Embed Size (px)
Citation preview

Donaldson et al. Progress in Biomaterials 2013, 2:3http://www.progressbiomaterials.com/content/2/1/3
ORIGINAL RESEARCH Open Access
An integrated experimental and modelingapproach to propose biotinylated PLGAmicroparticles as versatile targeting vehicles fordrug deliveryOlivia Donaldson, Zuyi Jacky Huang and Noelle Comolli*
Abstract
Polymeric microparticles with covalently attached biotin are proposed as versatile targeting vehicles for drugdelivery. The proposed microparticles made of 85/15 poly (lactic-co-glycolic acid) (PLGA) will have biotin availableon the outside of the particle for the further attachment with an avidin group. Taking advantage of biotin’s highaffinity for avidin, and avidin’s well-known chemistry, the particle has the potential to be easily coated with a varietyof targeting moieties. This paper focuses on the design and resulting effect of adding biotin to PLGA microparticlesusing an integrated experimental and modeling approach. A fluorescent-tagged avidin (488-streptavidin) was usedto confirm the presence and bioavailability of biotin on the outside of the particles. For the purpose of this study,bovine serum albumin (BSA) was used as a model therapeutic drug. Microparticles were created using two differenttypes of polyvinyl alcohol 88 and 98 mol% hydrolyzed, which were then analyzed for their size, morphology, andencapsulation capacity of BSA. Release studies performed in vitro confirmed the slow release of the BSA over a28-day period. Based on these release profiles, a release kinetics model was used to further quantify the effect ofbiotinylation of PLGA microparticles on their release characteristics by quantitatively extracting the effective drugdiffusivity and drug desorption rate from the release profiles. It was found that the biotinylation of the PLGAmicroparticles slowed down both the drug desorption and drug diffusion process, which confirmed thatbiotinylated PLGA microparticles can be used for controlled drug release. The presented technology, as well as theproposed integrated experimental and modeling approach, forms a solid foundation for future studies using acell-specific ligand that can be attached to avidin and incorporated onto the microparticles for targeted delivery.
BackgroundPolymeric microparticles have been widely researchedfor their ability to serve as controlled drug deliveryvehicles Brannon-Peppas (1995; Cleland 1997; Shive andAnderson 1997). The main goals of these vehicles are toprovide improved drug disposition Putney (1998), pro-tection from metabolic degradation Dziubla et al. (2005),and increased circulation time Putney (1998). The newgoal in these designs, however, is to take them a stepfurther and incorporate a method for targeting specificcells Brannon-Peppas and Blanchette (2004; Fung andSaltzman 1997). It is well known that the side effects of
* Correspondence: [email protected] University, 800 East Lancaster Avenue, Villanova, PA 19085, USA
© 2013 Donaldson et al.; licensee Springer. ThiAttribution License (http://creativecommons.orin any medium, provided the original work is p
drugs stem from drug interactions in non-targeted cells,such as severe anemia experienced by cancer patientsBalkwill (2004; Pegram et al. 1997) and those on antiviralmedications (such as HIV and HCV treatments) Sayceet al. (2010). A method to target these cells directlywould not only increase the potency of the drugs, butalso drastically improve the quality of life for thepatients on these treatments.While biodegradable polymeric microparticles have
been investigated for the controlled release of anticancertherapeutics Fung and Saltzman (1997; Datta et al. 2006;Folger et al. 2006), and to a limited extent, for antiviraldrugs Datta et al. (2006), the challenge of effectivetargeting still remains. There are few markers that arespecific only to tumor cells, but rather most are simply
s is an Open Access article distributed under the terms of the Creative Commonsg/licenses/by/2.0), which permits unrestricted use, distribution, and reproductionroperly cited.

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 2 of 10http://www.progressbiomaterials.com/content/2/1/3
upregulated and therefore more prevalent Balkwill(2004; Pegram et al. 1997). The challenge of identifyinga sole marker to a tumor cell or virus is one that biologistand biochemist are still researching. With an evolving fieldof possible targets and ligands, the challenge for the engi-neers then is to create a robust mechanism for incorpor-ation of these new ligands to a polymeric delivery vehicle.Polymers most commonly used for microparticle drug
release include poly(lactic acid) (PLA), poly(glycolic acid)(PGA) and their copolymer poly(lactic-co-glycolic acid)(PLGA) Anderson and Shive (1997; Cao and Shoichet1998; Panyam et al. 2003). In order to obtain the desiredrelease of the drug for the specific delivery, the amount ofglycolic acid can be increased to increase the degradationrate, and therefore speed the release time. The PLGA usedwas an 85/15 mixture of lactic to glycolic acid. Thebiodegradation of PLGA occurs through a homogenoushydrolytic chain cleavage mechanism, in which both thesurface and the bulk polymer degrade at similar ratesAnderson and Shive (1997). The breakdown of PLGA ispurely through hydrolysis and does not need the assistanceof an enzyme Muthu (2009).In order to target specific cell lines, a robust targeting
strategy is proposed. Taking advantage of the affinity ofbiotin with avidin, a strong non-covalent bond can easilybe created by adding biotin to the surface of the PLGA.Biotin strongly binds to avidin and streptavidin via acombination of van der Waals and hydrophobic interac-tions. In perfect conditions, a single molecule of avidinwould bind to four molecules of biotin. This high affinitymakes it possible to have site-specific microparticles thathave predetermined antibodies attached to the avidinDatta et al. (2006; Moro et al. 1997). This platformwould allow for various avidin-antibody complexes to beconnected to the biotin microparticle in order to make amulti-faceted drug delivery system. Since this takesadvantage of the same mechanism many biochemicalassays use, the chemistry of avidin attachment to anantibody, or antibody fragment, is already well knownMoro et al. (1997; Diamandis and Christopoulos 1991;Kocbek et al. 2007). This platform allows both targeteddelivery (via the PEG-biotin-avidin), as well as controlledrelease, and biochemical protection of the drug duringthe delivery via the PLGA.Previous research has shown the potential in using
polymeric microparticles with a similar linkage but usingthe reverse order (avidin linked to the polymer) Parket al. (2011). The proposed method is used as a simplermethod for attaching the conjugate covalently to thepolymer while controlling the length of the ‘tethering’arm spacing the conjugate and the polymer. Theproposed tethering arm in this case will be a short chainpolyethylene glycol (PEG) that can be increased ordecreased in length as needed. The attachment of
biotin-PEG to a nanoparticle of PLGA was previouslydone using a more complex chemistry by Weiss et al.,for the proposed use of rapid fluorescent tagging of thenanoparticles Weiss et al. (2007). Although these particleswere evaluated for their ability to attach to biotin, therelease of drug was not investigated.In order to better understand the effect that biotin has
on the polymeric microparticle, along with the usualin vitro characterization (drug encapsulation, morphology,release rates), release kinetics will further be modeledfrom experimental data to extract important quantitativeinformation that is essential for the comparative study ofthe proposed PLGA microparticles. Specifically, drugrelease from polymeric microparticles undergoes twomain phases: (1) the induction (or burst) phase in whichan initial burst of protein release is observed due to thedesorption of proteins from the surface of mesoporeswithin microparticles and the outer surface of microparti-cles; (2) the diffusion phase in which the macromoleculardrug contained in the occlusions of microparticles diffusethrough the pores that are formed during the hydration,degradation and erosion of microparticles. Accordingly,the drug desorption rate determines the dynamics of theinitial drug burst, while the diffusion rate determines thesubsequent drug release. In this work, a theoretical modelof macromolecular drug release presented by Batyckyet al. (1997), as shown in Equation (1), is used to quantifydrug desorption rate and effective drug diffusivity fromdrug release profiles. Thus, the effect of polyvinyl alcohol(PVA) surfactants as well as the attachment of biotin tothe polymeric microparticles on the drug release processcan be quantified.The mass fraction of released drug, frelease, is determined
by the following equation:
frelease ¼ φburstd 1� e�kdt� �
þ 1� φburstd
� �1� 6
π2
X1j¼1
�e�j2π2―D�d t�tdð Þ=r20
j2
!
ð1Þ
where φburstd is the mass fraction of drug involved in theburst phase, kd is the drug desorption rate constant, �D�
d isthe effective drug diffusivity, td is the drug induction timethat allows for the coalescence of micropores and the pas-sage of the macromolecular drug out from the occlusionsthrough the coalescing micropores in microparticles, and r0is the initial microparticle radius. The first term of Equation1 represents the burst phase during which the proteinsfrom the surface of mesopores within microparticles andthe outer surface of microparticles are released and duringwhich the micropores within microparticles coalesce forthe further release of the encapsulated drug. Following theburst phase is the diffusion phase that is described by the

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 3 of 10http://www.progressbiomaterials.com/content/2/1/3
second term of Equation 1 in a Fickian-release manner.The time evolution of released mass, frelease predicted fromEquation 1, will be compared to the experimental releasedprofile. The values of the parameters that are important forcharacterizing drug released process, including φburstd , kd,and td, are then determined by fitting the model given inEquation 1 to the experimental data. Therefore, Equation 1is used as a soft-sensor in this work for quantitatively moni-toring the drug desorption rate and effective drug diffusivitythat cannot be directly determined from the release profilesby eye inspection. These parameters can be used as quanti-tative criteria for the selection of PLGA microparticles fordrug delivery. The primary goal of this paper is to evaluatethe effect of the biotinylation of the PLGA microparticleson their morphology and release characteristics.
Materials and methodsMaterialsPLGA was purchased from SurModics, located inBirmingham, AL, USA. The EZ-LinkWTFPA-PEG3-Biotin,488-streptavidin, potassium nitrate, and micro bicinchoni-nic acid (BCA) protein assay kit were all obtained fromThermo Fisher Scientific (Waltham, MA, USA). A biotinquantification kit was bought from Pierce Biotechnology(Rockford, IL, USA). The ethyl acetate, dichloromethane(DCM), and dimethyl sulfoxide (DMSO) used in thepreparation of the microparticle, as well as bovine serumalbumin (BSA) and phosphate buffered solution (PBS) werepurchased from Sigma-Aldrich (St. Louis, MO, USA). Thesodium azide was purchased from Acros Organics (Geel,Belgium). The PVA was brought from Polysciences, Inc.(Warrington, PA, USA).
Biotinylation of PLGAThe PLGA and DMSO were combined in a 10:1 ratio andvortexed until the PLGA dissolved. EZ-Link TFPA-PEG3-Biotin was attached to PLGA in a 20-fold molar excess ofbiotin (10 mg/mL in DMSO). The amount of biotin wasdetermined using the following equation:
Vbiotin ¼ 1; 000� mPLGAMWbiotin
MWPLGAMbiotinCbiotinð2Þ
where mPLGA is the mass of PLGA; MWbiotin andMWPLGA are the molecular weight of biotin and PLGA,respectively; Mbiotin is the mole of excess of biotin; Cbiotin
is the concentration of biotin.The mixture was then photoactivated using UV light for
30 min. After quenching the reaction with approximately15 mL of deionized (DI) water, the solution was thencentrifuged using the Sorvall Legend RT Plus Centrifuge(Thermo Scientific) at 14,000 rpm for approximately7 h at room temperature. The samples were stored at4°C until use.
Quantification of biotinA biotin quantification kit was used to compare theabsorbance of a sample to a positive control, biotinylatedhorseradish peroxidase (HRP). To begin the analysis, aPLGA-biotin pellet was dissolved in ethyl acetate. 40-hydrocyazobenzene-2-carboxylic acid (HABA)-avidinwas then added to both the control and sample. Theplate was shaken for approximately 60 s, and thedisplaced HABA was measured using a BioTek ELx800UV/Vis microplate reader at a wavelength of 490 nm.The ratio of biotin to PLGA was determined using therecorded absorbance values. All results are presented as theaverage of triplicate samples with the standard deviation.
PLGA microparticle synthesisThe water-in-oil-in-water method is a common emulsiontechnique that was performed at room temperature.Briefly, 150 μL of phosphate buffered saline (pH 7.4) withvarying amounts of dissolved protein was added to 2 mL ofthe oil phase (10 mg/mL PLGA in ethyl acetate), and theemulsion was created by adding energy to the solution byhomogenizing for 60 s. The primary emulsion wasstabilized with the addition of bovine serum albumin(1 mg/mL) to the internal aqueous phase. The primaryemulsion was then quickly added to 300 mL of an externalaqueous phase (5 wt.% PVA). The emulsion was stabilizedthrough stirring at 500 rpm and the presence of the PVA(either 88 or 98 mol% hydrolyzed). The microparticleshardened while stirring overnight and the ethyl alcoholwas evaporated. The microparticles were collected viacentrifugation at 14,000 rpm for 90 min. Afterwards, thesupernatant was removed and the microparticles wereresuspended in DI water. The microparticles were washedtwo more times and centrifuged at 14,000 rpm at respect-ively 90 and 30 min. The microparticles were allowed todry and either used immediately or kept at 4°C until use.
Fluorescent imagingPLGA-biotin and non-biotinylated PLGA particles wereanalyzed under fluorescent imaging. Approximately 5 mgof particles were suspended in 1 mL of DI water in anamber microcentrifuge tube. A 5 μL of 488-streptavidinwas added to the solution and it was stored in a dark loca-tion for at least 90 min. The tube was then centrifuged at13,300 rpm for 15 min at room temperature. Microparticleswere then washed, removing the supernatant, and the pelletwas resuspended in a small amount of DI water. Thesample was centrifuged at 13,300 rpm for 15 min and thesupernatant was removed. The sample was then placed ona glass slide with a cover slip and viewed on a Leica DM2000 microscope (Leica Microsystems, USA). Imageswere captured and viewed using a Q imaging Retiga-SRVcamera and QCapture Pro 6.0 software (Q Imaging, Surrey,British Columbia, Canada).

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 4 of 10http://www.progressbiomaterials.com/content/2/1/3
Encapsulation efficiencyProtein encapsulation was evaluated by dissolving aknown weight of particles (5 mg) in 2 mL DCM. Thedissolved particles were mixed with 3 mL of DI waterand the solvent-water mixture was shaken overnight at200 rpm. This provided sufficient time for the protein tobe extracted into the water phase. A sample was takenfrom the water phase and the concentration was foundusing a BCA protein assay (used per manufacturer’sinstructions). The BCA assay is a colorimetric assaybased on bicinchoninic acid and measures the total proteincontent in a sample. Negative controls of the particle madewith no protein present at all were also performed toensure that the presence of the degraded lactic acid did notaffect the concentration readings.
In vitro release of model drugsProtein release from the microparticles was evaluatedin vitro using a known mass of dried microparticles in30 mL of PBS (with 0.01% NaN3 to prevent bacterialgrowth). All studies were set so sink conditions wouldbe maintained, specifically, that at no point would themaximum released concentration of protein be greaterthan 10% the saturation limit for that protein in PBS.Samples of the release medium were removed at designatedtimes using a sample probe with an inline 0.45-μm filter toprevent removal of the microparticles during sampling.Equal volumes of fresh PBS were back-flushed through thefilter to ensure a constant volume throughout the study aswell as to ensure that any microparticles trapped in thesample probe would be flushed back into the samplecontainer. Samples were kept at -20°C until analysis. Con-centrations were found using the BCA protein assay at 490nm, per manufacturer’s instructions.
Particle morphologyUsing completely dried microparticles, the size and surfacemorphology of the particles were observed using the scan-ning electron microscope. A fraction of the microparticlewas taken and placed on a small metal stage, fitted withdouble-sided carbon tape. The sample was then coated. Thesample was placed in a Hitachi S-570 scanning electronmicroscope (Hitachi America Ltd., Brisbane, CA, USA) forobservation under vacuum. The size and distribution of theparticles was determined. Using dissolved microparticles,the size of the particle was observed using the Hitachi 7600transmission electron microscope. Six microliters of thesample was placed on a carbon graph and allowed to dry.The dried sample was sputter coated with a conductivemetal and placed in the microscope for examination.
Particle sizeUsing completely dried microparticles, the size andpolydispersity of the particles were observed using the
particle size analyzer. A portion of the microparticle wassuspended in 4.5 mL of 10 mM of KNO3. The suspensionwas approximately 10 mg to 4 mL. The sample was thenplaced in a Brookhaven 90 plus particle size analyzer(Brookhaven Instruments Corporation, Holtsville, NY,USA) for examination. The KNO3 was used instead ofPBS since the salt solution was necessary to allow for thelaser scattering that is needed.
Quantifying drug desorption rate constant and effectivedrug diffusivity from drug release profilesWhile the BSA release percentage can be determinedfrom the release profile directly, the release kineticsparameters such as the effective diffusion rate and thedrug desorption cannot be directly determined by eyeinspection from experimental data. Thus, release kineticsparameters such as drug desorption rate (kd) and effective
drug diffusivity―D�
d in Equation 1 were estimated by fittingfrelease predicted by Equation 1 to experimental drug re-lease profiles (‘In vitro release of model drugs’ section) viathe following procedure:
1. φburstd and td , which correspond to the mass andtime for drug release in the burst phase respectively,were determined from the inflection point of drugrelease profiles, as the inflection point indicates theswitch of drug release from the burst phase to thediffusion phase.
2. kd and �D�d were determined via a nonlinear least
squares approach, which can be represented byEquation 3. This computation was performed byminimizing an objective function consisting of thesum of the squares over N measurements of thedifferences between the experimental data frelease andthe model-predicted output frelease. MATLAB(Mathworks Inc., Natick, MA, USA) routine fminconwas used for solving this parameter estimationproblem:
Minkd ;�D
�d
XNi¼1
f̂ release ið Þ � frelease ið Þh i2
subject to
frelease ¼ φburstd 1� e�kdt� �þ 1� φburstd
� �1� 6
π2
X1j¼1
�e�j2π2 �D�d t�tdð Þ=r20j2
!:
ð3Þ
Results and discussionThe first step in microparticle production was tosynthesize biotinylated PLGA. To do this, TFPA-PEG3-

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 5 of 10http://www.progressbiomaterials.com/content/2/1/3
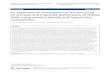
Biotin was attached using a UV-initiated reaction. Inorder to confirm the attachment of the biotin on thepolymer, a biotin quantification kit was used. The weightof biotin in relation to polymer was calculated and foundto be approximately 0.87 ± 0.37. Once the polymer wasconfirmed to have biotin, microparticles were madeusing a water-in-oil-in-water (W/O/W) method. Toensure that the biotin was available to avidin on theoutside of the polymer microparticle, a fluorescent assaywas performed. Microparticles were combined withstreptavidin that was tagged with a green fluorophore(Alexa 488; Life Technologies Corporation, Carlsbad,CA, USA). After incubation, the microparticles werecollected, washed, and immediately viewed using a LeicaDM 2000 microscope. Presence of green fluorophore(white in Figure 1, Alexa 488 + biotin-PLGA microparti-cles) around the microparticle indicates that the biotinnot only attached to the perimeter, but also was still bio-logically available for the streptavidin post-microparticleprocessing. A negative control (Figure 1, Alexa 488 +PLGA microparticles), confirms that the streptavidin isnot merely sticking to the polymer surface, but rather tothe biotin available on the surface.Once the microparticle synthesis viability was confirmed,
the size and polydispersity were confirmed using a Broo-khaven 90 plus particle size analyzer (via dynamic lightscattering). Microparticles were made with and withoutbiotin, as well as with and without the model drug (BSA).Microparticles were also made using two different types ofPVA (the surfactant for the secondary emulsion step, 88and 98 mol% hydrolyzed) in an attempt to optimize themicroparticle synthesis. Analysis of the samples (Table 1)found that the addition of biotin to the PLGA causes an in-crease in particle size for both 88 and 98 mol% hydrolyzedPVA. This trend is expected since the presence of the biotinmakes the polymer larger as well as more hydrophobic.
Figure 1 Fluorescent imaging confirms presence of biotin on the outthe surface of the PLGA microparticles was confirmed by incubating biotinseveral times prior to visualization under the fluorescent microscope. Left imfluorophore (white) indicates the biotin was present and capable of attach
The biontinylated PLGA particle containing no BSAcreated a smaller distribution in the molecular masses ofthe samples. This could be a result of the biotin itself help-ing to stabilize the emulsion. Interestingly, the additionof BSA without biotin present increased the particle size;however, the addition of BSA into the biotinylated PLGAparticles actually showed a decrease in size. This decreasein size was unexpected but may indicate a specific reac-tion of the BSA and biotin during the W/O/W emulsionand may not be critical when using the actual cancer thera-peutic drug. The particles were created using both 88 and98 mol% PVA, and it was found that particles generatedfrom the 98 mol% PVA are larger than the particles madefrom the 88 mol% PVA. The 98 mol% PVA is roughly tentimes larger than the 88 mol% PVA particles. The polydis-persity of the 98 mol% PVA microparticles was larger thanthe 88 mol% PVA microparticles as well, indicating that theincrease in hydrophilicity in the 98 mol% PVA did not pro-vide an increased stabilizing effect on the emulsion as com-pared to the 88 mol% PVA.Microparticles were synthesized using either 88 or 98
mol% PVA as a surfactant as well as with and without thebiotin attached to the PLGA. The resulting microparticlesize and polydispersity were determined via dynamic lightscattering. Results are presented as the average of n = 4samples ± standard deviation.In order to evaluate the morphology of the microparti-
cles, both a Hitachi S-4800 scanning electron microscope(SEM) and a Hitachi 7600 transmission electron micro-scope (TEM) were used to view samples of the biotinylatedand plain PLGA microparticles. SEM and TEM images ofboth 88 and 98 mol% PVA methods are shown in Figure 2.The TEM and SEM images confirm that the addition of thebiotin to the PLGA does not change the spherical morph-ology of the microparticles. The microparticles with andwithout biotin do not have any morphological differences
side of microparticles. The presence and bioavailability of biotin onylated microparticles with streptavidin-488. The particles were washedage, Alexa 488 + biotin-PLGA microparticles. The presence of the 488
ing the steptavidin. Right image, Alexa 488 + PLGA microparticles.

Table 1 Average size and polydispersity of differentmicroparticle formulations
Type(mol% PVA)
Average size(μm)
Average (PDI)polydispersity index
PLG no BSA (88) 7.81 ± 13.52 0.48 ± 0.55
PLGA BSA (88) 8.06 ± 23.12 0.36 ± 0.037
Biotin BSA (88) 1.83 ± 4.23 0.39 ± 0.18
Biotin no BSA (88) 21.24 ± 15.02 0.021 ± 0.015
PLGA No BSA (98) 21.99 ± 14.91 0.67 ± 0.39
PLGA BSA (98) 30.72 ± 49.99 0.50 ± 0.36
Biotin BSA (98) 16.47 ± 11.85 0.80 ± 0.36
Biotin No BSA (98) 37.97 ± 97.51 0.39 ± 0.52
Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 6 of 10http://www.progressbiomaterials.com/content/2/1/3
visible. The particles also have no visible morphologicalchanges (with the exception of size) when changing from88 to 98 mol% PVA during processing. The ability of themicroparticle to keep its spherical shape is an indicationthat the microparticle morphology is not changed by theaddition of the biotin. The agglomeration seen in some ofthe SEM is believed to be a result of the drying process forimaging and is not expected when the microparticles are insolution.
Figure 2 Microparticle size and morphology. Biotinylated microparticlesresulting microparticles were imaged using a SEM (top images) as well as aand size ranges determined via dynamic light scattering.
Once the particle size and morphology was characte-rized, the mass percentage of BSA encapsulated as wellas the encapsulation efficiency of the model drug wasdetermined (Table 2). It was found that for particlesmade from 88 mol% PVA, the PLGA particles had aslightly higher average encapsulation than the bioti-nylated particles. On the other hand, when the PVA waschanged to 98 mol%, the average encapsulation per-centage for the biotinylated particle was higher thanthe PLGA particle. The 88 mol% PVA, however, hadsimilar encapsulation of BSA between the plain PLGAand biotinylated PLGA microparticles. Although the 98mol% hydrolyzed PVA may be a better surfactant, thereis no clear trend apparent for the effect of biotin on theencapsulation of a drug; further optimization should bedone using the actual therapeutic. It was determinedthat the 98 mol% PVA more effectively stabilized themicroparticle during hardening, allowing for the higherencapsulation (shown in Table 2) of BSA. If the particlecan be successfully stabilized during the hardening step,it should not swell and allow water in, or drug out.If higher encapsulation corresponds to the larger size, sincemore internal aqueous phase is retained, the microparticles
were synthesized using either 88 or 98 mol% PVA as a surfactant. TheTEM (bottom images). Images confirmed the spherical morphology

Table 2 BSA loading for different microparticleformulations
Type(mol% PVA)
Average massencapsulationpercentage
Averageencapsulationefficiency
PLG (88) 4.12 3.21
Biotin (88) 3.30 1.78
PLGA (98) 3.02 4.35
Biotin (98) 12.26 15.57
Figure 3 BSA release from microparticles synthesized with 88mol% PVA. BSA release from microparticles synthesized using 88mol% PVA (with and without biotin) in vitro over a 28-day period.Both the biotinylated and non-biotinylated microparticles controlledthe release of the BSA with a minimal initial burst. Samples werequantified using the micro BCA and are presented as the average ofn = 4. Error bars indicate the standard deviation. No difference wasfound between the two sets of microparticles release profiles.
Figure 4 BSA release from microparticles synthesized with 98mol% PVA. BSA release from microparticles synthesized using 98mol% PVA (with and without biotin) in vitro over a 28-day period.Both the biotinylated and non-biotinylated microparticles controlledthe release of the BSA with a minimal initial burst, with no realdifference in their release profiles. Samples were quantified usingthe micro BCA and are presented as the average of n = 4. Error barsindicate the standard deviation.
Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 7 of 10http://www.progressbiomaterials.com/content/2/1/3
should be larger. The movement of drug and water in andout during this phase is a common problem during har-dening, since the osmotic pressure will readily drive mole-cules through the oil phase (polymer in solvent) until itfully hardens. This flux creates the largest challenge indesigning microparticle with a high drug loading.Microparticles were synthesized using either 88 or 98
mol% PVA as a surfactant as well as with and without thebiotin attached to the PLGA. The resulting microparticle’sloading of BSA was quantified dissolving the microparticleand extracting the BSA which was quantified using amicro BCA assay. The BSA loading is presented as boththe percentage of the particle mass that is BSA as well asthe percentage of the initial BSA that was actually loadedinto the microparticle.Once the amount of drug encapsulated in each type of
microparticle was determined, the rate of release of themodel drug could be evaluated. In vitro release studieswere performed under sink condition in PBS at 37°C forall four types of microparticles. Using a micro BCAassay, the mass of BSA released was determined. Thecumulative percentage of BSA released over time wascalculated and is shown in Figures 3 and 4. It wasdetermined that the release of BSA from the biotinylatedand PLGA particle, made from 88 mol% PVA, followedthe same trend (Figure 3) over a 28-day period. Thisindicates that the presence of biotin on the surface ofthe microparticle does not alter the release characteris-tics of the microparticles. The same result was found formicroparticles made using 98 mol% PVA (Figure 4). Forthe 28-day period, approximately 80% of the model drugis released for both biotinylated and non-biotinylatedmicroparticles. Comparing Figures 3 and 4, there is noapparent effect of the change in surfactant on release, asexpected. The change in the surfactant should mainlychange the microparticles stability during formation,leading to potential changes in morphology and encap-sulation capacity.In order to further quantify the effect of the biotinylation
of the PLGA microparticles on the drug release characteris-tics, the effective drug diffusivity and drug desorption ratethat directly characterize the drug release process arefurther determined from the release profiles. Specifically,
the model given by Equation 1 is fitted to the releaseprofiles presented in Figures 3 and 4 via the approachshown in ‘Quantifying drug desorption rate constant andeffective drug diffusivity from drug release profiles’ section.
The values of the parameters―D�
d , kd, φburstd and td in

Figure 5 Comparison of experimental data to BSA release profiles. Comparison of experimental data to BSA release profiles, predicted bythe model shown in Equation 1 with estimated parameters �D�
d , kd, ϕdburst, and td. The RMSD values for microparticles PLGA-BSA (88 mol% PVA),
biotin-BSA (88 mol% PVA), PLGA-BSA (98 mol% PVA), and biotin-BSA (98 mol% PVA) are 2.68%, 4.05%, 2.37%, and 4.55%, respectively. Error barsindicate the standard deviation.
Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 8 of 10http://www.progressbiomaterials.com/content/2/1/3
Equation 1 are estimated. The fitting result is presented inFigure 5, which shows that the drug release profilespredicted by the estimated model fit the experimental datawell for all types of microparticles under investigation. Inparticular, the predicted drug release profiles pass throughmost error bars shown in the data. The root-mean-squaredeviation of prediction (RMSD) for microparticles PLGA-BSA (88 mol% PVA), biotin-BSA (88 mol% PVA), PLGA-BSA (98 mol% PVA), and biotin-BSA (98 mol% PVA) iscalculated as 2.68%, 4.05%, 2.37%, and 4.55%, respectively.As shown in Figure 5, the model predicts that more BSA isreleased by the plain PLGA microparticles than the biotiny-lated PLGA microparticles, and that PLGA microparticlesmade of a lower percent of PVA release slightly more BSA.This is in a good agreement with the trends shown in therelease profiles. A conclusion drawn from these observa-tions is that the deviation of the model prediction from theexperimental data is within a reasonably small scale, andthat values of parameters �D�
d , kd, φburstd , and td estimated
Table 3 Parameter estimation results
Type(mol% PVA)
�D�d
(cm2s�1)ϕdburst Kd
(day−1)td(day)
88 PLGA BSa 1.5 × 10−13 0.03 7.60 0.25
Biotin BSA 9.87 × 10−14 0.16 8.10 0.25
98 PLGA BSa 1.38 × 10−13 0.28 8.63 0.25
Biotin BSA 8.37 × 10−14 0.22 8.50 0.25
Values of the mass fraction of drug involved in the burst phase (φburstd ), drugdesorption rate constant (kd), effective drug diffusivity ( �D�
d ), and druginduction time (td) are determined from the drug release profiles.
from release profiles properly characterize the drug releasedynamics of all microparticles under investigation.Table 3 shows the corresponding estimated values of
parameters �D�d , kd, φburstd , and td. It can be seen from
Table 3 that �D�d decreases by a factor of 0.65 when
microparticles made of 88 mol% PVA were biotinylated.A similar decreasing ratio (i.e., 0.61) is observed in thevalue of �D�
d for biotinylated microparticles that are madeof 98 mol% PVA. This means that the attachment ofbiotin to microparticles slightly slows down the drugdiffusion and thus the drug release process. This isexpected since the presence of the biotin on the outersurface acts as another layer of diffusion barrier. Thevalue of �D�
d decreases by a factor of approximately 0.90when 98 mol% PVA instead of 88 mol% PVA is used tomake microparticles. This implies that increasing themole percentage of PVA can slightly slow down drug re-lease but to a limited degree. The decrease in drugrelease at higher mole percentage of PVA may be due toits tendency to remain on the surface of the particleseven after hardening. In addition to influencing drugdiffusion process, the attachment of biotin and the molepercentage of PVA also affect drug release in the burstphase, in which proteins are desorbed from the outer sur-face of microparticles. Table 3 shows that the attachment ofbiotin reduces the value of φd
burst, the mass fraction of drugthat is desorbed during the burst stage. The authors believethis is due to the fact that the attachment of biotin reducesthe amount of BSA trapped on the surface during forma-tion due to steric hindrance. Since the attachment of biotincannot change the desorption pattern of proteins from the

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 9 of 10http://www.progressbiomaterials.com/content/2/1/3
outer surface, it does not affect drug desorption rate con-stant (kd) and drug induction time (td). It can also be seenfrom Table 3 that the mole percentage of PVA has a minoreffect on protein desorption during the burst phase. Thisresult, along with the result that the increasing mol% ofPVA did not have a drastic effect on the drug loading and/or particle morphology, indicates that the higher mole per-centage of PVA is not providing a more stable emulsionduring the solvent hardening stage of microparticle syn-thesis. This also indicates that the increase in PVA molepercentage does not improve the emulsion process, since itcannot guarantee that more of the drug will remain withinthe microparticle during encapsulation.
ConclusionsPolymeric microparticles created through a water-in-oil-in-water double emulsion effectively demonstrated acontrolled release of a model drug. The presence ofbiotin on the outside of the polymeric microparticleswas confirmed using fluorescent imaging. The micro-particle synthesis was optimized through the use of PVAconsisting of various mole percentages. The effect of thedifferent PVA surfactant on microparticle synthesis deter-mined that 88 and 98 mol% PVA created similar particlesthat only differed in size and slightly (approximately 10%)in encapsulation. It was determined that increasing themole percentage of PVA created a more stable emulsionduring the hardening phase, allowing for higher encapsu-lation efficiencies of the model drug (BSA). The releasestudies found that the attachment of biotin to the PLGAmicroparticle had only a minor effect on the release trendduring the 28-day period. The microparticles still exhibiteda controlled release over the 28 days with minimal burstand, therefore, are still believed to be effective as carriersfor therapeutic drugs. A release kinetics model was used tofurther quantify the effective drug diffusivity and drugdesorption rate, revealing that the attachment of biotin tomicroparticles slowed down both drug desorption and drugdiffusion processes, while the mole percentage of PVA onlyhas a minor effect on drug release rate.The presence of the biotin on the microparticle, overall,
did not have a negative effect on the microparticles abilityto entrap and control the release of the model drug. Thisindicates that the microparticles can be further investi-gated for their ability to target using a moiety specific tobreast cancer (or other types). This moiety will beattached to avidin and combined with the microparticleusing the biotin exposed on the microparticle’s surface.Until that specific moiety is identified, the effect of theavidin-targeting moiety on the microparticle cannot beevaluated. The authors believe that this robust linkagesystem will be valuable compared to other targetingstrategies, since the simple chemistry will allow for linkageof a variety of different moieties. Therefore, the same
technology as well as the proposed integrated experimentaland modeling approach can be used to target eithermultiple types of cancer cells, or to include multipletargeting antigens for the same cell on one microparticle.
Competing interestsThe authors declare that they have no competing interests.
Authors’ contributionsOD carried out the experimentation and drafted the manuscript. ZHcompleted the modeling and related calculations and helped draft thatsection of the paper. NC conceived of the study, and participated in itsdesign, coordination and preparation of the manuscript. All authors read andapproved the final manuscript.
AcknowledgmentsThe authors would like to thank the following students for their aid inrunning the experiments: Colleen Clark, Elizabeth Andrews, Lucille Bell, ErinWagner, Kaitlin Worden, Will Swalchik, and Sherrie Ann Martin. The authorswould also like to thank the Delaware Valley Section of the InternationalSociety of Pharmaceutical Engineers for partial funding of this research. NCand ZH gratefully acknowledge the financial support from VillanovaUniversity SRF/RSG 2012–2013.
Received: 3 July 2012 Accepted: 6 January 2013Published: 13 February 2013
ReferencesAnderson JM, Shive MS (1997) Biodegradation and biocompatability of PLA
and PLGA microspheres. Adv Drug Del Rev 28:5–24Balkwill F (2004) The significance of cancer cell expression of the chemokine
receptor CXCR4. Sem Cancer Biol 14(3):171–179Batycky RP, Hanes J, Langer R, Edwards DA (1997) a theoretical model of
erosion and macromolecular drug release from biodegradingmicrospheres. J Pharm Sci 87(12):1464–1477
Brannon-Peppas L (1995) Recent advances on the use of biodegradablemicroparticles and nanoparticles in controlled drug delivery. Int J Pharm116(1):1–9
Brannon-Peppas L, Blanchette JO (2004) Nanoparticle and targeted systemsfor cancer therapy. Adv Drug Deliv Rev 56(11):1649–1659
Cao X, Shoichet MS (1998) Biodegradation and biocompatibility of PLA andPLGA microspheres. Biomaterials 20:329–339
Cleland JL (1997) Protein delivery from biodegradable microspheres. In:Sanders L (ed) Protein delivery physical systems. Kluwer Academic,Hingham, pp 1–25
Datta S, Ray RD, Nath A, Bhattacharyya D (2006) Recognition basedseparation of HIV-Tat protein using avidin-biotin interaction in modifiedmicrofiltration membranes. J Membr Sci 280:298–310
Diamandis EP, Christopoulos TK (1991) The biotin-(strept)avidin system:principles and applications in biotechnology. Clin Chem 37(5):625–636
Dziubla TD, Karim A, Muzykantov VR (2005) Polymer nanocarriers protectingactive enzyme cargo against proteolysis. J Control Release102(2):427–439
Folger F, Noonpakdee W, Loretz B, Joojuntr S, Salvenmoser W, Thaler M,Bernkop-Schnürch A (2006) Inhibition of malarial topoisomerase II inPlasmodium falciparum by antisense nanoparticles. Int J Pharm319(1–2):139–146
Fung LK, Saltzman WM (1997) Polymeric implants for cancer chemotherapy.Adv Drug Del Rev 26(2–3):209–230
Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J (2007) Targeting cancercells using PLGA nanoparticles surface modified with monoclonalantibody.J Control Release 120(1–2):18–26
Moro M, Pelagi M, Fulci G, Paganelli G, Dellabona P, Casorati G, Siccardi AG,Corti A (1997) Tumor cell targeting with antibody-avidin complexes andbiotinylated tumor necrosis factor alpha. Cancer Res 57(10):1922–1928
Muthu M (2009) Nanoparticles based on PLGA and its co-polymer: anoverview. Asian J Pharm 3:266–273
Panyam J, Dali M, Sahoo SK, Ma W, Chakravarthi SS, Amidon GL, Levy RJ,Labhasetwar V (2003) Polymer degradation and in vitro release of a

Donaldson et al. Progress in Biomaterials 2013, 2:3 Page 10 of 10http://www.progressbiomaterials.com/content/2/1/3
model protein from poly(d, l-lactide-co-glycolide) nano- andmicroparticles.J Control Release 92(1–2):173–187
Park J, Mattessich T, Jay SM, Agawu A, Saltzman WM, Fahmy TM (2011)Enhancement of surface ligand display on PLGA nanoparticles withamphiphilic ligand conjugates. J Control Release 156(1):109–115
Pegram MD, Finn RS, Arzoo K, Beryt M, Pietras RJ, Slamon DJ (1997) Theeffect of HER-2/neu overexpression on chemotherapeutic drug sensitivityin human breast and ovarian cancer cells. Oncogene 15(5):537–547
Putney S (1998) Encapsulation of proteins for improved delivery. Curr OpinChem Biol 2:548–552
Sayce AC, Miller JL, Zitzmann N (2010) Targeting a host process as anantiviral approach against dengue virus. Trends Microbiol 18(7):323–330
Shive MS, Anderson JMS (1997) Biodegradation and biocompatibility of PLAand PLGA microspheres. Adv Drug Deliv Rev 28(1):5–24
Weiss B, Scheider M, Muys L, Taetz S, Neumann D, Schaefer UF, Lehr CM(2007) Coupling of biotin-(poly(ethylene glycol))amine to poly(d, l-lactide-co-glycolide) nanoparticles for versatile surface modification. BioconjugChem 18(4):1087–1094
doi:10.1186/2194-0517-2-3Cite this article as: Donaldson et al.: An integrated experimental andmodeling approach to propose biotinylated PLGA microparticles asversatile targeting vehicles for drug delivery. Progress in Biomaterials 20132:3.
Submit your manuscript to a journal and benefi t from:
7 Convenient online submission
7 Rigorous peer review
7 Immediate publication on acceptance
7 Open access: articles freely available online
7 High visibility within the fi eld
7 Retaining the copyright to your article
Submit your next manuscript at 7 springeropen.com