Embed Size (px)
Citation preview

© 2005 Nature Publishing Group
O P I N I O N
Neurofibromatosis type 1 — a model for nervous system tumour formation?Joshua B. Rubin and David H. Gutmann
and mast cells. Neurofibromas have been subdivided into discrete (dermal) neurofi-bromas, which are associated with a single peripheral nerve, and diffuse (plexiform) neurofibromas, which involve multiple nerve fascicles. Molecular analyses of neurofibro-mas have shown that only the Schwann cells in these tumours exhibit biallelic inactivation of the NF1 gene and complete loss of expres-sion of neurofibromin (the protein encoded by NF1; see below). The other cellular ele-ments are genetically heterozygous for an inactivating NF1 mutation and therefore maintain neurofibromin expression3–5.
Cutaneous (dermal) neurofibromas typically arise during the second decade of life, are often associated with the onset of puberty, and continue to increase in num-ber and size throughout adulthood. One of the curious features of these neurofibromas is their growth pattern, which is character-ized by an initial period of growth, followed by clinical quiescence for years or decades. On the basis of this pattern, it has been hypothesized that the neoplastic cellular elements within the tumour might be pro-liferating in response to a limiting supply of mitogen. In addition, the presence of other cell types within the tumour indicates that these stromal elements might directly influ-ence neurofibroma formation and growth by providing crucial mitogenic signals to NF1–/– Schwann cells.
Most peripheral nerve sheath tumours in individuals with NF1 are benign tumours with no lifetime risk of malig-nant transformation. However, the other subtype of neurofibroma (plexiform neu-rofibroma) can develop into cancer (malig-nant peripheral nerve sheath tumour; MPNST)6. Plexiform neurofibromas are thought to represent congenital tumours. They are often detected at birth and seem to enlarge more rapidly during the first decade of life. These tumours are similar to the discrete (cutaneous) neurofibroma, but are more extensive and often involve spinal or cranial nerve roots rather than
Abstract | Neurofibromatosis type 1 (NF1) is a common genetic condition in which affected individuals develop benign and malignant nervous system tumours. Genetically engineered mouse (GEM) models of these NF1-associated nervous system tumours recapitulate several of the unique clinical aspects of the disease. Moreover, these Nf1 GEM models allow for a direct examination of the earliest stages of tumour evolution, including the contributions that Nf1+/– cellular elements and cooperating genetic changes make to facilitate the transition from the pre-neoplastic to the neoplastic state and, in some cases, to promote malignant progression.
Tumour-predisposition syndromes provide a unique opportunity to identify and study genes that when dysfunctional lead to the development of cancer. These heritable syndromes represent nature’s ultimate mutagenesis experiment and clearly establish the responsible target genes as critical growth regulators in the patho-genesis of specific human cancers. One of the most common inherited tumour-predisposition syndromes is neurofibro-matosis type 1 (NF1), which affects 1 in 3,000 individuals worldwide1. As seen in other tumour-predisposition syndromes, individuals with NF1 are born with one mutated (non-functional) and one wild-type (functional) NF1 gene in every cell of their body. This NF1 heterozygous
condition is, by itself, insufficient for tumorigenesis; inactivation of the remain-ing wild-type NF1 allele in an individual cell (for example, a Schwann cell) is the rate-limiting step for subsequent tumour formation (for example, neurofibroma)2. In the NF1 tumour-predisposition syn-drome, nearly all individuals will develop a nervous system tumour sometime dur-ing their lifetime. In the nervous system, tumours arise from glial cells associated with peripheral nerves (Schwann cells) or astrocytes in the brain (astrocytomas or gliomas). Peripheral nerve sheath tumours composed of neoplastic Schwann cells typi-cally develop in early adulthood, whereas low-grade astrocytomas in the central nervous system arise almost exclusively during childhood. In addition to nervous system tumours, individuals with NF1 also manifest specific learning disabilities, pigmentary abnormalities (café-au-lait macules, freckling in the armpit and groin regions, and iris hamartomas), distinctive bony defects, cardiovascular abnormalities and other cancers (pheochromocytoma and leukaemia). The clinical features of NF1 are shown in BOX 1.
Nervous system tumours in NF1 Neurofibromas are benign, grade I (under World Health Organization (WHO) clas-sification) tumours composed of neoplastic Schwann cells and non-neoplastic stromal cells, including fibroblasts, perineurial cells
NATURE REVIEWS | CANCER VOLUME 5 | JULY 2005 | 557
PERSPECTIVES

© 2005 Nature Publishing Group
1 2,818
9 a 23 a 48 a
IRA-1 IRA-2GRD
small cutaneous nerves. Approximately 10% of individuals with NF1 will develop an MPNST, a clinically aggressive, often fatal cancer, which is poorly responsive to chemotherapy or radiation. Some MPNSTs arise in pre-existing plexiform neurofi-bromas, but many arise without clinical or radiographic evidence of a precursor benign tumour7,8. Many studies have exam-ined the genetic changes associated with MPNST, both in the context of NF1 and in sporadic tumours. Loss of NF1 gene expression is observed in both NF1-associ-ated and sporadic MPNSTs, and seems to be a common unifying molecular feature of these cancers4. However, expression profiling experiments have not identi-fied a unique molecular signature that distinguishes NF1-associated tumours from sporadic MPNSTs9. The next most frequent genetic changes identified were homozygous deletion of CDKN2A, which
encodes p16 (INK4A) and p14 (ARF); TP53 loss; and inactivation of CDKN1B, which encodes p27 (KIP1)10–13.
Children with NF1 are prone to the development of WHO grade I pilocytic astrocytomas primarily involving the optic nerves, chiasm and hypothalamus, with less frequent involvement of the brainstem14. NF1-associated optic-pathway astrocyto-mas typically develop in children less than 10 years of age, grow most rapidly in early childhood (ages 2–5), and then enter into a quiescent phase during adolescence and adulthood. Overall, these astrocytomas exhibit more indolent clinical behaviour compared with their sporadic counter-parts15,16. Remarkably, some of these low-grade NF1-associated astrocytomas have been reported to spontaneously regress after childhood17. Histological examina-tion of these tumours shows the pres-ence of new blood vessels and infiltrating macrophage-like cells (microglia). As the mainstay of clinical management of these tumours is chemotherapy and not surgery, limited pathological or molecular studies have been performed. In the few stud-ies that have been done, NF1 inactivation and loss of neurofibromin expression have been found in all NF1-associated pilocytic astrocytomas examined, but other genetic changes observed in high-grade gliomas are not seen18,19. Interestingly, in contrast to MPNST, NF1 inactivation is not found in the histologically identical pilocytic astrocy-tomas that occur in the general population, indicating that a different genetic mecha-nism underlies the development of sporadic pilocytic astrocytoma20.
High-grade gliomas are more frequently observed in adults with NF1 than in the general population, and tend to occur earlier in life21. Despite this increased inci-dence, high-grade gliomas in adults with NF1 are rare and do not arise from pre-existing pilocytic astrocytomas. Molecular analyses of these uncommon tumours have demonstrated NF1 inactivation as well as homozygous CDKN2A inactivation22.
Aside from NF1 inactivation, which is not observed in sporadic high-grade gliomas, the other genetic changes in NF1-associ-ated high-grade brain tumours are similar to their sporadic counterparts.
Mechanisms of NF1 growth regulationThe NF1 gene was identified by posi-tional cloning in 1990 and demonstrated to encode a cytoplasmic protein, termed neurofibromin23,24 (FIG. 1). Neurofibromin is a large 220–250-kDa protein contain-ing three alternatively spliced exons (9a, 23a and 48a). These alternatively spliced exons might reflect tissue-specific or dif-ferentiation-regulated RNA splicing events. Expression of neurofibromin containing exon 9a seems to be restricted to specific neuronal populations in the central nervous system25, whereas genetic ablation of exon 23a in mice results in learning disabilities similar to Nf1+/– mice, but no tumour devel-opment26. Further study of the expression and function of these neurofibromin iso-forms might provide insight into the roles of neurofibromin in specific tissues.
Inspection of the amino-acid sequence of neurofibromin revealed one central, 400-amino-acid region with sequence similarity to proteins found in Drosophila melanogaster, other vertebrates and yeast that function as negative regulators of RAS27. These GTPase-activating proteins (GAPs) accelerate the conversion of active GTP-bound RAS to its inactive GDP-bound form, inactivating RAS and reducing RAS-mediated growth signalling. The intrinsic GTPase activity of wild-type, but not oncogenic, RAS can be stimulated by the GAP-related domain (GRD) of neurofibromin28–30, such that loss of neurofibromin results in hyperactiva-tion of RAS31–34. Since dysregulated RAS activity is mitogenic for primary cells and transforming for established cell lines, the NF1 gene has been hypothesized to function as a tumour suppressor by inhibiting RAS activity and suppressing RAS-mediated cell growth. In addition to the GRD, there are sequences flanking the GRD in mam-malian neurofibromin that are shared with yeast GAP-like (IRA) proteins and that are important in yeast for IRA RAS-GAP func-tion (IRA domains). It is not clear whether neurofibromin growth regulation requires these IRA homology domains, as the growth advantage observed in Nf1-deficient cells can be reversed by re-expression of the Nf1 GRD alone35. In mouse fibroblasts, the normal function of neurofibromin seems to include the regulation of RAS and its downstream effectors, AKT (also known as
Box 1 | Clinical features of NF1
Established diagnostic criteria for neurofibromatosis type 1 (NF1)• Multiple café-au-lait macules• Multiple skinfold freckles• Iris hamartomas (Lisch nodules)• Multiple discrete neurofibromas or one
plexiform (diffuse) neurofibroma• Optic pathway glioma• Distinctive bony abnormalities
• First-degree relative with NF1
Other associated features of NF1• Specific learning disabilities• Unidentified bright objects on brain
magnetic resonance imaging• Short stature• Cardiovascular abnormalities• Malignant peripheral nerve sheath
tumours• Leukaemia• Pheochromocytoma
Figure 1 | Structure of the NF1 gene product, neurofibromin. Neurofibromin is a 2,818-amino acid protein with 3 alternatively spliced exons (9a, 23a and 48a). These alternatively spliced exons might reflect tissue-specific or differentiation-regulated RNA splicing events. There are two regions flanking the GAP-related domain (GRD) that show sequence similarity with the yeast IRA proteins (IRA-1 and IRA-2 domains) that function as negative regulators of RAS. The switch between active GTP-bound RAS to its inactive GDP bound form is stimulated by the GRD of neurofibromin.
558 | JULY 2005 | VOLUME 5 www.nature.com/reviews/cancer
P E R S P E C T I V E S

© 2005 Nature Publishing Group
AC
RAS
NF1
cAMPPDEAKT
ERK1/2
mTOR/S6K CDC42/RAC1/PAK
RAP1PI3K
GPCR
RTK
protein kinase B) and ERK1/2 (extracellular signal-regulated kinase 1/2), and modula-tion of mitogenic responses downstream of both receptor tyrosine kinases and G-protein-coupled receptors36.
The consequences of increased RAS activation in neurofibromin-deficient cells is manifested by increased activation of vari-ous RAS effectors, including RAF, ERK1/2, phosphatidylinositol 3-kinase (PI3K) and p21-activated kinase (PAK)37–42 (FIG. 2). The activation of these pathways contributes to the increased proliferation of Nf1–/– Schwann cells and astrocytes, as well as the increased proliferation and motility of Nf1+/– mast cells, Schwann cells and astrocytes38,39,43,44. In
haematopoietic cells, loss of neurofibromin not only results in increased PI3K activation of AKT, but also hyperactivation of mitogen-activated protein kinase through increased PAK activity. These results indicate that there might be a significant amount of crosstalk among these RAS downstream pathways, which might synergize to augment cell growth and survival. Recent studies in Nf1–/– astrocytes have shown that neurofibromin loss results in activation of only one RAS isoform (KRAS), despite abundant expres-sion of all three isoforms in astrocytes. In addition, activated KRAS, but not activated HRAS, accounts for the unique biological and signalling abnormalities associated with Nf1 loss in vitro and can substitute for Nf1 loss in astrocytes during optic glioma formation in vivo64. Moreover, loss of neuro-fibromin in astrocytes leads to hyperactiva-tion of the mammalian target of rapamycin (mTOR)–S6-kinase (S6K) pathway, leading to increased cell proliferation and protein translation72. This mTOR–S6K hyperactiva-tion is seen in both genetically engineered Nf1 mouse and human NF1-associated optic glioma, and rapamycin treatment selectively inhibits Nf1–/– astrocyte proliferation in vitro. The observation that KRAS and mTOR–S6K pathway activation result from NF1 loss pro-vides new potential therapeutic targets for NF1-associated tumours.
The regulation of RAS and its vari-ous downstream effectors is not the only function of neurofibromin. Restoration of RAS regulation in D. melanogaster does not correct the small body size defect that results from Nf1 inactivation45,46. Moreover, reconstitution of neurofibromin in NF1-deficient melanoma cells inhibits their growth without affecting RAS-GTP levels47, and whereas restoration of the neurofibro-min GRD in Ira-deficient yeast can restore GTPase regulation28, some neurofibromin GRD mutants retain GTPase activity, but lack other properties associated with yeast IRA function (for example, heat-shock sensitivity)48. Collectively, these results indicate that neurofibromin might have growth-regulatory properties beyond its ability to function as a RAS-GAP.
One of the additional functions ascribed to neurofibromin is regulation of adenylyl-cyclase activity and intracellular cyclic-AMP generation. In D. melanogaster, the body size abnormality and defects in learning and memory that result from Nf1 loss were corrected by protein kinase A activation, implicating the adenylyl-cyclase pathway45,46. Additional experiments demonstrated that in D. melanogaster, neurofibromin is required
for activation of adenylyl cyclase in response to defined mitogens, including PACAP (pituitary adenylyl-cyclase-activating pep-tide)49. Similarly, neurofibromin-deficient astrocytes fail to activate adenylyl cyclase in response to PACAP50. However, the role of neurofibromin in cAMP regulation does not seem to be limited to the activation of ade-nylyl cyclase, as resting levels of cAMP are increased in both Nf1–/– Schwann cells51 and astrocytes50. This baseline increase of cAMP could be related to RAS-dependent regula-tion of phosphodiesterases by ERK2 REF. 52, but this has not been formally examined in Nf1-deficient cells. Interestingly, expres-sion of the Nf1 GRD can partially restore the normal adenylyl-cyclase response to PACAP, indicating that sequences within the GRD might be responsible for mediating adenylyl-cyclase regulation in astrocytes50. It should be noted that oncogenic RAS expression in astrocytes has no effect on intracellular cAMP levels, arguing against a direct relationship between neurofibromin RAS-GAP function and adenylyl-cyclase regulation50. Collectively, these data sup-port the notion that some of the functions of neurofibromin might be independent of its RAS-GAP activity.
Mouse models One of the challenges to understanding the pathogenesis of NF1-associated nervous system tumours is the lack of information regarding the transition from the pre-neo-plastic (hyperproliferative) state to frank neoplasia. In this respect, most studies of benign NF1-associated nervous system tumours have focused on surgically excised neurofibromas, which have passed through their pre-neoplastic stage (hyperplasia) and represent fully evolved tumours. Moreover, in the case of neurofibromas, many of these tumours are removed after their period of most rapid growth, preserving only their fossil history and providing few insights into the active processes that facilitate neoplastic transformation. Similarly, the few studies on pilocytic astrocytomas in NF1 have focused on the genetic changes in these fully developed brain tumours.
With the advent of genetically engineered mouse models of NF1-associated tumours, we now have the opportunity to carefully analyse the transition from hyperplasia to neoplasia, and to examine the cellular and genetic conditions that contribute to tumour formation. Over the past several years, several groups have developed clinically relevant models of NF1-associated nervous system tumours using both conventional and
Figure 2 | Signalling pathways that are regulated by neurofibromin. Neurofibromin (NF1) modulates intracellular signalling that originates from both receptor tyrosine kinases (RTKs) and G-protein-coupled receptors (GPCR). Among the direct, physiological functions of neurofibromin seems to be the inhibition of RAS and activation of adenylyl cyclase (AC). Neurofibromin loss is associated with increased activation of RAS and its downstream mediators including phosphatidylinositol 3-kinase (PI3K), AKT, mammalian target of rapamycin (mTOR), S6 kinase (S6K), MEK (mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase) and ERK1/2 as well as RAC1 and PAK1 (p21-activated kinase 1). Neurofibromin loss is also associated with deficient generation of cAMP downstream of Gαs activation, but increased baseline cAMP levels. The former effect is likely to contribute to the hyperactivation of ERK1/2 through the loss of RAC1 inhibition, while the latter could be the result of RAS-dependent inhibition of phosphodiesterases (PDEs). As illustrated, there is probably significant crosstalk between signalling pathways modulated by neurofibromin.
NATURE REVIEWS | CANCER VOLUME 5 | JULY 2005 | 559
P E R S P E C T I V E S
© 2005 Nature Publishing Group
NF1+/–
NF1+/–
NF1 loss
Mast cells
CDKN2A orTP53 inactivation
CDKN2A or TP53
inactivation
Schwann cell
Schwann cellhyperproliferation Benign neurofibroma
Malignant nerve sheath tumour
Peripheral nervous system
NF1 loss
Brain
CDKN2A orTP53 inactivation
Astroglial cell
Astroglialhyperproliferation Low-grade astrocytoma
High-grade astrocytoma
Central nervous system
NF1+/–
NF1+/–
NF1+/–
NF1+/–
conditional Nf1 gene inactivation in vivo. Whereas mice lacking neurofibromin are not viable, Nf1+/– mice (genetically similar to humans with NF1) are healthy at birth but succumb to leukaemia and pheochromocy-toma by 15–18 months of age53. Analysis of the tumours from these Nf1+/– mice demon-strates homozygous Nf1 gene inactivation and loss of neurofibromin expression, con-sistent with Knudson’s two-hit hypothesis. Unfortunately, these mice develop neither neurofibromas nor astrocytomas.
To circumvent the embryonic lethality associated with homozygous Nf1 inactivation, Parada and colleagues generated mice bearing
conditional Nf1 alleles (Nf1flox/flox mice), which could be rendered neurofibromin-deficient in specific cell types by Cre-mediated excision54. By crossing Nf1flox/flox mice with transgenic mice in which Cre expression is regulated by the Schwann cell-specific KROX20 pro-moter (Nf1flox/flox; KROX20-Cre mice)55 or by using Nf1–/– embryonic Schwann cells56, the consequence of complete loss of neuro-fibromin expression in Schwann cells could be analysed. Such loss of neurofibromin in vitro or in vivo was associated with a modest growth advantage, but no evidence of tumour formation55. Similarly, when Nf1flox/flox mice were crossed with transgenic mice in which
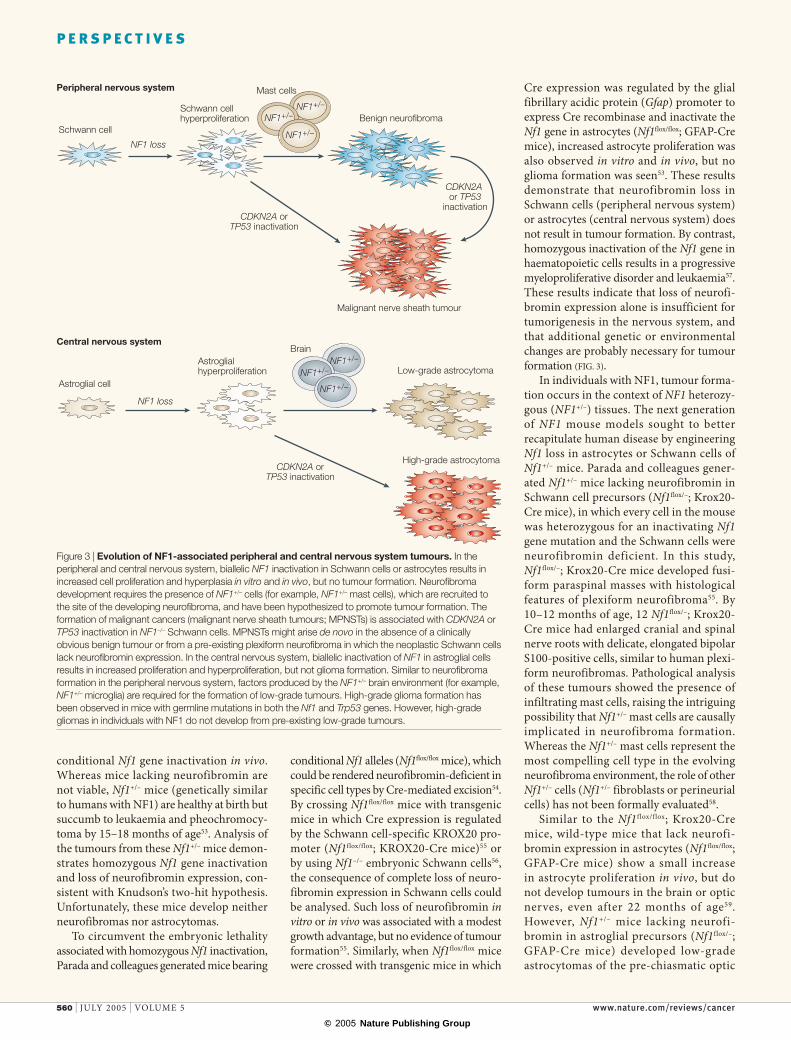
Cre expression was regulated by the glial fibrillary acidic protein (Gfap) promoter to express Cre recombinase and inactivate the Nf1 gene in astrocytes (Nf1flox/flox; GFAP-Cre mice), increased astrocyte proliferation was also observed in vitro and in vivo, but no glioma formation was seen53. These results demonstrate that neurofibromin loss in Schwann cells (peripheral nervous system) or astrocytes (central nervous system) does not result in tumour formation. By contrast, homozygous inactivation of the Nf1 gene in haematopoietic cells results in a progressive myeloproliferative disorder and leukaemia57. These results indicate that loss of neurofi-bromin expression alone is insufficient for tumorigenesis in the nervous system, and that additional genetic or environmental changes are probably necessary for tumour formation (FIG. 3).
In individuals with NF1, tumour forma-tion occurs in the context of NF1 heterozy-gous (NF1+/–) tissues. The next generation of NF1 mouse models sought to better recapitulate human disease by engineering Nf1 loss in astrocytes or Schwann cells of Nf1+/– mice. Parada and colleagues gener-ated Nf1+/– mice lacking neurofibromin in Schwann cell precursors (Nf1flox/–; Krox20-Cre mice), in which every cell in the mouse was heterozygous for an inactivating Nf1 gene mutation and the Schwann cells were neurofibromin deficient. In this study, Nf1flox/–; Krox20-Cre mice developed fusi-form paraspinal masses with histological features of plexiform neurofibroma55. By 10–12 months of age, 12 Nf1flox/–; Krox20-Cre mice had enlarged cranial and spinal nerve roots with delicate, elongated bipolar S100-positive cells, similar to human plexi-form neurofibromas. Pathological analysis of these tumours showed the presence of infiltrating mast cells, raising the intriguing possibility that Nf1+/– mast cells are causally implicated in neurofibroma formation. Whereas the Nf1+/– mast cells represent the most compelling cell type in the evolving neurofibroma environment, the role of other Nf1+/– cells (Nf1+/– fibroblasts or perineurial cells) has not been formally evaluated58.
Similar to the Nf1f lox/f lox; Krox20-Cre mice, wild-type mice that lack neurofi-bromin expression in astrocytes (Nf1flox/flox; GFAP-Cre mice) show a small increase in astrocyte proliferation in vivo, but do not develop tumours in the brain or optic nerves, even after 22 months of age59. However, Nf1+/– mice lacking neurofi-bromin in astroglial precursors (Nf1flox/–; GFAP-Cre mice) developed low-grade astrocytomas of the pre-chiasmatic optic
Figure 3 | Evolution of NF1-associated peripheral and central nervous system tumours. In the peripheral and central nervous system, biallelic NF1 inactivation in Schwann cells or astrocytes results in increased cell proliferation and hyperplasia in vitro and in vivo, but no tumour formation. Neurofibroma development requires the presence of NF1+/– cells (for example, NF1+/– mast cells), which are recruited to the site of the developing neurofibroma, and have been hypothesized to promote tumour formation. The formation of malignant cancers (malignant nerve sheath tumours; MPNSTs) is associated with CDKN2A or TP53 inactivation in NF1–/– Schwann cells. MPNSTs might arise de novo in the absence of a clinically obvious benign tumour or from a pre-existing plexiform neurofibroma in which the neoplastic Schwann cells lack neurofibromin expression. In the central nervous system, biallelic inactivation of NF1 in astroglial cells results in increased proliferation and hyperproliferation, but not glioma formation. Similar to neurofibroma formation in the peripheral nervous system, factors produced by the NF1+/– brain environment (for example, NF1+/– microglia) are required for the formation of low-grade tumours. High-grade glioma formation has been observed in mice with germline mutations in both the Nf1 and Trp53 genes. However, high-grade gliomas in individuals with NF1 do not develop from pre-existing low-grade tumours.
560 | JULY 2005 | VOLUME 5 www.nature.com/reviews/cancer
P E R S P E C T I V E S

© 2005 Nature Publishing Group
NF1+/–
Proliferation Chemoattractants
a
Proliferation
b
NF1+/–Proliferation
c
Mitogens
NF1–/–
NF1–/–
NF1–/–
nerve and chiasm by 2 months of age, implicating the Nf1+/– brain environment in the process of gliomagenesis60. In these mice, fusiform optic nerve and chiasm masses were detected, which contained hyperproliferating GFAP-immunoreactive cells with large atypical hyperchromatic nuclei, similar to optic nerve gliomas in children with NF1. Although there is evi-dence for increased astrocyte proliferation within the optic nerve by 3 weeks of age (pre-neoplastic stage) in Nf1flox/–; GFAP-Cre mice, nuclear changes typical of low-grade glioma were not seen until 7–8 weeks of
age61. In addition, brain magnetic resonance imaging demonstrated abnormal water dif-fusion in the optic nerves only at the tumour stage (7–8 weeks of age), and not in the pre-neoplastic tissue at 3 weeks of age, indicating that only neoplasia, but not the pre-neo-plastic state, was detected radiographically. Moreover, infiltrating activated microglia and new blood vessel formation were found in the pre-neoplastic optic nerves at 3 weeks of age before the development of an obvious tumour. These results raise the possibility that Nf1+/– cells might participate in the transition from the pre-neoplastic stage to frank neoplasia during the formation of an optic-pathway glioma.
Based on the observation that nearly 50% of human MPNST and a smaller proportion of NF1-associated high-grade glioma exhibit TP53 gene mutations, malig-nant NF1-associated tumours have been modelled in mice that are heterozygous for germline mutations in both the Nf1 and Trp53 genes. Similar to what is seen in the human disease, these mice develop MPNST and gliosarcoma (high-grade glioma)62. We hypothesize that loss of p53 function in astrocytes or Schwann cells is crucial in the development of these nervous system malig-nancies, as individuals with the Li–Fraumeni inherited cancer syndrome, resulting from germline TP53 mutation, have an increased risk of developing a range of malignancies (including soft tissue sarcoma, which is similar to MPNST, and high-grade glioma). Not all NF1-associated nervous system malignancies have loss of p53 function, but it seems likely that changes in other growth-regulatory genes (for example CDKN1B or CDKN2A) account for cancer development in the absence of TP53 loss. In the case of NF1-associated nervous system cancers, the growth advantage that is provided by neurofibromin loss and subsequent RAS hyperactivation is insufficient for the for-mation of high-grade tumours. Moreover, neither oncogenic KRAS expression in astrocytes or in Schwann cells leads to tumor formation, whereas RAS activation when combined with TP53 inactivation is sufficient for neoplastic transformation63,64. On the basis of these findings, neurofibro-min loss in the context of Trp53 inactiva-tion provides a mitogenic advantage that cooperates with disruption of p53 function to promote malignant tumour formation. This cooperativity could reflect either loss of p53-mediated cell-cycle regulation or disruption of the normal apoptotic (pro-grammed cell death) processes that are controlled by p53. Further studies will be
required to distinguish between these two non-mutually exclusive possibilities.
Molecular analyses of human NF1-associated MPNST and high-grade glioma have shown that one of the most frequent genetic changes is homozygous deletion of the CDKN2A locus that contains the INK4A and ARF (Arf/p19 in the mouse) tumour-suppressor genes. In one study examining the relationship between Nf1 and Arf inactivation, loss of one Nf1 allele did not greatly shorten the time to tumour formation in Arf –/– mice, and the tumours observed were characteristic of Arf –/– mice and not the tumours seen in individuals with NF1 REF. 65. However, many of these Nf1+/–;Arf –/– mice developed multiple tumours, a phenotype not seen in Arf –/– mice. Studies are in progress to determine whether inactivation of the entire CDKN2A locus facilitates high-grade glioma formation in Nf1 mutant mice.
In addition to these genetic changes, it is likely that there are modifying genes in the mouse (or human) genome that influ-ence tumour susceptibility. Recent studies have demonstrated a modifying effect of the mouse genetic strain on tumour phenotype when Nf1+/–;p53+/– mice were maintained on a C57Bl/6 genetic background, compared to Nf1+/–;p53+/– mice maintained on an sv129 genetic background62. These initial obser-vations led to the identification of a genetic locus on chromosome 11 in the mouse that probably functions to modify the tumour spectrum, and might explain why certain individuals with NF1 are more likely to develop one type of cancer, whereas other individuals, even within the same family, develop other tumour types66.
Underlying cellular mechanisms The study of tumour formation in the various Nf1 mouse models highlights the complexity of oncogenesis and, in particular, the necessity for cooperating genetic and environmental events for the transformation of hyperprolifer-ating, precancerous lesions into frank tumours (FIG. 4). Hyperproliferative, non-neoplastic lesions have been described in a wide spec-trum of diseases, including those related to the genesis of other paediatric cancers, and could provide insights into the biology of these early stages of oncogenesis. In this regard, a mouse model of medulloblastoma has been particularly informative. In this model, caused by a germline mutation in a single allele of the sonic hedgehog receptor gene patched (Ptc), abnormal foci of granule lineage cells — also known as hyperplastic rests — are found in the cerebellum well before the development of true neoplastic lesions67,68. These hyperplastic
Figure 4 | Interaction between stromal cells and hyperproliferating pre-neoplastic cells. Complete loss of NF1 (the gene that encodes neurofibromin) is necessary, but not sufficient, for efficient nervous system tumour formation. In the peripheral nervous system, loss of neurofibromin is an initiating step in neurofibroma formation. a | NF1–/– Schwann cells are hyperproliferative and secrete chemoattractants (for example, KIT ligand) and angiogenic factors (for example, the heparin-binding growth factor midkine). b | Secretion of these chemoattractants results in the recruitment of NF1+/– mast cells and endothelial cells to the hyperplastic lesions, and in doing so, alters the developing tumour microenvironment. c | Recruited NF1+/– mast cells secrete mitogens or survival factors that facilitate transformation and result in neurofibroma formation. A similar scenario might also occur in the central nervous system where hyperproliferating NF1–/– astrocytes require additional factors (for example, mitogens or survival factors) from the surrounding NF1+/– brain for glioma formation to occur. The combination of factors produced by NF1+/– microglia as well as mitogens localized to regions of NF1-associated optic glioma formation, such as the chemokine CXCL12, synergize to promote tumorigenesis.
NATURE REVIEWS | CANCER VOLUME 5 | JULY 2005 | 561
P E R S P E C T I V E S

© 2005 Nature Publishing Group
rests retain Ptc expression, but show increased and sustained proliferation beyond the normal developmental period, and possess a unique pattern of gene expression that is related to, but not identical to, either normal granule precursor cells or medulloblastoma cells. In particular, these hyperplastic cells showed altered expression of genes related to differen-tiation, migration and survival73. While most Ptc+/– mice harbor these precancerous lesions, only 16% develop frank medulloblastoma. The relationship between these hyperplastic lesions and the development of medulloblas-toma remains unclear. It is possible that these rests represent a reservoir of tumour precur-sor cells which are uniquely susceptible to the acquisition of additional cellular and genetic changes necessary for tumour formation.
Aggregates of hyperproliferating, imma-ture glial cells are also found in the optic nerves of Nf1flox/–; GFAP-Cre mice before optic glioma formation61. These hyperpro-liferative lesions contain cells that express protein markers (such as nestin and brain lipid-binding protein) normally found in neural stem cells, but are distinguished from frank tumours by a lack of nuclear atypia. It is conceivable that these immature glial cells represent specialized progenitor cells capable of serving as a reservoir of susceptible targets for additional genetic or cellular changes important in the process of tumorigenesis. Recent studies have demonstrated that brain tumour progenitor (stem) cells can be isolated from a wide range of human brain tumours, including medulloblastoma, pilocytic astrocy-toma and glioblastoma multiforme69,70. These cancer-associated stem cells are self-renewing and can generate neurons, oligodendrocytes and astrocytes. Remarkably, cancer stem cells isolated from human medulloblastoma and glioblastoma multiforme specimens can fully reconstitute the original tumour phenotype when implanted into rodent brains69. Whether similar tumour stem cells can arise from hyperplastic rests and what factors influence their development remains unclear. Further studies of these progenitor cell populations in tumour predisposition mouse models are likely to yield important insights into the role(s) of these cells in nervous system tumorigenesis.
Tumour environment is one of the most important contributing factors critical for neoplastic transformation in NF1 REFS 55,60. Multiple possibilities can be envisioned for the interaction between tumour precur-sor cells and their environment. First, the interaction could recapitulate normal stro-mal regulation of proliferation and survival such as occurs during development. In this
regard, the proper developmental context conferred by the tumour environment might explain the rarity of retinoblastoma formation after retinal development is com-plete. Alternatively, it is possible that the interaction between tumour and stroma is bi-directional, with the tumour precursor cells altering the normal stroma to support tumour development. In NF1-associated nervous system tumours (neurofibroma and low-grade glioma), there are data to indicate that the recruitment of Nf1+/– stromal ele-ments (new blood vessels, microglia/mast cells and fibroblasts) into the pre-neoplastic tumour region could result in the elabora-tion of mitogenic factors that facilitate the neoplastic transformation of Nf1–/– Schwann cells or astrocytes39. In this bi-directional interaction model, the Nf1–/– pre-neoplastic cells alter the tumour microenvironment by inducing the production of additional factors important for tumour formation.
The earliest descriptions of neurofibro-mas included reference to their cellular heterogeneity and the presence of numer-ous infiltrating immune system (mast) cells. The pathogenetic importance of these infiltrating mast cells is underscored by observations made in Nf1 mouse tumour models. As mentioned above, Schwann cell inactivation of the Nf1 gene results in the development of mostly hyperplasia, whereas the presence of an Nf1+/– peripheral nerve environment results in robust neurofibroma formation associated with the infiltration of mast cells55. This model has provided both genetic support for the Schwann cell origin of neurofibromas and established that the Nf1+/– cellular context provides other factor(s) necessary for tumour forma-tion. In the developing neurofibroma, the mast cell has been proposed as the critical Nf1+/– stromal cell that facilitates tumour formation. In these studies, Yang and col-leagues have shown that Nf1–/– Schwann cells secrete mast cell chemotactic factors, includ-ing KIT ligand (KITL). In response to these factors, Nf1+/– mast cells show a positive che-motactic response, which may result in the recruitment of Nf1+/– mast cells to regions of Nf1–/– Schwann cell proliferation, to facilitate Nf1–/– Schwann cell neoplastic transforma-tion mediated by mast cell-derived growth factors39. Moreover, loss of neurofibromin function and increased RAS activation in Schwann cells, results in hypersensitiv-ity to the mitogenic effects of these local growth factors37, and significantly increases the proliferation of these Nf1–/– Schwann cells. Additionally, Nf1 inactivation leads to alterations in intracellular cAMP levels
that result in the stabilization of cyclin D, further promoting Schwann cell prolifera-tion51. Collectively, the interplay between Nf1+/– mast cell growth-factor production and Nf1–/– Schwann cell hypersensitivity to these mitogens produces a favourable scenario for tumorigenesis.
The formation of low-grade astrocyto-mas along the optic pathway in NF1 also seems to require an interaction between Nf1–/– astrocytes and the surrounding Nf1+/– brain60. It can be surmised from the predilection for tumour formation along the optic pathway that local environmental factors specifically support Nf1–/– astrocy-toma formation. These tumours frequently contain infiltrating microglia, bone mar-row-derived cells of the macrophage-mono-cyte lineage. Some insights into the basis for the anatomical localization of tumour formation along the optic pathway derive from the observation that the chemokine, CXCL12 (SDF1α), is normally expressed by neurons as well as vascular endothelial cells in regions where NF1-associated glio-mas form, including the supra-chiasmatic portion of the hypothalamus, the gan-glion cell layer of the retina, and along the optic nerve. In addition, we have observed high levels of CXCL12 receptor expression in human optic nerve glioma specimens and found that Nf1-deficient astrocytes exhibit a unique hypersensitivity to CXCL12 in vitro71. These preliminary results indicate that tumour formation in the optic nerve might be dictated by the normal expression pattern of chemokines, like CXCL12, to which Nf1–/– astrocytes are hypersensitive. We postulate that optic glioma formation reflects the combined effects of the NF1+/– cellular environment and the regional pattern of CXCL12 expression during childhood.
The identification of candidate factors derived from the tumour environment that influence tumour formation provides a means to use tumour precursor cells from these mouse models in assays to define the early steps between Nf1 gene inactivation and the acquisition of the fully transformed neoplastic state. We hypothesize that these factors represent additional targets for thera-pies aimed at preventing tumour formation as well as therapies aimed at ameliorating tumour growth.
Future directionsThe availability of mouse models of NF1-associated nervous system tumours affords a unique opportunity to begin to dissect the events important in the molecular and cellular pathogenesis of tumour formation.
562 | JULY 2005 | VOLUME 5 www.nature.com/reviews/cancer
P E R S P E C T I V E S

© 2005 Nature Publishing Group
With the availability of Nf1 mouse models, we are now uniquely positioned to analyse the early pre-neoplastic events that facilitate tumorigenesis and to decipher the cellular influences that promote neoplastic transfor-mation. Similarly, with an improved under-standing of the growth-promoting pathways crucial for tumour formation and progression, it becomes possible to develop tailored thera-pies that specifically target crucial molecules involved in Nf1–/– Schwann cell or astrocyte growth control. Finally, these mouse models provide a platform to evaluate new therapies for neurofibromas and astrocytomas in pre-clinical trials prior to the treatment of patients as well as to define radiological and protein biomarkers for tumour progression.
Joshua B. Rubin is in the Departments of Pediatrics and Anatomy and Neurobiology, Washington University School of Medicine
and St. Louis Children’s Hospital, St. Louis Missouri 63110, USA.
David H. Gutmann is in the Department of Neurology, Washington University School of Medicine and St. Louis Children’s Hospital,
St. Louis Missouri 63110, USA.
Correspondence to D.H.G. e-mail: [email protected]
doi: 10.1038/nrc1653
1. Friedman, J. M., Gutmann, D. H., MacCollin, M. M. & Riccardi, V. M. (eds) Neurofibromatosis: Phenotype, Natural History & Pathogenesis (Johns Hopkins Univ. Press, Baltimore, Maryland, 1999).
2. Knudson, A. G. Two genetic hits (more or less) to cancer. Nature Rev. Cancer 1, 157–162 (2001).
3. Sherman, L. S., Atit, R., Rosenbaum, T., Cox, A. D. & Ratner, N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J. Biol. Chem. 275, 30740–30745 (2000).
4. Perry, A., Roth, K. A., Banerjee, R., Fuller, C. E. & Gutmann, D. H. NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. Am. J. Pathol. 159, 57–61 (2001).
5. Rutkowski, J. L., Wu, K., Gutmann, D. H., Boyer, P. J. & Legius, E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum. Mol. Genet. 9, 1059–1066 (2000).
6. Ferner, R. E. & Gutmann, D. H. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 62, 1573–1577 (2002).
7. King, A. A., Debaun, M. R., Riccardi, V. M. & Gutmann, D. H. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am. J. Med. Genet. 93, 388–392 (2000).
8. Korf, B. R. Plexiform neurofibromas. Am. J. Med. Genet. 89, 31–37 (1999).
9. Watson, M. A. et al. Gene expression profiling reveals unique molecular subtypes of Neurofibromatosis type I-associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol. 14, 297–303 (2004).
10. Perry, A. et al. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J. Neuropathol. Exp. Neurol. 61, 702–709 (2002).
11. Kourea, H. P., Cordon-Cardo, C., Dudas, M., Leung, D. & Woodruff, J. M. Expression of p27kip and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: the emerging role of p27kip in malignant transformation of neurofibromas. Am. J. Pathol. 155, 1885–1891 (1999).
12. Kourea, H. P., Orlow, I., Scheithauer, B. W., Cordon-Cardo, C. & Woodruff, J. M. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am. J. Pathol. 155, 1855–1860 (1999).
13. Nielsen, G. P. et al. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am. J. Pathol. 155, 1879–1884 (1999).
14. Listernick, R., Charrow, J. & Gutmann, D. H. Intracranial gliomas in neurofibromatosis type 1. Am. J. Med. Genet. 89, 38–44 (1999).
15. King, A., Listernick, R., Charrow, J., Piersall, L. & Gutmann, D. H. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am. J. Med. Genet. A 122, 95–99 (2003).
16. Thiagalingam, S., Flaherty, M., Billson, F. & North, K. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology 111, 568–577 (2004).
17. Parsa, C. F. et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch. Ophthalmol. 119, 516–529 (2001).
18. Gutmann, D. H., Donahoe, J., Brown, T., James, C. D. & Perry, A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol. Appl. Neurobiol. 26, 361–367 (2000).
19. Li, J., Perry, A., James, C. D. & Gutmann, D. H. Cancer-related gene expression profiles in NF1-associated pilocytic astrocytomas. Neurology 56, 885–890 (2001).
20. Wimmer, K., Eckart, M., Meyer-Puttlitz, B., Fonatsch, C. & Pietsch, T. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. J. Neuropathol. Exp. Neurol. 61, 896–902 (2002).
21. Gutmann, D. H. et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology 59, 759–761 (2002).
22. Gutmann, D. H. et al. Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology 61, 1397–1400 (2003).
23. Wallace, M. R. et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249, 181–186 (1990).
24. Viskochil, D. et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62, 187–192 (1990).
25. Gutmann, D. H., Zhang, Y. & Hirbe, A. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann. Neurol. 46, 777–782 (1999).
26. Costa, R. M. et al. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nature Genet. 27, 399–405 (2001).
27. Xu, G. F. et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62, 599–608 (1990).
28. Xu, G. F. et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63, 835–841 (1990).
29. Martin, G. A. et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63, 843–849 (1990).
30. Ballester, R. et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63, 851–859 (1990).
31. Basu, T. N. et al. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356, 713–715 (1992).
32. Bollag, G. et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nature Genet. 12, 144–148 (1996).
33. DeClue, J. E. et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69, 265–273 (1992).
34. Kim, H. A., Rosenbaum, T., Marchionni, M. A., Ratner, N. & DeClue, J. E. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene 11, 325–335 (1995).
35. Hiatt, K. K., Ingram, D. A., Zhang, Y., Bollag, G. & Clapp, D. W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1–/– cells. J. Biol. Chem. 276, 7240–7245 (2001).
36. Cichowski, K., Santiago, S., Jardim, M., Johnson, B. W. & Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 17, 449–454 (2003).
37. Tang, Y. et al. A role for Pak protein kinases in Schwann cell transformation. Proc. Natl Acad. Sci. USA 95, 5139–5144 (1998).
38. Ingram, D. A. et al. Hyperactivation of p21ras and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J. Exp. Med. 194, 57–69 (2001).
39. Yang, F. C. et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/– mast cells. J. Clin. Invest. 112, 1851–1861 (2003).
40. Donovan, S., See, W., Bonifas, J., Stokoe, D. & Shannon, K. M. Hyperactivation of protein kinase B and ERK have discrete effects on survival, proliferation, and cytokine expression in Nf1-deficient myeloid cells. Cancer Cell 2, 507–514 (2002).
41. Lau, N. et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J. Neuropathol. Exp. Neurol. 59, 759–767 (2000).
42. Zhang, Y. Y. et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J. Exp. Med. 187, 1893–1902 (1998).
43. Bajenaru, M. L. et al. Neurofibromatosis 1 (NF1) heterozygosity results in a cell-autonomous growth advantage for astrocytes. Glia 33, 314–323 (2001).
44. Gutmann, D. H. et al. Heterozygosity for the neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes. Hum. Mol. Genet. 10, 3009–3016 (2001).
45. The, I. et al. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 276, 791–794 (1997).
46. Guo, H. F., The, I., Hannan, F., Bernards, A. & Zhong, Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276, 795–798 (1997).
47. Johnson, M. R., Look, A. T., DeClue, J. E., Valentine, M. B. & Lowy, D. R. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP. Ras. Proc. Natl Acad. Sci. USA 90, 5539–5543 (1993).
48. Gutmann, D. H. et al. Analysis of the neurofibromatosis type 1 (NF1) GAP-related domain by site-directed mutagenesis. Oncogene 8, 761–769 (1993).
49. Waschek, J. A. Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev. Neurosci. 24, 14–23 (2002).
50. Dasgupta, B., Dugan, L. L. & Gutmann, D. H. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J. Neurosci. 23, 8949–8954 (2003).
51. Kim, H. A., Ratner, N., Roberts, T. M. & Stiles, C. D. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J. Neurosci. 21, 1110–1116 (2001).
52. Houslay, M. D. & Baillie, G. S. The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways. Biochem. Soc. Trans. 31, 1186–1190 (2003).
53. Jacks, T. et al. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nature Genet. 7, 353–361 (1994).
54. Zhu, Y. et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 15, 859–876 (2001).
55. Zhu, Y., Ghosh, P., Charnay, P., Burns, D. K. & Parada, L. F. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296, 920–922 (2002).
56. Kim, H. A., Ling, B. & Ratner, N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol. Cell. Biol. 17, 862–872 (1997).
57. Le, D. T. et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 103, 4243–4250 (2004).
58. Atit, R. P., Crowe, M. J., Greenhalgh, D. G., Wenstrup, R. J. & Ratner, N. The Nf1 tumor suppressor regulates mouse skin wound healing, fibroblast proliferation, and collagen deposited by fibroblasts. J. Invest. Dermatol. 112, 835–842 (1999).
59. Bajenaru, M. L. et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol. 22, 5100–5113 (2002).
NATURE REVIEWS | CANCER VOLUME 5 | JULY 2005 | 563
P E R S P E C T I V E S

© 2005 Nature Publishing Group
60. Bajenaru, M. L. et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 63, 8573–8577 (2003).
61. Bajenaru, M. L., Garbow, J. R., Perry, A., Hernandez, M. R. & Gutmann, D. H. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann. Neurol. 57, 119–127 (2005).
62. Reilly, K. M., Loisel, D. A., Bronson, R. T., McLaughlin, M. E. & Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nature Genet. 26, 109–113 (2000).
63. Ridley, A. J., Paterson, H. F., Noble, M. & Land, H. Ras-mediated cell cycle arrest is altered by nuclear oncogenes to induce Schwann cell transformation. EMBO J. 7, 1635–1645 (1988).
64. Dasgupta, B., Li, W., Perry, A., Gutmann, D. H. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 65, 236–245 (2005).
65. King, D., Yang, G., Thompson, M. A. & Hiebert, S. W. Loss of neurofibromatosis-1 and p19ARF cooperate to induce a multiple tumor phenotype. Oncogene 21, 4978–4982 (2002).
66. Reilly, K. M. et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc. Natl Acad. Sci. USA 101, 13008–13013 (2004).
67. Kim, J. Y. et al. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev. Biol. 263, 50–66 (2003).
68. Goodrich, L. V., Milenkovic, L., Higgins, K. M. & Scott, M. P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 (1997).
69. Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
70. Singh, S. K. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 (2003).
71. Woerner, B. et al. in Ninth Annual Meeting of the Society for Neuro-Oncology (ed. Bigner, D. D.) 323 (Duke Univ. Press, Toronto, Ontario Canada, 2004).
72. Dasgupta, B., Yi, Y., Chen, D. Y., Weber, J. D. & Gutmann, D. H. Proteomic analysis reveals hyperactivation of the mTOR pathway in NF1-associated human and mouse brain tumors. Cancer Res. 65, 2755-2760 (2005).
73. Oliver T.G. et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 132, 2425-2439 (2005)
AcknowledgementsThe authors thank our Neurofibromatosis Center Group at Washington University, especially A. Perry, J. Weber, M. Watson and J. Garbow, as well as R. Wechsler-Reya for critical reading of the manuscript. D.H.G. is supported by grants from the Department of Defense, National Institute of Neurological Disorders and Strokes, and James S. McDonnell Foundation. J.R. is a scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine and receives additional support from the National Institute of Child Health and Human Development, The American Cancer Society and Hope Street Kids. We also acknowledge the generous support from Schnuck Markets, Inc.
Competing interests statementThe authors declare no competing financial interests.
Online links
DATABASESThe following terms in this article are linked online to:Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneCDNK2A | NF1 National Cancer Institute: http://cancer.govNational Cancer Institute: http://cancer.govleukaemia | pheochromocytoma OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIMneurofibromatosis type 1
FURTHER INFORMATIONGutmann lab: http://www.neuro.wustl.edu/neurogenetics/lab.htmlRubin lab: http://peds.wustl.edu/research/labs/Rubin_Joshua_BAccess to this interactive links box is free online.
O P I N I O N
Cancer in xeroderma pigmentosum and related disorders of DNA repairJames E. Cleaver
Abstract | Nucleotide-excision repair diseases exhibit cancer, complex developmental disorders and neurodegeneration. Cancer is the hallmark of xeroderma pigmentosum (XP), and neurodegeneration and developmental disorders are the hallmarks of Cockayne syndrome and trichothiodystrophy. A distinguishing feature is that the DNA-repair or DNA-replication deficiencies of XP involve most of the genome, whereas the defects in CS are confined to actively transcribed genes. Many of the proteins involved in repair are also components of dynamic multiprotein complexes, transcription factors, ubiquitylation cofactors and signal-transduction networks. Complex clinical phenotypes might therefore result from unanticipated effects on other genes and proteins.
Xeroderma pigmentosum (XP) is the arche-type of an expanding family of nucleotide-excision repair (NER) diseases that includes XP itself, the XP variant (XP-V), Cockayne syndrome (CS), cerebro oculofacial skel-etal syndrome (COFS), a mild ultraviolet (UV)-light-sensitive syndrome, trichothi-odystrophy (TTD), and some diseases with combined symptoms of XP/CS and XP/TTD1. These diseases have complex over-lapping symptoms associated with cancer, developmental delay, immunological defects, neurodegeneration, retinal degeneration and ageing. They represent a continuum with skin cancer alone at one extreme and neuro-degeneration and developmental disorders at the other BOX 1.
The total number of genes directly involved in NER is estimated to be around 40 REF. 2, but only about a dozen of these genes have been found to be deregulated in NER-related human diseases. The remainder represent either essential genes that would be lethal if mutated or that might have such mild clinical disease that they blend into the population of ‘sun-sensitive’ individu-als. The molecular defects in repair and the clinical symptoms of XP as compared with CS and TTD enable us to understand how
certain pathways for processing DNA dam-age lead to cancer, whereas others lead to more complex disorders. Before the NER genes were cloned and identified, patients were assigned to complementation groups, such as XP-A or XP-D, by cell-fusion experiments1. Once the mutated genes were identified and sequenced, they were named after the complementation group with which they associate.
Many reviews have described the molecular mechanisms of NER and the amazing varieties of damage recogni-tion and DNA manipulation that are required for the recognition and removal of DNA damage represented by the UV-light-induced dipyrimidine pho-toproducts, the 5–5, 6–6 cyclobutane pyrimidine dimers (CPDs) and the pyri-midine–pyrimidone dimers, and other large carcinogen or oxidative products in DNA3–6 (FIG. 1). I want to summarize these mechanisms briefly in terms of the function of the XP genes and address broad general questions about the comparison between XP and CS. These include two contrasting questions, the considerations of which are particularly informative. Why do patients with mutations in genes specific for global genomic repair (GGR) or bypass replication predominantly develop cancer but rarely neurodegeneration? And why do patients with mutations in genes specific for repair of damage in transcribed genes not get cancer but suffer from developmental and neurodegenerative disorders?
Nucleotide-excision repairThe question of how DNA damage is rec-ognized can be viewed from two different perspectives: one question asks ‘which pro-teins interact directly with damaged bases in DNA’, and the other asks ‘which protein acts first in this damage-response pathway’ (FIG. 1). Many of the NER proteins interact directly with damaged DNA, including XPE, XPC–HR23B, XPA, replication protein A (RPA) and the transcription factor TFIIH3 TABLE 1. Which proteins act first depends on the transcriptional activity of the DNA.
564 | JULY 2005 | VOLUME 5 www.nature.com/reviews/cancer
P E R S P E C T I V E S





























