Embed Size (px)
Citation preview

Neuromuscular Disorders in Critically IllPatients: Review and Update
David Lacomis, MD
AbstractNeuromuscular disorders that are diagnosed in the
intensive care unit (ICU) usually cause substantial
limb weakness and contribute to ventilatory dys-
function. Although some lead to ICU admission,
ICU-acquired disorders, mainly critical illness my-
opathy (CIM) and critical illness polyneuropathy
(CIP), are more frequent and are associated with
considerable morbidity. Approximately 25% to
45% of patients admitted to the ICU develop CIM,
CIP, or both. Their clinical features often overlap;
therefore, nerve conduction studies and electro-
myography are particularly helpful diagnostically,
and more sophisticated electrodiagnostic studies
and histopathologic evaluation are required in
some circumstances. A number of prospective
studies have identified risk factors for CIP and
CIM, but their limitations often include the inability
to separate CIM from CIP. Animal models reveal
evidence of a channelopathy in both CIM and CIP,
and human studies also identified axonal degener-
ation in CIP and myosin loss in CIM. Outcomes are
variable. They tend to be better with CIM, and some
patients have longstanding disabilities. Future
studies of well-characterized patients with CIP and
CIM should refine our understanding of risk
factors, outcomes, and pathogenic mechanisms,
leading to better interventions.
Key Words: critical illness myopathy, critical illness
polyneuropathy, intensive care unit, myopathy,
polyneuropathy, neuromuscular disorders
( J Clin Neuromusc Dis 2011;12:197–218)
HISTORY
The study of neuromuscular disorders
in critically ill patients has been evolving over
the past 50 years. Patients with polio were the
first to have neuromuscular weakness that
often caused ventilatory dysfunction leading
to admission to the earliest intensive care
units (ICUs) that consisted of negative pres-
sure ventilators. As modern ICUs arose and
polio was mostly eradicated, patients with
other ‘‘traditional’’ neuromuscular disorders
such as Guillain Barre syndrome (GBS) and
myasthenia gravis with ‘‘crisis’’ more com-
monly benefited from ICU treatment of
ventilatory dysfunction or airway collapse,
and mortality rates declined. In the 1980s, it
became evident that some patients, who were
in the ICU for treatment of medical and
surgical conditions, developed diffuse weak-
ness, often with ventilatory failure.
Bolton and colleagues first reported the
clinical, electrodiagnostic, and histopatho-
logic features of ICU patients with newly
acquired weakness primarily in the setting of
sepsis—defined as suspected or proven infec-
tion with a systemic inflammatory response
syndrome (SIRS)1,2—and multiorgan failure,
culminating in their seminal reports of critical
illness polyneuropathy (CIP).3–5 In Bolton’s
comments on the discovery of CIP, he credits
Osler’s 1892 description of ‘‘rapid loss of
flesh’’ with prolonged sepsis as the first
possible observation of this association,6,7
and he notes reports of polyneuropathy (PN)
after circulatory shock8 and burns.9 Neverthe-
less, it was really Bolton, Zochodne, and
colleagues who identified CIP and brought
attention to the field of ICU-acquired neuro-
muscular weakness.3–5
While Bolton and colleagues were
studying CIP, there were also reports of single
cases10–14 and eventually series15–21 of adult
and pediatric10 patients who developed acute
myopathy during treatment of status asthma-
ticus. Later, a similar acute myopathy was
noted to follow organ transplantation22–27 and
to occur in association with many other
critical illness states in children as well as
adults.28–35 Terminology was highly variable
Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
From the Departments ofNeurology and Pathology(Neuropathology), University ofPittsburgh School of Medicine,Pittsburgh, PA.
Reprints: David Lacomis, MD,UPMC Presbyterian, 200 LothropStreet, F878, Pittsburgh, PA15213 (e-mail:[email protected]).
Copyright � 2011 byLippincott Williams & Wilkins
Review Article 197

initially, but eventually the name critical
illness myopathy (CIM) was accepted.36 Also,
during the same period, it was noted that rare
ICU patients developed prolonged neuromus-
cular junction (NMJ) blockade after receiving
paralytic drugs in high doses or for prolonged
periods in association with persisting drug
metabolites and organ failure.37–41 Some of
these patients also had myopathy or neurop-
athy.41–44 As treatment regimens shifted from
the use of paralytic agents to sedatives such as
propofol, persistent NMJ blockade has largely
disappeared, whereas CIP and CIM persist.
Zochodne et al also noted that biopsy
or autopsy specimens from some of their
patients with CIP exhibited muscle necrosis
consistent with a component of myopathy.4
Since the last review of neuromuscular
weakness in the ICU in this journal,45 there
has been a substantial increase in reports that
the disorders mentioned, especially CIM and
CIP, occur together. The coexistence of
CIM and CIP certainly complicates diagnosis.
Lumping CIM and CIP together, when they
cannot be differentiated, has also made it
more difficult to interpret the results of
clinical research on these two conditions.
This article provides an overview of the
approach to diagnosing neuromuscular dis-
orders in the ICU while providing a detailed
update on the ICU-acquired conditions,
mainly CIM and CIP.
Spectrum of NeuromuscularDisorders in the Intensive Care Unit
Given the lack of prospective data, the
spectrum and relative frequency of occur-
rence of all neuromuscular disorders diag-
nosed in the ICU is uncertain. One
retrospective, electromyography (EMG)-
based series performed on 92 patients over
4 years noted that 28% of patients who
underwent EMG in the ICU because of
weakness had ‘‘traditional’’ neuromuscular
disorders that led to ICU admission. Of these,
GBS was most common, but motor neuron
disease, myasthenia gravis, and rarely pre-
existing myopathy were reported. The major-
ity had newly acquired myopathy, mostly CIM,
and most of the rest had CIP. Neuromuscular
junction blockade was rarely noted.46 Other
disorders causing neuromuscular weakness
that sometimes leads to ICU admission
resulting from ventilatory failure or airway
collapse47–55 are shown in Table 1. These con-
ditions may be important causes of prolonged
ventilator dependency.56 In addition, some
pre-existing mild or subclinical neuromuscu-
lar disorders, including myasthenia gravis or
Lambert-Eaton myasthenic syndrome, may be
unmasked in the ICU by either the critical
illness or treatment with medications such as
aminoglycosides and magnesium.
Approach to the Weak IntensiveCare Unit Patient
Clinical and Laboratory Features
Like with all neurologic evaluations,
localization of the disease process is first
attempted from the history and examination.
TABLE 1. Neuromuscular Causes of Weaknessin the Intensive Care Unit
MyopathyCritical illness myopathyRhabdomyolysis (toxins, infection, and so on)Cachectic myopathyPolymyositis, dermatomyositisMuscular dystrophiesAcid maltase deficiencyMitochondrial myopathyHypophosphatemic myopathyToxic myopathy, eg, hydroxychloroquine
Peripheral neuropathyCritical illness polyneuropathyGuillian Barre syndromesVasculitic polyneuropathyPorphyriaParaneoplastic polyneuropathyToxic polyneuropathyNutritional polyneuropathy
Neuromuscular junction disordersProlonged neuromuscular junction blockadeMyasthenia gravisLambert-Eaton myasthenic syndromeBotulismTick paralysis
Motor neuron disordersAmyotrophic lateral sclerosis and variantsSpinal muscular atrophyPoliomyelitis syndromes, eg, West Nile virusPostpolio syndrome
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
198
� 2011 Lippincott Williams & Wilkins

Limitations may include encephalopathy,
tracheal intubation with limited patient com-
munication, and an unreliable sensory exam-
ination. The presence of encephalopathy
should not dissuade one from searching for
a neuromuscular disorder, especially when
the encephalopathy is improving and weak-
ness is not. Even hyperreflexia does not
exclude a coexisting lower motor neuron
disturbance in these complex patients. Fur-
thermore, a pre-existing neuromuscular dis-
order, typically a PN, may make diagnosis of
a new neuromuscular disturbance even more
challenging.
In the ICU, newly acquired central
nervous system (CNS) disorders are rarely
found to be the cause of acute flaccid
quadriparesis with airway or ventilatory
dysfunction. Considerations include high
cervical myelopathy or brainstem disorders
that may be the result of autoimmune de-
myelination or myelinolysis or from vascular
diseases, including hemorrhage or infarction
from thrombosis or hypoperfusion.
If present in CNS disorders, pupillary or
ocular motility abnormalities may help local-
ize the process to the brainstem. If a sensory
level and sphincter dysfunction are found,
a spinal cord lesion should be sought. In the
absence of these findings, magnetic reso-
nance imaging of the brain or spinal cord
may still be worth obtaining in patients with
risk factors for vascular diseases or for osmotic
or autoimmune demyelination.
In diagnosing acute lower motor neuron
disorders in the ICU, analysis of the following
clinical features is most helpful: presence and
pattern of weakness, pattern of tendon reflex
alteration, fasciculations, and sensory find-
ings. Acute motor neuron diseases such as
poliomyelitis syndrome from West Nile virus
may also have features of systemic illnesses,
including fever, fatigue, and headache.57
Occasionally, patients with chronic motor
neuron diseases are first diagnosed in the ICU
if they present with ventilatory or bulbar
dysfunction. They may or may not have subtle
pre-existing symptoms, including limb and
bulbar weakness. Fasciculations in the
tongue, paraspinal, intercostal, or limb
muscles along with atrophy and upper motor
neuron signs (in amyotrophic lateral sclerosis)
are most diagnostically helpful if present.
When limb weakness is present, it is often
asymmetric and not ‘‘length-dependent.’’ This
pattern of weakness with fasciculations and
without sensory dysfunction is not seen in
other groups of neuromuscular disorders
occurring in the ICU.
Neuromuscular junction disorders are
rarely first diagnosed in the ICU. As men-
tioned, they may be ‘‘unmasked’’ by critical
illness or by drugs such as magnesium and
aminoglycosides, which interfere with NMJ
transmission. Myasthenia gravis sometimes
presents with ‘‘crisis’’ and is diagnosed in the
ICU. Many of these patients have ptosis and
pupil-sparing ophthalmoparesis with facial,
bulbar, and proximal or generalized limb
weakness that is fatigable. Tendon reflexes
are usually normal. Although acetylcholine
receptor binding antibodies are often present
and anti-MuSK antibodies occur in many
‘‘seronegative’’ patients, finding a decrement
on 2- to 3-Hz repetitive stimulation (discussed
later) is a more timely way of supporting the
diagnosis and is more objective and possibly
more specific than a positive edrophonium
test.58 Patients with Lambert- Eaton myas-
thenic syndrome almost never present with
ventilatory failure. They usually have proxi-
mal weakness, hyporeflexia, and milder
cranial muscle and autonomic involvement.
Voltage-gated calcium channel antibodies are
often present, and an underlying small cell
lung cancer may be found in approximately
half. Botulism, on the other hand, often leads
to ICU admission and can be a difficult
diagnosis without an obvious history of eating
home-canned or spoiled food. Pupils are
usually affected in addition to ventilatory,
bulbar, and limb muscles. Tendon reflexes
may be attenuated.
Prolonged NMJ blockade is now rarely
seen as a result of decreased use of high doses
or continuous infusions of NMJ-blocking agents.
Affected patients have tetraplegia and areflexia
with or without ophthalmoplegia.37–41
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
199
www.jcnmd.com

The polyneuropathies that occur in the
ICU usually do not present like the typical
chronic length-dependent sensorimotor neu-
ropathies seen in outpatients. They often
affect phrenic or cranial nerve function and
may be acute to subacute in onset. There are
axonal and demyelinating subtypes. The pro-
totype demyelinating PN, the acute inflam-
matory demyelinating polyneuropathy variant
of GBS, may lead to ICU admission; it rarely
develops in the ICU. The axonal neuropathies
that may be diagnosed in the ICU include CIP,
axonal variants of GBS, and porphyric as well
as vasculitic neuropathies. In theory, some
toxic and nutritional neuropathies may be
diagnosed in the ICU. The axonal variants of
GBS—acute motor axonal neuropathy and
acute motor sensory axonal neuropathy—
usually follow a diarrheal illness and present
similarly to acute inflammatory demyelinating
polyneuropathy clinically, but they are un-
common in North America. In all GBS variants,
the cerebrospinal fluid protein is usually
elevated.59
Vasculitic neuropathies usually begin
before ICU admission and present as mono-
neuritis multiplex or asymmetric axonal
sensorimotor polyneuropathies more often
than symmetric PN. Systemic vasculitis or
a predisposing connective tissue may be
present. Laboratory studies may reflect the
presence of an associated connective tissue
disease or systemic vasculitis, for example,
positive anti-c-antineutrophilic cytoplasmic,
or antinuclear antibody. However, laboratory
abnormalities may be absent or less specific
such as an elevated erythrocyte sedimentation
rate. Histopathologic evaluation of nerve
and muscle is typically used to confirm the
diagnosis.
Porphyria could theoretically present in
the ICU. It can be precipitated by stress or
medications and cause a diffuse, severe,
nonlength-dependent primarily motor axonal
neuropathy with dysautonomia. There may be
sensory symptoms without much sensory
loss. Associated features often include
abdominal pain, constipation, psychiatric
manifestations, and sometimes brainstem
dysfunction. Urine assay for porphobilinogen
is the initial screening test.60
Critical Illness PolyneuropathyThe major features of CIP are general-
ized or sometimes distal more than proximal
flaccid weakness, often with ventilatory
dysfunction from phrenic nerve involvement.
Patients may develop muscle atrophy fairly
soon after onset. Tendon reflexes are usually
attenuated or absent. There may be distal
sensory loss, but the sensory examination
may be limited, and motor findings are usually
predominant. Extraocular muscles are spared,
but mild facial weakness may occur. In
contrast to axonal GBS, cerebrospinal fluid
protein is usually normal with CIP.3,4,61,62
Laboratory features are usually consistent
with an underlying SIRS, sepsis, multiorgan
failure, or a combination.
Most myopathies that lead to ICU
admission cause proximal-predominant
weakness with normal tendon reflexes and
sensation. ICU admission may be caused by
diaphragm, cardiac, or sometimes airway
muscle involvement. Some muscular dystro-
phies, acid maltase deficiency, and mitochon-
drial myopathy may present in this fashion,
but usually their presence has been known
before ICU admission. Mitochondrial disor-
ders often affect extraocular muscles, and this
may be a clue to diagnosis; in addition, serum
lactate is sometimes elevated.
Polymyositis and dermatomyositis rarely
lead to ICU admission unless there is either
interstitial lung disease as in the antisynthe-
tase syndromes or cardiac involvement. Such
cardiac involvement is rare.63 Toxic myopa-
thies such as hydroxychloroquine myopathy
rarely cause ventilatory muscle weakness.
Histopathologic studies reveal a vacuolar
myopathy with complex lipid and curvilinear
inclusions.51 Serum creatine kinase (CK) is
usually elevated in the inflammatory myopa-
thies, many dystrophies, toxic myopathies,
and acid maltase deficiency, and it may be
elevated in some cases of mitochondrial
myopathy.63
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
200
� 2011 Lippincott Williams & Wilkins

Cachectic myopathy, which develops in
chronically and critically ill patients, presents
with muscle wasting and proximal-predomi-
nant weakness. Serum CK is normal. EMG
does not reveal fibrillation potentials; normal
or short duration motor unit potentials may be
present. Type 2 myofiber atrophy is seen
histologically.64,65
Acute necrotizing myopathy with or
without myoglobinuria sometimes occurs in
ICU patients. It is associated with generalized
weakness, very high CK levels, irritable
myopathy on EMG, and widespread necrosis
histopathologically.34,66 There may be electro-
diagnostic evidence of a persistent defect in
NMJ transmission.34 Acute necrotizing myop-
athy in the ICU may be a subset of CIM,
because associated loss of thick filaments has
been described rarely66 or it may be consid-
ered a separate entity probably related to
certain medications, especially NMJ-blocking
agents, or possibly from sepsis. This syndrome
is distinct from the more typical rhabdomyol-
ysis related to other exogenous agents in
which there is muscle pain and tenderness
with variable degrees of weakness and normal
or only mildly ‘‘myopathic’’ EMGs.67 It should
be noted that some degree of subclinical
muscle necrosis is probably present in many
septic patients.68
Critical Illness MyopathyCIM presents with flaccid, generalized,
or proximal weakness, often with failure to
wean from mechanical ventilation. There are
normal or reduced tendon reflexes.16–18,20,22,29
Rarely is the weakness asymmetric69 or
upper extremity predominant.70 Facial
weakness may be present,22,29 and extra-
ocular muscle weakness is rare.71 There may
be loss of muscle bulk.28 Sensation is
normal, but it may impossible to test; thus,
there is often clinical overlap with CIP.
Creatine kinase levels are elevated in up to
50% or more of patients with CIM studied
retrospectively. In a prospective study in
status asthmaticus, CK levels were elevated
in 19 of 25 (76%), and peaked 4 days after
intravenous (IV) corticosteroid exposure
(median, 1576 IU/L; range, 66–7430 IU/L);
elevations lasted up to 16 days.20 Therefore,
if patients are not evaluated during the first
2 weeks after onset, the CK elevation may
be missed.
Electrodiagnostic Approach in theIntensive Care Unit
Because of the limitations in neurologic
examination, electrodiagnostic testing is
usually crucial for accurately localizing
neuromuscular disorders in ICU patients.
Unfortunately, it is also more difficult to
perform EMGs in the ICU as a result of the
frequent occurrences of electrical interfer-
ence, edema, cool limbs, limited patient
cooperation, and reduced access to sites of
stimulation and recording resulting from
catheters and dressings. It may be useful to
address some of these issues briefly.
Hot packs can be used for warming cool
limbs. Shutting off unnecessary electrical
equipment and using an isolated outlet may
reduce electrical interference, but it is usually
insufficient. Increasing the low-frequency
filter during needle examination may also
allow identification of fibrillation potentials,
and the electromyographer can use sound to
identify fibrillation potentials and small, poly-
phasic motor unit potentials even if they are
hidden within the artifact. Sixty-Hertz artifact
may especially interfere with F-wave record-
ings and sensory responses. Averaging may
help identify sensory responses. Fortunately,
motor nerve conductions are usually less
affected.
Attention to electrical safety is also more
important in the ICU. Proper grounding is
necessary. Stimulation in a region of fluid
spill should be avoided to prevent current leak.
Patients with external pacemakers cannot
undergo nerve conduction studies (NCSs).
Patients with implanted pacemakers can, but
it may be prudent to avoid repetitive stimula-
tion.72 As another precaution, we have not
stimulated in the neck when intravenous
catheters are present in the internal jugular
or subclavian veins,72 but Bolton finds such
stimulation safe.73
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
201
www.jcnmd.com

If it is clinically helpful to determine if
a patient’s ventilatory dysfunction is the result
of neuromuscular weakness, phrenic NCSs
are very useful, and diaphragm EMG may
provide additional information. The best
techniques have been summarized and re-
ported by Bolton and colleagues73–75 with an
alternative phrenic nerve stimulation site
reported by Resman-Gaspersc and Podnar.76
Our routine ICU NCS usually include at
least one motor and sensory nerve from an
arm and leg. Choices may depend on avail-
ability of stimulation and recording sites. If
the sensory responses are abnormal or if PN
is more likely on clinical grounds, we try to
examine sural, superficial peroneal, median,
ulnar, and radial sensory responses in addition
to peroneal, tibial, median, and ulnar motor
nerves. In the absence of significant edema,
low sensory amplitudes usually indicate
a component of axonal neuropathy. If edema
is present in the legs only, the radial sensory
response may be used as a more reliable
indicator of diffuse sensory axon loss as may
be seen with CIP. The combination of low
motor and sensory amplitudes with normal
latencies and normal or slightly slow conduc-
tion velocities support the diagnosis of an
axonal sensorimotor PN. In the ICU, we
usually do not search for side-to-side asymme-
tries unless vasculitis is suspected clinically or
if there is intralimb disparity in amplitudes, for
example, low superficial peroneal with
normal sural sensory response.
CIP and the axonal sensorimotor variant
of GBS have features of a generalized senso-
rimotor axonal PN,3,4,61,62,77 whereas only the
motor amplitudes are reduced with acute
motor axonal neuropathy.59 In CIP, phrenic
motor amplitudes may be reduced, and
fibrillation potentials appear in the dia-
phragm.62 In the limbs, fibrillation potentials
are usually seen diffusely within 2 to 3 weeks
after onset, and findings of reinnervation are
seen later. Mild denervation findings may also
be seen in facial muscles.4 Early on, there is
decreased motor unit potential (MUP)
recruitment. Short-duration, polyphasic MUPs
may be seen during the early phase of
reinnervation. Especially in this early phase,
if sensory responses are spared, CIM needs to
be excluded. Direct muscle stimulation—
discussed later—or muscle biopsy may be
useful in further evaluating for CIM if necessary.
Typical demyelinating GBS has the
familiar features that may include conduction
block or temporal dispersion, prolonged
latencies, slowing of conduction velocity,
and low amplitudes. Upper extremity sensory
responses may be more affected than the sural
response.78 F-wave prolongation is usually
present but may be difficult to identify in the
ICU as a result of 60-Hz artifact.
Low motor responses are not only
typical of CIP; they are commonly encoun-
tered in ICU patients with diffuse neuromus-
cular weakness. In fact, monitoring the
peroneal motor amplitude has been advo-
cated as a screening method for evolving CIM
or CIP.79 If the sensory responses are normal
or relatively spared, and the low motor
responses affect several nerves, the most
common cause is CIM, but anterior horn cell
disease, motor axonopathy, NMJ disorders,
and other myopathies may be considered. We
perform 2- to 3-Hz repetitive stimulation of the
ulnar or median motor nerve to exclude
a defect in NMJ transmission, especially if
paralytic agents were administered or if
myasthenia gravis is suspected. If the rare
instances when Lambert-Eaton syndrome or
botulism is considered, we perform post-
exercise single shocks to assess for potentia-
tion of the compound muscle action
potentials (CMAPs); and, in patients who
cannot exercise, we perform 50 Hz stimula-
tion. In patients with anterior horn cell
disease, the needle examination usually
reveals fasciculation potentials along with
features of denervation, reinnervation, or
both in one or more body regions (bulbar,
cervical, thoracic, or lumbosacral
segments).80,81
In CIM, the characteristic electrodiag-
nostic findings are diffusely low CMAPs with
normal or minimally reduced sensory nerve
action potentials.18,22,28,29,32 It has also been
noted that slowing of muscle fiber conduction
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
202
� 2011 Lippincott Williams & Wilkins

results in a broad, long-duration CMAP.82–85
Although the CMAP duration has not been
examined in CIP, it has been reported to be
normal in neuropathy from diabetes.84 In
milder cases of CIM, CMAPs may be normal. In
all cases, distal latencies and conduction
velocities are normal as is the number of
motor units in a given muscle.86 Fibrillation
potentials are seen in at least half of examined
limb muscles and uncommonly in facial
muscles19,29 They may be identified within 1
week of disease onset.19 Electrical myotonia
in uncommon.22 If MUPs can be recorded,
they are of short duration and low amplitude
and typically recruit early. However, needle
examination may be difficult in these patients.
When MUPs cannot be assessed, direct
muscle stimulation—discussed subsequently—
may be useful.
Nerve conductions are usually normal in
other myopathies diagnosed in the ICU. If the
needle examination reveals complex repeti-
tive discharges or myotonia along with
myopathic (short duration, low amplitude)
MUP abnormalities, adult acid maltase de-
ficiency should be suspected. The paraspinal
muscles may be preferentially involved. Myo-
tonic discharges may be also present in
hydroxychloroquine myopathy.51 Inflamma-
tory and dystrophic myopathies are usually
associated with proximal-predominant or
diffuse fibrillation potential activity with
short-duration, low-amplitude MUPs. Muscle
histopathologic analysis and sometimes ge-
netic studies are required for definite di-
agnosis in most of these myopathies.
Direct Muscle StimulationWhen the routine electrophysiological
studies do not distinguish CIM from CIP or if
there is suspected overlap, direct muscle
stimulation (DMS) may be useful in differen-
tiating these entities. Rich and colleagues first
suspected that muscle membrane inexcitabil-
ity was the cause of the low CMAPs in CIM,
and they used DMS to prove it in humans87
and to confirm it in an animal model that also
revealed a sodium channelopathy.88 Slightly
different DMS techniques have been
described, and it should be noted that DMS
does have technical pitfalls, including the risk
of end-plate stimulation.83 DMS is usually
performed using a stimulating monopolar
needle electrode with a surface or subdermal
reference electrode placed away from the
motor end plate, usually in the distal part of
a muscle. The tibialis anterior is commonly
studied. After causing a muscle twitch, a re-
cording subdermal needle electrode pair or
a concentric needle is placed in the center of
the muscle proximal to the site of stimulation,
and a maximal direct muscle stimulated CMAP
(dmCMAP) is recorded.31,83 Alternatively,
Trojaborg et al have used a surface stimulating
electrode in their variation of DMS.33 In either
scenario, the recording electrode pair is kept
in place, and the appropriate nerve undergoes
surface stimulation, recording a nerve-evoked
CMAP (neCMAP). A neCMAP-to-dmCMAP
(nerve to muscle) ratio can be calculated,
and a value greater than 0.5, in the setting of
a low dmCMAP, suggests that there is a distur-
bance in muscle membrane excitability con-
sistent with CIM.31 A ratio of less than 0.5 is
consistent with neuropathy.31 Allen et al also
used DMS to demonstrate slowing of muscle
fiber conduction and then confirmed reduced
muscle fiber excitability using paired stimuli.83
Epidemiology of Critical IllnessMyopathy and Critical IllnessPolyneuropathy: Incidence andRisk Factors
The results of the first prospective study
of CIP were reported in 1991.5 Of 43 patients
in the ICU for more than 5 days with sepsis and
multiorgan failure, 70% had electrodiagnostic
evidence of axonal sensorimotor PN. Thirty-
five percent had clinical features of PN; 23%
were moderately severe to severe. There was
worsening PN with time in the ICU and
a correlation with hyperglycemia, hypoalbu-
minemia, and the number of invasive proce-
dures. Other studies revealed that 47% to 76%
of ICU patients had electrophysiological evi-
dence of CIP (Table 2).89–93 Zifko et al studied
132 ICU patients referred for NCS/EMG found
that 47% had CIP, and most had substantial
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
203
www.jcnmd.com
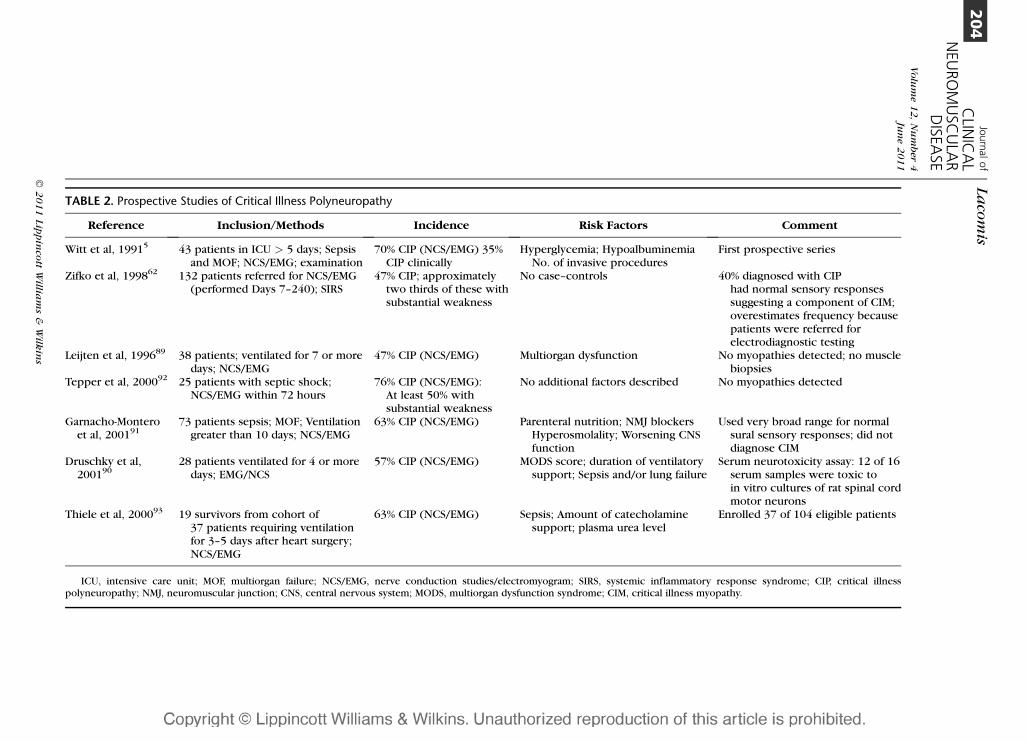
TABLE 2. Prospective Studies of Critical Illness Polyneuropathy
Reference Inclusion/Methods Incidence Risk Factors Comment
Witt et al, 19915 43 patients in ICU > 5 days; Sepsisand MOF; NCS/EMG; examination
70% CIP (NCS/EMG) 35%CIP clinically
Hyperglycemia; HypoalbuminemiaNo. of invasive procedures
First prospective series
Zifko et al, 199862 132 patients referred for NCS/EMG(performed Days 7–240); SIRS
47% CIP; approximatelytwo thirds of these withsubstantial weakness
No case–controls 40% diagnosed with CIPhad normal sensory responsessuggesting a component of CIM;overestimates frequency becausepatients were referred forelectrodiagnostic testing
Leijten et al, 199689 38 patients; ventilated for 7 or moredays; NCS/EMG
47% CIP (NCS/EMG) Multiorgan dysfunction No myopathies detected; no musclebiopsies
Tepper et al, 200092 25 patients with septic shock;NCS/EMG within 72 hours
76% CIP (NCS/EMG):At least 50% withsubstantial weakness
No additional factors described No myopathies detected
Garnacho-Monteroet al, 200191
73 patients sepsis; MOF; Ventilationgreater than 10 days; NCS/EMG
63% CIP (NCS/EMG) Parenteral nutrition; NMJ blockersHyperosmolality; Worsening CNSfunction
Used very broad range for normalsural sensory responses; did notdiagnose CIM
Druschky et al,200190
28 patients ventilated for 4 or moredays; EMG/NCS
57% CIP (NCS/EMG) MODS score; duration of ventilatorysupport; Sepsis and/or lung failure
Serum neurotoxicity assay: 12 of 16serum samples were toxic toin vitro cultures of rat spinal cordmotor neurons
Thiele et al, 200093 19 survivors from cohort of37 patients requiring ventilationfor 3–5 days after heart surgery;NCS/EMG
63% CIP (NCS/EMG) Sepsis; Amount of catecholaminesupport; plasma urea level
Enrolled 37 of 104 eligible patients
ICU, intensive care unit; MOF, multiorgan failure; NCS/EMG, nerve conduction studies/electromyogram; SIRS, systemic inflammatory response syndrome; CIP, critical illnesspolyneuropathy; NMJ, neuromuscular junction; CNS, central nervous system; MODS, multiorgan dysfunction syndrome; CIM, critical illness myopathy.
La
com
isJo
urn
alo
f
CLIN
ICA
LN
EUR
OM
USC
ULA
RD
ISEASE
Volu
me
12
,N
um
ber
4
Jun
e2
01
1
20
4
�2
01
1Lip
pin
cott
Willia
ms
&W
ilkin
s

weakness.62 Tepper et al showed that CIP
could be diagnosed as early as 72 hours after
admission for septic shock.92 Risk factors
included multiorgan failure,89,90 use of paren-
teral nutrition, NMJ blockers, hyperosmolal-
ity, and worsening CNS function.91 Patients
with CIP were on mechanical ventilation for
a longer periods, had longer lengths of stay,
and had higher mortality.
There are three prospective studies of
CIM (Table 3). Douglass et al reported the first
in 1992. They studied 25 ICU patients with
status asthmaticus; all received IV cortico-
steroids, and 23 received vecuronium. Se-
venty-six percent had elevated CK levels,
whereas 36% had clinical evidence of myop-
athy with weakness that was usually general-
ized. Myopathy was confirmed by EMG and
muscle biopsy in a subset, but myosin loss was
not described. There was some correlation
between doses of vecuronium and develop-
ment of myopathy.20
Campellone et al monitored 100 consec-
utive critically ill patients after liver trans-
plantation. All received IV corticosteroids and
NMJ blockers. Patients underwent electro-
diagnostic and histopathologic studies if they
had severe weakness with less than antigravity
muscle function, so milder cases were not
detected in this ill patient population. The
incidence of CIM was 7%. Five of seven
underwent muscle biopsies that revealed
patchy myonecrosis with myosin loss. Risk
factors for development of CIM were severity
of illness by Acute Physiology and Chronic
Health Evaluation II (APACHE II) scores and
renal failure requiring dialysis.22 Amaya-Villar
et al noted a 34.6% incidence of CIM in patients
with severe chronic obstructive pulmonary
disease exacerbations. Biopsies in a subset
confirmed CIM. Risk factors included total
corticosteroid dose and illness severity as well
as sepsis.21
A number of prospective studies have
evaluated the incidence of CIM, CIP, or both
(Table 4).94–99 As a result of differing meth-
odologies, they are somewhat difficult to
compare. Some evaluated the incidence of
neuromuscular disorders in a population at TA
BLE
3.Pro
spect
ive
Stu
die
sofC
ritica
lIll
ness
Myop
ath
y
Refe
ren
ce
Inclu
sio
n/M
eth
od
sIn
cid
en
ce
Ris
kF
acto
rsC
om
men
t
Do
ugla
sset
al,
19
92
20
25
pat
ien
tsst
atu
sas
thm
atic
us
CK
leve
ls;
exam
inat
ion
76
%ele
vat
ed
CK
36
%C
IMcli
nic
ally
All
receiv
ed
IVco
rtic
ost
ero
ids,
NM
Jb
lock
ers
and
oth
er
dru
gs
Myo
pat
hy
on
lyse
ries
Cam
pell
on
eet
al,
19
98
22
10
0p
atie
nts
po
stliver
tran
spla
nt;
eval
uat
ed
ifo
nve
nti
lato
rm
ore
than
7d
ays
or
inh
osp
ital
mo
reth
an1
4d
ays;
NC
S/E
MG
;m
usc
leb
iop
sy
7%
CIM
0%
CIP
Severi
tyo
fil
lness
;re
nal
fail
ure
Sele
cte
dfo
rse
vere
cas
es
becau
sep
atie
nts
were
alre
ady
weak
fro
mli
ver
fail
ure
;u
nd
ere
stim
ates
incid
en
ce
of
CIM
Am
aya-
Vil
lar
et
al,
20
05
21
26
pat
ien
tsw
ith
CO
PD
req
uir
ing
ven
tila
tio
nan
dh
igh
-do
seco
rtic
ost
ero
ids;
Need
leE
MG
atw
ean
ing
34
.6%
CIM
Illn
ess
seve
rity
;ra
teo
fse
psi
s;to
tal
do
seo
fco
rtic
ost
ero
ids
No
nerv
eco
nd
ucti
on
s;co
uld
no
tex
clu
de
CIP
.B
iop
syco
nfi
rmed
CIM
inth
ree
(myo
pat
hy
wit
hm
yosi
nlo
ssan
dT
ype
2fi
ber
atro
ph
y)M
yop
ath
yas
socia
ted
wit
hin
cre
ased
du
rati
on
of
ven
tila
tio
n
CK
,cre
atin
ekin
ase;
NC
S/E
MG
,n
erv
eco
nd
ucti
on
stu
die
s/ele
ctr
om
yogra
m;
CO
PD
,ch
ron
ico
bst
ructi
ve
pu
lmo
nar
yd
iseas
e;
CIM
,cri
tical
illn
ess
po
lyn
eu
rop
ath
y;
IV,
intr
aven
ou
s;N
MJ,
neu
rom
usc
ula
rju
ncti
on
.
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
205
www.jcnmd.com

risk. Others evaluated the cause of weakness
in ICU patients. In some studies, comprehen-
sive electrodiagnostic studies were performed
that allowed differentiation of CIM from CIP
or at least weighed the various contributions
of myopathy versus neuropathy. In other
studies, differentiation between myopathy
and neuropathy was not possible, and the
processes were lumped together. Some stud-
ies did not confirm CIM histopathologically.
They are summarized in Table 4, and some are
discussed subsequently.
Of note, De Jonghe et al performed
a large, multicenter study.95 Of the 95 in-
cluded patients, 25% were found to have
weakness. All had electrodiagnostic evidence
of sensorimotor axonal PN. Ten, who un-
derwent muscle biopsy, also had evidence of
myopathy, but myosin adenosine triphospha-
tase stains were not performed. This study is
the only one in the group in which risk factors
included corticosteroids.
Khan et al studied patients with sepsis
by NCS within 72 hours of ICU admission96
and examined them weekly, performing
needle EMG if there was weakness or if the
CMAPs were low. Sixty-three percent had
abnormal NCS at enrollment, and 10 of 48
(21%) were felt to have a neuromuscular
disease—mostly mixed CIM and CIP—during
serial evaluations. Muscle biopsies were not
performed.96 In a small study, Tennila et al also
performed early electrophysiological studies
on ICU Days 2 to 5 on nine ventilated ICU
patients with either SIRS or sepsis and multi-
organ deficiency syndrome. All had reduced
median and ulnar CMAPs; four of five had
fibrillation potentials. Motor and sensory
conduction velocities were normal. Sensory
amplitudes were not reported. These patients
were said to have neuromuscular ‘‘dysfunc-
tion.’’ It is not clear if this was CIP or CIM.100
de Letter et al also performed a large
prospective study, identifying neuromuscular
dysfunction in 33% of patients with various
disorders, including traumas, postsurgical
states, and medical conditions.97 Bednarik
et al performed a comprehensive study that
included early NCS (Day 3), quantitative EMG,
and DMS. Muscle and nerve biopsies were
performed in a subset. They completed
clinical and electrophysiological evaluations
over a 28-day period in 60 patients with
medical and surgical conditions as well as
cerebral infarction and intracranial hemor-
rhages. There was a 36.3% mortality rate. Of
27.9% of patients with clinical evidence of
a neuromuscular disorder, 40% had myopathy,
34% had neuropathy, and 26% had mixed
neuropathy and myopathy. In the subset that
underwent muscle biopsy, it is uncertain if
patients with myopathy had myosin loss. The
major risk factors are shown in Table 4.98
Most recently, Hough et al reviewed data
collected prospectively in ICU patients with
acute respiratory distress syndrome treated
with methylprednisolone versus placebo.
One hundred twenty-eight survived 60 days.
Chart review disclosed neuromuscular weak-
ness in 34%. Of those who underwent
electrodiagnostic testing, most had evidence
of CIM. The study had limitations, but there
was no definite relationship between de-
velopment of CIM and corticosteroid
treatment.101
Two large, prospective, randomized,
controlled studies that are not listed in Table
4 addressed the influence of intensive insulin
therapy in critically ill surgical and medical
patients. Critical illness neuromuscular disor-
ders were evaluated secondarily, and the
authors were unable to diagnose CIM or
clearly separate CIP from CIM.102–105 Van
den Berghe et al randomly treated intubated
surgical patients with either intensive IV
insulin therapy (765 subjects) or conventional
insulin dosing (783 subjects). There was a 44%
lower incidence of neuromuscular disease in
the intensively treated group (28.7% versus
51.9% in the conventionally treated group).104
The diagnoses of CIP were based purely on the
presence of positive sharp waves and fibrilla-
tion potentials. Motor unit potentials could
not be assessed, and NCS was not performed;
thus, myopathy could not be diagnosed.105
Next, this group performed a prospective
study of 1200 medical ICU patients; 767
stayed in the ICU for 3 days or longer.102
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
206
� 2011 Lippincott Williams & Wilkins
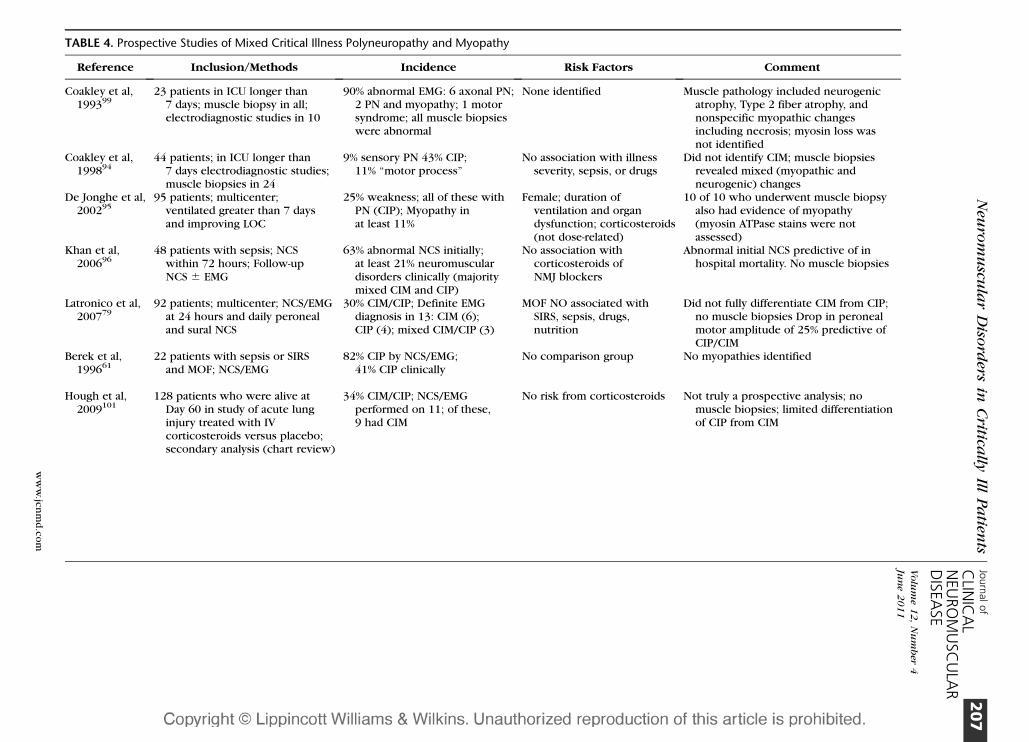
TABLE 4. Prospective Studies of Mixed Critical Illness Polyneuropathy and Myopathy
Reference Inclusion/Methods Incidence Risk Factors Comment
Coakley et al,199399
23 patients in ICU longer than7 days; muscle biopsy in all;electrodiagnostic studies in 10
90% abnormal EMG: 6 axonal PN;2 PN and myopathy; 1 motorsyndrome; all muscle biopsieswere abnormal
None identified Muscle pathology included neurogenicatrophy, Type 2 fiber atrophy, andnonspecific myopathic changesincluding necrosis; myosin loss wasnot identified
Coakley et al,199894
44 patients; in ICU longer than7 days electrodiagnostic studies;muscle biopsies in 24
9% sensory PN 43% CIP;11% ‘‘motor process’’
No association with illnessseverity, sepsis, or drugs
Did not identify CIM; muscle biopsiesrevealed mixed (myopathic andneurogenic) changes
De Jonghe et al,200295
95 patients; multicenter;ventilated greater than 7 daysand improving LOC
25% weakness; all of these withPN (CIP); Myopathy inat least 11%
Female; duration ofventilation and organdysfunction; corticosteroids(not dose-related)
10 of 10 who underwent muscle biopsyalso had evidence of myopathy(myosin ATPase stains were notassessed)
Khan et al,200696
48 patients with sepsis; NCSwithin 72 hours; Follow-upNCS 6 EMG
63% abnormal NCS initially;at least 21% neuromusculardisorders clinically (majoritymixed CIM and CIP)
No association withcorticosteroids ofNMJ blockers
Abnormal initial NCS predictive of inhospital mortality. No muscle biopsies
Latronico et al,200779
92 patients; multicenter; NCS/EMGat 24 hours and daily peronealand sural NCS
30% CIM/CIP; Definite EMGdiagnosis in 13: CIM (6);CIP (4); mixed CIM/CIP (3)
MOF NO associated withSIRS, sepsis, drugs,nutrition
Did not fully differentiate CIM from CIP;no muscle biopsies Drop in peronealmotor amplitude of 25% predictive ofCIP/CIM
Berek et al,199661
22 patients with sepsis or SIRSand MOF; NCS/EMG
82% CIP by NCS/EMG;41% CIP clinically
No comparison group No myopathies identified
Hough et al,2009101
128 patients who were alive atDay 60 in study of acute lunginjury treated with IVcorticosteroids versus placebo;secondary analysis (chart review)
34% CIM/CIP; NCS/EMGperformed on 11; of these,9 had CIM
No risk from corticosteroids Not truly a prospective analysis; nomuscle biopsies; limited differentiationof CIP from CIM
Neu
rom
uscu
lar
Diso
rders
inC
ritically
IllPa
tien
tsJo
urn
alo
f
CLIN
ICA
LN
EUR
OM
USC
ULA
RD
ISEASE
Volu
me
12
,N
um
ber
4
Jun
e2
01
1
20
7
ww
w.jc
nm
d.c
om

Patients who were in the ICU at and beyond
Day 7 were screened by weekly ‘‘electro-
neuromyography,’’ but the details of this
analysis were not provided. Again, there was
no separation of CIP from CIM. Blood glucose
levels, but not the insulin dose, independently
correlated with the risk of developing CIP.
There was also a protectant effect on the
CNS.105 A total of 50.5% of conventionally
treated patients developed neuromuscular
dysfunction, versus 38.9% in the intensively
treated group, a 20% reduction. Neuromus-
cular junction-blocking agents were a risk
factor for developing neuromuscular dysfunc-
tion, but corticosteroids were not. The
duration of mechanical ventilation was re-
duced in the intensively treated group.103 As
a note of caution, a recent study of intensive
treatment of hyperglycemia disclosed an
increased mortality rate.106
Two studies in which comprehensive
electrodiagnostic studies were performed on
weak patients provided interesting results.
Trojaborg et al evaluated 22 ICU patients who
underwent EMG for evaluation of weak-
ness.33 NCS, needle electrode examination,
DMS, quantitative EMG, and motor unit
number estimation were performed, and
nine underwent muscle biopsies. All were
found to have evidence of myopathy; a milder
PN was present in five patients, usually with
motor greater than sensory involvement.
Lefaucher et al107 performed a somewhat
similar study on 30 consecutive ICU patients
with moderate to severe weakness. They
underwent mechanical ventilation for 7 or
more days and were evaluated by NCS,
needle EMG, and DMS. The authors con-
cluded that 25 of 30 (83%) had evidence of
myopathy, whereas 16 (53%) had low
sensory nerve action potentials consistent
with a component of PN.
What is the take-home message of these
studies? First, neuromuscular weakness com-
monly occurs in ICU patients. Clinically
significant weakness occurs in at least 25%,
and subclinical neuromuscular dysfunction is
much more common. A systematic review of
CIP/CIM studies disclosed an incidence ofBed
nar
iket
al,
20
05
98
60
pat
ien
tsw
ith
two
or
mo
refa
ilin
go
rgan
s;E
xte
nsi
ve
ele
ctr
od
iagn
ost
icTest
ing
27
.9%
CIP
/CIM
(cli
nic
ally
);5
7.4
%C
IP/C
IM(N
CS/
EM
G)
Pre
sen
ce
and
du
rati
on
of
SIR
San
dse
veri
tyo
fM
OF;
no
co
rrela
tio
nw
ith
NM
Jb
lock
ers
or
co
rtic
ost
ero
ids
Degre
eo
fM
OF
and
du
rati
on
of
SIR
Sd
uri
ng
firs
tw
eek
was
pre
dic
tive
of
CIP
/CIM
.M
usc
leb
iop
sies
(11
pat
ien
ts):
myo
pat
hic
ch
ange
sin
all,
and
neu
roge
nic
chan
ges
in7
;n
erv
eb
iop
sies
(5):
axo
nlo
ss;
Did
no
td
iffe
ren
tiat
eC
IPfr
om
CIM
inm
ost
cas
es
de
Lett
er
et
al,
20
01
97
98
pat
ien
tso
nven
tila
tor
for
4o
rm
ore
day
s;C
lin
ical
exam
inat
ion
,N
CS/
EM
Go
nD
ays
4,
11
,an
d2
5m
inim
um
33
%C
IM/C
IPSI
RS
and
severi
tyo
fil
lness
;n
oin
cre
ased
risk
wit
hco
rtic
ost
ero
ids
or
NM
Jb
lock
ers
Did
no
td
iffe
ren
tiat
eC
IPfr
om
CIM
;n
om
usc
leb
iop
sies;
No
tab
leth
atau
tho
rsd
idn
ot
inclu
de
‘‘acu
teq
uad
rip
legic
myo
pat
hy
wit
hlo
sso
fth
ick
fila
men
ts’’
ICU
,in
ten
sive
car
eu
nit
;LO
C,l
eve
lofco
nsc
iou
sness
;NC
S,n
erv
eco
nd
ucti
on
stu
dy;
EM
G,e
lectr
om
yogra
ph
y;SI
RS,
syst
em
icin
flam
mat
ory
resp
on
sesy
nd
rom
e;M
OF,
mu
ltio
rgan
fun
cti
on
;IV
,in
trav
en
ou
s;P
N,
po
lyn
eu
rop
ath
y;C
IP,
cri
tical
illn
ess
po
lyn
eu
rop
ath
y;C
IM,
cri
tical
illn
ess
myo
pat
hy;
NM
J,n
eu
rom
usc
ula
rju
ncti
on
.
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
208
� 2011 Lippincott Williams & Wilkins

46% overall.108 Compared with CIP, it is often
more difficult to diagnose CIM without
a muscle biopsy or sophisticated electrophys-
iological testing. Therefore, it is difficult to
separate CIM and CIP in studies that not did
not use such measures, and CIM is probably
underrepresented. In studies of mixed CIM
and CIP in which these extensive studies were
performed, it seemed that CIM was the
predominant component.
The weight of evidence confirms that
SIRS109 and multiorgan failure are the major
risk factors for CIP, although some studies did
not note these associations.79,94 Hyperglyce-
mia is another. There is probably a correlation
with the severity of the underlying illness.
The likelihood of developing CIP increases
with the number of days in the ICU5;
however, it may occur within 72 hours of
onset of critical illness. There are no definite
associations with pharmacologic treatments
such as corticosteroids, but some debate
exists.
Risk factors for CIM are less certain as
a result of prospective study designs that
often did not differentiate CIM from CIP or
assess for myosin loss. It does appear that IV
corticosteroids and probably NMJ blockers
are risk factors, especially when myosin loss
is present. SIRS may be another. Earlier
uncontrolled studies in asthmatics associ-
ated CIM with the use of these drugs.10–
20,71,110–115 There may be a dose relationship
with NMJ blockers,21 but a dose relationship
with corticosteroids is uncertain. There have
been pathologically confirmed cases of CIM
with myosin loss in which neither cortico-
steroids nor NMJ blockers were adminis-
tered.116–118 These patients all had sepsis or
SIRS. In the reports of patients with status
asthmaticus, many had SIRS, but some had
only respiratory failure and did not meet
criteria for SIRS, suggesting SIRS is a risk
factor but not a prerequisite for CIM.18 In
addition, the severity of the underlying
illness does correlate with development of
myopathy (Table 3), and the presence of
renal failure also correlated in a transplant
population.22
Pathology and Pathogenesis
Critical Illness Polyneuropathy
The main pathologic lesion is axonal
degeneration of sensory and motor axons, but
nerve biopsies are sometimes normal.3,4,98,119
The cause of axonal degeneration is not
known, but peripheral nerve is considered
to be one of the tissues that is injured by SIRS
and multiorgan failure. A number of metabolic
derangements and release of cytokines such as
interleukins-1, -2, and -6, and tumor necrosis
factor-a likely culminate in axonal injury from
proinflammatory and vascular mediators as
reviewed by Bolton.65,120 There is some
supportive evidence of cytokines being pro-
duced by activated leukocytes in muscle
specimens from patients with CIP/CIM.121
Hyperglycemia, increased capillary perme-
ability, endothelial cell activation122 and
possibly hypoalbuminemia could also impair
delivery of oxygen and metabolic substrates to
the endoneurium. A humoral factor (identity
undetermined) has also been noted in patients
with CIP, but its significance is unknown and
warrants further investigation because it has
been shown be toxic to rat spinal cord
neurons.90,123,124
Several studies have also showed that
ICU patients may exhibit transient reductions
in motor and sensory responses consistent
with a reversible neuropathy without axonal
degeneration.125 A chronic sepsis animal
model has been produced by cecal ligature
and needle perforation. In this model, de-
creased sodium current was identified.126
Additional study of this model, in which there
are declines mixed sensory tail nerve record-
ings, revealed reduced excitability in dorsal
root sensory axons through intracellular
recordings. These experiments indicated that
sodium channels were inactivated,127 consis-
tent with the hypothesis that a potentially
reversible channelopathy also occurs early in
the course of CIP.
Critical Illness Myopathy
When CIM is suspected, a muscle biop-
sy may be obtained, especially if the
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
209
www.jcnmd.com

electrodiagnostic findings are inconclusive or
if the differential diagnosis includes toxic or
inflammatory myopathy. The major histopath-
ologic findings include myofiber atrophy,
which may affect Type 2 more than Type 1
fibers, along with myofibrillar disorganization.
Occasionally, all Type 2 fibers are atrophic,
regenerating, or both.128 A variable degree of
necrosis and regeneration may occur.29 Ex-
cess lipid deposits have also been noted.
However, the characteristic feature is the
loss of myosin-thick filaments.22,29,32,35,110,111,115
Suspicion for myosin loss is raised when there
is reduced or patchy reactivity on myosin
adenosine triphosphatase-reacted sections. It
may be present primarily in atrophic fibers,129
making it less noticeable. It can be subtle or
obvious resulting in core-like lesions (Fig. 1).
Myosin loss can be proven with immu-
nohistochemical staining for myosin, by
ultrastructural studies (Fig. 1D), and by
electrophoresis.130 Immunostains are more
variable depending on the myosin isoforms
that are examined.32 Myosin loss may be
related to a decreased transcription rate or
loss of myosin messenger RNA.32 Structural
proteins, aside from myosin, are mostly
unaffected.32,118,130 Myosin loss is often seen
along with features of myofibrillar disorgani-
zation, including abnormal basophilic stip-
pling with hematoxylin and eosin stains,
purplish staining with Gomori trichrome,
and irregular clumping of the reaction prod-
uct or core-like changes with NADH-TR
staining.
Additionally, evidence of abnormal deg-
radative pathway activation has been found.
There is upregulation of calpain118,131 along
with increased apoptosis.132 Results of ubiq-
uitin immunoreactivity are mixed.118,132
FIGURE 1. (A–C) Part of the spectrum of alterations in myosin-adenosine triphosphatase (ATPase)reactivity in critical illness myopathy (cryostat sections, pH 9.4). (A) There is a subtle, less thanexpected differentiation in stain intensity in Type 1 (light) and Type 2 (dark) fibers. (See normal inset inC for comparison.) Rare fibers have a mild patchy reduction in reactivity (see arrows), whereas a rarefiber (asterisk) has reduced reactivity that is also seen at pHs 4.3 and 4.6. (Other pHs are not shown.)There is also atrophy of Type 1 more than Type 2 fibers. (Bar = 30 mm.) (B) Type 2 fibers are atrophic,and many Type 1 and Type 2 fibers have a mild, patchy, and often central reduction in reactivity. (C)There are obvious core-like regions of absent myosin-ATPase reactivity in many fibers. (As mentioned,the inset reveals normal ATPase reactivity.) (D) An electron photomicrograph reveals a myofiber (top)with preservation of Z-bands and thin filaments with loss of the intervening A-bands containing thickfilaments. In comparison, an atrophic myofiber at the bottom has preservation of both thick and thinfilaments.
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
210
� 2011 Lippincott Williams & Wilkins

There is also upregulation of the transforming
growth factor-b/mitogen-activated protein
kinase pathway,133 and oxidative stress may
also play a role. It has been hypothesized that
loss of sarcolemmal nitric oxide synthase 1
leads to muscle fiber inexcitability by re-
ducing nitric oxide release at the muscle
membrane.134 In septic patients, an increase
in muscle nitric oxide synthase 2 mRNA and
protein has been associated with peroxyni-
trate formation and reduced contractile
strength.135
Myogenic differentiation factor D plays
an important role in regulating muscle
differentiation, and it may be involved in
CIM and in cachectic myopathy. Myogenic
differentiation factor D and other myogenic
regulatory factors influence the activity of
a number of muscle-specific genes, including
myosin light chain and myosin heavy chain.
Myogenic differentiation factor D is preferen-
tially expressed in fast twitch fibers, and it is
upregulated with denervation.136
Diaphragm dysfunction is presumably
present in patients with CIM and failure to
wean from mechanical ventilation, but a com-
prehensive histopathologic study of the hu-
man diaphragm has not been undertaken.
However, studies of diaphragm from patients
who had undergone mechanical ventilation
for 18 to 69 hours showed atrophy of slow-
and fast-twitch fibers, increased caspase
activation, decreased glutathione, and in-
creased activity in the ubiquitin proteosome
pathway. This study shows that the diaphragm
muscle is susceptible to proteolysis in critical
illness, disuse, or both.137
Animal Data
A rodent model using intraperitoneal
corticosteroids and denervation reproduces
the histologic, electrophysiological, and chan-
nelopathy features of CIM.138–140 It also
suggests that this combination leads to
selective depletion of myosin mRNA as
detected in the animal model by Mozaffar
et al141 and as shown in humans.32 Further-
more, a mouse neuropathy model suggests
that the state of innervation regulates myosin
isoforms.142 Activity levels also influence
myosin isoform expression.143 A recently
developed septic rat model uses limb immo-
bilization and systemic injections of Coryne-
bacterium resulting in loss of body weight,
muscle atrophy, reduced tetanic contraction,
and inflammation with probable myofiber
degeneration.144 Muscle specimens were not
assessed for myosin loss, and this could be
characterized as a model of necrotizing
myopathy.
Studies of the effects of high doses of
intramuscular methylprednisolone on rabbit
diaphragm muscle function revealed a decline
in diaphragm maximum muscle tension,
myofibrillar disarray, suppression of insulin
growth factor Type 1, and overexpression of
muscle atrophy F-box mRNA. The authors
suggested that there was activation of the
ubiquitin–proteasome pathway.145 A recent
review by Friedrich provides a comprehensive
discussion of all of the pathways that may be
affected in CIM.146
In summary, the animal models of CIM
support a role for corticosteroids as well as
neurogenic factors in leading to myosin loss
and channelopathy. Immobilization and in-
fection may play roles in muscle inflammation
and necrosis. In some patients, CIP could be
a neurogenic trigger for CIM. NMJ blockers or
myasthenia gravis147 could also serve as
neurogenic triggers as a result of motor end-
plate involvement. Furthermore, it is conceiv-
able that there are two categories of CIM, one
with myosin loss and one with myonecrosis
only, and that the risk factors differ. To confirm
that hypothesis, additional prospective hu-
man studies that include comprehensive
electrodiagnostic testing and muscle histopa-
thology will be necessary.
Treatment and OutcomesThere are no proven therapies that
reverse CIP or CIM. The underlying systemic
illness must be treated aggressively. As noted,
there is a 20% to 44% lower incidence of
neuromuscular disease in ICU patients who
are intensively treated for hyperglycemia,102,104
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
211
www.jcnmd.com

but there is concern about a possible in-
creased mortality rate with intensive treat-
ment of hyperglycemia.106 A prospective,
uncontrolled study of 33 patients with multi-
organ dysfunction syndrome (16 with Gram-
negative septicemia) suggested that 0.9 g/kg
of intravenous immunoglobulin given over 3
days may prevent CIP in patients with
septicemia or SIRS,148 but a larger prospec-
tive, controlled trial is necessary to make that
determination.
At least in CIM, injudicious use of IV
corticosteroids should be avoided, and seda-
tives should probably be used instead of NMJ
blockers when possible. Based on the work of
Latronico et al, patients at risk for CIM and CIP
could be monitored by serial peroneal motor
NCSs,79 and serial assessments of serum CK
may also predict development of CIM.20 Once
CIM is identified, corticosteroids should
probably be tapered or discontinued if
possible, but benefit from this intervention
has not been proven. Rechallenge with IV
corticosteroids should be avoided, if possible,
because CIM may recur.13 If there is associated
rhabdomyolysis, IV hydration with alkaline
diuresis is recommended to avoid renal
failure.149 Otherwise, treatment is largely
supportive. Like with all critically ill patients,
those with neuromuscular weakness should
be provided with adequate nutritional intake,
correction of underlying metabolic disorders
such as hypokalemia and hypophosphatemia,
and aggressive treatment of underlying in-
fections. Prophylaxis for deep venous throm-
bosis, pulmonary toilet, padding of pressure
points, frequent turning, physiotherapy, and
appropriate orthotics are recommended. Re-
habilitation may be required.150
Regarding outcomes, there is growing
evidence that persistent neuromuscular dys-
function is a common consequence of critical
illness. There is evidence of partial denerva-
tion on electrodiagnostic testing performed
up to 5 years after the critical illness.151 Most
survivors of adult respiratory distress syn-
drome, who were followed for 12 months
after discharge, were found to have muscle
wasting and weakness.152 Determining
whether these patients have CIM, CIP, or
another neuromuscular problem is a challenge.
In CIP, there is a high mortality rate (up
to 50%) resulting from the underlying disease.
In the acute period, abnormal NCS is pre-
dictive of in-hospital mortality.96,125 The
majority of survivors tend to recover partially
(severe PN) or fully (mild to moderate PN)
over months, and milder symptoms and signs
may ‘‘resolve’’ in weeks.153 Two-year follow
up of 19 patients with CIP, who had severe
enough weakness to be admitted to a re-
habilitation facility, revealed that 58% had full
recovery, 21% remained quadriplegic, 11%
had milder residual weakness, and 11%
died.154 Another study disclosed that 21% of
patients with CIP have severe residual handi-
caps at one year.89 Guarneri et al reported
worse outcomes in four patients with CIP
seen at 1 year. One recovered, one was
tetraplegic, and two had residual weak-
ness.155 Like with most axonopathies, distal
leg weakness and sensory disturbances are
the most common residual effects.156
Patients with CIM, who do not die of
their underlying disorder, usually recover over
weeks to months, and most recover
fully.18,29,150,155 However, there is consider-
able morbidity and increased medical costs
associated with CIM. For example, the mean
time to ambulation is approximately 8 weeks,
and in one study of patients with CIM
undergoing liver transplantation, the time in
the ICU was 49 6 36 days (mean 6 standard
deviation) versus 14 6 14 days for those
without CIM.22 Although patients with CIM
are generally in poorer health overall, failure
to wean from CIM is a major contributor to
prolonged ICU stays. However, CIM does
appear to have a better prognosis than CIP.155
Future DirectionsIn prospective human studies, it is
important to differentiate CIP from CIM as
best as possible using diagnostic criteria such
as those already suggested (Tables 5 and
6).36,65,157 Otherwise, clear identification of
risk factors is very difficult. Matched case–
control subjects are also necessary. It would
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
212
� 2011 Lippincott Williams & Wilkins

also be of interest to determine if patients with
CIM with myosin loss versus CIM with
necrosis and no myosin loss have different
risk factors such as corticosteroids or SIRS.
Although there is a tendency to avoid open
muscle biopsies in these patients, needle
muscle biopsies have been used effectively
in previous studies.29,32,94,99
Although there are considerable
amounts of data on disease mechanisms,
especially in CIM, further study is necessary
to identify specific factors that could be
subjected to therapy that blocks or reverses
symptoms. Such a treatment could be most
effective in the earlier stages when channel-
opathy may be predominant in both CIM and
CIP. Thus, it is also paramount that study
patients are monitored for onset by straight-
forward techniques such as serial peroneal
motor studies. It is desirable to have larger
TABLE 5. Diagnostic Criteria for Critical Illness Myopathy (CIM)
Major features:1. Sensory nerve amplitudes greater than 80% of the lower limit of normal (LLN) in two or more nerves;2. Needle electromyography with short duration, low amplitude motor unit potentials with early
or normal full recruitment with or without fibrillation potentials;3. Absence of a decremental response on repetitive nerve stimulation; and4. Muscle histopathologic findings of myopathy with myosin loss.
Supportive features:1. Compound muscle action potential (CMAP) amplitudes less than 80% LLN without
conduction block;2. Elevated serum creatine kinase (best assessed in first week of illness);3. Demonstration of muscle inexcitability by direct muscle stimulation; and4. Prolonged duration of CMAPs.
By definition, the patients are also critically ill.Definite CIM: All four major features.Probable CIM: Any three major features and one or more supportive features.Possible CIM: Either major features 1 and 3 or 2 and 3 and one or more supportive feature.
Modified from Lacomis D, Zochodne D, Bird SJ. Critical illness myopathy [Editorial]. Muscle Nerve.
2000;23:1785–1788.36
TABLE 6. Diagnostic Criteria for Critical Illness Polyneuropathy (CIP)
Major features:1. The patient is critically ill;2. Possible diffuse limb weakness, difficulty weaning from mechanical ventilation in the absence of
a nonneuromuscular etiology, or both; and3. Electrophysiological evidence of axonal motor and sensory polyneuropathy, including:
d Sensory and motor nerve amplitudes less than 80% of the lower limit of normal in two or morenerves
d Absence of conduction block or prolongation of F-wavesd Needle electromyography with reduced recruitment of normal motor unit potentials (MUPs)
(early), fibrillation potentials and reduced recruitment of long-duration, high-amplitude MUPs(after weeks)
Supportive features:1. Absence of a decremental response on repetitive nerve stimulation;2. Absence of myopathy with myosin loss on muscle biopsy;3. Presence of axonal degeneration on nerve biopsy; and4. A nerve-evoked muscle action potential-to-direct muscle stimulated continuous muscle action
potential (CMAP) (nerve to muscle) ratio of less than 0.5 with direct stimulation.Diagnosis of CIP should include all major features. If all of the elements of item 3 (electrophysiological
evidence) are not present, supportive features 1 and 4, 1 and 2, or 1 and 3 should be present.Modified from Bolton CF. Neuromuscular manifestations of critical illness. Muscle Nerve.2005;32:
140–16365 and Lacomis D. Neuromuscular weakness related to critical illness. In: Rose BD, ed.UpToDate.Wellesley, MA; 2007.157
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
213
www.jcnmd.com

prospective studies that assess outcomes and
interventions that may include specific and
innovative physiotherapy techniques such as
muscle stimulation.
All prospective studies of critical illness
states such as sepsis and lung injury should
include neuromuscular sequelae as a second-
ary end point. The role for such studies in
patients at risk for ICU-acquired neuromuscu-
lar disorders, the methods to use in these
studies, and the specific issues to address have
been thoughtfully discussed by Hough and
Needham.158
REFERENCES1. Russell JA. Management of sepsis. N Engl J Med.
2006;355:1699–1713.
2. Bone RC, Balk RA, Cerra FB, et al. Definitions forsepsis and organ failure and guidelines for the useof innovative therapies in sepsis. Chest. 1992;101:1644–1655.
3. Bolton CF, Gilbert JJ, Hahn AF, et al. Polyneuropathyin critically ill patients. J Neurol Neurosurg
Psychiatry. 1984;47:1223–1231.
4. Zochodne DW, Bolton CF, Wells GA, et al. Criticalillness polyneuropathy. A complication of sepsisand multiple organ failure. Brain. 1987;110:819–841.
5. Witt NJ, Zochodne DW, Bolton CF, et al. Peripheralnerve function in sepsis and multiple organ failure.Chest. 1991;99:176–184.
6. Bolton CF. The discovery of critical illness poly-neuropathy. Eur J Anaesthesiol. 2008;S42:66–67.
7. Osler W. The Principles and Practice of Medicine.New York: D Appleton; 1892.
8. Mertens HG. Disseminated neuropathy followingcoma. On the differentiation of so-called toxicpolyneuropathy. Nervenartz. 1961;32:71–79.
9. Henderson B, Koepke GH, Feller I. Peripheralpolyneuropathy among patients with burns. Arch
Phys Med Rehabil. 1971;52:149–151.
10. Kaplan PW, Rocha W, Sanders DB, et al. Acutesteroid-induced tetraplegia following status asthma-ticus. Pediatrics. 1986;78:121–123.
11. Knox AJ, Mascie-Taylor BH, Muers MF. Acutehydrocortisone myopathy in acute severe asthma.Thorax. 1986;41:411–412.
12. Van Marle W, Woods KL. Acute hydrocortisonemyopathy. BMJ. 1980;281:271–272.
13. Williams TJ, O’Hehir RE, Czarny D, et al. Acutemyopathy in severe acute asthma treated withintravenously administered corticosteroids. Am
Rev Respir Dis. 1988;137:460–463.
14. MacFarlane IA, Rosenthal FD. Severe myopathyafter status asthmaticus. Lancet. 1977;2:615.
15. Shee CD. Risk factors for hydrocortisone myopathyin acute severe asthma. Respir Med. 1990;84:229–233.
16. Blackie JD, Gibson P, Murree-Allen K, et al. Acutemyopathy in status asthmaticus. Clin Exp Neurol.1993;30:72–81.
17. Griffin D, Fairman N, Coursin D, et al. Acutemyopathy during treatment of status asthmaticuswith corticosteroids and steroidal muscle relaxants.Chest. 1992;102:510–514.
18. Leatherman JW, Fluegel WL, David WS, et al.Muscle weakness in mechanically ventilated pa-tients with severe asthma. Am J Respir Crit Care
Med. 1996;153:1686–1690.
19. Road J, Mackie G, Jiang TX, et al. Reversibleparalysis with status asthmaticus, steroids, andpancuronium: clinical electrophysiological corre-lates. Muscle Nerve. 1997;20:1587–1590.
20. Douglass JA, Tuxen DV, Horne M, et al. Myopathyin severe asthma. Am Rev Respir Dis. 1992;146:517–519.
21. Amaya-Villar R, Garnacho-Montero J, Garcia-Garmendia JL et al. Steroid-induced myopathy inpatients intubated due to exacerbation of chronicobstructive pulmonary disease. Intensive Care
Med. 2005;31:157–161.
22. Campellone JV, Lacomis D, Kramer DJ, et al. Acutemyopathy after liver transplantation. Neurology.1998;50:46–53.
23. Miro O, Salmeron JM, Masanes F, et al. Acutequadriplegic myopathy with myosin-deficient mus-cle fibres after liver transplantation: defining theclinical picture and delimiting the risk factors.Transplantation. 1999;67:1144–1151.
24. Subramony SH, Carpenter DE, Raju S, et al. Myo-pathy and prolonged neuromuscular blockade afterlung transplant. Crit Care Med. 1991:19:1580–1582.
25. Salviati L, Laverda AM, Zancan L, et al. Acutequadriplegic myopathy in a 17-month-old boy.J Child Neurol. 2000;15:63–66.
26. Chetaille P, Paut O, Fraisse A, et al. Acute myopathyof intensive care in child after heart transplantation.Can J Anaesth. 2000;47:342–346.
27. Perea M, Picon M, Miro O, et al. Acute quadriplegicmyopathy with loss of thick (myosin) filamentsfollowing heart transplantation. J Heart Lung
Transplant. 2001;20:1136–1141.
28. Hanson P, Dive A, Brucher JM, et al. Acutecorticosteroid myopathy in intensive care patients.Muscle Nerve. 1997;20:1371–1380.
29. Lacomis D, Giuliani MJ, Van Cott A, et al. Acutemyopathy of intensive care: clinical, electromyo-graphic, and pathological aspects. Ann Neurol.1996;40:645–654.
30. Faragher MW, Day, BJ, Dennett X. Critical caremyopathy: an electrophysiological and histologicalstudy. Muscle Nerve. 1996;19:516–518.
31. Rich MM, Bird SJ, Raps EC, et al. Direct musclestimulation in acute quadriplegic myopathy. Muscle
Nerve. 1997;20:665–673.
32. Larsson L, Li X, Edstrom L, et al. Acute quadriplegiaand loss of muscle myosin in patients treated withnondepolarizing neuromuscular blocking agentsand corticosteroids: mechanisms at the cellular andmolecular levels. Crit Care Med. 2000;28:34–45.
33. Trojaborg W, Weimer LH, Hays AP. Electrophysio-logic studies in critical illness associated weakness:
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
214
� 2011 Lippincott Williams & Wilkins

myopathy or neuropathy—a reappraisal. Clin
Neurophysiol. 2001;112:1586–1593.
34. Zochodne DW, Ramsay DA, Saly V, et al. Acutenecrotizing myopathy of intensive care: electro-physiological studies. Muscle Nerve. 1994;17:285–292.
35. Sander HW, Golden M, Danon MJ. Quadriplegicareflexic ICU illness: selective thick filament lossand normal nerve histology. Muscle Nerve. 2002:26;499–505.
36. Lacomis D, Zochodne D, Bird SJ. Critical illnessmyopathy [Editorial]. Muscle Nerve. 2000;23:1785–1788.
37. Segredo V, Caldwell JE, Matthay MA, et al. Persistentparalysis in critically ill patients after long-termadministration of vecuronium. N Engl J Med. 1992;327:524–528.
38. Segredo V, Matthay MA, Sharma ML, et al. Prolongedneuromuscular blockade after long-term adminis-tration of vecuronium in two critically ill patients.Anesthesiology. 1990;72:566–570.
39. Partridge BL, Abrams JH, Bazemore C, et al.Prolonged neuromuscular blockade after long-terminfusion of vecuronium bromide in the intensivecare unit. Crit Care Med. 1990;18:1177–1179.
40. Shanks AB, Long T, Aitkenhead AR. Prolongedneuromuscular blockade following vecuronium.Br J Anaesth. 1985;57:807–810.
41. Kupfer Y, Namba T, Kaldawi E, et al. Prolongedweakness after long-term infusion of vecuroniumbromide. Ann Intern Med. 1992;117:484–486.
42. Gooch JL, Suchyta MR, Balbierz, JM, et al. Pro-longed paralysis after treatment with neuromuscu-lar junction blocking agents. Crit Care Med. 1991,19:1125–1131.
43. Op de Coul AA, Lambregts PC, Koeman J, et al.Neuromuscular complications in patients givenPavulon (pancuronium bromide) during artificialventilation. Clin Neurol Neurosurg. 1985;87:17–22.
44. Barohn RJ, Jackson CE, Rogers SJ, et al. Prolongedparalysis due to nondepolarizing neuromuscularblocking agents and corticosteroids. Muscle Nerve.1994;17:647–654.
45. Campellone JV. Clinical approach to neuromuscularweakness in the critically ill patient. J Clin Neuro-
muscul Dis. 2000;1:151–158.
46. Lacomis D, Petrella JT, Giuliani MJ. Causes ofneuromuscular weakness in the intensive care unit:a study of ninety-two patients. Muscle Nerve. 1998;21:610–617.
47. Rosenow EC III, Engel AG. Acid maltase deficiencyin adults presenting as respiratory failure. Am J
Med. 1978;64:485–491.
48. Hughes RA, Bihari D. Acute neuromuscular re-spiratory paralysis. J Neurol Neurosurg Psychiatry.1993;56:334–343.
49. Kelly BJ, Luce JM. The diagnosis and managementof neuromuscular diseases causing respiratoryfailure. Chest. 1991;99:1485–1494.
50. O’Donohue WJ Jr, Baker JP, Bell GM, et al. Re-spiratory failure in neuromuscular disease. Manage-ment in a respiratory intensive care unit. JAMA.1976;235:733–735.
51. Abdel-Hamid H, Oddis CV, Lacomis D. Severehydroxychloroquine myopathy. Muscle Nerve.2008;38:1206–1210.
52. Souayah N, Sander HW, Menkes DL, et al. HepatitisC virus acute quadriparetic vasculitic neuropathyresponsive to cyclophosphamide. Neurol Neuro-
physiol Neurosci. 2006;18:5.
53. Newman JH, Neff TA, Ziporin P. Acute respiratoryfailure associated with hypophosphatemia. N Engl
J Med. 1997;296:1101–1103.
54. Bruno C, Sacco O, Santorelli FM, et al. Mitochon-drial myopathy and respiratory failure associatedwith a new mutation in the mitochondrial transferribonucleic acid glutamic acid gene. J Child Neurol.2003;18:300–303.
55. Gorson KC, Ropper AH. Nonpoliovirus poliomyeli-tis simulating Guillain-Barre syndrome. Arch Neu-
rol. 2001;58:1460–1464.
56. Spitzer AR, Giancarlo T, Maher L, et al. Neuromus-cular causes of prolonged ventilator dependency.Muscle Nerve. 1992;15:682–686.
57. Davis LE, DeBiasi R, Goade DE, et al. West Nile virusneuroinvasive disease. Ann Neurol. 2006;60:286–300.
58. Seybold ME. Diagnosis of myasthenia gravis. In:Engel AG, ed. Myasthenia Gravis and Myasthenic
Disorders. New York: Oxford University Press;1999:146–166.
59. McKhann GM, Cornblath DR, Griffin JW, et al.Acute motor axonal neuropathy: a frequent causeof acute flaccid paralysis in China. Ann Neurol.1993;33:333–342.
60. Albers JW, Fink JK. Porphyric neuropathy. Muscle
Nerve. 2004;30:410–422.
61. Berek K, Margreiter J, Willeit J, et al. Polyneuropa-thies in critically ill patients: a prospective evalua-tion. Intensive Care Med. 1996;22:849–855.
62. Zifko UA, Zipko HT, Bolton CF. Clinical andelectrophysiological findings in critical illness poly-neuropathy. J Neurol Sci. 1998;159:186–193.
63. Mastaglia FL, Garlepp MJ, Phillips BA, et al. Inflam-matory myopathies: clinical, diagnostic, and thera-peutic aspects. Muscle Nerve. 2003;27:407–425.
64. Bolton CF. Sepsis and the systemic inflammatoryresponse syndrome: neuromuscular manifestations.Crit Care Med. 1996;24:1408–1416.
65. Bolton CF. Neuromuscular manifestations of criticalillness. Muscle Nerve. 2005;32:140–163.
66. Ramsay DA, Zochodne DW, Robertson DM, et al.A syndrome of acute severe muscle necrosis inintensive care unit patients. J Neuropathol Exp
Neurol. 1993;52:387–398.
67. Al-Shekhlee A, Hachwi R, Jaberi MM, et al. Theelectromyographic features of acute rhabdomyoly-sis. J Clin Neuromuscul Dis. 2005;6:114–118.
68. Helliwell TR, Wilkinson A, Griffiths RD, et al.Muscle fibre atrophy in critically ill patients isassociated with the loss of myosin filaments and thepresence of lysosomal enzymes and ubiquitin.Neuropathol Appl Neurobiol. 1998;24:507–517.
69. Sun DY, Edgar M, Rubin M. Hemiparetic acutemyopathy of intensive care progressing to triplegia.Arch Neurol. 1997;54:1420–1422.
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
215
www.jcnmd.com

70. Vattemi G, Tonin P, Filosto M, et al. Reversibleupper limb muscle weakness with selective loss ofthick filaments. Neurology. 2003;61:863–864.
71. Sitwell LD, Weinshenker BG, Monpetit V, et al.Complete ophthalmoplegia as a complication ofacute corticosteroid- and pancuronium-associatedmyopathy. Neurology. 1991;41:921–922.
72. Al-Shekhlee A, Shapiro BE, Preston DC. Iatrogeniccomplications and risks of nerve conduction stud-ies and needle electromyography. Muscle Nerve.2003;27:517–526.
73. Bolton CF. Phrenic nerve conduction studies: tech-nical aspects and normative data. Muscle Nerve.2008;38:1658–1659.
74. Chen R, Collins S, Remtulla H, et al. Phrenic nerveconduction study in normal subjects. Muscle
Nerve. 1995;18:330–335.
75. Bolton CF. AAEM Minimonograph #40: clinicalneurophysiology of the respiratory system. Muscle
Nerve. 1993;16:809–818.
76. Resman-Gaspersc A, Podnar S. Phrenic nerve con-duction studies: technical aspects and normativedata. Muscle Nerve. 2008;37:36–41.
77. Chowdhury D, Arora A. Axonal Guillain-Barre syn-drome: a critical review. Acta Neurol Scand. 2001;103:267–277.
78. Van den Bergh PY, Pieret F. Electrodiagnostic cri-teria for acute and chronic inflammatory demyelin-ating polyradiculoneuropathy. Muscle Nerve. 2004;29:565–574.
79. Latronico N, Bertolini G, Guarneri B, et al. Simpli-fied electrophysiological evaluation of peripheralnerves in critically ill patients: the Italian multi-centre CRIMYNE study. Crit Care. 2007;11:R11.
80. Daube JR. Electrodiagnostic studies in amyotrophiclateral sclerosis and other motor neuron disorders.Muscle Nerve. 2000;23:1488–1502.
81. Makki AA, Benetar M. The electromyographic diag-nosis of amyotrophic lateral sclerosis: does theevidence support the El Escorial criteria? Muscle
Nerve. 2007;35:614–619.
82. Park EJ, Nishida T, Sufit RL, et al. Prolonged com-pound muscle action potential duration in criticalillness myopathy. Report of nine cases. J Clin
Neuromuscul Dis. 2004;5:176–183.
83. Allen DC, Arunachalam R, Mills KR. Critical illnessmyopathy: further evidence from muscle–fiberexcitability studies of an acquired channelopathy.Muscle Nerve. 2008;37:14–22.
84. Trojaborg W. Electrophysiologic techniques in criti-cal illness-associated weakness. J Neurol Sci. 2006:242:83–85.
85. Case Records of the Massachusetts General Hospi-tal. Weekly clinicopathological exercises. Case 11—1997. A 51-year-old man with chronic obstructivepulmonary disease and generalized muscle weak-ness. N Engl J Med. 1997:336:1079–1088.
86. Trojaborg W, Kaufmann P, Gooch CL. Motor unit esti-mate number in the anterior tibial muscle: normativedata versus findings in critically ill patients in intensivecare units. J Clin Neuromuscul Dis. 2002;3:139–142.
87. Rich MM, Teener JW, Raps EC, et al. Muscle iselectrically inexcitable in acute quadriplegic myop-athy. Neurology. 1996;46:731–736.
88. Rich MM, Pinter MJ, Kramer SD, et al. Loss ofelectrical excitability in an animal model of acutequadriplegic myopathy. Ann Neurol. 1998;43:171–179.
89. Leijten FS, De Weerd AW, Poortvliet DC. Criticalillness polyneuropathy in multiple organ dysfunc-tion syndrome and weaning from the ventilator.Intensive Care Med. 1996;22:856–861.
90. Druschky A, Herkert M, Radespiel-Troger M, et al.Critical illness polyneuropathy: clinical findings andcell culture assay of neurotoxicity assessed bya prospective study. Intensive Care Med. 2001;27:686–693.
91. Garnacho-Montero J, Madrazo-Osuna J, Garcıa-Garmendia JL, et al. Critical illness polyneuropathy:risk factors and clinical consequences. A cohortstudy in septic patients. Intensive Care Med. 2001;27:1288–1296.
92. Tepper M, Rakic S, Haas JA, et al. Incidence andonset of critical illness polyneuropathy in patientswith septic shock. Neth J Med. 2000;56:211–214.
93. Thiele RI, Jakob H, Hund E, et al. Sepsis andcatecholamine support are the major risk factorsfor critical illness polyneuropathy after open heartsurgery. Thorac Cardiovasc Surg. 2000;48:145–150.
94. Coakley JH, Nagendran K, Yarwood GD, et al.Patterns of neurophysiological abnormality inprolonged critical illness. Intensive Care Med.1998;24:801–807.
95. De Jonghe B, Sharshar T, Lefaucheur, JP, et al.Paresis acquired in the intensive care unit: a pro-spective multicenter study. JAMA. 2002;288:2859–2867.
96. Khan J, Harrison TB, Rich MM, et al. Earlydevelopment of critical illness myopathy andneuropathy in patients with severe sepsis. Neurol-
ogy. 2006;67:1421–1425.
97. de Letter MA, Schmitz PI, Visser LH, et al. Riskfactors for the development of polyneuropathy andmyopathy in critically ill patients. Crit Care Med.2001;29:2281–2286.
98. Bednarik J, Vondracek P, Dusek L, et al. Risk factorsfor critical illness polyneuromyopathy. J Neurol.2005;252:343–351.
99. Coakley JH, Nagendran K, Honavar M, et al.Preliminary observations on the neuromuscularabnormalities in patients with organ failure andsepsis. Intensive Care Med. 1993;19:323–328.
100. Tennila A, Salmi T, Pettila V, et al. Early signs ofcritical illness polyneuropathy in ICU patients withsystemic inflammatory response syndrome orsepsis. Intensive Care Med. 2000;26:1360–1363.
101. Hough CL, Steinberg KP, Thompson BT, et al.Intensive care unit-acquired neuromyopathy andcorticosteroids in survivors of persistent ARDS.Intensive Care Med. 2009;35:63–68.
102. Van den Berghe G, Wilmer A, Hermans G, et al.Intensive insulin therapy in the medical ICU. N
Engl J Med. 2006;354:449–461.
103. Hermans G, Wilmer A, Meersseman W, et al. Impactof intensive insulin therapy on neuromuscularcomplications and ventilator dependency in themedical intensive care unit. Am J Respir Crit Care
Med. 2007;175:480–489.
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
216
� 2011 Lippincott Williams & Wilkins

104. Van den Berghe G, Wouters P, Weekers F, et al.Intensive insulin therapy in the critically illpatients. N Engl J Med. 2001;345:1359–1367.
105. Van den Berghe G, Schoonheydt K, Becx P, et al.Insulin therapy protects the central and peripheralnervous system of intensive care patients. Neurol-
ogy. 2005;64:1348–1353.
106. The NICE-SUGAR Study Investigators. Intensiveversus conventional glucose control in critically illpatients. N Engl J Med. 2009;360:1283–1297.
107. Lefaucher JP, Nordine T, Rodriguez P, et al. Origin ofICU acquired paresis determined by direct musclestimulation. J Neurol Neurosurg Psychiatry. 2006;77:500–506.
108. Stevens RD, Dowdy DW, Michaels RK, et al.Neuromuscular dysfunction acquired in criticalillness: a systematic review. Intensive Care Med.2007;33:1876–1891.
109. American College of Chest Physicians/Society ofCritical Care Medicine Consensus Conference:definitions for sepsis and organ failure and guide-lines for the use of innovative therapies in sepsis.Crit Care Med. 1992;20:864–874.
110. Lacomis D, Smith TW, Chad DA. Acute myopathyand neuropathy in status asthmaticus: case reportand literature review. Muscle Nerve. 1993;16:84–90.
111. Danon MJ, Carpenter S. Myopathy with thickfilament (myosin) loss following prolonged paraly-sis with vecuronium during steroid treatment.Muscle Nerve. 1991;14:1131–1139.
112. De Smet Y, Jaminet M, Jaeger U, et al. Acutecorticosteroid myopathy in patient with asthma [inFrench]. Rev Neurol (Paris). 1991;147:682–685.
113. De Smet Y. Status asthmaticus. Acute myopathyinduced by cortisone and neuropathy duringresuscitation [in French]. Rev Neurol (Paris).1993;149:573–576.
114. Hirano M, Ott BR, Raps EC, et al. Acute quadriple-gic myopathy: a complication of treatment withsteroids, nondepolarizing blocking agents, or both.Neurology. 1992;42:2082–2087.
115. Waclawik AJ, Sufit RL, Beinlich BR, et al. Acutemyopathy with selective degeneration of myosinfilaments following status asthmaticus treated withmethylprednisolone and vecuronium. Neuromus-
cul Disord. 1992;2:19–26.
116. Hoke A, Rewcastle NB, Zochodne DW. Acutequadriplegic myopathy unrelated to steroids orparalyzing agents: quantitative EMG studies. Can J
Neurol Sci. 1999;26:325–239.
117. Deconinck N, Van Parijs V, Beckers-Bleukx G, et al.Critical illness myopathy unrelated to corticoste-roids or neuromuscular blocking agents. Neuro-
muscul Disord. 1998;8:186–192.
118. Showalter CJ, Engel AG. Acute quadriplegic myop-athy: analysis of myosin isoforms and evidence forcalpain-mediated proteolysis. Muscle Nerve. 1997;20:316–322.
119. Latronico N, Fenzi F, Recupero D, et al. Criticalillness myopathy and neuropathy. Lancet. 1996;347:1579–1582.
120. Bolton CF, Young GB, Zochodne DW. The neuro-logical complications of sepsis. Ann Neurol. 1993;33:94–100.
121. de Letter MA, van Doorn PA, Savelkoul HF, et al.Critical illness polyneuropathy and myopathy(CIPNM): evidence for local immune activationby cytokine-expression in the muscle tissue.J Neuroimmunol. 2000;106:206–213.
122. Fenzi F, Latronico N, Refatti N, et al. Enhancedexpression of E-selectin on the vascular endothe-lium of peripheral nerve in critically ill patientswith neuromuscular disorders. Acta Neuropathol.2003;106:75–82.
123. Hund E, Herkert M, Becker CM, et al. A humoralneurotoxic factor in sera of patients with criticalillness polyneuropathy. Ann Neurol. 1996;40:539.
124. Friedrich O, Fink RA, Hund E. Understandingcritical illness myopathy: approaching the patho-mechanism. J Nutr. 2005;135:1813S–1817S.
125. Khan J, Harrison TB, Rich MM. Mechanisms ofneuromuscular dysfunction in critical illness. Crit
Care Clin. 2008;24:165–177.
126. Rossignol B, Gueret G, Pennec JP, et al. Effects ofchronic sepsis on the voltage-gated sodium channelin isolated rat muscle fibers. Crit Care Med. 2007;35:351–357.
127. Novak KR, Nardelli P, Cope TC, et al. Inactivation ofsodium channels underlies reversible neuropathyduring critical illness in rats. J Clin Invest. 2009;119:1150–1158.
128. Gutmann L, Blumenthal D, Schochet SS. Acute typeII myofiber atrophy in critical illness. Neurology.1996;46:819–821.
129. Al-Lozi M, Pestronk A, Yee WC, et al. Rapidlyevolving myopathy with myosin-deficient fibers.Ann Neurol. 1994;35:273–279.
130. Matsumoto N, Nakamura T, Yasui, Y, et al. Analysisof muscle proteins in acute quadriplegic myopathy.Muscle Nerve. 2000;23:1270–1276.
131. Waclawik AJ, Selcen D, Keegan BM. A 46-year-oldwoman with severe weakness following acuterespiratory distress syndrome. Neurology. 2007;68:1529–1535.
132. Di Giovanni S, Mirabella M, D’Amico A, et al.Apoptotic features accompany acute quadriplegicmyopathy. Neurology. 2000;55:854–858.
133. Di Giovanni S, Molon A, Broccolini A, et al. Consti-tutive activation of MAPK cascade in acute quad-riplegic myopathy. Ann Neurol. 2004:55:195–206.
134. Capasso M, Di Muzio A, Pandolfi A, et al. Possiblerole for nitric oxide dysregulation in critical illnessmyopathy. Muscle Nerve. 2008;37:196–202.
135. Lanone S, Mebazaa A, Heymes C, et al. Muscularcontractile failure in septic patients. Role of theinducible nitric oxide synthase pathway. Am J
Respir Crit Care Med. 2000;162:2308–2315.
136. Legerlotz K, Smith HK. Role of MyoD in de-nervated, disused, and exercised muscle. Muscle
Nerve. 2008;38:1087–1100.
137. Levine S, Nguyen T, Talyor N, et al. Rapid disuseatrophy of diaphragm fibers in mechanicallyventilated humans. N Engl J Med. 2008;358:1327–1335.
138. Rouleau G, Karpati G, Carpenter S, et al. Glucocor-ticoid excess induces preferential depletion ofmyosin in denervated skeletal muscle fibers.Muscle Nerve. 1987;10:428–438.
Neuromuscular Disorders in Critically Ill Patients Journal of
CLINICALNEUROMUSCULARDISEASEVolume 12, Number 4
June 2011
217
www.jcnmd.com

139. Massa R, Carpenter S, Holland P, et al. Loss andrenewal of thick myofilaments in glucocorticoid-treated rat soleus after denervation and reinnerva-tion. Muscle Nerve. 1992;15:1290–1298.
140. Rich MM, Kramer SD, Barchi RL. Altered geneexpression in an animal model of acute quad-riplegic myopathy [Abstract]. Ann Neurol. 1998;40:461.
141. Mozaffar T, Haddal F, Zeng M, et al. Molecular andcellular defects of skeletal muscle in an animalmodel of acute quadriplegic myopathy. Muscle
Nerve. 2006;35:55–65.
142. Maggs AM, Huxley C, Hughes SM. Nerve-dependentchanges in skeletal muscle myosin heavy chain afterexperimental denervation and cross-reinnervationand in a demyelinating mouse model of Charcot-Marie-Tooth disease type 1A. Muscle Nerve. 2008;38:1572–1584.
143. Talmadge RJ. Myosin heavy chain isoform expres-sion following reduced neuromuscular activity:potential regulatory mechanisms. Muscle Nerve.2000;23:661–679.
144. Fink H, Helming M, Unterbuchner C, et al. Systemicinflammatory response syndrome increases immo-bility-induced neuromuscular weakness. Crit Care
Med. 2008;36: 910–916.
145. Sassoon CS, Zhu E, Pham HT, et al. Acute effects ofhigh-dose methylprednisolone on diaphragm mus-cle function. Muscle Nerve. 2008;38:1161–1172.
146. Friedrich O. Critical illness myopathy: sepsis-mediated failure of the peripheral nervous system.Eur J Anaesthesiol. 2008;42:73–82.
147. Panegyres PK, Squier M, Mills KR, et al. Acutemyopathy associated with large parenteral dose ofcorticosteroid in myasthenia gravis. J Neurol Neu-
rosurg Psychiatry. 1993;56:702–702.
148. Mohr M, Englisch L, Roth A, et al. Effects of earlytreatment with immunoglobulin on critical illnesspolyneuropathy following multiple organ failure
and Gram-negative sepsis. Intensive Care Med.1997;23:1144–1149.
149. Slater MS, Mullins RJ. Rhabdomyolysis and myoglo-binuric renal failure in trauma and surgical patients:a review. J Am Coll Surg. 1998;186:693–716.
150. Apte-Kakade S. Rehabilitation of patients withquadriparesis after treatment of status asthmaticuswith neuromuscular blocking agents and high-dosecorticosteroids. Arch Phys Med Rehabil. 1991;72;1024–1028.
151. Fletcher SN, Kennedy DD, Ghosh IR, et al. Persis-tent neuromuscular and neurophysiologic abnor-malities in long-term survivors of prolonged criticalillness. Crit Care Med. 2003;31:1012–1016.
152. Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratorydistress syndrome. N Engl J Med. 2003;348:683–693.
153. Bolton CF. Critical illness polyneuropathy. Muscle
Nerve. 1999;22:419–424.
154. de Seze M, Petit H, Wiart L. Critical illnesspolyneuropathy. A 2-year follow-up study in 19severe cases. Eur Neurol. 2000;43:61–69.
155. Guarneri B, Bertolini G, Latronico N. Long-termoutcome in patients with critical illness myopathyor neuropathy: the Italian multicentre CRIMYNEstudy. J Neurol Neurosurg Psychiatry. 2008;79:838–841.
156. Latronico N, Shehu I, Seghelini E. Neuromuscularsequelae of critical illness. Curr Opin Crit Care.2005;11:381–390.
157. Lacomis D. Neuromuscular weakness related tocritical illness. In: Rose BD, ed. UpToDate. Wellesley,MA; 2007.
158. Hough CL, Needham DM. The role of futurelongitudinal studies in ICU survivors: understand-ing determinants and pathophysiology of weaknessand neuromuscular dysfunction. Curr Opin Crit
Care. 2007;13:489–496.
LacomisJournal of
CLINICALNEUROMUSCULAR
DISEASEVolume 12, Number 4
June 2011
218
� 2011 Lippincott Williams & Wilkins

























![Renal Failure Critically Ill[1]](https://img.pdfslide.us/doc/110x75/577d26df1a28ab4e1ea26f52/renal-failure-critically-ill1.jpg)



