Embed Size (px)
Citation preview
Decomposition and Reaction of Polyvinyl Nitrate under Shock andThermal Loading: A ReaxFF Reactive Molecular Dynamics StudyMd Mahbubul Islam and Alejandro Strachan*
School of Materials Engineering and Birck Nanotechnology Center, Purdue University, West Lafayette, Indiana 47907, United States
*S Supporting Information
ABSTRACT: We use molecular dynamics (MD) simulations with thereactive force field ReaxFF to investigate the response of polyvinylnitrate (PVN), a high-energy polymer, to shock loading using theHugoniostat technique. We compare predictions from three widely usedReaxFF versions, and in all cases, we observe shock-induced, volume-increasing exothermic reactions following a short induction time forstrong enough insults. The three models predict NO2 dissociation to bethe first chemical, and relatively similar final product populations;however, we find significant differences in intermediate populationsindicating different reaction mechanisms due to discrepancies in therelative stability of various intermediate fragments. A time-resolvedspectral analysis of the reactive MD trajectories enables the first directcomparison of shock-induced chemistry between atomistic simulationsand experiments; specifically, ultrafast spectroscopy on laser shocked samples. The results from one of the ReaxFF versions are inexcellent agreement with the experiments both in terms of threshold shock strength required for the disappearance of NO2 peaksand in the time scale associated with the process. Such direct comparison between physical observables is an important steptoward a definite determination of detailed chemistry for high-energy density materials.
I. INTRODUCTION
Shock-induced chemistry of high-energy (HE) materials andthe shock to detonation transition involve complex and inter-related physical and chemical processes through which theenergy in the shockwave is transferred to chemical bonds withcharacteristic lengths of Ångstroms and vibrational periods offew femtoseconds. In many cases the microstructure of thematerial plays a key role in localizing the energy in the shock inspace, and these hotspots1,2 initiate chemical reactions,deflagration and eventually detonation. At the same time,inter- and intramolecular relaxation processes transfer energyfrom long wavelength, low-frequency modes of the shock tohigh-frequency bond vibrations, a process called up-pump-ing.3−5 To complicate matters, these processes occur underextreme conditions of temperature, pressure and strain rate andinvolve short time scales. It is, thus, not surprising that despitesignificant efforts in experiments and simulations a definite amolecular-level description of shock-induced chemistry is stilllacking even in relatively simple materials like nitromethane, aliquid at room temperature, or polyvinyl nitrate (PVN), anamorphous polymer.Recent experimental efforts are yielding insight and
quantitative information about the molecular level processesresponsible for shock-induced chemistry.6−10 For example,ultrafast spectroscopy study of laser shocked PVN enabled thedetection of chemical reactive events under shock loading,7
broadband multiplex vibrational sum-frequency generation(SFG) technique allows real-time observation of shock-front
interactions with molecular monolayers.11 The experiment onPVN12 revealed that 18 GPa shocks resulted in chemicalreactions within approximately 150 ps indicated by thedisappearance of a peak associated with NO2. Despite suchsignificant accomplishments, these experimental techniques arenot without limitations. For example, experiments do notenable a direct characterization of chemical mechanisms nor theidentification of all intermediates and products. Currentspatiotemporal and analytical resolution preclude in situevaluation of the detailed behavior of molecules behind adetonation front.8 On the simulation side, ab initio andquantum-based molecular dynamics (MD) simulations havebeen used to identify initiation mechanisms, reaction barriers,and rates associated with the decomposition of various energymaterials.13−20 However, the computationally intensity of thesesimulations limits the spatial and temporal scales achievable.Reactive force fields, like ReaxFF,21−23 provide a lesscomputationally intensive alternative and enable multimillionatom simulations and capturing the role of microstructure anddefects with scales approaching those in experiments.24,25
Recent efforts provide an atomic picture of the shock todeflagration transition.24 While these simulations provideunparalleled resolution in space and time, they involveapproximations whose effect on predictions remain poorly
Received: June 23, 2017Revised: September 5, 2017Published: September 6, 2017
Article
pubs.acs.org/JPCC
© XXXX American Chemical Society A DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX

understood. To different degrees, ab initio, tight-binding, andreactive potentials approximate atomic interactions and forces;in addition, the use of classical ionic dynamics is also anapproximation.We note that shock-induced chemistry is an extremely
challenging problem for an atomistic model as it needs tocapture two interdependent and complex processes: (i) Thefirst is the thermo-mechanics of shock loading, the amount ofenergy the shock deposits into the system and how this energyis distributed among different degrees of freedom via variety ofrelaxation processes: volumetric compression and heating aswell as plasticity, fracture, phase transformations, void collapseand interfacial sliding that can lead to energy localization andthe formation of hot spots. (ii) The model should also capturethe thermodynamics and kinetics of chemical decomposition atvarious conditions of pressure and temperature; including uni-and multimolecular processes. Given the complexity of theproblem, a definite understanding of shock-induced chemistrywill likely require a synergistic combination of experimental andtheoretical investigations26 where the experiments validate thesimulations and simulations help interpret the experimentalresults.In this paper, we use MD simulations with three different
versions of the ReaxFF15,27,28 force field to establish the initialdecomposition of PVN following shock loading. We build onthe successful use of ReaxFF in a family of high-energymolecular crystals RDX, HMX, PETN, and NM14,17,19,27,29,30
and apply it to an energetic polymer. This is motivated by theexistence of spectroscopic data on shocked PVN which enablesthe first direct comparison of ReaxFF predictions of shock-induced chemistry against experiments. Importantly, theamorphous nature of PVN simplifies the comparison betweenexperiments and simulations due to the lack of grain boundariesand other crystal defects that can significantly affect shockresponse. In addition, PVN is of interest as a binder in explosiveand propellant formulations and understanding its physics andchemistry can contribute to their safe operation. A high energydensity binder provides not just adhesion but also contributesto the total energy release.6 Despite the interest of the PVNpolymer in explosive applications, only a limited number ofexperiments have been performed to investigate the mecha-nistic details of its response to shock loading. Moreover, noatomistic simulation studies on PVN chemical dissociationmechanisms under shock and thermal loading conditions areavailable in literature. As such the detailed chemistry of PVNremains unknown. Therefore, we aim to provide a detailedcharacterization of the mechanical, chemical, and spectroscopicproperties of the PVN polymer. We compute shock Hugoniotsand investigate how shock and thermal loading dictates thechemical reaction pathways. We find that PVN chemicaldissociation initiates via the formation of NO2 and theactivation energy decreases with the increased system densities.For the first time, our simulations enabled a one-on-onecomparison of the reaction initiation time scales with theultrafast shock experiments via time-resolved spectra. Inaddition, each of the force fields predicted results are comparedwith the available literature to establish their performance inpredicting the mechanistic details of the PVN polymer.
II. SIMULATION METHODOLOGYII.A. ReaxFF and Force Field Description. ReaxFF is a
bond order (BO)31,32 empirical reactive force field which allowsthe description of chemical reactions during MD simulations. It
uses the concept of partial BOs between pairs of atoms todescribe covalent interactions including 2-, 3-, and 4-bodybonded interactions. BOs are a continuous, many body, andfunctions of the atomic coordinates. The nonbonded van derWaals and Coulomb interactions are calculated between everypair of atoms, within the respective cutoffs, regardless ofcovalent interactions.33,34 In the nonbonded energy expres-sions, shielding parameters are introduced to circumventexcessive repulsion at short distances, and a seventh ordertaper function is used to eliminate any energy discontinuity.35,36
The electronegativity equalization method (EEM)37 is utilizedto obtained environment dependent partial atomic charges thatare updated at every step during the MD simulations. For amore detailed description of the ReaxFF method, see refs21−23, 38, and 39.Over the past decade, a number of ReaxFF parameter sets
have been developed to describe high-energy density materials.The accurate prediction of the complex chemistry of thesematerials at extreme conditions of pressure and temperature aswell as the description of physical, thermodynamic, andspectroscopic properties are challenging. Therefore, severalversions are currently available, each emphasizing differentproperties. Importantly, the uncertainties associated withdifferent versions have not been characterized, and comparisonsacross different force fields are not common. Therefore, it is anevident necessity to systematically evaluate the performance ofthe available force fields in terms of their predictions ofmechanical, chemical and spectroscopic properties. In thisstudy, we used three widely used ReaxFF C/H/N/O forcefields to formulate a detailed chemical and mechanicaldescription of the PVN polymer. The original C/H/N/OReaxFF force field was developed and applied for RDX20,40 andsince then several updates have been proposed. The standardReaxFF C/H/N/O force field resulted in a less than idealdescription of lattice parameters and equations of states due tothe inadequate description of the London dispersion. Rom etal.27 incorporated additional inner-core repulsions for the vander Waals interactions to improve the description of latticeparameters and updated the previous C/H/N/O force field.40
We will denote this force field as ReaxFF-IW (inner-wall). Theadditional inner core repulsion has the following form
= −E c c r cexp( (1 / ))vdw IW ij( ) 1 2 3
Here c1, c2, and c3 are force field parameters, and rij is theinteratomic distance between atom i and j.To improve the bulk properties of the energetic materials
while, at the same time, maintaining the description of thechemical reactions of the unmodified ReaxFF− in the ReaxFF-lg force field, a low gradient (lg) attractive term wasincorporated to account for the long-range London dispersionusing following expression28
∑= −+<
EC
r dRij i j
N
ij eijlg
,
lg,ij6 6
Here rij and Reij are the distance and equilibrium vdW radiusbetween atoms i and j, respectively, and Clg,ij is the dispersionenergy correction parameter.It has been shown that the addition of the lg-parameters on
the ReaxFF-IW force field substantially improves the equationof states of energetic materials.28 We note that the differencebetween ReaxFF-IW and ReaxFF-lg force fields are only thelow-gradient parameters. More recently, Wood et al.15
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
B
developed an updated version of the C/H/N/O force fieldspecifically to improve the spectroscopic properties and thepostdetonation combustion chemistry (denoted here asReaxFF-2014).II.B. Structure Preparation. Amorphous PVN (molecular
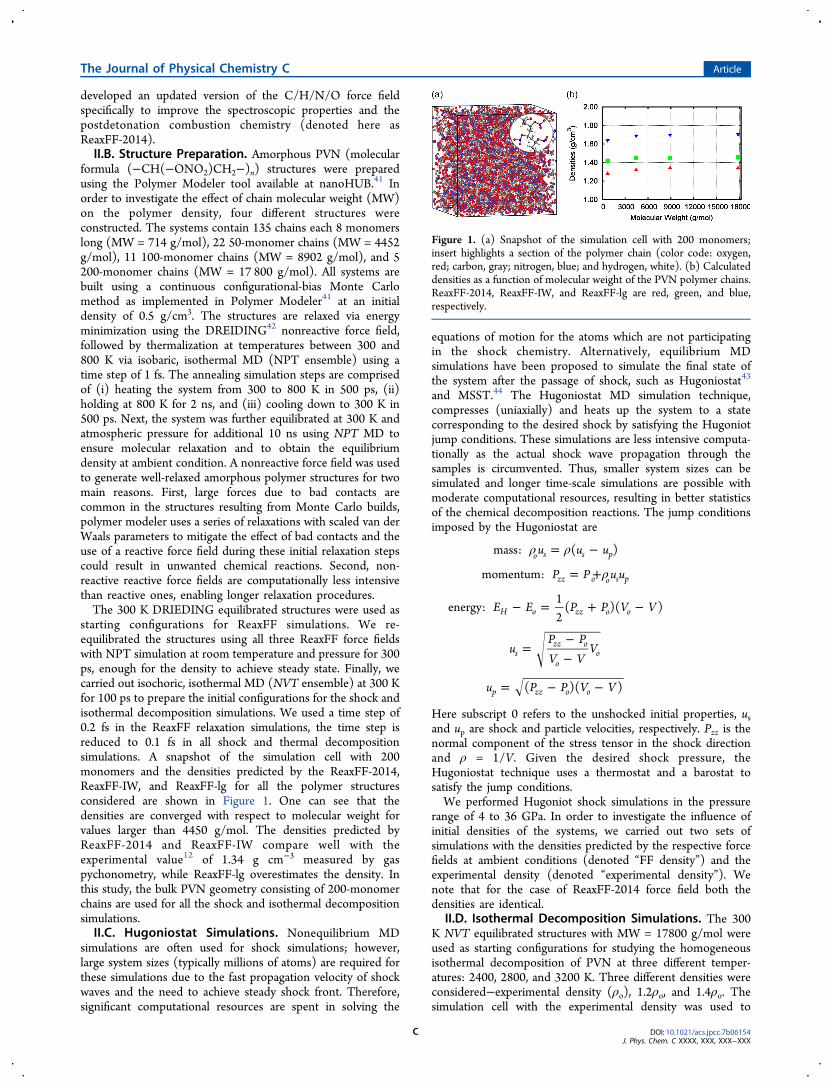
formula (−CH(−ONO2)CH2−)n) structures were preparedusing the Polymer Modeler tool available at nanoHUB.41 Inorder to investigate the effect of chain molecular weight (MW)on the polymer density, four different structures wereconstructed. The systems contain 135 chains each 8 monomerslong (MW = 714 g/mol), 22 50-monomer chains (MW = 4452g/mol), 11 100-monomer chains (MW = 8902 g/mol), and 5200-monomer chains (MW = 17 800 g/mol). All systems arebuilt using a continuous configurational-bias Monte Carlomethod as implemented in Polymer Modeler41 at an initialdensity of 0.5 g/cm3. The structures are relaxed via energyminimization using the DREIDING42 nonreactive force field,followed by thermalization at temperatures between 300 and800 K via isobaric, isothermal MD (NPT ensemble) using atime step of 1 fs. The annealing simulation steps are comprisedof (i) heating the system from 300 to 800 K in 500 ps, (ii)holding at 800 K for 2 ns, and (iii) cooling down to 300 K in500 ps. Next, the system was further equilibrated at 300 K andatmospheric pressure for additional 10 ns using NPT MD toensure molecular relaxation and to obtain the equilibriumdensity at ambient condition. A nonreactive force field was usedto generate well-relaxed amorphous polymer structures for twomain reasons. First, large forces due to bad contacts arecommon in the structures resulting from Monte Carlo builds,polymer modeler uses a series of relaxations with scaled van derWaals parameters to mitigate the effect of bad contacts and theuse of a reactive force field during these initial relaxation stepscould result in unwanted chemical reactions. Second, non-reactive reactive force fields are computationally less intensivethan reactive ones, enabling longer relaxation procedures.The 300 K DRIEDING equilibrated structures were used as
starting configurations for ReaxFF simulations. We re-equilibrated the structures using all three ReaxFF force fieldswith NPT simulation at room temperature and pressure for 300ps, enough for the density to achieve steady state. Finally, wecarried out isochoric, isothermal MD (NVT ensemble) at 300 Kfor 100 ps to prepare the initial configurations for the shock andisothermal decomposition simulations. We used a time step of0.2 fs in the ReaxFF relaxation simulations, the time step isreduced to 0.1 fs in all shock and thermal decompositionsimulations. A snapshot of the simulation cell with 200monomers and the densities predicted by the ReaxFF-2014,ReaxFF-IW, and ReaxFF-lg for all the polymer structuresconsidered are shown in Figure 1. One can see that thedensities are converged with respect to molecular weight forvalues larger than 4450 g/mol. The densities predicted byReaxFF-2014 and ReaxFF-IW compare well with theexperimental value12 of 1.34 g cm−3 measured by gaspychonometry, while ReaxFF-lg overestimates the density. Inthis study, the bulk PVN geometry consisting of 200-monomerchains are used for all the shock and isothermal decompositionsimulations.II.C. Hugoniostat Simulations. Nonequilibrium MD
simulations are often used for shock simulations; however,large system sizes (typically millions of atoms) are required forthese simulations due to the fast propagation velocity of shockwaves and the need to achieve steady shock front. Therefore,significant computational resources are spent in solving the
equations of motion for the atoms which are not participatingin the shock chemistry. Alternatively, equilibrium MDsimulations have been proposed to simulate the final state ofthe system after the passage of shock, such as Hugoniostat43
and MSST.44 The Hugoniostat MD simulation technique,compresses (uniaxially) and heats up the system to a statecorresponding to the desired shock by satisfying the Hugoniotjump conditions. These simulations are less intensive computa-tionally as the actual shock wave propagation through thesamples is circumvented. Thus, smaller system sizes can besimulated and longer time-scale simulations are possible withmoderate computational resources, resulting in better statisticsof the chemical decomposition reactions. The jump conditionsimposed by the Hugoniostat are
ρ ρ
ρ
= −
= +
− = + −
=−−
= − −
u u u
P P u u
E E P P V V
uP PV V
V
u P P V V
mass: ( )
momentum:
energy:12
( )( )
( )( )
o s s p
zz o o s p
H o zz o o
szz o
oo
p zz o o
Here subscript 0 refers to the unshocked initial properties, usand up are shock and particle velocities, respectively. Pzz is thenormal component of the stress tensor in the shock directionand ρ = 1/V. Given the desired shock pressure, theHugoniostat technique uses a thermostat and a barostat tosatisfy the jump conditions.We performed Hugoniot shock simulations in the pressure
range of 4 to 36 GPa. In order to investigate the influence ofinitial densities of the systems, we carried out two sets ofsimulations with the densities predicted by the respective forcefields at ambient conditions (denoted “FF density”) and theexperimental density (denoted “experimental density”). Wenote that for the case of ReaxFF-2014 force field both thedensities are identical.
II.D. Isothermal Decomposition Simulations. The 300K NVT equilibrated structures with MW = 17800 g/mol wereused as starting configurations for studying the homogeneousisothermal decomposition of PVN at three different temper-atures: 2400, 2800, and 3200 K. Three different densities wereconsidered−experimental density (ρo), 1.2ρo, and 1.4ρo. Thesimulation cell with the experimental density was used to
Figure 1. (a) Snapshot of the simulation cell with 200 monomers;insert highlights a section of the polymer chain (color code: oxygen,red; carbon, gray; nitrogen, blue; and hydrogen, white). (b) Calculateddensities as a function of molecular weight of the PVN polymer chains.ReaxFF-2014, ReaxFF-IW, and ReaxFF-lg are red, green, and blue,respectively.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
C
generate the volumetrically compressed systems by adjustingvolumes to the target densities using deformation simulations.The compressed systems were further relaxed using 300 K NVTsimulations for 10 ps. Next, the target temperatures werereached by rescaling the atomic velocities at a heating rate of1000 K/ps using NVT ensemble. The Nose−Hoover thermo-stat was used with a coupling constant of 20 fs. The rapidheating rate is comparable to the time scales of shock loading.Once the target temperatures were reached, the systems wereheld at the temperature with an NVT ensemble in the range of300−800 ps. The lower temperature cases required longersimulation time to attain steady-state. The isothermalsimulations allowed us to extract Arrhenius parameters tostudy reaction kinetics of the PVN polymer. The characteristicstime for the reaction rate calculations is considered as the timerequired to reach the half of the total exothermicity in eachcase.II.E. Analysis of MD Simulations. The MD simulation
trajectories were postprocessed to obtain power spectra usingthe following formula:15
∑ ∑ω βτ= | Δ |πω
= =
−− ΔP
Nm v n t( ) ( )e
j
N
jn
N
ji n t
1
3
0
12 2
where vj(t) are the atomic velocities at time t, β is defined as(kBT)
−1, T is the temperature, τ is the sampling period, mj is themass of atom j, Δt is the sampling rate, and N is the number offrames to be analyzed. We used fast Fourier transform toevaluate the power spectra. The atomic trajectories were
sampled at every 2.5 fs to resolve the high-frequency modesaccurately. The time-resolved spectra calculations were carriedout at 10 ps interval, and each case was averaged over 4000frames.The chemical bonds and molecular species were detected
using a conservative bond order cutoff of 0.30 for all thesimulations. The accuracy of on-the-fly species recognition wasimproved by sampling bond order values at every 0.50 fs andaveraged over 10 samples. This approach instead of usinginstantaneous bond orders also abate identification of spuriousbonds in high-density systems, typical for shocked compression.The molecular fractions represented in this study arenormalized by the total number of monomers.
III. SHOCK STATES AND EXOTHERMIC REACTIONS
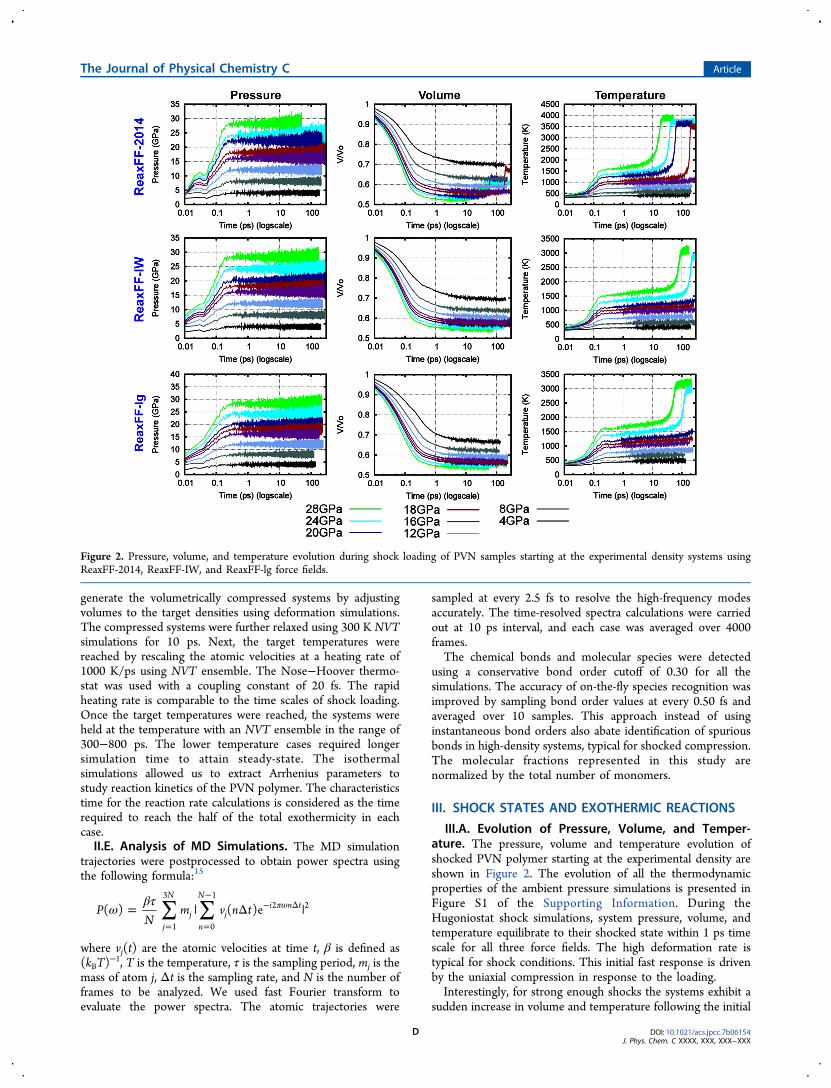
III.A. Evolution of Pressure, Volume, and Temper-ature. The pressure, volume and temperature evolution ofshocked PVN polymer starting at the experimental density areshown in Figure 2. The evolution of all the thermodynamicproperties of the ambient pressure simulations is presented inFigure S1 of the Supporting Information. During theHugoniostat shock simulations, system pressure, volume, andtemperature equilibrate to their shocked state within 1 ps timescale for all three force fields. The high deformation rate istypical for shock conditions. This initial fast response is drivenby the uniaxial compression in response to the loading.Interestingly, for strong enough shocks the systems exhibit a
sudden increase in volume and temperature following the initial
Figure 2. Pressure, volume, and temperature evolution during shock loading of PVN samples starting at the experimental density systems usingReaxFF-2014, ReaxFF-IW, and ReaxFF-lg force fields.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
D
compression and an induction time. This is due to theexothermic, volume-increasing reactions induced by a combi-nation of temperature and density increase due to the shock.During this phase, small molecules are produced (expansionstage). The simulations show a decrease in the induction timeand reaction-induced volume expansion with increasing shockstrength (and pressure). Interestingly, as can be seen in FigureS1, ReaxFF-lg force field does not exhibit exothermic reactionswhen shocked from its ambient pressure density for the shockstrengths up to 36 GPa, and time scales of 300 ps.The shock pressure−density data extracted from the shock
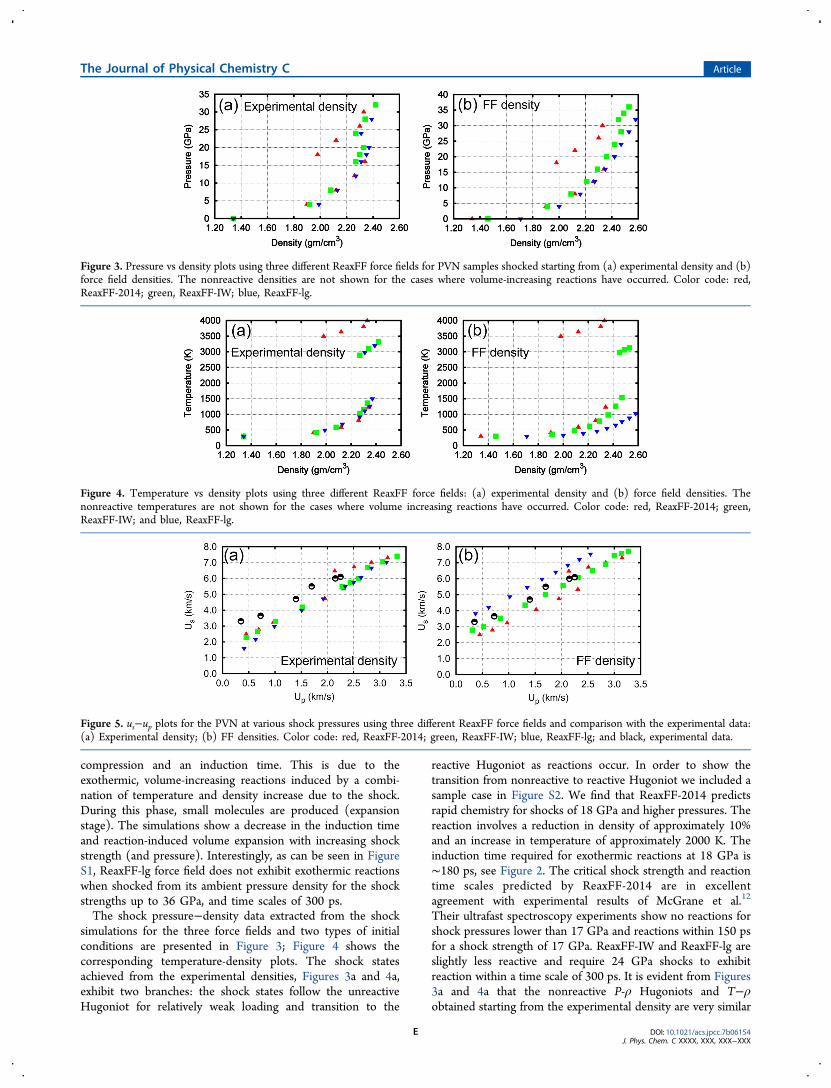
simulations for the three force fields and two types of initialconditions are presented in Figure 3; Figure 4 shows thecorresponding temperature-density plots. The shock statesachieved from the experimental densities, Figures 3a and 4a,exhibit two branches: the shock states follow the unreactiveHugoniot for relatively weak loading and transition to the
reactive Hugoniot as reactions occur. In order to show thetransition from nonreactive to reactive Hugoniot we included asample case in Figure S2. We find that ReaxFF-2014 predictsrapid chemistry for shocks of 18 GPa and higher pressures. Thereaction involves a reduction in density of approximately 10%and an increase in temperature of approximately 2000 K. Theinduction time required for exothermic reactions at 18 GPa is∼180 ps, see Figure 2. The critical shock strength and reactiontime scales predicted by ReaxFF-2014 are in excellentagreement with experimental results of McGrane et al.12
Their ultrafast spectroscopy experiments show no reactions forshock pressures lower than 17 GPa and reactions within 150 psfor a shock strength of 17 GPa. ReaxFF-IW and ReaxFF-lg areslightly less reactive and require 24 GPa shocks to exhibitreaction within a time scale of 300 ps. It is evident from Figures3a and 4a that the nonreactive P-ρ Hugoniots and T−ρobtained starting from the experimental density are very similar
Figure 3. Pressure vs density plots using three different ReaxFF force fields for PVN samples shocked starting from (a) experimental density and (b)force field densities. The nonreactive densities are not shown for the cases where volume-increasing reactions have occurred. Color code: red,ReaxFF-2014; green, ReaxFF-IW; blue, ReaxFF-lg.
Figure 4. Temperature vs density plots using three different ReaxFF force fields: (a) experimental density and (b) force field densities. Thenonreactive temperatures are not shown for the cases where volume increasing reactions have occurred. Color code: red, ReaxFF-2014; green,ReaxFF-IW; and blue, ReaxFF-lg.
Figure 5. us−up plots for the PVN at various shock pressures using three different ReaxFF force fields and comparison with the experimental data:(a) Experimental density; (b) FF densities. Color code: red, ReaxFF-2014; green, ReaxFF-IW; blue, ReaxFF-lg; and black, experimental data.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
E
for all the force fields. Interestingly, the more sensitive ReaxFF-2014 exhibits a higher exothermicity than the other two forcefields.Shock simulations starting from the ambient pressure
conditions for each force field exhibit larger discrepanciesamong each other; see Figures 3b and 4b. The ReaxFF-2014and -IW force fields predicted volume-increasing reactions andpressures of 18 and 30 GPa respectively, while no significantreactions occurred in the ReaxFF-lg simulations for shocks upto a pressure of 36 GPa. The lower reactivity in thesesimulations can be attributed to the smaller amount ofmechanical (PV) work done on the system during shockcompression. The equations of state in Figure 3b show asignificant difference in density for low pressures among thethree force fields, but this difference reduces with compression.Thus, the overestimation of density in the ReaxFF-IW and -lgforce fields resulted in a smaller amount of PV work andheating of the system as compared with the ReaxFF-2014simulations. As such the first rapid chemistry observed in theReaxFF-IW force field simulations is at a higher pressure of 30GPa compared to the 18 GPa in the case of ReaxFF-2014 forcefield.III.B. Shock Velocity−Particle Velocity Hugoniots. The
shock and particle velocity was calculated using Hugoniotrelations (section II.C). The shock-vs-particle velocity data forthe experimental density simulations are shown in Figure 5atogether with the nonreactive experimental curve from ref 10.The exothermic reactions lead to a second branch of the us−upplots and a sudden increase in the shock velocities. However,the slope of the reactive us−up curve is lower, and upon increasein shock pressure, the reactive and extrapolation of theunreactive us−up curves approach each other. This behaviorwas observed in nitromethane shock experiments.7 Thepredicted us−up curves slightly underestimate the experimentalresults, but overall they exhibit reasonable agreement. The shiftin the us−up plots is indicative to the chemical reactions. Whilethe nonreactive us−up data are analogous for all the force fields,
the differences in the us−up plots at the reactive zone is due tothe fact that ReaxFF-2014 predict chemical reactions at 18 GPain contrast to 24 GPa for the other two force fields. However, atthe pressure of 24 GPa or higher, us−up values from all the forcefields are in close agreement. The ReaxFF-2014 predicts thetransition to fast chemical decomposition at up = 2.2 km/s andus = 6.5 km/s, while in ReaxFF-IW and -lg simulations it occursat approximately up = 2.7 km/s and us = 6.8 km/s. The soundspeed (value of us at up = 0) predicted by the ReaxFF-2014,ReaxFF-IW, and ReaxFF-lg force fields starting from theexperimental densities are 1.75 ± 0.05, 1.55 ± 0.05, and 1.05 ±0.15 km/s, respectively. These values are smaller comparedwith the experimental data10 of 3.2 ± 0.3 km/s. However, theFF density simulations predict a relatively higher sound speedvalues, that is, 2.16 ± 0.02 and 3.14 ± 0.04 km/s for ReaxFF-IW and ReaxFF-lg, respectively.The effect of initial systems densities is evident in the results
presented in Figure 5b. In the FF density simulations, differentforce fields exhibit a wider variation. The computed shock-particle velocities follow the trend of densities predicted byeach of the force fields. The higher density resulted in astronger shock velocities. Therefore, the highest shockvelocities are predicted by the ReaxFF-lg force field, consistentwith the highest density predicted by this force field. The tworegimes in the us−up plots have been observed for ReaxFF-2014and ReaxFF-IW force fields. The ReaxFF-IW force fieldsimulations, the volume-increasing reactions occurred at up =3.0 km/s and us = 7.5 km/s. Since there is no experimental orcomputational data available for the reactive regimes of theshocked PVN polymer, we cannot make any comparison.
III.C. Shock-Induced Chemical Decomposition. Theprocesses of shock-induced decomposition of PVN can becategorized into three steps: (i) initial nonreactive compressionand heating when energy transfers from the shock wave to thematerial and (ii) a shock-induced onset of chemical reactions(iii) followed by rapid exothermic reactions. Therefore,investigation of chemical decomposition mechanisms is
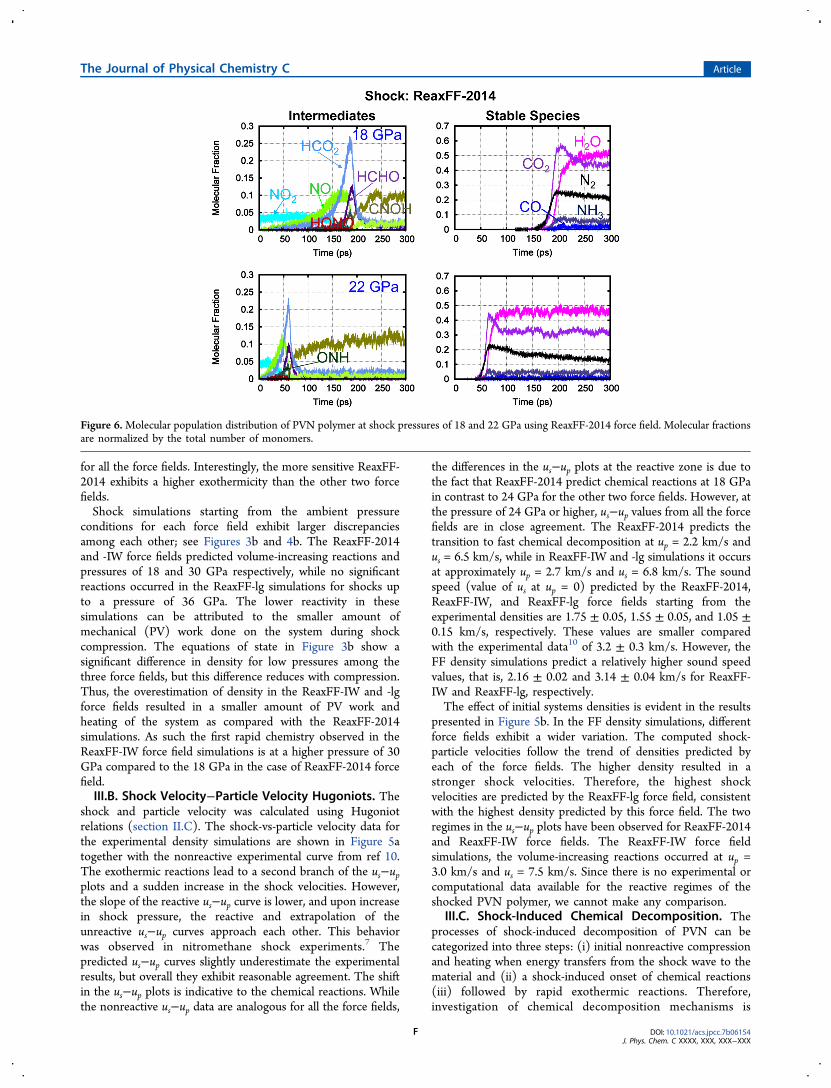
Figure 6.Molecular population distribution of PVN polymer at shock pressures of 18 and 22 GPa using ReaxFF-2014 force field. Molecular fractionsare normalized by the total number of monomers.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
F
required to elucidate the coupling between mechanical loadingand chemistry. The decomposition pathways are contingentupon the nature of the applied load, such as temperature andpressure conditions. With increasing shock strength, a largeramount of energy will be transferred to vibrational degrees offreedom of the molecules. If this results in sufficiently hightemperature, chemical reactions will be induced within shorttime scales.45 In this study, we are primarily interested inprobing the initial chemical reactions and subsequent pathwaystoward stable species formation.Possible reaction initiation mechanisms of PVN are NO2
fission, HONO elimination, C−C bond breaking, and hydrogen
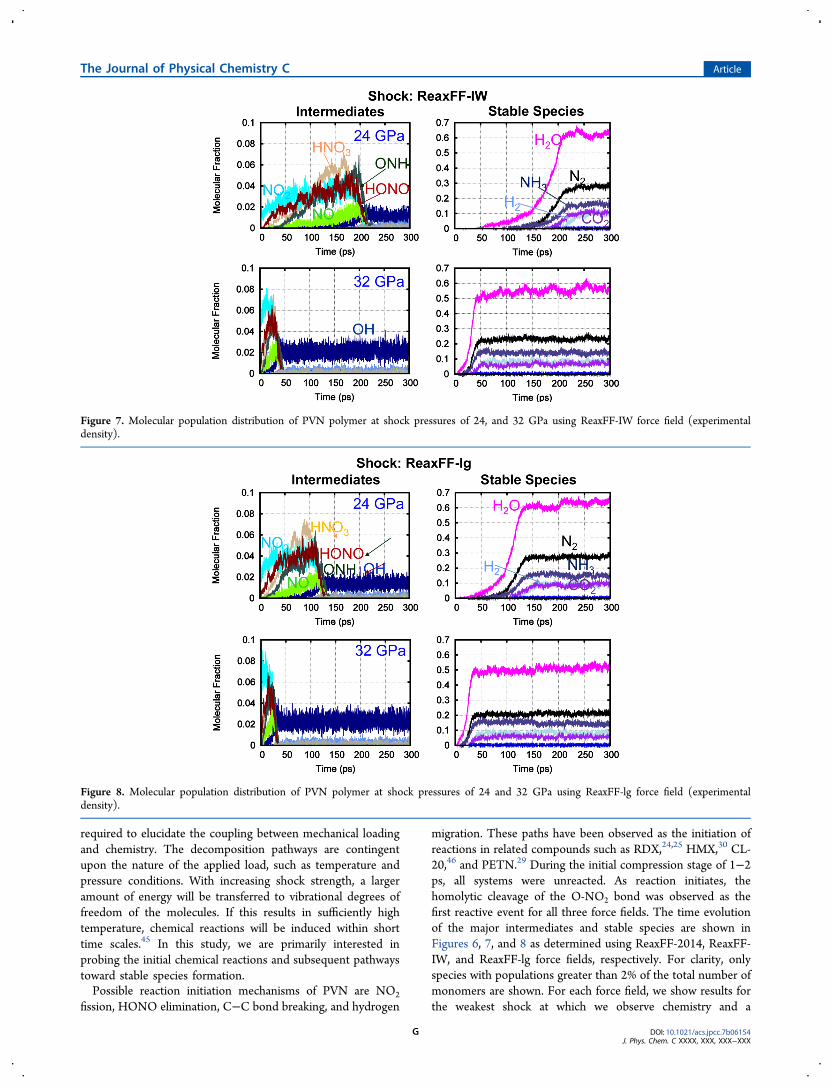
migration. These paths have been observed as the initiation ofreactions in related compounds such as RDX,24,25 HMX,30 CL-20,46 and PETN.29 During the initial compression stage of 1−2ps, all systems were unreacted. As reaction initiates, thehomolytic cleavage of the O-NO2 bond was observed as thefirst reactive event for all three force fields. The time evolutionof the major intermediates and stable species are shown inFigures 6, 7, and 8 as determined using ReaxFF-2014, ReaxFF-IW, and ReaxFF-lg force fields, respectively. For clarity, onlyspecies with populations greater than 2% of the total number ofmonomers are shown. For each force field, we show results forthe weakest shock at which we observe chemistry and a
Figure 7. Molecular population distribution of PVN polymer at shock pressures of 24, and 32 GPa using ReaxFF-IW force field (experimentaldensity).
Figure 8. Molecular population distribution of PVN polymer at shock pressures of 24 and 32 GPa using ReaxFF-lg force field (experimentaldensity).
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
G

stronger shock to highlight how pressure affects chemicaldecomposition. The rapid exothermic chemistry leads to thegeneration of final stable gaseous products, most notably H2O,CO2, N2, and NH3 for all the force fields.As expected, ReaxFF-IW and ReaxFF-lg that only differ in
their description of van der Waals interactions result in verysimilar chemical reactions. The final population of products issimilar in all three force fields; the main difference being thehigher population of CO2 in ReaxFF-2014. In addition, weobserve a significant amount of H2 formation in the ReaxFF-IWand -lg simulations. H2O was found as the most abundantspecies irrespective of simulation conditions and force fieldused. Unfortunately, we are not aware of any experimental dataon the distribution of final products of shocked PVN availablefor the comparison.The evolution of intermediate populations can shed light on
the reaction mechanisms and also explain differences in therelative amount of final products predicted by each of the forcefields. We observe that the primary intermediate moleculesreach a maximum and then react away to form final, exothermicproducts. The reaction initiation yields a significant amount ofNO2 and creates radical sites, followed by the generation ofother intermediates. In ReaxFF-2014, at 18 GPa shock pressure,the NO population increases steadily up to ∼180 ps, thensuddenly drops at the onset of rapid exothermic chemistry. Thepopulation of the most dominant intermediate, HCO2,increases to 25% of the total number of monomers. CH2O isgenerated at a relatively later stage. However, the decline of theboth HCO2 and CH2O occurs concurrently at the transition-to-fast exothermic reactions. ONH radicals are observed at timescales similar to CH2O, but in a lower quantity. The formationof the radicals proceeded via breaking of the backbone C−Cbond of the PVN polymer. The rapid chemistry leads to thegeneration of CNOH, and N2O2 intermediates and stable gasesare formed at a pronounced rate. At the onset point of rapidchemistry, ∼80% of the total C−C bonds of the PVN chainsare dissociated. In contrast to ReaxFF-2014, a large quantity ofHNO3, and HONO are produced in the ReaxFF-IW and -lgsimulations. HONO elimination has been observed in theprevious experiment,47 QM,16,17 and ReaxFF18,30,40 studies onenergetic materials.To better understand the population of products and
intermediate evolution for the three descriptions, we computedtheir formation energy using the three force fields andcompared them to experiments in Figure 9. ReaxFF-2014over stabilizes HCO2, CH2O, and CNOH radicals, compared tothe other two force fields and experimental data. Thisstabilization leads to the production of these species in largerquantities, while they are almost absent in other two versions.Similarly, the high stability of HNO3 predicted by ReaxFF-IWand -lg force fields causes it to be one of the most dominantintermediate species (see Figures 7 and 8). The formation ofHONO is endothermic in ReaxFF-2014: as such, this pathwayhas rarely been observed in ReaxFF-2014 simulations. Thus,our simulations show that the stability of intermediates andreaction paths have a direct effect on the final products, at leastduring the initial hundreds of picoseconds during decom-position. The major difference in the population of finalproducts across various force fields is the amount of CO2 andNH3. ReaxFF-2014 correctly stabilizes CO2 with respect to theother force fields. The higher preponderance of CO2production in ReaxFF-2014 simulations can also be attributedto the dissociation of the HCO2 radical pathway. The
distribution of the final products shows that ∼20% of Catoms in ReaxFF-2014 are contained in small productmolecules; this number if only 5% in the ReaxFF-lg andReaxFF-IW force fields. This indicates that a significant amountof carbon remains as intermediate species or other finalproducts. We further investigated the other carbon containingspecies, and they can be presented under the general formulasof HCxOy (x = 1−4, y = 2−5), H2CxOy (x = 1−4, y = 2−6),and H3CxOyN (x = 1−2, y = 2−3). A list of these representativespecies are included in the Table S1. We note that in oursimulations we have not observed any carbon cluster formation.One can also see that the distribution of both intermediates
and stable species indicates that the reaction initiationmechanisms are relatively insensitive to the magnitude of theapplied shock. Overall, the kinetics of the exothermic reactionsare relatively slower, and the exothermic heat release is lower inReaxFF-IW and -lg simulations compared to ReaxFF-2014.
III.D. Spectral Changes During Shock Simulations.Although detailed reaction mechanisms are inaccessible inshock experiments, ultrafast spectroscopy provides importantinsight into some of the reactions under shock loading.Importantly, spectroscopy information can be extracted directlyfrom MD simulations as discussed in section II.E and this canprovide a direct comparison between simulation results andexperimental measurements. We aim to compare reactive MDsimulations with published and ongoing spectral analysis for adirect validation to our simulations and, at the same time, tohelp interpret spectral data to detect various reaction pathwaysand associated time scales. Thus, we performed spectral analysisusing atomistic velocities at various shock conditions and forthe three force fields. The predicted per element spectra of asingle polymer chain using ReaxFF-2014 is shown in theSupporting Information, Figure S3a. This enables the assign-ment of peaks to vibrational modes. In order to compare withthe laser shock experiments by McGrane et al.,12 we consideredchanges in the N−O stretching modes as a measure of chemicalreactions in the system. The ReaxFF-2014 predicts NO2symmetric and asymmetric stretching modes are at 800 and1900 cm−1, respectively. These values are shifted from theexperimental values of 1270 and 1624 cm−1.12 However, thediscrepancy between experimental and our calculated vibra-tional modes does not significantly influence our comparisonwith experimental observations regarding the time scale orcharacter of reaction initiation. The C−H stretching mode is ingood agreement with literature data.
Figure 9. Formation energies of key intermediate and stable species ascalculated using three different ReaxFF force fields and comparisonwith the experimental data48. In these calculations, reference states areconsidered as follows: (i) atomic energies are used for the formationenergies of N2, O2, and H2, and (ii) N2, H2, O2, and graphite are usedas references for all other cases.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
H
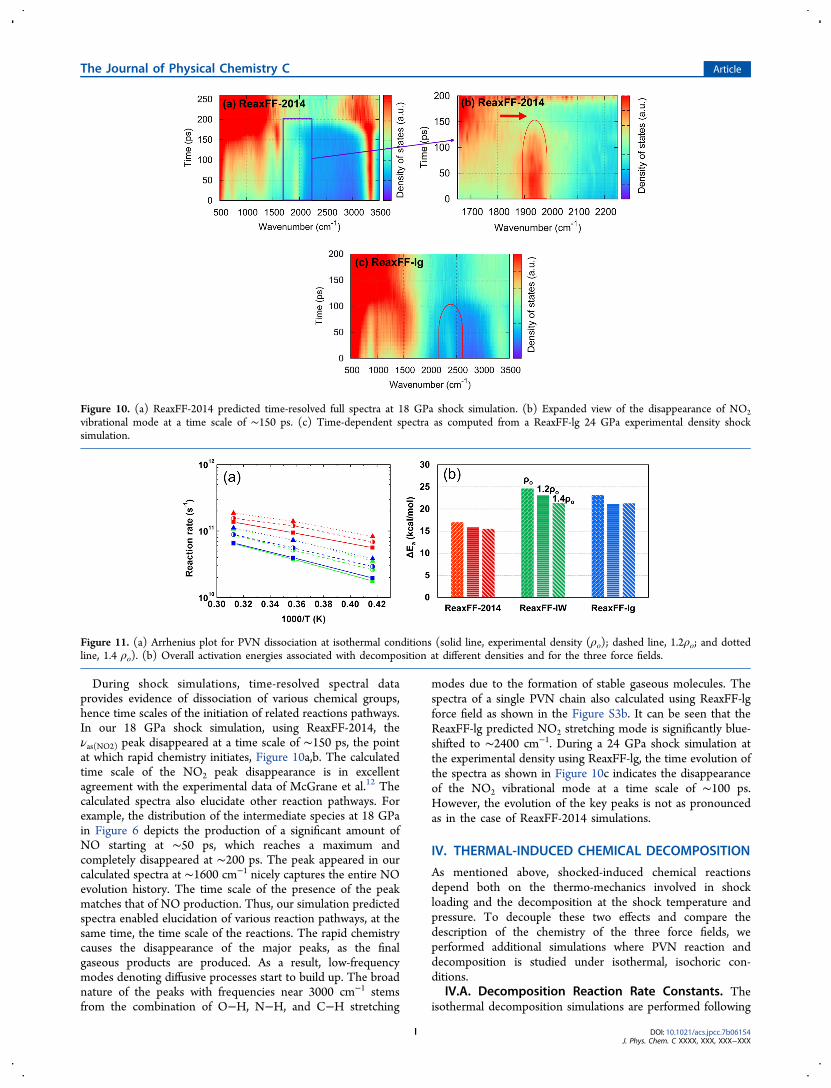
During shock simulations, time-resolved spectral dataprovides evidence of dissociation of various chemical groups,hence time scales of the initiation of related reactions pathways.In our 18 GPa shock simulation, using ReaxFF-2014, theνas(NO2) peak disappeared at a time scale of ∼150 ps, the pointat which rapid chemistry initiates, Figure 10a,b. The calculatedtime scale of the NO2 peak disappearance is in excellentagreement with the experimental data of McGrane et al.12 Thecalculated spectra also elucidate other reaction pathways. Forexample, the distribution of the intermediate species at 18 GPain Figure 6 depicts the production of a significant amount ofNO starting at ∼50 ps, which reaches a maximum andcompletely disappeared at ∼200 ps. The peak appeared in ourcalculated spectra at ∼1600 cm−1 nicely captures the entire NOevolution history. The time scale of the presence of the peakmatches that of NO production. Thus, our simulation predictedspectra enabled elucidation of various reaction pathways, at thesame time, the time scale of the reactions. The rapid chemistrycauses the disappearance of the major peaks, as the finalgaseous products are produced. As a result, low-frequencymodes denoting diffusive processes start to build up. The broadnature of the peaks with frequencies near 3000 cm−1 stemsfrom the combination of O−H, N−H, and C−H stretching
modes due to the formation of stable gaseous molecules. Thespectra of a single PVN chain also calculated using ReaxFF-lgforce field as shown in the Figure S3b. It can be seen that theReaxFF-lg predicted NO2 stretching mode is significantly blue-shifted to ∼2400 cm−1. During a 24 GPa shock simulation atthe experimental density using ReaxFF-lg, the time evolution ofthe spectra as shown in Figure 10c indicates the disappearanceof the NO2 vibrational mode at a time scale of ∼100 ps.However, the evolution of the key peaks is not as pronouncedas in the case of ReaxFF-2014 simulations.
IV. THERMAL-INDUCED CHEMICAL DECOMPOSITION
As mentioned above, shocked-induced chemical reactionsdepend both on the thermo-mechanics involved in shockloading and the decomposition at the shock temperature andpressure. To decouple these two effects and compare thedescription of the chemistry of the three force fields, weperformed additional simulations where PVN reaction anddecomposition is studied under isothermal, isochoric con-ditions.
IV.A. Decomposition Reaction Rate Constants. Theisothermal decomposition simulations are performed following
Figure 10. (a) ReaxFF-2014 predicted time-resolved full spectra at 18 GPa shock simulation. (b) Expanded view of the disappearance of NO2vibrational mode at a time scale of ∼150 ps. (c) Time-dependent spectra as computed from a ReaxFF-lg 24 GPa experimental density shocksimulation.
Figure 11. (a) Arrhenius plot for PVN dissociation at isothermal conditions (solid line, experimental density (ρο); dashed line, 1.2ρο; and dottedline, 1.4 ρο). (b) Overall activation energies associated with decomposition at different densities and for the three force fields.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
I
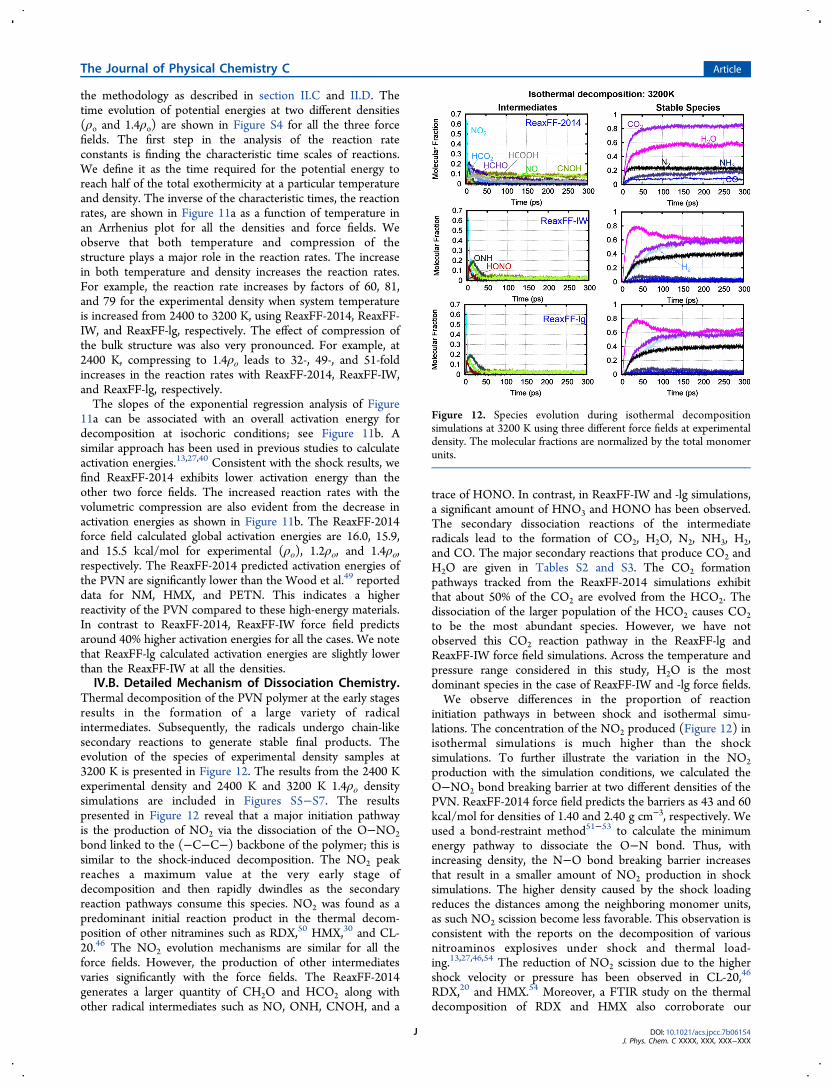
the methodology as described in section II.C and II.D. Thetime evolution of potential energies at two different densities(ρo and 1.4ρo) are shown in Figure S4 for all the three forcefields. The first step in the analysis of the reaction rateconstants is finding the characteristic time scales of reactions.We define it as the time required for the potential energy toreach half of the total exothermicity at a particular temperatureand density. The inverse of the characteristic times, the reactionrates, are shown in Figure 11a as a function of temperature inan Arrhenius plot for all the densities and force fields. Weobserve that both temperature and compression of thestructure plays a major role in the reaction rates. The increasein both temperature and density increases the reaction rates.For example, the reaction rate increases by factors of 60, 81,and 79 for the experimental density when system temperatureis increased from 2400 to 3200 K, using ReaxFF-2014, ReaxFF-IW, and ReaxFF-lg, respectively. The effect of compression ofthe bulk structure was also very pronounced. For example, at2400 K, compressing to 1.4ρο leads to 32-, 49-, and 51-foldincreases in the reaction rates with ReaxFF-2014, ReaxFF-IW,and ReaxFF-lg, respectively.The slopes of the exponential regression analysis of Figure
11a can be associated with an overall activation energy fordecomposition at isochoric conditions; see Figure 11b. Asimilar approach has been used in previous studies to calculateactivation energies.13,27,40 Consistent with the shock results, wefind ReaxFF-2014 exhibits lower activation energy than theother two force fields. The increased reaction rates with thevolumetric compression are also evident from the decrease inactivation energies as shown in Figure 11b. The ReaxFF-2014force field calculated global activation energies are 16.0, 15.9,and 15.5 kcal/mol for experimental (ρο), 1.2ρο, and 1.4ρο,respectively. The ReaxFF-2014 predicted activation energies ofthe PVN are significantly lower than the Wood et al.49 reporteddata for NM, HMX, and PETN. This indicates a higherreactivity of the PVN compared to these high-energy materials.In contrast to ReaxFF-2014, ReaxFF-IW force field predictsaround 40% higher activation energies for all the cases. We notethat ReaxFF-lg calculated activation energies are slightly lowerthan the ReaxFF-IW at all the densities.IV.B. Detailed Mechanism of Dissociation Chemistry.
Thermal decomposition of the PVN polymer at the early stagesresults in the formation of a large variety of radicalintermediates. Subsequently, the radicals undergo chain-likesecondary reactions to generate stable final products. Theevolution of the species of experimental density samples at3200 K is presented in Figure 12. The results from the 2400 Kexperimental density and 2400 K and 3200 K 1.4ρο densitysimulations are included in Figures S5−S7. The resultspresented in Figure 12 reveal that a major initiation pathwayis the production of NO2 via the dissociation of the O−NO2bond linked to the (−C−C−) backbone of the polymer; this issimilar to the shock-induced decomposition. The NO2 peakreaches a maximum value at the very early stage ofdecomposition and then rapidly dwindles as the secondaryreaction pathways consume this species. NO2 was found as apredominant initial reaction product in the thermal decom-position of other nitramines such as RDX,50 HMX,30 and CL-20.46 The NO2 evolution mechanisms are similar for all theforce fields. However, the production of other intermediatesvaries significantly with the force fields. The ReaxFF-2014generates a larger quantity of CH2O and HCO2 along withother radical intermediates such as NO, ONH, CNOH, and a
trace of HONO. In contrast, in ReaxFF-IW and -lg simulations,a significant amount of HNO3 and HONO has been observed.The secondary dissociation reactions of the intermediateradicals lead to the formation of CO2, H2O, N2, NH3, H2,and CO. The major secondary reactions that produce CO2 andH2O are given in Tables S2 and S3. The CO2 formationpathways tracked from the ReaxFF-2014 simulations exhibitthat about 50% of the CO2 are evolved from the HCO2. Thedissociation of the larger population of the HCO2 causes CO2to be the most abundant species. However, we have notobserved this CO2 reaction pathway in the ReaxFF-lg andReaxFF-IW force field simulations. Across the temperature andpressure range considered in this study, H2O is the mostdominant species in the case of ReaxFF-IW and -lg force fields.We observe differences in the proportion of reaction
initiation pathways in between shock and isothermal simu-lations. The concentration of the NO2 produced (Figure 12) inisothermal simulations is much higher than the shocksimulations. To further illustrate the variation in the NO2production with the simulation conditions, we calculated theO−NO2 bond breaking barrier at two different densities of thePVN. ReaxFF-2014 force field predicts the barriers as 43 and 60kcal/mol for densities of 1.40 and 2.40 g cm−3, respectively. Weused a bond-restraint method51−53 to calculate the minimumenergy pathway to dissociate the O−N bond. Thus, withincreasing density, the N−O bond breaking barrier increasesthat result in a smaller amount of NO2 production in shocksimulations. The higher density caused by the shock loadingreduces the distances among the neighboring monomer units,as such NO2 scission become less favorable. This observation isconsistent with the reports on the decomposition of variousnitroaminos explosives under shock and thermal load-ing.13,27,46,54 The reduction of NO2 scission due to the highershock velocity or pressure has been observed in CL-20,46
RDX,20 and HMX.54 Moreover, a FTIR study on the thermaldecomposition of RDX and HMX also corroborate our
Figure 12. Species evolution during isothermal decompositionsimulations at 3200 K using three different force fields at experimentaldensity. The molecular fractions are normalized by the total monomerunits.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
J

observation on the effect of pressure. It was found that anincrease in system pressure reduces NO2 scission.
50 To furtherunderstand the effect of shock vs thermal loading, weperformed isothermal simulations at equivalent thermodynam-ics conditions such as temperature, and density of the final stateof the shocked material. The comparison of the speciesevolution in the cases of equivalent shock and thermal loadingconditions are shown in Figures S8−S10 and a summary of thedistribution of the final gaseous products are presented inFigure S11. The details of the thermodynamic conditions aregiven in the captions of Figures S8−S10. From these figures, itis evident that the nature of the loading has a significantinfluence on the evolution of the intermediate species, as suchreaction pathways. We observe a pronounced effect ofmechano-chemistry due to the shock loading on the initialreaction pathways. The shock loading resulted in a decrease inthe amount of the intermediatesthose are evolved in areasonable quantitycompared to the thermal loading for allthe force fields. This implies that the PVN dissociates intomany smaller fragments, the individual quantities of thefragments were small thus we ignored them in the populationanalysis. However, despite the variation in the amount of theintermediates, the distribution of final gaseous products isalmost identical for both the simulation cases (see Figure S11).We observe that higher compression and temperature alters
the reaction pathways and final products distribution in theisothermal simulations. The increase in density reduces theamount of NO2 production for all the cases. In ReaxFF-2014simulations, the amount of CHNO radicals produced increaseswith the system pressure. At 3200 K simulations, ReaxFF-2014resulted in a reduction of CH2O and HCO2 and a substantialincrease in NO and H2O population. At 2400 K, irrespective ofthe system densities, the amount of CO2 produced is notablyhigher than the other stable products; however, with theincreasing temperature and density such as at 3200 K and 1.4ρo(see Figure S6), this dominance dwindles, and CO2 and H2Oare generated in a comparable quantity. This is due to thereduction of CO2 and an increase of H2O at highertemperatures. In contrary, higher temperature decreases theH2O population in both ReaxFF-IW and -lg simulations;however, the effect is less discernible at higher densities. Atexperimental density ReaxFF-IW and -lg simulations, CO2,H2O, and H2 reach at similar asymptotic quantities at 3200 K asa result of the reduced amount of H2O population. Thequantity of the H2 production exhibits an increasing trend withthe increasing system temperature. We note that H2 formationpathways are absent in ReaxFF-2014 force field simulations.One can also observe that the amount of N2 produced isinsensitive to the change in temperature and densities.
V. CONCLUSIONSIn conclusion, the first atomistic study of the chemicaldecomposition of PVN polymer under shock and thermalloading conditions provides a detailed description of mechan-ical, chemical, and spectroscopic properties. The predictedshock vs particle velocity Hugoniots using ReaxFF are inreasonable agreement with available experimental data. Moreimportantly, this study enabled the first direct comparisonbetween reactive MD simulations and experiments for thedecomposition of a material under shock loading. We foundthat ReaxFF-2014 predicts the onset of chemical reactions andthe time scale associated with the formation of NO2 in excellentagreement with shock experiments. The ReaxFF-IW and -lg
versions required stronger shocks for initiation. The combina-tion of reactive MD simulations and ultrafast spectroscopyopens the possibility of both the validation of ReaxFF atextreme conditions and can contribute to the interpretation ofthe experimental data relating changes in spectral features toatomic processes. Unfortunately, the narrow spectral region ofexisting experiments only enables a partial validation of thepredictions. Broad spectral analysis of laser-shocks, currentlyunder development, will enable a more complete validation ofatomistic simulations and a detailed understanding of chemicaldecomposition paths.Our investigation of the reaction initiation pathways reveals a
significant dependency on the loading type. NO2 formation ispredominantly found as a reaction initiation step, however, therelative amount of the species is contingent on the nature of theapplied load. The shock and volumetric compression results inthe reduction of NO2 production, consistent with observationsin other high energy explosives. The final product populationspredicted by the three force fields are similar, the maindifference being a higher population of CO2 in ReaxFF-2014.This can be attributed to the, correct, relative stability of thismolecule, see Figure 9, but also by the overstabilization of theHCO2 intermediate by ReaxFF-2014. While the comparisonexperimental spectroscopic studies with the MD simulationsindicates that ReaxFF-2014 provides a more accuratedescription of threshold for chemical initiation and its timescales, the lack of broadband data does not enable acomprehensive validation of the three force fields and thedetailed chemistry they predict. We expect such comparisons tobe possible in the near future.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jpcc.7b06154.
Further details of the temporal evolution of thermody-namics quantities during FF density shock simulations,per element spectra of a single polymer chain, evolutionof potential energies, and species in isothermaldecompositions (PDF)
■ AUTHOR INFORMATIONCorresponding Author*(A.S.) E-mail: [email protected] Mahbubul Islam: 0000-0003-4584-2204NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was support by the US Office of Naval Research,Multidisciplinary University Research Initiatives (MURI)Program, Contract N00014-16-1-2557. Program managers:Clifford Bedford and Kenny Lipkowitz.
■ REFERENCES(1) Tarver, C. M.; Chidester, S. K.; Nichols, A. L. Critical Conditionsfor Impact- and Shock-Induced Hot Spots in Solid Explosives. J. Phys.Chem. 1996, 100, 5794−5799.(2) McGuire, R. R.; Tarver, C. M. Chemical-Decomposition Models forthe Thermal Explosion of Confined Hmx, Tatb, Rdx, and Tnt Explosives;
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
K

UCRL-84986; CONF-810602-15; Lawrence Livermore National Lab.:Livermore, CA, 1981.(3) Dlott, D. D.; Fayer, M. D. Shocked Molecular Solids: Vibrationalup Pumping, Defect Hot Spot Formation, and the Onset of Chemistry.J. Chem. Phys. 1990, 92, 3798−3812.(4) Wen, X.; Tolbert, W. A.; Dlott, D. D. Multiphonon up-Pumpingand Molecular Hot Spots in Superheated Polymers Studied byUltrafast Optical Calorimetry. Chem. Phys. Lett. 1992, 192, 315−320.(5) Kim, H.; Dlott, D. D. Theory of Ultrahot Molecular Solids:Vibrational Cooling and Shock-induced Multiphonon up Pumping inCrystalline Naphthalene. J. Chem. Phys. 1990, 93, 1695−1709.(6) Moore, D. S.; McGrane, S. D. Comparative Infrared and RamanSpectroscopy of Energetic Polymers. J. Mol. Struct. 2003, 661−662,561−566.(7) Brown, K. E.; McGrane, S. D.; Bolme, C. A.; Moore, D. S.Ultrafast Chemical Reactions in Shocked Nitromethane Probed withDynamic Ellipsometry and Transient Absorption Spectroscopy. J. Phys.Chem. A 2014, 118, 2559−2567.(8) Dlott, D. D. New Developments in the Physical Chemistry ofShock Compression. Annu. Rev. Phys. Chem. 2011, 62, 575−597.(9) Bassett, W. P.; Dlott, D. D. High Dynamic Range EmissionMeasurements of Shocked Energetic Materials: Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine (HMX). J. Appl. Phys. 2016, 119,225103.(10) Moore, D. S.; McGrane, S. D.; Funk, D. J. UltrafastSpectroscopic Investigation of Shock Compressed Energetic PolymerFilms. AIP Conf. Proc. 2003, 706, 1285−1288.(11) Patterson, J. E.; Lagutchev, A.; Huang, W.; Dlott, D. D. UltrafastDynamics of Shock Compression of Molecular Monolayers. Phys. Rev.Lett. 2005, 94, 015501.(12) McGrane, S. D.; Moore, D. S.; Funk, D. J. Shock InducedReaction Observed via Ultrafast Infrared Absorption in Poly(VinylNitrate) Films. J. Phys. Chem. A 2004, 108, 9342−9347.(13) Rom, N.; Hirshberg, B.; Zeiri, Y.; Furman, D.; Zybin, S. V.;Goddard, W. A.; Kosloff, R. First-Principles-Based Reaction Kineticsfor Decomposition of Hot, Dense Liquid TNT from ReaxFFMultiscale Reactive Dynamics Simulations. J. Phys. Chem. C 2013,117, 21043−21054.(14) Shan, T.-R.; Wixom, R. R.; Mattsson, A. E.; Thompson, A. P.Atomistic Simulation of Orientation Dependence in Shock-InducedInitiation of Pentaerythritol Tetranitrate. J. Phys. Chem. B 2013, 117,928−936.(15) Wood, M. A.; van Duin, A. C.; Strachan, A. Coupled Thermaland Electromagnetic Induced Decomposition in the MolecularExplosive αHMX; A Reactive Molecular Dynamics Study. J. Phys.Chem. A 2014, 118, 885−895.(16) Schweigert, I. V. Quantum Mechanical Simulations ofCondensed-Phase Decomposition Dynamics in Molten RDX. J. Phys.Conf. Ser. 2014, 500, 052039.(17) Schweigert, I. V. Ab Initio Molecular Dynamics of High-Temperature Unimolecular Dissociation of Gas-Phase RDX and ItsDissociation Products. J. Phys. Chem. A 2015, 119, 2747−2759.(18) Joshi, K. L.; Chaudhuri, S. Reactive Simulation of the Chemistrybehind the Condensed-Phase Ignition of RDX from Hot Spots. Phys.Chem. Chem. Phys. 2015, 17, 18790−18801.(19) Riad Manaa, M.; Reed, E. J.; et al. Early Chemistry in Hot andDense Nitromethane: Molecular Dynamics Simulations. J. Chem. Phys.2004, 120, 10146−10153.(20) Strachan, A.; van Duin, A. C. T.; Chakraborty, D.; Dasgupta, S.;Goddard, W. A. Shock Waves in High-Energy Materials: The InitialChemical Events in Nitramine RDX. Phys. Rev. Lett. 2003, 91, 098301.(21) Van Duin, A. C.; Dasgupta, S.; Lorant, F.; Goddard, W. A.ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A2001, 105, 9396−9409.(22) Chenoweth, K.; van Duin, A. C. T.; Goddard, W. A. ReaxFFReactive Force Field for Molecular Dynamics Simulations ofHydrocarbon Oxidation. J. Phys. Chem. A 2008, 112, 1040−1053.(23) Senftle, T. P.; Hong, S.; Islam, M. M.; Kylasa, S. B.; Zheng, Y.;Shin, Y. K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M. J.; Aktulga,
H. M.; et al. The ReaxFF Reactive Force-Field: Development,Applications and Future Directions. Npj Comput. Mater. 2016, 2,15011.(24) Wood, M. A.; Cherukara, M. J.; Kober, E. M.; Strachan, A.Ultrafast Chemistry under Nonequilibrium Conditions and the Shockto Deflagration Transition at the Nanoscale. J. Phys. Chem. C 2015,119, 22008−22015.(25) Nomura, K.; Kalia, R. K.; Nakano, A.; Vashishta, P.; van Duin, A.C. T.; Goddard, W. A. Dynamic Transition in the Structure of anEnergetic Crystal during Chemical Reactions at Shock Front Prior toDetonation. Phys. Rev. Lett. 2007, 99, 148303.(26) Li, Y.; Vashishta, P.; et al. Multistage Reaction Pathways inDetonating High Explosives. Appl. Phys. Lett. 2014, 105, 204103.(27) Rom, N.; Zybin, S. V.; van Duin, A. C. T.; Goddard, W. A.;Zeiri, Y.; Katz, G.; Kosloff, R. Density-Dependent Liquid Nitro-methane Decomposition: Molecular Dynamics Simulations Based onReaxFF. J. Phys. Chem. A 2011, 115, 10181−10202.(28) Liu, L.; Liu, Y.; Zybin, S. V.; Sun, H.; Goddard, W. A. ReaxFF-Lg: Correction of the ReaxFF Reactive Force Field for LondonDispersion, with Applications to the Equations of State for EnergeticMaterials. J. Phys. Chem. A 2011, 115, 11016−11022.(29) Budzien, J.; Thompson, A. P.; Zybin, S. V. Reactive MolecularDynamics Simulations of Shock Through a Single Crystal ofPentaerythritol Tetranitrate. J. Phys. Chem. B 2009, 113, 13142−13151.(30) Zhou, T.; Song, H.; Liu, Y.; Huang, F. Shock Initiated Thermaland Chemical Responses of HMX Crystal from ReaxFF MolecularDynamics Simulation. Phys. Chem. Chem. Phys. 2014, 16, 13914−13931.(31) Tersoff, J. Empirical Interatomic Potential for Carbon, withApplications to Amorphous Carbon. Phys. Rev. Lett. 1988, 61, 2879−2882.(32) Brenner, D. W. Empirical Potential for Hydrocarbons for Use inSimulating the Chemical Vapor Deposition of Diamond Films. Phys.Rev. B: Condens. Matter Mater. Phys. 1990, 42, 9458−9471.(33) Ashraf, C.; van Duin, A. C. T. Extension of the ReaxFFCombustion Force Field toward Syngas Combustion and InitialOxidation Kinetics. J. Phys. Chem. A 2017, 121, 1051−1068.(34) Islam, M. M.; Zou, C.; van Duin, A. C. T.; Raman, S.Interactions of Hydrogen with the Iron and Iron Carbide Interfaces: AReaxFF Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2016, 18,761−771.(35) Van Duin, A. C.; Strachan, A.; Stewman, S.; Zhang, Q.; Xu, X.;Goddard, W. A. ReaxFFSiO Reactive Force Field for Silicon andSilicon Oxide Systems. J. Phys. Chem. A 2003, 107, 3803−3811.(36) Islam, M. M.; Ostadhossein, A.; Borodin, O.; Yeates, A. T.;Tipton, W. W.; Hennig, R. G.; Kumar, N.; van Duin, A. C. T. van.ReaxFF Molecular Dynamics Simulations on Lithiated Sulfur CathodeMaterials. Phys. Chem. Chem. Phys. 2015, 17, 3383−3393.(37) Mortier, W. J.; Ghosh, S. K.; Shankar, S. Electronegativity-Equalization Method for the Calculation of Atomic Charges inMolecules. J. Am. Chem. Soc. 1986, 108, 4315−4320.(38) Russo, M. F., Jr.; van Duin, A. C. T. Atomistic-Scale Simulationsof Chemical Reactions: Bridging from Quantum Chemistry toEngineering. Nucl. Instrum. Methods Phys. Res., Sect. B 2011, 269,1549−1554.(39) Islam, M. M.; Kolesov, G.; Verstraelen, T.; Kaxiras, E.; van Duin,A. C. T. eReaxFF: A Pseudoclassical Treatment of Explicit Electronswithin Reactive Force Field Simulations. J. Chem. Theory Comput.2016, 12, 3463−3472.(40) Strachan, A.; Kober, E. M.; van Duin, A. C.; Oxgaard, J.;Goddard, W. A., III Thermal Decomposition of RDX from ReactiveMolecular Dynamics. J. Chem. Phys. 2005, 122, 054502.(41) Haley, B. P.; Li, C.; Wilson, N.; Jaramillo, E.; Strachan, A.Atomistic Simulations of Amorphous Polymers in the Cloud withPolymerModeler. ArXiv Prepr. ArXiv150303894, 2015.(42) Mayo, S. L.; Olafson, B. D.; Goddard, W. A. DREIDING: ageneric force field for molecular simulations. J. Phys. Chem. 1990, 94,8897−8909.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
L

(43) Ravelo, R.; Holian, B. L.; Germann, T. C.; Lomdahl, P. S.Constant-Stress Hugoniostat Method for Following the DynamicalEvolution of Shocked Matter. Phys. Rev. B: Condens. Matter Mater.Phys. 2004, 70, 014103.(44) Reed, E. J.; Fried, L. E.; Joannopoulos, J. D. A Method forTractable Dynamical Studies of Single and Double ShockCompression. Phys. Rev. Lett. 2003, 90, 235503.(45) Tokmakoff, A.; Fayer, M. D.; Dlott, D. D. Chemical ReactionInitiation and Hot-Spot Formation in Shocked Energetic MolecularMaterials. J. Phys. Chem. 1993, 97, 1901−1913.(46) Xue, X.; Wen, Y.; Zhang, C. Early Decay Mechanism of Shockedε-CL-20: A Molecular Dynamics Simulation Study. J. Phys. Chem. C2016, 120, 21169−21177.(47) Maharrey, S.; Behrens, R. Thermal Decomposition of EnergeticMaterials. 5. Reaction Processes of 1,3,5-Trinitrohexahydro-S-Triazinebelow Its Melting Point. J. Phys. Chem. A 2005, 109, 11236−11249.(48) NIST WebBook http://webbook.nist.gov/ (accessed Jun 7,2017).(49) Wood, M. A.; Strachan, A. Nonequilibrium Reaction Kinetics inMolecular Solids. J. Phys. Chem. C 2016, 120, 542−552.(50) Oyumi, Y.; Brill, T. B. Thermal Decomposition of EnergeticMaterials 3. A High-Rate, in Situ, FTIR Study of the Thermolysis ofRDX and HMX with Pressure and Heating Rate as Variables. Combust.Flame 1985, 62, 213−224.(51) Rahaman, O.; van Duin, A. C.; Goddard, W. A., III; Doren, D. J.Development of a ReaxFF Reactive Force Field for Glycine andApplication to Solvent Effect and Tautomerization. J. Phys. Chem. B2011, 115, 249−261.(52) van Duin, A. C. T.; Damste,́ J. S. S. Computational ChemicalInvestigation into Isorenieratene Cyclisation. Org. Geochem. 2003, 34,515−526.(53) Ostadhossein, A.; Cubuk, E. D.; Tritsaris, G. A.; Kaxiras, E.;Zhang, S.; van Duin, A. C. T. van. Stress Effects on the InitialLithiation of Crystalline Silicon Nanowires: Reactive MolecularDynamics Simulations Using ReaxFF. Phys. Chem. Chem. Phys. 2015,17, 3832−3840.(54) Ge, N.-N.; Wei, Y.-K.; Ji, G.-F.; Chen, X.-R.; Zhao, F.; Wei, D.-Q. Initial Decomposition of the Condensed-Phase β-HMX underShock Waves: Molecular Dynamics Simulations. J. Phys. Chem. B 2012,116, 13696−13704.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b06154J. Phys. Chem. C XXXX, XXX, XXX−XXX
M





























