Embed Size (px)
Citation preview

Nucleoside Modifications
Mahesh K. Lakshman and Fumi Nagatsugi
www.mdpi.com/journal/molecules
Edited by
Printed Edition of the Special Issue Published in Molecules
molecules

Nucleoside Modifications Special Issue Editors Mahesh K. Lakshman Fumi Nagatsugi

Guest Editors Mahesh K. Lakshman Fumi Nagatsugi The City College and Tohoku University The City University of New York Japan USA Editorial Office MDPI AG St. Alban-Anlage 66 Basel, Switzerland This edition is a reprint of the Special Issue published online in the open access journal Molecules (ISSN 1420-3049) from 2014–2016 (available at: http://www.mdpi.com/journal/molecules/special_issues/Nucleoside_Modifications). For citation purposes, cite each article independently as indicated on the article page online and as indicated below: Author 1; Author 2; Author 3 etc. Article title. Journal Name. Year. Article number/page range. ISBN 978-3-03842-354-6 (Pbk) ISBN 978-3-03842-355-3 (PDF)
Articles in this volume are Open Access and distributed under the Creative Commons Attribution license (CC BY), which allows users to download, copy and build upon published articles even for commercial purposes, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book taken as a whole is © 2017 MDPI, Basel, Switzerland, distributed under the terms and conditions of the Creative Commons by Attribution (CC BY-NC-ND) license (http://creativecommons.org/licenses/by-nc-nd/4.0/).

iii
Table of Contents About the Guest Editors ............................................................................................................................ v
Preface to “Nucleoside Modifications” ................................................................................................... vii
Yong Liang and Stanislaw F. Wnuk
Modification of Purine and Pyrimidine Nucleosides by Direct C-H Bond Activation Reprinted from: Molecules 2015, 20(3), 4874–4901; doi: 10.3390/molecules20034874 http://www.mdpi.com/1420-3049/20/3/4874 ........................................................................................... 1
Kevin H. Shaughnessy
Palladium-Catalyzed Modification of Unprotected Nucleosides, Nucleotides, and Oligonucleotides Reprinted from: Molecules 2015, 20(5), 9419–9454; doi: 10.3390/molecules20059419 http://www.mdpi.com/1420-3049/20/5/9419 ........................................................................................... 25
Oleg Golubev, Tuomas Lönnberg and Harri Lönnberg
Formation of Mixed-Ligand Complexes of Pd2+ with Nucleoside 5'-Monophosphates and Some Metal-Ion-Binding Nucleoside Surrogates Reprinted from: Molecules 2014, 19(10), 16976–16986; doi: 10.3390/molecules191016976 http://www.mdpi.com/1420-3049/19/10/16976 ....................................................................................... 58
Sarah C. Zimmermann, Elizaveta O'Neill, Godwin U. Ebiloma, Lynsey J. M. Wallace, Harry P. De Koning and Katherine L. Seley-Radtke
Design and Synthesis of a Series of Truncated Neplanocin Fleximers Reprinted from: Molecules 2014, 19(12), 21200–21214; doi: 10.3390/molecules191221200 http://www.mdpi.com/1420-3049/19/12/21200 ....................................................................................... 67
Yasufumi Fuchi, Hideto Obayashi and Shigeki Sasaki
Development of New 1,3-Diazaphenoxazine Derivatives (ThioG-Grasp) to Covalently Capture 8-Thioguanosine Reprinted from: Molecules 2015, 20(1), 1078–1087; doi: 10.3390/molecules20011078 http://www.mdpi.com/1420-3049/20/1/1078 ........................................................................................... 79
Akkaladevi Venkatesham, Dhuldeo Kachare, Guy Schepers, Jef Rozenski, Mathy Froeyen and Arthur Van Aerschot
Hybridisation Potential of 1',3'-Di-O-methylaltropyranoside Nucleic Acids Reprinted from: Molecules 2015, 20(3), 4020–4041; doi: 10.3390/molecules20034020 http://www.mdpi.com/1420-3049/20/3/4020 ........................................................................................... 87
Kiet Tran, Michelle R. Arkin and Peter A. Beal
Tethering in RNA: An RNA-Binding Fragment Discovery Tool Reprinted from: Molecules 2015, 20(3), 4148–4161; doi: 10.3390/molecules20034148 http://www.mdpi.com/1420-3049/20/3/4148 ........................................................................................... 105
Yuichi Yoshimura, Satoshi Kobayashi, Hitomi Kaneko, Takeshi Suzuki and Tomozumi Imamichi
Construction of an Isonucleoside on a 2,6-Dioxobicyclo[3.2.0]-heptane Skeleton Reprinted from: Molecules 2015, 20(3), 4623–4634; doi: 10.3390/molecules20034623 http://www.mdpi.com/1420-3049/20/3/4623 ........................................................................................... 117

iv
Takuya Akisawa, Yuki Ishizawa and Fumi Nagatsugi
Synthesis of Peptide Nucleic Acids Containing a Crosslinking Agent and Evaluation of Their Reactivities Reprinted from: Molecules 2015, 20(3), 4708–4719; doi: 10.3390/molecules20034708 http://www.mdpi.com/1420-3049/20/3/4708 ........................................................................................... 127
Salvatore V. Giofrè, Roberto Romeo, Caterina Carnovale, Raffaella Mancuso, Santa Cirmi, Michele Navarra, Adriana Garozzo and Maria A. Chiacchio
Synthesis and Biological Properties of 5-(1H-1,2,3-Triazol-4-yl)isoxazolidines: A New Class of C-Nucleosides Reprinted from: Molecules 2015, 20(4), 5260–5275; doi: 10.3390/molecules20045260 http://www.mdpi.com/1420-3049/20/4/5260 ........................................................................................... 138
Kaustav Chakraborty, Swagata Dasgupta and Tanmaya Pathak
Carboxylated Acyclonucleosides: Synthesis and RNase A Inhibition Reprinted from: Molecules 2015, 20(4), 5924–5941; doi: 10.3390/molecules20045924 http://www.mdpi.com/1420-3049/20/4/5924 ........................................................................................... 151
Jolanta Brzezinska and Wojciech T. Markiewicz
Non-Nucleosidic Analogues of Polyaminonucleosides and Their Influence on Thermodynamic Properties of Derived Oligonucleotides Reprinted from: Molecules 2015, 20(7), 12652–12669; doi: 10.3390/molecules200712652 http://www.mdpi.com/1420-3049/20/7/12652 ......................................................................................... 166
Sakilam Satishkumar, Prasanna K. Vuram, Siva Subrahmanyam Relangi, Venkateshwarlu Gurram, Hong Zhou, Robert J. Kreitman, Michelle M. Martínez Montemayor, Lijia Yang, Muralidharan Kaliyaperumal, Somesh Sharma, Narender Pottabathini and Mahesh K. Lakshman
Cladribine Analogues via O6-(Benzotriazolyl) Derivatives of Guanine Nucleosides Reprinted from: Molecules 2015, 20(10), 18437–18463; doi: 10.3390/molecules201018437 http://www.mdpi.com/1420-3049/20/10/18437 ....................................................................................... 181
Alicja Stachelska-Wierzchowska, Jacek Wierzchowski, Agnieszka Bzowska and Beata Wielgus-Kutrowska
Site-Selective Ribosylation of Fluorescent Nucleobase Analogs Using Purine-Nucleoside Phosphorylase as a Catalyst: Effects of Point Mutations Reprinted from: Molecules 2016, 21(1), 44; doi: 10.3390/molecules21010044 http://www.mdpi.com/1420-3049/21/1/44 ............................................................................................... 203

v
About the Guest Editors Mahesh Lakshman obtained the B.Sc. and M.Sc. degrees from the University of Bombay (Mumbai), and MS and Ph.D. degrees from The University of Oklahoma. He completed postdoctoral work at the National Institutes of Health (NIDDK) developing the first total chemical synthesis approaches to site-specific DNA modification with stereochemically-defined polycyclic aromatic hydrocarbon metabolite adducts. After serving for a short while in industry, he returned to academia, joining the University of North Dakota and then relocating to The City College of New York (The City University of New York system). In addition to an active research program, funded by both the National Science Foundation and the National Institutes of Health, he has held several administrative positions such as Executive Officer for The City University of New York Ph.D. Program in Chemistry and as a Vice Chair of the Department of Chemistry and Biochemistry at The City College of New York. Professor Lakshman was recently inducted as a Fellow of Royal Society of Chemistry (UK).
Fumi Nagatsugi joined the Faculty of Pharmaceutical Sciences at Kyushu University as a research assistant in 1989. She received her PhD from the university in 1996 and performed postdoctoral research at the National Institutes of Aging (NIA) in 2001–2002. She was promoted to associate professor at Kyushu University in 2003. She moved to Tohoku University in 2006 as a professor. Her research interests are chemical biology and nucleic acid chemistry.


vii
Preface to “Nucleoside Modifications” Nucleosides are the fundamental components of genetic material, and are present in all living
organisms, and in viruses. By virtue of their ubiquity, they are highly important biomolecules. Nucleosides consist of a heterocyclic aglycone and a sugar unit. For several decades, the natural nucleoside structures have inspired the development of chemical and biochemical modifications, leading to new nucleoside-like entities via aglycone as well as saccharide modifications. As a result, modified nucleosides and nucleoside analogues have widespread utilities in biochemistry, biology, as pharmaceutical agents and as biological probes. There is a constant need for access to novel nucleoside analogues for a plethora of applications, prompting the development of new methodologies.
This book contains twelve original research articles that include such diverse topics as metal-complexation with nucleoside analogues, synthesis of flexible nucleoside analogues (fleximers), development of a new “clamp” for thioguanosine via hydrogen-bond interactions, synthesis and properties of nucleic acids containing a pyranose sugar, studies on small molecule binding to RNA via a tethering technique, synthesis of an isonucleoside developed on a dioxabicycloheptane scaffold, development of a peptide-nucleic acid for DNA crosslinking, synthesis of C-nucleoside analogues developed on a triazole linked to an isoxazolidine, development of acyclonucleoside-based RNAse A inhibitors, synthesis and studies of oligonucleosides containing acyclic analogues of polyaminonucleosides, development of new methodology for synthesis and structure–activity studies on cladribine and its analogues, and enzymatic ribosylation of 8-azaguanine and 2,6-diamino-8-azapurine. The book also contains two reviews on contemporary methods for modification of nucleosides. One is on the modification of purine and pyrimidine nucleosides by C–H bond activation and the other describes palladium-catalyzed modifications of unprotected nucleosides, nucleotides, and oligonucleotides.
Mahesh K. Lakshman and Fumi Nagatsugi Guest Editors


molecules
Review
Modification of Purine and Pyrimidine Nucleosidesby Direct C-H Bond Activation
Yong Liang and Stanislaw F. Wnuk *
Department of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA;[email protected]* Correspondence: [email protected]; Tel.: +1-305-348-6195; Fax: +1-305-348-3772
Academic Editor: Mahesh LakshmanReceived: 15 February 2015; Accepted: 13 March 2015; Published: 17 March 2015
Abstract: Transition metal-catalyzed modifications of the activated heterocyclic bases of nucleosidesas well as DNA or RNA fragments employing traditional cross-coupling methods have beenwell-established in nucleic acid chemistry. This review covers advances in the area of cross-couplingreactions in which nucleosides are functionalized via direct activation of the C8-H bond in purine andthe C5-H or C6-H bond in uracil bases. The review focuses on Pd/Cu-catalyzed couplings betweenunactivated nucleoside bases with aryl halides. It also discusses cross-dehydrogenative arylationsand alkenylations as well as other reactions used for modification of nucleoside bases that avoid theuse of organometallic precursors and involve direct C-H bond activation in at least one substrate.The scope and efficiency of these coupling reactions along with some mechanistic considerationsare discussed.
Keywords: C-H activation; cross-coupling; direct arylation; nucleosides; purines; pyrimidines
1. Introduction
Transition metal catalyzed traditional cross-coupling reactions have contributed significantlyto the formation of new carbon-carbon bonds and to the synthesis of biaryl compounds. With fewexceptions, the traditional Pd-catalyzed coupling reactions require two activated substrates, one isthe organometallic, alkene (Heck reaction), or terminal alkyne (Sonogashira reaction) and the other isthe halide or triflate [1,2]. The most often used Stille, Suzuki, Negishi, Kumada and Hiyama reactionsneed an organometallic (Sn, B, Zn, Mg, and Si) component and a halide or pseudohalide. Owing tothe high impact of these reactions in organic synthesis, natural product synthesis and pharmaceuticalapplications, the 2010 Nobel Prize in Chemistry was awarded jointly to Richard F. Heck, Ei-ichi Negishiand Akira Suzuki [3]. Pd-catalyzed cross-coupling reactions are carried out under mild conditions andcan be performed in the presence of most functional groups. The mechanisms in most cases followthree major steps of: (i) oxidative addition, (ii) transmetallation, and (iii) and reductive elimination [1,2].
Transition metal-catalyzed cross-coupling reactions which are based on direct C-Hfunctionalization have been recently developed [4–9]. These methodologies, which eliminate the use oforganometallic substrates, compete with traditional Pd-catalyzed cross-couplings in the developmentof new strategies for the formation of carbon-carbon bonds. These reactions require only oneactivated substrate (C-H activation) and sometimes even no activation is required for either substrate(double C-H activation). They are atom efficient and avoid the synthesis of often unstable activatedsubstrates. Major challenges associated with C-H functionalization reactions include: (i) the need fordeveloping regioselective activation of specific C-H bonds in the presence of other C-H bonds; (ii) lowchemoselectivity which means it is necessary to protect sensitive functional groups before performingthe coupling; and (iii) the necessity to work at high temperature needed to activate C-H bonds with
Molecules 2015, 20, 4874–4901 1 www.mdpi.com/journal/molecules

Molecules 2015, 20, 4874–4901
intrinsic low activity, which often causes decomposition of the substrates. Pd and Cu are two of themost common transition metal catalyst used for the C-H functionalization.
Transition metal-catalyzed approaches towards the synthesis of base-modified nucleosides canbe divided into five major categories as depicted in Figure 1. The first two approaches are based oncross-couplings between two activated components. One involves reactions between metal-activatednucleoside bases and halides (Figure 1, Path a) while the second employs couplings between halo (ortriflate) modified nucleoside bases and organometallics (Path b). These approaches were extensivelyreviewed [10–12] and are not discussed in this account. The next two approaches are based oncross-couplings between only one activated component and require C-H activation at the secondsubstrate. One involves reactions between C-H activated bond in nucleoside bases and halides (Path c),while the second employs couplings between halo-modified nucleoside bases and arenes, which, inturn, require selective C-H activation (Path d). The last approach involves cross-couplings betweentwo inactivated substrates [cross-dehydrogenative coupling (CDC) reactions; Path e]. Direct C-Hfunctionalization approaches (Paths c-e) alleviate some drawbacks associated with the synthesis ofmodified nucleosides employed in traditional Pd-catalyzed cross-coupling reactions (Paths a-b). Theyalso avoid usage of the toxic organotin components, which are problematic during biological studies,or the sometimes unstable organoboronic substrates.
Figure 1. Transition metal catalyzed cross-coupling approaches towards the synthesis of base-modified nucleosides.
Numerous C5 or C6 modified pyrimidine nucleosides and C2 or C8 modified purine nucleosideshave been synthesized in last 40 years employing the transition-metal assisted cross-couplingreactions [10]. Some of them show potent biological activity and/or are utilized as mechanistic orlabelling probes (Figure 2). For example, the (E)-5-(2-bromovinyl)-2′-deoxyuridine (1, BVDU) has beenfound to be a highly potent and selective anti-herpes agent [13]. The bicyclic furanopyrimidine-2-onenucleoside analogues bearing an aryl side chain 2 display remarkable antiviral potency against theVaricella-Zoster virus [14]. The 5-thienyl- 3 or 5-furyluridine 4 were used as molecular beaconsfor oligonucleotide labeling [15–18]. The 8-pyrenyl-2'-deoxyguanosine 5 serves as a probe forthe spectroscopic study of the reductive electron transfer through DNA [19,20]. Furthermore,
2

Molecules 2015, 20, 4874–4901
the 8-vinyl and 8-ethynyladenosines 6 show cytotoxic activity against tumor cell lines [21], whileoligodeoxynucleotides modified with the 8-alkynyl-dG possess thrombin inhibitory activity [22].
Figure 2. Selected base-modified pyrimidine and purine nucleosides.
2. Direct Activation of C8-H Bond in Purine and Purine Nucleosides
2.1. Cross-Coupling of Adenine Nucleosides with Aryl Halides
Hocek and coworkers reported the first example of direct arylation of adenosine 7 with arylhalides by selective activation of the C8-H bond which gave access to 8-arylated adenosine analogues 9.The cross-coupling occurred in the presence of a stoichiometric amount of CuI (3 equiv.) and a catalyticload of Pd(OAc)2 (5 mol %) in DMF at elevated temperature (100 ◦C/22 h or 150 ◦C/5 h) to produce 9
in 50%–68% yields (Scheme 1, Table 1 entry 1) [23]. The authors were able to improve the couplingconditions (e.g., shortening reaction time and lowering the reaction temperature), as compared to theirearlier work on C8-H arylation of purines and adenines [24,25] (vide infra), by addition of piperidine tothe reaction mixture. They assumed [23] that formation of dimethylamine, as a side product of theprolonged heating of the DMF solvent during the C8-H arylation of purines, favorable influenced therate of the arylation reaction, which is consistent with Fairlamb’s findings [26,27]. Consequently, theyfound that the addition of higher boiling secondary amine such as piperidine (4 equiv.) was beneficialto the coupling reactions. Couplings of 7 with aryl iodines also produced N6,8-diarylated byproducts11 in 12%–18% yield, whereas only 8-arylated products 9 were isolated when less reactive aryl brominewere employed.
Scheme 1. Pd-catalyzed direct C8-H arylation of adenosine 7 and 2'-deoxyadenosine 8 with aryl halides.
When 2'-deoxyadenosine 8 was subjected to this direct arylation protocol desired 8-arylatedproducts 10 were produced only after the temperature was lowered to 125 ◦C (31% after 5 h; entry 2).It is worth noting that this protocol was applicable to unprotected nucleosides and allowed for the firsttime the single-step introduction of the aryl group at the C8 position without the need to (i) halogenatenucleoside substrates, or (ii) use expensive arylboronic acids or toxic arylstannanes [10].
3
Molecules 2015, 20, 4874–4901
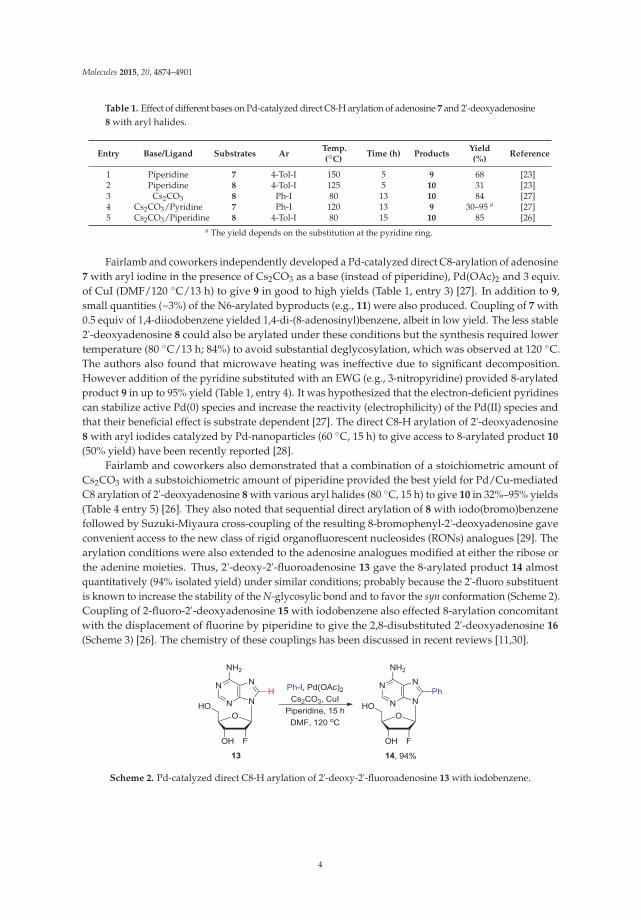
Table 1. Effect of different bases on Pd-catalyzed direct C8-H arylation of adenosine 7 and 2'-deoxyadenosine8 with aryl halides.
Entry Base/Ligand Substrates ArTemp.(◦C)
Time (h) ProductsYield(%)
Reference
1 Piperidine 7 4-Tol-I 150 5 9 68 [23]2 Piperidine 8 4-Tol-I 125 5 10 31 [23]3 Cs2CO3 8 Ph-I 80 13 10 84 [27]4 Cs2CO3/Pyridine 7 Ph-I 120 13 9 30–95 a [27]5 Cs2CO3/Piperidine 8 4-Tol-I 80 15 10 85 [26]
a The yield depends on the substitution at the pyridine ring.
Fairlamb and coworkers independently developed a Pd-catalyzed direct C8-arylation of adenosine7 with aryl iodine in the presence of Cs2CO3 as a base (instead of piperidine), Pd(OAc)2 and 3 equiv.of CuI (DMF/120 ◦C/13 h) to give 9 in good to high yields (Table 1, entry 3) [27]. In addition to 9,small quantities (~3%) of the N6-arylated byproducts (e.g., 11) were also produced. Coupling of 7 with0.5 equiv of 1,4-diiodobenzene yielded 1,4-di-(8-adenosinyl)benzene, albeit in low yield. The less stable2'-deoxyadenosine 8 could also be arylated under these conditions but the synthesis required lowertemperature (80 ◦C/13 h; 84%) to avoid substantial deglycosylation, which was observed at 120 ◦C.The authors also found that microwave heating was ineffective due to significant decomposition.However addition of the pyridine substituted with an EWG (e.g., 3-nitropyridine) provided 8-arylatedproduct 9 in up to 95% yield (Table 1, entry 4). It was hypothesized that the electron-deficient pyridinescan stabilize active Pd(0) species and increase the reactivity (electrophilicity) of the Pd(II) species andthat their beneficial effect is substrate dependent [27]. The direct C8-H arylation of 2'-deoxyadenosine8 with aryl iodides catalyzed by Pd-nanoparticles (60 ◦C, 15 h) to give access to 8-arylated product 10
(50% yield) have been recently reported [28].Fairlamb and coworkers also demonstrated that a combination of a stoichiometric amount of
Cs2CO3 with a substoichiometric amount of piperidine provided the best yield for Pd/Cu-mediatedC8 arylation of 2'-deoxyadenosine 8 with various aryl halides (80 ◦C, 15 h) to give 10 in 32%–95% yields(Table 4 entry 5) [26]. They also noted that sequential direct arylation of 8 with iodo(bromo)benzenefollowed by Suzuki-Miyaura cross-coupling of the resulting 8-bromophenyl-2'-deoxyadenosine gaveconvenient access to the new class of rigid organofluorescent nucleosides (RONs) analogues [29]. Thearylation conditions were also extended to the adenosine analogues modified at either the ribose orthe adenine moieties. Thus, 2'-deoxy-2'-fluoroadenosine 13 gave the 8-arylated product 14 almostquantitatively (94% isolated yield) under similar conditions; probably because the 2'-fluoro substituentis known to increase the stability of the N-glycosylic bond and to favor the syn conformation (Scheme 2).Coupling of 2-fluoro-2'-deoxyadenosine 15 with iodobenzene also effected 8-arylation concomitantwith the displacement of fluorine by piperidine to give the 2,8-disubstituted 2'-deoxyadenosine 16
(Scheme 3) [26]. The chemistry of these couplings has been discussed in recent reviews [11,30].
Scheme 2. Pd-catalyzed direct C8-H arylation of 2'-deoxy-2'-fluoroadenosine 13 with iodobenzene.
4

Molecules 2015, 20, 4874–4901
Scheme 3. Pd-catalyzed direct arylation of 2-fluoro-2'-deoxyadenosine 15 with iodobenzene.
These Pd-catalyzed/Cu-mediated methodologies were successfully applied to the synthesis ofnumerous 8-arylated purines and adenines. In 2006, Hocek and coworkers elaborated the originalprotocol for the efficient direct C8-H arylation of 6-phenylpurine analogues 17 using aryl iodides in thepresence of Cs2CO3 and CuI to give 18 (Scheme 4) [24]. This route required prolonged heating at hightemperature (160 ◦C/60 h) in DMF. Furthermore, it was essential to perform the coupling with strictexclusion of air to avoid formation of two byproducts, sometimes, in substantial yields (6%–54%). Onebyproduct was 19, which was formed by double arylation at C8 position and the ortho position of thephenyl ring at C6. The other byproduct was the 8,8'-bispurine dimer. Various 6,8,9-trisubstituted and2,6,8,9-tetrasubstituted purine analogues were synthesized using this approach in combination withSuzuki cross-coupling reaction and Cu-catalyzed N-arylation at 9 position [24,25]. These conditions(Cs2CO3 or piperidine) were successfully employed for the synthesis of 8,9-disubstituted adenines but6-N-(di)arylated byproducts were observed [24,25]. This protocol was also applied for direct C8-Harylation of adenines anchored to solid phase via 6-N amino group (in the presence of piperidine asbase) [31].
Scheme 4. Pd/Cu-mediated direct C8-H arylation of 6-phenylpurines with aryl halides.
Alami and coworkers developed a microwave-assisted direct C8 arylation of free-(NH2)9-N-protected adenine 20 with aryl halides catalyzed by Pd(OH)2/C (Pearlman's catalyst) in thepresence of CuI (Scheme 5) [32]. Reaction took only 15 min at 160 ◦C in NMP solvent when Cs2CO3
was used as base to give 21 in up to 90% yield. The application of Pd(OH)2/C catalyst allowed couplingwith aryl bromides and even less reactive aryl chlorides [33]. Sequential combination of C8-arylationwith ArCl and 6-N-arylation with ArBr or ArI (using Xantphos [34] instead of CuI) provides access todisubstituted adenines 22 [33].
Scheme 5. Microwave-assisted direct C8-H arylation of 9-N-benzyladenine with aryl halides.
5

Molecules 2015, 20, 4874–4901
Fairlamb and coworkers reported a detailed mechanism for the direct C8-arylation of adeninering with aryl halides mediated by Pd and Cu in the presence of Cs2CO3 [26,27]. The authors notedthat the use of a stoichiometric amount of Cu(I) is key to the direct arylation of the adenine ring andthat the process parallels the arylation of imidazole ring at the 2 position [35]. As depicted in Scheme 6,Cu(I) was proposed to assist the C-H functionalization process by an initial coordination to the adenineN7 atom. The subsequent base-assisted deprotonation leads to the formation of 8-cuprioadenineintermediate A or N-heterocyclic carbene like cuprates, which can then undergo a standard Pd(0)catalytic cycle for cross-coupling with aryl halides. This process resembles Sonogashira's reactionbetween alkynylcuprates and halides [26,27]. The requirement for excess of CuI was attributed to thehigh binding affinity of Cu(I) for both the substrate and presumably the 8-arylated product(s). Thedinucleoside copper(I) complex between 7-N and 6-NH atoms of the adenine have been identified asimportant intermediate [26].
Scheme 6. Proposed mechanism for the direct arylation at C8 position of adenosine [26,27].
2.2. Cross-Coupling of Inosine and Guanine Nucleosides with Aryl Halides
Guanosine 23 was found to be a poor substrate for the direct C8 arylation as indicated by thelow yield (15%) of 8-phenylated product 25 under the conditions (120 ◦C) which were effectivefor adenosine analogues (Scheme 7) [26]. Analogous arylation of 2'-deoxyguanosine 24 at lowertemperature (80 ◦C) yielded product 26 but only in 6% yield. The authors hypothesized that in thecase of guanine substrates, CuI-coordination most probably occurs at sites distal to C8 hamperingefficient arylation. A similar inhibitory effect, associated with the ionizable protons in the guaninemoiety was observed during the Suzuki couplings with 8-haloguanine nucleosides [36]. The authorsalso suggested that guanine-type nucleosides are poor substrates for direct C8-H arylation due to thelack of the "templating" role of exocyclic 6-amino group present in adenine nucleosides.
Scheme 7. Direct C8-H arylation of guanosine 23 and 2'-deoxyguanosine 24 with iodobenzene.
The Pd-catalyzed/Cu-mediated direct C8-H arylation of inosine 27 proceeded proficiently toafford 8-phenylated product 29 in good yield (60%) at 120 ◦C (Scheme 8) [26]. The analogous
6

Molecules 2015, 20, 4874–4901
functionalization of 2'-deoxyinosine 28, due to the stability of the glycosylic bond, had to be carriedout at lower temperature to give product 30 but in only 19% yield.
Scheme 8. Direct C8-H arylation of inosine 27 and 2'-deoxyinosine 28 with iodobenzene.
Recently, Pérez and coworkers synthesized the 8-arylated inosine analogues via amicrowave-assisted Pd/Cu-catalyzed direct C8-H arylation [37]. In order to increase the solubility ofthe nucleoside substrate, 2',3'-O-isopropylideneinosine 29 was employed to couple with iodopyridinesor aryl iodides, by adopting Fairlamb’s protocol [26,29], to produce 30 in only 1 h at 120 ◦C (Scheme 9).
Scheme 9. Microwave-assisted direct C8-H arylation of inosine 29 with aryl iodides.
2.3. Synthesis of Fused Purines via Inter- or Intramolecular Direct C8-H Arylation
Hocek and coworkers developed a direct C8-H arylation of 9-N-phenylpurine 31 for thesynthesis of fused purine analogues of type 32 with e-fusion (position 8 and 9 of purine ring).Thus, Pd-catalyzed intermolecular double direct C-H arylation of 6-methyl-9-N-phenylpurine 31
with 1,2-diiodobenzene gave 32 (R = CH3) in modest yield (35%). Alternatively, the sequential Suzukicoupling of 9-(2-bromophenyl)adenine 33 (R = NH2) with 2-bromophenylboronic acid 34 followed byintramolecular C8-H arylation also gave the desired product 32 (R = NH2) in moderate to high yieldswhich preserves the base-pairing and major groove facets of the intact adenine ring (Scheme 10) [38].However, attempted intramolecular oxidative coupling of 8,9-diphenyladenine failed to give 32.
Scheme 10. Pd-catalyzed cyclization of 9-N-arylpurines via C8-H activation.
7

Molecules 2015, 20, 4874–4901
The purines 37 with five or six-membered e-fused rings were also synthesized by intramolecularcyclizations of 9-(2-chlorophenylalkyl)purines 35 (n = 1 or 2; X = Cl) employing conditions developedby Fagnou [39] for direct arylation with aryl halides [Pd(OAc)2/tricyclohexylphoshine/K2CO3 inDMF] (Scheme 11) [38,40]. Domínguez and coworks reported the synthesis of five-membered ringanalogue 36 by Cu-catalyzed direct C8-H arylation of 9-(2-iodophenylmethyl)purines 35 (n = 1, X = I)in 58% yield [41].
Scheme 11. Pd/Cu-catalyzed intramolecular cyclization of 9-N-substituted purines via C8-H activation.
The five-, six- or seven-membered e-fused purines 39 have been prepared by the intramoleculardouble C-H activation (at C8 and ortho position of the phenyl ring) of 9-N-phenylalkylpurines 38 inthe presence of Pd catalyst and silver salt oxidant in high to excellent yields (Scheme 12) [42]. Theseven-, eight-, or nine-membered e-fused purines of type 40 were prepared by the one-pot coupling of38 with iodobenzene [42]. The reaction sequence was believed to be initiated by direct intermolecularC8-H arylation of 38 with iodobenzene followed the intramolecular cross-dehydrogenative-arylationbetween two phenyl rings to give product 40.
Scheme 12. Pd-catalyzed intramolecular cross-dehydrogenative arylation of 9-N-substituted purinesvia C8-H bond activation.
2.4. Miscellaneous Direct C8-H Functionalizations
You and co-workers reported intermolecular Pd/Cu-catalyzed regioselective C8-H cross-couplingof 1,3-diethyl xanthine 41 (R1 = R2 = Et, R3 = H) with electron-rich furans 42a and thiophenes 42b [43].For example coupling of 41 with 2-methylthiophene in the presence of catalytic amount of the coppersalt gave diheteroarene product of type 43 in 96%, indicating the tolerance of the free NH group atthe 9 position to the reaction conditions (Scheme 13). The differences in the electron density of twoheteroarene components was believed to facilitate the reactivity and selectivity in the two metalationsteps [44] of the catalytic cycle of this cross-dehydrogenative arylation reaction.
Scheme 13. Pd/Cu-catalyzed cross-dehydrogenative arylation of purines with heteroarenes.
8

Molecules 2015, 20, 4874–4901
The 8-alkenyl adenine analogues 45 have been synthesized via microwave-assisted direct C8-Halkenylation of 9-N-benzyladenines 20 with alkenyl bromides 44 (Scheme 14) [32]. AnalogousPd/Cu-mediated C8 alkenylations of 6-(benzylthio)-9-N-benzylpurines with styryl bromides providedaccess to 6,8,9-trisubstituted purines [45]. The optimized conditions (Pd/CuI/tBuOLi) were applicablefor the selective alkenylation of caffeine, benzimidazole and other aromatic azole heterocycles [45,46].These are significant developments since it was reported that 8-bromoadenosine was not a goodsubstrate for Mizoroki-Heck reaction [47] making modification at 8 position via direct functionalizationof C8-H bond a desirable transformation.
Scheme 14. Pd-catalyzed direct C8-H alkenylation of 9-N-benzyladenine with alkenyl halides.
Modification of biologically important 7-deazapurines by direct C-H activation have alsobeen explored (Scheme 15). Thus, regioselective Pd-catalyzed direct C8-H arylation of the6-phenyl-7-deazapurine analogue 46 (R1 = Bn) with aryl halides gave corresponding 8-arylatedproducts 46a albeit in low to moderate yields (0%–41%) [48]. Alternatively, Ir-catalyzed C-H borylationof 46 (R = Ph, R1 = Bn) followed by Suzuki coupling with aryl halides afforded 46a in high yields(79%–95%). Interestingly, Ir-catalyzed C-H borylation was not successful with purines suggestingthat the complexation of Ir catalyst to N7 nitrogen might be responsible for the lack of reactivity [48].The regioselective Pd/Cu-catalyzed direct C8-H amination of the 6-phenyl-7-deazapurine analogue46 (R1 = Bn) with N-chloro-N-sulfonamides provided the 8-amino-7-deazapurine analogues 46b [49].However, subjection of the 6-chloro-7-deazapurine 46 (R1 = Bn) to the similar coupling conditionsproduced a complex mixture. Remarkably, application of conditions, developed by Suna andco-workers for direct C5-H amination of uracils (see Scheme 33), to the same substrate 46 (R = Cl,R1 = Bn) provided 7-amino-7-deazapurine analogue 46c in 60% [50]. Cu-catalyzed direct C-Hsulfenylation of 6-substituted-7-deazapurines 46 (R1 = H) with aryl or alkyl disulfides provided7-aryl(or alkyl)sulfanyl products 46d (47%–96%) in addition to minor quantities of 7,8-bis(sulfanyl)byproducts [51].
Scheme 15. Transition-metal catalyzed direct C-H activation of 7-deazapurines.
9

Molecules 2015, 20, 4874–4901
3. N1-Directed Modifications of C6-Substituted Purine Nucleosides via ortho C-HBond Activation
The Cu-catalyzed direct C-H activation/intramolecular amination reaction of the 2',3',5'-tri-O-acetyl-6-N-aryladenosines 47 were employed for the synthesis of fluorescent polycyclic purineand purine nucleosides of type 48 (Scheme 16) [52]. It was found that addition of Ac2Osignificantly improved the reaction rate (2 h at 80 ◦C) when Cu(OTf)2 (5 mol %) was used as coppersource and PhI(OAc)2 was used as the oxidant. The 6-N-aryladenosines substrates 47 containingelectron-withdrawing groups in the benzene ring gave better yields (~85%–92%) than those bearingelectron donating groups (~45%–62%). The proposed catalytic cycle involves initial coordinationof Cu(OTf)2 to the 6-NH····N1 tautomer of substrate 47 at N1 position followed by electrophilicsubstitution yielding Cu(II) intermediate bridging N1 position of the purine ring and ortho position inthe aryl ring. Subsequent reductive elimination then provides the fused product 48.
Scheme 16. Cu-catalyzed intramolecular direct ortho C-H activation/amination of 6-N-aryladenosines.
Qu and coworkers developed a Pd-catalyzed strategy for the regioselective ortho monophenylationof 6-arylpurine nucleosides (e.g., 49) via (N1 purine nitrogen atom)-directed C−H activation using alarge excess (30 equiv.) of iodobenzene (Scheme 17) [53]. It was essential to perform the reaction underan inert N2 atmosphere at 120 ◦C in the presence of AcOH in order to synthesize 50 in excellent yield(85%). It was believed that AcOH might help (i) to overcome the poisoning of the catalyst attributedto the multiple nitrogens present in purine ring, and (ii) to facilitate the reductive elimination step.However, the phenylation reaction was not successful when PhI was replaced with PhBr or PhCl.
It is noteworthy that the conditions described by Qu and coworkers (Pd(OAc)2/AgOAc/AcOH)are selective for the arylation at the ortho site of the C6-phenyl ring, while the CuI-catalyzed coupling of49 with aryl halides in the presence of Pd(OAc)2/piperidine produced exclusively C8-arylated product51 [23], or a mixture of double arylated products 50/51 in addition to 8,8'-dimers when analogouspurine substrates 49 and Pd(OAc)2/Cs2CO3 were used [24,25].
Scheme 17. Direct C-H arylation at the ortho position in 6-arylpurine nucleosides vs. C8-H arylation.
10

Molecules 2015, 20, 4874–4901
Lakshman and coworkers reported direct arylation of 6-arylpurine nucleosides of type 52 by theRu-catalyzed C-H bond activation (Scheme 18) [54]. The coupling usually required only 2 equiv. ofaryl halides and K2CO3 or Cs2CO3 as a base to give mixture of the mono and diarylated ortho products53 and 54 in ratio of approximately of 2–7 to 1 with no 8-arylated byproducts detected. It is noteworthythat these conditions were applicable to acid-sensitive substrates such as 2'-deoxynucleosides asopposed to the previously described Pd-catalyzed arylation promoted by AcOH [53]. Both aryl iodidesand bromides gave arylated products in good yield, but aryl iodides proceeded with higher yieldsand better product ratio. Also, aryl halides bearing EWG gave higher yields compared to the onesbearing EDG.
Scheme 18. Ru-Catalyzed N1-directed ortho C-H arylation of 6-phenylpurine nucleosides.
A possible mechanism for this direct ortho-arylation was proposed involving purinyl N1-directedelectrophilic attack by the aryl/RuIV complex A on the ortho position of the C6-aryl ring atom of thesubstrate 52 (Scheme 19) [54]. Subsequent reductive-elimination of the five-membered Ru complex B
gave product 53. The 2-amino-6-arylpurine nucleosides were unreactive, indicating that the presenceof C2-amino group is critical and often inhibits the reactivity of purine nucleoside towards C-Hactivation reactions.
Scheme 19. Proposed mechanism for the Ru-catalyzed N1-directed C-H bond activation of 6-phenylpurinenucleoside [54].
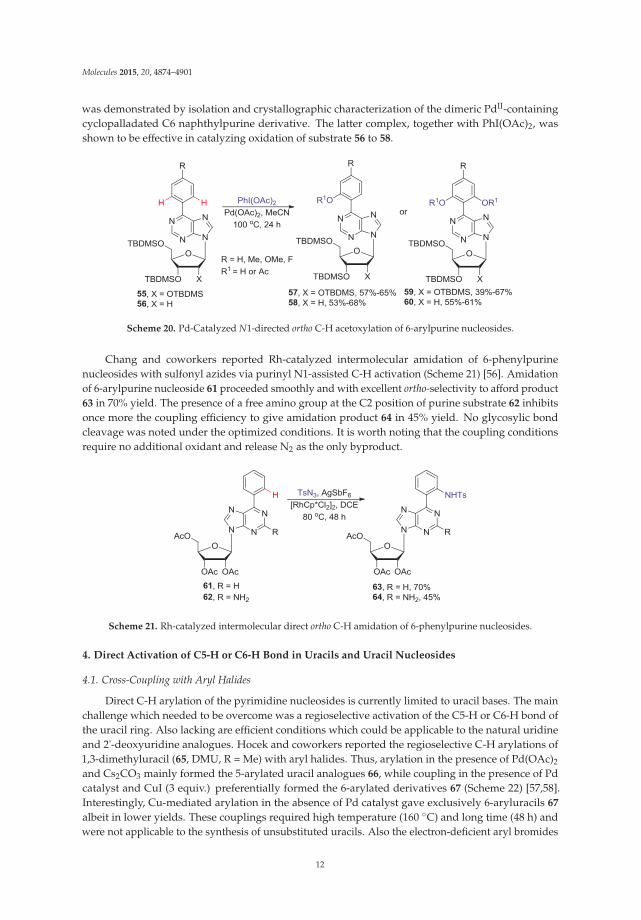
The Pd-catalyzed C-H bond activation and oxidation of the silyl protected C6-aryl ribonucleosidesof type 55, as well as the 2'-deoxy counterparts (e.g., 56), in the presence of PhI(OAc)2 as astoichiometric oxidant in MeCN provided access to monoacetoxylated products 57 or 58 in goodyields (Scheme 20) [55]. Increasing the loading of PhI(OAc)2 to 3 equiv gave mainly the diacetoxylatedproducts 59 or 60. The involvement of the N1-purinyl atom in this N-directed C-H bond activation
11
Molecules 2015, 20, 4874–4901
was demonstrated by isolation and crystallographic characterization of the dimeric PdII-containingcyclopalladated C6 naphthylpurine derivative. The latter complex, together with PhI(OAc)2, wasshown to be effective in catalyzing oxidation of substrate 56 to 58.
Scheme 20. Pd-Catalyzed N1-directed ortho C-H acetoxylation of 6-arylpurine nucleosides.
Chang and coworkers reported Rh-catalyzed intermolecular amidation of 6-phenylpurinenucleosides with sulfonyl azides via purinyl N1-assisted C-H activation (Scheme 21) [56]. Amidationof 6-arylpurine nucleoside 61 proceeded smoothly and with excellent ortho-selectivity to afford product63 in 70% yield. The presence of a free amino group at the C2 position of purine substrate 62 inhibitsonce more the coupling efficiency to give amidation product 64 in 45% yield. No glycosylic bondcleavage was noted under the optimized conditions. It is worth noting that the coupling conditionsrequire no additional oxidant and release N2 as the only byproduct.
Scheme 21. Rh-catalyzed intermolecular direct ortho C-H amidation of 6-phenylpurine nucleosides.
4. Direct Activation of C5-H or C6-H Bond in Uracils and Uracil Nucleosides
4.1. Cross-Coupling with Aryl Halides
Direct C-H arylation of the pyrimidine nucleosides is currently limited to uracil bases. The mainchallenge which needed to be overcome was a regioselective activation of the C5-H or C6-H bond ofthe uracil ring. Also lacking are efficient conditions which could be applicable to the natural uridineand 2'-deoxyuridine analogues. Hocek and coworkers reported the regioselective C-H arylations of1,3-dimethyluracil (65, DMU, R = Me) with aryl halides. Thus, arylation in the presence of Pd(OAc)2
and Cs2CO3 mainly formed the 5-arylated uracil analogues 66, while coupling in the presence of Pdcatalyst and CuI (3 equiv.) preferentially formed the 6-arylated derivatives 67 (Scheme 22) [57,58].Interestingly, Cu-mediated arylation in the absence of Pd catalyst gave exclusively 6-aryluracils 67
albeit in lower yields. These couplings required high temperature (160 ◦C) and long time (48 h) andwere not applicable to the synthesis of unsubstituted uracils. Also the electron-deficient aryl bromides
12

Molecules 2015, 20, 4874–4901
were poor substrates. In order to prepare the unprotected uracil derivatives, these protocols were alsoapplied to N-benzyl protected uracil derivatives. Subsequent debenzylation (e.g., 67, R = Bn) affordedefficiently 5- or 6-aryled uracil derivatives [57,58]. The experimental results indicated that differentmechanisms are involved in these diverse arylation reactions [57,58].
Scheme 22. Catalyst-controlled direct C5-H or C6-H arylation of uracil analogues with aryl halides.
Kim and coworkers reported the direct arylation of 1,3-dimetyluracil 65 with aryl bromides(including electron-deficient ones) in the presence of Pd(OAc)2, K2CO3 and PivOH (130 ◦C/12 h/DMFto give predominantly C5 arylated products 66 in up to 79% yield. A small amount of 6-arylatedisomers 67 were also observed [59]. However, application of this condition to 1-(tetrahydrofuran-2-yl)-3-benzyluracil 68 (a substrate having a glycosylic bond) resulted in severe decomposition.Nonetheless lowering the temperature to 100 ◦C (12 h) allowed the isolation of the 5-phenyluracilanalogue 69 as the sole product in 55% yield (Scheme 23) [59]. The intramolecular C6-H arylation ofthe uracil derivatives bearing a Morita-Baylis-Hillman adduct at the N1 position in the presence ofTBAB and Pd(OAc)2 provided a convenient access to the azepine scaffold [60].
Scheme 23. Pd-catalyzed direct C5-H arylation of 1-(tetrahydrofuran-2-yl)-3-benzyluracil.
Chien and coworkers further explored the base-dependence of direct activation of uracil C6-Hin the presence of Cu catalysts for the synthesis of 6-aryl uracil and 2-aryl-4-pyridone derivatives.The authors found that the CuBr-mediated arylation of DMU 65 (R = H) with aryl iodides (2 equiv.)in the presence of t-BuOLi in DMF at reflux gave the C6-arylated derivatives 71 (R1 = H) [61]. The5-substituted DMU analogues 70 (R1 = Me) afforded the 5,6-disubstituted derivatives 71 (R1 = Me) butin lower yields (Scheme 24). Both N1 and N3 positions in the uracil ring have to be protected for thereactions to proceed.
Scheme 24. Cu-mediated direct C6-H arylation of 1,3-disubstituteduracil 70 with aryl iodides.
Based on the literature reports, the mechanism for the direct arylation of uracil analogues (e.g.,DMU, 65) with aryl halides at either the C5 or C6 position in the presence of Pd or Cu/(Pd) are
13

Molecules 2015, 20, 4874–4901
summarized in Scheme 24. The coupling with only Pd(OAc)2 was proposed to proceed via anelectrophilic metalation-deprotonation (EMD) mechanism [5,9] (path a, complex A), and thus followthe regioselectivity of substitution at the C5 position to give 66 [59]. The formation of the C5 arylatedproducts was also suggested [57] to proceed via the concerted metalation-deprotonation (CMD)mechanism [9,62] (complex B). On the other hand, direct C-H arylation in the presence of CuI or incombination with Pd(OAc)2 was hypothesized to proceed via an Ullmann-type mechanism [27,63],which involves the formation of a carbanion by the abstraction of the most acidic C6-H [64–66] inthe uracil ring (e.g., 65) with base (path b). Subsequent cupration, transmetallation and reductiveelimination leading to the formation of C6 arylated uracil 67 [57,61]. Alternatively, addition of thecopper center of the arylcopper(III) complex C to the more nucleophilic C5-position in 65 followedby either base-promoted anti-elimination or Heck-type syn β-hydrogen elimination has been alsoproposed for C6-arylation [61]. Moreover, Heck-type carbopalladation of the 5,6-double bond of 65
with ArPdX species has been also considered for the arylation of uracil ring with aryl halides [59].
Scheme 25. Proposed mechanisms for the regioselective direct arylation of DMU [59,61].
4.2. Cross-Dehydrogenative Coupling with Arenes and Heteroarenes at C5 Position
Cross-couplings which required double C-H activation both at uracil and arene substrates havealso been developed. Thus, Do and Daugulis reported a highly regioselective CuI/phenantroline-catalyzed oxidative direct arylation of DMU 65 with arenes (e.g., 3-methylanisole 72) to giveC6-arylated uracil 73 as a sole product in 61% yield (130 ◦C, 48 h) (Scheme 26) [67]. The protocol usediodine as a terminal oxidant and required only a small excess of the arene (1.5–3.0 equiv.). This overallcross-dehydrogenative arylation was believed to proceed by in situ iodination of one of the couplingcomponents (e.g. 3-methylanisole 72 to give 4-iodo-3-methylanisole 72') followed by Cu-catalyzeddirect arylation at the most acidic bond in the DMU substrate (C6-H). Recently, an efficient (~70%–80%)and regioselective C-6 arylation of DMU 65 with arylboronic acids in the presence of Pd(OAc)2 andligand (1,10-phenanthroline) at 90 ◦C for 16 h has been also developed [68]. The coupling failed,however, when unsubstituted uracil or 2',3',5'-tri-O-acetyl-3-N-methyluridine was used as substrate orwhen the heteroaromatic boronic acids were employed [68].
Scheme 26. Sequential iodination/direct C6-H arylation of DMU 65 with arenes.
14

Molecules 2015, 20, 4874–4901
The Pd-catalyzed cross-dehydrogenative coupling (CDC) of DMU 65 with benzene or xylenes74 in the presence of PivOH and AgOAc at reflux was found to produce 6-aryluracil analogue 67
as the major product. Minor quantities of 5-arylated counterpart 66 and uracil dimeric byproductswere also formed (Scheme 27a) [59,69]. It is believed that the 6-arylation occurred via CMD processinvolving PdII(L)(OPiv) species. Deprotonation occurred at the more acid hydrogen at C6 of uracilring, followed by a second CMD process to give the 6-arylated uracil product 67. Interestingly, reactionof DMU 65 with mesitylene led only to the formation of 5,5- and 5,6-DMU dimers. Also, application ofthis protocol to 2',3',5'-tri-O-acetyl-3-N-benzyluridine 75 gave only the C5-C5 dimer 76 in 43% yield(Scheme 27b) [69].
Scheme 27. Pd-catalyzed cross-dehydrogenative coupling of uracils and uracil nucleosides.
The Pd-catalyzed cross-dehydrogenative heteroarylation between 1,3-dialkyluracils 77 andpyridine-N-oxides 78 (3 equiv.) substrates in the presence of Ag2CO3 at 140 ◦C for 12 h gave5-(2-pyridyl-N-oxide)uracils 79 in good-to-high yields (Scheme 28) [70]. As expected, the 3-substitutedpyridine-N-oxides gave products 83 with excellent regioselectivity at the less bulky site. Thecoupling, however, was not compatible with either 1,3-dibenzoyluracil or unprotected uracil substrates.Reduction of N-oxides 79 with PCl3 in toluene yielded the corresponding 5-uracil derivativessubstituted with a 2-pyridyl ring. The electrophilic palladation at the C5 of uracil and the coordinationof the palladium atom to N-oxide was believed to control the regioselectivity (at C5 of the uracil ringand C2 of pyridine oxide) of these double C-H activation cross-couplings.
Scheme 28. Synthesis of 5-(2-pyridyl-N-oxide)uracils 79 via cross-dehydrogenative arylation.
4.3. Cross-Dehydrogenative Alkenylation at C5 Position
The discovery of the potent antiviral activity of E-5-(2-bromovinyl)-2'-deoxyuridine (1, BVDU)led to the exploration of synthetic routes based on the oxidative coupling of uridine nucleosides
15

Molecules 2015, 20, 4874–4901
with alkenes. Such routes avoided the use of mercury that was central to Walker’s syntheticapproach involving the condensation of 5-mercurated 2'-deoxyuridine with methyl acrylate andradical decarboxylation-bromination sequence [71]. They also seem advantageous to othercoupling protocols which employ coupling between 5-halouracil nucleosides and organometallicsor methyl acrylate [10,72]. In 1987, Itahara reported the oxidative coupling of uracil nucleosides80 with maleimides 81 which gave 5-substituted coupling products of type 82 (Scheme 29) [73].Using stoichiometric amounts of Pd(OAc)2 was however necessary. The yields for uridine and2'-deoxyuridine substrates were lower (4%–22%) than those of the DMU substrate (~20%–50%). Theyields for DMU substrate slightly increased in the presence reoxidants such as AgOAc, Na2S2O8 orCu(OAc)2.
Scheme 29. Pd-mediated oxidative coupling of uridine and 2'-deoxyuridine with maleimides.
Also in 1987, Hirota and coworkers reported the oxidative coupling of uridine 80b and2'-deoxyuridine 80c with methyl acrylate or styrene using either stoichiometric amounts of Pd(OAc)2
in MeCN at ambient temperature or catalytic loading of Pd(OAc)2 in the presence of tert-butylperbenzoate as the reoxidant to generate 5-vinyl uridine analogues 84 (Scheme 30) [74]. The couplingsproceeded stereoselectively to give the trans isomers. The reaction conditions were compatible withunprotected and protected uridine substrates, though, coupling of 2',3'-di-O-isopropylideneuridinewith methyl acrylate produced also the 5',6-cyclouridine byproduct in 23% yield.
Scheme 30. Stoichiometric and catalytic oxidative coupling of uracil nucleosides with methyl acrylate.
Yun and Georg recently reported Pd-catalyzed cross-dehydrogenative coupling of 1,3-disubstituteduracils as well as protected uridine 85 and 2'-deoxyuridine derivatives 88 with tert-butyl acrylate inthe presence of AgOAc and PivOH in DMF at 60 ◦C/24 h to give 5-alkenyl products 87 and 89
in 66% and 75% yield, respectively (Scheme 31) [75]. The coupling occurred with the regio- (C5)and stereoselectivity (E-isomer). However, 3-N-methyl protection at the uracil ring was necessary.The coupling was postulated to occur via the electrophilic palladation pathway [76] at the C5position of the uracil ring followed by deprotonation with the pivalate anion to give palladatedintermediate. Coordination with the alkenes via transmetallation, and subsequent β-eliminationprovided 5-alkenyluracil derivatives.
16

Molecules 2015, 20, 4874–4901
Scheme 31. Pd-Catalyzed cross-dehydrogenative alkenylation of uracil nucleosides.
4.4. Miscellaneous Direct C-H Functionalizations
Pd-catalyzed direct C-H acetoxylation at the electron-rich C5 position of uracil nucleosides withPhI(OAc)2 under reasonably mild conditions (60 ◦C, 3–5 h) have been also developed (Scheme 32) [77].The acyl protected uridine 80a and silyl protected 2'-deoxyuridine 80d were compatible with theseconditions to give the corresponding 5-acetoxy products 90 in 55% and 25% yields, respectively. Thereaction was proposed to proceed via oxidative electrophilic palladation at the electron rich C5 positionto give the 5-palladauracil intermediate followed by oxidation to give Pd(IV) intermediate, whichyielded 5-acetoxyluridine via the reductive elimination of Pd(II).
Scheme 32. Pd-catalyzed acetoxylation of uracil nucleosides.
The Cu-catalyzed intermolecular C5-H amination of DMU 65 with 4-bromoaniline in thepresence of [hydroxy(tosyloxy)iodo]mesitylene has been recently developed (Scheme 33) [50]. Theregioselectivity of this C5 amination was proposed to be controlled via the formation of iodonium saltintermediate 91, which is consistent with C5 electrophilic aromatic substitution that is typical for uracilring. Subsequent Cu-catalyzed amination gave the 5-amino product 92 in 65% overall yield.
Majumdar and coworkers reported the synthesis of pyrrolo[3,2-d]pyrimidine derivatives of type94 by the intramolecular dehydrogenative coupling of the 5-amidouracils 93 via the selective activationof uracil C6-H bond in the presence of Cu(OTf)2 (Scheme 34) [78]. This coupling between Csp2-H (inthe uracil ring) and Csp3-H (in the side chain) bonds was not, however, successful when R2 = H. Theauthors suggested that coupling most probably involved single electron transfer (SET) processes andmight require a more stable tertiary radical on the side chain to proceed.
17

Molecules 2015, 20, 4874–4901
Scheme 33. Cu-catalyzed direct C5-H amination of 1,3-dimethyluracil 65.
Scheme 34. Intramolecular cross-dehydrogenative cyclization at C6 of uracil ring.
Recently, C5-H trifluoromethylation of DMU 65 by means of electrophilic, nucleophilic or radical“CF3” species in an effort to synthesize 6-aryl-5-(trifluoromethyl)uracils (e.g, 96) by direct activations ofboth C5-H and C6-H bonds in consecutive manner has been attempted (Scheme 35) [79]. Thus, reactionsof 65 with electrophilic (Umemoto's or Togni’s) or nucleophilic (Rupert’s) reagents in combination withPd or Cu catalyst either failed or led to the formation of 5,5- or 5,6-dimeric products or 5-CF3 productin low yields. However, radical trifluoromethylation of 65 with sodium trifluoromethanesulfinatein the presence of tert-butyl hydroperoxide provided 1,3-dimethyl-5-(trifluoromethyl)uracil 95 in67% yield [79]. The subsequent direct arylation at C6-H of 95 with 4-iodotoluene in the presence ofPd(OAc)2 and CuI/CsF afforded desired 5,6-disubstituted uracils 96 albeit in low yield (25%); probablybecause of the electron-withdrawing effect of the CF3 group at the 5 position on the pyrimidine ring.This C6-H arylation was not applicable to other aryl halides and often was accompanied by cleavageof the CF3 group due to hydrolysis followed by decarboxylation, especially when Cs2CO3 was usedas the base.
Scheme 35. Synthesis of 6-aryl-5-(trifluoromethyl)uracils by direct activation of C5-H and C6-H bonds.
5. Coupling of 5-Halouracil Nucleosides with Arenes and Heteroarenes
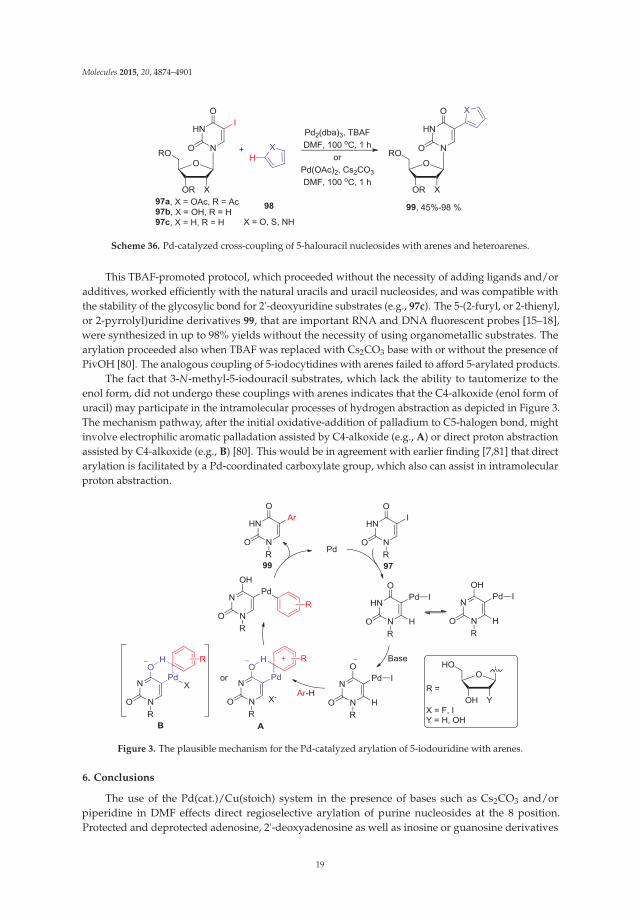
The lack of regioselectivity in direct activation of uracil derivatives (C5-H vs. C6-H) duringcross-couplings with aryl halides, and fact that coupling conditions are usually unsuitable forunprotected uracils and natural nucleosides, were recently overcome by switching the halidesubstituents from aryl halides to uracil ring and allowing to react of 5-halouridines with arenesinstead. Wnuk and coworkers found that the 5-iodouracil nucleosides 97 coupled with simplearenes or heteroaromatics 98 in the presence of Pd2(dba)3 and TBAF in DMF under milder condition(100 ◦C/1–2 h) to give the 5-arylated uracil nucleosides 99 in high yields (Scheme 36) [80].
18
Molecules 2015, 20, 4874–4901
Scheme 36. Pd-catalyzed cross-coupling of 5-halouracil nucleosides with arenes and heteroarenes.
This TBAF-promoted protocol, which proceeded without the necessity of adding ligands and/oradditives, worked efficiently with the natural uracils and uracil nucleosides, and was compatible withthe stability of the glycosylic bond for 2'-deoxyuridine substrates (e.g., 97c). The 5-(2-furyl, or 2-thienyl,or 2-pyrrolyl)uridine derivatives 99, that are important RNA and DNA fluorescent probes [15–18],were synthesized in up to 98% yields without the necessity of using organometallic substrates. Thearylation proceeded also when TBAF was replaced with Cs2CO3 base with or without the presence ofPivOH [80]. The analogous coupling of 5-iodocytidines with arenes failed to afford 5-arylated products.
The fact that 3-N-methyl-5-iodouracil substrates, which lack the ability to tautomerize to theenol form, did not undergo these couplings with arenes indicates that the C4-alkoxide (enol form ofuracil) may participate in the intramolecular processes of hydrogen abstraction as depicted in Figure 3.The mechanism pathway, after the initial oxidative-addition of palladium to C5-halogen bond, mightinvolve electrophilic aromatic palladation assisted by C4-alkoxide (e.g., A) or direct proton abstractionassisted by C4-alkoxide (e.g., B) [80]. This would be in agreement with earlier finding [7,81] that directarylation is facilitated by a Pd-coordinated carboxylate group, which also can assist in intramolecularproton abstraction.
Figure 3. The plausible mechanism for the Pd-catalyzed arylation of 5-iodouridine with arenes.
6. Conclusions
The use of the Pd(cat.)/Cu(stoich) system in the presence of bases such as Cs2CO3 and/orpiperidine in DMF effects direct regioselective arylation of purine nucleosides at the 8 position.Protected and deprotected adenosine, 2'-deoxyadenosine as well as inosine or guanosine derivatives
19

Molecules 2015, 20, 4874–4901
coupled efficiently under these conditions with aryl halides to give access to 8-arylated productsin up to 99% yields. The C8-H functionalization process is proposed to occur via 8-cupriopurineintermediates or N-heterocyclic carbene like cuprates, which subsequently can undergo a standardPd(0)-catalytic cycle for cross-coupling with aryl halides. The cross-dehydrogenative arylationprotocols which involve C8-H bond activation have been also developed for the synthesis of purinefused ring systems. Intramolecular N1-purinyl nitrogen directed C-H bond activation was utilizedfor direct ortho modification (arylation, amination or acetoxylation) of the aryl rings at C6 position ofpurine nucleosides.
In the case of pyrimidine nucleosides, direct arylation protocols have only been developed foruracil bases, which, in turn, usually require protection at the 1-N and 3-N positions. The biggestchallenge how to overcome regioselective C-H activation at the C5 or C6 position of uracil ring havebeen accomplished by employing different catalysts and ligands. For the cross-coupling with arylhalides, it was found that Pd-based catalytic systems were effective to promote C5 arylation, whereasPd/Cu or Cu-mediated systems affected C6 arylation with good selectivity. The C5 arylation isproposed to proceed via the electrophilic or concerted metalation-deprotonation mechanisms, whilethe C6 arylation likely to occur via a cuprate intermediates. Pd-catalyzed cross-dehydrogenativecoupling of uracil nucleosides with alkenes or arenes, in the presence of oxidants, were developedto give convenient access to 5-alkenyl/aryl derivatives. The 5-acetoxy and 5-amino derivatives werealso synthesized by direct activation of C5-H bond in uracil ring. No examples of couplings involvingdirect C-H activation of cytosine ring have been reported.
The transition-metal catalyzed syntheses of the modified nucleobases by direct C-H bondactivations have been substantially improved in the past decade. However, despite improvedcoupling efficiency and availability of milder reaction conditions applicable to the less stabledeoxynucleosides, application of the direct C-H functionalization approaches to nucleotides and/orshort (deoxy)oligonucleotides fragments still will require developing the conditions compatible bothwith solvent requirements for water-soluble nucleotides and stability of phosphate esters.
Acknowledgments: YL would like to thank Florida International University Graduate School (FIU) for theDissertation Year Fellowship.
Author Contributions: Study concept and design (YL, SFW). Drafting of the manuscript and preparation of theschemes (YL). Critical review of the manuscript (YL, SFW).
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Meijere, A.D.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH Verlag GmbH: Weinheim,Germany, 2008.
2. Kambe, N.; Iwasaki, T.; Terao, J. Pd-catalyzed cross-coupling reactions of alkyl halides. Chem. Soc. Rev. 2011,40, 4937–4947. [CrossRef] [PubMed]
3. The Nobel Prize in Chemistry Announcement. Available online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2010/# (accessed on 12 March 2015).
4. Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-metal-catalyzed direct arylation of (hetero)arenes by C-Hbond cleavage. Angew. Chem. Int. Ed. Engl. 2009, 48, 9792–9826. [CrossRef] [PubMed]
5. Fagnou, K. Mechanistic considerations in the development and use of azine, diazine and azole N-oxides inpalladium-catalyzed direct arylation. Top. Curr. Chem. 2010, 292, 35–56. [PubMed]
6. Su, Y.X.; Sun, L.P. Recent progress towards transition-metal-catalyzed direct arylation of heteroarenes.Mini Rev. Org. Chem. 2012, 9, 87–117. [CrossRef]
7. Lafrance, M.; Fagnou, K. Palladium-catalyzed benzene arylation: incorporation of catalytic pivalic acid as aproton shuttle and a key element in catalyst design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [CrossRef][PubMed]
20

Molecules 2015, 20, 4874–4901
8. Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. Establishment of broadly applicable reactionconditions for the Palladium-catalyzed direct arylation of heteroatom-containing aromatic compounds.J. Org. Chem. 2009, 74, 1826–1834. [CrossRef] [PubMed]
9. Joo, J.M.; Touré, B.B.; Sames, D. C−H Bonds as ubiquitous functionality: A general approach to complexarylated imidazoles via regioselective sequential arylation of all three C−H bonds and regioselectiveN-alkylation enabled by SEM-group transposition. J. Org. Chem. 2010, 75, 4911–4920. [CrossRef] [PubMed]
10. Agrofoglio, L.A.; Gillaizeau, I.; Saito, Y. Palladium-assisted routes to nucleosides. Chem. Rev. 2003, 103,1875–1916. [CrossRef] [PubMed]
11. De Ornellas, S.; Williams, T.J.; Baumann, C.G.; Fairlamb, I.J.S. (Eds.) Catalytic C-H/C-X bondfunctionalisation of nucleosides, nucleotides, nucleic acids, amino acids, peptides and proteins. In C-H andC-X Bond Functionalization: Transition Metal Mediation; The Royal Society of Chemistry: London, UK, 2013.
12. Shaughnessy, K. Palladium-catalyzed functionalization of unprotected nucleosides in aqueous media.Molecules 2015, in press.
13. De Clercq, E.; Descamps, J.; de Somer, P.; Barr, P.J.; Jones, A.S.; Walker, R.T. (E)-5-(2-Bromovinyl)-2'-deoxyuridine: A potent and selective anti-herpes agent. Proc. Natl. Acad. Sci. USA 1979, 76, 2947–2951.
14. McGuigan, C.; Barucki, H.; Blewett, S.; Carangio, A.; Erichsen, J.T.; Andrei, G.; Snoeck, R.; de Clercq, E.;Balzarini, J. Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidinenucleosides bearing an aryl side chain. J. Med. Chem. 2000, 43, 4993–4997. [CrossRef] [PubMed]
15. Greco, N.J.; Tor, Y. Simple fluorescent pyrimidine analogues detect the presence of DNA abasic sites. J. Am.Chem. Soc. 2005, 127, 10784–10785. [CrossRef] [PubMed]
16. Srivatsan, S.G.; Tor, Y. Using an emissive uridine analogue for assembling fluorescent HIV-1 TAR constructs.Tetrahedron 2007, 63, 3601–3607. [CrossRef] [PubMed]
17. Noé, M.S.; Ríos, A.C.; Tor, Y. Design, synthesis, and spectroscopic properties of extended and fusedpyrrolo-dC and pyrrolo-C analogs. Org. Lett. 2012, 14, 3150–3153. [CrossRef] [PubMed]
18. Wicke, L.; Engels, J.W. Postsynthetic on column RNA labeling via Stille coupling. Bioconjugate Chem. 2012,23, 627–642. [CrossRef]
19. Valis, L.; Mayer-Enthart, E.; Wagenknecht, H.A. 8-(Pyren-1-yl)-2′-deoxyguanosine as an optical probe forDNA hybridization and for charge transfer with small peptides. Bioorg. Med. Chem. Lett. 2006, 16, 3184–3187.[CrossRef] [PubMed]
20. Wanninger-Weiß, C.; Valis, L.; Wagenknecht, H.A. Pyrene-modified guanosine as fluorescent probe for DNAmodulated by charge transfer. Bioorg. Med. Chem. 2008, 16, 100–106. [CrossRef] [PubMed]
21. Manfredini, S.; Baraldi, P.G.; Bazzanini, R.; Marangoni, M.; Simoni, D.; Balzarini, J.; De Clercq, E. Synthesisand cytotoxic activity of 6-vinyl- and 6-ethynyluridine and 8-vinyl- and 8-ethynyladenosine. J. Med. Chem.1995, 38, 199–203. [CrossRef] [PubMed]
22. He, G.X.; Krawczyk, S.H.; Swaminathan, S.; Shea, R.G.; Dougherty, J.P.; Terhorst, T.; Law, V.S.; Griffin, L.C.;Coutré, S.; Bischofberger, N. N2- and C8-Substituted oligodeoxynucleotides with enhanced thrombininhibitory activity in vitro and in vivo. J. Med. Chem. 1998, 41, 2234–2242. [CrossRef] [PubMed]
23. Cerna, I.; Pohl, R.; Hocek, M. The first direct C-H arylation of purine nucleosides. Chem. Commun. 2007,4729–4730. [CrossRef]
24. Cerna, I.; Pohl, R.; Klepetárová, B.; Hocek, M. Direct C−H arylation of purines: Development of methodologyand its use in regioselective synthesis of 2,6,8-trisubstituted purines. Org. Lett. 2006, 8, 5389–5392. [CrossRef][PubMed]
25. Cerna, I.; Pohl, R.; Klepetárová, B.; Hocek, M. Synthesis of 6,8,9-tri- and 2,6,8,9-tetrasubstituted purines by acombination of the Suzuki cross-coupling, N-arylation, and direct C−H arylation reactions. J. Org. Chem.2008, 73, 9048–9054. [CrossRef] [PubMed]
26. Storr, T.E.; Baumann, C.G.; Thatcher, R.J.; De Ornellas, S.; Whitwood, A.C.; Fairlamb, I.J. S.Pd(0)/Cu(I)-mediated direct arylation of 2′-deoxyadenosines: Mechanistic role of Cu(I) and reactivitycomparisons with related purine nucleosides. J. Org. Chem. 2009, 74, 5810–5821. [CrossRef] [PubMed]
27. Storr, T.E.; Firth, A.G.; Wilson, K.; Darley, K.; Baumann, C.G.; Fairlamb, I.J. S. Site-selective direct arylation ofunprotected adenine nucleosides mediated by palladium and copper: Insights into the reaction mechanism.Tetrahedron 2008, 64, 6125–6137. [CrossRef]
21

Molecules 2015, 20, 4874–4901
28. Baumann, C.G.; De Ornellas, S.; Reeds, J.P.; Storr, T.E.; Williams, T.J.; Fairlamb, I.J.S. Formation andpropagation of well-defined Pd nanoparticles (PdNPs) during C–H bond functionalization of heteroarenes:are nanoparticles a moribund form of Pd or an active catalytic species? Tetrahedron 2014, 70, 6174–6187.[CrossRef]
29. Storr, T.E.; Strohmeier, J.A.; Baumann, C.G.; Fairlamb, I.J. S. A sequential direct arylation/Suzuki-Miyauracross-coupling transformation of unprotected 2'-deoxyadenosine affords a novel class of fluorescentanalogues. Chem. Commun. 2010, 46, 6470–6472. [CrossRef]
30. Ornellas, S.D.; Storr, T.E.; Williams, T.J.; Baumann, C.G.; Fairlamb, I.J. S. Direct C-H/C-X couplingmethodologies mediated by Pd/Cu or Cu: An examination of the synthetic applications and mechanisticfindings. Curr. Org. Synth. 2011, 8, 79–101. [CrossRef]
31. Vanková, B.; Krchnák, V.; Soural, M.; Hlavác, J. Direct C–H arylation of purine on solid phase and its use forchemical libraries synthesis. ACS Comb. Sci. 2011, 13, 496–500. [CrossRef] [PubMed]
32. Sahnoun, S.; Messaoudi, S.; Peyrat, J.F.; Brion, J.D.; Alami, M. Microwave-assisted Pd(OH)2-catalyzed directC−H arylation of free-(NH2) adenines with aryl halides. Tetrahedron Lett. 2008, 49, 7279–7283. [CrossRef]
33. Sahnoun, S.; Messaoudi, S.; Brion, J.D.; Alami, M. A site selective C-H arylation of free-(NH2) adenineswith aryl chlorides: Application to the synthesis of 6,8-disubstituted adenines. Org. Biomol. Chem. 2009, 7,4271–4278. [CrossRef] [PubMed]
34. Ngassa, F.N.; DeKorver, K.A.; Melistas, T.S.; Yeh, E.A.H.; Lakshman, M.K. Pd−Xantphos-Catalyzed DirectArylation of Nucleosides. Org. Lett. 2006, 8, 4613–4616. [CrossRef] [PubMed]
35. Bellina, F.; Cauteruccio, S.; Rossi, R. Palladium- and copper-mediated direct C-2 arylation ofazoles—including free (NH)-imidazole, -benzimidazole and -indole—Under base-free and ligandlessconditions. Eur. J. Org. Chem. 2006, 2006, 1379–1382. [CrossRef]
36. Western, E.C.; Shaughnessy, K.H. Inhibitory effects of the guanine moiety on Suzuki couplings of unprotectedhalonucleosides in aqueous media. J. Org. Chem. 2005, 70, 6378–6388. [CrossRef] [PubMed]
37. Gigante, A.; Priego, E.M.; Sánchez-Carrasco, P.; Ruiz-Pérez, L.M.; Vande Voorde, J.; Camarasa, M.J.;Balzarini, J.; González-Pacanowska, D.; Pérez-Pérez, M.J. Microwave-assisted synthesis of C-8 aryl andheteroaryl inosines and determination of their inhibitory activities against Plasmodium falciparum purinenucleoside phosphorylase. Eur. J. Med. Chem. 2014, 82, 459–465. [CrossRef] [PubMed]
38. Cerna, I.; Pohl, R.; Klepetárová, B.; Hocek, M. Intramolecular direct C-H arylation approach to fused purines.Synthesis of purino[8,9-f ]phenanthridines and 5,6-dihydropurino[8,9-a]isoquinolines. J. Org. Chem. 2010, 75,2302–2308. [CrossRef] [PubMed]
39. Campeau, L.C.; Parisien, M.; Jean, A.; Fagnou, K. Catalytic direct arylation with aryl chlorides, bromides,and iodides: Intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc. 2005, 128,581–590. [CrossRef]
40. Iaroshenko, V.O.; Ostrovskyi, D.; Miliutina, M.; Maalik, A.; Villinger, A.; Tolmachev, A.; Volochnyuk, D.M.;Langer, P. Design and synthesis of polycyclic imidazole-containing N-heterocycles based on C-Hactivation/cyclization reactions. Adv. Synth. Catal. 2012, 354, 2495–2503. [CrossRef]
41. Barbero, N.; SanMartin, R.; Dominguez, E. Ligand-free copper(I)-catalysed intramolecular direct C-Hfunctionalization of azoles. Org. Biomol. Chem. 2010, 8, 841–845. [CrossRef] [PubMed]
42. Meng, G.; Niu, H.Y.; Qu, G.R.; Fossey, J.S.; Li, J.P.; Guo, H.M. Synthesis of fused N-heterocycles via tandemC-H activation. Chem. Commun. 2012, 48, 9601–9603. [CrossRef]
43. Xi, P.; Yang, F.; Qin, S.; Zhao, D.; Lan, J.; Gao, G.; Hu, C.; You, J. Palladium(II)-catalyzed oxidative C−H/C−Hcross-coupling of heteroarenes. J. Am. Chem. Soc. 2010, 132, 1822–1824. [CrossRef] [PubMed]
44. Stuart, D.R.; Fagnou, K. The catalytic cross-coupling of unactivated arenes. Science 2007, 316, 1172–1175.[CrossRef] [PubMed]
45. Vabre, R.; Chevot, F.; Legraverend, M.; Piguel, S. Microwave-Assisted Pd/Cu-Catalyzed C-8 DirectAlkenylation of Purines and Related Azoles: An Alternative Access to 6,8,9-Trisubstituted Purines.J. Org. Chem. 2011, 76, 9542–9547. [CrossRef] [PubMed]
46. Sahnoun, S.; Messaoudi, S.; Brion, J.D.; Alami, M. Pd/Cu-catalyzed direct alkenylation of azole heterocycleswith alkenyl halides. Eur. J. Org. Chem. 2010, 2010, 6097–6102. [CrossRef]
47. Lagisetty, P.; Zhang, L.; Lakshman, M.K. Simple methodology for Heck arylation at C-8 of adeninenucleosides. Adv. Synth. Catal. 2008, 350, 602–608. [CrossRef]
22

Molecules 2015, 20, 4874–4901
48. Klecka, M.; Pohl, R.; Klepetarova, B.; Hocek, M. Direct C-H borylation and C-H arylation ofpyrrolo[2,3-d]pyrimidines: synthesis of 6,8-disubstituted 7-deazapurines. Org. Biomol. Chem. 2009, 7,866–868. [CrossRef] [PubMed]
49. Sabat, N.; Klecka, M.; Slavetinska, L.; Klepetarova, B.; Hocek, M. Direct C-H amination and C-Hchloroamination of 7-deazapurines. RSC Advances 2014, 4, 62140–62143.
50. Sokolovs, I.; Lubriks, D.; Suna, E. Copper-catalyzed intermolecular C–H amination of (hetero)arenes viatransient unsymmetrical λ3-iodanes. J. Am. Chem. Soc. 2014, 136, 6920–6928. [CrossRef] [PubMed]
51. Klecka, M.; Pohl, R.; Cejka, J.; Hocek, M. Direct C-H sulfenylation of purines and deazapurines. Org. Biomol.Chem. 2013, 11, 5189–5193. [CrossRef] [PubMed]
52. Qu, G.R.; Liang, L.; Niu, H.Y.; Rao, W.H.; Guo, H.M.; Fossey, J.S. Copper-catalyzed synthesis of purine-fusedpolycyclics. Org. Lett. 2012, 14, 4494–4497. [CrossRef] [PubMed]
53. Guo, H.M.; Jiang, L.L.; Niu, H.Y.; Rao, W.H.; Liang, L.; Mao, R.Z.; Li, D.Y.; Qu, G.R. Pd(II)-catalyzedortho arylation of 6-arylpurines with aryl iodides via purine-directed C−H activation: A new strategy formodification of 6-arylpurine derivatives. Org. Lett. 2011, 13, 2008–2011. [CrossRef] [PubMed]
54. Lakshman, M.K.; Deb, A.C.; Chamala, R.R.; Pradhan, P.; Pratap, R. Direct arylation of 6-phenylpurine and6-arylpurine nucleosides by Ruthenium-catalyzed C-H bond activation. Angew. Chem. Int. Ed. 2011, 50,11400–11404. [CrossRef]
55. Chamala, R.R.; Parrish, D.; Pradhan, P.; Lakshman, M.K. Purinyl N1-directed aromatic C–H oxidation in6-arylpurines and 6-arylpurine nucleosides. J. Org. Chem. 2013, 78, 7423–7435. [CrossRef] [PubMed]
56. Kim, J.Y.; Park, S.H.; Ryu, J.; Cho, S.H.; Kim, S.H.; Chang, S. Rhodium-catalyzed intermolecular amidationof arenes with sulfonyl azides via chelation-assisted C–H bond activation. J. Am. Chem. Soc. 2012, 134,9110–9113. [CrossRef] [PubMed]
57. Cernová, M.; Pohl, R.; Hocek, M. Switching the regioselectivity of direct C–H arylation of 1,3-dimethyluracil.Eur. J. Org. Chem. 2009, 22, 3698–3701. [CrossRef]
58. Cernová, M.; Cerná, I.; Pohl, R.; Hocek, M. Regioselective direct C–H arylations of protected uracils. Synthesisof 5- and 6-aryluracil bases. J. Org. Chem. 2011, 76, 5309–5319. [CrossRef] [PubMed]
59. Kim, K.H.; Lee, H.S.; Kim, J.N. Palladium-catalyzed direct 5-arylation of 1,3-dimethyluracil with arylbromides: An electrophilic metalation–deprotonation with electrophilic arylpalladium intermediate.Tetrahedron Lett. 2011, 52, 6228–6233. [CrossRef]
60. Lee, H.S.; Kim, K.H.; Kim, S.H.; Kim, J.N. Palladium-catalyzed synthesis of benzo[c]pyrimido[1,6-a]azepinescaffold from Morita–Baylis–Hillman adducts: Intramolecular 6-arylation of uracil nucleus. Tetrahedron Lett.2012, 53, 497–501. [CrossRef]
61. Cheng, C.; Shih, Y.C.; Chen, H.T.; Chien, T.C. Regioselective arylation of uracil and 4-pyridone derivativesvia copper(I) bromide mediated C–H bond activation. Tetrahedron 2013, 69, 1387–1396. [CrossRef]
62. Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the palladium-catalyzed (aromatic) C–H bondmetalation–deprotonation mechanism spanning the entire spectrum of arenes. J. Org. Chem. 2011, 77,658–668. [CrossRef] [PubMed]
63. Bellina, F.; Cauteruccio, S.; Rossi, R. Efficient and practical synthesis of 4(5)-aryl-1H-imidazoles and2,4(5)-diaryl-1H-imidazoles via highly selective palladium-catalyzed arylation reactions. J. Org. Chem.2007, 72, 8543–8546. [CrossRef] [PubMed]
64. Sievers, A.; Wolfenden, R. Equilibrium of formation of the 6-carbanion of UMP, a potential intermediate inthe action of OMP decarboxylase. J. Am. Chem. Soc. 2002, 124, 13986–13987. [CrossRef] [PubMed]
65. Yeoh, F.Y.; Cuasito, R.R.; Capule, C.C.; Wong, F.M.; Wu, W. Carbanions from decarboxylation of orotateanalogs: Stability and mechanistic implications. Bioorg. Chem. 2007, 35, 338–343. [CrossRef] [PubMed]
66. Amyes, T.L.; Wood, B.M.; Chan, K.; Gerlt, J.A.; Richard, J.P. Formation and stability of a vinyl carbanion atthe active site of orotidine 5'-monophosphate decarboxylase: pKa of the C-6 proton of enzyme-bound UMP.J. Am. Chem. Soc. 2008, 130, 1574–1575. [CrossRef] [PubMed]
67. Do, H.Q.; Daugulis, O. A general method for Copper-catalyzed arene cross-dimerization. J. Am. Chem. Soc.2011, 133, 13577–13586. [CrossRef] [PubMed]
68. Mondal, B.; Hazra, S.; Roy, B. Pd(II)-catalyzed regioselective direct arylation of uracil via oxidative Heckreaction using arylboronic acids. Tetrahedron Lett. 2014, 55, 1077–1081. [CrossRef]
69. Kim, K.H.; Lee, H.S.; Kim, S.H.; Kim, J.N. Palladium(II)-catalyzed oxidative homo-coupling of1,3-dimethyluracil derivatives. Tetrahedron Lett. 2012, 53, 1323–1327. [CrossRef]
23

Molecules 2015, 20, 4874–4901
70. Kianmehr, E.; Rezaeefard, M.; Rezazadeh Khalkhali, M.; Khan, K.M. Pd-catalyzed dehydrogenativecross-coupling of pyridine-N-oxides with uracils. RSC Adv. 2014, 4, 13764–13767. [CrossRef]
71. Jones, A.S.; Verhelst, G.; Walker, R.T. The synthesis of the potent anti-herpes virus agent, E-5-(2-bromovinyl)-2′-deoxyuridine and related compounds. Tetrahedron Lett. 1979, 20, 4415–4418. [CrossRef]
72. Ashwell, M.; Jones, A.S.; Kumar, A.; Sayers, J.R.; Walker, R.T.; Sakuma, T.; de Clercq, E. The synthesisand antiviral properties of (E)-5-(2-bromovinyl)-2'-deoxyuridine-related compounds. Tetrahedron 1987, 43,4601–4608. [CrossRef]
73. Itahara, T. Oxidative coupling of uracil derivatives with maleimides by Palladium acetate. Chem. Lett. 1986,15, 239–242. [CrossRef]
74. Hirota, K.; Isobe, Y.; Kitade, Y.; Maki, Y. A simple synthesis of 5-(1-alkenyl)uracil derivatives byPalladium-catalyzed oxidative coupling of uracils with olefins. Synthesis 1987, 1987, 495–496. [CrossRef]
75. Yu, Y.Y.; Georg, G.I. Dehydrogenative alkenylation of uracils via palladium-catalyzed regioselective C-Hactivation. Chem. Commun. 2013, 49, 3694–3696. [CrossRef]
76. Le Bras, J.; Muzart, J. Intermolecular dehydrogenative Heck reactions. Chem. Rev. 2011, 111, 1170–1214.77. Lee, H.S.; Kim, S.H.; Kim, J.N. Pd(II)-Catalyzed acetoxylation of uracil via electrophilic palladation.
Bull. Korean Chem. Soc. 2010, 31, 238–241. [CrossRef]78. Roy, B.; Hazra, S.; Mondal, B.; Majumdar, K.C. Cu(OTf)2-catalyzed dehydrogenative C–H activation under
atmospheric oxygen: An expedient approach to pyrrolo[3,2-d]pyrimidine derivatives. Eur. J. Org. Chem.2013, 2013, 4570–4577. [CrossRef]
79. Cernová, M.; Pohl, R.; Klepetárová, B.; Hocek, M. C-H trifluoromethylations of 1,3-dimethyluracil andreactivity of the products in C-H arylations. Heterocycles 2014, 89, 1159–1171. [CrossRef]
80. Liang, Y.; Gloudeman, J.; Wnuk, S.F. Palladium-catalyzed direct arylation of 5-halouracils and 5-halouracilnucleosides with arenes and heteroarenes promoted by TBAF. J. Org. Chem. 2014, 79, 4094–4103. [CrossRef][PubMed]
81. Lafrance, M.; Rowley, C.N.; Woo, T.K.; Fagnou, K. Catalytic intermolecular direct arylation ofperfluorobenzenes. J. Am. Chem. Soc. 2006, 128, 8754–8756. [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
24

molecules
Review
Palladium-Catalyzed Modification of UnprotectedNucleosides, Nucleotides, and Oligonucleotides
Kevin H. Shaughnessy
Department of Chemistry, The University of Alabama, Box 870336, Tuscaloosa, AL 35487-0336, USA;[email protected]; Tel.: +1-205-348-4435; Fax: +1-205-348-9104
Academic Editor: Mahesh LakshmanReceived: 24 March 2015; Accepted: 19 May 2015; Published: 22 May 2015
Abstract: Synthetic modification of nucleoside structures provides access to molecules of interestas pharmaceuticals, biochemical probes, and models to study diseases. Covalent modificationof the purine and pyrimidine bases is an important strategy for the synthesis of these adducts.Palladium-catalyzed cross-coupling is a powerful method to attach groups to the base heterocyclesthrough the formation of new carbon-carbon and carbon-heteroatom bonds. In this review,approaches to palladium-catalyzed modification of unprotected nucleosides, nucleotides, andoligonucleotides are reviewed. Polar reaction media, such as water or polar aprotic solvents,allow reactions to be performed directly on the hydrophilic nucleosides and nucleotides withoutthe need to use protecting groups. Homogeneous aqueous-phase coupling reactions catalyzed bypalladium complexes of water-soluble ligands provide a general approach to the synthesis of modifiednucleosides, nucleotides, and oligonucleotides.
Keywords: nucleosides; nucleotides; oligonucleotides; palladium; cross-coupling; aqueous-phase catalysis
1. Introduction
Nucleosides are one of the fundamental building blocks in biochemistry. There has beenlong-standing interest in the synthesis of non-natural analogs of nucleosides. The modified nucleosides,or their nucleotide or oligonucleotide analogs, have been widely explored as pharmaceuticallyactive compounds [1–4], in the study of carcinogenesis mechanisms [5–7], and to incorporate probefunctionality in DNA and RNA [8–11]. Nucleoside derivatives can be made through manipulationof the sugar moiety, replacement of the purine or pyrimidine heterocycle, or through covalentmodification of the natural heterocyclic base or their analogs. Functionalization of the canonicalnucleoside scaffolds and their close derivatives (Figure 1) provides a general approach to the synthesisof base-modified nucleosides [12–14].
Metal-catalyzed cross-coupling reactions are powerful methods to form C-C and C-heteroatombonds with aromatic and heteroaromatic structures [15]. These reactions can often be carried out undermild conditions and can be highly functional group tolerant. Palladium-catalyzed cross-couplingreactions have been widely used in the synthesis of base-modified nucleoside derivatives [16–22].Halogenated nucleoside derivatives can be prepared by standard electrophilic aromatic halogenationreactions. The halonucleosides can be coupled with a wide variety of nucleophilic reagents to formcarbon-carbon or carbon-heteroatom bonds (Figure 1). Amine-moieties on nucleoside rings can alsobe arylated. The rapid development of direct arylation methodologies has resulted in the reports ofseveral methods for the direct arylation of C-H bonds on nucleoside bases.
Molecules 2015, 20, 9419–9454 25 www.mdpi.com/journal/molecules

Molecules 2015, 20, 9419–9454
Figure 1. Typical functionalization sites in naturally occurring nucleosides and commonly usedunnatural analogs. Most common functionalization sites are bolded.
Although palladium-catalyzed cross-coupling reactions are well precedented for a wide range ofsubstrates, nucleosides present a number of challenges. Heterocycles are often challenging substratesin cross-coupling reactions because of their ability to coordinate to metal catalysts and deactivatethem. In addition, heterocycles are often electronically very different from more typical aromaticsubstrates, which can hinder steps in the catalytic cycle. For example, electron-rich halogenatedheterocycles, such as furans, thiophenes, and pyrroles, undergo slow oxidative addition to metalcomplexes. In addition to the challenges of cross-coupling highly functionalized heterocyclic substrates,the polar nature of nucleoside derivatives often results in them being poorly soluble in typical organicsolvents. One common approach is to protect the ribose alcohols as esters or silyl ethers to provide amore hydrophobic substrate (Scheme 1, Path A). This approach introduces two additional syntheticsteps, which results in decreased yields and poor atom economy. Protection strategies are typically noteffective for the more hydrophilic nucleotides and oligonucleotides.
Scheme 1. Synthetic approaches to palladium-catalyzed nucleoside modification.
A more attractive approach would be to use unprotected nucleoside, nucleotide, or oligonucleotidederivatives directly in cross-coupling reactions (Scheme 1, Path B). This can be accomplished usingpolar organic solvents, such as DMF, in the case of nucleosides. Water represents a more attractivesolvent for these types of reactions. Water alone or in combination with organic cosolvents effectively
26

Molecules 2015, 20, 9419–9454
dissolves nucleosides and nucleotide derivatives, which allows reactions to be carried out underhomogeneous conditions without the need for protection strategies. In this review, development ofmethods for functionalization of unprotected nucleosides using palladium-catalyzed cross-coupling inwater and polar organic solvents will be reviewed.
2. Cross-Coupling of Unprotected Nucleosides with Ligand-Free Palladium Catalysts
The earliest examples of cross-coupling of unprotected nucleosides were promoted by ligand-freepalladium salts in polar organic solvents. Mertes reported a Heck-type coupling of 5-(HgCl)dU withstyrene derivatives mediated by stoichiometric Li2PdCl4 in methanol to give 5-alkenyl-dU compounds(Scheme 2) [23]. The method was also applied to 5-(HgOAc)dUMP. Direct 5-alkenylation of uridineand 2′-deoxyuridine was achieved in modest yield (35%–57%) using catalytic Pd(OAc)2 with t-butylperbenzoate in acetonitrile (Scheme 3) [24].
Scheme 2. Pd-mediated coupling of 5-HgCldU and styrenes.
Scheme 3. Oxidative coupling of dU and methyl acrylate.
As palladium-catalyzed cross-coupling reactions developed into widely used syntheticmethodologies, significant effort has been devoted to developing highly active and general catalystsystems. Much of this effort has focused on developing supporting ligands that increase the reactivityand stability of the palladium center compared to ligand-free palladium catalysts. Ligand free-systemshave received renewed attention in recent years because they avoid the use of phosphines or relatedligands, which can often be a larger cost in the reaction than the palladium source [25]. Colloidal ornanoparticle palladium catalysts can often provide good levels of activity with aryl iodides andbromides. Recently, ligand-free palladium catalysts have been applied to coupling reactions ofunprotected nucleoside substrates.
2.1. Cross-Coupling with Organosilanes and Organoboranes
Hiyama reported the first examples of a modern cross-coupling of an unprotected halonucleosidewith an organometallic reagent. The coupling of 5-IdU with 1-alkenyldifluoromethylsilanes catalyzedby [Pd(allyl)Cl]2 was accomplished in THF [26]. A mixture of 1-alkenyl and 2-alkenyl products wereobtained (Scheme 4).
27

Molecules 2015, 20, 9419–9454
Scheme 4. Hiyama coupling of 5-IdU.
Suzuki coupling of 5-IdU with arylboronic acids has been successfully achieved using Na2PdCl4without a supporting ligand in water with microwave heating at 100 ◦C [27,28]. Moderate to excellentyields (33%–85%) were achieved using 0.050–1 mol % palladium with reactions times of one hour orless. The more reactive 6-IU underwent coupling at room temperature with a variety of aryl boronicacids using 10 mol % Na2PdCl4 (Scheme 5) [29]. To date there are no examples of coupling of lessreactive 8-bromopurine nucleosides using palladium without supporting ligands. Ligand-supportedcatalyst systems are generally required to activate electron-rich aryl bromides, such as 8-bromopurines.
Scheme 5. Ligand-free Suzuki coupling of 6-IdU.
2.2. Heck Coupling
Heck couplings catalyzed by palladium without supporting ligands are well precedented [30]. TheHeck coupling of 5-IdU and N-allyl trifluoroacetamide was achieved in 44% yield at room temperaturein water using excess palladium (10 equiv) [31]. Use of commercially available 5-IdU was preferable toprevious routes that relied on 5-mercurated uridine. Heck coupling of styrene derivatives and 5-IdUoccur in good yield using stoichiometric K2PdCl4 in DMF at 80 ◦C [32]. The reaction can be achievedusing catalytic amounts of palladium under similar conditions. Heck coupling of 5-IdU with acrylateswas achieved in modest to high yield (35%–90%) using Pd(OAc)2 (10 mol %) at 80 ◦C in water undermicrowave irradiation (Scheme 6) [33].
Scheme 6. Heck coupling of 5-IdU with a ligand-free palladium catalyst.
2.3. Direct Arylation via C-H Activation
The direct coupling of heterocycles with arenes has received significant interest in recent years [34,35]. Electron-rich arenes can be coupled with aryl halides under oxidative conditions with highselectivity for the cross-coupled product. The purine nucleosides are effective coupling partners witharyl iodides. 6-(4-Methoxyphenyl)purine ribonucleoside was coupled with aryl iodides in modest yield
28

Molecules 2015, 20, 9419–9454
(27%–50% yield) catalyzed by Pd(OAc)2 (5 mol %) using stoichiometric CuI in DMF at 125 ◦C [36]. Thecoupling reaction was specific for the electron-rich C8-position. The coupling protocol was also appliedto adenosine to give a mixture of C8- and N6-arylated products in 4–5:1 ratios (Scheme 7). Switchingthe base from piperidine to Cs2CO3 improved the selectivity for C8-arylation of adenosine underotherwise similar conditions [37,38]. The less stable 2′-deoxyadenosine gave significant depurination at125 ◦C. Lowering the reaction temperature 80 ◦C, allowed 8-ArdA derivatives to prepared in 84% yield.
Scheme 7. Oxidative coupling of 4-iodotoluene and adenosine.
Electron-deficient pyrimidines are more difficult to C-H activate than purines. By reversing thenature of the reaction it is possible to coupling halogenated pyrimidine nucleosides with electron-richheterocycles. Direct coupling of 5-IU and 5-IdU with furan, thiophene, and pyrrole was achieved using5 mol % Pd(dba)2 in the presence of TBAF in DMF (Scheme 8) [39]. Coupling occurred selectively togive 5-(2-heteroaryl)uridine or 2′-deoxyuridine derivatives.
Scheme 8. Coupling of 5-IdU and thiophene.
3. Cross-Coupling of Unprotected Nucleosides with Palladium Complexes ofHydrophobic Ligands
Palladium catalysts without supporting ligands are effective catalysts for cross-coupling ofiodopyrimidine nucleosides, but have not been effectively applied to coupling reaction of the lessreactive 8-halopurine derivatives. Phosphine and N-heterocyclic carbene ligands provide more activecatalyst systems that can effectively activate all classes of aryl halides. Typical supporting ligands, suchas triphenylphosphine, are highly hydrophobic. Using these catalyst species for coupling reactions ofunprotected nucleosides requires identifying solvent systems that can solubilize both the hydrophobiccatalyst and the hydrophilic nucleoside. Aprotic dipolar solvents, such as DMF and NMP, can beeffective solvents for these reactions. Alternatively, mixed aqueous-organic solvent systems can beused effectively. Hydrophobic catalysts can exhibit high activity in reactions run with water as thesolvent, even when all reagents are hydrophobic [40]. These "on-water" reactions represent a potentialfuture area of exploration for nucleoside coupling reactions.
3.1. Suzuki Couplings
Wagenknecht reported the first example of Suzuki coupling of an unprotected nucleoside [41,42].1-Pyrenylboronic acid was coupled with 5-IdU using Pd(PPh3)4 (10 mol %) in a solvent systemcomposed of THF/MeOH/H2O (2:1:2, Scheme 9). The coupled product was isolated in 70% yield.In comparison, coupling of acetyl-protected 5-IdU with 1-pyrenylboronic acid followed by deprotection
29

Molecules 2015, 20, 9419–9454
with NaOMe gave a 55% overall yield. Anthraquinone-labeled uridine was prepared by couplingof 9,10-dimethylanthracen-2-yl pinacolatoborane with 5-IdU catalyzed by Pd(PPh3)4 (5 mol %) inTHF/MeOH/H2O followed by oxidation to afford 5-(2-anthraquinonyl)dU in 50% yield over twosteps (Scheme 10) [43].
Scheme 9. Suzuki coupling of 5-IdU and 1-pyrenylboronic acid catalyzed by Pd(PPh3)4.
Scheme 10. Synthesis of anthraquinone-labeled uridine.
Other solvent systems have been used in the Pd/PPh3-catalyzed Suzuki coupling of unprotectedhalonucleosides. The synthesis of a novel spin-labeled uridine derivative began with the coupling of4-formylphenylboronic acid and 5-IdU catalyzed by Pd(PPh3)4 in methanol/water (Scheme 11) [44].The 5-aryldU derivative was then converted to 5-(2′-phenyl-4′,4′,5′,5′,-tetramethylimidazoline-3′-oxy-1′-oxyl)dU by a sequence of steps. A protected version of the spin-labeled nucleoside wasincorporated into oligonucleotides using the phosphoramidite method and used for EPR analysis ofDNA structures. Aqueous DMF was used for the Pd(PPh3)4-catalyzed coupling of arylboronic acidsand 5-IdU [45]. After monophosphorylation, the 5-aryldUMP derivatives were explored as potentialanti-tuberculosis agents.
Despite the insolubility of triphenylphosphine in water, Suzuki coupling of arylboronic acids and5-IdU have been successfully catalyzed by Pd(OAc)2/PPh3 with water as the only solvent at 120 ◦Cusing microwave irradiation [46]. Notably, the PPh3-based catalyst provided comparable yields to thoseobtained using the water-soluble phosphine sodium tri(2,4-dimethyl-5-sulfonatophenyl)phosphine(TXPTS).
Triphenylphosphine-based catalysts have also been applied to Suzuki arylation of unprotectedpurine nucleosides. Coupling of polycyclic arylboronic acids with 8-BrdG catalyzed by Pd(PPh3)4
in THF/MeOH/H2O (2:1:2) afforded 8-(1-pyrenyl)dG (65%), 8-(1-pyrenyl)dA (10%), and 8-(6-benzo[a]pyrenyl)dG (25%) [47,48]. A family of 8-arylA adducts were prepared using Pd(PPh3)4
in DME/H2O (2:1). The products were then converted to 5-amino-1-β-D-ribofuranosylimidazole-4-carboxamide derivatives (AICAR, Scheme 12), which were explored as potential AMP-activated
30

Molecules 2015, 20, 9419–9454
protein kinase activators [49]. Suzuki coupling of trans-β-styrylboronic acid with 8-BrdG catalyzed byPd(PPh3)4 in DMF affords 8-(trans-styrenyl)dG in 64% yield as a single alkene isomer [50]. The productwas used in the study of photochemical E to Z isomerization of the functionalized guanosine derivative.
Scheme 11. Synthesis of a spin-labeled uridine derivative via a Suzuki coupling.
Scheme 12. Suzuki coupling of 8-BrdA as first step in AICAR synthesis.
1,1′-Bis(diphenylphosphino)ferrocene (dppf) has also been applied as a ligand in the Suzukicoupling of unprotected nucleosides. Wagenknecht used PdCl2(dppf) (10 mol %) to catalyze thecoupling of 10-methylphenothiazin-3-ylboronate ester 1 with 5-IdU in THF/MeOH/H2O (2:1:2) toprovide the coupled product in 34% yield (Scheme 13) [51]. This method afforded 5-(2-pyrenyl)dUin 62% yield from 5-IdU and pinacol pyrene-2-boronate ester [52]. A route to BODIPY-modifieduridines starts with the PdCl2(dppf)-catalyzed coupling of 4-formylboronic acid with 5-IdU inwater/acetonitrile (2:1) to give the product in 76% yield (Scheme 14) [53]. The aldehyde was thencondensed with 2,4-dimethylpyrrole, followed by complexation with BF3 to afford the fluorescenturidine derivative. 2-Pyrenyl-dU was prepared in 65% by the Suzuki coupling of 2-pyrenylboronatepinacol ester with 5-IdU PdCl2(dppf) (11 mol %) in THF/MeOH/H2O (2:1:1) [47].
Scheme 13. Synthesis of 5-(10-methylphenothiazin-3-yl)dU.
31
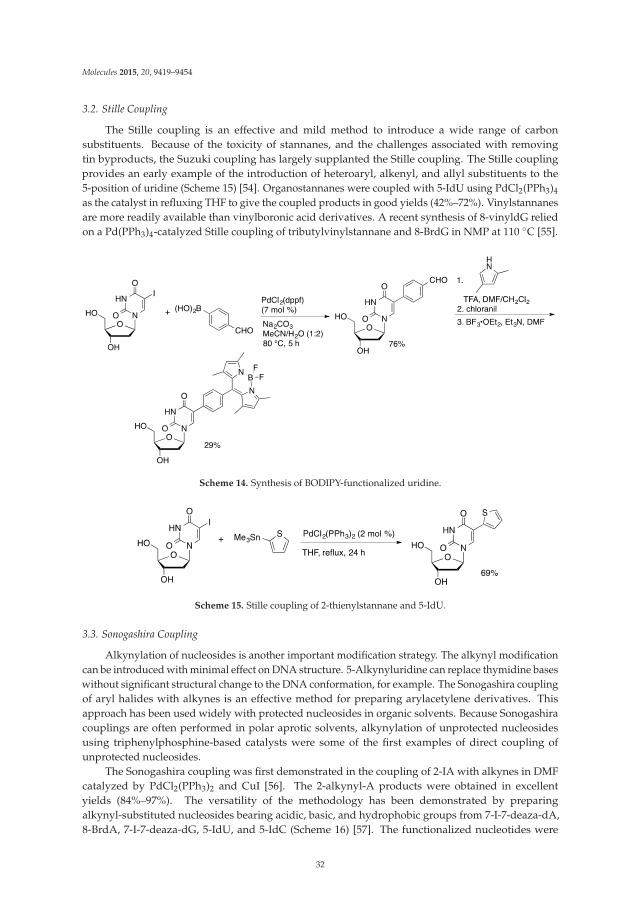
Molecules 2015, 20, 9419–9454
3.2. Stille Coupling
The Stille coupling is an effective and mild method to introduce a wide range of carbonsubstituents. Because of the toxicity of stannanes, and the challenges associated with removingtin byproducts, the Suzuki coupling has largely supplanted the Stille coupling. The Stille couplingprovides an early example of the introduction of heteroaryl, alkenyl, and allyl substituents to the5-position of uridine (Scheme 15) [54]. Organostannanes were coupled with 5-IdU using PdCl2(PPh3)4
as the catalyst in refluxing THF to give the coupled products in good yields (42%–72%). Vinylstannanesare more readily available than vinylboronic acid derivatives. A recent synthesis of 8-vinyldG reliedon a Pd(PPh3)4-catalyzed Stille coupling of tributylvinylstannane and 8-BrdG in NMP at 110 ◦C [55].
Scheme 14. Synthesis of BODIPY-functionalized uridine.
Scheme 15. Stille coupling of 2-thienylstannane and 5-IdU.
3.3. Sonogashira Coupling
Alkynylation of nucleosides is another important modification strategy. The alkynyl modificationcan be introduced with minimal effect on DNA structure. 5-Alkynyluridine can replace thymidine baseswithout significant structural change to the DNA conformation, for example. The Sonogashira couplingof aryl halides with alkynes is an effective method for preparing arylacetylene derivatives. Thisapproach has been used widely with protected nucleosides in organic solvents. Because Sonogashiracouplings are often performed in polar aprotic solvents, alkynylation of unprotected nucleosidesusing triphenylphosphine-based catalysts were some of the first examples of direct coupling ofunprotected nucleosides.
The Sonogashira coupling was first demonstrated in the coupling of 2-IA with alkynes in DMFcatalyzed by PdCl2(PPh3)2 and CuI [56]. The 2-alkynyl-A products were obtained in excellentyields (84%–97%). The versatility of the methodology has been demonstrated by preparingalkynyl-substituted nucleosides bearing acidic, basic, and hydrophobic groups from 7-I-7-deaza-dA,8-BrdA, 7-I-7-deaza-dG, 5-IdU, and 5-IdC (Scheme 16) [57]. The functionalized nucleotides were
32
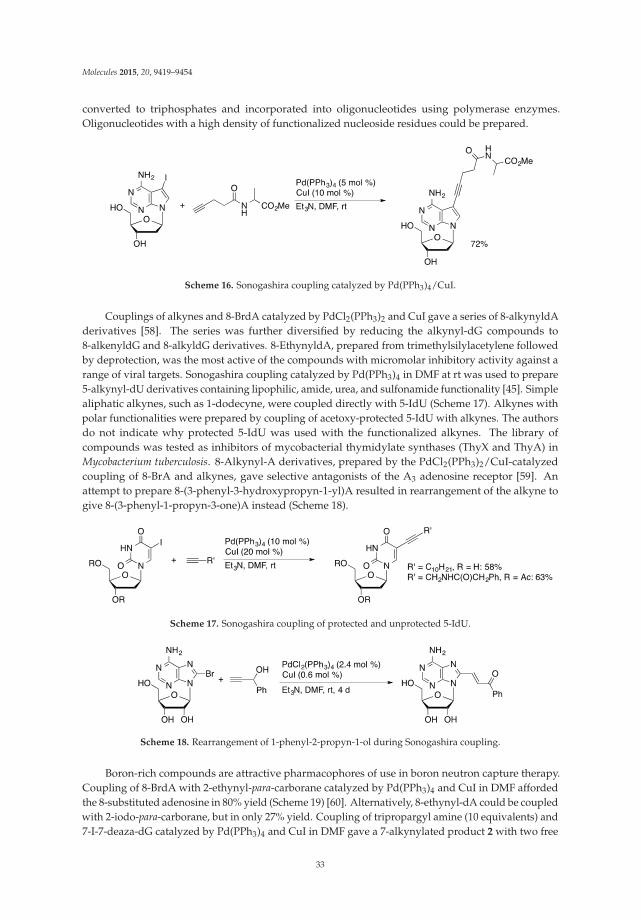
Molecules 2015, 20, 9419–9454
converted to triphosphates and incorporated into oligonucleotides using polymerase enzymes.Oligonucleotides with a high density of functionalized nucleoside residues could be prepared.
Scheme 16. Sonogashira coupling catalyzed by Pd(PPh3)4/CuI.
Couplings of alkynes and 8-BrdA catalyzed by PdCl2(PPh3)2 and CuI gave a series of 8-alkynyldAderivatives [58]. The series was further diversified by reducing the alkynyl-dG compounds to8-alkenyldG and 8-alkyldG derivatives. 8-EthynyldA, prepared from trimethylsilylacetylene followedby deprotection, was the most active of the compounds with micromolar inhibitory activity against arange of viral targets. Sonogashira coupling catalyzed by Pd(PPh3)4 in DMF at rt was used to prepare5-alkynyl-dU derivatives containing lipophilic, amide, urea, and sulfonamide functionality [45]. Simplealiphatic alkynes, such as 1-dodecyne, were coupled directly with 5-IdU (Scheme 17). Alkynes withpolar functionalities were prepared by coupling of acetoxy-protected 5-IdU with alkynes. The authorsdo not indicate why protected 5-IdU was used with the functionalized alkynes. The library ofcompounds was tested as inhibitors of mycobacterial thymidylate synthases (ThyX and ThyA) inMycobacterium tuberculosis. 8-Alkynyl-A derivatives, prepared by the PdCl2(PPh3)2/CuI-catalyzedcoupling of 8-BrA and alkynes, gave selective antagonists of the A3 adenosine receptor [59]. Anattempt to prepare 8-(3-phenyl-3-hydroxypropyn-1-yl)A resulted in rearrangement of the alkyne togive 8-(3-phenyl-1-propyn-3-one)A instead (Scheme 18).
Scheme 17. Sonogashira coupling of protected and unprotected 5-IdU.
Scheme 18. Rearrangement of 1-phenyl-2-propyn-1-ol during Sonogashira coupling.
Boron-rich compounds are attractive pharmacophores of use in boron neutron capture therapy.Coupling of 8-BrdA with 2-ethynyl-para-carborane catalyzed by Pd(PPh3)4 and CuI in DMF affordedthe 8-substituted adenosine in 80% yield (Scheme 19) [60]. Alternatively, 8-ethynyl-dA could be coupledwith 2-iodo-para-carborane, but in only 27% yield. Coupling of tripropargyl amine (10 equivalents) and7-I-7-deaza-dG catalyzed by Pd(PPh3)4 and CuI in DMF gave a 7-alkynylated product 2 with two free
33

Molecules 2015, 20, 9419–9454
alkynyl units (Scheme 20) [61]. Guanosine derivative 2 was converted to a protected phosphoramiditeand incorporated into oligonucleotides using solid phase synthetic techniques. The oligonucleotideswere then reacted with 1-azidomethylpyrene to give the doubly functionalized structure. Single strandoligonucleotides containing residue 2 (3) do not show excimer fluorescence, nor do double strand(ds) DNA containing only one strand with residue 2. In contrast, ds oligonucleotides containing twoappropriately placed 2 residues show strong excimer excitation.
Scheme 19. Sonogashira coupling of 8-BrdA with 2-ethynyl-para-carborane.
Scheme 20. Synthesis of doubly pyrene-substituted oligonucleotides.
3.4. Heck Couplings
In contrast to the other classic palladium-catalyzed C–C bond-forming reactions, there are limitedexamples of the Heck coupling of unprotected nucleosides. Baranger reported the coupling of2-IA with allylbenzene mediated by a stoichiometric amount of Pd(OAc)2/P(o-tolyl)3 in acetonitrile(Scheme 21) [62]. 2-(3-Phenyl-1-propenyl)adenosine (4) was isolated in 53% yield. Compound4 was then hydrogenated to give 2-(3-phenylpropyl)A. Palladium(PTA)2(saccharinate)2 (PTA =1,3,5-triaza-7-phosphaadamantane, Figure 2) is an effective precatalyst for the Heck coupling of 5-IdUand alkenes in acetonitrile to give 5-alkenylated dU derivatives in high yield (Scheme 22) [63]. Thesaccharinate complex was more active than other imidate PTA complexes (phthalimidate, maleimidate,or succinimidate).
34

Molecules 2015, 20, 9419–9454
Scheme 21. Heck coupling of 2-IA mediated by Pd(OAc)2/P(o-tolyl)3.
Scheme 22. Heck coupling of 5-IdU catalyzed by Pd(PTA)2(saccharinate)2.
4. Aqueous-Phase Cross-Coupling of Nucleosides, Nucleotides, and Oligonucleotides UsingHydrophilic Ligand-Supported Catalysts
Palladium-catalyzed cross-coupling reactions are typically performed in organic solvents usinghydrophobic supporting ligands, such as triphenylphosphine. For typical hydrophobic substrates,these homogeneous conditions typically offer optimal catalyst activity. Aqueous-biphasic usingwater-soluble transition metal catalysts offers a number of potential advantages over traditionalhomogeneous organic-phase catalysis [64–67]. Water is an attractive solvent as it is non-toxic,non-flammable, and a renewable resource. Separation of homogeneous catalysts from organicproducts is a common challenge, particularly in pharmaceutical processes [68]. The potential toconstrain the catalyst in the aqueous phase allows for easily separation from the organic products. Thestandard approach to design water-soluble ligands is to append hydrophilic functionality to commonlyused ligand structures, such as triphenylphosphine (Figure 2). Hydrophilic catalysts have primarilybeen applied in coupling of hydrophobic substrates. They also provide the opportunity to performhomogeneous coupling of hydrophilic substrates, such as biomolecules.
Figure 2. Hydrophilic ligands commonly applied in nucleoside cross-coupling reactions.
35

Molecules 2015, 20, 9419–9454
Casalnuovo [69] was the first to report the application of a water-soluble palladium/phosphinecatalyst for the cross-coupling of aryl halides. He showed that Pd(TPPMS)3 (TPPMS = sodiumdiphenyl(3-sulfonatophenyl)phosphine) provided an effective catalyst for Suzuki, Heck, andSonogashira couplings of aryl and heteroaryl halides in aqueous acetonitrile. In addition tohydrophobic substrates, examples of Heck and Sonogashira couplings of 5-IdU and 5-IdCMP werereported. By using a water-soluble catalyst system, cross-coupling of these hydrophilic substrateswas accomplished under homogeneous conditions. In the decade following Casalnuovo’s report,significant effort was devoted to developing new water-soluble phosphine ligands and their applicationto cross-coupling of water-insoluble substrates. In contrast, no examples of the application ofwater-soluble catalyst systems to nucleoside modification were reported in the decade followingCasalnuovo’s seminal paper.
4.1. Nucleosides
4.1.1. Suzuki Coupling
Methodology Development. The Shaughnessy group revisited the aqueous-phase cross-couplingof nucleosides with the goal of making this a general methodology for modification of bothpurine and pyrimidine nucleosides. Casalnuovo’s initial paper reported coupling of the morereactive iodopyrimidine nucleosides, whereas we had an interest in coupling the less reactive8-bromopurines, such as 8-Br(d)G and 8-Br(d)A. Catalysts derived from a range of hydrophilicphosphine ligands were screened for the ability to couple phenylboronic acid and 8-BrdG inaqueous acetonitrile [70]. The sterically demanding, electron-rich phosphine t-Bu-Pip-phos (4-(di-tert-butylphosphino)-N,N-dimethylpiperidinium chloride), which provides high activity catalysts forsimple aryl bromides [71], gave low conversion. Palladium in combination with TPPTS, providedan effective catalyst for this coupling despite being much less effective than t-Bu-Pip-phos in theaqueous-phase coupling of simple aryl halides.
The Pd(OAc)2/TPPTS catalyst system is a general method for Suzuki coupling of halogenatedpurine and pyrimidine nucleosides and 2′-deoxynucleosides. Good to excellent yields are affordedwith a range of aryl boronic acids and 5-IdU, 8-BrG, 8-BrdG, 8-BrA, and 8-BrdA (Scheme 23) [70].The methodology has also been extended to heteroarylboronic acids [72]. The order of reactivity ofhalonucleosides is 5-IdU > 8-BrdA >> 8-BrdG. The low reactivity of guanosine is believed to be due tocompetitive coordination of the deprotonated form of guanosine to palladium through the anionic N-1position [73].
Scheme 23. Pd(OAc)2/TPPTS-catalyzed Suzuki coupling of 8-BrdG.
The more sterically demanding TXPTS ligand provides a more active catalyst for Suzuki couplingof halonucleosides [70]. Using 10 mol % Pd/TXPTS, complete conversion of 8-BrdA to 8-PhdA wasachieved at room temperature in 30 minutes. In comparison, the TPPTS-derived catalyst required24 h to give 74% conversion to product. Even 8-BrdG gave 40% conversion to 8-PhdG after 18 h atroom temperature. Although TXPTS provides a more active catalyst, the Pd(OAc)2/TPPTS catalysthas become the standard system for these reactions. TPPTS is more widely commercially available and
36

Molecules 2015, 20, 9419–9454
costs about half of TXPTS on a per mole basis [74]. For challenging cases, TXPTS may prove to be anattractive alternative.
Organic cosolvents are not required in these reactions. Good yields can be achieved using water asthe only solvent [70,75,76]. In the case of the more reactive 5-IdU, the TPPTS ligand is not required [77].Good yields could be achieved with electron-rich arylboronic acids, but much lower yields wereobtained with electron-deficient boronic acids. Using TPPTS, good yields can be achieved with a broadrange of arylboronic acids in water alone.
Other hydrophilic ligands have been used in the Suzuki coupling of halonucleosides withboronic acids in aqueous media. Pd(PTA)2(phthalimidate)2 (PTA = 1,3,5-triaza-7-phosphaadamantane,Figure 2) is an effective precatalyst for the synthesis of 5-aryl-2′-deoxypyridimine derivatives from5-IdU and 5-IdC in water (Scheme 24) [78]. The phthalimidate complex gave higher yields thandihalide palladium-TPA complexes or the catalyst generated in situ from Pd(OAc)2 and PTA.Suzuki coupling of aryl- and alkenylboronic acids with 5-IdU can also be accomplished usingPd(OAc)2(2-aminopyrimidine-4,6-diolate)2 as the precatalyst (Scheme 25) [79]. These catalyst systemshave not been extended to the less reactive 8-halopurine nucleosides.
Scheme 24. Pd(PTA)2(phthalimidate)2-catalyzed Suzuki coupling of 5-IdU.
Scheme 25. Suzuki coupling of 5-IdU catalyzed by Pd-APD complexes.
Applications. The palladium/TPPTS methodology sparked interest in the direct functionalizationof unprotected nucleosides in aqueous reaction media. The homogeneous conditions have proven tobe more general than catalyst systems using hydrophobic ligands in water or polar organic solvents.This generality is particularly useful in the coupling of nucleotide and oligonucleotide substrates asdiscussed in Sections 4.2 and 4.3. The Pd/TPPTS catalyst system has been applied to the synthesis ofwide variety of modified nucleosides that incorporated fluorescent or electrochemically active reportergroups, coordination sites, or have potential pharmaceutical activity.
An early demonstration of the utility of the Pd/TPPTS catalyst system was the synthesis ofan amino acid-nucleoside adduct reported by Hocek [80]. The coupling of 8-BrA and 8-BrdA withphenylalanyl-4-boronic acid is achieved in 71 and 75% yield, respectively, in water-acetonitrile usingthe Pd/TPPTS catalyst system (Scheme 26). Notably, neither the nucleoside nor amino acid substratescontained protecting groups. In the coupling with 6-chloropurine nucleosides, improved yields
37

Molecules 2015, 20, 9419–9454
were obtained using microwave irradiation compared to traditional thermal heating [81]. The Hocekgroup has also reported the attachment of polypyridyl ligands to 8-BrdA [82], 7-I-7-deaza-dA [83],and 5-IdU [84] via Suzuki couplings using Pd/TPPTS (Scheme 27). High yields were achievedusing bipyridyl, phenanthryl, and terpyridyl boronic acids as either free ligands or preformedruthenium complexes. The ruthenium complex-modified nucleosides are of interest as luminescentand electroactive probes in oligonucleotides.
Scheme 26. Pd/TPPTS-catalyzed coupling of 8-bromoadenosines with phenylalanine.
Scheme 27. Attachment of a ruthenium(II) terpyridine complex to 7-I-7-deaza-dA.
The Pd/TPPTS catalyzed Suzuki coupling has been used to introduce a range of fluorescentprobes into nucleosides. This methodology has been used to attach five-membered ring heterocycles(2-pyrrolyl, 2-indolyl, 2-furyl, and 2-thiophenyl) to the 8 position of 2′-deoxyguanosine [85,86].The fluorescence of the resulting heterocyclic adducts is sensitive to hydrogen bonding with othernucleobases as well as the nucleoside conformation. The 8-(2-benzo[b]thienyl)-dG group serves as afluorescent reporter to probe for the preference for syn- or anti-conformations in duplex DNA [87].
Fluorescent pyrrolopyrimidopyrimidine [88] and pyrimidopyrimidoindole [89] nucleosides canbe prepared by aqueous-phase coupling of 5-IdC with N-Boc-protected 2-pyrrolylboronic acid orN-Boc-protected 2-indolylboronic acid (Scheme 28). The coupling is followed by condensation ofthe 6-amino group of the cytidine ring with the Boc moiety to give the fluorescent tri- or tetracyclicnucleoside analogs. Fluorescent 5-substituted uridine and 2-deoxyuridine analogs can be prepared bySuzuki coupling of 5-I(d)U with aryl or styrylboronic acid derivatives [90]. An alternative approach to5-styryluridine derivatives was achieved through the coupling of arylboronic acids with commerciallyavailable 5-(2-bromovinyl)uridine (BVDU, Scheme 29).
38
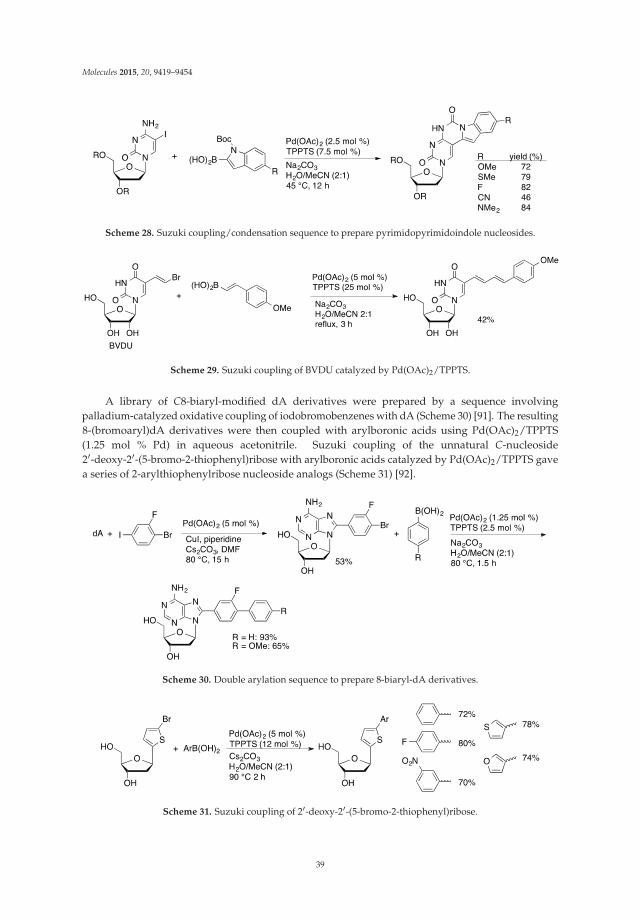
Molecules 2015, 20, 9419–9454
Scheme 28. Suzuki coupling/condensation sequence to prepare pyrimidopyrimidoindole nucleosides.
Scheme 29. Suzuki coupling of BVDU catalyzed by Pd(OAc)2/TPPTS.
A library of C8-biaryl-modified dA derivatives were prepared by a sequence involvingpalladium-catalyzed oxidative coupling of iodobromobenzenes with dA (Scheme 30) [91]. The resulting8-(bromoaryl)dA derivatives were then coupled with arylboronic acids using Pd(OAc)2/TPPTS(1.25 mol % Pd) in aqueous acetonitrile. Suzuki coupling of the unnatural C-nucleoside2′-deoxy-2′-(5-bromo-2-thiophenyl)ribose with arylboronic acids catalyzed by Pd(OAc)2/TPPTS gavea series of 2-arylthiophenylribose nucleoside analogs (Scheme 31) [92].
Scheme 30. Double arylation sequence to prepare 8-biaryl-dA derivatives.
Scheme 31. Suzuki coupling of 2′-deoxy-2′-(5-bromo-2-thiophenyl)ribose.
39

Molecules 2015, 20, 9419–9454
Suzuki coupling with Pd/TPPTS was used to introduce a photoresponsive chemical switch basedon a diarylethylene moiety in pyrimidine nucleosides. Suzuki coupling of boronate ester 5 with 5-IdUand 5-IdC afforded photoswitchable nucleosides that undergo reversible photochemical electrocycliccyclization under UV irradiation (Scheme 32) [93]. The modified nucleoside reverts to the open formunder visible light. The modified nucleosides could potentially be used to photochemically control thestructure and function of oligonucleotides.
Scheme 32. Synthesis of a photoswitchable cytidine derivative.
Nucleosides containing arylthiol moieties were prepared by Suzuki coupling of protected4-thiophenylboronic acid derivatives with 5-IdC and 7-I-7-deaza-dA using Pd(OAc)2/TPPTS inaqueous acetonitrile (Scheme 33) [94]. No conversion occurred in the presence of the free thiol group,but high yields were achieved with boronic acids containing protected thiols. The thiol-functionalizednucleosides can be incorporated into oligonucleotides and used to attach the DNA to gold surfaces.
Scheme 33. Synthesis of phenyl sulfide-substituted 7-deaza-dA derivatives.
Guanosine forms self-assembled tetrameric structures (G-tetrads) through intermolecularhydrogen bonding. In the presence of cations, these can further self assemble into G-quadruplexes.Structural modifications can be used to enhance this self assembly. Aqueous-phase Suzuki couplingwas used to prepare 8-(3- or 4-acetylphenyl)dG derivatives [95]. The acetylphenyl moiety enhancesG-tetrad formation by providing additional hydrogen bonding opportunities, while also extending thearomatic surface to improve noncovalent interactions.
8-Aryl-substituted derivatives of purine nucleosides are of interest as models of adducts formedin vivo during the metabolism of aromatic hydrocarbons. The motivation of our original studyof the aqueous-phase Suzuki coupling of 8-BrdG was to prepare adducts to study the effect ofthese modifications on DNA conformation [96]. The carcinogenesis of polyaromatic hydrocarbon isthought to involve covalent modification of guanosine during metabolism of these compounds in vivo.Suzuki coupling of 1-pyrenylboronic acid, 1-naphthylboronic acid, and 9-phenanthrenylboronic acidwith 8-BrdA gave good yields of the adducts using Pd(OAc)2/TPPTS (Scheme 34) [5]. The more
40

Molecules 2015, 20, 9419–9454
hindered 8-anthracenylboronic acid and its benzannulated analogs could not be coupled under theseconditions, however.
Scheme 34. Coupling of 8-BrdA with polyaromatic boronic acids.
Arylated nucleoside derivatives have been explored as potential pharmaceutically activecompounds with antiviral and anticancer activity. The Hocek group has used the Pd(OAc)2/TPPTScatalyst system to prepare a wide variety of natural and non-natural nucleoside derivatives throughaqueous-phase Suzuki couplings. A library of 6-heteroarylpurine nucleosides were prepared fromunprotected 6-cloropurine and 6-chloro-7-deazapurine nucleosides [97,98]. Good yields (40%–80%)were achieved with a variety of aryl and heteroaryl boronic acids. A similar library was preparedfrom 7-I-7-deaza-A [99]. A large library of 6-arylpurine nucleoside monophosphates were preparedby aqueous-phase Suzuki coupling of 6-chloropurine nucleoside with arylboronic acids followed byphosphorylation (Scheme 35) [100]. Further elaboration of 6-(3-bromophenyl)purine nucleoside witha subsequent Suzuki coupling afforded 6-biarylpurine nucleosides. The compounds were tested fortheir ability to inhibit 2′-deoxynucleoside 5′-phosphate N-hydrolase 1 (DNPH1), which is a potentialanticancer target.
The Hocek group has also used aqueous-phase Suzuki couplings to prepare arylated nucleosidederivatives in which the ribose unit has been modified. Good yields are obtained in coupling ofaryl and heteroarylboronic acids with 7-iodo-7-deazaadenine arabinoside (6) [101], 7-iodo-7-deaza-2′-C-methyladenosine (7) [101], 7-iodo-7-deazaadenine 2′-deoxy-2′-fluoroarabinoside (8) [101],6-chloro-7-deazapurine 2′-deoxy-2′-fluororibinoside (9) [102], and 7-iodo-7-deazaadenine 2′-deoxy-2′,2′-difluoro-β-D-erythro-pentofuranoside (10, Figure 3) [102]. The sugar modifications have littleeffect on the Suzuki coupling reaction. A family of 6-substituted-7-aryl-7-deazapurinenucleosides(13) was prepared starting from a protected 6-chloro-7-iodo-7-deazapurine nucleoside derivative (11,Scheme 36) [103]. Selective nucleophilic aromatic substitution at the 6-chloro position followed bydeprotection provides the 7-iodo precursors (12). These were coupled with aryl or heteroarylboronicacids using Pd(OAc)2/TPPTS.
4.1.2. Sonogashira Coupling
Palladium-catalyzed alkynylation of unprotected nucleosides with hydrophobic palladium/phosphinecatalysts is well precedented. Recently, aqueous-phase Sonogashira couplings using hydrophilicligands has received increasing attention. The Hocek group used Sonogashira couplings, in additionto Suzuki couplings, to introduce polypyridyl ligands to 8-BrdA (Scheme 37) [82], 7-I-7-deaza-dA [83],and 5-IdU [84] using the Pd/TPPTS catalyst system in combination with catalytic CuI in DMF. Highyields were achieved with bipyridyl alkynes (82%–96%). In contrast to the Suzuki coupling, lowyields were obtained in the coupling of ruthenium-coordinated analogs of the bipyridinyl alkynes(0%–57%). Decomposition of the alkyne-substituted ruthenium complexes competed with the desiredcross-coupling under the Sonogashira reaction conditions.
41
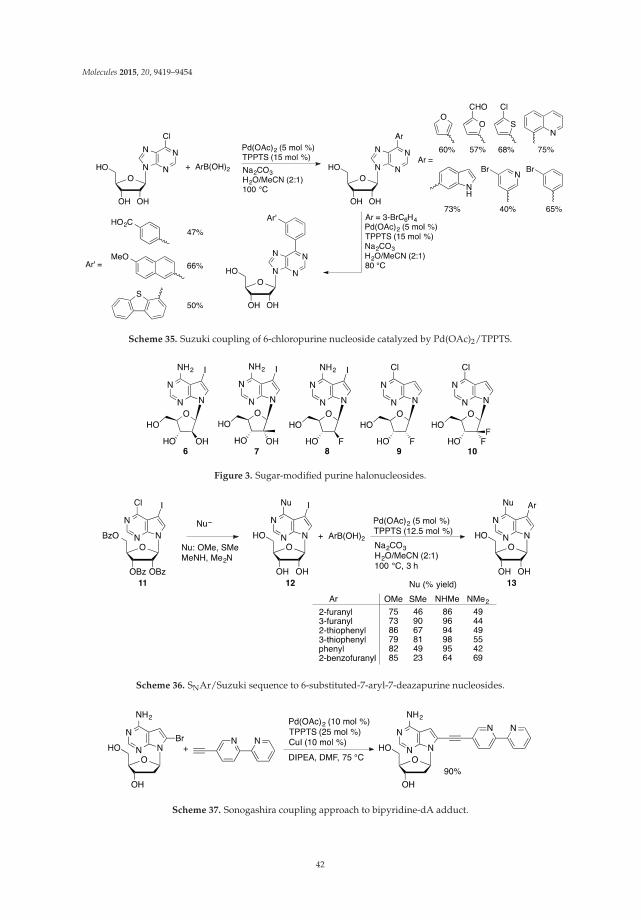
Molecules 2015, 20, 9419–9454
Scheme 35. Suzuki coupling of 6-chloropurine nucleoside catalyzed by Pd(OAc)2/TPPTS.
Figure 3. Sugar-modified purine halonucleosides.
Scheme 36. SNAr/Suzuki sequence to 6-substituted-7-aryl-7-deazapurine nucleosides.
Scheme 37. Sonogashira coupling approach to bipyridine-dA adduct.
42

Molecules 2015, 20, 9419–9454
Sonogashira coupling with Pd(OAc)2/TPPTS/CuI is effective for the coupling of 7-I-7-deaza-dAand 5-IdU with propargyl esters or amides of bile acids (Scheme 38) [104]. Yields of isolated productsranged from 31%–90%. The bile acid nucleoside adducts are of interest for oligonucleotide amphiphiles.
Scheme 38. Synthesis of bile acid-dC adducts through aqueous-phase Sonogashira coupling.
The more sterically demanding TXPTS ligand provides a more active catalyst for the Sonogashiracoupling of 8-halopurines and 5-IdU than the catalyst derived from TPPTS [105,106]. In the couplingof 5-IdU with phenylacetylene catalyzed by TPPTS, Pd(OAc)2 and CuI in aqueous acetonitrile reached80% conversion after six hours at 50 ◦C. Under the same conditions, 90% conversion was achieved inone hour using TXPTS as the ligand. The Pd(OAc)2/TXPTS system was effective for the coupling ofaryl and alkyl acetylenes with 8-Br(d)A and 8-BrdG (Scheme 39). In the case of 5-IdU, the Sonogashirareaction was followed by nucleophilic attack of the C6-O on the alkyne resulting in formation offurano [2,3]-pyrimidin-2-one byproducts (14, Scheme 40).
Scheme 39. Pd/TXPTS-catalyzed Sonogashira coupling of 8-bromopurine nucleosides.
Scheme 40. Pd/TXPTS-catalyzed Sonogashira coupling of 5-IdU.
4.1.3. Heck Couplings
There are relatively few examples of Heck couplings of unprotected nucleosides reported withhydrophilic ligands. The Shaughnessy group reported the Heck coupling of 5-IdU with styreneand conjugated enones [107]. The Pd(OAc)2/TPPTS gave good yields of 5-alkeynyl products, butsimilar yields were obtained when no ligand was used. More sterically demanding (TXPTS) orelectron-rich ligands (t-Bu-Amphos) did not improve the catalyst performance. The Pd(OAc)2/TPPTScatalyst was successfully used in the synthesis of 8-styryl-dG starting from 8-BrdG at 80 ◦C in 2:1water/acetonitrile [55].
43

Molecules 2015, 20, 9419–9454
4.2. Nucleotides
Modified nucleosides are often prepared as precursors to nucleotides, which can be enzymaticallyincorporated into oligonucleotides. Selective 5′-phosphorylation of modified nucleosides representsone common approach to preparing base-modified nucleotides. Phosphorylation of lipophilic modifiednucleosides can be difficult to achieve in high yield and good selectivity for the 5′-oxygen [108].An alternative approach is to perform the cross-coupling reaction directly on the halogenatednucleotide. Water-soluble catalyst systems have proven to be effective in performing cross-couplingreactions with nucleoside mono-, di-, and triphosphate substrates. In contrast, there are noreported examples of the use of catalysts derived from hydrophobic ligands in the direct reaction ofhalogenated nucleotides.
4.2.1. Suzuki Coupling
The Pd/TPPTS catalyst system that is widely used for nucleoside Suzuki couplings is alsoeffective in couplings with halogenated nucleotide derivatives. Early examples of Suzuki couplingsof halonucleotides were reported by the Wagner [109] and Hocek groups [81,110]. Both groups useda catalyst derived from a palladium(II) salt and TPPTS (Scheme 41). Notably, the reaction could becarried out at a high pH (0.3 M CO3
2−) at an elevated temperature (80–120 ◦C) without decompositionof the nucleotide.
Scheme 41. Pd/TPPTS-catalyzed Suzuki coupling of 8-bromoguanosine phosphates.
Hocek reported similar conditions for the synthesis of 8-MedATP and 8-PhdATP from8-BrdATP [111]. The methyl-substituted nucleoside was prepared using methylboronic acid, which isa rare example of the use of an alkylboron reagent in coupling reactions with nucleoside derivatives(Scheme 42). The resulting modified nucleosides could be incorporated into oligonucleotidesusing polymerase enzymes. This methodology was used to prepared 5-ArdUTP, 5-ArdCTP, and7-Ar-7-deazadATP derivatives with 3-nitrophenyl and 3-aminophenyl aryl groups [112]. Themodified nucleotides were incorporated into oligonucleotide sequences. The modifications serveas electrochemical labels in oligonucleotides that are sensitive to the local sequence.
Scheme 42. Methylation of 8-BrATP.
These conditions have been applied to the synthesis of a variety of functionalized nucleotidesthat were then incorporated into oligonucleotides using polymerase enzymes. Alkylsulfanylphenyl-modified nucleotides were prepared modes yields (10%–50%) by coupling of the thioether-functionalized boronic acids with 7-I-7-deaza-dATP and 5-ICTP using the Pd/TPPTS catalyst
44

Molecules 2015, 20, 9419–9454
system [94]. The resulting functionalized nucleotides were enzymatically incorporated into oligonucleotides.The functionalized oligonucleotides were studied electrochemically and found to associate at the goldelectrode surface.
Benzofurane has been proposed as a novel electrochemically active probe functionality forthe study of oligonucleotides. The benzofurane moiety was attached to 7-deaza-dATP and dCTPvia an aqueous-phase Suzuki coupling catalyzed by Pd/TPPTS [113]. Low yields (10%–22%)were obtained for the Suzuki coupling of the halonucleoside triphosphate substrates (Scheme 43).Suzuki coupling of the halonucleoside followed by phosphorylation gave a higher overall yield(17%–52%) of the benzofurane nucleotide adducts. Electrochemically active benzofurane moietieswere attached to nucleotides in incorporated into oligonucleotides in parallel with nitrophenyl- andaminophenyl-modified nucleotides. The three electroactive groups could be addressed independentlywithout apparent interference.
Scheme 43. Synthesis of benzofurane-dATP.
Solvatochromatic and pH-sensitive dual fluorescent 19F-NMR probes were attached to7-I-7-deaza-dATP and 5-IdUTP using an aqueous-phase Suzuki coupling (Scheme 44) [114]. Thenucleotides were incorporated into oligonucleotides where they showed environment-dependentfluorescence properties. The Pd/TPPTS catalyst was used to prepare 5-formylthiophen-2-yl-modifiednucleotides, which were then incorporated into oligonucleotides via primer extension or polymerasechain reaction protocols (Scheme 45) [115]. The aldehyde-modified oligonucleotides were thenconjugated with amine derivatives through the formation of imine linkages. This methodologywas used for selective staining of the aldehyde-containing oligonucleotides.
Scheme 44. Synthesis of pH-sensitive dual fluorescent 19F-NMR probe nucleotides.
The aqueous-phase Suzuki coupling protocol has also been applied to the synthesis ofdinucleotides and nucleotide-sugar conjugates. A series of arylboronic acids were coupled with5-I-UDP glucoside (15) in water catalyzed by Na2PdCl4/TPPTS (1 mol %) to give 5-arylated products16 in 40%–64% yield (Scheme 46) [116]. The corresponding 5-Br-UDP-glucose substrate gave noconversion under identical conditions. Analysis of the nucleotide conformation by NOE spectroscopyshowed that 5-Br-UDP-glucose preferred an anti-configuration that places the bromide over the ribosering, potentially making it less accessible. In contrast, 5-I-UDP-glucose preferred a syn-conformationin which the iodide is more readily accessible to the catalyst. It should also be noted that C–I bonds
45
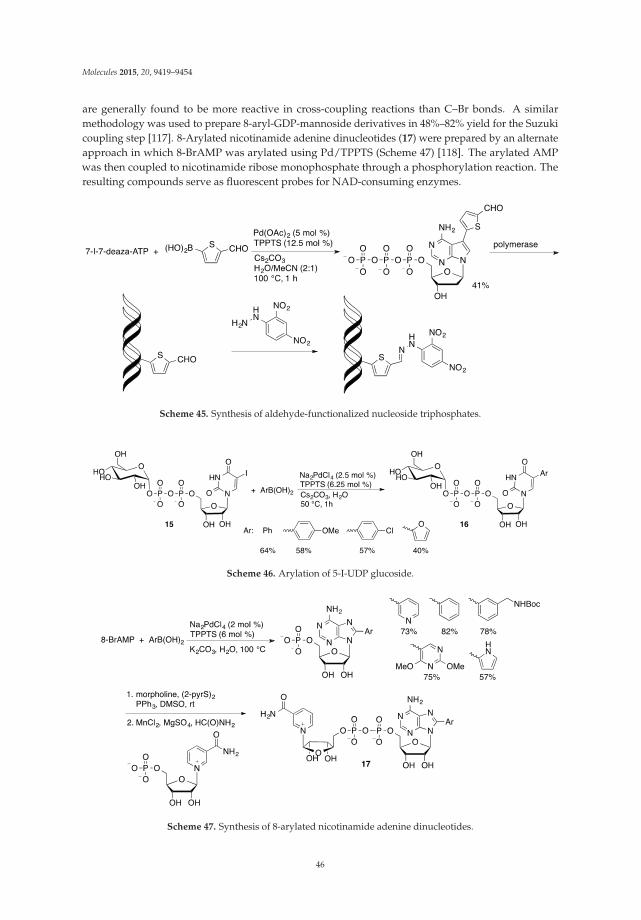
Molecules 2015, 20, 9419–9454
are generally found to be more reactive in cross-coupling reactions than C–Br bonds. A similarmethodology was used to prepare 8-aryl-GDP-mannoside derivatives in 48%–82% yield for the Suzukicoupling step [117]. 8-Arylated nicotinamide adenine dinucleotides (17) were prepared by an alternateapproach in which 8-BrAMP was arylated using Pd/TPPTS (Scheme 47) [118]. The arylated AMPwas then coupled to nicotinamide ribose monophosphate through a phosphorylation reaction. Theresulting compounds serve as fluorescent probes for NAD-consuming enzymes.
Scheme 45. Synthesis of aldehyde-functionalized nucleoside triphosphates.
Scheme 46. Arylation of 5-I-UDP glucoside.
Scheme 47. Synthesis of 8-arylated nicotinamide adenine dinucleotides.
46

Molecules 2015, 20, 9419–9454
4.2.2. Sonogashira Coupling
Sonogashira coupling of alkynes with halogenated nucleotides is an effective method to introduceprobe species. Burgess was the first to demonstrate this methodology in the synthesis of fluorescentdye-modified uridine triphosphates (18, Scheme 48) [108]. Fluorescein-based dyes with alkynesubstituents were coupled with 5-I-dUTP using a preformed Pd/TPPTS catalyst and CuI at roomtemperature with triethylamine as the base in phosphate buffer. The preformed catalyst was generatedby mixing Na2PdCl4, TPPTS, NaBH4 and water at room temperature followed by removal of waterand recovery of the resulting solid. Alkynylation of the nucleotide substrate was necessary toproduce the desired compounds. In contrast, attempted phosphorylation of fluorescein-modified dUwas unsuccessful.
Scheme 48. Sonogashira coupling of 5-I-dUTP and ethynylfluorescein.
Phenylalanyl-modified nucleotide triphosphates can be prepared in 60%–67% yield by theSonogashira coupling of 4-ethynylphenylalanine with halonucleotides using a catalyst derivedfrom Pd(OAc)2, TPPTS, and CuI in aqueous acetonitrile. This method was also used to prepareethynylferrocene-modified nucleotide triphosphates (Scheme 49) [119]. Oligonucleotides preparedfrom the ferrocene-functionalized nucleotides are electrochemical probes of DNA binding. Bile acidconjugates of nucleotides were prepared in moderate yields (32%–57%) by coupling of propargyl bileacid amides with iodonucleotide triphosphates [104].
Scheme 49. Synthesis of ferrocenyl-modified 7-deaza-dATP.
4.2.3. Heck Coupling
To date there is only one example of a Heck coupling of a halonucleotide. Hocek reportedthe coupling of butyl acrylate with a range of 5-iodopurine and 7-iodo-7-deazapurine nucleosidemono- and triphosphates using Pd(OAc)2 and TPPTS in water/acetonitrile at 80 ◦C (Scheme 50) [120].The yields with nucleotides were modest (14%–55%) compared to the high yields obtained with thecorresponding nucleosides (81%–98%). The monophosphates gave higher yields than triphosphates.Although modest yields were obtained in the Heck coupling of nucleotides, the yields were similar tothose obtained when the Heck coupling was carried out on the nucleoside followed by phosphorylation.
47

Molecules 2015, 20, 9419–9454
Cytidine gave low yields in the Heck coupling and no conversion was obtained with cytidine mono-or triphosphate. The resulting alkene-modified nucleotides could be successfully incorporated intooligonucleotides by primer extension methods.
Scheme 50. Aqueous-phase Heck coupling of 7-iodo-7-deazaguanosine monophosphate.
4.3. Oligonucleotides
Palladium-catalyzed coupling reactions provide effective ways to attach a wide variety of moietiesto nucleoside or nucleotide structures. The modified nucleosides can in many cases be effectivelyincorporated into oligonucleotides using primer extension, PCR, or solid-phase DNA synthesismethods. Although these approaches to modified oligonucleotides have been demonstrated, significantlimitations to these approaches have been observed. For example, C8-arylated purine nucleosides aresignificantly more prone to acidic hydrolysis of the glycosidic bond and are more sensitive to oxidationthan dA or dG [121]. As a result, they often are not compatible with solid-phase DNA synthesistechniques. A variety of modified nucleotide derivatives have been incorporated into oligonucleotidesthrough enzymatic polymerase approaches, but steric limitations can limit the effectiveness of thisapproach [110,111].
A more general approach to preparing oligonucleotides with modified bases would be to buildthe nucleotide containing halogenated residues at the desired locations, followed by post-syntheticpalladium-catalyzed coupling of the halonucleosides. The successful coupling of nucleotides suggeststhat the oligonucleotide backbone should be stable in the coupling reaction. An oligonucleotiderepresents a significantly more complex substrate than a simple nucleotide monomer, however.Successful development of this approach would allow a variety of modified oligonucleotides tobe prepared from a common precursor containing a halogenated base residue at the desired positionon the oligonucleotide.
Manderville reported the first successful example of the post-synthetic cross-coupling ofa halogenated oligonucleotide [121]. Solution-phase coupling of oligonucleotides containing asingle 8-bromoguanosine residue with arylboronic acids was performed with Pd(OAc)2/TPPTS inwater/acetonitrile with Na2CO3 as base (Scheme 51). Under optimized conditions, a guanine richdecanucleotide containing 8-BrdG (19) was arylated with 2-hydroxyphenylboronic acid to give 20 in87% yield. In contrast, an attempt to prepare the same 8-arylguanine-containing decanucleotideby traditional solid-phase DNA synthetic methods resulted in the formation of a mixture ofproducts. The major products were truncated oligomers that did not incorporate the 8-arylguanosineresidue. Subsequent studies showed that a maximum of two bases could be incorporated after the8-arylguanosine residue in the oligomerization process. The post-synthetic coupling approach wasapplied to the synthesis of oligonucleotides with up to 15 bases containing a single 8-BrdG residue.
The post-synthetic approach was applied to the synthesis of oligonucleotides containingdiarylethylene photoswitches [93]. Oligonucleotides (15- and 19-mers) containing 5-IdC or 5-IdUresidues were coupled with boronic acid 5 with Pd(OAc)2/TPPTS in aqueous acetonitrile at 120 ◦C togive the coupled oligonucleotides in modest yields (16%–35%). The hindered boronic acid (5) is a muchmore challenging substrate than those used by Manderville. Coupling of oligonucleotides containing5-IdU with vinylboronic acids occurs in high yields (49%–95%) using a preformed palladium complexof 2-aminopyrimidine-4,6-diolate (APD) as the precatalyst (Scheme 52) [79]. With the less-hindered
48
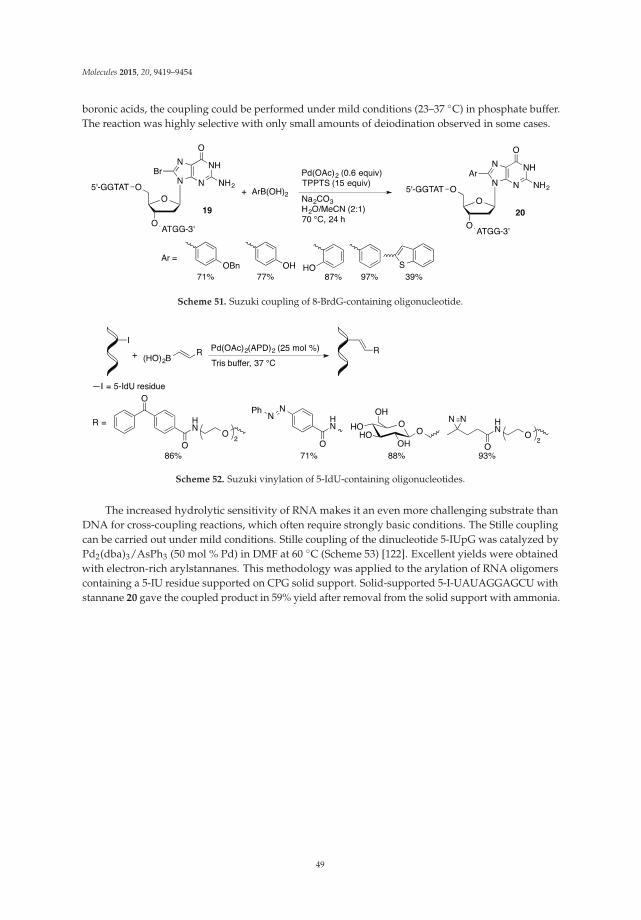
Molecules 2015, 20, 9419–9454
boronic acids, the coupling could be performed under mild conditions (23–37 ◦C) in phosphate buffer.The reaction was highly selective with only small amounts of deiodination observed in some cases.
Scheme 51. Suzuki coupling of 8-BrdG-containing oligonucleotide.
Scheme 52. Suzuki vinylation of 5-IdU-containing oligonucleotides.
The increased hydrolytic sensitivity of RNA makes it an even more challenging substrate thanDNA for cross-coupling reactions, which often require strongly basic conditions. The Stille couplingcan be carried out under mild conditions. Stille coupling of the dinucleotide 5-IUpG was catalyzed byPd2(dba)3/AsPh3 (50 mol % Pd) in DMF at 60 ◦C (Scheme 53) [122]. Excellent yields were obtainedwith electron-rich arylstannanes. This methodology was applied to the arylation of RNA oligomerscontaining a 5-IU residue supported on CPG solid support. Solid-supported 5-I-UAUAGGAGCU withstannane 20 gave the coupled product in 59% yield after removal from the solid support with ammonia.
49

Molecules 2015, 20, 9419–9454
Scheme 53. Stille coupling of 5-I-UG.
Although only a few examples have been reported to date, the post-functionalization ofoligonucleotides offers an exciting opportunity to prepare modified RNA and DNA structures. To date,only the Suzuki and Stille coupling of halogenated oligonucleotides has been demonstrated. Extensionof this methodology to other important coupling reactions, such as the Sonogashira coupling, willfurther expand the usefulness of this transformation.
5. Conclusions
Palladium-catalyzed cross-coupling has developed into a highly effective method for themodification of unprotected nucleosides through carbon–carbon bond-forming reactions. Through theuse of polar media, such as water, polar aprotic solvents, or combinations of the two, the hydrophilicnucleosides can be solubilized and converted to the desired adducts without the need to convert thenucleoside to a more lipophilic form. The ability to directly functionalize the nucleoside increases theoverall yield and atom efficiency of the synthesis by avoiding the protection/deprotection sequence.In addition, protection strategies are generally not possible with the more hydrophilic nucleotide andoligonucleotide substrates. Catalysts supported with hydrophilic ligands provide the most generalcatalysts for modification of nucleoside derivatives. In aqueous solvents, hydrophilic catalysts areeffective with nucleoside, nucleotides (mono- and triphosphates), and oligonucleotides. The abilityto directly couple oligonucleotides is particularly noteworthy. Firstly, the ability to couple highlycomplex biomacromolecules is a true testament to the power and flexibility of palladium-catalyzedcross-coupling. Furthermore, traditional oligomerization strategies are often not compatible withmodified nucleoside derivatives, so the ability to perform cross-coupling directly on oligonucleotidesmay be the only route to these materials.
Significant progress has been made in the area of palladium-catalyzed coupling of unprotectednucleosides, but there are still challenges left to be conquered. Although many of the classicC–C bond-forming coupling reactions have been demonstrated with unprotected nucleosides,examples of carbon-heteroatom bond formations remain unknown. In contrast, metal-catalyzedcarbon-heteroatom coupling reactions of unprotected nucleosides are well precedented [17]. In general,Buchwald-Hartwig-type coupling reactions are less effective in aqueous solvent systems, althoughrecent examples have been reported [123–125]. Developing these classes of coupling reactions
50

Molecules 2015, 20, 9419–9454
with unprotected nucleosides would provide access to new classes of nucleoside derivatives. Thedevelopment of direct coupling reactions of arenes through C–H bond activation represents anotherattractive area of development. Heterocycles are common substrates for these types of reactions.In addition, the nucleobase heterocycles provide the opportunity for directed C–H functionalizationreactions. Finally, further development of direct functionalization of oligonucleotides containinghalogenated base residues will provide a route to prepare libraries of oligonucleotides containingmodified bases. To date these reactions have been demonstrated with Suzuki and Stille couplings.Extension of these reactions to other classes of cross-coupling reactions would significantly increasethe flexibility of these methodologies.
Acknowledgments: Support for our work in this area by the National Science Foundation (CHE-0124255) isgratefully acknowledged.
Conflicts of Interest: The authors declare no conflict of interest.
References and Notes
1. De Clercq, E. The unabated synthesis of new nucleoside analogs with antiviral potential: A tribute to MorrisJ. Robins. Nucleos. Nucleot. Nucl. Acids 2009, 28, 586–600. [CrossRef]
2. Robak, T. New Purine Nucleoside Analogs for Acute Lymphoblastic Leukemia. Clin. Cancer Drugs 2014,1, 2–10. [CrossRef]
3. Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside andnucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [CrossRef][PubMed]
4. Sofia, M.J.; Chang, W.; Furman, P.A.; Mosley, R.T.; Ross, B.S. Nucleoside, Nucleotide, and Non-NucleosideInhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2012, 55, 2481–2531.[CrossRef] [PubMed]
5. Dai, Q.; Xu, D.; Lim, K.; Harvey, R.G. Efficient Syntheses of C8-Aryl Adducts of Adenine and GuanineFormed by Reaction of Radical Cation Metabolites of Carcinogenic Polycyclic Aromatic Hydrocarbons withDNA. J. Org. Chem. 2007, 72, 4856–4863. [CrossRef] [PubMed]
6. Champeil, E.; Pradhan, P.; Lakshman, M.K. Palladium-catalyzed synthesis of nucleoside adducts frombay-and fjord-region diol epoxides. J. Org. Chem. 2007, 72, 5035–5045. [CrossRef] [PubMed]
7. Lakshman, M.K.; Gunda, P. Palladium-catalyzed synthesis of carcinogenic polycyclic aromatic hydrocarbonepoxide-nucleoside adducts: The first amination of a chloro nucleoside. Org. Lett. 2003, 5, 39–42. [CrossRef][PubMed]
8. Tanpure, A.A.; Pawar, M.G.; Srivatsan, S.G. Fluorescent Nucleoside Analogs: Probes for InvestigatingNucleic Acid Structure and Function. Isr. J. Chem. 2013, 53, 366–378. [CrossRef]
9. Toseland, C.P.; Webb, M.R. Fluorescent nucleoside triphosphates for single-molecule enzymology. In SingleMolecule Enzymology: Methods and Protocols; Mashanov, G.I., Batters, C., Eds.; Spring Science+Business Media,LLC: New York, NY, USA, 2011; Volume 778, pp. 161–174.
10. Matarazzo, A.; Hudson, R.H.E. Fluorescent adenosine analogs: A comprehensive survey. Tetrahedron 2015,71, 1627–1657. [CrossRef]
11. Dodd, D.W.; Hudson, R.H.E. Intrinsically fluorescent base-discriminating nucleoside analogs. Mini-Rev.Org. Chem. 2009, 6, 378–391. [CrossRef]
12. Thomsen, N.M.; Vongsutilers, V.; Gannett, P.M. Synthesis of C8-Aryl Purines, Nucleosides, andPhosphoramidites. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 155–176. [CrossRef] [PubMed]
13. Kore, A.R.; Charles, I. Recent developments in the synthesis and applications of C5-substituted pyrimidinenucleosides and nucleotides. Curr. Org. Chem. 2012, 16, 1996–2013. [CrossRef]
14. Kore, A.R.; Yang, B.; Srinivasan, B. Recent Developments in the Synthesis of Substituted Purine Nucleosidesand Nucleotides. Curr. Org. Chem. 2014, 18, 2072–2107. [CrossRef]
15. Colacot, T.J. New Trends in Cross-Coupling: Theory and Applications; Royal Society of Chemistry: London,UK, 2015; p. 864.
16. Agrofoglio, L.A.; Gillaizeau, I.; Saito, Y. Palladium-assisted routes to nucleosides. Chem. Rev. 2003, 103,1875–1916. [CrossRef] [PubMed]
51

Molecules 2015, 20, 9419–9454
17. Lakshman, M.K. Synthesis of biologically important nucleoside analogs by palladium-catalyzed C–N bondformation. Curr. Org. Synth. 2005, 2, 83–112. [CrossRef]
18. Hocek, M.; Fojta, M. Cross-coupling reactions of nucleoside triphosphates followed by polymeraseincorporation. Construction and applications of base-functionalized nucleic acids. Org. Biomol. Chem.2008, 6, 2233–2241. [CrossRef] [PubMed]
19. De Ornellas, S.; Williams, T.J.; Baumann, C.G.; Fairlamb, I.J.S. Catalytic C-H/C-X bond functionalisationof nucleosides, nucleotides, nucleic acids, amino acids, peptides and proteins. In C-H and C-X BondFunctionalization; Ribas, X., Ed.; Royal Society of Chemistry: Cambridge, UK, 2013; Volume 11, pp. 409–447.
20. Hervé, G.; Sartori, G.; Enderlin, G.; MacKenzie, G.; Len, C. Palladium-catalyzed Suzuki reaction in aqueoussolvents applied to unprotected nucleosides and nucleotides. RSC Adv. 2014, 4, 18558–18594. [CrossRef]
21. Hervé, G.; Len, C. Heck and Sonogashira couplings in aqueous media—Application to unprotectednucleosides and nucleotides. Sust. Chem. Proc. 2015, 3, 1–14. [CrossRef]
22. Hervé, G.; Len, C. Aqueous microwave-assisted cross-coupling reactions applied to unprotected nucleosides.Front. Chem. 2015, 3. [CrossRef]
23. Bigge, C.F.; Kalaritis, P.; Deck, J. R.; Mertes, M.P. Palladium-catalyzed coupling reactions of uracil nucleosidesand nucleotides. J. Am. Chem. Soc. 1980, 102, 2033–2038. [CrossRef]
24. Hirota, K.; Isobe, Y.; Kitade, Y.; Maki, Y. A simple synthesis of 5-(1-alkenyl)uracil derivatives by palladiumcatalyzed oxidative coupling of uracils with olefins. Synthesis 1987, 5, 495–496. [CrossRef]
25. Deraedt, C.; Astruc, D. “Homeopathic” Palladium Nanoparticle Catalysis of Carbon–Carbon CouplingReactions. Acc. Chem. Res. 2014, 47, 494–503. [CrossRef] [PubMed]
26. Matsuhashi, H.; Hatanaka, Y.; Kuroboshi, M.; Hiyama, T. Synthesis of 5-substituted pyrimidine nucleosidesthrough a palladium-catalyzed cross-coupling of alkylhalosilanes. Heterocycles 1996, 42, 375–384. [CrossRef]
27. Gallagher-Duval, S.; Hervé, G.; Sartori, G.; Enderlin, G.; Len, C. Improved microwave-assisted ligand-freeSuzuki-Miyaura cross-coupling of 5-iodo-2′-deoxyuridine in pure water. New J. Chem. 2013, 37, 1989–1995.[CrossRef]
28. Kumar, P.; Hornum, M.; Nielsen, L.J.; Enderlin, G.; Andersen, N.K.; Len, C.; Hervé, G.; Sartori, G.; Nielsen, P.High-Affinity RNA Targeting by Oligonucleotides Displaying Aromatic Stacking and Amino Groups inthe Major Groove. Comparison of Triazoles and Phenyl Substituents. J. Org. Chem. 2014, 79, 2854–2863.[CrossRef] [PubMed]
29. Enderlin, G.; Sartori, G.; Hervé, G.; Len, C. Synthesis of 6-aryluridines via Suzuki-Miyaura cross-couplingreaction at room temperature under aerobic ligand-free conditions in neat water. Tetrahedron Lett. 2013, 54,3374–3377. [CrossRef]
30. De Vries, J.G. A unifying mechanism for all high-temperature Heck reactions. The role of palladium colloidsand anionic species. Dalton Trans. 2006, 421–429.
31. Sakthivel, K.; Barbas, C.F., III. Expanding the Potential of DNA for Binding and Catalysis: HighlyFunctionalized dUTP Derivatives That Are Substrates for Thermostable DNA Polymerases. Angew. Chem.Int. Ed. 1998, 37, 2872–2875. [CrossRef]
32. Ding, H.; Greenberg, M.M. Hole Migration is the Major Pathway Involved in Alkali-Labile Lesion Formationin DNA by the Direct Effect of Ionizing Radiation. J. Am. Chem. Soc. 2007, 129, 772–773. [CrossRef] [PubMed]
33. Hervé, G.; Len, C. First ligand-free, microwave-assisted, Heck cross-coupling reaction in pure water ona nucleoside—Application to the synthesis of antiviral BVDU. RSC Adv. 2014, 4, 46926–46929. [CrossRef]
34. Hussain, I.; Singh, T. Synthesis of Biaryls through Aromatic C–H Bond Activation: A Review of RecentDevelopments. Adv. Synth. Catal. 2014, 356, 1661–1696. [CrossRef]
35. Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Cross-Coupling of Heteroarenes by C–H Functionalization:Recent Progress towards Direct Arylation and Heteroarylation Reactions Involving Heteroarenes ContainingOne Heteroatom. Adv. Synth. Catal. 2014, 356, 17–117. [CrossRef]
36. Cerna, I.; Pohl, R.; Hocek, M. The first direct C–H arylation of purine nucleosides. Chem. Commun. 2007, 45,4729–4730. [CrossRef]
37. Storr, T.E.; Firth, A.G.; Wilson, K.; Darley, K.; Baumann, C.G.; Fairlamb, I.J.S. Site-selective direct arylation ofunprotected adenine nucleosides mediated by palladium and copper: Insights into the reaction mechanism.Tetrahedron 2008, 64, 6125–6137. [CrossRef]
52

Molecules 2015, 20, 9419–9454
38. Storr, T.E.; Baumann, C.G.; Thatcher, R.J.; De Ornellas, S.; Whitwood, A.C.; Fairlamb, I.J.S.Pd(0)/Cu(I)-Mediated Direct Arylation of 2′-Deoxyadenosines: Mechanistic Role of Cu(I) and ReactivityComparisons with Related Purine Nucleosides. J. Org. Chem. 2009, 74, 5810–5821. [CrossRef] [PubMed]
39. Liang, Y.; Gloudeman, J.; Wnuk, S.F. Palladium-Catalyzed Direct Arylation of 5-Halouracils and 5-HalouracilNucleosides with Arenes and Heteroarenes Promoted by TBAF. J. Org. Chem. 2014, 79, 4094–4103. [CrossRef][PubMed]
40. Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [CrossRef] [PubMed]41. Amann, N.; Pandurski, E.; Fiebig, T.; Wagenknecht, H.A. Electron injection into DNA: Synthesis and
spectrscopic properties of pyrenyl-modified oligonucleotides. Chem. Eur. J. 2002, 8, 4877–4883. [CrossRef][PubMed]
42. Amann, N.; Wagenknecht, H.A. Preparation of pyrenyl-modified nucleosides via Suzuki-Miyauracross-coupling reactions. Synlett 2002, 5, 687–691. [CrossRef]
43. Jacobsen, M.F.; Ferapontova, E.E.; Gothelf, K.V. Synthesis and electrochemical studies of ananthraquinone-conjugated nucleoside and derived oligonucleotides. Org. Biomol. Chem. 2009, 7, 905–908.[CrossRef] [PubMed]
44. Okamoto, A.; Inasaki, T.; Saito, I. Synthesis and ESR studies of nitronyl nitroxide-tetheredoligodeoxynucleotides. Tetrahedron Lett. 2005, 46, 791–795. [CrossRef]
45. Kögler, M.; Vanderhoydonck, B.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Herman, J.; Louat, T.; Parchina, A.;Sibley, C.; Lescrinier, E.; et al. Synthesis and Evaluation of 5-Substituted 2′-deoxyuridine MonophosphateAnalogs As Inhibitors of Flavin-Dependent Thymidylate Synthase in Mycobacterium tuberculosis. J. Med.Chem. 2011, 54, 4847–4862. [CrossRef] [PubMed]
46. Fresneau, N.; Hiebel, M.A.; Agrofoglio, L.A.; Berteina-Raboin, S. Efficient synthesis of unprotectedC-5-aryl/heteroaryl-2′-deoxyuridine via a Suzuki-Miyaura reaction in aqueous media. Molecules 2012,17, 14409–14417. [CrossRef] [PubMed]
47. Mayer, E.; Valis, L.; Huber, R.; Amann, N.; Wagenknecht, H.A. Preparation of pyrene-modified purineand pyrimidine nucleosides via Suzuki-Miyaura cross-couplings and characterization of their fluorescentproperties. Synthesis 2003, 15, 2335–2340.
48. Valis, L.; Wagenknecht, H.A. Synthesis and optical properties of the C-8 adduct of Benzo[a]pyrene anddeoxyguanosine. Synlett 2005, 15, 2281–2284.
49. Kohyama, N.; Katashima, T.; Yamamoto, Y. Synthesis of novel 2-aryl AICAR derivatives. Synthesis 2004, 17,2799–2804.
50. Ogasawara, S.; Saito, I.; Maeda, M. Synthesis and reversible photoisomerization of photo-switchablenucleoside, 8-styryl-2′-deoxyguanosine. Tetrahedron Lett. 2008, 49, 2479–2482. [CrossRef]
51. Wagner, C.; Wagenknecht, H.A. Reductive electron transfer in phenothiazine-modified DNA is dependenton the base sequence. Chem. Eur. J. 2005, 11, 1871–1876. [CrossRef] [PubMed]
52. Wanninger-Weiß, C.; Wagenknecht, H.A. Synthesis of 5-(2-pyrenyl)-2′-deoxyuridine as a DNA modificationfor electron-transfer studies: The critical role of the position of the chromophore attachment. Eur. J. Org. Chem.2008, 2008, 64–71. [CrossRef]
53. Ehrenschwender, T.; Wagenknecht, H.A. Synthesis and spectroscopic characterization of BODIPY-modifieduridines as potential fluorescent probes for nucleic acids. Synthesis 2008, 2008, 3657–3662. [CrossRef]
54. Hassan, M.E. Palladium-catalyzed cross-coupling reaction of organostannanes with nucleoside halides.Coll. Czech. Chem. Commun. 1991, 56, 1944–1947. [CrossRef]
55. Holzberger, B.; Strohmeier, J.; Siegmund, V.; Diederichsen, U.; Marx, A. Enzymatic synthesis of 8-vinyl- and8-styryl-2′-deoxyguanosine modified DNA—Novel fluorescent molecular probes. Bioorg. Med. Chem. Lett.2012, 22, 3136–3139. [CrossRef] [PubMed]
56. Matsuda, A.; Satoh, K.; Tanaka, H.; Miyasaka, T. Introduction of carbon substituents at C-2 position of purinenucleosides. Nucleic Acids Symp. Ser. 1983, 12, 5–8. [PubMed]
57. Jäger, S.; Rasched, G.; Komreich-Leshem, H.; Engesser, M.; Thum, O.; Famulok, M. A versatile toolboxfor variable DNA functionalization at high density. J. Am. Chem. Soc. 2005, 127, 15071–15072. [CrossRef][PubMed]
58. Sági, G.; Ötvös, L.; Ikeda, S.; Andrei, G.; Snoeck, R.; De Clercq, E. Synthesis and antiviral activities of8-alkynyl-, 8-alkenyl-, and 8-alkyl-2′-deoxyadenosine analogs. J. Med. Chem. 1994, 37, 1307–1311. [CrossRef][PubMed]
53

Molecules 2015, 20, 9419–9454
59. Volpini, R.; Costanzi, S.; Lambertucci, C.; Vittori, S.; Klotz, K.N.; Lorenzen, A.; Cristalli, G. Introductionof Alkynyl Chains on C-8 of Adenosine Led to Very Selective Antagonists of the A3 Adenosine Receptor.Bioorg. Med. Chem. Lett. 2001, 11, 1931–1934. [CrossRef] [PubMed]
60. Olejniczak, A.; Wojtczak, B.; Lesnikowski, Z.J. 2′-Deoxyadenosine Bearing Hydrophobic CarboranePharmacophore. Nucleos. Nucleot. Nucl. Acids 2007, 26, 1611–1613. [CrossRef]
61. Seela, F.; Ingale, S.A. “Double Click” Reaction on 7-Deazaguanine DNA: Synthesis and Excimer Fluorescenceof Nucleosides and Oligonucleotides with Branched Side Chains Decorated with Proximal Pyrenes.J. Org. Chem. 2010, 75, 284–295. [CrossRef] [PubMed]
62. Zhao, Y.; Baranger, A.M. Design of an adenosine analogue that selectively improves the affinity of a mutantU1A protein for RNA. J. Am. Chem. Soc. 2003, 125, 2480–2488. [CrossRef] [PubMed]
63. Ardhapure, A.V.; Sanghvi, Y.S.; Kapdi, A.R.; García, J.; Sanchez, G.; Lozano, P.; Serrano, J.L. Pd-imidatecomplexes as recyclable catalysts for the synthesis of C5-alkenylated pyrimidine nucleosides via Heckcross-coupling reaction. RSC Adv. 2015, 5, 24558–24563. [CrossRef]
64. Li, C.J. Organic reactions in aqueous media with a focus on carbon-carbon bond formations: A decadeupdate. Chem. Rev. 2005, 105, 3095–3165. [CrossRef] [PubMed]
65. Shaughnessy, K.H. Beyond TPPTS: New approaches to the development of efficient palladium-catalyzedaqueous-phase cross-coupling reactions. Eur. J. Org. Chem. 2006, 2006, 1827–1835. [CrossRef]
66. Polshettiwar, V.; Decottignies, A.; Len, C.; Fihri, A. Suzuki-Miyaura Cross-Coupling Reactions in AqueousMedia: Green and Sustainable Syntheses of Biaryls. ChemSusChem 2010, 3, 502–522. [CrossRef] [PubMed]
67. Shaughnessy, K.H. Greener approaches to cross-coupling. In New Trends in Cross-Coupling: Theory andApplication; Colacot, T.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 645–696.
68. Garrett, C.E.; Prasad, K. The art of meeting palladium specifiations in active pharmaceutical ingredientsproduced by Pd-catalyzed reactions. Adv. Synth. Catal. 2004, 346, 889–900. [CrossRef]
69. Casalnuovo, A.L.; Calabrese, J.C. Palladium-catalyzed alkylation in aqueous media. J. Am. Chem. Soc. 1990,112, 4324–4330. [CrossRef]
70. Western, E.C.; Daft, J.R.; Johnson, E.M., II; Gannett, P.M.; Shaughnessy, K.H. Efficient, one-step Suzukiarylation of unprotected halonucleosides using water-soluble palladium catalysts. J. Org. Chem. 2003, 68,6767–6774. [CrossRef] [PubMed]
71. Shaughnessy, K.H.; Booth, R.S. Sterically demanding, water-soluble alkylphosphines as ligands for highactivity Suzuki coupling of aryl bromides in aqueous solvents. Org. Lett. 2001, 3, 2757–2759. [CrossRef][PubMed]
72. Hobley, G.; Gubala, V.; Rivera-Sánchez, M.D.C.; Rivera, J.M. Synthesis of 8-heteroaryl-2′-deoxyguanosinederivatives. Synlett 2008, 2008, 1510–1514. [CrossRef] [PubMed]
73. Western, E.C.; Shaughnessy, K.H. Inhibitory effects of the guanine moiety on the Suzuki couplings ofunprotected halonucleosides in aqueous media. J. Org. Chem. 2005, 70, 6378–6388. [CrossRef] [PubMed]
74. Both ligands are available from STREM Chemical Co: TPPTS (15–8007, $79/mmol), TXPTS (15–7860,$149/mmol).
75. Collier, A.; Wagner, G.K. Suzuki-Miyaura cross-coupling of unprotected halopurine nucleosides inwater-influence of catalyst and cosolvent. Synth. Commun. 2006, 36, 3713–3721. [CrossRef]
76. Sartori, G.; Enderlin, G.; Hervé, G.; Len, C. Highly effective synthesis of C-5-substituted 2′-deoxyuridineusing Suzuki-Miyaura cross-coupling in water. Synthesis 2012, 44, 767–772. [CrossRef]
77. Sartori, G.; Hervé, G.; Enderlin, G.; Len, C. New, efficient approach for the ligand-free Suzuki-Miyaurareaction of 5-iodo-2′-deoxyuridine in water. Synthesis 2013, 45, 330–333. [CrossRef]
78. Kapdi, A.; Gayakhe, V.; Sanghvi, Y.S.; García, J.; Lozano, P.; da Silva, I.; Pérez, J.; Serrano, J.L. New watersoluble Pd-imidate complexes as highly efficient catalysts for the synthesis of C5-arylated pyrimidinenucleosides. RSC Adv. 2014, 4, 17567–17572. [CrossRef]
79. Lercher, L.; McGouran, J.F.; Kessler, B.M.; Schofield, C.J.; Davis, B.G. DNA Modification under MildConditions by Suzuki-Miyaura Cross-Coupling for the Generation of Functional Probes. Angew. Chem.Int. Ed. 2013, 52, 10553–10558. [CrossRef]
80. Capek, P.; Hocek, M. Efficient one-step synthesis of optically pure (adenin-8-yl)phenylalanine nucleosides.Synlett 2005, 19, 3005–3007.
54

Molecules 2015, 20, 9419–9454
81. Capek, P.; Pohl, R.; Hocek, M. Cross-coupling reactions of unprotected halopurine bases, nucleosides,and nucleoside triphosphates with 4-boronophenylalanine in water. Synthesis of (purin-8-yl)- and(purin-6-yl)phenylalanines. Org. Biomol. Chem. 2006, 4, 2278–2284. [CrossRef] [PubMed]
82. Vrábel, M.; Pohl, R.; Klepetárová, B.; Votruba, I.; Hocek, M. Synthesis of 2′-deoxyadenosine nucleosidesbearing bipyridine-type ligands and their Ru-complexes in position 8 through cross-coupling reactions.Org. Biomol. Chem. 2007, 5, 2849–2857. [CrossRef] [PubMed]
83. Vrábel, M.; Pohl, R.; Votruba, I.; Sajadi, M.; Kovalenko, S.A.; Ernsting, N.P.; Hocek, M. Synthesisand photophysical properties of 7-deaza-2′-deoxyadenosines bearing bipyridine ligands and theirRu(II)-complexes in position 7. Org. Biomol. Chem. 2008, 6, 2852–2860. [CrossRef] [PubMed]
84. Kalachova, L.; Pohl, R.; Hocek, M. Synthesis of 2′-deoxyuridine and 2′-deoxycytidine nucleosides bearingbipyridine and terpyridine ligands at position 5. Synthesis 2009, 2009, 105–112. [CrossRef]
85. Rankin, K.M.; Sproviero, M.; Rankin, K.; Sharma, P.; Wetmore, S.D.; Manderville, R.A. C8-Heteroaryl-2′-deoxyguanosine Adducts as Conformational Fluorescent Probes in the NarI Recognition Sequence.J. Org. Chem. 2012, 77, 10498–10508. [CrossRef] [PubMed]
86. Schlitt, K.M.; Millen, A.L.; Wetmore, S.D.; Manderville, R.A. An indole-linked C8-deoxyguanosine nucleosideacts as a fluorescent reporter of Watson-Crick versus Hoogsteen base pairing. Org. Biomol. Chem. 2011, 9,1565–1571. [CrossRef] [PubMed]
87. Manderville, R.A.; Omumi, A.; Rankin, K.M.; Wilson, K.A.; Millen, A.L.; Wetmore, S.D. Fluorescent C-linkedC8-aryl-guanine probe for distinguishing syn from anti structures in duplex DNA. Chem. Res. Toxicol. 2012,25, 1271–1282. [CrossRef] [PubMed]
88. Miyata, K.; Mineo, R.; Tamamushi, R.; Mizuta, M.; Ohkubo, A.; Taguchi, H.; Seio, K.; Santa, T.;Sekine, M. Synthesis and Fluorescent Properties of Bi- and Tricyclic 4-N-Carbamoyldeoxycytidine Derivatives.J. Org. Chem. 2007, 77, 102–108. [CrossRef]
89. Mizuta, M.; Seio, K.; Miyata, K.; Sekine, M. Fluorescent Pyrimidopyrimidoindole Nucleosides: Controlof Photophysical Characterizations by Substituent Effects. J. Org. Chem. 2007, 72, 5046–5055. [CrossRef][PubMed]
90. Segal, M.; Fischer, B. Analogs of uracil nucleosides with intrinsic fluorescence (NIF-analogs): Synthesis andphotophysical properties. Org. Biomol. Chem. 2012, 10, 1571–1580. [CrossRef] [PubMed]
91. Storr, T.E.; Strohmeier, J.A.; Baumann, C.G.; Fairlamb, I.J.S. A sequential direct arylation/Suzuki-Miyauracross-coupling transformation of unprotected 2′-deoxyadenosine affords a novel class of fluorescent analogs.Chem. Commun. 2010, 46, 6470–6472. [CrossRef]
92. Bárta, J.; Pohl, R.; Klepetárová, B.; Ernsting, N.P.; Hocek, M. Modular Synthesis of 5-SubstitutedThiophen-2-yl C-2′-Deoxyribonucleosides. J. Org. Chem. 2008, 73, 3798–3806. [CrossRef] [PubMed]
93. Cahová, H.; Jäschke, A. Nucleoside-Based Diarylethene Photoswitches and Their Facile Incorporation intoPhotoswitchable DNA. Angew. Chem. Int. Ed. 2013, 52, 3186–3190. [CrossRef]
94. Macícková-Cahová, H.; Pohl, R.; Horáková, P.; Havran, L.; Špacek, J.; Fojta, M.; Hocek, M.Alkylsulfanylphenyl Derivatives of Cytosine and 7-Deazaadenine Nucleosides, Nucleotides and NucleosideTriphosphates: Synthesis, Polymerase Incorporation to DNA and Electrochemical Study. Chem. Eur. J. 2011,17, 5833–5841. [CrossRef] [PubMed]
95. Gubala, V.; Betancourt, J.E.; Rivera, J.M. Expanding the Hoogsteen Edge of 2′-Deoxyguanosine:Consequences for G-Quadruplex Formation. Org. Lett. 2004, 6, 4735–4738. [CrossRef] [PubMed]
96. Vongsutilers, V.; Phillips, D.J.; Train, B.C.; McKelvey, G.R.; Thomsen, N.M.; Shaughnessy, K.H.; Lewis, J.P.;Gannett, P.M. The conformational effect of para-substituted C8-arylguanine adducts on the B/Z-DNAequilibrium. Biophys. Chem. 2011, 154, 41–48. [CrossRef] [PubMed]
97. Nauš, P.; Kuchar, M.; Hocek, M. Cytostatic and antiviral 6-arylpurine ribonucleosides IX. synthesis andevaluation of 6-substituted 3-deazapurine ribonucleosides. Coll. Czech. Chem. Comm. 2008, 73, 665–678.[CrossRef]
98. Nauš, P.; Pohl, R.; Votruba, I.; Džubák, P.; Hajdúch, M.; Ameral, R.; Birkuš, G.; Wang, T.; Ray, A.S.;Mackman, R.; et al. 6-(Het)aryl-7-Deazapurine Ribonucleosides as Novel Potent Cytostatic Agents.J. Med. Chem. 2010, 53, 460–470. [CrossRef] [PubMed]
99. Bourderioux, A.; Nauš, P.; Perlíková, P.; Pohl, R.; Pichová, I.; Votruba, I.; Džubák, P.; Konecný, P.; Hajdúch, M.;Stray, K.M.; et al. Synthesis and Significant Cytostatic Activity of 7-Hetaryl-7-deazaadenosines. J. Med. Chem.2011, 54, 5498–5507. [CrossRef] [PubMed]
55

Molecules 2015, 20, 9419–9454
100. Amiable, C.; Paoletti, J.; Haouz, A.; Padilla, A.; Labesse, G.; Kaminski, P.A.; Pochet, S. 6-(Hetero)Arylpurinenucleotides as inhibitors of the oncogenic target DNPH1: Synthesis, structural studies and cytotoxic activities.Eur. J. Med. Chem. 2014, 85, 418–437. [CrossRef] [PubMed]
101. Nauš, P.; Perlíková, P.; Bourderioux, A.; Pohl, R.; Slavetínská, L.; Votruba, I.; Bahador, G.; Birkuš, G.; Cihlár, T.;Hocek, M. Sugar-modified derivatives of cytostatic 7-(het)aryl-7-deazaadenosines: 2′-C-methylribonucleosides,2′-deoxy-2′-fluoroarabinonucleosides, arabinonucleosides and 2′-deoxyribonucleosides. Bioorg. Med. Chem.2012, 20, 5202–5214. [CrossRef] [PubMed]
102. Perlíková, P.; Jornet Martínez, N.; Slavetínská, L.; Hocek, M. Synthesis of 2′-deoxy-2′-fluororibo- and2′-deoxy-2′,2′-difluororibonucleosides derived from 6-(het)aryl-7-deazapurines. Tetrahedron 2012, 68,8300–8310. [CrossRef]
103. Nauš, P.; Caletková, O.; Konecny, P.; Džubak, P.; Bogdanova, K.; Kolár, M.; Vrbková, J.; Slavetinská, L.;Tloušt’ová, E.; Perlíková, P.; et al. Synthesis, Cytostatic, Antimicrobial, and Anti-HCV Activity of6-Substituted 7-(Het)aryl-7-deazapurine Ribonucleosides. J. Med. Chem. 2014, 57, 1097–1110. [CrossRef][PubMed]
104. Ikonen, S.; Macicková-Cahová, H.; Pohl, R.; Šanda, M.; Hocek, M. Synthesis of nucleoside and nucleotideconjugates of bile acids, and polymerase construction of bile acid-functionalized DNA. Org. Biomol. Chem.2010, 8, 1194–1201. [CrossRef] [PubMed]
105. Cho, J.H.; Prickett, C.D.; Shaughnessy, K.H. Efficient Sonogashira Coupling of Unprotected Halonucleosidesin Aqueous Solvents Using Water-Soluble Palladium Catalysts. Eur. J. Org. Chem. 2010, 3678–3683. [CrossRef]
106. Cho, J.H.; Shaughnessy, K.H. Aqueous-phase Sonogashira alkynylation to synthesize 5-substitutedpyrimidine and 8-substituted purine nucleosides. Curr. Prot. Nucleic Acid Chem. 2012, 49. [CrossRef]
107. Cho, J.H.; Shaughnessy, K.H. Aqueous-Phase Heck Coupling of 5-Iodouridine and Alkenes underPhosphine-Free Conditions. Synlett 2011, 20, 2963–2966.
108. Thoresen, L.H.; Jiao, G.S.; Haaland, W.C.; Metzker, M.L.; Burgess, K. Rigid, conjugated, fluoresceinatedthymidine triphosphates: Syntheses and polymerase mediated incorporation into DNA analogs. Chem. Eur. J.2003, 9, 4603–4610. [CrossRef] [PubMed]
109. Collier, A.; Wagner, G. A facile two-step synthesis of 8-arylated guanosine mono- and triphosphates (8-arylGXPs). Org. Biomol. Chem. 2006, 4, 4526–4532. [CrossRef] [PubMed]
110. Capek, P.; Cahová, H.; Pohl, R.; Hocek, M.; Gloeckner, C.; Marx, A. An efficient method for the construction offunctionalized DNA bearing amino acid groups through cross-coupling reactions of nucleoside triphosphatesfollowed by primer extension or PCR. Chem. Eur. J. 2007, 13, 6196–6203. [CrossRef] [PubMed]
111. Cahová, H.; Pohl, R.; Bednárová, L.; Nováková, K.; Cvacka, J.; Hocek, M. Synthesis of 8-bromo, 8-methyl-and 8-phenyl-dATP and their polymerase incorporation into DNA. Org. Biomol. Chem. 2008, 6, 3657–3660.[CrossRef] [PubMed]
112. Cahová, H.; Havran, L.; Brázdilová, P.; Pivonková, H.; Pohl, R.; Fojta, M.; Hocek, M. Aminophenyl- andnitrophenyl-labeled nucleoside triphosphates: Synthesis, enzymatic incorporation, and electrochemicaldetection. Angew. Chem. Int. Ed. 2008, 47, 2059–2062. [CrossRef]
113. Balintová, J.; Plucnara, M.; Vidláková, P.; Pohl, R.; Havran, L.; Fojta, M.; Hocek, M. Benzofurazane as a NewRedox Label for Electrochemical Detection of DNA: Towards Multipotential Redox Coding of DNA Bases.Chem. Eur. J. 2013, 19, 12720–12731. [CrossRef] [PubMed]
114. Riedl, J.; Pohl, R.; Rulíšek, L.; Hocek, M. Synthesis and Photophysical Properties of Biaryl-SubstitutedNucleosides and Nucleotides. Polymerase Synthesis of DNA Probes Bearing Solvatochromic andpH-Sensitive Dual Fluorescent and 19F-NMR Labels. J. Org. Chem. 2012, 77, 1026–1044. [CrossRef] [PubMed]
115. Raindlová, V.; Pohl, R.; Šanda, M.; Hocek, M. Direct Polymerase Synthesis of Reactive Aldehyde-Functionalized DNA and Its Conjugation and Staining with Hydrazines. Angew. Chem. Int. Ed. 2010,49, 1064–1066. [CrossRef]
116. Pesnot, T.; Wagner, G.K. Novel derivatives of UDP-glucose: Concise synthesis and fluorescent properties.Org. Biomol. Chem. 2008, 6, 2884–2891. [CrossRef] [PubMed]
117. Collier, A.; Wagner, G.K. A fast synthetic route to GDP-sugars modified at the nucleobase. Chem. Commun.2008, 2, 178–180. [CrossRef]
118. Pergolizzi, G.; Butt, J.N.; Bowater, R.P.; Wagner, G.K. A novel fluorescent probe for NAD-consuming enzymes.Chem. Commun. 2011, 47, 12655–12657. [CrossRef]
56

Molecules 2015, 20, 9419–9454
119. Brázdilová, P.; Vrábel, M.; Pohl, R.; Pivonková, H.; Havran, L.; Hocek, M.; Fojta, M. Ferrocenylethynylderivatives of nucleoside triphosphates: Synthesis, incorporation, electrochemistry, and bioanalyticalapplications. Chem. Eur. J. 2007, 13, 9527–9533. [CrossRef] [PubMed]
120. Dadová, J.; Vidlákova, P.; Pohl, R.; Havran, L.; Fojta, M.; Hocek, M. Aqueous Heck Cross-CouplingPreparation of Acrylate-Modified Nucleotides and Nucleoside Triphosphates for Polymerase Synthesis ofAcrylate-Labeled DNA. J. Org. Chem. 2013, 78, 9627–9637. [CrossRef] [PubMed]
121. Omumi, A.; Beach, D.G.; Baker, M.; Gabryelski, W.; Manderville, R.A. Postsynthetic Guanine Arylation ofDNA by Suzuki-Miyaura Cross-Coupling. J. Am. Chem. Soc. 2011, 133, 42–50. [CrossRef] [PubMed]
122. Krause, A.; Hertl, A.; Muttach, F.; Jäschke, A. Phosphine-Free Stille-Migita Chemistry for the Mild andOrthogonal Modification of DNA and RNA. Chem. Eur. J. 2014, 20, 16613–16619. [CrossRef] [PubMed]
123. Hirai, Y.; Uozumi, Y. C-N and C-S bond forming cross coupling in water with amphiphilic resin-supportedpalladium complexes. Chem. Lett. 2011, 40, 934–935. [CrossRef]
124. Lipshutz, B.H.; Ghorai, S.; Abela, A.R.; Moser, R.; Nishikata, T.; Duplais, C.; Krasovskiy, A.; Gaston, R.D.;Gadwood, R.C. TPGS-750-M: A Second-Generation Amphiphile for Metal-Catalyzed Cross-Couplings inWater at Room Temperature. J. Org. Chem. 2011, 76, 4379–4391. [CrossRef] [PubMed]
125. Tardiff, B.J.; Stradiotto, M. Buchwald-Hartwig Amination of (Hetero)aryl Chlorides by EmployingMor-DalPhos under Aqueous and Solvent-Free Conditions. Eur. J. Org. Chem. 2012, 3972–3977. [CrossRef]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
57

molecules
Article
Formation of Mixed-Ligand Complexes of Pd2+ withNucleoside 5'-Monophosphates and SomeMetal-Ion-Binding Nucleoside Surrogates
Oleg Golubev, Tuomas Lönnberg and Harri Lönnberg *
Department of Chemistry, University of Turku, FIN-20014 Turku, Finland; [email protected] (O.G.);[email protected] (T.L.)* Author to whom correspondence should be addressed; [email protected]; Tel.: +358-2-333-6770;
Fax: +358-2-333-6700.
External Editors: Mahesh K. Lakshman and Fumi NagatsugiReceived: 16 September 2014; in revised form: 8 October 2014; Accepted: 17 October 2014; Published:22 October 2014
Abstract: Formation of mixed-ligand Pd2+ complexes between canonical nucleoside 5'-monophosphatesand five metal-ion-binding nucleoside analogs has been studied by 1H-NMR spectroscopy to testthe ability of these nucleoside surrogates to discriminate between unmodified nucleobases byPd2+-mediated base pairing. The nucleoside analogs studied included 2,6-bis(3,5-dimethylpyrazol-1-yl)-,2,6-bis(1-methylhydrazinyl)- and 6-(3,5-dimethylpyrazol-1-yl)-substituted 9-(β-D-ribofuranosyl)purines1–3, and 2,4-bis(3,5-dimethylpyrazol-1-yl)- and 2,4-bis(1-methylhydrazinyl)-substituted 5-(β-D-ribofuranosyl)-pyrimidines 4–5. Among these, the purine derivatives 1-3 bound Pd2+ much more tightlythan the pyrimidine derivatives 4, 5 despite apparently similar structures of the potential coordinationsites. Compounds 1 and 2 formed markedly stable mixed-ligand Pd2+ complexes with UMP and GMP,UMP binding favored by 1 and GMP by 2. With 3, formation of mixed-ligand complexes was retardedby binding of two molecules of 3 to Pd2+.
Keywords: Pd2+ complexes; nucleosides; NMR; mixed-ligand complexes
1. Introduction
It has been well established that linear-coordinating Hg2+ and Ag+ ions may stabilize TTand CC mismatches within oligonucleotide duplexes while square-planar-coordinating Cu2+ ionis able to bridge various modified metal-ion-binding bases on opposite strands [1–3]. Muchless is known about discrimination between canonical nucleobases by oligonucleotide probesincorporating metal-ion-binding surrogate bases. Cu2+ and Zn2+ ions have been shown to enhancehybridization of 2'-O-methyl oligoribonucleotides containing a 2,6-bis(3,5-dimethylpyrazol-1-yl)purinebase with complementary unmodified 2'-O-methyl oligoribonucleotides [4]. The magnitude of duplexstabilization does not, however, depend only on the identity of the opposite base, but also on theflanking sequences.
Square-planar-coordinating Pd2+ ion is known to exhibit exceptionally high affinity to nucleic acidbases [5–7]. Accordingly, Pd2+ complexes are interesting candidates for base moiety discrimination,although so far no convincing examples of Pd2+-mediated base-pairing at oligonucleotide level areavailable. Only some indications of recognition of thymine within an oligodeoxyribonucleotide by2,6-bis(3,5-dimethylpyrazol-1-yl)purine in the presence of Pd2+ have been observed [8]. We havepreviously tried to evaluate the formation of mixed-ligand Pd2+ complexes between some metal ionbinding nucleoside analogs and pyrimidine nucleosides [9]. Owing to severe solubility problems, theresults obtained remain scanty. To learn more about the discrimination power of Pd2+ complexes,
Molecules 2014, 19, 16976–16986 58 www.mdpi.com/journal/molecules

Molecules 2014, 19, 16976–16986
we now report on NMR studies concerning formation of mixed-ligand Pd2+ complexes betweenmetal-ion-binding nucleosides 1–5 (Figure 1) and six nucleoside 5'-monophosphates (NMPs, Figure 2).
Figure 1. Structures of the metal-ion-binding nucleosides 1–5 used in the present study.
Figure 2. Structures of nucleoside 5'-monophosphates used in the present study.
2. Results and Discussion
2.1. Compounds Employed
The preparation of metal-ion-binding nucleosides 1–5 has been described previously [9,10].Among the NMPs employed, UMP, CMP, AMP, GMP and IMP were commercial products and the
59

Molecules 2014, 19, 16976–16986
preparation of the 5'-monophosphate of nebularine (NeMP), i.e., unsubstituted 9-(β-D-ribofuranosyl)purine, has been described previously [11].
2.2. Pd2+ Complexes of Modified Nucleosides (1–5)
Interaction of the modified nucleosides 1–5 with Pd2+ was studied first. For this purpose, K2PdCl4was added portionwise into a 5.0 mmol·L−1 solution of the nucleoside in phosphate buffered D2O(0.12 mol·L−1, pD 7.6, 25 ◦C) keeping the nucleoside concentration constant. After each addition a1H-NMR spectrum was recorded. Table 1 records the chemical shifts of the signals referring to the Pd2+
complexes formed.Upon addition of K2PdCl4 to 2,6-bis(3,5-dimethylpyrazol-1-yl)purine riboside (1), the intensity
of the H8 singlet of 1 at 8.59 ppm gradually decreased and two new pairs of singlets (8.85 and 8.83and 8.63 and 8.61) appeared (Figure S1 in Supplementary Files). When 0.5 equiv. of K2PdCl4 hadbeen added, the H8 singlet at 8.59 had almost entirely disappeared and a new singlet at 8.71 appeared.On approaching 1.0 equiv. addition of K2PdCl4, the two pairs of singlets weakened while the singlet at8.71 became more intense (Figure S2 in Supplementary Files). Corresponding changes occurred in theanomeric proton region. Formation of the two pairs of singlets at 8.85 and 8.83 and 8.63 and 8.61 wasaccompanied by the appearance of three anomeric proton doublets at 5.98 (J 7.6), 5.89 (J 4.4 Hz) and5.84 (J 4.8 Hz), the first one being twice as intense as the latter ones. A doublet at 6.19 (J 4.2 Hz), inturn, appeared parallel to the singlet at 8.71. Accordingly, at low concentration of K2PdCl4, a 2:1 (1:Pd)complex is formed and on increasing the concentration of K2PdCl4, the 1:1 complex predominates.
Table 1. Chemical shifts for the aromatic and anomeric protons of the modified nucleosides 1–5 andtheir Pd2+ complexes in D2O at pD 7.6 (0.12 M phosphate buffer, 25 ◦C).
Compd. Aromatic Proton Shifts Anomeric Proton Shifts
1 s 8.59(H8), s 6.19 and 6.11(H4'') d 6.15 (J 5.2)(1)Pd s 8.71(H8) a d 6.19 (J 4.2)(1)2Pd s 8.85 and 8.83 and 8.63 and 8.61(H8) b d5.98(J 7.6), d 5.89 (J 4.4), d 5.84(J 4.8)
2 s 7.86(H8) d 5.84 (J 5.3)(2)Pd s 8.06(H8) d 5.86 c
3 s 8.81(H2), s 8.63(H8), s 6.23(H4'') d 6.16 (J 5.6)
(3)Pds 8.92(H2/8), s 8.70(H2/8), s 6.40(H4'') d 6.19 (J 3.8)s 8.84(H2/8), s 8.42(H2/8), s 6.38(H4'') d 6.19 (J 3.8)
4 s 9.03(H6), s 6.12 and 6.07(H4'') d 5.06 (J 5.4)(4)Pd d e
5 s 7.92(H6) s 5.60a H4'' signals at 6.20–6.45 overlap with the corresponding signals of (1)2Pd. b Several H4'' signals at 6.20–6.64.c Overlaps with H1' of 2. d The disappearance of H6 singlet of uncomplexed 4 was accompanied by appearance of6 new singlets at 9.28, 9.22, 9.20, 8.93, 8.63 and 8.60. e The disappearance of the H1' doublet of 4 was accompaniedby appearance of 3 new signals at 5.73, 5.39 and 5.30.
The 1:1 complex, exhibiting only one set of 1H-NMR signals, most likely is a (1)PdCl+ complex,the metal ion being coordinated to N1 of the purine base and N2 atoms of the pyrazolyl substituents.When the concentration of 1 is high compared to that of K2PdCl4, the chlorido ligand is replaced withanother molecule of 1 which undergoes either N1 or N7 binding. N7 binding appears more likely, sincethis site is sterically less hindered than the N1 site flanked by the 3,5-dimethylpyrazol-1-yl groups,and since a reasonably large (0.26 and 0.24 ppm) downfield shift of the H8 signal is observed [7,12–15].The H8 resonances of both modified purine bases engaged in the complex appear as two singlets, mostlikely due to the fact that two mutual orientations of the ligands are possible: the sugar moieties maybe situated on the same or opposite sides of the plane of Pd2+ and the purine bases.
6-(3,5-Dimethylpyrazol-1-yl)purine riboside (3) bound Pd2+ much more weakly. Only half of 3
was complexed at an equimolar 5 mmol·L−1 concentration (Figure S3 in Supplementary Files). Twocomplexes were formed in parallel, evidently due to almost as efficient binding to N1 and N7 in
60

Molecules 2014, 19, 16976–16986
addition to binding to the pyrazolyl N2 atom. The markedly lower affinity compared to 1 lendssubstantial additional evidence for the assumption that both pyrazolyl groups of 1 participate inbinding of Pd2+.
Another important observation is that replacement of aromatic 3,5-dimethylpyrazol-1-yl groupswith aliphatic 1-methylhydrazinyl groups markedly weakens the binding of Pd2+. Only half of2,6-bis(1-methylhydrazinyl)purine riboside (2) was engaged in complex formation at 5.0 mmol·L−1
concentration of K2PdCl4 and 2, although a tridentate coordination, as with 1, apparently is possible(Figure S4 in Supplementary Files). However, binding to the terminal amino groups of the hydrazinylsubstituents is evidently impeded by the fact that the lone electron pair of the nitrogen atomsparticipates in the π-electron resonance of the purine ring, which lowers the electron density atthe potential donor atoms. With 1 the situation is different, since the lone electron pair of the N2atoms of the pyrazolyl substituents is not delocalized but the N2 atoms are pyridine type nitrogens.Additionally, hydrogen bonding of the NH2 group to N1 gives an expectedly moderately stable fivemembered structure, which may still retard the complexing ability of 2. A marked broadening of thesignals took place at high concentrations of K2PdCl4 and unidentified broad signals appeared, whichmay well refer to formation of polymeric complexes.
Quite unexpectedly, 2,4-bis(3,5-dimethylpyrazol-1-yl)-5-(β-D-ribofuranosyl)pyrimidine (4) alsoturned out to bind Pd2+ very weakly in spite of the fact that the expected binding site, viz. the N2atoms of the two pyrazolyl groups and the intervening N3 of the pyrimidine ring, appears very similarto the binding site in 1. At 5.0 mmol·L−1 concentration of both K2PdCl4 and 4, more than 50% of4 was complexed, but instead of a single clearly recognizable tridentate complex, several species incomparable amounts were formed (Figure S5 in Supplementary Files). Presumably, steric repulsionbetween the ribosyl group and the 5-methyl substituent of the pyrazolyl group at C4 prevents thisgroup to adopt a coplanar orientation with the pyrimidine and the other pyrazolyl ring required fortridentate binding of Pd2+ (Figure 3). In other words, owing to this repulsion, the N2 side of theprazolyl group is turned away from the vicinity of the N3 binding site.
2,4-Bis(1-methylhydrazinyl)-5-(β-D-ribofuranosyl)pyrimidine (5) binds Pd2+ even more weaklythan 4. In fact, no signals referring to complex formation could be detected upon addition of K2PdCl4into a 5 mmol·L−1 solution of 5. As with 2, involvement of the lone electron pair of the N2 atomof the 1-methylhydrazinyl groups in the π-electron resonance of the heteroaromatic ring makes thehydrazinyl amino groups poor donor atoms, but does not explain why binding of Pd2+ to 5 is evenweaker than binding to 2. Tentatively, the presence of the bulky ribofuranosyl group next to one of thehydrazinyl groups still sterically retards the binding of Pd2+.
Figure 3. Semi-empirical (PM6) minimized structure for 2,4-bis(3,5-dimethylpyrazol-1-yl)-5-(β-D-ribofuranosyl)pyrimidine (4, left) and the structure allowing tridentate binding to the N2atoms of the 3,5-dimethylpyrazolyl groups and the intervening N3 of the pyrimidine ring (right). Inthe latter structure the steric repulsion is much more pronounced than in the former.
61
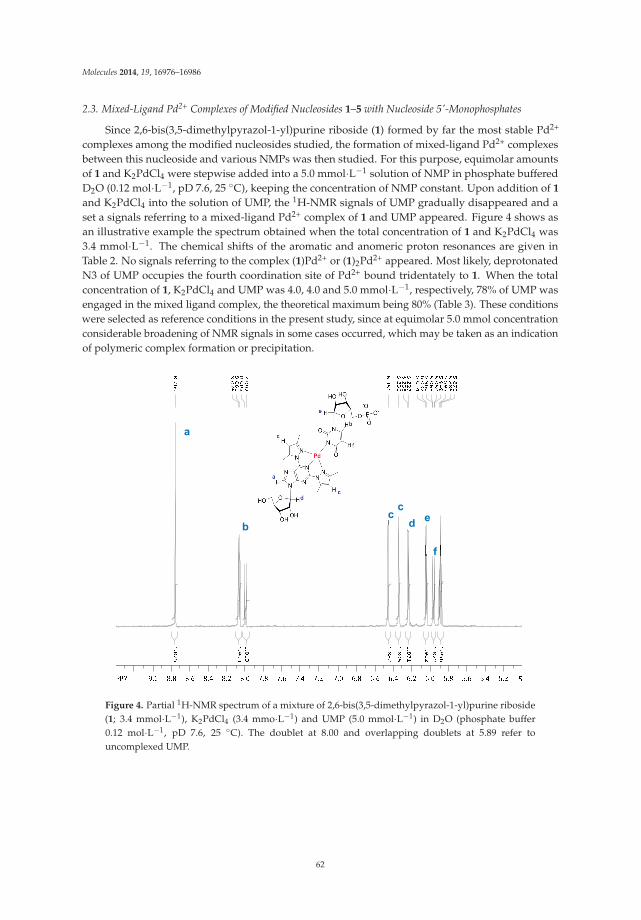
Molecules 2014, 19, 16976–16986
2.3. Mixed-Ligand Pd2+ Complexes of Modified Nucleosides 1–5 with Nucleoside 5'-Monophosphates
Since 2,6-bis(3,5-dimethylpyrazol-1-yl)purine riboside (1) formed by far the most stable Pd2+
complexes among the modified nucleosides studied, the formation of mixed-ligand Pd2+ complexesbetween this nucleoside and various NMPs was then studied. For this purpose, equimolar amountsof 1 and K2PdCl4 were stepwise added into a 5.0 mmol·L−1 solution of NMP in phosphate bufferedD2O (0.12 mol·L−1, pD 7.6, 25 ◦C), keeping the concentration of NMP constant. Upon addition of 1
and K2PdCl4 into the solution of UMP, the 1H-NMR signals of UMP gradually disappeared and aset a signals referring to a mixed-ligand Pd2+ complex of 1 and UMP appeared. Figure 4 shows asan illustrative example the spectrum obtained when the total concentration of 1 and K2PdCl4 was3.4 mmol·L−1. The chemical shifts of the aromatic and anomeric proton resonances are given inTable 2. No signals referring to the complex (1)Pd2+ or (1)2Pd2+ appeared. Most likely, deprotonatedN3 of UMP occupies the fourth coordination site of Pd2+ bound tridentately to 1. When the totalconcentration of 1, K2PdCl4 and UMP was 4.0, 4.0 and 5.0 mmol·L−1, respectively, 78% of UMP wasengaged in the mixed ligand complex, the theoretical maximum being 80% (Table 3). These conditionswere selected as reference conditions in the present study, since at equimolar 5.0 mmol concentrationconsiderable broadening of NMR signals in some cases occurred, which may be taken as an indicationof polymeric complex formation or precipitation.
a
b ccd e
f
Figure 4. Partial 1H-NMR spectrum of a mixture of 2,6-bis(3,5-dimethylpyrazol-1-yl)purine riboside(1; 3.4 mmol·L−1), K2PdCl4 (3.4 mmo·L−1) and UMP (5.0 mmol·L−1) in D2O (phosphate buffer0.12 mol·L−1, pD 7.6, 25 ◦C). The doublet at 8.00 and overlapping doublets at 5.89 refer touncomplexed UMP.
62
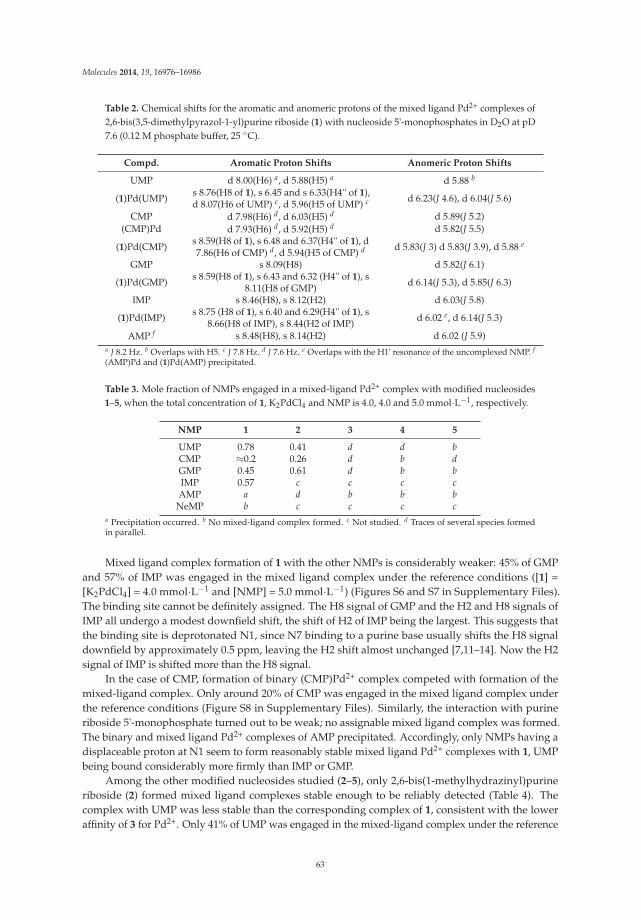
Molecules 2014, 19, 16976–16986
Table 2. Chemical shifts for the aromatic and anomeric protons of the mixed ligand Pd2+ complexes of2,6-bis(3,5-dimethylpyrazol-1-yl)purine riboside (1) with nucleoside 5'-monophosphates in D2O at pD7.6 (0.12 M phosphate buffer, 25 ◦C).
Compd. Aromatic Proton Shifts Anomeric Proton Shifts
UMP d 8.00(H6) a, d 5.88(H5) a d 5.88 b
(1)Pd(UMP) s 8.76(H8 of 1), s 6.45 and s 6.33(H4'' of 1),d 8.07(H6 of UMP) c, d 5.96(H5 of UMP) c d 6.23(J 4.6), d 6.04(J 5.6)
CMP d 7.98(H6) d, d 6.03(H5) d d 5.89(J 5.2)(CMP)Pd d 7.93(H6) d, d 5.92(H5) d d 5.82(J 5.5)
(1)Pd(CMP) s 8.59(H8 of 1), s 6.48 and 6.37(H4'' of 1), d7.86(H6 of CMP) d, d 5.94(H5 of CMP) d d 5.83(J 3) d 5.83(J 3.9), d 5.88 e
GMP s 8.09(H8) d 5.82(J 6.1)
(1)Pd(GMP) s 8.59(H8 of 1), s 6.43 and 6.32 (H4'' of 1), s8.11(H8 of GMP) d 6.14(J 5.3), d 5.85(J 6.3)
IMP s 8.46(H8), s 8.12(H2) d 6.03(J 5.8)
(1)Pd(IMP) s 8.75 (H8 of 1), s 6.40 and 6.29(H4'' of 1), s8.66(H8 of IMP), s 8.44(H2 of IMP) d 6.02 e, d 6.14(J 5.3)
AMP f s 8.48(H8), s 8.14(H2) d 6.02 (J 5.9)a J 8.2 Hz. b Overlaps with H5. c J 7.8 Hz. d J 7.6 Hz. e Overlaps with the H1' resonance of the uncomplexed NMP. f
(AMP)Pd and (1)Pd(AMP) precipitated.
Table 3. Mole fraction of NMPs engaged in a mixed-ligand Pd2+ complex with modified nucleosides1–5, when the total concentration of 1, K2PdCl4 and NMP is 4.0, 4.0 and 5.0 mmol·L−1, respectively.
NMP 1 2 3 4 5
UMP 0.78 0.41 d d bCMP ≈0.2 0.26 d b dGMP 0.45 0.61 d b bIMP 0.57 c c c cAMP a d b b b
NeMP b c c c ca Precipitation occurred. b No mixed-ligand complex formed. c Not studied. d Traces of several species formedin parallel.
Mixed ligand complex formation of 1 with the other NMPs is considerably weaker: 45% of GMPand 57% of IMP was engaged in the mixed ligand complex under the reference conditions ([1] =[K2PdCl4] = 4.0 mmol·L−1 and [NMP] = 5.0 mmol·L−1) (Figures S6 and S7 in Supplementary Files).The binding site cannot be definitely assigned. The H8 signal of GMP and the H2 and H8 signals ofIMP all undergo a modest downfield shift, the shift of H2 of IMP being the largest. This suggests thatthe binding site is deprotonated N1, since N7 binding to a purine base usually shifts the H8 signaldownfield by approximately 0.5 ppm, leaving the H2 shift almost unchanged [7,11–14]. Now the H2signal of IMP is shifted more than the H8 signal.
In the case of CMP, formation of binary (CMP)Pd2+ complex competed with formation of themixed-ligand complex. Only around 20% of CMP was engaged in the mixed ligand complex underthe reference conditions (Figure S8 in Supplementary Files). Similarly, the interaction with purineriboside 5'-monophosphate turned out to be weak; no assignable mixed ligand complex was formed.The binary and mixed ligand Pd2+ complexes of AMP precipitated. Accordingly, only NMPs having adisplaceable proton at N1 seem to form reasonably stable mixed ligand Pd2+ complexes with 1, UMPbeing bound considerably more firmly than IMP or GMP.
Among the other modified nucleosides studied (2–5), only 2,6-bis(1-methylhydrazinyl)purineriboside (2) formed mixed ligand complexes stable enough to be reliably detected (Table 4). Thecomplex with UMP was less stable than the corresponding complex of 1, consistent with the loweraffinity of 3 for Pd2+. Only 41% of UMP was engaged in the mixed-ligand complex under the reference
63

Molecules 2014, 19, 16976–16986
conditions ([K2PdCl4] = [3] = 4.0 mmol·L−1, [NMP] = 5.0 mmol·L−1] (Figure S9 in SupplementaryFiles). For comparison, the observed 78% engagement of UMP in the mixed ligand complex with1 was close to the theoretical maximum, 80%. Ternary complexes (2)Pd2+(CMP) and (2)Pd2+(GMP)were, in turn, formed even slightly more readily than the corresponding complexes of 1; 26% ofCMP and 61% of GMP were engaged in a mixed ligand complex under the reference conditions(Figures S10 and S11 in Supplementary Files). In fact, GMP was now bound slightly more firmly thanUMP. The marked downfield shift of the H8 resonance of GMP (0.66 ppm) suggests N7 coordination.Interaction with AMP appeared rather weak, and several species were formed in parallel. Uponmixing of 6-bis(3,5-dimethylpyrazol-1-yl)purine riboside (3) and K2PdCl4 with UMP, CMP or GMP,so complicated mixtures were formed that assignment of any single mixed-ligand complex wasimpossible. With AMP, no complexes were formed. As discussed above, the pyrimidine derivatives 4
and 5 did not form stable Pd2+ complexes. Expectedly, they did not form an assignable mixed-ligandcomplex with any of the NMPs studied. The only species that could be assigned referred to binaryPd2+ complexes of NMPs.
Table 4. Chemical shifts for the aromatic and anomeric protons of the mixed ligand Pd2+ complexes of2,6-bis(1-methylhydrazinyl)purine riboside (2) with nucleoside 5'-monophosphates in D2O at pD 7.6(0.12 M phosphate buffer).
Compd. Aromatic Proton Shifts Anomeric Proton Shifts
(2)Pd(UMP) s 8.13(H8 of 2), d 7.81(H6 of UMP)b, d 5.77(H5 of UMP) b m 5.91–5.95 a
(2)Pd(CMP) s 8.14(H8 of 2), d 8.11(H6 of CMP)c, d 6.13(H5 of CMP) c m 5.90–5.95 a
(2)Pd(GMP) s 8.04(H8 of 2), s 8.75(H8 of GMP) br s 5.81 d
(2)Pd(AMP) e ea The H1' resonances of both ligands overlap. b J 7.7 Hz. c J 7.6 Hz. d Overlaps with the H1' resonance of GMP. e
Could not be reliably assigned.
3. Experimental Section
3.1. General Information
The 1H-NMR spectra were recorded on Bruker Avance 500- or 400-MHz NMR spectrometers usingMe4Si as an external standard. The chemical shifts, δ, are given in ppm and the coupling constants, J,in Hz. HR-ESI-MS spectra were obtained by a Bruker Daltonics MicrOTOF-Q instrument.
3.2. 1H-NMR Spectroscopic Studies of the Interaction of K2PdCl4 with Nucleosides 1–5
To a solution of nucleoside 1–5 (5.0 mmol·L−1) in a phosphate buffer in D2O (0.12 mol·L−1,pH 7.2), K2PdCl4 was added portionwise keeping the concentration of nucleoside constant. Aftereach addition, a 1H-NMR spectrum was recorded at 25 ◦C and the signals appearing in the regionof aromatic and anomeric protons (chemical shift > 5 ppm) were carefully integrated. The speciesdistribution at different concentrations was calculated on the basis of the relative intensities of theresonances of their aromatic and anomeric protons.
3.3. 1H-NMR Spectroscopic Studies of the Formation of Mixed-Ligand Pd2+ Complexes of Nucleosides 1–5with NMPs
To a solution of NMP (5.0 mmol·L−1) in a phosphate buffer in D2O (0.12 mol·L−1, pD 7.6), a1:1 mixture of K2PdCl4 and one of nucleosides 1–5 in the same buffer was portionwise added. Theconcentration of K2PdCl4 and the nucleoside (1–5) was in this manner varied from zero to 5 mmol·L−1,while the concentration of NMP was kept constant (5.0 mmol·L−1). After each addition, a 1H-NMRspectrum was recorded at 25 ◦C and the signals appearing in the region of aromatic and anomeric
64

Molecules 2014, 19, 16976–16986
protons (chemical shift > 5 ppm) were carefully integrated. The species distribution at differentconcentrations was calculated on the basis of the relative intensities of the resonances of their aromaticand anomeric protons.
4. Conclusions
In spite of the apparent similarity of tridentate coordination sites in purine ribosides 1 and2, on the one hand, and in pyrimidine C-ribosides 4 and 5, on the other hand, only the purinederivatives turned out to be able to form stable Pd2+ complexes. Evidently the bulky ribosylgroup at C5 of the pyrimidine ring sterically prevents the donor atom of the C4-substituentfrom adopting an orientation allowing tridentate binding. Among the purine derivatives,2,6-bis(3,5-dimethylpyrazol-1-yl)purine riboside (1) forms mixed-ligand Pd2+ complexes with NMPsmuch more readily than its 2,6-bis(1-methylhydrazinyl) counterpart, probably due to participation ofthe lone electron pair of the terminal amino groups of the latter in the purine π-electron resonance.UMP is recognized most efficient, followed by IMP, GMP and CMP, in this order. Interestingly,interaction with unsubstituted purine riboside 3'-monophosphate is very weak. Mixed-ligandcomplexes with AMP precipitate. Bidentately coordinating 6-(3,5-dimethylpyrazol-1-yl)purine ribosidegive a complicated product mixture upon mixing with K2PdCl4 and any of the canonical NMPs.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/10/16976/s1.
Acknowledgments: Financial aid from the Foundation of Turku University is gratefully acknowledged.
Author Contributions: O.G., T.L. and H.L. designed the research together. O.G. performed the experimentalmeasurements. O.G., T.L. and H.L. participated in analyzing the data and writing the paper. H.L. prepared thefinal version. O.G., T.L. and H.L. read and approved the final manuscript.
Conflicts of Interest: The authors declare no conflict of interest
References
1. Takezawa, Y.; Shinoya, M. Metal-mediated DNA base pairing: Alternatives to hydrogen-bondedWatson-Crick base pairs. Acc. Chem. Res. 2012, 45, 2066–2076. [CrossRef]
2. Müller, J. Metal-ion-mediated base pairs in nucleic acids. Eur. J. Inorg. Chem. 2008, 3749–3763.3. Clever, G.H.; Kaul, C.; Carell, T. DNA-metal base pairs. Angew. Chem. Int. Ed. 2007, 46, 6226–6236. [CrossRef]4. Taherpour, S.; Lönnberg, H.; Lönnberg, T. 2,6-Bis(functionalized) purines as metal-ion-binding surrogate
nucleobases that enhance hybridization with unmodified 2'-O-methyl oligoribonucleotides. Org. Biomol.Chem. 2013, 11, 991–1000. [CrossRef]
5. Shehata, M.R.; Shoukry, M.M.; Ali, S. Mono- and binuclear complexes involving [Pd(N,N-dimethylethylenediamine)(H2O)2]2+, 4,4'-bipiperidine and DNA constituents. J. Coord. Chem. 2012, 65,1311–1323. [CrossRef]
6. Kiss, A.; Farkas, E.; Sovago, I.; Thormann, B.; Lippert, B. Solution equilibria of the ternary complexes of[Pd(dien)Cl]+ and [Pd(terpy)Cl]+ with nucleobases and N-acetyl amino acids. J. Inorg. Biochem. 1997, 68,85–92. [CrossRef]
7. Kim, S.H.; Martin, R.B. Stabilities and 1H-NMR studies of (diethylenetriamine)Pd(II) and (1,1,4,7,7-pentamethyldien)Pd(II) with nucleosides and related ligands. Inorg. Chim. Acta 1984, 91, 11–18. [CrossRef]
8. Taherpour, S.; Lönnberg, T. Metal ion chelates as surrogates of nucleopbases for the recognition of nucleicacid sequences: The Pd2+ complex of bis(3,5-dimethylpyrazol-1-yl)purine riboside. J. Nucleic Acids 2012.[CrossRef]
9. Golubev, O.; Lönnberg, T.; Lönnberg, H. Metal-ion-binding analogs of ribobucleosides: Preparation andformation of ternary Pd2+ and Hg2+ complexes with natural pyrimidine nucleosides. Helv. Chim. Acta 2013,96, 1658–1669. [CrossRef]
10. Taherpour, S.; Golubev, O.; Lönnberg, T. Metal-ion-mediated base pairing between natural nucleobases andbidentate 3,5-dimethylpyrazolyl-substituted purine ligands. J. Org. Chem. 2014, 79, 8990–8999. [CrossRef]
65

Molecules 2014, 19, 16976–16986
11. Golubev, O.; Lönnberg, T.; Lönnberg, H. Interaction of Pd2+ complexes of 2,6-disubstituted pyridines withnucleoside 5'-monophosphates. J. Inorg. Biochem. 2014, 139, 21–29. [CrossRef]
12. Scheller, K.H.; Scheller-Krattiger, V.; Martin, R.B. Equilibria in solutions of nucleosides, 5-nucleotides anddienPd2+. J. Am. Chem. Soc. 1981, 103, 6833–6839. [CrossRef]
13. Häring, U.K.; Martin, R.B. Complexes of (ethylenediamine)Pd(II) with inosine, guanosine, adenosine andtheir phosphates. J. Inorg. Nucl. Chem. 1976, 38, 1915–1918. [CrossRef]
14. Matczak-Jon, E.; Jezowska-Trzebiatowska, B.; Kozlowski, H. Interaction of Pd(II) glycyl-L-histidine complexwith cytidine and GMP. Proton and carbon-13 nmr studies. J. Inorg. Biochem. 1980, 12, 143–156. [CrossRef]
15. Jezowska-Trzebiatowska, B.; Kozlowski, H.; Wolowiec, S. Coordination of Gly-Typ·Pd(II) complex to GMPnucleotide. Acta Biochim. Pol. 1980, 27, 99–109.
Sample Availability: Sample Availability: Samples of the compounds are not available.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
66

molecules
Communication
Design and Synthesis of a Series of TruncatedNeplanocin Fleximers
Sarah C. Zimmermann 1, Elizaveta O’Neill 1, Godwin U. Ebiloma 2, Lynsey J. M. Wallace 2,Harry P. De Koning 2 and Katherine L. Seley-Radtke 1,*
1 Department of Chemistry & Biochemistry, University of Maryland, Baltimore County, 1000 Hilltop Circle,Baltimore, MD 21250, USA
2 Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences,University of Glasgow, 120 University Place, Glasgow G12 8TA, UK
* Correspondence: [email protected]; Tel.: +1-410-455-8684; Fax: +1-410-455-2608
External Editor: Mahesh K. LakshmanReceived: 23 October 2014; in revised form: 8 December 2014; Accepted: 9 December 2014; Published:16 December 2014
Abstract: In an effort to study the effects of flexibility on enzyme recognition and activity, we havedeveloped several different series of flexible nucleoside analogues in which the purine base is splitinto its respective imidazole and pyrimidine components. The focus of this particular study wasto synthesize the truncated neplanocin A fleximers to investigate their potential anti-protozoanactivities by inhibition of S-adenosylhomocysteine hydrolase (SAHase). The three fleximers testeddisplayed poor anti-trypanocidal activities, with EC50 values around 200 μM. Further studies of thecorresponding ribose fleximers, most closely related to the natural nucleoside substrates, revealedlow affinity for the known T. brucei nucleoside transporters P1 and P2, which may be the reason forthe lack of trypanocidal activity observed.
Keywords: fleximer; carbocyclic nucleosides; 3-deazaneplanocin A; SAHase; trypanomiasis
1. Introduction
Modified nucleosides, in particular carbocyclic nucleosides, are potent inhibitors of S-adenosylhomocysteine hydrolase (SAHase) [1]. SAHase is a critical enzyme that hydrolyzes S-adenosylhomocysteine, the byproduct of biomethylations that utilize S-adenosylmethionine (SAM) [2,3].By inhibiting SAHase, an excess of SAH is produced, which in turn exhibits potent inhibitory effectson methyltransferases [4]. Thus, inhibition of SAHase leads to incomplete methylation of nucleic acids,phospholipids, proteins, and other small molecules, disrupting various biochemical pathways [5]. As aresult, carbocyclic nucleosides have proven useful in a number of chemotherapeutic applications [6–8].
Neplanocin A (NpcA, Figure 1) and aristeromycin (Ari) are both naturally occurringcarbocyclic adenosine analogues that have shown significant antiviral, antiparasitic and anticancerproperties [4,7,9,10]. Unfortunately, NpcA and Ari both exhibit deleterious cytotoxicity due tointracellular conversion to their triphosphate forms by adenosine kinase as well as their recognitionand metabolism by adenosine deaminase [11–13]. Removal of the 4'-CH2OH from Ari and NpcA, asshown in the truncated analogues shown in Figure 1 (R = H), significantly lowers the cytotoxicity [14].
Interestingly, nucleosides with base modifications such as 3-deazaadenosine have also beenfound to act as substrates, with similar Km’s found for adenosine and 3-deazaadenosine [7,10,15].To date, the truncated 3-deaza analogues of Ari and NpcA (truncated DZNepA, Figure 1) lackingthe 4'-hydroxy-methyl group have both exhibited greater levels of inhibition than their parentcounterparts [5,6,13,16].
Molecules 2014, 19, 21200–21214 67 www.mdpi.com/journal/molecules

Molecules 2014, 19, 21200–21214
Figure 1. Neplanocin A (NpcA) and analogues and the target NcpA fleximers (1–3).
More importantly, these compounds have also shown potent inhibition against chloroquine-resistant and chloroquine-susceptible strains of P. falciparum [5]. In protozoan parasites, methylationof the four nucleosides present in the “cap-four” terminal end of mRNA requires SAM as the methyldonor. This cap structure is important for RNA recognition and stability, is highly conserved acrossalmost all protozoan species, and is critical for replication [17–19]. Thus, inhibition of SAHase resultsin an accumulation of SAH, causing methylations to cease, which then disrupts the methylation of thecap structure, thereby providing an important target for the development of potential antiparasiticchemotherapeutics [5].
The Seley-Radtke group has long been interested studying the effects of flexibility on thenucleobase. This flexibility is achieved by “splitting” the purine base into its respective imidazoleand pyrimidine (or pyridine) components, which remain connected by a single carbon-carbon bondbetween the two heteroaromatic moieties. This connectivity allows for free rotation, while still retainingthe elements essential for base pairing and molecular recognition [20–28]. This modification has ledto enhanced enzyme binding and recognition, as well as the ability to overcome point mutations inenzyme binding sites [29,30]. These analogues have also been studied for their potential therapeuticproperties [20–28]. Interestingly, when the fleximer analogues of adenosine (Flex-A), inosine (Flex-I)and guanosine (Flex-G) were studied in SAHase, which is a flexible enzyme, Flex-A and Flex-I actedas substrates, whereas Flex-G proved to be an inhibitor [22]. This is significant because it is, to ourknowledge, the only report of a G-nucleoside inhibiting an adenosine metabolizing enzyme. It hasbeen postulated that this is due to an intramolecular hydrogen bond between the pyrimidine and the5'-OH of the sugar, which then positions the amino group into the binding site where the amino groupon adenosine would normally reside, thus essentially creating an adenosine mimic [22].
Historically, a number of nucleoside analogues have been evaluated for trypanocidalactivity [31–34]. For example, Cai et al. showed that the antiviral drug ribavirin was an inhibitorof Trypanosoma cruzi SAHase [33]. Additionally, 7-deaza-5'-noraristeromycin was shown to be a potentinhibitor of four strains of Trypanosoma brucei [34]. To further explore the potential of base flexibilityand antiparasitic activity, we combined the fleximer base with the carbocyclic nucleoside scaffold, todetermine whether the flexible base motif would enhance the biological results previously observedwith carbocyclic analogues such as NpcA and Ari. Thus, a series of 3-deaza fleximers (compounds 1–3,Figure 1) were designed and synthesized to evaluate their anti-parasitic properties.
2. Results and Discussion
2.1. Chemistry
As shown in Scheme 1, cyclopentenol 5 was available from known literature proceduresstarting from D-cyclopentenone 4 [35], which can be obtained following stereospecific reduction
68

Molecules 2014, 19, 21200–21214
to the “down” hydroxyl using Luche reduction conditions [36]. Alcohol 5 was then coupled to4,5-diiodoimidazole [29] using standard Mitsunobu [37] conditions to give 6. Initially the Mitsunobureaction was attempted with diisopropylazodicarboxylate (DIAD) and triphenylphosphine (TPP)in dichloromethane at room temperature to yield 5, however only in a 12% yield. Attempts atheating the reaction only served to give additional side products, as well as to lower the yieldeven further. Changing the solvent to THF increased the solubility of the diiodoimidazole andsubsequently resulted in an improved yield of 40%. Unfortunately, contaminates from the byproduct,triphenylphospine oxide (TPPO), still proved to be problematic during purification. Altering thephosphine reagent to DPPE (1,2-bis(diphenylphosphino)ethane) drastically improved the ease inpurification. Other coupling methods were also tried, such as using Hendrickson’s “POP” reagent,bis(triphenyl)oxodiphosphonium trifluoromethanesulfonate [38], or using bases such as NaH orK2CO3 [39] to form the imidazole nucleophile, proved unsuccessful when compared to the Mitsunobucoupling using DPPE.
Scheme 1. Synthesis of compound 6. Reagents and Conditions: a. CeCl3·7H2O, MeOH, NaBH4; b. DPPE,DIAD, 4,5-diiodoimidazole, THF, rt.
Next, as shown below in Scheme 2, removal of the 5-iodo group of 6 to give compound 7 wasachieved via selective deiodination using ethyl magnesium bromide (EtMgBr) followed by quenchingwith water. Coupling to the pyridine ring was then accomplished using Stille [40] coupling.
N
N
OO
I
I
6
N
N
OO
I
N
N
OO
NCl
N
N
OO
NR
9: R=NH210: R=OH
7 8
a b c
N
N
OHOH
NCl
N
N
OHOH
NR
2 1: R=NH23: R=OH
d d
Scheme 2. Synthesis of compounds 1–3. Reagents and Conditions: a. EtMgBr, THF, 0 ◦C; b. 3-tributyltin-2-chloropyridine, Pd(PPh3)4, Cu(I) Br, 1,4-dioxane, reflux; c. for compound 9: (i) hydrazine neat, 80 ◦C;(ii) TiCl3; for compound 10: concentrated acetic acid neat, 110 ◦C; d. TFA/H2O (1/1) in THF.
69

Molecules 2014, 19, 21200–21214
The 3-tributyltin-2-chloropyridine was prepared from the commercially available 3-bromo-2-chloropyridine. Stille coupling of 7 with the 3-tributyltin-2-chloropyridine provided 8 in a 23%yield, however when copper (I) bromide was used, the yield improved to 71%. Following Stillecoupling, transformation of the chloro group into the exocyclic amine group was necessary. Standardprocedures using MeOH/NH3 or converting the chloro to an azide using sodium or lithium azideproved unsuccessful. A literature search revealed a palladium-assisted method developed by Hartwigusing sodium t-butoxide in ammonia saturated 1,4-dioxane [41]. Unfortunately this method alsoproved unsuccessful.
Related to this latter route, Buchwald developed a similar method, where the catalyst is made insitu using a more common phosphine ligand [42]. This method seemed promising since one of theexamples utilized 2-chloropyridine, which was successfully converted in a 96% yield [28], but it tooproved to be unsuccessful. Use of NaNH2 in ammonia was also tried but the conditions proved to betoo harsh and decomposition ensued [43]. Another approach involved converting the chloro groupusing hydrazine followed by reduction. Initial attempts at reducing the hydrazine employed zinc inacetic acid, but this resulted in a complex mixture that could not be purified. Using titanium chloride(TiCl3) [44] proved to be successful, although there was evidence of some isopropylidene deprotectedproduct(s) as well as protected products, thus treatment of the mixture with dilute TFA in THF gavethe desired final product 1.
Next, deaminated compound 10 was obtained from 8 using concentrated acetic acid at hightemperature. Although this conversion also led to partial deprotection of the isopropylidene on the 2'-and 3'-hydroxyls, the protected pyridine 10 was the major product. Subsequent deprotection of theisopropylidene of 10 led to the fleximer inosine 3.
2.2. Trypanosomiasis Screening
The three NpcA fleximers (1–3) were tested for trypanocidal activity against the laboratoryTrypanosoma brucei brucei strain Lister 427, using a standard protocol based on the fluorescent format of23 doubling dilutions, starting at 500 μM, in 96-well plates. All three fleximers tested displayed verysimilar activities against this strain, with EC50 values around 200 μM; in contrast, the control drugpentamidine displayed activity in the low nM range (Table 1), consistent with previous results [45,46].
Table 1. Trypanosomiasis results.
Compound Average EC50 (μM)
1 216 ± 212 212 ± 313 287 ± 24
pentamidine 0.0044 ± 0.0001
EC50 = concentration of drug required to give a 50% response. Data are the average of three independent experimentsand SEM.
We considered that the relatively low activity might be related to a lack of recognition of thesemolecules by the T. brucei nucleoside transporters. We therefore investigated whether fleximers ingeneral display reduced uptake kinetics in these parasites, compared to their fixed-ring counterparts(Figure 2). Using the fleximers [21] most closely related to the original nucleoside substrates, it is clearfrom Table 2 that fleximers indeed show low affinity for the known T. brucei nucleoside transporters P1and P2 [47].
70

Molecules 2014, 19, 21200–21214
-9 -8 -7 -6 -5 -4 -30
25
50
75
100
125
Guanosine
Flex-AFlex-IFlex-GAdenosineInosine
log[Inhibitor] (M)
Aden
osin
e Up
take
(per
cent
of c
ontro
l)
Figure 2. Transport of 0.1 μM [3H]-adenosine by Trypanosoma brucei brucei bloodstream form parasites.
Table 2. Comparison of affinity of purine nucleosides and corresponding fleximers for T. brucei transporters.
P1 Ki (μM) P2 Ki (μM)
Nucleoside 1 Fleximer δ(ΔG0) Nucleoside 1 Fleximer δ(ΔG0)
Adenosine 0.36 ± 0.05 35 ± 11 11.4 0.91 ± 0.29 37 ± 3 9.2Guanosine 1.8 ± 0.3 251 ± 75 12.2 >500 >500
Inosine 0.44 ± 0.10 387 ± 30 16.8 >500 >500
Data are the average inhibition constants (Ki) and SEM of at least three independent experiments; Values foradenosine are Michaelis-Menten constants (Km). 1 values were taken from previous findings of De Koning [47] andincluded here for comparison; δ(ΔG0) is the difference in Gibbs free energy of interaction of the nucleoside and thefleximer with the transporter, given in kJ/mol.
Transport, mediated by the P1 nucleoside transporter, was measured in the presence or absence ofvarious concentrations of nucleosides (filled symbols) or their corresponding fleximers (open symbols),in the presence of 100 μM adenine to block potential adenosine transport through the P2 transporter.Data shown are the average and SEM of triplicate determinations in a single experiment, representativeof three independent experiments with essentially identical outcomes.
It is thus clear that the fleximers generally display about two orders of magnitude less affinityfor the T. brucei nucleoside transporters than the corresponding nucleosides, limiting cellular uptakeas there are no other nucleoside uptake mechanisms in these parasites than the P1 and P2 systems,although P1 consists of a cluster of multiple genes with slightly divergent sequences [48,49]. Inaddition, the truncated NpcA fleximers lack a 4'-hydroxymethyl group and an equivalent of the purineN3 residue, and both required for high affinity for P1 [50]. Moreover, the P2 transporter does notrecognize any oxopurine nucleoside analogues [47]. The loss of approximately 10 kJ/mol in Gibbsfree energy for the fleximer-transporter interaction may in part be due to the increased entropy in theorientation of the fleximer orientation in the binding pocket, as well as the slightly larger volume ofthe base. We thus conclude that the low effectiveness of the Npc fleximers is at least partially due tounfavorable interactions with the parasite’s nucleoside transporters. As important differences existbetween nucleoside transporters of even closely related pathogenic parasites including Trypanosomacongolense [51] and Leishmania species [48], it would be worthwhile to follow this study with a widerscreening of anti-parasite activity for a diverse panel of protozoa.
71

Molecules 2014, 19, 21200–21214
3. Experimental Section
3.1. General Information
All chemicals were obtained from commercial sources and used without further purificationunless otherwise noted. Anhydrous DMF, MeOH, DMSO and toluene were purchased from FisherScientific (Pittsburgh, PA, USA). Anhydrous THF, acetone, CH2Cl2, CH3CN and ether were obtainedusing a solvent purification system (mBraun Labmaster 130, MBRAUN, Stratham, NH, USA).3-Bromo-2-chloropyridine was obtained from Sigma-Aldrich (St. Louis, MO, USA). Melting pointsare uncorrected. NMR solvents were purchased from Cambridge Isotope Laboratories (Andover, MA,USA). All 1H- and 13C-NMR spectra were obtained on a JEOL ECX 400 MHz NMR, operated at 400and 100 MHz respectively, and referenced to internal tetramethylsilane (TMS) at 0.0 ppm. The spinmultiplicities are indicated by the symbols s (singlet), d (doublet), dd (doublet of doublets), t (triplet),q (quartet), m (multiplet), and b (broad). Reactions were monitored by thin-layer chromatography(TLC) using 0.25 mm Whatman Diamond silica gel 60-F254 precoated plates. Column chromatographywas performed using silica gel (63–200 μm) from Dynamic Adsorptions Inc. (Norcross, GA, USA), andeluted with the indicated solvent system. Yields refer to chromatographically and spectroscopically(1H- and 13C-NMR) homogeneous materials. High resolution mass spectra were recorded at theJohns Hopkins Mass Spectrometry Facility (Baltimore, MD, USA) using fast atom bombardmentfor ionization.
3.2. Synthesis
Preparation of (4R,5R)-4,5-O-isopropylidene-2-cyclopenten-1-ol (5): (4R,5R)-4,5-O-isopropylidene-2-cyclopentenone 4 [35] (4.63 g, 0.03 mol) was dissolved in dry methanol (20 mL) at room temperature.CeCl3·7H2O was added to the reaction followed by the portionwise addition of NaBH4 (1.36 g, 0.04mol). Once TLC analysis showed the complete disappearance 4, the reaction was extracted into ethylacetate (50 mL) and washed with water (10 mL). The organic layer was dried over MgSO4 and thesolvent was removed under rotary evaporation. The crude oil was used in the following reactionwithout further purification.
Preparation of (1′R,2′S,3′R)-1-[(2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl]-4,5-diiodoimidazole (6):Thoroughly dried 5 (0.78 g, 0.05 mol) was dissolved in dry THF (100 mL) under N2. Ethylenebis(diphenylphosphine) (2.0 g, 0.05 mol) and 4,5-diiodoimidazole (3.2 g, 0.01 mol) added to the reaction,followed by the dropwise addition of diisopropyl azodicarboxylate. The reaction was allowed to stirfor 48 h and then the solvent was removed using reduced pressure. The crude material was purifiedby silica gel column chromatography hexanes–ethyl acetate (2:1) to yield a yellow waxy solid (0.95 g,0.02 mol, 41% yield). 1H-NMR (CDCl3): δ 1.32 (s, 3H), 1.45 (s, 3H), 4.44 (d, 1H, J = 5.5 Hz), 5.24 (d, 1H,J = 1.4 Hz), 5.31 (m, 1H), 5.90 (dt, 1H, J = 4.6, 5.9 Hz), 6.34 (dt, 1H, J = 1.8 Hz, 5.9 Hz), 7.38 (s, 1H).13C-NMR (CDCl3): δ 25.9, 27.4, 70.8, 82.7, 84.2, 84.3, 97.1, 112.6, 128.9, 139.0, 139.4. HRMS calculatedfor C11H12I2N2O2 [M+H]+ 458.9066, found 458.9073.
Preparation of (1′R,2′S,3′R)-1-[(2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl]-4-iodoimidazole (7): Dried 6
(1.83 g, 0.004 mol) was dissolved in anhydrous THF under N2. The reaction was dropped to 0 ◦C,and ethyl magnesium bromide (3.0 M, 1.3 mL, 0.004 mol) was added dropwise to the reaction. After1 h the reaction was quenched with saturated NH4Cl (5 mL) and then the solvent was removed.The crude mixture was dissolved in ethyl acetate (50 mL) and washed with water (20 mL) and thendried over MgSO4. The solvent was removed under reduced pressure and purified using silica gelchromatography petroleum ether–ethyl acetate (2:1) to yield a yellow oil (1.12 g, 0.003 mol, 85% yield).1H-NMR (CDCl3): δ 1.28 (s, 3H), 1.40 (s, 3H), 4.44 (d, 1H, J = 5.5 Hz), 5.15 (d, 1H, J = 1.4 Hz), 5.30 (dq,1H, J = 1.8, 5.5 Hz), 5.87 (dt, 1H, J = 0.9, 5.5 Hz), 6.22 (dt, 1H, J = 1.8, 5.9 Hz), 6.89 (d, 1H, J = 1.4 Hz),7.34 (d, 1H, J = 1.4 Hz). 13C-NMR (CDCl3): δ 25.7, 27.3, 68.3, 82.7, 84.2, 84.9, 112.5, 123.2, 129.8, 137.5,138.1. HRMS calculated for C11H13IN2O2 [M+H]+ 333.0100, found 333.0010.
72

Molecules 2014, 19, 21200–21214
Preparation of 3-tributylstannyl-2-chloropyridine: Commercially available 3-bromo-2-chloropyridine(0.30 g, 0.002 mol) was dissolved in anhydrous THF (20 mL) under N2. Ethyl magnesium bromide(3.0 M, 0.5 mL, 0.002 mol) was added dropwise at room temperature. The reaction was allowed to stirfor 2 h, and then tributyltin chloride (0.42 mL, 0.002 mol) was added and the reaction was left to stirovernight. The reaction was concentrated in vacuo and then purified on silica gel chromatographyhexanes-ethyl acetate (15:1) to yield a colorless oil (0.40 g, 0.001 mol, 64% yield). 1H-NMR (CDCl3):δ 1.13 (m, 5 H), 1.30 (m, 13H), 1.56 (m, 6 H), 1.65 (m, 3 H), 7.13 (dd, 1H, J = 4.6, 7.8 Hz), 7.67 (dd, 1H,J = 1.8, 4.6 Hz), 8.27 (dd, 1H, J = 1.8, 7.8 Hz). 13C-NMR (CDCl3): δ 26.9, 27.0, 27.3, 27.9, 28.9, 29.0, 29.1,122.2, 139.5, 146.9, 147.1, 147.2, 149.6, 159.2.
Preparation of (1′R,2′S,3′R)-3-[((2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl)-(imidazol-4-yl)]-2-chloropyridine(8): Intermediate 7 (0.34 g, 0.001 mol) and 2-chloro-3-(tributylstannyl)pyridine (3.50 g, 0.007 mol) weredissolved in 1,4-dioxane under N2. Pd(PPh3)4 (0.05 g, 0.04 mmol) and CuBr (0.08 g, 0.5 mmol) wereadded to the reaction and the reaction and was refluxed at 120 ◦C for 12 h. The reaction was cooledand filtered through a pad of Celite. The filtrate was diluted in ethyl acetate (20 mL) and washed witha saturated solution of NH4Cl (20 mL), water (20 mL), brine (20 mL) and then dried over MgSO4. Theorganic solvent was removed under reduced pressure and the crude material was purified using 5%MeOH in CH2Cl2 to yield a yellow oil (0.23 g, 0.7 mmol, 71% yield). 1H-NMR (CDCl3): δ 1.36 (s, 3H),1.48 (s, 3H), 4.58 (d, 1H, J = 5.5 Hz), 5.28 (d, 1H, J = 1.4 Hz), 5.40 (dt, 1H, J = 0.9, 4.6 Hz), 6.00 (dd, 1H,J = 1.2, 5.5 Hz), 6.32 (dt, 1H, J = 1.8, 5.5 Hz), 7.30 (dd, 1H, J = 4.6, 7.7 Hz), 7.55 (d, 1H, J = 1.4 Hz), 7.67(d, 1H, J = 0.9 Hz), 8.26 (dd, 1H, J = 1.8, 4.6 Hz), 8.50 (dd, 1H, J = 1.8, 7.8 Hz). 13C-NMR (CDCl3): δ25.7, 27.3, 68.5, 84.4, 85.1, 112.6, 118.5, 122.8, 129.4, 130.1, 132.1, 135.9, 136.9, 137.7, 137.9, 147.2. HRMScalculated for C16H16ClN3O2 [M+H 35Cl]+ 318.1009, [M+H 37Cl]+ 320.0980, found, 318.1001, 320.0976.
Preparation of (1′R,2′S,3′R)-3-[((2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl)-(imidazol-4-yl)]-2-pyrimidone(10): Analogue 9 (0.1 g, 0.3 mmol) was dissolved in concentrated acetic acid in a sealed glass tube andheated to 120 ◦C overnight. The acetic acid was evaporated and the crude material was extracted intoethyl acetate. Silica gel chromatography using ethyl acetate-acetone-methanol (6:1:1) yielded an offwhite solid (0.05 g, 0.2 mmol, 56% yield). 1H-NMR (DMSO-d6): δ 1.24 (s, 3H), 1.32 (s, 3H), 4.41 (d,1H, J = 6.9 Hz), 5.05 (d, 1H, J = 4.6 Hz), 5.98 (d, 1H, J = 5.9 Hz), 6.12 (dt, 1H, J = 2.3, 5.9 Hz), 6.26 (t,1H, J = 6.4 Hz), 7.24 (d, 1H, J = 4.6 Hz), 7.65 (d, 1H, J = 0.9 Hz), 7.79 (d, 1H, J = 1.4 Hz), 8.07 (dd, 1H,J = 1.8, 7.3 Hz), 11.78 (bs, 1H). 13C-NMR (DMSO-d6): δ 25.5, 27.3, 67.4, 73.0, 78.9, 105.8, 117.6, 125.1,131.0, 131.6, 132.9, 133.9, 136.7, 136.8, 136.9, 160.7. HRMS calculated for C16H17N3O3 [M+H]+ 300.1348,found 300.1342.
Preparation of (1′R,2′S,3′R)-3-[(2′,3′-Dihydroxy)-4′-cyclopenten-1′-yl]-(imidazol-4-yl)-2-amino-pyridine (1):Compound 8 (80 mg, 0.25 mmol) was refluxed in hydrazine (2 mL) for 1 h. The solvent wasremoved under reduced pressure and the residue dissolved in THF (5 mL). Titanium (III) chloride(1.7 mmol, 0.5 mL, 20% in 3% HCl) was neutralized using NaOH (0.5 mL, 20%), and 0.6 mL of thesolution was added dropwise to the reaction. The mixture was refluxed at 70 ◦C for 4 h, cooled toroom temperature, and brought to pH > 10 using NaOH (20%) while cooling in an ice-bath. Thesolvents were removed under vacuum, and the product was extracted with CH2Cl2 (10 mL × 5).The organic layer was dried over MgSO4. The solvents were evaporated, and the crude material,3-[((2', 3'-O-isopropylidene)-4'-cyclopenten-1'-yl)-imidazol-4-yl]-2-aminopyridine (9), was used directlyin preparation of 1. Crude 9 was dissolved in THF (5 mL) and TFA:H2O (1 mL:1 mL) was addeddropwise. This was allowed to stir overnight at room temperature. The solvent was evaporated andco-evaporated with ethanol (3 × 5 mL) to yield an off-white solid (0.03 g, 0.1 mmol, 46% yield over2 steps). 1H-NMR (DMSO-d6): δ 3.93 (m, 1H), 4.45 (m, 1H), 4.96 (m, 1H), 5.08 (m, 1H), 5.14 (d, 1H,J = 6.9 Hz), 5.96 (dd, 1H, J = 1.4, 6.4 Hz), 6.11 (dt, 1H, J = 2.3, 6.4 Hz), 6.54 (dd, 1H, J = 5.0, 7.3 Hz), 6.95(bs, 2H), 7.59 (d, 1H, J = 0.9 Hz), 7.70 (dd, 1H, J = 1.8, 7.4 Hz), 7.78 (d, 1H, J = 1.0 Hz), 7.81 (m, 1H).13C-NMR (DMSO-d6): δ 67.6, 73.1, 78.6, 112.2, 112.5, 115.1, 132.9, 133.7, 136.2, 136.9, 140.1, 146.5, 156.5HRMS calculated for C13H14N4O2 [M+H]+ 259.1195, found 259.1193.
73

Molecules 2014, 19, 21200–21214
Preparation of (1′R,2′S,3′R)-3-[(2′,3′-Dihydroxy)-4′-cyclopenten-1′-yl]-(imidazol-4-yl)-2-chloro-pyridine (2):Intermediate 8 (0.16 g, 0.5 mmol) was dissolved in THF (5 mL) and TFA:H2O (1 mL:1 mL) was addeddropwise. This was allowed to stir overnight at room temperature. The solvent was evaporated andco-evaporated with ethanol (3 × 5 mL). Column chromatography in 10% MeOH in CH3CN returnedan off-white solid (0.12 g, 0.4 mmol, 86% yield). 1H-NMR (DMSO-d6): δ 3.90 (m, 1H), 4.42 (m, 1H), 4.98(m, 1H), 5.08 (m, 1H), 5.17 (d, 1H, J = 6.4 Hz), 6.00 (dd, 1H, J = 1.4, 5.9 Hz), 6.11 (dt, 1H, J = 2.3, 5.9Hz), 7.43 (dd, 1H, J = 4.6, 7.8 Hz), 7.77 (s, 1H), 7.80 (s, 1H), 8.22 (dd, 1H, J = 2.3, 4.6 Hz), 7.81 (dd, 1H,J = 1.8, 7.8 Hz). 13C-NMR (DMSO-d6): δ 67.6, 73.0, 78.9, 119.0, 123.8, 129.9, 132.7, 135.4, 137.1, 137.6,137.9, 146.5, 147.5. HRMS calculated for C13H12ClN3O2 [M+H 35Cl]+ 278.0696, [M+H 37Cl]+ 280.0667,found, 278.0689, 280.0668.
Preparation of (1′R,2′S,3′R)-3-[(2′,3′-Dihydroxy)-4′-cyclopenten-1′-yl]-imidazol-4-yl)-2-hydroxypyridine (3):Intermediate 10 (0.08 g, 0.3 mmol) was dissolved in THF (5 mL) and TFA:H2O (1 mL:1 mL)was added dropwise. This was allowed to stir overnight at room temperature. The solventwas evaporated and co-evaporated with ethanol (3 × 5 mL). Column chromatography in ethylacetate–acetone–methanol–water (6:1:1:0.5) produced an off-white solid (0.03 g, 0.1 mmol, 43% yield).1H-NMR (DMSO-d6): δ 3.82 (d, 1H, J = 4.6 Hz), 4.43 (bs, 1H), 4.95 (m, 1H), 5.05 (m, 1H), 5.11 (d, 1H,J = 7.4 Hz), 5.97 (dd, 1H, J = 1.4, 5.9 Hz), 6.12 (dt, 1H, J = 2.8, 5.9 Hz), 6.29 (t, 1H, J = 6.9 Hz), 7.23 (d, 1H,J = 4.6 Hz), 7.65 (d, 1H, J = 0.9 Hz), 7.80 (d, 1H, J = 1.4), 8.08 (dd, 1H, J = 2.3, 7.3 Hz), 11.73 (bs, 1H).13C-NMR (DMSO-d6): δ 67.6, 73.1, 78.6, 112.2, 112.5, 115.1, 132.9, 133.7, 136.2, 136.9, 140.1, 146.5, 156.5HRMS calculated for C13H13N3O3 [M+H]+ 260.1035, found 260.1033.
3.3. Anti-Trypanosome Activity
In vitro activity against Trypanosoma brucei was determined using the Alamar blue (resazurin)assay for cell viability exactly as described [52]. Briefly, serial dilutions of test compounds were madein 96-well plates by serial passage of 100 μL of test compound (usually at 2 mM) to 100 μL of HMI9medium containing 10% fetal bovine serum (Invitrogen), using 2 rows, with the negative control valuesobtained from wells with 100 μL of medium without test compound. Serial dilutions with pentamidineisethionate (Sigma) were used as positive control. To each well, 100 μL of medium, containing 104
culture-adapted bloodstream T. b. brucei (strain Lister 427), was added and the plates were incubatedat 37 ◦C for 48 h after which 20 μL 5 mM resazurin solution was added. Following a further incubationof 24 h at 37 ◦C, fluorescence was determined in a FLUOstar OPTIMA (BMG Labtech, Aylesbury, UK)fluorimeter with excitation and emission filters at 544 nm and 620 nm, respectively. EC50 values (theeffective concentration reducing specific fluorescence by 50%) were calculated by nonlinear regressionusing the Prism 5 software package (GraphPad, La Jolla, CA, USA).
3.4. Transport Assays
Transport assays with bloodstream forms of T. b. brucei were performed exactly as describedpreviously [53,54]. Briefly, transport was initiated by the addition of 100 μL of T. b. brucei bloodstreamforms (107 cells/mL in assay buffer [53]) to 100 μL of [2,8,5'-3H]-adenosine (PerkinElmer, Waltham,MA, USA; 54.4 Ci/mmol) pre-mixed with up to 1 mM of test inhibitor in assay buffer. After exactly10 s the mixture was centrifuged through an oil layer in a microfuge (13,000× g) and the microfugetubes were flash-frozen in liquid nitrogen. Pellets were cut off and collected in scintillation tubes; aftersolubilisation in 2% SDS and addition of scintillation fluid, radioactivity was determined in a liquidscintillation counter. Inhibition data were fitted to a sigmoidal curve with variable slope (GraphPadPrism 5.0), allowing for the determination of EC50 values, from which inhibition constants (Ki) werecalculated using the Cheng-Prusoff equation, and Gibbs Free Energy using ΔG0 = −RTln (Ki), asdescribed [52].
74

Molecules 2014, 19, 21200–21214
4. Conclusions
The strategy of the work presented herein was to potentially synthesize new and more potentinhibitors of SAHases, thereby disrupting mRNA capping in protozoa as a strategy towards newantiparasitic therapeutics. To this end, characteristics of known SAHase inhibitors such as neplanocinand Aristeromycin were combined, and the nucleoside analogue was given enhanced flexibility usingthe “fleximer” approach, and added specificity by omitting the N3 equivalent nitrogen residue in thepyrimidine half of the fleximer base group. In addition, the 4'-CH2OH moiety was omitted to reducegeneral cytotoxicity [10,14]. The data, however, show that the resulting 3-deazaneplanocin fleximers(1–3) displayed only moderate activity in a standardized anti-protozoal test, against Trypanosoma brucei,despite the possibility of this species being vulnerable to inhibition of SAHase [55].
We have previously shown that the trypanocidal action of nucleoside and nucleobase analoguesis either enabled or limited by their rate of uptake by specific transport proteins [52–54,56,57], andtherefore investigated the effect of the fleximer modification on nucleoside transport. We foundthat the introduction of this modification of the purine ring reduces affinity, and thus presumablytranslocation rates, for both of the transport systems expressed in bloodstream T. brucei, and concludethat the lack of suitable transporters for these molecules causes (or at least contributes to) theobserved lack of trypanocidal potency. However, we have also shown that purine transportersin other protozoan parasites, e.g., Toxoplasma gondii [58], Plasmodium falciparum [59], Leishmaniadonovani [48], and Trichomonas vaginalis (Natto and De Koning, unpublished data) all have very differentsubstrate-specificity characteristics. Further studies with additional parasites, and the optimization ofthe inhibitors for enhanced uptake by the parasites, are in progress.
Acknowledgments: This work was supported by the National Institutes of Health (NIH) (R01 CA97634 to KSR,NIH T32 GM066706 CBI Fellowship to SCZ) and Wellcome Trust (grant to HPdK). We are also grateful to PhilMortimer (Johns Hopkins Mass Spectrometry Facility) for his invaluable assistance with the HRMS analysis. Wealso thank Therese Ku for her help with compound characterizations and editorial assistance.
Author Contributions: KLS was responsible for the oversight of the project and supervision of the students, SCZand EO, while HdK was responsible for supervision of GUE and LW. KLS and SCZ designed the synthesis; SCZand EO performed the synthesis and characterization of the compounds; GUE, LW and HdK were responsible forthe parasite testing and adenosine uptake assays. All authors read and approved the final manuscript.
Conflicts of Interest: The authors have no conflict of interest.
References
1. Tseng, C.K.; Marquez, V.E.; Fuller, R.W.; Goldstein, B.M.; Haines, D.R.; McPherson, H.; Parsons, J.L.;Shannon, W.M.; Arnett, G.; Hollingshead, M.; et al. Synthesis of 3-deazaneplanocin A, a powerful inhibitorof S-adenosylhomocysteine hydrolase with potent and selective in vitro and in vivo antiviral activities.J. Med. Chem. 1989, 32, 1442–1446. [CrossRef]
2. Cantoni, G. The centrality of S-adenosylhomocysteinase in the regulation of the biological utilizationof S-adenosylmethionine. In Biological Methylation and Drug Design; Borchardt, R.T., Creveling, C.R.,Ueland, P.M., Eds.; Humana Press: Clifton, NJ, USA, 1986; pp. 227–238.
3. Cantoni, G.L.; Scarano, E. The formation of S-adenosylhomocysteine in enzymatic transmethylation reactions.J. Am. Chem. Soc. 1954, 76, 4744. [CrossRef]
4. Borchardt, R.T.; Keller, B.T.; Patel-Thombre, U.; Neplanocin, A. A potent inhibitor of S-adenosylhomocysteinehydrolase and of vaccinia virus multiplication in mouse L929 cells. J. Biol. Chem. 1984, 259, 4353–4358.[PubMed]
5. Bujnicki, J.M.; Prigge, S.T.; Caridha, D.; Chiang, P.K. Structure, evolution, and inhibitor interaction ofS-adenosyl-L-homocysteine hydrolase from Plasmodium falciparum. Proteins 2003, 52, 624–632. [CrossRef][PubMed]
6. Chiang, P.K. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 1998, 77,115–134. [CrossRef] [PubMed]
7. De Clercq, E. John Montgomery’s legacy: Carbocyclic adenosine analogues as SAH hydrolase inhibitorswith broad-spectrum antiviral activity. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1395–1415.
75

Molecules 2014, 19, 21200–21214
8. Marquez, V.E. Carbocyclic nucleosides. Adv. Antivir. Drug Des. 1996, 2, 89–146.9. Borchardt, R.T.; Wu, Y.-S. S-Aristeromycinyl-L-homocysteine, a potent inhibitor of S-adenosylmethionine-
dependent transmethylations. J. Med. Chem. 1976, 19, 197–198. [CrossRef] [PubMed]10. Guranowski, A.; Montgomery, J.A.; Cantoni, G.L.; Chiang, P.K. Adenosine analogues as substrates and
inhibitors of S-adenosylhomocysteine hydrolase. Biochemistry 1981, 20, 110–115. [CrossRef] [PubMed]11. Van Brummelen, A.C.; Olszewski, K.L.; Wilinski, D.; Llinas, M.; Louw, A.I.; Birkholtz, L.M. Co-inhibition
of Plasmodium falciparum S-adenosylmethionine decarboxylase/ornithine decarboxylase revealsperturbation-specific compensatory mechanisms by transcriptome, proteome, and metabolome analyses.J. Biol. Chem. 2009, 284, 4635–4646. [CrossRef] [PubMed]
12. Wolfe, M.S.; Borchardt, R.T. S-adenosyl-L-homocysteine hydrolase as a target for antiviral chemotherapy.J. Med. Chem. 1991, 34, 1521–1530. [CrossRef] [PubMed]
13. Hasobe, M.; Liang, H.; Ault-Riche, D.B.; Borcherding, D.R.; Wolfe, M.S.; Borchardt, R.T. (1'R,2'S,3'R)-9-(2',3'-Dihydroxycyclopentan-1'-yl)-adenine and -3-deaza-adenine: Analogs of aristeromycin which exhibitpotent antiviral activity with reduced cytotoxicity. Antivir. Chem. Chemother. 1993, 4, 245–248.
14. Wolfe, M.S.; Lee, Y.; Bartlett, W.J.; Borcherding, D.R.; Borchardt, R.T. 4'-Modified analogs of aristeromycin andneplanocin A: Synthesis and inhibitory activity toward S-adenosyl-L-homocysteine hydrolase. J. Med. Chem.1992, 35, 1782–1791. [CrossRef] [PubMed]
15. Richards, H.H.; Chiang, P.K.; Cantoni, G.L. Adenosylhomocysteine hydrolase. Crystallization of the purifiedenzyme and its properties. J. Biol. Chem. 1978, 253, 4476–4480. [PubMed]
16. Hasobe, M.; McKee, J.G.; Borcherding, D.R.; Borchardt, R.T. 9-(trans-2',trans-3'-Dihydroxycyclopent-4'-enyl)-adenine and -3-deazaadenine: Analogs of neplanocin A which retain potent antiviral activity but exhibitreduced cytotoxicity. Antimicrob. Agents Chemother. 1987, 31, 1849–1851. [CrossRef] [PubMed]
17. Ruan, J.P.; Shen, S.; Ullu, E.; Tschudi, C. Evidence for a capping enzyme with specificity for the trypanosomespliced leader RNA. Mol. Biochem. Parasitol. 2007, 156, 246–254. [CrossRef] [PubMed]
18. Mair, G.; Ullu, E.; Tschudi, C. Cotranscriptional Cap 4 Formation on the Trypanosoma brucei spliced leaderRNA. J. Biol. Chem. 2000, 275, 28994–28999. [CrossRef] [PubMed]
19. Zamudio, J.R.; Mittra, B.; Campbell, D.A.; Sturm, N.R. Hypermethylated cap 4 maximizes Trypanosomabrucei translation. Mol. Microbiol. 2009, 72, 1100–1110. [CrossRef] [PubMed]
20. Seley, K.L.; Zhang, L.; Hagos, A. “Fleximers”. Design and synthesis of two novel split nucleosides. Org. Lett.2001, 3, 3209–3210. [CrossRef] [PubMed]
21. Seley, K.L.; Zhang, L.; Hagos, A.; Quirk, S. “Fleximers”. Design and synthesis of a new class of novelshape-modified nucleosides. J. Org. Chem. 2002, 67, 3365–3373. [CrossRef] [PubMed]
22. Seley, K.L.; Quirk, S.; Salim, S.; Zhang, L.; Hagos, A. Unexpected inhibition of S-adenosyl-L-homocysteinehydrolase by a guanosine nucleoside. Bioorg. Med. Chem. Lett. 2003, 13, 1985–1988. [CrossRef] [PubMed]
23. Polak, M.; Seley, K.L.; Plavec, J. Conformational properties of shape modified nucleosides—Fleximers. J. Am.Chem. Soc. 2004, 126, 8159–8166. [CrossRef] [PubMed]
24. Seley, K.L.; Salim, S.; Zhang, L. “Molecular chameleons”. Design and synthesis of C-4-substituted imidazolefleximers. Org. Lett. 2005, 7, 63–66. [CrossRef] [PubMed]
25. Seley, K.L.; Salim, S.; Zhang, L.; O’Daniel, P.I. “Molecular chameleons”. Design and synthesis of a secondseries of flexible nucleosides. J. Org. Chem. 2005, 70, 1612–1619. [CrossRef] [PubMed]
26. Zimmermann, S.C.; Sadler, J.M.; Andrei, G.; Snoeck, R.; Balzarini, J.; Seley-Radtke, K.L. Carbocyclic 5'-nor“reverse” fleximers. Design, synthesis, and preliminary biological activity. Med. Chem. Commun. 2011, 2,650–654. [CrossRef]
27. Wauchope, O.R.; Velasquez, M.; Seley-Radtke, K. Synthetic routes to a series of proximal and distal 2'-deoxyfleximers. Synthesis (Stuttg.) 2012, 44, 3496–3504. [CrossRef]
28. Zimmermann, S.C.; Sadler, J.M.; O’Daniel, P.I.; Kim, N.T.; Seley-Radtke, K.L. “Reverse” carbocyclic fleximers:Synthesis of a new class of adenosine deaminase inhibitors. Nucleosides Nucleotides Nucleic Acids 2013, 32,137–154. [CrossRef] [PubMed]
29. Quirk, S.; Seley, K.L. Identification of catalytic amino acids in the human GTP fucose pyrophosphorylaseactive site. Biochemistry 2005, 44, 13172–13178. [CrossRef] [PubMed]
30. Quirk, S.; Seley, K.L. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry2005, 44, 10854–10863. [CrossRef] [PubMed]
76

Molecules 2014, 19, 21200–21214
31. Williamson, J.; Scott-Finnigan, T.J. Trypanocidal activity of antitumor antibiotics and other metabolicinhibitors. Antimicrob. Agents Chemother. 1978, 13, 735–744. [CrossRef] [PubMed]
32. Sufrin, J.R.; Rattendi, D.; Spiess, A.J.; Lane, S.; Marasco, C.J., Jr.; Bacchi, C.J. Antitrypanosomal activity ofpurine nucleosides can be enhanced by their conversion to O-acetylated derivatives. Antimicrob. AgentsChemother. 1996, 40, 2567–2572. [PubMed]
33. Cai, S.; Li, Q.S.; Borchardt, R.T.; Kuczera, K.; Schowen, R.L. The antiviral drug ribavirin is a selective inhibitorof S-adenosyl-L-homocysteine hydrolase from Trypanosoma cruzi. Bioorg. Med. Chem. 2007, 15, 7281–7287.[CrossRef] [PubMed]
34. Seley, K.L.; Schneller, S.W.; Rattendi, D.; Bacchi, C.J. (+)-7-deaza-5'-noraristeromycin as an anti-trypanosomalagent. J. Med. Chem. 1997, 40, 622–624. [CrossRef] [PubMed]
35. Yang, M.; Ye, W.; Schneller, S.W. Preparation of carbocyclic S-adenosylazamethionine accompanied bya practical synthesis of (−)-aristeromycin. J. Org. Chem. 2004, 69, 3993–3996. [CrossRef] [PubMed]
36. Luche, J.-L. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J. Am.Chem. Soc. 1978, 100, 2226–2227. [CrossRef]
37. Swamy, K.C.; Kumar, N.N.; Balaraman, E.; Kumar, K.V. Mitsunobu and related reactions: Advances andapplications. Chem. Rev. 2009, 109, 2551–2651. [CrossRef] [PubMed]
38. Moussa, Z. The Hendrickson ‘POP’ reagent and analogues thereof: Synthesis, structure, and application inorganic synthesis. ARKIVOC 2012, 432–490.
39. Kim, J.-H.; Kim, H.O.; Lee, K.M.; Chun, M.W.; Moon, H.R.; Jeong, L.S. Asymmetric synthesis ofhomo-apioneplanocin A from D-ribose. Tetrahedron 2006, 62, 6339–6342. [CrossRef]
40. Hassan, J.; Sevignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl-aryl bond formation one century after thediscovery of the Ullmann reaction. Chem. Rev. 2002, 102, 1359–1469. [CrossRef] [PubMed]
41. Vo, G.D.; Hartwig, J.F. Palladium-catalyzed coupling of ammonia with aryl chlorides, bromides, iodides,and sulfonates: A general method for the preparation of primary arylamines. J. Am. Chem. Soc. 2009, 131,11049–11061. [CrossRef] [PubMed]
42. Huang, X.; Buchwald, S.L. New ammonia equivalents for the Pd-catalyzed amination of aryl halides.Org. Lett. 2001, 3, 3417–3419. [CrossRef] [PubMed]
43. Benkeser, R.A.; Buting, W.E. The preparation of aromatic amines with sodium amide in liquid ammonia.J. Am. Chem. Soc. 1952, 74, 3011–3014. [CrossRef]
44. Zhang, Y.; Tang, Q.; Luo, M. Reduction of hydrazines to amines with aqueous solution of titanium(iii)trichloride. Org. Biomol. Chem. 2011, 9, 4977–4982. [CrossRef] [PubMed]
45. Matovu, E.; Stewart, M.L.; Geiser, F.; Brun, R.; Maser, P.; Wallace, L.J.; Burchmore, R.J.; Enyaru, J.C.;Barrett, M.P.; Kaminsky, R.; et al. Mechanisms of arsenical and diamidine uptake and resistance inTrypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [CrossRef] [PubMed]
46. Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Aguinaga Andres, D.; Natto, M.J.; Teka, I.A.;McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter forpentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to thesedrugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [CrossRef] [PubMed]
47. De Koning, H.P.; Jarvis, S.M. Adenosine transporters in bloodstream forms of Trypanosoma brucei brucei:Substrate recognition motifs and affinity for trypanocidal drugs. Mol. Pharmacol. 1999, 56, 1162–1170.[PubMed]
48. De Koning, H.P.; Bridges, D.J.; Burchmore, R.J.S. Purine and pyrimidine transport in pathogenic protozoa:From biology to therapy. FEMS Microbiol. Rev. 2005, 29, 987–1020. [CrossRef] [PubMed]
49. Al-Salabi, M.I.; Wallace, L.J.; Luscher, A.; Maser, P.; Candlish, D.; Rodenko, B.; Gould, M.K.; Jabeen, I.;Ajith, S.N.; de Koning, H.P. Molecular interactions underlying the unusually high adenosine affinity of anovel Trypanosoma brucei nucleoside transporter. Mol. Pharmacol. 2007, 71, 921–929. [CrossRef] [PubMed]
50. De Koning, H.P. Transporters in African trypanosomes: Role in drug action and resistance. Int. J. Parasitol.2001, 31, 512–522. [CrossRef] [PubMed]
51. Munday, J.C.; Rojas Lopez, K.E.; Eze, A.A.; Delespaux, V.; van den Abbeele, J.; Rowan, T.; Barrett, M.P.;Morrison, L.J.; de Koning, H.P. Functional expression of TcoAT1 reveals it to be a P1-type nucleosidetransporter with no capacity for diminazene uptake. Int. J. Parasitol. Drugs Drug Resist. 2013, 3, 69–76.[CrossRef] [PubMed]
77

Molecules 2014, 19, 21200–21214
52. Wallace, L.J.; Candlish, D.; de Koning, H.P. Different substrate recognition motifs of human and trypanosomenucleobase transporters. Selective uptake of purine antimetabolites. J. Biol. Chem. 2002, 277, 26149–26156.[CrossRef] [PubMed]
53. Natto, M.J.; Wallace, L.J.; Candlish, D.; Al-Salabi, M.I.; Coutts, S.E.; de Koning, H.P. Trypanosoma brucei:Expression of multiple purine transporters prevents the development of allopurinol resistance. Exp. Parasitol.2005, 109, 80–86. [CrossRef] [PubMed]
54. Ali, J.A.; Creek, D.J.; Burgess, K.; Allison, H.C.; Field, M.C.; Maser, P.; de Koning, H.P. Pyrimidinesalvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines.Mol. Pharmacol. 2013, 83, 439–453. [CrossRef] [PubMed]
55. Parker, N.B.; Yang, X.; Hanke, J.; Mason, K.A.; Schowen, R.L.; Borchardt, R.T.; Yin, D.H. Trypanosoma cruzi:Molecular cloning and characterization of the S-adenosylhomocysteine hydrolase. Exp. Parasitol. 2003, 105,149–158. [CrossRef] [PubMed]
56. Geiser, F.; Luscher, A.; de Koning, H.P.; Seebeck, T.; Maser, P. Molecular pharmacology of adenosine transportin Trypanosoma brucei: P1/P2 revisited. Mol. Pharmacol. 2005, 68, 589–595. [PubMed]
57. Vodnala, S.K.; Lundbäck, T.; Yeheskieli, E.; Sjöberg, B.; Gustavsson, A.-L.; Svensson, R.; Olivera, G.C.;Eze, A.A.; de Koning, H.P.; Hammarström, L.G.J.; et al. Structure-activity relationships of syntheticcordycepin analogues as experimental therapeutics for African Trypanosomiasis. J. Med. Chem. 2013,56, 9861–9873. [CrossRef] [PubMed]
58. De Koning, H.P.; Al-Salabi, M.I.; Cohen, A.M.; Coombs, G.H.; Wastling, J.M. Identification andcharacterisation of high affinity nucleoside and nucleobase transporters in Toxoplasma gondii. Int. J.Parasitol. 2003, 33, 821–831. [CrossRef] [PubMed]
59. Quashie, N.B.; Dorin-Semblat, D.; Bray, P.G.; Biagini, G.A.; Doerig, C.; Ranford-Cartwright, L.C.; deKoning, H.P. A comprehensive model of purine uptake by the malaria parasite Plasmodium falciparum:Identification of four purine transport activities in intraerythrocytic parasites. Biochem. J. 2008, 411, 287–295.[CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples of the compounds are not available from the authors at this time.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
78

molecules
Article
Development of New 1,3-DiazaphenoxazineDerivatives (ThioG-Grasp) to CovalentlyCapture 8-Thioguanosine
Yasufumi Fuchi, Hideto Obayashi and Shigeki Sasaki *
Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582,Japan; [email protected] (Y.F.); [email protected] (H.O.)* Correspondence: [email protected]; Tel.: +81-92-642-6615
Academic Editor: Fumi NagatsugiReceived: 9 December 2014; Accepted: 7 January 2015; Published: 9 January 2015
Abstract: The derivatives of 8-thioguanosine are thought to be included in the signal transductionsystem related to 8-nitroguanosine. In this study, we attempted to develop new 1,3-diazaphenoxazine(G-clamp) derivatives to covalently capture 8-thioguanosine (thioG-grasp). It was expected that thechlorine atom at the end of the linker would be displaced by the nucleophilic attack by the sulfuratom of 8-thioguanosine via multiple hydrogen-bonded complexes. The thioG-grasp derivative witha propyl linker reacted efficiently with 8-thioguanosine to form the corresponding adduct.
Keywords: oxidative damage; oxidized nucleoside; 8-oxoguanosine; 8-nitroguanosine; 8-thioguanosine
1. Introduction
Reactive oxygen species (ROS) and reactive nitrogen oxide species (RNOS) are the chemical sourcesof oxidative stress, and they oxidize nucleic acids to produce the 8-oxoguanosine and 8-nitroguanosinederivatives [1–3]. The oxidized nucleosides/nucleotides are highly mutagenic and are regarded asbiomarkers of oxidative stress [4]. On the other hand, recent studies have revealed that nitrated-guanosinederivatives also play important roles as signal messengers; 8-nitroguanosine-3′,5′-cyclic monophosphate(8-nitro-cGMP) is generated from guanosine- 5′-triphosphate (GTP) in response to the productionof peroxynitrite (ONOO−) and reacts with the sulfhydryl groups of proteins [5], H2S/HS− [6] orpersulfide [7] to form 8-adduct-cGMP (S-guanylation) or 8-thio-cGMP. These metabolic cycles haveled to the proposal of a new signaling pathway mediated by 8-nitro-cGMP. 8-Thio-cGMP may be returnedto cGMP by ROS such as hydrogen peroxide in cells, but its biochemical role is not well understood. Thus,selective molecules that can form a covalently bonded complex with the 8-thioG derivative are requiredto further understand their biological functions. However, there is no specific recognition compound for8-thioguanosine derivatives. In this study, we reported new 1,3-diazaphenoxazine nucleoside derivatives(thioG-grasp) that exhibit an efficient covalent capture of 8-thioguanosine via the formation of multiplehydrogen-bonded complexes.
We have focused on the development of recognition molecules for the 8-oxidized guanosinederivative based on the tricyclic cytosine analog “G-clamp” [8–12], and have reported the“8-oxoG-clamp” derivatives for the selective fluorescent detection of 8-oxo-dG [13–15]. Mostrecently, a new 1,3-diazaphenoxazine nucleoside derivative bearing a thiol arm, nitroG-grasp (1), hasdemonstrated the efficient capture of 8-nitroguanosine via multiple hydrogen-bonded complexes [16].The displacement reactivity of nitroG-grasp (1) depends on the thiol pKa and the length of the alkyllinker between the urea and the thiol group. For the new capture molecules for 8-thioguanosine, webased the design on a hydrogen bonded complex, such as that between the 8-nitroguanine portionand the 1,3-diazaphenoxazine portion connecting the urea-linker (Figure 1A). A nucleophilic attack
Molecules 2015, 20, 1078–1087 79 www.mdpi.com/journal/molecules

Molecules 2015, 20, 1078–1087
of the 8-sulfur atom on the chloride leaving group was expected to form the corresponding covalentbond. The urea-type linker (X=NH) and the carbamate-type linker (X=O) were anticipated to form asuitable hydrogen bond with the thioenolate form or with the thioamide form, respectively (Figure 1B,nitroG-grasp, 2a–c, 3). In this study, we report the synthesis of 8-thioG-grasp derivatives and evaluatedtheir reactivity with 8-thioguanosine.
Figure 1. Molecular design of the selective capture molecules for 8-nitroG and 8-thioG. dR and Rrepresent 3′,5′-diOTBS-2′-deoxyribosyl and triacetyl ribosyl groups, respectively.
2. Results and Discussion
2.1. Chemistry
In this study, 2′,3′,5′-tri-O-acetyl-8-thioguanosine (tri-Ac-8-thioG) was used as a substrate andwas synthesized from 2′,3′,5′-tri-O-acetyl-8-bromoguanosine according to the literature [17]. The8-thioG-grasp derivatives were synthesized through the reaction between the imidazole linker unit 5
and the amino group of the 3′,5′-O-diTBDMS-G-clamp unit 4 as a common intermediate (Scheme 1).The chloroalkylamine or chloropropanol were treated with carbonylimidazole in CH3CN in thepresence of triethylamine to form the corresponding imidazole intermediates 5, which reacted with 4
to produce the desired 8-thioG-grasp derivatives. N-(3-Chloropropyl)-2-(pyrene-1-yl) acetamide 6 wassynthesized from 1-pyrene-acetic acid as a control compound with no complexation site for 8-thioG.
Scheme 1. The synthesis of the 8-thioG-grasp derivatives.
2.2. Reaction of 8-ThioG-Grasp with 8-ThioG
The reaction of the 8-thioG-grasp derivatives with triAc-8-thioG was performed at 50 ◦C inCH3CN in the presence of Et3N, and the reaction progress was monitored by HPLC. An equimolar
80

Molecules 2015, 20, 1078–1087
mixture of 8-thioG-grasp (2b) and triAc-8-thioG formed an adduct in a nearly quantitative yield as asingle product within 3 h (Figure 2).
This product was isolated, and its structure was confirmed as depicted in 7b by 1H-NMR, ESI-MS,and HMBC (see the Supporting Information). The HMBC spectrum indicated the distinct correlationbetween the C8 of the 8-thioguanine unit and the methylene protons next to the sulfur atom of 7b.
2.3. Comparison of the Reactivity between 8-ThioG-Grasp and the Control Compound
The time courses of the reaction between the 8-thioG-grasp derivatives 2a–c, 3 and triAc-8-thioGare compared in Figure 3. 2b and 2c exhibited efficient reactivity, and a relatively slow reaction wasobserved with 2a. In the reaction of 2a, the formation of the amino-oxazoline ring (8) as a byproductdecreased the adduct yield (Figure 3).
In contrast, the carbamate type 3 showed a significantly decreased efficiency compared with thecompounds 2 with the urea-type linker. This is of great interest because 8-thioketo tautomer of 8-thioGis stable in neutral organic solvents [18] and forms more stable complexes with the carbamate type 9
than with the urea type 10 (Ks in CHCl3, 9: 7.6 × 106 M−1 vs. 10: 1.7 × 106 M−1) (Figure 4). Accordingly,it is reasonably explained that the 7N-H of 8-thioG is deprotonated by Et3N to form the 8-thioenolate,thereby facilitating hydrogen-bonded complexes with the urea-type compounds (2b and 2c) suchas shown in Figure 1B to exhibit efficient reactivity. As the pKa value of 7N-H of 8-thioguanosineis around 8.5 (see the Supporting Information), Et3N is a suitable base for its deprotonation. Noadduct was formed with N-(3-chloropropyl)-2-(pyrene-1-yl) acetamide (6), a control without a bindingsite, emphasizing the contribution of the hydrogen-bonded complexation of 2b and 2c for efficientreactivity. Among the urea-type 8-thioG-grasp derivatives, 2a produced the adduct in a low yield.The HPLC monitoring showed that 2a formed the byproduct 8 as a major peak, which structure wasdetermined by H-NMR, ESI-MS and 2D-HMBC, as shown in Figure 3. The amino-oxazoline wasformed by intramolecular displacement, which was faster than the intermolecular nucleophilic attackfrom 8-thioG. It should be emphasized that the displacement of the sulfur atom of 8-thioG in thecomplex with 2b or 2c is favorable compared with the intramolecular amino-oxazoline ring formationbecause this type of amino-oxazoline ring formation was not observed for 2b or 2c.
3h
8-thioG 2b 7b
Figure 2. Reaction of 8-thioG-grasp with tri-Ac-8-thioG monitored by HPLC. dR and R represent3′,5′-diOTBS-2′-deoxyribosyl and triacetyl ribosyl groups, respectively. The reaction was performedusing 0.4 mM each of 2b and 8-thioG in the presence of 20 mM Et3N in CH3CN at 50 ◦C. HPLCconditions: column: Xbridge C8 3.5 μm, 3.0 mm × 100 mm; solvents: (A) 0.1 M TEAA buffer at pH 7.0and (B) CH3CN, A/B = 20:80; flow rate: 0.5 mL/min; monitored by UV at 254 nm.
81

Molecules 2015, 20, 1078–1087
(A) (B)
Figure 3. (A) Comparison of the time courses of the reactions with 8-thioG-grasp (2a–c and 3) and thecontrol compound 6. (B) Byproduct 8 formed via the intramolecular reaction of 2a. Product yields wereobtained at the indicated time points by HPLC analysis, as described in the footnote to Figure 2. dRrepresents the 3′,5′-diOTBS-2′-deoxyribosyl group.
Ks= 7.6 × 106 M 1 Ks= 1.7 × 106 M 1
Figure 4. Proposed complexation of 8-thioG with the carbamate or the urea type. dR and R represent3′, 5′-diOTBS-2′-deoxyribosyl and triacetyl ribosyl groups, respectively.
3. Experimental Section
3.1. General Information
The reagents and solvents were purchased from commercial suppliers and were used withoutpurification. The 1H- and 13C- NMR spectra were recorded on a Bruker Avance III spectrometer. The2D-NMR spectra were measured on a Varian Inova 500 instrument. The IR spectra were recorded on aPerkin Elmer Spectrum One FT-IR spectrometer. The ESI-HRMS spectra were measured on an AppliedBiosystems Mariner Biospectrometry Workstation using neurotensin, angiotensin I, bradykinin andpicolinic acid as the internal standards.
82

Molecules 2015, 20, 1078–1087
3.2. Chemistry
3.2.1. General Synthesis of 8-ThioG-Grasp Derivatives
1,1′-Carbonyldiimidazole (5 equiv.) was added to a solution of chloroalkylamine hydrochloride(5 equiv.) in anhydrous CH3CN (0.05 M in G-clamp unit) under an argon atmosphere. The reactionmixture was stirred at room temperature for 30 min, followed by the addition of the G-clamp unit4 (1 equiv.) and Et3N (8 equiv.). After stirring overnight at room temperature, saturated aqueousNaHCO3 solution was added to the reaction mixture, which was extracted with CHCl3. The organiclayer was washed with brine, dried over Na2SO4 and evaporated in vacuo. The resulting residue waspurified by silica gel column chromatography (CH2Cl2/MeOH =100:0 to 95:5) to give 2a–c and 3 aslight yellow foams.
1-(2-((3-((2R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-tetrahydrofuran-2-yl)-2-oxo-2,10-dihydro-3H-benzo[b]pyrimido[4,5-e][1,4]oxazin-9-yl)oxy)ethyl)-3-(2-chloroethyl)urea. (C2-8-ThioG-grasp, 2a): 2a was synthesized using G-clamp unit 4 and 2-chloroethylamine in 48% as a lightyellow foam. 1H-NMR (400 MHz, CD3OD) δ (ppm) 7.62 (1H, s), 6.82 (1H, t, J = 8.6 Hz), 6.60 (1H, d,J = 8.6 Hz), 6.35 (1H, d, J = 8.6 Hz), 6.17 (1H, t, J = 6.4 Hz), 4.46 (1H, dt, J = 6.4, 3.4 Hz), 4.05 (2H, t,J = 4.9 Hz), 3.96–3.93 (2H, m), 3.82 (1H, dd, J = 12.1, 3.4 Hz), 3.57 (2H, t, J = 6.0 Hz), 3.54 (2H, t, J = 5.2Hz), 3.45 (2H, t, J = 6.4 Hz), 2.33 (1H, ddd, J = 13.4, 6.1, 4.6 Hz), 2.11 (1H, dt, J = 13.4, 6.4 Hz), 0.97 (9H,s), 0.91 (9H, s,), 0.18 (3H, s), 0.16 (3H, s), 0.11 (6H, s). 13C-NMR (125 MHz, CDCl3) δ (ppm) 158.53,152.89, 146.88, 143.06, 127.52, 124.49, 122.97, 115.06, 108.21, 107.54, 87.75, 86.00, 70.84, 69.22, 62.32, 42.09,41.89, 39.33, 29.28, 26.04, 25.71, 18.49, 17.94, −4.58, −4.90, −5.45, −5.52. IR (cm−1): 2929, 1673, 1557,1473. HR ESI-MS (m/z): Calcd. for [C32H53ClN5O7Si2]+: 710.3167 ([M+H]+), found 710.3176.
1-(2-((3-((2R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-tetrahydrofuran-2-yl)-2-oxo-2,10-dihydro-3H-benzo[b]pyrimido[4,5-e][1,4]oxazin-9-yl)oxy)ethyl)-3-(3-chloropropyl)urea. (C3-8-ThioG-grasp, 2b): 2b was synthesized using G-clamp unit 4 and 3-chloropropylamine in 46% as alight yellow foam. 1H-NMR (400 MHz, CD3OD) δ (ppm) 7.62 (1H, s), 6.82 (1H, t, J = 8.2 Hz), 6.60 (1H,dd, J = 8.4, 1.2 Hz), 6.35 (1H, dd, J = 8.2, 1.2 Hz), 6.17 (1H, t, J = 6.4 Hz), 4.47 (1H, dt, J = 5.7, 3.7 Hz),4.05 (2H, t, J = 5.2 Hz), 3.95 (1H, t, J = 3.1 Hz), 3.94 (1H, t, J = 3.1 Hz), 3.82 (1H, dd, J = 12.2, 3.4 Hz), 3.57(2H, t, J = 6.7 Hz), 3.53 (2H, t, J = 5.2 Hz), 3.27 (2H, t, J = 6.7 Hz), 2.33 (1H, ddd, J = 13.2, 6.2, 4.0 Hz),2.12 (1H, dt, J = 13.2, 6.4 Hz), 1.92 (2H, quin, J = 6.4 Hz), 0.97 (9H, s), 0.91 (9H, s,), 0.18 (3H, s), 0.16(3H, s), 0.11 (6H, s). 13C-NMR (125 MHz, CDCl3) δ (ppm) 158.99, 152.94, 152.67, 146.79, 143.04, 127.61,124.38, 122.87, 115.26, 108.27, 107.74, 87.71, 85.94, 70.82, 69.45, 62.31, 42.74, 41.88, 39.39, 37.45, 33.21,26.04, 25.71, 18.48, 17.93, −4.58, −4.90, −5.45, −5.52. IR (cm−1): 2931, 1670, 1558, 1499. HR ESI-MS(m/z): Calcd. for [C33H55ClN5O7Si2]+: 724.3323 ([M+H]+), found 724.3340.
1-(2-((3-((2R,4S,5R)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydro-furan-2-yl)-2-oxo-2,10-dihydro-3H-benzo[b]pyrimido[4,5-e][1,4]oxazin-9-yl)oxy)ethyl)-3-(4-chloro-butyl)urea. (C4-8-ThioG-grasp, 2c): 2c was synthesized using G-clamp unit 4 and 4-chlorobutylamine [19] in 82% as alight yellow foam. 1H-NMR (400 MHz, CD3OD) δ (ppm) 7.62 (1H, s), 6.81 (1H, t, J = 8.6 Hz), 6.59 (1H,d, J = 8.6 Hz), 6.24 (1H, t, J = 8.2 Hz), 6.16 (1H, t, J = 6.1 Hz), 4.46 (1H, br), 4.03 (2H, t, J = 5.2 Hz), 3.94(2H, dd, J = 11.6, 2.4 Hz), 3.82 (1H, d, J = 9.5 Hz), 3.61 (2H, t, J = 6.4 Hz) 3.53 (2H, t, J = 6.4 Hz), 3.12 (2H,t, J = 6.7 Hz), 2.35–2.29 (1H, m), 2.11 (1H, dt, J = 13.1, 6.4 Hz), 1.88–1.73 (4H, m), 0.97 (9H, s), 0.91 (9H,s,), 0.18 (3H, s), 0.16 (3H, s), 0.11 (6H, s). 13C-NMR (125 MHz, CD3OD) δ (ppm) 161.16, 156.45, 155.65,148.10, 144.31, 129.56, 125.02, 123.52, 109.17, 108.64, 89.33, 87.51, 72.88, 70.17, 63.73, 45.49, 42.80, 40.43,40.32, 31.10, 28.78, 26.60, 26.25, 19.37, 18.86, −4.45, −4.65, −5.25, −5.30. IR (cm−1): 2953, 1670, 1558.HR ESI-MS (m/z): calcd. for [C34H57ClN5O7Si2]+, 738.3480 ([M+H]+); found 738.3452.
(2-((3-((2R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydro-furan-2-yl)-2-oxo-2,10-dihydro-3H-benzo[b]pyrimido[4,5-e][1,4]oxazin-9-yl)oxy)ethyl)-3-chloropropyl carbamate.(C3 (O)-8-ThioG-grasp, 3): 3 was synthesized using G-clamp unit 4 and 3-chloropropanol in 82% as a
83

Molecules 2015, 20, 1078–1087
light yellow foam. 1H-NMR (400 MHz, CD3OD) (ppm) 7.64 (1H, s), 6.83 (1H, t, J = 8.2 Hz), 6.61 (1H,dd, J = 8.2, 0.9 Hz), 6.37 (1H, dd, J = 8.2, 0.9 Hz), 6.18 (1H, t, J = 6.1 Hz), 4.47 (1H, dt, J = 5.8, 4.0 Hz),4.19 (2H, t, J = 6.1 Hz), 4.06 (1H, t, J = 5.2 Hz), 3.96 (1H, t, J = 2.75 Hz), 3.94 (1H, t, J = 3.1 Hz), 3.83 (1H,dd, J = 12.2, 3.4 Hz), 3.63 (2H, t, J = 6.7 Hz), 3.52 (2H, t, J = 5.2 Hz), 3.27 (2H, t, J = 6.7 Hz), 2.34 (1H,ddd, J = 13.4, 6.1, 4.3 Hz), 2.12 (1H, dt, J = 13.4, 6.1 Hz), 2.06 (2H, quin, J = 6.1 Hz), 0.97 (9H, s), 0.92(9H, s,), 0.18 (3H, s), 0.17 (3H, s), 0.11 (6H, s). 13C-NMR (125 MHz, CDCl3) δ (ppm) 156.62, 152.69,152.02, 146.63, 143.16, 127.26, 124.14, 122.85, 115.58, 108.41, 107.15, 87.63, 86.02, 70.71, 68.38, 62.26, 41.89,41.38, 40.39, 32.05, 26.04, 25.72, 18.48, 17.94, −4.56, −4.90, −5.46, −5.52. IR (cm−1): 2953, 1679, 1556,1474. HR ESI-MS (m/z): calcd. for [C33H54ClN4O8Si2]+, 725.3163 ([M+H]+); found 725.3142.
3.2.2. Synthesis of 8-ThioG Adduct 7b
Et3N (100 μL, 0.72 mmol) was added to a solution of 2b (11 mg, 0.015 mmol) and tri-Ac-8-thioG(14 mg, 0.031 mmol) in anhydrous CH3CN (1 mL) under an argon atmosphere. The reaction mixturewas stirred at 50 ◦C for 12 h, and evaporated in vacuo. The resulting residue was purified by silica gelcolumn chromatography (CHCl3/MeOH =70:1) to give 5b as a colorless oil (9 mg, 50%). 1H-NMR(400 MHz, CD3OD) δ (ppm) 7.51 (1H, s), 6.70 (1H, t, J = 8.2 Hz), 6.41 (1H, d, J = 8.6 Hz), 6.18 (1H, t,J = 4.9 Hz), 6.15 (1H, d, J = 6.1 Hz), 6.04–6.03 (1H, m), 5.84 (1H, d, J = 4.6 Hz), 5.77 (1H, t, J = 6.1 Hz),4.51 (1H, dd, J = 7.6, 2.4 Hz), 4.41–4.31 (3H, m), 3.89–3.85 (4H, m), 3.76 (1H, d, J = 9.5 Hz), 3.60 (2H,br), 3.29–3.21 (4H, m), 2.29 (1H, ddd, J = 13.0, 6.5 Hz), 2.13–2.06 (1H, m), 1.96 (2H, br), 0.93 (9H,s), 0.91 (9H, s,), 0.14 (3H, s), 0.13 (3H, s), 0.09 (6H, s) 13C-NMR (125 MHz, CD3OD) δ (ppm) 172.40,171.44, 171.20, 161.36, 160.17, 156.20, 156.01, 155.09, 155.04, 148.62, 147.36, 144.32, 129.27, 125.38, 123.73,117.51, 115.86, 107.64, 88.03, 87.47, 81.00, 72.63, 71.79, 64.20, 42.70, 40.58, 39.76, 31.34, 29.52, 26.66, 26.34,20.42, 19.37, 18.88, −4.29, −4.54, −5.14. IR (cm−1): 2928, 1749, 1684. HR ESI-MS (m/z): calcd. for[C49H73N10O15SSi2]+, 1129.4511 ([M+H]+); found 1129.4469.
3.2.3. N-(3-Chloropropyl)-2-(pyren-1-yl)acetamide (6)
3-Chloropropylamine hydrochloride (12 mg, 0.093 mmol), 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (EDC, 17 mg, 0.089 mmol), 4,4-dimethylaminopyridine (DMAP,1 mg 0.008 mmol) and Et3N (50 μL, 0.359 mmol) were added to a solution of 1-pyreneacetic acid (20mg, 0.077 mmol) in anhydrous CH2Cl2 (1 mL) under an argon atmosphere. The reaction mixture wasstirred at room temperature for 7 h, quenched with aqueous saturated NH4Cl solution, and extractedwith AcOEt. The organic layer was washed with brine, dried over Na2SO4 and evaporated in vacuo.The resulting residue was purified by silica gel column chromatography (CH2Cl2) to give 6 as a paleyellow solid (13 mg, 50%). 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.20 (2H, d, J = 7.6 Hz), 8.15 (3H, d,J = 7.9 Hz), 8.08 (1H, d, J = 9.2 Hz), 8.04 (1H, d, J = 9.2 Hz), 8.02 (1H, t, J = 7.9 Hz), 7.88 (1H, d, J = 7.9Hz), 4.28 (2H, s), 3.31 (2H, t, J = 6.4 Hz), 3.25 (2H, q, J = 6.4 Hz), 1.78 (2H, quin, J = 6.4 Hz). 13C-NMR(125 MHz, CDCl3) δ (ppm) 171.23, 131.28, 131.16, 130.76, 129.53, 128.50, 128.16, 127.69, 127.27, 126.28,125.60, 125.48, 125.20, 125.12, 124.62, 122.76, 42.22, 42.08, 37.28, 31.86. IR (cm−1): 3293, 1646, 1544. HRESI-MS (m/z): calcd. for [C21H19ClNO]+, 336.1150 ([M+H]+); found 336.1161.
3.2.4. Determination of the Structure of the Byproduct 8
To a solution of 2a (70 mg, 0.099 mmol) in MeOH (4 mL) was added NaHCO3 (90 mg, 1.07 mmol)under an argon atmosphere. The reaction mixture was refluxed for 25 h, filtered, and evaporated invacuo. The resulting residue was purified by silica gel column chromatography (CHCl3/Acetone =100:0to 50:50) to give 8 as light yellow foams (26 mg, 39%). 1H-NMR (400 MHz, CD3OD) δ (ppm) 7.60 (1H,s), 6.81 (1H, t, J = 8.2 Hz), 6.58 (1H, d, J = 8.2 Hz), 6.35 (1H, d, J = 8.2 Hz), 6.17 (1H, t, J = 6.4 Hz), 4.47(1H, dt, J = 6.1, 3.5 Hz), 4.35 (2H, t, J = 8.2 Hz), 4.07 (2H, J = 4.9 Hz), 3.96–3.93 (2H, m), 3.82 (1H, dd,J = 12.2, 3.1 Hz), 3.78 (2H, t, J = 8.2 Hz), 3.55 (2H, t, J = 5.2 Hz), 2.33 (1H, ddd, J = 13.3, 6.1, 4.1 Hz),2.12 (1H, dt, J = 13.3, 6.3 Hz), 0.97 (9H, s), 0.91 (9H, s,), 0.18 (3H, s), 0.16 (3H, s), 0.11 (6H, s). 13C-NMR(125 MHz, CD3OD) δ (ppm) 164.20, 156.51, 155.66, 147.98, 144.31, 129.55, 124.95, 123.42, 117.14, 109.25,
84

Molecules 2015, 20, 1078–1087
108.51, 89.34, 87.51, 72.97, 69.64, 69.37, 63.77, 43.22, 42.77, 29.54, 26.60, 26.26, 19.36, 18.85, −4.46, −4.65,−5.26, −5.30. IR (cm−1): 2931, 1669, 1557, 1497, 1473. HR ESI-MS (m/z): calcd. for [C32H52N5O7Si2]+ ,674.3400 ([M+H]+); found 674.3438.
3.2.5. General Procedure of Reaction Monitoring by HPLC
Reaction was initiated by the addition of Et3N (20 mM) to a solution of 8-thioG-grasp derivative(0.4 mM) and triAc-8-thioG) (0.4 mM) in CH3CN at 50 ◦C. The reaction progress was monitored byHPLC at 0.5, 1, 3 and 5 h. Product yield with time course of reaction were obtained from reverse-phaseHPLC analysis. (Column: Xbridge C8 3.5 μm, 3.0 × 100 mm; Solvent: A: 0.1 M TEAA buffer at pH 7.0,B: CH3CN, A/B= 20: 80; Flow rate: 0.5 mL/min; monitored by UV detector at 254 nm).
4. Conclusions
In this study, we designed new recognition molecules to covalently capture 8-thioguanosinebased on the G-clamp skeleton by introducing chloroalkyl urea linker. 8-ThioG-grasp 2b and 2c withchloropropyl urea and chlorobutyl linker exhibited the most efficient reactivity for tri-Ac-8-thioG.It has been shown from the comparison with control compounds that the multiple hydrogen-bondedcomplexes contribute to the efficient reactivity. There is increasing interest in 8-thioguanosinederivatives for their biological roles in signal transduction pathways [7], the 8-thioG-grasp derivativesare expected to be a potential platform to develop specific molecules. For example, an 8-thioG-graspderivative with a phosphate binding unit are expected to covalently trap phosphate derivatives of8-thioguanosine and interfere their biological functions. Systematic studies are now ongoing in thisline in our group, which will be reported in due course.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/01/1078/s1.
Acknowledgments: We are grateful for support provided by a Grant-in-Aid for Challenging Exploratory Research(No. 26670004) from the Japan Society for the Promotion of Science (JSPS). S.S. and Y.F. also acknowledge theAstellas Foundation for Research on Metabolic Disorders and a Research Fellowship for Young Scientists fromJSPS, respectively.
Author Contributions: Y. Fuchi and H. Obayashi performed chemistry experiments and analyzed the data.Y. Fuchi and S. Sasaki designed the capture molecules, and wrote the paper.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [CrossRef][PubMed]
2. Yermilov, V.; Rubio, J.; Ohshima, H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitroand its rapid removal from DNA by depurination. FEBS Lett. 1995, 376, 207–210. [CrossRef] [PubMed]
3. Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker ofcellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [CrossRef] [PubMed]
4. Maeda, H. The link between infection and cancer: Tumor vasculature, free radicals, and drug delivery totumors via the EPR effect. Cancer Sci. 2013, 104, 779–789. [CrossRef] [PubMed]
5. Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.;Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclicmonophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [CrossRef] [PubMed]
6. Nishida, M.; Sawa, T.; Kitajima, N.; Ono, K.; Inoue, H.; Ihara, H.; Motohashi, H.; Yamamoto, M.; Suematsu, M.;Kurose, H.; et al. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration.Nat. Chem. Biol. 2012, 8, 714–724. [CrossRef] [PubMed]
7. Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.;Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redoxsignaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [CrossRef] [PubMed]
85

Molecules 2015, 20, 1078–1087
8. Lin, K.; Matteucci, M.D. A Cytosine analogue capable of clamp-like binding to a guanine in helical nucleicacids. J. Am. Chem. Soc. 1998, 120, 8531–8532. [CrossRef]
9. Flanagan, W.M.; Wolf, J.J.; Olson, P.; Grant, D.; Lin, K.Y.; Wangner, R.W.; Matteucci, M.D. A cytosine analogthat confers enhanced potency to antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1999, 96, 3513–3518.[CrossRef] [PubMed]
10. Maier, M.A.; Leeds, J.M.; Balow, G.; Springer, R.H.; Bharadwaj, R.; Manoharan, M. Nuclease resistanceof oligonucleotides containing the tricyclic cytosine analogues phenoxazine and 9-(2-aminoethoxy)-phenoxazine (“G-clamp”) and origins of their nuclease resistance properties. Biochemistry 2002, 41, 1323–1327.[CrossRef] [PubMed]
11. Wilds, C.J.; Maier, M.A.; Tereshko, V.; Manoharan, M.; Egli, M. Direct observation of a cytosine analoguethat forms five hydrogen bonds to guanosine: guanidino G-clamp. Angew. Chem. Int. Ed. 2002, 41, 115–117.[CrossRef]
12. Holmes, S.C.; Arzumanov, A.A.; Gait, M.J. Steric inhibition of human immunodeficiency virus type-1Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2′-O-methyl G-clampribonucleoside analogues. Nucleic Acids Res. 2003, 31, 2759–2768. [CrossRef] [PubMed]
13. Nakagawa, O.; Ono, S.; Li, Z.; Tsujimoto, A.; Sasaki, S. Specific fluorescent probe for 8-Oxoguanosine.Angew. Chem. Int. Ed. Engl. 2007, 119, 4584–4587. [CrossRef]
14. Li, Z.; Nakagawa, O.; Koga, Y.; Taniguchi, Y.; Sasaki, S. Synthesis of new derivatives of 8-oxoG-clamp forbetter understanding the recognition mode and improvement of selective affinity. Bioorg. Med. Chem. 2010,18, 3992–3998. [CrossRef] [PubMed]
15. Koga, Y.; Fuchi, Y.; Nakagawa, O.; Sasaki, S. Optimization of fluorescence property of the 8-oxodGclampderivative for better selectivity for 8-oxo-2′-deoxyguanosine. Tetrahedron 2011, 67, 6746–6752. [CrossRef]
16. Fuchi, Y.; Sasaki, S. Efficient covalent capture of 8-nitroguanosine via a multiple hydrogen-bonded complex.Org. Lett. 2014, 16, 1760–1763. [CrossRef] [PubMed]
17. Holmes, E.; Robins, K. Purine nucleosides. VII. Direct bromination of adenosine, deoxyadenosine, guanosine,and related purine nucleosides. J. Am. Chem. Soc. 1964, 86, 1242–1245. [CrossRef]
18. Strassmaier, T.; Karpen, J.W. Novel N7- and N1-substituted cGMP derivatives are potent activators of cyclicnucleotide-gated channels. J. Med. Chem. 2007, 50, 4186–4194. [CrossRef] [PubMed]
19. Song, A.; Walker, S.G.; Parker, K.A.; Sampson, N.S. Antibacterial studies of cationic polymers with alternating,random, and uniform backbones. ACS Chem. Biol. 2011, 6, 590–599. [CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples are available from authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
86

molecules
Article
Hybridisation Potential of1',3'-Di-O-methylaltropyranoside Nucleic Acids
Akkaladevi Venkatesham †, Dhuldeo Kachare †, Guy Schepers, Jef Rozenski, Mathy Froeyen andArthur Van Aerschot *
Medicinal Chemistry, Rega Institute for Medical Research, KU Leuven, Minderbroedersstraat 10,Leuven BE-3000, Belgium; [email protected] (A.V.); [email protected] (D.K.);[email protected] (G.S.); [email protected] (J.R.);[email protected] (M.F.);* Correspondence: [email protected]; Tel.: +32-16-372-624† These authors contributed equally to this work.
Academic Editor: Mahesh K. LakshmanReceived: 22 January 2015; Accepted: 24 February 2015; Published: 3 March 2015
Abstract: In further study of our series of six-membered ring-containing nucleic acids, different1',3'-di-O-methyl altropyranoside nucleoside analogs (DMANA) were synthesized comprising all fourbase moieties, adenine, cytosine, uracil and guanine. Following assembly into oligonucleotides (ONs),their affinity for natural oligonucleotides was evaluated by thermal denaturation of the respectiveduplexes. Data were compared with results obtained previously for both anhydrohexitol (HNAs) and3'-O-methylated altrohexitol modified ONs (MANAs). We hereby demonstrate that ONs modifiedwith DMANA monomers, unlike some of our previously described analogues with constrained6-membered hexitol rings, did not improve thermodynamic stability of dsRNA complexes, mostprobably in view of an energetic penalty when forced in the required 1C4 pairing conformation.Overall, a single incorporation was more or less tolerated or even positive for the adenine congener,but incorporation of a second modification afforded a slight destabilization (except for A), while afully modified sequence displayed a thermal stability of −0.3 ◦C per modification. The selectivityof pairing remained very high, and the new modification upon incorporation into a DNA strand,strongly destabilized the corresponding DNA duplexes. Unfortunately, this new modification doesnot bring any advantage to be further evaluated for antisense or siRNA applications.
Keywords: modified oligonucleotides; hexitol nucleic acids; pairing behaviour; hybridisation;constrained oligonucleotides
1. Introduction
Gene silencing has become a standard technique for studying gene functions or in trying to obtaintherapeutic effects and theoretically can be attained by interfering with transcription (via formation oftriple stranded complexes [1] or translation processes. The latter can be obtained via steric blockingantisense oligonucleotides (ASOs) or via mRNA cleavage of double stranded complexes with RNAseHactivating ASOs [2–4]. However, the vast majority of researchers nowadays have turned to the useof RNA interference (RNAi)-based strategies, which has recently become the technique of choice tosilence gene expression in mammalian cell culture and is envisaged as a first choice for therapeutictreatment as well [5–7]. However, we need to point out that at the moment only one aptamer [8] andone antisense oligonucleotide have been effectively FDA approved for gene silencing [9], and alsoexon-skipping oligonucleotides are receiving considerable attention [10].
In cell culture in general unmodified siRNAs are highly efficient, however for in vivo applicationsome chemical modifications are warranted to stabilise the siRNAs and to increase their selectivity
Molecules 2015, 20, 4020–4041 87 www.mdpi.com/journal/molecules

Molecules 2015, 20, 4020–4041
and to promote delivery [11,12]. In the past, we and others have studied a wide variety of strategiesfor both ASO and siRNA modification as reviewed several times [13–15].
Our group has been very successful in increasing the affinity for RNA using modified buildingblocks based on 6-membered hexitol rings which resulted in the hexitol nucleic acids series [16–19].However, the LNA monomers of the Wengel group [20] consistently showed the strongest affinity forRNA, and the series comprises many alternative structures [21]. Overall, both our hexitol nucleic acidsand the LNA series of compounds take on a pre-organized conformation, fitting the A-form of dsRNAand rationalizing the strong hybridization characteristics noticed.
Herein, hexitol nucleic acids (HNA, Figure 1) are composed of 2,3-dideoxy-D-arabino-hexitol unitswith a nucleobase situated in the 2-(S)-position (in cis to the hydroxymethyl substituent as in naturalnucleosides). Addition of a supplementary hydroxyl at the 3'-α-position resulted in D-altritol nucleicacid (ANA, Figure 1) analogs with increased affinity for RNA strands [18,22]. More recently, we finallyreported on the 3'-O-methylated ANA congeners (MANA, Figure 1) resulting in a further increaseof 0.5 ◦C/modification when evaluating melting temperatures (Tm) upon hybridisation to RNA [23].Herein, both the heterocyclic base and the 3'-O-methyl moiety are located in an axial position witha 4C1 conformation. However, assembly of the hexitol series of nucleosides is long-routed startingwith synthesis of the 1,5-anhydrohexitol ring. In view of the positive results obtained for the MANAseries of congeners and the abundance of cheap α-D-methylglucoside, we now planned to prepareand evaluate “bis-methylated altritol nucleosides”, or more correctly di-O-methylated altropyranosidenucleic acids (DMANA, Figure 1).
O
HO HO
Base
HNA
O
HO HO
Base
OH
ANA
O
HO HO
Base
O
DMANA
O
HO HO
Base
OCH3
MANA
O
CH3 CH3
Figure 1. Structures of the different hexitol based nucleic acids (HNA, ANA and MANA) as discussedabove and the newly envisaged structure DMANA, based on methyl-altropyranoside.
2. Results and Discussion
2.1. Chemical Synthesis of the New Building Blocks
The 1'-O-methylglycosidic protected analogues 4, 6 and 9 (Scheme 1) were obtained startingfrom ubiquitous methyl glucopyranoside 1, which in three steps was converted to 2 in 51.4% overallyield according to literature procedures [24]. Herein, regioselective epoxide ring opening of 2 withthe sodium salts of uracil or adenine in DMF at 120–130 ◦C afforded the corresponding altrohexitolderivatives 3 and 7 in 66%–85% yield. Chemoselective methylation of 7 and 3 was accomplished usingNaH, MeI in dry THF at low temperature for 1 h to afford the methylated nucleosides 4 and 8 in 65%and 75% yield, respectively. The selective O- vs. N-methylation mainly depends upon the dielectricconstant of the solvent [23,25] and the stability of sodium-enolate chelation [26]. Hence, low dielectricconstant and high chelation stabilizing capacity of THF afforded a higher O-selectivity comparedto DMF. The one pot conversion of compound 3 to the triazolide derivative [27] using 1,2,4-triazole,POCl3, and triethylamine, and subsequent treatment with aqueous ammonia/dioxane (1:1) at ambienttemperature for 18 h yielded the cytidine derivative (5) with 58% (overall yield for 2 steps). Baseprotection of 5 and 8 using benzoylchloride in pyridine at rt for 3 h afforded 6 and 9 in 83 and 88%yield, respectively.
Due to the scalability, solubility and reproducibility of the reaction it proved advantageousto use the guanine derivative 10 for the epoxide ring opening reaction. The latter was obtainedvia Mitsunobu reaction of N2-acetylguanine [28] and 2-(trimethylsilyl)ethanol in analogy with the
88

Molecules 2015, 20, 4020–4041
previously described protocol for O6-[2-(p-nitrophenyl)ethyl]guanine [29]. Selective epoxide ringopening was accomplished with the lithium salt of 10 (Scheme 2) in DMF at 130 ◦C and afforded41% of 11 along with 26% of recovered 10. Remarkably, the acetyl protection was lost in 11 upon theprolonged heating in DMF. Further chemoselective methylation using NaH (60%) and MeI in DMFand DCM at low temperature for 4 h gave 12 in 94% yield. Deprotection of 12 was done by 1 M TBAFin THF at rt for 2 h to yield 75% of 13. The more base labile N2-dimethylformamidine (dmf) [30–32]group was introduced using N,N-dimethylformamide diethylacetal in methanol under reflux for 12 hto afford 14 with 85% yield.
Scheme 1. Synthetic scheme of the protected DMANA congeners for uracil, cytosine and adenine.Reagents and Conditions: (i) (a) C6H5CHO, ZnCl2, 72 h (66%); (b) 6 eq. CH3C6H4SO2Cl, pyridine, 60 ◦C,72 h (78%); (c) 5.3 M in MeOH, CH2Cl2, rt, 12 h (99%); (ii) NaH (60%), DMF, 20 ◦C, 12 h (33.3% of 3 alongwith 40.1% recovery of 2 and 75% of 7); (iii) NaH (60%), CH3I, THF, 0 ◦C, 1 h (64.7%); (iv) (a) POCl3,1H-1,2,4-triazole, Et3N, pyridine, rt, 2 h; (b) 1,4 dioxane, aq. NH3, rt, 12 h (58.17% overall 2 steps); (v)Benzoyl chloride, pyridine, rt, 3 h (87.6% of 6 and 83% of 9; (vi) NaH (60%), CH3I, THF, −78 ◦C, 4.5 hto −30 ◦C, 1 h (75%).
Scheme 2. Synthetic scheme for the protected guanine containing analog. Reagents and Conditions:(i) LiH, DMF, 130 ◦C, 18 h (42% of 11 along with 26% recovery of 10); (ii) NaH (60%), CH3I, DMF, DCM,−30 ◦C to −20 ◦C, 4 h (94%); (iii) TBAF, THF, rt, 2 h (75%); (iv) Me2NCH(OEt)2, MeOH, reflux for12 h (85%).
89

Molecules 2015, 20, 4020–4041
Deprotection of the benzylidene protecting group under mild conditions using AcOH:H2O (3:1)at 45 ◦C for 12 h afforded 15a–d in 50%–94% yield (Scheme 3), which was followed by classicaldimethoxytritylation and phosphitylation. Hereto, the nucleosides 15a–d were selectively protectedat the 6'-OH by reaction with DMTrCl in pyridine at rt for 3 h to furnish the correspondingprotected derivatives 16a–d in 74%–93% yield. Phosphitylation of 16a–d at the 4'-OH with2-cyanoethyl N,N-diisopropylchlorophosphoramidite in anhydrous CH2Cl2 at 0 ◦C for 1.5 h affordedthe corresponding phosphoramidite building blocks 17a–d in 65%–93% yield, to be used for oligomerassembly. Assembly of all oligonucleotides and purification was carried out as described before [33].
O
O OPh
O
Base
O
4: B=U; 6: B=CBz
9: B=ABz; 14: B=Gdmf
O
HO HO O
Base
O
O
DMTrO HO O
Base
O
15a: B=U; 15b: B=CBz
15c: B=ABz; 15d: B=Gdmf16a: B=U; 16b: B=CBz
16c: B=ABz; 16d: B=Gdmf
(i) (ii)
O
DMTr O O
Base
OP 17a: Base=U; 17b: Base=CBz
17c: Base=ABz; 17d: Base=Gdmf
(iii)
CEON(ipr)2
Scheme 3. Scheme for assembly of the different phosporamidites. Reagents and Conditions: (i) AcOH:H2O (3:1), 45 ◦C, 12 h (50% to 94%); (ii) DMTrCl, pyridine, rt, 3 h (74% to 93%); (iii) (iPr)2N(OCE)PCl,DIPEA, DCM, 0 ◦C, 1.5 h (65% to 93%).
2.2. Oligonucleotide Affinity Measurements
First, following incorporation of a single DMANA modification into an dsRNA nonamer sequence[5'-GCGU-X*-UGCG/5'-CGCAYACGC], the respective affinities for complementary RNA were studiedin a 0.1 M NaCl buffer and were compared with the melting temperatures (Tm) of previously studiedsix-membered ring structures substituting for the ribose ring (Table 1). Within the context of all fournatural bases, the HNA (anhydrohexitol) and ANA (altritol) substitutions proved advantageous andconsiderably stabilized the RNA helix. Where ANA modifications were either slightly less stabilizing(for pyrimidines) or more stabilizing (as with purines) versus HNA modifications, methylation ofthe 3'-hydroxyl moiety of ANA congeners further improved the affinity for RNA systematicallywith approximately 0.5 ◦C. However, converting the hexitol into a methyl hexopyranoside viaattachment of a second “methoxy substituent” at the 1'-position as in our DMANA constructs, wipedout the advantage which was gained before in using constrained hexitol moieties. The obtainedaffinities of these DMANA containing constructs more or less matched those of the correspondingfully complementary RNA sequences. Incorporation of a single DMANA building block slightlydestabilized the RNA duplex for substitution of a pyrimidine, but on contrast slightly stabilized theduplex in case of guanine and to a larger extent upon substitution of adenine within this sequencecontext (Table 1 and Figure 2, base matches).
As on average the DMNA pairing affinity to RNA was not advantageous nor reallydetrimental, we further studied their mismatch behavior within the same RNA nonamer constructs(5'-GCGU-X*-UGCG/5'-CGCAYACGC; Figure 2) in comparison to different constructs. The graphicalchart displays the destabilization in relation to the respective matched sequence as obtained forDMANA building blocks in comparison to discrimination properties for HNA and MANA ([23] andwithin dsRNA duplexes. Analogous discrimination properties were noted, and especially for dmanaC
90
Molecules 2015, 20, 4020–4041
very selective pairing to guanosine was obtained with −22 ◦C to −24 ◦C of destabilization for thedifferent mismatches. However, no overall advantage can be seen in terms of either pairing selectivityor universal base pairing capabilities. Hence, normal WC pairing can be assumed.
Table 1. Tm values of complementary RNA duplexes [5'-GCGU-X*-UGCG/5'-CGCAYACGC].
X* Structure Tm (◦C) X* Structure Tm (◦C)
U*
RNA 50.4 ± 0.0
G*
RNA 60.4 ± 0.0HNA 53.4 ± 0.1 HNA 62.4 ± 0.1ANA 53.0 ± 0.2 ANA 62.9 ± 0.1
MANA 53.8 ± 0.2 MANA 63.4 ± 0.1DMANA 50.6 ± 0.2 DMANA 61.6 ± 0.1
C*
RNA 60.8 ± 0.1
A*
RNA 52.5 ± 0.1HNA 62.0 ± 0.0 HNA 55.0 ± 0.2ANA 60.9 ± 0.1 ANA 56.5 ± 0.1
MANA 61.4 ± 0.0 MANA 57.0 ± 0.1DMANA 60.1 ± 0.2 DMANA 55.5 ± 0.1
Conditions: as determined in 100 mM NaCl buffer containing 20 mM KH2PO4 and 0.1 mM EDTA, pH 7.5, witha duplex concentration of 4 μM. Annotations U*, C*, A* and G* denote either a RNA, HNA, ANA, MANA orDMANA monomer respectively, versus the natural complementary base Y.
-35
-30
-25
-20
-15
-10
-5
0
5
RNA
MANA
DMANA
HNA
Figure 2. Base pairing selectivity for different 6-membered ring analogues. Conditions: graphicaloverview of the (de)stabilization of the matched and mismatched nonamer sequences following a singleincorporation of a sugar modified nucleoside (MANA in red, DMANA in green and HNA in purple)wherein the selectivity of pairing is shown by destabilization of the respective mismatches. The pairingselectivity for RNA is shown in blue.
This picture was confirmed with a second substitution within different nonamer dsRNA sequences(Table 2), with the largest destabilization noted for dmanaC building blocks (−1.8 ◦C/modification)while still an increase in stability of 1.1 ◦C/modification was seen with incorporation of 2 dmanaAblocks. The stabilizing effects of the HNA and 3'-methylated ANA constructs (MANA) are included
91

Molecules 2015, 20, 4020–4041
for comparative reasons. We therefore decided to prepare a fully modified DMANA octamer andhybridized it to the complementary RNA sequence (Table 2, bottom). Where the natural RNA duplexdisplayed a Tm of 40.6 ◦C, the DMANA construct still paired albeit with slightly lower affinity (−0.3◦C/modification). The different hexitol constructs on the other hand strongly increased the affinityfor the RNA complement as documented before [23,34]. It hence can be concluded that the DMANAanalogues are different from the previous hexitol series of compounds with a constrained 6-memberedring conformation fit for pairing to RNA.
Table 2. Thermal stability for RNA duplexes containing a double modification.
Sequences X* Tm (◦C) ΔTm/Modification (◦C)
5'-GCU*GUGU*CG-3'
RNA 55.6 ± 0.4 ReferenceMANA 62.6 ± 0.2 3.5
DMANA 54.5 ± 0.1 −0.5HNA 60.2 ± 0.1 2.3
5'-GCC*AUAC*CG-3'
RNA 57.1 ± 0.1 ReferenceMANA 59.3 ± 0.1 1.1
DMANA 53.4 ± 0.1 −1.8HNA 58.2 ± 0.1 0.6
5'-GCG*UUUG*CG-3'
RNA 51.3 ± 0.2 ReferenceMANA 54.3 ± 0.1 1.5
DMANA 51.1 ± 0.1 −0.1HNA 53.0 ± 0.2 0.8
5'-GCA*CUCA*CG-3'
RNA 57.1 ± 0.1 ReferenceMANA 63.5 ± 0.1 3.2
DMANA 59.3 ± 0.1 1.1HNA 62.1 ± 0.1 2.5
5'-G*C*G*U*A*G*C*G*-3'
RNA 40.6 ± 0.1 ReferenceMANA 61.1 ± 0.3 2.6
DMANA 38.1 ± 0.2 −0.3ANA 59.6 [34] 2.4HNA 52.0 [34] 1.4
Conditions: Tm as determined in 100 mM NaCl buffer containing 20 mM KH2PO4 and 0.1 mM EDTA, pH 7.5, with aduplex concentration of 4 μM. U*, C*, A* and G* denote either a RNA, MANA, DMANA, ANA or a HNA monomer,respectively. At the bottom the results are given for the fully modified strands paired to the complementaryRNA strand.
Finally, incorporation of a single DMANA building block into dsDNA 13-mer sequences[5'-CACCGX*TGCTACC-3'/3'-GTGGCYACGATGG-5'] was evaluated at 0.1 M salt concentration(Table 3) for both match and mismatch sequences. However, a single DMANA incorporation alreadyafforded respectively 9 ◦C (for dmanaU and dmanaC), 5 ◦C (for dmanaA) or 6 ◦C (for dmanaG)of destabilization for the different matched pairs within this sequence context. A slightly betterresult could be expected if we could compare the dmanaT construct instead of dmanaU in view ofthe stabilizing effect of a 5-methyl substituent on the base, but this still would have resulted in adestabilization of 7 to 8 ◦C. The selectivity of pairing is adequate but gives a mixed picture, withselectivity being dependent probably on the base and the sequence context. In view of the fairly strongdestabilization versus DNA sequences it is clear however that these DMANA blocks do not have aDNA like conformation.
92

Molecules 2015, 20, 4020–4041
Table 3. Hybridization studies following incorporation into a DNA strand.
Y A T G C
X* (X) Tm ΔTm Tm ΔTm Tm ΔTm Tm ΔTm
U* 47.7 - 40.9 −6.8 41.7 −6.0 38.3 −9.4T 57.1 - 46.7 −10.4 50.3 −6.8 44.2 −12.9
A* 41.9 −10.8 52.7 - 45.3 −7.4 41.4 −11.3A 46.6 −10.7 57.3 - 52.9 −4.4 44.4 −12.9C* 41.8 −10.3 40.8 −11.3 52.1 - 38.5 −13.6C 44.4 −16.5 45.6 −15.3 60.9 - 40.6 −20.3G* 42.9 −11.0 48.9 −5.0 46.0 −7.9 53.9 -G 51.6 −8.4 51.0 −9.0 53.5 −6.5 60.0 -
Conditions: Tm values are provided for a single incorporation of a DMANA (X*) modification into a mixedDNA sequence [5'-CACCGX*TGCTACC-3'/3'-GTGGCYACGATGG-5'] for the match and the different mismatchsequences (Y) at 0.1 M salt and 4 M of duplex concentration.
2.3. Discussion
The MedChem group of the Rega Institute has already been elaborating for many years onnucleoside analogues with a 6-membered ring system substituting for ribose for various applications.Especially the analogues with a 1,5-anhydrohexitol ring having the base at the C2' in syn orientationwith the remaining hydroxymethyl substituent (HNA, ANA and MANA) turned out to be wellpre-organized for pairing with RNA, and are thus strongly stabilizing for RNA duplexes. Severalbiological studies have been undertaken with both HNA and ANA [22,35,36] and interesting resultswere obtained more recently regarding their use as xeno-nucleic acids [37,38]. Highest affinities so farhowever were obtained with MANA building blocks [23]. We therefore started to study the influenceof DMANA analogues carrying an additional methoxy substituent on the 6-membered ring system.However, as shown by the various Tm studies, incorporation of the new modification does not furtherincrease the affinity for RNA, but overall rather tends to slightly destabilize dsRNA complexes, whilestrongly destabilizing DNA upon incorporation. Therefore the modification still resembles moreclosely RNA monomers with their 3'-endo conformation as in RNA duplexes.
These findings are further corroborated by inserting in silico a modified building block into adsRNA. As can be seen in Figure 3, no steric hindrance occurs when substituting a DMANA residuehaving two OMe groups at the sugar 1' and 3' positions for a uridine into an RNA duplex for whichthe model of Mooers was used [39]. The HNA sugar conformation upon pairing with RNA is 1C4
having an axial base orientation. Likewise, for the DMANA structure incorporated into the dsRNA,both methoxy substituents likewise are oriented axially with no apparent steric clashes. However, it iswell known that apart from forces like the anomeric effect, substituents in 6-membered rings prefer theequatorial orientation to avoid 1,3-diaxial interactions. Energy calculations for the monomers usingAmber force field [40] indeed show a slight preference for having the base and both methoxy groupsin an all-equatorial configuration (with 4'-OH and 6'-CH2OH in axial orientation), opposite to whatis found for HNA and ANA building blocks with an axial oriented heterocyclic base. Increasing thenumber of OMe substituents therefore may destabilize the 1C4 chair conformation giving preferenceto 4C1, which is less compatible with the RNA duplex. Hence, the energy penalty to preserve theDMANA monomer in a 1C4 conformation to allow for pairing within a dsRNA strand, might upsetthe entropic gain as expected of a pre-organized monomer.
93

Molecules 2015, 20, 4020–4041
Figure 3. RNA duplex following insertion of a DMANA modification (DMANA modification in purple;picture generated using Chimera (UCSF Chimera—a visualization system for exploratory research andanalysis [41]).
Table 4 indeed shows the largest energy difference for both chair conformations for DMANAconstructs. This energetic penalty for a forced change in conformation could be the basis of the reducedfitness of DMANA analogues for pairing with RNA.
Table 4. Energy calculations for monomers with different scaffold.
Base and OMeOrientation
Axial EquatorialBase and OMe
OrientationAxial Equatorial
HNA −205.31 −205.79 MANA −195.22 −200.35ANA −200.58 −198.19 DMANA −187.62 −194.82Chair 1C4
4C1 Chair 1C44C1
Conditions: Final energies in kcal/mol are shown following minimization using Amber force field. Nucleotides allhave a thymine base, uncharged residues (phosphate groups protonated), with parametrization via antechamber,Gaff force field, 5000 steps of energy minimization, born solvation energy model.
94

Molecules 2015, 20, 4020–4041
3. Experimental Section
3.1. General
All chemicals including methylglucopyranoside were provided by Sigma-Aldrich (Diegem,Belgium) or Acros Organics (Geel, Belgium) and were of the highest quality. 1H and 13C-NMRspectra were determined with a 300, 500 and 600 MHz Varian Gemini apparatus (currently AgilentTechnologies, Santa Clara, CA, USA) with tetramethylsilane as internal standard for the 1H NMRspectra (s = singlet, d = doublet, dd = double doublet, t = triplet, br. s = broad signal, m = multiplet)and the solvent signal; CD3OD-d4 (δ = 48.9 ppm), DMSO-d6 (δ = 39.6 ppm) or CDCl3 (δ = 76.9 ppm)for the 13C-NMR spectra. Exact mass measurements were performed with a quadrupole/orthogonalacceleration time-of-flight tandem mass spectrometer (qTOF2, Micromass, Manchester, UK) fitted witha standard electrospray ionization (ESI) interface. All solvents were carefully dried or bought as such.
3.2. 1',5';2',3'-Dianhydro-4',6'-O-benzylidene-1'-O-methyl-D-allopyranoside (2)
To a solution of the 4',6'-O-benzylidene-2',3'-ditosyl intermediate (10.0 g, 17 mmol) in CH2Cl2(80 mL) was added NaOMe (5.3 M in MeOH, 12.8 mL, 67.8 mmol). After stirring for overnight at roomtemperature, the reaction mixture was concentrated and the residue was dissolved in CH2Cl2 (150 mL),and washed twice with brine (50 mL). The aqueous layer was again extracted with CH2Cl2 (2 × 80 mL).Combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to get pureepoxide 2 (4.45 g, 99%). 1H-NMR (300 MHz, CDCl3): δ 7.60–7.33 (m, 5H, Ar-H), 5.60 (d, J = 6.1 Hz,1H, Ph-CH), 4.92 (dd, J = 6.1, 2.7 Hz, 1H, 1'-H), 4.32–4.22 (m, 1H, 6'-He), 4.18–4.05 (m, 1H, 5-H), 3.98(dd, J = 9.0, 6.2 Hz, 1H, 4-H), 3.71 (m, 1H, 6-Ha), 3.58–3.45 (m, 5H, 2'-H, O-Me). 13C-NMR (75 MHz,CDCl3): δ 137.11 (Ar-Ci); 129.19; 128.28 (Ar-Cp+o); 126.27 (Ar-Cm); 102.71 (Ph-C); 95.27 (C-1'); 77.83(C-4'); 68.86 (C-6'); 59.99 (C-5'); 55.82 (OMe); 53.07 (C-3'); 50.66 (C-2'); HRMS calcd. for C14H16O5Na+
[M+Na]+ 287.0890, found 287.0891.
3.3. 4',6'-O-Benzylidene-1'-O-methyl-2'-deoxy-2'-(uracil-l-yl)-D-altropyranoside (3)
To a solution of uracil (4.62 g, 38.6 mmol) in dry DMF (30 mL) was added NaH (60% dispersionin oil, 1.29 g, 41.2 mmol). The reaction mixture was heated at 120 ◦C under an argon atmosphere for1 h and to this reaction mixture epoxide 2 (3.4 g, 12.9 mmol) in dry DMF (20 mL) was added andstirring was continued for overnight at same temperature. The reaction mixture was then cooledand evaporated to dryness. The residue was dissolved in ethyl acetate (150 mL) and the organiclayer was washed with a saturated aqueous NaHCO3 solution (2 × 50 mL). The aqueous layer wasagain extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine(2 × 50 mL), dried over Na2SO4, and concentrated under vacuo and purification by silica gel columnchromatography (elution with 2% MeOH in DCM) afforded 3 (1.61 g, 33%) as a white foam whilerecovering a large part of the starting epoxide 2 (1.94 g, 40% yield). 1H-NMR (500 MHz, CDCl3): δ 7.75(d, J = 8.1 Hz, 1H, 6-H), 7.49–7.41 (m, 2H, Ar-H), 7.39–7.30 (m, 3H, Ar-H), 5.77 (d, J = 8.1 Hz, 1H, 5-H),5.62 (s, 1H, Ph-CH), 4.83 (s, 1H, 1'-H), 4.81 (s, 1H, 2'-H), 4.48 – 4.38 (m, 2H, 5'-H, 6'-He), 4.16 (brs, 1H,3'-H), 3.81 (t, J = 9.9 Hz, 1H, 6'-Ha), 3.69 (dd, J = 9.9, 2.4 Hz, 1H, 4'-H), 3.41–3.51 (m, 4H, OMe, -OH).13C-NMR (125 MHz, CDCl3): δ 163.08 (C-4); 150.60 (C-2); 141.14 (C-6); 136.81 (Ar-Ci); 129.26 (Ar-Cp);128.28 (Ar-Cm); 126.18 (Ar-Co); 102.91 (C-5); 102.27 (Ph-C); 98.99 (C-1'); 75.61 (C-3'); 68.94 (C-4'); 67.02(C-6'); 58.19 (C-5'); 57.91 (OMe); 55.97(C-2'). HRMS calcd. for C18H20N2O7Na+ [M+Na]+ 399.1163,found 399.1156.
3.4. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(uracil-l-yl)-D-altropyranoside (4)
To a solution of 3 (1.51 g, 4.0 mmol) in dry THF (15 mL) was added NaH (60% dispersion inoil, 404 mg, 12.04 mmol) at 0 ◦C and the reaction mixture was stirred at 0 ◦C for 1h under argonatmosphere. Methyl iodide (0.37 mL, 6.02 mmol) in dry THF (1 mL) was added and stirring wascontinued for another 1 h at the same temperature. The reaction mixture was quenched with 5 mL
95

Molecules 2015, 20, 4020–4041
MeOH. The solution was concentrated and dissolved in ethyl acetate (150 mL) and washed withsaturated aqueous NaHCO3 (150 mL). The aqueous layer was again extracted with ethyl acetate(3 × 50 mL). The combined the organic layers were dried over Na2SO4, filtered, concentrated undervacuo, and purification by silica gel column chromatography (elution with 2% MeOH in DCM) affordedthe dimethylated nucleoside 4 (1.01 g, 65%). 1H-NMR (500 MHz, CDCl3): δ 9.41 (s, 1H, N3-H), 7.80 (d,J = 8.2 Hz, 1H, 6-H), 7.47–7.35 (m, 5H, Ar-H), 5.80 (dd, J = 8.1, 1.7 Hz, 1H, 5-H), 5.55 (s, 1H, Ph-CH),4.90 (d, J = 1.7 Hz, 1H, 1'-H), 4.80 (s, 1H, 2'-H), 4.48–4.36 (m, 2H, 6'-He, 3'-H), 3.77 (t, J = 10.3 Hz, 1H,6'-Ha),3.72–3.67 (m, 2H, 4'-H, 5'-H), 3.62 (s, 3H, OMe), 3.46 (s, 3H, OMe). 13C-NMR (125 MHz, CDCl3):δ 162.98 (C-4); 150.39 (C-2); 141.18 (C-6); 137.06 (Ar-Ci); 129.20 (Ar-Cp); 128.28 (Ar-Cm); 126.21 (Ar-Co);102.86 (C-5); 102.48 (Ph-C); 98.84 (C-1'); 76.06 (C-3'); 75.95 (C-4'); 69.11 (C-5'); 59.59 (C-6'); 58.56 (OMe);55.94 (OMe); 55.81(C-2'). HRMS calcd. for C19H23N2O7 [M+H]+ 391.1500, found 391.1494.
3.5. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2-deoxy-2'-(cytosin-1-yl)-D-altropyranoside (5)
A solution of triazole (1.05 g, 15.2 mmol) and phosphorus oxychloride (0.3 mL, 3.18 mmol) wasprepared in pyridine (8 mL) at 0 ◦C. Triethylamine (2.03 mL, 14.5 mmol) was added dropwise at 0 ◦Cand the solution was stirred 30 min. The uracil derivative 4 (0.650 g, 1.66 mmol) dissolved in drypyridine (8 mL) was added at 0 ◦C and the solution was stirred for 2 h at room temperature andconcentrated and co-evaporated with toluene (2 × 20 mL). The crude product was dissolved withDCM (150 mL) and washed twice with brine (60 mL). The aqueous layer was extracted with DCM(30 mL). Combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated. Theresidue was dissolved in 1,4-dioxane (30 mL), cooled to 0 ◦C and aqueous ammonia 25% (13 mL) wereadded. The solution was left overnight at RT. The solution was evaporated and co-evaporated withtoluene (3 × 20 mL). The residue was purified by silica gel column chromatography (elution with 10%MeOH/dichloromethane) and afforded the dimethylated cytidine analog 5 (377 mg, 58%) as a whitefoam. 1H-NMR (500 MHz, CD3OD): δ 7.91 (d, J = 7.5 Hz, 1H, 6-H), 7.50–7.42 (m, 2H, Ar-H), 7.38–7.26(m, 3H, Ar-H), 5.95 (d, J = 7.5 Hz, 1H, 5-H), 5.63 (s, 1H, Ph-CH), 4.93 (s, 1H, 2'-H), 4.91 (d, J = 2.1 Hz,1H, 1'-H), 4.34–4.30 (m, 1H, 6'-He), 4.28 (dd, J = 9.9, 5.3 Hz, 1H, 5'-H), 3.84 (dd, J = 11.0, 2.3 Hz, 1H,6'-Ha), 3.81 (dd, J = 6.6, 2.3 Hz, 1H, 4'-H), 3.67 (t, J = 2.3 Hz, 1H, 3'-H), 3.57 (s, 3H, OMe), 3.42 (s, 3H,OMe). 13C-NMR (125 MHz, CD3OD): δ 167.49 (C-4); 158.15 (C-2); 144.10 (C-6), 139.09 (Ar-Ci), 129.97(Ar-Cp), 129.07 (Ar-Cm), 127.44 (Ar-Co), 103.41 (Ph-C), 100.28 (C-5), 96.46 (C-1'), 77.40 (C-3'), 76.86(C-4'), 70.05 (C-6'), 59.93 (C-5'), 59.13 (OMe), 57.45 (OMe), 55.84 (C-2'). HRMS calcd. for C19H24N3O6
+
[M+H]+ 390.1654, found 390.1653.
3.6. 4',6'-O-Benzylidene-1',3'-O-methyl-2'-deoxy-2'-(N6-benzoylcytosin-1-yl)-D-altropyranoside (6)
The analog 5 (0.35 g, 0.9 mmol) was co-evaporated with dry pyridine (6 mL), dissolved in drypyridine (4 mL), and cooled at 0 ◦C. Benzoyl chloride (0.314 mL, 2.7 mmol) was added and the solutionwas allowed to come to RT. The solution was stirred for 3 h at RT. The mixture was cooled to 0 ◦C andwater (0.25 mL) was added. Then, aqueous ammonia 25% (2 mL) was added and the solution wasstirred for 30 min at RT. The volatiles were removed under reduced pressure and co-evaporated, eachtime with toluene (3 × 5 mL). The residue was adsorbed on silica by co-evaporation from DCM andpurified by silica column chromatography (elution with 2% MeOH/DCM) affording the protectednucleoside 6 (370 mg, 83% yield) as a white foam. 1H-NMR (300 MHz, CDCl3): δ 8.24 (d, J = 7.5 Hz,1H, 6-H), 7.94 (d, J = 7.3 Hz, 2H, Bz-H), 7.70–7.33 (m, 9H, 5-H, Ar-H, Bz-H), 5.56 (s, 1H, Ph-CH), 5.12(d, J = 1.6 Hz, 1H, 1'-H), 4.90 (s, 1H, 2'-H), 4.55–4.37 (m, 2H, 5'-H, 6'-He), 3.86–3.71 (m, 2H, 6'-Ha, 3'-H),3.74 (dd, J = 9.5, 3.1 Hz, 1H, 4'-H), 3.68 (s, 3H, OMe), 3.49 (s, 3H, OMe). 13C-NMR (75 MHz, CDCl3): δ167.76 (PhCONH); 162.14 (C-4); 137.11 (C-2); 133.34 (C-6); 129.18, 129.08, 128.28, 127.60, 126.23 (Ar-C,Bz-C); 102.48 (Ph-C); 99.00 (C-5); 97.14 (C-1'), 78.35 (C-3'), 75.77 (C-4'); 75.33 (C-5'); 69.20 (C-6'); 59.51(OMe); 58.72 (OMe); 56.03 (C2'). HRMS calcd. for C26H28N3O7
+ [M+H]+ 494.1922, found 494.1919.
96

Molecules 2015, 20, 4020–4041
3.7. 4',6'-O-Benzylidene-1'-O-methyl-2'-deoxy-2'-(adenin-9-yl)-D-altropyranoside (7)
To a solution of adenine (3.22 g, 23.85 mmol) in dry DMF (40 mL) was added NaH (60% dispersionin oil, 890 mg, 22.26 mmol). The reaction mixture was heated at 90 ◦C under argon atmosphere for1 h, after which the epoxide 2 (2.1 g, 7.95 mmol) in dry DMF (15 mL) was added and stirring wascontinued overnight at 120 ◦C. The reaction mixture was quenched with MeOH (15mL) and followingevaporation the residue was partitioned between EtOAc and saturated aqueous NaHCO3 solution.The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 50 mL). Thecombined the organic layers were dried over Na2SO4, filtered, concentrated in vacuo, and purificationby normal silica gel column chromatography (elution with 3% MeOH in DCM) afforded compound7 (2.37 g, 75%). 1H-NMR (300 MHz, CDCl3): δ 8.30 (s, 1H, 8-H), 8.18 (s, 1H, 2-H), 7.50–7.29 (m, 5H,Ar-H), 6.00 (s, 2H, -NH2), 5.55 (s, 1H, Ph-CH), 5.15 (s, 1H, 1'-H), 5.07 (s, 1H, 2'-H), 4.64− 4.52 (m, 1H,5'-H), 4.46 (dd, J = 9.9, 4.7 Hz, 1H, 6'-He), 4.33 (brs, 1H, 3'-H), 3.86 (t, J = 9.9 Hz, 1H, 6'-Ha), 3.73 (d,J = 8.3 Hz, 1H, 4'-H), 3.54 (s, 3H, OMe). 13C-NMR (75 MHz, CDCl3): δ 155.60 (C-6); 153.44 (C-2); 149.77(C-4); 138.45 (C-8); 136.86 (C-5); 129.28, 128.26, 126.15, 118.89 (Ar-C); 102.35 (Ph-C); 99.66 (C-1'); 75.87(C-5'); 69.14 (C-3'); 67.03 (C-4'); 58.52 (C-6'); 57.18 (OMe); 56.13 (C-2'). HRMS calcd. for C19H22N5O5
+
[M+H]+ 400.1615, found 400.1613.
3.8. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(adenin-9-yl)-D-altropyranoside (8)
The adenine analog 7 (2.36 g, 5.9 mmol) was co-evaporated with dry DMF (40 mL).The foamwas dissolved in dry DMF (70 mL), cooled to −78 ◦C and NaH (330 mg, 8.28 mmol) was added andthe mixture was stirred at −78 ◦C for 30 min under argon. Subsequently, methyl iodide (0.590 mL,9.46 mmol) was dissolved in dry DCM (20 mL) and the solution was added drop wise over 30 minto the reaction mixture at the same temperature. Stirring was continued for another 4.5 h at −78 ◦Cand at −30 ◦C for an additional 1 h before quenching of the reaction with MeOH (10 mL). Thesolution was warmed to RT and concentrated to dryness under vacuum. The residue was dissolvedin ethyl acetate (150 mL) and washed with aqueous saturated NaHCO3 (130 mL). The organic layerwas then washed with brine (130 mL), and the combined aqueous layers were again extracted withethyl acetate (2 × 150 mL). The combined organic layers were dried over Na2SO4, filtered, andconcentrated. Purification by silica column chromatography (0%–3% MeOH/DCM) afforded themethylated nucleoside 8 (1.84 g, 75.4%). 1H-NMR (500 MHz, CDCl3): δ 8.39 (s, 1H, 8-H), 8.20 (s, 1H,2-H), 7.47–7.40 (m, 2H, Ar-H), 7.37–7.31 (m, 3H, Ar-H), 5.92 (s, 2H, -NH2), 5.49 (s, 1H, Ph-CH), 5.15 (d,J = 2.5 Hz, 1H, 1'-H), 5.07 (s, 1H, 2'-H), 4.54 (td, J = 10.1, 5.3 Hz, 1H, 5'-H), 4.44 (dd, J = 10.5, 5.3 Hz, 1H,6-He), 3.88 (t, J = 2.6 Hz, 1H, 3'-H), 3.83 (t, J = 10.5 Hz, 1H, 6'-Ha), 3.79 (dd, J = 9.8, 2.5 Hz, 1H, 4'-H),3.71 (s, 3H, OMe), 3.52 (s, 3H, OMe). 13C-NMR (125 MHz, CDCl3): δ 155.57 (C-6); 153.54 (C-2); 149.99(C-4); 138.56 (C-8); 137.11, 129.15, 128.26, 126.16 (Ar-C); 119.10 (C-5); 102.48 (Ph-C); 99.47 (C-1'); 76.25(C-5'); 76.19 (C-3'); 69.26 (C-4'); 60.06 (C-6'); 58.81 (OMe); 56.06 (OMe); 55.05 (C-2'). HRMS calcd. forC20H24N5O5
+ [M+H]+ 414.1772, found 414.1767.
3.9. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(N6-benzoyladenin-9-yl)-D-altropyranoside (9)
The obtained analog 8 (1.59 g, 3.85 mmol) was co-evaporated with dry pyridine (2 mL), dissolvedin dry pyridine (16 mL), and cooled at 0 ◦C. Benzoyl chloride (1.34 mL, 11.54 mmol) was added andthe solution was allowed to come to RT. The solution was stirred 3 h at RT. The mixture was cooled to0 ◦C and water (4 mL) was added. Then, aqueous ammonia 25% (8 mL) was added and the solutionwas stirred 30 min at RT. The volatiles were removed under reduced pressure and co-evaporated, eachtime with toluene (3 × 25 mL). The residue was adsorbed on silica by co-evaporation and purified bysilica column chromatography (elution with 2% MeOH/DCM) affording the protected nucleoside 9
(1.70 g, 83% yield) as a white foam. 1H-NMR (500 MHz, CDCl3): δ 9.43 (s, 1H, NH), 8.78 (s, 1H, 8-H),8.43 (s, 1H, 2-H), 8.05 (d, J = 7.5 Hz, 2H, H-Bz), 7.66–7.29 (m, 8H, Bz-H, Ar-H), 5.49 (s, 1H, Ph-CH),5.22 (d, J = 2.1 Hz, 1H, 1'-H), 5.09 (s, 1H, 2'-H), 4.55 (td, J = 10.0, 5.3 Hz, 1H, 5'-H), 4.44 (dd, J = 10.4,
97

Molecules 2015, 20, 4020–4041
5.2 Hz, 1H, 6'-He), 3.88 (brs, 1H, 3'-H), 3.85–3.76 (m, 2H, ,6'-Ha, 4'-H), 3.71 (s, 3H, OMe), 3.52 (s, 3H,OMe). 13C-NMR (125 MHz, CDCl3): δ 164.86 (C=O); 152.77 (C-6); 152.06 (C-2); 149.78 (C-4); 141.06(C-8); 137.01, 133.35, 132.75, 129.09, 128.72, 128.19, 128.00, 126.11 (Ar-C, Bz-C); 122.75 (C-5); 102.43(Ph-C); 99.19 (C-1'); 76.11 (C-5'); 76.08 (C-3'); 69.16 (C-4'); 59.98 (C-6'); 58.83 (OMe); 56.01 (OMe); 55.13(C-2'). HRMS calcd. for C27H28N5O6
+ [M+H]+ 518.2034, found 518.2031.
3.10. 4',6'-O-Benzylidene-1'-O-methyl-2'-deoxy-2'-(2-amino-6-(2-trimethylsilyl-ethoxy)purin-9-yl)-D-altropyranoside (11)
A mixture of 4.5 g (15.4 mmol) of 10 and 0.266 g (33.5 mmol) of lithium hydride in 60 mL of dryDMF was stirred under nitrogen at 90 ◦C for 2 h. After addition of 2.6 g (9.8 mmol) of the epoxide 2
dissolved in 20 mL of dry DMF, stirring was continued for 18 h at 130 ◦C. The reaction mixture wascooled and evaporated to dryness. The residue was dissolved in ethyl acetate (100 mL) and washedwith brine (100 mL). A small part of the base precipitated during extraction and was filtered off andkept for recycling. The aqueous layer was again extracted with EtOAc (3 × 50 mL). The combinedorganic layers were dried over Na2SO4, filtered, concentrated under vacuo, and purification by silica gelcolumn chromatography (elution with 3% MeOH in DCM) afforded 11 (2.11 g, 42%) while recoveringa large point of the starting 10 (1.5 g, 26%). 1H-NMR (300 MHz, CDCl3): δ 8.03 (s, 1H, 8-H), 7.39 (m,5H, Ar-H), 5.55 (s, 1H, Ph-CH), 5.27 (d, J = 1.8 Hz, 1H, 1'-H), 5.04 (s, 1H, 2'-H), 4.83–4.60 (m, 3H, -NH2,5'-H), 4.59–4.38 (m, 3H, 6'-He, 6'-Ha, 3'-H), 4.36–4.29 (m, 1H, 4'-H), 3.89–3.73 (m, 2H, OCH2-), 3.55 (s,3H, OMe), 1.21–1.10 (m, 9H, -SiC3H9). 13C-NMR (75 MHz, CDCl3): δ 161.74 (C-6); 159.43 (C-2); 152.79(C-4); 137.10 (C-8); 136.53 (Ar-Ci), 129.53 (Ar-Cp), 128.34 (Ar-Cm), 126.46 (Ar-Co); 115.06 (C-5); 102.58(Ph-C); 100.05 (C-1'); 76.14 (C-4'); 69.25 (C-6'); 66.10 (C-5'); 65.09 (C-3'); 58.63 (OCH2-); 57.19 (OMe);56.40 (C-2'); 17.49 (CH2Si); −1.46 (-SiC3H9).
3.11. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(2-amino-6-(2-trimethylsilylethoxy)-purin-9-yl)-D-altropyranoside (12)
The obtained foam 11 (2.11 g, 4.1 mmol) was dried overnight under vacuum. The foam wasdissolved in dry DMF (42 mL), cooled to −30 ◦C and NaH (1.3 g, 32 mmol) was added and themixture was stirred at −30 ◦C for 1 h under argon. Subsequently, methyl iodide (1 mL, 16 mmol) wasdissolved in dry DCM (10.5 mL) and the solution was added drop wise over 30 min to the reactionmixture at −30 ◦C which was stirred further at −30 ◦C for 1 h. The reaction was finally running3 h more at −30 ◦C to −20 ◦C before quenching with 10 mL of MeOH for 20 min. The solution wasconcentrated and dissolved in DCM (50 mL) and washed with brine (50 mL). The aqueous layer wasagain extracted with DCM (3 × 50 mL). The combined organic layers were dried over Na2SO4, filteredand concentrated under vacuo, and purification by silica gel column chromatography (elution with 2%MeOH in DCM) afforded methylated nucleoside 12 (2.03 g, 94%) as a white foam. 1H-NMR (500 MHz,CDCl3): δ 7.98 (s, 1H, 8-H), 7.49–7.40 (m, 1H, 2H, Ar-H), 7.38–7.30 (m, 3H, Ar-H), 5.47 (s, 1H, Ph-CH),5.01 (s, 1H, 1'-H), 4.98–4.92 (m, 1H, 5'-H), 4.66–4.55 (m, 1H, 3'-H), 4.53–4.46 (m, 1H, 4'-H), 4.41 (dd,J = 10.4, 5.3 Hz, 1H, 6'-Ha), 1.26 (t, J = 12.1 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 161.59 (C-6); 159.61(C-2); 153.62 (C-4); 137.21 (C-8); 136.93 (Ar-Ci); 129.07 (Ar-Cp); 128.21 (Ar-Cm); 126.16 (Ar-Co); 115.15(C-5); 102.42 (Ph-C); 99.45 (C-1'); 76.26 (C-4'); 76.06 (C-6'); 69.26 (C-5'); 65.04 (C-3'); 59.98 (OCH2-); 58.73(OMe); 55.97 (OMe); 54.82 (C-2'); 17.54 (CH2Si); −1.41 (3C, Si(CH3)3). HRMS calcd. For C25H36N5O6Si+
[M+H]+ 530.2429, found 530.2430.
3.12. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(guanin-9-yl)-D-altropyranoside (13)
A 1 M solution of tetrabutylammonium fluoride in dry THF (15.10 mL) was added to the guaninederivative 12 (2.0 g , 3.77 mmol) and the mixture was stirred at room temperature under N2 for 2 hafter which water (15 mL) was added. The pH was adjusted to 5 with acetic acid and the mixture wasevaporated. The residue was purified by silica gel column chromatography (elution with 10% MeOHin DCM) affording 13 (1.25 g, 75%). 1H-NMR (500 MHz, CDCl3): δ 11.99 (s, 1H, NH), 7.86 (s, 1H, 8-H),
98

Molecules 2015, 20, 4020–4041
7.42 (m, 2H, Ar-H), 7.31 (m, 3H, Ar-H), 6.73 (s, H, 2H, 2-NH2), 5.49 (s, 1H, Ph-CH), 5.07 (s, 1H, 1'-H),4.81 (s, 1H, 2'-H), 4.50–4.42 (m, 1H, 5'-H), 4.37 (dd, J = 10.1, 5.0 Hz, 1H, 6'-He), 3.78 (s, 3H, 3'-H, 6-Ha,4'-H), 3.58 (s, 3H, OMe), 3.53 (s, 3H, OMe). 13C-NMR (125 MHz, CDCl3): δ 159.03 (C-6); 154.05 (C-2);151.58 (C-4); 137.20 (Ar-Ci); 135.42 (C-8); 129.08, 128.22, 126.19 (Ar-Cm+o+p); 116.24 (C-5); 102.40 (Ph-C);99.23 (C-1'); 76.11 (C-4'); 76.04 (C-6'); 69.19 (C-5'); 59.76 (C-4'); 58.68 (OMe); 55.98 (OMe); 54.79 (C-2').HRMS calcd. For C20H24N5O6
+ [M+H]+ 430.1721, found 430.1711.
3.13. 4',6'-O-Benzylidene-1',3'-di-O-methyl-2'-deoxy-2'-(N2-(dimethylamino)methylene-guanin-9-yl))-D-altropyranoside (14)
An amount of 13 (0.66 g, 1.54 mmol) was co evaporated three times with pyridine, dissolvedin 30 mL of dry MeOH (20 mL) and N,N-dimethylformamide diethyl acetal (0.824 mL, 6.16 mmol)was added. The mixture was stirred at reflux for 2 h under argon, after which it was evaporated andco-evaporated with toluene (3 × 30 mL). The residue was purified by silica gel column chromatography(2%–5% MeOH/dichloromethane) affording the analog 14 (0.634 g, 85%). 1H-NMR (500 MHz, CDCl3):δ 9.78 (s, 1H, N1-H), 8.65 (s, 1H, N=CH-N), 7.97 (s, 1H, 8-H), 7.45 (m, 2H, Ar-H), 7.40–7.30 (m, 3H,Ar-H), 5.51 (s, 1H, Ph-CH), 5.02 (s, 1H, 2'-H), 4.99 (d, J = 2.2 Hz, 1H, 1'-H), 4.50 (td, J = 10.4, 5.3 Hz, 1H,5'-H), 4.41 (dd, J = 10.4, 5.3 Hz, 1H, 6-He), 3.90–3.80 (m, 3H, 3'-H, 6-Ha, 4'-H), 3.68 (s, 3H, OMe), 3.50 (s,3H, OMe), 3.18 (s, 3H, NMe), 3.15 (s, 3H, NMe). 13C-NMR (125 MHz, CDCl3): δ 158.12 (N=CH-N);157.97 (C-6); 157.04 (C-2); 150.18 (C-4); 137.20 (C-8); 136.08, 129.05, 128.19, 126.14 (Ar-C); 119.85 (C-4);102.39 (Ph-CH); 99.31 (C-1'); 76.47 (C-4'); 76.12 (C-6'); 69.17 (C-3'); 60.14 (C-5'); 58.67 (OMe); 55.89(OMe); 55.13 (C-2'); 41.44 (NMe); 35.23 (NMe). HRMS calcd. for C23H29N6O6
+ [M+H]+ 485.2143, found485.2145.
3.14. 1',3'-Di-O-methyl-2'-deoxy-2'-(uracil-1-yl)-D-altropyranoside (15a)
Compound 4 (0.9 g, 2.31 mmol) was dissolved in 60 mL of AcOH-H2O (3:1) at rt. The reactionmixture was slowly heated at 45 ◦C and reaction progress was monitored using TLC. After 12 h, themixture was concentrated and co evaporated with toluene (30 mL). The crude residue was purificationby flash silica gel column chromatography (elution with 5% MeOH in DCM) afforded compound 15a
(0.66 g, 94%). 1H-NMR (600 MHz, DMSO-d6): δ 11.15 (s, 1H, NH), 7.71 (s, 1H, 6-H), 5.56 (d, J = 5.5 Hz,1H, 5-H), 4.93 (d, J = 3.9 Hz, 1H, 1'-H), 4.88 (t, J = 5.0 Hz, 1H, 2'-H), 4.84–4.60 (m, 1H, 3'-H), 4.03 (dd,J = 9.8, 5.5 Hz, 1H, 5'-H), 3.77 (q, J = 5.5 Hz, 1H, 4'-H), 3.65–3.53 (m, 2H, 6'-H), 3.26, (s, 3H, OMe), 3.22(s, 3H, OMe). 13C-NMR (150 MHz, DMSO-d6): δ 170.83 (C-4); 163.61 (C-2); 151.66 (C-6); 101.62 (C-5);99.07 (C-1'); 77.52 (C-5'); 76.08 (C-3); 63.67 (C-4'); 61.12 (C-6'); 60.23 (OMe); 56.65 (OMe); 55.39 (C-2').HRMS calcd. For C12H19N2O7
+ [M+H]+ 325.1006, found 325.1005.
3.15. 1',3'-Di-O-methyl-2'-deoxy-2'-(N6-benzoylcytosin-1-yl)-D-altropyranoside (15b)
Compound 6 (0.36 g, 0.73 mmol) was dissolved in 24.1 mL of AcOH:H2O (3:1) at rt. The reactionmixture was slowly heated at 45 ◦C and further treated as per the synthesis of 15a affording thenucleoside analog 15b (0.28 g, 50%). 1H-NMR (300 MHz, CD3OD): 8.23–7.94 (m, 3H, 6-H, Bz-H),7.68–7.51 (m, 4H, 5-H, Bz-H), 5.26 (brs, 1H, 1'-H), 4.87–4.81 (m, 1H, 2'-H), 4.23–4.09 (m, 1H, 5'-H), 4.03(q, 1H, J = 5.5 Hz, 4'-H), 3.80–3.66 (m, 2H, 3'-H, 6'-He), 3.30 (s, 6H, 2OMe), 3.26–3.20 (m, 1H, 6'-Ha).13C-NMR (150 MHz, CD3OD): 169.07 (PhCONH), 164.69 (C-4), 158.61 (C-2), 134.65 (C-6), 134.13, 130.47,129.84, 129.59, 129.19 (Bz-C), 100.35 (C-5), 98.59 (C-1'), 64.94 (C-3'), 64.74 (C-5'), 63.46 (C-4'), 62.47(C-6'), 57.75 (OMe), 56.05 (OMe), 49.85 (C-2'). HRMS calcd. for C19H24N3O7
+ [M+H]+ 406.1609, found406.1608.
3.16. 1',3'-Di-O-methyl-2'-deoxy-2'-(N6-benzoyladenin-9-yl)-D-altropyranoside (15c)
Following the procedure for 15a, compound 9 (1.65 g, 3.19 mmol) was treated 82.85 mL ofAcOH:H2O (3:1) at rt. The reaction mixture was slowly heated at 45 ◦C and reaction progress was
99

Molecules 2015, 20, 4020–4041
monitored using TLC. After 12 h, the mixture was concentrated and co evaporated with toluene, andafter purification afforded (0.78 g, 57%) of 15c.
1H-NMR (300 MHz, CD3OD): δ 8.76 (s, 1H, 8-H), 8.52 (s, 1H, 2-H), 8.13 (d, J = 7.4 Hz, 2H, Bz-H),7.77–7.53 (m, 3H, Bz-H), 5.39 (d, J = 5.5 Hz, 1H, 1'-H), 4.88–4.82 (m, 1H, 2'-H), 4.31 (dd, J = 8.8, 3.7 Hz,1H, 5'-H), 4.26–4.21 (m, 1H, 3'-H), 4.13 (q, J = 5.5 Hz, 1H, 4'-H), 4.00–3.9 (m, 2H, 6'-H), 3.42 (s, 3H, OMe),3.36 (s, 3H, OMe). 13C-NMR (75 MHz, DMSO-d6): δ 165.20 (C=O); 152.25 (C-6); 150.77 (C-2); 149.68(C-4); 143.85 (C-8); 132.92 (C-5); 131.89, 127.95, 127.91, 125.07 (Bz-C); 97.91 (C-1'); 77.10 (C-5'); 75.44(C-3'); 62.37 (C-4'); 59.78 (C-6'); 55.75 (OMe); 55.53 (OMe); 54.53 (C-2'). HRMS calcd. for C20H24N5O6
+
[M+H]+ 430.1721, found 430.1716.
3.17. 1',3'-Di-O-methyl-2'-deoxy-2'-(N2-(dimethylamino)methylene-guanin-9-yl))-D-altro-pyranoside (15d)
Compound 14 (0.58 g, 1.2 mmol) was dissolved in 30 mL of AcOH:H2O (3:1) at rt. The reactionmixture was slowly heated at 45 ◦C and reaction progress was monitored using TLC. After 12 h, themixture was concentrated and co-evaporated with toluene. The crude residue was purified by flashsilica gel column chromatography (elution with 10% MeOH in DCM) afforded compound 15d (0.4 g,81%). 1H-NMR (500 MHz, CD3OD): δ 8.65 (s, 1H, N=CH-N), 7.85 (s, 1H, 8-H), 5.21 (d, J = 5.3 Hz, 1H,2'-H), 4.64–4.57 (m, 1H, 1'-H), 4.10 (m, 2H, 4'-H, 3'-H), 4.00 (dd, J = 8.8, 4.1 Hz, 1H, 5'-H), 3.84 (d, J = 4.6Hz, 2H, 6'-H), 3.33 (s, 3H, OMe), 3.30 (s, 3H, OMe), 3.16 (s, 3H, OMe), 3.07 (s, 3H, OMe). 13C-NMR (125MHz, CD3OD): δ 160.31 (N=C-N); 159.83 (C-6); 158.79 (C-2); 152.31 (C-4); 140.37 (C-8); 120.48 (C-5);100.37 (C-1'); 78.41 (C-5'); 76.47 (C-3'); 65.24 (C-4'); 62.66 (C-6'); 57.94 (OMe); 57.74 (OMe); 56.17 (C-2');41.53 and 35.34 (NMe2). HRMS calcd. for C16H25N6O6
+ [M+H]+ 397.1830, found 397.1827.
3.18. 6'-O-Dimethoxytrityl-1',3'-di-O-methyl-2'-deoxy-2'-(uracil-1-yl)-D-altropyranoside (16a)
To a solution of the nucleoside 15a (0.4 g, 1.32 mmol) in anhydrous pyridine (10 mL) and underargon atmosphere, dimethoxytrityl chloride (0.49 g, 1.45 mmol) was added under stirring on an icebath. After stirring at room temperature for 3 h, 5% aqueous NaHCO3 solution (1 mL) was added, thereaction solvent was evaporated, diluted with CH2Cl2 (50 mL) and washed with 5% aqueous NaHCO3
(2 × 30 mL). The aqueous layers were back extracted once with 30 mL CH2Cl2. The combined organiclayer was dried (Na2SO4), evaporated under reduced pressure and the crude residue was purifiedby flash chromatography (CH2Cl2/MeOH 96:4) affording the corresponding dimethoxytritylatednucleoside 16a (0.745 g, 93% yield) as a white foam. 1H-NMR (125 MHz, CDCl3): δ 9.31 (brs, 1H,NH), 7.48–7.43 (m, 2H, 6-H, Ar-H), 7.37–7.33 (m, 4H, Ar-H), 7.31–7.26 (m, 2H, Ar-H), 7.24–7.19 (m, 1H,Ar-H), 6.85–6.82 (m, 4H, Ar-H), 5.69 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 5.30 (brs, 1H, 1'-H), 4.94 (brs, 1H,2'-H), 4.08–3.95 (m, 2H, 5'-H, 3'-H), 3.79 (s, 3H, OMe), 3.78–3.77 (m, 4H, OMe, 4'-H), 3.50–3.42 (brs, s,6H, 2OMe), 3.42–3.40 (m, 2H, 6'-H). 13C-NMR (125 MHz, CDCl3): δ 163.28 (C-4); 158.50 (C-2); 150.68(C-6); 144.85, 135.84, 130.09, 130.06, 128.10, 127.80, 126.81, 113.11 (Ar-C); 102.35 (C-5); 98.36 (C-1'); 86.20(CTr-O); 77.14 (C-5'), 76.87 (C-3'); 64.05 (C-4'); 62.88 (C-6'); 57.93 (OMe); 55.69 (OMe); 55.19 (2OMe, C-2').HRMS calcd. for C33H36N2O9Na+ [M+Na]+ 627.23186, found 627.2311.
3.19. 6'-O-Dimethoxytrityl-1',3'-di-O-methyl-2'-deoxy-2'-(N6-benzoylcytosin-1-yl)-D-altropyranoside (16b)
Compound 16b (0.346 g, 87% yield) was synthesized from compound 15b (0.228 g, 0.56 mmol)using dimethoxytrityl chloride (0.09 g, 1.29 mmol) in anhydrous pyridine (10 mL) according to theprocedure used for the synthesis of compound 16a. 1H-NMR (500 MHz, CDCl3) : δ 8.75 (brs, 1H, NH),7.90 (d, J = 7.3 Hz, 2H, 6-H, Ar-H), 7.64–7.45 (m, 6H, Ar-H), 7.40–7.19 (m, 8H, 5-H, Ar-H), 6.86 (d,J = 8.8 Hz, 4H, Ar-H), 5.29 (s, 1H, 1'-H), 5.03 (brs, 1H, 2'-H), 4.11–3.95 (m, 2H, 5'-H, 3'-H), 3.83–3.77(m, 7H, 2OMe, 4'-H), 3.60–3.37 (m, 8H, 6'-H, 2OMe), 2.50 (s, 1H, 4-OH). 13C-NMR (126 MHz, CDCl3):δ 166.13 (C=0), 162.13 (C-4), 158.47 (C-4), 144.71 (C-6), 136.02, 133.17, 130.03, 129.01, 128.20, 127.79,127.50, 126.80, 113.10 (Ar-C), 98.06 (C-5), 96.85 (C-1'), 86.11 (CTr-O), 77.13 (C-5'), 76.88 (C-3'), 65.62 (C-4'),62.73 (C-6'), 58.04 (OMe), 55.72 (OMe), 55.18 (C-2, 2OMe). HRMS calcd. For C40H42N3 O9
+ [M+H]+
708.29208, found 708.2909.
100

Molecules 2015, 20, 4020–4041
3.20. 6'-O-Dimethoxytrityl-1',3'-di-O-methyl-2'-deoxy-2'-(N6-benzoyladenin-9-yl)-D-altropyranoside (16c)
Compound 16c (0.77 g, 91% yield) was synthesized from compound 15c(0.5 g, 1.16 mmol) usingdimethoxytrityl chloride (0.44 g, 1.27 mmol) in anhydrous pyridine (10 mL) according to the procedureused for the synthesis of compound 16a. 1H-NMR (500 MHz, CDCl3): δ 9.15 (brs, 1H, 2-H), 8.12 (s,1H, 8-H),8.03 (d, J = 8.0 Hz, 2H, Ar-H), 7.60 (t, J = 7.4 Hz, 1H, Ar-H), 7.52–7.49 (m, 3H, Ar-H), 7.40 (d,J = 8.8 Hz, 4H, Ar-H), 7.34–7.21 (m, 5H, Ar-H), 6.85 (d, J = 8.8 Hz, 3H, Ar-H), 5.29 (t, J = 1.1 Hz, 1H,1'-H), 5.20 (d, J = 4.8 Hz, 1H, 2'-H), 4.79–4.68 (m, 1H, 6'-He), 4.27 (q, J = 5.5 Hz, 1H, 5'-H), 4.18 (dd,J = 7.9, 3.8 Hz, 1H, 4'-H), 4.07 (t, J = 4.7 Hz, 1H, 3'-H), 3.79 (s, 6H, OMe), 3.53 – 3.47 (m, 2H, 6'-Ha, OH),3.41 (s, 3H, OMe), 3.29 (s, 3H, OMe). 13C-NMR (125 MHz, CDCl3): δ 164.63 (C-6); 158.52 (Ar-C); 152.46(C-2); 151.78 (C-4); 149.56 (C-8); 144.66, 143.58, 135.84, 133.67, 132.75, 130.05, 128.84, 128.17, 127.85,126.84, 123.11, 113.16 (Ar-C); 98.31 (C-1'); 86.33 (CTr-O); 76.26 (C-5'); 72.89 (C-3'); 64.68 (C-4'); 62.90(C-6'); 57.89 (OMe); 56.87 (OMe); 55.94 (OMe); 55.19 (OMe); 53.38 (C-2'). HRMS calcd. for C41H42N5O8
+
[M+H]+ 732.30332, found 732.3030.
3.21. 6'-O-Dimethoxytrityl-1',3'-di-O-methyl-2'-deoxy-2'-(N2-(dimethylamino)methylene-guanin-9-yl))-D-altropyranoside (16d)
Compound 16d (0.51 g, 74% yield) was synthesized from compound 15d (0.39 g, 0.974 mmol)using dimethoxytrityl chloride (0.362 g, 1.07 mmol) in anhydrous pyridine (10 mL) according to theprocedure used for the synthesis of compound 16a. 1H-NMR (500 MHz, CDCl3): δ 9.20 (s, 1H, NH),8.61 (d, J = 4.2 Hz, 1H, N=CH-N), 8.52 (s, 1H, 8-H), 7.79 (s, 1H, Ar-H), 7.68 (tt, J = 7.7, 1.8 Hz, 1H, Ar-H),7.52–7.48 (m, 2H, Ar-H), 7.41–7.36 (m, 4H, Ar-H), 7.33–7.20 (m, 5H, Ar-H), 6.87–6.82 (m, 4H, Ar-H),5.29 (s, 1H, 1'-H), 5.09 (d, J = 3.1 Hz, 1H, 2'-H), 4.79 (dd, J = 6.4, 3.1 Hz, 1H, 3'-H), 4.17–4.11 (m, 1H,5'-H), 4.01 (brs, 1H, -OH), 3.88 (dd, J = 6.4, 4.0 Hz, 1H, 4'-H), 3.52 (dd, J = 10.2, 2.9 Hz, 1H, 6-He), 3.46(s, 3H, OMe), 3.44 (s, 3H, OMe), 3.35 (dd, J = 10.2, 5.5 Hz, 1H, 6'-Ha), 3.05 (s, 3H, OMe), 2.93 (s, 3H,OMe). 13C-NMR (125 MHz, CDCl3): δ 158.50 (N=CH-N); 157.92 (MMTr); 157.78 (C-6); 156.55(C-2);150.39(C-4); 149.81 (C-8); 144.84 (Ar-C), 137.33, 135.98, 135.87, 130.06, 130.02, 128.10, 127.85, 126.81,123.70, 120.37, 113.13 (Ar-C); 99.83 (C-1'); 86.17 (CTr-O); 78.04 (C-5'); 71.31 (C-3'); 64.79 (C-4'); 63.87(C-6'); 58.37 (OMe); 55.65 (OMe); 54.56 (C-2'); 41.12 and 35.12 (-NMe2). HRMS calcd. for C37H43N6O8
+
[M+H]+ 699.3142, found 699.3121.
3.22. General Procedure for Nucleoside Phosphitylation
To a solution of the dimethoxytritylated nucleoside 16a (0.73 g, 1.2 mmol) in anhydrous CH2Cl2(6 mL) at 0 ◦C and under argon atmosphere, freshly dried diisopropylethylamine (0.063 mL, 3.6 mmol)and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.040 mL, 1.8 mmol) were added. Thereaction mixture was stirred at 0 ◦C for 90 min after which completeness of the reaction was indicatedby TLC. Saturated NaHCO3 solution (2 mL) was added, the solution was stirred for another10 min andpartitioned between CH2Cl2 (50 mL) and aqueous NaHCO3 (30 mL). The organic layer was washedwith brine (3 × 30 mL) and the aqueous phases were back extracted with CH2Cl2 (30 mL). Aftersolvent evaporation, the resulting oil was purified by flash chromatography (hexane/acetone/TEA =62/36/2). The yellow solid was then dissolved in CH2Cl2 (2 mL) and precipitated twice in cold hexane(160 mL, −30 ◦C) to afford the desired corresponding phosphoramidite nucleoside 17a (0.908 g, 93%yield) as a white powder. The obtained product was dried under vacuum and stored overnight underargon at −20 ◦C. 31P-NMR (CDCl3): δ = 150.84. HRMS calcd. for C42H54N4O10P1
+ [M+H]+ 805.35773,found 805.3557; 31P-NMR (CDCl3): δ = 150.84.
Compound 17b (0.34 g, 79% yield) was synthesized from compound 16b (0.34 g, 0.47 mmol), drydiisopropylethylamine (0.025 mL, 1.42 mmol), 2-cyanoethyl-N,N-diisopropylchloro-phosphoramidite(0.016 mL, 0.71 mmol) and anhydrous CH2Cl2 (10 mL) according to procedure used for the synthesisof compound 17a. 31P-NMR (CDCl3): δ = 150.96. HRMS calcd. for C49H59N5O10P1 [M+H]+ 908.39993,found 908.3981.
101

Molecules 2015, 20, 4020–4041
Compound 17c (0.83 g, 85% yield) was synthesized from compound 16c (0.76 g, 1.03 mmol), drydiisopropylethylamine (0.054 mL, 3.09 mmol), 2-cyanoethyl-N,N-diisopropylchloro-phosphoramidite(0.034 mL, 1.54 mmol) and anhydrous CH2Cl2 (10 mL) according to procedure used for the synthesis ofcompound 17a. 31P-NMR (CDCl3): δ = 150.287 and 151.231. HRMS calcd. for C50H59N7O9P1
+ [M+H]+
932.41116, found 932.4103.Compound 17d (0.42 g, 65% yield) was synthesized from compound 16d (0.5 g, 0.71 mmol), dry
diisopropylethylamine (0.037 mL, 2.13 mmol), 2-cyanoethyl-N,N-diisopropylchloro-phosphoramidite(0.025 mL, 1.15 mmol) and anhydrous CH2Cl2 (10 mL) according to procedure used for the synthesis ofcompound 17a. 31P-NMR (CDCl3): δ = 150.065 and 151.206. HRMS calcd. for C46H60N8O9P1
+ [M+H]+
899.42206, found 899.4240.
4. Conclusions
A new nucleoside analogue scaffold for incorporation into oligonucleotides was developed andall four monomers with the natural heterocyclic bases have been prepared and evaluated on theirhybridization potential with natural DNA and RNA. While it was anticipated that the constraintimposed by the 6-membered ring structure could afford the entropic advantage as seen with HNA andANA constructs, an entropic penalty to preserve the 1C4 conformation required for pairing with RNAannulated the affinity gain which one could expect from a pre-organized structure.
Acknowledgments: This work was supported by a FWO (Flemish Scientific Research) grant G.0784.11 andKU Leuven financial support (GOA/10/13). Mass spectrometry was made possible by the support of theHercules Foundation of the Flemish Government (grant 20100225-7). We are indebted to Chantal Biernaux forfinal typesetting.
Author Contributions: A.V. and D.K. synthesized and analyzed the different monomers; G.S. assembledoligonucleotides and carried out Tm analysis; J.R. carried out all mass spectrometric analysis; M.F. performed themodeling experiments; A.V.A. conceived and supervised research and wrote the paper.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Helene, C. The anti-gene strategy: Control of gene expression by triplex-forming-oligonucleotides.Anti-Cancer Drug Des. 1991, 6, 569–584.
2. Wagner, R.W. Gene inhibition using antisense oligodeoxynucleotides. Nature 1994, 372, 333–335. [CrossRef][PubMed]
3. Opalinska, J.B.; Gewirtz, A.M. Nucleic-acid therapeutics: Basic principles and recent applications. Nat. Rev.Drug. Discov. 2002, 1, 503–514. [CrossRef] [PubMed]
4. Castanotto, D.; Stein, C.A. Antisense oligonucleotides in cancer. Curr. Opin. Oncol. 2014, 26, 584–589.[CrossRef] [PubMed]
5. Dean, N.M.; Bennett, C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003, 22,9087–9096. [CrossRef] [PubMed]
6. Yang, W.Q.; Zhang, Y. RNAi-mediated gene silencing in cancer therapy. Expert Opin. Biol. Ther. 2012, 12,1495–1504. [CrossRef] [PubMed]
7. Gao, S.; Yang, C.; Jiang, S.; Xu, X.-N.; Lu, X.; He, Y.-W.; Cheung, A.; Wang, H. Applications of RNAinterference high-throughput screening technology in cancer biology and virology. Protein Cell 2014, 5,805–815. [CrossRef] [PubMed]
8. Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targetedcancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [CrossRef] [PubMed]
9. Jiang, K. Biotech Comes to Its “Antisenses” after Hard-Won Drug Approval. (Archived by WebCite®athttp://www.webcitation.org/6WNb4M1rP). Available online: http://blogs.nature.com/spoonful/2013/02/biotech-comes-to-its-antisenses-after-hard-won-drug-approval.html (accessed on 6 January 2015).
10. Lu, Q.L.; Cirak, S.; Partridge, T. What Can We Learn From Clinical Trials of Exon Skipping for DMD?Mol. Ther. Nucl. Acids 2014, 3, e152. [CrossRef]
102

Molecules 2015, 20, 4020–4041
11. Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.;Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration ofmodified siRNAs. Nature 2004, 432, 173–178. [CrossRef] [PubMed]
12. De Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progressreport on siRNA-based therapeutics. Nat. Rev. Drug. Discov. 2007, 6, 443–453.
13. Herdewijn, P. Heterocyclic modifications of oligonucleotides and antisense technology. Antisense Nucl. Acid.Drug Dev. 2000, 10, 297–310. [CrossRef]
14. Herdewijn, P. Conformationally restricted carbohydrate-modified nucleic acids and antisense technology.Biochim. Biophys. Acta 1999, 1489, 167–179. [CrossRef] [PubMed]
15. Manoharan, M. RNA interference and chemically modified small interfering RNAs. Curr. Opin. Chem. Biol.2004, 8, 570–579. [CrossRef] [PubMed]
16. Van Aerschot, A.; Verheggen, I.; Hendrix, C.; Herdewijn, P. 1,5-Anhydrohexitol nucleic acids, a newpromising antisense construct. Angew. Chem. Int. Ed. Engl. 1995, 34, 1338–1339. [CrossRef]
17. Hendrix, C.; Rosemeyer, H.; Verheggen, I.; Seela, F.; van Aerschot, A.; Herdewijn, P. 1',5'-Anhydrohexitololigonucleotides: Synthesis, base pairing and recognition by regular oligodeoxyribonucleotides andoligoribonucleotides. Chem. Eur. J. 1997, 3, 110–120. [CrossRef]
18. Allart, B.; Khan, K.; Rosemeyer, H.; Schepers, G.; Hendrix, C.; Rothenbacher, K.; Seela, F.; Van Aerschot, A.;Herdewijn, P. D-Altritol nucleic acids (ANA): Hybridisation properties, stability, and initial structuralanalysis. Chem. Eur. J. 1999, 5, 2424–2431. [CrossRef]
19. Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; vanAerschot, A.; Herdewijn, P. Cyclohexene nucleic acids (CeNA): Serum stable oligonucleotides that activateRNase H and increase duplex stability with complementary RNA. J. Am. Chem. Soc. 2000, 122, 8595–8602.[CrossRef]
20. Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinitynucleic acid recognition. Chem. Commun. 1998, 455–456. [CrossRef]
21. Veedu, R.N.; Wengel, J. Locked nucleic acids: Promising nucleic acid analogs for therapeutic applications.Chem. Biodivers. 2010, 7, 536–542. [CrossRef] [PubMed]
22. Fisher, M.; Abramov, M.; van Aerschot, A.; Xu, D.; Juliano, R.L.; Herdewijn, P. Inhibition of MDR1 expressionwith altritol-modified siRNAs. Nucl. Acids Res. 2007, 35, 1064–1074. [CrossRef] [PubMed]
23. Chatelain, G.; Schepers, G.; Rozenski, J.; van Aerschot, A. Hybridization potential of oligonucleotidescomprising 3'-O-methylated altritol nucleosides. Mol. Divers. 2012, 16, 825–837. [CrossRef] [PubMed]
24. Richtmyer, N.K.; Hudson, C.S. Crystalline α-methyl-D-altroside and some new derivatives of D-altrose1.J. Am. Chem. Soc. 1941, 63, 1727–1731. [CrossRef]
25. Teste, K.; Colombeau, L.; Hadj-Bouazza, A.; Lucas, R.; Zerrouki, R.; Krausz, P.; Champavier, Y.Solvent-controlled regioselective protection of 5'-O-protected thymidine. Carbohydr. Res. 2008, 343, 1490–1495.[CrossRef] [PubMed]
26. Lucas, R.; Teste, K.; Zerrouki, R.; Champavier, Y.; Guilloton, M. Chelation-controlled regioselective alkylationof pyrimidine 2'-deoxynucleosides. Carbohydr. Res. 2010, 345, 199–207. [CrossRef] [PubMed]
27. Divakar, K.J.; Reese, C.B. 4-(1,2,4-Triazol-1-yl) and 4-(3-nitro-1,2,4-triazol-1-yl)-1-(beta-D-2,3,5-tri-O-acetylarabinofuranosyl)pyrimidin-2(1H)-ones—Valuable intermediates in the synthesis of derivatives of1-(beta-D-arabinofuranosyl)cytosine (Ara-C). J. Chem. Soc. Perk. Trans. 1 1982, 1171–1176. [CrossRef]
28. Pulido, D.; Sánchez, A.; Robles, J.; Pedroso, E.; Grandas, A. Guanine-containing DNA minor-groove binders.Eur. J. Org. Chem. 2009, 2009, 1398–1406. [CrossRef]
29. Jenny, T.F.; Schneider, K.C.; Benner, S.A. N-2-Isobutyrl-O-6-[2-(para-nitrophenyl)ethyl]guanine—A newbuilding block for the efficient synthesis of carbocyclic guanosine analogs. Nucleos. Nucleot. 1992, 11,1257–1261. [CrossRef]
30. Mcbride, L.J.; Kierzek, R.; Beaucage, S.L.; Caruthers, M.H. Amidine protecting groups for oligonucleotidesynthesis 16. J. Am. Chem. Soc. 1986, 108, 2040–2048. [CrossRef]
31. Vu, H.; Mccollum, C.; Jacobson, K.; Theisen, P.; Vinayak, R.; Spiess, E.; Andrus, A. Fast Oligonucleotidedeprotection phosphoramidite chemistry for DNA-synthesis. Tetrahedron Lett. 1990, 31, 7269–7272. [CrossRef]
32. Theisen, P.; McCollum, C.; Andrus, A. N-6-Dialkylformamidine-2'-deoxyadenosine phosphoramidites inoligodeoxynucleotide synthesis—Rapid deprotection of oligodeoxynucleotides. Nucleos. Nucleot. 1993, 12,1033–1046. [CrossRef]
103

Molecules 2015, 20, 4020–4041
33. D’Alonzo, D.; van Aerschot, A.; Guaragna, A.; Palumbo, G.; Schepers, G.; Capone, S.; Rozenski, J.;Herdewijn, P. Synthesis and base pairing properties of 1',5'-anhydro-L-hexitol nucleic acids (L-HNA). Chem.Eur. J. 2009, 15, 10121–10131. [CrossRef] [PubMed]
34. Van Aerschot, A.; Meldgaard, M.; Schepers, G.; Volders, F.; Rozenski, J.; Busson, R.; Herdewijn, P. Improvedhybridisation potential of oligonucleotides comprising O-methylated anhydrohexitol nucleoside congeners.Nucl. Acids Res. 2001, 29, 4187–4194. [CrossRef] [PubMed]
35. Vandermeeren, M.; Preveral, S.; Janssens, S.; Geysen, J.; Saison-Behmoaras, E.; Van Aerschot, A.; Herdewijn, P.Biological activity of hexitol nucleic acids targeted at Ha-ras and intracellular adhesion molecule-1 mRNA.Biochem. Pharmacol. 2000, 59, 655–663. [CrossRef] [PubMed]
36. Fisher, M.; Abramov, M.; Van Aerschot, A.; Rozenski, J.; Dixit, V.; Juliano, R.L.; Herdewijn, P. Biologicaleffects of hexitol and altritol-modified siRNAs targeting B-Raf. Eur. J. Pharmacol. 2009, 606, 38–44. [CrossRef][PubMed]
37. Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.;Peak-Chew, S.Y.; McLaughlin, S.H.; et al. Synthetic genetic polymers capable of heredity and evolution.Science 2012, 336, 341–344. [CrossRef] [PubMed]
38. Taylor, A.I.; Pinheiro, V.B.; Smola, M.J.; Morgunov, A.S.; Peak-Chew, S.; Cozens, C.; Weeks, K.M.;Herdewijn, P.; Holliger, P. Catalysts from synthetic genetic polymers. Nature 2015, 518, 427–430. [CrossRef][PubMed]
39. Mooers, B.H.; Singh, A. The crystal structure of an oligo(U):pre-mRNA duplex from a trypanosome RNAediting substrate. RNA 2011, 17, 1870–1883. [CrossRef] [PubMed]
40. Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package.Wires Comput. Mol. Sci. 2013, 3, 198–210. [CrossRef]
41. Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSFChimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612.[CrossRef] [PubMed]
Sample Availability: Sample Availability: Only small amounts of the different amidites are still available fromthe authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
104

molecules
Article
Tethering in RNA: An RNA-Binding FragmentDiscovery Tool
Kiet Tran 1, Michelle R. Arkin 2 and Peter A. Beal 1,*
1 Department of Chemistry, University of California, One Shields Ave, Davis, CA 95616, USA;[email protected]
2 Small Molecule Discovery Center, Department of Pharmaceutical Chemistry, University of California,San Francisco, CA 94158, USA; [email protected]
* Correspondence: [email protected]; Tel.: +1-530-752-4132
Academic Editors: Mahesh K. Lakshman and Fumi NagatsugiReceived: 5 December 2014; Accepted: 17 February 2015; Published: 4 March 2015
Abstract: Tethering has been extensively used to study small molecule interactions with proteinsthrough reversible disulfide bond forming reactions to cysteine residues. We describe the adaptationof Tethering to the study of small molecule binding to RNA using a thiol-containing adenosineanalog (ASH). Among 30 disulfide-containing small molecules screened for efficient Tetheringto ASH-bearing RNAs derived from pre-miR21, a benzotriazole-containing compound showedprominent adduct formation and selectivity for one of the RNAs tested. The results of this screendemonstrate the viability of using thiol-modified nucleic acids to discover molecules with bindingaffinity and specificity for the purpose of therapeutic compound lead discovery.
Keywords: Tethering; miRNA; nucleoside analog
1. Introduction
The structural complexity and functional diversity of cellular RNAs make them attractive targetsfor high affinity small molecule ligands [1,2]. However, a significant challenge in the developmentof RNA-binding molecules is the difficulty in optimizing lead compounds from initial screeninghits. This is due, at least in part, to difficulties in characterizing structures of RNA targets bound toweakly interacting lead compounds [3,4]. Here we describe the application of the Tethering screeningmethod to the discovery of RNA-interacting fragments. In the Tethering method, lead molecules arecovalently bound to the target such that future structural studies are not complicated by weak fragmentinteractions or multiple binding modes. Tethering has been successfully used in the generation ofpotent enzyme and protein-protein interaction inhibitors, but has yet to be applied to RNA [5]. Forprotein targets, Tethering involves the introduction of individual cysteine mutations surroundingthe site of interest followed by screening against libraries of disulfide-containing fragments [6]. Thedisulfide exchange reactions allow identification of ligands since the reversible protein-fragmentinteractions increase the local concentration of the small molecules at the targeted site. Fragmentshaving affinity for the protein, apart from the disulfide bond, are efficiently conjugated at equilibrium.Hits are then detected by mass spectrometry of the mixture. Fragment-target covalent conjugates canthen be structurally characterized and guide the improvement of affinity and conversion to noncovalentinhibitors by subsequent modification [7]. To adapt Tethering to the discovery of RNA-binding ligands,one must introduce a thiol near the site of interest in the RNA. This can be achieved using thiol-bearingnucleoside analogs. In this initial report on Tethering for RNA, we endeavored to discover fragmentsthat bind to a biosynthetic precursor to a biologically important miRNA, miR21.
Molecules 2015, 20, 4148–4161 105 www.mdpi.com/journal/molecules

Molecules 2015, 20, 4148–4161
MicroRNAs (miRNAs) are endogenously encoded ~23 nt RNA molecules that control geneexpression at the post-transcriptional level [8]. Hundreds of human miRNAs are known witheach miRNA regulating multiple targets. Furthermore, altered miRNA expression correlates withhuman disease states, including cancer [9]. Indeed, overexpression of certain miRNAs is knownto cause malignant phenotypes and these miRNAs are targets for the development of cancertherapeutics. Among these is miR-21, whose expression is dramatically upregulated in many solidtumors [10]. MiRNAs are biosynthesized from hairpin precursors via the action of two duplex-specificendonucleases, Drosha and Dicer [11]. Given this biosynthetic pathway, small molecules that bindto functionally significant sites on miRNA precursors should block their conversion to the maturemiRNA. Several recent reports highlight this attractive approach that will undoubtedly benefit fromthe discovery of new RNA-targeting compounds that have the requisite properties to be useful startingpoints for drug discovery efforts [12–15]. Here we use Tethering to identify molecular fragments withaffinity for pre-miR-21 near the Dicer cleavage sites. We envision such fragments providing startingstructures for the development of inhibitors of the biosynthesis of miR-21.
2. Results and Discussion
2.1. Synthesis of N6-(2-(triphenylmethylthio)ethyl)adenosine Phosphoramidite and Reactivity ofModified RNAs
To introduce a thiol into RNA screening targets, we synthesized a derivative of adenosine (ASH)that bears an ethane thiol substituent on N6 (Figure 1). This modification does not interfere withWatson-Crick hydrogen bonding and directs the reactive thiol into the major groove of an RNA duplex.Verdine and colleagues described the synthesis of RNA bearing this modification using a convertiblenucleoside approach [16]. We chose to synthesize the S-trityl protected phosphoramidite, which hadnot been previously reported (Scheme 1). While many methods could be used to introduce a protectedthiol moiety into a ribonucleoside [17,18], our previous work showed that a trityl-protected thiol groupin RNA could be easily deprotected with AgNO3(aq) [19].
Figure 1. Pre-miR21 sequence-based RNAs bearing the thiol tether. (A) Sequence and secondarystructure for pre-miR21. Underlined sequence is mature miR21; (B) Corresponding truncated pre21RNA1 and pre21 RNA2 31-nucleotide oligomers (left) containing the adenosine modification ASH
(right). Two non-native G-C base pairs (blue) have been added to the duplex end for stability.
First, the tri-O-acetyl protected 6-chloropurine nucleoside was allowed to react with S-tritylprotected 2-aminoethanethiol in an SNAr reaction to give the N6-alkylated adenosine analog.Removal of the acyl protecting groups with methanolic ammonia yielded free nucleoside 1.Protection of the 5' and 2' hydroxyls of the ribose moiety was accomplished with DMTr-Cl andTBDMS-Cl to yield compounds 2 and 3, respectively. Finally, the 3'-OH was allowed to react withβ-cyanoethyl-(N,N-diisopropyl)chlorophosphite to give the corresponding phosphoramidite 4 whichwas then used to incorporate the nucleoside analog via solid phase synthesis into two different RNAs
106

Molecules 2015, 20, 4148–4161
mimicking the hairpin loop of pre-miR21 (pre21 RNA 1 and pre21 RNA 2, Figure 1). These two RNAsdiffer in the positioning of the reactive adenosine analog near the Dicer cleavage sites on oppositesides of the duplex. The modified RNAs still containing S-trityl protecting groups were purified viadenaturing polyacrylamide gel electrophoresis.
Scheme 1. Synthesis of N6-(2-(triphenylmethylthio)ethyl)-adenosine phosphoramidite. Reagents andconditions: (i) (a) S-trityl-2-aminoethanethiol, DMF (b) NH3(sat) in MeOH, 72%; (ii) DMTr-Cl, DMAP,pyridine, 88%; (iii) TBDMS-Cl, N(Et)3, DMAP, THF, 43%; (iv) 2-cyanoethyl-(N,N-diisopropylamino)chlorophosphite, DIPEA, CH2Cl2, 82%.
Removal of the trityl protecting group on the sulfur was necessary before subjecting the RNA toTethering. This was accomplished using aqueous silver nitrate giving the RNA free thiol. Deprotectedpre21 RNA 2 (free thiol) and cystamine were then used to determine the appropriate conditions neededfor the disulfide screen (Figure 2). Detection of reaction products was performed with ESI-MS and therelative abundances of species in a sample were determined from the areas of the peaks. The resultclearly showed that the thiol-containing oligonucleotide can form a disulfide linkage with cystamineunder the experimental conditions in Figure 2A, to give a product peak observed at 10,030 amu in themass spectrum shown. The percent abundance of the cysteamine adduct was 33% in the presenceof 0.1 mM β-mercaptoethanol (BME). The concentration of BME to be used in the screen should behigh enough such that the majority of the small molecules would not conjugate efficiently with theRNA. Another experiment was performed using pre21 RNA 2 to determine the concentration ofBME necessary to completely inhibit cystamine conjugation. The results in Figure 2B showed that noadduct could be detected at a concentration of 1.0 mM BME. Therefore, the screen was performed at aconcentration of 0.5 mM BME.
107

Molecules 2015, 20, 4148–4161
Figure 2. Testing the reactivity of a thiol-modified RNA. (A) Reaction of pre21 RNA 2 free-thiol withcystamine; (B) ESI-MS spectra of the disulfide conjugation reaction in 0.1 mM BME (left) and 1.0 mMBME (right).
2.2. RNA Tethering Screen with pre21 RNA 1 and pre21 RNA 2
A small library of thirty disulfide-containing small molecules, all containing anN,N-dimethylethylamino (N,N-DMA) moiety for solubility purposes, was obtained from theSmall Molecule Discovery Center, UCSF for the Tethering screen (Figure 3) [20]. Each compoundat 0.12 mM concentration was allowed to react with 20 μM of thiol-bearing RNA in 0.1 M TEAAbuffer (pH 7.2) for 1 h at room temperature. The resulting sixty reaction mixtures were then analyzedby ESI-MS and percent relative abundance of the target adduct was used to assess small moleculebinding efficiency. The results of the screen are shown in Figure 4, Supplementary Table S1 andSupplementary Table S2. In addition, the ESI-MS data for an example screening reaction is shownin Figure 5 with compound A05. The majority of the compounds in the library showed minimal tono target adduct formation (Figure 4). This was expected considering the concentration of reducingagent used. However, compounds A04 and A05 gave >30% target adduct formation for pre21 RNA 1under the reducing conditions of the Tethering screen suggesting a binding interaction between theRNAs and these compounds. For compound A04, efficient conjugation was not specific to pre21RNA 1 as pre21 RNA 2 also gave a high conjugate yield. This is perhaps not surprising considering thestructure of A04 (Figure 3). 2-Phenylquinolines are known RNA ligands that bind by an intercalationmechanism [21]. Such a binding mode is possible at multiple sites on these RNAs consistent withefficient reaction independent of Tethering site. While this result is not desirable for the developmentof selective ligands, it does provide confirmation of the validity of this screening method as a means ofdiscovering RNA-interacting ligands. In contrast to A04, compound A05 gave a high conjugate yield(31%) with pre21 RNA 1 and a lower yield (13%) with pre21 RNA 2 suggesting a selective binding sitenear the bulged adenosine for the benzotriazole appendage of A05 (Figures 4 and 5). Furthermore,when the reaction with A05 was carried out with an unrelated thiol-modified 16 bp duplex, a lowconjugate yield was observed (<5%) while compound A04 produced conjugate with this RNA in38% yield (Supplementary Information). Interestingly, the benzotriazole structure of A05 can beconsidered a base analog with the potential to stack and hydrogen bond like natural nucleobases.
108

Molecules 2015, 20, 4148–4161
Indeed, tetrabromobenzotriazole is a known inhibitor of nucleotide-dependent enzymes where theinhibitor interacts with nucleotide-binding sites [22]. A recent NMR study of the structure of this partof the pre-miR21 RNA indicates the bulged A is extrahelical and partially associated with a groove ofthe adjacent duplex [23]. It is possible that the benzotriazole of A05 contacts the RNA groove at thislocation. However, at this point, we cannot rule out the possibility that our covalent modificationstrategy altered the RNA structure, placing the bulged A in a different position. Nevertheless, ifthe binding site for the tethered benzotriazole is present in the native RNA, it can provide a usefulstarting point for the development of selective pre-miR21 ligands. Additional structural studies willbe necessary to fully define this binding site.
Figure 3. Structures of the thirty disulfide-containing small molecules used in RNA Tethering screen.
109

Molecules 2015, 20, 4148–4161
Figure 4. Results of an RNA Tethering screen with disulfide containing small molecules and pre21RNA 1 and pre21 RNA 2. Plotted is the % yield of the target adduct vs. compound used (see Figure 3for structures of disulfides and Experimental Section for reaction conditions.) No bar indicates noproduct detected in that reaction by ESI-MS.
(A)
Compound A05 adduct Observed Mass = 10175.7 Da
Compound A05 adduct Observed Mass = 10176.2 Da
9840
9980
1012
0
1026
0
1040
0
9840
9980
1012
0
1026
0
1040
0
Mass (Da) Mass (Da)
(B)
Figure 5. Selectivity was observed during the screen. (A) RNA Tethering reaction of pre21 RNA 1 andpre21 RNA 2 with compound A05; (B) Corresponding ESI-MS spectra for these reactions (pre21 RNA 1(left) and pre21 RNA 2 (right)).
110

Molecules 2015, 20, 4148–4161
3. Experimental Section
3.1. General Procedures
All reagents and solvents were purchased from Sigma/Aldrich or Fischer Scientific and were usedwithout additional purification, unless noted otherwise. Reactions using dry solvents were performedunder an atmosphere of argon, in oven-dried glassware. Solvents were dried in a solvent purificationsystem that passes solvent through two columns of dry neutral alumina. Column chromatographywas conducted using silica gel (Sorbent Technologies, 60–200 mesh) and analytical TLC was performedon glass plates coated with 0.25 mm silica gel using UV for visualization. 1H and 13C magneticresonance spectra were recorded with Varian VNMRS 600, Varian Mercury 300 spectrometers andreferenced to the residual solvent peak. 31P magnetic resonance spectra was recorded with a VarianMercury 300 spectrometer. The abbreviations s, t, m, brs, dd, d stand for singlet, triplet, multiplet,broad singlet, doublet of doublets and doublet. High-resolution ESI mass spectra were obtained at theUniversity of California, Davis Mass spectrometry facility, on a Thermo Electron LTQ-Orbitrap HybridMass Spectrometer.
N6-[2-(triphenylmethylthio)ethyl]-9-(β-D-ribofuranosyl)adenine (1):
2',3',5'-Tri-O-acetyl-9-(β-D-ribofuranosyl)-6-chloropurine (100 mg, 0.218 mmol) was dissolvedin 1.0 mL anhydrous DMF and treated with S-trityl-protected 2-aminoethanethiol [24] (161 mg, 2.3equiv) in 1.0 mL DMF. After stirring for 18 h, NH3/MeOH (5 mL, saturated solution) was added,and stirring continued for a further 15 h. The crude was then diluted with 10 mL CH2Cl2, extractedtwice with saturated NaCl, and dried over Na2SO4. The crude mixture was then concentrated underreduced pressure, absorbed onto silica gel, and purified by column chromatography (0% → 6% MeOHin CH2Cl2), yielding a pale yellow solid (85 mg, 72%). 1H-NMR (300 MHz, CD3OD) δ 8.25 (s, 1H), 8.13(s, 1H), 7.40-7.07 (m, 15H), 5.96 (d, J = 6.4 Hz, 1H), 5.49 (s, 1H), 4.76 (t, J = 6.0 Hz, 1H), 4.33 (d, J = 2.6Hz, 1H), 4.18 (s, 1H), 3.90 (d, J = 12.5 Hz, 1H), 3.75 (d, J = 12.7 Hz, 1H), 3.49 (s, 1H), 2.54 (t, J = 6.5 Hz,2H), 2.39 (dd, J = 14.5, 5.6 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 154.53, 152.15, 144.54, 139.92, 146.79,129.49, 127.86, 127.20, 126.68, 91.10, 87.65, 73.63, 73.64, 72.59, 67.92, 39.22, 31.67, 29.64, 25.54. ESIHRMS:calcd for C31H31N5O4S (M+H)+: 570.2169, obsd 570.2168.
N6-[2-(triphenylmethylthio)ethyl]-9-[5-O-(4,4’-dimethoxytrityl)-β-D-ribofuranosyl]adenine (2):
DMTrCl (229 mg, 1.1 eq) and DMAP (37 mg, 0.5 eq) were added to a solution of 1 (350 mg,0.61 mmol) in 5 mL anhydrous pyridine. The reaction mixture was stirred at RT for 16 h.
111

Molecules 2015, 20, 4148–4161
Co-evaporation with 20 mL tolune and 20 mL CH3CN was performed to remove pyridine andtoluene, respectively. Afterwards, the mixture was diluted with CH2Cl2 (15 mL) and washed withsaturated aqueous NaHCO3 (2 × 10 mL). The organic layer was dried (Na2SO4), concentrated underreduced pressure and purified by column chromatography (1% Et3N in CH2Cl2). Excess Et3N incolumn fractions were removed via azeotrope formation with CH3CN, yielding a white solid (467 mg,88%). 1H-NMR (300 MHz, CD2Cl2) δ 8.22 (s, 1H), 8.03 (s, 1H), 7.45–7.19 (m, 24H), 6.79 (m, 4H), 6.26(d, J = 5.4 Hz, 1H), 5.99 (d, J = 5.2 Hz, 1H), 4.73 (t, J = 4.8 Hz, 1H), 4.40 (d, J = 4.5 Hz 1H), 3.75 (s, 6H),3.47–3.29 (m, 4H), 2.62–2.51 (m, 2H), 2.31 (t, J = 6.4 Hz, 2H). 13C-NMR (75 MHz, CD2Cl2) δ 158.57,152.29, 144,72, 144.59, 138.04, 135.54, 135.38, 129.91, 129.87, 129.49, 129.47, 127,90, 127.83, 127.75, 126.74,126.66, 90.84, 86.32, 86.11, 76.02, 72.61, 66.76, 63.55, 55.14, 38.06, 31.85, 29.67, 22.92. ESIHRMS: calcd forC52H49N5O6S (M+H)+: 872.3476, obsd: 872.3473.
N6-[2-(triphenylmethylthio)ethyl]-9-[2-O-(tert-butyldimethylsilyl)-5-O-(4,4’-dimethoxytrityl)-β-D-ribofuranosyl]adenine (3):
In a flame-dried 25 mL round-bottom flask, TBDMSCl (93 mg, 1.2 eq), freshly distilledtriethylamine (145 uL, 2.0 eq) and DMAP (32 mg, 0.5 eq) were added to a solution 5'-O-DMTr protectedderivative 2 (450 mg, 0.516 mmol, 1 eq) in 1 mL anhydrous THF. The reaction mixture was stirred atRT for 24 h. Upon completion of the reaction, the mixture was diluted with EtOAc (10 mL), filteredout white precipitate, and washed with 5% aqueous NaHCO3 (2 × 10 mL). The organic layer wasdried (Na2SO4) and concentrated under reduced pressure. The crude reaction mixture was purified bycolumn chromatography (15% → 30% (CH3)2CO in CH2Cl2) to afford a white foam (152 mg, 43%).1H-NMR (300 MHz, CDCl3) δ 8.30 (s, 1H), 8.06 (s, 1H), 7.59–7.32 (m, 24H), 6.94 (d, J = 8.8 Hz, 4H), 6.09(d, J = 5.0 Hz, 1H), 5.97 (s, 1H), 5.10 (s, 1H), 4.42 (d, J = 4.2 Hz, 1H), 4.32 (d, J = 4.2 Hz, 1H), 3.89 (s, 6H),3.54 (m, 4H), 2.78 (d, J = 4.6 Hz, 1H), 2.68 (t, J = 6.6 Hz, 2H), 2.13 (s, 1H), 0.97 (s, 9H), 0.12 (s, 3H), 0.00(s, 3H). 13C-NMR (75 MHz, CD2Cl2) δ 158.59, 144.80, 144.72, 138.73, 135.65, 135.62, 130.01, 130.00,129.47, 128.01, 127.81, 127.77, 126.75, 126.61, 113.02, 88.37, 86.40, 83.81, 75.26, 71.35, 63.40, 55.12, 29.62,25.29, 17.73. ESIHRMS: calcd for C58H63N5O6SSi (M + H)+: 986.4341, obsd: 986.4370.
N6-[2-(triphenylmethylthio)ethyl]-9-[2-O-(tert-butyldimethylsilyl)-5-O-(4,4’-dimethoxytrityl)-β-D-ribofuranosyl]adenine 3'-(2-Cyanoethyl)-N,N-diisopropylphosphoramidite (4):
Dry diisopropylethylamine (70 uL, 6 eq) and 2-cyanoethyl-(N,N-diisopropylamino)chlorophosphite(29 uL, 2 eq) were added to a solution of 3 (74 mg, 0.075 mmol, 1 eq) in 1.0 mL dry DCM.
112

Molecules 2015, 20, 4148–4161
The reaction mixture was stirred for 1 h. Upon completion of the reaction, the mixture was dilutedwith EtOAc (5 mL) and washed with 5% (w/v) aqueous NaHCO3 (2 × 5 mL). The organic layer wasdried (Na2SO4) and concentrated under reduced pressure. Purification by column chromatography(30%–40% EtOAc in Hexane) yielded a white foam (62 mg, 82%). 31P-NMR (121 MHz, CD2Cl2)δ 151.68, 150.24. ESIHRMS: calcd for C67H80N7O7PSSi (M+H)+: 1186.5420, obsd: 1186.5474.
3.2. RNA Synthesis, Purification and S-Tr Deprotection
RNA oligonucleotides were synthesized on an ABI 394 synthesizer (DNA/Peptide Core Facility,University of Utah) at 200 nmol and 1 umol scale using 5'-DMTr, 2'-OTBDMS protected β-cyanoethylphosphoramidites. Conditions for synthesis, cleavage and standard deprotection are identical to thosepreviously described [25]. Crude RNA synthesis products were purified by urea-polyacrylamide gelelectrophoresis (19%), and desalted with Sep-Pak cartridges, as previously described. The S-Trityl RNAwas stored as a lyophilized pellet at −70 ◦C. Identification and purity was determined by ESI-MS.
Prior to S-trityl deprotection [19], the RNA was allowed to fold into its native secondary structureby heating at 95 ◦C for 30 min. then slow cooling in the heat block until the sample has reachedroom temperature. To remove the S-Trityl protecting group, purified RNA (1.2 nmol) was dissolvedin aqueous TEAA buffer (triethylammonium acetate, 15 uL, 0.1 M, pH 6.5) and treated with AgNO3
(2 μL, 1 M in H2O). After 30 min of gentle agitation by vortex at room temperature, DTT was added(2 μL, 1 M H2O), and the reaction was allowed to proceed for a further 10 min. The mixture wasdiluted with aqueous 0.1 M TEAA (pH 6.5) and centrifuged (16,000× g, 4 ◦C, 5 min) to separatethe precipitate. The supernatant was collected. The pellet was washed 3 times with 50 uL aqueous0.1 M TEAA, to minimize the loss of the RNA. Each wash was followed by vigorous vortex agitation.The combined supernatants (~170 μL) were loaded in to a 3000 MWCO centrifugal concentrator(Microcon-3, Millipore) and centrifuged (11,000× g, 4 ◦C, approx. 30 min). The concentrated crudesample was then washed once with 150 μL of 0.1 M TEAA and centrifuged (11,000× g, 4 ◦C, approx.30 min). A final wash was performed with 100 μL of nuclease-free water and centrifuged (11,000× g,4 ◦C, approx. 30 min). After the buffer exchange into water, the deprotected RNA was quantified by theabsorbance of the solution at 260 nm. Yields ranged from 340 pmol (28%) to 940 pmol (76%). ESI-MSconfirmed quantitative conversion to the free thiol-RNA, and the purity of the product. The deprotectedRNA was stored in aliquots of water at −20 ◦C if not used immediately for experimentation.
STr-protected pre21 RNA 1: 5'-GGU GUU GCX UGU UGA AUC UCA UGG CAA CAC C-3'X = ASH
Calcd mass (monoisotopic): 10,195.76Obsd mass (ESI-MS): 10,193.56
Free thiol pre21 RNA 1: 5'- GGU GUU GCX UGU UGA AUC UCA UGG CAA CAC C -3'X = ASH
Calc mass (monoisotopic): 9954.5Obsd mass (ESI-MS): 9955.5
STr-protected pre21 RNA 2: 5'-GGU GUU GCA UGU UGA AUC UCA UGG CXA CAC C-3'X = ASH
Calcd mass (monoisotopic): 10,195.8Obsd mass (ESI-MS): 10,194.3
Free thiol pre21 RNA 2: 5'- GGU GUU GCX UGU UGA AUC UCA UGG CXA CAC C-3'X = ASH
Calc mass (monoisotopic): 9954.5
113

Molecules 2015, 20, 4148–4161
Obsd mass (ESI-MS): 9955.1
3.3. Mass Spectrometry Analysis of RNAs
Mass spectra were obtained on a Thermo Electron LTQ-Orbitrap Hybrid Mass Spectrometer.Free-thiol RNAs were desalted and purified prior to conjugate formation. Conjugate reaction sampleswere analyzed crude. The samples were run with a mobile phase consisting of 50% MeOH/H2Ow/0.2% triethylamine. Mass spectra were recorded in negative ionization mode. A 9 kDa DNAstandard was used to tune the instrument before each run. Spectra were deconvoluted using ProMassand MassLynx software.
3.4. Conjugation Reaction Procedure
Free-thiol RNA (~200 pmol) was dissolved in 8 μL of 0.1 M TEAA (triethylammonium acetate,pH 7.2). Freshly prepared β-mercaptoethanol (1 μL, 0.5 M) was then added to the solution andpipet-mixed. The disulfide compound (1 μL, 1.25 mM in H2O) was added to the reaction mixtureimmediately. The reaction was allowed to proceed for 1 h at room temperature under gentle agitation.Samples were then analyzed with ESI-MS immediately or stored at −20 ◦C.
3.5. Calculation of Reaction Product Abundances
Abundances were calculated using the area of the peaks in the deconvoluted spectrum of eachsample reaction. All peaks were accounted for in each spectrum. The noteworthy peaks in eachspectrum are as follows: (1) free thiol; (2) N,N-dimethylethylamino adduct (N,N-DMA); (3) product.Given their structural similarities, these species were assumed to have equivalent ionization efficiencies.However, due to possible variations in ionization behavior, quantification of conjugates in this mannermay not always reflect exact yields. At an RNA concentration of 20 μM, no RNA dimers were observedin the spectra.
% abundance = (area of species peak/sum of the areas of all peaks in spectrum) × 100
4. Conclusions
We have demonstrated that oligonucleotides bearing the ASH nucleoside analog can effectivelyconjugate with disulfide-containing small molecules under varying reducing conditions. Amongthe library of 30 disulfide-containing compounds screened, a small molecule bearing a benzotriazolemoiety displayed not only prominent yield of the adduct, but also selectivity for pre21 RNA 1 wherethe ASH analog replaces a bulged adenosine near the Dicer cleavage site. The result suggests a modeof binding to the RNA outside of the disulfide linkage and may provide a starting point for thedevelopment of pre-miRNA21 ligands.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/03/4148/s1.
Acknowledgments: This work was supported by the University of California Cancer Research CoordinatingCommittee and a grant from the National Institutes of Health to PAB (R01GM061115). We also thank WilliamJewell, Core Mass Spectrometry Facility, UC Davis for technical assistance and Adam Renslo, Small MoleculeDiscovery Center, Department of Pharmaceutical Chemistry, UCSF for providing screening compounds.
Author Contributions: Peter A. Beal designed the research, analyzed the data, and wrote the paper; Kiet Tranperformed the research, analyzed the data, and wrote the paper. Michelle Arkin assisted in the design of theresearch and editing of the paper. All authors read and approved the final transcript.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Gallego, J.; Varani, G. Targeting RNA with Small-Molecule Drugs: Therapeutic Promise and ChemicalChallenges. Acc. Chem. Res. 2001, 34, 836–843. [CrossRef] [PubMed]
114

Molecules 2015, 20, 4148–4161
2. Guan, L.; Disney, M.D. Recent Advances in Developing Small Molecules Targeting RNA. ACS Chem. Biol.2012, 7, 73–86. [CrossRef] [PubMed]
3. Griffey, R.H.; Sannes-Lowery, K.A.; Drades, J.J.; Venkatraman, M.; Swayze, E.E.; Hofstadler, S.A.Characterization of Low-Affinity Complexes between RNA and Small Molecule Using ElectrosprayIonization Mass Spectrometry. J. Am. Chem. Soc. 2000, 122, 9933–9938. [CrossRef]
4. Jimenez-Moreno, E.; Gomez-Pinto, I.; Corzana, F.; Santan, A.G.; Revuelta, J.; Bastida, A.; Jimenez-Barbero, J.;Gonzalez, C.; Asensio, J.L. Chemical Interrogation of Drug/RNA Complexes: From Chemical Reactivity toDrug Design. Angew. Chem. Int. Ed. 2013, 52, 3148–3151. [CrossRef]
5. Hardy, J.A.; Lam, J.; Nguyen, J.; O’Brien, T.; Wells, J.A. Discovery of an allosteric site in the caspases.Proc. Natl. Acad. Sci. USA 2004, 101, 12461–12466. [CrossRef] [PubMed]
6. Yang, W.; Fucini, R.V.; Fahr, B.T.; Randal, M.; Lind, K.E.; Lam, M.B.; Lu, W.; Lu, Y.; Cary, D.R.;Romanowski, M.J.; et al. Fragment-based Discovery of Nonpeptidic BACE-1 Inhibitors Using Tethering.Biochemistry 2009, 48, 4488–4496. [CrossRef] [PubMed]
7. Erlanson, D.A.; Braisted, A.C.; Raphael, M.; Stroud, R.M.; Gordon, E.M.; Wells, J.A. Site-directed liganddiscovery. Proc. Natl. Acad. Sci. USA 2000, 97, 9367–9372. [CrossRef] [PubMed]
8. Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [CrossRef][PubMed]
9. Volinia, S.; Calin, G.; Liu, C.-G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Ferracin, M.;Prueit, R.L. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl.Acad. Sci. USA 2006, 103, 2257–2261. [CrossRef] [PubMed]
10. Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-celllymphoma. Nature 2010, 467, 86–91. [CrossRef] [PubMed]
11. Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [CrossRef][PubMed]
12. Chirayil, S.; Chirayil, R.; Luebke, K.J. Discovering ligands for a microRNA precursor with peptoidmicroarrays. Nucl. Acids Res. 2009, 37, 5486–5497. [CrossRef] [PubMed]
13. Velagapudi, S.P.; Gallo, S.M.; Disney, M. Sequence-based Design of Bioactive Small Molecules that TargetPrecursor MicroRNAs. Nat. Chem. Biol. 2014, 10, 291–299. [CrossRef] [PubMed]
14. Bose, D.; Gopal, J.; Suryawanshi, H.; Agarwala, P.; Pore, S.K.; Banerjee, R.; Maiti, S. The Tuberculosis DrugStreptomycin as a Potential Cancer Therapeutic: Inhibition of miR-21 Function by Directly Targetting ItsPrecursor. Angew. Chem. Int. Ed. 2012, 51, 1019–1023. [CrossRef]
15. Maiti, M.; Nauwelaerts, K.; Herdewijn, P. Pre-mircoRNA binding aminoglycosides and antitumor drugs asinhibitors of Dicer catalyzed microRNA processing. Bioorg. Med. Chem. Lett. 2012, 22, 1709–1711. [CrossRef][PubMed]
16. Allerson, C.; Swaine, L.; Verdine, G.L. A Chemical Method for Site-Specific Modification of RNA:The Convertible Nucleoside Approach. J. Am. Chem. Soc. 1997, 119, 7423–7433. [CrossRef]
17. Porcher, S.; Myeyyappan, M.; Pitsch, S. Spontaneous Aminoacylation of a RNA Sequence Containinga 3'-Terminal 2'-Thioadenosine. Helvetica Chimica Acta 2005, 88, 2897–2910. [CrossRef]
18. Geiermann, A.; Polacek, N.; Micura, R. Native Chemical Ligation of Hydrolysis-Resistant 3'-Peptidyl-RNAMimics. J. Am. Chem. Soc. 2011, 133, 19068–19071. [CrossRef] [PubMed]
19. Peacock, H.; Bachu, R.; Beal, P.A. Covalent stabilization of a small molecule-RNA complex. Bioorg. Med.Chem. Lett. 2011, 21, 5002–5005. [CrossRef] [PubMed]
20. Burlingame, M.A.; Tom, C.T.; Renslo, A.R. Simple One-Pot Synthesis of Disulfide Fragments for Use inDisulfide-Exchange Screening. ACS Comb. Sci. 2011, 13, 205–208. [CrossRef] [PubMed]
21. Zhao, M.; Janda, L.; Nguyen, J.; Strekowski, L.; Wilson, W.D. The Interaction of Substituted 2-phenylquinolineIntercalators with Poly(A) • poly(U): Classical and Threading Intercalation Modes of RNA. Biopolymers 2004,34, 61–73. [CrossRef]
22. Battistutta, R.; de Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selectiveinhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10,2200–2206. [CrossRef] [PubMed]
23. Chirayil, S.; Wu, Q.; Amezcua, C.; Leubke, K.J. NMR Characterization of an Oligonucleotide Model of theMiR-21 Pre-Element. PLoS One 2014, 9, e108231. [CrossRef] [PubMed]
115

Molecules 2015, 20, 4148–4161
24. Li, M.; Yamato, K.; Ferguson, J.S.; Gong, B. Sequence-Specific Associaion in Aqueous Media by IntegratingHydrogen Bonding and Dynamic Covalent Interactions. J. Am. Chem. Soc. 2006, 128, 12628–12629. [CrossRef][PubMed]
25. Hamm, M.L.; Piccirilli, J.A. Incorporation of 2'-deoxy-2'-mercaptocytidine into Oligonucleotides viaPhosphoramidite Chemistry. J. Org. Chem. 1997, 62, 3415–3420. [CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples are not available from authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
116

molecules
Article
Construction of an Isonucleoside on a2,6-Dioxobicyclo[3.2.0]-heptane Skeleton
Yuichi Yoshimura 1,*, Satoshi Kobayashi 1, Hitomi Kaneko 1, Takeshi Suzuki 1 andTomozumi Imamichi 2
1 Faculty of Pharmaceutical Sciences, Tohoku Pharmaceutical University, 4-4-1 Komatsushima, Aoba-ku,Sendai 981-8558, Japan; [email protected] (S.K.); [email protected] (H.K.);[email protected] (T.S.)
2 Laboratory of Human Retrovirology, Leidos Biochemical Research Inc., Frederick National Laboratory forCancer Research, Frederick, MD 21702, USA; [email protected]
* Correspondence: [email protected]; Tel./Fax: +81-22-727-0144
Academic Editors: Mahesh K. Lakshman and Fumi NagatsugiReceived: 2 February 2015; Accepted: 4 March 2015; Published: 12 March 2015
Abstract: We have built a new isonucleoside derivative on a 2,6-dioxobicyclo[3.2.0]heptaneskeleton as a potential anti-HIV agent. To synthesize the target compound, an acetal-protecteddihydroxyacetone was first converted to a 2,3-epoxy-tetrahydrofuran derivative. Introduction of anazide group, followed by the formation of an oxetane ring, gave a pseudosugar derivative with a2,6-dioxobicyclo[3.2.0]heptane skeleton. The desired isonucleoside was obtained by constructing apurine base moiety on the scaffold, followed by amination.
Keywords: nucleoside; bicyclo; oxetane ring; conformation
1. Introduction
Since the discovery of 3'-azidothymidine (AZT), much attention has been paid to the developmentof effective chemotherapeutic agents against the human immunodeficiency virus (HIV), a causativeagent for AIDS [1,2]. More than 20 anti-HIV drugs have now been approved and are clinically usedfor the treatment of AIDS. Among them, nucleoside reverse transcriptase inhibitors (NRTIs) play acritical role in the treatment of AIDS patients. In the most successful regimen for AIDS referred toas ART (Anti-Retroviral Therapy), a cocktail of anti-HIV drugs, including NRTIs, non-nucleosidereverse transcriptase inhibitors (NNRTIs), and protease inhibitors (PIs) [3], is used. Although ARTgreatly contributes to increasing the lifespan of patients, drug-resistant strains of the virus are still aserious problem [4,5]. Therefore, new drugs that are effective against the resistant virus strains areconstantly needed.
Most NRTIs belong to a category of dideoxynucleosides, e.g., zalcitabine (ddC) [6] and didanosine(ddI) [6]. AZT [7] and lamivudine [8] are 3'-substituted dideoxynucleoside derivatives, and abacavir isa carbocyclic analogue of dideoxynucleoside. Only tenofovir [9], which is a nucleoside phosphonate(Figures 1 and 2), is different. From the viewpoint of designing new anti-HIV agents, nucleosidesconstructed on a novel scaffold are expected to have antiviral activity against the resistant virus strainsand may avoid cross-resistance to the known NRTIs.
Molecules 2015, 20, 4623–4634 117 www.mdpi.com/journal/molecules
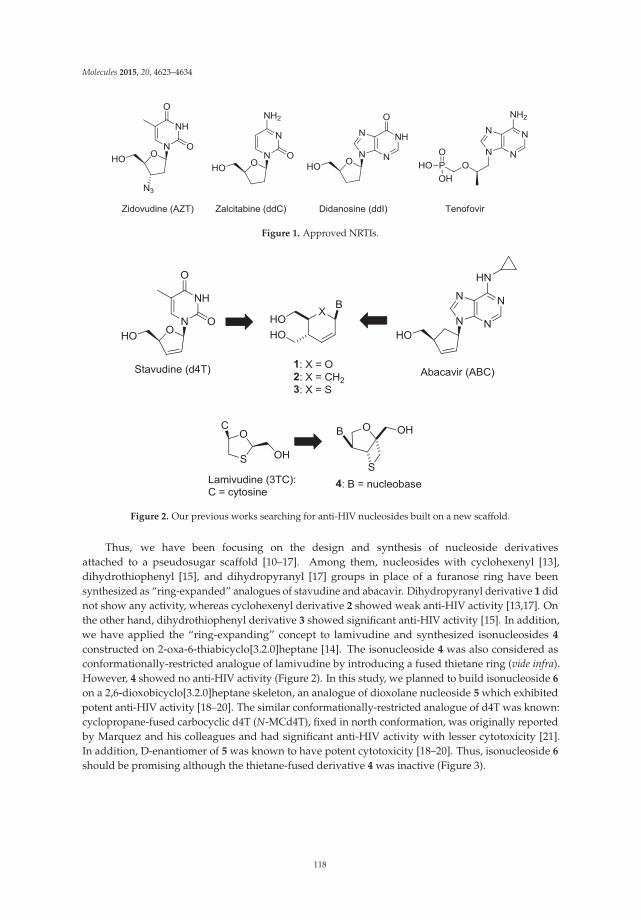
Molecules 2015, 20, 4623–4634
Figure 1. Approved NRTIs.
Figure 2. Our previous works searching for anti-HIV nucleosides built on a new scaffold.
Thus, we have been focusing on the design and synthesis of nucleoside derivativesattached to a pseudosugar scaffold [10–17]. Among them, nucleosides with cyclohexenyl [13],dihydrothiophenyl [15], and dihydropyranyl [17] groups in place of a furanose ring have beensynthesized as “ring-expanded” analogues of stavudine and abacavir. Dihydropyranyl derivative 1 didnot show any activity, whereas cyclohexenyl derivative 2 showed weak anti-HIV activity [13,17]. Onthe other hand, dihydrothiophenyl derivative 3 showed significant anti-HIV activity [15]. In addition,we have applied the “ring-expanding” concept to lamivudine and synthesized isonucleosides 4
constructed on 2-oxa-6-thiabicyclo[3.2.0]heptane [14]. The isonucleoside 4 was also considered asconformationally-restricted analogue of lamivudine by introducing a fused thietane ring (vide infra).However, 4 showed no anti-HIV activity (Figure 2). In this study, we planned to build isonucleoside 6
on a 2,6-dioxobicyclo[3.2.0]heptane skeleton, an analogue of dioxolane nucleoside 5 which exhibitedpotent anti-HIV activity [18–20]. The similar conformationally-restricted analogue of d4T was known:cyclopropane-fused carbocyclic d4T (N-MCd4T), fixed in north conformation, was originally reportedby Marquez and his colleagues and had significant anti-HIV activity with lesser cytotoxicity [21].In addition, D-enantiomer of 5 was known to have potent cytotoxicity [18–20]. Thus, isonucleoside 6
should be promising although the thietane-fused derivative 4 was inactive (Figure 3).
118
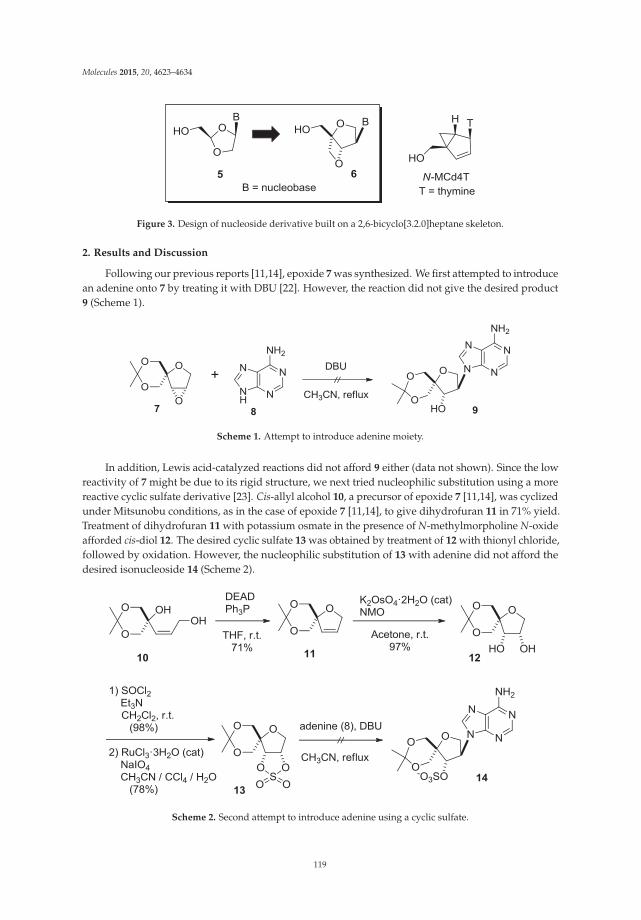
Molecules 2015, 20, 4623–4634
Figure 3. Design of nucleoside derivative built on a 2,6-bicyclo[3.2.0]heptane skeleton.
2. Results and Discussion
Following our previous reports [11,14], epoxide 7 was synthesized. We first attempted to introducean adenine onto 7 by treating it with DBU [22]. However, the reaction did not give the desired product9 (Scheme 1).
Scheme 1. Attempt to introduce adenine moiety.
In addition, Lewis acid-catalyzed reactions did not afford 9 either (data not shown). Since the lowreactivity of 7 might be due to its rigid structure, we next tried nucleophilic substitution using a morereactive cyclic sulfate derivative [23]. Cis-allyl alcohol 10, a precursor of epoxide 7 [11,14], was cyclizedunder Mitsunobu conditions, as in the case of epoxide 7 [11,14], to give dihydrofuran 11 in 71% yield.Treatment of dihydrofuran 11 with potassium osmate in the presence of N-methylmorpholine N-oxideafforded cis-diol 12. The desired cyclic sulfate 13 was obtained by treatment of 12 with thionyl chloride,followed by oxidation. However, the nucleophilic substitution of 13 with adenine did not afford thedesired isonucleoside 14 (Scheme 2).
Scheme 2. Second attempt to introduce adenine using a cyclic sulfate.
119

Molecules 2015, 20, 4623–4634
Therefore, we revised our plan to synthesize an isoadenosine constructed on a2,6-dioxobicyclo[3.2.0]heptane scaffold, and the revised scheme is shown in Scheme 3 in a retrosyntheticmanner. Instead of the direct introduction of adenine, we decided to build the adenine ring on the2,6-dioxobicyclo[3.2.0]heptane pseudosugar skeleton in a stepwise manner. According to this plan,compound 16 was thought to be a suitable intermediate for preparing 15 since it can be transformedto 6 by the formation of an imidazole ring, followed by amination. Fused oxetane derivative 16 canbe obtained from dimesylate 17. Finally, epoxide 7, described above, was selected as the startingcompound because it can be converted to 17 by the selective cleavage of the oxirane ring with an azideanion (Scheme 2).
Scheme 3. Revised retrosynthesis of isoadenosine 6 built on a 2,6-dioxobicyclo[3.2.0]heptane skeleton.
First, regioselective cleavage of the oxirane ring of 7 with sodium azide in 2-methoxyethanol underreflux conditions gave the desired azide-alcohol 18 as a single regioisomer in 66% yield. It is obviousthat the nucleophilic azide anion attacked from the less hindered side since similar regioselectiveepoxide opening was observed in our previous report [11,14]. After benzoylation of the hydroxyl group,the acetal group of 19 was removed by using acidic hydrolysis, and the resulting diol was mesylatedto give dimesylate 20 in good yield. Deprotection of the benzoyl group and the subsequent formationof an oxetane ring were achieved by treating 20 with sodium methoxide under reflux conditions togive mesylate 21 in 72% yield. The structure of 21 was unambiguously determined by comparison of1D NMR spectrum with that of 2-oxa-6-thiabicyclo[3.2.0]heptane skeleton [14] after converting it tobenzoate 22 by treatment with benzoic acid in the presence of cesium fluoride. In 1H-NMR spectra of22, the peaks corresponding to the methyl groups of the dimesylate were absent, and only the peakscorresponding to the benzoyl group in the range of 8.1–7.4 ppm were present. In addition, one ofthe methylene protons at the 2-position was observed as a doublet at 4.42 ppm, meaning that thecoupling with H-3 disappeared. This indicates that the conformation around the tetrahydrofuran ringchanges and becomes fixed, which causes a loss of coupling between one pair of H-2 and H-3 protons.A similar correlation between conformation and couplings in 1H-NMR spectra has been reported forthe 2-oxa-6-thiabicyclo[3.2.0]heptane skeleton [14]. Moreover, in the mass spectrum of the compound,a molecular ion peak was observed at m/z = 276, further supporting the assignment of the structure.Finally, 22 was deprotected to afford azido-alcohol 23 in 88% yield (Scheme 4).
120

Molecules 2015, 20, 4623–4634
Scheme 4. Synthesis of isoadenosine 6.
Azido-alcohol 23 was reduced by catalytic hydrogenation to give key intermediate 16, which wastreated with 5-amino-4,6-dichloropyrimidine and diisopropylethylamine in refluxing n-butanol [23] togive diaminopyrimidine derivative 15 in 58% yield from 23. Formation of the imidazole ring of 15 wasaccomplished by treatment with orthoethyl formate under acidic conditions [24] to give 6-chloropurinenucleoside 24. Finally, the isoadenosine was built on the 2,6-dioxobicyclo[3.2.0]heptane scaffold 6 byheating 24 with methanolic ammonia in a sealed tube in 69% yield (Scheme 4). Isoadenosine 6 did notshow any significant activity against HIV even at a concentration of 100 μM.
3. Experimental Section
General Information
Melting points are uncorrected. NMR spectra were recorded at 400 MHz (1H), 100 MHz (13C)using CDCl3 as a solvent. As an internal standard, tetramethylsilane was used for CDCl3. Mass spectrawere obtained by EI or FAB mode. Silica gel for chromatography was Silica Gel 60N (spherical, neutral,100–210 μm, Kanto Chemical Co. Inc., Tokyo, Japan). When the reagents sensitive to moisture wereused, the reaction was performed under argon atmosphere.
8,8-Dimethyl-1,7,9-trioxaspiro[4,5]dec-3-ene (11). To a solution of PPh3 (2.49 g, 9.48 mmol) in THF (10 mL)was added DEAD (4.31 mL, 9.48 mmol) and the mixture was stirred at room temperature for 5 min.To this mixture, a solution of 10 [11,14] (1.04 g, 5.58 mmol) in THF (10 mL) was added. The mixturewas stirred at room temperature for 1 h. After the solvent was removed under reduced pressure, theresidue was purified by silica gel column chromatography (hexane–ethyl acetate = 19:1) to give 11
(677 mg, 71%) as a white crystal. mp 47–49 ◦C; 1H-NMR (CDCl3) δ 1.45 (3H, s), 1.48 (3H, s), 3.76 (2H, d,
121

Molecules 2015, 20, 4623–4634
J = 11.6 Hz), 3.81 (2H, d, J = 11.6 Hz), 4.71 (2H, t, J = 1.9 Hz), 5.84-5.87 (1H, m), 6.02 (1H, d, J = 6.3 Hz);13C-NMR (CDCl3) δ 22.6, 24.6, 66.7, 74.9, 98.0, 128.1, 129.1; IR (neat) 2924.2, 2853.1, 1724.1, 1215.9, 758.3cm−1; FAB-MS (m/z) 155 [M−15]+; Anal. Calcd for C9H14O3; C, 62.84; H, 8.32. Found; C, 62.78; H, 8.44.
(3S*,4S*)-8,8-Dimethyl-1,7,9,-trioxaspiro[4.5]decane-3,4-diol (12). To a solution of 11 (73 mg, 0.43 mmol)and NMO (0.22 mL) in acetone (4 mL), was added a solution of K2OsO4·2H2O (1 mg, 0.004 mmol)in H2O (0.4mL) at 0 ◦C. After stirred at room temperature for 60 h, Na2S2O3·5H2O (125 mg) wasadded and the mixture was stirred at room temperature for 30 min. After the whole mixture wasdried over Na2SO4, the solid materials were removed by suction and washed with ethyl acetate. Thecombined filtrate was concentrated under reduced pressure. The residue was purified by silica gelcolumn chromatography (CHCl3–MeOH = 19:1) to give 12 (84 mg, 97%). 1H-NMR (CDCl3) δ 1.43 (3H,s), 1.49 (3H, s), 3.59 (1H, dd, J = 11.6, 1.9 Hz), 3.80 (1H, d, J = 9.7 Hz), 3.82–3.87 (2H, m), 3.94 (1H, dd,J = 9.7, 4.9 Hz), 4.14 (1H, dd, J = 11.6, 1.9 Hz), 4.22 (1H, d, J = 5.3 Hz) 4.34 (1H, q, J = 5.0 Hz); 13C-NMR(CDCl3) δ 21.0, 25.8, 63.3, 66.5, 71.0, 71.4, 74.9, 76.7, 98.4; IR (KBr) 3306.4, 2953.6, 2741.6, 1452.2, 524.19cm−1; EI-MS (m/z): 204 [M+1]+; HRMS Calcd for C9H15N3O4: 204.0998, Found: 204.0992.
(3S*,4S*)-8,8-Dimethyl-1,7,9,-trioxaspiro[4.5]decane-3,4-cyclicsulfate (13). To a solution of 12 (410 mg,2.01 mmol) and Et3N (672 μL, 4.82 mmol) in CH2Cl2 (10 mL), was added dropwise a solution of SOCl2(113 μL, 1.55 mmoL) in CH2Cl2 (10 mL) at 0 ◦C. After stirred at room temperature for 15 min, themixture was washed with water. The water layer was extracted with CHCl3 twice and the combinedorganic layer was washed with brine, then dried over Na2SO4. After filtration, the residue was passedthrough a short silica gel column (eluate: hexane–ethyl acetate = 1:1). After the solvents were removedunder reduced pressure, the residue was dissolved in CCl4–CH3CN–H2O (2:2:3, 3 mL). To this solution,were added RuCl3·3H2O (2.7 mg) and NaIO4 (73 mg, 0.34 mmol) at 0 ◦C. The mixture was stirred at thesame temperature for 1.5 h. After diluted with ether, the mixture was washed with water, sat.NaHCO3
and brine, then dried over Na2SO4. After filtration, the solvents were removed under reduce pressure,the residue was purified by silica gel column chromatography (hexane-ethyl acetate = 6:1) to give 13
(82 mg, 77%). 1H-NMR (CDCl3) δ1.43 (3H, s), 1.50 (3H, s), 3.56 (1H, dd, J = 12.1, 2.4 Hz), 3.82 (1H, d,J = 12.1 Hz), 3.91 (1H, d, J = 12.1 Hz), 3.97 (1H, dd, J = 12.6, 4.4 Hz), 4.06 (1H, dd, J = 12.1, 2.4 Hz), 4.28(1H, d, J = 12.6 Hz), 5.42 (1H, d, J = 6.3 Hz), 5.48 (1H, t, J = 5.1 Hz); 13C-NMR (CDCl3) δ 19.6, 27.1, 61.0,62.4, 69.7, 79.0, 83.3, 84.5, 99.2; IR (KBr) 3000.8, 2892.3, 1699.7, 1380.9, 1089.9 cm−1; EI-MS (m/z): 267[M+1]+; HRMS Calcd for C9H15N3O4: 266.0460, Found: 266.0467.
(3R*,4S*)-3-Azido-8,8-dimethyl-1,7,9,-trioxaspiro[4.5]decan-4-ol (18). A mixture of 7 [11,14] (433 mg,2.33 mmol) and NaN3 (752 mg 11.6 mmol) in 2-methoxyethanol (26 mL) was kept at 100 ◦C for5 h. After the solvent was removed under reduced pressure, the residue was dissolved in ethylacetate. After washed with water and brine, the organic layer was dried over Na2SO4. After filtration,the solvents were removed under reduce pressure, the residue was purified by silica gel columnchromatography (hexane–ethyl acetate = 5:1) to give 18 (351 mg, 66%). 1H-NMR (CDCl3) δ 1.40 (3H,s), 1.49 (3H, s), 3.69-3.80 (3H, m), 3.84 (1H, d, J = 11.6 Hz), 4.01–4.08 (3H, m), 4.38 (1H, t, J = 1.9, 2.4Hz); 13C-NMR (CDCl3) δ 19.5, 27.4, 62.3, 66.0, 67.6, 69.4, 78.2, 79.1, 98.5; IR (neat) 3419.0, 2104.8, 1086.6,831.7 cm−1; EI-MS (m/z): 229 [M+1]+; HRMS Calcd for C9H15N3O4: 229.1063, Found: 229.1062.
(3R*,4S*)-3-Azido-8,8-dimethyl-1,7,9,-trioxaspiro[4.5]decan-4-yl benzoate (19). To a solution of 18 (432 mg,1.88 mmol), Et3N (0.59 mL, 4.24 mmol), and DMAP (23 mg, 0.19 mmol) in CH2Cl2 (15 mL) was addedbenzoyl chloride (0.40 mL, 3.39 mmol) and the mixture was stirred at room temperature for 6.5 h.The reaction was quenched by addition of MeOH, and the whole was stirred at room temperaturefor 10 min. The mixture was diluted with CH2Cl2 and washed with water and brine, then dried overNa2SO4. After filtration, the solvents were removed under reduce pressure, the residue was purifiedby silica gel column chromatography (hexane–ethyl acetate = 4:1) to give 19 (598 mg, 95%). 1H-NMR(CDCl3) δ 1.37 (3H, s), 1.47 (3H, s), 3.84-3.91 (3H, m), 3.99 (1H, dd, J = 1.4, 10.6 Hz), 4.07 (1H, dd, J = 1.4,10.6 Hz), 4.18-4.25 (2H, m) 5.47 (1H, d, J = 1.0 Hz), 7.48 (2H, t, J = 7.2 Hz), 7.62 (1H, J = 7.2 Hz), 8.03
122

Molecules 2015, 20, 4623–4634
(2H, J = 7.2 Hz); 13C-NMR (CDCl3) δ 22.4, 24.2, 62.1, 65.0, 66.2, 69.9, 78.9, 79.2, 98.4, 128.6, 129.0, 129.6,133.6, 165.3; IR (neat) 2993.1, 2107.2, 1725.4, 1267.4, 1091.3, 711.5 cm−1; EI-MS (m/z): 333 [M]+; HRMSCalcd for C16H19N3O5: 333.1325, Found: 333.1336.
(3S*,4R*)-4-Azido-2,2-bis((methylsulfonyloxy)methyl)tetrahydrohuran-3-yl benzoate (20). A mixture of19 (1.01 g, 3.04 mmol) in 80% AcOH (80 mL) was stirred at room temperature for 5 h. After thesolvent was removed under reduced pressure, the residue was co-evaporated with EtOH five timesto remove residual AcOH. The resulting crude product was dissolved in CH2Cl2 (40 mL). To thismixture, were added, MsCl (1.19 mL, 15.18 mmol), Et3N (2.14 mL, 15.18 mmol), and DMAP (38 mg,0.30 mmol). After stirred at room temperature for 1 h, the mixture was diluted with CH2Cl2 washedwith 5%HCl, sat.NaHCO3 and brine. The separated organic layer was dried over Na2SO4. Afterfiltration, the solvents were removed under reduce pressure, the residue was purified by silica gelcolumn chromatography (hexane–ethyl acetate = 2:1) to give 20 (1.05 g, 77%). 1H-NMR (CDCl3) δ 3.00(3H, s), 3.12 (3H, s), 3.94 (1H, dd, J = 5.80, 4.37 Hz), 4.33~4.48 (6H, m), 5.48 (1H, d, J = 3.4 Hz), 7.50(2H, t, J = 7.7 Hz), 7.64 (1H, t, J = 8.0 Hz), 8.04 (2H, d, J = 7.3 Hz); 13C-NMR (CDCl3) δ 37.7, 65.5, 65.8,67.4, 70.3, 77.2, 78.9, 82.8, 128.1, 128.8, 129.9, 134.2, 165.2; IR (near) 2110.7, 1728.8, 1360.0, 1267.0 cm−1;FAB-MS (m/z): 450 [M+1]+; HRMS Calcd for C15H20N3O9S2: 450.0641, Found: 450.0631.
((1R*,4R*,5S*)-4-Azido-2,6-dioxabicyclo[3.2.0]heptan-1-yl)methyl methanesulfonate (21). A mixture of 20
(36 mg, 0.08 mmol) and NaOCH3 (4.6 mg, 0.08 mmol) in MeOH (2 mL) was kept at 75 ◦C overnight.After the solvent was removed under reduced pressure, the residue was dissolved in CHCl3 andwashed with water and brine, then dried over Na2SO4. After filtration, the solvents were removedunder reduce pressure, the residue was purified by silica gel column chromatography (hexane–ethylacetate = 2:1) to give 21 (14 mg, 72%). 1H-NMR (CDCl3) δ 3.09 (3H, s), 4.05 (1H, d, J = 3.4 Hz), 3.40 (1H,d, J = 11.1 Hz), 4.29~4.77 (4H, m), 4.76 (1H, d, J = 8.2 Hz), 5.06 (1H, s); 13C-NMR (CDCl3) δ 37.8, 63.6,67.4, 71.9, 77.1, 84.4, 89.9; IR (neat) 2098.2, 1360.4, 1175.0, 960.2, 815.5 cm−1; FAB-MS (m/z): 250 [M+1]+;HRMS Calcd for C7H12N3O5S: 250.0498, Found: 250.0490.
((1R*,4R*,5S*)-4-Azido-2,6-dioxabicyclo[3.2.0]heptan-1-yl)methyl benzoate (22). A mixture of CsF (332 mg,2.19 mmol) and PhCOOH (267 mg, 2.19 mmol) in DMF (40 mL) was stirred at room temperature for20 min. To this mixture was added a solution of 21 (182 mg, 0.73 mmol) in DMF (20 mL). After stirredat 60 ◦C overnight, the mixture was partitioned between EtOAc and H2O. The separated water layerwas extracted with EtOAc, and the organic layer was washed with sat.NaHCO3, brine, then driedover Na2SO4. After filtration, the filtrated was concentrated under reduced pressure. The residue waspurified by silica gel column chromatography (hexane-ethyl acetate = 5:1) to give 22 (170 mg, 86%).1H-NMR (CDCl3) δ 4.08 (1H, d, J = 3.4 Hz), 4.42 (1H, d, J = 10.6 Hz), 4.52 (2H, dd, J = 10.6, 3.4 Hz), 4.60(2H, s), 4.83 (1H, d, J = 7.7 Hz), 5.15 (1H, s), 7.61 (2H, t, J = 15.5 Hz) 7.59 (1H, t, J = 17.4 Hz), 8.10 (2H, d,J = 9.7 Hz); 13C-NMR (CDCl3) δ 63.1, 63.8, 71.5, 77.8, 85.1, 90.4, 128.4, 129.4, 129.7, 133.3, 166.1; IR (neat)2099.7, 1723.1, 1284.3, 713.4 cm−1; FAB-MS (m/z); 276 [M+1]+; HRMS Calcd for C13H14N3O4: 276.0984,Found: 276.0977.
((1S*,4R*,5S*)-4-Azido-2,6-dioxa-bicyclo[3.2.0]heptan-1-yl)methanol (23). A mixture of 22 (184 mg, 0.67mmol) and NaOCH3 (19 mg, 0.33 mmol) in MeOH (15 mL) was stirred at room temperature. Afterthe mixture was neutralized with AcOH (19 μL), the solvents were removed under reduced pressureand the residue was purified by silica gel column chromatography (hexane–ethyl acetate = 4:1) togive 23 (90 mg, 79%). 1H-NMR (CDCl3) δ 3.91 (2H, d, J = 6.3 Hz), 4.04 (1H, d, J = 3.9 Hz), 4.36 (1H, d,J = 10.6 Hz), 4.48 (2H, dd, J = 10.6, 3.4 Hz), 4.48 (1H, d, J = 8.2 Hz), 4.68 (1H, d, J = 8.2 Hz), 5.05 (1H, s);13C-NMR (CDCl3) δ 62.3, 64.0, 71.5, 77.6, 86.9, 90.2; IR (neat) 3431.5, 2101.6, 1248.2, 870.88 cm−1; EI-MS(m/z): 171 [M]+; HRMS Calcd for C6H9N3O3: 171.0644, Found: 171.0643.
((1S*,4R*,5S*)-4-(5-Amino-6-chloropyrimidin-4-ylamino)-2,6-dioxa-bicyclo[3.2.0]heptan-1-yl)methanol (15).A mixture of 23 (69 mg, 0.40 mmol) and Pd(OH)2 (6.2 mg, 0.04 mmol) in MeOH (5 mL) was stirredat room temperature overnight under H2 atmosphere. After insoluble materials were removed by
123

Molecules 2015, 20, 4623–4634
filtration, the solvents were removed under reduced pressure. The resulting crude product wasdissolved in n-BuOH (3 mL). To this mixture, were added 5-amino-4,6-dichloropyrimidine (140.1 mg,0.86 mmol) and i-Pr2NEt (298 μL, 1.71 mmol). The mixture was kept under reflux overnight. Afterthe solvents were removed under reduced pressure, the residue was purified by silica gel columnchromatography (chloroform–methanol = 19:1) to give 15 (64 mg, 58%). 1H-NMR (CD3OD) δ3.67 (2H,q, J = 12.6 Hz), 4.24 (1H, d, J = 10.1 Hz), 4.38 (1H, d, J = 7.7 Hz), 4.45 (1H, d, J = 4.4 Hz), 4.50 (1H, d,J = 10.1, 4.4 Hz), 4.63 (1H, d, J = 7.2 Hz) 4.90 (1H, s), 7.72 (1H, s); 13C NMR (CD3OD) δ 58.1, 58.3, 62.7,73.1, 88.3, 92.1, 125.5, 138.9, 147.3, 153.3; IR (KBr) 3381.3, 2926.7, 1578.6, 1056.3 cm−1; EI-MS (m/z): 272[M]+; HRMS Calcd for C10H13ClN4O3: 272.0676, Found: 272.0673.
((1S*,4R*,5S*)-4-(6-Chloro-9H-purin-9-yl)-2,6-dioxa-bicyclo[3.2.0]heptan-1-yl)methanol (24). To a solutionof 15 (18 mg, 0.07 mmol) in DMF (0.5mL), were added orthoethyl formate (0.7 mL, 4.21 mmol)and conc HCl (2 μL, 0.024 mmol) at 0 ◦C. After the mixture was stirred at room temperature, thesolvents were removed under reduced pressure. The residue was dissolved in 0.5 M aqHCl (1 mL)and the mixture was stirred at room temperature for 1 h. The mixture was neutralized with 1MaqNaOH (0.5 mL) and concentrated under reduced pressure. The residue was extracted with asolution of chloroform–methanol = 1:1. After the insoluble materials were removed by filtration, thesolvents were removed under reduced pressure. The residue was purified by pTLC (developed bychloroform-methanol = 5:1) to give 24 (14.9 mg, 81%). 1H-NMR (CDCl3) δ 4.12 (2H, q, J = 12.6 Hz),4.59 (1H, d, J = 11.6 Hz), 4.66 (1H, d, J = 9.7 Hz), 4.68 (1H, d, J = 9.7 Hz), 4.66 (1H, d, J = 9.7 Hz), 4.90(1H, dd, J = 11.1, 4.8 Hz), 5.24 (1H, s), 5.38 (1H, d, J = 4.4 Hz), 8.68 (1H, s), 8.76 (1H, s); 13C-NMR(CDCl3-CD3OD = 19 : 1) δ 58.6, 61.2, 71.9, 77.2, 87.8, 90.9, 130.7, 144.6, 150.7, 151.2, 151.8; IR (KBr)3401.4, 2931.1, 1597.4, 1567.4, 1056.6 cm−1; EI-MS (m/z): 282 [M]+; HRMS Calcd for C11H11ClN4O3:282.0520, Found: 282.0506.
((1S*,4R*,5S*)-4-(6-Amino-9H-purin-9-yl)-2,6-dioxa-bicyclo[3.2.0]heptan-1-yl)methanol (6). Compound 24
(29.4 mg, 0.10 mmol) was dissolved in sat. methanolic ammonia (7 mL) and the mixture was kept at 100◦C for 21 h in a glass sealed tube. After the solvents were removed under reduced pressure, the residuewas purified by pTLC (developed by chloroform–methanol = 5:1) to give 6 (18.8 mg, 69%). 1H-NMR(CDCl3-CD3OD = 17:3) δ 3.91 (1H, d, J = 12.6 Hz), 4.00 (1H, d, J = 12.1 Hz), 4.57 (1H, d, J = 10.6 Hz),4.64 (1H, d, J = 7.7 Hz), 4.70 (1H, d, J = 7.7 Hz), 4.87 (1H, dd, J = 11.1, 4.8 Hz), 5.19 (1H, s), 5.25 (1H, d,J = 4.8 Hz), 8.25 (1H, s), 8.28 (1H, s); 13C-NMR (CDCl3–CD3OD = 17:3) δ 29.5, 58.3, 61.3, 72.1, 87.6, 91.0,118.2, 139.1, 148.9, 152.6, 155.3; IR (KBr) 3192.2, 2409.9, 1660.5, 1615.0, 1054.9 cm−1; EI-MS (m/z): 263[M]+; HRMS Calcd for C11H13N5O3: 263.1018, Found: 263.1021.
4. Conclusions
We constructed an isoadenosine derivative on a 2,6-dioxobicyclo[3.2.0]heptane scaffold. Sinceour initial attempt to synthesize 6 by directly introducing the adenine moiety was not successful, wesynthesized it by the de novo synthesis of an adenine ring on a pseudosugar moiety. However, thisunique adenosine analogue showed no activity against HIV. Previously, we have reported that neitherthymine nor adenine analogues 4 built on a 2-oxa-6-thiabicyclo[3.2.0]heptane skeleton inhibit HIV [14].The structural rigidities of these analogues and isoadenosine 6 due to the introduction of fused thietaneand oxetane rings, respectively, appear to inhibit anti-HIV activity. In particular, phosphorylation atthe 5'-hydroxyl group would be inhibited since deoxynucleoside kinase recognizes the puckering ofsugars [25]. Thus, we are currently preparing new substituted nucleoside derivatives based on 4 and 6,and the results will be reported elsewhere.
Acknowledgments: This work was supported in part by a Grant-in-Aid for Scientific Research (No. 24590143,Y.Y.) from JSPS and by a grant of Strategic Research Foundation Grant-aided Project for Private Universities fromMinistry of Education, Culture, Sport, Science, and Technology, Japan (MEXT), 2010–2014.
Author Contributions: YY and TI designed research; SK, HK and TS performed the synthesis of compounds andTI assayed anti-HIV activity. YY wrote the paper. All authors read and approved the final manuscript.
124

Molecules 2015, 20, 4623–4634
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Cihlar, T.; Ray, A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine.Antivir. Res. 2010, 85, 39–58. [CrossRef] [PubMed]
2. Mehellou, Y.; de Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where dowe go? J. Med. Chem. 2010, 53, 521–538. [CrossRef] [PubMed]
3. WHO. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIVInfection. Available online: http://www.who.int/hiv/pub/guidelines/arv2013/download/en/ (accessedon 30 June 2013).
4. Meadows, D.C.; Gervey-Hague, J. Current developments in HIV chemotherapy. ChemMedChem 2006, 1, 16–29.[CrossRef] [PubMed]
5. Imamichi, T. Action of anti-HIV drugs and resistance: Reverse transcriptase inhibitors and protease inhibitors.Curr. Pharm. Des. 2004, 10, 4039–4053. [CrossRef] [PubMed]
6. Mitsuya, H.; Broder, S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophicvirus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2',3'-dideoxynucleosides. Proc. Natl.Acad. Sci. USA 1986, 83, 1911–1915. [CrossRef] [PubMed]
7. Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.;Barry, D.W.; Broder, S. 3'-Azido-3'-deoxythymidine (BW A509U): An antiviral agent that inhibits theinfectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associatedvirus in vitro. Proc. Natl. Acad. Sci. USA 1985, 82, 7096–7100. [CrossRef] [PubMed]
8. Schinazi, R.F.; Chu, C.K.; Peck, A.; McMillan, A.; Mathis, R.; Cannon, D.; Jeong, L.S.; Beach, J.W.; Choi, W.B.;Yeola, S.; et al. Activities of the four optical isomers of 2',3'-dideoxy-3'-thiacytidine (BCH-189) against humanimmunodeficiency virus type 1 in human lymphocytes. Antimicrob. Agents Chemother. 1992, 36, 672–676.[CrossRef]
9. Balzarini, J.; Holy, A.; Jindrich, J.; Naesens, L.; Snoeck, R.; Schols, D.; de Clercq, E. Differentialantiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates:Potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 1993, 37, 332–338. [CrossRef] [PubMed]
10. Yoshimura, Y.; Yamazaki, Y.; Kawahata, M.; Yamaguchi, K.; Takahata, H. Design and synthesis of a novelring-expanded 4'-Thio-apio-nucleoside derivatives. Tetrahedron Lett. 2007, 48, 4519–4522. [CrossRef]
11. Yoshimura, Y.; Asami, K.; Matsui, H.; Tanaka, H.; Takahata, H. New synthesis of (±)-isonucleosides. Org. Lett.2006, 8, 6015–6018. [CrossRef] [PubMed]
12. Yoshimura, Y.; Yamazaki, Y.; Saito, Y.; Takahata, H. Synthesis of 1-(5,6-dihydro-2H-thiopyran-2-yl)uracil by aPummerer-type thioglycosylation reaction: The regioselectivity of allylic substitution. Tetrahedron 2009, 65,9091–9102. [CrossRef]
13. Yoshimura, Y.; Ohta, M.; Imahori, T.; Imamichi, T.; Takahata, H. A new entry to carbocyclic nucleosides:Oxidative coupling reaction of cycloalkenylsilanes with a nucleobase mediated by hypervalent iodinereagent. Org. Lett. 2008, 10, 3449–3452. [CrossRef] [PubMed]
14. Yoshimura, Y.; Asami, K.; Imamichi, T.; Okuda, T.; Shiraki, K.; Takahata, H. Design and synthesis ofisonucleosides constructed on a 2-oxa-6-thiabicyclo[3.2.0]heptane scaffold. J. Org. Chem. 2010, 75, 4161–4171.[CrossRef] [PubMed]
15. Yoshimura, Y.; Yamazaki, Y.; Saito, Y.; Natori, Y.; Imamichi, T.; Takahata, H. Synthesis of5-thiodidehydropyranylcytosine derivatives as potential anti-HIV agents. Bioorg. Med. Chem. Lett. 2011, 21,3313–3316. [CrossRef] [PubMed]
16. Kiran, Y.B.; Wakamatsu, H.; Natori, Y.; Takahata, H.; Yoshimura, Y. Design and synthesis of a nucleoside anda phosphonate analogue constructed on a branched-threo-tetrofuranose skeleton. Tetrahedron Lett. 2013, 54,3949–3952. [CrossRef]
17. Kan-no, H.; Saito, Y.; Omoto, S.; Minato, S.; Wakamatsu, H.; Natori, Y.; Imamichi, T.; Takahata, H.;Yoshimura, Y. Synthesis of a dihydropyranonucleoside using an oxidative glycosylation reaction mediatedby hypervalent iodine. Synthesis 2014, 46, 879–886. [CrossRef]
125

Molecules 2015, 20, 4623–4634
18. Chu, C.K.; Ahn, S.K.; Kim, H.O.; Beach, J.W.; Alves, A.J.; Jeong, L.S.; Islam, Q.; van Roey, P.; Schinazi, R.F.Asymmetric synthesis of enantiomerically pure (−)-(1'R,4'R)-dioxolane-thymine and its anti-HIV activity.Tetrahedron Lett. 1991, 32, 3791–3794. [CrossRef]
19. Kim, H.O.; Schinazi, R.F.; Nampalli, S.; Shanmuganathan, K.; Cannon, D.L.; Alves, A.J.; Jeong, L.S.;Beach, J.W.; Chu, C.K. 1,3-dioxolanylpurine nucleosides (2R,4R) and (2R,4S) with selective anti-HIV-1activity in human lymphocytes. J. Med. Chem. 1993, 36, 30–37. [CrossRef] [PubMed]
20. Kim, H.O.; Schinazi, R.F.; Shanmuganathan, K.; Jeong, L.S.; Beach, J.W.; Nampalli, S.; Cannon, D.L.; Chu, C.K.L-beta-(2S,4S)- and L-alpha-(2S,4R)-dioxolanyl nucleosides as potential anti-HIV agents: Asymmetricsynthesis and structure-activity relationships. J. Med. Chem. 1993, 36, 519–528. [CrossRef]
21. Choi, Y.; George, C.; Comin, M.J.; Brachi, J.J.; Kim, H.S.; Jacobsen, K.A.; Balzarini, J.; Mitsuya, H.; Boyer, P.L.;Hughes, S.H.; et al. A conformationally locked analogue of the anti-HIV agent stavudine. An importantcorrelation between pseudorotation and maximum amplitude. J. Med. Chem. 2003, 46, 3292–3299. [CrossRef][PubMed]
22. D’Alonzo, D.; van Aerschot, A.; Guaragna, A.; Palumbo, G.; Schepers, G.; Capone, S.; Rozenski, J.;Herdewijn, P. Synthesis and base pairing properties of 1',5'-anhydro-L-hexitol nucleic acids (L-HNA).Chem. Eur. J. 2009, 15, 10121–10131. [CrossRef] [PubMed]
23. Bera, S.; Nair, V. A new general synthesis of isomeric nucleosides. Tetrahedron Lett. 2001, 42, 5813–5815.[CrossRef]
24. Quadrelli, P.; Piccanello, A.; Mella, M.; Corsaro, A.; Pistarà, V. From cyclopentadiene to isoxazoline-carbocyclic nucleosides: A rapid access to biological molecules through aza-Dielse Alder reactions.Tetrahedron 2008, 64, 3541–3547. [CrossRef]
25. Comin, M.J.; Vu, B.C.; Boyer, P.L.; Liao, C.; Hughes, S.H.; Marquez, V.E. D-(+)-iso-methanocarbathymidine:A high-affinity substrate for herpes simplex virus 1 thymidine kinase. ChemMedChem 2008, 3, 1129–1134.[CrossRef]
Sample Availability: Sample Availability: Sample of the final compound is available from the authors. About theother compounds, please contact the authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
126

molecules
Article
Synthesis of Peptide Nucleic Acids Containinga Crosslinking Agent and Evaluation ofTheir Reactivities
Takuya Akisawa, Yuki Ishizawa and Fumi Nagatsugi *
Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku,Sendai-shi, Miyagi 980-8577, Japan; [email protected] (T.A.); [email protected] (Y.I.)* Correspondence should be addressed; [email protected]; Tel./Fax: +81-22-217-5633.
Academic Editor: Mahesh K. LakshmanReceived: 26 January 2015; Accepted: 9 March 2015; Published: 13 March 2015
Abstract: Peptide nucleic acids (PNAs) are structural mimics of nucleic acids that form stablehybrids with DNA and RNA. In addition, PNAs can invade double-stranded DNA. Due to thesecharacteristics, PNAs are widely used as biochemical tools, for example, in antisense/antigenetherapy. Interstrand crosslink formation in nucleic acids is one of the strategies for preparing astable duplex by covalent bond formation. In this study, we have synthesized PNAs incorporating4-amino-6-oxo-2-vinylpyrimidine (AOVP) as a crosslinking agent and evaluated their reactivities fortargeting DNA and RNA.
Keywords: peptide nucleic acid (PNA); crosslink; antisense; invasion
1. Introduction
Peptide nucleic acids (PNAs) are synthetic nucleic acid analogs, in which the sugar phosphatebackbone is replaced with N-(2-aminoethyl)glycine [1] (Figure 1). The hybrid complexes between PNAand DNA or RNA exhibit an extraordinary thermal stability due to the lack of a negatively-chargedbackbone. Additionally, PNAs are chemically stable in comparison to DNA over a wide range oftemperatures and pH values and are resistant to nucleases and proteases [2]. These characteristics of thePNAs provide a variety of applications in therapeutic approaches [3–5], including use as biochemicaltools [6]. The PNA was used as an antisense to interfere with dimerization of the HIV-1 RNA transcriptand to inhibit the template switching process in HIV-1 [7]. Many modifications have been madeto the PNA backbone [8] and the nucleobases [9–11] for improving PNA’s properties, such as theircellular uptake and solubility. GPNA is a backbone-modified PNA constructed of guanidine and is cellpermeable. The GPNA designed to target the epidermal growth factor receptor (EGFR) induced potentantitumor effects due to its antisense effect [12]. A higher cellular uptake of the PNA was also achievedby conjugation with cysteine and lysine at the N-terminal position, which exhibited the anti-miRNAeffect for miR-122 [13]. The most remarkable property of PNAs is their ability to invade the secondarystructure of nucleic acids, for example, the DNA duplex [14] and the G-quadruplex [15]. This invasionability of the PNAs raises the possibility their use to control gene expression at the DNA level withhigh efficiency. Strand invasion requires a high stability of the PNA-DNA complex due to competitionfrom the displaced DNA strand. One strategy for increasing the stability of the PNA-DNA complex isby forming a covalent bond between the PNA and the target DNA.
Molecules 2015, 20, 4708–4719 127 www.mdpi.com/journal/molecules
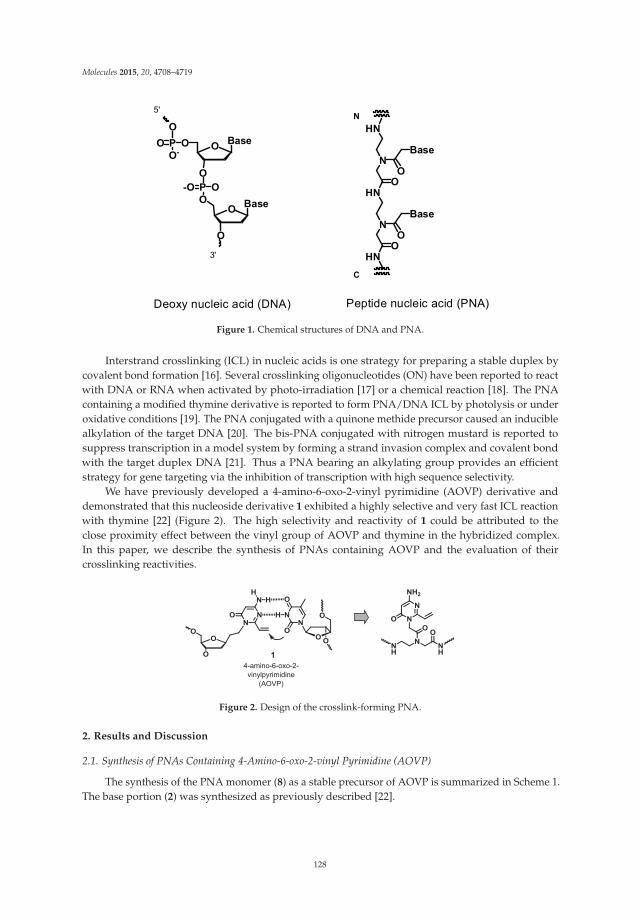
Molecules 2015, 20, 4708–4719
BaseOOPOO-
O
OP-OO
O
O
O
Base
HN
N
HN
N
HN
O
Base
O
O
Base
O
5'
3'
N
C
Deoxy nucleic acid (DNA) Peptide nucleic acid (PNA)
Figure 1. Chemical structures of DNA and PNA.
Interstrand crosslinking (ICL) in nucleic acids is one strategy for preparing a stable duplex bycovalent bond formation [16]. Several crosslinking oligonucleotides (ON) have been reported to reactwith DNA or RNA when activated by photo-irradiation [17] or a chemical reaction [18]. The PNAcontaining a modified thymine derivative is reported to form PNA/DNA ICL by photolysis or underoxidative conditions [19]. The PNA conjugated with a quinone methide precursor caused an induciblealkylation of the target DNA [20]. The bis-PNA conjugated with nitrogen mustard is reported tosuppress transcription in a model system by forming a strand invasion complex and covalent bondwith the target duplex DNA [21]. Thus a PNA bearing an alkylating group provides an efficientstrategy for gene targeting via the inhibition of transcription with high sequence selectivity.
We have previously developed a 4-amino-6-oxo-2-vinyl pyrimidine (AOVP) derivative anddemonstrated that this nucleoside derivative 1 exhibited a highly selective and very fast ICL reactionwith thymine [22] (Figure 2). The high selectivity and reactivity of 1 could be attributed to theclose proximity effect between the vinyl group of AOVP and thymine in the hybridized complex.In this paper, we describe the synthesis of PNAs containing AOVP and the evaluation of theircrosslinking reactivities.
1 4-amino-6-oxo-2-vinylpyrimidine
(AOVP)
Figure 2. Design of the crosslink-forming PNA.
2. Results and Discussion
2.1. Synthesis of PNAs Containing 4-Amino-6-oxo-2-vinyl Pyrimidine (AOVP)
The synthesis of the PNA monomer (8) as a stable precursor of AOVP is summarized in Scheme 1.The base portion (2) was synthesized as previously described [22].
128

Molecules 2015, 20, 4708–4719
NH
N
NH2
O Br NH
N
NH2
O SC8H17N
N
NH2
O SC8H17O
OtBu
N
N
NHPac
O SC8H17O
OtBu
N
N
NHPac
O SC8H17O
HO
FmocHNHN
O
OtBu
FmocHN NO
OtBu
N
N
NHPac
O SC8H17O CH2Cl2, rt., 2 h,
93%
PacCl
HBTU, HOBt, DIPEA
2 3 4
5
6
LiH,tert-Butyl bromoacetateDMF/dioxanert., 2 h, 85%
dry-pyridinert., 90 min, 93%
CH2Cl2rt., 2 h
1)Tributylvinyl Tin,(PPh3)2PdCl2DMF, 100 oC, 2 h
2)C8H17SH 25 oC, 1 day 58% (2 steps)
TFA
DMF, rt., 1.5 h, 87%
FmocHN NO
OH
N
N
NHPac
O SC8H17O
TFA
87
Scheme 1. Synthesis of the AOVP PNA monomer.
The N1 position of 2 was alkylated with t-butyl bromoacetate to yield the t-butyl ester 3 in 85%yield. The 4-amino group of 3 was protected with a phenoxyacetyl group, and subsequent deprotectionof the t-butyl group with trifluoroacetic acid (TFA) afforded the acid 5, which was coupled with theFmoc/t-Bu PNA backbone 6 using HBTU/HOBT/DIPEA to form the monomer 7, which was thendeprotected of the t-butyl ester by treatment with TFA to produce the PNA monomer 8.
We designed the PNA sequence in order to target the template RNA included in the telomeraseribonucleoprotein based on a previous report, in which a quinone methide cross-linking agent wassuccessfully incorporated [20]. It was also reported that non-covalent binding of the PNA to thetemplate RNA of the telomerase efficiently inhibited the telomerase activity in cell cultures [23].Therefore, we expected that our crosslinking agent in PNA would further improve the inhibitoryactivity for the telomerase. In this study, we synthesized two sequences that contained AOVP in themiddle position or in the N-terminal position.
Generally, the C- or N-terminal position of the PNA is conjugated with lysine (Lys) to increaseits solubility in water. Our previous study showed that the vinyl group of AOVP in the PNA causedan intramolecular reaction with the amino group of Lys conjugated at the C terminal position. In thisstudy, glycine (Gly) and two arginines (Arg) were conjugated with the PNA at the C-terminal position.The solid-phase synthesis of the PNA oligomer was manually performed on the NovaSin TGR resinusing Fmoc strategies. The N-terminal position of the PNA probe was modified with an acetyl group toavoid side reactions between the vinyl group in AOVP and the amino group at the N-terminal position.The resin was treated with potassium carbonate (K2CO3) in MeOH for the deprotection of the phenoxyacetyl group. Each synthesized PNA was cleaved from the resin using water/triisopropylsilane/TFA(2.5%/2.5%/95%) and purified by reverse phase HPLC to give the desired PNA1 (Scheme 2).
The sulfide-protected PNA1 was smoothly converted to PNA2 by oxidation with magnesiummonoperoxyphthalate (MMPP), followed by treatment with an aqueous NaOH solution to generatethe vinyl group of PNA3, as shown in Figure 3.
The structure of PNA3 was confirmed by MALDI-TOF mass spectra measurements. The presenceof the vinyl group in PNA3 was further supported by the fact that the treatment of PNA3 with anaqueous NaSMe solution generated the sulfide derivatives PNA4.
129

Molecules 2015, 20, 4708–4719
Scheme 2. Synthesis of the PNAs containing the stable precursors of AOVP.
MMPP, 0.8 eq
30 min
NaOH MeS- Na+
Time (min.)0 10 20 30
Time (min.)0 10 20 30
Time (min.)0 10 20 30
Time (min.)0 10 20 30
PNA2a: R1=G3T2AG, R2=Ac-GT2PNA2b: R1=GT2AG3T2AG, R2=CH3
PNA1b PNA2b PNA3b PNA4b
PNA3a: R1=G3T2AG, R2=Ac-GT2PNA3b: R1=GT2AG3T2AG, R2=CH3
PNA4a: R1=G3T2AG, R2=Ac-GT2PNA4b: R1=GT2AG3T2AG, R2=CH3
Column: Nacalai Tesque COSMOSIL 5C18-AR (4.6 250 mm)Solvent: A: H2O containing 0.1% TFA B: CH3CN containing 0.1% TFA, B conc. 10% to 60% for 50 min, UV-monitor: 254 nm, column heater: 50 C.
PNA1a: R1=G3T2AG, R2=Ac-GT2PNA1b: R1=GT2AG3T2AG, R2=CH3
Figure 3. Synthesis of the PNAs containing AOVP and analysis of these reactions by HPLC.
2.2. Evaluation of the Crosslinking Reactivity of the PNA Containing AOVP
The crosslinking reaction was investigated under neutral conditions using the reactive PNA3 andthe target DNA (Y = dG, dA, dC, dT) or RNA (Y = rG, rA, rC, U) labelled with fluorescein at the 5' end.
After 24 h, the reaction mixtures were analyzed by gel electrophoresis with 20% denaturing gel.The crosslinked products were identified as the less mobile bands (Figure 4). The crosslink yieldswere calculated from the ratio of the crosslinked product to the total amount of the remaining singlestranded DNA1/RNA1 and crosslinked product. PNA3a, which contained AOVP in the middleposition, did not produced significant adducts to any targets (Figure 4A,B). Conversely the crosslinkproduct was observed in the reaction between PNA3b containing AOVP at the terminal positionand DNA2 (Y=T) (Figure 4C). The crosslinking reaction of PNA3b did not efficiently occur with anRNA target (Figure 4D). The lower mobility bands in the reaction between PNA3b and DNA2 (Y=T)was extracted from the denaturing gel and proved to be the crosslinked adduct by MALDI-TOF MSmeasurements (calcd.10,071.8 found 10,072.3).
130
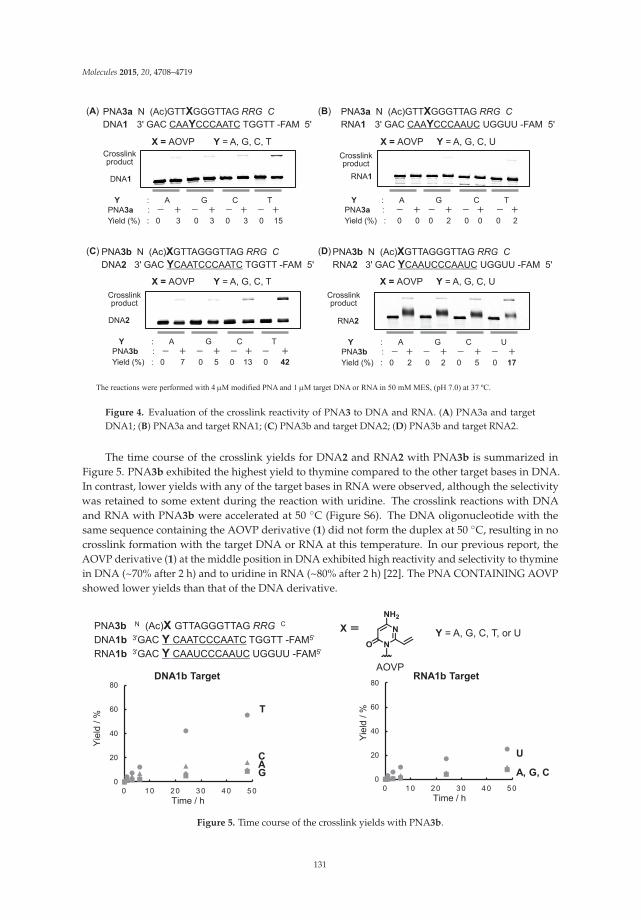
Molecules 2015, 20, 4708–4719
Y : A G C TPNA3a : Yield (%) : 0 3 0 3 0 3 0 15
Crosslink product
DNA1
PNA3a N (Ac)GTTXGGGTTAG RRG CDNA1 3' GAC CAAYCCCAATC TGGTT -FAM 5'
Y = A, G, C, TX = AOVP
PNA3a N (Ac)GTTXGGGTTAG RRG CRNA1 3' GAC CAAYCCCAAUC UGGUU -FAM 5'
Y = A, G, C, UX = AOVP
Y : A G C TPNA3a : Yield (%) : 0 0 0 2 0 0 0 2
RNA1
Crosslink product
PNA3b N (Ac)XGTTAGGGTTAG RRG CDNA2 3' GAC YCAATCCCAATC TGGTT -FAM 5'
Y : A G C TPNA3b : Yield (%) : 0 7 0 5 0 13 0 42
Crosslink product
DNA2
PNA3b N (Ac)XGTTAGGGTTAG RRG CRNA2 3' GAC YCAAUCCCAAUC UGGUU -FAM 5'
Y : A G C UPNA3b : Yield (%) : 0 2 0 2 0 5 0 17
RNA2
Crosslink product
Y = A, G, C, TX = AOVP Y = A, G, C, UX = AOVP
(A) (B)
(C) (D)
The reactions were performed with 4 M modified PNA and 1 M target DNA or RNA in 50 mM MES, (pH 7.0) at 37 ºC.
Figure 4. Evaluation of the crosslink reactivity of PNA3 to DNA and RNA. (A) PNA3a and targetDNA1; (B) PNA3a and target RNA1; (C) PNA3b and target DNA2; (D) PNA3b and target RNA2.
The time course of the crosslink yields for DNA2 and RNA2 with PNA3b is summarized inFigure 5. PNA3b exhibited the highest yield to thymine compared to the other target bases in DNA.In contrast, lower yields with any of the target bases in RNA were observed, although the selectivitywas retained to some extent during the reaction with uridine. The crosslink reactions with DNAand RNA with PNA3b were accelerated at 50 ◦C (Figure S6). The DNA oligonucleotide with thesame sequence containing the AOVP derivative (1) did not form the duplex at 50 ◦C, resulting in nocrosslink formation with the target DNA or RNA at this temperature. In our previous report, theAOVP derivative (1) at the middle position in DNA exhibited high reactivity and selectivity to thyminein DNA (~70% after 2 h) and to uridine in RNA (~80% after 2 h) [22]. The PNA CONTAINING AOVPshowed lower yields than that of the DNA derivative.
0
20
40
60
80
0 10 20 30 40 50
T
CAG
Time / h
Yiel
d / %
0
20
40
60
80
0 10 20 30 40 50Time / h
U
A, G, C
Yiel
d / %
PNA3b N (Ac)X GTTAGGGTTAG RRG C
DNA1b 3'GAC Y CAATCCCAATC TGGTT -FAM5'
RNA1b 3'GAC Y CAAUCCCAAUC UGGUU -FAM5'
Y = A, G, C, T, or U
AOVP
X
DNA1b Target RNA1b Target
Figure 5. Time course of the crosslink yields with PNA3b.
131

Molecules 2015, 20, 4708–4719
Table 1. Melting temperatures of PNA4 with DNA and RNA.
PNA4a PNA4b
Target Tm (◦C) ΔTm/◦C Target Tm (◦C) ΔTm/◦C
DNA1 (Y=G) 69.7 −7.6 DNA2 (Y=G) 79.2 −1.1DNA1 (Y=A) 68.0 −9.3 DNA2 (Y=A) 79.2 −1.1DNA1 (Y=C) 66.1 −11.2 DNA2 (Y=C) 78.6 −1.7DNA1 (Y=T) 68.1 −9.2 DNA2 (Y=T) 77.9 −2.4RNA1 (Y=G) 71.3 −11.7 RNA2 (Y=G) 83.2 −1.3RNA1 (Y=A) 72.9 −10.1 RNA2 (Y=A) 84.1 −0.4RNA1 (Y=C) 69.1 −13.9 RNA2 (Y=C) 83.2 −1.3RNA1 (Y=U) 69.1 −13.9 RNA2 (Y=U) 82.9 −1.6
UV melting profiles measured using 1.5 μM each of the strands in 50 mM MES buffer, pH 7.0; PNA5a:GTTAGGGTTAG-RRG, PNA5b AGTTAGGGTTAG-RRG; Tm (◦C) for PNA5a/DNA1 (Y=T): 77.3, PNA5a/RNA1(Y=U): 83.0, PNA5b/DNA2 (Y=T): 80.3, PNA5a/DNA2 (Y=U): 84.5.
To explain the difference in the crosslink yields, the thermal stability was estimated by the meltingtemperature (Tm) of the duplex between PNA4 containing the stable precursor of AOVP and thetarget DNA or RNA. The Tm values for PNA4a/DNA1 or RNA1 were 8–14 ◦C lower than thoseof PNA5 containing adenosine instead of AOVP (Table 1). Conversely, no significant difference inthe Tm values was observed between PNA4b/DNA, RNA and PNA5b/DNA, RNA. These resultsindicated that the incorporation of AOVP in the middle site may destabilize the duplex and that theincorporation of AOVP at the terminal position does not affect the thermal stability of the PNA/DNA,RNA hetero-duplex. It may be assumed that the lower reactivity of PNA3a might be attributed to thelower stability of the duplex. However, despite a similar stability of the duplexes formed with PNA3b
or RNA, the reactivity of PNA3b with DNA was higher than that with RNA. Accordingly, the lowerthermal stability of the duplex might not be the major cause of the lower reactivity of PNA containingAOVP. As the crosslink reactivity of AOVP is highly dependent on the close proximity between thereactants, a small difference in the local structure around AOVP and the target base might become amajor factor of the reactivity (Figure S7). Further study about the local structure of the PNA containingAOVP is also needed for a rational explanation of the higher reactivity of PNA3b with DNA than thatwith RNA.
3. Experimental Section
3.1. General
All air-sensitive reactions were carried out under argon in oven-dried glassware using standardsyringe and septa techniques, unless otherwise noted. The 1H- and 13C-NMR spectra (500 MHz for 1Hand 125 MHz for 13C) were recorded on a Bruker spectrometer (Bruker BioSpin K.K., Kanagawa, Japan).Chemical shifts (δ) are reported in parts per million (ppm) and are referenced to the solvent CDCl3(7.26 ppm), DMSO (2.50 ppm), for 1H-NMR and CDCl3 (77.16 ppm), DMSO (39.52 ppm) for 13C-NMR.Multiplicity and qualifier abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet,quint. = quintet, m = multiplet, br = broad. Coupling constants (J) are reported in hertz (Hz). Highresolution mass spectra were obtained using electrospray ionization (ESI). MALDI-TOF mass spectrawere measured by using an Autoflex Speed mass spectrometer (Bruker Daltonics, Kanagawa, Japan)with the laser at 337 nm in negative or positive mode using 3-hydroxypicolinic acid or gentisic acid asthe matrix. Thin-layer chromatography was performed on Merck 60 F254 pre-coated silica gel plates(EMD Millipore Co., Darmstadt, Germany). Merck 60 F254 pre-coated silica gel on glass in a thicknessof 0.9 mm was used for preparative TLC. Column chromatography was performed on silica gel (SilicaGel 60 N; 40–100 μm, or 100–210 μm, Kanto Chemical Co., Inc, Tokyo, Japan). The ultraviolet-visible(UV-vis) absorption spectra were recorded on a Beckman Coulter DU800 (Beckman Coulter K.K., Tokyo,Japan). PNA synthesis was carried out by Fmoc-SPPS. High performance liquid chromatography
132

Molecules 2015, 20, 4708–4719
(HPLC) was performed using Nacalai Tesque (Kyoto, Japan) Cosmosil 5C18AR (4.6 or 10 × 250 mm)as the column, a JASCO PU-2089 as the pump, JASCO 2075 for the UV monitoring, and JASCO 2067as the column oven (all Jasco Co., Tokyo, Japan). pH measurements were measured performed witha Mettler Toledo (Schwerzenbaha, switzerland) Seven Easy pH meter using a 8220BNWP electrode(Thermo Scientific, Waltham, MA, USA). UV-melting experiments were performed on a BeckmanCoulter DU800. Densitometric analysis of the gel was carried out on 20% denaturing polyacrylamidegel plates, and visualized and quantified using a FLA-5100 Fluor Imager (Fujifilm Co., Tokyo, Japan).Commercially available reagents were obtained from Wako Pure Chemical Industries Ltd. (Osaka,Japan) or Kanto Chemical Co., Inc. and used without further purification. DNA and RNA oligomerswere purchased from Japan Bio Services Co., Ltd. (Saitama, Japan), buffers and salts were fromNacalai Tesque.
3.2. Chemistry
3.2.1. Synthesis of 1-(tert-Butoxycarbonylmethyl)-4-amino-6-oxo-2-(2-octylthioethyl)pyrimidine (3)
Compound 2 (300 mg, 1.1 mmol) and lithium hydride (84 mg, 11 mmol) were dissolved in drydioxane (20 mL) and dry DMF (10 mL), and the reaction mixture was stirred at room temperature.After 1.5 h, tert-butyl bromoacetate (233 μL, 1.6 mmol) was added and stirred at room temperature.After 1.5 h, the reaction mixture was diluted with 50 mL EtOAc and washed with sat. NH4Cl aq.(50 mL), and brine (50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated underreduced pressure. The residue was purified by column chromatography (hexane/EtOAc = 1:1) to give3 as a colorless oil (362 mg, 0.91 mmol 87%). 1H-NMR (CDCl3): δ 5.35 (s, 1H), 4.69 (s, 2H), 4.67 (s, 2H),2.92–2.89 (m, 2H), 2.80–2.76 (m, 2H), 2.53 (t, J = 7.5 Hz, 2H), 1.59 (quin, J = 7.5Hz, 2H), 1.48 (s, 9H),1.38–1.27 (m, 10H), 0.88 (t, J = 7.5 Hz, 3H). 13C-NMR (CDCl3): δ 167.1, 162.8, 161.3, 160.3, 85.5, 83.1,44.5, 35.2, 32.7, 31.9, 29.8, 29.3, 29.0, 28.5, 28.1, 22.7, 14.2. HRMS (ESI-MS) m/z calcd for C20H35N3O3S[M+H]+ 398.24719, found 398.24711.
3.2.2. Synthesis of 1-(tert-Butoxycarbonylmethyl)-4-phenoxyacetylamino-6-oxo-2-(2-octylthioethyl)pyrimidine (4)
To a solution of 3 (130 mg, 0.33 mmol) in dry pyridine (5 mL) was added phenoxyacetyl chloride(68 μL, 4.9 mmol), and the reaction mixture was stirred at room temperature. After 1.5 h, the reactionmixture was diluted with 50 mL EtOAc and washed with water (50 mL), and brine (50 mL). Theorganic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residuewas purified by column chromatography (CH2Cl2/EtOAc=9:1 to 4:1) to give 4 as a white solid (169 mg,0.32 mmol, 93%). 1H-NMR (CDCl3): δ 8.49 (s, 1H), 7.37–7.33 (m, 2H), 7.25 (s, 1H), 7.08–7.05 (m, 1H),7.00–6.98 (m, 2H), 4.70 (s, 2H), 4.59 (s, 2H), 2.93–2.90 (m, 2H), 2.85–2.82(m, 2H), 2.54 (t, J = 7.5 Hz, 2H),1.59 (quin, J = 7.5Hz, 2H), 1.48 (s, 9H), 1.39–1.26 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C-NMR (CDCl3):δ 167.3, 166.4, 162.9, 160.5, 157.0, 152.8, 130.1, 122.8, 115.1, 97.0, 83.5, 67.7, 45.0, 35.1, 32.8, 31.9, 29.8,29.3, 29.0, 28.5, 28.1, 22.7, 14.2. HRMS (ESI-MS) m/z calcd for C28H41N3O5S [M+H]+ 532.28397, found532.28398.
3.2.3. Synthesis of 1-(Carbonylmethyl)-4-phenoxyacetylamino-6-oxo-2-(2-octylthioethyl)pyrimidine (5)
To a solution of 4 (200 mg, 0.38 mmol) in CH2Cl2 (1 mL) was added TFA (9 mL) and the reactionmixture was stirred for 1 h at room temperature. The solvent was removed under reduced pressure andthe residue was diluted with 5 mL diethyl ether. The solution was added to hexane (30 mL) to causeprecipitation. The resulting precipitates were collected by filtration and washed with hexane/diethylether (9:1) to give a pure sample of 5 as a white powder (131 mg, 0.28 mmol, 89%). 1H-NMR (DMSO):δ 10.4 (s, 1H), 7.30 (t, J = 8.0 Hz, 2H), 6.98–6.92 (m, 3H), 6.86 (s, 1H), 4.81 (s, 2H), 4.75 (s, 2H), 2.96 (t,J = 7.0 Hz, 2H), 2.88 (t, J = 7.0 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.52 (quin, J = 7.5 Hz, 2H), 1.34–1.24 (m,
133

Molecules 2015, 20, 4708–4719
10H), 0.85 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO): δ 169.1, 168.3, 162.1, 161.0, 157.7, 153.8, 129.5, 121.1,114.4, 94.7, 66.6, 44.3, 34.2, 31.2, 31.1, 29.0, 28.6, 28.5, 28.2, 27.3, 22.0, 13.9. HRMS (ESI-MS) m/z calcd forC24H33N3O5S [M+H]+ 476.22137, found 476.22136.
3.2.4. Synthesis of tert-Butyl-N-[2-(N-9-fluorenylmethoxycarbonyl)aminoethyl]-N-[[4-phenoxyacetyl-amino-6-oxo-2-(2-octylthioethyl)pyrimidin-1-yl]acetyl]glycinate (7)
Compound 5 (100 mg, 0.21 mmol) was dissolved in dry DMF (5 mL). HBTU (159 mg, 0.42 mmol),HOBt (57 mg, 0.42 mmol), and DIPEA (146 μL, 0.84 mmol) were added and stirred for 5 min at roomtemperature. The reaction mixture was added to a solution of 6 (82 mg, 0.21 mmol) in dry DMF(5 mL) and stirred at room temperature. After 1.5 h, the reaction mixture was diluted with 50 mLEtOAc, and washed with sat.NaHCO3 (2 × 50 mL) and brine (50 mL). The organic layer was driedover Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by columnchromatography (hexane/EtOAc=2:1) to give 7 as a white foam (140 mg, 0.16 mmol, 87%). 1H-NMR(DMSO): δ 10.42 (s, 1H), 7.81 (d, J = 7.5 Hz, 2H), 7.68 (t, J = 7.0 Hz, 2H), 7.44–7.39 (m, 2H), 7.33–7.29(m, 4H), 6.96–6.92 (m, 3H), 6.85 (s, 1H), 5.07 and 4.86 (br s, 2H), 4.81 (s, 2H), 4.35–4.20 (m, 4H), 3.95(s, 1H), 3.51–3.12 (m, 4H), 2.86 (br s, 4H), 1.48–1.39 (m, 11H), 1.29–1.20 (m, 10H), 0.84–0.82 (m, 3H).13C-NMR (DMSO): δ 168.6, 168.3, 168.2, 167.9, 166.9, 166.6, 162.0, 161.50, 161.45, 157.7, 156.4, 156.1,153.7, 143.8, 142.6, 140.7, 139.4, 137.4, 129.5, 128.9, 127.6, 127.2, 127.0, 125.1, 121.3, 121.1, 120.1, 120.0,114.4, 109.6, 94.6, 82.0, 80.9, 66.6, 65.6, 65.4, 50.0, 49.0, 47.3, 46.7, 43.7, 43.3, 38.0, 34.0, 31.2, 31.09, 31.05,29.0, 28.6, 28.5, 28.24, 28.21, 27.6, 27.4, 27.3, 22.0, 13.9. HRMS (ESI-MS) m/z calcd for C47H59N5O8S[M+Na]+ 876.39766, found 876.39760.
3.2.5. Synthesis of N-[2-(N-9-Fluorenylmethoxycarbonyl)aminoethyl]-N-[[4-phenoxyacetyl-amino-6-oxo-2-(2-octylthioethyl)pyrimidin-1-yl]acetyl]glycine (8)
To a solution of 7 (40 mg, 47 μmol) in CH2Cl2 (1 mL) was added TFA (9 mL) and the resultingmixture was stirred at room temperature for 1.5 h. The solvent was removed and the residue wasdiluted with 5 mL diethyl ether. The solution was added to hexane (30 mL) to cause the precipitation.The resulting precipitates were collected by filtration and washed with hexane/diethyl ether (9/:1) togive a pure sample of 8 as a white powder (34 mg, 41 μmol, 93%). 1H-NMR (DMSO): δ 10.42 and 10.41(s, 1H), 7.88 (d, J = 7.5 Hz, 2H), 7.68 (t, J = 7.5 Hz, 2H), 7.46–7.39 (m, 2H), 7.33–7.39 (m, 4H), 6.98–6.92(m, 3 H), 6.85 (s, 1H), 5.06 and 4.89 (br s, 2H), 4.81 (s, 2H), 4.34–4.21 (m, 4H), 4.00 (s, 1H), 3.52–3.13 (m,4H), 2.85 (br s, 4H), 1.46–1.45 (m, 2H), 1.27–1.19 (m, 10H), 0.84–0.80 (m, 3H). 13C-NMR (DMSO): δ 170.9,170.2, 168.3, 167.0, 166.6, 162.0, 161.6, 161.5, 157.7, 156.4, 156.1, 153.7, 143.9, 140.7, 129.5, 127.6, 127.0,125.1, 121.1, 120.1, 114.4, 94.6, 66.6, 65.6, 65.5, 49.2, 47.9, 47.2, 46.7, 43.7, 43.4, 38.0, 34.04, 33.96, 31.2,31.12, 31.06, 29.0, 28.60, 28.57, 28.2, 27.44, 27.35, 22.0, 13.9. HRMS (ESI-MS) m/z calcd for C43H51N5O8S[M+Na]+ 820.33506, found 820.33508.
3.2.6. Synthesis of PNA1
Modified PNAs were synthesized on Novasyn TGR resin by the Fmoc-SPPS procedure. Afteracetylation of the N-terminal amino group, the resin was treated with 0.1 M K2CO3 in MeOH containing20% H2O for 3 h for deprotection of the Pac group in the precursor of AOVP. The resulting oligomer wascleaved from the resin by treatment with a cleaving cocktail containing water/triisopropylsilane/TFA(12.5:12.5:475 μL for 20 mg of resin) for 90 min. The crude mixture was eluted and precipitatedin diethyl ether, dissolved in water containing 0.1% TFA, purified by reversed-phase HPLC, andcharacterized by MALDI-TOF mass spectrometry. The concentration of each oligomer was determinedby UV absorption at 260 nm in water using the following extinction coefficients: 115,800 M−1 cm−1
(PNA1a), 131,000 M−1 cm−1 (PNA1b). MALDI-TOF MS (m/z) PNA1a: calcd 3647.5, found 3,647.4,PNA1b: calcd 3922.7, found 3922.3.
134

Molecules 2015, 20, 4708–4719
3.2.7. Synthesis of PNA3
To a solution of PNA1 (0.1 mM, 5 μL, 0.5 nmol) was added a solution of magnesiummonoperoxyphthalate (MMPP) (0.5 mM, 0.6 μL, 0.3 nmol) in carbonate buffer adjusted to pH 10at room temperature. After 30 min, a solution of NaOH (0.1 M, 0.5 μL, 50 nmol) was added, and themixture was left for an additional 1 h to give PNA3. MALDI-TOF MS (m/z) PNA3a: calcd 3500.4,found 3500.4, PNA3b: calcd 3776.6, found 3776.0.
3.2.8. Synthesis of PNA4
A solution of sodium thiomethoxide (1 M, 0.5 μL, 0.5 μmol) was added to the above mentionedmixture of PNA3. The mixture was left for 1h and purified by HPLC to give PNA4. MALDI-TOF MS(m/z) PNA4a: calcd 3549.4, found 3549.9, PNA4b: calcd 3824.6, found 3824.5.
3.3. General Procedure for the Crosslinking Reaction
The crosslinking reaction was performed with 4 μM of PNA3 and 1 μM of the target DNA orRNA labeled by fluorescein at the 5'-end in a buffer of 50 mM MES (pH 7.0). The reaction mixturewas incubated at 37 ◦C. An aliquot of the reaction mixture was collected at each indicated time andquenched by the addition of loading dye (formamide [~100% v/v], 0.5 M EDTA [20 μL: 1 mM], xylenecyanol [0.05% w/v], and bromophenol blue [0.05% w/v]). The cross-linked products were analyzed bydenaturing 20% polyacrylamide gel electrophoresis containing urea (7 M) with TBE buffer [PNA3a] ordenaturing 20% polyacrylamide gel electrophoresis containing urea (7 M) and 10% v/v formamide at50 ◦C with TBE buffer [PNA3b]. The labeled bands were visualized and quantified using a FLA-5100Fluor Imager.
3.4. Melting Temperature (Tm) Measurements
UV-melting experiments were performed on a Beckman Coulter DU800. For the hetero-duplexformation study, equimolar amounts of each oligonucleotide and PNA were dissolved in 50 mM MESbuffer (pH 7.0) to provide final concentrations of 1.5 μM. The solutions were heated to 90 ◦C for 5 minand allowed to cool to room temperature slowly. The melting profiles were recorded at 260 nm from20 to 90 ◦C at a scan rate of 1 ◦C/min.
4. Conclusions
In this study, we have synthesized PNAs containing AOVP as a crosslinking agent and evaluatedtheir reactivities toward the targets DNA and RNA. The PNA incorporating AOVP at the terminalposition exhibited a highly selective crosslinking reactivity to the thymine in DNA and lower reactivityto any bases in RNA. This PNA is expected to provide a new probe for sequence specific PNA-DNAinteractions for a regulation of the gene expression. The application of the PNA-incorporatedAOVP for crosslinking reactions with the duplex DNA by forming an invasion complex is currentlyunder investigation.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/03/4708/s1.
Acknowledgments: This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas(“Chemical Biology of Natural Products: Target ID and Regulation of Bioactivity”) and a Grant-in-Aid for ScientificResearch (B) from the Japan Society for the Promotion of Science (JSPS). This work was also supported in part bythe Management Expenses Grants National Universities Corporations from the Ministry of Education, Science,Sports and Culture of Japan (MEXT).
Author Contributions: F. Nagatsugi was responsible for supervision of the students, T. Akisawa and Y. Ishizawa.T. Akisawa and Y. Ishizawa performed the synthesis of the PNAs and the evaluation of their reactivities.F. Nagatsugi and T. Akisawa wrote the paper.
Conflicts of Interest: The authors declare no conflict of interest.
135

Molecules 2015, 20, 4708–4719
References
1. Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by StrandDispacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500. [CrossRef] [PubMed]
2. Demidov, V.V.; Potaman, V.N.; Frankkamenetskii, M.D.; Egholm, M.; Buchard, O.; Sonnichsen, S.H.;Nielsen, P.E. Stability of Peptide Nucleic-Acids in Human Serum and Cellular-Extracts. Biochem. Pharmacol.1994, 48, 1310–1313. [CrossRef] [PubMed]
3. Nielsen, P.E. Peptide Nucleic Acids (PNA) in Chemical Biology and Drug Discovery. Chem. Biodivers. 2010,7, 786–804. [CrossRef] [PubMed]
4. Nielsen, P.E. Targeted Gene Repair Facilitated by Peptide Nucleic Acids (PNA). Chembiochem 2010, 11,2073–2076. [CrossRef] [PubMed]
5. Nielsen, P.E. Gene Targeting and Expression Modulation by Peptide Nucleic Acids (PNA). Curr. Pharm. Des.2010, 16, 3118–3123. [CrossRef] [PubMed]
6. Shakeel, S.; Karim, S.; Ali, A. Peptide nucleic acid (PNA)—A review. J. Chem. Technol. Biotechnol. 2006, 81,892–899. [CrossRef]
7. Parkash, B.; Ranjan, A.; Tiwari, V.; Gupta, S.K.; Kaur, N.; Tandon, V. Inhibition of 5'-UTR RNA ConformationalSwitching in HIV-1 Using Antisense PNAs. PLoS One 2012, 7, e49310. [CrossRef] [PubMed]
8. Sugiyama, T.; Kittaka, A. Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycineBackbone. Molecules 2013, 18, 287–310. [CrossRef]
9. Xia, X.; Piao, X.; Bong, D. Bifacial Peptide Nucleic Acid as an Allosteric Switch for Aptamer and RibozymeFunction. J. Am. Chem. Soc. 2014, 136, 7265–7268. [CrossRef] [PubMed]
10. Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-formingpeptide nucleic acids for enhanced recognition of RNA duplexes. Nucl. Acids Res. 2014, 42, 4008–4018.[CrossRef] [PubMed]
11. Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A Simple Cytosine to G-ClampNucleobase Substitution Enables Chiral gamma-PNAs to Invade Mixed-Sequence Double-Helical B-formDNA. Chembiochem 2008, 9, 2388–2391. [CrossRef] [PubMed]
12. Thomas, S.M.; Sahu, B.; Rapireddy, S.; Bahal, R.; Wheeler, S.E.; Procopio, E.M.; Kim, J.; Joyce, S.C.;Contrucci, S.; Wang, Y.; et al. Antitumor Effects of EGFR Antisense Guanidine-Based Peptide NucleicAcids in Cancer Models. ACS Chem. Biol. 2013, 8, 345–352. [CrossRef] [PubMed]
13. Torres, A.G.; Fabani, M.M.; Vigorito, E.; Williams, D.; Al-Obaidi, N.; Wojciechowski, F.; Hudson, R.H.E.;Seitz, O.; Gait, M.J. Chemical structure requirements and cellular targeting of microRNA-122 by peptidenucleic acids anti-miRs. Nucleic Acids Res. 2012, 40, 2152–2167. [CrossRef] [PubMed]
14. Nielsen, P.E. Peptide nucleic acid. A molecule with two identities. Acc. Chem. Res. 1999, 32, 624–630.[CrossRef]
15. Gupta, A.; Lee, L.-L.; Roy, S.; Tanious, F.A.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Strand Invasion of DNAQuadruplexes by PNA: Comparison of Homologous and Complementary Hybridization. Chembiochem 2013,14, 1476–1484. [CrossRef] [PubMed]
16. Nagatsugi, F.; Sasaki, S. Synthesis of Reactive Oligonucleotides for Gene Targeting and Their Application toGene Expression Regulation. Bull.Chem. Soc. Jpn. 2010, 83, 744–755. [CrossRef]
17. Fujimoto, K.; Yamada, A.; Yoshimura, Y.; Tsukaguch, T.; Sakamoto, T. Details of the Ultrafast DNAPhoto-Cross-Linking Reaction of 3-Cyanovinylcarbazole Nucleoside: Cis-Trans Isomeric Effect and theApplication for SNP-Based Genotyping. J. Am. Chem. Soc. 2013, 135, 16161–16167. [CrossRef] [PubMed]
18. Op de Beeck, Madder, A. Unprecedented C-Selective Interstrand Cross-Linking through in Situ Oxidation ofFuran-Modified Oligodeoxynucleotides. J. Am. Chem. Soc. 2011, 133, 796–807.
19. Kim, Y.; Hong, I.S. PNA/DNA interstrand cross-links from a modified PNA base upon photolysis oroxidative conditions. Bioorg.Med. Chem. Lett. 2008, 18, 5054–5057. [CrossRef] [PubMed]
20. Liu, Y.; Rokita, S.E. Inducible Alkylation of DNA by a Quinone Methide-Peptide Nucleic Acid Conjugate.Biochemistry 2012, 51, 1020–1027. [CrossRef] [PubMed]
21. Zhilina, Z.V.; Ziemba, A.J.; Nielsen, P.E.; Ebbinghaus, S.W. PNA-nitrogen mustard conjugates are effectivesuppressors of HER-2/neu and biological tools for recognition of PNA/DNA interactions. BioconjugateChem. 2006, 17, 214–222. [CrossRef]
136

Molecules 2015, 20, 4708–4719
22. Hattori, K.; Hirohama, T.; Imoto, S.; Kusano, S.; Nagatsugi, F. Formation of highly selective and efficientinterstrand cross-linking to thymine without photo-irradiation. Chem. Commun. 2009, 45, 6463–6465.[CrossRef]
23. Hamilton, S.E.; Pitts, A.E.; Katipally, R.R.; Jia, X.Y.; Rutter, J.P.; Davies, B.A.; Shay, J.W.; Wright, W.E.;Corey, D.R. Identification of determinants for inhibitor binding within the RNA active site of humantelomerase using PNA scanning. Biochemistry 1997, 36, 11873–11880. [CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples of the compounds (2–4) are available from the authors. On theother compounds, please contact with the authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
137

molecules
Article
Synthesis and Biological Properties of5-(1H-1,2,3-Triazol-4-yl)isoxazolidines: A New Classof C-Nucleosides
Salvatore V. Giofrè 1,*, Roberto Romeo 1,*, Caterina Carnovale 1,†, Raffaella Mancuso 2,†,Santa Cirmi 1,†, Michele Navarra 1,†, Adriana Garozzo 3,† and Maria A. Chiacchio 4,†
1 Dipartimento Scienze del Farmaco e Prodotti per la Salute, Università di Messina, Via S.S. Annunziata,Messina 98168, Italy; [email protected] (C.C.); [email protected] (S.C.); [email protected] (M.N.)
2 Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Via P. Bucci, 12/C, Arcavacata diRende (CS) 87036, Italy; [email protected]
3 Dipartimento di Scienze Bio-mediche, Università di Catania, Via Androne 81, Catania 95124, Italy;[email protected]
4 Dipartimento Scienze del Farmaco, Università di Catania, Viale A. Doria 6, Catania 95125, Italy;[email protected]
* Correspondence: [email protected] (S.V.G.); [email protected] (R.R.); Tel.: +39-090-356230 (S.V.G. & R.R.);Fax: +39-090-676-6562 (S.V.G. & R.R)
† These authors contributed equally to this work.
Academic Editors: Mahesh K. Lakshman and Fumi NagatsugiReceived: 4 February 2015; Accepted: 13 March 2015; Published: 24 March 2015
Abstract: A novel series of C-nucleosides, featuring the presence of a 1,2,3-triazole ring linkedto an isoxazolidine system, has been designed as mimetics of the pyrimidine nucleobases. Anantiproliferative effect was observed for compounds 17a and 17b: the growth inhibitory effect reachesthe 50% in HepG2 and HT-29 cells and increases up to 56% in the SH-SY5Y cell line after 72 h ofincubation at a 100 μM concentration.
Keywords: vinyl triazoles; C-Nucleosides; 1,3-dipolar cycloaddition; antiproliferative activity; microwave
1. Introduction
Structural modification of natural N-nucleosides on either the sugar unit or the nucleobase hasled to the discovery of a variety of new therapeutic agents, which includes antiviral and anticanceragents [1–9]. In this context, the family of C-nucleosides constitutes a group of molecules characterizedby the link of the sugar unit to a carbon atom of the pyrimidine/purine nucleobase: some ofthese compounds have been reported to exhibit significant antibacterial, antiviral and antitumoralactivities [10].
While natural and synthetic N-nucleosides are vulnerable to enzymatic and acid-catalyzedhydrolysis of nucleosidic bond, the C-analogues are much more stable. Many natural and syntheticC-nucleosides, also containing modified heterocyclic bases, biologically active, have been described inliterature. Natural antibiotics formycins [11] (in particular FA, FB, and oxoformycin B OFB) have beenknown since the early 1960s to possess antibiotic and cytotoxic properties; their antiparasitic activity(antimalarial, antischisostoma, etc.) was unraveled later. Pseudouridine is the most abundant naturalC-nucleoside present in most tRNA and rRNA structures [12], where it has been shown to stabilizeRNA duplex [13,14]. Showdomycin [15] possesses well known antibiotic and cytotoxic properties,involving inhibition of nucleoside transport into the cell [16]. Pyrazofurin [17] and tiazofurin [18]have been shown to possess a wide range of medicinal properties, including antibiotic, antiviral, andantitumor activity (Figure 1).
Molecules 2015, 20, 5260–5275 138 www.mdpi.com/journal/molecules

Molecules 2015, 20, 5260–5275
Figure 1. Examples of C-Nucleosides.
As part of ongoing efforts to identify new antiviral/antitumor agents, we were interested inthe investigation of N,O-nucleosides, where the ribose moiety has been replaced by an isoxazolidinesystem, as mimetic of the sugar unit [19–23]. Phosphonated carbocyclic 2'-oxa-3'-azanucleosides 1 haveshown to be potent inhibitors of RT of different retroviruses, following incubation with human PBMCscrude extract [24–26]; truncated phosphonated azanucleosides 2 are able to inhibit HIV and HTLV-1viruses at concentration in the nanomolar range [27]; truncated phosphonated N,O-psiconucleosides 3
inhibit HIV in vitro infection with low or absent cytotoxicity (Figure 2) [28].
Figure 2. Phosphonated N,O-nucleosides.
New base-modified N,O-nucleosides have been also synthesized. N,O-pseudouridine 4 [29]and N,O-tiazofurin 5 [30] are characterized by a C-C bond between the isoxazolidine and theheterocyclic systems.
Recently, 1,2,3-triazolyl N,O-nucleosides have been designed: 3-hydroxymethyl-5-(1H-1,2,3-triazol)isoxazolidine 6 are able to inhibit proliferation of follicular and anaplastic human thyroid cancercell lines, with IC50 values ranging from 3.87 to 8.76 mM (Figure 3) [31]. In the same context, novel1,2,3-triazole-appended N,O-nucleoside analogs 7 were developed [32]: some of these compoundsshow a good anticancer activity against the follicular (FTC-133), the anaplastic (8305C) human thyroidcancer cell lines, and especially on the U87MG human primary glioblastoma cell line (Figure 3).
139

Molecules 2015, 20, 5260–5275
Figure 3. Examples of N,O-Nucleosides.
According to these promising results, we have developed a novel series of C-nucleosides featuredby the presence of a 1,2,3-triazole ring, linked to an isoxazolidine system, as mimetic of the pyrimidinenucleobases. We report in this paper the synthesis of 5-(1H-1,2,3-triazol-4-yl)isoxazolidines 8 and theirbiological activities as antiviral and/or antitumoral agents.
2. Results and Discussion
C-Vinyl triazoles 12a–g have been prepared according to Scheme 1 [33]. Thus, 3-butyn-1-ol 10 wasreacted with a variety of azides 9a–g, by click chemistry reactions, performed in H2O/tert-BuOH (1:1)in the presence of sodium ascorbate, copper(II) sulfate and TEA at room temperature. The obtainedtriazole derivatives have been tosylated and then converted into vinyl triazoles 12a–g by reaction withpotassium tert-butoxide in tert-butanol (78%–90% yields).
Scheme 1. Synthesis of C-Vinyl triazoles 12a–g. Reagents and conditions: (a) CuSO4·5H2O, sodiumascorbate, TEA, 4 h, rt; (b) tosyl chloride, TEA, CH2Cl2, rt, 12 h; (c) t-BuOK, t-BuOH, 40 ◦C, 12 h.
The 1,3-dipolar cycloaddition of 1-substituted-4-vinyl-1,2,3-triazoles 12a–g with C-[(tert-butyldiphenylsilyl)oxy]-N-methylnitrone 13 [34,35], at 150 W, 80 ◦C for 2 h in CHCl3, proceededwith a good yield and a complete regioselectivity to give a mixture of trans/cis isoxazolidines 14a–g
and 15a–g in a 1:1.3 relative ratio (global yield 80%–85%). Removal of the TBDPS protecting group wasaccomplished under standard conditions, by treating the diastereomeric mixture with TBAF in THF, toafford the triazolyl nucleosides 16a–f and 17a–f, which were separated by silica gel chromatography(Scheme 2, Table 1).
The diastereomeric ratio of the products was determined by 1H NMR spectroscopy of the crudereaction mixture, whereas the relative configuration was assigned by NOEDS spectra. In particular,in the cis derivative 17a, chosen as model compound, a positive NOE effect observed for H-4' andH-5'b (the downfield resonance of protons at C-5', 2.91 ppm) upon irradiation of H-1'(δ = 5.25 ppm),is clearly indicative of their cis relationship. Analogously, irradiation of H-4'(δ = 3.25 ppm) in thesame compound gives rise to an enhancement in the signals corresponding to H-1' and H-5'b (δ = 5.25and 2.91 ppm, respectively). On the contrary, no NOE effect was detected between H-4' and H-1' incompound 16a.
The absence of cis/trans diastereoselectivity can be rationalized by assuming that the E-endo attackof the dipolarophile on the nitrone, which leads to cis adducts, competes efficiently with the E-exo
140
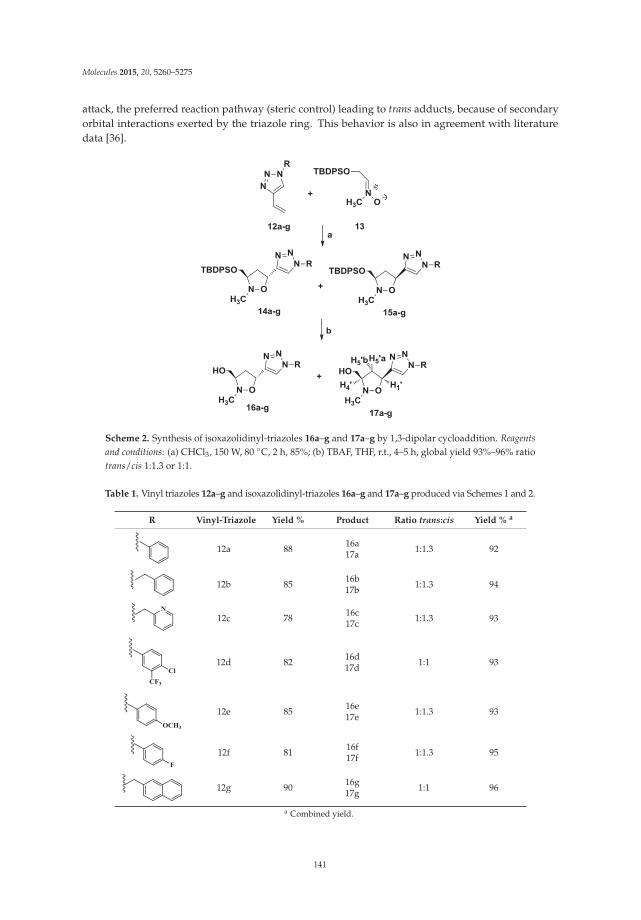
Molecules 2015, 20, 5260–5275
attack, the preferred reaction pathway (steric control) leading to trans adducts, because of secondaryorbital interactions exerted by the triazole ring. This behavior is also in agreement with literaturedata [36].
Scheme 2. Synthesis of isoxazolidinyl-triazoles 16a–g and 17a–g by 1,3-dipolar cycloaddition. Reagentsand conditions: (a) CHCl3, 150 W, 80 ◦C, 2 h, 85%; (b) TBAF, THF, r.t., 4–5 h, global yield 93%–96% ratiotrans/cis 1:1.3 or 1:1.
Table 1. Vinyl triazoles 12a–g and isoxazolidinyl-triazoles 16a–g and 17a–g produced via Schemes 1 and 2.
R Vinyl-Triazole Yield % Product Ratio trans:cis Yield % a
12a 88 16a17a 1:1.3 92
12b 85 16b17b 1:1.3 94
12c 78 16c17c 1:1.3 93
12d 82 16d17d 1:1 93
12e 85 16e17e 1:1.3 93
12f 81 16f17f 1:1.3 95
12g 90 16g17g 1:1 96
a Combined yield.
141

Molecules 2015, 20, 5260–5275
3. Experimental Section
3.1. General Information
Solvents and reagents were used as received from commercial sources. Melting points weredetermined with a Kofler apparatus (Fisher Scientific, Loughborough, UK). Elemental analyseswere performed with a Perkin–Elmer elemental analyzer (PerkinElmer, Waltham, MA, USA). NMRspectra (1H-NMR recorded at 500 MHz, 13C-NMR recorded at 125 MHz) were obtained with aVarian instrument (Agilent Technologies, Palo Alto, CA, USA), and data are reported in ppmrelative to tetramethylsilane. Thin-layer chromatographic separations were carried out on Mercksilica gel 60-F254 precoated aluminum plates (Merk, Darmstadt, Germany). Flash chromatographywas carried out using Merck silica gel (200–400 mesh). Preparative separations were carried outusing an Büchi C-601 MPLC instrument (BUCHI Italia S.r.l., Milano, Italy) using Merck silicagel 0.040–0.063 mm, and the eluting solvents were delivered by a pump at the flow rate of3.5–7.0 mL/min. C-[(tert-Butyldiphenylsilyl)oxy]-N-methyl nitrone was prepared according todescribed procedures [34,35]. Benzyl/alkyl and aromatic azides were synthesized according toliterature procedures [32].
3.2. General 1,3-Dipolar Cycloaddition Procedure
A solution of 12a (0.50 g, 2.92 mmol) and nitrone 13 (1.10 g, 3.50 mmol) in CHCl3 (5 mL) was putin a sealed tube and irradiated under microwave conditions at 150 W, 80 ◦C, for 2 h. The removalof the solvent in vacuo afforded a crude material which, after flash chromatography purification byusing as eluent a mixture of cyclohexane/ethyl acetate 7:3, gave the unseparable mixture (trans/cis)of compound 14a and 15a, as yellow oil, that was used for the next reaction, yield 1.24 g (85%).The 1H-NMR spectrum of the crude reaction mixture shows the presence of trans and cis isomers in1:1.3 ratio, respectively. Compounds 14b–g and 15b–g were prepared by the 1,3-dipolar cycloadditionprocedure in 80%–85% yield as yellow oil and then used for the next reaction.
3.3. General Desilylation of the Hydroxymethyl Group Procedure: Synthesis of 16 and 17
A solution of compounds 14a and 15a (1.24 g, 2.49 mmol) and TBAF (0.90 mL, 3.73 mmol) infreshly distilled THF (30 mL) was stirred until desilylation was completed (TLC, 4–5 h). Volatiles wereflash evaporated, and the residue was purified by MPLC (CH2Cl2/MeOH, 98:2) to afford 17a–g firsteluted isomer (trans) and 16a–g second eluted isomer (cis) in 92% global yield.
((3RS,5RS)-2-Methyl-5-(1-phenyl-1H-1,2,3-triazol-4-yl)isoxazolidin-3-yl) methanol (16a): White solid, 52%yield, mp = 57–59 ◦C. 1H-NMR (CDCl3): δ = 8.00 (s, 1H), 7.70 (d, J = 7.7 Hz, 2H), 7.50 (t, J = 7.8 Hz,2H), 7.42 (t, J = 7.4 Hz, 1H), 5.52 (dd, J = 8.5, 6.6 Hz, 1H), 3.66–3.52 (m, 2H), 3.31–3.19 (m, 1H), 2.94 (dt,J = 12.8, 8.5 Hz, 2H), 2.81 (s, 3H), 2.36 (ddd, J = 12.8, 6.6, 4.8, 1H). 13C-NMR (CDCl3): δ = 148.50, 137.07,129.86, 128.95, 120.66, 119.84, 71.06, 69.53, 63.42, 44.94, 36.62; Anal. Calcd for C13H16N4O2: C, 59.99; H,6.20; N, 21.52; found C, 60.04; H, 6.27; N, 21.60.
((3RS,5SR)-2-Methyl-5-(1-phenyl-1H-1,2,3-triazol-4-yl)isoxazolidin-3-yl) methanol (17a): White solid, 40%yield, mp = 100–102 ◦C. 1H-NMR (CDCl3): δ = 7.97 (s, 1H), 7.71 (d, J = 7.8 Hz, 2H), 7.51 (t, J = 7.8 Hz,2H), 7.43 (t, J = 7.4 Hz, 1H), 5.25 (t, J = 8.0 Hz, 1H), 3.68 (ddd, J = 17.2, 11.4, 4.8 Hz, 2H), 3.25–3.18 (m,1H), 2.98–2.85 (m, 1H), 2.83 (s, 3H), 2.62 (ddd, J = 12.7, 8.0, 5.0 Hz, 1H). 13C-NMR (CDCl3): δ = 148.15,137.06, 129.89, 128.99, 120.72, 120.49, 72.39, 69.36, 62.35, 45.70, 36.78; Anal. Calcd for C13H16N4O2: C,59.99; H, 6.20; N, 21.52; found C, 60.08; H, 6.26; N, 21.48.
((3RS,5RS)-5-(1-Benzyl-1H-1,2,3-triazol-4-yl)-2-methylisoxazolidin-3-yl) methanol (16b): Compound 16b
was prepared by the general desilylation procedure in 53.1% yield as yellow oil. 1H-NMR (CDCl3):δ = 7.46 (s, 1H), 7.38–7.30 (m, 3H), 7.26–7.23 (m, 2H), 5.51–5.43 (m, 2H), 5.40 (dd, J = 8.2, 7.0 Hz, 1H),3.55–3.46 (m, 2H), 3.23–3.14 (m, 1H), 2.84 (dt, J = 12.8, 8.4 Hz, 1H), 2.74 (s, 3H), 2.26 (ddd, J = 12.8, 6.7,
142

Molecules 2015, 20, 5260–5275
4.7, 1H), 2.15 (bs, 1H). 13C-NMR (CDCl3): δ = 147.87, 134.48, 129.21, 128.88, 128.24, 121.55, 71.00, 69.48,63.37, 54.32, 44.90, 36.44; Anal. Calcd for C14H18N4O2: C, 61.30; H, 6.61; N, 20.42; found C, 61.36; H,6.67; N, 20.45.
((3RS,5SR)-5-(1-Benzyl-1H-1,2,3-triazol-4-yl)-2-methylisoxazolidin-3-yl) methanol (17b): Compound 17b
was prepared by the general desilylation procedure in 40.9% yield as white solid, mp = 117–119 ◦C.1H-NMR (CDCl3): δ = 7.43 (s, 1H), 7.39–7.33 (m, 3H), 7.28–7.24 (m, 2H), 5.54–5.44 (m, 2H), 5.12 (t,J = 7.9 Hz, 1H), 3.63 (ddd, J = 17.2, 11.4, 4.8, 2H), 3.10–3.18 (m, 1H), 2.82 (dt, J = 12.7, 8.3, 1H), 2.76(s, 3H), 2.52 (ddd, J = 12.7, 7.9, 5.0, 1H). 13C-NMR (CDCl3): δ = 147.72, 134.52, 129.24, 128.92, 128.26,122.10, 72.35, 69.28, 62.31, 54.31, 45.55, 36.77; Anal. Calcd for C14H18N4O2: C, 61.30; H, 6.61; N, 20.42;found C, 61.38; H, 6.68; N, 20.47.
((3RS,5RS)-2-Methyl-5-(1-(pyridin-2-ylmethyl)-1H-1,2,3-triazol-4-yl) isoxazolidin-3-yl)methanol (16c):Compound 16c was prepared by the general desilylation procedure in 52.6% yield as yellow oil.1H-NMR (CDCl3): δ = 7.71 (s, 1H), 7.65 (td, J = 7.8, 1.7 Hz, 1H), 7.23 (dd, J = 7.0, 5.1 Hz, 1H), 7.16 (d,J = 7.8 Hz, 1H), 5.65–5.54 (m, 2H), 5.41 (dd, J = 8.5, 6.8, 1H), 3.58–3.48 (m, 2H), 3.23–3.15 (m, 1H), 2.84(dt, J = 12.8, 8.5, 1H), 2.74 (s, 3H), 2.39 (bs, 1H), 2.28 (ddd, J = 12.8, 6.5, 4.8 Hz, 1H). 13C-NMR (CDCl3):δ = 154.31, 149.82, 147.92, 137.49, 123.55, 122.58, 122.37, 70.93, 69.50, 63.32, 55.68, 44.86, 36.42; Anal.Calcd for C13H17N5O2: C, 56.71; H, 6.22; N, 25.44; found C, 56.76; H, 6.27; N, 25.48.
((3RS,5SR)-2-Methyl-5-(1-(pyridin-2-ylmethyl)-1H-1,2,3-triazol-4-yl) isoxazolidin-3-yl)methanol (17c):Compound 17c was prepared by the general desilylation procedure in 40.4% yield as yellow solid,mp = 76–78 ◦C. 1H-NMR (CDCl3): δ = 8.66–8.56 (m, 1H), 7.72 (s, 1H), 7.70 (dt, J = 7.7, 1.7 Hz, 1H),7.33–7.26 (m, 1H), 7.23 (d, J = 7.8 Hz, 1H), 5.73–5.59 (m, 2H), 5.18 (t, J = 7.8 Hz, 1H), 3.66 (ddd, J = 17.2,11.4, 4.8 Hz, 2H), 3.17 (s, 1H), 2.93–2.81 (m, 1H), 2.80 (s, 3H), 2.62–2.52 (m, 1H). 13C-NMR (CDCl3):δ = 154.31, 149.82, 137.49, 123.55, 122.58, 122.37, 70.93, 69.50, 63.32, 55.68, 44.86, 36.42; Anal. Calcd forC13H17N5O2: C, 56.71; H, 6.22; N, 25.44; found C, 56.78; H, 6.30; N, 25.50.
((3RS,5RS)-5-(1-(4-Chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)-2-methyl isoxazolidin-3-yl)methanol(16d): Compound 16d was prepared by the general desilylation procedure in 46.5% yield as yellowsolid, mp = 94–96 ◦C. 1H-NMR (CDCl3): δ = 8.07 (s, 1H), 8.06 (d, J = 2.6 Hz, 1H), 7.86 (dd, J = 8.7, 2.6Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 5.49 (dd, J=8.7, 6.3 Hz, 1H), 3.58 (d, J = 5.7 Hz, 2H), 3.26–3.18 (m, 1H),2.94 (dt, J = 12.8, 8.7, 1H), 2.79 (s, 3H), 2.35 (ddd, J = 12.8, 6.3, 4.9, 1H). 13C-NMR (CDCl3): δ = 149.50,135.51, 133.11, 132.57, 130.06 (q, J = 32.4 Hz), 124.38, 123.24, 121.07, 119.73, 119.64 (q, J = 5.5 Hz), 70.94,69.55, 63.25, 44.92, 36.78, 29.78; Anal. Calcd for C14H14ClF3N4O2: C, 46.36; H, 3.89; N, 15.45; found C,46.41; H, 3.93; N, 15.48.
((3RS,5SR)-5-(1-(4-Chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)-2-methyl isoxazolidin-3-yl)methanol(17d): Compound 17d was prepared by the general desilylation procedure in 46.5% yield as yellowthick oil. 1H-NMR (CDCl3): δ = 8.08 (d, J = 2.5 Hz, 1H), 8.00 (s, 1H), 7.89 (dd, J = 8.6, 2.5 Hz, 1H),7.69 (d, J = 8.6 Hz, 1H), 5.26 (t, J = 7.9, 1H), 3.69 (ddd, J=17.1, 11.4, 4.7, 2H), 3.25–3.18 (s, 1H), 2.90 (dt,J = 12.8, 8.0, 1H), 2.84 (s, 3H), 2.66 (ddd, J = 12.8, 8.0, 5.0, 1H). 13C-NMR (CDCl3): δ = 149.02, 135.55,134.94, 133.22, 132.77, 130.11, 127.85, 124.51, 119.77 (q, J = 5.3), 72.29, 69.31, 62.28, 45.62, 36.84; Anal.Calcd for C14H14ClF3N4O2: C, 46.36; H, 3.89; N, 15.45; found C, 46.40; H, 3.95; N, 15.50.
((3RS,5RS)-5-(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)-2-methylisoxazolidin -3-yl)methanol (16e):Compound 16e was prepared by the general desilylation procedure in 52.6% yield as white solid,mp = 57–59 ◦C. 1H-NMR (CDCl3): δ = 7.91 (s, 1H), 7.56 (dd, J =8.9, 1.3 Hz, 2H), 6.96 (dd, J = 8.9, 1.3 Hz,2H), 5.48 (dd, J = 8.3, 6.8 Hz, 1H), 3.82 (s, H), 3.62–3.53 (m, 2H), 3.25–3.18 (m, 1H), 2.94–2.85 (m, 1H),2.78 (s, 3H), 2.37–2.29 (m, 1H). 13C-NMR (CDCl3): δ = 159.89, 148.21, 130.45, 122.21, 120.01, 114.81,70.99, 69.53, 63.38, 55.67, 44.91, 36.64; Anal. Calcd for C14H18N4O3: C, 57.92; H, 6.25; N, 19.30; found C,57.96; H, 6.28; N, 19.27.
143

Molecules 2015, 20, 5260–5275
((3RS,5SR)-5-(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)-2-methyl isoxazolidin-3-yl)methanol (17e): Compound17e was prepared by the general desilylation procedure in 40.4% yield as white solid, mp = 103–105 ◦C.1H-NMR (CDCl3): δ = 7.88 (s, 1H), 7.60 (d, J = 8.9 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 5.24 (t, J = 7.8 Hz,1H), 3.85 (s, 3H), 3.67 (ddd, J=17.1, 11.4, 4.8 Hz, 2H), 3.22–3.15 (m, 1H), 2.97–2.86 (m, 1H), 2.82 (s, 3H),2.76 (bs, 1H), 2.60 (ddd, J = 12.7, 7.9, 5.0 Hz, 1H). 13C-NMR (CDCl3): δ = 160.00, 147.89, 130.51, 122.36,120.66, 114.89, 72.39, 69.35, 55.74, 45.68, 36.79; Anal. Calcd for C14H18N4O3: C, 57.92; H, 6.25; N, 19.30;found C, 57.99; H, 6.30; N, 19.33.
((3RS,5RS)-5-(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)-2-methylisoxazolidin-3-yl)methanol (16f): Compound16f was prepared by the general desilylation procedure in 53.7% yield as white solid, mp = 68–70 ◦C.1H-NMR (CDCl3): δ = 7.96 (s, 1H), 7.71–7.64 (m, 2H), 7.20 (t, J = 8.5 Hz, 2H), 5.51 (dd, J = 8.5, 6.6 Hz,1H), 3.58 (d, J = 5.7 Hz, 2H), 3.31–3.18 (m, 1H), 2.94 (dt, J = 12.8, 8.5 Hz, 1H), 2.80 (s, 3H), 2.36 (ddd,J = 12.8, 6.2, 4.9, 1H). 13C-NMR (CDCl3): δ = 163.56, 161.57, 148.73, 133.37, 122.65 (d, J = 8.7 Hz), 120.00,116.84 (d, J = 23.2 Hz), 71.04, 69.51, 63.38, 44.93, 36.65; Anal. Calcd for C13H15FN4O2: C, 56.11; H, 5.43;N, 20.13; found C, 56.14; H, 5.49; N, 20.17.
((3RS,5SR)-5-(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)-2-methylisoxazolidin-3-yl)methanol (17f): Compound17f was prepared by the general desilylation procedure in 41.3% yield as white solid, mp = 102–104◦C. 1H-NMR (CDCl3): δ = 7.93 (s, J = 1.0, 1H), 7.73–7.63 (m, 2H), 7.24–7.14 (m, 2H), 5.24 (t, J = 7.8 Hz,1H), 3.68 (ddd, J = 17.2, 11.5, 4.8 Hz, 2H), 3.24–3.17 (m, 1H), 2.97–2.86 (m, 1H), 2.82 (s, 3H), 2.66–2.56(m, 1H). 13C-NMR (CDCl3): δ = 163.58, 161.59, 148.26, 133.34, 122.70 (d, J = 8.5 Hz), 120.67, 116.86 (d,J = 23.2 Hz), 72.30, 69.38, 62.32, 45.68, 36.79; Anal. Calcd for C13H15FN4O2: C, 56.11; H, 5.43; N, 20.13;found C, 56.15; H, 5.47; N, 20.19.
((3RS,5RS)-2-Methyl-5-(1-(naphthalen-2-ylmethyl)-1H-1,2,3-triazol-4-yl) isoxazolidin-3-yl)methanol (16g):Compound 16g was prepared by the general desilylation procedure in 48.0% yield as white solid,mp = 95–97 ◦C. 1H-NMR (CDCl3): δ = 7.83–7.80 (m, 3H), 7.74 (s, 1H), 7.51 (dd, J = 6.2, 3.3 Hz, 2H), 7.48(s, 1H), 7.33 (dd, J = 8.4, 1.4 Hz, 1H), 5.70–5.56 (m, 2H), 5.41 (dd, J = 8.3, 7.0 Hz, 1H), 3.51 (d, J = 6.2Hz, 2H), 3.21–3.17 (m, 1H), 2.84 (dt, J = 12.8, 8.3 Hz, 1H), 2.74 (s, 3H), 2.31–2.22 (m, 1H), 1.91 (bs, 1H).13C-NMR (CDCl3): δ = 148.00, 133.32, 131.83, 129.32, 128.07, 127.90, 127.67, 126.87, 125.51, 71.06, 69.44,63.40, 54.57, 44.89, 36.40; Anal. Calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27; found C, 66.68; H,6.25; N, 17.30.
((3RS,5RS)-2-Methyl-5-(1-(naphthalen-2-ylmethyl)-1H-1,2,3-triazol-4-yl) isoxazolidin-3-yl)methanol (17g):Compound 17g was prepared by the general desilylation procedure in 48.0% yield as white solid,mp = 115–117 ◦C. 1H-NMR (CDCl3): δ = 7.88–7.79 (m, 3H), 7.74 (s, J = 6.8 Hz, 1H), 7.55–7.48 (m, 2H),7.46 (s, 1H), 7.34 (d, J = 8.3 Hz, 1H), 5.72–5.60 (m, 2H), 5.12 (t, J = 7.9 Hz, 1H), 3.62 (ddd, J = 17.2,11.5, 4.8, 2H), 3.08–3.16 (m, 1H), 2.90–2.76 (m, 1H), 2.75 (s, 3H), 2.56–2.47 (m, 1H). 13C-NMR (CDCl3):δ = 147.79, 133.29, 131.85, 129.29, 128.03, 127.89, 127.62, 126.87, 126.84, 125.49, 122.15, 72.35, 69.27, 62.28,54.50, 45.54, 36.80; Anal. Calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27; found C, 66.70; H, 6.27;N, 17.32.
3.4. Biological Tests
3.4.1. Antiviral Activity
Compounds 16–17 were evaluated for their ability to inhibit the replication of a variety of DNAand RNA viruses, using the following cell-based assays: (a) Vero cell cultures: poliovirus 1, humanechovirus 9, coxsackievirus B4, adenovirus type 2, herpes simplex type 1 (HSV-1), herpes simplextype 2 (HSV-2); (b) human embryonic lung fibroblast cells (MRC-5): cytomegalovirus (CMV: VR-538);(c) African green monkey kidney cells (BS-C-1): varicella-zoster virus (VZV). Acyclovir was used asthe reference compounds. Unfortunately, no inhibitory activity against any virus was detected for theevaluated compounds (data not shown).
144

Molecules 2015, 20, 5260–5275
3.4.2. Antiproliferative Activity
The antiproliferative activity of all the synthesized C-Nucleosides was evaluated. An antiproliferativeeffect was observed for compounds 17a and 17b, while other derivatives show a IC50 in the range150–200 μM. In particular, as shown in Figures 4A and 5A cell culture incubation with increasingconcentration of 17a and 17b, ranging from 1 μM to 100 μM for 24, 48 and 72 h, reduced the proliferationin all the cancer cell lines with a similar trend.
BrdU
inco
rpor
atio
n (%
)
0
20
40
60
80
100
120
24 h 48 h 72 h
*********
0 1 10 100
17a ( M)
50
******
24 h 48 h 72 h
**
******
0 1 10 100
17a ( M)
50
***
*
24 h 48 h 72 h
***
****
******
0 1 10 100
17a ( M)
50
***
***
ASH-SY5YHT-29HepG2
LDH
rele
ase
0
1
2
3
4
5
0 10 50 100
17a ( M)
***
**
1 0 10 50 100
17a ( M)
***
**
1 0 10 50 100
17a ( M)
***
**
1
B
Figure 4. Compound 17a reduces cell proliferation and induce LDH release. HepG2, HT-29 andSH-SY5Y cells were exposed to increased concentration of 17a compound for 24, 48 and 72 h. (A)Proliferation rate assessed by BrdU assay. (B) Cytotoxic effect assessed in terms of LDH releaseafter 24 h of exposure. Each value is the mean ± S.E.M. of three experiments performed eight times(BrdU) or in triplicate (LDH) and repeated three different times. * p < 0.05 vs. ctrl, ** p < 0.01 vs. ctrl,*** p < 0.001 vs. ctrl.
In particular, data of the BrdU assay shows that the growth inhibitory effect induced by 17a
reaches the 50% in both HepG2 and HT-29 cells and increases up to 56% in the SH-SY5Y cell line(p < 0.001 vs. control) after 72 h of incubation at a 100 μM concentration. Reduction of cell proliferationwas also observed when the cells were exposed for 48 (p < 0.001 vs. control) and 24 hours (p < 0.01vs. control in HepG2 and HT-29 cells and p < 0.001 in SH-SY5Y cell line; Figure 4A) to 100 μMconcentration of the compound 17a.
Lesser antiproliferative effect was observed treating the cells with compound 17a at concentrationsof 50 and 10 μM (expecially for longer time of exposure). Similar results were obtained by thecompound 17b (56% of cell growth inhibition for the HepG2, 62% for the HT-29 and the SH-SY5Ycell lines at the 100 μM concentration for 72 h; Figure 5A). Results of the BrdU assay show very closealignment with those of MTT test performed after 24, 48 and 72 h of incubation with both 17a and 17b
(see Supplementary Materials Figures S1A and S2A).
145

Molecules 2015, 20, 5260–5275
LDH
rele
ase
0
1
2
3
4
5
0 10 50 100
17b ( M)
***
**
1
A
B
SH-SY5YHT-29HepG2Br
dU in
corp
orat
ion
(%)
0
20
40
60
80
100
120
24 h 48 h72 h
*********
0 1 10 100
17b ( M)
50
*****
24 h 48 h 72 h
******
0 1 10 100
17b ( M)
50
********
**
24 h 48 h72 h
******
****
0 1 10 100
17b ( M)
50
*****
0 10 50 100
17b ( M)
***
**
1 0 10 50 100
17b ( M)
***
***
1
Figure 5. Compound 17b inhibit cell growth and determine LDH release. (A) Proliferation rate assessedby BrdU assay after 24–72 h of treatment. (B) Cytotoxic activities evaluated by the test of LDH after24 h of incubation. Each value is the mean ± S.E.M. of three experiments performed in eight times(BrdU) or in triplicate (LDH) and repeated three different times. * p < 0.05 vs. ctrl, ** p < 0.01 vs. ctrl,*** p < 0.001 vs. ctrl.
3.4.3. Cytoxicity Evaluation
The cytotoxic effect induced by 17a and 17b was evaluated through either LDH and trypanblue dye exclusion assays. Figures 4B and 5B show that both 17a and 17b caused increase of theLDH release at 50 and 100 μM concentration in all the investigated cell lines. Furthermore, LDHrelease was accompanied by a significant increase of cell death, as detected by the trypan blue test (seeSupplementary Materials Figures S1B and S2B), underlying the cytotoxic activity of the compounds17a and 17b.
3.4.4. Cell Culture and Treatments
HepG2 (hepatocellular carcinoma), HT-29 (colorectal adenocarcinoma) and SH-SY5Y(neuroblastoma) cells were obtained originally from ATCC (Rockville, MD, USA). These cell lines,maintained in RPMI supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodiumpyruvate, 2 mM glutamine, penicillin/streptomycin (100 units/mL and 100 μg/mL, respectively),were grown at 37 ◦C and 5% CO2 conditions. All the reagents for cell cultures were from Gibco (LifeTechnologies, Monza, Italy). For biological investigations, 100 mM stock solutions were prepareddissolving the tested compounds in dimethyl sulfoxide (DMSO). Small aliquots were stored at −20 ◦Cand were diluted in culture media to the final concentration, ranging from 1 to 100 μM, just prior theuse. The highest DMSO concentration used in this study (0.1%) did not have any appreciable effect oncell proliferation or cytotoxicity.
146

Molecules 2015, 20, 5260–5275
3.4.5. Antiproliferative Activity
The antiproliferative activity of all the synthesized compounds was evaluated by either BrdU andMTT assays. For the first assay, the cells were seeded in 96-well plates at a density of 10 × 104 cell/well(HT-29 cells and SH-SY5Y) and 12 × 104 (HepG2) and allowed to stand overnight. Then, the culturemedium was replaced with clean media containing the tested compounds at a concentrations rangingfrom 1 to 100 μM or with media with equivalent dilutions of DMSO (control cultures). After 24, 48 or72 h incubation, BrdU assay was performed as described [37]. BrdU is a uridine derivative, structuralanalog of thymidine, which can be used as a marker for proliferation, because it is incorporated intoDNA during the synthesis-phase of the cell cycle as a substitute for thymidine. Cells marked by BrdUincorporation may be detected after addition of goat anti-mouse IgG-peroxidase conjugated secondaryantibody. Results are expressed as percentages of the absorbance measured in control cells.
Cell growth was also detected by the MTT assay as reported with modification [38]. The cellswere seeded onto 96-well plates at a density of 5 × 103 cells/well (HT-29 cells and SH-SY5Y) or6 × 103 cells/well (HepG2). The following day, cells were treated with the test compounds asdescribed above. At the end of the exposure time, the plates were centrifuged at 1200 rpm for 10 min,the supernatants were replaced with clean medium without phenol red containing 0.5 mg/mL of3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, Milan, Italy),and the plates were returned in the incubator for 4 h. Then, the solutions were removed from the wellsand crystals of formazan (MTT metabolic product) were solubilised by 100 μL HCl/isopropanol 0.1 Nlysis buffer. The latter were spectrophotometrically quantified at a wavelength of 595 nm (iMark™microplate reader, Bio-Rad Laboratories, Milan, Italy). Results of the cell proliferation assays areexpressed as percentages in untreated cultures. The tests were performed in triplicate (BrdU) or eighttimes (MTT) and repeated three different times.
3.4.6. Detection of Cell Viability
Lactate dehydrogenase is a stable cytosolic enzyme which is released into the surrounding culturemedium when the plasma membrane is damaged, and can be considered a marker of cytotoxicity.The released LDH can be quantified by a coupled enzymatic reaction. First, LDH catalyzes theconversion of lactate to pyruvate via reduction of NAD+ to NADH. Second, diaphorase uses NADHto reduce a tetrazolium salt to a red formazan product. Therefore, the level of formazan formationis directly proportional to the amount of released LDH in the medium. HepG2, HT-29 and SH-SY5Ycells were seeded in 96-well plates in a number of 15 × 103 cells/well. The following day, cells wereincubated with the synthesized C-Nucleosides for 24 h. Then the LDH concentration was measuredas reported [39] by using commercial LDH kit (CytoTox 96® Non-Radioactive Cytotoxicity Assay,Promega, Milan, Italy), according to the manufacturer’s protocol. LDH levels are extrapolated as thevalues detected in control cells, which are arbitrarily expressed as 1.
Cell viability in presence of the tested compounds was assessed also by the trypan blue dye (0.4%w/v) exclusion test [40] and cell death was reported as the percentage of stained (non-viable) vs. totalcells counted. All the experiments were carried out in triplicate and repeated three times.
3.4.7. Statistical Analysis
Data were expressed as mean ± S.E.M. and statistically evaluated for differences using one-wayanalysis of variance (ANOVA), followed by Turkey-Kramer multiple comparisons test (GraphPADSoftware for Science, GraphPad Software, Inc., La Jolla, CA, USA).
4. Conclusions
We report in this paper the synthesis of 5-(1H-1,2,3-triazol-4-yl)isoxazolidines, a novel series ofC-nucleosides, featured by the presence of a 1,2,3-triazole ring linked to an isoxazolidine system, asmimetic of the pyrimidine nucleobases. The synthesized compounds have been evaluated for their
147

Molecules 2015, 20, 5260–5275
ability to inhibit the replication of a variety of DNA and RNA viruses: unfortunately, no inhibitoryactivity against any virus was detected for the evaluated compounds. The antiproliferative activityof all the synthesized C-Nucleosides was also tested. An antiproliferative effect was observed forcompounds 17a and 17b: the induced growth inhibitory effect reaches the 50% in HepG2 and HT-29cells and increases up to 56% in the SH-SY5Y cell line (p < 0.001 vs. control) after 72 h of incubation at a100 μM concentration.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/04/5260/s1.
Acknowledgments: We gratefully acknowledge the Italian Ministry of Education, Universities, and Research(MIUR), the Universities of Messina and Catania, and the Interuniversity Consortium for InnovativeMethodologies and Processes for Synthesis (CINMPIS) for partial financial support.
Author Contributions: SVG and RR, designed research; MN, SC, AG performed biological data; SVG, RR, CC,RM and MAC performed research and analyzed the data; SVG and RR wrote the paper. All authors read andapproved the final manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. De Clercq, E. The history of antiretrovirals: Key discoveries over the past 25 years. Rev. Med. Virol 2009, 19,287–299.
2. De Clercq, E. Antiviral Drug Strategies; Wiley-VCH: Weinheim, Germany, 2011.3. Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer.
Chem. Rev. 2009, 109, 2880–2893. [CrossRef] [PubMed]4. Vanek, V.; Budesinsky, M.; Rinnova, M.; Rosenberg, I. Prolinol-based nucleoside phosphonic acids:
New isosteric conformationally flexible nucleotide analogues. Tetrahedron 2009, 65, 862–876. [CrossRef]5. Ishitsuka, H.; Shimma, N. Capecitabine Preclinical Studies: From Discovery to Translational Research.
In Modified Nucleosides in Biochemistry, Biotechnology and Medicine; Herdewijn, P., Ed.; Wiley-VCH: Weinheim,Germany, 2008; pp. 587–600.
6. Kumamoto, H.; Topalis, D.; Broggi, J.; Pradere, U.; Roi, V.; Berteina-Raboin, S.; Nolan, S.P.; Deville-Bonne, D.;Andrei, G.; Snoeck, R.; et al. Preparation of acyclo nucleoside phosphonate analogues based oncross-metathesis. Tetrahedron 2008, 64, 3517–3526.
7. Jung, K.-H.; Marx, A. Synthesis of 4'-C-modified 2'-Deoxyribonucleoside Analogues and Oligonucleotides.Curr. Org. Chem. 2008, 12, 343–354. [CrossRef]
8. Littler, E.; Zhou, X.-X. Deoxyribonucleic Acid Viruses: Antivirals for Herpesviruses and Hepatitis B Virus.In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford UK, 2006; Volume 7,pp. 295–327.
9. Thottassery, J.V.; Westbrook, L.; Someya, H.; Parker, W.B. c-Abl-independent p73 stabilization duringgemcitabine- or 4'-thio-beta-D-arabinofuranosylcytosine-induced apoptosis in wild-type and p53-nullcolorectal cancer cells. Mol. Cancer Ther. 2006, 5, 400–410. [CrossRef] [PubMed]
10. Stambasky, J.; Hocek, M.; Kocovsky, P. C-Nucleosides: Synthetic Strategies and Biological Applications.Chem. Rev. 2009, 109, 6729–6764. [CrossRef] [PubMed]
11. Bzowska, A. Formycins and their Analogues: Purine Nucleoside Phosphorylase Inhibitors and TheirPotential Application in Immunosuppression and Cancer. In Modified Nucleosides: in Biochemistry,Biotechnology and Medicine; Herdewijn, P., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 473–510.
12. Desaulniers, J.P.; Chang, Y.C.; Aduri, R.; Abeysirigunawardena, S.C.; Santa Lucia, J.; Chow, C.S. Pseudouridinesin rRNA helix 69 play a role in loop stacking interactions. Org. Biomol. Chem. 2008, 6, 3892–3895. [CrossRef][PubMed]
13. Matsuura, S.; Shiratori, O.; Katagiri, K. Antitumor activity of showdomycin. J. Antibiot. 1964, 17, 234–237.[PubMed]
14. Nakagawa, Y.; Kano, H.; Tsukuda, Y.; Koyama, H. Structure of a new class of C-nucleoside antibiotic,showdomycin. Tetrahedron Lett. 1967, 42, 4105–4109. [CrossRef] [PubMed]
15. Uehara, Y.I.; Rabinowicz, J. Showdomycin and its reactive moiety, maleimide. A comparison in selectivetoxicity and mechanism of action in vitro. Biochem. Pharmacol. 1980, 29, 2199–2204.
148

Molecules 2015, 20, 5260–5275
16. Shaban, M.A., E; Nasr, A.Z. The Chemistry of C-Nucleosides and Their Analogs I: C-Nucleosides of HeteroMonocyclic Bases. Adv. Heterocycl. Chem. 1997, 68, 223–432.
17. Sallam, M.A.E.; Luis, F.F.; Cassady, J.M. Studies on Epimeric-D-arabino-and-D-ribo-tetritol-1-yl-2-phenyl-2H-1,2,3-triazoles. Synthesis and Anomeric Configuration of 4-(α- and β-D-Erythrofuranosyl)-2-phenyl-2H-1,2,3-Triazole C-Nucleoside Analogs. Nucleos. Nucleot. 1998, 17, 769–783.
18. Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chemical Synthesis of Heterocyclic−Sugar NucleosideAnalogues. Chem. Rev. 2010, 110, 3337–3370. [CrossRef] [PubMed]
19. Romeo, G.; Giofré, S.V.; Piperno, A.; Romeo, R.; Chiacchio, M.A. Synthesis of N,O-homonucleosides withhigh conformational freedom. ARKIVOC 2009, viii, 168–176. [CrossRef]
20. Romeo, R.; Giofrè, S.V.; Iaria, D.; Sciortino, M.T.; Ronsisvalle, S.; Chiacchio, M.A.; Scala, A. Synthesis of5-Alkynyl Isoxazolidinyl Nucleosides. Eur. J. Org. Chem. 2011, 28, 5690–5695.
21. Romeo, R.; Giofrè, S.V.; Macchi, B.; Balestrieri, E.; Mastino, A.; Merino, P.; Carnovale, C.; Romeo, G.;Chiacchio, U. Truncated Reverse Isoxazolidinyl Nucleosides: A New Class of Allosteric HIV-RT Inhibitors.ChemMedChem 2012, 7, 565–569. [CrossRef] [PubMed]
22. Romeo, R.; Giofrè, S.V.; Garozzo, A.; Bisignano, B.; Corsaro, A.; Chiacchio, M.A. Synthesis and biologicalevaluation of furopyrimidine N,O-nucleosides. Bioorg. Med. Chem. 2013, 21, 5688–5693. [CrossRef] [PubMed]
23. Chiacchio, U.; Rescifina, A.; Iannazzo, D.; Piperno, A.; Romeo, R.; Borrello, L.; Sciortino, M.T.; Balestrieri, E.;Macchi, B.; Mastino, A.; et al. Phosphonated Carbocyclic 2'-Oxa-3'-azanucleosides as New AntiretroviralAgents. J. Med. Chem. 2007, 50, 3747–3750.
24. Chiacchio, U.; Balestrieri, E.; Macchi, B.; Iannazzo, D.; Piperno, A.; Rescifina, A.; Romeo, R.; Saglimbeni, M.;Sciortino, M.T.; Valveri, V.; et al. Synthesis of Phosphonated Carbocyclic 2'-Oxa-3'-aza-nucleosides: NovelInhibitors of Reverse Transcriptase. J. Med. Chem. 2005, 48, 1389–1394.
25. Chiacchio, U.; Iannazzo, D.; Piperno, A.; Romeo, R.; Romeo, G.; Rescifina, A.; Saglimbeni, M. Synthesis andbiological evaluation of phosphonated carbocyclic 2'-oxa-3'-aza-nucleosides. Bioorg. Med. Chem. 2006, 14,955–959. [CrossRef] [PubMed]
26. Romeo, R.; Carnovale, C.; Giofrè, S.V.; Monciino, G.; Chiacchio, M.A.; Sanfilippo, C.; Macchi, B.Enantiomerically Pure Phosphonated Carbocyclic 2'-Oxa-3'-Azanucleosides: Synthesis and BiologicalEvaluation. Molecules 2014, 19, 14406–14416. [CrossRef] [PubMed]
27. Piperno, A.; Giofrè, S.V.; Iannazzo, D.; Romeo, R.; Romeo, G.; Chiacchio, U.; Rescifina, A.; Piotrowska, D.G.Synthesis of C-4' Truncated Phosphonated Carbocyclic 2'-Oxa-3'-azanucleosides as Antiviral Agents.J. Org. Chem. 2010, 75, 2798–2805. [CrossRef] [PubMed]
28. Romeo, R.; Carnovale, C.; Giofrè, S.V.; Romeo, G.; Macchi, B.; Frezza, C.; Merino-Merlo, F.; Pistarà, V.;Chiacchio, U. Truncated phosphonated C-1'-branched N,O-nucleosides: A new class of antiviral agents.Bioorg. Med. Chem. 2012, 20, 3652–3657. [CrossRef] [PubMed]
29. Chiacchio, U.; Corsaro, A.; Mates, J.; Merino, P.; Piperno, A.; Rescifina, A.; Romeo, G.; Romeo, R.; Tejero, T.Isoxazolidine analogues of pseudouridine: A new class of modified nucleosides. Tetrahedron 2003, 59,4733–4738. [CrossRef]
30. Chiacchio, U.; Rescifina, A.; Saita, M.G.; Iannazzo, D.; Romeo, G.; Mates, J.A.; Tejero, T.; Merino, P. Zinc(II)triflate-controlled 1,3-dipolar cycloadditions of C-(2-thiazolyl)nitrones: application to the synthesis of a novelisoxazolidinyl analogue of tiazofurin. J. Org. Chem. 2005, 70, 8991–9001. [CrossRef] [PubMed]
31. Romeo, R.; Giofrè, S.V.; Carnovale, C.; Campisi, A.; Parenti, R.; Bandini, L.; Chiacchio, M.A. Synthesis andBiological Evaluation of 3-Hydroxymethyl-5-(1H-1,2,3-Triazol) Isoxazolidines. Bioorg. Med. Chem. 2013, 21,7929–7937. [CrossRef] [PubMed]
32. Romeo, R.; Giofrè, S.V.; Carnovale, C.; Chiacchio, M.A.; Campisi, A.; Mancuso, R.; Cirmi, S.; Navarra, M.Synthesis and Biological Activity of Triazole-Appended N,O-Nucleosides. Eur. J. Org. Chem. 2014, 25,5442–5447. [CrossRef]
33. Nulwala, H.; Takizawa, K.; Odukale, A.; Anzar Khan, A.; Thibault, R.J.; Taft, B.R.; Lipshutz, B.H.; Hawker, C.J.Synthesis and Characterization of Isomeric Vinyl-1,2,3-triazole Materials by Azide−Alkyne Click Chemistry.Macromolecules 2009, 42, 6068–6074. [CrossRef]
34. Iannazzo, D.; Piperno, A.; Pistarà, V.; Rescifina, A.; Romeo, R. Modified nucleosides. A general anddiastereoselective approach to N,O-psiconucleosides. Tetrahedron 2002, 58, 581–587.
149

Molecules 2015, 20, 5260–5275
35. Romeo, R.; Carnovale, C.; Giofrè, S.V.; Chiacchio, M.A.; Garozzo, A.; Amata, E.; Romeo, G.; Chiacchio, U.C-5'-Triazolyl-2'-oxa-3'-aza-4'a-carbanucleosides: Synthesis and biological evaluation. Beilstein J. Org. Chem.2015, 11, 328–334. [CrossRef]
36. Merino, P.; Revuelta, J.; Tejero, T.; Chiacchio, U.; Rescifina, A.; Romeo, G. A DFT study on the 1,3-dipolarcycloaddition reactions of C-(methoxycarbonyl)-N-methyl nitrone with methyl acrylate and vinyl acetate.Tetrahedron 2003, 59, 3581–3592. [CrossRef]
37. Visalli, G.; Ferlazzo, N.; Cirmi, S.; Campiglia, P.; Gangemi, S.; Pietro, A.D.; Calapai, G.; Navarra, M. Bergamotjuice extract inhibits proliferation by inducing apoptosis in human colon cancer cells. Anticancer Agents Med.Chem. 2014, 14, 1402–1413. [CrossRef] [PubMed]
38. Navarra, M.; Celano, M.; Maiuolo, J.; Schenone, S.; Botta, M.; Angelucci, A.; Bramanti, P.; Russo, D.Antiproliferative and pro-apoptotic effects afforded by novel Src-kinase inhibitors in human neuroblastomacells. BMC Cancer 2010, 10, 602. [CrossRef] [PubMed]
39. Romeo, R.; Navarra, M.; Giofrè, S.V.; Carnovale, C.; Cirmi, S.; Lanza, G.; Chiacchio, M.A. Synthesis andbiological activity of new arenediyne-linked isoxazolidines. Bioorg. Med. Chem. 2014, 22, 3379–3385.[CrossRef] [PubMed]
40. Delle Monache, S.; Sanità, P.; Trapasso, E.; Ursino, M.R.; Dugo, P.; Russo, M.; Ferlazzo, N.; Calapai, G.;Angelucci, A.; Navarra, M. Mechanisms Underlying the Anti-Tumoral Effects of Citrus bergamia Juice.PLoS ONE 2013, 8, e61484. [CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
150

molecules
Article
Carboxylated Acyclonucleosides: Synthesis andRNase A Inhibition
Kaustav Chakraborty, Swagata Dasgupta * and Tanmaya Pathak *
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India;[email protected]* Correspondence: [email protected] (S.D.); [email protected] (T.P.);
Tel.: +91-3222-283-306 (S.D.); +91-3222-283-342 (T.P.)
Academic Editor: Mahesh LakshmanReceived: 17 December 2014; Accepted: 20 March 2015; Published: 3 April 2015
Abstract: Strategically designed carboxylated acyclonucleosides have been probed as a new class ofRNase A inhibitors. Several experimental and theoretical studies have been performed to compilerelevant qualitative and quantitative information regarding the nature and extent of inhibition. Theinhibition constant (Ki) values were determined using a UV-based kinetics experiment. The changesin the secondary structure of the enzyme upon binding with the inhibitors were obtained fromcircular dichroism studies. The binding constants for enzyme-inhibitor interactions were determinedwith the help of fluorescence spectroscopy. Docking studies were performed to reveal the possiblebinding sites of the inhibitors within the enzyme. The cytosine analogues were found to possessbetter inhibitory properties in comparison to the corresponding uracil derivatives. An incrementin the number of carboxylic acid groups (-COOH) in the inhibitor backbone was found to result inbetter inhibition.
Keywords: acyclonucleosides; RNase A; inhibition; kinetics; docking
1. Introduction
Ribonucleases are a family of digestive enzymes that degrade RNA [1]. Effective inhibitionof their enzymatic activity has become a topic of growing interest with the realization that thedetrimental biological activities manifested by certain members of this family [2–6] are criticallydependent upon their ribonucleolytic activity [7,8]. Ribonuclease A (RNase A) is a representativemember of this family [9,10] that works at the juncture of the transcription and translation processes,thereby maintaining the cellular RNA levels. Structural homology among the various membersof this family [11–13] permits the use of Ribonuclease A (RNase A) as a model system to explorestructure–activity relationships.
Recent reports from our laboratory have revealed that modified nucleoside carboxylic acidsmanifest RNase A inhibitory properties in a competitive, reversible manner [14–21]. Thesemolecules have an added advantage over the reported phosphate- or pyrophosphate-based nucleotideinhibitors [22–31] as the polyionic nature of the latter hampers their migration through the cellmembrane [32]. The active site of RNase A consists of several subsites made up of polar amino acidresidues for specific recognition [9,33] (Figure 1). The cleavage of the phosphodiester bond of RNA atthe P1 subsite involves the two His residues (His 12 and His 119) participating in a conjugate acid-basemechanism [34,35]. At physiological pH, the carboxylic acid (-COOH) group(s) in the inhibitor remainsdeprotonated, and interacts electrostatically with the protonated His and Lys residues present at theribonucleolytic site [36]. This perturbation of the protonating/deprotonating environment of the P1
subsite results in the inhibition of RNase A.
Molecules 2015, 20, 5924–5941 151 www.mdpi.com/journal/molecules

Molecules 2015, 20, 5924–5941
Following the same hypothesis, it was expected that carboxylated acyclonucleosides may elicitsimilar inhibitory properties because of their in-built structural features. The absence of the rigid ribosering further enhances the flexibility of these molecules, and generates additional information on theimportance of the furanoside ring. We report the synthesis of several uracil- and cytosine-basedmodified acyclonucleosides followed by the exploration of their RNase A inhibitory properties.The nucleobases have been carefully selected, as the B1 subsite of RNase A shows preferentialrecognition towards pyrimidine bases. The acidity of the molecules has been increased via stepwiseincorporation of carboxylic acid groups in the molecular framework to study the resulting effect ontheir inhibition capacities.
Figure 1. Key residues of the active site of RNase A.
2. Results and Discussion
2.1. Synthesis of Nucleosides
The uracil-glycine conjugate 3 was synthesized according to a literature reported procedure [37](Scheme 1). Syntheses of other modified uracil derivatives were achieved via coupling of the knownuracil-1-acetic acid 1 [37] with suitable secondary amines, followed by hydrolysis and/or deprotectionas required (Scheme 1). The synthesis of the uracil-diethanolamine conjugate 5 [38] was achieved bycoupling 1 with the tertbutyldimethyl silyl (TBDMS)-protected diethanolamine, and deprotecting theTBDMS groups of the coupled product 4 with a catalytic amount of CH3COCl in methanol (Scheme 1).TBDMS-protected N-(2-hydroxyethyl)glycine ethyl ester, on EDC-HOBT-mediated coupling with 1,generated compound 6. Deprotection of the TBDMS group produced compound 7, which wasconverted to the corresponding acid 8 via hydrolysis (Scheme 1). Again, 1 was coupled with diethyliminodiacetate to afford the corresponding diester 9. The diacid derivative 10 was produced from 9 bybase-mediated hydrolysis (Scheme 1).
152

Molecules 2015, 20, 5924–5941
N
NH
O
O
HO2C
NH
U
O
CO2Et NH
U
O
CO2H
NRO
U
O
ORN
OH
U
O
OH
NHTBDMSO CO2Et
NRO
U
O
CO2Et NOH
U
O
CO2H
N
U
O
CO2EtEtO2CN
U
O
CO2HHO2C
1
1. N-hydroxy succinimide DCC, DMF, 0 oC-rt, 2h 2. Glycine ethyl ester rt, 18h
2 (82%)
2 N NaOHMeOH, rt, 4h
3 (96%)U-acid
NH(CH2CH2OTBDMS)2 TEA, EDC.HCl, HOBT DMF, 0 oC-rt, Overnight
4 R = TBDMS (66%) 5 (81%)
CH3COCl MeOH, rt, 2h
U-ol-ol
TEA, EDC.HCl, HOBTDMF, 0 oC-rt, Overnight
6 R = TBDMS (62%)
7 R = H (55%)
8 (62%)U-ol-acid
1 N NaOHMeOH, 0 oC-rt, 1h
CH3COCl MeOH, rt, 2h
NH(CH2CO2Et)2TEA, EDC.HCl, HOBTDMF, 0 oC-rt, Overnight
1 N NaOHMeOH, 0 oC-rt, 1h
9 (49%) 10 (74%)U-di-acid
ref. [37]
Scheme 1. Synthesis of uracil-based modified acyclonucleosides.
Syntheses of the corresponding cytosine-based molecules were achieved using the reportedcytosine derivative 11 [39] as the starting material (Scheme 2). EDC-HOBT-mediated coupling ofglycine ethyl ester to compound 11 produced the coupled product 12. The deprotection of thetertbutyloxy carbonyl (BOC) groups of 12 by TFA resulted in the formation of compound 13. Thehydrolysis of the ester group provided the desired acid derivative 14 (Scheme 2). Compound 11, oncoupling with TBDMS-protected diethanolamine, generated the coupled derivative 15. TFA treatmentof compound 15 afforded the deprotected cytosine-diethanolamine conjugate 16 (Scheme 2). Again,coupling of the TBDMS-protected N-(2-hydroxyethyl)glycine ethyl ester with compound 11, followedby hydrolysis and deprotection of the resulting ester 17, afforded the corresponding hydroxy acidderivative 18 (Scheme 2). Finally, the diacid 21 was obtained from compound 11 following a similarsequence of steps (Scheme 2). Compound 11, on coupling with diethyl iminodiacetate, producedthe coupled product 19. Compound 19 was transformed to compound 20 by treatment with TFA.Base-mediated hydrolysis of 20 afforded the desired diacid derivative 21.
153
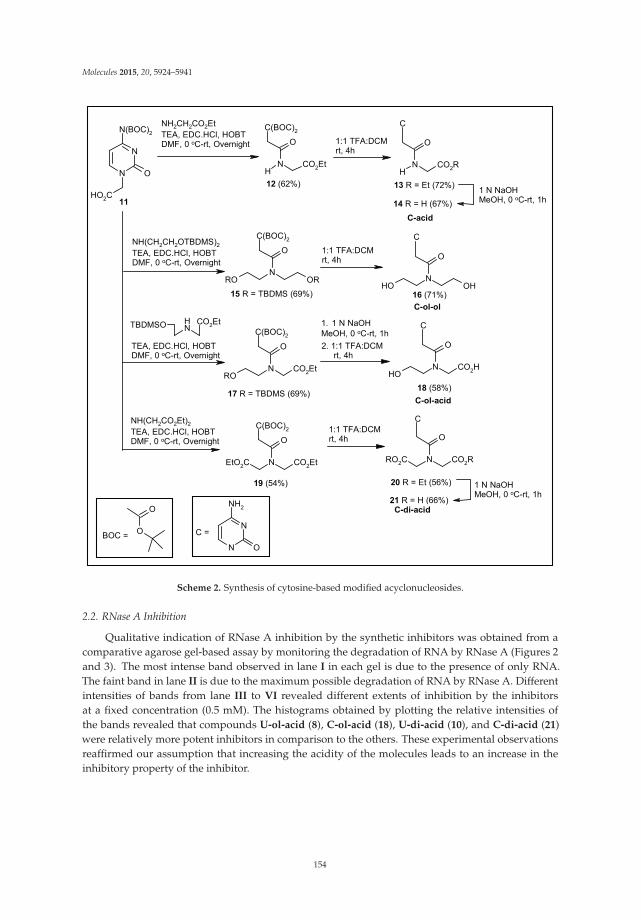
Molecules 2015, 20, 5924–5941
NHTBDMSO CO2Et
N
N
O
N(BOC)2
HO2C
NH
C(BOC)2
O
CO2Et NH
O
CO2R
NRO
C(BOC)2
O
OR NOH
O
OH
NRO
C(BOC)2
O
CO2Et
N
C(BOC)2
O
CO2EtEtO2CN
O
CO2RRO2C
NOH
O
CO2H
O
O
N
N
O
NH2
NH(CH2CH2OTBDMS)2 TEA, EDC.HCl, HOBT DMF, 0 oC-rt, Overnight
TEA, EDC.HCl, HOBTDMF, 0 oC-rt, Overnight
NH(CH2CO2Et)2TEA, EDC.HCl, HOBTDMF, 0 oC-rt, Overnight
11
12 (62%)
1:1 TFA:DCMrt, 4h
13 R = Et (72%)
14 R = H (67%)1 N NaOHMeOH, 0 oC-rt, 1h
15 R = TBDMS (69%)
1:1 TFA:DCMrt, 4h
16 (71%)C-ol-ol
2. 1:1 TFA:DCM rt, 4h
17 R = TBDMS (69%)
1. 1 N NaOHMeOH, 0 oC-rt, 1h
1:1 TFA:DCM rt, 4h
19 (54%) 20 R = Et (56%)
18 (58%)C-ol-acid
21 R = H (66%)
1 N NaOHMeOH, 0 oC-rt, 1h
C-di-acid
C-acid
BOC = C =
NH2CH2CO2Et TEA, EDC.HCl, HOBT DMF, 0 oC-rt, Overnight
C
C
C
C
Scheme 2. Synthesis of cytosine-based modified acyclonucleosides.
2.2. RNase A Inhibition
Qualitative indication of RNase A inhibition by the synthetic inhibitors was obtained from acomparative agarose gel-based assay by monitoring the degradation of RNA by RNase A (Figures 2and 3). The most intense band observed in lane I in each gel is due to the presence of only RNA.The faint band in lane II is due to the maximum possible degradation of RNA by RNase A. Differentintensities of bands from lane III to VI revealed different extents of inhibition by the inhibitorsat a fixed concentration (0.5 mM). The histograms obtained by plotting the relative intensities ofthe bands revealed that compounds U-ol-acid (8), C-ol-acid (18), U-di-acid (10), and C-di-acid (21)were relatively more potent inhibitors in comparison to the others. These experimental observationsreaffirmed our assumption that increasing the acidity of the molecules leads to an increase in theinhibitory property of the inhibitor.
154
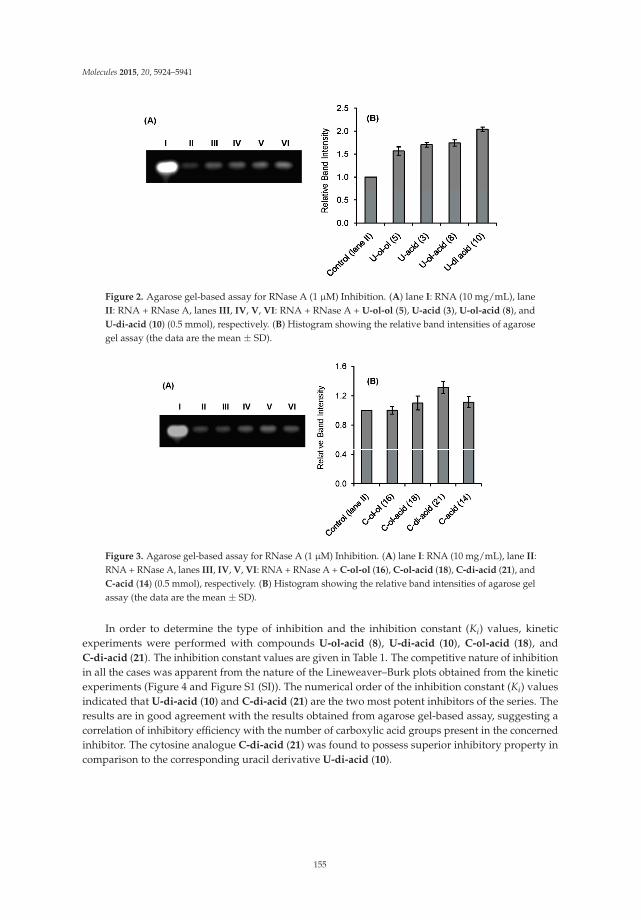
Molecules 2015, 20, 5924–5941
Figure 2. Agarose gel-based assay for RNase A (1 μM) Inhibition. (A) lane I: RNA (10 mg/mL), laneII: RNA + RNase A, lanes III, IV, V, VI: RNA + RNase A + U-ol-ol (5), U-acid (3), U-ol-acid (8), andU-di-acid (10) (0.5 mmol), respectively. (B) Histogram showing the relative band intensities of agarosegel assay (the data are the mean ± SD).
Figure 3. Agarose gel-based assay for RNase A (1 μM) Inhibition. (A) lane I: RNA (10 mg/mL), lane II:RNA + RNase A, lanes III, IV, V, VI: RNA + RNase A + C-ol-ol (16), C-ol-acid (18), C-di-acid (21), andC-acid (14) (0.5 mmol), respectively. (B) Histogram showing the relative band intensities of agarose gelassay (the data are the mean ± SD).
In order to determine the type of inhibition and the inhibition constant (Ki) values, kineticexperiments were performed with compounds U-ol-acid (8), U-di-acid (10), C-ol-acid (18), andC-di-acid (21). The inhibition constant values are given in Table 1. The competitive nature of inhibitionin all the cases was apparent from the nature of the Lineweaver–Burk plots obtained from the kineticexperiments (Figure 4 and Figure S1 (SI)). The numerical order of the inhibition constant (Ki) valuesindicated that U-di-acid (10) and C-di-acid (21) are the two most potent inhibitors of the series. Theresults are in good agreement with the results obtained from agarose gel-based assay, suggesting acorrelation of inhibitory efficiency with the number of carboxylic acid groups present in the concernedinhibitor. The cytosine analogue C-di-acid (21) was found to possess superior inhibitory property incomparison to the corresponding uracil derivative U-di-acid (10).
155

Molecules 2015, 20, 5924–5941
Table 1. Inhibition constants (Ki) of the inhibitors.
Inhibitor Ki * (μM)
U-ol-acid (8) 454 ± 9C-ol-acid (18) 356 ± 7U-di-acid (10) 301 ± 15C-di-acid (21) 235 ± 9
* The data are the mean ± SD.
Figure 4. Lineweaver–Burk plot for inhibition of RNase A by C-di-acid (21) of 0.15 (�), 0.05 (�), or0 ( ) mM, with 2',3'-cCMP concentrations of 0.75–0.52 mM and RNase A concentration of 9.8 μM.
Inhibitors of RNase A are known to perturb the secondary structure of the enzyme uponbinding [40–44]. A model drug–protein interaction study showed that 3'-azido-3'-deoxythymidineincreases the α-helix content of RNase A [40]. Similar increments were observed with 3'-O-carboxyesters of thymidine, which inhibited RNase A in reversible competitive mode [42]. Therefore, theprobable changes in the secondary structure of the enzyme by the inhibitors were monitored byobserving the CD spectra of RNase A in the absence or presence of compounds U-di-acid (10) andC-di-acid (21) (Figure 5). Both the inhibitors induced moderate changes in the secondary structureof the enzyme, which was reflected in the enhanced α-helix content. The α-helix content in nativeRNase A was 22.7%, which was found to increase upon binding with U-di-acid (10) and C-di-acid (21)to 24.7% and 29.2%, respectively.
The fluorescence emission intensity of RNase A due to the presence of six Tyr residues is found todecrease upon interactions with small molecule inhibitors [40–44]. The emission spectrum of RNaseA in presence of U-di-acid (2.10) and C-di-acid (2.21) at 25 ◦C showed quenching of the fluorescenceintensity of Tyr residues (Figure S2, (SI)). The fluorescence quenching study was used to calculatethe binding parameters for enzyme-inhibitor interactions. Binding constants (Kb) calculated from theexperiment were found to be in the order of 105 for C-di-acid (21) and 104 for U-di-acid (10) (Table S1,(SI)), indicating strong binding of the inhibitors with the enzyme.
156

Molecules 2015, 20, 5924–5941
Figure 5. CD spectra of RNase A in absence or presence of U-di-acid (10) and C-di-acid (21).
To gain an insight into the probable binding sites, protein-ligand docking studies were undertaken.The docked conformations shown in Figure 6 revealed that the nucleobase-amino acid conjugateswere in close proximity to the amino acid residues of the P1 subsite, resulting in a competitive modeof inhibition, as observed from the kinetic study. The carboxylic acid (-COOH) groups, in each case,were positioned close to His12 and His 119, probably engaged in hydrogen bonding with the residues.For the cytosine-based inhibitors, the N3 and –NH2 groups of the nucleobase were near to severalamino acid residues (Arg39, Lys41, Val43), thus increasing the possibility of polar interactions betweenthem. Such interactions were absent for the uracil-derived inhibitors. These extra interactions probablycontribute to the experimentally observed better inhibitory potency of the cytosine inhibitors. Apartfrom the interactions with the active site residues, one of the carboxylic acid (-COOH) groups ofU-di-acid (10) was found to be within hydrogen bonding distance of Oε1 of Gln11. Similarly, one–COOH group of C-di-acid (21) was engaged in hydrogen bonding with Phe120. Such favorableinteractions of the –COOH groups may possibly result in the better inhibition capacity of the inhibitors.The docked enzyme-inhibitor complexes also revealed that the nucleobase in the acyclic structure isnot in close proximity to the amino acid residues of the pyrimidine binding subsite B1 (Thr45, Asp83,Phe120). It can, therefore, be assumed that the ribose ring in the nucleosides may have a role inrecognition of the inhibitors by the enzyme. A detailed account of all the interactions between theinhibitors and the enzyme has been provided in Table S2 (SI).
A further clarification regarding the possible binding sites of the inhibitors was obtained bycalculating the changes in accessible surface area (ΔASA) of the interacting residues between the freeand complexed forms of the enzyme (Table 2). The measurements revealed that the accessible surfacearea of the amino acid residues of the P1 subsite (His12, His 119 and Lys 41) was largely affected uponbinding with the inhibitors. The observed results correlate well with the results of the docking study,suggesting that the inhibitors do bind to the P1 subsite of RNase A.
Table 2. Changes in accessible surface area (ΔASA) of the interacting residues between the uncomplexedand complexed forms of RNase A.
Amino AcidResidue
ASA (Å2) inRNase A
ΔASA (Å2) for Different Inhibitors
U-ol-Acid (8) C-ol-Acid (18) U-di-Acid (10) C-di-Acid (21)
Lys 7 88.03 42.88 41.75 29.62 43.82His 12 12.64 7.09 7.76 9.21 7.53Arg 39 142.03 25.81 29.72 5.06 30.79Lys 41 36.39 26.80 31.58 33.00 31.31
His 119 85.05 33.62 28.29 26.69 26.64
157

Molecules 2015, 20, 5924–5941
Figure 6. Docked poses of (A) C-di-acid (21) (yellow) and U-di-acid (10) (red), and (B) U-ol-acid (8)(green) and C-ol-acid (18) (brown) with RNase A (1FS3).
3. Experimental Section
3.1. General Methods
All reagents and fine chemicals were purchased from commercial suppliers and were usedwithout further purification. Column chromatography was performed with silica gel (230–400 mesh).Solvents were dried and distilled following standard methods. TLC was carried out on precoatedplates (Merck silica gel 60, f254). 1H and 13C-NMR for the compounds were recorded at 200 and 50MHz, respectively, using a Bruker NMR instrument. For 1H and 13C-NMR spectra in D2O, CH3CN wasused as internal standard. Chemical shifts are reported in parts per million (ppm, δ scale). Methylenecarbons have been identified using DEPT spectrum. Melting points were determined in open-endcapillary tubes. Bovine pancreatic RNase A, RNA (Torula utilis), and 2',3'-cCMP were purchased fromcommercial suppliers. UV-vis measurements were made using a UV-vis spectrophotometer (Shimadzu2450). Concentrations of the solutions were estimated spectrophotometrically using the following data:ε278.5 = 9800 M−1·cm−1 (RNase A) [45] and ε268 = 8500 M−1·cm−1 (2',3'-cCMP) [22]. CD measurementswere carried out on a Jasco-810 automatic recording spectrophotometer. Fluorescence measurementswere carried out using a Horiba Jobin Yvon Fluoromax-4 Spectrofluorimeter.
3.2. General Procedure for Carboxylic Acid–Amine Coupling Reaction
To a well-stirred solution of carboxylic acid (1, 11) (1 mmol) in DMF (5 mL) at 0 ◦C was addedEDC-HCl (1.2 mmol) followed by triethylamine (1.2 mmol) and the stirring was further continued atthis temperature. After 10 min, HOBT (1.2 mmol) was added and the reaction mixture was allowed towarm back to room temperature. Amine (1 mmol) was added to the resulting solution and the stirringwas allowed to continue overnight. The reaction mixture was then poured into a brine solution (50 mL)and extracted with EtOAc (3 × 20 mL). Organic extracts were pooled together, dried over anhydrousNa2SO4, filtered, and the filtrate was evaporated under reduced pressure. The resulting residue waspurified by column chromatography over silica gel (EtOAc/petroleum ether) to obtain pure couplingproducts (4, 6, 9, 12, 15, 17, 19).
3.3. General Procedure for Deprotection of –TBDMS Group
To a well-stirred solution of compound (4, 6) (1 mmol) in methanol (10 mL), was added CH3COCl(cat). The resulting solution was allowed to stir for 2 h at room temperature. The solvent wasevaporated under reduced pressure and the resulting residue was purified by column chromatographyover silica gel (DCM/MeOH) to obtain pure products (5, 7).
158

Molecules 2015, 20, 5924–5941
3.4. General Procedure for the Deprotection of –BOC Group/Simultaneous Deprotection of –BOC Groupand –TBDMS Group
The compound (12, 15, 19) (1 mmol) was dissolved in 1:1 TFA/DCM (5 mL) and the resultingsolution was stirred at room temperature for 4 h. The solvent was evaporated under reduced pressureand the resulting residue was purified by column chromatography over silica gel (DCM/MeOH) toobtain pure products (13, 16, 20).
3.5. General Procedure for Ester Hydrolysis
To a well-stirred solution of the compound (7, 9, 13, 20) (1 mmol) in methanol (5 mL) at 0 ◦Cwas added 1 N NaOH solution (2 mL) dropwise. The resulting solution was allowed to warm backup to room temperature and stirred for 1 h. After evaporation of methanol under reduced pressure,the resulting residue was dissolved in water (10 mL) and neutralized with acidic amberlyte. Theresulting mixture was filtered, and the filtrate was evaporated under reduced pressure. The resultingresidue was purified by column chromatography over silica gel (DCM/MeOH) to obtain pure products(8, 10, 14, 21).
N,N-Bis-[2-(tert-butyldimethylsilyloxy)-ethyl]-2-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetamide (4):Compound 1 (0.68 g, 3.99 mmol) was converted to compound 4 (1.28 g, 66%) following the generalprocedure in Section 3.2. Rf = 0.4 [30% EtOAc in pet ether]. White solid. M.P: 122–124 ◦C. 1H-NMR(200 MHz, CDCl3): δ 0.02 (s, 6H), 0.06 (s, 6H), 0.86 (s, 9H), 0.88 (s, 9H), 3.47 (t, J = 5.5 Hz, 2H), 3.59 (t,J = 5.1 Hz, 2H), 3.71–3.80 (m, 4H), 4.66 (s, 2H), 5.69 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 9.28 (bs,1H). 13C-NMR (50 MHz, CDCl3): δ −5.3, −5.2, 18.3, 18.5, 26.0, 26.1, 48.1 (CH2), 49.2 (CH2), 51.1 (CH2),61.0(CH2), 61.5 (CH2), 102.1, 145.4, 151.2, 163.9, 167.0. HRMS (ESI+): m/z calcd for C22H44N3O5Si2(M+H)+: 486.2820; found: 486.2827.
2-(2,4-Dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-N,N-bis-(2-hydroxy-ethyl) acetamide (5): Compound 5 [38](0.39 g, 81%) was obtained from compound 4 (0.9 g, 1.85 mmol) following the general procedure inSection 3.3. White solid. Rf = 0.4 [15% MeOH in DCM]. 1H-NMR (200 MHz, DMSO-d6): δ 3.31–3.58 (m,8H), 4.67 (s, 2H), 4.75–4.80 (m, 1H), 5.01–5.05 (m, 1H), 5.54 (d, J = 7.8 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H),11.23 (bs, 1H).
{[2-(tert-Butyldimethylsilyloxy) ethyl]-[2-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl-acetyl] amino}acetic acidethyl ester (6): Compound 1 (0.93 g, 5.47 mmol) was converted to compound 6 (1.40 g, 62%) followingthe general procedure in Section 3.2. Rf = 0.3 [40% EtOAc in pet ether]. White solid. M.P: 171–172 ◦C.The compound was obtained as a mixture of rotamers as indicated by the NMR spectra. 1H-NMR(200 MHz, CDCl3): δ 0.01 (s, 6H), 0.06 (s, 6H), 0.86 (s, 9H), 0.87 (s, 9H), 1.20–1.32 (m, 3H), 3.52–3.56(m, 2H), 3.70–3.81 (m, 2H), 4.10–4.24 (m, 4H), 4.30 (s, 2H), 4.46 (s, 2H), 4.71 (s, 2H), 5.68–5.73 (m, 1H),7.13–7.26 (m, 1H), 9.39 (bs, 1H), 9.51 (bs, 1H). 13C-NMR (50 MHz, CDCl3): δ −5.3, 14.3, 18.4, 26.0, 26.1,47.9 (CH2), 48.6 (CH2), 50.8 (CH2), 61.4 (CH2), 61.6 (CH2), 62.2 (CH2), 102.3, 145.4, 151.1, 163.9, 167.5,168.9, 172.9. HRMS (ESI+): m/z calcd for C18H32N3O6Si (M+H)+: 414.2060; found: 414.2088.
[[2-(2,4-Dioxo-3,4-dihydro-2H-pyrimidin-1-yl-acetyl]-(2-hydroxyethyl)-amino] acetic acid ethyl ester (7):Compound 7 (0.48 g, 55%) was generated from compound 6 (1.21 g, 2.90 mmol) following the generalprocedure in Section 3.3. Rf = 0.4 [10% MeOH in DCM]. Light yellow gum. The compound wasobtained as a mixture of rotamers as indicated by the NMR spectra. 1H-NMR (200 MHz, DMSO-d6):δ 1.14–1.26 (m, 3H), 3.34–3.58 (m, 4H), 4.01–4.18 (m, 4H), 4.34 (s, 2H), 4.53 (s, 2H), 4.74 (s, 2H), 5.53–5.57(m, 1H), 7.34–7.45 (m, 1H), 11.25 (bs, 1H). 13C-NMR (50 MHz, DMSO-d6): δ 14.0, 48.0 (CH2), 49.9 (CH2),50.7 (CH2), 58.3 (CH2), 58.9 (CH2), 60.5 (CH2), 61.03(CH2), 100.6, 146.5, 151.0, 163.9, 167.4, 167.5, 169.1,169.4. HRMS (ESI+): m/z calcd for C12H17N3O6Na (M+Na)+: 322.1015; found: 322.1003.
[[2-(2,4-Dioxo-3,4-dihydro-2H-pyrimidin-1-yl-acetyl]-(2-hydroxyethyl) amino] acetic acid (8): Compound7 (0.4 g, 1.34 mmol) was converted to compound 8 (0.22 g, 62%) following the general procedure inSection 3.5. Rf = 0.2 [20% MeOH in DCM]. White solid. M.P: 177–179 ◦C. The compound was obtained
159

Molecules 2015, 20, 5924–5941
as a mixture of rotamers as indicated by the NMR spectra. 1H-NMR (200 MHz, D2O): δ 3.54–3.82 (m,4H), 4.19 (s, 2H), 4.37 (s, 2H), 4.57 (s, 2H), 4.90 (s, 2H), 5.80–5.87 (m, 1H), 7.50–7.56 (m, 1H). 13C-NMR(50 MHz, D2O): δ 49.2 (CH2), 50.0 (CH2), 50.6 (CH2), 51.0 (CH2), 59.3 (CH2), 59.4 (CH2), 102.2, 148.1,152.6, 167.2, 170.0, 173.3. HRMS (ESI+): m/z calcd for C10H13N3O6Na (M+Na)+: 294.0702; found:294.0707.
{[2-(2,4-Dioxo-3,4-dihydro-2H-pyrimidin-1-yl-acetyl] ethoxycarbonylmethylamino}acetic acid ethyl ester (9):Compound 1 (0.52 g, 3.05 mmol) was transformed to compound 9 (0.51 g, 49%) following the generalprocedure in Section 3.2. Rf = 0.5 [70% EtOAc in pet ether]. White solid. M.P: 140–142 ◦C. 1H-NMR(200 MHz, CDCl3): δ 1.22–1.34 (m, 6H), 4.12–4.31 (m, 8H), 4.61 (s, 2H), 5.73 (d, J = 8.0 Hz, 1H), 7.20(d, J = 7.8 Hz, 1H), 9.09 (bs, 1H). 13C-NMR (50 MHz, CDCl3): δ 14.3, 47.7 (CH2), 49.0 (CH2), 50.2(CH2), 61.8 (CH2), 62.5 (CH2), 102.6, 145.2, 151.0, 163.7, 167.6, 168.6. HRMS (ESI+): m/z calcd forC14H19N3O7Na (M+Na)+: 364.1121; found: 364.1109.
{Carboxymethyl-[2-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl) acetyl] amino}acetic acid (10): Compound9 (0.42 g, 1.23 mmol) was converted to compound 10 (0.26 g, 74%) following the general procedurein Section 3.5. Rf = 0.2 [30% MeOH in DCM]. White solid. M.P: 118–120 ◦C 1H-NMR (200 MHz,DMSO-d6): δ 4.00 (s, 2H), 4.25 (s, 2H), 4.62 (s, 2H), 5.55 (d, J = 7.2 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 11.28(s, 1H). 13C-NMR (50 MHz, DMSO-d6): δ 47.7 (CH2), 48.4 (CH2), 49.2 (CH2), 100.7, 146.4, 151.0, 163.8,167.7, 170.3. HRMS (ESI+): m/z calcd for C10H11N3O7Na (M+Na)+: 308.0495; found: 308.0475.
{[4-[Bis(1,1-dimethylethoxy)carbonyl]amino-2-oxo-2H-pyrimidin-1-yl] acetylamino}acetic acid ethyl ester (12):Compound 11 (0.39 g, 1.05 mmol) was converted to compound 12 (0.30 g, 62%) following the generalprocedure in Section 3.2. Rf = 0.5 [60% EtOAc in pet ether]. White solid. M.P: 118–120 ◦C. 1H-NMR(200 MHz, CDCl3): δ 1.18–1.25 (m, 3H), 1.52 (s, 18H), 3.95 (d, J = 5.6 Hz, 2H), 4.12 (q, J = 7.2 Hz, 2H),4.58 (s, 2H), 7.08 (d, J = 7.2 Hz, 1H), 7.69–7.76 (m, 1H). 13C-NMR (50 MHz, CDCl3): δ 14.1, 27.7, 41.4(CH2), 52.8 (CH2), 61.4 (CH2), 85.0, 96.7, 149.0, 149.4, 152.6, 155.5, 162.7, 167.1, 169.4. HRMS (ESI+):m/z calcd for C20H30N4O8Na (M+Na)+: 477.1961; found: 477.1975.
[2-(4-Amino-2-oxo-2H-pyrimidin-1-yl) acetylamino] acetic acid ethyl ester (13): Compound 12 (0.28 g,0.62 mmol) was converted to compound 13 (0.11 g, 72%) following the general procedure in Section 3.4.Rf = 0.3 [5% MeOH in DCM]. White solid. M.P: 155–158 ◦C. 1H-NMR (200 MHz, DMSO-d6): δ 1.17 (t,J = 7.2 Hz, 3H), 3.86 (d, J = 5.8 Hz, 2H), 4.07 (q, J = 7.2 Hz, 2H), 4.42 (s, 2H), 5.82 (d, J = 7.2 Hz, 1H), 7.65(d, J = 7.4 Hz, 1H), 7.90 (bs, 1H), 8.09 (bs, 1H), 8.61 (t, J = 5.8 Hz, 1H). 13C-NMR (50 MHz, DMSO-d6):δ 14.5, 41.2 (CH2), 50.9 (CH2), 61.0 (CH2), 93.8, 148.8, 153.5, 164.4, 168.0, 170.0. HRMS (ESI+): m/z calcdfor C10H15N4O4 (M+H)+: 255.1093; found: 255.1076.
[2-(4-Amino-2-oxo-2H-pyrimidin-1-yl) acetylamino] acetic acid (14): Compound 13 (0.09 g, 0.35 mmol) wastransformed to compound 14 (0.05 g, 67%) following the general procedure in Section 3.5. Rf = 0.3 [20%MeOH in DCM]. Yellowish white solid. M.P: > 200 ◦C. 1H-NMR (200 MHz, DMSO-d6): δ 3.61–3.95 (m,2H), 4.54 (s, 2H), 6.22 (d, J = 7.2 Hz, 1H), 7.99 (d, J = 7.4 Hz, 1H), 8.77 (s, 1H), 8.94 (s, 1H), 10.17 (s, 1H).13C-NMR (50 MHz, DMSO-d6): δ 40.8 (CH2), 50.4 (CH2), 93.1, 147.4, 150.6, 160.1, 166.4, 170.8. HRMS(ESI+): m/z calcd for C8H11N4O4 (M+H)+: 227.0780; found: 227.0784.
[4-[Bis(1,1-dimethylethoxy)carbonyl]amino-2-oxo-2H-pyrimidin-1-yl]-N,N-bis-[2(tertbutyldimethyl silyloxy)ethyl]acetamide (15): Compound 15 (0.55 g, 69%) was obtained from compound 11 (0.43 g, 1.16 mmol)following the general procedure in Section 3.2. Rf = 0.3 [25% EtOAc in pet ether]. Colourless gum.1H-NMR (200 MHz, CDCl3): δ −0.03 (s, 6H), 0.01 (s, 6H), 0.81 (s, 9H), 0.83 (s, 9H), 1.49 (s, 18H), 3.43 (t,J = 5.6 Hz, 2H), 3.64–3.74 (m, 6H), 4.70 (s, 2H), 7.00 (d, J = 7.4 Hz, 1H), 7.52 (d, J = 7.4 Hz, 1H). 13C-NMR(50 MHz, CDCl3): δ −5.4, 18.2, 18.3, 26.0, 27.7, 49.3 (CH2), 49.5 (CH2), 51.1 (CH2), 61.0 (CH2), 61.2(CH2), 84.8, 96.1, 149.1, 149.6, 155.1, 162.6, 167.1. HRMS (ESI+): m/z calcd for C32H61N4O8Si2 (M+H)+:685.4028; found: 685.4038.
160

Molecules 2015, 20, 5924–5941
2-(4-Amino-2-oxo-2H-pyrimidin-1-yl)-N,N-bis-(2-hydroxyethyl) acetamide (16): Compound 15 (0.48 g,0.70 mmol) was transformed to compound 16 (0.13 g, 71%) following the general procedure inSection 3.4. Rf = 0.3 [15% MeOH in DCM]. White solid. M.P: 150 ◦C (decomposed). 1H-NMR(200 MHz, DMSO-d6): δ 2.99–3.04 (m, 1H), 3.34–3.68 (m, 8H), 4.65 (s, 2H), 5.78 (d, J = 7.2 Hz, 1H),7.51 (d, J = 7.2 Hz, 1H), 7.75 (bs, 2H). 13C-NMR (50 MHz, DMSO-d6): δ 48.8 (CH2), 49.3 (CH2), 49.9(CH2), 58.7 (CH2), 59.0 (CH2), 93.2, 148.1, 154.4, 164.8, 167.3. HRMS (ESI+): m/z calcd for C10H17N4O4
(M+H)+: 257.1250; found: 257.1255.
{[4-[Bis(1,1-dimethylethoxy)carbonyl]amino-2-oxo-2H-pyrimidin-1-yl-acetyl]-[2-(tert-butyldimethylsilyloxy)ethyl] acetic acid ethyl ester (17): Compound 11 (0.46 g, 1.24 mmol) was converted to compound 17 (0.52g, 69%) following the general procedure in Section 3.2. Rf = 0.4 [40% EtOAc in pet ether]. Colorlessgum. The compound was obtained as a mixture of rotamers as indicated by the NMR spectra.1H-NMR (200 MHz, CDCl3): δ −0.03–0.01 (m, 6H), 0.81 (s, 9H), 0.83 (s, 9H), 1.15–1.27 (m, 3H), 1.49 (s,18H), 3.33–3.80 (m, 4H), 4.05–4.18 (m, 4H), 4.40 (s, 2H), 4.49 (s, 2H), 4.75 (s, 2H), 6.97–7.03 (m, 1H),7.54–7.62 (m, 1H). 13C-NMR (50 MHz, CDCl3): δ −5.4, 14.2, 18.2, 25.9, 27.7, 49.0 (CH2), 49.3 (CH2), 51.1(CH2), 51.2 (CH2), 61.2 (CH2), 61.6 (CH2), 61.8 (CH2), 62.0 (CH2), 84.8, 96.2, 149.0, 149.1, 149.5, 155.0,162.7, 162.8, 167.4, 167.5, 168.8, 169.7. HRMS (ESI+): m/z calcd for C28H49N4O9Si (M+H)+: 613.3269;found: 613.3278.
[[2-(4-Amino-2-oxo-2H-pyrimidin-1-yl) acetyl]-(2-hydroxyethyl) amino] acetic acid (18): Compound 17
(0.40 g, 0.65 mmol) was hydrolyzed to corresponding acid following the general procedure inSection 3.5. The crude residue obtained was subjected to TFA treatment as mentioned in the generalprocedure in Section 3.4, and purified by column chromatography over silica gel to obtain 18 (0.10 g,58%). Rf = 0.2 [30% MeOH in DCM]. White solid. M.P: 184–188 ◦C. The compound was obtained as amixture of rotamers, as indicated by the NMR spectra. 1H-NMR (200 MHz, DMSO-d6): δ 3.35–3.59(m, 4H), 4.01 (s, 2H), 4.24 (s, 2H), 4.54 (s, 2H) 4.75 (s, 2H), 5.84 (d, J = 7.0 Hz, 1H), 7.55 (d, J = 7.0 Hz,1H), 7.97 (bs, 2H). 13C-NMR (50 MHz, DMSO-d6): δ 47.9 (CH2), 49.1 (CH2), 49.9 (CH2), 58.9 (CH2),93.4, 148.6, 152.8, 163.6, 167.6, 170.7, 171.1. HRMS (ESI+): m/z calcd for C10H15N4O5 (M+H)+: 271.1042;found: 271.1039.
{[4-[Bis(1,1-dimethylethoxy)carbonyl]amino-2-oxo-2H-pyrimidin-2-yl-acetyl] ethoxycarbonylmethylamino}acetic acid ethyl ester (19): Compound 19 (0.29 g, 54%) was generated from compound 11 (0.37 g,1.00 mmol) following the general procedure in Section 3.2. Rf = 0.4 [50% EtOAc in pet ether]. Colorlessgum. 1H-NMR (200 MHz, CDCl3): δ 1.17–1.30 (m, 6H), 1.51 (s, 18H), 4.07–4.25 (m, 6H), 4.32 (s, 2H),4.67 (s, 2H), 7.05 (d, J = 7.4 Hz, 1H), 7.62 (d, J = 7.4 Hz, 1H). 13C-NMR (50 MHz, CDCl3): δ 14.2, 27.8,49.1 (CH2), 49.4 (CH2), 50.5 (CH2), 61.6 (CH2), 62.1 (CH2), 85.0, 96.5, 149.0, 149.5, 155.1, 162.8, 167.9,168.7, 168.9. HRMS (ESI+): m/z calcd for C24H37N4O10 (M+H)+: 541.2510; found: 541.2516.
{[2-(4-Amino-2-oxo-2H-pyrimidin-1-yl) acetyl] ethoxycarbonylmethylamino}acetic acid ethyl ester (20):Compound 19 (0.27 g, 0.50 mmol) was converted to compound 20 (0.09 g, 56%) following the generalprocedure in Section 3.4. Rf = 0.3 [3% MeOH in DCM]. White solid. M.P: 152–154 ◦C. 1H-NMR(200 MHz, DMSO-d6): δ 1.14–1.27 (m, 6H), 4.00–4.22 (m, 6H), 4.39 (s, 2H), 4.80 (s, 2H), 6.10 (d, J = 7.6 Hz,1H), 7.81 (d, J = 7.6 Hz, 1H), 9.52 (bs, 1H), 9.67 (bs, 1H). 13C-NMR (50 MHz, DMSO-d6): δ 14.0, 48.5(CH2), 49.1 (CH2), 60.6 (CH2), 61.1 (CH2), 93.5, 148.2, 150.2, 160.6, 167.3, 168.5, 168.8. HRMS (ESI+):m/z calcd for C14H21N4O6 (M+H)+: 341.1461; found: 341.1452.
{[2-(4-Amino-2-oxo-2H-pyrimidin-1-yl)-acetyl] carboxymethylamino}acetic acid (21): Compound 20 (0.07 g,0.20 mmol) was transformed to compound 21 (0.04 g, 66%) following the general procedure inSection 3.5. Rf = 0.15 [40% MeOH in DCM]. Eluent: 30%–50% MeOH in DCM. White solid. M.P:190–193 ◦C. 1H-NMR (200 MHz, D2O): δ 4.23 (s, 2H), 4.39 (s, 2H), 4.89 (s, 2H), 6.24 (d, J = 7.4 Hz, 1H),7.79 (d, J = 7.8 Hz, 1H). 13C-NMR (50 MHz, DMSO-d6): δ 48.8 (CH2), 49.3 (CH2), 49.9 (CH2), 93.4, 148.4,152.2, 163.5, 167.8, 170.4, 170.7. HRMS (ESI+): m/z calcd for C10H13N4O6 (M+H)+: 285.0835; found:285.0852.
161

Molecules 2015, 20, 5924–5941
3.6. Comparative Agarose Gel-Based Assay
Inhibition of RNase A was assayed qualitatively by the degradation of RNA in an agarose gel.In this method, 20 μL of RNase A (1 μM) was mixed with 20 μL (0.5 mM) of compounds U-ol-ol (5),C-ol-ol (16), U-acid (3), C-acid (14), U-ol-acid (8), C-ol-acid (18), U-di-acid (10), and C-di-acid (21)separately to a final volume of 50 μL and the resulting solutions incubated for 3 h. Twenty-microliteraliquots of the incubated mixtures were then mixed with 20 μL of RNA solution (10.0 mg/mL RNA,freshly dissolved in RNase free water) and incubated for another 30 min. Then 10 μL of sample buffer(containing 10% glycerol and 0.025% bromophenol blue) were added to this mixture and 15 μL fromeach solution were extracted and loaded onto a 1.1% agarose gel. The gel was run using a 0.04-MTris-Acetic acid-EDTA (TAE) buffer (pH 8.0). The residual RNA was visualized by ethidium bromidestaining under UV light.
3.7. Inhibition Kinetics with RNase A
A quantitative account of RNase A inhibition by the individual inhibitors was obtained by aUV spectroscopic method described by Anderson and co-workers [22]. The assay was performedin a 0.1-M Mes-NaOH buffer, pH 6.0 containing 0.1 M NaCl using 2',3'-cCMP as the substrate.The inhibition constants were calculated from initial velocity data using a Lineweaver–Burk plot.The slopes from the Lineweaver–Burk double reciprocal plot were plotted against the correspondinginhibitor concentrations to get inhibition constants (Ki).
3.8. Circular Dichroism Measurements
Circular Dichroism (CD) was performed in order to monitor the changes in the secondary structureof the enzyme as a result of interaction with the inhibitors. In this method, 200 μL of RNase A (30 μM)were mixed separately with 200 μL of U-di-acid (10) (30 μM) and C-di-acid (21) (30 μM) and incubatedfor 3 h. From the resulting solutions, an aliquot of 300 μL was used for CD measurements, takinga 1 mm path length quartz cell. The spectra were recorded in the range of 190–240 nm with a scanrate of 50 nm/min. Three scans were accumulated for each spectrum. The secondary structure wasdetermined using an online server, DICHROWEB [46].
3.9. Fluorescence Spectroscopy
Fluorescence quenching study was performed to garner an idea of the binding affinities ofU-di-acid (10) and C-di-acid (21) towards RNase A. The emission spectra were recorded from 290 to400 nm with excitation at 275 nm [47] using a 5 nm slit width. The interaction between the ligandsand RNase A was investigated by titration of 3 mL solution of RNase A with successive addition ofthe respective ligands (0–15 μM) in a 20-mM phosphate buffer of pH 7.0. Binding constants (Kb) werecalculated using double-logarithm plot [48].
3.10. FlexX Docking
The crystal structure of RNase A (PDB entry 1FS3) was downloaded from the Protein DataBank [49]. The 3D structures of the inhibitors were generated in Sybyl6.92 (Tripos Inc., St. Louis,MO, USA). Minimum energy conformations were obtained with the help of the MMFF94 force fieldusing MMFF94 charges with a gradient of 0.005 kcal/mole by 1000 iterations with all other defaultparameters. The ligands were docked with the protein using FlexX software. The ranking of thegenerated solutions was performed using a scoring function that estimates the free binding energy(ΔG) of the protein-ligand complex considering various types of molecular interactions [50]. Dockedconformations were visualized using PyMol [51].
162

Molecules 2015, 20, 5924–5941
3.11. Accessible Surface Area Calculations
Accessible surface area of uncomplexed RNase A and its docked complexes were calculated usingthe program NACCESS. The structures obtained from the FlexX analysis were used for the calculation.The change in ASA for a particular residue X was calculated using: ΔASAX = ASAX
RNase A −ASAX
RNase A + inhibitor.
4. Conclusions
Strategically designed carboxylated acyclonucleosides have been established as moderate RNaseA inhibitors. The experimental outcome points towards the possible contribution of a sugar ring inRNase A inhibition. Cytosine analogues have been proven to have better inhibitory properties thanthe corresponding uracil derivatives. C-di-acid (21) emerged as the most potent inhibitor of the series,having an inhibition constant (Ki) value of 235 μM. It was observed that an increment in the numberof carboxylic acid groups resulted in better inhibitory properties. However, the absence of the rigidribose ring in the molecules significantly affects the inhibition properties of the synthetic nucleosides.These findings should act as a guideline for future design of inhibitors for RNase A and other membersof the ribonuclease superfamily.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/04/5924/s1.
Acknowledgments: The authors thank the Department of Biotechnology, Ministry of Science and Technology,New Delhi for funding (project no. SB/S1/OC-30/2014). Kaustav Chakraborty thanks D. Tripathy and S. Ghoshfor their help in some of the experiments and the Council for Scientific and Industrial Research, New Delhi fora fellowship.
Author Contributions: S.D.G and T.P conceived the idea and designed the study. K.C performed the syntheticand biological experiments, and analyzed the data. K.C, S.D.G, and T.P wrote the manuscript together. All authorsread and approved the final version of the article.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. D’Alessio, G. The superfamily of vertebrate-secreted ribonucleases. In Ribonucleases; Nicholson, A.W., Ed.;Springer: Heidelberg, Germany, 2011; pp. 1–34.
2. Loverix, S.; Steyaert, J. Ribonucleases: from prototypes to therapeutic targets. Curr. Med. Chem. 2003, 10,779–785. [CrossRef] [PubMed]
3. Viola, M.; Libra, M.; Callari, D.; Sinatra, F.; Spada, D.; Noto, D.; Emmanuele, G.; Romano, F.;Averna, M.; Pezzino, F.M.; et al. Bovine seminal ribonuclease is cytotoxic for both malignant and normaltelomerase-positive cells. Int. J. Oncol. 2005, 27, 1071–1077. [PubMed]
4. Zrinski, R.T.; Dodig, S. Eosinophil cationic protein-current concept and controversies. Biochem. Med. 2011,21, 111–121. [CrossRef]
5. Li, S.; Ibaragi, S.; Hu, G. Angiogenin as a molecular target for the treatment of prostate cancer. Curr. CancerTher. Rev. 2011, 7, 83–90. [CrossRef] [PubMed]
6. Fang, E.F.; Ng, T.B. Ribonucleases of different origins with a wide spectrum of medicinal applications.Biochim. Biophys. Acta 2011, 1815, 65–74. [PubMed]
7. Leland, P.A.; Schultz, L.W.; Kim, B.; Raines, R.T. Ribonuclease A variants with potent cytotoxic activity.Proc. Natl. Acad. Sci. USA 1998, 95, 10407–10412. [CrossRef] [PubMed]
8. Leland, P.A.; Staniszewski, K.E.; Park, C.; Kelemen, B.R.; Raines, R.T. The ribonucleolytic activity ofangiogenin. Biochemistry 2002, 41, 1343–1350. [CrossRef] [PubMed]
9. Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1065. [CrossRef] [PubMed]10. Marshall, G.; Feng, J.A.; Kuster, D.J. Back to the future: ribonuclease A. Biopolymers 2007, 90, 259–277.
[CrossRef]11. Shapiro, R.; Weremowicz, S.; Riordan, J.F.; Vallee, B.L. Ribonucleolytic activity of angiogenin: Essential
histidine, lysine, and arginine residues. Proc. Natl. Acad. Sci. USA 1987, 84, 8783–8787. [CrossRef] [PubMed]
163

Molecules 2015, 20, 5924–5941
12. Hamann, K.J.; Barker, R.L.; Loegering, D.A.; Pease, L.R.; Gleich, G.L. Sequence of human eosinophil-derivedneurotoxin cDNA: Identity of deduced amino acid sequence with human nonsecretory ribonucleases. Gene1989, 83, 161–167. [CrossRef] [PubMed]
13. Gagné, D.; Charest, L.; Morin, S.; Kovrigin, E.L.; Doucet, N. Conservation of flexible residue clusters amongstructural and functional enzyme homologues. J. Biol. Chem. 2012, 287, 44289–44300. [CrossRef] [PubMed]
14. Maiti, T.K.; De, S.; Dasgupta, S.; Pathak, T. 3'-N-Alkylamino-3'-deoxy-ara-uridines: A new class of potentialinhibitors of ribonuclease A and angiogenin. Bioorg. Med. Chem. 2006, 14, 1221–1228. [CrossRef]
15. Leonidas, D.D.; Maiti, T.K.; Samanta, A.; Dasgupta, S.; Pathak, T.; Zographos, S.E.; Oikonomakos, N.G.The binding of 3'-N-piperidine-4-carboxyl-3'-deoxy-ara-uridine to ribonuclease A in the crystal. Bioorg.Med. Chem. 2006, 14, 6055–6066. [CrossRef] [PubMed]
16. Debnath, J.; Dasgupta, S.; Pathak, T. Inhibition of ribonuclease A by nucleoside-dibasic acid conjugates.Bioorg. Med. Chem. 2009, 17, 6491–6496. [CrossRef] [PubMed]
17. Debnath, J.; Dasgupta, S.; Pathak, T. Comparative inhibitory activity of 3'- and 5'-functionalized nucleosideson ribonuclease A. Bioorg. Med. Chem. 2010, 18, 8257–8263. [CrossRef] [PubMed]
18. Samanta, A.; Dasgupta, S.; Pathak, T. 5'-modified pyrimidine nucleosides as inhibitors of ribonuclease A.Bioorg. Med. Chem. 2009, 17, 6491–6496. [CrossRef] [PubMed]
19. Datta, D.; Samanta, A.; Dasgupta, S.; Pathak, T. 3'-Oxo-, amino-, thio-, and sulfone-acetic acid modifiedthymidines: Effect of increased acidity on ribonuclease A inhibition. Bioorg. Med. Chem. 2013, 21, 4634–4645.[CrossRef] [PubMed]
20. Datta, D.; Samanta, A.; Dasgupta, S.; Pathak, T. Synthesis of 5'-carboxymethylsulfonyl-5'-deoxyribonucleosidesunder mild hydrolytic conditions: A new class of acidic nucleosides as inhibitors of ribonuclease A. RSC Adv.2014, 4, 2214–2218. [CrossRef]
21. Datta, D.; Dasgupta, S.; Pathak, T. Ribonuclease A inhibition by carboxymethylsulfonyl-modified xylo- andarabinopyrimidines. ChemMedChem 2014, 9, 2138–2149. [CrossRef] [PubMed]
22. Anderson, D.G.; Hammes, G.G.; Walz, F.G. Binding of phosphate ligands to ribonuclease A. Biochemistry1968, 7, 1637–1645. [CrossRef] [PubMed]
23. Walz, F.G. Kinetic and equilibrium studies on the interaction of ribonuclease A and 2'-deoxyuridine3'-phosphate. Biochemistry 1971, 10, 2156–2162. [CrossRef] [PubMed]
24. Russo, N.; Shapiro, R.; Vallee, B.L. 5'-Diphosphoadenosine-3'-phosphate is a potent inhibitor of bovinepancreatic ribonuclease A. Biochem. Biophys. Res. Commun. 1997, 231, 671–674. [CrossRef] [PubMed]
25. Leonidas, D.D.; Shapiro, R.; Irons, L.I.; Russo, N.; Acharya, K.R. Crystal structures of ribonuclease Acomplexes with 5'-diphosphoadenosine 3'-phosphate and 5'-diphosphoadenosine 2'-phosphate at 1.7 Åresolution. Biochemistry 1997, 36, 5578–5588. [CrossRef] [PubMed]
26. Leonidas, D.D.; Shapiro, R.; Irons, L.I.; Russo, N.; Acharya, K.R. Towards rational design of ribonuclease inhibitors:High resolution crystal structure of a ribonuclease A complex with a potent 3',5'-pyrophosphate-linkeddinucleotide inhibitor. Biochemistry 1999, 38, 10287–10297. [CrossRef] [PubMed]
27. Russo, N.; Shapiro, R. Potent inhibition of mammalian ribonucleases by 3',5'-pyrophosphate-linkednucleotides. J. Biol. Chem. 1999, 274, 14902–14908. [CrossRef] [PubMed]
28. Leonidas, D.D.; Chavali, G.B.; Oikonomakos, N.G.; Chrysina, E.D.; Kosmopoulou, M.N.; Vlassi, M.;Frankling, C.; Acharya, K.R. High-resolution crystal structures of ribonuclease A complexed with adenylicand uridylic nucleotide inhibitors. Implications for structure-based design of ribonucleolytic inhibitors.Protein Sci. 2003, 12, 2559–2574. [CrossRef] [PubMed]
29. Kumar, K.; Jenkins, J.L.; Jardine, A.M.; Shapiro, R. Inhibition of mammalian ribonucleases by endogenousadenosine dinucleotides. Biochem. Biophys. Res. Commun. 2003, 300, 81–86. [CrossRef] [PubMed]
30. Jenkins, C.L.; Thiyagarajan, N.; Sweeney, R.Y.; Guy, M.P.; Kelemen, B.R.; Acharya, K.R.; Raines, R.T. Bindingof non-natural 3'-nucleotides to ribonuclease A. FEBS J. 2005, 272, 744–755. [CrossRef] [PubMed]
31. Hatzopoulos, G.N.; Leonidas, D.D.; Kardakaris, R.; Kobe, J.; Oikonomakos, N.G. The binding of IMP toribonuclease A. FEBS J. 2005, 272, 3988–4001. [CrossRef] [PubMed]
32. Yakovlev, G.I.; Mitkevich, V.A.; Makarov, A.A. Ribonuclease inhibitors. Mol. Biol. 2006, 40, 867–874.[CrossRef]
33. Nogués, M.V.; Vilanova, M.; Cuchillo, C.M. Bovine pancreatic ribonuclease A as a model of an enzyme withmultiple substrate binding sites. Biochim. Biophys. Acta 1995, 1253, 16–24. [CrossRef] [PubMed]
164

Molecules 2015, 20, 5924–5941
34. Findlay, D.; Herries, D.G.; Mathias, A.P.; Rabin, B.R.; Ross, C.A. The active site and mechanism of action ofbovine pancreatic ribonuclease. Nature 1961, 190, 781–784. [CrossRef] [PubMed]
35. Cuchillo, C.M.; Nogués, M.V.; Raines, R.T. Bovine pancreatic ribonuclease: Fifty years of the first enzymaticreaction mechanism. Biochemistry 2011, 50, 7835–7841. [CrossRef] [PubMed]
36. Silverman, R.B. The Organic Chemistry of Drug Design and Drug Action, 2nd ed.; Elsevier: San Diego, CA, USA,2004; p. 126.
37. Liu, X.; Chen, R. Synthesis of novel phosphonotripeptides containing uracil or thymine group. PhosphorusSulfur Silicon Relat. Elem. 2001, 176, 19–28. [CrossRef]
38. Rezazgui, O.; Boëns, B.; Teste, K.; Vergaud, J.; Trouillas, P.; Zerrouki, R. One-pot and catalyst-free amidationof ester: A matter of non-bonding interactions. Tetrahedron Lett. 2011, 52, 6796–6799. [CrossRef]
39. Porcheddu, A.; Giacomelli, G.; Piredda, I.; Carta, M.; Nieddu, G. A practical and efficient approach to PNAmonomers compatible with Fmoc-mediated solid-phase synthesis protocols. Eur. J. Org. Chem. 2008, 34,5786–5797. [CrossRef]
40. Gaudreau, S.; Novetta-dellen, A.; Neault, J.F.; Diamantoglou, S.; Tajmir-riahi, H.A. 3'-azido-3'-deoxythymidine binding to ribonuclease A: Model for drug-protein interaction. Biopolymers 2003, 72,435–441. [CrossRef] [PubMed]
41. Ghosh, K.S.; Maiti, T.K.; Mandal, A.; Dasgupta, S. Copper complexes of (-)-epicatechin gallate and(-)-epigallactocatechin gallate act as inhibitors of ribonuclease A. FEBS Lett. 2006, 580, 4703–4708. [CrossRef][PubMed]
42. Ghosh, K.S.; Debnath, J.; Dutta, P.; Sahoo, B.K.; Dasgupta, S. Exploring the potential of 3'-O-carboxy estersof thymidine as inhibitors of ribonuclease A and angiogenin. Bioorg. Med. Chem. 2008, 16, 2819–2828.[CrossRef] [PubMed]
43. Dutta, S.; Basak, A.; Dasgupta, S. Synthesis and ribonuclease A inhibition activity of resorcinol andphloroglucinol derivatives of catechin and epicatechin: Importance of hydroxyl groups. Bioorg. Med. Chem.2010, 18, 6538–6546. [CrossRef] [PubMed]
44. Tripathy, D.R.; Roy, A.S.; Dasgupta, S. Complex formation of rutin and quercetin with copper alters themode of inhibition of ribonuclease A. FEBS Lett. 2011, 585, 3270–3276. [CrossRef] [PubMed]
45. Sela, M.; Anfinsen, C.B. Some spectrophotometric and polarimetric experiments with ribonucleases.Biochim. Biophys. Acta 1957, 24, 229–235. [CrossRef] [PubMed]
46. Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses fromcircular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, 668–673. [CrossRef]
47. Garcia-Borron, J.C.; Escribano, J.; Jimenez, M.; Iborra, J.L. Quantitative determination of tryptophanyl andtyrosyl residues of proteins by second-derivative fluorescence spectroscopy. Anal. Biochem. 1982, 125,277–285. [CrossRef] [PubMed]
48. Jiang, M.; Xie, M.X.; Zheng, D.; Liu, Y.; Li, X.Y.; Cheng, X. Spectroscopic studies on the interaction of cinnamicacid and its hydroxyl derivatives with human serum albumin. J. Mol. Struct. 2004, 692, 71–80. [CrossRef]
49. Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E.The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [CrossRef] [PubMed]
50. Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incrementalconstruction algorithm. J. Mol. Biol. 1996, 261, 470–489. [CrossRef] [PubMed]
51. DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2004. Availableonline: http://pymol.sourceforge.net/.
Sample Availability: Sample Availability: Samples are not available from authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
165

molecules
Article
Non-Nucleosidic Analogues of Polyaminonucleosidesand Their Influence on Thermodynamic Properties ofDerived Oligonucleotides
Jolanta Brzezinska and Wojciech T. Markiewicz *
Institute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland;[email protected]* Correspondence: [email protected]; Tel.: +48-61-852-8503 (ext. 180); Fax: +48-61-852-0532
Academic Editors: Mahesh K. Lakshman and Fumi NagatsugiReceived: 17 March 2015; Accepted: 9 July 2015; Published: 13 July 2015
Abstract: The rationale for the synthesis of cationic modified nucleosides is higher expected nucleaseresistance and potentially better cellular uptake due to an overall reduced negative charge based oninternal charge compensation. Due to the ideal distance between cationic groups, polyamines areperfect counterions for oligodeoxyribonucleotides. We have synthesized non-nucleosidic analoguesbuilt from units that carry different diol structures instead of sugar residues and functionalized withpolyamines. The non-nucleosidic analogues were attached as internal or 5′-terminal modifications inoligodeoxyribonucleotide strands. The thermodynamic studies of these polyaminooligonucleotideanalogues revealed stabilizing or destabilizing effects that depend on the linker or polyamine used.
Keywords: polyaminonucleoside analogue; non-nucleosidic analogue; spermine; putrescine;oligonucleotide; duplex DNA; thermodynamic stability
1. Introduction
The polyanionic character of nucleic acids and their synthetic fragments is a barrier for theirintroduction into cells. Many laboratories try to improve nucleic acid delivery to cells by synthesis ofcationic bioconjugates [1–3], bioconjugate analogues with reduced polyanionic character [4–6], or agreat variety of polymeric transfection media [7–12]. On the other hand, there are studies to elucidatea specific mode of small cationic molecular drugs interactions with biomolecules (e.g., nucleic acids,proteins) useful in developing their more active analogues [13]. Among this clinically important classof compounds are antibiotics carrying aminosugar residues [14].
Chemically modified oligonucleotides have been utilized as indispensable materials for DNA genetherapy [15,16], gene regulation [17,18], chip technology [19,20] and recent nanotechnology [21–23]because of their hybridization affinity for target DNA and/or RNA molecules [24]. The polyanioniccharacter of antisense and siRNA oligonucleotides is a major cause of insufficient cellular uptake andside effects such as binding to serum proteins. The combination of nucleotides with aminoalkyl chainsgreatly enhances the variety of possible structures as well as their potential application [24]. Thus,oligonucleotides possessing cationic functionalities in addition to the anionic phosphate backbonehave been shown to exhibit promising properties [25–28]. Polyamines, putrescine, spermine andspermidine, are involved in the regulation of gene function [29,30]. In vitro, they stabilize DNA andRNA duplexes [31,32], especially ones with imperfect base pairing [33]. The distance between aminogroups of three and four carbon atoms is practically the same as the distance between phosphateanions in the backbone of DNA making polyamines the perfect compounds for creating “zwitterionic”oligonucleotides [27]. It is known that polyamines interact with DNA and RNA in different ways. Theelectrostatic binding is performed via water molecules, by hydrogen bonding with polar functional
Molecules 2015, 20, 12652–12669 166 www.mdpi.com/journal/molecules

Molecules 2015, 20, 12652–12669
groups or with hydrophobic surfaces of nucleobases. There have been numerous studies aimedat determining these interactions using NMR imaging, circular dichroism, Raman spectroscopy,IR spectroscopy, X-ray crystallography and differential scanning calorimetry. In spite of theseextensive studies, precise mechanisms for the interaction between polyamine and DNA is still not fullyunderstood in vivo [34].
The non-nucleosidic analogs of polyaminonucleosides have not been examined so far. Therefore,our aim was to elaborate versatile procedures for synthesis of non-nucleosidic polyamine derivativesand learn their properties within oligonucleotide chains. The choice of carbon chain skeletons ofanalogues (Figure 1) was based on the extent of their commercial availability.
OCH3HO
HO O
O
OHOOH
OH
HOO
Figure 1. Substrates for the non-nucleosidic polyaminonucleoside analogues.
The obtained polyamine building blocks were incorporated at the 5′-end and internal positionswithin an oligodeoxyribonucleotide chain and the resulting conjugates were evaluated for theirhybridization properties.
2. Results and Discussion
2.1. Chemistry
The synthesis of the non-nucleosidic polyamine derived phosphoramidite building blockscontaining different linkers is shown in Scheme 1.
We decided to use 2,2-bis(hydroxymethyl)-propionic acid (bis-MPA, 1) that was earlier applied indendrimer synthesis [35,36].
Bis-MPA (1) was protected with benzaldehyde [37] as it is more lipophilic than a isopropylidenegroup preferred to ease the final workup procedure and increase yield when using an excess ofpolar polyamines [38,39]. The protection of hydroxyl groups in bis-MPA gives rise to formationof two stereoisomers in the ratio ca 2:1, however, further reactions were carried out withoutseparation of isomers. Several approaches for activation of carboxyl function (2) were checked.Reactions with 1,4-butylamine as a model amine in the presence of carbonyldiimidazole or2-chloro-4,6-dimethoxy-1,3,5-triazine gave the expected n-butylamide derivative (results not shown).The best results were obtained when activation was performed with thionyl chloride in the presence ofpyridine and traces of DMF. Thus, reaction with molar excess of putrescine (1,4-diaminobutane) atambient conditions led to N-(4-aminobutyl)-5-methyl-2-phenyl-1,3-dioxane-5-carboxyamide 2 in 60%yield (Scheme 1).
167

Molecules 2015, 20, 12652–12669
O
NNHRO
O
O
NNHRDMTO
R1O
iii
ivii
3 R = H
4 R = TFA
O
OO
OH
7 (S) or 7a (R)
viO
OO
NHH
NN NHR
iii8 (S) or 8a (R) R = H
9 (S) or 9a (R) R= TFA
v5 R = TFA, R1= H
6 R = TFA, R1= P(N-iPr2)OCH2CH2CN
R1O
DMTOO
NHH
NN NHR
v10 (S) or 10a (R) R = TFA, R1= H
11 (S) or 11a (R) R = TFA, R1= P(N-iPr2)OCH2CH2CN
vii
O
O
R2O
viii12 R2= H
13 R2= TBDMS
OR2
O
NH
NN NHRix
iii14 R = H, R2= TBDMS
15 R = TFA, R2= TBDMS
OR1
O
NH
NN NHR
v16 R = TFA, R1= H
17 R = TFA, R1= P(N-iPr2)OCH2CH2CN
R
R
R
R
R
R
R
R
x
H H
DMTO
HO
i2
1 bis-MPA
O
OHO
O
Scheme 1. Synthetic routes to non-nucleosidic polyaminonucleoside analogues: (i) benzaldehyde,p-TsOH, DMF; (ii) SOCl2, putrescine, Et3N, DCM; (iii) (F3CCO)2O, pyridine; (iv) (a) 6 M HCl, reflux,(b) DMTCl, pyridine; (v) (iPr2N)2POCH2CH2CN, 5-ethylthio-1H-tetrazole, DCM; (vi) spermine,MeOH; (vii) (a) p-TsOH, MeOH, (b) DMTCl, pyridine; (viii) TBDMSCl, DMAP, imidazole, CH3CN;(ix) spermine, MW, 40 min; (x) (a) TEAHF, THF/dioxane, (b) DMTCl, pyridine.
We also synthesized the analogs based on the enantiomers (7) and (7a) which were coupledwith excess of spermine in dry MeOH. Due to the lack of a chromophore in synthesized compounds,a reaction control was performed using TLC plates stained with a solution of fluorescein (free acid) inacetonitrile. Spermine derivatives (8, 8a) were isolated by several extractions with large amounts oforganic solvent and used in the next step without additional purification. The configuration of thebranching point in these linkers is retained throughout the synthetic procedure. After protection ofthe amine function with trifluoroacetyl groups, 1,3-dioxolane was hydrolyzed to prepare polyaminenon-nucleosidic derivatives for selective 4,4′-dimethoxytritylation. In order to avoid transacetylation
168

Molecules 2015, 20, 12652–12669
of trifluoroacetyl groups during the isopropylidene cleavage of 9 we used p-TsOH instead aceticacid. Otherwise, amino groups would be at least in part protected with Ac instead of TFA and theirdeprotection with ammonia would not be possible under the conditions of the final deprotection ofoligonucleotides. Commercially available precursor α-hydroxy-γ-butyrolactone (12) was used as astarting material to obtain a polyaminonucleoside analogue (17). This compound provides a threecarbon distance between the phosphate groups after introduction into the oligodeoxyribonucleotidestrand. It also contains a chiral carbon center with a secondary hydroxyl group and the carbonylgroup corresponding to the 3′-hydroxyl and 2′-carbon of nucleosidic residues, respectively. Thelactone-spermine conjugate was subsequently obtained after protection of the secondary hydroxylgroup of the lactone (13) with lipophilic and a bulky t-butyldimethylsilyl (TBDMS) group. Thus, thiseased the isolation of the product (14) resulting from the coupling reaction with spermine (Scheme 1).The reaction of protected lactone 13 and unprotected spermine was performed by microwave synthesisto achieve the desired product 14 with 64% yield, even when both reagents were used in equimolar ratio.To eliminate side reactions during the condensation step of DNA synthesis, the protection of putrescineand spermine amino functions with trifluoroacetyl groups was ensured (as previously described [40].For all non-nucleosidic analogue yields of trifluoroacetylation ranged from 58% (spermine derivatives)up to 70% (putrescine derivative) (Scheme 1). Conversion to the corresponding phosphoramiditederivative was preceded by removal of protecting groups of hydroxyl function with HCl (4) and p-TsOH(9, 9a) acids or Et3N*3HF (15). The protection 5′-OH-groups with a 4,4′-dimethoxytrityl group wasused in a last step of the 3′-OH reaction with 2-cyanoethyl-N,N,N′,N′-tetraisopropylaminophosphaneand 5-(ethylthio)-1H-tetrazole as the activator. However, due to similar physical properties, theseparation of the products from impurities was difficult by silica gel column chromatography. Thiswas resolved by precipitation from hexane. The spermine derivatives of (R) and (S) enantiomersof glyceric acid gained an additional chiral center as phosphoramidites (11, 11a). In the case of thepolyamine derivative of α-hydroxy-γ-butyrolactone, opening of the lactone ring 14 led to a mixture ofenantiomers. Thus, after phosphitylation, compound 17 as a mixture of enantiomers was obtained. Allamidites were lyophilized and were stable during long-term storage at −20 ◦C.
2.2. Oligonucleotides
The phosphoramidites (6, 11, 11a, 17) were used for synthesis of polyamine analogs ofoligodeoxyribonucleotides (Figure 2) using a 12-mer as a reference sequence (Table 1). The couplingefficiencies of these phosphoramidites were 53%–70% as determined by measuring detritylation. Themodified oligodeoxyribonucleotides were obtained after a standard deprotection procedure, and theirstructures were confirmed by the MALDI-TOF (Table 1). The oligodeoxyribonucleotides ON5–ON6
and ON9 were used as pure enantiomers and ON4 and ON8 as mixtures of diastereoisomers.
Table 1. Sequences of oligodeoxyribonucleotides and MALDI-TOF MS data.
No. Sequence 5′ → 3′ a m/z [M − H]−
Calcd Found
ON1 CTC AAG CAA GCT 3614.42 3613.08ON2 CTC ACA TGC GCG 3606.40 3605.54ON3 X0 CTC AAG CAA GCT 3994.42 3993.19ON4 X1 CTC ACA TGC GCG 3973.12 3972.23ON5 X2 CTC ACA TGC GCG 3959.54 3959.24ON6 X3 CTC ACA TGC GCG 3959.54 3959.57ON7 CTC AAG X0 CAA GCT 3994.42 3993.25ON8 CTC ACA X1 TGC GCG 3973.12 3973.12ON9 CTC ACA X2 TGC GCG 3959.54 3957.45
a R = H for X0–X3 in ON3–ON6 and R = 3′-end oligo for X0–X2 in ON7–ON9.
169

Molecules 2015, 20, 12652–12669
NHR1O
OOR
RO
R1O
O
R1O
RO O
HNH
NH
HN NH2
NH
HN NH2
X0
X1
X2
R1O
RO O
HNH
NH
HN NH2
NH
NH2
R = H or 3'-end oligoR1= 5'-end oligo
X3
Figure 2. Non-nucleosidic polyamino-nucleoside analogues units in oligodeoxyribonucleotide strands.
2.3. Stability of Duplexes with Modified Oligodeoxyribonucleotides
The influence of conjugated polyamines on the stability of the DNA duplexes was studied usingUV melting spectra. It was shown previously that polyamine conjugation to oligodeoxyribonucleotidesresults in stabilizing of DNA duplexes and triplexes [2–4,38,39,41–43]. Recently, we described theNMR structure of a DNA duplex carrying a single spermine modified deoxycytidine unit [34]. Thismodification moderately stabilizes the DNA duplex (Figure 3, dCSp vs. RF1) and does not perturb theDNA structure.
The 5′-end dangling modifications in oligodeoxyribonucleotide duplexes usually increase duplexstability and do not show large sequence dependence [44,45]. Since the terminal unpaired nucleotidesare not involved in base pairing, stacking, electrostatic, perhaps to some extent, hydrophobicinteractions are responsible for the thermodynamic effects of the 5′- and 3′-dangling ends. Thenearest-neighborhood model assumes the duplex region for a dangling end to have the same calculatedthermodynamic stability as a matching blunt-end duplex [32,33].
Thus, the stability was increased when X1 and X3 (Scheme 1) were incorporated at the 5′
position (Table 1, entry ON4 and ON6), but for incorporations of X0 and X2 (Scheme 1) a moderatedestabilizing effect was observed (Table 1, entry ON3 and ON5). Despite lack of a nucleobase andhigher conformational flexibility at the 5′-end of the strand the observed stabilities are rather typicalfor 5′-end dangling units as described in the literature. Thermodynamic data (Table 2, Figure 1) showthat X3 results in ΔΔG (−0.3 kcal/mol) similar to unpaired nucleotide (−0.1 to −0.5 kcal/mol) at the5'-end. The change of ΔG for X1 is even higher (ΔΔG = −0.64 kcal/mol). These results suggest thatthe lack of a nucleobase does not affect the thermal stability of the duplexes. This might indicate thatincreased duplex stability is provided by the three positive charges in the spermine chain. In the caseof the putrescine residue, X0, which introduces one positive charge only, a small decrease of duplexstability was observed. (Figure 1, ON3). One can conclude that the small decrease in duplex stability
170

Molecules 2015, 20, 12652–12669
observed for ON5 (Figure 1) containing spermine attached to the glycerol isomer (X2) is caused by thestructure and configuration of this particular linker.
Figure 3. Changes in free energy (ΔG) of polyamine modified duplexes: RF-RF3- reference duplexes,dCSp—spermine modified (dCSp = 4-N-[4,9,13-triazatridecan-1-yl]-2′-deoxycytidine) duplex [34]; ON3,ON7—non-nucleosidic putrescine analogues (Table 2); ON4–ON6 and ON8–ON9—non-nucleosidicspermine analogues (Table 2).
Table 2. DNA duplex stability of non-nucleosidic polyamine-modified oligodeoxyribonucleotides a.
OligoAverage of Curve Fits TM
−1 vs. log (CT/4) Plots
−ΔH◦[kcal/mol]
−ΔS◦ [eu]−ΔG◦
37
[kcal/mol]TM
[◦C]−ΔH◦
[kcal/mol]−ΔS◦ [eu]
−ΔG◦37
[kcal/mol]TM
[◦C]ΔΔG◦
37
[kcal/mol]ΔTM
b
[◦C]
A. Thermodynamic parameters of references duplexes
ON1 c 67.8 ± 9.0 185.6 ± 27.8 10.27 ± 0.44 55.1 72.6 ± 2.0 200.6 ± 6.2 10.45 ± 0.08 54.7ON2 d 115.9 ± 4.8 322.1 ± 14.2 16.00 ± 041 64.6 108.5 ± 1.8 300.1 ± 5.4 15.46 ± 0.12 64.8
B. Thermodynamic parameters of duplexes with the polyamine analog as a dangling end
ON3 c 63.3 ± 6.8 171.5 ± 20.9 10.12 ± 0.37 55.7 69.09 ± 3.1 189.4 ± 9.7 10.33 ± 0.13 55.0 +0.12 −0.3ON4 d 131.8 ± 10.4 368.8 ± 31.0 17.47 ± 0.80 65.1 114.0 ± 8.3 315.9 ± 24.9 16.10 ± 0.64 65.4 −0.64 +0.6ON5 d 97.8 ± 9.4 268.4 ± 28.1 14.57 ± 0.68 64.8 98.9 ± 5.1 271.9 ± 15.4 14.59 ± 0.36 64.5 +0.87 −0.3ON6 d 106.1 ± 4.7 292.4 ± 14.0 15.43 ± .0.37 65.4 110.9 ± 3.9 307.0 ± 11.7 15.76 ± 0.27 65.1 −0.3 +0.3
C. Thermodynamic parameters of duplexes with the polyamine analog as a bulge
ON7 c 62.7 ± 16.7 178.2 ± 53.8 7.50 ± 0.20 41.9 59.0 ± 4.2 166.5 ± 13.7 7.38 ± 0.08 41.5 +3.07 −13.2ON8 d 71.6 ± 6.0 194.4 ± 18.3 11.31 ± 0.35 59.2 82.0 ± 2..0 226.3 ± 6.2 11.83 ± 0.10 58.4 +3.63 −6.4ON9 d 81.2 ± 11.5 225.0 ± 35.6 11.45 ± 0.48 57.0 70 ± 6.4 192.1 ± 20.0 11.03 ± 0.25 58.1 +4.43 −6.7
a Solutions are 100 mM NaCl, 20 mM sodium cacodylate, 0.5 mM Na2EDTA, pH 7; b Calculated for 10−4 M totalstrand concentration; c Complementary strand 5′-AGC TTG CTT GAG-3′; d Complementary strand 5′-CGC GCATGT GAG-3′.
Next, we investigated the influence of polyamine derivatives X0, X1 and X2 on duplex stability,inserted as bulges in the middle of the sequence (Table 1, entry ON7–ON9). The changes of duplexstability caused by single nucleotide bulges differ and depend on the type of flanking bases [46,47].Incorporation of X0–X2 resulted in a lowering of melting temperature independently of polyamineresidue. However, the melting data suggest that the duplex formation occurs with the proper W-Cbase pairing despite of a slight increase in free energy for ON7–ON9 (Table 2, ΔΔG ca. 3–4.5 kcal/mol).
171

Molecules 2015, 20, 12652–12669
Moreover, the effect in ON7 where the putrescine bulge (X0) is flanked by GC/CG pairs is practicallythe same as for ON8 and ON9 (spermine bulge, X1 and X2) flanked by AT/TA pairs. This can beattributed to the stronger stabilizing effect of a spermine residue when compared to putrescine—threepositive charges vs. one. Yet, even three positive charges of a spermine residue do not neutralize thebulge effect as such. The observed changes of ΔG for the studied duplexes (ON7–ON9) are in therange observed for duplexes with a single bulge (ΔΔG, 2–6 kcal/mol) [46,47].
Linkers X1 and X2 differ in length by one carbon and this additional carbon in X1 makes thislinker somehow less rigid, allowing for more favorable placement of the polyamine residue. Thus, theenergy gain of 1.5 kcal/mol in ON4 when compared to ON5. Therefore, ON4 duplex is more stablethan the unmodified duplex. When the same modified linkers (X1 and X2) are inserted as bulges ina more spatially “demanding environment”, the difference of free energies ΔΔG◦
37 of ON8 (+3.63)and ON9 (+4.43) is smaller (0.8 kcal/mol). The influence of linker structure is less profound when themodification is inserted in the middle of chain.
Linkers X2 and X3 carrying a spermine residue differ only in the configuration of carbon (S andR respectively). Comparison of ΔΔG◦
37 of ON6 (−0.3 kcal/mol) and ON5 (+0.87 kcal/mol) suggeststhat X3 allows more favorable placement of polyamine residue This seems to indicate that in this casethe spermine residue attached to a glycerol linker with R-configuration (X3) is closer to the duplexcharged surface.
We would like to conclude that the overall effect of DNA modification with polyamine analoguesseems to offer a convenient way to modify nucleic acids properties. An attachment of polyamineresidues via various open chain linkers allows to maintain the general scheme of base pairing.Moreover, the structure and configuration of the linkers seems to have a higher influence on stabilitywhen placed at the 5′-end of DNA duplexes.
3. Experimental Section
3.1. General Methods
All reagents were of analytical grade, obtained from commercial resources and used withoutfurther purification. For synthesis, solvents with quality pro analysis were used. Solvents were driedand distilled following standard methods and kept over molecular sieve. All reactions were carried atroom temperature unless described otherwise. Column chromatography was performed with silicagel (Merck KGaA, Darmstadt, Germany, 200–630 mesh) and TLC was carried out on precoated plates(Merck silica gel 60, F254). All NMR spectra were recorded at 298 K on Bruker AVANCE II (400 MHz, 1H,13C) and Varian Unity (300 MHz, 31P) spectrometers (Bruker BioSpin GmbH, Rheinstetten, Germanyand Varian, Inc., Palo Alto, CA, USA). Chemical shifts (δ) are reported in parts per million (ppm).J values are given in Hz. Mass spectra were recorded on the MicroTofQ mass spectrometer withelectrospray ionization (ESI) sources (ESI source voltage of 3.2 kV, nebulization with nitrogen at 0.4 bar,dry gas flow of 4.0 L/min at temperature 220 ◦C) and Bruker Autoflex MALDI-TOF (Bruker DaltonikGmbH, Bremen, Germany). Microwave reactions was performed in domestic microwave oven (800 W,Amica, Wronki, Poland). Some NMR (Figures S1–S16) and MS (Figures S17–S22) spectra are availablein Supplementary.
3.2. Synthesis of Monomers
Benzylidene-2,2-bis(oxymethyl)propionic acid (2) [37]. Benzaldehyde (4.2 mL, 40.7 mmol) was added toa well-stirred solution of 2,2-bis(hydroxymethyl)-propionic acid (1) (5 g, 37 mmol) in DMF (30 mL),followed by catalytic p-TsOH (0.355 g, 1.85 mmol) with stirring at room temperature for 4 days. Thereaction was quenched with NH4OH/EtOH (1 mL, 1:1). The solvent was evaporated and the residuedissolved in DCM (100 mL) and washed with NaHCO3 (2 × 100 mL). Organic extracts were dried overanhydrous MgSO4 and filtered then evaporated under reduced pressure. The residue was purified byrecrystallization from DCM to obtain product 2 as mixture of two isomers (77%). 1H-NMR (CDCl3);
172

Molecules 2015, 20, 12652–12669
isomer A: 7.28–7.46 (m, 5H, Ph), 5.45 (s, 1H, PhCHO2), 4.65 (d, 2H, J = 11.7 Hz, OCH2CCO, 3.67 (d,2H, J = 11.7 Hz, OCH2CCO), 1.02 (s, 3H, CH3); isomer B: 7.28–7.51 (m, 5H, Ph), 5.41 (s, 1H, PhCHO2),4.14–4.06 (q, 4H, J = 11.23 Hz, J = 9.27 Hz, 2CH2), 1.57 (s, 3H, CH3). 13C-NMR (CDCl3): 178.89 (C=O),137.58 (C, Ph), 129.08 (CH, Ph), 128.23 (CH, Ph), 128.23 (CH, Ph), 101.86 (CH), 73.43 (CH2), 42.17 (CMe),17.78 (CH3). MS (ESI) m/z: calcd for C12H14O4 221.0819 [M − H]−, found 221.182.
N-(4-Aminobutyl)-5-methyl-2-phenyl-1,3-dioxane-5-carboxamide (3). Thionyl chloride (1.26 mL, 17.8 mmol)was added dropwise to ice-cooled solution of 2 (2 g, 8.9 mmol) in 45 mL DCM containing 10% ofpyridine and 3 drops of DMF. The mixture was stirred for 2 h at room temperature, and the excessthionyl chloride was removed by several co-evaporations with a mixture of DCM and toluene. Thebrown residue was dissolved in DCM (46 mL) and the temperature was lowered to −10 ◦C. Putrescine(4.47 mL, 44.5 mmol) and triethylamine (1.4 mL) in DCM (2 mL) was added after 15 min. The mixturewas stirred for 1h at room temperature, and the reaction was quenched with saturated aqueousNaHCO3 (3 mL). The mixture was extracted with DCM (2 × 100 mL). The combined organic extractswere dried over anhydrous MgSO4 and concentrated under vacuum and purified by chromatographywith 4% MeOH in DCM as the eluent to give 3 (0.910 g, 60% yield) as a yellow oil. 1H-NMR (CDCl3):7.34–7.43 (m, 5H, Ph), 7.03 (t, 1H, NHCO), 5.48 (s, 1H, PhCHO2), 4.33 (d, 2H, J = 12 Hz, OCH2CCO),3.78 (d, 2H, J = 12 Hz, OCH2CCO), 3.33 (q, 2H, J = 6.3 Hz, J = 5.8 Hz, CONHCH2), 3.21 (q, 2H, J = 6.3 Hz,J = 5.8 Hz, NH2CH2, putrescine), 1.5–1.56 (m, 4H, 2 × CH2, putrescine), 1.04 (s, 3H, CH3); 13C-NMR(CDCl3): δ (ppm) 178.89 (C=O), 137.45 (C, Ph), 128.7 (CH, Ph), 128.03 (CH, Ph), 101.22 (CH), 75.07(CH2), 47.6 (CMe), 47.6 (CH2, putrescine), 26.53 (CH2, putrescine), 18.03 (CH3); MS (ESI) m/z: calcdfor C16H24N2O3 293.186 [M + H]+, found 293.2037.
5-Methyl-2-phenyl-N-(4-(2,2,2-trifluoroacetamid)butyl)-1,3-dioxane-5-carboxamide (4) 3 (0.430 g, 1.47 mmol)was co-evaporated with pyridine (3 × 5 mL), dissolved in pyridine (15 mL), followed by the additionof N-methylimidazole (0.16 mL, 1.46 mmol). Then, trifluoroacetic anhydride (0.593 mL, 4.41 mmol)was added dropwise to the mixture cooled at 0 ◦C. The mixture was stirred for 30 min and thenpoured into saturated aqueous NaHCO3 (30 mL), and extracted with DCM (3 × 50 mL). The combinedorganic extracts were dried over anhydrous Na2SO4 and concentrated under vacuum. The residue waspurified by chromatography using DCM as the eluent to give 4 (0.500 g, 52% yield) as an oil. 1H-NMR(CDCl3): 7.33–7.45 (m, 5H, Ph), 7.03 (t, 1H, NHCO), 5.5 (s, 1H, PhCHO2), 4.38 (d, 2H, J = 11.7 Hz,OCH2CCO), 3.78 (d, 2H, J = 11.7 Hz, OCH2CCO), 3.2–3.4 (m, 4H, 2NHCH2), 1.49–1.54 (m, 4H, CH2,putrescine), 1.06 (s, 3H, CH3). 13C-NMR (CDCl3): 182.83 (C=O), 158.9 (C=O), 137.45 (C, Ph), 128.7 (CH,Ph), 128.03 (CH, Ph), 125.53 (C, CF3), 101.22 (CH), 75.07 (CH2), 47.4 (CMe), 45.3 (CH2, putrescine),26.53 (CH2, putrescine), 18.03 (CH3); 19F NMR 1.78 (s, 3F, CF3); MS (ESI) m/z: calcd for C18H23F3N2O4
389.1683 [M + H]+, found 389.1627.
3-(4,4′-Dimethoxytrityl)-2-(hydroxymethyl)-2-methyl-N-(4-(2,2,2-trifluoroacetamido)butyl)propanamide (5).4 (0.240 g, 0.61 mmol) was dissolved in a mixture conc. aq HCl/EtOH (1:5, v/v) and refluxed for72 h. The excess of HCl was removed by several co-evaporations with a mixture of methanol andtoluene and finally a brown oil with was dried by co-evaporation with anhydrous pyridine (2 × 5 mL)and dissolved in anhydrous pyridine (2.4 mL). To this solution 4,4′-dimethoxytrityl chloride (0.243 g,0.72 mmol) was added and the reaction was quenched after 3 h by adding saturated aqueous NaHCO3.The resulting solution was extracted with DCM and the combined organic extracts were washed withbrine, dried over Na2SO4 and concentrated under vacuum. The residual yellow oil was purified bychromatography with 5% MeOH in DCM as the eluent to give 0.18 g (49%) 5 as a white foam. 1H-NMR(CDCl3): δ (ppm) 7.42–7.2 (m, 10H, Ph, NHCO), 7.04 (t, 1H, NHCO), 6.89 (m, 4H, Ph), 3.82 (s, 3H,OCH3), 3.83–3.62 (m, 4H, OCH2CCO), 3.43–3.23 (m, 4H, 2NHCH2), 1.4–1.8 (m, 4H, 2CH2, putrescine),1.2 (s, 3H, CH3). 13C-NMR (CDCl3): δ (ppm) 174.4 (C=O), 158.5 (2C, COCH3), 158.4 (C=O, TFA), 144.6(C, Ph), 135.5 (C, Ph), 130.0 (CH, Ph), 128.3 (CH, Ph), 128.0 (CH, Ph), 127.8 (CH, Ph), 126.8 (C, CF3),113.1 (CH, Ph), 86.2 (C), 66.9 (CH2), 65.5 (CH2), 55.1 (CH3), 47.5 (C, CMe), 43.2 (CH2, putrescine), 24.5(CH2, putrescine), 18.7 (CH3); MS (ESI) m/z: calcd for C32H37F3N2O6 603.2676 [M + H]+, found 603.261.
173

Molecules 2015, 20, 12652–12669
3-[(4,4′-Dimethoxytrityl)-2-(hydroxymethyl)-2-methyl-N-(4-(2,2,2-trifluoroacetamido)butyl)propanamide]phosphoramidite (6). 2-Cyanoethyl-N,N,N,N-tetraisopropylphosphoramidite (0.9 mL,0.3 mmol) was added to the solution of 5 (0.143 g, 0.234 mmol) and 5-(ethylthio)-1H-tetrazole (0.0273 g,0.21 mmol) in dichloromethane (1.2 mL) and the mixture was stirred at room temperature. After 2h, TLC revealed complete reaction. The mixture was diluted with dichloromethane, washed withsaturated sodium bicarbonate solution and the organic extracts were dried over Na2SO4. The productwas purified by silica gel chromatography with benzene–Et3N (10%) and lyophilisation from benzeneto give 6 (134 mg, 70% yield) as a white powder. 31P-NMR (C6H6): δ (ppm) 148.30; 148.54. MS (ESI)m/z: calcd for, C41H54F3N4O7P [M + H]+ 803.3755, found 803.371.
(S)-N-(4,9,13-Triazatridecan-1-yl)-2,2-dimethyl-1,3-dioxolane-4-carboxyamide (8). The methyl (S)-2,2-dimethyl-1,3-dioxalane-4-carboxylate (7) (0.45 mL, 3 mmol), spermine (2.4 g, 12 mmol) were dissolvedin anhydrous methanol (1 mL) and the resulting solution was stirred at room temperature for 48–72h at 25–35 ◦C. The solvent was evaporated under reduced pressure and the residue was purified bycolumn chromatography over silica gel (MeOH/MeNH2/H2O) to give 8 (720 mg, 70% yield) as yellowoil. 1H-NMR (CDCl3): δ (ppm) 7.22 (m, 1H, NHCO), 4.45 (q, 1H, J = 5.37 Hz, J = 2.44 Hz, COCH), 4.25(t, 1H, J =7.8, OCHCO), 4.05 (q, 1H, J = 5.37 Hz, J = 3.41 Hz, COCH), 3.38–3.24 (m, 2H, CONHCH2),2.53–2.65 (m, 6H, CH2), 2.05 (2H, NH), 1.66 (m, 2H, spermine), 1.49 (m, 2H, spermine), 1.44 (s, 3H,CH3), 1.44 (s, 3H, CH3); 13C-NMR (CDCl3): δ (ppm) 171.3 (C=O), 110.6 (OCO), 74.8 (C), 67.6 (C), 49.1(CH2), 46.8 (CH2), 37.1 (CH2), 28.6 (CH2), 27.1 (CH2), 26.0 (CH2), 24.8 (CH3). MS (ESI) m/z: calcd forC16H34N4O3 [M + K]+ 369.2262, found 370.2801.
(R)-N-(4,9,13-Triazatridecan-1-yl)-2,2-dimethyl-1,3-dioxolane-4-carboxyamide (8a). The methyl (R)-2,2-dimethyl-1,3-dioxalane-4-carboxylate (7a) (0.45 mL, 3 mmol) was converted into compound 8a
following the above procedure (566 mg, 55% yield). 1H-NMR (CDCl3): δ (ppm) 7.19 (m, 1H, NHCO),4.45 (q, 1H, J = 5.37 Hz, J = 2.44 Hz, COCH), 4.26 (t, 1H, J = 7.8 Hz, OCHCO), 4.05 (q, 1H, J = 5.37 Hz,J = 3.41 Hz, COCH), 3.43–3.3 (m, 2H, CONHCH2), 2.74–2.59 (m, 6H, CH2), 1.76 (m, 2H, spermine), 1.63(m, 2H, spermine), 1.47 (s, 3H, CH3), 1.37 (s, 3H, CH3); 13C-NMR (CDCl3): δ (ppm) 171.3 (C=O), 110.6(OCO), 74.8 (C), 67.6 (C), 49.1 (CH2), 46.8 (CH2), 37.1 (CH2), 28.6 (CH2), 27.1 (CH2), 26.0 (CH2), 24.8(CH3). MS (ESI) m/z: calcd for C16H34N4O3 [M + H]+ 331.2704, found 331.2701.
(S)-N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-2,2-dimethyl-1,3-dioxolane-4-carboxyamide (9).8 (0.200 g, 0.6 mmol) was co-evaporated with anhydrous pyridine (3 × 5 mL) and redissolved inpyridine (6 mL). Trifluoroacetic anhydride (0.5 mL, 3.6 mmol) was added dropwise to a cooledand stirred solution at 0 ◦C. The solution was warmed to room temperature and stirred for 30 min,quenched with NaHCO3 and extracted with CH2Cl2. The combined organic extracts were washedwith saturated aqueous NaCl, dried over Na2SO4 and concentrated under vacuum. The residual oilwas purified by chromatography with 2% MeOH in DCM as the eluent to give 9 (0.2 g, 54% yield)as a white foam. 1H-NMR (CDCl3): δ (ppm) 7.09 (m, 1H, NHCO), 6.72 (m, 1H, NHCO), 4.46 (q, 1H,J = 5.34 Hz, J = 3.78 Hz, COCH), 4.07 (q, 1H, J = 5.34 Hz, J = 3.78 Hz, COCH), 4.28 (t, 1H, J = 7.64Hz, OCHCO), 3.49–3.25 (m, 8H, NHCH2CH2, spermine), 1.86–1.78 (m, 2H, spermine), 1.61 (m, 2H,spermine), 1.49 (s, 3H, CH3), 1.39 (s, 3H, CH3). 19F-NMR: 6.98–7.22 (m, 9F, 3CF3). MS (ESI) m/z: calcdfor C22H30F9N4O6 [M − H]− 617.2022, found 617.3021.
(R)-N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-2,2-dimethyl-1,3-dioxolane-4-carboxyamide (9a).8a (0.350 g, 1.05 mmol) was converted into 9a (0.368 g, 58% yield) following procedure for 9. 1H-NMR(CDCl3): δ (ppm) 7.17 (m, 1H, NHCO), 6.76 (m, 1H, NHCO), 4.47 (q, 1H, J = 7.2 Hz, J = 3.3 Hz,COCH), 4.07 (m, 1H, COCH), 4.29 (t, 1H, J = 8 Hz, OCHCO), 3.48–3.26 (m, 8H, NHCH2CH2, spermine),1.88–1.82 (m, 2H, spermine), 1.62 (m, 2H, spermine), 1.49 (s, 3H, CH3), 1.39 (s, 3H, CH3). 19F-NMR:6.98–7.22 (m, 9F, 3CF3). MS (ESI) m/z calcd for C22H31F9N4O3 [M + K]+ 657.1737, found 657.175.
(S)-N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-3-(4,4′-dimethoxytrityl)-3-hydroxypropanamide(10). To a solution of 9 (0.500 g, 0.75 mmol) in MeOH (4 mL), p-toluenosulfonic acid (0.03 g, 0.15 mmol)
174

Molecules 2015, 20, 12652–12669
was added at room temperature. The mixture was stirred for ca 30 h at room temperature, andsolvent was evaporated. The mixture was diluted with ethyl acetate and washed with aq. saturatedNaHCO3. The combined organic extracts were dried over Na2SO4 and concentrated under vacuum.The resulting crude was co-evaporated with pyridine (4 × 5 mL) and dissolved in pyridine (2.5 mL).4,4′-Dimethoxytrityl chloride (0.330 g, 0.975 mmol) was added in portions at room temperature. Themixture was stirred for 3 h at room temperature, and the reaction was quenched with methanol (1 mL)and saturated aq. NaHCO3 (10 mL). The mixture was extracted with DCM. The combined organicextracts were dried over MgSO4 and concentrated under vacuum. The resulting orange oil waspurified by chromatography with 2%–3% MeOH in CH2Cl2 (with 0.2% of pyridine by vol.) as theeluent to give 10 (0.494 g, 69% yield) as a white solid. 1H-NMR (CDCl3): δ (ppm) 7.39–6.8 (m, 15H,DMT, NHCO), 4.19–4.13 (m, 1H, OCHCO), 3.75 (s, 6H, 2OCH3), 3.51–3.22 (m, 14H), 1.99–175 (m, 4H,spermine), 1.68–1.55 (m, 4H, spermine). 13C-NMR (CDCl3): δ (ppm) 171.9 (C), 171.7 (C), 158.7 (C),144.4 (C), 135 (C), 129.9 (CH), 127,9 (CH), 127 (CH), 125.8 (C), 121 (C), 113.2 (CH), 86.8 (C), 77.2 (CH),67.7 (CH2), 55.2 (CH3), 46.3 (CH2), 36.1 (CH2), 35.1 (CH2), 29.7 (CH2), 27.1 (CH2). MS (ESI) m/z: calcdfor C40H45F9N4O8 [M + Na]+ 903.2991, found 903.2938; calcd for C40H45F9N4O8 [M + K]+ 919.2731,found 919.266.
(R)-N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-3-(4,4′-dimethoxytrityl)-3-hydroxypropanamide(10a). 9a (0.350 g, 1.05 mmol) was converted into 10a (0.383 g, 54% yield) following procedure for 10.1H-NMR (CDCl3): δ (ppm) 7.39–6.8 (m, 15H, DMT, NHCO), 4.2–4.12 (m, 1H, OCHCO), 3.75 (s, 6H,2OCH3), 3.48–3.18 (m, 14H), 1.91–1.74 (m, 4H, spermine), 1.63–1.51 (m, 4H, spermine). MS (ESI) m/z:calcd for C40H45F9N4O8 [M − H]− 879.3015, found 879.3028.
2-(S)-[(4,4′-Dimethoxytrityl)-3-(hydroxymethyl)-N-((2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane)propanamide]phosphoramidite (11). The compound 10 was converted into compound 11 followingprocedure for 6. Except purification: crude phosphoramidite was trice precipitated from hexane andfinally lyophilized from benzene to give 11 (0.210 g, 52% yield) as a white light solid. 31P-NMR (C6H6):δ (ppm) 151.89; 151.68; 148.99; 148.68; 148.56; 146.81. MS (ESI) m/z: calcd for C49H62F9N6NaO9P[M + Na]+ 1103.4070, found 1103.4575; calcd for C49H62F9KN6O9P [M + K]+ found 1119.4509.
2-(R)-[(4,4′-Dimethoxytrityl)-3-(hydroxymethyl)-N-((2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane)propanamide]phosphoramidite (11a). 10a (0.200 g, 0.226 mmol) was converted into compound 11a
(0.128 g, 52% yield) according to procedure for 11. 1H-NMR (CDCl3): δ (ppm) 7.14–7.38 (m, 9H, DMT),6.78 (m, 4H, DMT), 4.8 (m, 1H, COCH), 4.65 (m, 1H, OCHCO), 3.73 (s, 6H, 2OCH3), 3.12–3.55 (m, 16H,CH2OH, spermine, CH2OP) 2.4–2.78 (m, 5H, 2CH, iPr, CH2CN), 1.41–1.92 (m, 8H, spermine) 1.17–1.23(m, 12H, iPr). 31P-NMR (C6H6): δ (ppm) 151.76; 151.42; 149.24; 149.05; 148.96; 148.83. MS (ESI) m/z:calcd for C49H62F9N6O9P [M − H]− 1079.4094, found 1079.63.
3-(tert-Butyldimethylsilyloxy)butyrolactone (13) [48]. tert-Butyldimethylchlorosilane (1.50 g, 9.63 mmol),imidazole (0.470 g, 3.21 mmol) and N,N-dimethylaminopyridine (0.392 g, 3.21 mmol) were addedto a solution of α-hydroxy-γ-butyrolactone (12) (0.25 mL, 3.21 mmol) in anhydrous CH3CN (16 mL).The reaction mixture was stirred at room temperature for 16 h. Then, solvent was evaporated underreduced pressure. The residue was partitioned between saturated aq. NaHCO3 and Et2O (3 × 50 mL).The organic extracts were washed with brine, dried over MgSO4. The crude residue was purified bycolumn chromatography (eluent hexane/Et2O 4:1→2:1) to give compound 12 as colourless oil (0.485 g,70% yield). 1H-NMR (CDCl3): δ (ppm) 4.35–4.4 (m, 2H, CH2O), 4.15–4.2 (dt, 1H, OCH), 2.16–2.5 (m,1H, OCHCH2), 0.9 (s, 9H, 3CH3, t-Bu), 0.16 (s, 3H, CH3CSi), 0.14 (s, 3H, CH3CSi); 13C-NMR (CDCl3):δ (ppm) 175.86 (C=O), 68.19 (CH), 64.72 (CH2), 33.3 (CH2), 25.61 (CH3), 18.19 (SiC(CH3)3), 5.5 (CH3);MS (ESI) m/z: calcd for C10H20O3Si [M − H]− 215.1103, found 215.0295.
N-(4,9,13-Triazatridecan-1-yl)-2-(tert-butylodimethylsiloxy)-butyramide (14). A solution of 13 (0.216 g,1 mmol) and spermine (0.202 g, 1 mmol) in a small (10 mL) Erlenmeyer flask was kept in microwaveoven for 45 min. The flask was cooled down and the mixture was diluted with DCM (20 mL), washed
175

Molecules 2015, 20, 12652–12669
with NaHCO3 (3 × 30 mL), brine (30 mL), and organic layers dried over MgSO4. After evaporation,the residue was chromatographed using MeOH/H2O/MeNH2 as the eluent to give 14 (0.266 g, 64%yield) as oil. 1H-NMR (CDCl3): δ (ppm) 7.02 (t, 1H, NHCO), 4.25 (t, 1H, J = 6.01 Hz, COCHOSi), 3.71(q, 2H, J = 5.89 CH2OH), 3.38–3.32 (m, 2H, spermine), 2.65–2.58 (m, 4H), 1.99–1.94 (m, 2H, OCHCH2),1.76 (m, 2H, spermine), 1.63 (m, 2H, spermine), 0.91 (s, 9H, 3CH3, t-Bu), 0.11 (s, 3H, CH3CSi), 0.08 (s,3H, CH3CSi). MS (ESI) m/z: calcd for C20H47N4O3Si [M + H]+ 419.3417, found 419.3539.
N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-2-(tert-butyldimethylsiloxy)-butyramide (15). 14 (0.266 g,0.64 mmol) was co-evaporated with anhydrous pyridine (3 × 5 mL) and redissolved in pyridine (6mL). Trifluoroacetic anhydride (0.517 mL, 3.83 mmol) was added dropwise at 0 ◦C. The solution waswarmed to room temperature and stirred for 30 min., quenched with NaHCO3 and extracted withDCM (3 × 30mL). The combined organic extracts were washed with brine, dried over MgSO4 andpurified by chromatography with 2%–3% MeOH in DCM as the eluent to give 15 (0.297 g, 66% yield) asan oil. 1H-NMR (CDCl3): δ (ppm) 7.09 (bs, 1H, NHCO), 6.83 (bs, 1H, NHCO), 4.27 (t, 1H, COCHOSi),3.73 (q, 2H, J = 5.89 Hz, CH2OH), 3.47–3.15 (m, 6H), 2.01–1.75 (m, 4H), 1.63 (m, 2H, spermine), 0.93 (s,9H, 3CH3, t-Bu), 0.12 (s, 3H, CH3CSi), 0.10 (s, 3H, CH3CSi). 19F-NMR 6.98–7.22 (m, 9F, CF3). MS (ESI)m/z: calcd for C26H43F9N4O6Si [M − H]− 705.2730, found 705.2809.
N-[Tris(2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane]-4-(4,4′-dimethoxytrityl)-butyramide (16). TEAHF [49,50]solution (1 M in THF/dioxane, 1.08 mmol, 1 mL) was added at room temperature to a solution of15 (0.254 g, 0.36 mmol) in THF (1 mL) and kept overnight at room temperature. The reaction wasquenched with NH4Cl solution and extracted with ethyl acetate (3 × 15 mL). The combined extractswere dried over MgSO4, concentrated under vacuum. The residue was co-evaporated with anhydrouspyridine (4 × 5 mL) and redissolved in pyridine (2 mL). 4,4′-Dimethoxytrityl chloride (1.2 eq) wasadded in portions at room temperature. The mixture was stirred for 3 h at room temperature, and thereaction was quenched with methanol (1 mL) and saturated aqueous NaHCO3 (10 mL). The mixturewas extracted with DCM. The combined organic extracts were dried over MgSO4, concentrated undervacuum and the resulting orange oil was purified by chromatography with 2%–3% MeOH in DCM(with 0.2% of pyridine) as the eluent to give 16 (0.231 g, 72% yield ) as a white solid. 1H-NMR (CDCl3):δ (ppm) 7.19–7.36 (m, 9H, DMT), 6.78 (d, 4H, J = 8.6 Hz, DMT), 4.21 (dt, 1H, COCHOH), 3.74 (s,6H, 2OCH3), 3.0–3.44 (m, 14H, CH2OH, spermine), 2.18–2.19 (m, 2H, OCHCH2), 1.56–1.88 (m, 8H,spermine). MS (ESI) m/z: calcd for C40H46F9N4O8 [M + K]+ 933.2887, found 933.279.
2-(4,4′-Dimethoxytrityl)-4-(hydroxymethyl)-N-((2,2,2-trifluoroacet-1-yl)-4,9,13-triazatridecane)-butyramide]phosphoramidite (17). 17 was synthesized according to procedure described for 11. (0.155 g, 55% yield).1H-NMR (CDCl3): δ (ppm) 6.76–7.4 (m, 13H, DMT), 4.22 (dt, 1H, COCHOH), 3.74 (s, 6H, 2OCH3),3.12–3.55 (m, 16H, CH2OH, CONHCH2 (spermine), CH2OP), 2.56–2.62 (m, 2H, 2CH, i-Pr), 2.24–2.42(m, 4H, OCHCH2, CH2CN), 1.57–1.84 (m, 8H, spermine), 1.1–1.2 (m, 12H, 4CH3, i-Pr). 31P-NMR(C6H6): δ (ppm) 151.73; 151.59; 148.25; 147.83. MS (ESI) m/z: calcd for C50H64F9N6O9P [M + K]+
1133.3966, found 1133.303; calcd for C50H64F9N6NaO9P [M + Na]+ 1117.4226, found 1117.45.
3.3. Oligonucleotide Preparation
The DNA dodecamers (5′-CTC AAG CAA GCT-3′, 5′-AGC TTG CTT GAG-3′, 5′-CTC ACATGC GCG-3′, 5′-CGC GCA TGT GAG-3′) were synthesized using DNA synthesizer Gene AssemblerPlus from Pharmacia-LKB (Uppsala, Sweden) or K & A Laborgerate GbR DNA/RNA (Frankfurtam Main, Germany), using standard phosphoramidite chemistry. The non-nucleosidic analogues(6, 11, 11a, 17) were inserted at positions marked with an X as listed in the Table 1. For the modifiedphosphoramidites, two-fold excess of phosphoramidites (in comparison to the standard protocol) anda prolonged coupling step of 10 min were used. The oligomers were cleaved from the CPG-supportwith 32% aqueous ammonia (room temperature, 1 h). The deprotection under standard conditionsusing concentrated aqueous ammonia at 55 ◦C overnight allowed for removal of all protecting groups,including trifluoroacetyls [3,38]. The oligomers were purified by the TLC on Merck 60 F254 TLC plates
176

Molecules 2015, 20, 12652–12669
with n-propanol/aqueous ammonia/water solution (55:35:10, by vol.) as an eluent. The productband (least mobile) was cut out, eluted with water and desalted with Waters Sep-pak C-18 cartridges.First, the solution containing the oligonucleotide was loaded onto the cartridge and the column wasflushed with 10 mM ammonium acetate (10 mL). In the next step, the oligonucleotides were eluted byflushing the cartridge with 5 mL of 30% acetonitrile/water solution. The fraction with the product wasevaporated to dryness and the purity of oligonucleotides was monitored using HPLC and confirmed(by MALDI-TOF spectrometry Autoflex, Bruker). Oligonucleotides were also purified in a subsequentreverse phase HPLC step (UFLC system with LC-20AD pump; C(18)2 100 Å column (15 cm × 4.6 mm);starting from 0.01 M triethylammonium acetate (pH 7.0) up to CH3CN:CH3COOEt3N (40%/60%).Identity was confirmed by mass spectroscopy on MALDI-TOF (Autoflex, Bruker Daltonik GmbH,Bremen, Germany). The isolated yields of modified oligonucleotides were at the range of 23%–57%:26%–57% and 23%–26% for ON3–ON6 and ON7–ON9 respectively.
3.4. Thermodynamic Analysis
UV melting profiles of the DNA duplexes were obtained in a buffer containing 100 mMsodium chloride, 20 mM sodium cacodylate, 0.5 mM Na2EDTA, pH 7.0. Duplexes were used in the10−3–10−6 M concentration range. Single strand concentrations were calculated from absorbance above80 ◦C with single strand extinction coefficients approximated by the nearest-neighbor model [51]. Thetemperature range, in a heating-cooling cycle, was 0–90 ◦C with a temperature gradient of 1 ◦C/min.Thermal-induced transitions of each mixture were monitored at 260 nm with a Beckman DU 650spectrophotometer with a temperature controller. The thermodynamic parameters were determinedfrom fits of data acc. to a two-state model with the MeltWin 3.5 software [52].
4. Conclusions
Nucleic acids that carry polycationic modifications have many therapeutic and biotechnologicaladvantages. Our studies corroborate that non-nucleosidic analogues of polyaminooligonucleosidesmaintain affinity and easily form duplexes with complementary strands. In some cases, this mayincrease the stability of modified complexes of nucleic acids. These new properties based on therationale of charge masking do not change the scheme of the secondary structure of nucleic acids.Thus, they can ease transferring of nucleic acids past cellular barriers.
Supplementary Materials: Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/07/12652/s1.
Acknowledgments: This work was supported by the Polish Ministry of Science and Higher Education.
Author Contributions: W.T.M. designed research and supervised the project, J.B. performed all experiments.Both authors analyzed the data, wrote the manuscript and approved the final version of the manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery.Nat. Rev. Drug Discov. 2005, 4, 581–593. [CrossRef] [PubMed]
2. Thomas, R.M.; Thomas, T.; Wada, M.; Sigal, L.H.; Shirahata, A.; Thomas, T.J. Facilitation of the cellularuptake of a triplex-forming oligonucleotide by novel polyamine analogues: Structure-activity relationships.Biochemistry 1999, 38, 13328–13337. [CrossRef] [PubMed]
3. Prakash, T.P.; Barawkar, D.A.; Vaijayanti, K.; Ganesh, K.N. Synthesis of site-specific oligonucleotide-polyamine conjugates. Bioorg. Med. Chem. Lett. 1994, 4, 1733–1738. [CrossRef]
4. Horn, T.; Chaturvedi, S.; Balasubramaniam, T.N.; Letsinger, R.L. Oligonucleotides with alternatinganionic and cationic phosphoramidate linkages: Synthesis and hybridization of stereo-uniform isomers.Tetrahedron Lett. 1996, 37, 743–746. [CrossRef]
177

Molecules 2015, 20, 12652–12669
5. Letsinger, R.L.; Singman, C.N.; Histand, G.; Salunkhe, M. Cationic oligonucleotides. J. Am. Chem. Soc. 1988,110, 4470–4471. [CrossRef]
6. Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; Berg, A.V.D.; Hagopian, J.C.; Springer, A.D.;Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversiblecharge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1261. [CrossRef][PubMed]
7. Boussif, O.; Delair, T.; Brua, C.; Veron, L.; Pavirani, A.; Kolbe, H.V. Synthesis of polyallylamine derivativesand their use as gene transfer vectors in vitro. Bioconjug. Chem. 1999, 10, 877–883. [CrossRef] [PubMed]
8. Ahmed, M.; Bhuchar, N.; Ishihara, K.; Narain, R. Well-controlled cationic water-soluble phospholipidpolymer-DNA nanocomplexes for gene delivery. Bioconjug. Chem. 2011, 22, 1228–1238. [CrossRef] [PubMed]
9. Zhang, S.; Zhao, B.; Jiang, H.; Wang, B.; Ma, B. Cationic lipids and polymers mediated vectors for delivery ofsiRNA. J. Control. Release 2007, 123, 1–10. [CrossRef] [PubMed]
10. Piest, M.; Engbersen, J.F.J. Effects of charge density and hydrophobicity of poly(amido amine)s for non-viralgene delivery. J. Control. Release 2010, 148, 83–90. [CrossRef] [PubMed]
11. Patil, S.P.; Yi, J.W.; Bang, E.K.; Jeon, E.M.; Kim, B.H. Synthesis and efficient siRNA delivery ofpolyamine-conjugated cationic nucleoside lipids. Med. Chem. Commun. 2011, 2, 505–508. [CrossRef]
12. Geihe, E.I.; Cooley, C.B.; Simon, J.R.; Kiesewetter, M.K.; Edward, J.A.; Hickerson, R.P.; Kaspar, R.L.;Hedrick, J.L.; Waymouth, R.M.; Wender, P.A. Designed guanidinium-rich amphipathic oligocarbonatemolecular transporters complex, deliver and release siRNA in cells. Proc. Natl. Acad. Sci. USA 2012, 109,13171–13176. [CrossRef] [PubMed]
13. Hamilton, P.L.; Arya, D.P. Natural product DNA major groove binders. Nat. Prod. Rep. 2012, 29, 134–143.[CrossRef] [PubMed]
14. Xi, H.; Davis, E.; Ranjan, N.; Xue, L.; Hyde-Volpe, D.; Arya, D.P. Thermodynamics of nucleic acid “shapereadout” by an aminosugar. Biochemistry 2011, 50, 9088–9113. [CrossRef] [PubMed]
15. Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class ofpharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [CrossRef] [PubMed]
16. Davis, S.; Lollo, B.; Freier, S.; Esau, C. Improved targeting of miRNA with antisense oligonucleotides. NucleicAcids Res. 2006, 34, 2294–2304. [CrossRef] [PubMed]
17. Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substratesenhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [CrossRef] [PubMed]
18. Cekaite, L.; Furset, G.; Hovig, E.; Sioud, M. Gene Expression Analysis in Blood Cells in Response toUnmodified and 2′-Modified siRNAs Reveals TLR-dependent and Independent Effects. J. Mol. Biol. 2007,365, 90–108. [CrossRef] [PubMed]
19. Raddatz, S.; Mueller-Ibeler, J.; Kluge, J.; Wäss, L.; Burdinski, G.; Havens, J.R.; Onofrey, T.J.; Wang, D.;Schweitzer, M. Hydrazide oligonucleotides: New chemical modification for chip array attachment andconjugation. Nucleic Acids Res. 2002, 30, 4793–4802. [CrossRef] [PubMed]
20. Yim, S.C.; Park, H.G.; Chang, H.N.; Cho, D.Y. Array-based mutation detection of BRCA1 using directprobe/target hybridization. Anal. Biochem. 2005, 337, 332–337. [CrossRef] [PubMed]
21. Wengel, J. Nucleic acid nanotechnology-towards Angstrom-scale engineering. Org. Biomol. Chem. 2004, 2,277–280. [CrossRef] [PubMed]
22. Shu, W.; Liu, D.; Watari, M.; Riener, C.K.; Strunz, T.; Welland, M.E.; Balasubramanian, S.; McKendry, R.A.DNA molecular motor driven micromechanical cantilever arrays. J. Am. Chem. Soc. 2005, 127, 17054–17060.[CrossRef] [PubMed]
23. Mangraviti, A.; Tzeng, S.Y.; Kozielski, K.L.; Wang, Y.; Jin, Y.; Gullotti, D.; Pedone, M.; Buaron, N.; Liu, A.;Wilson, D.R.; et al. Polymeric Nanoparticles for Nonviral Gene Therapy Extend Brain Tumor Survival in Vivo.ACS Nano 2015, 9, 1236–1249. [CrossRef] [PubMed]
24. Saneyoshi, H.; Tamaki, K.; Ohkubo, A.; Seio, K.; Sekine, M. Synthesis and hybridization properties of2′-O-(tetrazol-5-yl)ethyl-modified oligonucleotides. Tetrahedron 2008, 64, 4370–4376. [CrossRef]
25. Urban, E.; Noe, C.R. Structural modifications of antisense oligonucleotides. Il Farmaco 2003, 58, 243–258.[CrossRef]
26. Debart, F.; Abes, S.; Deglane, G.; Moulton, H. M.; Clair, P.; Gait, M.J.; Vasseur, J.J.; Lebleu, B. Chemicalmodifications to improve the cellular uptake of oligonucleotides. Curr. Top. Med. Chem. 2007, 7, 727–737.[CrossRef] [PubMed]
178

Molecules 2015, 20, 12652–12669
27. Winkler, J.; Saadat, K.; Díaz-Gavilán, M.; Urban, E.; Noe, C.R. Oligonucleotide-polyamine conjugates:Influence of length and position of 2′-attached polyamines on duplex stability and antisense effect. Eur. J.Med. Chem. 2009, 44, 670–677. [CrossRef] [PubMed]
28. Rahman, S.M.A.; Baba, T.; Kodama, T.; Islam, M.A.; Obika, S. Hybridizing ability and nuclease resistanceprofile of backbone modified cationic phosphorothioate oligonucleotides. Bioorg. Med. Chem. 2012, 20,4098–5102. [CrossRef] [PubMed]
29. Bachrach, U. Polyamines and cancer: Minireview article. Amino Acids 2004, 26, 307–309. [CrossRef] [PubMed]30. Childs, A.C.; Mehta, D.J.; Gerner, E.W. Polyamine-dependent gene expression. Cell. Mol. Life Sci. 2003, 60,
1394–1406. [CrossRef] [PubMed]31. Venkiteswaran, S.; Vijayanathan, V.; Shirahata, A.; Thomas, T.; Thomas, T.J. Antisense recognition of the
HER-2 mRNA: Effects of phosphorothioate substitution and polyamines on DNA·RNA, RNA·RNA, andDNA·DNA duplex stability. Biochemistry 2005, 44, 303–312. [CrossRef] [PubMed]
32. Antony, T.; Thomas, T.; Shirahata, A.; Thomas, T.J. Selectivity of polyamines on the stability of RNA-DNAhybrids containing phosphodiester and phosphorothioate oligodeoxyribonucleotides. Biochemistry 1999, 38,10775–10784. [CrossRef] [PubMed]
33. Hou, M.H.; Lin, S.B.; Yuann, J.M.; Lin, W.C.; Wang, A.H.J.; Kan, L. Effects of polyamines on the thermalstability and formation kinetics of DNA duplexes with abnormal structure. Nucleic Acids Res. 2001, 29,5121–5128. [CrossRef] [PubMed]
34. Brzezinska, J.; Gdaniec, Z.; Popenda, L.; Markiewicz, W.T. Polyaminooligonucleotide: NMR structure ofduplex DNA containing a nucleoside with spermine residue, N-[4,9,13-triazatridecan-1-yl]-2′-deoxycytidine.Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1163–1170. [CrossRef] [PubMed]
35. Malkoch, M.; Schleicher, K.; Drockenmuller, E.; Hawker, C.J.; Russell, T.P.; Wu, P.; Fokin, V.V. StructurallyDiverse Dendritic Libraries: A Highly Efficient Functionalization Approach Using Click Chemistry.Macromolecules 2005, 38, 3663–3678. [CrossRef]
36. Asanuma, H.; Liang, X.; Komiyama, M. meta-Aminoazobenzene as a thermo-insensitive photo-regulator ofDNA-duplex formation. Tetrahedron Lett. 2000, 41, 1055–1058. [CrossRef]
37. Ihre, H.; Padilla De Jesús, O.L.; Fréchet, J.M.J. Fast and convenient divergent synthesis of aliphatic esterdendrimers by anhydride coupling. J. Am. Chem. Soc. 2001, 123, 5908–5917. [CrossRef] [PubMed]
38. Markiewicz, W.T.; Godzina, P.; Markiewicz, M.; Astriab, A. Synthesis of A PolyaminooligonucleotideCombinatorial Library. Nucleos. Nucleot. 1998, 17, 1871–1880. [CrossRef]
39. Godzina, P.; Adrych-Rozek, K.; Markiewicz, W.T. Synthetic Oligonucleotide Combinatorial Libraries. 3.Synthesis of Polyaminonucleosides. Nucleos. Nucleot. 1999, 18, 2397–2414. [CrossRef]
40. Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons, Inc.:Hoboken, NJ, USA, 2007.
41. Sund, C.; Puri, N.; Chattopadhyaya, J. The Chemistry of C-Branched Spermine Tethered Oligo-DNAs andTheir Properties in Forming Duplexes and Triplexes. Nucleos. Nucleot. 1997, 16, 755–760. [CrossRef]
42. Sund, C.; Puri, N.; Chattopadhyaya, J. Synthesis of C-branched spermine tethered oligo-DNA and thethermal stability of the duplexes and triplexes. Tetrahedron 1996, 52, 12275–12290. [CrossRef]
43. Moriguchi, T.; Sakai, H.; Suzuki, H.; Shinozuka, K. Spermine moiety attached to the C-5 position ofdeoxyuridine enhances the duplex stability of the phosphorothioate DNA/complementary DNA andshows the susceptibility of the substrate to RNase H. Chem. Pharm. Bull. (Tokyo) 2008, 56, 1259–1263.[CrossRef] [PubMed]
44. Breslauer, K.J.; Frank, R.; Blöcker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence.Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [CrossRef] [PubMed]
45. Bommarito, S.; Peyret, N.; SantaLucia, J. Thermodynamic parameters for DNA sequences with danglingends. Nucleic Acids Res. 2000, 28, 1929–1934. [CrossRef] [PubMed]
46. Longfellow, C.E.; Kierzek, R.; Turner, D.H. Thermodynamic and spectroscopic study of bulge loops inoligoribonucleotides. Biochemistry 1990, 29, 278–285. [CrossRef] [PubMed]
47. Minetti, C.A.; Remeta, D.P.; Dickstein, R.; Breslauer, K.J. Energetic signatures of single base bulges:Thermodynamic consequences and biological implications. Nucleic Acids Res. 2009, 38, 97–116. [CrossRef][PubMed]
179

Molecules 2015, 20, 12652–12669
48. Dauben, W.G.; Hendricks, R.T.; Pandy, B.; Wu, S.C.; Zhang, Xiaoming; Luzzio, M.J. Stereoselectiveintramolecular cyclopropanations: Enantioselective syntheses of 1α,25-dihydroxyvitamin D3 A-ringprecursors. Tetrahedron Lett. 1995, 36, 2385–2388. [CrossRef]
49. Markiewicz, W.T.; Biała, E.; Adamiak, R.W.; Grzeskowiak, K.; Kierzek, R.; Kraszewski, A.; Stawinski, J.;Wiewiórowski, M. Further studies on oligoribonucleotide synthesis. Nucleic Acids Symp. Ser. 1980, 115–127.
50. Markiewicz, W.T.; Biała, E.; Kierzek, R. Application of the Tetraisopropyldisiloxane-1,3-diyl Group in theChemical Synthesis of Oligoribonucleotides. Bull. Pol. Acad. Sci. Chem. 1984, 32, 433–451.
51. Borer, P.N.; Dengler, B.; Tinoco, I.; Uhlenbeck, O.C. Stability of ribonucleic acid double-stranded helices.J. Mol. Biol. 1974, 86, 843–853. [CrossRef]
52. McDowell, J.A.; Turner, D.H. Investigation of the structural basis for thermodynamic stabilities of tandemGU mismatches: Solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing.Biochemistry 1996, 35, 14077–14089. [CrossRef] [PubMed]
Sample Availability: Sample Availability: Samples are not available from authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
180

molecules
Article
Cladribine Analogues via O6-(Benzotriazolyl)Derivatives of Guanine Nucleosides
Sakilam Satishkumar 1,†, Prasanna K. Vuram 1,†, Siva Subrahmanyam Relangi 2,†,Venkateshwarlu Gurram 2, Hong Zhou 3, Robert J. Kreitman 3,Michelle M. Martínez Montemayor 4, Lijia Yang 1, Muralidharan Kaliyaperumal 2,Somesh Sharma 2, Narender Pottabathini 2 and Mahesh K. Lakshman 1,*
1 Department of Chemistry, The City College and The City University of New York, 160 Convent Avenue,New York, NY 10031, USA; [email protected] (S.S.); [email protected] (P.K.V.);[email protected] (L.Y.)
2 Discovery and Analytical Services, GVK Biosciences Pvt. Ltd., 28A IDA Nacharam, Hyderabad 500076,India; [email protected] (S.S.R.); [email protected] (V.G.);[email protected] (M.K.); [email protected] (S.S.);[email protected] (N.P.)
3 Clinical Immunotherapy Section, Laboratory of Molecular Biology, National Cancer Institute,National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892, USA; [email protected] (H.Z.);[email protected] (R.J.K.)
4 Department of Biochemistry, Universidad Central del Caribe-School of Medicine, P. O. Box 60327, Bayamón,PR 00960, USA; [email protected]
* Correspondence: [email protected]; Tel.: +1-212-650-7835; Fax: +1-212-650-6107† These authors contributed equally to this work.
Academic Editor: Fumi NagatsugiReceived: 17 August 2015; Accepted: 22 September 2015; Published: 9 October 2015
Abstract: Cladribine, 2-chloro-2′-deoxyadenosine, is a highly efficacious, clinically used nucleosidefor the treatment of hairy cell leukemia. It is also being evaluated against other lymphoidmalignancies and has been a molecule of interest for well over half a century. In continuation ofour interest in the amide bond-activation in purine nucleosides via the use of (benzotriazol-1yl-oxy)tris(dimethylamino)phosphonium hexafluorophosphate, we have evaluated the use ofO6-(benzotriazol-1-yl)-2′-deoxyguanosine as a potential precursor to cladribine and its analogues.These compounds, after appropriate deprotection, were assessed for their biological activities, andthe data are presented herein. Against hairy cell leukemia (HCL), T-cell lymphoma (TCL) and chroniclymphocytic leukemia (CLL), cladribine was the most active against all. The bromo analogue ofcladribine showed comparable activity to the ribose analogue of cladribine against HCL, but wasmore active against TCL and CLL. The bromo ribose analogue of cladribine showed activity, butwas the least active among the C6-NH2-containing compounds. Substitution with alkyl groups atthe exocyclic amino group appears detrimental to activity, and only the C6 piperidinyl cladribineanalogue demonstrated any activity. Against adenocarcinoma MDA-MB-231 cells, cladribine and itsribose analogue were most active.
Keywords: cladribine; nucleoside; guanosine; benzotriazole; (benzotriazol-1yl-oxy)-tris(dimethylamino)phosphonium hexafluorophosphate; BOP
1. Introduction
Cladribine, 2-chloro-2′-deoxyadenosine, has been a molecule of interest for well over fivedecades. The history of this compound dates back to 1960, when it was used in the synthesis of2′-deoxyguanosine and 2′-deoxyinosine [1]. A decade later, cladribine was shown to be a poor
Molecules 2015, 20, 18437–18463 181 www.mdpi.com/journal/molecules

Molecules 2015, 20, 18437–18463
substrate for adenosine deaminase that underwent phosphorylation by deoxycytidine kinase, finallyresulting in the triphosphate, and inhibiting DNA synthesis rather than RNA synthesis [2,3]. Later still,it was shown to be a substrate for deoxyguanosine kinase, which is responsible for phosphorylation ofpurine nucleosides in mitochondria [3]. Several mechanisms have been proposed by which cladribinecan cause mitochondrial damage and apoptotic cell death [4–8].
In contemporary medicine, cladribine is used in the treatment of lymphoid malignancies, mostnotably for its efficacy against hairy cell leukemia [9]. Cladribine is also being evaluated against severalother indolent lymphoid malignancies, also in combination with other drug candidates [10,11].
The synthesis of cladribine has primarily relied on three major methods: (a) glycosylationreactions of a nucleobase with a sugar [12–18], and its variations; (b) deoxygenation of the C2′ hydroxylgroup of a suitable nucleoside derivative [12,15,19,20]; (c) enzymatic glycosyl transfer reactions [21–24]; and(d) conversion of readily available nucleoside precursors (some utilizing nucleosides for glycosyltransfer reactions) [21,22,24–26]. Each of these methods has been used with varying levels ofconvenience and success. Among the many approaches, one convenient method is the selectivedisplacement of a leaving group (chloride or aryl sulfonate) from the C6 position of a suitable purinenucleoside precursor. Despite the availability of this selective SNAr displacement, we find that noother N6-substituted cladribine analogues have been synthesized by such a method. Because of ourinterest in broadening the utilities of O6-(benzotriazol-1-yl)purine nucleoside derivatives, we electedto evaluate the synthesis of N6-substituted cladribine analogues via amide-bond activation of guaninenucleosides with (benzotriazol-1yl-oxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP).
2. Results and Discussion
For the modification of the nucleobases of inosine, and 2′-deoxyinosine (3 examples), BOPhad been used for in situ activation of the amide moieties in these substrates, followed by SNArdisplacement with amines [27,28]. It was proposed that reaction of the amide group with BOPproceeded via a phosphonium intermediate, which could be directly captured by a reactive amine [27].On the other hand, with less reactive amines, the O-(benzotriazol-1-yl) intermediate can be formed bycompetitive capture of the phosphonium intermediate by the benzotriazol-1-yloxy anion [27].
In 2007, we first reported the isolation of O6-(benzotriazol-1-yl)inosine and -2′-deoxyinosineby amide-bond activation with BOP, in the absence of a nucleophile [29], an observation that waslater reconfirmed by others [30,31]. These electrophilic nucleosides, which are stable to storage, areexceptionally good partners in SNAr reactions with oxygen, nitrogen, and sulfur nucleophiles [29].Subsequently, we demonstrated that O6-(benzotriazol-1-yl)inosine and -2′-deoxyinosine can alsobe prepared via the use of PPh3/I2 and 1-hydroxybenzotriazole [32]. The amide-bond activationprotocol was then modified to tether the O6-(benzotriazol-1-yl) nucleosides onto a polymer support forhigh-throughput type of applications [33]. Interestingly, the amide bond activation when applied to theurea functionality of O6-benzyl-protected 2′-deoxyxanthosine did not yield the O2-(benzotriazol-1-yl)derivative but rather terminated in an isolable and synthetically useful phosphonium salt [34]. Wealso showed that guanosine and 2′-deoxyguanosine undergo facile reactions with BOP, and thatO6-(benzotriazol-1-yl) guanine nucleosides are effective substrates for SNAr reactions as well [35]. Inthe combined course of these investigations we had ascertained plausible operative mechanisms ofthese amide-activation reactions, results that were later applied to a one-pot etherification protocol forpurine nucleosides and pyrimidines [36].
Considering only the nucleoside modification literature, our BOP-mediated amide-activationmethodology has found wide application [37–43]. Of specific interest to our current work was areport wherein some of our results on guanosine derivatives were reevaluated [44]. In additionto 2′,3′,5′-tri-O-(t-butyldimethylsilyl)guanosine, which we had originally investigated, five othercompounds were also studied: unprotected 2′-deoxyguanosine, its 2′,3′,5′-triacetyl and the2′,3′-isopropylidene derivatives, and two 2′,3′-isopropylidiene 5′-carboxamides [44]. These productswere subsequently tested in diazotization-halogenation reactions, en route to 2-chloro and 2-iodo
182

Molecules 2015, 20, 18437–18463
adenosine analogues [44]. 2,6-Dihalopurine ribonucleosides are more readily accessible forthis purpose, as compared to 2′-deoxy derivatives. Thus, we were intrigued by the fact thatdiazotization-halogenation reactions of O6-(benzotriazol-1yl)-2′-deoxyguanosine have not beenreported. This prompted use to investigate the chlorination and bromination of this compound,and in this context we decided to evaluate the synthesis of cladribine and other cladribine analoguesvia such an approach.
In 2001, diazotization reactions leading to cladribine and other halo derivatives have beenperformed on unprotected 2,6-diaminopurine 2′-deoxyribonucleoside [45]. However, no substituentsother than NH2 were introduced into the C6 position, and the synthesis of the precursor is notparticularly convenient. For the present study, we anticipated the need for saccharide protection, andboth acetyl and t-butyldimethysilyl (TBS) protecting groups were considered. Between these, TBSwas selected because acetyl groups can be susceptible to cleavage with amines, which could be acomplicating problem in the method development stage. Thus, nucleosides 1a and 1b were silylatedto give the corresponding products 2a and 2b, which were converted to the O6-(benzotriazol-1-yl)guanosine derivatives 3a and 3b, respectively (Scheme 1).
Scheme 1. Preparation of O6-(benzotriazol-1-yl) guanosine derivatives and the chlorination reaction.
We opted for diazotization-chlorination conditions using t-BuONO/TMSCl, a reagentcombination initially introduced for nucleoside modification in 2003 [46]. We [47] and others [44]have previously used non-aqueous conditions for halogenation at the C2 position of purinenucleoside derivatives. Under these conditions, reactions of substrate 3a proceeded modestly.However, the obtained product was contaminated with ~30% of the C2 protio O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine (Table 1, entries 1 and 2). With the combination oft-BuONO/TMSCl/(BnNEt3)+Cl−, no C2 protio product was observed, but only a low product yieldwas obtained (entry 3).
These results compelled us to consider other conditions. SbCl3 and SbBr3 have previously beenused for the halogenation of nucleosides [48,49] Thus, the next series of experiments involved SbCl3,(BnNEt3)+Cl−, and combinations of these reagents (entries 4–11). Whereas most experiments yieldedonly 35%–39% of product 4a, a reasonable yield improvement was observed in entry 9. It was notedthat efficient filtration of the reaction mixture after workup is critical to obtaining a good productrecovery due to the pasty nature of the mixture (see the Experimental Section). On larger scales, betterproduct recoveries were observed (entries 10 and 11). By comparison, diazotization/chlorination ofthe ribose derivative 3b proceeded well with both t-BuONO/SbCl3 (entry 12) and t-BuONO/TMSCl(entry 13), with the latter providing a better yield of compound 4b. No C2 protio product was apparentin the reactions of precursor 3b.
183

Molecules 2015, 20, 18437–18463
Table 1. Evaluation of conditions for the diazotization/chlorination of O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyguanosine.
Entry Conditions Scale Yield
1 t-BuONO (2.6 equiv), TMSCl (2.6 equiv), CH2Cl2, −10 ◦C to 0 ◦C, 2 h 73 μmol of 3a 43% a
2 t-BuONO (10 equiv), TMSCl (5 equiv), CH2Cl2, 0 ◦C, 1 h 73 μmol of 3a 58% a
3 t-BuONO (10 equiv), TMSCl (0.4 equiv), (BnNEt3)+Cl− (10 equiv), CH2Cl2, −10 ◦C to 0 ◦C, 2 h 49 μmol of 3a 26%4 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), ClCH2CH2Cl, −10 ◦C to −15 ◦C, 1.5 h 49 μmol of 3a 35%5 t-BuONO (10 equiv), (BnNEt3)+Cl− (10 equiv), CH2Cl2, −78 ◦C and then rt, 1 h 49 μmol of 3a 39%6 t-BuONO (20 equiv), SbCl3 (0.4 equiv), (BnNEt3)+Cl− (20 equiv), CH2Cl2, −10 ◦C to 0 ◦C, 5 h 49 μmol of 3a 39%7 t-BuONO (10 equiv), (BnNEt3)+Cl− (10 equiv), (Me3Si)2NH (10 equiv), CH2Cl2, −10 ◦C to 0 ◦C, 4 h 49 μmol of 3a 35%8 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 ◦C, 2 h 49 μmol of 3a 39% b
9 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 ◦C to −15 ◦C, 2 h 73 μmol of 3a 51%10 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 ◦C, 2 h 0.14 mmol of 3a 60%11 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 ◦C to −15 ◦C, 2 h 1.6 mmol of 3a 65%12 t-BuONO (3.5 equiv), SbCl3 (1.4 equiv), CH2Cl2, −10 ◦C to −15 ◦C, 3.5 h 0.27 mmol of 3b 61%13 t-BuONO (10 equiv), TMSCl (5 equiv), CH2Cl2, 0 ◦C, 1 h 2.69 mmol of 3b 68%
a Yield was calculated on the basis of the molecular weight of compound 4a. However, by 1H-NMR (500 MHz,CDCl3), the chromatographically homogenous product band was observed to contain a 2:1 ratio of compound 4aand the C2 protio O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine. In addition to these, aminor uncharacterized nucleoside byproduct was also formed; b Compound 3a dissolved in CH2Cl2 was added tothe mixture of reagents in CH2Cl2.
The next stage in the chemistry involved SNAr reactions at the C6 positions of substrates 4a
and 4b. Cladribine and its ribose analogue were prepared by reactions with aqueous ammonia(see the Experimental Section for details). In order to prepare other analogues, reactions wereconducted with 1.5 equiv each of methylamine, dimethylamine, pyrrolidine, piperidine, morpholine,and N,N,N′-trimethylethylenediamine (products, reaction times, and yields are shown in Figure 1).
Figure 1. Structures of the products, reaction times, and yields obtained in the SNAr reactions.
Most reactions proceeded smoothly and in good to high yields. Methylamine (2 M in THF)was used for the synthesis of 6a and 6b, whereas a 40% aqueous solution of dimethylaminewas used for the synthesis of 7a and 7b. As we have previously shown, water is not generallydetrimental to reactions of these benzotriazolyl purine nucleosides [35]. In reactions of 4a and 4b withN,N,N′-tri-methylethylenediamine, yields were lower. In each ~10%–15% of compounds 7a and 7b
were isolated as byproducts. The source of dimethylamine is currently unknown but its origin can belinked to N,N,N′-trimethylethylenediamine.
184

Molecules 2015, 20, 18437–18463
Finally, for biological testing, desilylation was performed. Because we did not anticipatedecomposition of starting materials or products, we only tested the use of KF (2 equiv/silyl group) inMeOH at 80 ◦C (Scheme 2). The results of the desilylation reactions are shown in Table 2.
Scheme 2. Desilylation of the protected nucleosides.
Table 2. Structures of the products, reaction times, and yields of the desilylated compounds.
Entry R Reaction Times/KF Equiv Yields
1 24 h with 4 equiv KF 12a: 72%2 24 h with 6 equiv KF 12b: 74%
3 24 h with 4 equiv KF 13a: 85%4 16 h with 6 equiv KF 13b: 75%
5 24 h with 4 equiv KF 14a: 82%6 16 h with 6 equiv KF 14b: 75%
7 20 h with 4 equiv KF 15a: 81%8 16 h with 6 equiv KF 15b: 85%
9 24 h with 4 equiv KF 16a: 71%10 16 h with 6 equiv KF 16b: 84%
11 26 h with 4 equiv KF 17a: 74%12 16 h with 6 equiv KF 17b: 78%
13 24 h with 4 equiv KF 18a: 84%14 24 h with 6 equiv KF 18b: 65%
Because there is currently no known method for the diazotization/bromination of compound3a, we investigated a route similar to that for chlorination. Results from these experiments are listedin Table 3. What is notable with the diazotization/bromination, in contrast to the chlorination, wasthat use of 3.5 equiv of t-BuONO led to incomplete conversion over 2 h at −10 to −15 ◦C. Addition ofanother 3.5 equiv of t-BuONO then led to complete conversion over an additional 1 h.
A similar conversion of 3b led to the ribose analogue 19b in a comparable 64% yield (Table 3,entry 3). Compound 19a was then converted to the bromo analogue of cladribine as shown in Scheme 3.SNAr displacement with aqueous NH3 proceeded in 83% yield producing compound 20a, which wasfinally desilylated with KF in anhydrous MeOH at 80 ◦C (28 h) to yield 2-bromo-2-deoxyadenosine(21a) in 66% yield. Corresponding conversions of the ribose analog 19b, via intermediate 20b, gavecompound 21b. Yields for these conversions were comparable to the deoxyribose series.
185
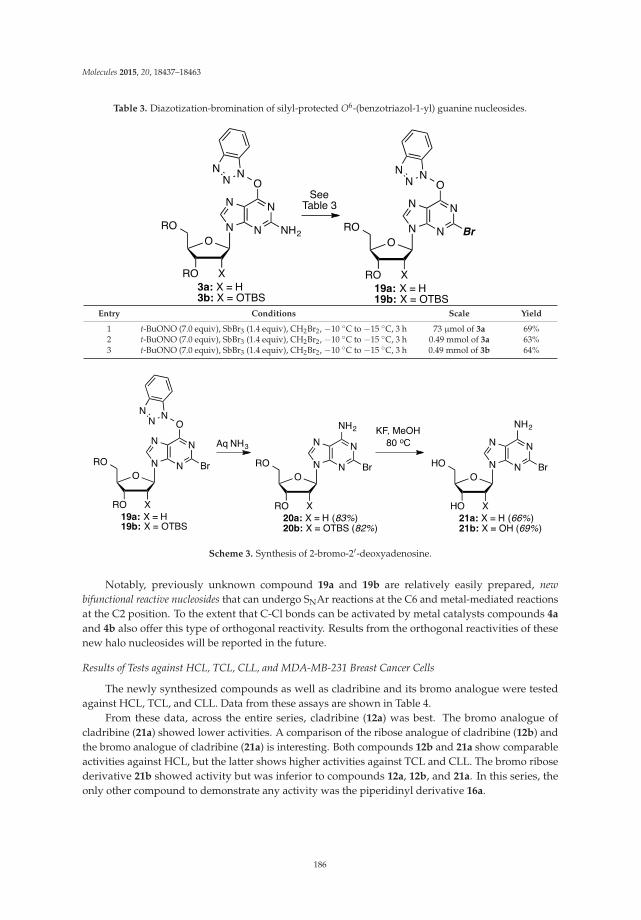
Molecules 2015, 20, 18437–18463
Table 3. Diazotization-bromination of silyl-protected O6-(benzotriazol-1-yl) guanine nucleosides.
Entry Conditions Scale Yield
1 t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 ◦C to −15 ◦C, 3 h 73 μmol of 3a 69%2 t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 ◦C to −15 ◦C, 3 h 0.49 mmol of 3a 63%3 t-BuONO (7.0 equiv), SbBr3 (1.4 equiv), CH2Br2, −10 ◦C to −15 ◦C, 3 h 0.49 mmol of 3b 64%
Scheme 3. Synthesis of 2-bromo-2′-deoxyadenosine.
Notably, previously unknown compound 19a and 19b are relatively easily prepared, newbifunctional reactive nucleosides that can undergo SNAr reactions at the C6 and metal-mediated reactionsat the C2 position. To the extent that C-Cl bonds can be activated by metal catalysts compounds 4a
and 4b also offer this type of orthogonal reactivity. Results from the orthogonal reactivities of thesenew halo nucleosides will be reported in the future.
Results of Tests against HCL, TCL, CLL, and MDA-MB-231 Breast Cancer Cells
The newly synthesized compounds as well as cladribine and its bromo analogue were testedagainst HCL, TCL, and CLL. Data from these assays are shown in Table 4.
From these data, across the entire series, cladribine (12a) was best. The bromo analogue ofcladribine (21a) showed lower activities. A comparison of the ribose analogue of cladribine (12b) andthe bromo analogue of cladribine (21a) is interesting. Both compounds 12b and 21a show comparableactivities against HCL, but the latter shows higher activities against TCL and CLL. The bromo ribosederivative 21b showed activity but was inferior to compounds 12a, 12b, and 21a. In this series, theonly other compound to demonstrate any activity was the piperidinyl derivative 16a.
186

Molecules 2015, 20, 18437–18463
Table 4. IC50 values (μM) of the cladribine and its analogues on HCL, TCL, and CLL a.
CompoundHCL TCL CLL
ATP 3H-Leu ATP 3H-Leu ATP 3H-Leu
12a 0.065 0.090 0.020 0.039 0.004 0.01112b 0.81 0.85 4.81 9.40 1.33 0.8813a >33 >33 >33 >33 >33 >3313b >32 >32 >32 >32 ND b >3214a >32 >32 >32 >32 >32 >3214b >30 >30 >30 >30 >30 >3015a >29 >29 >29 >29 >29 >2915b >28 >28 >28 >28 >28 >2816a 11.5 16.3 9.6 9.0 4.3 5.416b >27 >27 >27 >27 ND b ND b
17a >28 >28 >28 >28 >28 >2817b >27 >27 >27 >27 >27 >2718a >27 >27 >27 >27 >27 >2718b >26 >26 >26 >26 >26 >2621a 0.76 0.96 0.13 0.16 0.13 0.1421b 1.8 7.2 8.6 10.2 2.0 4.0
a One patient per disease type; b ND = not determined.
The compounds were also tested against adenocarcinoma MDA-MB-231 breast cancer cells. Thesecells were treated with three-fold serial dilutions of the compounds ranging from 0–1.8 mM for 24 h.Viable cells were fixed with cold methanol and nuclei were stained with 0.4% propidium iodide(PI). Cell viability was calculated as the percentage of surviving cells after treatment as measured bydifferences in fluorescence units between treated and untreated wells (Figure 2).
IC50 values were obtained from dose response curve fittings using the non-linear regressionfunction of GraphPad Prism® (La Jolla, CA, USA). Dashed horizontal line represents 50% cell viability.Columns represent means ± SEM of at least three independent experiments. Significant differencesare described with * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 compared to control.
From the IC50 values shown in Table 5, the two compounds that emerged as most promising werecladribine 12a and its ribose analogue 12b, followed by the bromo derivatives 21a and 21b, whichwere about 10 times less active.
Table 5. IC50 values (mM) of the compounds synthesized on MDA-MB-231 breast cancer cells.
2′-Deoxyribose Series Ribose Series
12a 13a 14a 15a 16a 17a 18a 21a 12b 13b 14b 15b 16b 17b 18b 21b0.05 2.15 6.28 2.54 4.18 2.32 1.88 0.64 0.06 2.39 1.70 1.54 2.45 2.57 2.91 0.55
187
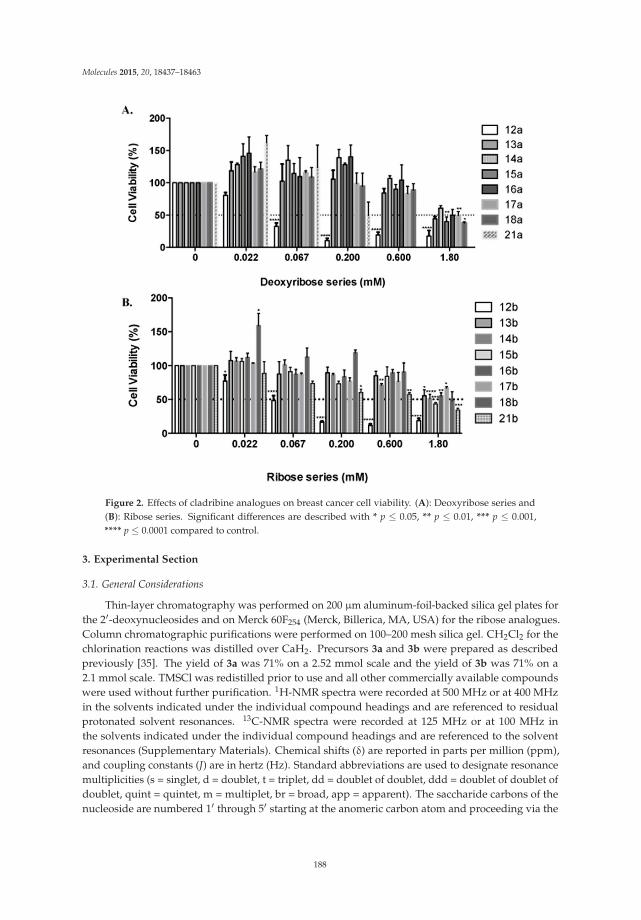
Molecules 2015, 20, 18437–18463
Figure 2. Effects of cladribine analogues on breast cancer cell viability. (A): Deoxyribose series and(B): Ribose series. Significant differences are described with * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001,**** p ≤ 0.0001 compared to control.
3. Experimental Section
3.1. General Considerations
Thin-layer chromatography was performed on 200 μm aluminum-foil-backed silica gel plates forthe 2′-deoxynucleosides and on Merck 60F254 (Merck, Billerica, MA, USA) for the ribose analogues.Column chromatographic purifications were performed on 100–200 mesh silica gel. CH2Cl2 for thechlorination reactions was distilled over CaH2. Precursors 3a and 3b were prepared as describedpreviously [35]. The yield of 3a was 71% on a 2.52 mmol scale and the yield of 3b was 71% on a2.1 mmol scale. TMSCl was redistilled prior to use and all other commercially available compoundswere used without further purification. 1H-NMR spectra were recorded at 500 MHz or at 400 MHzin the solvents indicated under the individual compound headings and are referenced to residualprotonated solvent resonances. 13C-NMR spectra were recorded at 125 MHz or at 100 MHz inthe solvents indicated under the individual compound headings and are referenced to the solventresonances (Supplementary Materials). Chemical shifts (δ) are reported in parts per million (ppm),and coupling constants (J) are in hertz (Hz). Standard abbreviations are used to designate resonancemultiplicities (s = singlet, d = doublet, t = triplet, dd = doublet of doublet, ddd = doublet of doublet ofdoublet, quint = quintet, m = multiplet, br = broad, app = apparent). The saccharide carbons of thenucleoside are numbered 1′ through 5′ starting at the anomeric carbon atom and proceeding via the
188

Molecules 2015, 20, 18437–18463
carbon chain to the primary carbinol carbon atom. The purinyl proton is designated as H-8 and thesaccharide protons are designated on the basis of the carbon atom they are attached to.
O6-(Benzotriazol-1-yl)-2-chloro-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (4a):A mixture of compound 3a (1.0 g, 1.63 mmol) and SbCl3 (520.6 mg, 2.28 mmol) in dry CH2Cl2(16.3 mL) was cooled to −15 ◦C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO(0.678 mL, 5.70 mmol) was added dropwise and the mixture was stirred at −10 to −15 ◦C for 3.5 h, atwhich time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold,saturated aqueous NaHCO3 (25 mL) with stirring. The mixture was filtered using vacuum (note: useof vacuum for this filtration is critical for maximizing product recovery) and the residue was washedwith CH2Cl2 (25 mL). The organic layer was separated and the aqueous layer was back extracted withCH2Cl2 (2 × 15 mL). The combined organic layer was washed with water (15 mL) and brine (15 mL),dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crudematerial on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, followed by20% EtOAc in hexanes gave 0.67 g (65% yield) of compound 4a as a white foam. Rf (SiO2 and 30%EtOAc in hexanes) = 0.55. 1H-NMR (500 MHz, CDCl3): δ 8.47 (s, 1H, H-8), 8.09 (d, J = 8.3 Hz, 1H,Ar-H), 7.52–7.41 (m, 3H, Ar-H), 6.45 (t, J = 5.8 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′), 4.01 (m, 1H, H-4′),3.89 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~6.2 Hz,1H, H-2′), 2.49–2.48 (app quint, Japp ~5.8 Hz, 1H, H-2′), 0.88 (s, 18H, t-Bu), 0.08 and 0.07 (2s, 12H,SiCH3). 13C-NMR (125 MHz, CDCl3): δ 159.2, 154.9, 152.6, 144.4, 143.5, 129.0, 128.8, 125.0, 120.7, 119.1,108.6, 88.4, 85.3, 71.6, 62.6, 41.6, 26.1, 25.9, 18.5, 18.1, −4.5, −4.7, −5.2, −5.3. HRMS (TOF) calcd forC28H43ClN7O4Si2 [M + H]+ 632.2598, found 632.2583.
2-Chloro-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (5a): To a solution of compound 4a (126.0 mg,0.2 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (32 μL) was added, and the mixture wasstirred at room temperature for 1.5 h. The mixture was diluted with EtOAc (5 mL) and washed with5% aqueous NaCl (5 mL). The organic layer was separated and the aqueous layer was back extractedwith EtOAc (5 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporatedunder reduced pressure. The crude material was chromatographed on a silica gel column by sequentialelution with 20% EtOAc in hexanes followed by 5% MeOH in CH2Cl2 to afford 85.0 mg (82% yield)of compound 5a as a white solid. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.40. 1H-NMR (500 MHz,CDCl3): δ 8.12 (s, 1H, H-8), 6.54 (br s, 2H, NH2), 6.38 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′),3.99 (app q, Japp ~3.4 Hz, 1H, H-4′) 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 3.0, 11.2 Hz, 1H,H-5′), 2.61 (app quint, Japp ~6.4 Hz, 1H, H-2′), 2.44–2.39 (ddd, J = 3.1, 6.3, 13.2 Hz, 1H, H-2′), 0.90 (s,18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 156.2, 154.0, 150.4, 139.5,118.7, 87.9, 84.5, 71.7, 62.6, 41.3, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd forC22H40ClN5O3Si2Na [M + Na]+ 536.2250, found 536.2252.
3.2. General Procedure for the Synthesis of Cladribine Analogues
To a solution of compound 4a (126.0 mg, 0.2 mmol) in 1,2-DME (2 mL) the appropriate amine(1.5 equiv) was added, and the mixture was stirred at room temperature. The mixture was dilutedwith EtOAc (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a) and washed 5%aqueous NaCl (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The organiclayer was separated and the aqueous layer was back extracted with EtOAc (5 mL for compounds 6a,9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The combined organic layer was dried over anhydrousNa2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silicagel column; see individual compound headings for details.
2-Chloro-N6-methyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (6a): Compound 6a (85.0 mg, 80%yield) was obtained as a white, foamy solid after chromatography on a silica gel column by sequentialelution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) =0.26. 1H-NMR (500 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.37 (t, J = 6.4 Hz, 1H, H-1′), 6.17 (br s, 1H, NH),
189

Molecules 2015, 20, 18437–18463
4.60 (m, 1H, H-3′), 3.97 (app q, Japp ~3.4 Hz, 1H, H-4′), 3.87 (dd, J = 4.1, 11.2 Hz, 1H, H-5′), 3.75 (dd,J = 3.0, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~6.4 Hz, 1H, H-2′), 2.42–2.37 (ddd, J = 2.8, 6.1, 12.7 Hz,1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.9,154.5, 149.2; 138.7, 119.1, 87.9, 84.4, 71.8, 62.7, 41.2, 27.5, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5.HRMS (TOF) calcd for C23H42ClN5O3Si2Na [M + Na]+ 550.2407, found 550.2419.
2-Chloro-N6,N6-dimethyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (7a): Compound 7a (91.1 mg,84% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column bysequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30%EtOAc in hexanes) = 0.70. 1H-NMR (500 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′),4.58 (m, 1H, H-3′), 3.95 (m, 1H, H-4′), 3.83 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.73 (dd, J = 2.9, 11.2 Hz, 1H,H-5′), 3.50 (br s, 6H, N(CH3)2), 2.58 (app quint, Japp ~6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 0.88 (s,18H, t-Bu), 0.08 and 0.06 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.2, 153.8, 151.4, 137.2,119.6, 87.9, 84.4, 72.1, 62.9, 41.1, 26.1, 26.0, 25.9, 25.8, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF)calcd for C24H45ClN5O3Si2 [M + H]+ 542.2744, found 542.2749.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (8a):Compound 8a (93.3 mg, 82% yield) was obtained as a colorless, thick gum after chromatography on asilica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes.Rf (SiO2 and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t,J = 6.6 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.13 (br s, 2H, pyrrolidinyl NCH), 3.96 (m, 1H, H-4′), 3.83 (dd,J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.79–3.73 (br m, 2H, pyrrolidinyl NCH),2.61 (app quint, Japp ~6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 2.10–2.0 (br m, 2H, pyrrolidinyl CH),1.96–1.85 (br m, 2H, pyrrolidinyl CH), 0.90 (s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR(125 MHz, CDCl3): δ 154.3, 153.5, 151.0, 137.8, 119.8, 88.1, 84.4, 72.3, 63.1, 49.1, 41.08, 26.4, 26.2, 25.9,24.3, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd. for C26H47ClN5O3Si2 [M + H]+ 568.2900,found 568.2906.
2-Chloro-6-(piperidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (9a):Compound 9a (99.0 mg, 85% yield) was obtained as a white, foamy solid after chromatography on asilica gel column by sequential elution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2
and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t, J = 6.6 Hz,1H, H-1′), 4.59 (m, 1H, H-3′), 4.45–3.73 (br m, 4H, piperidinyl N(CH2)2), 3.96 (app q, Japp ~3.6 Hz, 1H,H-4′), 3.83 (dd, J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 2.59 (app quint, Japp
~6.3 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 3.3, 6.2, 13.3 Hz, 1H, H-2′), 1.68 (br m, 6H, piperidinyl CH), 0.90(s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 153.8, 151.4, 136.7, 119.0,109.9, 87.8, 84.2, 72.0, 62.8, 46.3, 40.9, 26.1, 25.9, 25.7, 24.6, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS(TOF) calcd for C27H49ClN5O3Si2 [M + H] + 582.3057 found 582.3074.
2-Chloro-6-(morpholin-4-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (10a):Compound 10a (97.4 mg, 83% yield) was obtained as a colorless, thick gum after chromatographyon a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc inhexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.68. 1H-NMR (500 MHz, CDCl3): δ 7.98 (s, 1H, H-8),6.37 (t, J = 6.3 Hz, 1H, H-1′), 4.58 (m, 1H, H-3′), 4.55–3.98 (br m, 4H, morpholinyl N(CH2)2), 3.96 (m,1H, H-4′), 3.84 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.78 (t, J = 4.6 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd,J = 2.9, 11.2 Hz, 1H, H-5′), 2.56 (app quint, Japp ~6.5 Hz, 1H, H-2′), 2.40–2.36 (m, 1H, H-2′), 0.89 (s, 18H,t-Bu), 0.08 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.0, 153.9, 151.8, 137.6, 119.3,88.0, 84.4, 72.0, 67.1, 62.9, 43.5 (br), 41.3, 26.1, 26.0, 25.9, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF)calcd for C26H47ClN5O4Si2 [M + H]+ 584.2850, found 584.2855.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (11a): Compound 11a (77.0 mg, 64% yield) was obtained as a pale-yellow, thickgum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes,
190

Molecules 2015, 20, 18437–18463
20% EtOAc in hexanes, and 10% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10.1H-NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′),4.28–3.84 (br m, 2H, NCH2), 3.99 (app q, Japp ~3.7 Hz, 1H, H-4′), 3.85 (dd, J = 4.4, 11.2 Hz, 1H, H-5′),3.78 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.47 (br s, 3H, NCH3), 2.65–2.59 (m, 2H, NCH2 and 1H, H-2′),2.41–2.37 (ddd, J = 4.2, 6.2, 13.1 Hz, 1H, H-2′), 2.34 (s, 6H, N(CH3)2), 0.92 and 0.91 (s, 18H, t-Bu), 0.11,0.083, and 0.079 (3s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.9, 153.8, 151.4, 137.2, 119.4, 87.9,84.4, 72.0, 62.9, 57.0, 48.6, 45.7, 41.0, 36.9, 26.0, 25.8, 18.4, 8.0, −4.6, −4.8, −5.4, −5.5. HRMS (TOF) calcdfor C27H52ClN6O3Si2 [M + H]+ 599.3322 found 599.3313.
O6-(Benzotriazol-1-yl)-2-chloro-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (4b): To asolution of t-BuONO (2.78 g, 26.91 mmol) in CH2Cl2 (197 mL) was added TMSCl (1.46 g, 13.45 mmol)at 0 ◦C. To this mixture a solution of 3b (2 g, 2.69 mmol) in CH2Cl2 (197 mL) was added dropwise,and then the reaction mixture was stirred at 0 ◦C for 1 h at which time the reaction was completed asindicated by TLC. The reaction mixture was diluted with CH2Cl2 (200 mL), washed with saturatedNaHCO3 (100 mL), H2O (100 mL), and brine (100 mL). The organic layer was dried over anhydrousNa2SO4 and evaporated under reduced pressure. Purification of the crude material on a silica gelcolumn sequentially eluted with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes gave 1.4 g(68% yield) of compound 4b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.55. 1H-NMR(400 MHz, CDCl3): δ 8.56 (s, 1H, H-8), 8.14 (d, J = 8.0 Hz, 1H, Ar-H), 7.55–7.45 (m, 3H, Ar-H), 6.05 (d,J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.34 (t, J = 4.4 Hz, 1H, H-3′), 4.18–4.15 (m, 1H, H-4′),4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.88 (3s, 27H,t-Bu), 0.15, 0.11, 0.02, and −0.11 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 159.0, 154.9, 152.6,144.4, 143.4, 128.9, 128.7, 124.9, 120.6, 119.0, 108.5, 89.4, 85.3, 71.2, 62.0, 26.1, 25.8, 25.6, 18.5, 18.0, 17.9,−4.3, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57ClN7O5Si3 [M + H]+ 762.3412, found762.3427.
2-Chloro-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (5b): To a solution of compound 4b (500 mg,0.655 mmol) in 1,2-DME (8 mL), 28%–30% aqueous ammonia (64 μL) was added and the mixturewas stirred at room temperature for 1.5 h. The reaction mixture was diluted with EtOAc (25 mL) andwashed 5% aqueous NaCl (25 mL). The organic layer was separated and the aqueous layer was backextracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 andevaporated under reduced pressure. The crude material was chromatographed on a silica gel columnby sequential elution with 20% EtOAc in hexanes and 5% MeOH in EtOAc to afford 310 mg (75% yield)of compound 5b as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz,CDCl3): δ 8.12 (s, 1H, H-8), 6.17 (br s, 2H, NH2), 5.93 (d, J = 4.8 Hz, 1H, H-1′), 4.69 (t, J = 4.6 Hz, 1H,H-2′), 4.32 (t, J = 4.6 Hz, H-3′), 4.14–4.06 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.6 Hz, 1H, H-5′), 3.80 (dd,J = 2.8, 11.2 Hz, 1H, H-5′), 0.94, 0.91, and 0.84 (3s, 27H, t-Bu), 0.14, 0.09, −0.01, and −0.17 (4s, 18H,SiCH3). 13C-NMR (100 MHz, CDCl3): δ 156.1, 154.0, 150.7, 140.2, 118.9, 88.9, 85.4, 75.5, 71.7, 62.3, 26.0,25.8, 25.7, 18.5, 18.0, 17.9, −4.3, −4.5, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd. for C28H55ClN5O4Si3[M + H]+ 644.3245 found 644.3217.
3.3. General Procedure for the Synthesis of Ribose Analogues of Cladribine
To a solution of compound 4b (500 mg, 0.655 mmol) in 1,2-DME (8 mL) was added the appropriateamine (1.5 equiv) and the mixture was stirred at room temperature. The mixture was diluted withEtOAc (25 mL) and washed with 5% aqueous NaCl (15 mL). The organic layer was separated and theaqueous layer was back extracted with EtOAc (15 mL). The combined organic layers were dried overanhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographedon a silica gel column. See individual compound headings for details.
2-Chloro-N6-methyl-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (6b): Compound 6b (340 mg, 80% yield)was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequentialelution with 10% EtOAc in hexanes, 20% EtOAc in hexanes, and 50% EtOAc in hexanes. Rf (SiO2 and
191

Molecules 2015, 20, 18437–18463
EtOAc) = 0.20. 1H-NMR (400 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.04 (br s, 1H, NH), 5.91 (d, J = 5.2 Hz,1H, H-1′), 4.72 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.12–4.10 (m, 1H, H-4′), 4.06 (dd,J = 5.2, 11.6 Hz, 1H, H-5′), 3.79 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.17 (s, 3H, CH3), 0.94, 0.92, and 0.81 (3s,27H, t-Bu), 0.13, 0.06, −0.02, and −0.19 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 154.5,139.3, 119.3, 88.8, 85.4, 75.3, 71.5, 65.2, 62.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.8, 14.0, 11.0, −4.4, −4.7, −5.1,−5.4, −5.7. HRMS (TOF) calcd for C29H57ClN5O4Si3 [M + H]+ 658.3401, found 658.3416.
2-Chloro-N6,N6-dimethyl- 2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (7b): Compound 7b (360 mg, 81%yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column bysequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc inhexanes) = 0.40. 1H-NMR (400 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H, H-1′), 4.77 (t,J = 4.8 Hz, 1H, H-2′), 4.31 (t, J = 3.8 Hz, 1H, H-3′), 4.04–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.6, 11.2 Hz, 1H,H-5′), 3.78 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.58 (br s, 6H, CH3), 0.94, 0.92, and 0.82 (3s, 27H, t-Bu), 0.13,0.10, −0.02, and −0.18 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 153.7, 151.4, 137.9, 119.7,88.8, 85.4, 74.8, 72.0, 62.5, 38.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.4. HRMS (TOF)calcd for C30H59ClN5O4Si3 [M + H]+ 672.3558, found 672.3561.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (8b): Compound8b (390 mg, 85% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gelcolumn by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and20% EtOAc in hexanes) = 0.30. 1H-NMR (400 MHz, CDCl3): δ 7.90 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H,H-1′), 4.81 (t, J = 5.0 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.14–4.08 (m, 1H, H-4′ and br m, 2H,pyrrolidinyl NCH), 4.06 (dd, J = 5.6, 11.2 Hz, 1H, H-5′), 3.77–3.73 (m, 3H, H-5′ and pyrrolidinyl NCH),2.04 (br m, 2H, pyrrolidinyl CH), 1.98 (br m, 2H, pyrrolidinyl CH), 0.93, 0.89, and 0.81 (3s, 27H, t-Bu),0.12, 0.11, −0.03, and −0.20 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 154.0, 153.4, 151.0, 138.0,119.9, 88.7, 85.5, 74.6, 72.2, 62.6, 48.9, 47.6, 26.0, 25.8, 25.7, 18.4, 18.1, 17.9, −4.4, −4.6, −4.7, −5.0, −5.3.HRMS (TOF) calcd for C32H61ClN5O4Si3 [M + H]+ 698.3714 found 698.3731.
2-Chloro-6-(piperidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (9b): Compound9b (375 mg, 80% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gelcolumn by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20%EtOAc in hexanes) = 0.50. 1H-NMR (400 MHz, CDCl3): δ 7.91 (s, 1H, H-8), 5.89 (d, J = 5.6 Hz, 1H, H-1′),4.79 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.22 (br s, 4H, piperidinyl NCH2), 4.11–4.09(m, 1H, H-4′), 4.06 (dd, J = 5.2, 10.8 Hz, 1H, H-5′), 3.77 (dd, J = 2.8, 10.8 Hz, 1H, H-5′), 1.69 (br m, 6H,piperidinyl CH2), 0.94, 0.90, and 0.84 (3s, 27H, t-Bu), 0.12, 0.10, −0.02, and −0.17 (4s, 18H, SiCH3).13C-NMR (100 MHz, CDCl3): δ 153.9, 153.8, 151.6, 137.6, 119.4, 88.8, 85.3, 74.7, 71.9, 62.5, 29.6, 25.8,25.7, 24.6, 22.6, 18.4, 18.0, 17.9, 14.0, −4.3, −4.7, −5.0, −5.3. HRMS (TOF) calcd for C33H63ClN5O4Si3[M + H]+ 712.3871, found 712.3875.
2-Chloro-6-(morpholin-4-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (10b): Compound10b (430 mg, 90% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gelcolumn by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20%EtOAc in hexanes) = 0.47. 1H-NMR (400 MHz, CDCl3): δ 7.98 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′),4.73 (t, J = 4.8 Hz, 1H, H-2′), 4.31–4.20 (t, J = 4.2 Hz, 1H, H-3′and br m, 4H, morpholinyl N(CH2)2),4.12–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.83 (t, J = 5.0 Hz, 4H, morpholinylCH2OCH2), 3.77 (dd, J = 3.2, 11.2 Hz, 1H, H-5′), 0.94, 0.92, and 0.83 (3s, 27H, t-Bu), 0.13, 0.10, −0.01,and −0.16 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 153.9, 153.7, 151.8, 138.1, 119.4, 88.8,85.3,77.3,77.0,76.6,75.0, 71.8, 66.9, 62.3, 45.60, 29.6, 29.3, 26.0,25.8, 25.7, 22.6, 18.4, 18.0, 17.8,14.0, −4.3,−4.7, −5.0, −5.4. HRMS (TOF) calcd for C32H61ClN5O5Si3 [M + H]+ 714.3664 found 714.3668.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (11b): Compound 11b (280 mg, 58%) was obtained as a pale-yellow, thick gum afterchromatography on a silica gel column by sequential elution with 10% MeOH in CH2Cl2, 20% MeOH
192

Molecules 2015, 20, 18437–18463
in CH2Cl2, and 30% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10. 1H-NMR (500 MHz,CDCl3): δ 7.97 (s, 1H, H-8), 5.90 (d, J = 4.8 Hz, 1H, H-1′), 4.72 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 3.8 Hz,1H, H-3′), 4.15–4.10 (m, 1H, H-4′ and br s, 2H, NCH2), 4.07 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.78 (dd,J = 2.8, 11.2 Hz, 1H, H-5′ and br m, 2H, NCH2), 2.59 (br s, 2H, NCH2), 2.32 (s, 6H, N(CH3)2), 0.94, 0.92,and 0.82 (3s, 27H, t-Bu), 0.13, 0.10, −0.01, and −0.15 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3):δ 154.8, 153.7, 151.4, 137.9, 119.5, 88.8, 85.2, 75.0, 71.9, 62.4, 45.7, 29.6, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9,−4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C33H66ClN6O4Si3 [M + H]+ 729.4136, found 729.4157.
3.4. General Procedure for the Desilylation of Cladribine Analogues
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group) wasadded. The mixture was heated at 80 ◦C for 20–26 h, cooled, and silica gel was added. The mixturewas evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packedsilica gel column. The products were obtained by elution with appropriate solvents (see the individualcompound headings for details).
2-Chloro-2′-deoxyadenosine (12a): Prepared from compound 5a (60.0 mg, 0.117 mmol) and KF (27.0 mg,0.467 mmol) in MeOH (1.17 mL). Chromatography on a silica gel column sequentially eluted with 5%MeOH in EtOAc and 10% MeOH in EtOAc gave 24.0 mg (72% yield) of compound 12a as an off-whitesolid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (500 MHz, CD3OD): δ 8.28 (s, 1H, H-8), 6.36(t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.74(dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd, J = 2.9, 5.9, 13.2Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.3, 155.3, 151.4, 141.9, 119.8, 89.9, 87.0, 73.0, 63.6,41.6. HRMS (TOF) calcd for C10H12ClN5O3Na [M + Na]+ 308.0521, found 308.0523.
2-Chloro-N6-methyl-2′-deoxyadenosine (13a) [50]: Prepared from compound 6a (80.0 mg, 0.154 mmol)and KF (36.0 mg, 0.618 mmol) in MeOH (1.54 mL). Chromatography on a silica gel column sequentiallyeluted with 2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 39.0 mg (85% yield) of compound 13a
as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (500 MHz, CD3OD): δ 8.18(s, 1H, H-8), 6.34 (t, J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.4, 12.2 Hz,1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.05 (br s, 3H, NCH3), 2.76 (app quint, Japp ~ 6.8 Hz,1H, H-2′), 2.42–2.38 (ddd, J = 2.9, 5.8, 13.7 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 157.3, 155.5,150.1, 141.2, 120.4, 89.9, 87.0, 73.0, 63.7, 41.6, 27.8. HRMS (TOF) calcd for C11H14ClN5O3Na [M + Na]+
322.0677, found 322.0682.
2-Chloro-N6,N6-dimethyl-2′-deoxyadenosine (Cladribine, 14a): Prepared from compound 7a (80.0 mg,0.147 mmol) and KF (34.3 mg, 0.590 mmol) in MeOH (1.4 mL). Chromatography on a silica gel columnsequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 38.0 mg (82%yield) of compound 14a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.29. 1H-NMR(500 MHz, CD3OD): δ 8.16 (s, 1H, H-8), 6.35 (t, J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H,H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.70–3.10 (br m, 6H,N(CH3)2), 2.74 (app quint, Japp ~6.8 Hz, 1H, H-2′), 2.41–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′).13C-NMR (125 MHz, CD3OD): δ 156.4, 154.6, 152.2, 140.0, 120.72, 89.8, 86.9, 73.0, 63.7, 41.5, 39.0 (br).HRMS (TOF) calcd for C12H16ClN5O3Na [M + Na]+ 336.0834, found 336.0823.
2-Chloro-6-(pyrrolidin-1-yl)-9-(2-deoxy-β-D-ribofuranosyl)purine (15a): Prepared from compound 8a
(60.0 mg, 0.105 mmol) and KF (24.5 mg, 0.422 mmol) in MeOH (1.0 mL). Chromatography on asilica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAcgave 29.1 mg (81% yield) of compound 15a as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.19.1H-NMR (500 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 6.36 (t, J = 7.1 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′),4.14–4.07 (br m, 2H, pyrrolidinyl NCH), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 3.4, 12.7 Hz, 1H, H-5′), 3.74 (dd,J = 3.4, 12.2 Hz, 1H, H-5′), 3.71–3.62 (br m, 2H, pyrrolidinyl NCH), 2.75 (app quint, Japp ~6.7 Hz, 1H,H-2′), 2.40–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′), 2.15–2.06 (br m, 2H, pyrrolidinyl CH), 2.04–1.92
193

Molecules 2015, 20, 18437–18463
(br m, 2H, pyrrolidinyl CH). 13C-NMR (125 MHz, CD3OD): δ 155.0, 154.7, 151.7, 140.6, 120.8, 89.9, 87.0,73.0, 63.7, 50.2, 49.6, 41.6, 27.3, 25.2. HRMS (TOF) calcd for C14H19ClN5O3 [M + H]+ 340.1171, found340.1149.
2-Chloro-6-(piperidin-1-yl)-9-(2-deoxy-β-D-ribofuranosyl)purine (16a): Prepared from compound 9a
(70.0 mg, 0.120 mmol) and KF (28.0 mg, 0.481 mmol) in MeOH (1.20 mL). Chromatography on asilica gel column sequentially eluted with EtOAc and 2% MeOH in EtOAc gave 30.0 mg (71% yield) ofcompound 16a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.46. 1H-NMR (500 MHz,CD3OD): δ 8.15 (s, 1H, H-8), 6.34 (t, J = 6.6 Hz, 1H, H-1′), 4.55 (m, 1H, H-3′), 4.18 (br s, 4H, piperidinylN(CH2)2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′),2.74 (app quint, Japp ~6.8 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 2.7, 5.9, 13.5 Hz, 1H, H-2′), 1.74 (br m,2H, piperidinyl CH2), 1.65 (br m, 4H, piperidinyl CH2). 13C-NMR (125 MHz, CD3OD): δ 155.3, 154.9,152.7, 139.6, 120.5, 89.8, 86.8, 73.0, 63.7, 47.8, 41.6, 27.3, 25.7. HRMS (TOF) calcd for C15H20ClN5O3Na[M + Na]+ 376.1147, found 376.1148.
2-Chloro-6-(morpholin-4-yl)-9-(2-deoxy-β-D-ribofuranosyl)purine (17a): Prepared from compound 10a
(80.0 mg, 0.137 mmol) and KF (31.8 mg, 0.548 mmol) in MeOH (1.4 mL). Chromatography on a silicagel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave36.1 mg (74% yield) of compound 17a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21.1H-NMR (500 MHz, CD3OD): δ 8.22 (s, 1H, H-8), 6.37 (t, J = 7.1 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′),4.40–4.12 (br m, 4H, morpholinyl N(CH2)2), 4.04 (m, 1H, H-4′), 3.83 (dd, J = 2.9, 12.2 Hz, 1H, H-5′),3.79 (t, J = 4.9 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.74 (app quint,Japp ~6.7 Hz, 1H, H-2′), 2.41–2.37 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD):δ 155.2, 154.7, 152.7, 140.3, 120.6, 89.8, 86.8, 72.9, 68.0, 63.6, 47.0 (br), 41.5. HRMS (TOF) calcd forC14H19ClN5O4 [M + H]+ 356.1120, found 356.1107.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(2-deoxy-β-D-ribofuranosyl)purine (18a): Preparedfrom compound 11a (70.0 mg, 0.117 mmol) and KF (27.0 mg, 0.467 mmol) in MeOH (1.17 mL).Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20% MeOHin EtOAc gave 36.0 mg (84% yield) of compound 18a as a white, foamy solid. Rf (SiO2 and 10% MeOHin EtOAc) = 0.10. 1H-NMR (500 MHz, CD3OD): δ 8.14 (s, 1H, H-8), 6.35 (t, J = 6.9 Hz, 1H, H-1′), 4.56(m, 1H, H-3′), 4.12 (br s, 2H, NCH2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.74 (dd,J = 3.9, 12.2 Hz, 1H, H-5′), 3.48 (br s, 3H, NCH3), 2.76–2.71 (br m, 3H, NCH2 and 1H, H-2′), 2.42–2.41(m, 1H, H-2′), 2.39 (s, 6H, N(CH3)2). 13C-NMR (125 MHz, CD3OD): δ 156.4, 154.7, 152.5, 140.1, 120.8,89.8, 86.8, 73.0, 63.7, 57.5, 45.8, 41.6, 37.6, (one broadened resonance could not be identified). HRMS(TOF) calcd for C15H24ClN6O3 [M + H]+ 371.1598, found 371.1584.
3.5. General Procedure for the Desilylation of Ribose Cladribine Analogues
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group)was added. The mixture was heated at 80 ◦C for 24 h, cooled, and silica gel was added. The mixturewas evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packedsilica gel column. The products were obtained by elution with appropriate solvents (see the individualcompound headings for details).
2-Chloroadenosine (12b): Prepared from compound 5b (100 mg, 0.155 mmol) and KF (54.0 mg, 0.93mmol) in MeOH (1.55 mL). Chromatography on a silica gel column sequentially eluted with 5% MeOHin EtOAc and 10% MeOH in EtOAc gave 35.0 mg (74% yield) of compound 12b as an off-white solid.Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (400 MHz, CD3OD): δ 8.27 (s, 1H, H-8), 5.92(d, J = 6.0 Hz, 1H, H-1′), 4.72 (t, J = 5.6, 1H, H-2′), 4.32 (dd, J = 2.8, 5.2 Hz, 1H, H-3′), 4.14 (m, 1H,H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz,DMSO-d6): δ 174.5, 156.7, 152.9, 150.3, 140.0, 118.1, 87.4, 85.7, 73.8, 70.3, 61.3, 25.3. HRMS (TOF) calcdfor C10H13ClN5O4 [M + H]+ 302.0651, found 302.0627.
194

Molecules 2015, 20, 18437–18463
2-Chloro-N6-methyladenosine (13b): Prepared from compound 6b (100 mg, 0.152 mmol) and KF (52.9 mg,0.912 mmol) in MeOH (1.52 mL). Chromatography on a silica gel column sequentially eluted with2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 36.0 mg (75% yield) of compound 13b as a white,foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CD3OD): δ 8.20 (s, 1H,H-8), 5.90 (d, J = 6.0 Hz, 1H, H-1′), 4.68 (t, J = 5.6 Hz, 1H, H-2′), 4.31 (dd, J = 2,8, 4.8 Hz, 1H, H-3′), 4.14(m, 1H, H-4′), 3.96 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.07 (br s, 3H,NCH3). 13C-NMR (100 MHz, DMSO-d6): δ 155.5, 153.2, 149.2, 139.7, 118.6, 87.3, 85.6, 73.6, 70.6, 70.3,61.6, 61.3, 53.9, 27.1. HRMS (TOF) calcd for C11H15ClN5O4 [M + H]+ 316.0807, found 316.0808.
2-Chloro-N6,N6-dimethyladenosine (14b): Prepared from compound 7b (100 mg, 0.148 mmol) and KF(51.8 mg, 0.892 mmol) in MeOH (1.48 mL). Chromatography on a silica gel column sequentially elutedwith 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 36.8 mg (75% yield) of compound14b as an off-white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.32. 1H-NMR (400 MHz,CD3OD): δ 8.17 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.31 (dd, J = 2.8,4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 12.4 Hz, 1H,H-5′), 3.60–3.49 (br m, H, N(CH3)2). 13C-NMR (100 MHz, DMSO-d6): δ 154.4, 152.5, 151.1, 138.6, 118.5,87.2, 85.6, 73.6, 70.2, 61.2, 37.5 (br). HRMS (TOF) calcd for C12H17ClN5O4 [M + H]+ 330.0964, found330.0964.
2-Chloro-6-(pyrrolidin-1-yl)-9-(β-D-ribofuranosyl)purine (15b): Prepared from compound 8b (100 mg,0.143 mmol) and KF (49.8 mg, 0.858 mmol) in MeOH (1.43 mL). Chromatography on a silica gel columnsequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 43.3 mg (85%yield) of compound 15b as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.38. 1H-NMR (400 MHz,CD3OD): δ 8.18 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.32 (dd, J = 2.8,4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 4.13–4.10 (br m, 2H, pyrrolidinyl NCH), 3.90 (dd, J = 2.8, 12.8Hz, 1H, H-5′), 3.74 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.72–3.65 (br m, 2H, pyrrolidinyl NCH), 2.13–2.07(br m, 2H, pyrrolidinyl CH), 2.05–1.95 (br m, 2H, pyrrolidinyl CH). 13C-NMR (100 MHz, DMSO-d6):δ 152.7, 152.7, 150.7, 139.1, 118.7, 87.2, 85.6, 73.7, 70.2, 61.2, 48.6, 47.3, 25.6, 23.6. HRMS (TOF) calcd forC14H19ClN5O4 [M + H]+ 356.1120, found 356.1127.
2-Chloro-6-(piperidin-1-yl)-9-(β-D-ribofuranosyl)purine (16b): Prepared from compound 9b (100 mg, 0.140mmol) and KF (48.9 mg, 0.842 mmol) in MeOH (1.40 mL). Chromatography on a silica gel columnsequentially eluted with EtOAc and 2% MeOH in EtOAc gave 43.6 mg (84% yield) of compound 16b
as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.41. 1H-NMR (400 MHz, CD3OD):δ 8.13 (s, 1H, H-8), 5.87 (d, J = 6.0 Hz, 1H, H-1′), 4.64 (t, J = 5.4 Hz, 1H, H-2′), 4.27 (dd, J = 2.8, 5.2 Hz,1H, H-3′), 4.19 (br s, 4H, piperidinyl N(CH2)2), 4.11 (m, 1H, H-4′), 3.89 (dd, J = 2.8, 12.2 Hz, 1H, H-5′),3.73 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 1.74 (br m, 2H, piperidinyl CH2), 1.64 (br m, 4H, piperidinylCH2). 13C-NMR (100 MHz, DMSO-d6): δ 153.1, 152.6, 151.4, 138.5, 118.2, 87.2, 85.6, 73.6, 70.4, 70.2, 70.1,61.2, 44.8 (br), 25.6, 25.2, 23.9, 14.0. HRMS (TOF) calcd for C15H21ClN5O4 [M + H]+ 370.1277, found370.1302.
2-Chloro-6-(morpholin-4-yl)-9-(β-D-ribofuranosyl)purine (17b): Prepared from compound 10b (100 mg,0.139 mmol) and KF (48.7 mg, 0.839 mmol) in MeOH (1.40 mL). Chromatography on a silica gel columnsequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 40.6 mg (78%yield) of compound 17b as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21. 1H-NMR(400 MHz, CD3OD): δ 8.21 (s, 1H, H-8), 5.92 (d, J = 6.0 Hz, 1H, H-1′), 4.66 (t, J = 5.6 Hz, 1H, H-2′), 4.32(dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.30–4.24 (br m, 4H, morpholinyl N(CH2)2), 4.14 (m, 1H, H-4′), 3.90 (dd,J = 2.8, 12.4 Hz, 1H, H-5′), 3.80 (t, J = 4.8 Hz, 4H, morpholinyl CH2OCH2), 3.76 (dd, J = 2.8, 12.4 Hz,1H, H-5′). 13C-NMR (100 MHz, DMSO-d6): δ 153.3, 152.5, 151.6, 139.0, 118.3, 87.3, 85.6, 85.4, 73.7, 70.4,70.2, 66.0, 61.4, 61.1, 48.5, 45.0 (br). HRMS (TOF) calcd for C14H19ClN5O5 [M + H]+ 372.1069, found372.1056.
195

Molecules 2015, 20, 18437–18463
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(β-D-ribofuranosyl)purine (18b): Prepared fromcompound 11b (100 mg, 0.137 mmol) and KF (47.7 mg, 0.822 mmol) in MeOH (1.37 mL).Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20%MeOH in EtOAc gave 34.5 mg (65% yield) of compound 18b as a white, foamy solid. Rf (SiO2 and 15%MeOH in EtOAc) = 0.10. 1H-NMR (400 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 5.92 (d, J = 6 Hz, 1H, H-1′),4.67 (t, 1H, J = 5.6 Hz, 1H, H-2′), 4.34 (dd, J = 3.2, 5.2 Hz 1H, H-3′), 4.14 (d, J = 2.4 Hz, 3H, H-4′), 3.90(dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.49 (br s, 3H, NCH3), 2.83 (t, 2H,NCH2, J = 6.8 Hz ), 2.47 (s, 6H, N(CH3)2). 13C-NMR (100 MHz, CD3OD): δ 156.1, 154.7, 152.3, 140.5,120.7, 90.8, 87.7, 75.3, 72.3, 63.2, 57.0, 49.6, 49.4, 49.2, 49.0, 48.7, 48.5, 48.3, 45.5, 37.4. HRMS (TOF) calcdfor C15H24ClN6O4 [M + H]+ 387.1542, found 387.1513.
O6-(Benzotriazol-1-yl)-2-bromo-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (19a):A mixture of compound 3a (300.0 mg, 0.489 mmol) and SbBr3 (247.7 mg, 0.685 mmol) in dry CH2Br2
(4.9 mL) was cooled to −15 ◦C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO(203.8 μL, 1.713 mmol) was added dropwise and the mixture was stirred at −10 ◦C to −15 ◦C for2 h. Because TLC indicated the presence of starting material, another aliquot of t-BuONO (203.8 μL,1.713 mmol) was added and the reaction proceeded for 1 h at −15 ◦C, at which time TLC indicated thereaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3
(5 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtrationis critical for maximizing product recovery) and the residue was washed with CH2Cl2 (5 mL). Theorganic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 5 mL). Thecombined organic layer was washed with water (5 mL) and brine (5 mL), dried over anhydrousNa2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gelcolumn sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave208.5 mg (63% yield) of compound 19a as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.60.1H-NMR (500 MHz, CDCl3): δ 8.45 (s, 1H, H-8), 8.13 (d, J = 8.3 Hz, 1H, Ar-H), 7.56–7.45 (m, 3H, Ar-H),6.47 (t, J = 5.9 Hz, 1H, H-1′), 4.63 (m, 1H, H-3′), 4.03 (m, 1H, H-4′), 3.90 (dd, J = 3.4, 11.2 Hz, 1H, H-5′),3.78 (dd, J = 1.5, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~6.2 Hz, 1H, H-2′), 2.49 (app quint, Japp ~5.8Hz, 1H, H-2′), 0.91 (s, 18H, t-Bu), 0.11 and 0.09 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 158.7,154.9, 144.3, 143.6, 142.6, 129.1, 128.9, 125.1, 120.8, 119.5, 108.7, 88.5, 85.4, 71.7, 62.7, 41.8, 26.2, 25.9,18.6, 18.2, −4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C28H43BrN7O4Si2 [M + H]+ 676.2093, found676.2078.
2-Bromo-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (20a): To a solution of compound 19a (135.3mg, 0.20 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (48.6 μL) was added, and the mixturewas stirred at room temperature for 45 min. The mixture was diluted with EtOAc (15 mL) and washedwith 5% aqueous NaCl (10 mL). The organic layer was separated and the aqueous layer was backextracted with EtOAc (15 mL). The combined organic layer was dried over anhydrous Na2SO4 andevaporated under reduced pressure. The crude material was chromatographed on a silica gel columnby sequential elution with hexanes, 20% EtOAc in hexanes and 40% EtOAc in hexanes to afford 93.1 mg(83% yield) of compound 20a as a white foam. Rf (SiO2 and EtOAc) = 0.50. 1H-NMR (500 MHz, CDCl3):δ 8.08 (s, 1H, H-8), 6.63 (br s, 2H, Ar-NH2), 6.37 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′), 3.98 (m,1H, H-4′), 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.75 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp
~6.3 Hz, 1H, H-2′), 2.43–2.38 (m, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR(125 MHz, CDCl3): δ 156.3, 150.4, 144.9, 139.6, 119.3, 88.2, 84.7, 71.9, 62.9, 41.4, 26.1, 25.9, 18.6, 18.2,−4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C22H41BrN5O3Si2 [M + H]+ 558.1926, found 558.1902.
2-Bromo-2′-deoxyadenosine (21a): As described in the general desilylation procedures, compound 21a
was prepared from compound 20a (80.0 mg, 0.143 mmol) and KF (33.2 mg, 0.572 mmol) in MeOH(1.4 mL), over 28 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAcin hexanes, EtOAc, and 10% MeOH in EtOAc gave 31.1 mg (66% yield) of compound 21a as awhite/off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.40. 1H-NMR (500 MHz, CD3OD): δ 8.25
196

Molecules 2015, 20, 18437–18463
(s, 1H, H-8), 6.36 (t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz,1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd,J = 2.9, 5.9, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.1, 151.4, 145.9, 141.7, 120.3, 89.9,86.9, 72.9, 63.7, 41.6. HRMS (TOF) calcd for C10H12BrN5O3Na [M + Na]+ 352.0016, found 352.0021.
O6-(Benzotriazol-1-yl)-2-bromo-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (19b):A mixture of compound 3b (500.0 mg, 0.67 mmol) and SbBr3 (340.8 mg, 0.94 mmol) in dry CH2Br2 (8.3mL) was cooled to −15 ◦C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (280 μL,2.352 mmol) was added dropwise and the mixture was stirred at −10 ◦C to −15 ◦C for 2 h. BecauseTLC indicated the presence of starting material, another aliquot of t-BuONO (280 μL, 2.352 mmol) wasadded and the reaction was allowed to progress for 1 h at −15 ◦C, at which time TLC indicated thereaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3
(10 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration iscritical for maximizing product recovery) and the residue was washed with CH2Cl2 (15 mL). Theorganic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 15 mL). Thecombined organic layer was washed with water (10 mL) and brine (10 mL), dried over anhydrousNa2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gelcolumn sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave350 mg (64% yield) of compound 19b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.80.1H-NMR (400 MHz, CDCl3): δ 8.54 (s, 1H, H-8), 8.15 (d, J = 8.4 Hz, 1H, Ar-H), 7.58–7.45 (m, 3H, Ar-H),6.05 (d, J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.33 (t, J = 4.6 Hz, 1H, H-3′), 4.17 (m, 1H,H-4′), 4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.85 (3s,27H, t-Bu), 0.16, 0.11, −0.03, and −0.09 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 158.5, 154.8,144.2, 143.4, 142.5, 128.8, 128.7, 124.8, 120.6, 119.4, 108.5, 89.5, 85.2, 76.1, 71.1, 61.9, 29.6, 26.1, 25.8, 25.6,18.5, 18.0, 17.9, −4.2, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57BrN7O5Si3 [M + H]+
806.2907, found 806.2912.
2-Bromo-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-adenosine (20b): To a solution of compound 19b (200 mg,0.247 mmol) in 1,2-DME (4 mL), 28%–30% aqueous ammonia (60 μL) was added, and the mixture wasstirred at room temperature for 45 min. The mixture was diluted with EtOAc (20 mL) and washedwith 5% aqueous NaCl (20 mL). The organic layer was separated and the aqueous layer was backextracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 andevaporated under reduced pressure. The crude material was chromatographed on a silica gel columnby sequential elution with hexanes, 20% EtOAc in hexanes, and 40% EtOAc in hexanes to afford140 mg (82% yield) of compound 20b as a white foam. Rf (SiO2 and EtOAc) = 0.55. 1H-NMR (400 MHz,CDCl3): δ 8.10 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′), 5.77 (br s, 2H, Ar-NH2), 4.69 (t, J = 4.6 Hz, 1H,H-2′), 4.32 (t, J = 4.2 Hz, 1H, H-3′), 4.13 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.2 Hz, 1H, H-5′), 3.80 (dd,J = 2.8, 11.2 Hz, 1H, H-5′), 0.95, 0.93, and 0.83 (3s, 27H, t-Bu), 0.14, 0.11, −0.00, and −0.15 (4s, 18H,SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.8, 150.5, 144.6, 140.0, 119.4, 110.0, 89.0, 85.3, 75.4, 71.7, 62.3,26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd for C28H55BrN5O4Si3[M + H]+ 688.2740, found 688.2728.
2-Bromoadenosine (21b): As described in the general desilylation procedures, compound 21b wasprepared from compound 20b (100 mg, 0.145 mmol) and KF (50.5 mg, 0.870 mmol) in MeOH (1.9 mL),over 24 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes,EtOAc, and 10% MeOH in EtOAc which gave 34.9 mg (69% yield) of compound 21b as a pale yellowsolid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.39. 1H-NMR (400 MHz, CD3OD): δ 8.25 (s, 1H, H-8),5.92 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (d, J = 5.6 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.15(m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR(100 MHz, CD3OD): δ 156.5, 150.2, 144.1, 139.8, 118.4, 87.2, 85.7, 73.6, 70.3, 61.3. HRMS (TOF) calcd forC10H13BrN5O4 [M + H]+ 346.0145, found 346.0154.
197

Molecules 2015, 20, 18437–18463
3.6. Protocols for Tests against HCL, TCL, and CCL
Blood was obtained in sodium heparin tubes from patients as part of protocols with consentforms approved by the investigators review board (IRB) of the National Cancer Institute. The bloodwas diluted 1:1 with PBS without calcium or magnesium, layered over 15 mL Ficoll in 50 mL tubes,and centrifuged to obtain mononuclear cells. Patients with high lymphocytosis had leukemic cells>80%–90% pure after Ficoll. The cells were viably frozen in 7.5% DMSO in leucine-poor media (LPM,88% leucine-free RPMI, 2% RPMI, and 10% FBS) in cryovials and stored under liquid nitrogen. LPMalso contained penicillin, streptomycin, glutamine, gentamycin, and doxycycline. To assay, thawed cellswere washed, suspended in LPM, added to 96-well round-bottom plates (15 μL/well), and treated with15 μL of purine analogues diluted in LPM. The aliquots were incubated 3 days, then treated with 10 μLof either ATP (CellTiter-Glo, Promega, Madison, WI, USA) or {3H}-leucine (Perkin-Elmer, Waltham,MA, USA) diluted in leucine-free RPMI. After 30 min of ATP, the plate is read for bioluminescence.After 6 h of {3H}-leucine, the cells were liberated by freeze-thaw, harvested on to glass-fiber filters,counted either by a beta scintillation counter. The number of cells cultured in 30 μL aliquots forATP assay was 20,000, 20,000, and 100,000 for HCL, TCL, and CLL, respectively. The cell number for{3H}-leucine assay was 60,000, 60,000 and 200,000 for HCL, TCL, and CLL, respectively. HCL andTCL cells were pulsed with 1 μCi of {3H}-leucine, while CLL cells were pulsed with 1.5–2 μCi/well.The IC50 was the calculated concentration needed for 50% inhibition, defined as the ATP uptake or{3H}-leucine incorporation corresponding to halfway between that of control (cells with LPM alone)and that of cycloheximide 10 μg/mL. Reported IC50 values were the means of 3 triplicate experiments.
3.7. Protocols for Tests against Breast Cancer Cells
MDA-MB-231 breast cancer (BC) cells were obtained from the American Type Culture Collection(ATCC, Manassas, VA, USA), and were cultured at 37 ◦C in 5% CO2 using culture mediumrecommended by the supplier. Each tested compound was dissolved in sterile DMSO, then diluted(4.8–57.7 mM) in sterile 1X PBS for a final DMSO concentration of 0.1%. Then, 2 × 105 cells/well, wereseeded and cultured for 24 h as described [51,52]. Afterwards, the media was changed to 5% FBS for1 h and cells were treated in duplicate with dilutions of each treatment (0–1.8 mM) for 24 h. Cellswere fixed (cold methanol), and nuclei stained (0.4% propidium iodide, (PI)) (Sigma-Aldrich, St Louis,MO, USA), and measured using a GloMax® Microplate Reader (Promega, Madison, WI, USA). Cellviability was calculated as percent of surviving cells after treatment relative to vehicle wells. The IC50
was obtained from dose response curve fittings using the non-linear regression function of GraphPadPrism® version 6.0b for Mac (GraphPad Software, San Diego, CA, USA) [53].
4. Conclusions
We have demonstrated, for the first time, the synthesis of O6-(benzotriazol-1-yl)-2-chloro-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine (4a) by diazotization-chlorinationof O6-(benzotriazol-1-yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyguanosine (3a) with t-BuONO andSbCl3 in CH2Cl2. This procedure afforded better yields than the chlorination using t-BuONOand Me3SiCl. This compound and its ribose analogue, O6-(benzotriazol-1-yl)-2-chloro-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-D-ribofuranosyl]purine, both undergo smooth reactions with ammonia,and primary, and secondary amines to produce cladribine (12a), its N-modified analogues (13a–18a),and the corresponding ribose derivatives (12b–18b), after a simple desilylation with KF in MeOH.These compounds were tested against HCL, TCL, and CLL, but none of the new compounds was moreactive than cladribine itself. The bromo as well as ribose analogues of cladribine displayed activitybut the bromo analogue of cladribine was more active against TCL and CLL as compared to boththe ribose equivalent and the bromo ribose analogue of cladribine. The compound containing boththe bromine atom and a ribose ring was least active among the compounds possessing a primaryamino group at the C6 position. Thus, it appears that a free amino group at this location is critical to
198

Molecules 2015, 20, 18437–18463
the activity of cladribine. Interestingly, the C6 piperidinyl analog of cladribine showed low activity.Tests against MDA-MB-231 breast cancer cells showed that only cladribine and its ribose analogueshowed some activity. The bromo analogues were about 10 times less active and all others showed nopotential. Despite the lack of a major improvement in the activity of cladribine, or the identification ofnew compounds with activity against breast cancer, this work has provided a route to four doublyfunctionalizable nucleoside derivatives. The orthogonal reactivities of these compounds, i.e., SNAr atthe C6 position and metal catalysis at the C2 position, can be used for development of novel nucleosideanalogues. We anticipate pursuing further work along these lines in the future.
Supplementary Materials: Supplementary can be accessed at: http://www.mdpi.com/1420-3049/20/10/18437/s1.
Acknowledgments: This work was supported by National Science Foundation Grant CHE-1265687 to MKL,in part by National Institutes of Health Grant SC3GM111171 to MMMM from the National Institute on GeneralMedical Sciences, and Title V PPOHA US Department of Education P031M105050 to Universidad Central delCaribe. Infrastructural support at CCNY was provided by National Institute on Minority Health and HealthDisparities Grant G12MD007603, while that at UCC was provided by G12MD007583. Siva Subrahmanyam Relangi,Venkateshwarlu Gurram, Muralidharan Kaliyaperumal, Somesh Sharma, and Narender Pottabathini gratefullyacknowledge support from GVKBIO and Siva Subrahmanyam Relangi thanks Anna Venkateswara Rao (KoneruLakshmaiah University, Guntur (Andhra Pradesh), India) for his support.
Author Contributions: Sakilam Satishkumar, Prasanna K. Vuram, and Siva Subrahmanyam Relangi had equalcontributions to the synthesis portion of this work. Sakilam Satishkumar and Prasanna K. Vuram (City College ofNew York) performed optimization of the synthetic procedures, executed syntheses of the deoxyribose series,performed spectroscopic analyses of the compounds, and produced a part of the experimental section. Messrs.Relangi and Venkateshwarlu Gurram (GVKBIO) executed syntheses of the ribose series, performed spectroscopicanalyses of the compounds, and produced a part of the experimental section. Hong Zhou and Robert J. Kreitman(National Cancer Institute) tested the compounds against HCL, TCL, and CLL. Michelle M. Martínez Montemayortested the compounds against breast cancer cell lines. Lijia Yang (City College of New York) performed HRMSanalysis of all compounds synthesized in Lakshman’s laboratories. Narender Pottabathini (GVKBIO) wasresponsible for the oversight of the work performed in his laboratories. Mahesh K. Lakshman (City College ofNew York) conceived and designed the research, assisted with data analysis, and wrote a significant portion ofthis manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References and Notes
1. Venner, H. Synthese der den natürlichen entsprechenden 2-desoxy-nucleoside des adenins, guanins, undhypoxanthins. Chem. Ber. 1960, 63, 100–110. [CrossRef]
2. Carson, D.A.; Kaye, J.; Seegmiller, J.E. Lymphospecific toxicity in adenosine deaminase deficiency and purinenucleoside phosphorylase deficiency: Possible role of nucleosidase kinase(s). Proc. Natl. Acad. Sci. USA 1977,74, 5677–5681. [CrossRef] [PubMed]
3. Carson, D.A.; Wasson, D.B.; Kaye, J.; Ullman, B.; Martin, D.W., Jr.; Robins, R.K.; Montgomery, J.A.Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblastsin vitro and toward murine L1210 leukemia in vivo. Proc. Natl. Acad. Sci. USA 1980, 77, 6865–6869. [CrossRef][PubMed]
4. Wang, L.; Karlsson, A.; Arnér, E.S.J.; Eriksson, S. Substrate specificity of mitochondrial 2′-deoxyguanosinekinase. Efficient phosphorylation of 2-chlorodeoxyadenosine. J. Biol. Chem. 1993, 268, 22847–22852. [PubMed]
5. Sjöberg, A.H.; Wang, L.; Eriksson, S. Substrate specificity of human recombinant mitochondrialdeoxyguanosine kinase with cytostatic and antiviral purine and pyrimidine analogs. Mol. Pharmacol.1998, 53, 270–273. [PubMed]
6. Genini, D.; Adachi, S.; Chao, Q.; Rose, D.W.; Carrera, C.J.; Cottam, H.B.; Carson, D.A.; Leoni, L.M.Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damagingthe DNA and by directly affecting the mitochondria. Blood 2000, 96, 3537–3543. [PubMed]
7. Marzo, I.; Pérez-Galán, P.; Giraldo, P.; Rubio-Félix, D.; Anel, A.; Naval, J. Cladribine induces apoptosisin human leukaemia cells by caspase-dependent and -independent pathways acting on mitochondria.Biochem. J. 2001, 359, 537–546. [CrossRef] [PubMed]
199

Molecules 2015, 20, 18437–18463
8. Klöpfer, A.; Hasenjäger, A.; Belka, C.; Schulze-Osthoff, K.; Dörken, B.; Daniel, P.T. Adenine deoxynucleotidesfludarabine and cladribine induce apoptosis in a CD95/Fas receptor, FADD and caspase-8-independentmanner by activation of the mitochondrial cell death pathway. Oncogene 2004, 23, 9408–9418. [CrossRef][PubMed]
9. Goodman, G.R.; Burian, C.; Koziol, J.A.; Saven, A. Extended follow-up of patients with hairy cell leukemiaafter treatment with cladribine. J. Clin. Oncol. 2003, 21, 891–896. [CrossRef] [PubMed]
10. Sigal, D.S.; Miller, H.J.; Schram, E.D.; Saven, A. Beyond hairy cell: The activity of cladribine in otherhematologic malignancies. Blood 2010, 116, 2884–2896. [CrossRef] [PubMed]
11. The US National Institutes of Health Clinical Trials. Available online: https://www.clinicaltrials.gov/ct2/results?term=cladribine&Search=Search (accessed on 18 June 2015).
12. Ikehara, M.; Tada, H. Studies of nucleosides and nucleotides. XVIV. Purine cyclonucleosides. I.8,2′-Cyclonucleoside derived from 2-chloro-8-mercapto-9-β-D-xylofuranosyladenine. J. Am. Chem. Soc. 1965,87, 606–610. [CrossRef] [PubMed]
13. Christensen, L.F.; Broom, A.D.; Robins, M.J.; Bloch, A. Synthesis and biological activity of selected2,6-disubstituted-(2-deoxy-α- and -β-D-erythro-pentofuranosyl)purines. J. Med. Chem. 1972, 15, 735–739.[CrossRef]
14. Kazimierczuk, Z.; Cottam, H.B.; Revankar, G.R.; Robins, R.K. Synthesis of 2′-deoxytubercidin, 2′-deoxyadenosine,and related 2′-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure. J. Am.Chem. Soc. 1984, 106, 6379–6382. [CrossRef]
15. Lioux, T.; Gosselin, G.; Mathé, C. Azido/tetrazole tautomerism in 2-azidoadenine β-D-pentofuranonucleosidederivatives. Eur. J. Org. Chem. 2003, 3997–4002. [CrossRef]
16. Zhong, M.; Nowak, I.; Robins, M.J. Regiospecific and highly stereoselective coupling of 6-(substituted-imidazol-1-yl)purines with 2-deoxy-3,5-di-O-(p-toluoyl)-α-D-erythro-pentofuranosyl chloride. Sodium-saltglycosylation in binary solvent mixtures: Improved synthesis of cladribine. J. Org. Chem. 2006, 71, 7773–7779.[CrossRef] [PubMed]
17. Yang, F.; Zhu, Y.; Yu, B. A dramatic concentration effect on the stereoselectivity of N-glycosylation for thesynthesis of 2′-deoxy-β-ribonucleosides. Chem. Commun. 2012, 48, 7097–7099. [CrossRef] [PubMed]
18. Henschke, J.P.; Zhang, X.; Huang, X.; Mei, L.; Chu, G.; Hu, K.; Wang, Q.; Zhu, G.; Wu, M.; Kuo, C.; Chen, Y.A stereoselective process for the manufacture of a 2′-deoxy-β-D-ribonucleoside using the Vorbrüggenglycosylation. Org. Process Res. Dev. 2013, 17, 1419–1429. [CrossRef]
19. Xu, S.; Yao, P.; Chen, G.; Wang, H. A new synthesis of 2-chloro-2′-deoxyadenosine (cladribine), CdA).Nucleosides Nucleotides Nucleic Acids 2011, 30, 353–359. [CrossRef] [PubMed]
20. Sakakibara, N.; Kakoh, A.; Maruyama, T. First synthesis of [6-15N]-cladribine using ribonucleoside asa starting material. Heterocycles 2012, 85, 171–182.
21. Mikhailopulo, I.A.; Zinchenko, A.I.; Kazimierczuk, Z.; Barai, V.N.; Bokut, S.B.; Kalinichenko, E.N. Synthesis of2-chloro-2′-deoxyadenosine by microbiological transglycosylation. Nucleosides Nucleotides 1993, 12, 417–422.[CrossRef]
22. Barai, V.N.; Zinchenko, A.I.; Eroshevskaya, L.A.; Kalinichenko, E.N.; Kulak, T.I.; Mikhailopulo, I.A.A universal biocatalyst for the preparation of base- and sugar-modified nucleosides via an enzymatictransglycosylation. Helv. Chim. Acta 2002, 85, 1901–1908. [CrossRef]
23. Komatsu, H.; Araki, T. Efficient chemo-enzymatic syntheses of pharmaceutically useful unnatural2′-deoxynucleosides. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1127–1130. [CrossRef] [PubMed]
24. Taran, S.A.; Verevkina, K.N.; Esikova, T.Z.; Feofanov, S.A.; Miroshnikov, A.I. Synthesis of 2-chloro-2′-deoxyadenosine by microbiological transglycosylation using a recombinant Escherichia coli strain.Appl. Biochem. Microbiol. 2008, 44, 162–166. [CrossRef]
25. Janeba, Z.; Francom, P.; Robins, M.J. Efficient syntheses of 2-chloro-2′-deoxyadenosine (cladribine) from2′-deoxyguanosine. J. Org. Chem. 2003, 68, 989–992. [CrossRef] [PubMed]
26. Peng, Y. A practical synthesis of 2-chloro-2′-deoxyadenosine (cladribine) from 2′-deoxyadenosine.J. Chem. Res. 2013, 37, 213–215. [CrossRef]
27. Wan, Z.-K.; Wacharasindhu, S.; Binnun, E.; Mansour, T. An efficient direct amination of cyclic amides andcyclic ureas. Org. Lett. 2006, 8, 2425–2428. [CrossRef] [PubMed]
28. Wan, Z.-K.; Binnun, E.; Wilson, D.P.; Lee, J. A highly facile and efficient one-step synthesis of N6-adenosineand N6-2′-deoxyadenosine derivatives. Org. Lett. 2005, 7, 5877–5880. [CrossRef] [PubMed]
200

Molecules 2015, 20, 18437–18463
29. Bae, S.; Lakshman, M.K. O6-(Benzotriazol-1-yl)inosine derivatives: Easily synthesized, reactive nucleosides.J. Am. Chem. Soc. 2007, 129, 782–789. [CrossRef] [PubMed]
30. Wan, Z.-K.; Wacharasindhu, S.; Levins, C.G.; Lin, M.; Tabei, K.; Mansour, T.S. The scope and mechanism ofphosphonium-mediated SNAr reactions in heteocyclic amides and ureas. J. Org. Chem. 2007, 72, 10194–10210.[CrossRef] [PubMed]
31. Ashton, T.D.; Scammells, P.J. Microwave-assisted direct amination: Rapid access to multi-functionalizedN6-substituted adenosines. Aust. J. Chem. 2008, 61, 49–58. [CrossRef]
32. Bae, S.; Lakshman, M.K. Unusual deoxygenation and reactivity studies related to O6-(benzotriazol-1yl)inosine derivatives. J. Org. Chem. 2008, 73, 1311–1319. [CrossRef] [PubMed]
33. Bae, S.; Lakshman, M.K. A novel polymer supported approach to nucleoside modification. J. Org. Chem.2008, 73, 3707–3713. [CrossRef] [PubMed]
34. Bae, S.; Lakshman, M.K. Synthetic utility of an isolable nucleoside phosphonium salt. Org. Lett. 2008, 10,2203–2206. [CrossRef] [PubMed]
35. Lakshman, M.K.; Frank, J. A simple method for C-6 modification of guanine nucleosides. Org. Bimol. Chem.2009, 7, 2933–2940. [CrossRef] [PubMed]
36. Kokatla, H.P.; Lakshman, M.K. One-pot etherification of purine nucleosides and pyrimidines. Org. Lett.2010, 12, 4478–4881. [CrossRef] [PubMed]
37. Lakshman, M.K.; Singh, M.K.; Parrish, D.; Balachandran, R.; Day, B.W. Azide-tetrazole equilibrium ofC-6 azidopurine nucleosides and their ligation reactions with alkynes. J. Org. Chem. 2010, 75, 2461–2473.[CrossRef] [PubMed]
38. Lakshman, M.K.; Kumar, A.; Balachandran, R.; Day, B.W.; Andrei, G.; Snoeck, R.; Balzarini, J. Synthesisand biological properties of C-2 triazolylinosine derivatives. J. Org. Chem. 2012, 77, 5870–5883. [CrossRef][PubMed]
39. Ghosh, S.; Greenberg, M.M. Synthesis of cross-linked DNA containing oxidized abasic site analogues. J. Org.Chem. 2014, 79, 5948–5957. [CrossRef] [PubMed]
40. Devine, S.M.; May, L.T.; Scammells, P.J. Design, synthesis, and evaluation of N6-substituted2-aminoadenosine-5′-N-methylcarboxamides as A3 adenosine receptor agonists. Med. Chem Commun.2014, 5, 192–196. [CrossRef]
41. Krüger, S.; Meier, C. Synthesis of site-specific damaged DNA strands by 8-(acetylarylamino)-2′-deoxyguanosine adducts and effects on various DNA polymerases. Eur. J. Org. Chem. 2013, 2013, 1158–1169.[CrossRef]
42. Van der Henden van Noort, G.J.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V. Synthesis ofnucleotidylated poliovirus VPg proteins. J. Org. Chem. 2010, 75, 5733–5736. [CrossRef] [PubMed]
43. Printz, M.; Richert, C. Pyrenylmethyldeoxyadenosine: A 3′-cap for universal DNA hybridization probes.Chem. Eur. J. 2009, 15, 3390–3402. [CrossRef] [PubMed]
44. Devine, S.M.; Scammells, P.J. Synthesis and utility of 2-halo-O6-(benzotriazol-1-yl)-functionalized purinenucleosides. Eur. J. Org. Chem. 2011, 1092–1098. [CrossRef]
45. Sampath, U.; Bartlett, L.; (Reliable Biopharmaceutical, Inc., St. Louis, MO, USA). Process for the Productionof 2-halo-6-aminopurine Derivatives. US Patent 6,252,061 B1, 26 June 2001.
46. Francom, P.; Robins, M.J. Nucleic acid related compounds. 118. Nonaqueous diazotization of aminopurinederivatives. Convenient access to 6-halo and 2,6-dihalopurine nucleosides and 2′-deoxynucleosides withacyl or silyl halides. J. Org. Chem. 2003, 68, 666–669. [CrossRef] [PubMed]
47. Pottabathini, N.; Bae, S.; Pradhan, P.; Hahn, H.-G.; Mah, H.; Lakshman, M.K. Synthesis and reactions of2-chloro and 2-tosyloxy 2′-deoxyinosine derivatives. J. Org. Chem. 2005, 70, 7188–7195. [CrossRef] [PubMed]
48. Robins, M.J.; Uznanski, B. Nucleic acids related compounds. 34. Non-aqueous diazotization with tert-butylnitrite. Introduction of fluorine, chlorine, and bromine at C-2 of purine nucleosides. Can. J. Chem. 1981, 59,2608–2611. [CrossRef]
49. Francom, P.; Janeba, Z.; Shibuya, S.; Robins, M.J. Nucleic acid related compounds. 116. Nonaqueousdiazotization of aminopurine nucleosides. Mechanistic considerations and efficient procedures withtert-butyl nitrite or sodium nitrite. J. Org. Chem. 2002, 67, 6788–6796. [CrossRef] [PubMed]
50. Kazimierczuk, Z.; Vilpo, J.A.; Seela, F. Base-modified nucleosides related to 2-chloro-2′-deoxyadenosine.Helv. Chim. Acta 1992, 75, 2289–2297. [CrossRef]
201

Molecules 2015, 20, 18437–18463
51. Martínez-Montemayor, M.M.; Rosario-Acevedo, R.; Otero-Franqui, E.; Cubano, L.A.; Dharmawardhane, S.F.Ganoderma lucidum (Reishi) inhibits cancer cell growth and expression of key molecules in inflammatorybreast cancer. Nutr. Cancer 2011, 63, 1085–1094.
52. Suarez-Arroyo, I.J.; Rosario-Acevedo, R.; Aguilar-Perez, A.; Clemente, P.L.; Cubano, L.A.; Serrano, J.;Schneider, R.J.; Martinez-Montemayor, M.M. Anti-tumor effects of Ganoderma lucidum (Reishi) ininflammatory breast cancer in in vivo and in vitro models. PLoS ONE 2013, 8, e57431. [CrossRef] [PubMed]
53. GraphPad Software. Available online: http://www.graphpad.com.
Sample Availability: Sample Availability: Limited amounts of compounds 12a–18a and 12b–18b may be availablefrom the authors.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).
202

Article
Site-Selective Ribosylation of Fluorescent NucleobaseAnalogs Using Purine-Nucleoside Phosphorylase as aCatalyst: Effects of Point Mutations
Alicja Stachelska-Wierzchowska 1,*, Jacek Wierzchowski 1, Agnieszka Bzowska 2 andBeata Wielgus-Kutrowska 2
1 Department of Physics and Biophysics, University of Varmia & Masuria in Olsztyn, 4 Oczapowskiego St.,10-719 Olsztyn, Poland; [email protected]
2 Division of Biophysics, Institute of Experimental Physics, University of Warsaw, Zwirki i Wigury 93,02-089 Warsaw, Poland; [email protected] (A.B.); [email protected] (B.W.-K.)
* Correspondence: [email protected]; Tel.: +48-89-523-3406; Fax: +48-89-523-3861
Academic Editor: Mahesh K. LakshmanReceived: 13 November 2015 ; Accepted: 9 December 2015 ; Published: 28 December 2015
Abstract: Enzymatic ribosylation of fluorescent 8-azapurine derivatives, like 8-azaguanine and2,6-diamino-8-azapurine, with purine-nucleoside phosphorylase (PNP) as a catalyst, leads to N9,N8, and N7-ribosides. The final proportion of the products may be modulated by point mutationsin the enzyme active site. As an example, ribosylation of the latter substrate by wild-type calf PNPgives N7- and N8-ribosides, while the N243D mutant directs the ribosyl substitution at N9- andN7-positions. The same mutant allows synthesis of the fluorescent N7-β-D-ribosyl-8-azaguanine.The mutated form of the E. coli PNP, D204N, can be utilized to obtain non-typical ribosides of8-azaadenine and 2,6-diamino-8-azapurine as well. The N7- and N8-ribosides of the 8-azapurinescan be analytically useful, as illustrated by N7-β-D-ribosyl-2,6-diamino-8-azapurine, which is agood fluorogenic substrate for mammalian forms of PNP, including human blood PNP, while theN8-riboside is selective to the E. coli enzyme.
Keywords: fluorescent nucleosides; enzymatic ribosylation; 8-azapurines; purine nucleosidephosphorylase
1. Introduction
Purine-nucleoside phosphorylase (PNP, E.C.2.4.2.1) is a key enzyme of purine metabolism,important, inter alia, for the proper activity of the immune system in mammals [1–3]. Potentinhibitors of PNP, like immucillin H (forodesine) and some analogs [4], are clinically approvedto treat lymphomas [4–6], and others are considered as potential anti-parasitic and anti-malarialdrugs [4,7]. Bacterial forms of PNP are of interest because they can be used as a suicidal-gene in cancerchemotherapy [8]. Besides this, PNP is used as a catalyst in the gram-scale preparative ribosylation ofpurines and purine analogs [9–11], thanks to the reverse (synthetic) pathway of the phosphorolyticprocess. Nucleoside analogues are widely applied as pharmaceuticals and as biochemical probes forenzymological studies [5–7,9–13].
In the preceding papers [14,15], we have demonstrated that enzymatic ribosylation of some8-azapurines leads not only to the canonical nucleoside analogs, but also to non-typical, and highlyfluorescent ribosides, ribosylated at the N7- and N8-positions (see Figure 1 below). Now we presenta kinetic analysis of these processes, with application of several wild and mutated forms of PNP ascatalysts. We will demonstrate a considerable selectivity of the 8-azapurine ribosylation sites withvarious PNP forms, and a remarkable sensitivity of the ribosylation process to point mutations at the
Molecules 2016, 21, 44 203 www.mdpi.com/journal/molecules

Molecules 2016, 21, 44
critical active site residue. Finally, we present an example of potential analytical application of theobtained compounds to blood PNP determination.
Figure 1. Ribosylation of 8-azaguanine with α-D-ribose-1-phosphate: possible reaction products.The purine numbering is maintained for simplicity. Only the major tautomeric form of 8-azaguanine(N(9)H) is shown.
2. Results and Discussion
In comparison with natural purines, their 8-aza analogues are not as good PNP substrates, buttheir ribosides are highly fluorescent and therefore can be utilized as probes in enzymology or clinicalinvestigations [11]. Our aim was to identify those forms of PNP which can be used as catalyst in theeffective and selective enzymatic syntheses of these ribosides.
We have investigated bacterial (E. coli) and mammalian (calf) forms of PNP, as the most widelyaccessible, and representing two main classes of the enzyme, as well as their mutated forms, whichhas been previously shown to express altered activity of the phosphorolytic process [1]. In particular,the N243D mutant of the calf and human enzymes is known to catalyze the phosphorolysis not only ofGuo and Ino (as do the wild forms), but also of Ado [1,16]. We expected that the analogous mutant ofthe E. coli PNP, D204N, obtained recently [17], will be also interesting as a potential catalyst.
In the ribosylation reactions, we used α-D-ribose-1-phosphate, prepared enzymatically(see Section 3 for details), as a ribosyl donor [14]. Additionally, we have also measured kineticsof phosphorolysis of 8-azapurine ribosides in the phosphate buffer.
2.1. Ribosylation of 8-Azaguanine and 2,6-Diamino-8-azapurine
Ribosylation of 8-azaguanine (8-azaGua) and 2,6-diamino-8-azapurine (8-azaDaPur) was followedspectrophotometrically, and the reaction products analyzed using HPLC separation coupled withspectrophotometric and fluorimetric detectors. The products were identified by comparing theirspectral properties with those published earlier [18–20]. The non-typical ribosides of both substrates arespectrally distinct from the N9-nucleosides in that the UV spectra of the formers are quite red-shifted,allowing relatively easy detection at 315 nm. Typical elution profile from the C8-column is shownon Figure 2.
204
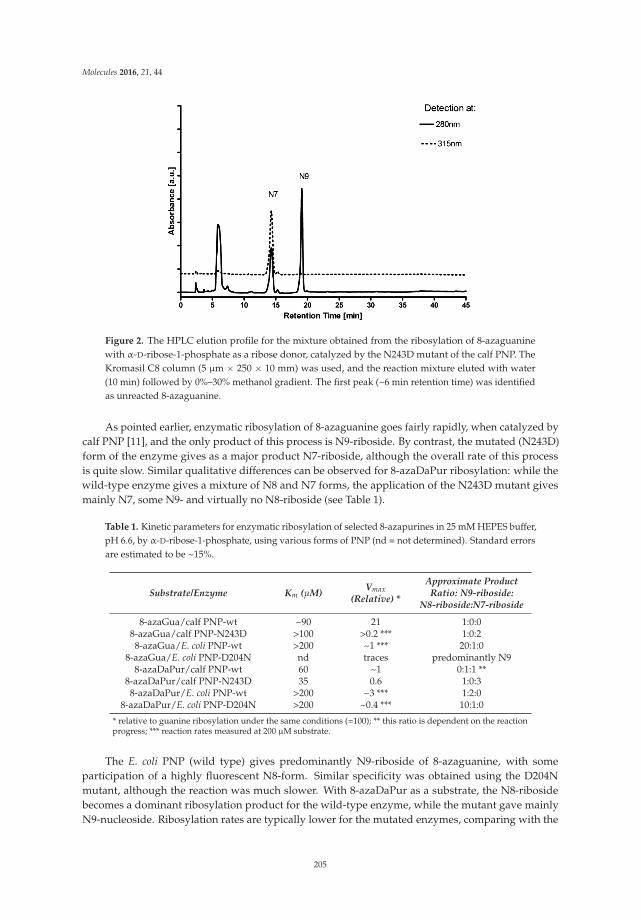
Molecules 2016, 21, 44
Figure 2. The HPLC elution profile for the mixture obtained from the ribosylation of 8-azaguaninewith α-D-ribose-1-phosphate as a ribose donor, catalyzed by the N243D mutant of the calf PNP. TheKromasil C8 column (5 μm ˆ 250 ˆ 10 mm) was used, and the reaction mixture eluted with water(10 min) followed by 0%–30% methanol gradient. The first peak (~6 min retention time) was identifiedas unreacted 8-azaguanine.
As pointed earlier, enzymatic ribosylation of 8-azaguanine goes fairly rapidly, when catalyzed bycalf PNP [11], and the only product of this process is N9-riboside. By contrast, the mutated (N243D)form of the enzyme gives as a major product N7-riboside, although the overall rate of this processis quite slow. Similar qualitative differences can be observed for 8-azaDaPur ribosylation: while thewild-type enzyme gives a mixture of N8 and N7 forms, the application of the N243D mutant givesmainly N7, some N9- and virtually no N8-riboside (see Table 1).
Table 1. Kinetic parameters for enzymatic ribosylation of selected 8-azapurines in 25 mM HEPES buffer,pH 6.6, by α-D-ribose-1-phosphate, using various forms of PNP (nd = not determined). Standard errorsare estimated to be ~15%.
Substrate/Enzyme Km (μM) Vmax(Relative) *
Approximate ProductRatio: N9-riboside:
N8-riboside:N7-riboside
8-azaGua/calf PNP-wt ~90 21 1:0:08-azaGua/calf PNP-N243D >100 >0.2 *** 1:0:28-azaGua/E. coli PNP-wt >200 ~1 *** 20:1:0
8-azaGua/E. coli PNP-D204N nd traces predominantly N98-azaDaPur/calf PNP-wt 60 ~1 0:1:1 **
8-azaDaPur/calf PNP-N243D 35 0.6 1:0:38-azaDaPur/E. coli PNP-wt >200 ~3 *** 1:2:0
8-azaDaPur/E. coli PNP-D204N >200 ~0.4 *** 10:1:0
* relative to guanine ribosylation under the same conditions (=100); ** this ratio is dependent on the reactionprogress; *** reaction rates measured at 200 μM substrate.
The E. coli PNP (wild type) gives predominantly N9-riboside of 8-azaguanine, with someparticipation of a highly fluorescent N8-form. Similar specificity was obtained using the D204Nmutant, although the reaction was much slower. With 8-azaDaPur as a substrate, the N8-ribosidebecomes a dominant ribosylation product for the wild-type enzyme, while the mutant gave mainlyN9-nucleoside. Ribosylation rates are typically lower for the mutated enzymes, comparing with the
205

Molecules 2016, 21, 44
wild types. The Km values, determined by standard methods, are lower for the calf enzymes (Table 1),while the E. coli PNP was not saturated under the applied conditions.
The E. coli PNP is known to ribosylate adenine and 8-azaadenine [1,11]. In both cases, theN9-ribosides were the only detectable ribosylation products. We have found that the D204N mutantreacting with the latter substrate does produce also a non-typical product with red-shifted UV spectra,tentatively identified as N8-riboside (data not shown).
The altered specificity of the PNP mutants towards some of the purine analogs has been observedin other laboratories [21]. It can be related to differences in binding modes of 8-azapurine bases in thePNP active site, including binding in the “upside-down” position (rotation by 180 deg when comparedwith standard binding mode). In fact, such an “upside-down” binding mode was already observedin the crystal structure of calf PNP complexed with N7-acycloguanosine inhibitor [22]. The alteredbinding geometry can possibly be associated also with prototropic tautomerism or proton affinityalterations [23–26]. This illustrates remarkable plasticity of the mammalian and bacterial PNP activesite, which may be profited from in designing new more potent and better membrane-permeable PNPinhibitors—potential pharmaceutical agents [1–7].
There is a considerable progress in enzymatic synthesis of nucleoside analogues on a gramscale using PNP as a catalyst [9,10,27–30]. This work demonstrates that in some substrates thereis a possibility to alter ribosylation specificity of PNP by point mutations, an effect noted also byother authors [21]. Although the non-typical ribosides are less biologically active than their N9-βisomers, they sometimes exhibit interesting spectral properties and could be applied as fluorescentprobes [14,15,21].
2.2. Phosphorolysis of 8-Azaguanine and 2,6-Diamino-8-azapurine Ribosides
On the basis of the micro-reversibility principle, it can be expected that mutations in the activesite of PNP can alter the phosphorolytic processes as well. We have therefore purified the 8-azapurineribosides and examined them as potential PNP substrates in the phosphate buffer, with resultssummarized in Table 2. The phosphorolytic reactions were followed spectrophotometrically and, ifpossible, also fluorimetrically, and the results are summarized in Table 2.
Table 2. Kinetic parameters for enzymatic phosphorolysis of selected ribosides in 25 mM phosphatebuffer, pH 6.5, at 25 ˝C, catalyzed by various forms of PNP. Errors about 15%.
Substrate Enzyme (wt = Wild Type) Km (μM) Vmax (Relative) *
N7-β-D-ribosyl-Gua ** calf PNP-wt 27 0.6N7-β-D-ribosyl-Gua ** E. coli-wt ~450 33N7-ribosyl-8-azaGua calf PNP-wt nd >0.13N7-ribosyl-8-azaGua calf PNP-N243D nd >1.5
N7-ribosyl-8-azaDaPur calf PNP-wt 52 ~20N7-ribosyl-8-azaDaPur calf PNP-N243D >50 >60N7-ribosyl-8-azaDaPur E. coli PNP-wt ~80 ~1.7N7-ribosyl-8-azaDaPur E. coli PNP-D204N nd ~0.7 ***N8-ribosyl-8-azaDaPur E. coli PNP-wt 7 1.1N8-ribosyl-8-azaDaPur E. coli PNP-D204N nd ~1.6 ***N9-ribosyl-8-azaDaPur E. coli PNP-wt ~20 ~0.02
* relative to guanosine phosphorolysis under the same conditions (=100); ** data from ref. [1]; *** rate with40 μM substrate.
8-Azaguanosine was reported to be a very weak substrate for mammalian PNP [11]. Similarly,only traces of activity of the calf PNP towards N7-riboside were detected. One possible reason canbe unfavorable equilibrium of the phosphorolytic process, which for N9-riboside was estimated tobe ~300 in favor of nucleoside synthesis, compared to ~50 for natural purines [11]. The mutated formof the calf enzyme is somewhat more active (Table 2).
As mentioned earlier, there is a considerable specificity in the phosphorolytic pathway in bothcalf and E. coli PNP in relation to 8-azaDaPur ribosides [15]. There was no detectable activity towards
206

Molecules 2016, 21, 44
N9-riboside with the calf PNP, and only residual with the E. coli enzyme. By contrast, the N8-ribosideis quite effectively phosphorolysed by the wild type E. coli PNP, but not by the mutant (see Table 2).Worth noting is low Km value for this process (Table 2), contrasting with very high Km observed in thesynthetic reaction (see Table 1).
The N7-riboside of 8-azaDaPur seems to be quite rapidly and specifically phosphorolysed bycalf PNP [15]. The mutation at Asn243 markedly accelerates this process (see Table 2), while virtuallyno activity towards N8- or N9-ribosides was detected using both wild and mutated types of PNP.This finding is somewhat surprising in view of the fact that N8-ribosides are produced in the reverse(synthetic) reaction catalyzed by wild (but not the mutated) form of calf PNP (see previous sections).
Although the spectrum of non-typical substrates of mammalian, and especially bacterial forms ofPNP is broad [1,9,11], recent years brought much more examples of applications of these enzymes tosynthesis and phosphorolysis of biologically interesting nucleoside analogs [10,27–30]. But the exactprediction of the substrate preferences of various forms is at present difficult [31], we think that largevariability of natural PNP and possibility of mutations in the active site can offer new potentiallyuseful catalyst for synthetic procedures in nucleoside chemistry.
2.3. Fluorescence of 8-Azaguanine and 2,6-Diamino-8-azapurine Ribosides and Potential Applications
Fluorescent nucleoside analogues are widely studied because of potential applications inenzymology [11,32,33]. It has been reported that many 8-azapurines and their nucleosides exhibitmeasurable fluorescence in aqueous medium at room temperature, which was in several instancesapplied to mechanistic and analytical studies [11,34,35]. The highest yields of fluorescence are observedin 8-azaxanthine and 2,6-diamino-8-azapurine, as well as in some N-alkyl derivatives of these [11,36].Nucleosides of adenine and hypoxanthine are more fluorescent than the parent bases [13], andN8-ribosides of many 8-azapurines, including N8-ribosyl-8-azaguanine and N8-ribosyl-8-azaDaPur,exhibit yields comparable to the best fluorophores known [11].
2.3.1. Fluorescence of 2,6-Diamino-8-azapurine Ribosides
Ribosylation of 2,6-diamino-8-azapurine leads to at least three fluorescent ribosides (Table 3). TheN9-riboside is very highly fluorescent, but its fluorescence is generally similar, in terms of λmax anddecay time, to that of the free base [11]. By contrast, fluorescence of N7- and N8-ribosides is red-shiftedby ~70 nm and thanks to this shift their cleavage via phosphorolysis (or hydrolysis) leads to a highfluorogenic effect at ~360 nm, which can be analytically useful (see next section).
Table 3. Ionization constants (pKa values) and spectral parameters for neutral and ionic forms ofthe selected 8-azapurine ribosides and free bases. The UV spectral data are compiled from theliterature [11,18,20] and fluorescence parameters determined in this and previous [11,15] works.
Compound pKaForm *(pH)
UV Absorption Fluorescence
λmax (nm) εmax (M´1¨ cm´1) λmax (nm) φ τ (ns)
9-β-D-ribofuranosyl-8-azaDaPur 2.9 n (7) 285 10,800 368 0.9 6c (2) 283 8100 360 nd ** nd **
8-β-D-ribofuranosyl-8-azaDaPur 4.9 n (7) 313 8200 430 0.41 10.6c (2.7) 264 13,200 430 nd ** nd **
7-β-D-ribofuranosyl-8-azaDaPur 3.95 n (7) 314 ~5500 420 0.063 1.5; 0.45c (2) 258 ~12,000 420 nd ** nd **
8-azaDaPur (free base) 3.7; 7.7 n (6) 280 8500 365 0.40 7.5; 0.2
9-β-D-ribofuranosyl-8-azaGua 8.05 n (5) 256 12,900 347 ~0.01 ~0.1ma (10) 278 11,700 362 0.55 5.6
7-β-D-ribofuranosyl-8-azaGua 7.4 n (5) 302 4900 410 ~0.04 nd **ma (10) 304 5100 420 ~0.03 nd **
8-azaGua (free base) 6.5 n (4.5) 249 11,200 395 0.05–0.33 6.2
* n—neutral form; c—cation; ma—monoanion; ** nd—not determined.
207

Molecules 2016, 21, 44
It is of interest that protonation of N7- and N8-ribosides, which leads to a significant blue shift inthe UV spectra (Table 3), apparently does not alter the observed fluorescence band. This is undoubtedlydue to a large shift in the acid-base equilibrium in the excided state (pK*), as compared to ground-state(pKa), so upon excitation the protonated 8-azapurine moiety undergoes rapid deprotonation. Thisprocess is observed also in acidified alcohols, where dual fluorescence can be observed, especially forthe N8-riboside (data not shown), an analogous effect reported earlier for the N8-methyl derivative [36].
It must be stressed that the strong fluorescence of 8-azaDaPur and its ribosides is sensitive to bufferconcentration, isotope exchange and other environmental factors [36], partially related to excited-stateproton transfer reactions and relatively long fluorescence decay times (Table 3). This creates somedifficulty in analytical applications, which can be overcome by using internal concentration standards(e.g., purified products of enzymatic reactions at standardized concentrations).
2.3.2. Blood PNP Determination Using Fluorogenic Ribosides
It has been reported earlier that 7-β-D-ribofuranosyl-8-azaDaPur is a highly fluorogenic substratefor calf PNP in phosphate buffer [15]. Human PNP is similar to the calf enzyme [1], and we havetherefore verified a possibility to detect PNP activity in human blood using the same substrate.The experiment was run with 1000-fold diluted, lysed human blood in 25 mM phosphate, and ina phosphate-free 20 mM HEPS buffer, pH 6.6, as a control. Ca. 16 μM N7-riboside was used asa substrate. Virtually no fluorescence change was observed in the phosphate-free buffer (data notshown), but in the presence of phosphate the appearance of an emission band at 365 nm indicated freebase production (Figure 3, Table 3).
Figure 3. Fluorescence changes during the incubation of 16 μM N7-ribosyl-8-azaDaPur in 1000-dilutedblood in the presence of 25 mM phosphate. Spectra, excited at 300 nm, were recorded every 5 min, andafter 60 min an aliquot of the purified calf PNP was added, and fluorescence recorded after next 3 min(dotted line, 5-fold diminished relative to remaining curves). Note that the first (lowest) curve reflectsblood fluorescence background, and minimum at 415 nm is due to the re-absorption by hemoglobin.
The apparent Km for this process was rather high, ~90 μM (data not shown), and the presentedfluorogenic effect (Figure 3) can undoubtedly be enhanced by optimizing assay conditions. Althoughphosphorolysis of 7-β-D-ribofuranosyl-8-azaDaPur is slow in comparison to the best PNP substrates,like guanosine or 7-methylguanosine [11], the fluorogenic effect is large thanks to high fluorescenceyield of the product (~0.4), and the present assay, in its optimized version, may be more sensitive andsimpler than other assays proposed for this enzyme [1,37].
208

Molecules 2016, 21, 44
Of special interest is high selectivity of the fluorogenic ribosides of 8-azaDaPur to various formsof PNP, namely, mammalian (N7-ribosides) and bacterial (N8-ribosides), particularly in view of recentapplications of bacterial (E. coli) PNP as a suicidal-gene in cancer chemotherapy [8]. These two formsof PNP can likely be assayed selectively in the same blood sample, with no pre-purification necessary.
3. Experimental Section
Recombinant E. coli and calf spleen PNP, as well as mutated form of these, were obtained andpurified as described elsewhere [17,38]. Stock solutions (0.1–1.7 mM per subunit) were stored frozenat ´20 ˝C and diluted prior to experiments. 2,6-Diamino-8-azapurine sulfate, N7-methylguanosine(m7Guo) and 8-azaguanine were from Sigma-Aldrich (St. Louis, MO, USA), the latter wasre-crystallized as a monosodium salt. N7-methylguanosine was used without further purification.
Fluorescence was measured on a Varian Eclipse instrument (Varian Corp., Palo Alto, CA, USA),and UV absorption kinetic experiments on a Cary 300 (Varian). All buffers were of analytical gradeand displayed no fluorescence background.
α-D-Ribose-1-phosphate (100 mM solution in 100 mM HEPES buffer, pH ~7.2) was preparedenzymatically from the N7-methylguanosine and inorganic phosphate, using the modified procedureof Krenitsky et al. [39]. The recombinant calf PNP (~2 μg per 1 mL reaction volume) was used as catalyst,and the reaction progress was monitored fluorimetrically [40]. Alternatively, the N243D mutant canbe used as a more selective towards m7Guo [41]. The second reaction product, N7-methylguanine,was removed in nearly 97% by spontaneous crystallization and filtration. The phosphorylated ribosesolution was stored at ´20 ˝C and assayed using previously described fluorimetric method [37]. It wasfound that 1-year storage caused hydrolysis of not more than 20% of the compound.
Synthetic reactions were carried out on a milligram scale as previously described [14], typically in1 mL volume, in HEPES buffer. Enzyme concentrations were 1–10 μg/mL, and ribose-1-phosphate~5 mM. After 24 h reaction mixtures were frozen.
Reaction products were analyzed by the analytical reverse-phase HPLC on a UFLC system fromShimadzu (Kyoto, Japan) equipped with UV (diode-array) detection at 280 nm and 315 nm. Thecolumn used was a Kromasil reversed-phase analytical C8 column (250 ˆ 4.6 mm, 5-μm particle size).For product separation, an analogous semi-preparative column was used. The eluent was deionizedwater (10 min), followed by a linear gradient from 0% to 30% methanol (60 min). The reactions werecarried out at pH ~6.5, and samples containing 8-azaguanine ribosides were acidified to ~5 prior to theHPLC analysis.
Kinetic parameters of the enzymatic reactions were calculated using linear regression analysis ofthe double-reciprocal plots. Substrate concentration in the kinetic experiments ranged typically from1 to ~200 μM, and enzyme concentrations were adjusted so that the reaction rates were in the range0.1 to ~5 uM/min. Fluorescence measurements were conducted in semi-micro cuvettes (pathlength0.4 cm, volumen 1 mL) to diminish the inner filter effect.
Acknowledgments: This work has been supported by the Ministry of Higher Education of Poland, grant #NN301 044939 and by the University of Varmia & Masuria in Olsztyn, internal grant #17.610.006-110. The authorsare indebted to Goran Mikleusevic for a sample of the D204N mutated form of the E. coli PNP and to MariuszSzabelski for determination of fluorescence lifetimes.
Author Contributions: A.S.-W. and J.W. are responsible for kinetic and spectroscopic measurements, and A.S.-W.additionally for HPLC analysis. Enzyme cloning and purifications has been done by A.M.B. and B.W.K. Thispaper was written jointly by all the authors.
Conflicts of Interest: The authors declare no conflict of interest.
Abbreviations
PNP, purine-nucleoside phoshorylase; 8-azaGua, 8-azaguanine; 8-azaDaPur, 2,6-diamino-8-azapurine.
209

Molecules 2016, 21, 44
References
1. Bzowska, A.; Kulikowska, E.; Shugar, D. Purine nucleoside phosphorylases: Properties, functions, andclinical aspects. Pharmacol. Ther. 2000, 88, 349–425. [CrossRef]
2. Grunebaum, E.; Cohen, A.; Roifman, C.M. Recent advances in understanding and managing adenosinedeaminase and purine nucleoside phosphorylase deficiencies. Curr. Opin. Allergy Clin. Immunol. 2013, 13,630–638. [CrossRef] [PubMed]
3. Edwards, P.N. A kinetic, modeling and mechanistic re-analysis of thymidine phosphorylase and somerelated enzymes. J. Enzym. Inhib. Med. Chem. 2006, 21, 483–499. [CrossRef] [PubMed]
4. Taylor Ringia, E.A.; Schramm, V.L. Transition states and inhibitors of the purine nucleoside phosphorylasefamily. Curr. Top. Med. Chem. 2005, 5, 1237–1258. [CrossRef] [PubMed]
5. Robak, P.; Robak, T. Older and new purine nucleoside analogs for patients with acute leukemias.Cancer Treat. Rev. 2013, 39, 851–861. [CrossRef] [PubMed]
6. Al-Kali, A.; Gandhi, V.; Ayoubi, M.; Keating, M.; Ravandi, F. Forodesine: Review of preclinical and clinicaldata. Future Oncol. 2010, 6, 1211–1217. [CrossRef] [PubMed]
7. De Jersey, J.; Holý, A.; Hocková, D.; Naesens, L.; Keough, D.T.; Guddat, L.W. 6-oxopurinephosphoribosyltransferase: A target for the development of antimalarial drugs. Curr. Top. Med. Chem. 2011,11, 2085–2102. [CrossRef] [PubMed]
8. Portsmouth, D.; Hlavaty, J.; Renner, M. Suicide genes for cancer therapy. Mol. Asp. Med. 2007, 28, 4–41.[CrossRef] [PubMed]
9. Mikhailopulo, I.A.; Miroshnikov, A.I. Biologically important nucleosides: Modern trends in biotechnologyand application. Mendeleev Commun. 2011, 21, 57–68. [CrossRef]
10. Zhou, X.; Mikhailopulo, I.A.; Cruz Bournazou, M.N.; Neubauer, P. Immobilization of thermostablenucleoside phosphorylases on MagReSyn� epoxide microspheres and their application for the synthesis of2,6-dihalogenated purine nucleosides. J. Mol. Catal. B Enzym. 2015, 115, 119–127. [CrossRef]
11. Wierzchowski, J.; Antosiewicz, J.M.; Shugar, D. 8-Azapurines as isosteric purine fluorescent probes fornucleic acid and enzymatic research. Mol. BioSyst. 2014, 10, 2756–2774. [CrossRef] [PubMed]
12. Giorgi, I.; Scartoni, V. 8-Azapurine nucleus: A versatile scaffold for different targets. Mini Rev. Med. Chem.2009, 9, 1367–1378. [CrossRef] [PubMed]
13. Wierzchowski, J.; Wielgus-Kutrowska, B.; Shugar, D. Fluorescence emission properties of 8-azapurinesand their nucleosides, and application to the kinetics of the reverse synthetic reaction of PNP.Biochim. Biophys. Acta 1996, 1290, 9–17. [CrossRef]
14. Stachelska-Wierzchowska, A.; Wierzchowski, J.; Wielgus-Kutrowska, B.; Mikleuševic, G. Enzymatic synthesisof highly fluorescent 8-azapurine ribosides using purine-nucleoside phosphorylase reverse reaction: Variableribosylation sites. Molecules 2013, 18, 12587–12598. [CrossRef] [PubMed]
15. Wierzchowski, J.; Stachelska-Wierzchowska, A.; Wielgus-Kutrowska, B.; Mikleuševic, G. Two fluorogenicsubstrates for purine-nucleoside phosphorylase, selective for mammalian and bacterial forms of the enzyme.Anal. Biochem. 2014, 446, 25–27. [CrossRef] [PubMed]
16. Stoeckler, J.D.; Poirot, A.F.; Smith, R.M.; Parks, R.E., Jr.; Ealick, S.E.; Takabayashi, K.; Erion, M.D. Purinenucleoside phosphorylase. 3. Reversal of purine base specificity by site-directed mutagenesis. Biochemistry1997, 36, 11749–11756. [CrossRef] [PubMed]
17. Mikleuševic, G.; Štefanic, Z.; Narczyk, M.; Wielgus-Kutrowska, B.; Bzowska, A.; Luic, M. Validation of thecatalytic mechanism of Escherichia coli purine nucleoside phosphorylase by structural and kinetic studies.Biochimie 2011, 93, 1610–1622. [CrossRef] [PubMed]
18. Montgomery, J.A.; Shortnacy, A.T.; Secrist, J.A., III. Synthesis and biological evaluation of2-fluoro-8-azaadenosine and related compounds. J. Med. Chem. 1983, 26, 1483–1489. [CrossRef] [PubMed]
19. Seela, F.; Lampe, S. 8-Aza-21-deoxyguanosine and related 1,2,3-triazolo[4,5-d]pyrimidine 21-deoxyribofuranosides.Helv. Chim. Acta 1993, 72, 2388–2397. [CrossRef]
20. Elliott, R.D.; Montgomery, J.A. Analogues of 8-azaguanosine. J. Med. Chem. 1976, 19, 1186–1191. [CrossRef]21. Ye, W.; Paul, D.; Gao, L.; Seckute, J.; Sangaiah, R.; Jayaraj, K.; Zhang, Z.; Kaminski, P.A.; Ealick, S.E.;
Gold, A.; et al. Ethenoguanines undergo glycosylation by nucleoside 21-deoxyribosyltransferases atnon-natural sites. PLoS ONE 2014, 9, e115082. [CrossRef] [PubMed]
210

Molecules 2016, 21, 44
22. Luic, M.; Koellner, G.; Shugar, D.; Saenger, W.; Bzowska, A. Calf spleen purine nucleoside phosphorylase:Structure of its ternary complex with an N(7)-acycloguanosine inhibitor and a phosphate anion.Acta Crystallogr. D57 2001, 57, 30–36. [CrossRef]
23. Kierdaszuk, B.; Modrak-Wójcik, A.; Wierzchowski, J.; Shugar, D. Formycin A and its N-methyl analogues,specific inhibitors of E. coli purine nucleoside phosphorylase: Induced tautomeric shift on binding to enzyme,and enzyme Ñ ligand fluorescence resonance energy transfer. Biochim. Biophys. Acta 2000, 1476, 109–128.[CrossRef]
24. Pyrka, M.; Maciejczyk, M. Theoretical study of tautomeric equillibria of 2,6–diamino-8-azapurine and8-aza-isoguanine. Chem. Phys. Lett. 2015, 627, 30–35. [CrossRef]
25. Wierzchowski, J.; Bzowska, A.; Stepniak, K.; Shugar, D. Interactions of calf spleen purine nucleosidephosphorylase with 8-azaguanine, and a bisubstrate analogue inhibitor: Implications for the reactionmechanism. Z. Naturforsch. 2004, 59, 713–725. [CrossRef]
26. Gasik, Z.; Shugar, D.; Antosiewicz, J.M. Resolving differences in substrate specificities between humanand parasite phosphoribosyltransferases via analysis of functional groups of substrates and receptors.Curr. Pharm. Des. 2013, 19, 4226–4240. [CrossRef] [PubMed]
27. Zhou, X.; Szeker, K.; Jiao, L.Y.; Oestreich, M.; Mikhailopulo, I.A.; Neubauer, P. Synthesis of 2,6-dihalogenatedpurine nucleosides by thermostable nucleoside phosphorylases. Adv. Synth. Catal. 2015, 357, 1237–1244.[CrossRef]
28. Fateev, I.V.; Kharitonova, M.I.; Antonov, K.V.; Konstantinova, I.D.; Stepanenko, V.N.; Esipov, R.S.; Seela, F.;Temburnikar, K.W.; Seley-Radtke, K.L.; Stepchenko, V.A.; et al. Recognition of Artificial Nucleobases by E. coliPurine Nucleoside Phosphorylase versus its Ser90Ala Mutant in the Synthesis of Base-Modified Nucleosides.Chem. Eur. J. 2015, 21, 13401–13419. [CrossRef] [PubMed]
29. Calleri, E.; Cattaneo, G.; Rabuffetti, M.; Serra, I.; Bavaro, T.; Massolini, G.; Speranza, G.; Ubiali, D.Flow-Synthesis of Nucleosides Catalyzed by an Immobilized Purine Nucleoside Phosphorylase fromAeromonas hydrophila: Integrated Systems of Reaction Control and Product Purification. Adv. Synth. Catal.2015, 357, 2520–2528. [CrossRef]
30. Calleri, E. Immobilized purine nucleoside phosphorylase from Aeromonas hydrophila as an on-line enzymereactor for biocatalytic applications. J. Chromatogr. B 2014, 968, 79–86. [CrossRef] [PubMed]
31. Štefanic, Z.; Mikleuševic, G.; Narczyk, M.; Wielgus-Kutrowska, B.; Bzowska, A.; Luic, M. Still a longway to fully understanding the molecular mechanism of Escherichia coli Purine Nucleoside Phosphorylase.Croat. Chem. Acta 2013, 86, 117–127. [CrossRef]
32. Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent analogs of biomolecular building blocks: Design, properties,and applications. Chem. Rev. 2010, 110, 2579–2619. [CrossRef] [PubMed]
33. Pollum, M.; Martínez-Fernández, L.; Crespo-Hernández, C.E. Photochemistry of nucleic acid bases and theirthio- and aza-analogues in solution. Top. Curr. Chem. 2015, 355, 245–355. [PubMed]
34. Liu, L.; Cottrell, J.W.; Scott, L.G.; Fedor, M.J. Direct measurement of the ionization state of an essentialguanine in the hairpin ribozyme. Nat. Chem. Biol. 2009, 5, 351–357. [CrossRef] [PubMed]
35. Cottrell, J.W.; Scott, L.G.; Fedor, M.J. The pH dependence of hairpin ribozyme catalysis reflects ionization ofan active site adenine. J. Biol. Chem. 2011, 286, 17658–17664. [CrossRef] [PubMed]
36. Wierzchowski, J.; Medza, G.; Szabelski, M.; Stachelska-Wierzchowska, A. Properties of 2,6-diamino-8-azapurine, a highly fluorescent purine analog and its N-alkyl derivatives: Tautomerism and excited-stateproton transfer reactions. J. Photochem. Photobiol. A 2013, 265, 49–57. [CrossRef]
37. Wierzchowski, J.; Ogiela, M.; Iwanska, B.; Shugar, D. Selective fluorescent and fluorogenic substrates forpurine-nucleoside phosphorylases from various sources, and direct fluorimetric determination of enzymelevels in human and animal blood. Anal. Chim. Acta 2002, 472, 63–74. [CrossRef]
38. Breer, K.; Girstun, A.; Wielgus-Kutrowska, B.; Staron, K.; Bzowska, A. Overexpression, purification andcharacterization of functional calf purine nucleoside phosphorylase (PNP). Protein Expr. Purif. 2008, 61,122–130. [CrossRef] [PubMed]
39. Krenitsky, T.A.; Koszalka, G.W.; Tuttle, J.V. Purine nucleoside synthesis, an efficient method employingnucleoside phosphorylases. Biochemistry 1981, 20, 3615–3621. [CrossRef] [PubMed]
40. Kulikowska, E.; Bzowska, A.; Wierzchowski, J.; Shugar, D. Properties of two unusual, andfluorescent substrates of purine-nucleoside phosphorylase: 7-methylguanosine and 7-methylinosine.Biochim. Biophys. Acta 1986, 874, 355–366. [CrossRef]
211

Molecules 2016, 21, 44
41. Wielgus-Kutrowska, B. Division of Biophysics, Institute of Experimental Physics; University of Warsaw: Warsaw,Poland, Unpublished Work.
Sample Availability: Samples of the compounds are available from the authors.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
212

MDPI AG
St. Alban-Anlage 66
4052 Basel, Switzerland
Tel. +41 61 683 77 34 Fax +41 61 302 89 18
http://www.mdpi.com
Molecules Editorial Office
E-mail: [email protected]
http://www.mdpi.com/journal/molecules


MDPI AG St. Alban-Anlage 66 4052 Basel Switzerland
Tel: +41 61 683 77 34 Fax: +41 61 302 89 18
www.mdpi.com ISBN 978-3-03842-355-3





























