Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY, Nov. 2011, p. 4623–4632 Vol. 31, No. 220270-7306/11/$12.00 doi:10.1128/MCB.05715-11Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Nucleosome Disruption by DNA Ligase III-XRCC1 PromotesEfficient Base Excision Repair�‡
Ian D. Odell,1 Joy-El Barbour,1† Drew L. Murphy,2 Julie A. Della-Maria,3 Joann B. Sweasy,1,2
Alan E. Tomkinson,3 Susan S. Wallace,1 and David S. Pederson1*Department of Microbiology and Molecular Genetics, University of Vermont, Burlington, Vermont 054051; Departments ofTherapeutic Radiology and Human Genetics, Yale University School of Medicine, New Haven, Connecticut 065202; andDepartment of Radiation Oncology, Greenebaum Cancer Center, University of Maryland, Baltimore, Maryland 212013
Received 27 May 2011/Returned for modification 2 July 2011/Accepted 3 September 2011
Each day, approximately 20,000 oxidative lesions form in the DNA of every nucleated human cell. The baseexcision repair (BER) enzymes that repair these lesions must function in a chromatin milieu. We have determinedthat the DNA glycosylase hNTH1, apurinic endonuclease (APE), and DNA polymerase � (Pol �), which catalyze thefirst three steps in BER, are able to process their substrates in both 601- and 5S ribosomal DNA (rDNA)-basednucleosomes. hNTH1 formed a discrete ternary complex that was displaced by the addition of APE, suggesting anorderly handoff of substrates from one enzyme to the next. In contrast, DNA ligase III�-XRCC1, which completesBER, was appreciably active only at concentrations that led to nucleosome disruption. Ligase III�-XRCC1 was alsoable to bind and disrupt nucleosomes containing a single base gap and, because of this property, enhanced both itsown activity and that of Pol � on nucleosome substrates. Collectively, these findings provide insights into rate-limiting steps that govern BER in chromatin and reveal a unique role for ligase III�-XRCC1 in enhancing theefficiency of the final two steps in the BER of lesions in nucleosomes.
Reactive oxygen species (ROS), generated as by-products ofnormal aerobic cellular metabolism or from exposure to exog-enous agents, such as gamma irradiation, generate approxi-mately 20,000 DNA damage events per day in each nucleatedhuman cell. The DNA lesions produced include numerousoxidative base damages, apurinic/apyrimidinic (AP) sites, andsingle-strand DNA breaks (6). Base excision repair (BER)enzymes recognize and replace oxidized bases with the corre-sponding undamaged bases. In its simplest (“short-patch”)form, BER entails four enzymatic steps (1, 10, 21, 23, 51, 53)(Fig. 1A), beginning with the recognition and excision of adamaged base by either a mono- or bifunctional DNA glyco-sylase. Bifunctional glycosylases first cleave the glycosidic bondbetween the damaged base and the deoxyribose and thencleave the phosphodiester bond 3� of the resulting AP site. APendonuclease (APE) removes a residual moiety to generate asingle nucleotide gap, with a 3�-OH group that can be filled byDNA polymerase � (Pol �). Finally, DNA ligase III-�(LigIII�), in association with XRCC1, catalyzes the formationof a phosphodiester bond between the 3�-OH of the newlyadded nucleotide and the adjacent downstream 5�-phosphate.
The nucleosomes that package most of the nuclear DNA ineukaryotes provide only minimal protection from ROS (14,31); a small degree of protection from hydroxyl radicals is
evident in DNA segments where the minor groove faces intothe histone octamer (20), and histones themselves may act as asink for ROS, thereby reducing the frequency of free-radical-inflicted DNA damage (28). Clearly, however, nucleosomalDNA is vulnerable to oxidative damage that must be madeavailable to BER enzymes. Chromatin remodeling agents andhistone chaperones facilitate most processes involving chroma-tin, and the other DNA repair pathways—nucleotide excisionrepair, mismatch repair, nonhomologous end-joining and ho-mologous recombination-mediated repair—are all thought torequire local disruption of nucleosomes (e.g., see references 18and 38). As detailed in Discussion, we and others have re-ported that at least some steps in BER can occur withoutrequiring or inducing nucleosome disruption (3, 22, 32, 35–37,43). However, there is not yet a clear consensus on whetherBER in its entirety, or a subset of specific steps in BER,requires disruption of nucleosomes; nor is it clear which if anyof the known chromatin remodeling factors facilitate BER invivo.
To better define the rate-limiting steps in BER of lesions innucleosomes and elucidate mechanisms that govern BER inchromatin, we have conducted a series of studies using purifiedhuman BER enzymes and well-defined nucleosome substrates.We reported earlier that DNA glycosylases in both families ofbifunctional DNA glycosylases can initiate BER of lesions innucleosomes, without irreversibly disrupting or altering thetranslational position of the lesion containing nucleosomes (37,43). As well, we provided evidence that spontaneous, partialunwrapping of DNA from the histone octamer enables DNAglycosylases to bind and process lesions that would ordinarilybe sterically occluded (43). We have now reconstituted theentire BER reaction with nucleosomes containing discretelypositioned oxidative lesions or BER intermediates. As we re-ported earlier for DNA glycosylases, both APE and Pol � were
* Corresponding author. Mailing address: Department of Microbi-ology and Molecular Genetics, Stafford Building, Room 302, 95 Car-rigan Drive, University of Vermont, Burlington, VT 05405. Phone:(802) 656-8586. Fax: (802) 656-8749. E-mail: [email protected].
† Present address: Department of Molecular and Cell Biology, Uni-versity of California, Berkeley, CA 94720.
‡ Supplemental material for this article may be found at http://mcb.asm.org/.
� Published ahead of print on 19 September 2011.
4623
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
able to process their substrates without irreversibly altering thenucleosome or its translational position, although the efficiencyof these enzymes varied depending on the helical orientationof their substrates relative to the histone octamer. In contrast,DNA ligase III� complexed with XRCC1 (LigIII�-XRCC1)exhibited very little activity on intact nucleosomes at low con-centrations, regardless of the helical orientation of its sub-strate. High concentrations of LigIII�-XRCC1 resulted inconsiderable ligation activity but also led to nucleosome dis-ruption. LigIII�-XRCC1 proved able to bind and disruptnucleosomes containing either a nick or a single base gap,independently of any chromatin remodeling agents. It isthrough this activity that LigIII�-XRCC1 enhanced not only itsown activity on nucleosomal substrates but that of Pol � aswell.
MATERIALS AND METHODS
Construction of DNA containing BER lesions and intermediates. The 147-bpcore of the 601 nucleosome positioning sequence and 184-bp DNA containingthe Lytechinus variegatus 5S ribosomal DNA (rDNA) (Lv5S) nucleosome posi-tioning sequence, each containing a single, discretely positioned thymine glycol(Tg) residue, were prepared as previously described (43). Briefly, synthetic oli-gomers Tg-out, Tg-in, or Tg-in (601) (see Table SA1 in the supplemental ma-terial) were end labeled with [�-32P]ATP, annealed to their respective templates,and extended with (exo-) Klenow enzyme (New England BioLabs). The resultingdouble-stranded DNAs were gel purified and assembled into nucleosomes, asdescribed below. Nucleosome length Lv5S DNA containing an AP site wasprepared in the same manner, but with oligomers AP-out or AP-in, whichcontain tetrahydrofuran in place of Tg. To prepare gap- or nick-containing DNAfragments, the DNA oligomers Out (3�) and In (3�) were 5�-end labeled with[�-32P]ATP, annealed to equimolar amounts of Lv5S template, and extendedwith (exo-) Klenow enzyme (New England BioLabs) to create 154- and 149-nucleotide-long DNA segments. The extension reactions were stopped with 10
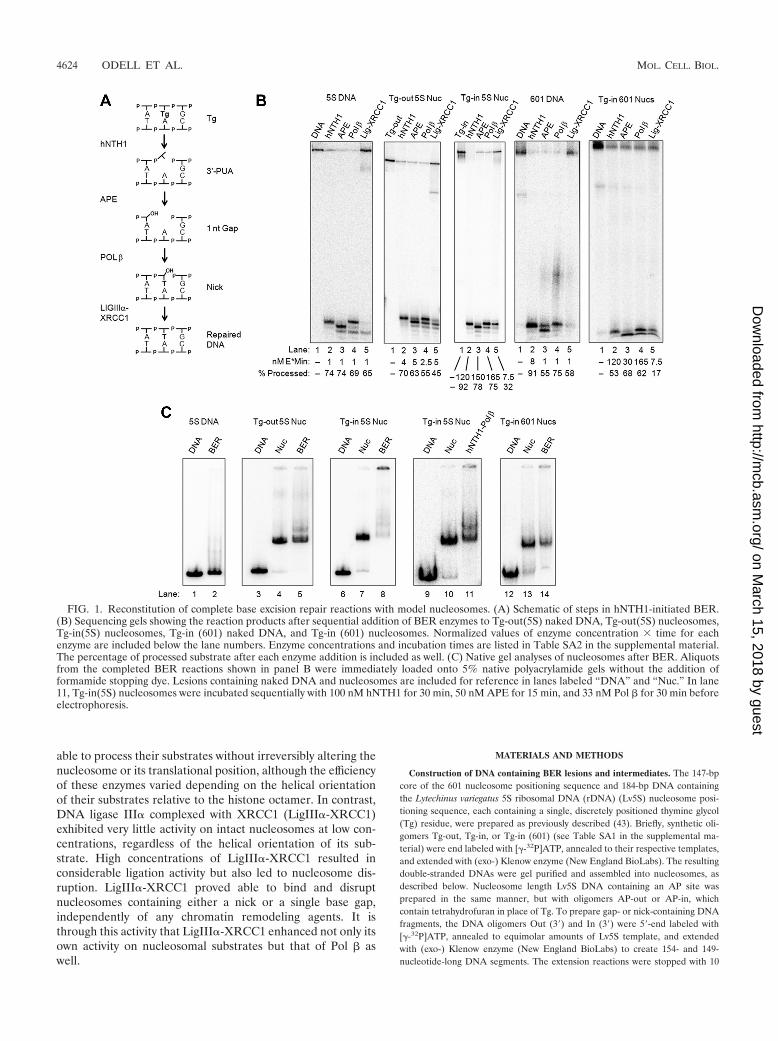
FIG. 1. Reconstitution of complete base excision repair reactions with model nucleosomes. (A) Schematic of steps in hNTH1-initiated BER.(B) Sequencing gels showing the reaction products after sequential addition of BER enzymes to Tg-out(5S) naked DNA, Tg-out(5S) nucleosomes,Tg-in(5S) nucleosomes, Tg-in (601) naked DNA, and Tg-in (601) nucleosomes. Normalized values of enzyme concentration � time for eachenzyme are included below the lane numbers. Enzyme concentrations and incubation times are listed in Table SA2 in the supplemental material.The percentage of processed substrate after each enzyme addition is included as well. (C) Native gel analyses of nucleosomes after BER. Aliquotsfrom the completed BER reactions shown in panel B were immediately loaded onto 5% native polyacrylamide gels without the addition offormamide stopping dye. Lesions containing naked DNA and nucleosomes are included for reference in lanes labeled “DNA” and “Nuc.” In lane11, Tg-in(5S) nucleosomes were incubated sequentially with 100 nM hNTH1 for 30 min, 50 nM APE for 15 min, and 33 nM Pol � for 30 min beforeelectrophoresis.
4624 ODELL ET AL. MOL. CELL. BIOL.
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

mM EDTA and then mixed with an equimolar amount of the appropriate 32P5�-end-labeled upstream oligomers (Gap-out, Gap-in, Nick-out, or Nick-in) andannealed to create a full-length DNA fragment containing a single, discretelypositioned gap or nick.
DNA containing a 3�-phospho-�,�-unsaturated aldehyde (3�-PUA) group wasprepared by incubating Tg-containing naked DNA with excess hNTH1 for 30min at 37°C. hNTH1 was removed by phenol-chloroform extraction, and theDNA was ethanol precipitated and suspended in the appropriate volume forassembly into nucleosomes.
Nucleosome reconstitution. Xenopus histones H2A, H2B, H3, and H4 wereexpressed in Escherichia coli and assembled into octamers, and the octamerswere purified, all as described by Luger et al. and Dyer et al. (12, 30). Toassemble nucleosomes, we added an �1.25-fold molar excess of purified octamerto a mixture of one part end-labeled substrate DNA and nine parts unlabeledlesion-free DNA. The resulting mixture was introduced into a button dialysischamber constructed from a 0.2-ml PCR tube, as described by Thastrom et al.(50), and dialyzed slowly from 2 M NaCl in HED buffer (25 mM HEPES [pH8.0], 1 mM EDTA, and 1 mM dithiothreitol [DTT]) to HED buffer lacking NaCl.Tg-out(5S) and Tg-in(5S) nucleosomes in the experiments shown in Fig. 3A to Cwere reconstituted by octamer transfer as described in reference 43. To quantifyreconstitution efficiency, nucleosomes were fractionated through 5% native poly-acrylamide gels in 1/4� Tris-borate-EDTA (TBE) buffer and visualized by eitherautoradiography or phosphorimaging.
Expression and purification of BER enzymes. hNTH1, Pol � wild type (wt),T304I, and E309K were expressed and purified as previously described (34, 37).A manuscript describing the coexpression and purification of ligase III� withXRCC1 is in preparation. The expression and purification of APE are describedin the supplemental material.
Enzyme assays. All enzyme concentrations reported in the text and figuresrefer to total protein concentration, except in the case of hNTH1, where theactive fraction was determined as described by Blaisdell and Wallace (5). En-zymes were freshly diluted into ice-cold BER reaction buffer (25 mM NaHEPESNaOH [pH 8.0], 100 mM NaCl, 5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, and0.1 mg/ml bovine serum albumin [BSA]) containing 20 �M dTTP, unless other-wise indicated in the figure legend, and added to substrates in reaction buffer, asindicated in the text and figure legends. The dilution and reaction buffers usedwith LigIII�-XRCC1 included 1 mM ATP. Final substrate concentrations in allreactions were 4 nM, with 36 nM unlabeled lesion-free nucleosomes or nakedDNA, and all reactions were conducted at 37°C, unless otherwise indicated. Tomonitor enzyme activity, aliquots from BER reactions were stopped by theaddition of 4 volumes 0.1 N NaOH, 90% formamide, and 0.1% bromophenolblue and 0.1% xylene cyanol or, in the case of hNTH1 reactions, the same bufferminus the 0.1 N NaOH. Reaction products were resolved on 12% or 15%sequencing gels. To assess the fate and integrity of lesion-containing nucleo-somes, aliquots from BER reactions were loaded immediately onto 5% nativepolyacrylamide gels and separated by electrophoresis in 1/2� TBE buffer; at nopoint were these samples exposed to formamide or other denaturants.
XRCC1 pulldown and Western blot assays. To assess interactions betweenXRCC1 and either Pol � or selected Pol � mutants, we added 33 nM purified Pol� in nickel buffer (50 mM Tris [pH 7.6], 75 mM KCl, 0.1% IGEPAL CA-630(Sigma-Aldrich), 1 mM DTT, 10 mM imidazole) to 100 �g of XRCC1-containingwhole-cell extract, prepared from the 88Tag Pol ��/� mouse embryonic fibro-blast cell line (13, 48), as previously described (25). After 30 min on ice, theXRCC1-Pol � mixtures were mixed with 15 �l of Ni-nitrilotriacetic acid (NTA)agarose (Qiagen). The resin was collected by centrifugation and washed, and theresin-associated protein was recovered by suspension in SDS-PAGE loadingbuffer. Proteins were then fractionated through a 10% SDS-PAGE gel, trans-ferred to nitrocellulose, and incubated with an anti-XRCC1 antibody (AbCamnumber ab9147). The blots were incubated with a horseradish peroxidase-con-jugated secondary antibody (1:5,000 dilution), and XRCC1-antibody conjugateswere visualized using a Bio-Rad Molecular Imager ChemiDoc XRS.
RESULTS
Assembly of model nucleosome substrates. To examine theimpact of nucleosomes on each step in BER, we assembledlesion-containing nucleosomes with either of two DNA frag-ments that each form well-positioned nucleosomes (11, 15, 41,43, 45, 46, 49). The first of these contained a single thymineglycol (Tg) embedded in either of two sites within the L.variegatus 5S rDNA gene (Lv5S gene). Tg-out(5S) nucleo-
somes contained a Tg whose minor groove faces outward fromthe histone octamer, while Tg-in(5S) nucleosomes contained aTg whose minor groove faces toward the histone octamer. Weshowed previously that neither lesion alters the preferred he-lical positioning of DNA in the nucleosome (43). We showedas well that, in both the major and minor translational variantsthat form with 5S DNA, these lesions reside within the nucleo-some (37, 43). We also assembled nucleosomes using the syn-thetic 601 DNA segment that Widom and colleagues selectedfor its capacity to form an even more stable, positioned nucleo-some (45, 49). The nucleosome Tg-in (601) contained an in-ward-facing Tg residue 47 nucleotides from the dyad axis, a siteclose to that of the Tg residue in Tg-in(5S). To examine indi-vidual steps in BER, we also assembled nucleosomes withsubstrates for enzymes that act subsequently to DNA glycosy-lases (an unsaturated aldehyde for APE, gapped DNA for Pol�, and nicked DNA for LigIII�-XRCC1).
Reconstitution of complete base excision repair reactionswith nucleosomes. Figure 1B shows the DNA products formedat each step in reactions where we added, sequentially,hNTH1, APE, Pol �, and LigIII�-XRCC1 to Tg-out(5S) andTg-in (601) naked DNAs and to Tg-out(5S), Tg-in(5S), andTg-in (601) nucleosomes. As illustrated in Fig. 1A, hNTH1 firstcleaves the N-glycosylic bond between the damaged base (inthis case, thymidine glycol) and its associated sugar residue andthen cleaves the phosphodiester bond 3� to the AP site, gen-erating the faster-migrating product evident in lanes 2 of Fig.1B. Next, APE removes the 3�-unsaturated aldehyde, generat-ing a faster-migrating primer with a 3�-OH evident in lanes 3 ofFig. 1B. As shown in lanes 4 of Fig. 1B, Pol � was able toextend this primer by 1 or more nucleotides. Finally, under theconditions used, LigIII�-XRCC1 was able to seal most of thesingle nucleotide extension products in naked DNA and abouthalf of those in the 5S nucleosomes to generate the full-lengthrepaired DNA (lanes 5 of Fig. 1B). However, LigIII�-XRCC1largely failed to act on nicks in the 601-based nucleosomes(lane 5 in the right-most panel of Fig. 1B). We address thereasons for the differing ligase results in a later section.
To determine the impact of BER on nucleosome integrity,we immediately electrophoresed aliquots from the completedBER reactions on 5% native polyacrylamide gels (without theaddition of formamide stopping dye). Figure 1C shows nakedDNA and nucleosome templates before and immediately afterthe BER reactions (lanes 1, 3, 4, 6, 7, 9, 10, 12, and 13 versuslanes 2, 5, 8, 11, and 14). The presence of a major band with themobility of an intact nucleosome in lane 5, together with theabsence of a band with the mobility of naked DNA and noevidence of a high-molecular-weight aggregate, suggested thatlesions in most of the Tg-out(5S) nucleosomes had been fullyrepaired without irreversible disruption of the nucleosome.Also evident in lane 5 of Fig. 1C is a supershifted band thatlikely reflects a residual ternary complex that formed betweennucleosomes and Pol �. In contrast, the virtual absence of anucleosome band together with the prominent high-molecular-weight aggregate in lane 8 of Fig. 1C suggested that Tg-in(5S)nucleosomes had been disrupted at some point during BER.Further investigation revealed that nucleosomes remainlargely intact during the hNTH1, APE, and Pol � reactions(lane 11), indicating that LigIII�-XRCC1 was responsible forthe nucleosome disruption observed in lane 8. Additionally,
VOL. 31, 2011 BASE EXCISION REPAIR IN NUCLEOSOMES 4625
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
BER reactions with Tg-in (601) nucleosomes demonstratedlittle disruption (lane 14), which correlates with the small pro-portion (�17%) of 601 molecules that were ligated in thesereactions.
Lesion orientation plays a major role in the efficiency of thefirst three steps of hNTH1-initiated BER but not the final step.In the experiments shown in Fig. 1, we deliberately adjustedenzyme concentrations and reaction times to achieve roughlyequivalent levels of repair for all substrates. The relative “en-zyme-times-time” (E � t) values given in Table SA2 in thesupplemental material and at the bottom of each lane in Fig.1B show that it was necessary to use increasing enzymeamounts and longer reaction times as we progressed fromnaked 5S DNA to naked 601 DNA to Tg-out nucleosomes toTg-in nucleosomes. As these values indicate, hNTH1 was lessable to excise Tg from Tg-in (601) DNA than from 5S DNA-based DNA templates, possibly because we had positioned theTg residue such that its minor groove resided on the inner sideof the intrinsically curved 601 DNA (29). E � t amounts alsohad to be increased 2.5- to 5-fold relative to the correspondingnaked DNAs to repair Tg-out(5S) nucleosomes. Thus, nucleo-somes moderately inhibit BER even for optimally orientedlesions. Conditions yielding far higher E � t values wereneeded for repair of Tg-in(5S) nucleosomes, indicating thathelical orientation of substrates is a critical determinant ofrepair efficiency. It should be noted, however, that enzymeconcentrations used to initiate BER of lesion-containingnucleosomes in vitro can substantially affect the extent of repairobserved for lesions that, nominally, are sterically occluded.As we reported previously, increasing the concentration ofhNTH1 to its estimated concentration in vivo resulted in rel-atively efficient excision of occluded lesions (37, 43).
As indicated in Fig. 1, the E � t values for each of thesubsequent steps in BER increased in the same fashion as thatdescribed above for hNTH1, with the progression from nakedDNA to nucleosomal substrates. To more precisely relate theefficiency of each enzyme on nucleosome substrates to that onnaked DNA, we estimated initial reaction velocities from re-actions where enzyme concentrations were adjusted to main-tain single turnover conditions during the first 30 s of thereaction. These values, listed in Table 1, correlate closely toE � t values shown in Fig. 1B.
To examine the effect of substrate orientation on each en-
zyme in more detail, we conducted additional kinetic studies,this time choosing enzyme concentrations that would facilitatecomparisons between different nucleosomal substrates (Fig. 2).We prepared DNA and nucleosomes containing each of the
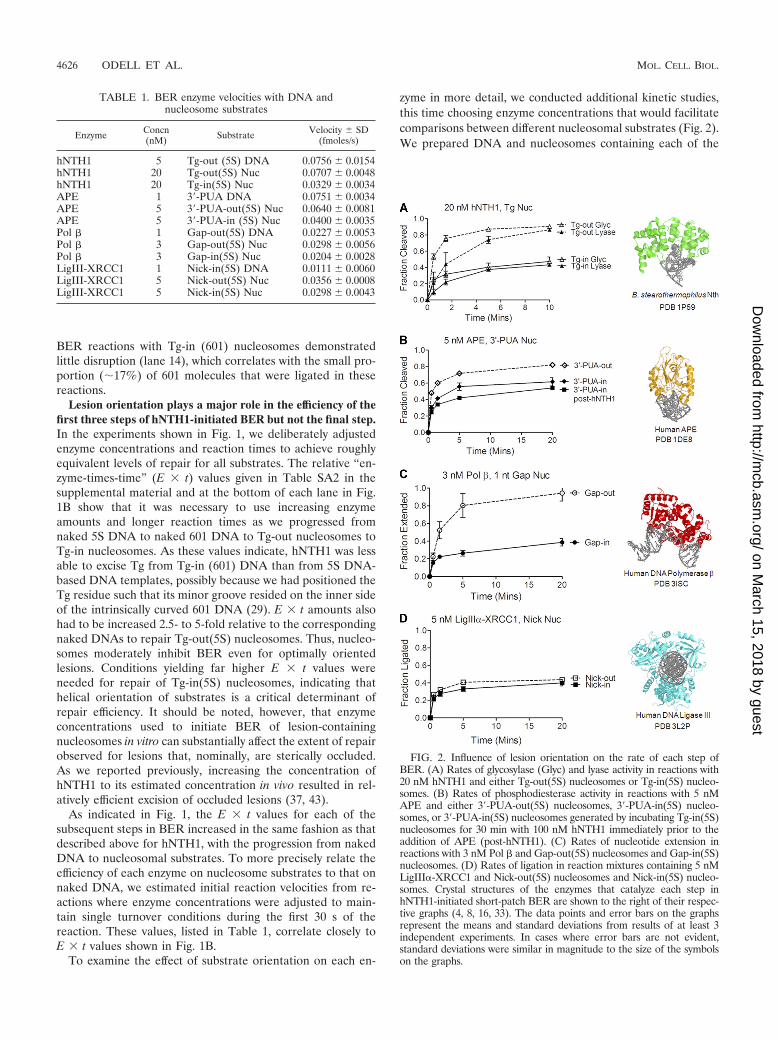
FIG. 2. Influence of lesion orientation on the rate of each step ofBER. (A) Rates of glycosylase (Glyc) and lyase activity in reactions with20 nM hNTH1 and either Tg-out(5S) nucleosomes or Tg-in(5S) nucleo-somes. (B) Rates of phosphodiesterase activity in reactions with 5 nMAPE and either 3�-PUA-out(5S) nucleosomes, 3�-PUA-in(5S) nucleo-somes, or 3�-PUA-in(5S) nucleosomes generated by incubating Tg-in(5S)nucleosomes for 30 min with 100 nM hNTH1 immediately prior to theaddition of APE (post-hNTH1). (C) Rates of nucleotide extension inreactions with 3 nM Pol � and Gap-out(5S) nucleosomes and Gap-in(5S)nucleosomes. (D) Rates of ligation in reaction mixtures containing 5 nMLigIII�-XRCC1 and Nick-out(5S) nucleosomes and Nick-in(5S) nucleo-somes. Crystal structures of the enzymes that catalyze each step inhNTH1-initiated short-patch BER are shown to the right of their respec-tive graphs (4, 8, 16, 33). The data points and error bars on the graphsrepresent the means and standard deviations from results of at least 3independent experiments. In cases where error bars are not evident,standard deviations were similar in magnitude to the size of the symbolson the graphs.
TABLE 1. BER enzyme velocities with DNA andnucleosome substrates
Enzyme Concn(nM) Substrate Velocity SD
(fmoles/s)
hNTH1 5 Tg-out (5S) DNA 0.0756 0.0154hNTH1 20 Tg-out(5S) Nuc 0.0707 0.0048hNTH1 20 Tg-in(5S) Nuc 0.0329 0.0034APE 1 3�-PUA DNA 0.0751 0.0034APE 5 3�-PUA-out(5S) Nuc 0.0640 0.0081APE 5 3�-PUA-in (5S) Nuc 0.0400 0.0035Pol � 1 Gap-out(5S) DNA 0.0227 0.0053Pol � 3 Gap-out(5S) Nuc 0.0298 0.0056Pol � 3 Gap-in(5S) Nuc 0.0204 0.0028LigIII-XRCC1 1 Nick-in(5S) DNA 0.0111 0.0060LigIII-XRCC1 5 Nick-out(5S) Nuc 0.0356 0.0008LigIII-XRCC1 5 Nick-in(5S) Nuc 0.0298 0.0043
4626 ODELL ET AL. MOL. CELL. BIOL.
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
intermediates of hNTH1-initiated BER: Tg, 3�-PUA, singlenucleotide gap, and nicked DNA (see Materials and Methods).We placed each of these substrates at the positions corre-sponding to those in the Tg-out(5S) and Tg-in(5S) nucleo-somes. As expected, hNTH1 excised lesions from Tg-out(5S)nucleosomes more efficiently than from Tg-in(5S) nucleo-somes; as well, the lyase activity of hNTH1 was slower than itsglycosylase activity regardless of lesion orientation (filled ver-sus open triangles in Fig. 2A). These results are consistent withthe largely one-sided binding of DNA glycosylase to DNA (Fig.2A). Figure 2B shows that, like hNTH1, APE interacts largelywith one side of the DNA helix. This led us to predict that ittoo would be able to bind directly to optimally oriented sub-strates; this proved correct, as shown in the graph in Fig. 2B.The binding of Pol � to gapped DNA induces DNA to bendaway from the prospective site of nucleotide insertion, asshown in Fig. 2C. An outward-facing gap in a nucleosomemight to some extent be appropriately “prebent” and therebyfacilitate the initial binding of Pol �. As with hNTH1 and APE,Pol � was not as active in nucleosomes as it was on naked DNAbut nonetheless was significantly more active on outward- thanon inward-facing gaps (Fig. 2C).
Unlike the first three steps in BER, the efficiency with whichLigIII� (in complex with XRCC1) ligated nicks in nucleo-somes was unaffected by the helical orientation of the nick(Fig. 2D). Increasing the concentration of LigIII�-XRCC1 by10-fold increased the rate of ligation for both inward- andoutward-facing nicks (see Fig. SA1 in the supplemental mate-rial). This degree of enhancement was reminiscent of the en-hanced processing of inward-facing lesions that are accessibleonly when transiently exposed due to partial unwrapping ofDNA from the histone octamer. Structural studies, which showthat LigIII� fully encircles its substrate, as depicted in Fig. 2D,suggest that LigIII� can bind substrates in nucleosomes onlyduring an episode of partial DNA unwrapping. Unlike hNTH1,APE, and Pol �, however, high concentrations of LigIII�-XRCC1 appear to drive nucleosome disruption (see “Sub-strate handoff to DNA ligase III� is accompanied by progres-sive nucleosome disruption” below).
Substrate handoff from hNTH1 to APE. Several workershave proposed that enzyme substrates produced during BER
are handed off from one enzyme to the next, such that a DNAglycosylase, for example, would remain bound to its productuntil it was displaced by APE; in this fashion, processing in-termediates would remain sequestered during repair (39, 52,54). We used two different assays to determine if enzymehandoff occurs during the processing of lesions in nucleo-somes. The underlying premise behind the first kinetic assaywas that binding of hNTH1 to its inward-facing product innucleosomes would prevent nearby DNA from rewrappingaround the histone octamer and thereby facilitate the subse-quent binding of APE. If this occurs, however, the kinetic assaywe used lacked the necessary sensitivity, as the rate of APE-mediated cleavage of an inward-facing 3�-PUA formed in situby treating Tg-containing nucleosomes with hNTH1 was ap-proximately the same as that seen when APE was added tonucleosomes that had been assembled with preformed 3�-PUA(Fig. 2B). An alternative possibility is that handoff does nothave a kinetic signature but may still occur as a means ofprotecting substrates during repair. Accordingly, we decided toexamine the dynamics of the ternary complexes that form withlesion-containing nucleosomes during BER.
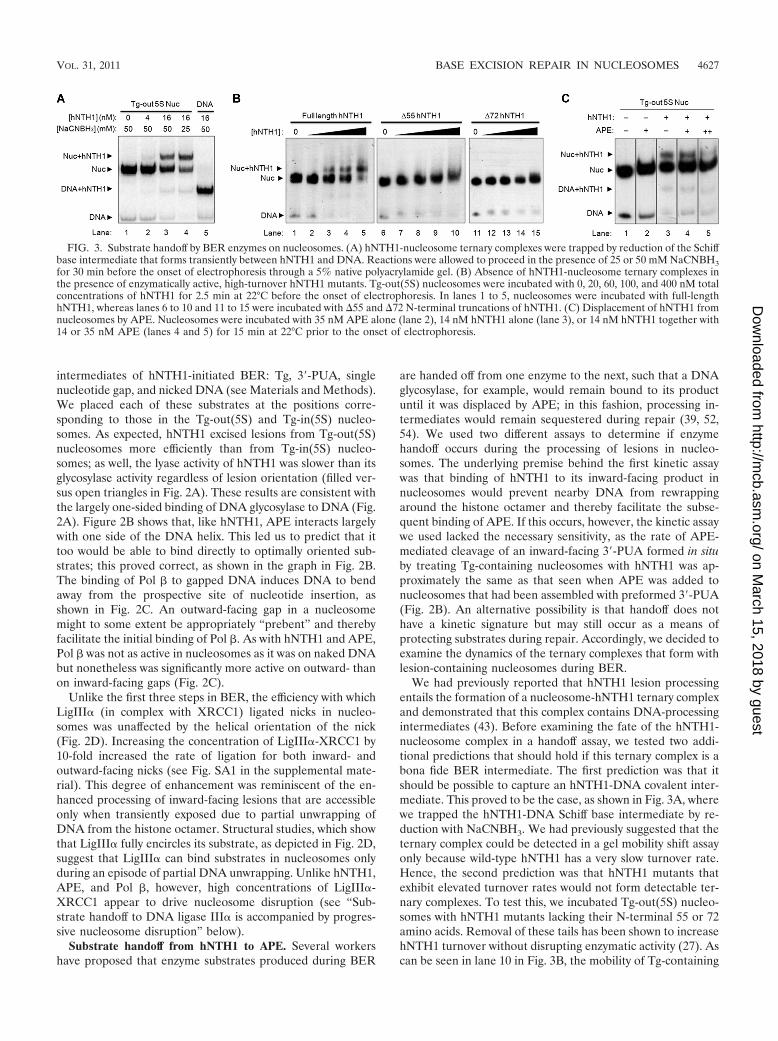
We had previously reported that hNTH1 lesion processingentails the formation of a nucleosome-hNTH1 ternary complexand demonstrated that this complex contains DNA-processingintermediates (43). Before examining the fate of the hNTH1-nucleosome complex in a handoff assay, we tested two addi-tional predictions that should hold if this ternary complex is abona fide BER intermediate. The first prediction was that itshould be possible to capture an hNTH1-DNA covalent inter-mediate. This proved to be the case, as shown in Fig. 3A, wherewe trapped the hNTH1-DNA Schiff base intermediate by re-duction with NaCNBH3. We had previously suggested that theternary complex could be detected in a gel mobility shift assayonly because wild-type hNTH1 has a very slow turnover rate.Hence, the second prediction was that hNTH1 mutants thatexhibit elevated turnover rates would not form detectable ter-nary complexes. To test this, we incubated Tg-out(5S) nucleo-somes with hNTH1 mutants lacking their N-terminal 55 or 72amino acids. Removal of these tails has been shown to increasehNTH1 turnover without disrupting enzymatic activity (27). Ascan be seen in lane 10 in Fig. 3B, the mobility of Tg-containing
FIG. 3. Substrate handoff by BER enzymes on nucleosomes. (A) hNTH1-nucleosome ternary complexes were trapped by reduction of the Schiffbase intermediate that forms transiently between hNTH1 and DNA. Reactions were allowed to proceed in the presence of 25 or 50 mM NaCNBH3for 30 min before the onset of electrophoresis through a 5% native polyacrylamide gel. (B) Absence of hNTH1-nucleosome ternary complexes inthe presence of enzymatically active, high-turnover hNTH1 mutants. Tg-out(5S) nucleosomes were incubated with 0, 20, 60, 100, and 400 nM totalconcentrations of hNTH1 for 2.5 min at 22°C before the onset of electrophoresis. In lanes 1 to 5, nucleosomes were incubated with full-lengthhNTH1, whereas lanes 6 to 10 and 11 to 15 were incubated with �55 and �72 N-terminal truncations of hNTH1. (C) Displacement of hNTH1 fromnucleosomes by APE. Nucleosomes were incubated with 35 nM APE alone (lane 2), 14 nM hNTH1 alone (lane 3), or 14 nM hNTH1 together with14 or 35 nM APE (lanes 4 and 5) for 15 min at 22°C prior to the onset of electrophoresis.
VOL. 31, 2011 BASE EXCISION REPAIR IN NUCLEOSOMES 4627
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
nucleosomes was slightly retarded by high concentrations ofthe �55 truncation mutant, possibly due to nonspecific inter-actions between nucleosomes and the enzyme. However, atlower enzyme concentrations, only full-length hNTH1 formeda discrete ternary complex with Tg-containing nucleosomes.
Figure 3C shows the impact of adding APE to hNTH1-nucleosome ternary complexes. At APE concentrations in ex-cess of hNTH1, hNTH1 was displaced from the ternary com-plex; this did not depend on the presence of Mg2, whichrenders APE catalytically active. This result supports a modelin which hNTH1 remains associated with its product until it isdisplaced by APE.
In the experiment shown in Fig. 3C, the APE-mediateddisplacement of hNTH1 from nucleosomes was not followedby formation of a detectable APE-containing ternary complex.It was possible that an APE-nucleosome complex formed buteither could not be resolved or was not stable enough to bedetected in a gel assay. As shown in Fig. SA2 in the supple-mental material, changing assay conditions allowed us to de-tect a probable APE-containing ternary complex. However, itslow stability and abundance made it impossible to conductdefinitive studies of handoff from APE to Pol �.
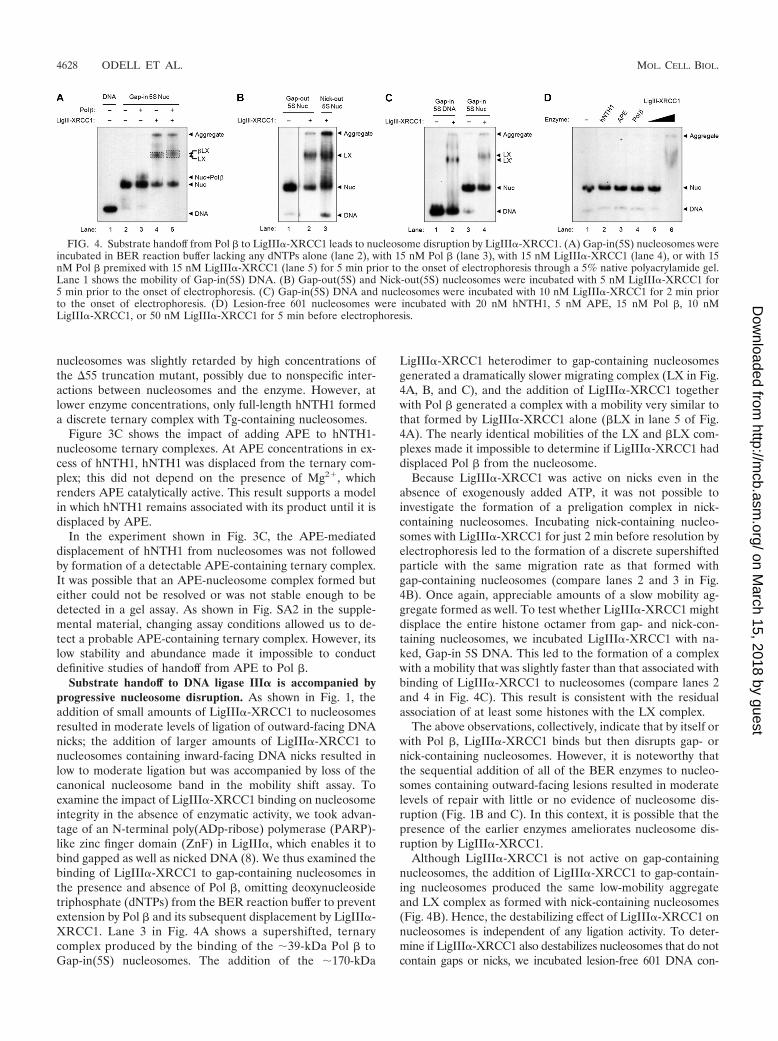
Substrate handoff to DNA ligase III� is accompanied byprogressive nucleosome disruption. As shown in Fig. 1, theaddition of small amounts of LigIII�-XRCC1 to nucleosomesresulted in moderate levels of ligation of outward-facing DNAnicks; the addition of larger amounts of LigIII�-XRCC1 tonucleosomes containing inward-facing DNA nicks resulted inlow to moderate ligation but was accompanied by loss of thecanonical nucleosome band in the mobility shift assay. Toexamine the impact of LigIII�-XRCC1 binding on nucleosomeintegrity in the absence of enzymatic activity, we took advan-tage of an N-terminal poly(ADp-ribose) polymerase (PARP)-like zinc finger domain (ZnF) in LigIII�, which enables it tobind gapped as well as nicked DNA (8). We thus examined thebinding of LigIII�-XRCC1 to gap-containing nucleosomes inthe presence and absence of Pol �, omitting deoxynucleosidetriphosphate (dNTPs) from the BER reaction buffer to preventextension by Pol � and its subsequent displacement by LigIII�-XRCC1. Lane 3 in Fig. 4A shows a supershifted, ternarycomplex produced by the binding of the �39-kDa Pol � toGap-in(5S) nucleosomes. The addition of the �170-kDa
LigIII�-XRCC1 heterodimer to gap-containing nucleosomesgenerated a dramatically slower migrating complex (LX in Fig.4A, B, and C), and the addition of LigIII�-XRCC1 togetherwith Pol � generated a complex with a mobility very similar tothat formed by LigIII�-XRCC1 alone (�LX in lane 5 of Fig.4A). The nearly identical mobilities of the LX and �LX com-plexes made it impossible to determine if LigIII�-XRCC1 haddisplaced Pol � from the nucleosome.
Because LigIII�-XRCC1 was active on nicks even in theabsence of exogenously added ATP, it was not possible toinvestigate the formation of a preligation complex in nick-containing nucleosomes. Incubating nick-containing nucleo-somes with LigIII�-XRCC1 for just 2 min before resolution byelectrophoresis led to the formation of a discrete supershiftedparticle with the same migration rate as that formed withgap-containing nucleosomes (compare lanes 2 and 3 in Fig.4B). Once again, appreciable amounts of a slow mobility ag-gregate formed as well. To test whether LigIII�-XRCC1 mightdisplace the entire histone octamer from gap- and nick-con-taining nucleosomes, we incubated LigIII�-XRCC1 with na-ked, Gap-in 5S DNA. This led to the formation of a complexwith a mobility that was slightly faster than that associated withbinding of LigIII�-XRCC1 to nucleosomes (compare lanes 2and 4 in Fig. 4C). This result is consistent with the residualassociation of at least some histones with the LX complex.
The above observations, collectively, indicate that by itself orwith Pol �, LigIII�-XRCC1 binds but then disrupts gap- ornick-containing nucleosomes. However, it is noteworthy thatthe sequential addition of all of the BER enzymes to nucleo-somes containing outward-facing lesions resulted in moderatelevels of repair with little or no evidence of nucleosome dis-ruption (Fig. 1B and C). In this context, it is possible that thepresence of the earlier enzymes ameliorates nucleosome dis-ruption by LigIII�-XRCC1.
Although LigIII�-XRCC1 is not active on gap-containingnucleosomes, the addition of LigIII�-XRCC1 to gap-contain-ing nucleosomes produced the same low-mobility aggregateand LX complex as formed with nick-containing nucleosomes(Fig. 4B). Hence, the destabilizing effect of LigIII�-XRCC1 onnucleosomes is independent of any ligation activity. To deter-mine if LigIII�-XRCC1 also destabilizes nucleosomes that do notcontain gaps or nicks, we incubated lesion-free 601 DNA con-
FIG. 4. Substrate handoff from Pol � to LigIII�-XRCC1 leads to nucleosome disruption by LigIII�-XRCC1. (A) Gap-in(5S) nucleosomes wereincubated in BER reaction buffer lacking any dNTPs alone (lane 2), with 15 nM Pol � (lane 3), with 15 nM LigIII�-XRCC1 (lane 4), or with 15nM Pol � premixed with 15 nM LigIII�-XRCC1 (lane 5) for 5 min prior to the onset of electrophoresis through a 5% native polyacrylamide gel.Lane 1 shows the mobility of Gap-in(5S) DNA. (B) Gap-out(5S) and Nick-out(5S) nucleosomes were incubated with 5 nM LigIII�-XRCC1 for5 min prior to the onset of electrophoresis. (C) Gap-in(5S) DNA and nucleosomes were incubated with 10 nM LigIII�-XRCC1 for 2 min priorto the onset of electrophoresis. (D) Lesion-free 601 nucleosomes were incubated with 20 nM hNTH1, 5 nM APE, 15 nM Pol �, 10 nMLigIII�-XRCC1, or 50 nM LigIII�-XRCC1 for 5 min before electrophoresis.
4628 ODELL ET AL. MOL. CELL. BIOL.
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
taining nucleosomes with each BER enzyme individually. Theaddition of hNTH1, APE, or Pol � to nucleosomes had no impacton their mobilities (lanes 1 to 4 of Fig. 4D), indicating that theternary complexes shown in Fig. 1, 3, and 4 are substrate specific.Likewise, the addition of 10 nM LigIII�-XRCC1 to nucleosomes,an amount similar to that used in earlier experiments, producedneither a nucleosome supershift nor an aggregate (lane 5 of Fig.4D). Thus, the above-described effects of LigIII�-XRCC1 onnucleosomes are also substrate specific. Only upon addition of a5-fold-higher concentration of LigIII�-XRCC1 did we observe alow mobility smear along with an aggregate that failed to migrateinto the gel (lane 6 of Fig. 4D). Of note, the high LigIII�-XRCC1concentration used here represents a 1.25-fold molar excess ofenzyme over total nucleosomes in the reaction (50 nM and 40nM, respectively). Therefore, at high concentrations, LigIII�-XRCC1 can bind and disrupt nucleosomes indiscriminately; how-ever, purified XRCC1 alone was not sufficient to cause nucleo-some disruption, as shown in Fig. SA3 in the supplementalmaterial.
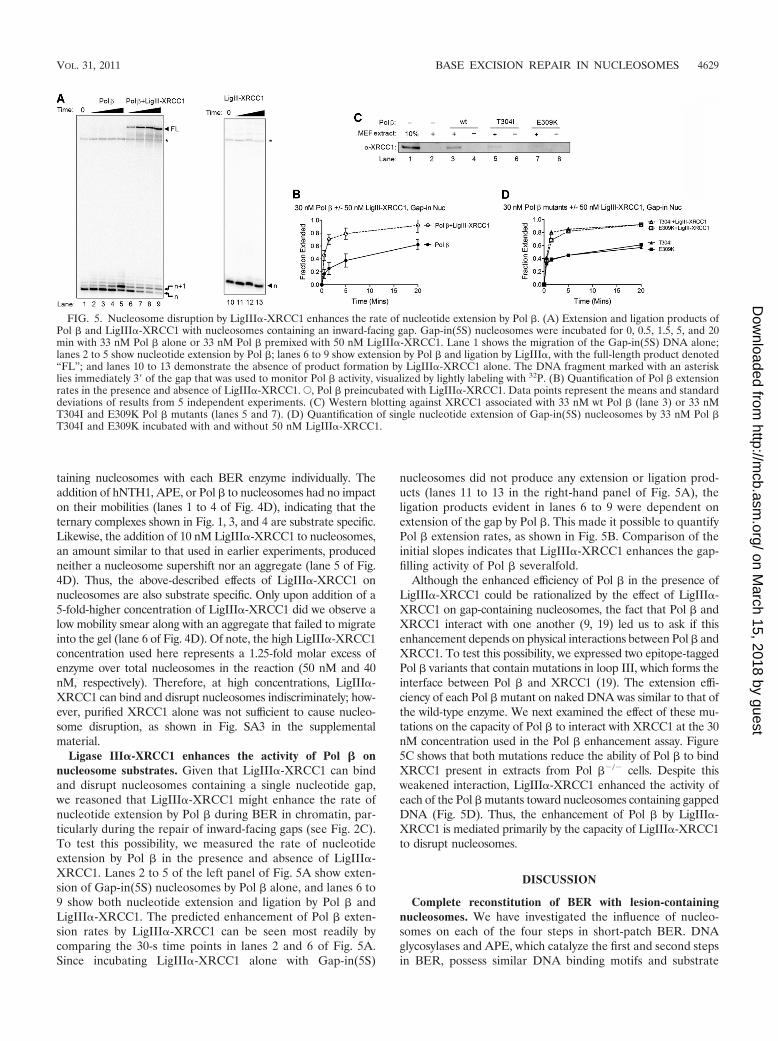
Ligase III�-XRCC1 enhances the activity of Pol � onnucleosome substrates. Given that LigIII�-XRCC1 can bindand disrupt nucleosomes containing a single nucleotide gap,we reasoned that LigIII�-XRCC1 might enhance the rate ofnucleotide extension by Pol � during BER in chromatin, par-ticularly during the repair of inward-facing gaps (see Fig. 2C).To test this possibility, we measured the rate of nucleotideextension by Pol � in the presence and absence of LigIII�-XRCC1. Lanes 2 to 5 of the left panel of Fig. 5A show exten-sion of Gap-in(5S) nucleosomes by Pol � alone, and lanes 6 to9 show both nucleotide extension and ligation by Pol � andLigIII�-XRCC1. The predicted enhancement of Pol � exten-sion rates by LigIII�-XRCC1 can be seen most readily bycomparing the 30-s time points in lanes 2 and 6 of Fig. 5A.Since incubating LigIII�-XRCC1 alone with Gap-in(5S)
nucleosomes did not produce any extension or ligation prod-ucts (lanes 11 to 13 in the right-hand panel of Fig. 5A), theligation products evident in lanes 6 to 9 were dependent onextension of the gap by Pol �. This made it possible to quantifyPol � extension rates, as shown in Fig. 5B. Comparison of theinitial slopes indicates that LigIII�-XRCC1 enhances the gap-filling activity of Pol � severalfold.
Although the enhanced efficiency of Pol � in the presence ofLigIII�-XRCC1 could be rationalized by the effect of LigIII�-XRCC1 on gap-containing nucleosomes, the fact that Pol � andXRCC1 interact with one another (9, 19) led us to ask if thisenhancement depends on physical interactions between Pol � andXRCC1. To test this possibility, we expressed two epitope-taggedPol � variants that contain mutations in loop III, which forms theinterface between Pol � and XRCC1 (19). The extension effi-ciency of each Pol � mutant on naked DNA was similar to that ofthe wild-type enzyme. We next examined the effect of these mu-tations on the capacity of Pol � to interact with XRCC1 at the 30nM concentration used in the Pol � enhancement assay. Figure5C shows that both mutations reduce the ability of Pol � to bindXRCC1 present in extracts from Pol ��/� cells. Despite thisweakened interaction, LigIII�-XRCC1 enhanced the activity ofeach of the Pol � mutants toward nucleosomes containing gappedDNA (Fig. 5D). Thus, the enhancement of Pol � by LigIII�-XRCC1 is mediated primarily by the capacity of LigIII�-XRCC1to disrupt nucleosomes.
DISCUSSION
Complete reconstitution of BER with lesion-containingnucleosomes. We have investigated the influence of nucleo-somes on each of the four steps in short-patch BER. DNAglycosylases and APE, which catalyze the first and second stepsin BER, possess similar DNA binding motifs and substrate
FIG. 5. Nucleosome disruption by LigIII�-XRCC1 enhances the rate of nucleotide extension by Pol �. (A) Extension and ligation products ofPol � and LigIII�-XRCC1 with nucleosomes containing an inward-facing gap. Gap-in(5S) nucleosomes were incubated for 0, 0.5, 1.5, 5, and 20min with 33 nM Pol � alone or 33 nM Pol � premixed with 50 nM LigIII�-XRCC1. Lane 1 shows the migration of the Gap-in(5S) DNA alone;lanes 2 to 5 show nucleotide extension by Pol �; lanes 6 to 9 show extension by Pol � and ligation by LigIII�, with the full-length product denoted“FL”; and lanes 10 to 13 demonstrate the absence of product formation by LigIII�-XRCC1 alone. The DNA fragment marked with an asterisklies immediately 3� of the gap that was used to monitor Pol � activity, visualized by lightly labeling with 32P. (B) Quantification of Pol � extensionrates in the presence and absence of LigIII�-XRCC1. E, Pol � preincubated with LigIII�-XRCC1. Data points represent the means and standarddeviations of results from 5 independent experiments. (C) Western blotting against XRCC1 associated with 33 nM wt Pol � (lane 3) or 33 nMT304I and E309K Pol � mutants (lanes 5 and 7). (D) Quantification of single nucleotide extension of Gap-in(5S) nucleosomes by 33 nM Pol �T304I and E309K incubated with and without 50 nM LigIII�-XRCC1.
VOL. 31, 2011 BASE EXCISION REPAIR IN NUCLEOSOMES 4629
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

recognition mechanisms; both enzymes bind to one face of theDNA helix and flip the substrate out of the DNA duplex andinto their active sites (16, 33, 47). These shared structuralfeatures may explain why APE and representative enzymesfrom the two major families of bifunctional DNA glycosylasesare capable of directly binding to optimally oriented substratesin nucleosomes (as inferred from the efficient processing ofoutward-facing substrates). Pol �, which carries out the thirdstep in BER, also was more active on outward- than inward-facing substrates. This result suggests that Pol � also can binddirectly to optimally oriented determinants in nucleosomalDNA even though, in its enzymatically active configuration,Pol � bends DNA opposite the gap by 90°. The severity of thisbend suggests that DNA near the site of the gap must lift awayfrom the histone octamer as Pol � takes on an active config-uration. These same geometric considerations may account forwhy FEN1, which bends the DNA opposite a DNA flap by 90to 100°, is more active on nucleosomal substrates than onnaked DNA (24).
To explain how DNA glycosylases, APE, and Pol � are allable to process nominally occluded substrates in nucleosomes(albeit at enzyme concentration � time values 6- to 66-foldhigher than were needed to process sterically accessible sub-strates), we have systematically tested (in this study and inreferences 37 and 43) known mechanisms that could makesubstrates in nucleosomes accessible to BER enzymes, usinggel mobility shift assays to rule out enzyme-induced disruptionof nucleosomes, restriction endonuclease accessibility assays torule out shifts in nucleosome position, and high-resolutionDNase I footprinting to rule out lesion- or enzyme-inducedchanges in helical positioning. Because nucleosomes assem-bled with 5S DNA exhibit limited but potentially significantlevels of positional heterogeneity that might have escaped de-tection in our positioning assays, we also conducted parallelstudies of BER of lesions in more robustly positioned 601nucleosomes. Results in all cases were in good agreement withone another, which strengthens the conclusion that processingof occluded substrates is not due to positional heterogeneity.
The single remaining known mechanism for exposure ofoccluded lesions in nucleosomes, which our results favor, is thebinding of BER enzymes to their substrates when they aretransiently exposed through spontaneous partial unwrappingof DNA from the histone octamer. The low concentration andshort (10- to 50-ms) duration of the unwrapped state (26) canaccount for why elevated enzyme concentrations are needed toprocess occluded lesions. It is important here to note thatwe observed appreciable processing of occluded lesions byhNTH1 at concentrations no higher than its in vivo concentra-tion (37). Inconsistent reporting of reaction conditions make itdifficult to determine if different active enzyme concentrationscontributed to the wide variation in results among certainearlier studies of BER on chromatin substrates. A secondvariable that may account for much of the variation reported inearlier studies of BER in model nucleosomes is the distancebetween the lesion and the dyad axis of the nucleosome. Ingeneral, DNA glycosylases (and in some instances downstreamenzymes as well) were substantially more active on substrates�40 nt or more from the dyad axis than on substrates locatedat or near the dyad axis (compare, e.g., this study and refer-ences 36, 37, and 43 to references 3, 32, and 35). For inward-
facing lesions, this result conforms to a prediction of the DNAunwrapping hypothesis, in that the probability of lesion expo-sure due to DNA unwrapping diminishes with increasing prox-imity of the lesion to the center of the nucleosome (2, 22, 43).Differences in the probability of substrate exposure may notfully account for why Pol � exhibited good activity in our studybut virtually none at all in an earlier study (3); it may also beharder for Pol � to bend DNA substrates near the center of thenucleosome into an active conformation.
Substrate handoff during BER. It has been hypothesizedthat the individual steps in BER involve recognition of a prod-uct-enzyme complex by the next enzyme in the pathway inorder to avoid the cytotoxic or mutagenic effects of unrepairedBER intermediates (44, 54). Although we did not detect thekinetic signature that we hypothesized might be associatedwith substrate handoff, we did observe a succession of discreteternary complexes that form between enzymes and nucleo-somes during BER. The hNTH1 and Pol � complexes wereeasier to detect than the APE-containing complex, suggestingthat the complexes differ in stability or half-life. Nevertheless,the fact that we were able to detect even a trace of thesecomplexes in a gel mobility shift assay, without use of priorcross-linking, suggests that the lifetimes of these complexesexceed the time it takes to process oxidative lesions in vivo.Thus, our results support the idea that BER enzymes displaceone another in an orderly fashion, thereby ensuring that sub-strate intermediates remain sequestered until repair is com-plete.
LigIII�-XRCC1-driven disruption of nucleosomes duringBER. DNA ligase III was recently reported to be essential forrepair of DNA in mitochondria but not in nuclei (17). Thisprobably reflects the participation of DNA ligase I in long-patch BER, which can take the place of short-patch BER in theevent of defects in LigIII (42). Structural studies, showing thatboth LigI and LigIII fully encircle their DNA substrates (8, 40),led us to predict that LigIII�-XRCC1 would be unable todirectly bind nucleosomal DNA. Consistent with this predic-tion, the low to moderate levels of activity that LigIII�-XRCC1exhibited toward nicks in nucleosomes was independent of thehelical orientation of the nick. Thus, it is likely that partialDNA unwrapping is an obligatory first step in the binding ofLigIII�-XRCC1 to nucleosomal substrates. This probably alsoaccounts for the capacity of LigI to act on nucleosomal sub-strates, albeit with an �10-fold-lower efficiency than that seenon naked DNA (7). Increasing concentrations of LigIII�-XRCC1 resulted in nucleosome disruption. LigIII�-XRCC1appeared to more readily disrupt 5S DNA-containing nucleo-somes than 601 nucleosomes, which probably accounts for themore efficient ligation of nicked DNA substrates in the 5Snucleosomes. LigIII�-XRCC1 also significantly enhanced ex-tension of occluded gaps by Pol �, most likely via LigIII�-XRCC1-driven disruption of gap-containing nucleosomes. In-terestingly, LigI is unable to bind DNA gaps because it lacksthe ZnF of LigIII and thus may not enhance the activity of Pol� in the same manner.
The severely restricted activity of Pol � on gaps near thedyad axis of nucleosomes suggests that nucleosome movementor disruption prior to the gap-filling step is generally requiredto complete BER in vivo. It is possible as well that the fate ofnucleosomes during earlier steps in BER differs with lesion
4630 ODELL ET AL. MOL. CELL. BIOL.
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

position: nucleosomes in this study remained intact during thefirst three steps in BER, but rare DNA unwrapping events thatexpose and allow binding of DNA glycosylases or APE tosubstrates near the dyad axis may be sufficiently destabilizing asto result in nucleosome disruption. In any event, the pooractivity of Pol � on gaps at or near the dyad axis of thenucleosomes underscores the importance of our finding thatLigIII�-XRCC1 can disrupt gap-containing nucleosomes.LigIII�-XRCC1 by itself may prove sufficient to ensure com-pletion of BER in vivo. Whether known histone chaperones orchromatin remodeling agents participate as well remains to bedetermined.
ACKNOWLEDGMENTS
We thank the UVM DNA Analysis Facility for DNA sequencingand phosphorimagery support and April Averill and Lauren Harveyfor the expression and purification of hNTH1, APE, and wild-typeDNA Pol �.
This research was supported in part by a grant from the NSF (MCB-0821941) to D. S. Pederson; a grant from the NCI (P01-CA098993) toS. S. Wallace, J. B. Sweasy, and D. S. Pederson; and grants from theNIH (R01 ES012512 and P01 CA92584) to A. E. Tomkinson.
We have no financial conflicts of interest.
REFERENCES
1. Almeida, K. H., and R. W. Sobol. 2007. A unified view of base excision repair:lesion-dependent protein complexes regulated by post-translational modifi-cation. DNA Repair 6:695–711.
2. Anderson, J. D., and J. Widom. 2000. Sequence and position-dependence ofthe equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol.296:979–987.
3. Beard, B. C., S. H. Wilson, and M. J. Smerdon. 2003. Suppressed catalyticactivity of base excision repair enzymes on rotationally positioned uracil innucleosomes. Proc. Natl. Acad. Sci. U. S. A. 100:7465–7470.
4. Beard, W. A., D. D. Shock, V. K. Batra, L. C. Pedersen, and S. H. Wilson.2009. DNA polymerase beta substrate specificity: side chain modulation ofthe “A-rule.” J. Biol. Chem. 284:31680–31689.
5. Blaisdell, J. O., and S. S. Wallace. 2007. Rapid determination of the activefraction of DNA repair glycosylases: a novel fluorescence assay for trappedintermediates. Nucleic Acids Res. 35:1601–1611.
6. Breen, A. P., and J. A. Murphy. 1995. Reactions of oxyl radicals with DNA.Free Radic. Biol. Med. 18:1033–1077.
7. Chafin, D. R., J. M. Vitolo, L. A. Henricksen, R. A. Bambara, and J. J. Hayes.2000. Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J.19:5492–5501.
8. Cotner-Gohara, E., et al. 2010. Human DNA ligase III recognizes DNA endsby dynamic switching between two DNA bound states. Biochemistry 49:6165–6176.
9. Cuneo, M. J., and R. E. London. 2010. Oxidation state of the XRCC1N-terminal domain regulates DNA polymerase beta binding affinity. Proc.Natl. Acad. Sci. U. S. A. 107:6805–6810.
10. David, S. S., V. L. O’Shea, and S. Kundu. 2007. Base-excision repair ofoxidative DNA damage. Nature 447:941–950.
11. Dong, F., J. C. Hansen, and K. E. van Holde. 1990. DNA and proteindeterminants of nucleosome positioning on sea urchin 5S rRNA gene se-quences in vitro. Proc. Natl. Acad. Sci. U. S. A. 87:5724–5728.
12. Dyer, P. N., et al. 2004. Reconstitution of nucleosome core particles fromrecombinant histones and DNA. Methods Enzymol. 375:23–44.
13. Engelward, B. P., et al. 1997. Base excision repair deficient mice lacking theAag alkyladenine DNA glycosylase. Proc. Natl. Acad. Sci. U. S. A. 94:13087–13092.
14. Enright, H., W. J. Miller, R. Hays, R. A. Floyd, and R. P. Hebbel. 1996.Preferential targeting of oxidative base damage to internucleosomal DNA.Carcinogenesis 17:1175–1177.
15. Flaus, A., K. Luger, S. Tan, and T. J. Richmond. 1996. Mapping nucleosomeposition at single base-pair resolution by using site-directed hydroxyl radi-cals. Proc. Natl. Acad. Sci. U. S. A. 93:1370–1375.
16. Fromme, J. C., and G. L. Verdine. 2003. Structure of a trapped endonucleaseIII-DNA covalent intermediate. EMBO J. 22:3461–3471.
17. Gao, Y., et al. 2011. DNA ligase III is critical for mtDNA integrity but notXrcc1-mediated nuclear DNA repair. Nature 471:240–244.
18. Groth, A., W. Rocha, A. Verreault, and G. Almouzni. 2007. Chromatinchallenges during DNA replication and repair. Cell 128:721–733.
19. Gryk, M. R., et al. 2002. Mapping of the interaction interface of DNApolymerase beta with XRCC1. Structure 10:1709–1720.
20. Hayes, J. J., T. D. Tullius, and A. P. Wolffe. 1990. The structure of DNA ina nucleosome. Proc. Natl. Acad. Sci. U. S. A. 87:7405–7409.
21. Hegde, M. L., T. K. Hazra, and S. Mitra. 2008. Early steps in the DNA baseexcision/single-strand interruption repair pathway in mammalian cells. CellRes. 18:27–47.
22. Hinz, J. M., Y. Rodriguez, and M. J. Smerdon. 2010. Rotational dynamics ofDNA on the nucleosome surface markedly impact accessibility to a DNArepair enzyme. Proc. Natl. Acad. Sci. U. S. A. 107:4646–4651.
23. Huffman, J. L., O. Sundheim, and J. A. Tainer. 2005. DNA base damagerecognition and removal: new twists and grooves. Mutat. Res. 577:55–76.
24. Huggins, C. F., et al. 2002. Flap endonuclease 1 efficiently cleaves baseexcision repair and DNA replication intermediates assembled into nucleo-somes. Mol. Cell 10:1201–1211.
25. Lang, T., M. Maitra, D. Starcevic, S. X. Li, and J. B. Sweasy. 2004. A DNApolymerase beta mutant from colon cancer cells induces mutations. Proc.Natl. Acad. Sci. U. S. A. 101:6074–6079.
26. Li, G., M. Levitus, C. Bustamante, and J. Widom. 2005. Rapid spontaneousaccessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12:46–53.
27. Liu, X., S. Choudhury, and R. Roy. 2003. In vitro and in vivo dimerization ofhuman endonuclease III stimulates its activity. J. Biol. Chem. 278:50061–50069.
28. Ljungman, M., and P. C. Hanawalt. 1992. Efficient protection against oxi-dative DNA damage in chromatin. Mol. Carcinog. 5:264–269.
29. Lowary, P. T., and J. Widom. 1998. New DNA sequence rules for high affinitybinding to histone octamer and sequence-directed nucleosome positioning.J. Mol. Biol. 276:19–42.
30. Luger, K., T. J. Rechsteiner, and T. J. Richmond. 1999. Preparation ofnucleosome core particle from recombinant histones. Methods Enzymol.304:3–19.
31. McGhee, J. D., and G. Felsenfeld. 1979. Reaction of nucleosome DNA withdimethyl sulfate. Proc. Natl. Acad. Sci. U. S. A. 76:2133–2137.
32. Menoni, H., et al. 2007. ATP-dependent chromatin remodeling is requiredfor base excision repair in conventional but not in variant H2A.Bbd nucleo-somes. Mol. Cell. Biol. 27:5949–5956.
33. Mol, C. D., T. Izumi, S. Mitra, and J. A. Tainer. 2000. DNA-bound structuresand mutants reveal abasic DNA binding by APE1 and DNA repair coordi-nation. Nature 403:451–456.
34. Murphy, D. L., J. Jaeger, and J. B. Sweasy. 2011. A triad interaction in thefingers subdomain of DNA polymerase beta controls polymerase activity.J. Am. Chem. Soc. 133:6279–6287.
35. Nakanishi, S., R. Prasad, S. H. Wilson, and M. Smerdon. 2007. Differentstructural states in oligonucleosomes are required for early versus late stepsof base excision repair. Nucleic Acids Res. 35:4313–4321.
36. Nilsen, H., T. Lindahl, and A. Verreault. 2002. DNA base excision repair ofuracil residues in reconstituted nucleosome core particles. EMBO J. 21:5943–5952.
37. Odell, I. D., K. Newick, N. H. Heintz, S. S. Wallace, and D. S. Pederson.2010. Non-specific DNA binding interferes with the efficient excision ofoxidative lesions from chromatin by the human DNA glycosylase, NEIL1.DNA Repair 9:134–143.
38. Palomera-Sanchez, Z., and M. Zurita. 2011. Open, repair and close again:chromatin dynamics and the response to UV-induced DNA damage. DNARepair 10:119–125.
39. Parikh, S. S., C. D. Mol, D. J. Hosfield, and J. A. Tainer. 1999. Envisioningthe molecular choreography of DNA base excision repair. Curr. Opin.Struct. Biol. 9:37–47.
40. Pascal, J. M., P. J. O’Brien, A. E. Tomkinson, and T. Ellenberger. 2004.Human DNA ligase I completely encircles and partially unwinds nickedDNA. Nature 432:473–478.
41. Pennings, S., G. Meersseman, and E. M. Bradbury. 1991. Mobility of posi-tioned nucleosomes on 5S rDNA. J. Mol. Biol. 220:101–110.
42. Petermann, E., C. Keil, and S. L. Oei. 2006. Roles of DNA ligase III andXRCC1 in regulating the switch between short patch and long patch BER.DNA Repair 5:544–555.
43. Prasad, A., S. S. Wallace, and D. S. Pederson. 2007. Initiation of baseexcision repair of oxidative lesions in nucleosomes by the human, bifunc-tional DNA glycosylase NTH1. Mol. Cell. Biol. 27:8442–8453.
44. Prasad, R., D. D. Shock, W. A. Beard, and S. H. Wilson. 2010. Substratechanneling in mammalian base excision repair pathways: passing the baton.J. Biol. Chem. 285:40479–40488.
45. Schalch, T., S. Duda, D. F. Sargent, and T. J. Richmond. 2005. X-raystructure of a tetranucleosome and its implications for the chromatin fibre.Nature 436:138–141.
46. Simpson, R. T., and D. W. Stafford. 1983. Structural features of a phasednucleosome core particle. Proc. Natl. Acad. Sci. U. S. A. 80:51–55.
47. Slupphaug, G., et al. 1996. A nucleotide-flipping mechanism from the struc-ture of human uracil-DNA glycosylase bound to DNA. Nature 384:87–92.
48. Sobol, R. W. 2007. DNA polymerase beta null mouse embryonic fibroblastsharbor a homozygous null mutation in DNA polymerase iota. DNA Repair6:3–7.
VOL. 31, 2011 BASE EXCISION REPAIR IN NUCLEOSOMES 4631
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

49. Thastrom, A., et al. 1999. Sequence motifs and free energies of selectednatural and non-natural nucleosome positioning DNA sequences. J. Mol.Biol. 288:213–229.
50. Thastrom, A., P. T. Lowary, and J. Widom. 2004. Measurement of histone-DNA interaction free energy in nucleosomes. Methods 33:33–44.
51. Wallace, S. S., V. Bandaru, S. D. Kathe, and J. P. Bond. 2003. The enigmaof endonuclease VIII. DNA Repair 2:441–453.
52. Waters, T. R., P. Gallinari, J. Jiricny, and P. F. Swann. 1999. Humanthymine DNA glycosylase binds to apurinic sites in DNA but is displaced byhuman apurinic endonuclease 1. J. Biol. Chem. 274:67–74.
53. Wilson, D. M., III, T. M. Sofinowski, and D. R. McNeill. 2003. Repairmechanisms for oxidative DNA damage. Front. Biosci. 8:D963–D981.
54. Wilson, S. H., and T. A. Kunkel. 2000. Passing the baton in base excisionrepair. Nat. Struct. Biol. 7:176–178.
4632 ODELL ET AL. MOL. CELL. BIOL.
on March 15, 2018 by guest
http://mcb.asm
.org/D
ownloaded from





























