Embed Size (px)
Citation preview

Mutual Recognition and Decentralised Procedure
UnternehmensstrategieAblaufplanung
Probleme
Dr. Ulrich Granzerwww.granzer.biz

Regulatorische Aspekte und Strategien
Strategien für die Verfahrenswahl
Dialogstrategien mit Behörden: Scientific Advice
Auswahlkriterien des RMS und CMS
Dossier und Sachverständigengutachten
EU Zulassungsstrategien als Teil der globalen Entwicklung
User Testing
Environmental Risk Assessment
Pharmacovigilanzsysteme

Kriterienkatalog für eine neue Substanz: Verfahrenswahl
Bekanntheitsgrad der Indikation– „Je neuer desto zentraler“
– immer zentral: HIV und virale Erkrankungen, Krebs, Diabetes, Neurodegenerative Erkrankungen (z.B. Alzheimer), Orphans, Biotechs
Medizinische Schule in der EU– Indikationsspezifische Unterschiede
– Länderspezifische Besonderheiten
Paediatric Regulation
KONSEQUENZ: NEUE SUBSTANZEN IMMER ZENTRAL

Das Finden des „richtigen“ RMSKriterien
Flexibilität, wenn „es schwierig“ wird
Antizipation der europäischen Situation– Bisherige Verfahren (Track Record)
– Rolle in Gremien (z.B. Efficacy WP, ICH)
Verteidigung der Erstzulassung– „Anwalt“ der Firma
– kritisch-positive Zusammenarbeit mit der Firma

Das Finden des „richtigen“ RMSweitere Kriterien
Indikationsgebiet– Spezialisierung von Behörden
– Expertenwissen in Behörden
– Assoziationen zwischen Behörden
– Medizinische Praxis in Europa• Nord - Süd - Ost - West - Unterschiede
• Unterschiede zwischen Einzelländern

Weitere Punkte
Concerned Member States– Welche?
– Gibt es in einem Staat besondere Vorbehalte?
– Kann man von einer soliden Mehrheit der CMSs ausgehen? (Gefahr der Arbitration/Referrals)
Ziel: Aufbau eines Kriterienkatalogs mit vordefinierter Gewichtung – Vermeidung einer „Bauchentscheidung“
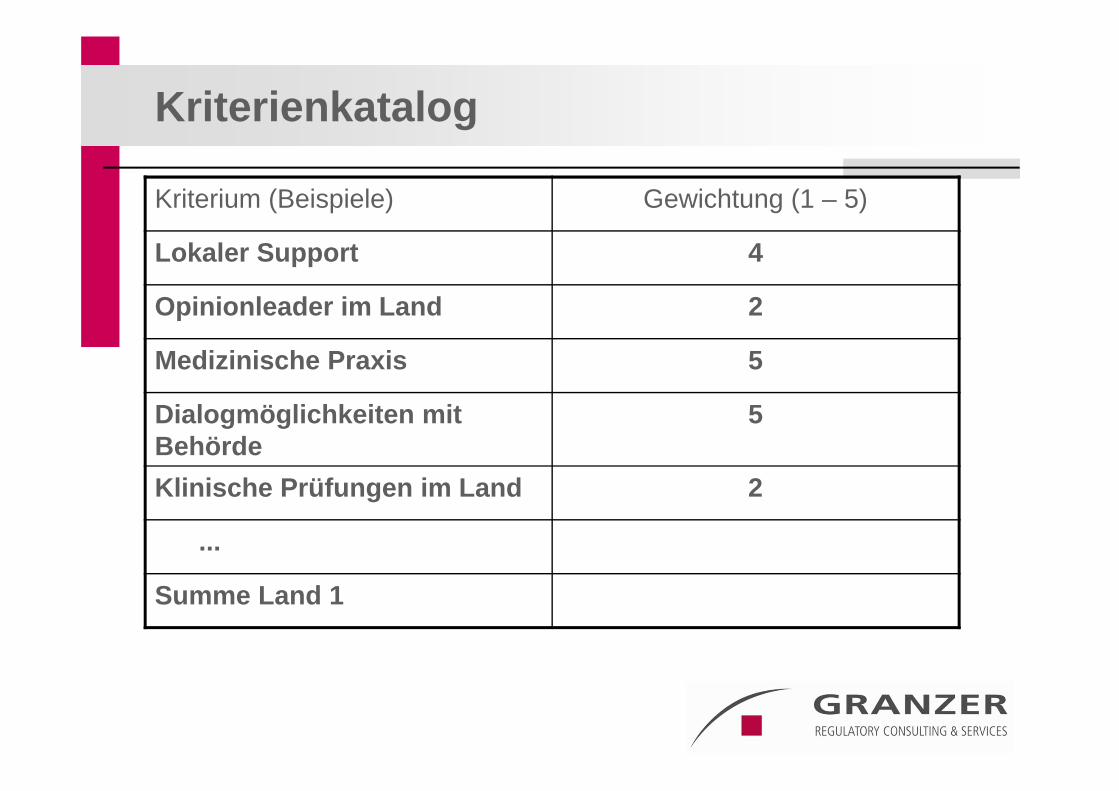
Kriterienkatalog
Kriterium (Beispiele) Gewichtung (1 – 5)
Lokaler Support 4
Opinionleader im Land 2
Medizinische Praxis 5
Dialogmöglichkeiten mit Behörde
5
Klinische Prüfungen im Land 2
...
Summe Land 1

Prinzipiell geeignete Produkte für die dezentralen Verfahren
Früher:– Neue Moleküle in bekannten Indikationen, die nicht im
Brennpunkt einer Lobbying – Gruppe stehen
Jetzt:– Neue Wirkmechanismen in bekannten Indikationen– Generika

Paediatric development
PIP and its implications for the choice of the procedure
Regulation states – “Extension of the duration of the supplementary protection
certificate
For new medicines and for products covered by a patent or a Supplementary Protection Certificate (SPC), the six-month SPC extension will be granted”
– if all the measures included in the agreed paediatric investigation plan are complied with,
– if relevant information on the results of studies is included in product information
– if the product is authorised in all Member States


Behördendialog
Welche Inhalte?– Briefing Document, Präsentation, jeweils mit konkretem Ziel
Wann, Wie oft?– Vor Phase III
– Vor Einreichung
– Während des Verfahrens, wann immer ein konkreter Grund vorliegt
Wer mit Wem?– Experte mit Experte in moderiertem und gut vorbereitetem
Gespräch

Ziele des Behördendialogs
Vorstellung des Produktes
Kennen lernen der Experten
Erfahren, wo eventuelle Problemfelder liegen
Aufbau eines Vertrauensverhältnisses zwischen pharmazeutischem Unternehmer und Behörde

How to obtain knowledge about authorities: Scientific Advice
Questions regarding SciAdv– When to go? Development plan
– How often?
– Topics?
– Topics to be avoided?
– How „binding“ is a scientific advice?
– What happens if the advice from different authorities differs?

Comparison EU/FDA
FDA: – Pre-IND Meeting
– End of phase II Meeting
– Pre-NDA/pre-BLA Meeting
EU:– Before start of clinical development
– Before phase III
– Before submission

EMA Guidance Cover letter (1-3 pages)
Name of company Contact Person details (Telephone; Fax; E-mails) Description of the product Trade Name (if available) INN (if available) Company’s code Pharmacological classification (ATC code if available) Eligibility for centralised procedure Type of request: SA or PA, Initial or Follow-Up Area of advice: Quality/Pre-Clinical/Clinical/Significant benefit (for
protocol assistance)

EMA Guidance
Fee payment (for SA)
Fee waiver/reduction (Protocol Assistance)
Justification for request
Intended Indication(s) to be supported by the development at time of MAA
Mention of previous Scientific Advice received (National and/or EU Authorities, Other Relevant International Authorities)
Detailed Table of Contents; containing full listing of annexes and references

Briefing document including the Questions and Company's positions
The questions are ordered and numbered sequentially to address specific scientific issues (order: quality/biotech/pre-clinical/clinical issues/significant benefit).
Each question is followed by a separate company’s position including a justification(s) of the company strategy for each topic.
The company’s position should be summarised after each question in the briefing document.
Overall objectives: Get advice and learn whether a working relationship can easily be established

Die Verfahren

Verfahrensablauf
Die Rolle des BfArM als „Reference Member State”
Assessment Reports
Klärungs- und Dialogphase
Arbitration – gewünscht oder gefürchtet?
SPCs
Der Entscheidungsfindungsprozeß
Übersetzung
Spezifische nationale Anforderungen
20
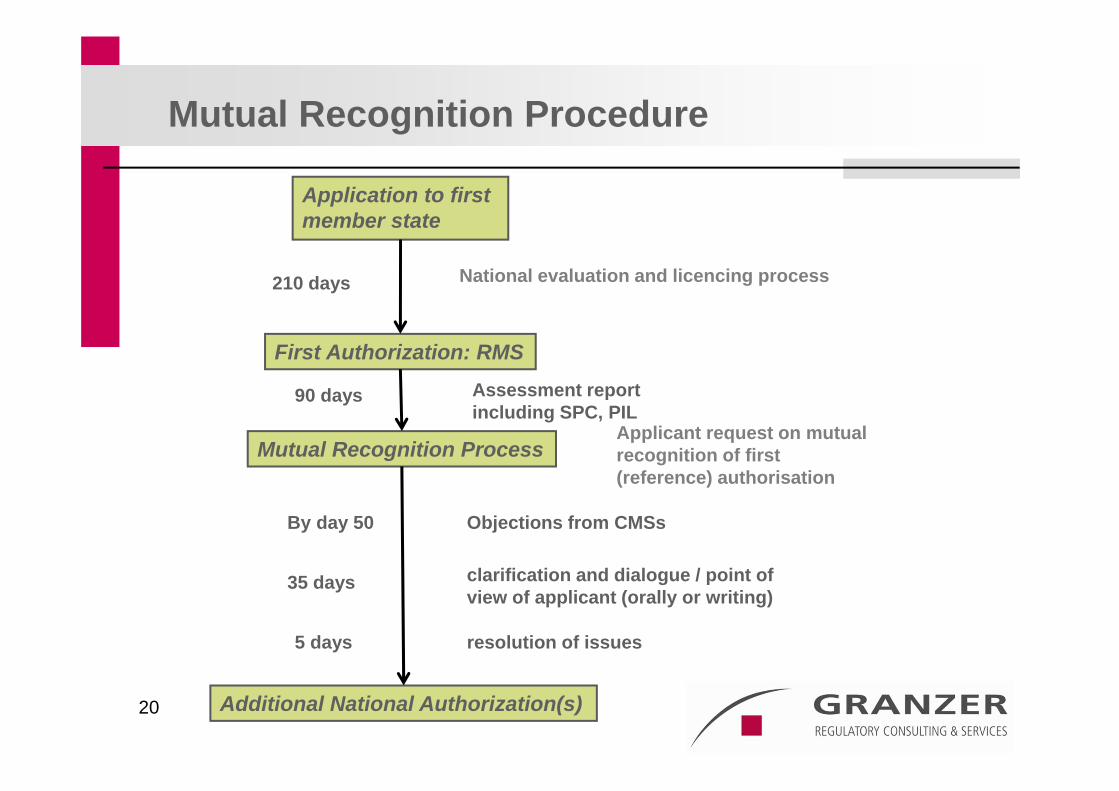
Application to first member state
Assessment reportincluding SPC, PIL
First Authorization: RMS
210 days
Applicant request on mutual recognition of first(reference) authorisation
Mutual Recognition Process
Objections from CMSsBy day 50
clarification and dialogue / point ofview of applicant (orally or writing)
resolution of issues
Additional National Authorization(s)
35 days
National evaluation and licencing process
Mutual Recognition Procedure
90 days
5 days

21
Application to all member statesRMS to start review
CMSs send comments to RMS and Applicant Consultation between RMS, CMS and Applicant (5 days)
PAR to CMSs and Applicant
70 days
Day 105: Clock stop orpositive close of procedure
RMS updates PrAR to prepare draft AR (DAR)
Day 106: Submission of the response
RMS evaluation of the dossier: Preliminary Assessment Report(PAR)
Decentralized Procedure
30 days
Preparation of response document by applicant3 – 6 months
Day 120: Consensus?End of procedure
Day 120: No consensus?Follow on process

Mutual Recognition und dezentrales Verfahren -Unterschiede
MR– MA vorhanden
– Assessment Report durch RMS
– Gegenseitige Anerkennung innerhalb von 90 Tagen nach Start
– Basis: Assessment Report
Dezentral– Keine MA vorhanden
– Application an alle Staaten, in denen eine Zul. angestrebt wird
– RMS erstellt Draft Assessment Report innerhalb der ersten Phase des Verfahrens

Mutual Recognition und dezentrales Verfahren -Unterschiede
MR– Bei Problemen:
Coordination Group
– 60 Tage zur Lösung
– Falls die Unstimmigkeiten nicht gelöst werden: CHMP
Dezentral– Bei Unstimmigkeiten:
Coordination Group
– 60 Tage zur Lösung
– Falls die Unstimmigkeiten nicht gelöst werden: CHMP

Rücknahmen
Rücknahme vs potentielle Arbitration– “Seriousness” of risks to public health
• Label changes
• Einführung von Kontraindikationen
• Anzahl der CMSs, die serious risks beschreiben
• Diskussion second wave vs “Augen zu und durch” Strategie
– Wann?• Bis direkt vor Erhalt des Draft Assessment Reports

BfArM als RMS
Beratungsgespräch(e) vor Einreichung Festlegung einer für beide Seiten verbindlichen
Zeitschiene „Europäisches“ Assessment
– Mängelschreiben mit besonderer Berücksichtigung möglicher nationaler Besonderheiten
Dialog bereits während der Phase I der Bearbeitung– Hinweis auf gravierende Probleme

Assessment Reports
Kritisch genug Eindeutige, positive Stellungnahme bei positiver
Entscheidung Eingehen auf europäische „Problemfelder“
– Beispiel: QT – Streckenveränderungen im EKG– Antibiotikaproblematik in der EU– Verschieden medizinische Schulen
Unterstützung durch pharm. Unternehmer: – kritische Overviews (nur die sind gute Overviews)

Assessment reports und SPC
Unterstützung („Justification“) der Schlüsselstellen der SPC1. Indikation
2. Kontraindikationen
3. Vorsichtsmaßnahmen bei der Anwendung
4. Nebenwirkungen: CAVE US Data Sheet
Alle anderen Punkte sind weniger bedeutsam

MRP

Tag 50 bis Tag 90
„Serious risks to public health“– Die Fragen müssen zufriedenstellend beantwortet werden
RMS als wichtigster Ansprechpartner – Diskussion der Antwortentwürfe
– Kommentierung von Fragen der CMS‘s
– Direkter Dialog RMS und CMS(s)
– Austausch aller relevanten Informationen
– Kollegen in der Behörde als Partner

Tag 85 - 89
Finale – Finalisierung der SPC
• kann man mit der SPC das Produkt vermarkten?
• Konkurrenznachteile?
– Alle Entscheidungsträger in der Firma anwesend?– Diskussion potentieller Entscheidung mit RMS– Information von RMS über Entscheidung vor Info an CMS– Verhandlungsführer in Firma mit ausreichend Erfahrung
essentiell

Wenn alles gut gegangen ist
Information über Abschluss des Verfahrens an CMS‘s und Antragsteller
Übersetzung der SPC in alle Landessprachen– Zeitpunkt der Übersetzung innerhalb der Firma
– Geschwindigkeit der Zulassungserteilung: abhängig von der Bereitstellung der Übersetzung und der PIL
Referral aus der Sicht des Betroffenen
CPMP Meetings in wingdings

Referral
Läuft wie ein zentrales Verfahren Dauert einige Monate Entscheidung immer für die ganze EU, aber
gerichtet an die Mitgliedsstaaten– Wo kein Zulassungsantrag aktiv ist, gibt es nach
Arbitration keine Zulassung
Kriterien für die Einleitung von Seiten des Antragstellers– Geschwindigkeit: Repeat use oder Referral: Entscheidung
vor Tag 120 dezentral, Auswahl der CMS im MR Verfahren– Situation (Mehrheitsverhältnisse) im CHMP

Risiko: Referral
Geschwindigkeit– Definierte clock stops, Zeit bei der Kommission und dem
Standing Committee
Effizienz– Bezug nur auf "serious risks"
– Keine neuen Probleme im Referral Verfahren
Status: Das Verfahren ist übersichtlicher geworden, die Zeiten werden eingehalten, neue Probleme werden (meist) nicht aufgeworfen


EU Risk Management Plan

Vier Hauptpunkte
Risk detection
Risk assessment
Risk minimisation
Risk communication

Gesetzestext
ensuring that any request from the competent authorities for the provision of additional information necessary for the evaluation of the risks and benefits of a medicinal product is answered fully and promptly, including the provision of information regarding the volume of sales or prescriptions for themedicinal product concerned
providing the competent authorities with any other information relevant to the evaluation of the risks and benefits of a medicinal product particularly information concerning post-authorisation safety studies.

EU Risk Management Plan (EU-RMP)
The description of a risk management system should be submitted in the form of an EU-RMP.
Part I– A Safety Specification
– A Pharmacovigilance Plan
Part II– An evaluation of the need for risk minimisation activities
– and (only) if there is a need for additional (ie non- routine) risk minimisation activities
– A risk minimisation plan

Wann muß ein Plan erstellt werden?
Zu jeder Zeit möglich– Einreichung zur Zulassung
• Alle neuen Arzneimittel!
• Biosimilars
• Signifikante neue Darreichungsform/Indikation
– Während der ersten Vermarktungsphase• Signal
– Wiedereinführung nach Marktrücknahme• Spezielle Sicherheitsvorkehrungen
– Immer, wenn es von einer Behörde gefordert wird• Beispiele: Referral/Arbitration

EU Risk Management Plan (EU-RMP)
The description of a risk management system should be submitted in the form of an EU-RMP.
Part I– A Safety Specification
– A Pharmacovigilance Plan
Part II– An evaluation of the need for risk minimisation activities
– and (only) if there is a need for additional (ie non- routine) risk minimisation activities
– A risk minimisation plan

The risk minimisation plan should list the safety concerns for which risk minimisation activities are proposed

Risk Minimisation Plan: Aufgaben
Definition der potentiellen Risiken– Alle Indikationseinschränkungen
• Kontra - Indikationen
• Vorsichtsmaßnahmen bei der Anwendung
• Schwangerschaft
• Bedienen von Maschinen
– Nebenwirkungen– Abhängigkeiten– Wechselwirkungen– Klasseneffekte

Wechselwirkungen
Häufigster Grund für Rücknahmen
Fast immer Cytochrom P 450

Umsetzung
Risikokommunikation– Arzt/Apotheker
• Werbematerial
• Außendienstmitarbeiterschulung
• Dear Dr Letter
– Patient• Gebrauchsanweisung
• Schulungsmaterial für den Arzt zur Abgabe an den Patienten

Überwachung der Effekte der Umsetzung
Umfragen zur Risikokommunikation
Epidemiologische Studien (Beispiel GPRD/ HMO‘s)
Anwendungsbeobachtungen
Phase IV Studien
Signaldetektion aus PMS

Auflagen und Committments
Unterschied– Auflage: Condition for Approval
– Committment: „Freiwillige Verpflichtung, eine Leistung zu erbringen“
• Häufig verwendetes Tool
• Oft Phase IV Studien
• Manchmal Patientenregister (z.B. bei Orphans)

Bedeutung für die Firma
Ohne RMP keine Zulassung!
Standard für Neuzulassungen in neuen Therapiegebieten und neuen Produktklassen
Marketingtool oder Verkaufsverhinderer– Vorlage der Werbematerialien bei lokaler Behörde
– Rechtzeitige Abstimmung
– Planung innerhalb der Firma• Globale vs nationale Planung


ERA
Wird bei neuen Substanzen immer verlangt
Soll die Zulassung nicht behindern
Umweltbundesamt als Behörde im Hintergrund
Abstufung je nach Eintragsmenge in die Umwelt– Abhängigkeit vom geschätzten Umsatz

Erfahrung mit dem DCP
Das Verfahren funktioniert, wenn es losgegangen ist
RMS kann positiv und negativ sein
Referral bei Divergenzen: – Einzelbehörde kann Verfahren verlangen
– 60 (90) Tage Extra
– CMD kann diskutieren, nicht entscheiden
Slots– Timing für Slots
– Auswahl der Behörde aufgrund nicht vorhandener Kapazität
– Fragen an Dr Bachmann: Wie slottet es denn so?
– Lösungsmöglichkeiten???

Clinical Overview

Clinical Overview
The overview will be the most crucial document for a successful submission. Thus, the main items and issues should be named as clearly as possible and a “story” should be told throughout the document

Overview - content
Medical need for treatment
Introduction and exec summary– Indication sought
– Summary of the main clinical trials
– Brief discussion that the benefit risk ratio is positive

Efficacy
PK/PD profile
Efficacy overview – Description of the population treated
– Rationale on the medical need and a clear description of the patients benefit of the treatment
– Critical description of the clinical trials
– Justification of duration of treatment

Safety
Overview on the safety of the drug class – Drug drug interactions seen as well as some information
on in vitro and animal experiments• Cytochrome P450 subclasses
• Most important reason for withdrawals in the last 20 years
• QT prolongation

Safety
– SAE’s, AE’s: Description and valuation• Signals of substance specific issues
• Target organ toxicity from animal experiments
– Special patient populations• Elderly
• Children
• Hepatic and renal impairment

Overall benefit/risk evaluation
This is the chapter where the SPC has to be defended based on the items and issues discussed above– The rationale for the indication should be given based on
the objectives of the trials• Phase III trials determine the indication
• Only what has been shown and proven will be approved
• Patient populations excluded may become contra-indications

Overall benefit/risk evaluation
The dosing should be explained for all strengths
The side effects should be listed from trials (and from the field – post launch experience)
Precautions and contra indications need to be explained and justified

Aim of the overview
Explain and defend the SmPC and the US and Japanese Data Sheets– All headlines and paras to be dealt with
– Clear opinion of the expert
– Final statement: Benefit risk evaluation leads to a positive conclusion – the new drug should be approved


From Potential Serious Risk to Public Health to a Decision of the European Commission - Industry Experiences
Dr Ulrich GranzerGranzer Regulatory Consulting & Services
Munich, Germany

63
European Marketing Authorisation
Scope

64
MRP and DCP
Scope

65
Scope of MRP/DCP:
New active substances (if not mandatory for the centralised procedure)
Generic medicinal products to authorised reference medicinal products
Informed consent
Well established use (WEU; bibliographic applications)
line extensions to national authorisations
known substances in new combination
New indications/new pharmaceutical forms for known substances

66
Article 17 of Directive 2001/83/EC
“1. ...
Applications for marketing authorizations in two or more Member States in respect of the same medicinal product shall be submitted in accordance with Articles 27 to 39.
2. Where a Member State notes that another marketing authorization application for the same medicinal product is being examined in another Member State, the Member State concerned shall decline to assess the application and shall advise the applicant that Articles 27 to 39 apply.”

67
Article 18 of Directive 2001/83/EC
“Where a Member State is informed in accordance with Article 8(3)(1) that another Member State has authorized a medicinal product which is the subject of a marketing authorization application in the Member State concerned, it shall reject the application unless it was submitted in compliance with Articles 27 to 39.”
68
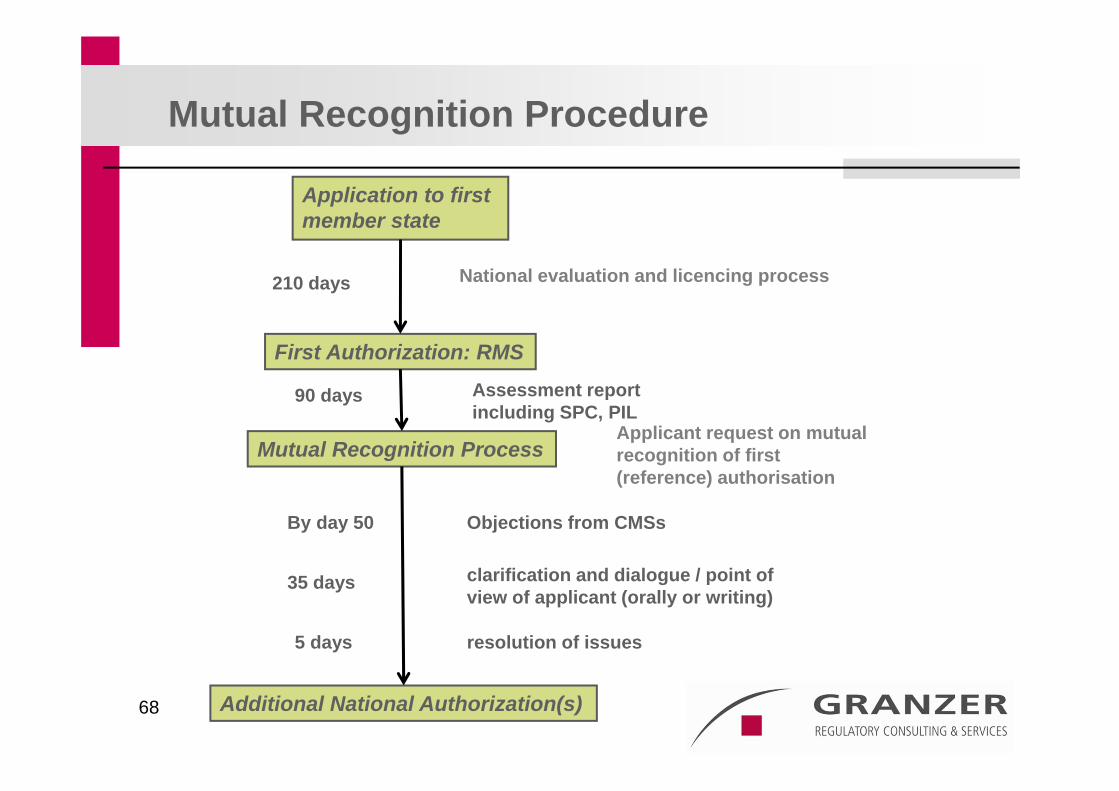
Application to first member state
Assessment reportincluding SPC, PIL
First Authorization: RMS
210 days
Applicant request on mutual recognition of first(reference) authorisation
Mutual Recognition Process
Objections from CMSsBy day 50
clarification and dialogue / point ofview of applicant (orally or writing)
resolution of issues
Additional National Authorization(s)
35 days
National evaluation and licencing process
Mutual Recognition Procedure
90 days
5 days

Decentralised Procedure
69

70
Application to all member statesRMS to start review
CMSs send comments to RMS and Applicant Consultation between RMS, CMS and Applicant (5 days)
PAR to CMSs and Applicant
70 days
Day 105: Clock stop orpositive close of procedure
RMS updates PrAR to prepare draft AR (DAR)
Day 106: Submission of the response
RMS evaluation of the dossier: Preliminary Assessment Report(PAR)
Decentralized Procedure
30 days
Preparation of response document by applicant3 – 6 months
Day 120: Consensus?End of procedure
Day 120: No consensus?Follow on process
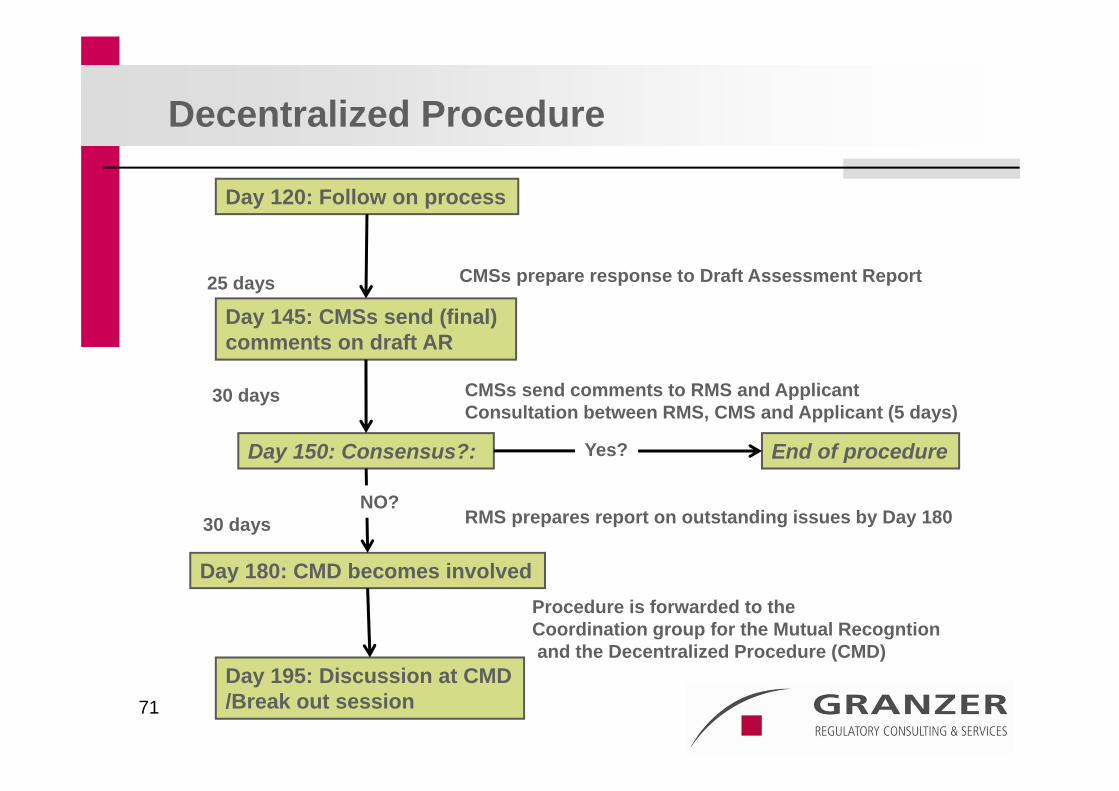
71
Day 120: Follow on process
CMSs send comments to RMS and Applicant Consultation between RMS, CMS and Applicant (5 days)
Day 145: CMSs send (final) comments on draft AR
25 days
Day 150: Consensus?:
Procedure is forwarded to the Coordination group for the Mutual Recogntionand the Decentralized Procedure (CMD)
Day 180: CMD becomes involved
CMSs prepare response to Draft Assessment Report
Decentralized Procedure
30 days
RMS prepares report on outstanding issues by Day 18030 days
Day 195: Discussion at CMD/Break out session
End of procedureYes?
NO?
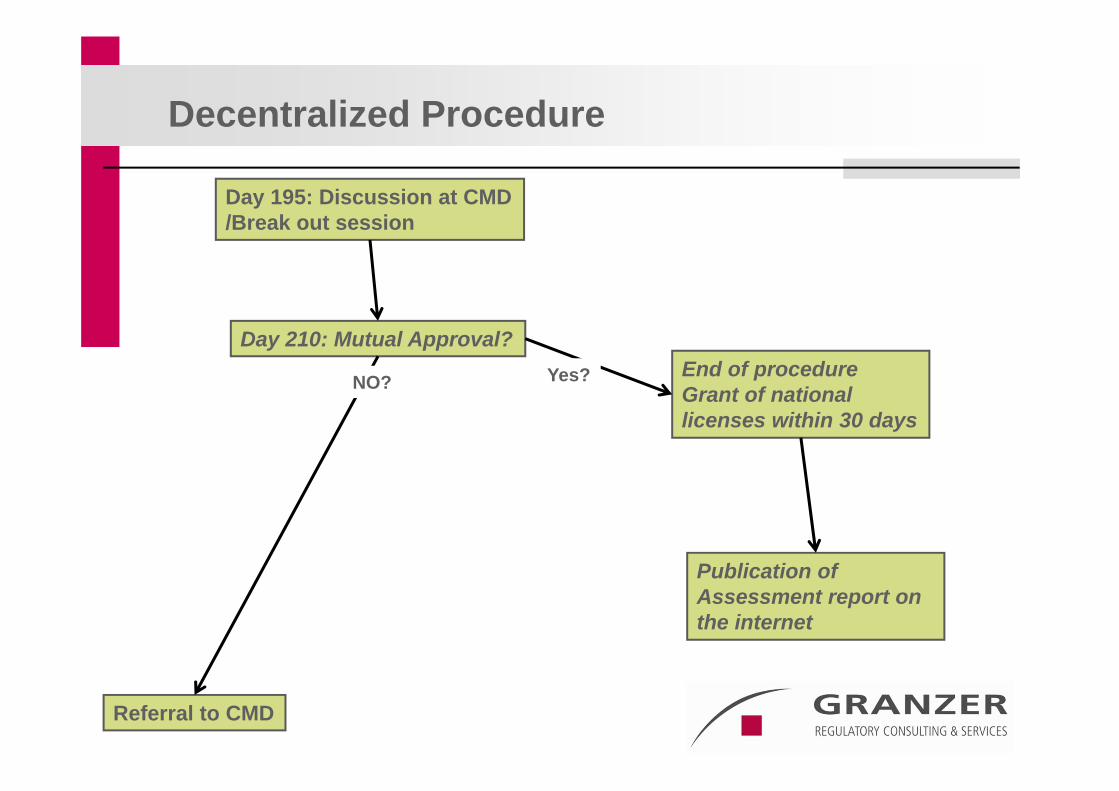
Day 195: Discussion at CMD/Break out session
Day 210: Mutual Approval?
Referral to CMD
Decentralized Procedure
End of procedureGrant of national licenses within 30 days
Yes?NO?
Publication ofAssessment report on the internet
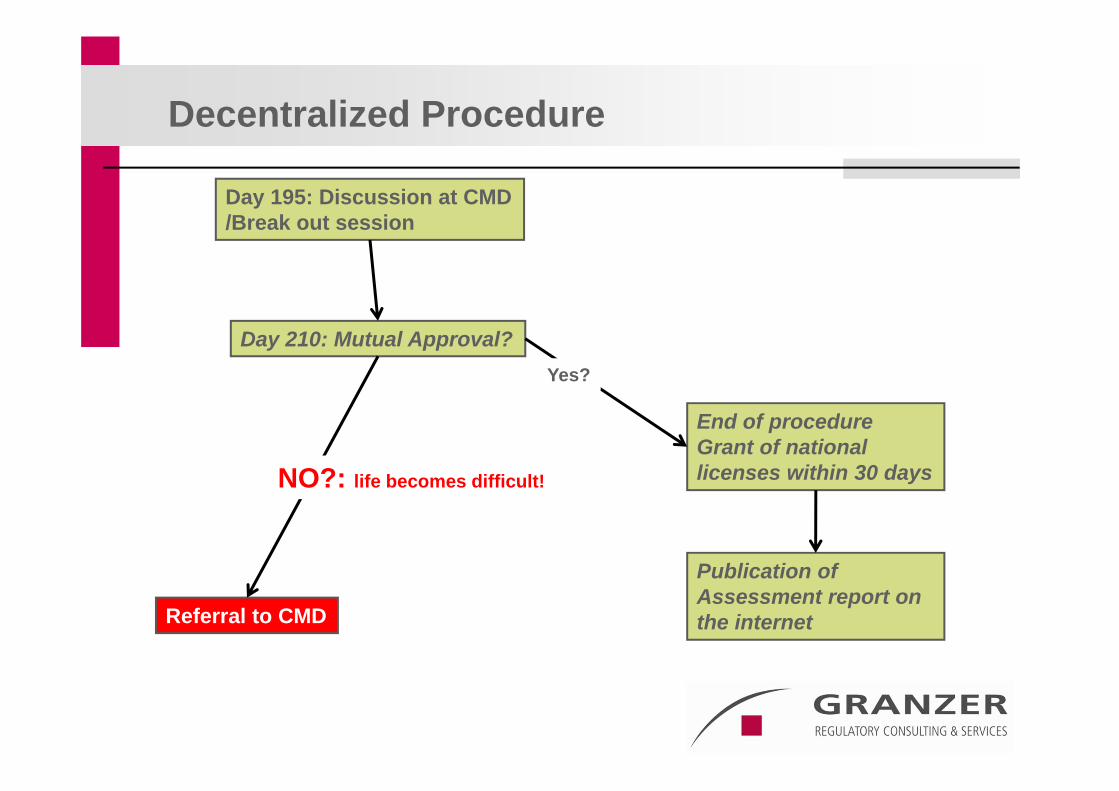
Day 195: Discussion at CMD/Break out session
Day 210: Mutual Approval?
Referral to CMD
Decentralized Procedure
End of procedureGrant of national licenses within 30 days
Yes?
NO?: life becomes difficult!
Publication ofAssessment report on the internet

Public Assessment report on the internet
Information on the product
Information on the reasons for apporval
Justification of indications and other major sections of SPC (Summary of Product Characteristics)
Description of dossier content: Preclinical and clinical– Good source for competitors
Deletion of commercially confidential information– Definition may differ between applicant and authority

75
CMD(h)-Referral in DCP and MRP

CMD-Referral - Trigger:
Disagreement between MS concerned by the application at the end of MRP (Day 90) or DCP (Day 210) based on potential serious risk to public health
What does this mean?– If all are positive: Case closed
– If all are negative but the applicant: CASE CLOSED
One MS has to be positive to trigger a referral

77
Day 0 RMS starts procedure: Draft LoQ to MSs for comments
Day 10 final LoQ to the Applicant
Day~20 1st CMD-Meeting: Discussion of case
Day 25 Response from Applicant on LoQ
10 days for preparation of response (day 11 – day 20)– No new information
– Usually no new arguments
– Improvement of statements if possible
– Cooperation with RMS, if RMS is positive
– Cooperation with CMS, if at least one CMS is positive
60 Days CMD-Procedure (CMD-SOP)

Day 35 Updated AR of the RMS to CMD
Day 45 MSs position on response to LoQ
Day~50 Discussions at 2nd CMD-Meeting; Hearing of written comments
Day 60 close of procedure with two options:
consensus or referral to CHMP

79
Withdrawal of Applications
The applicant has the right to withdraw the application until the last day of the MRP or DCP.
However, if the applicant has withdrawn the application from a MS because a potential serious risk to public health was raised by this MS, the application will be automatically forewarded for discussion to the CMD(h)
A withdrawal will not help the applicant!
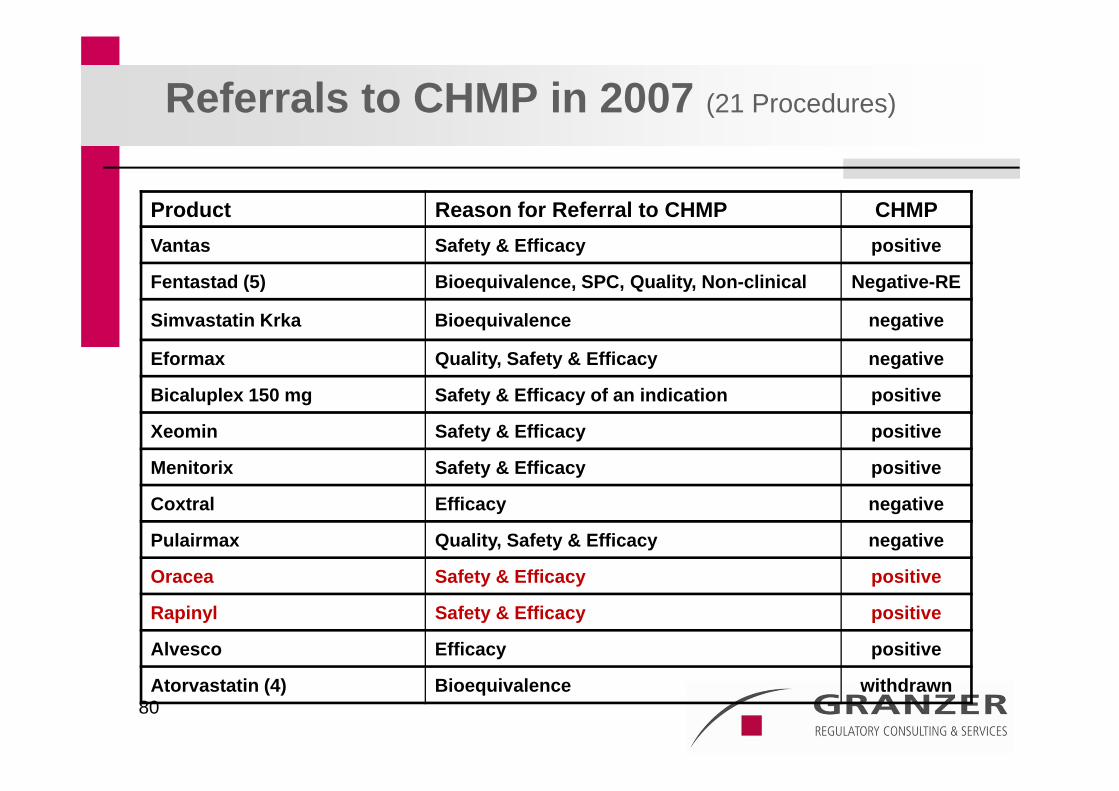
80
Referrals to CHMP in 2007 (21 Procedures)
Product Reason for Referral to CHMP CHMPVantas Safety & Efficacy positive
Fentastad (5) Bioequivalence, SPC, Quality, Non-clinical Negative-RE
Simvastatin Krka Bioequivalence negative
Eformax Quality, Safety & Efficacy negative
Bicaluplex 150 mg Safety & Efficacy of an indication positive
Xeomin Safety & Efficacy positive
Menitorix Safety & Efficacy positive
Coxtral Efficacy negative
Pulairmax Quality, Safety & Efficacy negative
Oracea Safety & Efficacy positive
Rapinyl Safety & Efficacy positive
Alvesco Efficacy positive
Atorvastatin (4) Bioequivalence withdrawn

The next round
CHMP Referral

Referral under Article 29(4) of Council Directive 2001/83/EC, as amended

Why does it come to referrals, an example?
Feedback from CHMP– Summary of major issues resolved during the CMD(h) procedure
between day 0 and day 60
– Summary of the final coordination group discussion and remaining unresolved scientific issues
– Risk:benefit concerns
– In conclusion the overall risk:benefit for “Interesting Product” has not been sufficiently demonstrated by the submitted documentation and the RMS concludes that the product is not approvable
Proposed list of questions– To be addressed by applicant in the OE and in writing

The “life” example
Start of Procedure 20 September 2007
Responses to LoQ: 03 December 2007
Restart of the procedure: 25 December 2007
Assessment Report: 09 January 2008
Comments from CHMP: 14 January 2008
Oral Explanation/Opinion: CHMP January 2008

The outcome
The new Rapporteur and the Applicant convinced the CHMP to give a consensus vote, which was positive.
The RMS eventually became positive as well
The eventual Rapporteur played a key role in the entire procedure and was open for discussions with the company at any stage of the procedure starting already in the pre-CMD phase

Meaning for the applicant
Eventual approval
Repeat use procedure with member states not involved in the DCP should lead to approval without Objections as CHMP and EU Commission have been positive
Time consuming compared to “regular” DCP

Overall Conclusion:
Science has to be right
All steps need a very thorough and detailed preparation
The responses and, in particular, the OE need a lot of work
Someone with the right experience needs to be part of the applicant’s team:
– Experience in• Oral Explanations
• Preparation of response documents
• Discussion and sometimes negotiations with regulatory authorities at any stage of procedure

THANK YOU






























