Embed Size (px)
Citation preview

Dynamic Article LinksC<Journal ofMaterials Chemistry
Cite this: J. Mater. Chem., 2011, 21, 3477
www.rsc.org/materials PAPER
Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online / Journal Homepage / Table of Contents for this issue
Metal-catalyzed graphitic nanostructures as sorbents for vapor-phaseammonia†
Jeffrey W. Long,*a Matthew Laskoski,b Gregory W. Peterson,c Teddy M. Keller,b Katherine A. Pettigrewa
and Bryan J. Schindlerd
Received 21st September 2010, Accepted 15th December 2010
DOI: 10.1039/c0jm03167d
Activated carbons have long been used as substrates for the filtration of vapor-phase molecules, often
with metal salts or oxides added to improve their sorption capacities for specific agents, but their real-
world performance and applicability may be hindered by such factors as long-term stability and complex
processing. En route to a new class of carbon-based sorbents, we have developed solid-state synthetic
methods to produce bulk carbonaceous solids based on the pyrolysis of thermoset solids containing low
concentrations (<1 wt%) of metal precursors (based on either Ni, Fe, or Co) that decompose in situ to
catalyze the formation of a complex graphitic nanostructure. Selective combustion of residual
amorphous carbon from the pyrolyzed solid generates a mesoporous network that facilitates diffusional
transport of gas-phase molecules to the interior surfaces of the solid, and also converts the entrained
metals to their respective metal oxide forms. We examine the ammonia-sorption properties of a series of
these graphitic nanostructured compositions, and demonstrate that ammonia uptake is primarily
determined by the type of residual metal oxide, with the Co-containing carbonaceous solid providing the
best ammonia-sorption capacity (1.76 mol kg�1). Thermal reduction of the Co-containing material
drastically decreases its ammonia-sorption capacity, showing that the oxide form (in this case Co3O4) of
the entrained metal nanoparticles is most active for ammonia filtration. The effects of the carbon–oxygen
functionalities on the nanostructured graphitic surfaces for ammonia sorption are also discussed.
Introduction
Nanostructured carbons are often used as sorbents for toxic
small molecules, such as ammonia, which presents particular
challenges for filtration because of its high vapor pressure.1,2 In
the case of carbonaceous materials, oxidation of the carbon
surface improves the adsorption of ammonia by the addition of
functionalities (carboxylic, hydroxyl), whereby hydrogen
bonding, complexation or acid–base chemistry can occur.3–7 For
example, Seredych and Bandosz oxidized graphite using fuming
nitric acid and found enhanced reactivity towards ammonia from
the resultant carboxylic functional groups;8 their results
aCode 6170, Surface Chemistry Branch, Naval Research Laboratory,Washington, DC, 20375, USA. E-mail: [email protected] 6120, Materials Chemistry Branch, Naval Research Laboratory,Washington, DC, 20375, USAcUS Army Research, Development and Engineering Command EdgewoodChemical Biological Center Research & Technology Directorate, CBRFiltration Team, Aberdeen Proving Ground, MD, 21010, USAdSAIC Gunpowder Branch, PO Box 68, Aberdeen Proving Ground, MD,21010, USA
† Electronic supplementary information (ESI) available: additionalX-ray diffraction, supplementary pore-size distribution plots,water-sorption isotherms, expanded versions of ammonia-breakthroughplots, and additional information on ammonia-breakthrough testingapparatus. See DOI: 10.1039/c0jm03167d
This journal is ª The Royal Society of Chemistry 2011
indicated the formation of ammonium ions through Brønsted
activity and amides through bond-breaking chemistry. The
addition of metal salts or oxides to carbon sorbents also
improves their uptake for ammonia via complexation, as in the
case of metal chloride salts,9 or by acid–base interactions, as
shown for molybdenum and tungsten oxides.10 Although effec-
tive for improving filtration performance, the methods used to
incorporate these active metal components add complexity and
cost to the production of the final product, and the spatial
distribution and long-term stability of the metal impregnants
may also be difficult to control.
Over the past several years, we have developed a general solid-
state synthetic approach for the production in large quantity,
high yield, and moldable forms of multi-walled carbon nano-
tubes (MWNTs), segmented or bamboo-type nanotubes, and/or
graphitic nanofibers/nanoribbons.11–16 This general method is
based on in situ generation of low concentrations of metal
nanoparticles (typically <2 weight%) via the thermal decompo-
sition of melt-processable organometallic compounds and/or
metal salts in the presence of an excess amount of a polymer-
izable aromatic carbon precursor. For pyrolysis temperatures
exceeding 700 �C, the resulting carbonaceous solids are typically
composites of interpenetrating graphitic and amorphous carbon
phases, in which the ratio of crystalline-to-amorphous carbon is
determined by such factors as the type of transition-metal
J. Mater. Chem., 2011, 21, 3477–3484 | 3477

Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
catalyst (Fe, Ni, or Co), the concentration of the catalyst, the
chemical structure of the polymeric precursor, and the carbon-
ization temperature/time profile.
We recently described a simple thermal oxidation procedure to
selectively combust and remove the entrained amorphous carbon
phase, thereby revealing a highly porous solid comprising
graphitic nanostructures and residual metal oxides from the
graphitization catalyst.17 We further demonstrated that the
interior surfaces of the graphitic nanostructure are highly
accessible to gas-phase molecules such as ammonia. Using the
design flexibility afforded by the solid-state synthetic approach
and subsequent selective-combustion purification, we are
exploring and optimizing these carbonaceous materials for
a variety of applications, including electrochemical energy
storage, hydrogen storage, and as sorbents for the filtration of
vapor-phase toxins. In many such cases, the incorporated metals
used to catalyze the formation of the graphitic nanostructure
during the initial synthesis may also serve as functional compo-
nents in the final product, particularly for applications that
involve the sorption or catalysis of small molecules.
In the present report, we examine the ammonia sorption
properties of a particular series of graphitic nanostructured
compositions developed at the NRL.13,17 Among the composi-
tions containing Ni, Fe, or Co nanoparticulate graphitization
catalysts, the Co-containing composition exhibits the highest
ammonia-sorption capacity. We further show that the oxide
form of the Co-nanoparticle graphitization catalyst that is
generated during the selective-combustion purification process
promotes more effective ammonia adsorption compared to the
same composition in which the metal oxide (Co3O4) has been
thermally reduced back to Co metal. The effects of the surface
carbon–oxygen functionalities present in the calcined form of
these carbonaceous materials are also discussed with respect to
their impact on ammonia sorption.
Experimental
Synthetic procedures
The synthesis and characterization of carbonaceous solids
prepared by the carbonization of thermoset resins based on
nickel(cyclooctadiene) (Ni(COD)2), dicobalt octacarbonyl
(Co2(CO)8) and diiron nonocarbonyl (Fe2(CO)9) and 1,2,4,5-
tetrakis(phenylethynyl)benzene (TPEB, 1) have been described in
previous publications.13 Briefly, the metal catalyst precursors
(Ni(COD)2, (Co2(CO)8) or (Fe2(CO)9) and 1 were dissolved in a
1 to 20 molar ratio in a common solvent such as methylene
chloride. The resulting solutions were stirred for 1 h and the
solvent was removed to yield dark solids. A circular Al planchet
was treated with Teflon mold release and the individual dark
products were melted and heated at 220 �C until the polymeri-
zation reaction was complete, which typically occurred within
1 h. The resulting polymeric solids were cooled, removed from
the mold, placed in a tube furnace, heated at 0.3 �C min�1 from
room temperature to 1000 �C under an argon atmosphere, and
cooled to room temperature at a rate of 0.5 �C min�1 to yield the
nanostructured carbonaceous solid.
Following the carbonization step, the resulting solids were
heated in a muffle furnace under static air to selectively combust
3478 | J. Mater. Chem., 2011, 21, 3477–3484
or remove any entrained amorphous carbon, using calcination
conditions (set temperature and time held at the temperature)
determined by separate thermal analysis experiments. The
calcination conditions varied according to the graphitization
catalyst used: Ni-catalyzed, 480 �C for 20 h; Fe-catalyzed, 460 �C
for 16 h; Co-catalyzed, 420 �C for 16 h. For the purposes of this
manuscript, the calcined versions of these materials are desig-
nated as ‘‘Ni-TPEB-calc’’, ‘‘Fe-TPEB-calc’’, and ‘‘Co-TPEB-calc,
respectively. Portions of the Co-TPEB-calc material were sub-
jected to an additional thermal treatment in a Lindberg tube
furnace under flowing 10% H2/90% argon (total flow rate ¼220 mL min�1), ramping at 5 �C min�1 to set points of either
300 �C or 700 �C, and holding at the set points for 2 h.
Materials characterization
Nitrogen-sorption porosimetry was performed with a Micro-
meritics ASAP2010 Accelerated Surface Area and Porosity
Analyzer; all samples were degassed under vacuum at 150 �C for
24 h prior to measurements. Temperature-programmed reduction
(TPR) experiments were performed using a Micromeritics
Autochem II Chemisorption Unit coupled with a Pfeiffer Vacuum
ThermoStar� Residual Gas Analyzer (RGA) attachment (with
internal quadrupole mass spectrometer). Samples for TPR
measurements were first degassed at 150 �C in UHP He (99.999%)
for 2 h, then ramped at 10 �C min�1 in 10% H2/90% argon to
700 �C, while the composition of the eluting gas stream was
monitored by a thermal conductivity detector in the chemisorp-
tion unit and at selected mass-to-charge (m/z) channels with the
RGA.
X-Ray diffraction analysis was performed with a Bruker D8
powder diffractometer, using CuKa radiation from a rotating
anode X-ray source. Transmission electron microscopy (TEM)
studies were performed on a JEOL 2200FS equipped with
a Gatan CCD camera; a Noran System Six EDS was used to
evaluate the polyhedral carbon structures with visible lattice
fringes. Powders of the nanostructured carbonaceous materials
were analyzed by brushing a small amount onto holey-carbon
film supports. Multiple areas were imaged to ensure that the
results obtained were representative of the whole sample. Scan-
ning electron microscopy (SEM) studies were performed on
a Zeiss Model Supra 55 Electron Microscope. X-Ray photo-
electron spectroscopy measurements were performed using
a Thermo Scientific K-Alpha X-ray photoelectron spectrometer
with a monochromatic Al-Ka X-ray source. Samples were
analyzed as powders that were filled into the wells of a Cu
mounting plate (no signals originating from the sample stage
were observed). A flood gun was used in all experiments to
minimize any charging effects with the powdered samples.
Breakthrough testing
Ammonia breakthrough experiments were conducted on the
carbonaceous powders using a micro-breakthrough push system;
for a full description, see the work by Peterson et al.18 Briefly,
a ballast was charged with a mixture of ammonia and air; this
pressurized stream was then mixed with a dry or humid diluent
stream at a rate necessary to achieve an ammonia concentration
of 1000 mg m�3 (�1430 ppm). The respective powders were
This journal is ª The Royal Society of Chemistry 2011

Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
packed in a 4 mm ID glass fritted tube to a height of 4 mm. All
samples (�25 mg) were dried at 150 �C for 1 h and humid
samples were then pre-humidified for 2 h. The effluent stream
was constantly monitored using a Hewlett Packard 5890 Series II
gas chromatograph equipped with a photoionization detector
and a 10.6 eV lamp. Test conditions are specified in Table S1
(ESI†). Expanded versions of the breakthrough curves, focusing
on the initial ammonia breakthrough of the respective sample
beds, are also shown in the ESI†.
Results and discussion
Structural characteristics of calcined metal–TPEB compositions
The microscale and nanoscale structures of the metal–TPEB
compositions examined in this study are determined mainly by
the morphology of graphitic networks that develop during the
solid-state carbonization process, as catalyzed by the low
concentrations of incorporated metal nanoparticles that form
in situ by decomposition of the respective organometallic
precursors. As described previously for Ni-TPEB-based carbo-
naceous solids,17 selective-combustion of the amorphous carbon
component reveals the underlying graphitic nanostructure, and
results in a porous solid that remains monolithic. Qualitatively
similar structures are observed for the related Fe-TPEB-calc and
Fig. 1 (a and b) SEM characterization for the Co-TPEB samples (low
and high resolution); (c and d) TEM characterization for the Co-TPEB
samples (low and high resolution); (e and f) TEM characterization for the
Fe-TPEB samples (low and high resolution).
This journal is ª The Royal Society of Chemistry 2011
Co-TPEB-calc materials. When observed by SEM (Fig. 1a and
b), the calcined solids exhibit interpenetrating, aperiodic
networks of solid and disordered voids with feature sizes ranging
from �10 to 200 nm. The mesopore/macropore network that
forms via combustion of the amorphous carbon serves a critical
function in providing a facile pathway for the infiltration of gas-
phase molecules to the interior graphitic surfaces, as demon-
strated previously for the dynamic sorption of ammonia on the
Ni-TPEB-calc solid.17 A closer inspection of the solid domains
using TEM (Fig. 1c–f) reveals a complex graphitic nanostructure
comprising cockleshelled,19 spherical, tubular, and ribbon-like
morphologies, with feature sizes ranging from several nano-
metres to �20 nm with different curvatures and lengths, for both
the Fe-TPEB-calc and Co-TPEB-calc materials. The areas of
higher, sharper contrast that are observed dispersed throughout
the graphitic nanostructure (Fig. 1c and e) are assigned to the
metal nanoparticles (in this case Co or Fe) that form in situ during
the carbonization process, and exhibit a wide range of particle
sizes, from a few nm to�100 nm. Due to the embedded nature of
the metal nanoparticles within the carbonaceous matrix, it was
difficult to obtain a more quantitative particle-size analysis.
X-Ray diffraction measurements confirm that these metal
nanoparticles are converted to their respective metal oxide
forms—NiO,17 a-Fe2O3 (ESI†), and Co3O4—during the calci-
nation process. The nanoparticulate metals and corresponding
metal oxides are incorporated at relatively low loadings. For
example, prior analysis of the Ni-TPEB-calc material revealed
a residual NiO content of 2.0 wt%.17 In the present case,
elemental analysis was also performed on the Co-TPEB-calc and
Fe-TPEB-calc materials, with the Co-TPEB-calc analog having
a Co content of 3.0 wt%, corresponding to 4.1 wt% Co3O4, while
the Fe-TPEB-calc material contained 1.3 wt% Fe and 1.9 wt%
Fe2O3. The metal/metal oxide content of the final product is
ultimately a result of both the initial metal content of precursor
mixture and the subsequent mass loss that occurs during
carbonization and calcination. The adventitious formation of
highly dispersed metal oxide nanoparticles in the purified
graphitic nanomaterials produced by our process may ultimately
be beneficial for the filtration of small molecules such as
ammonia, via acid–base interactions10 or potentially through
catalytic mechanisms that proceed at the metal oxide surfaces. In
the following sections, we examine how the oxidation state of the
metal in a series of Co-TPEB carbon nanomaterials impacts their
performance for ammonia filtration.
The effectiveness of a sorbent depends not only on surface
functionality, but also on the nature of the pore–solid architec-
ture. Nitrogen-sorption porosimetry was performed on the series
of calcined solids to assess such characteristics as specific surface
area, pore volume, and pore-size distribution. A summary of N2-
sorption metrics for the Ni-, Fe-, and Co-TPEB-calc materials is
shown in Table 1. The Ni-TPEB-calc material exhibits relatively
modest specific surface area (140 m2 g�1), consistent with the
larger feature size of the graphitic nanostructure in that mate-
rial,17 relative to the Fe-TPEB-calc and Co-TPEB-calc versions
(see Fig. 1), which exhibit total specific surface areas of 360 and
500 m2 g�1, respectively. The Co-TPEB-calc material is further
distinguished by significant surface area and pore volume located
in micropores (<2 nm), which may also enhance its adsorption
for small-molecule gases.20 The origin of the microporosity in the
J. Mater. Chem., 2011, 21, 3477–3484 | 3479

Table 1 Metrics derived from nitrogen-sorption porosimetry
Carbonsample
BET surfacearea/m2 g�1
Microporeareaa/m2 g�1
Cumulative porevolumeb/cm3 g�1
Microporevolumea/cm3
g�1
Ni-TPEB-calc
140 18 0.37 0.01
Fe-TPEB-calc
360 50 0.86 0.02
Co-TPEB-calc
500 340 0.60 0.16
Co-TPEB-calc-300-red
470 320 0.68 0.15
Co-TPEB-calc-700-red
530 360 0.71 0.17
a Calculated using the t-plot method. b Calculated using the Barrett–Joyner–Halenda model.
Fig. 3 Ammonia-breakthrough curves for various carbon sorbents
under (a) dry and (b) humid conditions.
Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
Co-TPEB-calc material, which is not observed in the Ni-TPEB-
calc or Fe-TPEB-calc analogs, is not presently understood, but
may arise from some open tubular structures, which are difficult
to observe by TEM in this complex graphitic nanostructure.
Pore-size distribution plots were also generated from the N2-
sorption data (see Fig. 2). All materials exhibited a broad range
of pore sizes from �2 to 40 nm, but with much of the pore
volume concentrated in pores <10 nm, a size range that should
still facilitate diffusional transport of gas-phase molecules to the
interior surfaces of these materials. Low-pressure isotherm data
for the Co-TPEB-calc material were also fitted using the MP
method to model the structure of the micropores (see ESI†).
Ammonia-sorption on calcined metal–TPEB compositions as
a function of their residual metal nanoparticles
Ammonia breakthrough curves for the Co-TPEB-calc, Fe-
TPEB-calc, and Ni-TPEB-calc compositions are illustrated in
Fig. 3, corresponding to dry and humid challenge conditions,
respectively. Testing was conducted on equivalent volumes of
each sample, and the breakthrough curves are plotted on a rela-
tive time scale to account for differences in mass used for each
Fig. 2 Pore-size distribution plots for the Ni-TPEB-calc, Fe-TPEB-calc,
and Co-TPEB-calc compositions, derived using a DFT model and
assuming a slit-shaped pore geometry.
3480 | J. Mater. Chem., 2011, 21, 3477–3484
test. Under dry conditions ammonia begins breaking through on
both the Fe-TPEB-calc and Ni-TPEB-calc analogs almost
immediately, and exhibits very sharp curves to saturation indi-
cating very limited adsorption/reaction capacities (see Fig. 3a).
The Co-TPEB-calc analog, on the other hand, has a delay in
ammonia breakthrough and also shows a somewhat elongated
curve, which may indicate significant interaction between
ammonia and the metal oxide nanoparticles and/or graphitic
surfaces. The pore structure may also play a role in ammonia
removal, with the Co-TPEB-calc composition having signifi-
cantly higher pore volumes in the micropore range. In all cases,
much of the adsorbed ammonia eventually elutes after the
challenge is terminated and the sample is purged with clean air,
indicating that the ammonia sorption is most likely due to
a specific physisorption interaction that is not yet understood.
Among the series of materials investigated, the Co-TPEB-calc
composition provides the best ammonia removal by far, with
a calculated capacity of 1.76 mol kg�1 as compared to 0.51 and
0.19 mol kg�1 for the Fe-TPEB-calc and Ni-TPEB-calc materials,
respectively (see Table 2). By comparison, the dynamic
ammonia-sorption capacities of typical off-the-shelf activated
carbons (with specific surface areas >1000 m2 g�1) are on the
order of 0.1 mol kg�1,6,18 while boiling activated carbons in
concentrated acid to introduce acidic carbon–oxygen surface
functionalities can extend their sorption capacities to 2.4 mol
kg�1.6 Petit and Bandosz investigated activated carbons
impregnated with acidic tungsten and molybdenum oxides, and
reported dynamic ammonia-sorption capacities as high as
1.5 mol kg�1.for carbons containing 10.7 wt% Mo.10
This journal is ª The Royal Society of Chemistry 2011

Table 2 Summary of ammonia-breakthrough results
SampleRelativehumidity Mass/mg
Loading to saturation
(mol kg�1) (mg g�1)
Ni-TPEB-calc 0% 22.5 0.19 3.280% 19.5 0.30 5.1
Fe-TPEB-calc 0% 15.3 0.51 8.780% 13.6 0.62 10.6
Co-TPEB-calc 0% 9.4 1.76 30.080% 10.2 0.81 13.8
Co-TPEB-calc-300-red 0% 11.8 0.83 14.180% 10.8 1.48 25.2
Co-TPEB-calc-700-red 0% 4.6 0.19 3.280% 10.0 0.69 11.8
Fig. 4 Temperature-programmed reduction of the Co-TPEB-calc
material heated at 10 �C min�1 under a 10% H2/90% Ar gas flow. The
response of the TCD is shown on the left y-axis and detector currents
from the various RGA channels are shown on the right y-axis.
Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
In the present case, the significantly higher ammonia-sorption
capacity of the Co-TPEB-calc composition cannot solely be
attributed to its modestly higher specific surface area (500 m2 g�1
vs. 360 m2 g�1 for the Fe-TPEB-calc material). Although there are
variations in the total metal/metal oxide content among this
series, the Co-TPEB-calc composition still exhibits the highest
capacity beyond what would be expected for its modestly higher
metal oxide content. We have observed similarly enhanced
ammonia-sorption capacities with other Co-containing graphitic
nanostructures, but derived from alternative carbon precursors
such as Novolac resins.21 Cobalt oxides (specifically Co3O4) have
been reported as effective ammonia-oxidation catalysts at
elevated temperatures (>700 �C),22 but it is unclear whether such
mechanisms are operational at room temperature. The ammonia
capacities under dry conditions for this series do generally follow
the trend of isoelectric points for the respective metal oxides,
ranging from �10 for NiO to 9 for a-Fe2O3 to 7.5 for Co3O423–25
which may indicate that the acid–base character of the metal
oxide is a factor for ammonia sorption. Regardless, the enhanced
capacity of the Co-TPEB-calc material does not derive simply
from ammonia sorption at the surfaces of the incorporated metal
oxide nanoparticle. Normalizing the molar capacity of ammonia
to the known Co3O4 molar concentration would yield a NH3-to-
Co3O4 molar ratio of 3.4, far beyond a theoretical monolayer
adsorption, particularly considering the particle size and dis-
persity (�5 to 20 nm) of the Co3O4. Further work is underway to
identify the specific adsorption mechanisms for the Co-TPEB-
calc composition and related nanostructured graphitic materials.
Under humid conditions (Fig. 3b), the Co-TPEB-calc material
again outperforms the other two analogs in breakthrough
capacity. The Fe-TPEB-calc composition actually has the best
initial rate of ammonia removal, yet the Co-containing analog has
substantial capacity as seen by the shape of the curve at higher
C/C0 values, indicative of specific interaction of the ammonia
either with the cobalt oxide, as observed under dry conditions, or
through hydrogen-bonding with adsorbed moisture. The Co-
TPEB-calc material adsorbs the most moisture (�12 wt%) at 80%
relative humidity compared to the Fe-TPEB-calc (�9 wt%) and
Ni-TPEB-calc (�2 wt%) analogs (water-sorption isotherms for all
three carbon materials are shown in the ESI†). The higher mois-
ture content in the Co-TPEB-calc sorbent should facilitate better
ammonia removal via dissolution in the adsorbed water layer. All
samples show significant desorption after feed termination, indi-
cating that the strength of ammonia sorption is modest.
This journal is ª The Royal Society of Chemistry 2011
While the Co-TPEB-calc material provides the best ammonia
removal of the samples tested, it is also the only sample for which
ammonia capacity decreases with increasing humidity. Typically,
ammonia removal capacity increases with increasing relative
humidity on activated carbons due to the relatively high solu-
bility of ammonia in water. In the case of the Co-TPEB-calc
sorbent, competitive adsorption between water and ammonia at
the active cobalt oxide sites may result in somewhat lower
ammonia capacities in humid conditions, overshadowing the
expected solubility effects. The ammonia-sorption behavior
under humid conditions may also be affected by the formation of
ammonium ions in the adsorbed water layer when acidic surface
sites are present, as noted in the work by Le Leuch and
Bandosz.26
Tuning the oxygen functionalities of metal and carbon
components for the Co-containing solids
With the Co-TPEB-calc composition identified as the most effec-
tive sorbent among those studied for ammonia filtration, we
selected this material for further investigation, with the goal of
elucidating the origins of its superior ammonia-sorption proper-
ties. The Co-TPEB-calc solid possesses a complex structure in
which multiple factors, such as specific adsorption or catalytic
interactions of the incorporated Co3O4 nanoparticles or the pres-
ence of significantly greater micropore volume may distinguish this
material from the Fe-TPEB-calc and Ni-TPEB-calc analogs.
Carbon–oxygen functionalities on the surface of these graphitic
nanostructures may also play a role in the ammonia-sorption
properties of these materials via acid–base interactions, as
described previously for activated carbon6 and graphite oxide.7,27,28
In order to deconvolute these various substrate effects with
respect to the filtration of ammonia, we applied a series of
thermal-reduction treatments to the Co-TPEB-calc material with
the goal of selectively reducing Co-oxide and/or carbon–oxygen
functionalities, while otherwise maintaining a consistent pore–
solid architecture. Temperature-programmed reduction (TPR)
measurements were first performed on the Co-TPEB-calc
powder (see Fig. 4) to determine the appropriate treatment
conditions. The composition of the evolving gases during the
J. Mater. Chem., 2011, 21, 3477–3484 | 3481

Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
TPR experiment was monitored using both a non-selective
thermal conductivity detector (TCD), and a residual gas analyzer
(RGA) that was tuned to track evolved H2O, CO, and CO2.
The initial TPR peak that is observed at �260 �C in both the
TCD response and in the RGA channel for H2O (m/z ¼ 18) is
consistent with the reduction of Co3O4, which may proceed
through a CoO intermediate between 300 and 400 �C, en route to
forming Co metal nanoparticles.29,30 Additional reduction is
noted between 400 and 700 �C in the form of a large convoluted
peak shape in the TCD response that is in part coincident with
a large RGA peak for CO (m/z ¼ 28) and a smaller peak for CO2
(m/z¼ 44). The detection of CO and CO2 at these temperatures is
consistent with the desorption of surface-sited carbon–oxygen
functionalities, as has been previously noted for activated
carbon.31–33 The temperature range of the prominent peak in this
region, at �600 �C, and the preferential evolution of CO relative
to CO2 are consistent with decomposition of phenolic and/or
carbonyl functionalities from the graphitic surface.
On the basis of the results of the TPR analysis, we chose two
temperatures to thermally reduce portions of the Co-TPEB-calc
carbon: 300 �C to reduce the Co3O4 phase under 10% H2/90%
argon gas flow; and 700 �C to desorb surface-sited carbon–
oxygen functionalities. The resulting samples are denoted at ‘‘Co-
TPEB-calc-300-red’’ and ‘‘Co-TPEB-calc-700-red’’, respectively.
Powder X-ray diffraction measurements confirmed that the
crystalline phase of the incorporated cobalt species in the initial
Co-TPEB-calc form is Co3O4, as evidenced by the diffraction
peaks at 32 and 37� 2 � q (see Fig. 5). These peaks are greatly
reduced after thermal reduction at 300 �C, suggesting that the
Co3O4 converts to another phase (one that does not strongly
diffract), but clear diffraction evidence for Co metal is only
observed for the sample reduced at 700 �C. The persistence of the
graphitic peak with comparable peak width confirms that the
Fig. 5 Powder X-ray diffraction for a series of Co-TPEB carbon samples.
Also shown are reference patterns for graphite, Co3O4, and Co metal.
3482 | J. Mater. Chem., 2011, 21, 3477–3484
underlying graphitic nanostructure is not markedly altered by
these thermal reduction treatments.
The TPR results suggest that carbon–oxygen functionalities
are ultimately removed from these materials, particularly for the
Co-TPEB-calc-700-red material. X-Ray photoelectron spec-
troscopy, which is a well-established method for characterizing
the surface chemistry of carbonaceous materials,34 was used to
monitor changes in surface carbon–oxygen surface functionality
of the Co-TPEB series as a function of thermal treatment.
Deconvolution of the C1s spectrum for the initial Co-TPEB-calc
material reveals a wide range of carbon–oxygen functionalities,
including carboxylate and carbonyl groups, but with phenol/
hydroxyl moieties (C1s peak at 285.8 eV) being the most prom-
inent (see Fig. 6).
Because the TPEB precursor to these materials contains no
oxygen, we posit that the oxygen functionalities observed for the
Co-TPEB-calc material are generated during the calcination/
purification step, which involves heating in air at 420 �C,
conditions that may partially oxidize the graphitic surfaces. The
presence of carbon–oxygen functionalities, particularly those
that are either weakly acidic (phenol) or strongly acidic
(carboxylic acid) should promote the adsorption of
ammonia,3,6,22 although we note that the quantity of adventitious
carbon–oxygen functionalities in the present materials is far less
than those previously reported carbon-based sorbents that are
intentionally oxidized. Considering only the total integration of
Fig. 6 X-Ray photoelectron spectra in the C1s region for the series of
Co-TPEB materials. Deconvoluted peaks are shown with dashed lines,
which are also labeled according with their respective assignments to
particular carbon functionalities.
This journal is ª The Royal Society of Chemistry 2011
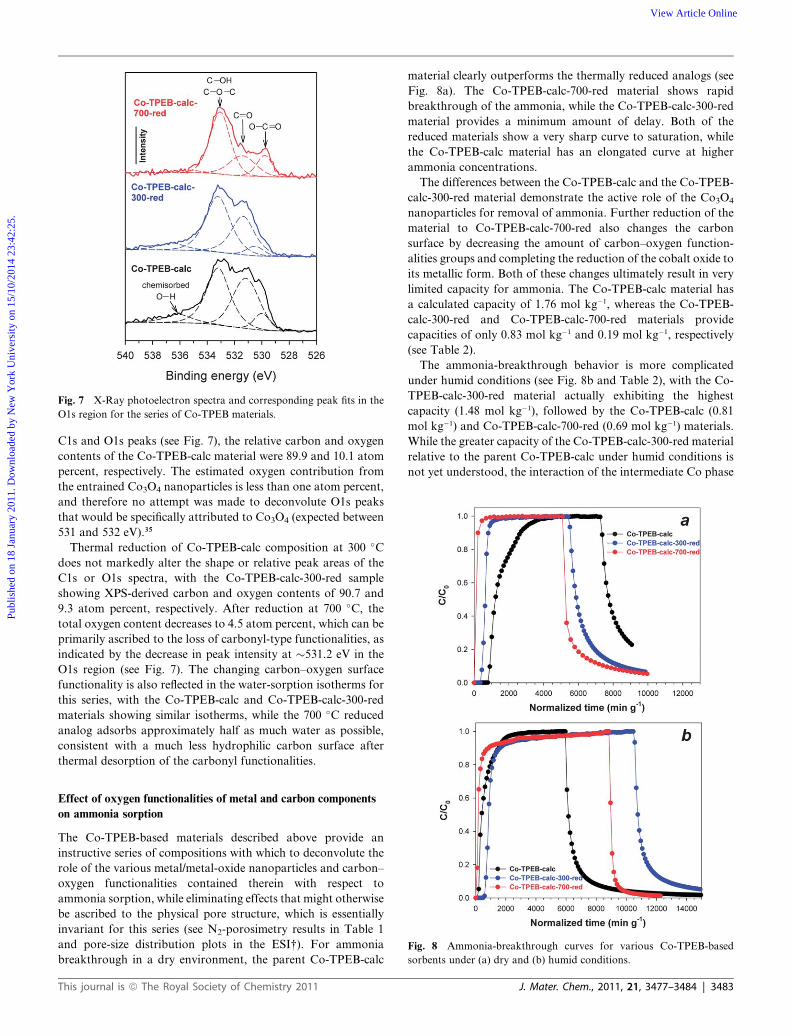
Fig. 7 X-Ray photoelectron spectra and corresponding peak fits in the
O1s region for the series of Co-TPEB materials.
Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
C1s and O1s peaks (see Fig. 7), the relative carbon and oxygen
contents of the Co-TPEB-calc material were 89.9 and 10.1 atom
percent, respectively. The estimated oxygen contribution from
the entrained Co3O4 nanoparticles is less than one atom percent,
and therefore no attempt was made to deconvolute O1s peaks
that would be specifically attributed to Co3O4 (expected between
531 and 532 eV).35
Thermal reduction of Co-TPEB-calc composition at 300 �C
does not markedly alter the shape or relative peak areas of the
C1s or O1s spectra, with the Co-TPEB-calc-300-red sample
showing XPS-derived carbon and oxygen contents of 90.7 and
9.3 atom percent, respectively. After reduction at 700 �C, the
total oxygen content decreases to 4.5 atom percent, which can be
primarily ascribed to the loss of carbonyl-type functionalities, as
indicated by the decrease in peak intensity at �531.2 eV in the
O1s region (see Fig. 7). The changing carbon–oxygen surface
functionality is also reflected in the water-sorption isotherms for
this series, with the Co-TPEB-calc and Co-TPEB-calc-300-red
materials showing similar isotherms, while the 700 �C reduced
analog adsorbs approximately half as much water as possible,
consistent with a much less hydrophilic carbon surface after
thermal desorption of the carbonyl functionalities.
Fig. 8 Ammonia-breakthrough curves for various Co-TPEB-based
sorbents under (a) dry and (b) humid conditions.
Effect of oxygen functionalities of metal and carbon components
on ammonia sorption
The Co-TPEB-based materials described above provide an
instructive series of compositions with which to deconvolute the
role of the various metal/metal-oxide nanoparticles and carbon–
oxygen functionalities contained therein with respect to
ammonia sorption, while eliminating effects that might otherwise
be ascribed to the physical pore structure, which is essentially
invariant for this series (see N2-porosimetry results in Table 1
and pore-size distribution plots in the ESI†). For ammonia
breakthrough in a dry environment, the parent Co-TPEB-calc
This journal is ª The Royal Society of Chemistry 2011
material clearly outperforms the thermally reduced analogs (see
Fig. 8a). The Co-TPEB-calc-700-red material shows rapid
breakthrough of the ammonia, while the Co-TPEB-calc-300-red
material provides a minimum amount of delay. Both of the
reduced materials show a very sharp curve to saturation, while
the Co-TPEB-calc material has an elongated curve at higher
ammonia concentrations.
The differences between the Co-TPEB-calc and the Co-TPEB-
calc-300-red material demonstrate the active role of the Co3O4
nanoparticles for removal of ammonia. Further reduction of the
material to Co-TPEB-calc-700-red also changes the carbon
surface by decreasing the amount of carbon–oxygen function-
alities groups and completing the reduction of the cobalt oxide to
its metallic form. Both of these changes ultimately result in very
limited capacity for ammonia. The Co-TPEB-calc material has
a calculated capacity of 1.76 mol kg�1, whereas the Co-TPEB-
calc-300-red and Co-TPEB-calc-700-red materials provide
capacities of only 0.83 mol kg�1 and 0.19 mol kg�1, respectively
(see Table 2).
The ammonia-breakthrough behavior is more complicated
under humid conditions (see Fig. 8b and Table 2), with the Co-
TPEB-calc-300-red material actually exhibiting the highest
capacity (1.48 mol kg�1), followed by the Co-TPEB-calc (0.81
mol kg�1) and Co-TPEB-calc-700-red (0.69 mol kg�1) materials.
While the greater capacity of the Co-TPEB-calc-300-red material
relative to the parent Co-TPEB-calc under humid conditions is
not yet understood, the interaction of the intermediate Co phase
J. Mater. Chem., 2011, 21, 3477–3484 | 3483

Publ
ishe
d on
18
Janu
ary
2011
. Dow
nloa
ded
by N
ew Y
ork
Uni
vers
ity o
n 15
/10/
2014
23:
42:2
5.
View Article Online
formed at 300 �C and ammonia dissolved in the adsorbed water
layer may play a role. The decrease in the capacity upon further
reduction to Co-TPEB-calc-700-red material can be attributed to
the loss of both the acidic carbon–oxygen sites on the graphitic
surfaces and also cobalt oxide being reduced to its metallic state.
Conclusions
Nanostructured graphitic solids produced via solid-state
methods exhibit promising performance for the adsorption of
gas-phase ammonia. Our preliminary studies demonstrate that
the Co-catalyzed carbonaceous solid provides the highest
ammonia-sorption capacities, facilitated by the residual Co3O4
nanoparticles dispersed throughout the graphitic nanostructure,
while acidic carbon–oxygen surface functionalities play a smaller
role in the ammonia-uptake mechanism. Future efforts will focus
on improving the strength of ammonia sorption on these and
related materials, and on developing a more thorough under-
standing of the relevant adsorption mechanisms. The findings of
this preliminary investigation will be used to translate the
advantages of our solid-state synthetic approach toward the
production of large-scale, moldable, solid forms of nano-
structured graphitic compositions in new materials that are
specifically designed for the filtration of vapor-phase toxins, such
as ammonia.
Acknowledgements
Financial support for this project was provided by the Defense
Threat Reduction Agency and the Office of Naval Research. The
authors thank Dr Jean M. Wallace (Nova Research, Inc.,
Alexandria, VA) for assistance with the TPR experiments, and
Dr Debra R. Rolison (Naval Research Laboratory) for helpful
discussions in the preparation of this manuscript.
References
1 G. L. Bridger and R. D. Sinner, Ind. Eng. Chem., 1953, 45, 581–582.2 C. L. Mangun, R. D. Braatz, J. Economy and A. J. Hall, Ind. Eng.
Chem. Res., 1999, 38, 3499–3504.3 H. Tamon and M. Okazaki, Carbon, 1996, 34, 741–746.4 S. J. Park and S. Y. Jin, J. Colloid Interface Sci., 2005, 286, 417–419.5 B. J. Kim and S. J. Park, J. Colloid Interface Sci., 2007, 311, 311–314.6 C.-C. Huang, H.-S. Li and C.-H. Chen, J. Hazard. Mater., 2008, 159,
523–527.
3484 | J. Mater. Chem., 2011, 21, 3477–3484
7 M. Seredych and T. J. Bandosz, J. Phys. Chem. C, 2007, 111, 15596–15604.
8 M. Seredych and T. J. Bandosz, Langmuir, 2010, 26, 5491–5498.9 C. Petit, C. J. Karwacki, G. W. Peterson and T. J. Bandosz, J. Phys.
Chem. C, 2007, 111, 12705–12714.10 C. Petit and T. J. Bandosz, Environ. Sci. Technol., 2008, 42, 3033–
3039.11 T. M. Keller and S. B. Qadri, Chem. Mater., 2004, 16, 1091–1097.12 T. M. Keller, S. B. Qadri and C. A. Little, J. Mater. Chem., 2004, 14,
3063–3070.13 T. M. Keller, M. Laskoski, M. Osofsky and S. B. Qadri, Phys. Status
Solidi A, 2008, 205, 1585–1591.14 T. M. Keller, M. Laskoski and S. B. Qadri, J. Phys. Chem. C, 2007,
111, 2514–2519.15 M. Laskoski, S. B. Qadri and T. M. Keller, Carbon, 2007, 45, 443–
448.16 V. O. Nyamori, S. D. Mhlanga and N. J. Coville, J. Organomet.
Chem., 2008, 693, 2205–2222.17 J. W. Long, M. Laskoski, T. M. Keller, K. A. Pettigrew, S. B. Qadri,
T. M. Zimmerman and G. W. Peterson, Carbon, 2010, 48, 501–508.18 G. W. Peterson, G. W. Wagner, A. Balboa, J. Mahle, T. Sewell and
C. J. Karwacki, J. Phys. Chem. C, 2009, 113, 13906–13917.19 O. P. Krivoruchko, N. I. Maksimova, V. I. Zaikovskii and
A. N. Salanov, Carbon, 2000, 38, 1075–1082.20 C. Nguyen and D. D. Do, J. Phys. Chem. B, 1999, 103, 6900–6908.21 J. W. Long, M. Laskoski, T. M. Keller and G. W. Peterson, Naval
Research Laboratory and Edgewood Chemical/Biological Center,unpublished results, 2010.
22 K. Schmidt-Szalowski, K. Krawczyk and J. Petryk, Appl. Catal., A,1998, 175, 147–157.
23 J. A. Lewis, J. Am. Ceram. Soc., 2004, 83, 2341–2359.24 M. Gunnarsson, M. Rasmusson, S. Wall, E. Ahlberg and J. Ennis,
J. Colloid Interface Sci., 2001, 240, 448–458.25 C. Pirovano and S. Trasatti, J. Electroanal. Chem., 1984, 180, 171–
184.26 L. M. Le Leuch and T. J. Bandosz, Carbon, 2007, 45, 568–578.27 C. Petit, M. Serdych and T. J. Bandosz, J. Mater. Chem., 2009, 19,
9176–9185.28 M. Seredych, A. V. Tamashuasky and T. J. Bandosz, Adv. Funct.
Mater., 2010, 20, 1670–1679.29 O. A. Bulavchenko, S. V. Cherpanova, V. V. Malakhov,
L. S. Dovlitova, A. V. Ishchenko and S. V. Tsybulya, Kinet. Catal.,2009, 50, 192–198.
30 H. Zhang, C. Lancelot, W. Chu, J. Hong, A. Y. Khodakov,P. A. Chernavskii, J. Zheng and D. Tong, J. Mater. Chem., 2009,19, 9241–9249.
31 Y. Otake and R. G. Jenkins, Carbon, 1993, 31, 109–121.32 J. L. Figueredo, M. F. R. Pereira, M. M. A. Freitas and
J. J. M. �Orfao, Carbon, 1999, 37, 1379–1389.33 M. Almarri, X. Ma and C. Song, Energy Fuels, 2009, 23, 3940–3947.34 S. Biniak, G. Szyma�nski, J. Siedlewski and A. �Swiatkowski, Carbon,
1997, 35, 1799–1810.35 V. M. Jim�enez, A. Fern�andez, J. P. Espin�os and A. R. Gonz�ales,
J. Electron Spectrosc. Relat. Phenom., 1995, 71, 61–67.
This journal is ª The Royal Society of Chemistry 2011





























