Embed Size (px)
Citation preview

EXPERIMENTAL CELL RESEARCH 228, 65–75 (1996)ARTICLE NO. 0300
Modulation of Protein Kinase C Activity and Calcium-Sensitive IsoformExpression in Human Myeloid Leukemia Cells by Bryostatin 1:Relationship to Differentiation and Ara-C-Induced Apoptosis
STEVEN GRANT,*,†,1 AMY J. TURNER,* ALEX J. FREEMERMAN,* ZHILIANG WANG,*LORA KRAMER,* AND W. DAVID JARVIS*
Departments of *Medicine and of †Pharmacology and Toxicology, Medical College of Virginia, Richmond, Virginia 23298
tion of total assayable PKC activity and cPKC expres-sion by bryostatin 1 are insufficient, by themselves, toPrevious studies have shown that pretreatment ofaccount for potentiation of leukemic cell apoptosis, athuman myeloid leukemia cells (HL-60) with the pro-least under conditions in which differentiation occurs.tein kinase C (PKC) activator bryostatin 1 potentiatesThey also provide further evidence that a reciprocalara-C-induced apoptosis. To test the hypothesis thatand highly schedule-dependent relationship exists be-this capacity stems from down-regulation of PKC ac-tween leukemic cell differentiation and drug-inducedtivity and/or Ca2/-dependent (group-I; cPKC) isoformapoptosis. q 1996 Academic Press, Inc.expression, comparisons were made between the ef-
fects of this agent and the stage-2 tumor promotermezerein under conditions favoring either cellular dif-ferentiation or drug-induced apoptosis. Twenty-four-
INTRODUCTIONhour pretreatment of HL-60 cells with 10 nM bryo-statin 1, which does not induce differentiation in thiscell line, led to a profound reduction in membrane and In hematopoietic cell systems, there is evidence thatcytosolic PKC activity, decreased expression of cPKC apoptosis is regulated at the level of signal transduc-isoforms (a, bI , bII , g), and a marked increase in ara-C tion pathways, particularly those involving the Ca2/-induced apoptosis. In contrast, 10 nM mezerein, which and lipid-dependent serine–threonine kinase, proteininduces HL-60 cell differentiation, was less effective in kinase C (PKC) [1]. PKC consists of at least nine iso-down-regulating membrane and cytosolic PKC activ- forms, subdivided into the conventional (or Ca2/-depen-ity as well as a, bI , and g cPKC isoform expression, dent, group-I; cPKC), novel (group-II), and atypicaland failed to potentiate ara-C-related apoptosis. The (group-III) subfamilies [2]. It has been proposed thateffects of bryostatin 1 were dominant to those of me- one of the functions of PKC is to oppose apoptoticzerein, in that the combination resulted in down-regu- events. For example, tumor-promoting PKC activatorslation of PKC activity and expression and potentiation such as phorbol 12-myristate 13-acetate (PMA) protectof ara-C-induced apoptosis, but not cellular matura-
hematopoietic cells from growth-factor deprivation-in-tion. However, coadministration of the Ca2/ ionophoreduced apoptosis [3]. Conversely, PKC inhibitors are po-A23187 (250 nM) restored bryostatin 1’s differentiatingtent inducers of apoptosis in hematopoietic cells [4, 5].ability while antagonizing its capacity to augmentTogether, these findings suggest a protective functionapoptosis, despite failing to reverse bryostatin 1-in-for PKC in the regulation of hematopoietic cell death.duced down-regulation of PKC activity and cPKC iso-In addition to direct effects of PKC, a leukemic cell’sform expression. Furthermore, pretreatment of differ-susceptibility to apoptosis is influenced by cellular mat-entiation-responsive monocytic leukemia cells (U937)uration [6]. The nature of the relationship between leu-with bryostatin 1 substantially reduced PKC activitykemic cell apoptosis and differentiation remains theand cPKC isoform expression, but exerted minimal ef-
fects on ara-C-related apoptosis. In contrast, exposure subject of debate. For example, induction of leukemicof U937 cells to bryostatin 1 after ara-C dramatically cell differentiation (e.g., by retinoic acid or PMA) leadsincreased apoptosis, a phenomenon that did not occur to the appearance of apoptotic features, often as a rela-in differentiation-unresponsive HL-60 cells. Collec- tively late event [7, 8]. On the other hand, leukemictively, these observations indicate that down-regula- cell maturation has been shown to oppose drug-induced
apoptosis [9, 10]. Attempts to define the role of PKCin the regulation of leukemic cell death are further1 To whom correspondence and reprint requests should be sent atcomplicated by inherent difficulties in distinguishingthe Division of Hematology/Oncology, Medical College of Virginia,
MCV Station Box 230, Richmond VA, 23298. Fax: (804) 828-8079. between direct effects of PKC activation versus indirect
65 0014-4827/96 $18.00Copyright q 1996 by Academic Press, Inc.
All rights of reproduction in any form reserved.
AID ECR 3294 / 6i14$$$$$1 09-13-96 08:18:32 eca AP: Exp Cell

66 GRANT ET AL.
viously described [23]. The human monocytic leukemic cell line U937actions stemming from induction of leukemic cell differ-was isolated from the peripheral blood of a patient with diffuse histio-entiation [11].cytic lymphoma as reported earlier [24]. Cells were cultured in RPMIIn a previous communication, we reported that pre- 1640 medium supplemented with sodium pyruvate, MEM essential
treatment of human leukemia cells (HL-60) with bry- vitamins, L-glutamate, penicillin and streptomycin, and 10% heat-inactivated fetal calf serum (Hyclone, Logan, UT). They were main-ostatin 1, a macrocyclic lactone PKC activator now intained in a 377C, 5% CO2, fully humidified incubator, passed twicephase I clinical evaluation [12], augmented apoptosisweekly, and routinely examined for mycoplasma contamination us-induced by subsequent exposure to 1-[b-D-arabino-ing the Gen-Probe kit (Gen-Probe, La Jolla, CA).
furanosyl]cytosine (ara-C) [13]. Bryostatin 1 has Drugs and chemicals. 1-b-D-Arabinofuranosylcytosine (free base)been shown to induce differentiation in some HL-60 was purchased from Sigma Chemicals (St. Louis, MO) and main-sublines [14, 15], but not in others [15, 16], including tained as a dry powder at 0207C. It was reformulated in PBS prior
to use. Bryostatin 1 was provided by the Cancer Treatment andthe one used in this laboratory. In contrast, the stage-Evaluation Program, NIH, and stored desiccated at 0207C. It was2 tumor promoter mezerein induces HL-60 cell dif-formulated in sterile dimethylsulfoxide (DMSO; Sigma), and subse-ferentiation but fails to potentiate ara-C-related quently diluted in RPMI medium so that the final concentration of
apoptosis [17]. Furthermore, the capacity of bryo- DMSO was in all cases£0.05%. Mezerein was purchased from Sigmastatin 1 to facilitate ara-C-induced cell death has Chemicals, formulated in DMSO, and stored at 0207C in light-pro-
tected vials. Calcium ionophore (A23187) was purchased from Calbio-been temporally related to down-regulation of totalchem (La Jolla, CA), stored as a dry powder at 0207C, and reformu-assayable PKC activity [17]. These findings, alonglated in sterile water before use.with evidence of a similar action by PKC inhibitors
Experimental format. Logarithmically growing cells (Ç3–41 105
[18], have given rise to the hypothesis that the capac- cells/ml) were suspended in plastic T-flasks and exposed to the desig-ity of bryostatin 1 to enhance ara-C-related apoptosis nated agent for 24 h in a 377C, 5% CO2 fully humidified incubator.
At the end of this interval, ara-C was added to the flasks to achievemay be related to its ability to induce profound down-a final concentration of 1005 M. The flasks were placed back in theregulation of PKC, and perhaps to its failure to initi-incubator for an additional 6 h, after which the cells were transferredate a differentiation program.to a conical centrifuge tube and pelleted at 400g for 6 min at 47C.
To address these issues, an attempt has been made In each experiment, the sides of the flasks were scraped with a rubberto compare the effects of mezerein and bryostatin 1 policeman to ensure that both adherent and nonadherent cells were
assayed. Alternatively, in some experiments, cells were exposed towith respect to leukemic cell PKC activity and group-10 mM ara-C for 6 h, washed 13 to remove drug, and resuspendedI isoform expression. Our studies have focused onin medium containing bryostatin 1 for an additional 24 h. Cells wereCa2/-dependent isoforms, because these species are enumerated using a Model ZBI Coulter counter (Hialeah, FL), and
the most abundantly expressed in leukemic cells [19] cell numbers were normalized prior to the studies of apoptosis, PKCand have been implicated in HL-60 cell differentia- activity and expression, and differentiation, as described below.tion [20]. Second, we have recently reported that Agarose gel electrophoresis. The degree of internucleosomal DNA
fragmentation in cell samples was assessed qualitatively throughcoadministration of the Ca2/ ionophore A23187 withagarose gel electrophoresis as previously described [17]. Followinga very low concentration of bryostatin 1 (i.e., 0.5 nM )drug exposure, pelleted cells (2 1 107 cells/condition) were lysed inpartially restores bryostatin 1’s differentiation ca- 0.1% Nonidet-P40, 10 mM Tris–HCl, 25 mM EDTA, pH 7.4, con-
pacity, but antagonizes potentiation of ara-C-related taining 200 mg/ml proteinase K (Calbiochem), and incubated for 24apoptosis [21]. Studies employing this model system, h at 567C. The lysates were then centrifuged at 48,000g for 45 min,
and the supernatants adjusted to 200 mg/ml ribonuclease A (Sigma)as well as a human myeloid leukemia cell line (U937)for 4 h at 377C. DNA unassociated with intact chromatin residing insusceptible to bryostatin 1-induced maturation [22],the supernatants was then resolved by agarose gel electrophoresisprovide an opportunity to compare the effects of bryo- at 6 V/cm in 11 TBE buffer (45 mM Tris–borate, 10 mM EDTA, pH
statin 1 on cellular PKC activity and isoform expres- 8.0) for 3 h on 1.8% agarose gels impregnated with ethidium bromide[17]. Each lane was loaded with a volume of cell lysate correspondingsion under conditions favoring either differentiationto 2 1 106 cells, rather than with a fixed quantity of DNA. To permitor drug-induced apoptosis. Our results indicate thatestimation of fragment size, 100-bp DNA reference preparationsin these leukemic cell lines, a hierarchy exists inwere run in parallel.
which bryostatin 1-mediated down-regulation of PKCQuantitative analysis of DNA fragmentation. A previously de-
predominates. They also demonstrate that the ca- scribed method was used to quantify the extent of low molecularpacity of bryostatin 1 pretreatment to facilitate weight DNA fragmentation in leukemic cells undergoing apoptosis
[17]. We have found the degree of such fragmentation to correlateara-C-related apoptosis is opposed by differentiationclosely with the percentage of cells exhibiting apoptotic morphologicevents, despite persistent and profound reductions infeatures, as well as with the qualitative results of agarose gel electro-total cellular PKC activity. Last, our data reveal that phoresis. Briefly, logarithmically growing cells were exposed to the
a highly schedule-dependent potentiation of ara-C- indicated agents and maintained in a 377C, 5% CO2, fully humidifiedrelated apoptosis occurs in leukemic cells (U937) sus- incubator. At the end of the exposure interval, the cells were pelleted,
washed twice in cold phosphate-buffered saline, and the pellets (5 1ceptible to the differentiating actions of bryostatin 1.106 cells/condition) lysed for 24 h in 5 mM Tris–HCl, 20 mM EDTA,pH 8.0, containing 0.1% Triton X-100. The lysates were then centri-MATERIALS AND METHODSfuged at 48,000g at 47C, after which the pellets were discarded andthe supernatant was diluted in 3 mM NaCl, 10 mM Tris–HCl, 1 mMCells. The human promyelocytic leukemia cell line HL-60 was
derived from a patient with acute promyelocytic leukemia as pre- EDTA, pH 8.0, containing 1.0 mg/ml bisbenzamide trihydrochloride
AID ECR 3294 / 6i14$$$$$1 09-13-96 08:18:32 eca AP: Exp Cell

67EFFECTS OF BRYOSTATIN 1 IN MYELOID LEUKEMIA CELLS
(Hoechst-33258; Sigma Chemicals). The presence of nonsedimenting 100,000g, and the supernatant, representing the cytosolic fraction,assayed for PKC activity using a commercially available kit provideddouble-stranded DNA, unbound to intact chromatin, was determined
by measurement of net fluorescence in each sample (lex Å 365, lem by GIBCO. In addition, pellets from the preceding step were homoge-nized in the same buffer containing 0.5% Triton X-100. After incuba-Å 460), utilizing a Hoefer TKO microfluorimeter. Highly purified calf
thymus DNA is used as a reference standard. tion on ice for 30 min followed by centrifugation for 2 min at 12,000g,the supernatant, representing the membrane fraction, was assayedas described below. For both fractions, normalized quantities of pro-Differentiation Studiestein were added to an assay mixture containing mixed micelles ofphosphatidylserine and PMA in suspension, after which the reactionCell adherence. Following exposure to the designated agents forwas initiated by addition of 2.5 1 1005 Ci/ml [g-32P]ATP, 2 1 100572 h, the density of cells in suspension was determined utilizing aM nonisotopic ATP, and 5 1 1005 M synthetic peptide substratehemacytometer. The sides of the flasks were then scraped with a(acetylated myelin basic protein N-terminal peptide AcMB4–14). Afterrubber policeman to permit detachment of adherent cells, and thea 5-min incubation at 307C, aliquots of the reaction mixture werecells dispersed prior to repeat density determinations. The percent-transferred to phosphocellulose filters, and the reaction was termi-age of adherent cells was expressed relative to the total cell popula-nated by immersion of disks in cold 1% (v/v) phosphoric acid. Thetion as previously described [17].disks were washed thoroughly, and radioactivity was quantified byCD11b expression. CD11b, a marker of monocytic/macrophageconventional liquid scintillography.differentiation [25], was monitored in drug-treated cells utilizing flow
Statistical analysis. The significance of differences between ex-cytometry. Following drug treatment, cell concentrations were ad-perimental conditions was determined utilizing the Student’s t testjusted to 5 1 106 cells/ml with PBS, and 100 ml of the suspensionfor unpaired observations.combined with 10 ml of fluorescein isothyocyanate-labeled anti-
CD11b antibody (Becton–Dickinson, Mountain View, CA). After gen-tle agitation, cells were incubated for 20 min at 47C in the dark.
RESULTSAfter incubation, cells were analyzed using a Becton–DickinsonFACScan flow cytometer and Verity Winlist software (Verity Soft-ware, Topsham, ME). An isotype-matched antibody control was used Previous studies from this laboratory have demon-with each run. strated that pretreatment of HL-60 cells for 24 h with
Cell morphology and apoptosis. Cytocentrifuge preparations the stage-2 tumor promoter mezerein, unlike bryo-were stained with Wright-Giemsa and viewed by light microscopy to
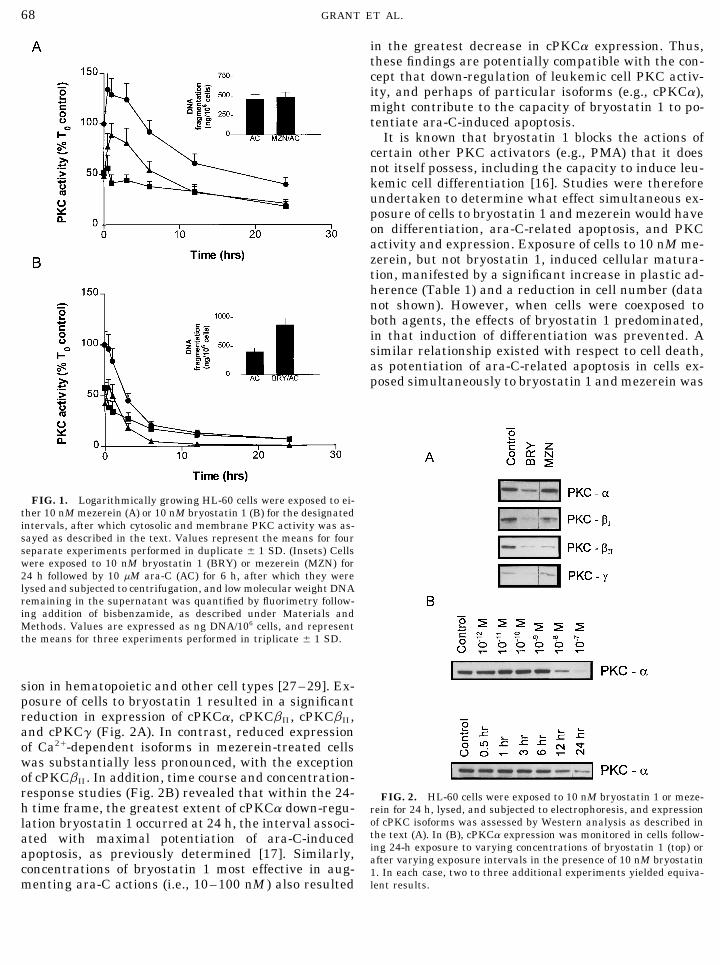
statin 1, fails to potentiate ara-C-induced apoptosisevaluate features of cellular differentiation as well as apoptosis (i.e.,[17]. A time course analysis of cytosolic and membranecell shrinkage, nuclear condensation, formation of apoptotic bodies,
etc.) as previously described [16, 17]. For the latter studies, the per- PKC activity in cells exposed to 10 nM mezerein orcentage of apoptotic cells was determined by evaluating §500 cells/ bryostatin 1 is shown in Fig. 1. Immediately followingcondition in triplicate. We have previously reported that the inci- mezerein treatment, an increase in membrane and adence of apoptosis as determined by these morphological criteria
decrease in cytosolic activity was noted, reflecting en-correlates very closely with the degree of low molecular weight DNAzyme translocation. Subsequently, a decline in activityfragmentation assayed quantitatively by spectrofluorometry, and
qualitatively with the amount of internucleosomal DNA fragmenta- was observed in both compartments, although at thetion determined by agarose gel electrophoresis [17, 26]. end of the 24-h incubation period approximately 50%
Western analysis. Expression of cPKC a, b1 , bII , and g protein of total activity persisted. As depicted in the inset, thiswas determined by Western analysis using minor modifications of a exposure did not potentiate low molecular weight DNApreviously described method [21]. Following treatment, whole-cell
fragmentation in cells subsequently treated with 10pellets (1 1 107 cells/condition) were washed twice in PBS, resus-mM ara-C for 6 h. Equivalent results were obtainedpended in 50 ml PBS, lysed by the addition of 50 ml 21 Laemmli (11
is 30 mM Tris-base, pH 6.8, 2% SDS, 2.88 mM b-mercaptoethanol, when cells were examined for morphologic features of10% glycerol), and briefly sonicated. Homogenates were quantified apoptosis (data not shown). Cells incubated with 10using Coomassie protein assay reagent (Pierce, Rockford, IL). Equal nM bryostatin 1 also displayed a modest early increaseamounts of protein (20 mg) were boiled for 10 min, separated by
in membrane PKC activity, accompanied by a declineSDS–PAGE (5% stacker and 10% resolving) and electroblotted tonitrocellulose. The blots were stained in 0.1% amido black and de- in cytosolic activity. However, the decline in activity instained in 5% acetic acid to ensure transfer and equal loading. After both compartments was more rapid and profound thanblocking in PBS–Tween (PBS-T; 0.05%) and 5% milk for 1 h at 227C, that observed with mezerein, and at the end of the 24-the blots were incubated in fresh blocking solution with an appro-
h incubation period, less than 10% of total PKC activitypriate dilution of primary antibody (PKCa 1:5000, PKCb1 1:3000,persisted. As depicted in the inset, this exposure wasPKCbII 1:3000, PKCg 1:500; all Santa Cruz Biotechnology, Santa
Cruz, CA) for 4 h at 227C. Blots were washed 3 1 5 min in PBS-T associated with a 100% increase in DNA fragmentationand then incubated with a 1:2000 dilution of horseradish peroxidase- (as well as in the percentage of apoptotic cells—dataconjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA) not shown) following treatment with ara-C.for 1 h at 227C. Blots were again washed 3 1 5 min in PBS-T and
The actions of bryostatin 1 and mezerein (10 nMthen developed by enhanced chemiluminescence (Pierce).each, 24-h exposure) were then compared with respectPKC activity. A minor modification of a previously describedto effects on PKC isoform expression (Fig. 2). Thesemethod [17] was employed to assay membrane and cytosolic PKC
activity in HL-60 cells exposed to bryostatin 1 or mezerein. Briefly, studies focused on group-I isoforms (a, bI , bII , and g),pelleted cells were homogenized in 2 1 1002 M Tris, 5 1 1004 M since induction of HL-60 cell differentiation has beenEDTA, 5 1 1004 M EGTA, pH 7.5, containing 25 mg/ml protease associated with increased expression of this PKC sub-inhibitors (aprotinin, leupeptin), with 35 1-s strokes of a motorized
family, particularly PKCa [20]; moreover, bryostatin 1micropestle homogenizer (Glas-Col) at setting 35. The homogenatewas incubated on ice for 30 min and centrifuged for 30 min at is known to be a potent down-regulator of PKCa expres-
AID ECR 3294 / 6i14$$$$$2 09-13-96 08:18:32 eca AP: Exp Cell
68 GRANT ET AL.
in the greatest decrease in cPKCa expression. Thus,these findings are potentially compatible with the con-cept that down-regulation of leukemic cell PKC activ-ity, and perhaps of particular isoforms (e.g., cPKCa),might contribute to the capacity of bryostatin 1 to po-tentiate ara-C-induced apoptosis.
It is known that bryostatin 1 blocks the actions ofcertain other PKC activators (e.g., PMA) that it doesnot itself possess, including the capacity to induce leu-kemic cell differentiation [16]. Studies were thereforeundertaken to determine what effect simultaneous ex-posure of cells to bryostatin 1 and mezerein would haveon differentiation, ara-C-related apoptosis, and PKCactivity and expression. Exposure of cells to 10 nM me-zerein, but not bryostatin 1, induced cellular matura-tion, manifested by a significant increase in plastic ad-herence (Table 1) and a reduction in cell number (datanot shown). However, when cells were coexposed toboth agents, the effects of bryostatin 1 predominated,in that induction of differentiation was prevented. Asimilar relationship existed with respect to cell death,as potentiation of ara-C-related apoptosis in cells ex-posed simultaneously to bryostatin 1 and mezerein was
FIG. 1. Logarithmically growing HL-60 cells were exposed to ei-ther 10 nM mezerein (A) or 10 nM bryostatin 1 (B) for the designatedintervals, after which cytosolic and membrane PKC activity was as-sayed as described in the text. Values represent the means for fourseparate experiments performed in duplicate { 1 SD. (Insets) Cellswere exposed to 10 nM bryostatin 1 (BRY) or mezerein (MZN) for24 h followed by 10 mM ara-C (AC) for 6 h, after which they werelysed and subjected to centrifugation, and low molecular weight DNAremaining in the supernatant was quantified by fluorimetry follow-ing addition of bisbenzamide, as described under Materials andMethods. Values are expressed as ng DNA/106 cells, and representthe means for three experiments performed in triplicate { 1 SD.
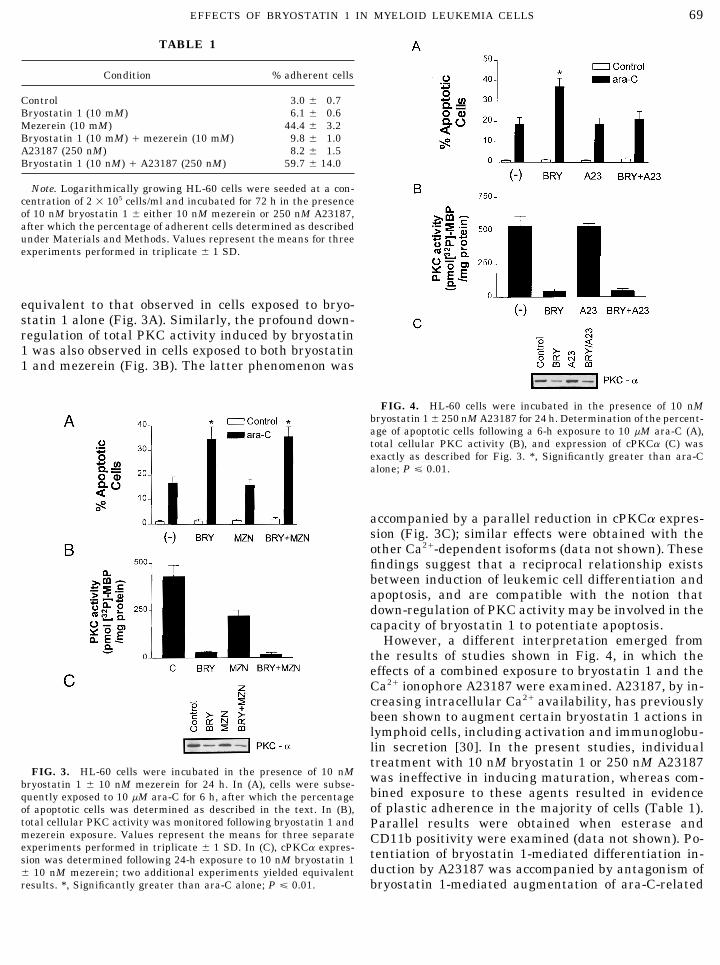
sion in hematopoietic and other cell types [27–29]. Ex-posure of cells to bryostatin 1 resulted in a significantreduction in expression of cPKCa, cPKCbII , cPKCbII ,and cPKCg (Fig. 2A). In contrast, reduced expressionof Ca2/-dependent isoforms in mezerein-treated cellswas substantially less pronounced, with the exceptionof cPKCbII . In addition, time course and concentration-response studies (Fig. 2B) revealed that within the 24- FIG. 2. HL-60 cells were exposed to 10 nM bryostatin 1 or meze-h time frame, the greatest extent of cPKCa down-regu- rein for 24 h, lysed, and subjected to electrophoresis, and expression
of cPKC isoforms was assessed by Western analysis as described inlation bryostatin 1 occurred at 24 h, the interval associ-the text (A). In (B), cPKCa expression was monitored in cells follow-ated with maximal potentiation of ara-C-induceding 24-h exposure to varying concentrations of bryostatin 1 (top) orapoptosis, as previously determined [17]. Similarly, after varying exposure intervals in the presence of 10 nM bryostatin
concentrations of bryostatin 1 most effective in aug- 1. In each case, two to three additional experiments yielded equiva-lent results.menting ara-C actions (i.e., 10–100 nM ) also resulted
AID ECR 3294 / 6i14$$$$$2 09-13-96 08:18:32 eca AP: Exp Cell
69EFFECTS OF BRYOSTATIN 1 IN MYELOID LEUKEMIA CELLS
TABLE 1
Condition % adherent cells
Control 3.0 { 0.7Bryostatin 1 (10 mM) 6.1 { 0.6Mezerein (10 mM) 44.4 { 3.2Bryostatin 1 (10 mM) / mezerein (10 mM) 9.8 { 1.0A23187 (250 nM) 8.2 { 1.5Bryostatin 1 (10 nM) / A23187 (250 nM) 59.7 { 14.0
Note. Logarithmically growing HL-60 cells were seeded at a con-centration of 2 1 105 cells/ml and incubated for 72 h in the presenceof 10 nM bryostatin 1 { either 10 nM mezerein or 250 nM A23187,after which the percentage of adherent cells determined as describedunder Materials and Methods. Values represent the means for threeexperiments performed in triplicate { 1 SD.
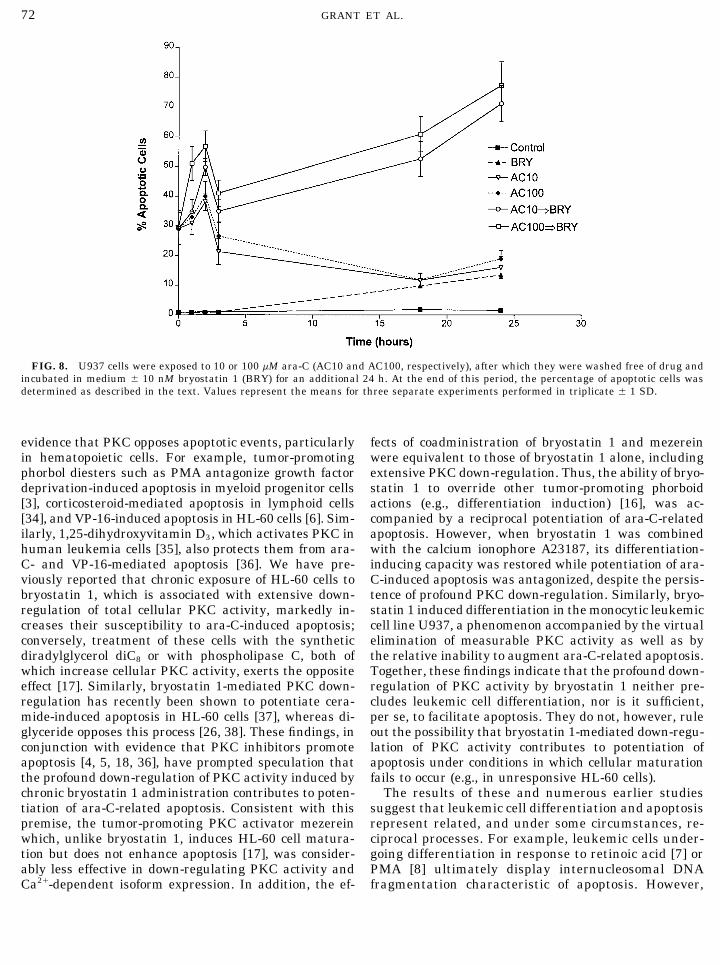
equivalent to that observed in cells exposed to bryo-statin 1 alone (Fig. 3A). Similarly, the profound down-regulation of total PKC activity induced by bryostatin1 was also observed in cells exposed to both bryostatin1 and mezerein (Fig. 3B). The latter phenomenon was
FIG. 4. HL-60 cells were incubated in the presence of 10 nMbryostatin 1{ 250 nM A23187 for 24 h. Determination of the percent-age of apoptotic cells following a 6-h exposure to 10 mM ara-C (A),total cellular PKC activity (B), and expression of cPKCa (C) wasexactly as described for Fig. 3. *, Significantly greater than ara-Calone; P £ 0.01.
accompanied by a parallel reduction in cPKCa expres-sion (Fig. 3C); similar effects were obtained with theother Ca2/-dependent isoforms (data not shown). Thesefindings suggest that a reciprocal relationship existsbetween induction of leukemic cell differentiation andapoptosis, and are compatible with the notion thatdown-regulation of PKC activity may be involved in thecapacity of bryostatin 1 to potentiate apoptosis.
However, a different interpretation emerged fromthe results of studies shown in Fig. 4, in which theeffects of a combined exposure to bryostatin 1 and theCa2/ ionophore A23187 were examined. A23187, by in-creasing intracellular Ca2/ availability, has previouslybeen shown to augment certain bryostatin 1 actions inlymphoid cells, including activation and immunoglobu-lin secretion [30]. In the present studies, individualtreatment with 10 nM bryostatin 1 or 250 nM A23187
FIG. 3. HL-60 cells were incubated in the presence of 10 nM was ineffective in inducing maturation, whereas com-bryostatin 1 { 10 nM mezerein for 24 h. In (A), cells were subse-bined exposure to these agents resulted in evidencequently exposed to 10 mM ara-C for 6 h, after which the percentageof plastic adherence in the majority of cells (Table 1).of apoptotic cells was determined as described in the text. In (B),
total cellular PKC activity was monitored following bryostatin 1 and Parallel results were obtained when esterase andmezerein exposure. Values represent the means for three separate CD11b positivity were examined (data not shown). Po-experiments performed in triplicate { 1 SD. In (C), cPKCa expres- tentiation of bryostatin 1-mediated differentiation in-sion was determined following 24-h exposure to 10 nM bryostatin 1
duction by A23187 was accompanied by antagonism of{ 10 nM mezerein; two additional experiments yielded equivalentresults. *, Significantly greater than ara-C alone; P £ 0.01. bryostatin 1-mediated augmentation of ara-C-related
AID ECR 3294 / 6i14$$$$$2 09-13-96 08:18:32 eca AP: Exp Cell

70 GRANT ET AL.
The effects of bryostatin 1 were then examined withrespect to ara-C-related apoptosis in this cell line (Fig.6). In contrast to HL-60 cells, which do not undergoapoptosis in response to this agent [17], preincubationof U937 cells with 10 nM bryostatin 1 alone for 24 hslightly increased the percentage of apoptotic cells (Fig.6A). However, pretreatment of U937 cells with bryo-statin 1 did not result in a dramatic increase inapoptosis following a subsequent 6-h exposure to 10 mMara-C, as observed in the HL-60 cell line. Qualitativeassessment of internucleosomal DNA fragmentationconfirmed that bryostatin 1, while inducing a smallamount of internucleosomal DNA fragmentation by it-self (Fig. 6B, lane 3), did not increase ara-C-relatedDNA damage in this cell line (Fig. 6B, lane 5). Thus,the ability of bryostatin 1 to trigger a differentiationprogram in U937 cells was accompanied by a reducedcapacity to augment ara-C-induced apoptosis comparedto HL-60 cells.
PKC activity and expression was then monitored inU937 cells exposed to bryostatin 1 (Fig. 7). Although
FIG. 5. Logarithmically growing U937 cells were seeded at 2 1 10 nM bryostatin 1 exerted marginal effects on ara-C-105 cells/ml in the presence of 10 nM bryostatin 1 (BRY) or PMA
induced apoptosis in these cells, it was highly effectiveand cell density determinations were obtained at 24-h intervals (A).in down-regulating total cellular PKC activity, as wereAlternatively, cell adherence was determined at 72 h as described
in the text (B). Values represent the means for three separate experi-ments performed in triplicate { 1 SD.
apoptosis (Fig. 4A), further supporting the notion thata reciprocal relationship exists between leukemic celldifferentiation and apoptosis. However, coadministra-tion of A23187 did not reverse the profound down-regu-lation of total PKC activity induced by bryostatin 1(Fig. 4B), despite restoring bryostatin 1’s differentia-tion-inducing capacity and opposing its ability to aug-ment ara-C-related apoptosis. Similarly, A23187 didnot reverse bryostatin 1-mediated down-regulation ofcPKCa expression (Fig. 4C), nor did it prevent bryo-statin 1 from reducing expression of other group-I iso-forms (not shown). Collectively, these findings indicatethat bryostatin 1-mediated down-regulation of PKC ac-tivity (or cPKC expression) is insufficient, by itself, topotentiate ara-C-related apoptosis under conditions inwhich cellular maturation occurs.
To gain further insights into the relationship be-tween bryostatin 1’s effects on leukemic cell differentia-tion, ara-C-related apoptosis, and PKC activity, paral-
FIG. 6. U937 cells were incubated in the presence of 10 nM bryo-lel studies were conducted in human monocytic leuke-statin 1 for 24 h, followed by 10 mM ara-C for 6 h, after which themia cells (U937). The results of a recent report indicate percentage of apoptotic cells (means for three experiments performed
that a relatively high bryostatin 1 concentration (e.g., in triplicate { 1 SD) was determined as described previously (A). In200 nM ) induces a partial differentiation program in (B), DNA from cells exposed sequentially to 10 nM bryostatin 1 and
10 mM ara-C was subjected to agarose gel electrophoresis. Lanes:this cell line [22]. Consistent with these findings, 101, 100-bp reference ladder; 2, control; 3, bryostatin 1; 4, ara-C; 5,nM bryostatin 1 significantly inhibited the growth ofbryostatin 1 followed by ara-C. Lane 6 corresponds to DNA from cellsU937 cells and led to an increase in cell adherence (Fig. exposed to 10 mM ara-C for 6 h, washed, and incubated for 24 h in
5), although it was clearly less effective in this regard fresh medium; lane 7 corresponds to DNA from cells exposed to ara-C (6 h) followed by 10 nM bryostatin 1 (24 h).than an equivalent concentration of PMA.
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell

71EFFECTS OF BRYOSTATIN 1 IN MYELOID LEUKEMIA CELLS
apoptotic cells was followed over the next several hoursby a decline, presumably reflecting the combined ef-fects of dissolution of dying cells and the dilutionalinfluence of regrowth of unaffected cells. However, avery marked difference was noted at later time points.For example, ara-C-pretreated cells incubated in ab-sence of bryostatin 1 exhibited a progressive decline inthe percentage of apoptotic cells, reaching a level of18.9 { 3.4% at 24 h. In contrast, the large majority ofcells exposed to bryostatin 1 following ara-C exhibitedpronounced apoptotic features at the end of the 24-hinterval (77.3 { 7.9%; P£ 0.001). Similarly, the degreeof internucleosomal DNA fragmentation in cells ini-tially exposed to 10 mM ara-C for 6 h, washed, and thenincubated in fresh medium for an additional 24 h wasquite modest (Fig. 6B, lane 6), whereas the same cellsincubated in bryostatin 1-containing medium exhibitedextensive DNA damage (Fig. 6B, lane 7). Photomicro-graphs of cells taken at 24 h further emphasize this
FIG. 7. U937 cells were exposed to the designated concentrations difference (Fig. 9). Control cells (Fig. 9A) displayedof bryostatin 1 for 24 h, after which total cellular PKC activity as-minimal evidence of apoptosis. Following treatmentsayed as described under Materials and Methods (A). Values repre-with bryostatin 1, occasional apoptotic cells and cellsent the means for three separate experiments performed in tripli-
cate. In (B), cPKC isoform expression was monitored by Western fragments were noted, although the majority of cellsanalysis following a 24-h exposure to 10 nM bryostatin 1. cPKCg appeared unaffected (Fig. 9B). Similarly, relatively in-was present in low abundance in these cells, and was not detectable frequent apoptotic cells were seen 24 h following ara-under any conditions.
C exposure (Fig. 9C). It should be noted that ara-C-pretreated cells appeared somewhat larger than con-trols, possibly reflecting the residual effects of ara-Cconcentrations of 1 and 100 nM (Fig. 7A). Down-regula-
tion of Ca2/-dependent isoform expression by 10 nM exposure. In striking contrast, pretreatment of U937cells with ara-C followed by bryostatin 1 resulted in,bryostatin 1 was also observed in U937 cells (Fig. 7B).
These results are analogous to those observed in HL- aside from a marked reduction in cell number, the ap-pearance of classical apoptotic features in the large60 cells exposed to the combination of bryostatin 1 and
A23187 (Fig. 4) in that (a) evidence of differentiation majority of the remaining cells (Fig. 9D). We have pre-viously reported that incubation of HL-60 cells withwas noted despite a profound reduction in total PKC
activity and (b) down-regulation of PKC was not accom- bryostatin 1 after, rather than before, ara-C treatmentdoes not augment ara-C-related cytoxicity [33]. Consis-panied by a dramatic potentiation of ara-C-induced
apoptosis. Thus, these findings provide further support tent with these results, and in contrast to those ob-tained in the U937 line, exposure of HL-60 cells tofor the concept that down-regulation of PKC, by itself,
is insufficient to augment ara-C-mediated apoptosis in bryostatin 1 following a 6-h incubation with 10 mM ara-C had no discernible effect on either the percentage ofhuman leukemia cells, at least under conditions in
which cellular maturation occurs. apoptotic cells or the degree of internucleosomal DNAfragmentation (data not shown). Collectively, theseFinally, in view of reports that administration of a
differentiation stimulus following a cytotoxic agent fa- findings suggest that the factors responsible for theability of bryostatin 1 to induce differentiation in U937cilitates apoptosis [31, 32], the effect of the reverse se-
quence was examined (Fig. 8). U937 cells were exposed but not HL-60 cells (a) allow pretreatment to potentiateara-C-related apoptosis in a differentiation-unrespon-to 10 (or 100) mM ara-C for 6 h, washed free of drug,
and incubated in medium { 10 nM bryostatin 1, and sive line (HL-60) and (b) facilitate apoptosis when bryo-statin 1 is administered after ara-C in a maturation-the percentage of apoptotic cells was monitored over
the ensuing 24 h. Cells exposed to bryostatin 1 alone competent line (U937).for 24 h exhibited a modest degree of apoptosis (Ç12%).Within the first 2 h, ara-C-pretreated cells displayed DISCUSSIONan initial increase in apoptosis whether or not theywere subsequently exposed to bryostatin 1, although The results of this study indicate that under conditions
in which leukemic cell maturation is induced, down-regu-the presence of the latter agent potentiated this process(e.g., 49.8 { 3.2% versus 34.9 { 4.3% apoptotic cells at lation of PKC by bryostatin 1 is not, by itself, sufficient
to enhance ara-C-mediated apoptosis. There is abundant2 h; P £ 0.02). The initial increase in percentage of
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell
72 GRANT ET AL.
FIG. 8. U937 cells were exposed to 10 or 100 mM ara-C (AC10 and AC100, respectively), after which they were washed free of drug andincubated in medium { 10 nM bryostatin 1 (BRY) for an additional 24 h. At the end of this period, the percentage of apoptotic cells wasdetermined as described in the text. Values represent the means for three separate experiments performed in triplicate { 1 SD.
evidence that PKC opposes apoptotic events, particularly fects of coadministration of bryostatin 1 and mezereinwere equivalent to those of bryostatin 1 alone, includingin hematopoietic cells. For example, tumor-promoting
phorbol diesters such as PMA antagonize growth factor extensive PKC down-regulation. Thus, the ability of bryo-statin 1 to override other tumor-promoting phorboiddeprivation-induced apoptosis in myeloid progenitor cells
[3], corticosteroid-mediated apoptosis in lymphoid cells actions (e.g., differentiation induction) [16], was ac-companied by a reciprocal potentiation of ara-C-related[34], and VP-16-induced apoptosis in HL-60 cells [6]. Sim-
ilarly, 1,25-dihydroxyvitamin D3, which activates PKC in apoptosis. However, when bryostatin 1 was combinedwith the calcium ionophore A23187, its differentiation-human leukemia cells [35], also protects them from ara-
C- and VP-16-mediated apoptosis [36]. We have pre- inducing capacity was restored while potentiation of ara-C-induced apoptosis was antagonized, despite the persis-viously reported that chronic exposure of HL-60 cells to
bryostatin 1, which is associated with extensive down- tence of profound PKC down-regulation. Similarly, bryo-statin 1 induced differentiation in the monocytic leukemicregulation of total cellular PKC activity, markedly in-
creases their susceptibility to ara-C-induced apoptosis; cell line U937, a phenomenon accompanied by the virtualelimination of measurable PKC activity as well as byconversely, treatment of these cells with the synthetic
diradylglycerol diC8 or with phospholipase C, both of the relative inability to augment ara-C-related apoptosis.Together, these findings indicate that the profound down-which increase cellular PKC activity, exerts the opposite
effect [17]. Similarly, bryostatin 1-mediated PKC down- regulation of PKC activity by bryostatin 1 neither pre-cludes leukemic cell differentiation, nor is it sufficient,regulation has recently been shown to potentiate cera-
mide-induced apoptosis in HL-60 cells [37], whereas di- per se, to facilitate apoptosis. They do not, however, ruleout the possibility that bryostatin 1-mediated down-regu-glyceride opposes this process [26, 38]. These findings, in
conjunction with evidence that PKC inhibitors promote lation of PKC activity contributes to potentiation ofapoptosis under conditions in which cellular maturationapoptosis [4, 5, 18, 36], have prompted speculation that
the profound down-regulation of PKC activity induced by fails to occur (e.g., in unresponsive HL-60 cells).The results of these and numerous earlier studieschronic bryostatin 1 administration contributes to poten-
tiation of ara-C-related apoptosis. Consistent with this suggest that leukemic cell differentiation and apoptosisrepresent related, and under some circumstances, re-premise, the tumor-promoting PKC activator mezerein
which, unlike bryostatin 1, induces HL-60 cell matura- ciprocal processes. For example, leukemic cells under-going differentiation in response to retinoic acid [7] ortion but does not enhance apoptosis [17], was consider-
ably less effective in down-regulating PKC activity and PMA [8] ultimately display internucleosomal DNAfragmentation characteristic of apoptosis. However,Ca2/-dependent isoform expression. In addition, the ef-
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell

73EFFECTS OF BRYOSTATIN 1 IN MYELOID LEUKEMIA CELLS
FIG. 9. U937 cells were exposed to 10 mM ara-C for 6 h, washed free of drug, and subsequently incubated for an additional 24 h inmedium { 10 nM bryostatin 1. Cytospin preparations were obtained and stained with Wright-Giemsa, and representative fields werephotographed at 4001 original magnification. (A) Control, (B) bryostatin 1, (C) ara-C, (D) ara-C followed by bryostatin 1.
induction of leukemic cell differentiation (e.g., by PKCa, has been implicated in HL-60 cell differentia-tion [20, 44]; (b) bryostatin 1 potently down-regulatesDMSO, IL-6, or PMA) reduces cellular susceptibility
to apoptosis induced by 5-azacytidine, camptothecin, PKCa [27–29]; and (c) maturation of HL-60 cells alongthe macrophage lineage requires sustained activationTGF-b1 , and VP-16, among other agents [6, 39–41].
Furthermore, leukemic cells displaying impaired mat- of cPKCa [45]. However, coadministration of A23187did not prevent bryostatin 1 from down-regulatinguration programs undergo apoptosis when exposed to
differentiation signals [42]. A possible explanation for cPKCa expression (or other cPKC isoforms), despitetriggering a differentiation response and antagonizingthese findings is that apoptosis may represent an alter-
native pathway traversed by cells unable to undergo a the increase in ara-C-related apoptosis. Similarly, bry-ostatin 1 markedly reduced cPKC isoform expressionnormal differentiation program [43]. It therefore ap-
peared reasonable to postulate that the ability of bryo- in U937 cells, an event accompanied by induction ofcellular maturation and a relative failure to potentiatestatin 1 to potentiate ara-C-mediated apoptosis might
stem from its capacity to disrupt normal differentiation ara-C-induced apoptosis. Thus, these findings indicatethat bryostatin 1’s ability to down-regulate cPKC iso-events [16]. Of these, attention centered on bryostatin
1’s actions on the conventional PKC isoforms since (a) form expression neither precludes cellular maturation,nor is it sufficient, by itself, to enhance drug-inducedup-regulation of members of this family, particularly
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell

74 GRANT ET AL.
apoptosis. It should be noted that the basis for the tions associated with either cellular maturation or po-tentiation of ara-C-induced apoptosis. These findingsunique actions of bryostatin 1 remains obscure, and
may involve specific patterns of isoform activation [46], also demonstrate that interventions triggering leuke-mic cell differentiation (e.g., administration of A23187)isoform translocation [47], or perturbations in expres-
sion of nonconventional PKC isoforms [48]. The possi- oppose potentiation of ara-C-mediated apoptosis bybryostatin 1, despite failure to prevent profound reduc-bility that one or more of these factors may play a role
in bryostatin 1’s unique ability to potentiate apoptosis tions in total PKC activity and expression of Ca2/-de-pendent isoforms. Thus, while bryostatin 1’s ability towill require further investigation.
The observation that exposure of U937 cells to bryo- down-regulate PKC activity in a differentiation-resis-tant cell line (e.g., HL-60) may be necessary for en-statin 1 after, but not before, ara-C resulted in a dra-
matic potentiation of apoptosis is consistent with the hancement of apoptosis, such an action is not by itselfsufficient to augment this process, at least under condi-results of several earlier reports. For example, treat-
ment of murine erythroleukemia cells with HMBA tions in which cellular maturation occurs. Finally, theobservation that bryostatin 1 dramatically promotesafter 5-FU exposure has been shown to produce a
marked increase in apoptosis [49]. Similarly, treatment apoptosis in a differentiation-competent line (U937)only when administered after ara-C may have implica-of human myeloid leukemia cells with vitamin D3 [50]
and all-trans-retinoic acid [51] after ara-C exposure led tions for the rational design of combination regimensemploying the former compound in conjunction withto potentiation of DNA damage and cytotoxicity. More
recently, Bhatia et al. reported that induction of differ- cytotoxic agents in leukemia therapy.entiation in HL-60 cells by n-butyrate or retinoic acidfollowing exposure to DNA-damaging agents such as This work was supported by RO1 CA63753 and RO3 CA66990
from the NIH, and by the Bone Marrow Transplantation Researchcamptothecin and nitrogen mustard produced a dra-Laboratory of the Massey Cancer Center. A.J.F. is the recipient of amatic increase in apoptosis [32]. While the mechanismCancer Biology Fellowship Award supported by USPHS Trainingunderlying this phenomenon is unclear, it has been Grant CA09564.
postulated that DNA-damaging agents accentuate theDNA fragmentation that ordinarily occurs early in the
REFERENCEScourse of leukemic cell maturation [52, 53]. Conse-quently, this phenomenon may represent another in- 1. McConkey, D. J., and Orrenius, S. (1994) Trends Cell Biol. 4,stance in which cells prevented from completing a nor- 370–375.mal differentiation program are forced to proceed along 2. Hug, H., and Saue, T. (1993) Biochem. J. 291, 329–343.an alternative, apoptotic pathway. Such a model pro- 3. Lotem, J., Cragoe, E., and Sachs, L. (1991) Blood 78, 953–960.vides a framework for understanding cell line- and se- 4. Bertrand, R., Solary, E., O’Connor, P., Kohn, K. W., and Pom-
mier, Y. (1994) Exp. Cell. Res. 211, 314–321.quence-dependent effects of bryostatin 1 and ara-C. For5. Jarvis, W. D., Turner, A. J., Povirk, L. F., Traylor, R. S., andexample, in this and in a previous communication [33],
Grant, S. (1994) Cancer Res. 54, 1707–1714.we observed that exposure of HL-60 cells to bryostatin6. Solary, E., Bertrand, R., Kohn, K., and Pommier, Y. (1993)1 following ara-C did not modify apoptosis or cytotoxic-
Blood 81, 1359–1368.ity, presumably reflecting bryostatin 1’s inability to7. Martin, S. J., Bradley, J. G., and Cotter, T. G. (1990) Clin. Exp.trigger a differentiation program in this line. Con-
Immunol. 79, 448–453.versely, pretreatment of these cells with bryostatin 18. Solary, E., Bertrand, R., and Pommier, Y. (1994) Leukemia 8,may, by disrupting normal differentiation events, facil- 792–797.
itate activation of an alternative, apoptotic pathway. 9. Lotem, J., and Sachs, L. (1993) Cell Growth Differ. 4, 41–47.In U937 cells, which are partially differentiation re- 10. Del Bino, G., Li, X., Traganos, F., and Darzynkiewicz, Z. (1994)sponsive, bryostatin 1 pretreatment would be expected Leukemia 8, 281–282.to reduce cellular susceptibility to drug-mediated 11. Nishikawa, M., and Shirakawa, S. (1992) Leuk. Lymphoma 8,apoptosis, as observed elsewhere [6, 40, 41]. In con- 201–211.trast, the sequence ara-C followed by differentiation 12. Jayson, G. C., Crowther, D., Prendiville, J., McGown, A. T.,
Scheid, C., Stern, P., Young, R., Brenchley, P., Chang, J.,induction by bryostatin 1 in this cell line would be pre-Owens, S., and Pettit, G. R. (1995) Br. J. Cancer 72, 461–468.dicted to promote apoptosis, analogous to results ob-
13. Grant, S., Jarvis, D., Swerdlow, P., Turner, A., Traylor, R., Wal-tained in other systems [31, 32, 49–52]. Thus, the ca-lace, H., Lin, P-S., Pettit, G. R., and Gewirtz, D. A. (1992) Cancerpacity of bryostatin 1 to modulate ara-C-related Res. 52, 6270–6278.
apoptosis may be both sequence and cell line depen-14. Stone, R. M., Sariban, E., Pettit, G. R., and Kufe, D. W. (1988)
dent. Blood 72, 208–213.In summary, the results of the present study indicate 15. Kraft, A. S., Williams, F., Pettit, G. R., and Lilly, M. B. (1989)
that in human leukemia cells, a hierarchy exists in Cancer Res. 49, 1287–1293.which bryostatin 1’s ability to down-regulate PKC ac- 16. Kraft, A. S., Smith, J. B., and Berkow, R. L. (1986) Proc. Natl.
Acad. Sci. USA 83, 1334–1338.tivity and cPKC isoform expression occurs under condi-
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell

75EFFECTS OF BRYOSTATIN 1 IN MYELOID LEUKEMIA CELLS
17. Jarvis, W. D., Gewirtz, D. A., Povirk, L., Turner, A., Traylor, 35. Obeid, L. M., Okazaki, T., Karolak, L. A., and Hannun, Y. A.(1990) J. Biol. Chem. 265, 2370–2374.R., Pettit, G. R., and Grant, S. (1994) Biochem. Pharmacol. 47,
839–852. 36. Xu, H-M., Tepper, C. G., Jones, J. B., Fernandez, C. E., andStudzinski, G. P. (1993) Exp. Cell Res. 209, 367–374.18. Grant, S., Turner, A., Bartimole, T. M., Nelms, P., Joe, V. C.,
and Jarvis, W. D. (1994) Oncol. Res. 6, 87–99. 37. Jarvis, W. D., Fornari, F. A., Traylor, R. S., Martin, H. S.,Kramer, L. B., Erkulla, K., Bittman, R., and Grant, S. (1996)19. Komada, F., Nishikawa, M., Uemura, Y., Morita, K., Hidaka,J. Biol. Chem. 271, 8275–8284.H., and Shirakawa, S. (1991) Cancer Res. 51, 4271–4278.
38. Jarvis, W. D., Fornari, F. A., Kolesnick, R. N., Browning, J. L.,20. Makowske, M., Ballester, R., Cayre, Y., and Rosen, O. (1988)Gewirtz, D. A., and Grant, S. (1994) J. Biol. Chem. 269, 31685–J. Biol. Chem. 263, 3402–3410.31692.
21. Grant, S., Rao, A., Freemerman, A. J., Turner, A. J., Kornstein,39. Gorczyca, W., Gong, J., Ardelt, B., Traganos, F., and Darzynkie-M. J., Chelliah, J., and Jarvis, W. D. (1995) Mol. Cell. Differ.
wicz, Z. (1993) Cancer Res. 53, 3186–3192.3, 337–359.40. Del Bino, G., Li, X., Traganos, F., and Darzynkiewicz, Z. (1994)
22. Asiedu, C., Biggs, J., Lilly, M., and Kraft, A. S. (1995) Cancer Leukemia 8, 281–288.Res. 55, 3716–3720.
41. Selvakumaran, M., Reed, J. C., Liebermann, D., and Hoffman,23. Gallagher, R., Collins, S., Trujillo, J., McCredie, K., Ahearn, B. (1994) Blood 84, 1036–1042.
M., Tsai, S., Metzgar, R., Aulakh, G., Ting, R., Ruscetti, F., and 42. de Vente, J., Kiley, S., Garris, T., Bryant, W., Hooker, J., Pose-Gallo, R. (1979) Blood 54, 713–733. kany, K., Parker, P., Cook, P., Fletcher, D., and Ways, D. K.
24. Sundstrom, C., and Nillson, K. (1976) Int. J. Cancer 17, 565– (1995) Cell Growth Differ. 6, 371–382.577. 43. Hoffmann, B., and Liebermann, D. A. (1994) Oncogene 9, 1807–
25. Hass, R., Bartels, H., Topley, N., Hadam, M., Kohler, L., and 1812.Goppelt-Strube, M. (1989) Eur. J. Cell Biol. 48, 282–293. 44. Edashige, K., Sato, E. F., Akimaru, K., Kasai, M., and Utsumi,
K. (1992) Arch. Biochem. Biophys. 299, 200–205.26. Jarvis, D. W., Kolesnick, R. W., Fornari, F. A., Traylor, R. S.,Gewirtz, D. A., and Grant, S. (1994) Proc. Natl. Acad. Sci. USA 45. Aihara, H., Asaoka, Yoshida, K., and Nishizuka, Y. (1991) Proc.91, 73–77. Natl. Acad. Sci. USA 88, 11062–11066.
46. Szallasi, Z., Smith, C. B., Pettit, G. R., and Blumberg, P. M.27. Isakov, N., Galron, D., Mustelin, T., Pettit, G. R., and Altman,(1994) J. Biol. Chem. 269, 2118–2124.A. J. (1993) Immunology 150, 1195–1204.
47. Hocevar, B. A., and Fields, A. P. (1991) J. Biol. Chem. 266, 28–28. Kennedy, M. J., Prestigiacomo, L. J., Tyler, G., May, W. S., and33.Davidson, N. E. Cancer Res. 52, 1278–1283.
48. Szallasi, Z., Denning, M. F., Smith, C. B., Dlugosz, A. A., Yuspa,29. Basu, A., and Lazo, J. S. (1992) Cancer Res. 52, 3119–3124.S. H., Pettit, G. R., and Blumberg, P. M. (1994) Mol. Pharmacol.
30. Drexler, H. G., Gignac, S. M., Jones, R. A., Scott, C. S., Pettit, 46, 840–850.G. R., and Hoffbrand, A. V. (1989) Blood 74, 1747–1754.
49. Huang, Y., and Waxman, S. (1994) Mol. Cell. Differ. 2, 83–100.31. Studzinski, G. P., Bhandal, A. K., and Brelvi, Z. S. (1986) J. 50. Studzinski, G. P., Reddy, K. B., Hill, H. Z., and Bhandal, A. K.
Natl. Cancer Inst. 76, 641–648. (1991) Cancer Res. 51, 3451–3455.32. Bhatia, U., Traganos, F., and Darzynkiewicz, Z. (1995) Cell 51. Yang, G. S., Minden, M. D., and McCulloch, E. A. (1993) Leuke-
Growth Differ. 6, 937–944. mia 7, 1012–1019.33. Grant, S., McCrady, C., Boise, L., Westin, E., and Pettit, G. R. 52. McMahon, G., Alsina, J. T., and Levy, S. B. (1984) Proc. Natl.
(1991) Biochem. Pharmacol. 42, 853–867. Acad. Sci. USA 81, 7461–7463.53. Pommier, Y., and Colburn, N. (1992) Cancer Res. 52, 1907–34. McConkey, D. J., Hartzell, P., Jondal, M., and Orrenius, S.
(1989) J. Biol. Chem. 264, 13999–13402. 1915.
Received March 21, 1996Revised version received July 5, 1996
AID ECR 3294 / 6i14$$$$$3 09-13-96 08:18:32 eca AP: Exp Cell





























