Embed Size (px)
Citation preview

QJMed, 1994; 87:385-401
Original papers
Hereditary complement factor I deficiency
T.J. VYSE, P.J. SPATH1, K.A. DAVIES, B.J. MORLEY, P. PHILIPPE2,P. ATHANASSIOU, C M . GILES and M.J. WALPORT
From the Rheumatology Unit, RPMS, Hammersmith Hospital, London, UK,^ZLB, Central Laboratory, Blood Transfusion Service, SRC, Berne, Switzerland,and2Regional Hospital Delemont, Detemont, Switzerland
Received 17 March 1994; Accepted 13 April 1994
QJM
Summary
We describe four cases (from three families) ofhereditary factor I deficiency, bringing the totalnumber of cases now reported to 23. In one familythere are two affected siblings: one has sufferedrecurrent pyogenic infections; the other is asympto-matic. In the second family, the patient had recur-rent pyogenic infections and a self-limiting vasculiticillness; in the third family, the patient suffered recur-rent pyogenic and neisserial infections. All fourpatients had markedly reduced concentrations of C3in the serum (family 1 propositus: 28%; family 1asymptomatic sibling: 15%; family 2: 3 1 % ; and
family 3: 31 % normal human serum) which was inthe form of C3b. Low lgG2 levels may occur inprimary C3 deficiency, and a reduction in lgG2
concentration to 1.14 g/l (normal: 1.30-5.90 g/l)was found in the patient from family 2. Using radio-ligand binding assays, we demonstrated increasedbinding of C3b to erythrocytes in a patient withfactor I deficiency. This C3b could not be cleavedby autologous serum but could be cleaved by normalserum or purified factor I. We review and comparethe published cases of C3, factor H and factor Ideficiency.
IntroductionComplement factor I (Fl) is a serine esterase that actsto control the amplification loop of the alternativepathway of the complement cascade. Hereditarydeficiency of Fl is a rare autosomal recessive condi-tion. In the absence of Fl, the amplification loop ofthe alternative pathway is activated in an uncon-trolled fashion, so that there is consumptive loss ofcomplement C3.1- t Secondary depletion of C3 isalso caused by genetic deficiency of complementcomponent factor H (FH), which is a cofactor for Fl.The study of these hereditary deficiencies is importantbecause of the evidence that they provide in relationto the roles of the complement system in vivo.
Furthermore, it was the investigation of the physi-ology of the complement system in the first case ofFl deficiency1"* that generated much of the earlyevidence for the existence of the amplification loopof the alternative pathway.5
The clinical effects of C3, Fl and FH deficiencyare similar: a propensity to suffer recurrent pyogenicinfections and an increased incidence of glomerulo-nephritis and SLE-like illness. The clinical con-sequences of C3 deficiency reflect the role playedby the complement system in the opsonization ofpathogens and the clearance of immune complexes.6
Although there are subtle differences in the effects
Address correspondence to Professor M.J. Walport, Rheumatology Unit, RPMS, Hammersmith Hospital, Du Cane Road,London W12 ONN
O Oxford University Press 1994

386 T.J. Vyseetal.
of these three conditions on the complement system,the predominant influence on the clinical outcomein all three states is that of C3 deficiency.
To date, 19 patients with Fl deficiency have beenreported.7"19 We report an additional four patientsfrom three families. In one family, there were twohomozygously affected siblings; one of whom suf-fered recurrent pyogenic infections, the other beinghealthy. In the other two families there were single,homozygous, symptomatic individuals. We reviewthe clinical features of Fl deficiency, and thencompare them with the clinical consequences of FHand C3 deficiencies. The clinical features of all thereported cases of these hereditary deficiencies aretabulated (see Table 1) and their pathogenesis andtreatment are briefly discussed.
Patients
Family 1
A 14-year-old boy presented with a history of recur-rent pyogenic infections that began at the age of 18months. At this time he developed a septic arthritisof the left shoulder from which Staph. epidermidiswas cultured. At the age of 5 years he sufferedorbital cellulitis, and over the next 5 years he hadrecurrent episodes of sinusitis. When 8 years old, anabscess on the side of his neck was drained. At theage of 13 he had a single episode of meningococcalmeningitis. He has subsequently had a number ofminor respiratory tract infections. Physical examina-tion was unremarkable. There was no family historyof immunodeficiency or autoimmune disease, andno consanguinity, although both parents originatefrom western Scotland. He has one sibling, a sisterwho is 5 years older, who has neither suffered fromany major episodes of infection with pyogenic organ-isms, nor had recurrent sinusitis or otitis media.
Investigation of both siblings revealed a normalfull blood count and basic biochemistry profile. Theimmunoglobulin concentrations of both siblings werewithin the normal ranges: the affected male's valueswere: IgG 10.5 g/l (normal: 7.2-16.2); IgA 1.3 g/1(0.8-3.9); IgM 2.3 g/l (0.5-3.5); his sister's valueswere: IgG 9.5 g/l (7.2-16.2); IgA 1.1 g/l (0.8-3.9);IgM 1.5 g/l (0.5-3.5). The IgG subtype concentrationsgave no evidence of selective isotype deficiency: theaffected male's concentrations were: IgG, 9.05 g/l(3.2-10.2); lgG2 2.97 g/l (1.2-6.6); lgG3 1.25 g/l(0.2-1.9); lgG4 0.20 g/l (0.10-1.3); the IgG subtypelevels of the asymptomatic sister were as follows:IgG, 10.05 g/l (3.2-10.2); IgG, 2.73 g/l (1.2-6.6);lgG3 1.55 g/l (0.2-1.9); lgG4 0.20 g/l (0.10-1.3).Antinuclear antibodies (ANA) (measured by indirectimmunofluorescence on Hep-2 cells) and rheumatoid
factor (measured by Latex agglutination) were notdetected. His complement system was investigatedtogether with that of all four family members (seeTable 2). The results demonstrate the diagnosis of Fldeficiency in the patient and in his asymptomaticelder sister: Fl was undetectable in both siblings, aswas factor B (FB); there was no detectable alternatepathway activity (APH50); and both FH and proper-din levels were reduced. Both parents had approxi-mately half the normal concentration of circulating Fl.
The patient is being treated with prophylacticphenoxymethylpenicillin, 250 mg bd. He was vac-cinated with Pneumovax and mounted a significantlgG2 response: pre-immunization 1:16; post-immunization 1 :110; control 1 :160 (measured byDr Kumaratne, Immunology Dept, Dudley RoadHospital, Birmingham). He is now aged 19 yearsand has had no further significant infective episodes.His sister, who is 24 years old, also remains well.
Family 2
The affected male in this pedigree has suffered fromrecurrent otitis media since infancy. He had oneepisode of idiopathic keratitis when aged 5 years.Because of recurrent sinusitis, surgery to the nasalseptum was performed at age 14 and it was notedthat he was allergic to both penicillin and sulphon-amides. When he was 16 years old, he suffered fourbouts of left-sided bronchopneumonia together withsinusitis. Str. pneumoniae and Staph. aureus werecultured from the sputum. The following year abronchogram was performed, no lesion was demon-strated, but an episode of bronchopneumonia wasprecipitated. When aged 20, a sixth episode ofbronchopneumonia occurred. Five years later, hehad a self-limiting illness characterized by hepatitis,pneumonitis, myositis, possible meningitis, and pur-pura, with histological evidence of a microangi-opathic vasculitis on skin biopsy. During the nextten years, he had five additional episodes of broncho-pneumonia.
At the age of 36 he was fully investigated, andthe diagnosis of Fl deficiency was established. Hehad a normal full blood count and biochemistry. HisANA and rheumatoid factor were negative. Theconcentrations of immunoglobulin isotypes werewithin the normal ranges, however, analysis of IgGsubtypes: IgG, 11.49 g/l (3.55-11.25); lgG2 1.14 g/l(1.30-5.90); lgG3 0.44 g/l (0.15-1.05); lgG4 0.54 g/l(0.10-1.10), indicated that the lgG2 concentrationwas diminished. Analysis of the complement systemconfirmed the diagnosis of Fl deficiency (see Table 3).Additional family members were also studied (seeFigure 1) indicating the inheritance of the traitthroughout the family. This family is Swiss/Germanin origin, there is no known consanguinity. The

Table 1a Hereditary Factor I deficiency: twenty-three patients from nineteen families (separated by lines)
Patient Age Sex Nationality Consanguinity Age of(years) Race onset
Infections Other complications
Klinefelter'ssyndrome XXY
Urticaria
Notes
Became C3 Coombs negativefor 14 days and hadundetectable C3b and 17 daysafter infusion of normal plasmaC3 and C5 levels raised for 12days but FB levels maintained foronly 3 days after infusion ofpurified Fl
Reference
(1)(2)
(3)(4)
1 25 M USA 1 year Haemorrhagic measles
Recurrent otitis mediaRecurrent sinusitisInguinal abscessAuricular abscess: C. diphtheriaeSepticaemia x 2 : Str. pyogenes
N. meningitidisPneumonia: H. influenzae
2 11 F English noCaucasoid
4 months Meningitis x 4:Str. pneumoniae x 1N. meningitidis x 2Otitis media
(7)
o
II"
8-1'5
? F USA ?
4 ?
Meningitis x 3 H. influenzae bN. meningitidis
3 months Pneumonia x 1
(8)
5 3 M Germany 6 months Gastroenteritis
Recurrent otitis mediaSepticaemiaUTI x 3
IgM rheumatoid Abnormal FH mobilityfactor positive (by immunoelectrophoresis)
probably due to its binding to C3bANA negative Infusion of plasma (FFP):
C5 and FH elevated above pre-infusion levels for up to 1 monthFB increased for 6 h onlyNo complications after 22 infusionsof FFP
(9)
28 M CanadaCaucasian
No Infancy Recurrent otitis, bronchitis, andmastoiditisPneumonia x 4 Serum sickness-likePleural empyema: Str. pneumoniae illness following theBronchiectasis administration ofMeningitis penicillin
5 of 10 siblings died in infancy
3 from sepsis, ?FI status
IgM rheumatoid factor (1 :1280)ANA negative
(10)
00

Table la
Patient
7
(continued)
Age Sex(years)
19 F
NationalityRace
DenmarkCaucasoid
Consanguinity
No
Age ofonset
19 years
Infections
Otitis mediaMeningitis: N. meningitidis CpBHerpes zoster
Other complications Notes Reference
(11)(12)
8 15/12 F USA 3 weeks Septicaemia: Str. pneumoniaePneumonia: Str. pneumoniaeOtitis media?Osteomyelitis
low IgA (0.36 g/1) Infusion of Fl:C3 and FB increased for 4 daysFH and CH50 increased for 14 daysFl remained undetectable
(13)
0000
9 9 ? France?
? Recurrent bronchitis, otitis mediaand mastoiditis
Reduced erythrocyte CR1Normal range 300-1320
(14)
10 ? ? France ?
11 ? France
9 years Arthritis: septic ?agent
11 years Arthritis: N. meningitidis
Reduced erythrocyte CR1Normal range 300-1320patients: 10-115; 11-182Defective CRT- and CR3-dependentphagocytosis
(14)
12
13
14
15
37
37
27
?
M
F
M
M
?Caucasoid
Caucasoid
Tunisian
No
No
No
Childhood Recurrent otitis, sinusitisPneumonia x 8Meningitis x 5:N. meningitidis (GpB x 1, W135
x l )Septicaemia with DIC x 2
37 years Fatal systemic vasculitis: cutaneousleucocytoclastic lesionsHaematuria and proteinuriaDeep venous thrombosisPerivenous encephalomyelitis(diagnosed at post-mortem)
Childhood Recurrent otitis media andcutaneous abscesses
Vasculitic illnessfollowed theadministration ofpenicillin forpharyngitisCryoglobulinaemiaANA not reported
Asymptomatic
Treatment with FFP: (15)C3 increase 16 daysNative FB increase 4 daysFall in C4 and rise in C4d duration- 2 daysFl half-life estimated 29 hAnaphylaxis with 8th and 9th FFPinfusions
Very low, but detectable levels of (15)Fl, C3d and C4d also detectable(?from blood transfusion)
(16)
In
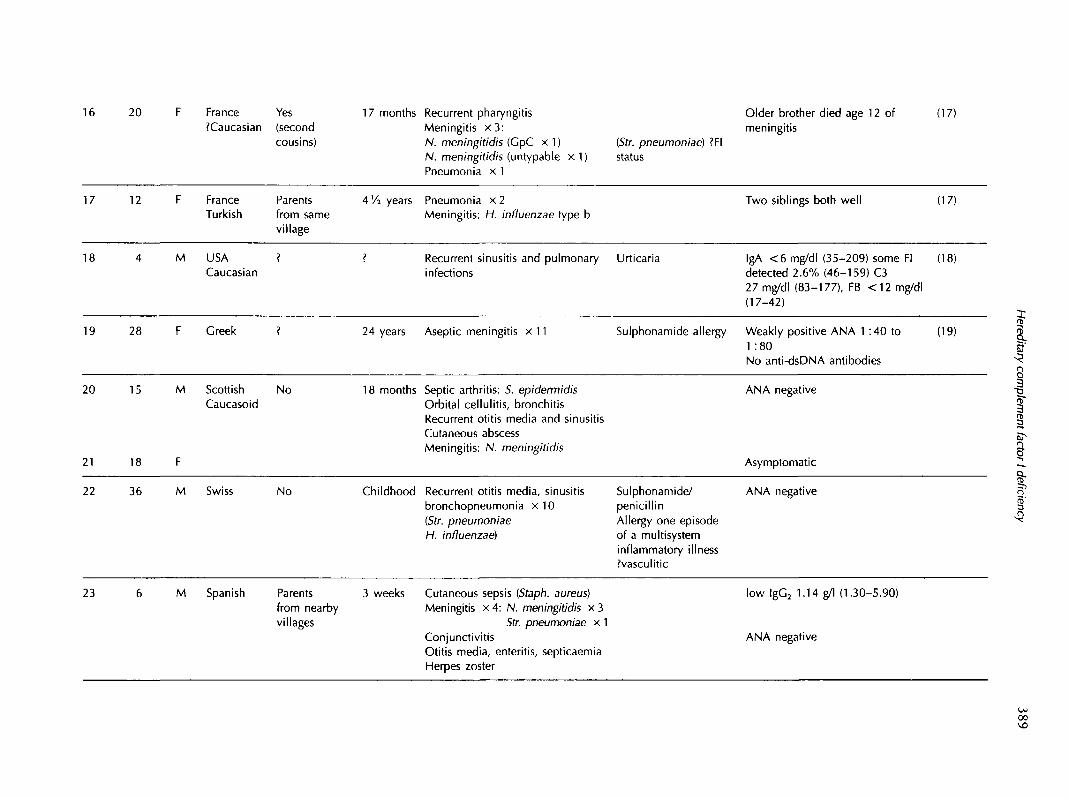
20 France Yes?Caucasian (second
cousins)
17 months Recurrent pharyngitisMeningitis x 3 :N. meningitidis (GpC x 1)N. meningitidis (untypable x 1)Pneumonia x 1
(Str. pneumoniae) ?FIstatus
Older brother died age 12 ofmeningitis
(17)
17 12 F FranceTurkish
Parentsfrom samevillage
4 Vi years Pneumonia x 2Meningitis: H. influenzae type b
Two siblings both well (17)
18 M USA ?Caucasian
Recurrent sinusitis and pulmonary Urticariainfections
IgA <6 mg/dl (35-209) some Fidetected 2.6% (46-159) C327 mg/dl (83-177), FB <12 mg/dl(17-42)
(18)
19 28 F Greek ? 24 years Aseptic meningitis x 11 Sulphonamide allergy Weakly positive ANA 1 :40 to1 :80No anti-dsDNA antibodies
(19) I1?5
20
21
22
15
18
M Scottish NoCaucasoid
18 months Septic arthritis: 5. epidermidisOrbital cellulitis, bronchitisRecurrent otitis media and sinusitisCutaneous abscessMeningitis: N. meningitidis
ANA negative
Asymptomatic
I3'43
36 M Swiss No Childhood Recurrent otitis media, sinusitisbronchopneumonia x 1 0[Str. pneumoniaeH. influenzae)
Sulphonamide/penicillinAllergy one episodeof a multisysteminflammatory illness?vasculitic
ANA negative
23 M Spanish Parentsfrom nearbyvillages
3 weeks Cutaneous sepsis (Staph. aureus)Meningitis x 4: N. meningitidis x 3
Str. pneumoniae x 1ConjunctivitisOtitis media, enteritis, septicaemiaHerpes zoster
low lgG2 1.14 g/l (1.30-5.90)
ANA negative
00
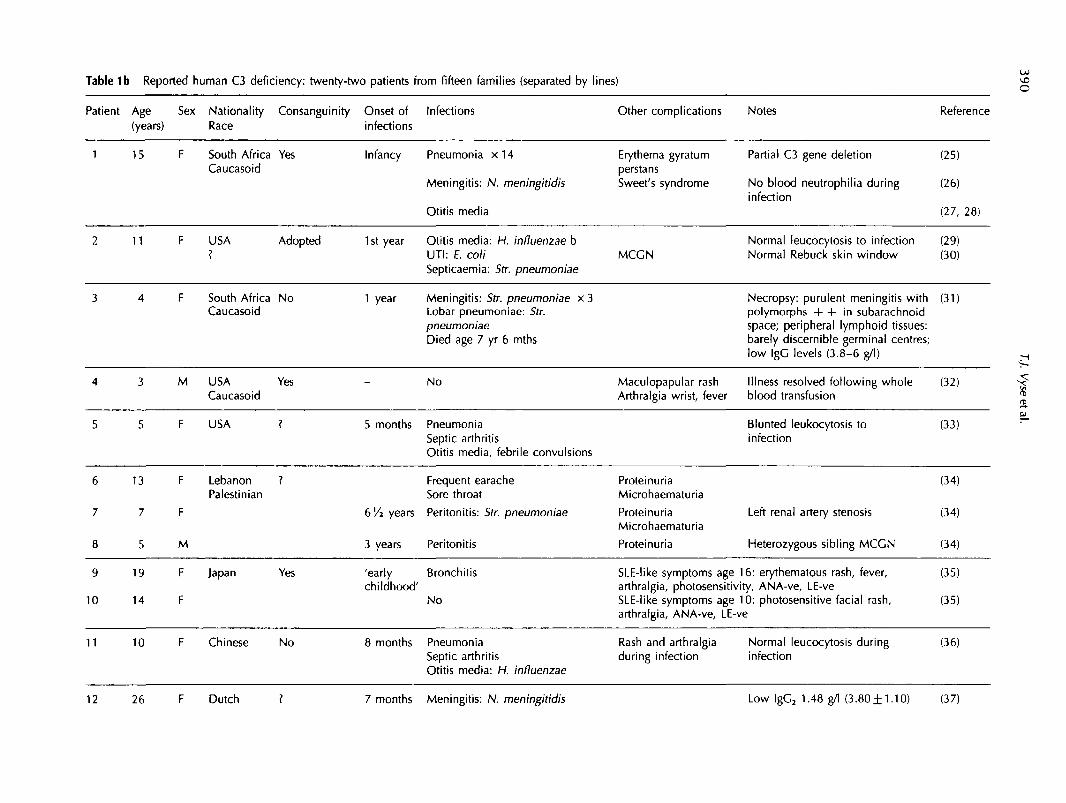
Table 1b Reported human C3 deficiency: twenty-two patients from fifteen families (separated by lines) o
Patient Age Sex Nationality Consanguinity Onset of Infections(years) Race infections
Other complications Notes Reference
1 15 F South Africa YesCaucasoid
Infancy Pneumonia x 1 4
Meningitis: N. meningitidis
Otitis media
Erythema gyratumperstansSweet's syndrome
MCGN
Partial C3 gene deletion
No blood neutrophilia duringinfection
Normal leucocytosis to infectionNormal Rebuck skin window
(25)
(26)
(27, 28)
(29)(30)
11 F USA Adopted 1st year Otitis media: H. influenzae bOtts edaUTI: E. coliSepticaemia: Str. pneumoniae
South Africa NoCaucasoid
6
7
8
9
10
13
7
5
19
14
F
F
M
F
F
LebanonPalestinian
Japan
1 year Meningitis: Str. pneumoniae x 3Lobar pneumoniae: Str.pneumoniaeDied age 7 yr 6 mths
Necropsy: purulent meningitis withpolymorphs + + in subarachnoidspace; peripheral lymphoid tissues:barely discernible germinal centres;low IgC levels (3.8-6 g/l)
(31)
4
5
3
5
M
F
USACaucasoid
USA
Yes
?
-
5 months
No
PneumoniaSeptic arthritisOtitis media, febrile convulsions
MaculopapularArthralgia wrist,
rashfever
Illness resolved following wholeblood transfusion
Blunted leukocytosis toinfection
(32)
(33)
Frequent earacheSore throat
bVi years Peritonitis: Str. pneumoniae
3 years Peritonitis
ProteinuriaMicrohaematuria
ProteinuriaMicrohaematuria
Proteinuria
Left renal artery stenosis
Heterozygous sibling MCCN
(34)
(34)
(34)
I
Yes 'early Bronchitischildhood'
No
SLE-like symptoms age 16: erythematous rash, fever, (35)arthralgia, photosensitivity, ANA-ve, LE-veSLE-like symptoms age 10: photosensitive facial rash, (35)arthralgia, ANA-ve, LE-ve
11 10 F Chinese No 8 months PneumoniaSeptic arthritisOtitis media: H. influenzae
Rash and arthralgiaduring infection
Normal leucocytosis during (36)infection
12 26 F Dutch ? 7 months Meningitis: N. meningitidis Low lgC2 1.48 g/l (3.80± 1.10) (37)

13
14
19
16
Meningitis: Str. pneumoniae'sepsis': S. aureus
years Meningitis: Str. pneumoniaeOtitis media
8 months OsteomyelitisOtitis media
Transientmaculopapular rashduring infectiousepisodes
Transientmaculopapular rashduring infectiousepisodes MCGNtype I
Low lgC2 0.11 g/1 (3.80±1.10)
Low lgC2 0.44 g/l (3.80±1.10)Administration of FFP of no benefit
(64)
15 M Laos No 5 months Lobar pneumoniaMeningitis: Str. pneumoniae x 2
MCGN Acute administration of FFP notassociated with renal deterioration
(38)
(39)
16 12 Kuwait
Dysfunctional C3 molecules
1 47 F Swedish
2 42 F
No
Nil MicrohematuriaNephrotic syndromeRenal failureMCGN Type I
Recurrent otitis mediaMeningitis N. meningitidis Gp YNone SLE
Weak ANA, IgG and IgM atdermo-epidermal junction+ ve ANA, anti-dsDNA antibody,anti-centromere antibody
(40)
17
18
19
20
21
22
6
10
23
14
19
7
M
M
M
F
M
F
Brazil
England
Japan
N. Zealand/Israel
Yes
Yes
Yes
?
3 months
4
Childhood
Meningitis: N. meningitidisBronchopneumonia x 4Otitis media, osteomyelitis
Otitis mediaURTI: Str. pyogenes
Meningitis
PneumoniaMeningitis: N. meningitidis
x 3
x 2
Transient erythemamultiforme at time ofinfection
IgA nephropathy
Lupus-like illness
Normal leucocytosis to infection
First cousin of patient 19
Asthma, rhinitis, otherwise healthy
(41)
(41)
(42)
(42)
(43)
(50)
8Iri8
I

Table 1c Hereditary Factor H deficiency: twelve patients from six families
Patient Age Sex Nationality Consanguinity Age of Infections(years) Race onset
Other features Notes Reference
1 8/12 M Indian
M
Yes 8 months Otitis media: H. influenzae(first cousins)
Haemolytic-uraemicsyndrome (HUS)
Asymptomatic
Very low factor H detected (6% (44)NHS)Renal biopsy characteristic of HUS
5 M Algerian Yes 14 months Recurrent otitis and bronchitis(first cousins) followed by haematuria
GlomerulonephritisMCGN-like
6/12 M 4 months Otitis media and bronchitisSepticaemia: E. coliUTI: Proteus sp.Persistent haematuria
GlomerulonephritisMCGN-like
FH 12% NHS in both cases renalbiopsy appearance similar in both:intramembranous dense depositsdetected atypicalimmunofluorescence (IF): anti-C3staining in mesangium andcapillary walls, not in the basementmembrane H antigen and C5b-9neoantigens in the areas of C3staining
(45)
I2-EU
Spain No N. meningitidis
N. meningitidis
MCGN
MCGN
MCGN
No factor H antigen detectable inall sibsReduced C5 ( < 1 0 % NHS) and C9(10% NHS)
No details of renal biopsy given
All ANA negative
(46)
11
Italy Yes None
11 years
AsthmaHaematuria
SLE with nephritisAnti-dsDNAantibodies
FH undetectable in all 3 sibsHeterozygous C2 deficiency
Heterozygous C2 deficiency Renalbiopsy: diffuse proliferativenephritis 6 of 25 glomeruli withcrescents C3 ( 3 + ) and IgG (1 + )in mesangium by IF Responded toprednisolone (2 mg/kg/day)
(47)
10 M Asymptomatic Normal C2

11 15 Danish No 10 years Meningitis x 2:N. meningitidis Cp B
No detectable FH (48)Depressed C5 (7% NHS) and C7( < 0 . 5 % NHS) ?C7 deficiency too
12 49 Netherlands Meningitis: N. meningitidis Gp X SLE/lupus-like illness No details of autoantibodies (49)
Partial factor H deficiency
1 17 M USA ?Polish
11 years Henoch-SchonleinpurpuraThrombocytopeniaand splenomegaly
Three other asymptomatic family (51)members
(52)
F USA ?Anglo-Irish
63 years ?Recurrent UTIs
38 M
33
26 M
30 years
36 years
26 years
Hypertension
IgA nephropathyleading to end-stagerenal failure
IgA nephropathy(mild)
Proteinuria
(52)
Diagnosis on renal biopsy
Renal biopsy performed (sister ofpatient 2)
Brother of patient 2
1I"
IF USA No 18 months Recurrent respiratory tractinfections
Haemolytic-uraemicsyndrome x 3
Died age 2 yrs
Null allele at C4A and C4BAbnormal C3 variant with reducedtotal C3
(53)
OJ

394 T.J. Vyseetal.
Table 2 Complement profile of family 1
MotherFatherPropositusSister
MotherFatherPropositusSister
Fl
4174--
C2
84967790
All results expressed as-, undetectable.
FH
961554642
C3
84992815
FB
7171--
C4
7114760
105
P
51523829
C5
137919948
APH50
5045--
CH50
92102--
% normal human serum.
Table 3 Complement profiles of the propositi from family2 and family 3
Fl FH FB P C4BP APH50 FD100
Propositus 2 - 53 17 34 107 - 82Propositus 3 - 42 9 38 72 - 68
C2 C3 C4 C5 C7 CH50
Propositus 2 ND 31 159 53 30 47Propositus 3 ND 31 124 34 36 40
-, undetectable; ND, not determined.All results expressed as % normal human serum.
patient is now 43 years old and is well apart fromoccasional febrile illnesses which respond rapidly tothe administration of antibiotics.
Family 3
A 6-year-old presented with a history of recurrentpyogenic infections which began at the age of 3weeks with a cutaneous abscess and omphalitis dueto Staph. aureus. At 3 months, he developed asecond abscess, and at 6 months he started havingpersistent folliculitis. When 4 months old, he becamesepticaemic with Str. pneumoniae, and at 9 monthshe had a further pyrexial illness. At 14, 15 and 16months of age he had an abscess on the right cheek,enteritis, and purulent conjunctivitis, respectively.When 19 months old he suffered a pyrexial illnesswith erythematous rash that responded to penicillin.Four months later, he had two episodes of meningitisin rapid succession; the first due to N. meningitidisand the second due to Str. pneumoniae. The follow-ing month he had otitis media and then septicaemiawith Str. pneumoniae. Between the ages of 3 and 6years the patient suffered from otitis media, bron-chitis, enteritis, purulent pharyngitis, and aphthousstomatitis. When 6 years old, he had an episode ofmeningococcal septicaemia after which complementstudies were performed (see Table 2) and the dia-gnosis of Fl deficiency was made. The immunoglob-ulin concentrations were: IgG 8.24 g/1 (6.86-14.9);IgA 1.5 g/l (0.45-3.09); IgM 0.74 g/l (0.41-3.00) andIgG subtypes were: IgG, 8.07 g/l (3.55-11.25); lgG2
2.67 g/l (1.30-5.90); lgG3 0.26 g/l (0.15-1.05); lgG4
0.26 g/I (0.10-1.10). The concentration of Fl in otherfamily members is shown in Figure 2. Although thepropositus and his parents now live in Switzerland,the family was originally from northern Spain, andthe parents of the Fl-deficient boy were from neigh-bouring villages. There is no history of consanguinity,however.
The patient was vaccinated with Pneumovax and
II
I l l 107( ) 110 57
IV
Figure 1. The pedigree shows the inheritance of factor I deficiency in the family of Swiss/German origin (family 2). Thepropositus is marked with an arrow. The number to the left of the individuals is the factor I concentration in the serum ofthat individual expressed as a percentage of the value of normal human serum: normal range 75%—135%.

Hereditary complement factor I deficiency 395
NT NT NT3
NT4 r^i
NTQ
II
III
Figure 2. The pedigree shows the inheritance of factor I deficiency in the family of Spanish origin (family 3). The propositusis marked with an arrow. The number to the left of the individuals is the factor I concentration in the serum of thatindividual expressed as a percentage of the value of normal human serum: normal range 75%-135%. NT signifies anindividual whose Fl concentration was not tested.
maintained on prophylactic penicillin for 3 years.During this period he had intermittent bouts ofbronchitis and sinusitis, and when 8 years old,varicella-zoster. He is now 15, and having withdrawnprophylactic penicillin of his own volition for 3years, has remained relatively well with occasionalpyrexial illnesses but no septicaemic episodes.
Methods
Complement assays
Antigenic and functional assays
Serum concentrations of the complement proteinswere assayed by radial immunodiffusion using poly-clonal antisera (sheep anti-C2, anti-C3, anti-C4,anti-C5, anti-FI, anti-FH, and anti-properdin: TheBinding Site, Birmingham). Functional complementassays were performed as follows:20 classical path-way activity (CH50) was assessed using a haemolyticassay with sheep red blood cells sensitized withrabbit antibody (Tissue Culture Services,Buckingham), in a 1.5% CFD/agarose gel (Oxoid,Basingstoke); alternative pathway activity (APH50)was measured by haemolytic assay with guinea pigerythrocytes (Tissue Culture Services, Buckingham)in a 1.5% agarose gel in the presence of 7 j iMMgCI2 and I O U M ethyleneglycol tetraacetic acid(EGTA). The FD100 is an assessment of factor D (FD)activity. It was measured in the same way as APH50,except that the guinea pig erythrocytes were sus-pended in FD-depleted (by affinity chromatography)serum. The investigation of the state of circulatingC3 in the Fl-deficient members of family 1 wascarried out by crossed immunoelectrophoresisaccording to the method of Laurell, as described inreference 20.
Assays for cell-bound C3 and CR1
The binding of C3b and its degradation products onthe red cell surface was analysed by radioligandbinding assay as previously described.21 Threeanti-C3 monoclonal antibodies were used (clones 3,4 and 9, kindly provided by Prof. P. Lachmann,Cambridge) which recognize C3d, C3c and C3g,respectively.22 Erythrocyte CR1 numbers were alsomeasured by radioligand binding assay using themonoclonal antibody El 1 (kindly provided by DrNancy Hogg, ICRF, London). The binding of thisantibody to the receptor is not affected by the ligationof CR1 with C3b.23
The purified Fl and FH reagents, used in the ligandbinding studies, were prepared by affinity chromato-graphy using specific monoclonal antibodiescoupled to cyanogen-bromide-activated Sepharose(Pharmacia): anti-FI: MRC OX21; anti-FH: MRCOX23 (Serotec, Oxford).24
ResultsComplement estimation
The complement profiles of the four homozygouslyaffected patients from the three families are given inTables 2 and 3. In none of them was Fl detected inthe circulation. The complement profiles of thesepatients were similar. They had very low or undetect-able levels of factor B (FB), and no detectablealternative pathway activity (APH50). The C3 con-centrations were also very low. Crossed immuno-electrophoresis of serum from the two affected sib-lings from family 1, indicated that 90% of the C3was in the form of C3b (data not shown). Thereduced concentrations of FH and properdin in allthe propositi to approximately one half of their

396 TJ. Vyse etal.
normal value have been documented in all ninecases of Fl deficiency in which these componentshave been assessed.4'9'10'13151719 In one report,15
normal FH and properdin levels were probably theresult of a prior infusion of blood. Reduced levels ofthe terminal pathway components C5 and C7 werefound in the affected individuals from families 2 and3 and the sister of the propositus from family 1 (C5only). A reduction in C5 has been reported in sevenprevious cases,5'910'13'15'17 and a reduction in C7 infour cases.5'10'17 In none of the reports in whichthese two complement components were measuredwere normal values recorded.
The results of the radioligand binding studiesperformed on family 1 (see Table 4) indicate that theamount of C3b present on the erythrocyte surfacewas increased ten-fold in both homozygotes, asdemonstrated by the binding of the two monoclonalantibodies to the C3d and C3c portions of C3b. Noincrease in binding was observed in the heterozyg-ously-affected parents. The antibody specific for C3g,clone 9, recognises a neoantigen on C3 revealed bythe action of Fl. No significant binding was foundwith this antibody to erythrocytes from normal oraffected individuals.
Following the incubation of erythrocytes from theFl-deficient patient with autologous serum, there wasno change in the pattern of binding observed withthe three different anti-C3 monoclonal antibodies(see Figure 3). The incubation of the patient's erythro-cytes with either normal serum, purified Fl, or amixture of Fl and FH, resulted in a similar outcomeas regards monoclonal antibody binding: an increasein clone 9 binding (indicating conversion of C3b toiC3b and C3dg by Fl); no apparent change in thebinding of clone 3 (which binds to epitopes expressedin both C3b and in its breakdown products thatremain surface-bound); but a fall in clone 4 binding(this antibody binds C3c which is cleaved from iC3bby Fl and released into the fluid phase).
Review of the cases of hereditary Fl, FH andC3 deficiencies
All the reported cases of hereditary Fl, FH and C3deficiencies are outlined in Table 1. There are 22
MoAb:Hclone9 W clone 3 • clone 4
300
encc!o"coCO
.a
200
100
1 1'Fl-def. normalserum serum
Fl FH + Fl
Figure 3. The treatment of erythrocytes from the propositusof family 1 with either normal serum or purified factor I( + /— factor H) resulted in a 2-3 fold increase in thebinding of the monoclonal clone 9 (which binds toneoepitopes in C3g). The concentration of factor I usedwas 10 Hg/ml and the concentration of factor H used was50 Hg/ml. There was no change in clone 3 binding (whichbinds to C3b and its degradation products), and a fall inclone 4 binding (which binds to the C3c portion of C3).
patients with homozygous C3 deficiency,25"43 23with Fl deficiency,1"4'7-19 and 12 who lack FH.44^19
Of these, all but one case of homozygous C3deficiency is symptomatic;43 two cases of Fl16
(family 1, this report) and FH deficiency44'47 areasymptomatic. In addition, we tabulate one familyin which complete C3 deficiency has resulted fromthe inheritance of one null allele and one dysfunc-tional molecule;50 six instances of partial FH defi-ciency are also summarized.51"53
Discussion
The most consistent clinical feature of these threeinherited complement deficiencies is susceptibilityto infection. This is predominantly due to pyogenicorganisms and Neisseria meningitidis. Infection withN. meningitidis was documented in 11/22 cases ofFl deficiency, in 4/12 cases of FH deficiency, and in
Table 4 Assessment of C3 and CR1 numbers on erythrocytes (family 1)
C3 fragment.. C3d C3c C3g CR1Monoclonal antibody... (clone 3) (clone 4) (clone 9) (E11)
MotherFatherProbandSisterNormal
"The number of erythrocyte CR1 is subject to a genetic influence, and hence an absolute normal range has limited meaning.
417447551645
595257362193
1524292731
908645311339300-1300*

Hereditary complement factor I deficiency 397
4/22 cases of C3 deficiency. The efficacy of comple-ment as an opsonin is indicated by the recurrentpyogenic infections sustained by C3-deficient indi-viduals. In both Fl and C3 deficiency, defectiveopsonization and killing of microbes has been dem-onstrated with phagocytes from affected individualsusing in vitro assays.4'14'37'39 Moreover, the defectiveopsonization and bactericidal activity of leucocyteswere improved, in vitro, if either infusions of FFPwere given,39 or the missing complement componentwas replaced, either Fl4 or C3.37
Isolated hereditary deficiency of the terminal path-way components increases susceptibility to neisserialinfection, implying that the membrane attack com-plex is important in the eradication of these organ-isms.54 The increased incidence of neisserialinfections in C3 deficiency is presumably due to thereduced efficiency of the terminal pathway. Low C5and C7 levels were found in the propositi fromfamilies 2 and 3 in this paper, confirming observa-tions from other reports of Fl deficiency.5'9'10'13'15'17
A reduction in the concentration of C5 has beenreported in nine of the ten symptomatic cases of FHdeficiency,45"46 and was normal in only one case.44
In this latter case there was a low level of circulatingFH detected, and no instance of neisserial infectionwas recorded in the propositus. C5 is consumed inFH and Fl deficiency because of the C5 convertaseactivity (C3bBbC3b) derived from the alternativepathway C3 convertase (C3bBbP) which is presentin excess. An additional factor in the predispositionof these patients to infection may be that opsoniz-ation of the pathogens by antibody and C3 to formimmune complexes facilitates their delivery to thesplenic macrophage system.55 Therefore, C3-, FH-and Fl-deficient individuals may be functionallyhyposplenic. The infections typically commence inearly childhood: the median age of the first pyogenicinfection in Fl deficiency was 17 months; in FHdeficiency it was 14 months; and in primary C3deficiency it was 8 months. Evidence from the threesymptomatic cases presented in this paper suggeststhat the frequency of infection declines with age. Apossible explanation of this phenomenon is that asthe immunological memory of the adaptive immunesystem expands with increasing age, so the role ofthe innate immune system becomes less important.
It is apparent from the data in Table 1 that thereis a marked contrast in the incidence of renal diseasein Fl deficiency compared to the incidence in C3and FH deficiencies. Clomerulonephritis has notbeen described in any of the reported cases of Fldeficiency, including the four examples reported inthis paper. In C3 and FH deficiencies, there isfrequent evidence of some renal involvement. FHdeficiency seems to have the strongest associationwith renal disease. From a total of 12 patients, seven
had definite nephritis: five of these had mesangio-capillary glomerulonephritis (MCGN),45'46 one hadthe haemolytic-uraemic syndrome,44 and anotherhad crescentic nephritis47 (this patient aJso +iadheterozygous C2 deficiency). In C3 deficiency, glom-erulonephritis was recorded in 8/20 patients and infour of these was of MCGN-type.30-37"39'40 In two ofthese cases,37'40 it was specified to be type 1 MCGN,of which the characteristic feature is subendothelialdeposits. The exact mechanism by which nephritisdevelops in these two C3 deficiency states is unclear.That C3 and FH deficiency patients suffer an MCGN-like nephritis is interesting because of the associationof this disease with the C3 depletion found inconjunction with a C3 nephritic factor. The C3nephritic factor is usually associated with type II(dense-deposit) MCGN. The association between C3deficiency and MCGN-like nephritis is furtherstrengthened by the observation that dogs withhereditary C3 deficiency develop a nephritis whosehistological pattern is that of MCGN.57
The absence of Fl produces marked abnormalitiesof the complement system (see Table 5 for a compar-ison of the effects of C3, FH and Fl deficiency), andyet has not been associated with glomerulonephritis.Thus there is no straightforward relationship betweenthe derangement of the complement system as meas-ured in vitro and the predisposition to disease. In Fldeficiency, only three cases of 'immune complex'illnesses have been described; one being the multisy-stem inflammatory disorder that occurred in thepropositus from family 2. Another patient succumbedto a fatal vasculitic syndrome,15 and the third had aserum sickness-like syndrome following the adminis-tration of penicillin.10 The association of immunecomplex disease with recurrent infection raises thepossibility that the two are related. Recurrent infec-tions, by stimulating an acute-phase response, maytheoretically exacerbate an immune-mediatedinflammatory process; alternatively, microbial patho-gens may act as the antigenic source for immunecomplex disease. In one case of FH deficiency,45 inwhich there was MCGN-like glomerulonephritis,recurrent episodes of otitis media and bronchitiswere followed by episodic haematuria. In six patientswith hereditary C3 deficiency, transient maculopapu-lar skin rashes have developed during infectiveepisodes.2532-36-37'41 In one study37 circulatingimmune complexes (identified by C1q binding assay)were present at the time when a rash developed intwo patients. Skin biopsy revealed local depositionof IgG, IgM, C1q, but not C3, by immunofluores-cence. It is of note that in the three cases of Fldeficiency in which 'immune complex' type illnessdeveloped (the case from family 2, and thosereported in references 10 and 15) the illness followedthe administration of antibiotic. The complement

398 T.J. Vyseetal.
system is also an important effector mechanism inthe inflammatory response. It is therefore possiblethat if some residual C3 activity remained the con-sequences of immune complex deposition/formationwould be aggravated. In one instance of FH defi-ciency,45 in which there was some FH antigendetected (12% normal), two siblings both developedMCGN-like glomerulonephritis. Renal biopsies wereperformed and subjected to immunofluorescence.The results indicated that C3 was deposited in themesangium and capillary walls, and that FH andC5b-9 neoantigens were present in a similar distribu-tion to C3. The localization of C5b-9 neoantigenssuggests that there was formation of the C5 con-vertase and that complement was playing an activepart in the inflammatory process.
There may be an increased incidence of nephritisin cases of partial (heterozygous) complement defi-ciency. Two families are summarized in Table 1 inwhich partial FH deficiency occurs with IgA nephro-pathy.51-52 There is one instance of IgA nephropathyin a patient with homozygous C3 deficiency.42 Nodisease has been associated with heterozygous Fldeficiency. In the three families reported, there wereno unusual clinical problems in any of the heterozyg-ous Fl-deficient individuals. In addition, in one familyin which partial Fl deficiency occurred in conjunc-tion with C1 -inhibitor deficiency,57 the clinical mani-festations of the C1-inhibitor deficiency were notaltered by the coexistence of heterozygous Fl defi-ciency.
A reduced concentration of lgG2 was found in thepropositus from family 3. Low lgG2 concentrationswere found in one family to be associated withhomozygous C3 deficiency37 together with a reducedantibody response to pneumococcal capsular poly-saccharide (which is lgG2-dependent).M Antibodyisotypes have not been studied in FH deficiency,and in family 1, one of the cases of Fl deficiencyreported here, normal concentrations of IgG, andlgG2 were measured together with a normal lgG2
response to pneumococcal capsular polysaccharide.In one systematic investigation of IgG subclasses incomplement deficiencies,59 lower mean levels oflgG2 were found in primary C3 deficiency comparedto normals, and a marked reduction in lgG4 was aconsistent finding in classical pathway andC3-deficiency states. Using bacteriophage <f>X'\74 asa test antigen, two C3-deficient patients producednormal titres of IgM following primary and secondaryimmunization, but failed to make an isotype switchto IgG when the antigen was used at low dose.60 InC3, Fl, and FH deficiencies, normal antibodyresponses to antigens such as tetanus toxoid, dip-theria, and pertussis vaccine have been recorded.37'48
In one study61 which included one FH-deficientpedigree in Italy, high titres of antibody against
meningococcal polysaccharides A and C wereobserved, presumably as a consequence of naturalinfection. However, only a modest response wasthen generated by immunization with the meningoc-occal vaccine, Menpovax A + C.
The mechanism by which five patients (one with-out C3, and two without FH and Fl) are asymptomaticis not known. The explanation presumably lies inthe fact that within both the innate and adaptiveimmune systems there is a great deal of redundancy.Thus genetic variation at many loci may influencethe penetrance as well as the expressivity of thesemonogenic disorders. The asymptomatic cases of Fldeficiency were detected because their siblings wereclinically unwell. Because Fl deficiency may beasymptomatic, it is possible that a substantial propor-tion of cases are undetected. In one report15 twocases were identified in two non-consanguinousfamilies from the Danish island of Funen. The authorsestimate the minimum frequency for the deficientgene to be 0.002 using the Funen island data.However, in two large studies, one in 41 083 SwissArmy recruits,62 and a second in 145 640 blooddonors from Osaka, Japan,63 no instances of C3deficiency were identified.
The mainstay of treatment at the present time forthese inherited conditions is immunization againstpathogens to which affected individuals are particu-larly susceptible, Str. pneumoniae, H. influenzae b,N. meningitidis (vaccination with polysaccharideantigens types A and C is currently available),together with the administration of prophylactic anti-biotics.
An additional potential therapeutic option isreplacement of the deficient complement compon-ent. Replacement treatment using either fresh-frozenplasma (FFP) or purified protein has been used inC337-39,64 a n d F| deficiencies.1'91315 In both circum-stances, this form of treatment is limited by the highrate of turnover of the deficient protein. Moreover,there are two potential drawbacks: firstly, the replace-ment of a genetically-absent protein may stimulatean alloimmune response against it; secondly, thereconstitution of the complement system in the acutephase may exacerbate the underlying illness. FFPhas been administered in two cases of C3 deficiencyand MCGN with no evidence of improvement ordeterioration in renal function.14"39'64 In one of thesecases a renal biopsy was performed before and aftertwo months of infusion therapy without evidence ofchange in renal function, but there was some histo-logical improvement, with a reduction of staining forIgG and C4.64 The nephritis subsequently showed adefinite response to corticosteroid therapy. FFP muststill be used cautiously because of the observationthat in C3-deficient dogs, the nephritis was worsenedby replacement of C3.56

Hereditary complement factor I deficiency 399
The administration of FFP in Fl deficiency hasresulted in a rapid increase in FB and C3 levels.There was a transient rise in C3d and C4d concentra-tions, accompanied by a loss of C3 from the patient'sred ceHs together with a slight fall in C4 concentra-tion.1'9'13'15 No deterioration in clinical condition hasbeen reported in response to FFP administration,although in one case anaphylaxis occurred with theeighth and ninth FFP infusions which were thenhalted.15 After an FFP infusion, the decline in theserum concentration of Fl is paralleled by that of FB.It has been observed in three instances4'9'15 thatdespite the fall in Fl and FB levels there is aprolonged effect of Fl replacement on the C3 concen-tration. This starts to decline only after 14 days, atwhich time there is no antigenically detectable Fl,and FB has returned to its baseline level. Themechanism of this discrepancy is unclear. C2 defi-ciency has been successfully managed with regularFFP replacement therapy, and the clinical improve-ment, in arthralgia and skin rash for instance, hasbeen observed to last 4 -8 weeks, considerably longerthan the half-life of C2.65
In summary, the four cases of hereditary Fl defi-ciency described in this paper reflect the range ofclinical manifestations that can occur in this comple-ment deficiency. The spectrum of illness was fromone individual who was completely asymptomatic,to another patient who had recurrent pyogenicinfections starting in infancy and continuing until hewas diagnosed at the age of 36 years, together witha single episode of a multisystem vasculitic illness.The mechanism by which this variation in diseaseexpression is generated is not known, but it can notbe accounted for by any differential affects of Fldeficiency on the complement system that can bemeasured in the laboratory. The clinical con-sequences of primary C3 deficiency and FH defi-ciency are similar to those of Fl deficiency. Thereare some differences, however, notably the predis-position towards renal disease in C3 and FH defi-ciency that has not been found in Fl deficiency.
Acknowledgements
The authors wish to thank Liselotte Meyer-Haenniand Roland Zehnder for technical help in the comple-ment analysis of the two Swiss pedigrees. Much ofthe clinical data from these two pedigrees wasassembled by S. Jakob in preparation for his MDthesis which was submitted to the Medical Facultyof the University of Bern. For secretarial help, weacknowledge Ms. Crete Voegeli. We also wish tothank the physicians who originally referred thefactor-l-deficient individuals reported in this paperfor investigation. Family 1 was referred by Dr David
Webster, Clinical Research Centre, Harrow; family 2was referred by Dr B. Ott, Tiefenauspital, Bern; andfamily 3 was referred by Dr P. Imbach, Inselspital,Bern. Dr Kevin Davies is a Senior Clinical ResearchFellow funded by the Arthritis and RheumatismCouncil (ARC), Dr Tim Vyse is a Junior ClinicalResearch Fellow also funded by the ARC.
References1. Alper CA, Abramson N, Johnston JB, Jandl JH, Rosen FS.
Increased susceptibility to infection associated withabnormalities of complement-mediated functions and of thethird component of complement (C3). New Eng J Med 1970;282:349-52.
2. Abramson N, Alper CA, Lachmann PJ, Rosen FS, Jandl JH.Deficiency of C3 inactivator in man. J Immunol 1971;107:19-27.
3. Alper CA, Rosen FS, Lachmann PJ. Inactivator of the thirdcomponent of complement as an inhibitor in the properdinpathway. Proc Natl AcadSci USA 1972; 69:2910-13.
4. Ziegler JB, Alper CA, Rosen RS, Lachmann PJ, Sherington L.Restoration by purified C3b inactivator of complement-mediated function in vivo in a patient with C3b inactivatordeficiency. J Clin Invest 1975; 55:668-72.
5. Lachmann PJ, Nicol P. Reaction mechanism of thealternative pathway of complement fixation. Lancet 1973;1:465-7.
6. Lambris JD. The multifunctional role of C3, the thirdcomponent of complement. Immunol Today 1988;9:387-93.
7. Thompson RA, Lachmann PJ. A second case of human C3binhibitor (KAF) deficiency. Clin Exp Immunol 1977;27:23-9.
8. Eng RHK, Seligman SJ, Arnaout MA, Alper CA. Variableexpression of homozygous C3b inactivator deficiency. ClinRes 1978; 26:394 (Abstract).
9. Wahn V, Rother U, Rauterberg EW, Day NK, Laurell AB.C3b inactivator deficiency: association with an alpha-migrating Factor H. J Clin Immunol 1981; 1:228-33.
10. Solal-Celigny P, Laviolette M, HebertJ, Atkins PC, SiriosM,Brun C, Lehner-Netsch C, DelSge JM. O b inactivatordeficiency with immune complex manifestations. Clin ExpImmunol 1982; 47:197-205.
11. Teisner B, Brandslund I, Folkersen J, Rasmussen JM, PoulsenLO, Svehag S-E. Factor I deficiency and C3 nephritic factor:immunochemical findings and association with Neissenameningitidis infection in two patients. ScandJ Immunol1984; 20:291-7.
12. Rasmussen JM, Teisner B, Brandslund I, Svehag S-E. Afamily with complement factor I deficiency. ScsndJ Immunol 1986; 23:711-15.
13. Barrett DJ, Boyle MDP. Restoration of complement functionin vivoby plasma infusion in factor I (C3b inactivator)deficiency. J Pediatr 1984; 104:76-81.
14. Porteu F, Fischer A, Descamps-Latscha B, Halbwachs-Mecarelli L. Defective complement receptors (CR1 andCR3) on erythrocytes and leucocytes of factor I (C3binactivator) deficient patients. Clin Exp Immunol 1986;66:463-71.
15. Rasmussen JM, Teisner B, Jepsen HH, Svehag S-E,Knudsen F, Kirstein H, Buhl M. Three cases of factor I

400 T.J. Vyseetal.
deficiency: The effect of treatment with plasma. Clin ExpImmunol 1988; 74:131-6.
16. Maillet F, Weiss L, Chibani), Kazatchkine MD. Deficit enfacteur I, une proteine regulatrice du complement. PresseMed 1990; 19:762. |Fr.|
17. Floret D, Stamm D, Ponard D. Increased susceptibility toinfection in children with congenital deficiency of Factor I.Pediatr Infect Dis J 1991; 10:615-18.
18. Tottori DH, Hilman B, Daul CB. Concomitant factor I andIgA deficiencies. Ann Allergy 1992; 68:115. (abstract).
19. Bonnin AJ, Zeitz HJ, Cewurz A. Complement factor Ideficiency with recurrent aseptic meningitis. Arch InternA<fed1993; 153:1380-3.
20. Harrison RA, Lachmann PJ. Complement technology. In:Weir DM, Herzenberg LA, Blackzell C, eds. Handbook ofExperimental Immunology (4th edn). Oxford, BlackwellScientific Publications, 1986.
21. Walport MJ, Ross GD, Mackworth-Young C, Watson JV,Hogg N, Lachmann PJ. Family studies of erythrocytecomplement receptor type 1 levels: reduced levels inpatients with SLE are acquired not inherited. Clin ExpImmunol 1985; 59:547-54.
22. Lachmann P), Oldroyd RC, Milstein C, Wright BW. Threerat monoclonal antibodies to human C3. Immunology 1980;41:503-15.
23. Hogg N, Ross CD, Jones DB, Slusarenko M, Walport MJ,Lachmann PJ. Identification of an anti-monocytemonoclonal antibody that is specific for membranecomplement receptor type one (CR1). EurJ Immunol 1984;14:236-43.
24. Sim RB, Day AJ, Moffatt BE, Fontaine M. Complementfactor I and cofactors in control of complement systemconvertase enzymes. Methods Enzymol 1993; 223:14-35.
25. Alper CA, Colten HR, Rosen FS, Rabson AR, MacNab CM,Gear JSS. Homozygous deficiency of O in a patient withrepeated infections. Lancet 1972; 2:1179-81.
26 Alper CA, Colten HR, Gear JSS, Rabson AR, Rosen FS.Homozygous human C3 deficiency. The role of C3 inantibody production, Cis-induced vasopermeability, andcobra venom-induced passive hemolysis. ) Clin Invest 1976;57:222-9.
27. Weiss RM, Schulz EJ. Complement deficiency in Sweet'ssyndrome [letter). Br J Dermatol 1989; 121:413-15.
28. Botto M, Fong KY, So AK, Morley BJ, Barlow R, Routier R,Walport MJ. Homozygous hereditary C3 deficiency due to apartial gene deletion. Proc Natl Acad Sci USA 1992;89:4957-61.
29. Ballow M, Shira JE, Harden L, Yang Soo Young, Day NK.Complete absence of the third component of complement inman. J Clin Invest 1973; 56:703-10.
30. Berger M, Balow JE, Wilson CB, Frank MM. Circulatingimmune complexes and glomerulonephritis in a patient withcongenital absence of the third component of complement.N Engl J Med 1983; 308:1009-13.
31. Grace HJ, Brereton-Stiles GG, Vos GH, Schonland M. Afamily with partial and total deficiency of complement C3. 5AfrMedJ 1976; 50:139-40.
32. Osofsky SG, Thompson BH, Lint TF, Gewurz H. Hereditarydeficiency of the third component of complement in a childwith fever, skin rash, and arthralgias: response to transfusionof whole blood. J Pediatr 1977; 90:180-6.
33. Davis III AE, Davis IV JS, Rabson AR, Osofsky SG, ColtenHR, Rosen FS, Alper CA. Homozygous C3 deficiency:
detection of C3 by radioimmunoassay. Clin ImmunolImmunopathoh977; 8:543-50.
34. Pussell BA, Bourke E, Nayef M, Morris S, Peters DK.Complement deficiency and nephritis. Lancet 1980;1:675-7.
35. Sano Y, Nishimukai H, Kitamura H, Nagaki K, Inai S,Hamasaki Y, Maruyama I, Igata A. Hereditary deficiency ofthe third component of complement in two sisters withsystemic lupus erythematosus-like symptoms. ArthritisRheum 1981; 241255-60.
36. Hsieh KH, Lin CY, Lee TC. Complete absence of the thirdcomponent of complement in a patient with repeatedinfections. Clin Immunol Immunopathol 1981; 20:305-12.
37. Roord JJ, Daha M, Kuis W, Verburgh HA, Verhoef), ZegersBJM, Stoop JW. Inherited deficiency of the third componentof complement associated with recurrent pyogenicinfections, circulating immune complexes, and vasculitis ina Dutch family. Pediatrics 1983; 71:81-7.
38. Borzy MS, Houghton D. Mixed pattern immune complexdeposit glomerulonephritis in a child with inheriteddeficiency of the third component of complement. AmJ Kidney Dis 1985; 5:54-9.
39. Borzy MS, Gewurz A, Wolff L, Houghton D, Lovrien E.Inherited C3 deficiency with recurrent infections andglomerulonephritis. Am J Dis Child 1988; 142:79-83.
40. Cozma G, Aburumeih S, Malik-Cozma MC, Johny KV.CAPD in a patient with a complete absence of O . ClinNephrol 1987; 27:269 (Abstract).
41. Grumach AS, Vilela MM, Gonzalez CH, Starobinas N,Pereira AB, Dias-da-Silva W, Carneiro-Sampaio MMS.Inherited C3 deficiency of the complement system. Braz] Med Biol Res 1988; 21:247-57.
42. Imai K, Nakajima K, Eguchi K, Miyazaki M, Endoh H,Tomino Y, Nomoto Y, Sakai H, Hyodo Y. Homozygous C3deficiency associated with IgA nephropathy. Nephron 1991;59:148-52.
43. Peleg D, Harit-Bustan H, Katz Y, Peller S, Schlesinger M,Schonfeld S. Inherited C3 deficiency and meningococcaldisease in a teenager. Pediatr Infect Dis J 1992; 11:401 -4.
44. Thompson RA, Winterborn MH. Hypocomplementaemiadue to a genetic deficiency of betai H globulin. Clin ExpImmunol 1981; 46:110-19.
45. Levy M, Halbachs-Mecarelli L, Gubler M-C, Kohout G,Bensenouci A, Niaudet P, Hauptmann G, Lesavre P. Hdeficiency in two brothers with atypical intramembranousdeposit disease. Kidney Int 1986; 30:949-56.
46. Lopez-Larrea C, Diegez MA, Enguix A, Dominguez O,Marin B, Gomez E. A family deficiency of complementfactor H. Biochem Soc Trans 1987; 15:648-9.
47. Brai M, Misiano G, Maringhini S, Cutaja I, Hauptmann G.Combined homozygous factor H and heterozygous C2deficiency in an Italian family. J Clin Immunol 1988;8:50-6.
48. Nielsen HE, Christensen KC, Koch C, Thomsen BS,Heegaard NH, Tranum Jensen J. Hereditary, completedeficiency of complement factor H associated withrecurrent meningococcal disease. ScandJ Immunol 1989;30:711-18.
49. Fijen CA, Kuijper EJ, Hannema AJ, Sjoholm AG, van PuttenJP. Complement deficiencies in patients over ten years oldwith meningococcal disease due to uncommon serogroups.Lancet 1989; 2:585-8.
50. Nilsson UR, Nilsson B, Storm KE, Sjolin-Forsberg G,Hallgren R. Hereditary dysfunction of the third component

Hereditary complement factor I deficiency 401
of complement associated with an SLE-like syndrome andmeningococcal meningitis. Arthritis Rheum 1992;35:580-5.
51. McClean RH, Weinstem A, Chapitis J, Lowenstein M,Rothfield NF. Familial partial deficiency of the thirdcomponent of complement (C3) and thehypocomplementaemic vasculitis syndrome. Ami Med1980; 68:549-58.
52. Wyatt RJ, Julian BA, Weinstein A, Rothfield NF, McCleanRH. Partial H (betai H) deficiency with glomerulonephritisin two families. I Clin Immunol 1982; 2:110-17.
53. Roodhooft AM, McClean RH, Elst E, Van AckerKJ. Recurrent haemolytic uraemic syndrome and acquiredhypomorphic variant of the third complement componentPediatr Nephrol 1990; 4:597-9.
54. Ross SC, Densen P. Complement deficiency states andinfection: epidemiology, pathogenesis and consequences ofNeisserial and other infections in an immune deficiency.Medicine (Baltimore) 1984; 63:243-73.
55. Davies KA, Erlendsson K, Beynon HLC, Peters AM,Steinsson K, Valdimarsson H, Walport MJ. Splenic uptake ofimmune complexes in man is complement-dependent.) Immunol 1993; 151:3866-73.
56. Cork CL, Morris JM, Olson JL, Krakowka S, Swift AJ,Winkelstein JA. Membranoproliferative glomerulonephritisin dogs with genetically-determined deficiency of the thirdcomponent of complement. Chn Immunol Immunopathol1991;60:455-70S.
57. Spath PJ, Misiano G, Goetz G, Wurthrich B, Hauptmann G,Butler R. Heterozygous condition of factor I, C4A or C4B ina kindred with hereditary angioedema (HAE). Complement1985,2:73.
58. Hazlewood MA, Kumaratne DS, Webster ADB, Goodall M,Bird P, Daha M. An association between homozygous C3deficiency and low levels of anti-pneumococcalpolysaccharide. Clin Exp Immunol 1992; 87:404-9.
59. Bird P, Lachmann PJ. The regulation of IgG subclassproduction in man: low serum lgG4 in inheriteddeficiencies of the classical pathway of C3 activation, furJ Immunol 1988; 18:1217-22.
60. Ochs HD, Wedgwood RJ, Heller SR, Beatty PG.Complement, membrane glycoproteins, and complementreceptors: their role in regulation of the immune response.Clin Immunol Immunopathol 1986; 40:94-104.
61. Biselli R, Casapollo I, fJAmelio R, Salvato S, Matricardi PM,Brai M. Antibody response to meningococcalpolysaccharides A and C with complement defects. ScandJ Immunol 1993; 37:644-50.
62. HSssig Von A, Borel JF, Ammann P, Thoni M, Butler R.Essentialle hypokomplementSmie. Pathol Microbiol. 1964;27:542-7.
63. Fukumori Y, Yoshimura K, Ohnoki S, Yamaguchi H,Akagaki Y, Inai S. A high incidence of C9 deficiency amonghealthy blood donors in Osaka, Japan. Int Immunol 1988;1:85-9.
64. Roord JJ, van Dienn van Steenvoorde RAAM, SchuurmannHJ, Rijkers GT, Zegers BJM, Gmelig-Meyling FHJG, StoopJW. Membranoproliferative glomerulonephritis in a patientwith congenital deficiency of the third component ofcomplement: effect of treatment with plasma. Am J KidneyDis 1989; 13:413-17.
65. Steinsson K, Erlendsson K, Valdimarsson H. Successfulplasma infusion treatment of a patient with C2 deficiencyand systemic lupus erythematosus: clinical experience overforty-five months. Arthritis Rheum 1989; 32:906-13.





























