Embed Size (px)
Citation preview

BRIEF REPORT
Neonatal atypical hemolytic uremic syndromedue to methylmalonic aciduria and homocystinuria
Francesca Menni & Sara Testa & Sophie Guez &
Gabriella Chiarelli & Luisella Alberti & Susanna Esposito
Received: 29 January 2012 /Revised: 27 February 2012 /Accepted: 27 February 2012 /Published online: 25 March 2012# IPNA 2012
AbstractBackground Inborn errors of cobalamin (Cbl) absorptionand metabolism form a large group of rare diseases thatinclude Cbl-C disorder. Among the renal complications ofCbl-C disorder, atypical hemolytic uremic syndrome (HUS)is the least common and has been described only in a smallnumber of cases.Case-diagnosis/Treatment Four patients were admitted toour clinic after 15–30 days of life with vomiting associatedwith poor sucking, failure to thrive, lethargy and hypotonia.Examinations showed thrombocytopenia and microangio-pathic hemolytic anemia associated with renal damage. Theneonates had high blood homocysteine levels, increased uri-nary levels of both homocystine and methylmalonic acid,increased propionylcarnitine (C3) levels and an increased C3/acetylcarnitine ratio. Homozygosity for c.271-272dupA(p.Arg91LysfsX14) of theMMACHC genewas detected in threepatients, and heterozygosity for c.271-272dupA and c.666C >A(p.Tyr222X) in one patient, which confirmed the diagnosisof Cbl-C disorder. Treatment with parenteral hydroxycobalamin
in combination with folic acid and betaine gradually normal-ized the metabolic test findings and hematological and renalparameters after about 1 week.Conclusions Atypical HUS in neonates with Cbl-C disordermay be associated with mild to moderate renal involvementalso in early-onset disease, and early adequate therapy canreverse renal damage.
Keywords Cobalamin C disorder . Methylmalonicacidemia . Homocystinuria . Hemolytic uremic syndrome
Introduction
Inborn errors of cobalamin (Cbl) absorption and metabolismform a large group of rare diseases that include Cbl-Cdisorder [1], which is due to mutations in the MMACHCgene located on chromosome 1p34.1. Cbl-C impairs theconversion of cobalamin to its two metabolically activeforms (methycobalamin and adenosylcobalamin), thus lead-ing to the accumulation of methylmalonic acid (MMA) andhomocysteine (HC) in blood and tissues and the increasedurinary excretion of both compounds [1]. The clinical signsand symptoms related to Cbl-C disorder can appear in thefirst days or weeks of life, or significantly later in childhoodor adulthood. Older patients mainly suffer from neurologicalor psychiatric problems, rarely accompanied by signs andsymptoms of other organ involvement [2], whereas neonatesand infants have a multisystem disease with a more complexclinical picture [3]. Among the renal complications, atypicalhemolytic uremic syndrome (HUS) is the least common andhas been described in only very few cases [4–7]. Conse-quently, the clinical evolution of renal involvement, itsresponse to therapy, and the correlations between it andspecific genetic mutations have not been precisely defined.
F. Menni : S. Guez :G. Chiarelli : S. Esposito (*)Department of Maternal and Pediatric Sciences, FondazioneIRCCS Ca’ Granda Ospedale Maggiore Policlinico,Università degli Studi di Milano,Via Commenda 9,20122 Milan, Italye-mail: [email protected]
S. TestaNephrology Unit, Fondazione IRCCSCa’ Granda Ospedale Maggiore Policlinico,Milan, Italy
L. AlbertiLaboratory for Neonatal Screening, Buzzi Children’s Hospital,Milan, Italy
Pediatr Nephrol (2012) 27:1401–1405DOI 10.1007/s00467-012-2152-6

We have recently observed four cases of neonates withCbl-C disorder and atypical HUS and were able to treat andfollow them up them after diagnosis. As all of these patientsalso underwent a genetic evaluation of the MMACHC gene,we believe that a detailed description of these cases providesvaluable information that contributes to a better character-ization of Cbl-C disorder with atypical HUS in neonates.
Case report
The clinical and laboratory data of our patients are shown inTable 1. All four neonates had a low to normal birth weightafter an uneventful pregnancy. The symptoms occurred in allof the patients when they were between 15 and 30 days old,after an uncomplicated perinatal period. The common featuresat presentation were vomiting associated with poor sucking,failure to thrive, lethargy and hypotonia. Tests revealed meta-bolic acidosis with severe hyponatremia in two patients, andthrombocytopenia and microangiopathic hemolytic anemia(schistocytes in the peripheral blood, low haptoglobin and highlactate dehydrogenase levels) associated with evidence of renaldamage (microhematuria and proteinuria) in all four cases.Three cases were in class I (Injury) and one in class F (Failure)of the Risk, Injury, Failure, Loss, and End-Stage kidneydisease (RIFLE) classification of acute kidney injury [8].
The first three patients developed cardiovascular involve-ment with significantly reduced left ventricular function andwere transferred to our pediatric intensive care unit becauseof cardiac failure. Packed red cells and plasma transfusions,diuretics (furosemide, ethacrinic) and an angiotensin-converting enzyme inhibitor (captopril) were effective insupporting vital functions until metabolic-specific treatmentwas started. Patient 4 had also pancytopenia, increased C-reactive protein levels and a urine culture positive forEscherichia coli, and was treated for suspected sepsis. Noneof the patients required dialysis.
A metabolic disease was suspected because of the pres-ence of atypical HUS associated with a failure to thrive,lethargy and hypotonia. Plasma amino acid analysis showedhigh HC levels, and the urinary levels of both homocystineand MMA were increased. There was also an increase inpropionylcarnitine (C3) level and in the C3/C2 (acetylcarni-tine) ratio. Molecular analysis showed homozygosity forc.271-272dupA (p.Arg91LysfsX14) of the MMACHC genein three patients and heterozygosity for c.271-272dupA andc.666C > A(p.Tyr222X) in one patient, which confirmed thediagnosis of Cbl-C disorder.
Treatment with parenteral hydroxycobalamin (1 mg intra-muscular once daily) in combination with folic acid (5 mgorally once every 2 days) and betaine (250 mg/kg/day orallydivided in three doses every day) gradually normalized the
metabolic test findings, hematological parameters, and heartand renal function after about 1 week.
Fifteen months later, all of the patients had normal renaland cardiac function. Sucking has improved and the patientshave gained weight. The 12-month attainment of develop-mental milestones was only slightly delayed. The electroen-cephalograms of all four patients showed normal organizationwith minor abnormalities. However, despite the early diagno-sis and treatment, all of the patients developed nystagmus,although retinopathy was observed in only two cases.
Discussion
Although more than 500 patients with Cbl-C disorder havebeen described, fewer than 20 cases of secondary atypical HUShave been reported.Most of these involved neonates, infants orpatients aged less than 4 years; very few involved older child-ren. The infantile cases generally had a significantly worseprognosis than those with a later onset because most of thepatients died within the first 6 months of life despite hydrox-ycobalamin therapy and peritoneal dialysis. On the contrary,despite being clinically symptomatic in the first days of life,our patients had only mild to moderate renal insufficiency, didnot require dialysis and showed a very good response totherapy, with a rapid return to normal renal function and norecurrence of HUS. The difference in clinical outcomes be-tween the previously reported early-onset cases and our fourcases suggests that the prognosis of HUS associated with Cbl-C disorder is not age-related. However, as our follow-up is sofar limited to about 15 months and the possibility of laterrecurrences cannot be excluded, further evaluations are neces-sary before this conclusion can be definitely reached.
The pathogenesis of the thrombotic microangiopathy un-derlying the renal damage caused by HUS in patients withCbl-C disorder is not known, but it is thought that theincreased plasma levels of MMA and HC may play a sub-stantial role. HC modifies the antithrombotic properties ofthe vascular endothelium by impairing the nitric oxide-mediated inhibition of platelet aggregation, thus inducingthe binding of tissue plasminogen activator to the endothe-lium and increasing the endothelium expression of procoa-gulants [9]; moreover, the HC thiolactone metabolite of HCcan cause cell damage by inducing the intracellular accu-mulation of free radicals [10]. MMA can interfere withintrarenal mitochondrial metabolism [11]. However, as thereis no evidence that the plasma and urine values of HC andMMA in our patients, as well as in the literature, correlatewith the degree of renal insufficiency, other unknown fac-tors may play a role in conditioning renal damage in Cbl-Cdisorder. Furthermore, although Cbl-D disorder is alwayscharacterized by high plasma concentrations of HC andMMA, no case of HUS has ever been reported [12].
1402 Pediatr Nephrol (2012) 27:1401–1405
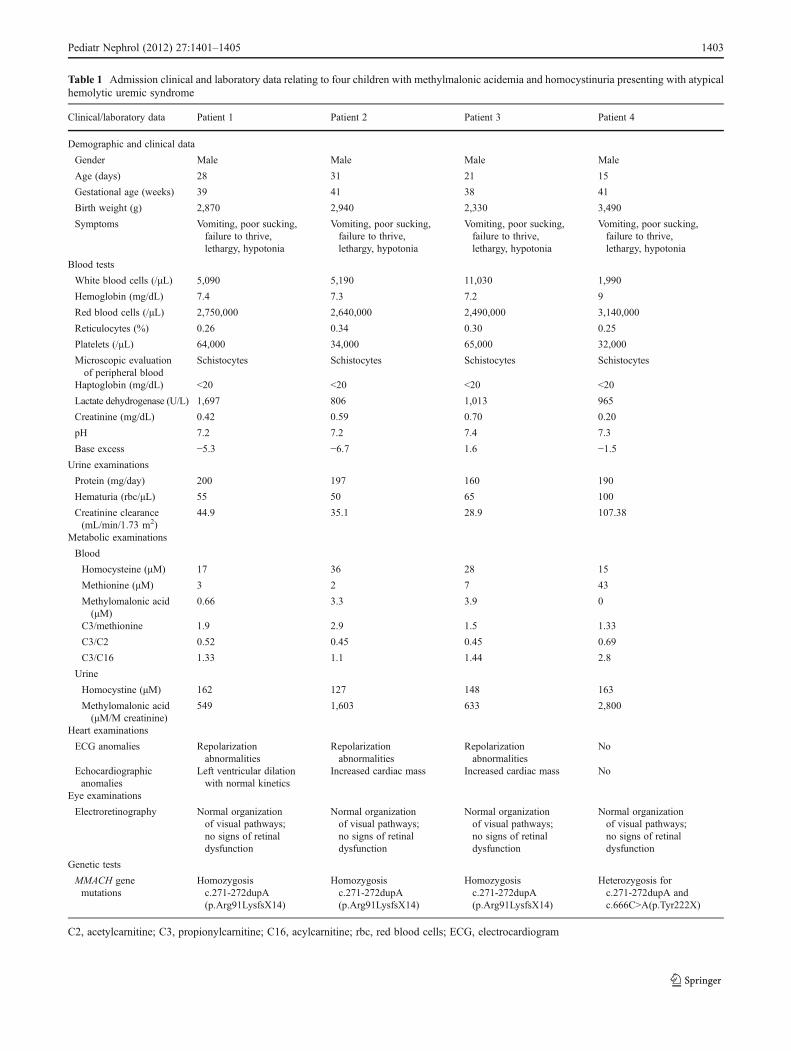
Table 1 Admission clinical and laboratory data relating to four children with methylmalonic acidemia and homocystinuria presenting with atypicalhemolytic uremic syndrome
Clinical/laboratory data Patient 1 Patient 2 Patient 3 Patient 4
Demographic and clinical data
Gender Male Male Male Male
Age (days) 28 31 21 15
Gestational age (weeks) 39 41 38 41
Birth weight (g) 2,870 2,940 2,330 3,490
Symptoms Vomiting, poor sucking,failure to thrive,lethargy, hypotonia
Vomiting, poor sucking,failure to thrive,lethargy, hypotonia
Vomiting, poor sucking,failure to thrive,lethargy, hypotonia
Vomiting, poor sucking,failure to thrive,lethargy, hypotonia
Blood tests
White blood cells (/μL) 5,090 5,190 11,030 1,990
Hemoglobin (mg/dL) 7.4 7.3 7.2 9
Red blood cells (/μL) 2,750,000 2,640,000 2,490,000 3,140,000
Reticulocytes (%) 0.26 0.34 0.30 0.25
Platelets (/μL) 64,000 34,000 65,000 32,000
Microscopic evaluationof peripheral blood
Schistocytes Schistocytes Schistocytes Schistocytes
Haptoglobin (mg/dL) <20 <20 <20 <20
Lactate dehydrogenase (U/L) 1,697 806 1,013 965
Creatinine (mg/dL) 0.42 0.59 0.70 0.20
pH 7.2 7.2 7.4 7.3
Base excess −5.3 −6.7 1.6 −1.5
Urine examinations
Protein (mg/day) 200 197 160 190
Hematuria (rbc/μL) 55 50 65 100
Creatinine clearance(mL/min/1.73 m2)
44.9 35.1 28.9 107.38
Metabolic examinations
Blood
Homocysteine (μM) 17 36 28 15
Methionine (μM) 3 2 7 43
Methylomalonic acid(μM)
0.66 3.3 3.9 0
C3/methionine 1.9 2.9 1.5 1.33
C3/C2 0.52 0.45 0.45 0.69
C3/C16 1.33 1.1 1.44 2.8
Urine
Homocystine (μM) 162 127 148 163
Methylomalonic acid(μM/M creatinine)
549 1,603 633 2,800
Heart examinations
ECG anomalies Repolarizationabnormalities
Repolarizationabnormalities
Repolarizationabnormalities
No
Echocardiographicanomalies
Left ventricular dilationwith normal kinetics
Increased cardiac mass Increased cardiac mass No
Eye examinations
Electroretinography Normal organizationof visual pathways;no signs of retinaldysfunction
Normal organizationof visual pathways;no signs of retinaldysfunction
Normal organizationof visual pathways;no signs of retinaldysfunction
Normal organizationof visual pathways;no signs of retinaldysfunction
Genetic tests
MMACH genemutations
Homozygosisc.271-272dupA(p.Arg91LysfsX14)
Homozygosisc.271-272dupA(p.Arg91LysfsX14)
Homozygosisc.271-272dupA(p.Arg91LysfsX14)
Heterozygosis forc.271-272dupA andc.666C>A(p.Tyr222X)
C2, acetylcarnitine; C3, propionylcarnitine; C16, acylcarnitine; rbc, red blood cells; ECG, electrocardiogram
Pediatr Nephrol (2012) 27:1401–1405 1403

The identification of the MMACHC gene has made itpossible to establish phenotype/genotype correlations inchildren with Cbl-C disorder. Homozygosity for c.271dupAhas been described in children with early-onset disease,frequently with acute metabolic decompensation [13],whereas subjects who are homozygous for c.394C>Treportedly show a later onset, a better treatment response,and the reversal of neurological manifestations [13].Moreover, the genotype also seems to be associated withethnicity because the c.271dupA mutation has beenmainly observed in Europeans, whereas most individualswith c.394C>T are of Middle Eastern origin [14]. Inter-estingly, patients who are compound heterozygous for thec.271dupA and c.394C>T mutations can manifest a dis-ease that is intermediate between the severe early-onsetform associated with homozygosity for c.271dupA andthe mild late-onset phenotype associated with homozy-gosity for c.394C>T [14].
Little data are currently available on the genetic charac-teristics of children with Cbl-C disorder and HUS. Sharmaet al. described a 12-year-old patient with late-onset HUSwho was found to have the c.271dupA mutation [7]. Inanother study of Italian and Portuguese patients, threechildren aged 2 and 5 months and 4 years, respectively,had c.271dupA/c.468_469delCT, c.3G>A/c.271dupA,and c.271dupA/c.394C>T [13]. In our patients, who wereborn in Italy of Italian parents, genetic tests demonstratedhomozygosity for c271dupA in three cases and heterozy-gosity for c.271dupA and c.666C>A in one case. Thisresult confirms that c271dupA is the most frequentlyfound mutation in children with Cbl-C disorder and thatit is frequently associated with a European origin andmainly early-onset disease. Moreover, given the presentingclinical picture of our patients and their very good response toparenteral hydroxycobalamin administration, homozygosityof this mutation cannot be considered a marker of renalinvolvement. Further studies are needed to verify whetherheterozygosity for some specific mutations is associated witha higher probability of severe kidney disease. It is thought thatthe MMACHC gene codes for a protein that plays a role inchemical reduction of the central cobalt atom of the cobalaminmolecule and that particular mutations can lead to differencesin mRNA stability or residual protein functioning that affectthe clinical course of the disease [14].
As reported by other authors, hydroxycobalamin com-bined with folic acid and betaine was effective in controllingthe renal alterations in all of our children. Unlike in previouscases [1], the treatment was also neurologically effectivebecause the developmental delay and neurological altera-tions were marginal more than 1 year after diagnosis. Theearly prescription of vitamins is probably the main reasonfor these results. However, despite treatment, the onset ofocular problems suggests the need for further studies that
can clarify their origin and whether the process starts inutero, and the possibility of their prevention or treatment.
Interestingly, our four cases of neonatal HUS due toCbl-C disorder were observed between September 1,2010, and October 30, 2010. We did not admit othercases of Cbl-C disorder without HUS in the same period,and we did not observe further cases in the following15 months. The high frequency of this finding from asingle region in a very short period is unusual, but we did notfind an environmental explanation for this observation in ourpatients or in the literature.
In conclusion, our cases of HUS in children with Cbl-Cdisorder indicate that renal involvement can be mild tomoderate in early-onset disease and that early and appropriatetherapy can reverse the renal damage in the acute phase.Further studies, also with the possibility of an early diagnosisof Cbl-C disorder based on detection of elevated C3 wheretandem mass spectrometry is used in neonatal screening [15],will allow a better understanding of phenotype/genotypecorrelations and establish the best way of reducing therisk of the development of neurological and ocularcomplications.
Acknowledgments We are grateful to the patients and their parents.Our sincere thanks go to the staff following up these patients:Fabio Mosca and Lorenza Pugni (neonatologists); Nicola Principiand Marta Cerutti (pediatricians); Faustina Lalatta and DonatellaMilani (geneticists); Alberto Edefonti, Gianluigi Ardissino andMirko Berlinghieri (pediatric nephrologists); Giovanna Chidiniand Cristiano Gandini (pediatric intensive care unit specialists);Silvia Osnaghi (ophthalmologist); Mariella Galli (pediatric cardiologist);Paola Vizziello (neuropsychiatrist); Simona Salera (dietitian). Weare also grateful to Carlo Corbetta and Katia Cavicchi for performing thelaboratory studies.
References
1. Martinelli D, Deodato F, Dionisi-Vici C (2011) Cobalamin Cdefect: natural history, pathophysiology, and treatment. J InheritMetab Dis 34:127–135
2. Tsai AC, Morel CF, Scharer G, Yang M, Lerner-Ellis JP,Rosenblatt DS, Thomas JA (2007) Late-onset combinedhomocystinuria and methylmalonic aciduria (cblC) and neuro-psychiatric disturbance. Am J Med Genet A 143A:2430–2434
3. Martinelli D, Dotta A, Massella L, Picca S, Di Pede A, Boenzi S,Aiello C, Dionisi-Vici C (2011) Cobalamin C defect presenting assevere neonatal hyperammonemia. Eur J Pediatr 170:887–890
4. Van Hove JL, Van Damme-Lombaerts R, Grünewald S, Peters H,Van Damme B, Fryns JP, Arnout J, Wevers R, Baumgartner ER,Fowler B (2002) Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am J Med Genet 111:195–201
5. Kind T, Levy J, Lee M, Kaicker S, Nicholson JF, Kane SA (2002)Cobalamin C disease presenting as hemolytic-uremic syndrome inthe neonatal period. J Pediatr Hematol Oncol 24:327–329
6. Sharma AP, Greenberg CR, Prasad AN, Prasad C (2007) Hemolyticuremic syndrome (HUS) secondary to cobalamin C (cblC) disorder.Pediatr Nephrol 22:2097–2103
1404 Pediatr Nephrol (2012) 27:1401–1405

7. Bouts AH, Roofthooft MT, Salomons GS, Davin JC (2010) CD46-associated atypical hemolytic uremic syndrome with uncommoncourse caused by cblC deficiency. Pediatr Nephrol 25:2547–2548
8. Cruz DN, Bagshaw SM, Ronco C, Ricci Z (2010) Acute kidneyinjury: classification and staging. Contrib Nephrol 164:24–32
9. Stamler JS, Osborne JA, Jaraki O, Rabbani LE, MullinsM SD,Loscalzo J (1993) Adverse vascular effects of homocysteine aremodulated by endothelium-derived relaxing factor and relatedoxides of nitrogen. J Clin Invest 91:308–318
10. McCully KS (1993) Chemical pathology of homocysteine. I.Atherogenesis. Ann Clin Lab Sci 23:477–493
11. Whelan DT, Ryan E, Spate M, Morris M, Hurley RM, Hill R (1979)Methylmalonic acidemia: 6 years’ clinical experience with two variantsunresponsive to vitamin B12 therapy. CanMedAssoc J 120:1230–1235
12. Miousse IR, Watkins D, Coelho D, Rupar T, Crombez EA, VilainE, Bernstein JA, Cowan T, Lee-Messer C, Enns GM, Fowler B,Rosenblatt DS (2009) Clinical and molecular heterogeneity inpatients with the cblD inborn error of cobalamin metabolism. JPediatr 154:551–556
13. Nogueira C, Aiello C, Cerone R, Martins E, Caruso U, Moroni I,Rizzo C, Diogo L, Leão E, Kok F, Deodato F, Schiaffino MC,Boenzi S, Danhaive O, Barbot C, Sequeira S, Locatelli M,Santorelli FM, Uziel G, Vilarinho L, Dionisi-Vici C (2008)Spectrum of MMACHC mutations in Italian and Portuguesepatients with combined methylmalonic aciduria and homocystinuria,cblC type. Mol Genet Metab 93:475–480
14. Lerner-Ellis JP, Anastasio N, Liu J, Coelho D, Suormala T, StuckiM, Loewy AD, Gurd S, Grundberg E, Morel CF, Watkins D,Baumgartner MR, Pastinen T, Rosenblatt DS, Fowler B (2009)Spectrum of mutations in MMACHC, allelic expression, andevidence for genotype phenotype correlations. Hum Mutat30:1072–1081
15. Weisfeld-Adams JD, Morrissey MA, Kirmse BM, Salveson BR,Wasserstein MP, McGuire PJ, Sunny S, Cohen-Pfeffer JL, YuC, Caggana M, Diaz GA (2010) Newborn screening and earlybiochemical follow-up in combined methylmalonic aciduria andhomocystinuria, cblC type, and utility of methionine as asecondary screening analyte. Mol Genet Metab 99:116–123
Pediatr Nephrol (2012) 27:1401–1405 1405
![REVIEW Open Access Proposed guidelines for the diagnosis ......also manifest as combined methylmalonic aciduria and homocystinuria (cblC, cblD, cblF and cblJ defects) [2,3]. MMA and](https://img.pdfslide.us/doc/110x75/60a702da0ccce350ab13ff02/review-open-access-proposed-guidelines-for-the-diagnosis-also-manifest-as.jpg)










![Three Main Causes of Homocystinuria: of Metabolism ... · most frequent causes are classical homocystinuria [deficiency of cystathionine beta-synthase (CBS)], methylmalonic aciduria](https://img.pdfslide.us/doc/110x75/5e951dcb19bd325819567b57/three-main-causes-of-homocystinuria-of-metabolism-most-frequent-causes-are.jpg)