Embed Size (px)
Citation preview

Pathogen receptor discovery with a microfluidic humanmembrane protein arrayYair Glicka,1, Ya’ara Ben-Aria,1, Nir Draymanb, Michal Pellacha, Gregory Neveuc,d, Jim Boonyaratanakornkitc,d,Dorit Avrahamia, Shirit Einavc,d, Ariella Oppenheimb, and Doron Gerbera,2
aMina and Everard Goodman Faculty of Life Sciences, Bar Ilan University, 5290002, Israel; bFaculty of Medicine, Hebrew University, Jerusalem, 9112001,Israel; cDepartment of Medicine, Division of Infectious Diseases and Geographic Medicine, Stanford University School of Medicine, Stanford, CA 94305;and dDepartment of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA 94305
Edited by Stephen R. Quake, Stanford University, Stanford, CA, and approved March 1, 2016 (received for review September 20, 2015)
The discovery of how a pathogen invades a cell requires one todetermine which host cell receptors are exploited. This determina-tion is a challenging problem because the receptor is invariably amembrane protein, which represents an Achilles heel in proteomics.We have developed a universal platform for high-throughput ex-pression and interaction studies of membrane proteins by creating amicrofluidic-based comprehensive human membrane protein array(MPA). The MPA is, to our knowledge, the first of its kind and offersa powerful alternative to conventional proteomics by enabling thesimultaneous study of 2,100 membrane proteins. We characterizeddirect interactions of a whole nonenveloped virus (simian virus 40),as well as those of the hepatitis delta enveloped virus large formantigen, with candidate host receptors expressed on the MPA. Se-lected newly discovered membrane protein–pathogen interactionswere validated by conventional methods, demonstrating that theMPA is an important tool for cellular receptor discovery and forunderstanding pathogen tropism.
pathogen–host interactions | membrane protein array |receptor discovery | integrated microfluidics
The human genome contains ∼21,000 distinct protein-codinggenes (1), out of which ∼5,360 code for membrane proteins
(2). Membrane proteins are critical for many cellular processes,such as signaling, transport, cell–cell communication, and alsointeraction with pathogens leading to various cellular responses.It is not surprising that 60% of drugs currently in the markettarget proteins at the cell surface (3). Mapping molecular inter-actions of membrane proteins is, therefore, of utmost importance.Pathogen–host recognition involves surface interactions regulatedby membrane proteins. Many interactions between membraneproteins and pathogens are unknown, partly because of the lowsensitivity and limited compatibility of current methodologies withmembrane proteins (4). These limitations pose a major obstacle inunderstanding pathogen tropism, a public health concern in viewof emerging diseases, e.g., severe acute respiratory syndrome andEbola. There is therefore a need for new approaches that wouldrecapitulate the pathogen–host molecular recognition, let alone inthe context of intact pathogens.Mapping protein–protein interaction (PPI) is a major chal-
lenge in proteomics. Many molecular interactions are transientand weak, leading to low yield of bound material and thus de-manding highly sensitive detection methods. Current methodsfor characterizing PPI networks suffer from several basic disad-vantages: low sensitivity, leading to high false negative rate; lowspecificity, leading to high false positive rate (5–7); low coverageof known interactions; and high variability, even in screens fromthe same species (4). Protein arrays could potentially overridesuch limitations (8), but suffer from a purification bottleneck andlimited functionality after deposition (9). These difficulties areeven more pronounced with membrane proteins. Membrane pro-teins are usually in low abundance; in addition, they are in-compatible with high-throughput methods (e.g., yeast two-hybrid)and are difficult to purify in functional form (e.g., protein ar-rays). One partial solution to these obstacles was to print DNA
and translate into proteins in situ (10, 11). This approach hasenabled the study of the Pseudomonas aeruginosa outer mem-brane protein for immunity (12).Combining integrated microfluidics with microarrays and
in vitro transcription and translation (TNT) systems may over-come all of the above mentioned difficulties (Fig. 1A) (13–16).The integrated microfluidic device allows smart liquid manage-ment in very low volumes, partitioning, and process integration(i.e., protein expression, immobilization, and interaction exper-iments). Microarray technology provides the means for pro-gramming thousands of different experiments (17). In vitro TNTexpression systems allow protein biosynthesis and are compatiblewith high throughput (18). Such systems are commercially avail-able and benefit from fast protein expression, low reaction vol-umes, and short reaction times and enable expression of syntheticproteins with inserted epitope tags. Adding microsomal mem-branes enable the correct folding of membrane proteins andsupport posttranslational modifications, such as glycosylation(9). In short, the microfluidic platform facilitates using in vitroTNT systems to produce a reliable membrane protein array(MPA) from DNA with high sensitivity, low material and proteinconsumption, and compatibility with membrane proteins.In this study, we used a microfluidic platform to combine
microarray technology, cell-free protein expression, and integratedmicrofluidics, allowing high-throughput screening of pathogenswith a human membrane proteome library. As a proof-of-concept,we screened two pathogens differing in structure and physiology.The first was simian virus 40 (SV40), a nonenveloped polyomaviruscontaining circular double-stranded DNA, which can inducemembrane invaginations, similarly to other polyomaviruses. Itcauses infections of the kidney and possibly also other tissues
Significance
In this work, we report, to our knowledge, the first in vitro toolfor host–pathogen screening that encompasses thousands offunctional insoluble proteins—primarily transmembrane pro-teins—immobilized within a microfluidic device. We discoveredpreviously unknown protein–pathogen interactions, and thenselected interactions were further validated by conventionalmethods. Considering the tremendous difficulty in discoveringpathogen receptors, this in vitro high-throughput approach isextremely important and efficient for receptor discovery andunderstanding pathogen tropism, with relevance to emerginghuman diseases.
Author contributions: Y.G., Y.B.-A., N.D., D.A., S.E., A.O., and D.G. designed research; Y.G.,Y.B.-A., N.D., G.N., J.B., and S.E. performed research; A.O. contributed new reagents/analytic tools; Y.G., Y.B.-A., N.D., M.P., G.N., J.B., D.A., S.E., A.O., and D.G. analyzed data;and Y.G., Y.B.-A., M.P., A.O., and D.G. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1Y.G. and Y.B.-A. contributed equally to this work.2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1518698113/-/DCSupplemental.
4344–4349 | PNAS | April 19, 2016 | vol. 113 | no. 16 www.pnas.org/cgi/doi/10.1073/pnas.1518698113
such as the mesothelium, and it has also been associated withcancer (19, 20). The second pathogen was the hepatitis delta virus(HDV). We screened the large-form delta antigen (L-HDAg), aprenylated protein essential for HDV virion assembly by allowinginteraction with the envelope proteins of the hepatitis B helpervirus (21, 22). Understanding interactions of both these viruseswith host membrane proteins may shed light on important patho-genic processes.We created a library of ∼2,700 synthetic genes, which we
arrayed and expressed within the microfluidic platform using acell-free protein expression system. Then, we screened the as-sembled MPA for interactions with each pathogen and identifiednew interactions. A bioinformatics approach was used to identifybiological processes associated with the newly found interactions.Specific interactions of interest were chosen for further valida-tion by either coimmunoprecipitation (Co-IP) or protein–frag-ment complementation assay of luciferase activity (PCA). Wesuccessfully demonstrated the effectiveness of using a micro-fluidic platform for performing a high-throughput screen ofpathogen–membrane protein interactions.
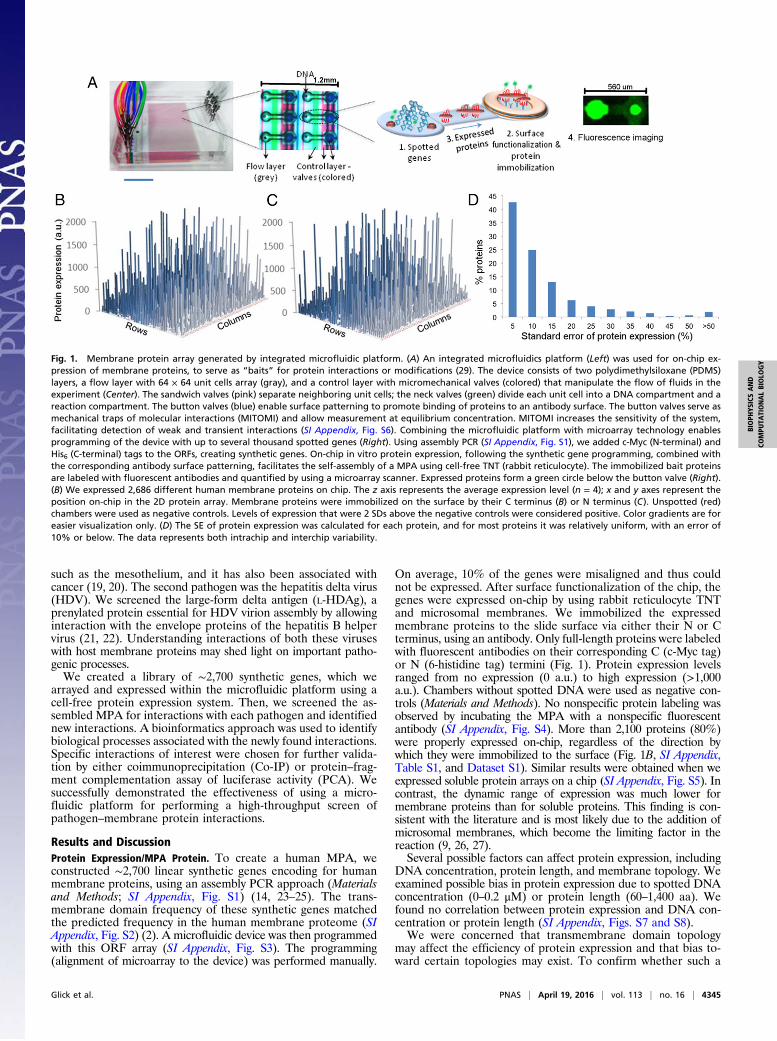
Results and DiscussionProtein Expression/MPA Protein. To create a human MPA, weconstructed ∼2,700 linear synthetic genes encoding for humanmembrane proteins, using an assembly PCR approach (Materialsand Methods; SI Appendix, Fig. S1) (14, 23–25). The trans-membrane domain frequency of these synthetic genes matchedthe predicted frequency in the human membrane proteome (SIAppendix, Fig. S2) (2). A microfluidic device was then programmedwith this ORF array (SI Appendix, Fig. S3). The programming(alignment of microarray to the device) was performed manually.
On average, 10% of the genes were misaligned and thus couldnot be expressed. After surface functionalization of the chip, thegenes were expressed on-chip by using rabbit reticulocyte TNTand microsomal membranes. We immobilized the expressedmembrane proteins to the slide surface via either their N or Cterminus, using an antibody. Only full-length proteins were labeledwith fluorescent antibodies on their corresponding C (c-Myc tag)or N (6-histidine tag) termini (Fig. 1). Protein expression levelsranged from no expression (0 a.u.) to high expression (>1,000a.u.). Chambers without spotted DNA were used as negative con-trols (Materials and Methods). No nonspecific protein labeling wasobserved by incubating the MPA with a nonspecific fluorescentantibody (SI Appendix, Fig. S4). More than 2,100 proteins (80%)were properly expressed on-chip, regardless of the direction bywhich they were immobilized to the surface (Fig. 1B, SI Appendix,Table S1, and Dataset S1). Similar results were obtained when weexpressed soluble protein arrays on a chip (SI Appendix, Fig. S5). Incontrast, the dynamic range of expression was much lower formembrane proteins than for soluble proteins. This finding is con-sistent with the literature and is most likely due to the addition ofmicrosomal membranes, which become the limiting factor in thereaction (9, 26, 27).Several possible factors can affect protein expression, including
DNA concentration, protein length, and membrane topology. Weexamined possible bias in protein expression due to spotted DNAconcentration (0–0.2 μM) or protein length (60–1,400 aa). Wefound no correlation between protein expression and DNA con-centration or protein length (SI Appendix, Figs. S7 and S8).We were concerned that transmembrane domain topology
may affect the efficiency of protein expression and that bias to-ward certain topologies may exist. To confirm whether such a
Fig. 1. Membrane protein array generated by integrated microfluidic platform. (A) An integrated microfluidics platform (Left) was used for on-chip ex-pression of membrane proteins, to serve as “baits” for protein interactions or modifications (29). The device consists of two polydimethylsiloxane (PDMS)layers, a flow layer with 64 × 64 unit cells array (gray), and a control layer with micromechanical valves (colored) that manipulate the flow of fluids in theexperiment (Center). The sandwich valves (pink) separate neighboring unit cells; the neck valves (green) divide each unit cell into a DNA compartment and areaction compartment. The button valves (blue) enable surface patterning to promote binding of proteins to an antibody surface. The button valves serve asmechanical traps of molecular interactions (MITOMI) and allow measurement at equilibrium concentration. MITOMI increases the sensitivity of the system,facilitating detection of weak and transient interactions (SI Appendix, Fig. S6). Combining the microfluidic platform with microarray technology enablesprogramming of the device with up to several thousand spotted genes (Right). Using assembly PCR (SI Appendix, Fig. S1), we added c-Myc (N-terminal) andHis6 (C-terminal) tags to the ORFs, creating synthetic genes. On-chip in vitro protein expression, following the synthetic gene programming, combined withthe corresponding antibody surface patterning, facilitates the self-assembly of a MPA using cell-free TNT (rabbit reticulocyte). The immobilized bait proteinsare labeled with fluorescent antibodies and quantified by using a microarray scanner. Expressed proteins form a green circle below the button valve (Right).(B) We expressed 2,686 different human membrane proteins on chip. The z axis represents the average expression level (n = 4); x and y axes represent theposition on-chip in the 2D protein array. Membrane proteins were immobilized on the surface by their C terminus (B) or N terminus (C). Unspotted (red)chambers were used as negative controls. Levels of expression that were 2 SDs above the negative controls were considered positive. Color gradients are foreasier visualization only. (D) The SE of protein expression was calculated for each protein, and for most proteins it was relatively uniform, with an error of10% or below. The data represents both intrachip and interchip variability.
Glick et al. PNAS | April 19, 2016 | vol. 113 | no. 16 | 4345
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
bias existed, we divided the proteins according to the number oftheir transmembrane domain. These ranged from 0 to 14 trans-membrane domains. The number of transmembrane domains didnot affect average expression levels, which hence were not affectedby protein topology (SI Appendix, Fig. S9). We also demonstratedthat both sides of the membrane proteins were accessible for in-teraction (SI Appendix, Fig. S10). We attributed this accessibilityto microsomal membrane instability (i.e., vesicle fusion) due tochanges in surface tension following protein expression (8, 22, 23).To demonstrate that the membrane proteins expressed using
the cell-free expression procedure were functional, selectedmembrane proteins with previously established ligand interac-tions were expressed via the same expression procedure andimmobilized onto a chip (SI Appendix, Fig. S11). Interactions of13 human receptors and their corresponding fluorescently la-beled ligand were examined, and 10 pairs (∼75%) demonstratedspecific interactions, indicating a likelihood that a significantproportion of the 2,700 membrane proteins were properly foldedand functional.
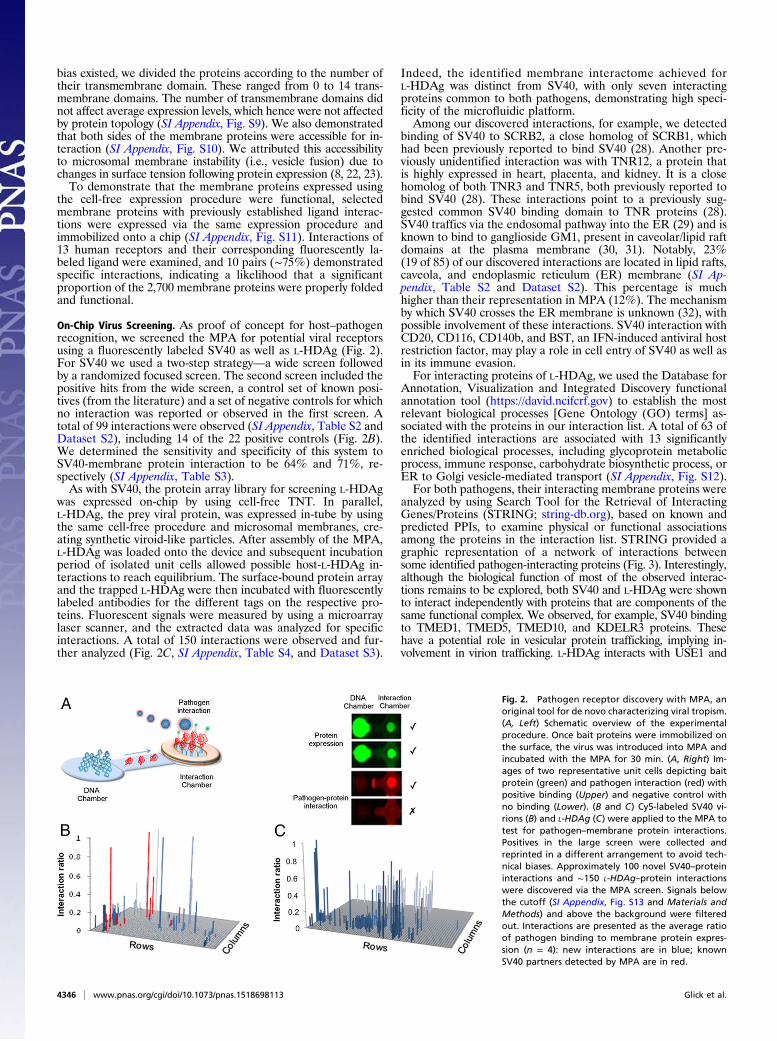
On-Chip Virus Screening. As proof of concept for host–pathogenrecognition, we screened the MPA for potential viral receptorsusing a fluorescently labeled SV40 as well as L-HDAg (Fig. 2).For SV40 we used a two-step strategy—a wide screen followedby a randomized focused screen. The second screen included thepositive hits from the wide screen, a control set of known posi-tives (from the literature) and a set of negative controls for whichno interaction was reported or observed in the first screen. Atotal of 99 interactions were observed (SI Appendix, Table S2 andDataset S2), including 14 of the 22 positive controls (Fig. 2B).We determined the sensitivity and specificity of this system toSV40-membrane protein interaction to be 64% and 71%, re-spectively (SI Appendix, Table S3).As with SV40, the protein array library for screening L-HDAg
was expressed on-chip by using cell-free TNT. In parallel,L-HDAg, the prey viral protein, was expressed in-tube by usingthe same cell-free procedure and microsomal membranes, cre-ating synthetic viroid-like particles. After assembly of the MPA,L-HDAg was loaded onto the device and subsequent incubationperiod of isolated unit cells allowed possible host-L-HDAg in-teractions to reach equilibrium. The surface-bound protein arrayand the trapped L-HDAg were then incubated with fluorescentlylabeled antibodies for the different tags on the respective pro-teins. Fluorescent signals were measured by using a microarraylaser scanner, and the extracted data was analyzed for specificinteractions. A total of 150 interactions were observed and fur-ther analyzed (Fig. 2C, SI Appendix, Table S4, and Dataset S3).
Indeed, the identified membrane interactome achieved forL-HDAg was distinct from SV40, with only seven interactingproteins common to both pathogens, demonstrating high speci-ficity of the microfluidic platform.Among our discovered interactions, for example, we detected
binding of SV40 to SCRB2, a close homolog of SCRB1, whichhad been previously reported to bind SV40 (28). Another pre-viously unidentified interaction was with TNR12, a protein thatis highly expressed in heart, placenta, and kidney. It is a closehomolog of both TNR3 and TNR5, both previously reported tobind SV40 (28). These interactions point to a previously sug-gested common SV40 binding domain to TNR proteins (28).SV40 traffics via the endosomal pathway into the ER (29) and isknown to bind to ganglioside GM1, present in caveolar/lipid raftdomains at the plasma membrane (30, 31). Notably, 23%(19 of 85) of our discovered interactions are located in lipid rafts,caveola, and endoplasmic reticulum (ER) membrane (SI Ap-pendix, Table S2 and Dataset S2). This percentage is muchhigher than their representation in MPA (12%). The mechanismby which SV40 crosses the ER membrane is unknown (32), withpossible involvement of these interactions. SV40 interaction withCD20, CD116, CD140b, and BST, an IFN-induced antiviral hostrestriction factor, may play a role in cell entry of SV40 as well asin its immune evasion.For interacting proteins of L-HDAg, we used the Database for
Annotation, Visualization and Integrated Discovery functionalannotation tool (https://david.ncifcrf.gov) to establish the mostrelevant biological processes [Gene Ontology (GO) terms] as-sociated with the proteins in our interaction list. A total of 63 ofthe identified interactions are associated with 13 significantlyenriched biological processes, including glycoprotein metabolicprocess, immune response, carbohydrate biosynthetic process, orER to Golgi vesicle-mediated transport (SI Appendix, Fig. S12).For both pathogens, their interacting membrane proteins were
analyzed by using Search Tool for the Retrieval of InteractingGenes/Proteins (STRING; string-db.org), based on known andpredicted PPIs, to examine physical or functional associationsamong the proteins in the interaction list. STRING provided agraphic representation of a network of interactions betweensome identified pathogen-interacting proteins (Fig. 3). Interestingly,although the biological function of most of the observed interac-tions remains to be explored, both SV40 and L-HDAg were shownto interact independently with proteins that are components of thesame functional complex. We observed, for example, SV40 bindingto TMED1, TMED5, TMED10, and KDELR3 proteins. Thesehave a potential role in vesicular protein trafficking, implying in-volvement in virion trafficking. L-HDAg interacts with USE1 and
Fig. 2. Pathogen receptor discovery with MPA, anoriginal tool for de novo characterizing viral tropism.(A, Left) Schematic overview of the experimentalprocedure. Once bait proteins were immobilized onthe surface, the virus was introduced into MPA andincubated with the MPA for 30 min. (A, Right) Im-ages of two representative unit cells depicting baitprotein (green) and pathogen interaction (red) withpositive binding (Upper) and negative control withno binding (Lower). (B and C) Cy5-labeled SV40 vi-rions (B) and L-HDAg (C) were applied to the MPA totest for pathogen–membrane protein interactions.Positives in the large screen were collected andreprinted in a different arrangement to avoid tech-nical biases. Approximately 100 novel SV40–proteininteractions and ∼150 L-HDAg–protein interactionswere discovered via the MPA screen. Signals belowthe cutoff (SI Appendix, Fig. S13 and Materials andMethods) and above the background were filteredout. Interactions are presented as the average ratioof pathogen binding to membrane protein expres-sion (n = 4): new interactions are in blue; knownSV40 partners detected by MPA are in red.
4346 | www.pnas.org/cgi/doi/10.1073/pnas.1518698113 Glick et al.

syntaxin 18 (STX18), both components of a complex involved inGolgi to ER retrograde transport that directly interact with eachother. L-HDAg also interacts with PDGFRA and PDGFRB,which upon ligand binding at the plasma membrane, form aheterodimer and initiate several signaling cascades, dependingon the nature of the bound ligand.
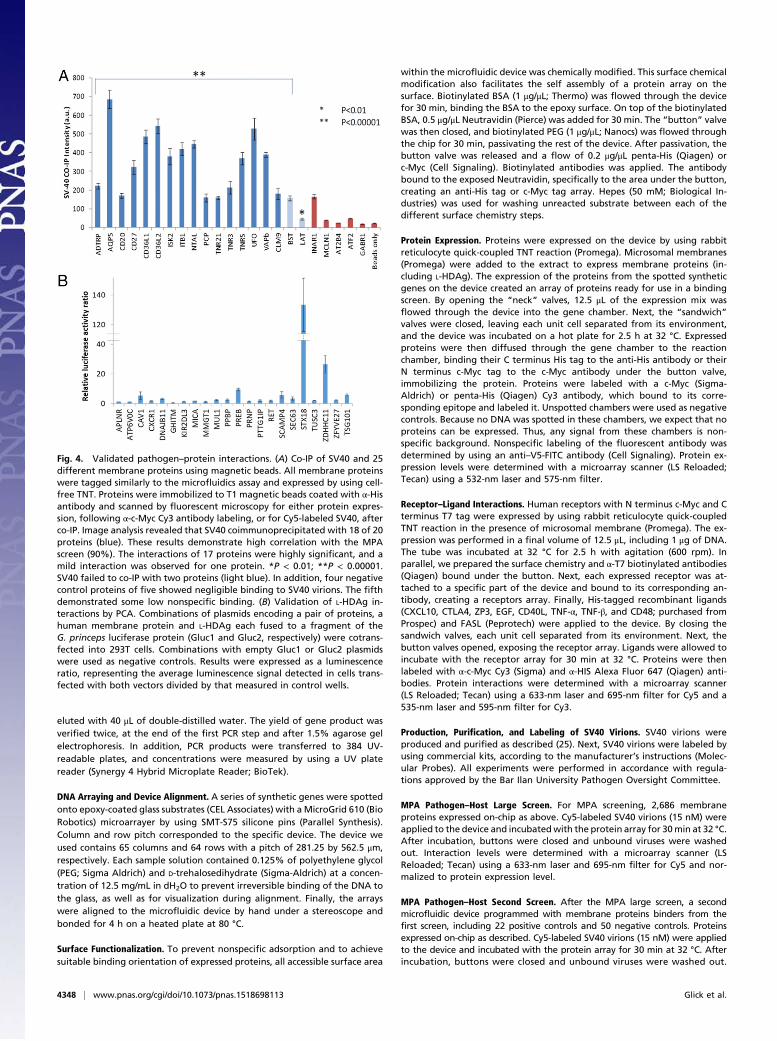
Validation of Proteomic Results. SV40 protein interactions werevalidated by co-IP, and we independently validated 25 interac-tions including negative controls. These interactions includedrepresentative proteins from different biological processes, someof which are observed in the STRING diagram (Fig. 3). Of 20interactions detected by MPA, 18 were verified by co-IP (Fig. 4Aand SI Appendix, Fig. S14), demonstrating very high correlation(90%) between the MPA-detected interactions and co-IP ex-periments. The failure of two proteins in co-IP with SV40 may beattributed to the higher sensitivity of our MPA. Interactions weremeasured relative to five negative controls, four of which binding
to SV40 was negligible. The cutoff for the positive signal wasthree SDs above the average of all five negative controls. Thebead analysis was performed by fluorescence microscopy becauseof the limited amount of membrane protein sample available,which further emphasized the advantage of using MPA.Validation of the L-HDAg proteomic results was performed
for selected proteins by PCA, because very high nonspecificbinding was observed with co-IP. Because of the role of L-HDAgin viral assembly and secretion, we selected identified host pro-teins that are involved in vesicular trafficking for further verifi-cation. We also selected several representative proteins fromother biological processes, several of which are represented inthe STRING analysis (Fig. 3). The luciferase activity of the 21L-HDAg–protein interactions we found was measured, and re-sults were expressed as normalized luminescence ratios. Seveninteractions gained a relative luciferase activity signal that was atleast three times greater than their respective negative controls(Fig. 4B). The lower success rate of L-HDAg validation comparedwith SV40 could be attributed to the differences in validationtechniques and/or that its lipid-bilayer envelope reduces the MPAspecificity. STX18, PREB, SCAMP4, and CAV-1 are proteins in-volved in vesicular trafficking. SEC63 is involved in translocationof proteins across the ER membrane, whereas DNAJB11 is in-volved in the folding of proteins entering the ER. ZDHHC11 is amember of the palmitoyltransferase family.In conclusion, we have presented the application of our hu-
man functional MPA toward discovery of pathogen–host inter-actions. MPA impact in pathogen tropism research is especiallyimportant for investigating emerging diseases. Our results dem-onstrated that the microfluidic affinity assay is a powerful toolfor identifying PPIs in general, and, more specifically, pathogen–transmembrane protein interactions. We estimate the function-ality of our MPA to be >65%, based on the number of validatedinteractions (35 of 54). The proteomics establishment of inter-twined relations between many of the identified interactingproteins provide new perspectives on viral behaviors during in-fection and pave the way for further research in various direc-tions. Furthermore, this platform may also serve for testing fordrug design, antibody specificities, or orphan receptors/ligands aswell as in a wide spectrum of other research areas that involvemembrane proteins.
Materials and MethodsMold Fabrication. The microfluidic devices were designed in a similar manneras described (14, 23, 33). Details are described in SI Appendix, SI Materialsand Methods).
Device Fabrication. The microfluidic devices were fabricated on silicone moldscasting silicone elastomer polydimethylsiloxane (SYLGARD 184, Dow Corn-ing). Details are described in SI Appendix, SI Materials and Methods.
Production of Human Synthetic Genes Library via Assembly PCR. Syntheticlinear human genes were generated by using two-step assembly PCR (SIAppendix, Fig. S1). As a template for the first PCR, we used a library of theHuman ORFome in gateway-donor GW223 plasmids (Open Biosystems). Ahigh-fidelity hot start DNA polymerase (Phusion II; FINNZYMES) was used forall PCR procedures. In the first PCR step, two epitope tags were added toeach protein, c-Myc at the N terminus and 6-His at the C terminus. The tagswere added by using the primers 5′GW223–c-Myc and 3′-GW223–His. A re-action mix with a total volume of 20 μL was prepared by using 0.8 units ofDNA polymerase for each reaction. The PCR assay was performed in a PCR96-well plate in 25 cycles with annealing temperature of 64 °C. The exten-sion time ranged from 45 to 300 s in 72 °C depending on the ORF length. Thefirst PCR product served as a template for the second PCR. In addition, twodifferent pairs of primers were used for the second PCR step by adding the5′ UTR (T7 promoter) and 3′ UTR (T7 terminator) for each gene. The reactionmixture with a total volume of 50 μL was prepared containing 1.5 units ofDNA polymerase. The first extension primer pair, containing 85 and 95 bp,was added to the mixture in a low concentration (2.5 nM). After 10 cycles,the second primer pair (5′ final and 3′ final) was added to the PCR mixture(0.2 μM) for an additional 25 cycles, completing the PCR process. The PCRproducts were filtered in multiwell 10k filter plates (AcroPrep; PALL) and
Fig. 3. Known and predicted PPI between SV40 (A) and L-HDAg (B) iden-tified pathogen-interacting proteins using STRING. Disconnected proteins(nodes) were excluded.
Glick et al. PNAS | April 19, 2016 | vol. 113 | no. 16 | 4347
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
eluted with 40 μL of double-distilled water. The yield of gene product wasverified twice, at the end of the first PCR step and after 1.5% agarose gelelectrophoresis. In addition, PCR products were transferred to 384 UV-readable plates, and concentrations were measured by using a UV platereader (Synergy 4 Hybrid Microplate Reader; BioTek).
DNA Arraying and Device Alignment. A series of synthetic genes were spottedonto epoxy-coated glass substrates (CEL Associates) with a MicroGrid 610 (BioRobotics) microarrayer by using SMT-S75 silicone pins (Parallel Synthesis).Column and row pitch corresponded to the specific device. The device weused contains 65 columns and 64 rows with a pitch of 281.25 by 562.5 μm,respectively. Each sample solution contained 0.125% of polyethylene glycol(PEG; Sigma Aldrich) and D-trehalosedihydrate (Sigma-Aldrich) at a concen-tration of 12.5 mg/mL in dH2O to prevent irreversible binding of the DNA tothe glass, as well as for visualization during alignment. Finally, the arrayswere aligned to the microfluidic device by hand under a stereoscope andbonded for 4 h on a heated plate at 80 °C.
Surface Functionalization. To prevent nonspecific adsorption and to achievesuitable binding orientation of expressed proteins, all accessible surface area
within the microfluidic device was chemically modified. This surface chemicalmodification also facilitates the self assembly of a protein array on thesurface. Biotinylated BSA (1 μg/μL; Thermo) was flowed through the devicefor 30 min, binding the BSA to the epoxy surface. On top of the biotinylatedBSA, 0.5 μg/μL Neutravidin (Pierce) was added for 30 min. The “button” valvewas then closed, and biotinylated PEG (1 μg/μL; Nanocs) was flowed throughthe chip for 30 min, passivating the rest of the device. After passivation, thebutton valve was released and a flow of 0.2 μg/μL penta-His (Qiagen) orc-Myc (Cell Signaling). Biotinylated antibodies was applied. The antibodybound to the exposed Neutravidin, specifically to the area under the button,creating an anti-His tag or c-Myc tag array. Hepes (50 mM; Biological In-dustries) was used for washing unreacted substrate between each of thedifferent surface chemistry steps.
Protein Expression. Proteins were expressed on the device by using rabbitreticulocyte quick-coupled TNT reaction (Promega). Microsomal membranes(Promega) were added to the extract to express membrane proteins (in-cluding L-HDAg). The expression of the proteins from the spotted syntheticgenes on the device created an array of proteins ready for use in a bindingscreen. By opening the “neck” valves, 12.5 μL of the expression mix wasflowed through the device into the gene chamber. Next, the “sandwich”valves were closed, leaving each unit cell separated from its environment,and the device was incubated on a hot plate for 2.5 h at 32 °C. Expressedproteins were then diffused through the gene chamber to the reactionchamber, binding their C terminus His tag to the anti-His antibody or theirN terminus c-Myc tag to the c-Myc antibody under the button valve,immobilizing the protein. Proteins were labeled with a c-Myc (Sigma-Aldrich) or penta-His (Qiagen) Cy3 antibody, which bound to its corre-sponding epitope and labeled it. Unspotted chambers were used as negativecontrols. Because no DNA was spotted in these chambers, we expect that noproteins can be expressed. Thus, any signal from these chambers is non-specific background. Nonspecific labeling of the fluorescent antibody wasdetermined by using an anti–V5-FITC antibody (Cell Signaling). Protein ex-pression levels were determined with a microarray scanner (LS Reloaded;Tecan) using a 532-nm laser and 575-nm filter.
Receptor–Ligand Interactions. Human receptors with N terminus c-Myc and Cterminus T7 tag were expressed by using rabbit reticulocyte quick-coupledTNT reaction in the presence of microsomal membrane (Promega). The ex-pression was performed in a final volume of 12.5 μL, including 1 μg of DNA.The tube was incubated at 32 °C for 2.5 h with agitation (600 rpm). Inparallel, we prepared the surface chemistry and α-T7 biotinylated antibodies(Qiagen) bound under the button. Next, each expressed receptor was at-tached to a specific part of the device and bound to its corresponding an-tibody, creating a receptors array. Finally, His-tagged recombinant ligands(CXCL10, CTLA4, ZP3, EGF, CD40L, TNF-α, TNF-β, and CD48; purchased fromProspec) and FASL (Peprotech) were applied to the device. By closing thesandwich valves, each unit cell separated from its environment. Next, thebutton valves opened, exposing the receptor array. Ligands were allowed toincubate with the receptor array for 30 min at 32 °C. Proteins were thenlabeled with α-c-Myc Cy3 (Sigma) and α-HIS Alexa Fluor 647 (Qiagen) anti-bodies. Protein interactions were determined with a microarray scanner(LS Reloaded; Tecan) using a 633-nm laser and 695-nm filter for Cy5 and a535-nm laser and 595-nm filter for Cy3.
Production, Purification, and Labeling of SV40 Virions. SV40 virions wereproduced and purified as described (25). Next, SV40 virions were labeled byusing commercial kits, according to the manufacturer’s instructions (Molec-ular Probes). All experiments were performed in accordance with regula-tions approved by the Bar Ilan University Pathogen Oversight Committee.
MPA Pathogen–Host Large Screen. For MPA screening, 2,686 membraneproteins expressed on-chip as above. Cy5-labeled SV40 virions (15 nM) wereapplied to the device and incubatedwith the protein array for 30min at 32 °C.After incubation, buttons were closed and unbound viruses were washedout. Interaction levels were determined with a microarray scanner (LSReloaded; Tecan) using a 633-nm laser and 695-nm filter for Cy5 and nor-malized to protein expression level.
MPA Pathogen–Host Second Screen. After the MPA large screen, a secondmicrofluidic device programmed with membrane proteins binders from thefirst screen, including 22 positive controls and 50 negative controls. Proteinsexpressed on-chip as described. Cy5-labeled SV40 virions (15 nM) were appliedto the device and incubated with the protein array for 30 min at 32 °C. Afterincubation, buttons were closed and unbound viruses were washed out.
Fig. 4. Validated pathogen–protein interactions. (A) Co-IP of SV40 and 25different membrane proteins using magnetic beads. All membrane proteinswere tagged similarly to the microfluidics assay and expressed by using cell-free TNT. Proteins were immobilized to T1 magnetic beads coated with α-Hisantibody and scanned by fluorescent microscopy for either protein expres-sion, following α-c-Myc Cy3 antibody labeling, or for Cy5-labeled SV40, afterco-IP. Image analysis revealed that SV40 coimmunoprecipitated with 18 of 20proteins (blue). These results demonstrate high correlation with the MPAscreen (90%). The interactions of 17 proteins were highly significant, and amild interaction was observed for one protein. *P < 0.01; **P < 0.00001.SV40 failed to co-IP with two proteins (light blue). In addition, four negativecontrol proteins of five showed negligible binding to SV40 virions. The fifthdemonstrated some low nonspecific binding. (B) Validation of L-HDAg in-teractions by PCA. Combinations of plasmids encoding a pair of proteins, ahuman membrane protein and L-HDAg each fused to a fragment of theG. princeps luciferase protein (Gluc1 and Gluc2, respectively) were cotrans-fected into 293T cells. Combinations with empty Gluc1 or Gluc2 plasmidswere used as negative controls. Results were expressed as a luminescenceratio, representing the average luminescence signal detected in cells trans-fected with both vectors divided by that measured in control wells.
4348 | www.pnas.org/cgi/doi/10.1073/pnas.1518698113 Glick et al.

Interaction levels were determined with a microarray scanner (LS Reloaded;Tecan) using a 633-nm laser and 695-nm filter for Cy5 and normalized toprotein expression level. A cutoff of 2 and 4 SDs were used for the SV40 (SIAppendix, Fig. S13) and the L-HDAg screens, respectively.
Human “synthetic genes” were created with c-Myc (N-terminal) and sixhistidine (C-terminal) tags and expressed in vitro as described above. The ex-pression was performed in a final volume of 12.5 μL, including 1 μg of DNA andincubated at 32 °C for 2.5 h with agitation (600 rpm). Expressed proteins werethen incubated either with anti–c-Myc-Cy3 antibody (Sigma-Aldrich) or withCy5-labeled SV40 virions (15 nM). Next, the proteins were immobilized to T1magnetic beads (Invitrogen) coated by α-His–biotinylated antibody (Qiagen).The beads were washed with PBS and scanned by Nikon Eclipse fluorescentmicroscope for either protein expression or for SV40 co-IP. Images were ana-lyzed by Nikon’s NIS elements software. Single beads’ median intensities weremeasured for either protein expression or SV40 co-IP. Statistical significancewas determined by calculating P values for bead intensities of each proteincompared with bead intensities of all negative controls.
L-HDAg Interaction Validation by PCA. Two engineered Gateway vectors,pGluc1-Nter-Gateway and pGluc2-Nter-Gateway, encode for two fragments ofthe Gaussia princeps luciferase protein. A total of 21 ORFs encoding the humanproteins were picked from the Human ORFeome library (34) (Open Biosystems)and inserted as described (35, 36) into the pGluc1-Nter-Gateway plasmid, andL-HDAg was inserted into pGluc2-Nter-Gateway (22, 37). A human membraneprotein and L-HDAg fused to each plasmid, respectively, or control emptyvectors were cotransfected into 293T or Huh-7.5 cells plated in 96-well plates intriplicates. At 24 h after transfection, cells were lysed and subjected to standardluciferase reporter gene assays by using the Renilla luciferase assays system(Promega). Results were expressed as luminescence ratio, which represents theaverage luminescence signal detected in cells transfected with both Gluc1 andGluc2, divided by the average of luminescence measured in negative controlwells transfected with Gluc1 and an empty Gluc2 vector and those transfectedwith Gluc2 and an empty Gluc1 vector. A combination with TSG101, which hasbeen identified as a L-HDAg partner in a small-scale microfluidic screen (33),was used as a positive control for the assay.
Image and Data Analysis. For MPA expression experiment, images were an-alyzed with GenePix7.0 (Molecular Devices). The image (cy3 channel) wasused to determine bait (on ChIP-expressed protein) expression level. Rowsand columns without DNA array were used to assess nonspecific binding oflabeled antibodies to the surface. Each row and column was then normalizedby subtracting the nonspecific baseline signal. A signal that was 2 SD abovethe average noise was considered successful protein expression.
For interaction two images (Cy3 and Cy5/GFP channels) were analyzedwithGenePix (Molecular Devices). Rows without DNA array were used to assessnonspecific binding to the surface. Columns with no prey were used to assessnonspecific binding of labeled antibodies to the surface proteins. Each rowand column was then normalized by subtracting the nonspecific baselinesignal. The “interaction ratio” (Cy5/Cy3 or GFP/Cy3) was calculated, and thehighest ratio was normalized to 1.
For each MPA-expressed protein, the L-HDAg interaction signal was calcu-lated as the average of median signals obtained from the respective qua-druplet. The average of median signals obtained from unit cells in the arraywith no spotted DNA was used to calculate a control background signal. Ahistogram of the L-HDAg interaction signal was plotted and allowed de-termination of a cutoff at 4 SD above the array control background signal asthe threshold above which the interactions were considered positive (SI Ap-pendix, Fig. S2). Each quadruplet above that threshold was examined, andinteractions with false signal that resulted from visible aggregates on the glassslide or inside the microfluidic device or high SD within the quadruplet-resulting inconsistent L-HDAg interaction signal were discarded.
ACKNOWLEDGMENTS. This work was supported by European ResearchCouncil Starter Grant 309600 (to D.G.); Israel Science Foundation Grant715/11 (to D.G.) and Israel Science Foundation Grant 291/12 (to A.O.);American Cancer Society Grant RSG-14-11 0-0 1-MPC (to S.E.); Doris DukeCharitable Foundation Grant 2013100 (to S.E.). G.N. was supported byChild Health Research Institute, Lucile Packard Foundation for Children’sHealth, and Stanford Clinical and Translational Science Award GrantUL1 TR000093.
1. Lander ES (2011) Initial impact of the sequencing of the human genome. Nature470(7333):187–197.
2. Almén MS, Nordström KJV, Fredriksson R, Schiöth HB (2009) Mapping the humanmembrane proteome: A majority of the human membrane proteins can be classifiedaccording to function and evolutionary origin. BMC Biol 7(1):50.
3. Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there?Nat Rev Drug Discov 5(12):993–996.
4. Jäger S, et al. (2012) Global landscape of HIV-human protein complexes. Nature481(7381):365–370.
5. Brückner A, Polge C, Lentze N, Auerbach D, Schlattner U (2009) Yeast two-hybrid, apowerful tool for systems biology. Int J Mol Sci 10(6):2763–2788.
6. Koegl M, Uetz P (2007) Improving yeast two-hybrid screening systems. Brief FunctGenomics Proteomics 6(4):302–312.
7. Yu H, et al. (2008) High-quality binary protein interaction map of the yeast inter-actome network. Science 322(5898):104–110.
8. Zhu H, et al. (2001) Global analysis of protein activities using proteome chips. Science293(5537):2101–2105.
9. Sachse R, et al. (2013) Synthesis of membrane proteins in eukaryotic cell-free systems.Eng Life Sci 13(1):39–48.
10. Ramachandran N, et al. (2004) Self-assembling protein microarrays. Science 305(5680):86–90.
11. He M, et al. (2008) Printing protein arrays from DNA arrays. Nat Methods 5(2):175–177.
12. Montor WR, et al. (2009) Genome-wide study of Pseudomonas aeruginosa outermembrane protein immunogenicity using self-assembling protein microarrays. InfectImmun 77(11):4877–4886.
13. Einav S, et al. (2008) Discovery of a hepatitis C target and its pharmacological in-hibitors by microfluidic affinity analysis. Nat Biotechnol 26(9):1019–1027.
14. Glick Y, Avrahami D, Michaely E, Gerber D (2012) High-throughput protein expressiongenerator using a microfluidic platform. J Vis Exp 23(66):e3849.
15. Zheng C, et al. (2012) High-throughput immunoassay through in-channel microfluidicpatterning. Lab Chip 12(14):2487–2490.
16. Pelham HR, Jackson RJ (1976) An efficient mRNA-dependent translation system fromreticulocyte lysates. Eur J Biochem 67(1):247–256.
17. Carr PA, et al. (2012) Enhanced multiplex genome engineering through co-operativeoligonucleotide co-selection. Nucleic Acids Res 40(17):e132.
18. Goshima N, et al. (2008) Human protein factory for converting the transcriptome intoan in vitro-expressed proteome. Nat Methods 5(12):1011–1017.
19. Gazdar AF, Butel JS, Carbone M (2002) SV40 and human tumours: Myth, associationor causality? Nat Rev Cancer 2(12):957–964.
20. Ewers H, et al. (2010) GM1 structure determines SV40-induced membrane in-vagination and infection. Nat Cell Biol 12(1):11–18, 1–12.
21. Chang FL, Chen PJ, Tu SJ, Wang CJ, Chen DS (1991) The large form of hepatitis deltaantigen is crucial for assembly of hepatitis delta virus. Proc Natl Acad Sci USA 88(19):8490–8494.
22. Glenn JS, Watson JA, Havel CM, White JM (1992) Identification of a prenylation site indelta virus large antigen. Science 256(5061):1331–1333.
23. Gerber D, Maerkl SJ, Quake SR (2009) An in vitro microfluidic approach to generatingprotein-interaction networks. Nat Methods 6(1):71–74.
24. Maerkl SJ, Quake SR (2007) A systems approach to measuring the binding energylandscapes of transcription factors. Science 315(5809):233–237.
25. Meier M, Sit RV, Quake SR (2013) Proteome-wide protein interaction measurementsof bacterial proteins of unknown function. Proc Natl Acad Sci USA 110(2):477–482.
26. Kubick S, Gerrits M, Merk H, Stiege W, Erdmann VA (2009) In vitro synthesis ofposttranslationally modified membrane proteins. Curr Topics Membr 63:25–49.
27. Fenz SF, Sachse R, Schmidt T, Kubick S (2014) Cell-free synthesis of membrane pro-teins: tailored cell models out of microsomes. Biochim Biophys Acta 1838(5):1382–1388.
28. Drayman N, et al. (2013) Pathogens use structural mimicry of native host ligands as amechanism for host receptor engagement. Cell Host Microbe 14(1):63–73.
29. Noach-Hirsh M, et al. (2015) Integrated microfluidics for protein modification dis-covery. Mol Cell Proteomics 14(10):2824–2832.
30. Campanero-Rhodes MA, et al. (2007) N-glycolyl GM1 ganglioside as a receptor forsimian virus 40. J Virol 81(23):12846–12858.
31. Tsai B, et al. (2003) Gangliosides are receptors for murine polyoma virus and SV40.EMBO J 22(17):4346–4355.
32. Walczak CP, Ravindran MS, Inoue T, Tsai B (2014) A cytosolic chaperone complexeswith dynamic membrane J-proteins and mobilizes a nonenveloped virus out of theendoplasmic reticulum. PLoS Pathog 10(3):e1004007–e1004007.
33. Ben-Ari Y, et al. (2013) Microfluidic large scale integration of viral-host interactionanalysis. Lab Chip 13(12):2202–2209.
34. Rual JF, et al. (2004) Human ORFeome version 1.1: a platform for reverse proteomics.Genome Res 14(10B):2128–2135.
35. Cassonnet P, et al. (2011) Benchmarking a luciferase complementation assay for de-tecting protein complexes. Nat Methods 8(12):990–992.
36. Neveu G, et al. (2012) Identification and targeting of an interaction between a ty-rosine motif within hepatitis C virus core protein and AP2M1 essential for viral as-sembly. PLoS Pathog 8(8):e1002845.
37. Glenn JS, Taylor JM, White JM (1990) In vitro-synthesized hepatitis delta virus RNAinitiates genome replication in cultured cells. J Virol 64(6):3104–3107.
Glick et al. PNAS | April 19, 2016 | vol. 113 | no. 16 | 4349
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
















![Welcome [med.stanford.edu]med.stanford.edu/.../aboutus/2017-MIPS-brochure.pdf · via sound (ultrasound, photoacoustic), magnetism (MRI or magnetic resonance imaging, MPI or magnetic](https://img.pdfslide.us/doc/110x75/5f0c64747e708231d4352ce6/welcome-med-med-via-sound-ultrasound-photoacoustic-magnetism-mri-or-magnetic.jpg)












