Embed Size (px)
Citation preview

Eur J Plast Surg (1991) 14:3941 European " ! -~1 ,Ill ,~nal of ~ l f l $ l l C
bur g - Y © Springer-Verlag 1991
Nasopalpebral lipoma-coloboma: a very rare autosomal dominant dysplasia-malformation syndrome K. Sevin, E. Yormuk and N. Savaci
Ankara University, Ankara Medical Faculty, Department of Plastic and Reconstructive Surgery, Cebeci, Ankara, Turkey
Summary . A case of nasopalpebral lipoma-coloboma syndrome is reported. The genetic significance of this autosomal dominant, dysplasia-malformation syndrome is emphasized and the surgical management is reviewed.
Key words: Nasopalpebral lipoma Eyelid coloboma - Telecanthus Autosomal dominant dysplasia - Mal- formation syndrome
Nasopalpebral lipoma-coloboma syndrome is a very rare, autosomal dominant dysplasia-malformation syn- drome. It is characterized by bilateral upper and lower lid colobomas, telecanthus, nasopalpebral lipomas and maxillary hypoplasia [7].
Case report
A 5-year-old boy was admitted with his mother who had the same facial malformation. On physical examination, bilateral symmetri- cal V-shaped colobomas were present, located at the junction of the inner and middle thirds of both upper and lower eyelids (Fig. 1 a, b). Ten years previously, the mother had excision of the lipomas, but the colobomas and telecanthus were still present. The upper lacrimal punctum was absent and lower punctum was mis- placed lateral to the coloboma. The eyelashes were normal, again being located lateral to the coloboma. The eyebrows were sparse in the medial third. The nasal bridge was depressed. Under the skin of both nasopalpebral regions, symmetrical subcutaneous masses having a soft consistency were palpated. The nasopalpebral lipomas produced a marked outward and lateral displacement of the inner canthi; these covered the medial segment of the irises. Fundoscopy showed normal discs and retinal vessels.
The auricles were normally shaped and placed. Mouth, lips, tongue and palate were normal. The midface was slightly short, and the forehead was broad. His limbs, genitalia and cardiovascu- lar system were normal. The phenotypic anomalies were found to be limited to the patient's face. The chromosome study showed normal 46,XY constitution.
Radiographic examination of the skull showed normal cranial sutures and intact orbital margins. Only a minimal deformity was
Request for reprints: Dr. K. Sevin, M.D., Assistant Professor, Tur- gutlu sok. 8/2, Gaziosmanpasa 06700, Ankara, Turkey
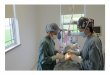
Fig. I a, b. Preoperative appearance of the case. a Frontal view of the patient and his mother, b The upper lid retracted to show the coloboma

40
75 77 80 67
36
21
3 Fig. 2a, b. Skull and facial bone X-rays. a PA; b Lateral
Fig. 3. In the family pedigree, the condition was inherited as an autosomal dominant, n, o=unaffected; B, e=affected
33 35 37 ox
observed in the shape of the bony margins of the orbits. The dis- tance between the lacrimal crests was within normal limits (29 ram) according to Tessier (Fig. 2a, b) [10]. In the family pedigree, the condition was found to be autosomal dominant (Fig. 3).
Operative technique
The "jumping-man" technique of Mustard6 was planned to correct telecanthus (Fig. 4a) [6]. The paranasal root skin was incised hori- zontally, and the excess skin and the underlying lipomatous mass were resected. These lipomas extended over the nasal bridge on both sides of the nose and the medial parts of the lid (Fig. 4b). The medial canthal ligaments were reinserted into the nasal bones using steel wires, and the skin flaps were rearranged in the standard fashion. The upper eyelid coloboma was excised and closed in two layers.
Histological examination of the excised tissue revealed lipoma.
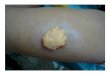
Fig. 4. a The operative plan. b The extra tissue of skin and fat were resected from the midline
Discussion
This autosomal dominant dysplasia-malformation syn- drome is characterized by 1) congenital symmetrical na- sopalpebral l ipomas; 2) bilateral symmetrical upper and lower eyelid colobomas; 3) midface hypoplasia; 4) broad nasal bridge and telecanthus; and 5) broad forehead and abnormal eyebrow distribution [7].
All of these features were present in this patient. Lid clefts occur in other complex facial syndromes such as mandibulofacial dysostosis [2], or frontonasal dysplasia [81.
In our case, the interorbital distance was within nor- mal limits which ruled out the existence of true bony hypertelorism. However, the l ipomas under the skin cov- ering the root of the nose extended symmetrically toward the medial canthi. This extension not only produced marked telecanthus but also covered the medial sides of the irises.
A broad forehead and abnormal eyebrow distribu- tion is evidence of a disturbance in upper face develop- ment during early embryogenesis [9]. All the structures of the lid margin are differentiated during the period of lid adhesion which extends f rom the tenth week to the sixth month ; after this, epithelial adhesion begins to break down [5]. Thus, any local factor interfering with the fusion or the breakdown of the adhesion may

41
cells, but they are very rare in the nasopalpebra l region. Thus, a m u t a n t gene m a y be responsible for the produc- t ion o f nasopalpebra l l ipomas [7].
The lacrimal puncture is frequently misplaced as also observed in this pat ient ; the puncture was no t relocated because there was no epiphora.
Different t rea tment techniques were reported by Berke [1], Hughes [3], Johnson [4] and Mustard~ [6], for the repair o f telecanthus. While m a n y authors stress the limitations o f surgical correct ion o f this condit ion, it usually results in significant cosmetic improvement . In this case, the results were gratifying bo th for the sur- geon and the family.
Addendum
The child was seen one year following surgery, and the result was sat isfactory (Fig. 5). I t came as a great sur- prise to see that a new addi t ion to the family had exactly the same deformi ty (Fig. 6).
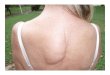
Fig. 5. Appearance one year after correction
Fig. 6. The mother with the patient and a new addition to the family
cause an interference with lid growth leading to colobo- mas and maldeve lopment o f other close structures.
L ipomas are considered developmental dysplasias o f the adipose tissue which usually consists o f normal fat
References
1. Berke RN (1953) A modified Kronlein operation. Trans Am Acad Ophthalmol 51:193
2. Franchesetti A, Klein D (1949) Mandibulo-facial dysostosis: new hereditary syndrome. Acta Ophthalmol 27:143
3. Hughes WL (1955) Surgical treatment of congenital palpebral phimosis. The V-Y operation. Arch Ophthalmol 54:586
4. Johnson CC (1956) Operations for epicanthus and blepharophi- mosis. Am J Ophthalmol 41:71
5. Mann IC (1957) Developmental abnormalities of the eye. Clowes and Sons, London, pp 371-379
6. Mustard~ JC (1963) Epicanthus and telecanthus. Br J Plast Surg 16:346
7. Penchaszadeh VB, Velasquez D, Arrivillaga R (1982) The naso- palpebral lipoma-coloboma syndrome : a new autosomal domi- nant dysplasia-malformation syndrome with congenital naso- palpebral lipomas, eyelid colobomas, telecanthus, and maxil- lary hypoplasia. Am J Med Gen 11 : 397
8. Sedano HO, Cohen MM Jr, Jirasek JE, Gorlin RJ (1970) Fron- tonasal dysplasia. J Pediatr 76:906
9. Smith DW, Cohen MM Jr (1973) Widow's peak scalp anomaly, origin and relevance to ocular hypertelorism. Lancet II : 1127
10. Tessier P (1976) Orbital hypertelorism: symposium on plastic surgery in the orbital region. Mosby, St Louis, 12:67