main disadvantage that the patient now has the
burden of a life-long prosthetic valve. For the
young who receive a mechanical prosthesis, this
requires anticoagulation with warfarin with its
1% to 2% per year risk of bleeding. All such
patients are subject to the risk of prosthetic valvefailure and endocarditis. Because of these dis-
advantages, myectomy–mitral-valve sparing
operations are always preferred.153
The exceptions are for patients with mitral
regurgitation due to structural mitral disease
above and beyond that caused by SAM: mitral
valvular or annular calcification with restricted
motion, severe unrepairable mitral prolapse, ordamage from endocarditis. Besides calcification,
a clue to the presence of structural mitral
regurgitation is a central or anteriorly directed
jet. Mitral regurgitation solely from SAM is
invariably posteriorly directed and always
improves after myectomy.
The most common serious complication of
myectomy is complete heart block that occurs in0% to 10% of patients.14,55,148 Ventricular septal
defect has been reported in 0% to 2%. Transient
heart block may disappear after the operative
day. As surgical patients invariably are given a
left bundle branch block, patients who have right
bundle branch block preoperatively are at higher
risk for heart block and a pacemaker. Resection
too close to the aortic valve may result inventricular septal defect. As this area plays no
role in the etiology of SAM, resection here
should be avoided, in preference for resection
lower down in the mid-septum. Perhaps the
largest morbidity of myectomy stems from
cardiopulmonary bypass and thoracotomy and
its associated risk of infection and stroke,
especially in the elderly.
Alcohol Septal Ablation
Percutaneous ASA offers the attractive promise
of septal reduction without cardiopulmonary

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 21
ARTICLE IN PRESS
bypass and its complications.154-160 After place-
ment of a temporary right ventricular pacing
lead, a small diameter balloon catheter is placed
in a selected left anterior descending (LAD)
septal branch, and after inflating the balloon,
angiographic contrast is injected to assure thatcontrast does not reflux back into the LAD.154
Diluted echo contrast is then injected during
transthoracic or transesophageal echocardio-
graphic imaging. In 8% of such injections,
contrast is seen to flow to structures where
alcohol injection would be disastrous: posterior
LV wall, RV free wall, mitral papillary muscles, or
the entire septum. With this information, theoperator searches for a septal branch that can be
demonstrated to just supply the upper septum,
preferably extending past the point of mitral
septal contact.161 One to 3 mL of absolute alcohol
is instilled into the septal branch. Optimally, a
controlled myocardial infarction occurs. This is
accompanied by typical chest pain, enzyme
elevation, and risk for potential lethal ventriculararrhythmia. The acute gradient reduction of ASA
is caused by reduced ejection acceleration from
the infarct and decreased hemodynamic force on
the mitral valve decreasing SAM in exactly the
same way as negatively inotropic medications.162
After recovery from the infarction, progressive
thinning of the septum occurs and flow is
directed away from the mitral valve, actingsynergistically with the persistent infarct-related
reduction in ejection acceleration.
Temporary right ventricular pacing is fre-
quently necessary because of heart block which
proves to be permanent in 7%.161 Almost all
patients develop right bundle branch, so com-
plete heart block is more frequent in those with
preprocedure left bundle branch block. Manip-ulation of the LAD is not without risk. Proce-
dure-related LAD dissection has been reported.
Reflux of alcohol back into the main LAD may
result in massive anterior infarction. Acute and
late progressive mitral regurgitation requiring
mitral valve replacement has been reported.163
Of concern is that an anterior infarction and
scar are produced by ASA and indeed are itsexplicit goal. There is concern expressed in the
literature that large scar may subject patients to
increased life-long risk of potentially lethal
ventricular arrhythmia.141 To date, there are
no long-term longitudinal studies of consecu-
tively operated patients available to address this
concern.
Alcohol septal ablation is not as effective as
surgical myectomy in reducing gradient and
alleviating symptoms.149,164 Another concern is
the perception that because ASA is a relativelyeasy procedure that it might be performed on
patients with mild symptoms, without adequate
medical trial, and by any interventional cardiol-
ogist experienced with angioplasty. But obs-
tructive HCM is a heterogeneous complex mul-
tifaceted disease; among HCM experts, there is
the universal opinion that ASA should only be
performed in centers committed and familiarwith overall HCM care, including myectomy. It
should not be performed without online expert
echocardiographic guidance.
It has been pointed out that the number of
ASA procedures that have been performed in
the 10 years since its introduction far out-
numbers the number of surgical myectomies
reported in the 30 years it has been performed.As ASA and myectomy have the same indica-
tions, there is the perception that ASA is being
applied to patients who are less symptomatic
than those who previously have been sent for
surgery.141
As with surgery, ASA should be reserved for
patients who are NYHA class III, who have had
no relief of their symptoms with maximalpharmacologic therapy, and who have persis-
tently high gradients (z50 mm Hg) at rest or
after physiologic provocation. Maximal phar-
macologic therapy should include a trial of
b-blockade combined with disopyramide. It
should not be used in NYHA class II patients.
We reserve it for patients who have comorbid
conditions that preclude surgery or for thosewho refuse thoracotomy. In our experience,
these circumstances are uncommon (b1 in
50 patients with obstruction).
Dual-Chamber Pacing
Dual-chamber pacing with complete ventricular
preexcitation through a short atrioventriculardelay significantly reduces outflow tract gra-
dients.165 However, therapeutic effect is often
incomplete; SAM persists with mean gradients of
30 to 55 mm Hg after 3 months of pacing.166,167
The mechanism by which pacing benefits SAM is

ARTICLE IN PRESS
MARK V. SHERRID22
ARTICLE IN PRESS
unclear at this time. It must be due to the
dyssynchrony caused by the right ventricular
pacing, or due to the short AV delay. One
hypothesis is that pacing might cause asynchro-
nous or paradoxical septal motion, widening the
outflow tract and decreasing Venturi forces.However, against this notion is that septal
paradox is only rarely seen. Jeanrenaud and
Kappenberger168 did find a modest decrease in
regional septal wall motion, but there was no
uniform correlation between the magnitude of
decreased septal motion and percent gradient
reduction. Also, as discussed above, a decrease in
Venturi forces can only play a minor role in SAMimprovement.32 Therefore, both direct observa-
tions and pathophysiology indicate that the
mechanism by which DDD pacing reduces SAM
is more complex than just a widening of the
outflow tract. An alternate hypothesis is the
negative inotropic pathway: LV dyssynchony
may cause decreased LV ejection acceleration
and decreases early forces on the mitral valve. Afinal hypothesis is that apical pacing may tense
the mitral subvalvular apparatus sooner than the
rest of the LV, so that when ejection drag forces
are applied to the mitral valve, excess slack has
already been mitigated. In all patients, truncation
of active transmitral filling (the Doppler A wave)
should be avoided by excessive shortening of the
AV delay.169 In our experience, AV delays of60 milliseconds or less are almost invariably
associated with such shortening. Atrioventricular
optimization with echocardiography is often
performed with the goal of gradient reduction
by complete electrocardiographic ventricular
capture, without A-wave truncation. Late gradi-
ent reduction is often higher than that observed
at acute testing.The benefit of pacing for symptom relief and
gradient reduction is less than that observed after
surgical septal myectomy.170 Initial enthusiasm
for symptom relief by DDD pacing with short AV
delay has been tempered by randomized clinical
trials.166,167,171-173 Although gradients are re-
lieved, overall exercise capacity and symptom
relief vary from patient to patient and benefit isunpredictable. In the M-Pathy randomized trial,
a consistent significant treatment effect could be
identified only in elderly patients older than
65 years. A significant placebo effect of pacing
has been found.166,172 But Gadler et al174 have
shown that when pacing is withdrawn by blinded
institution of AAI pacing, there is a dramatic and
prompt recrudescence of symptoms, only re-
lieved by reinstitution of DDD pacing. Another
recent series of pacing for obstruction in patients
older than 50 years has shown long-termbenefit.175 Despite the observation that some
patients derive benefit, DDD pacing cannot be
regarded as a primary treatment modality for
gradient reduction in younger and middle aged
patients with HCM because of its unpredictabil-
ity and because myectomy is more effective. As
device therapy is now common for SCD preven-
tion, many obstructed patients may receive aDDD pacemaker as a potentially useful adjunct
to their ICD implantation procedure.
Coexisting Hypertension orHyperlipidemia
The prevalence of systemic hypertension in our
HCM clinic is 30%, which is comparable to itsprevalence in the middle-aged population. This
poses diagnostic and therapeutic problems. It is
sometimes difficult to assess whether hypertro-
phy is primary or due to the hypertension. The
presence of SAM with obstruction, family mem-
bers with HCM, or extreme hypertrophy out of
proportion to mild hypertension suggests that
the hypertrophy is due to primary HCM.Hypertensive HCM is characterized by marked
hypertrophy accompanied by LV end diastolic
diameter of less than 42 mm and LV fractional
shortening greater than 45%.176
Vasodilators (angiotensin-converting enzymes,
angiotensin receptor blockers, dihydropyridine
calcium antagonists) for hypertension are con-
traindicated in obstructive HCM because theyworsen obstruction and symptoms and may
precipitate syncope. Often, when patients pres-
ent for evaluation at HCM centers, their symp-
toms are found to date from the week
vasodilators were started. Symptoms respond
almost immediately to stopping vasodilators.
Hypertension in obstructed patients is best
treated by increasing doses of b-blockers orverapamil and sparing use of thiazide diuretics.
Clonidine may be used, often given at night to
avoid daytime sedation. Rarely, intervention for
gradient may be necessary to both control
outflow gradient and allow vasodilator therapy

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 23
ARTICLE IN PRESS
of severe hypertension. In patients without a
provocable gradient or after successful surgical
septal myectomy, angiotensin inhibition or
blockade can be safely used.
Concomitant coronary artery disease increases
mortality in HCM and may be overlooked whenconservatively managing HCM.64 There should
be a low threshold for coronary angiography in
patients with angina. Hyperlipidemia should be
treated aggressively. There is no contraindication
to the use of nicotine patches or bupropion for
smoking cessation in HCM.
Transformation into Hypokinetic LVDysfunction
Less than 5% of patients with HCM progress to
LV systolic dysfunction with low ejection frac-tion. Exercise intolerance worsens in these
patients who may become symptomatic at rest
and often deteriorate rapidly and die from heart
failure.177 When this transformation occurs,
pharmacologic treatment is changed: negative
inotropes are stopped and angiotensin-convert-
ing enzyme inhibitors, digoxin, diuretics, and
b-blockers are begun. Because of a high risk forsudden arrhythmic death, ICD implantation
should be considered in all of these patients.
Biventricular pacing to improve heart-failure
symptoms is now available for patients with
LBBB. Patients with systolic LV dysfunction
should be referred for transplantation evaluation
when symptoms progress to NYHA Class 3-4.
Future Research
There is a need for basic understanding of the
mechanisms that transform a genotype-positive
individual into a patient with HCM. If these wereunderstood, means to prevent or modify patho-
logic hypertrophy might be found even without
correcting the mutation (which would be best).
More medications are needed for this condition
for both palliation of symptoms and preventing
disease progression. In particular, there is no
effective pharmacologic means to prevent fibrosis
and improve chamber compliance. For obstruc-tion, long-term study of the benefits and risks of
alcohol ablation must be compiled and compared
to myectomy. Current paradigms for sudden
death risk stratification lack predictive accuracy.
References
1. Teare D: Asymmetrical hypertrophy of the heart in
young adults. Br Heart J 20:1-18, 1958
2. Seidman JG, Seidman C: The genetic basis for
cardiomyopathy: from mutation identification tomechanistic paradigms. Cell 104:557-567, 2001
3. Mogensen J, Bahl A, McKenna WJ: Hypertrophiccardiomyopathy—the clinical challenge of manag-
ing a hereditary heart condition. Eur Heart J 24:
496-498, 2003
4. Watkins H, Rosenzweig A, Hwang DS, et al: Char-acteristics and prognostic implications of myosin
missense mutations in familial hypertrophic cardio-
myopathy. N Engl J Med 326:1108-1114, 1992
5. Van Driest SL, Ommen SR, Tajik AJ, et al: Yield of
genetic testing in hypertrophic cardiomyopathy.Mayo Clin Proc 80:739-744, 2005
6. Binder J, Ommen SR, Gersh BJ, et al: Echocardi-
ography-guided genetic testing in hypertrophic
cardiomyopathy: septal morphological features pre-dict the presence of myofilament mutations. Mayo
Clin Proc 4:459-467, 2006
7. Lever HM, Karam RF, Currie PJ, et al: Hypertrophic
cardiomyopathy in the elderly. Distinctions fromthe young based on cardiac shape. Circulation 79:
580-589, 19898. Marian AJ: Pathogenesis of diverse clinical and
pathological phenotypes in hypertrophic cardiomy-opathy. Lancet 355:58-60, 2000
9. Maron BJ, McKenna WJ, Danielson GK, et al:
American College of Cardiology/European Society
of Cardiology clinical expert consensus documenton hypertrophic cardiomyopathy. A report of the
American College of Cardiology Foundation Task
Force on Clinical Expert Consensus Documents and
the European Society of Cardiology Committeefor Practice Guidelines. J Am Coll Cardiol 42:
1687-1713, 2003
10. Maron BJ, Epstein SE: Hypertrophic cardiomyopa-thy: a discussion of nomenclature. Am J Cardiol
43:1242-1244, 1979
11. Wigle ED, Sasson Z, Henderson MA, et al: Hyper-
trophic cardiomyopathy. The importance of the siteand the extent of hypertrophy. A review. Prog
Cardiovasc Dis 28:1-83, 1985
12. Wigle ED: Hypertrophic cardiomyopathy: a 1987
viewpoint. Circulation 75:311-322, 198713. Maron BJ: Hypertrophic cardiomyopathy: a system-
atic review. JAMA 287:1308-1320, 2002
14. Ommen SR, Nishimura RA: Hypertrophic cardiomy-opathy. Curr Probl Cardiol 29:239-291, 2004
15. Eriksson MJ, Sonnenberg B, Woo A, et al: Long-term
outcome in patients with apical hypertrophic cardio-myopathy. J Am Coll Cardiol 39:638-645, 2002
16. Rickers C, Wilke NM, Jerosch-Herold M, et al: Utility
of cardiac magnetic resonance imaging in the
diagnosis of hypertrophic cardiomyopathy. Circula-tion 112:855-861, 2005
17. Maron BJ, Wolfson JK, Roberts WC, et al: Relation
between extent of cardiac muscle cell disorganiza-

ARTICLE IN PRESS
MARK V. SHERRID24
ARTICLE IN PRESS
tion and left ventricular wall thickness in hypertrophic
cardiomyopathy. Am J Cardiol 70:785-790, 1992
18. Maron BJ, Anan TJ, Roberts WC: Quantitativeanalysis of the distribution of cardiac muscle cell
disorganization in the left ventricular wall of patients
with hypertrophic cardiomyopathy. Circulation63:882-894, 1981
19. Varnava AM, Elliott PM, Baboonian C, et al: Hyper-
trophic cardiomyopathy: histopathological features
of sudden death in cardiac troponin T disease.Circulation 104:1380-1384, 2001
20. Shirani J, Pick R, Roberts WC, et al: Morphology
and significance of the left ventricular collagen
network in young patients with hypertrophic cardio-myopathy and sudden cardiac death. J Am Coll
Cardiol 35:36-44, 2000
21. St John Sutton MG, Lie JT, Anderson KR, et al:Histopathological specificity of hypertrophic ob-
structive cardiomyopathy. Myocardial fibre disarray
and myocardial fibrosis. Br Heart J 44:433-443, 1980
22. Tanaka M, Fujiwara H, Onodera T, et al: Quantitativeanalysis of myocardial fibrosis in normals, hyper-
tensive hearts, and hypertrophic cardiomyopathy.
Br Heart J 55:575-581, 1986
23. Factor SM, Butany J, Sole MJ, et al: Pathologicfibrosis and matrix connective tissue in the subaortic
myocardium of patients with hypertrophic cardio-
myopathy. J Am Coll Cardiol 17:1343-1351, 199124. Nihoyannopoulos P, Karatasakis G, Frenneaux M,
et al: Diastolic function in hypertrophic cardiomy-
opathy: relation to exercise capacity. J Am Coll
Cardiol 19:536-540, 199225. Nagueh SF, Lakkis NM, Middleton KJ, et al:
Changes in left ventricular diastolic function 6
months after nonsurgical septal reduction therapy
for hypertrophic obstructive cardiomyopathy. Cir-culation 99:344-347, 1999
26. Varnava AM, Elliott PM, Mahon N, et al: Relation
between myocyte disarray and outcome in hyper-
trophic cardiomyopathy. Am J Cardiol 88:275-279,2001
27. Maron BJ, Wolfson JK, Epstein SE, et al: Intramural
(bsmall vesselQ) coronary artery disease in hypertro-phic cardiomyopathy. J Am Coll Cardiol 8:545-557,
1986
28. Sherrid MV, Mahenthiran J, Casteneda V, et al:
Comparison of diastolic septal perforator flowvelocities in hypertrophic cardiomyopathy versus
hypertensive left ventricular hypertrophy. Am J
Cardiol 97:106-112, 2006
29. Maron BJ, Epstein SE, Roberts WC: Hypertrophiccardiomyopathy and transmural myocardial infarc-
tion without significant atherosclerosis of the extra-
mural coronary arteries. Am J Cardiol 43:1086-1102,1979
30. Maron MS, Olivotto I, Betocchi S, et al: Effect of left
ventricular outflow tract obstruction on clinical
outcome in hypertrophic cardiomyopathy. N Engl JMed 348:295-303, 2003
31. Sherrid MV, Chu CK, Delia E, et al: An echocardio-
graphic study of the fluid mechanics of obstruction
in hypertrophic cardiomyopathy. J Am Coll Cardiol
22:816-825, 1993
32. Sherrid MV, Gunsburg DZ, Moldenhauer S, et al:Systolic anterior motion begins at low left ventricular
outflow tract velocity in obstructive hypertrophic
cardiomyopathy. J Am Coll Cardiol 36:1344-1354,2000
33. Shah PM, Gramiak R, Kramer DH: Ultrasound
localization of left ventricular outflow obstruction in
hypertrophic obstructive cardiomyopathy. Circula-tion 40:3-11, 1969
34. Henry WL, Clark CE, Griffith JM, et al: Mechanism of
left ventricular outflow obstruction in patients with
obstructive asymmetric septal hypertrophy (idio-pathic hypertrophic subaortic stenosis). Am J Car-
diol 35:337-345, 1975
35. Schwammenthal E, Levine RA: Dynamic subaorticobstruction: a disease of the mitral valve suitable for
surgical repair? J Am Coll Cardiol 28:203-206, 1996
36. Murgo JP: Does outflow obstruction exist in hyper-
trophic cardiomyopathy? N Engl J Med 307:1008-1009, 1982
37. Criley JM, Siegel RJ: Has dobstructionT hindered our
understanding of hypertrophic cardiomyopathy?
Circulation 72:1148-1154, 198538. Sherrid MV, Gunsburg DZ, Pearle G: Mid-systolic
drop in left ventricular ejection velocity in obstructive
hypertrophic cardiomyopathy—the lobster claw ab-normality. J Am Soc Echocardiogr 10:707-712, 1997
39. Conklin HM, Huang X, Davies CH, et al: Biphasic left
ventricular outflow and its mechanism in hypertro-
phic obstructive cardiomyopathy. J Am Soc Echo-cardiogr 17:375-383, 2004
40. Barac I, Pilchik R, Shteerman E, et al: Midsystolic
drop in left ventricular ejection velocity in hypertro-
phic cardiomyopathy is caused by premature ter-mination and dyssynchrony of longitudinal
contraction. Doppler flow and tissue velocity study
of the blobster claw abnormalityQ (abstract). Circu-
lation 110:746, 2004 (Suppl III)41. Klues HG, Roberts WC, Maron BJ: Anomalous
insertion of papillary muscle directly into anterior
mitral leaflet in hypertrophic cardiomyopathy. Sig-nificance in producing left ventricular outflow ob-
struction. Circulation 84:1188-1197, 1991
42. Falicov RE, Resnekov L, Bharati S, et al: Mid-
ventricular obstruction: a variant of obstructivecardiomyopathy. Am J Cardiol 37:432-437, 1976
43. Nakamura T, Matsubara K, Furukawa K, et al:
Diastolic paradoxic jet flow in patients with hyper-
trophic cardiomyopathy: evidence of concealedapical asynergy with cavity obliteration. J Am Coll
Cardiol 19:516-524, 1992
44. Sasson Z, Yock PG, Hatle LK, et al: Dopplerechocardiographic determination of the pressure
gradient in hypertrophic cardiomyopathy. J Am Coll
Cardiol 11:752-756, 1988
45. Marwick TH, Nakatani S, Haluska B, et al: Provo-cation of latent left ventricular outflow tract gra-
dients with amyl nitrite and exercise in hypertrophic
cardiomyopathy. Am J Cardiol 75:805-809, 1995

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 25
ARTICLE IN PRESS
46. Klues HG, Maron BJ, Dollar AL, et al: Diversity of
structural mitral valve alterations in hypertrophic
cardiomyopathy. Circulation 85:1651-1660, 199247. Cape EG, Simons D, Jimoh A, et al: Chordal
geometry determines the shape and extent of
systolic anterior mitral motion: in vitro studies. JAm Coll Cardiol 13:1438-1448, 1989
48. Jiang L, Levine RA, King ME, et al: An integrated
mechanism for systolic anterior motion of the mitral
valve in hypertrophic cardiomyopathy based onechocardiographic observations. Am Heart J
113:633-644, 1987
49. Shah PM, Taylor RD, Wong M: Abnormal mitral
valve coaptation in hypertrophic obstructive cardio-myopathy: proposed role in systolic anterior motion
of mitral valve. Am J Cardiol 48:258-262, 1981
50. Levine RA, Vlahakes GJ, Lefebvre X, et al: Papillarymuscle displacement causes systolic anterior mo-
tion of the mitral valve. Experimental validation and
insights into the mechanism of subaortic obstruc-
tion. Circulation 91:1189-1195, 199551. Lefebvre XP, Yoganathan AP, Levine RA: Insights
from in-vitro flow visualization into the mechanism of
systolic anterior motion of the mitral valve in
hypertrophic cardiomyopathy under steady flowconditions. J Biomech Eng 114:406-413, 1992
52. Vogel S: Life in moving fluids. The physical biology of
flow. Princeton, NJ: Princeton University Press,1994, pp 81-155
53. Spirito P, Maron BJ: Patterns of systolic anterior
motion of the mitral valve in hypertrophic cardiomy-
opathy: assessment by two-dimensional echocardi-ography. Am J Cardiol 54:1039-1046, 1984
54. Maron BJ, Harding AM, Spirito P, et al: Systolic
anterior motion of the posterior mitral leaflet: a
previously unrecognized cause of dynamic sub-aortic obstruction in patients with hypertrophic
cardiomyopathy. Circulation 68:282-293, 1983
55. Sherrid MV, Chaudhry FA, Swistel DG: Obstructive
hypertrophic cardiomyopathy: echocardiography,pathophysiology, and the continuing evolution
of surgery for obstruction. Ann Thorac Surg 75:
620-632, 200356. Roberts CS, McIntosh CL, Brown Jr PS, et al:
Reoperation for persistent outflow obstruction in
hypertrophic cardiomyopathy. Ann Thorac Surg
51:455-460, 199157. Messmer BJ: Extended myectomy for hypertrophic
obstructive cardiomyopathy. Ann Thorac Surg
58:575-577, 1994
58. Schoendube FA, Klues HG, Reith S, et al: Long-termclinical and echocardiographic follow-up after sur-
gical correction of hypertrophic obstructive cardio-
myopathy wi th extended myectomy andreconstruction of the subvalvular mitral apparatus.
Circulation 92:II122- II127, 1995
59. Nakatani S, Schwammenthal E, Lever HM, et al:
New insights into the reduction of mitral valvesystolic anterior motion after ventricular septal
myectomy in hypertrophic obstructive cardiomyop-
athy. Am Heart J 131:294-300, 1996
60. Grigg LE, Wigle ED, Williams WG, et al: Trans-
esophageal Doppler echocardiography in obstruc-
tive hypertrophic cardiomyopathy: clarification ofpathophysiology and importance in intraoperative
decision making. J Am Coll Cardiol 20:42-52,
199261. Balaram SK, Sherrid MV, Derose Jr JJ, et al:
Beyond extended myectomy for hypertrophic car-
diomyopathy: the resection-plication-release (RPR)
repair. Ann Thorac Surg 80:217-223, 200562. McIntosh CL, Maron BJ, Cannon III RO, et al: Initial
results of combined anterior mitral leaflet plication
and ventricular septal myotomy-myectomy for relief
of left ventricular outflow tract obstruction inpatients with hypertrophic cardiomyopathy. Circu-
lation 86:II60- II67, 1992
63. Spirito P, Rapezzi C, Bellone P, et al: Infectiveendocarditis in hypertrophic cardiomyopathy: prev-
alence, incidence, and indications for antibiotic
prophylaxis. Circulation 99:2132-2137, 1999
64. Sorajja P, Ommen SR, Nishimura RA, et al: Adverseprognosis of patients with hypertrophic cardiomy-
opathy who have epicardial coronary artery disease.
Circulation 108:2342-2348, 2003
65. Spirito P, Bellone P, Harris KM, et al: Magnitude ofleft ventricular hypertrophy and risk of sudden death
in hypertrophic cardiomyopathy. N Engl J Med
342:1778-1785, 200066. Elliott PM, Gimeno J, Blanes R, et al: Relation
between severity of left-ventricular hypertrophy and
prognosis in patients with hypertrophic cardiomy-
opathy. Lancet 357:420-424, 200167. Maron BJ, Shen WK, Link MS, et al: Efficacy of
implantable cardioverter-defibrillators for the pre-
vention of sudden death in patients with hypertro-
phic cardiomyopathy. N Engl J Med 342:365-373,2000
68. Watkins H, McKenna WJ, Thierfelder L, et al:
Mutations in the genes for cardiac troponin T and
alpha-tropomyosin in hypertrophic cardiomyopathy.N Engl J Med 332:1058-1064, 1995
69. Maron BJ, Olivotto I, Spirito P, et al: Epidemiology of
hypertrophic cardiomyopathy-related death: revis-ited in a large non–referral-based patient population.
Circulation 102:858-864, 2000
70. Maron BJ, Roberts WC, Edwards JE, et al: Sudden
death in patients with hypertrophic cardiomyopathy:characterization of 26 patients with functional limi-
tation. Am J Cardiol 41:803-810, 1978
71. Elliott PM, Poloniecki J, Dickie S, et al: Sudden
death in hypertrophic cardiomyopathy: identificationof high risk patients. J Am Coll Cardiol 36:2212-
2218, 2000
72. Maron BJ, Roberts WC, Epstein SE: Sudden deathin hypertrophic cardiomyopathy: a profile of 78
patients. Circulation 65:1388-1394, 1982
73. Maron BJ, Casey SA, Poliac LC, et al: Clinical
course of hypertrophic cardiomyopathy in a regionalUnited States cohort. JAMA 281:650-655, 1999
74. McKenna WJ, Franklin RC, Nihoyannopoulos P,
et al: Arrhythmia and prognosis in infants, children

ARTICLE IN PRESS
MARK V. SHERRID26
ARTICLE IN PRESS
and adolescents with hypertrophic cardiomyopathy.
J Am Coll Cardiol 11:147-153, 1988
75. McKenna W, Deanfield J, Faruqui A, et al: Prognosisin hypertrophic cardiomyopathy: role of age and
clinical, electrocardiographic and hemodynamic
features. Am J Cardiol 47:532-538, 198176. Maron BJ, Mathenge R, Casey SA, et al: Clinical
profile of hypertrophic cardiomyopathy identified de
novo in rural communities. J Am Coll Cardiol
33:1590-1595, 199977. Cecchi F, Olivotto I, Montereggi A, et al: Hypertro-
phic cardiomyopathy in Tuscany: clinical course and
outcome in an unselected regional population. J Am
Coll Cardiol 26:1529-1536, 199578. Maron BJ, Olivotto I, Spirito P, et al: Epidemiology of
hypertrophic cardiomyopathy–related death: re-
vised in a large non–referral-based patient popula-tion. Circulation 102:858-864, 2000
79. Maron BJ, Spirito P: Impact of patient selection
biases on the perception of hypertrophic cardio-
myopathy and its natural history. Am J Cardiol 72:970-972, 1993
80. Fay WP, Taliercio CP, Ilstrup DM, et al: Natural
history of hypertrophic cardiomyopathy in the elder-
ly. J Am Coll Cardiol 16:821-826, 199081. Elliott PM, Sharma S, Varnava A, et al: Survival after
cardiac arrest or sustained ventricular tachycardia in
patients with hypertrophic cardiomyopathy. J AmColl Cardiol 33:1596-1601, 1999
82. Maron BJ, Lipson LC, Roberts WC, et al:
bMalignantQ hypertrophic cardiomyopathy: iden-
tification of a subgroup of families with unusuallyfrequent premature death. Am J Cardiol 41:
1133-1140, 1978
83. Monserrat L, Elliott PM, Gimeno JR, et al: Non-
sustained ventricular tachycardia in hypertrophiccardiomyopathy: an independent marker of sudden
death risk in young patients. J Am Coll Cardiol
42:873-879, 2003
84. Sadoul N, Prasad K, Elliott PM, et al: Prospectiveprognostic assessment of blood pressure re-
sponse during exercise in patients with hypertro-
phic cardiomyopathy. Circulation 96:2987-2991,1997
85. Olivotto I, Maron BJ, Montereggi A, et al: Prognostic
value of systemic blood pressure response during
exercise in a community-based patient populationwith hypertrophic cardiomyopathy. J Am Coll Car-
diol 33:2044-2051, 1999
86. Ciampi Q, Betocchi S, Lombardi R, et al: Hemody-
namic determinants of exercise-induced abnormalblood pressure response in hypertrophic cardiomy-
opathy. J Am Coll Cardiol 40:278-284, 2002
87. Cecchi F, Olivotto I, Montereggi A, et al: Prognosticvalue of non-sustained ventricular tachycardia and
the potential role of amiodarone treatment in
hypertrophic cardiomyopathy: assessment in an
unselected non–referral based patient population.Heart 79:331-336, 1998
88. Van Driest SL, Ellsworth EG, Ommen SR, et al:
Prevalence and spectrum of thin filament mutations
in an outpatient referral population with hypertrophic
cardiomyopathy. Circulation 108:445-451, 2003
89. Mogensen J, Murphy RT, Kubo T, et al: Frequencyand clinical expression of cardiac troponin I muta-
tions in 748 consecutive families with hypertrophic
cardiomyopathy. J Am Coll Cardiol 44:2315-2325,2004
90. Ackerman MJ, VanDriest SL, Ommen SR, et al:
Prevalence and age-dependence of malignant
mutations in the beta-myosin heavy chain andtroponin T genes in hypertrophic cardiomyopathy:
a comprehensive outpatient perspective. J Am Coll
Cardiol 39:2042-2048, 2002
91. Van Driest SL, Ackerman MJ, Ommen SR, et al:Prevalence and severity of bbenignQ mutations in the
beta-myosin heavy chain, cardiac troponin T, and
alpha-tropomyosin genes in hypertrophic cardiomy-opathy. Circulation 106:3085-3090, 2002
92. Marian AJ, Yu QT, Workman R, et al: Angiotensin-
converting enzyme polymorphism in hypertrophic
cardiomyopathy and sudden cardiac death. Lancet342:1085-1086, 1993
93. Lechin M, Quinones MA, Omran A, et al: Angioten-
sin-I converting enzyme genotypes and left ventric-
ular hypertrophy in patients with hypertrophiccardiomyopathy. Circulation 92:1808-1812, 1995
94. Saad EB, Saliba WI, Schweikert RA, et al: Non-
thoracotomy implantable defibrillator lead extrac-tion: results and comparison with extraction of
pacemaker leads. Pacing Clin Electrophysiol
26:1944-1950, 2003
95. Maron BJ, Shirani J, Poliac LC, et al: Sudden death inyoung competitive athletes. Clinical, demographic,
and pathological profiles. JAMA 276:199-204, 1996
96. Maron BJ, Isner JM, McKenna WJ, et al: 26th
Bethesda conference: recommendations for deter-mining eligibility for competition in athletes with
cardiovascular abnormalities. Task Force 3: hyper-
trophic cardiomyopathy, myocarditis and other
myopericardial diseases and mitral valve prolapse.Med Sci Sports Exerc 26:S261-S267, 1994
97. Maron BJ, Mitten MJ, Quandt EF, et al: Competitive
athletes with cardiovascular disease—the case ofNicholas Knapp. N Engl J Med 339:1632-1635,
1998
98. Lele SS, Thomson HL, Seo H, et al: Exercise
capacity in hypertrophic cardiomyopathy. Role ofstroke volume limitation, heart rate, and diastolic
filling characteristics. Circulation 92:2886-2894,
1995
99. Wigle ED, Rakowski H, Kimball BP, et al: Hypertro-phic cardiomyopathy. Clinical spectrum and treat-
ment. Circulation 92:1680-1692, 1995
100. Schwammenthal E, Nakatani S, He S, et al: Mech-anism of mitral regurgitation in hypertrophic cardio-
myopathy: mismatch of posterior to anterior leaflet
length and mobility. Circulation 98:856-865, 1998
101. Cecchi F, Olivotto I, Gistri R, et al: Coronarymicrovascular dysfunction and prognosis in
hypertrophic cardiomyopathy. N Engl J Med 349:
1027-1035, 2003

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 27
ARTICLE IN PRESS
102. Cannon III RO, Rosing DR, Maron BJ, et al:
Myocardial ischemia in patients with hypertrophic
cardiomyopathy: contribution of inadequate vaso-dilator reserve and elevated left ventricular filling
pressures. Circulation 71:234-243, 1985
103. Krams R, Kofflard MJ, Duncker DJ, et al: Decreasedcoronary flow reserve in hypertrophic cardiomyop-
athy is related to remodeling of the coronary
microcirculation. Circulation 97:230-233, 1998
104. Cannon III RO, Schenke WH, Maron BJ, et al:Differences in coronary flow and myocardial metab-
olism at rest and during pacing between patients
with obstructive and patients with nonobstructive
hypertrophic cardiomyopathy. J Am Coll Cardiol10:53-62, 1987
105. Nienaber CA, Hiller S, Spielmann RP, et al: Syncope
in hypertrophic cardiomyopathy: multivariate analy-sis of prognostic determinants. J Am Coll Cardiol
15:948-955, 1990
106. McKenna W, Harris L, Deanfield J: Syncope
in hypertrophic cardiomyopathy. Br Heart J 47:177-179, 1982
107. Frenneaux MP, Counihan PJ, Caforio AL, et al:
Abnormal blood pressure response during exercise
in hypertrophic cardiomyopathy. Circulation 82:1995-2002, 1990
108. Maron BJ, Casey SA, Hauser RG, et al: Clinical
course of hypertrophic cardiomyopathy with survivalto advanced age. J Am Coll Cardiol 42:882-888,
2003
109. Kass DA, Wolff MR, Ting CT, et al: Diastolic
compliance of hypertrophied ventricle is not acutelyaltered by pharmacologic agents influencing active
processes. Ann Intern Med 119:466-473, 1993
110. TenCate FJ, Serruys PW, Mey S, et al: Effects of
short-term administration of verapamil on left ven-tricular relaxation and filling dynamics measured by
a combined hemodynamic-ultrasonic technique in
patients with hypertrophic cardiomyopathy. Circu-
lation 68:1274-1279, 1983111. Sherrid M, Barac I: Pharmacologic treatment of
symptomatic hypertrophic cardiomyopathy, in
Maron B, (ed): Diagnosis and management of
hypertrophic cardiomyopathy. London: Blackwell-
Futura, 2004, pp 200-219
112. Bonow RO, Ostrow HG, Rosing DR, et al: Effects
of verapamil on left ventricular systolic anddiastolic function in patients with hypertrophic
cardiomyopathy: pressure-volume analysis with a
nonimaging scintillation probe. Circulation 68:
1062-1073, 1983113. Bonow RO: Effects of calcium-channel blocking
agents on left ventricular diastolic function in
hypertrophic cardiomyopathy and in coronary arterydisease. Am J Cardiol 55:172B-178B, 1985
114. Bonow RO, Rosing DR, Bacharach SL, et al: Effects
of verapamil on left ventricular systolic function and
diastolic filling in patients with hypertrophic cardio-myopathy. Circulation 64:787-796, 1981
115. Nishimura RA, Holmes DR, Tajik AJ: Failure of
calcium channel blockers to improve ventricu-
lar relaxation in humans. J Am Coll Cardiol 21:
182-188, 1993
116. Udelson JE, Bonow RO, O’Gara PT, et al: Vera-pamil prevents silent myocardial perfusion abnor-
malities during exercise in asymptomatic patients
with hypertrophic cardiomyopathy. Circulation 79:1052-1060, 1989
117. Gilligan DM, Chan WL, Joshi J, et al: A double-blind,
placebo-controlled crossover trial of nadolol and
verapamil in mild and moderately symptomatichypertrophic cardiomyopathy. J Am Coll Cardiol
21:1672-1679, 1993
118. Matsubara H, Nakatani S, Nagata S, et al: Salutary
effect of disopyramide on left ventricular diastolicfunction in hypertrophic obstructive cardiomyopa-
thy. J Am Coll Cardiol 26:768-775, 1995
119. Pollick C, Kimball B, Henderson M, et al: Disopyr-amide in hypertrophic cardiomyopathy: I. Hemody-
namic assessment after intravenous administration.
Am J Cardiol 62:1248-1251, 1988
120. Tsybouleva N, Zhang L, Chen S, et al: Aldosterone,through novel signaling proteins, is a fundamental
molecular bridge between the genetic defect and
the cardiac phenotype of hypertrophic cardiomyop-
athy. Circulation 109:1284-1291, 2004121. Senthil V, Chen SN, Tsybouleva N, et al: Prevention
of cardiac hypertrophy by atorvastatin in a trans-
genic rabbit model of human hypertrophic cardio-myopathy. Circ Res 97:285-292, 2005
122. Sherrid MV, Pearle G, Gunsburg DZ: Mechanism of
benefit of negative inotropes in obstructive hyper-
trophic cardiomyopathy. Circulation 97:41 - 47,1998
123. Flamm MD, Harrison DC, Hancock EW: Muscular
subaortic stenosis. Prevention of outflow obstruc-
tion with propranolol. Circulation 38:846 - 858,1968
124. Stenson RE, Flamm Jr MD, Harrison DC, et al:
Hypertrophic subaortic stenosis. Clinical and hemo-
dynamic effects of long-term propranolol therapy.Am J Cardiol 31:763-773, 1973
125. Rosing DR, Condit JR, Maron BJ, et al: Verapamil
therapy: a new approach to the pharmacologictreatment of hypertrophic cardiomyopathy: III.
Effects of long-term administration. Am J Cardiol
48:545-553, 1981
126. Rosing DR, Kent KM, Maron BJ, et al: Verapamiltherapy: a new approach to the pharmacologic
treatment of hypertrophic cardiomyopathy: II.
Effects on exercise capacity and symptomatic
status. Circulation 60:1208-1213, 1979127. Sherrid MV, Barac I, McKenna WJ, et al: Multicenter
study of the efficacy and safety of disopyramide in
obstructive hypertrophic cardiomyopathy. J Am CollCardiol 45:1251-1258, 2005
128. Pollick C: Muscular subaortic stenosis: hemody-
namic and clinical improvement after disopyramide.
N Engl J Med 307:997-999, 1982129. Sherrid M, Delia E, Dwyer E: Oral disopyramide
therapy for obstructive hypertrophic cardiomyopa-
thy. Am J Cardiol 62:1085-1088, 1988

ARTICLE IN PRESS
MARK V. SHERRID28
ARTICLE IN PRESS
130. Pollick C: Disopyramide in hypertrophic cardiomy-
opathy: II. Noninvasive assessment after oral ad-
ministration. Am J Cardiol 62:1252-1255, 1988131. Rosing DR, Idanpaan-Heikkila U, Maron BJ, et al:
Use of calcium-channel blocking drugs in hypertro-
phic cardiomyopathy. Am J Cardiol 55:185B-195B,1985
132. Rosing DR, Kent KM, Borer JS, et al: Verapamil
therapy: a new approach to the pharmacologic
treatment of hypertrophic cardiomyopathy: I. He-modynamic effects. Circulation 60:1201-1207, 1979
133. Epstein SE, Rosing DR: Verapamil: its potential
for causing serious complications in patients
with hypertrophic cardiomyopathy. Circulation 64:437-441, 1981
134. Pollick CGK, Blaschke TF, Nelson WL, et al: The
cardiac effects of d- and l-disopyramide in normalsubjects: a noninvasive study. Circulation 66:447-
453, 1982
135. Olivotto I, Maron BJ, Cecchi F: Clinical significance
of atrial fibrillation in hypertrophic cardiomyopathy.Curr Cardiol Rep 3:141-146, 2001
136. Olivotto I, Cecchi F, Casey SA, et al: Impact of atrial
fibrillation on the clinical course of hypertrophic
cardiomyopathy. Circulation 104:2517-2524, 2001137. Maron BJ, Olivotto I, Bellone P, et al: Clinical profile
of stroke in 900 patients with hypertrophic cardio-
myopathy. J Am Coll Cardiol 39:301-307, 2002138. Robinson K, Frenneaux MP, Stockins B, et al: Atrial
fibrillation in hypertrophic cardiomyopathy: a longi-
tudinal study. J Am Coll Cardiol 15:1279-1285,
1990139. Herweg B, Polosajian L, Vloka M, et al: Anatomic
substrate, procedural results and clinical outcome
of ultrasound-guided left atrial–pulmonary vein dis-
connection for treatment of atrial fibrillation. Am JCardiol 95:871-875, 2005
140. Williams WG, Wigle ED, Rakowski H, et al: Results
of surgery for hypertrophic obstructive cardiomyop-
athy. Circulation 76:V104-V108, 1987141. Maron BJ, Dearani JA, Ommen SR, et al: The case
for surgery in obstructive hypertrophic cardiomyop-
athy. J Am Coll Cardiol 44:2044-2053, 2004142. Ommen SR, Maron BJ, Olivotto I, et al: Long-term
effects of surgical septal myectomy on survival in
patients with obstructive hypertrophic cardiomyop-
athy. J Am Coll Cardiol 46:470-476, 2005143. McCully RB, Nishimura RA, Bailey KR, et al: Hyper-
trophic obstructive cardiomyopathy: preoperative
echocardiographic predictors of outcome after sep-
tal myectomy. J Am Coll Cardiol 27:1491-1496, 1996144. McCully RB, Nishimura RA, Tajik AJ, et al: Extent of
clinical improvement after surgical treatment of
hypertrophic obstructive cardiomyopathy. Circula-tion 94:467-471, 1996
145. Schulte HD, Borisov K, Gams E, et al: Management
of symptomatic hypertrophic obstructive cardiomy-
opathy—long-term results after surgical therapy.Thorac Cardiovasc Surg 47:213-218, 1999
146. Schulte HD, Bircks WH, Loesse B, et al: Prognosis
of patients with hypertrophic obstructive cardiomy-
opathy after transaortic myectomy. Late results up
to twenty-five years. J Thorac Cardiovasc Surg
106:709-717, 1993147. Schoendube FA, Klues HG, Reith S, et al: Surgical
correction of hypertrophic obstructive cardiomyop-
athy with combined myectomy, mobilisation andpartial excision of the papillary muscles. Eur J
Cardiothorac Surg 8:603-608, 1994
148. Heric B, Lytle BW, Miller DP, et al: Surgical
management of hypertrophic obstructive cardiomy-opathy. Early and late results. J Thorac Cardiovasc
Surg 110:195-206, 1995 [discussion 206-8]
149. Qin JX, Shiota T, Lever HM, et al: Outcome of
patients with hypertrophic obstructive cardiomyop-athy after percutaneous transluminal septal myo-
cardial ablation and septal myectomy surgery. J Am
Coll Cardiol 38:1994-2000, 2001150. Marwick TH, Stewart WJ, Lever HM, et al: Benefits
of intraoperative echocardiography in the surgical
management of hypertrophic cardiomyopathy. J Am
Coll Cardiol 20:1066-1072, 1992151. Ommen SR, Park SH, Click RL, et al: Impact of
intraoperative transesophageal echocardiography in
the surgical management of hypertrophic cardiomy-
opathy. Am J Cardiol 90:1022-1024, 2002152. Krajcer Z, Leachman RD, Cooley DA, et al: Mitral
valve replacement and septal myomectomy in
hypertrophic cardiomyopathy. Ten-year follow-upin 80 patients. Circulation 78:I35- I43, 1988
153. Roberts WC: Operative treatment of hypertrophic
obstructive cardiomyopathy. The case against mi-
tral valve replacement. Am J Cardiol 32:377-381,1973
154. Seggewiss H, Gleichmann U, Faber L, et al: Percu-
taneous transluminal septal myocardial ablation in
hypertrophic obstructive cardiomyopathy: acuteresults and 3-month follow-up in 25 patients. J Am
Coll Cardiol 31:252-258, 1998
155. Lakkis NM, Nagueh SF, Kleiman NS, et al: Echo-
cardiography-guided ethanol septal reduction forhypertrophic obstructive cardiomyopathy. Circula-
tion 98:1750-1755, 1998
156. Nagueh SF, Lakkis NM, He ZX, et al: Role ofmyocardial contrast echocardiography during non-
surgical septal reduction therapy for hypertrophic
obstructive cardiomyopathy. J Am Coll Cardiol
32:225-229, 1998157. Faber L, Seggewiss H, Fassbender D, et al: Percu-
taneous transluminal septal myocardial ablation in
hypertrophic obstructive cardiomyopathy: acute
results in 66 patients with reference to myocardialcontrast echocardiography. Z Kardiol 87:191-201,
1998
158. Faber L, Ziemssen P, Seggewiss H, et al: Targetingpercutaneous transluminal septal ablation for hy-
pertrophic obstructive cardiomyopathy by intrapro-
cedural echocardiographic monitoring. J Am Soc
Echocardiogr 13:1074-1079, 2000159. Gietzen FH, Leuner CJ, Obergassel L, et al: Role of
transcoronary ablation of septal hypertrophy in
patients with hypertrophic cardiomyopathy, New

ARTICLE IN PRESS
PATHOPHYSIOLOGY AND TREATMENT OF HYPERTROPHIC CARDIOMYOPATHY 29
ARTICLE IN PRESS
York Heart Association functional class III or IV, and
outflow obstruction only under provocable condi-
tions. Circulation 106:454-459, 2002160. Lakkis N, Plana JC, Nagueh S, et al: Efficacy of
nonsurgical septal reduction therapy in symptomat-
ic patients with obstructive hypertrophic cardiomy-opathy and provocable gradients. Am J Cardiol
88:583-586, 2001
161. Faber L, Seggewiss H, Welge D, et al: Echo-
guided percutaneous septal ablation for symptoma-tic hypertrophic obstructive cardiomyopathy: 7 years
of experience. Eur J Echocardiogr 5:347-355, 2004
162. Flores-Ramirez R, Lakkis NM, Middleton KJ, et al:
Echocardiographic insights into the mechanisms ofrelief of left ventricular outflow tract obstruction after
nonsurgical septal reduction therapy in patients with
hypertrophic obstructive cardiomyopathy. J Am CollCardiol 37:208-214, 2001
163. Fernandes VL, Nagueh SF, Wang W, et al: A
prospective follow-up of alcohol septal ablation for
symptomatic hypertrophic obstructive cardiomyop-athy—the Baylor experience (1996-2002). Clin Car-
diol 28:124-130, 2005
164. Nagueh SF, Ommen SR, Lakkis NM, et al: Compar-
ison of ethanol septal reduction therapy with surgi-cal myectomy for the treatment of hypertrophic
obstructive cardiomyopathy. J Am Coll Cardiol
38:1701-1706, 2001165. Fananapazir L, Epstein ND, Curiel RV, et al: Long-
term results of dual-chamber (DDD) pacing
in obstructive hypertrophic cardiomyopathy. Evi-
dence for progressive symptomatic and hemo-dynamic improvement and reduction of left
ventricular hypertrophy. Circulation 90:2731-2742,
1994
166. Maron BJ, Nishimura RA, McKenna WJ, et al:Assessment of permanent dual-chamber pacing as
a treatment for drug-refractory symptomatic
patients with obstructive hypertrophic cardiomyop-
athy. A randomized, double-blind, crossover study(M-PATHY). Circulation 99:2927-2933, 1999
167. Kappenberger L, Linde C, Daubert C, et al: Pacing in
hypertrophic obstructive cardiomyopathy. A ran-domized crossover study. PIC Study Group. Eur
Heart J 18:1249-1256, 1997
168. Jeanrenaud X, Kappenberger L: Regional wall
motion during pacing for hypertrophic obstructive
cardiomyopathy. Pacing Clin Electrophysiol 20:
1673-1681, 1997
169. Nishimura RA, Hayes DL, Ilstrup DM, et al: Effect ofdual-chamber pacing on systolic and diastolic
function in patients with hypertrophic cardiomyop-
athy. Acute Doppler echocardiographic and cathe-terization hemodynamic study. J Am Coll Cardiol
27:421-430, 1996
170. Ommen SR, Nishimura RA, Squires RW, et al:
Comparison of dual-chamber pacing versus septalmyectomy for the treatment of patients with hyper-
tropic obstructive cardiomyopathy: a comparison of
objective hemodynamic and exercise end points. J
Am Coll Cardiol 34:191-196, 1999171. Gadler F, Linde C, Daubert C, et al: Significant
improvement of quality of life following atrioventric-
ular synchronous pacing in patients with hypertro-phic obstructive cardiomyopathy. Data from 1 year
of follow-up. PIC study group. Pacing in cardiomy-
opathy. Eur Heart J 20:1044-1050, 1999
172. Linde C, Gadler F, Kappenberger L, et al: Place-bo effect of pacemaker implantation in obstruc-
tive hypertrophic cardiomyopathy. PIC Study
Group. Pacing in cardiomyopathy. Am J Cardiol
83:903-907, 1999173. Nishimura RA, Trusty JM, Hayes DL, et al: Dual-
chamber pacing for hypertrophic cardiomyopathy: a
randomized, double-blind, crossover trial. J Am CollCardiol 29:435-441, 1997
174. Gadler F, Linde C, Ryden L, et al: Rapid return of left
ventricular outflow tract obstruction and symptoms
following cessation of long-term atrioventricularsynchronous pacing for obstructive hypertrophic
cardiomyopathy. Am J Cardiol 83:553-557, 1999
175. Topilski ISJ, Keren G, Copperman I: Long-term
effects of dual-chamber pacing with periodic echo-cardiographic evaluation of optimal atrioventricular
delay in patients with hypertrophic cardiomyopathy
N50 years of age. Am J Cardiol 97:1769-1775,
2006176. Topol EJ, Traill TA, Fortuin NJ: Hypertensive hyper-
trophic cardiomyopathy of the elderly. N Engl J Med
312:277-283, 1985177. Spirito P, Maron BJ, Bonow RO, et al: Occurrence
and significance of progressive left ventricular wall
thinning and relative cavity dilatation in hypertrophic
cardiomyopathy. Am J Cardiol 60:123-129, 1987

![GENETIC BASIS OF HYPERTROPHIC CARDIOMYOPATHYThroughout the years, names such as idiopathic hypertrophic subaortic stenosis[5], muscular subaortic stenosis[6] and hypertrophic obstructive](https://img.pdfslide.us/doc/110x75/60571329c95e4748070a14f6/genetic-basis-of-hypertrophic-cardiomyopathy-throughout-the-years-names-such-as.jpg)



























