Embed Size (px)
Citation preview

J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
ª 2 0 1 7 P U B L I S H E D B Y E L S E V I E R O N B E H A L F O F AM E R I C A N C O L L E G E O F
C A R D I O L O G Y F O U N D A T I O N . T H I S I S A N O P E N A C C E S S A R T I C L E U N D E R
T H E C C B Y - N C - N D L I C E N S E ( h t t p : / / c r e a t i v e c o mm o n s . o r g / l i c e n s e s / b y - n c - n d / 4 . 0 / ) .
I S S N 2 4 5 2 - 3 0 2 X
h t t p : / / d x . d o i . o r g / 1 0 . 1 0 1 6 / j . j a c b t s . 2 0 1 7 . 0 4 . 0 0 4
PRECLINICAL RESEARCH
Matrix Signaling Subsequent toa Myocardial InfarctionA Proteomic Profile of Tissue Factor Microparticles
Derrick Akpalu,a Gale Newman, PHD,a Mark Brice, PHD,a Mike Powell, PHD,a Rajesh Singh, PHD,a
Alexander Quarshie, MD,b Elizabeth Ofili, MD,b James Fonger, MD,c Nic Chronos, MD,d David Feldman, MD, PHDe,f
VISUAL ABSTRACT
F
R
LfD
ti
5
R
5
re
Akpalu, D. et al. J Am Coll Cardiol Basic Trans Science. 2017;2(5):529–42.
rom the aDepartment of Microbiology, Biochemistry and Immunology, Morehouse School of M
esearch Center, Morehouse School of Medicine, Atlanta, Georgia; cCardioScout LLC, Atlanta,
ake Oconee, Eatonton, Georgia; eDivision of Cardiology University of Cincinnati Medical
epartment of Cardiology, Inselspital Hospital, University of Bern, Switzerland. This work w
onal Institutes of Health (NIH) and National Institute on Minority Health and Health
RO1HL84498-5, NIH/NIMHD 2S21MD000101NIMHD 8U54MD007588-04; the Minority Bio
esearch Initiative for Scientific Advancement program 5R25GM058268; the Research
G12MD007602; and NIH/NHLBI contract grant number R21HL092358. All authors have repor
levant to the contents of this paper to disclose.
HIGHLIGHTS
� The occurrence of an MI activates
production of TFMPs.
� We induced an MI in Yucatan miniswine
and collected plasma samples over a
6-month period post-MI.
� Experimental groups consisted of
infarcted but untreated animals and
infarcted animals treated with CRT plus
b-blocker.
� Using proteomic profiling, we confirm the
heterogeneity of TFMP protein content
with respect to physiological status of the
host temporally.
� Spatially, the contents of the TFMPs
provided information about multiple
entities supplemental to what we
obtained from assessing a set of
8 currently used cardiac biomarkers.
� The results from this study support
recommending TFMP protein content
profiling be used prospectively as a viable
investigative methodology for chronic
ischemic cardiomyopathy to help improve
our understanding of b-adrenergic
receptor signaling after an MI.
edicine, Atlanta, Georgia; bClinical
Georgia; dCardiology Care Clinic of
Center, Cincinnati, Ohio; and the
as supported by the following Na-
Disparities (NIMHD) grants: NIH
medical Research Support of the
Centers in Minority Institutions
ted that they have no relationships

ABBR EV I A T I ON S
AND ACRONYMS
ADRB1 = b1-adrenergic
receptor
ADRB2 = b2-adrenergic
receptor
AR = adrenergic receptor
ARRB1 = b1-arrestin
BB = b-blocker
cAMP = cyclic adenosine
monophosphate
CRT = cardiac
resynchronization therapy
EDV = end-diastolic volume
EF = ejection fraction
ELISA = enzyme-linked
immunosorbent assay
ESV = end-systolic volume
FACS = fluorescence-activated
cell sorting
GRK = G-protein receptor
kinase
HSP = heat shock protein
HUVEC = human umbilical vein
endothelial cell
LVAd MV = left ventricular
area around the mitral valve at
diastole
LVAs MV = left ventricular
area around the mitral valve at
systole
LVAd PM = left ventricular
area around the papillary
muscle at diastole
LVAs PM = left ventricular area
around the papillary muscle at
systole
MI = myocardial infarction
MP = microparticle
PCR = polymerase chain
reaction
TF = tissue factor
TFMP = tissue factor–bearing
microparticle
TnT = troponin T
All authors
institutions
visit the JA
Manuscript
Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
530
SUMMARY
at
an
CC
re
This study investigated the release and proteomic profile of tissue factor microparticles (TFMPs) prospectively
(up to 6 months) following a myocardial infarction (MI) in a chronic porcine model to establish their utility in
tracking cellular level activities that predict physiologic outcomes. Our animal groups (n ¼ 6 to 8 each)
consisted of control, noninfarcted (negative control); infarcted only (positive control); and infarcted animals
treated with cardiac resynchronization therapy (CRT) and a b-blocker (BB) (metoprolol succinate). The authors
found different protein profiles in TFMPs between the control, infarcted only group, and the CRT þ BB treated
group with predictive impact on the outward phenotype of pathological remodeling after an MI within and
between groups. This novel approach of monitoring cellular level activities by profiling the content of
TFMPs has the potential of addressing a shortfall of the current crop of cardiac biomarkers, which is the inability
to capture composite molecular changes associated with chronic maladaptive signaling in a spatial and temporal
manner. (J Am Coll Cardiol Basic Trans Science 2017;2:529–42) © 2017 Published by Elsevier on behalf of
American College of Cardiology Foundation. This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
A dvances in diagnosis and manage-ment of myocardial infarction (MI)have accounted for a decrease in
acute mortality fromMI (1,2). Much, however,remains to be understood about thecellular and molecular mechanisms of MIlongitudinally beyond the initial few daysand weeks.
Progressive chronic heart failure and thereduction in cardiac output after an MI causethe activation of neurohormonal responsesand perturbation in long-term adrenergicsignaling, which leads to changes in thesympathetic nervous system (3). b-adrenergicreceptor (AR) activation, in addition toincreasing acute cardiac performance, initi-ates multiple signaling cascades simulta-neously through G-protein receptor kinases(GRKs) and b-arrestin–mediated pathways.With an adaptive upregulation of GRK2, thereis a concordant increase in heart failurephenotype, in part mediated by the depletionof b-AR–mediated inotropic reserve (4–6).Additionally, chronic activation of the sym-pathetic nervous system leads to pathologicalremodeling, necrosis, and apoptosis (7). Two
important aspects in the treatment of chronic heartfailure with pathological remodeling include the use ofb-blockers (BBs) and cardiac resynchronization ther-apy (CRT) (4–6). Together these interventions improve
test they are in compliance with human studies committe
d Food and Drug Administration guidelines, including patien
: Basic to Translational Science author instructions page.
ceived November 17, 2016; revised manuscript received Febru
symptoms and enhance left ventricular function whileslowing down the progression of maladaptive remod-eling and improving morbidity and mortality inappropriately selected patients (4–6).
Previous investigations revealed an elevationof microparticle (MP) levels in patients with cardio-vascular diseases, specifically those with acutecoronary syndromes (8–11). MPs are small vesiclesreleased from the plasma membrane of cells such asplatelets, leukocytes, erythrocytes, endothelial cells,and muscle cells; they contain cell surface proteinsalong with cytoplasmic components of their cells oforigin (8,9,12–15). MPs produced as a result of humanatherosclerotic plaque formation possess tissue factor(TF) activity along with an outer membrane composedof phosphatidylserine for prohemostatic activity(8,9,11,12,16,17). In patients with various forms of car-diovascular disease, circulating MPs cause endothelialcell dysfunction (9,11,12,16,18–20) and act as a keydriver of atherosclerosis (9,12,13,15,21). In addition toensuring hemostasis, TF plays a cell signaling role bypromoting pleiotropic inflammatory responses. Hith-erto, the reports of the elevation of tissue factor (TF)microparticles (MPs) in patients have been of quanti-tative observations, with no studies describing theprotein content of TFMPs over the long term. This studyinvestigated the release and proteomic profile of TFMPsprospectively after an MI in a chronic porcine model.
The majority of the current models of post-MIsignaling are in smaller animal models with limited
es and animal welfare regulations of the authors’
t consent where appropriate. For more information,
ary 27, 2017, accepted April 4, 2017.

FIGURE 1 Study Schedule and Sample Collection Plan
CRT ¼ cardiac resynchronization therapy.
J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
531
duration. For a variety of reasons, investigatorstypically follow signaling for a few hours to severalweeks. Because humans usually do not developpathological remodeling or heart failure in days orweeks, the purpose of this study was to identifyadditional mechanisms involved in progressive heartfailure over time. An approach that enables theobservation of both spatial (membrane to the nu-cleus) and temporal signaling in a large animal modelwould enhance our understanding of the multifac-eted post-ischemic processes. Investigators haveestablished that signaling pathways seldom functionin isolation but are part of a matrix and involve theactivation of multiple pathways at different time in-tervals (22). Hence, we propose that evaluation ofdiscrete signaling pathways for a moment in timemight not adequately capture the complex signalingobserved in humans.
METHODS
ANIMAL MODEL OF MI. All animal studies wereapproved by the St. Joseph’s Translational ResearchInstitute and Morehouse School of Medicine Institu-tional Animal Care and Use Committee and complywith The Guide for the Care and Use of LaboratoryAnimals (8th edition, 2011). The animals were housedindividually and had access to food and water adlibitum to exceed all animal standards. Eight-month-old female Yucatan miniswine were infarcted by useof a collagen plug procedure (23). Two and a halfmonths after the infarct, animals were placed in 1 of 2experimental groups. Pigs in the first group (n ¼ 8)had a collagen plug–induced MI and survived butreceived no treatment, whereas pigs in the secondgroup (n ¼ 6) were MI survivors treated with CRT plusa BB (metoprolol). The third group of female controlpigs (n ¼ 8) had no infarction or treatments. Plasma
was collected at 2.5, 3.5, 4.5, and 6 months afterinfarction (Figure 1). All infarcted animals were killedat 6 months.
MI PROCEDURE. All animals were intubated, and theear or external jugular veinwas cannulated tomaintainadequate anesthesia throughout the procedure. Theanimals (weighing 45 to 55 kg each) were sedated withketamine combined with xylazine and maintainedon isoflurane (0% to 5%). Buprenorphine or morphinewas given perioperatively. Arterial pressure wasmonitored by accessing a femoral artery with anappropriately sized vascular introducer sheath.Heparin (150 U/kg) was administered, and 10 to 30 minlater, activated clotting time was checked to confirm itwas >300 s. An appropriately sized guiding catheterwas introduced and advanced to the ostium of the leftmain coronary artery, and contrast angiography wasperformed. An infusion catheter was advancedthrough the guiding catheter into the midportion ofthe left anterior descending coronary artery. At thediscretion of the operator, z2 ml of collagen wasinjected in 0.2-ml increments to occlude the majorityof the left anterior descending coronary artery.Additional buprenorphine was given for immediatepost-operative analgesia 1 to 2 times per day for 1 to5 days after infarction.
CRT AND BB THERAPY. Biventricular pacing deviceswere implanted as described previously (24). Briefly,pigs that had an MI were treated with CRT and BBtherapy (metoprolol succinate, Novartis, Basel,Switzerland) titrated to 2 mg/kg over several weeks,from 2.5 months post-infarction until termination at6 months. This dosing was intended to emulate thedosing used in MERIT-HF (Metoprolol CR/XL Ran-domized Intervention Trial in Congestive Heart Fail-ure) (25). To achieve a biventricular capture rate>95%, pacing was obtained by simultaneous left

Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
532
lateral wall (via coronary sinus) and right ventricularseptal wall pacing. Placement was ensured by fluo-roscopy at the time of placement, and fluoroscopywas used at post-operative 3 days and at 1 month toensure capture.
BLOOD SAMPLE COLLECTION, STORAGE, AND
NECROPSY. Blood samples (7 to 8 ml) were collectedinto ethylenediaminetetraacetic acid tubes (BDVacutainer Systems, Franklin Lakes, New Jersey).The plasma samples were centrifuged at 2,000 � g (20min) to remove any residual platelets, cell debris, andprecipitates. Platelet-poor plasma was stored frozenat �80�C in 1-ml aliquots until one-time use at anal-ysis. Plasma samples were subjected to no furtherfreeze-thaw cycles.
QUANTITATION ASSAYS (HIGH-SENSITIVITY
TROPONIN T, b1-AR, b2-AR, b-ARRESTIN-1, cAMP,
EPINEPHRINE, NOREPINEPHRINE, AND GRK2). Tomeasure b1-AR (ADRB1), b2-AR (ADRB2), b1-arrestin(ARRB1), and cyclic adenosine monophosphate(cAMP) levels, porcine hearts were harvested whenthe animals were killed 6 months after the infarction.Harvested cardiac tissue was rinsed with phosphate-buffered saline and quickly frozen in liquid nitro-gen. Frozen tissue was then placed in RNAlater RNAStabilization Reagent (Qiagen, Valencia, California)and stored at �80�C. Frozen ventricle samples werehomogenized in protein lysate buffer before beingcentrifuged for 20 min at 2,000 rpm. The supernatantwas assayed for ARRB1, ADRB1, ADRB2, and cAMPcontent by enzyme-linked immunosorbent assay(ELISA) (MyBioSource Inc., San Diego, California) inaccordance with the manufacturer’s protocol. Real-time polymerase chain reaction (PCR) was used tomeasure levels of GRK2 (Qiagen OneStep real-timePCR and HotStarTaq Plus Master Mix kit). Harvestedcardiac tissue samples were analyzed for fibrosis in-tensity using a commercially available trichromestain (Masson) kit following the assay proceduredetailed by Sigma-Aldrich (St. Louis, Missouri).
Plasma samples were collected and used to deter-mine the levels of troponin T (TnT) by ELISA (LifeSciences Advanced Technologies, Inc., St. Petersburg,Florida) following the manufacturer’s protocol. Con-centrations of epinephrine and norepinephrine weredetermined with plasma samples by ELISA at all 4time points post-infarction. (MyBioSource Inc.).
MP ISOLATION AND LABELING. Platelet-poor plasmawas centrifuged at 20,000 � g (30 min) to pellet theMPs. The MP pellet was washed by resuspension inHank’s balanced salt solution (Sigma-Aldrich). Thewashed pellet was incubated in 200 ml of bindingbuffer with saturating concentrations (20 ml) of
fluorescein isothiocyanate–conjugated annexin Vantibody (Clontech, Mountain View, California) andphycoerythrin-conjugated TF antibody (Abcam, Cam-bridge, Massachusetts) for 1 h on ice in the dark. Afterlabeling, the MPs were washed by resuspension withHank’s balanced salt solution to remove excess un-bound antibody by pelleting at 20,000 � g for 30 minand resuspending in 500 ml of Hank’s solution beforefluorescence-activated cell sorting (FACS) analysis.
FLOW CYTOMETRY SORTING OF MPs. Analysis ofTFMPs was performed with 2-color flow cytometry ona BD FACS Aria II equipped with the FACS DIVAsoftware, version 6.1.2 (BD Biosciences, San Jose,California). The MPs were analyzed with logarithmicmeasurements for forward scatter and side scatter.TFMPs were sorted based on double positivity for TFantigen and annexin V, which identifies TFMPs. Thesorted TFMPs were centrifuged at 100,000 � g torecover all MPs in the sample for proteomic analysis.
MASS SPECTROMETRY ANALYSIS. The samples wereacetone precipitated, rehydrated in trypsin digestbuffer (50 mmol/l ammonium bicarbonate), reducedin dithiothreitol 10 mmol/l at 56�C for 30 min, andalkylated with iodoacetic acid (15 mmol/l) for 30 minat room temperature in the dark. Samples weretrypsin digested 20 (ng/ml) for 4 h at 37�C, and beforeanalysis, they were acidified by formic acid to a con-centration of 0.1%. Spectra were collected withXcalibur 2.2 software (Thermo Fisher Scientific,Waltham, Massachusetts) using a threshold of 200counts/hits. Spectra were then searched with Prote-ome Discoverer 3.0 software (Thermo Fisher Scienti-fic). Each sample produced 1 or 2 significant hits asdetermined by false discovery rates of 1.0% and 5.0%using a corresponding reverse database.
Porcine peptide sequences detected were exportedfrom ProteoIQ software version 2.3.08 (NuSep, Inc.,Bogart, Georgia) into protein BLAST, Blastp (NationalCenter for Biotechnology Information) and werechecked against the human protein database todetermine porcine/human protein homologs. Onlyhuman proteins with >85% homology were used forthe analysis. This conversion was necessary becausePathway Studio (Elsevier, Atlanta, Georgia) only usesproteins from humans, mice, and rats in its analysis.The resultant homologous human protein Entrez IDnumbers were uploaded into Pathway Studio 10 forpathway analysis.
PATHWAY AND FUNCTIONAL ENRICHMENT ANALYSIS.
Pathway Studio version 10 (PS10) with Disease Fx andChem Fx cartridges was used to analyze the proteo-mic data between the different treatment groups andan analysis of cell processes, disease processes, and

J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
533
functional classes determined for the identified pro-teins. Functional enrichment analysis was conductedwith FunRich (26) version 2.1.2 (27).
STATISTICAL ANALYSIS. SigmaPlot version 12 wasused for the statistical analysis of MP counts, ELISA,and PCR data. Where nonparametric methods ofanalysis were applied, data are summarized usingmedian (1st quartile, 3rd quartile); otherwise data areexpressed as mean � SD. Comparisons were madeusing nonparametric tests, Kruskal-Wallis 1-wayanalysis of variance and Mann-Whitney rank sumtest, as appropriate. Dunn’s method was applied tothe probability values whenever multiple compari-sons arose. To compare TFMP count changes acrosstime within treatments groups, the Wilcoxonmatched-pairs signed ranks method was applied. The1-way repeated-measures analysis of variancemethod was applied to the TnT data with multiplepairwise comparison involving the Holm-Sidakmethod applied. All tests were 2-sided, withp < 0.05 considered significant.
RESULTS
HEMODYNAMIC, CARDIAC DIMENSION, AND
FIBROSIS INTENSITY OBSERVATIONS. Assessmentsof end-diastolic volume (EDV), end-systolic volume(ESV), and ejection fraction (EF) were made at base-line, 1 week post-infarction, and at study termination.Overall, after the MI, there were increases in EDV andESV with decreasing EF in both infarcted groups ofanimals. There were no statistically significant dif-ferences between groups at baseline or at 1 week post-infarction. Baseline EDVs for the CRTþBB–treatedanimals and for infarcted, untreated animals aver-aged 49.25 � 20.47 ml and 53.10 � 15.67 ml, respec-tively (p ¼ 0.722). Median ESV values for the treatedversus untreated animals were 17.86 ml (15.35 ml,21.223 ml) and 20.47 ml (12.49 ml, 25.43 ml), respec-tively (p ¼ 0.931). Baseline EF for CRTþBB–treatedversus infarcted, untreated animals averaged 59.78 �5.32% and 61.85 � 2.52%, respectively (p ¼ 0.448).
Post-infarction EDV values for CRTþBB–treatedversus infarcted, untreated animals averaged73.61 � 14.60 ml and 85.86 � 16.67 ml, respectively(p ¼ 0.205). At the same time point, ESV values forCRTþBB–treated versus infarcted, untreated animalsaveraged 50.67 � 11.96 ml versus 56.93 � 9.88 ml,respectively (p ¼ 0.375). The assessed EF for theCRTþBB–treated and infarcted, untreated groupsafter infarction averaged 27.22 � 5.18% and 24.15 �1.44%, respectively (p ¼ 0.238).
At termination of the study, there was a statisti-cally significant difference (p ¼ 0.046) in mean EDV
values between the 2 infarcted groups (CRTþBB 112 �35.35 ml vs. MI only 151.03 � 13.14 ml). At the sametime point, there was a noticeable trend of increasingESV in both groups; however, the increase in ESVvalues was comparatively more pronounced in theuntreated group than in the CRTþBB–treated animals.ESV values for the CRTþBB versus MI-only animals atthis time point were 70.78 � 28.03 ml versus 104.63 �18.95 ml (p ¼ 0.069). At study termination, EF valuesfor the CRTþBB–treated animals were relatively bet-ter than for the infarcted, untreated animals, with thedifference approaching statistical significance (p ¼0.0527) 3.5 months after treatment was initiated inthe treatment group. The values were 37.905 �6.036% versus 30.865 � 5.874% for the CRTþBB andMI-only groups, respectively.
Cardiac dimension measurements were conductedwith echocardiograms at baseline, 1 week post-infarction, and after necropsy. Predictably, after theinfarction, there was a noticeable increase in cardiacdimensions for all animals, as observed in left ven-tricular area around the mitral valve at diastole (LVAdMV) and systole (LVAs MV). Additionally, cardiacenlargement was observed in the area around thepapillary muscle at diastole (LVAd PM) and systole(LVAs PM). In all groups, there was an increase in thesecardiac measurements between the 1-week post-infarction assessment time point and study termina-tion. Comparison of the cardiac dimensions betweenthe 2 infarcted groups at study termination showed nostatistical differences with respect to LVAd MV, LVAsMV, and LVAs PM; however, there was a statisticallysignificant difference (p ¼ 0.009) in LVAd PM valuesbetween the CRTþBB–treated versus MI-only groups.
Extent of fibrosis in the harvested infarcted heartswas also determined on a scale of 1 to 4 (0 ¼ normal,1 ¼ minimal, 2 ¼ mild, 3 ¼ moderate, and 4 ¼ severe),normalized to samples from normal, uninfarctedporcine hearts. Seven cardiac regions were examined:right ventricle lateral wall; septum; border; infarct;normal; left atrium; and right atrium. Briefly, the re-sults revealed grade 3 to 4 fibrosis in the septum,border, and infarct regions of the hearts of bothinfarcted animal groups. In the right lateral wall, leftatrium, and right atrial regions of the harvested car-diac tissues, normal to mild fibrosis was observed.TFMP COUNTS. At 2.5 months post-infarction, therewas no significant difference in TFMP counts betweenthe experimental groups (Figure 2). The averageTFMP counts per 10,000 events at 2.5 months were1,806 � 423 for the control group, 1,281 � 909 for theinfarcted, untreated pigs, and 1,233 � 752 for theCRTþBB–treated group. Over the remaining timepoints, the counts of the infarcted, untreated animals

FIGURE 2 Flow Cytometric Enumeration of TFMP Counts per 10,000 Events
Tissue factor microparticles (TFMPs) increased in the infarcted animals over time, but
there were no statistically significant differences in the numbers of TFMPs.
BB ¼ b-blocker; CRT ¼ cardiac resynchronization therapy; MI ¼ myocardial infarction;
MP ¼ microparticles; TF ¼ tissue factor.
Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
534
continued to increase to 3,156 � 1,360, whereas thecounts of the treated animals tracked between 1,233 �752 and 2,173 � 1,405. Statistically, there were nosignificant differences in the numbers of MPs withinor between the groups at any time points.
TFMP PROTEOMIC PROFILE. Mass spectrometry re-sults showed that the proteomic content of TFMPschanged over time within and between groups. Forthe CRTþBB–treated animals, there were 177, 125, and137 proteins detected in the TFMPs collected at the3.5-, 4.5-, and 6-month time points, respectively. Inthe infarcted, untreated group, we observed 196, 207,and 65 proteins at the 3.5-, 4.5-, and 6-month timepoints, respectively. The control group animals had202 different proteins detected at the single timepoint assessed. The differences and similarities of theproteomic profiles were evaluated for functionalenrichment using the FunRich tool and the PathwayStudio software as further described in “MolecularFunction Analyses of TFMP proteins.”
On the basis of the sites of expression of theidentified proteins, TFMPs were determined to befrom heart muscle, plasma, blood vessels, and humanumbilical vein endothelial cells (HUVECs), as shownin Table 1. The observed high number of proteinsmapped to HUVECs is a consequence of the abun-dance of model systems involving HUVECs for theidentification of endothelial cell function in pub-lished literature (28–30). FunRich and other molecu-lar analysis tools rely on published literature for thegeneration of their output. This observation, howev-er, points to the relevance of these molecular entitiesin the post-MI state.
MOLECULAR FUNCTION ANALYSES OF TFMP
PROTEINS. To further characterize the identifiedTFMP proteins and to explore for temporal differ-ences, the standalone open-access tool FunRich andPathway Studios version 10 software were jointly usedto determine their molecular functions. Comparedwith the other 2 groups, the CRTþBB–treated group atthe 3.5-month time point had a 6-fold increase in thenumber of proteins associated with ATPase activityand a lower percentage of proteins functioning asextracellular matrix structural constituent (Figure 3).The control group had 2.5 times and 4 times as manyproteins related to ubiquitin-specific protease activityas did the infarcted, untreated animals and theCRTþBB–treated animals, respectively. With regardto G-protein–coupled receptor activity at the 3.5-month time point, the infarcted, untreated grouphad the most activity, with nearly a 10- and 5-foldincrease in the number of proteins compared withthe control and CRTþBB–treated animals,respectively.
At the 4.5-month time point, the TFMP proteinsdiffered in 3 main functional classes between theexperimental groups: structural constituents of thecytoskeleton, ATPase activity, and intracellularligand-gated ion channel activity (Figure 4). Therewas a considerable difference in the structural con-stituents of the cytoskeleton between the CRTþBB–treated animals and the control animals, in the ratioof z1:35 (Figure 4). The infarcted, untreated animalshad no detectible proteins functioning as structuralconstituents of the cytoskeleton. With regard toATPase activity, the CRTþBB group recorded thehighest percentage, with nearly a 2-fold increasecompared to the control group and z1.5 times thenumber observed in the infarcted, untreated group.The CRTþBB group had no detectable proteins func-tioning in intracellular ligand-gated ion channel ac-tivity, but there was a 4-fold increase in the infarcted,untreated group compared with the control group.
By the 6-month time point, CRTþBB treatment wasassociated with anz4-fold increase in ATPase proteincontent compared with controls, whereas theinfarcted, untreated animals had 3 times as manyATPase proteins as the control animals. There were noproteins associatedwith extracellular ligand-gated ionchannel activity detected in the CRTþBB group,although in the infarcted, untreated animals, therewas a 3-fold increase compared with the control group.The infarcted animals had higher levels of extracel-lular matrix structural constituents than the controlgroup animals (Figure 5). Additionally, the infarcted,untreated animals had a higher percentage of proteinsassociated with ubiquitin protease activity than the

TABLE 1 FunRich Analysis of Site of Expression of TFMP Protein
Treatment andSite of Expression
No. of Genesin Dataset
Uncorrectedp Value
(Hypergeometric Test)
Storey andTibshirani Method
q Value
Control
Heart muscle 72 0.15 0.07
Plasma 156 0.00 0.00
Blood vessels 8 0.00 0.00
HUVECs 163 0.00 0.00
Untreated 3.5 months
Heart muscle 77 0.01 0.00
Plasma 138 0.00 0.00
Blood vessels 10 0.00 0.00
HUVECs 143 0.01 0.00
Untreated 4.5 months
Heart muscle 76 0.13 0.09
Plasma 158 0.00 0.00
Blood vessels 7 0.00 0.00
HUVECs 172 0.00 0.00
Untreated 6 months
Heart muscle 29 0.03 0.09
Plasma 52 0.00 0.02
Blood vessels 2 0.07 0.12
HUVECs 52 0.03 0.09
CRTþBB 3.5 months
Heart muscle 62 0.20 0.09
Plasma 142 0.00 0.00
Blood vessels 9 0.00 0.00
HUVECs 141 0.00 0.00
CRTþBB 4.5 months
Heart muscle 50 0.04 0.01
Plasma 106 0.00 0.00
Blood vessels 1 0.58 0.07
HUVECs 98 0.00 0.00
CRTþBB 6 months
Heart muscle 51 0.09 0.01
Plasma 112 0.00 0.00
Blood vessels 8 0.00 0.00
HUVECs 109 0.00 0.00
BB ¼ b-blocker; CRT ¼ cardiac resynchronization therapy; HUVECs ¼ human umbilical veinendothelial cells; TFMP ¼ tissue factor–bearing microparticles.
J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
535
CRTþBB–treated group (1:8 ratio) and an z50% in-crease compared with the control group animals.TFMP PROTEIN CONTENT AND POST-MI DISEASE STATUS.
To further understand the association between diseasestatus and TFMP protein content, Pathway Studioanalysis was conducted. Evaluations were based onfunctional associations with known active cell pro-cesses post-MI, such as apoptosis, oxidative stress,and inflammation, with adjustments made for theadministration of the BBmetoprolol where applicable.
At the 3.5-month time point (SupplementalFigure 1), TFMP proteins in the treated group wereassociated with cardiac cell antiapoptosis and facili-tation of cardiomyocyte function. These TFMP pro-teins included sarcoplasmic/endoplasmic reticulumCa2þ-ATPase, SERCA proteins (ATP2A2), ephrin, andhexokinase 1 and 2 (HK I, HK II). Another cardiac cell–enhancing factor detected in the treated group butnot the infarcted, untreated group or control groupwas guanylate cyclase A. Conversely, at the 3.5-month time point, hypoxia-driven factors such ashypoxia-inducible factor-1a, hypoxia-induciblefactor-2a, and FLT-1 (Fms-related tyrosine kinase-1)were detected in the infarcted, untreated animals.
At the 4.5-month time point, repair and structuraltissue formation proteins were found in the CRTþBB–treated group (Supplemental Figure 2). These proteinswere keratin, desmin, actin, vimentin, myosin, and b-catenin. Compared with the treated pigs, there werefewer myosin subtypes observed in the infarcted, un-treated group. Additionally, proteins that enhancecardiac cell performance, such as sodium/potassium-transporting ATPase subunit alpha-1 and -2 (ATP1A1,ATP1A2), and proteins of ryanodine receptors 2 and 3(RYR2, RYR3) were observed in the untreated group.Furthermore, in both infarcted groups, factors that canpromote inflammation were detected; however, inaddition to thrombin (F2), TF, and putative serineproteases (PRSS), which were detected in the treatedgroup, nuclear factor-kappa-B1 (NF-KB1) and ADAMmetallopeptidase domain 17 (ADAM17) were detectedin the infarcted, untreated group.
By the 6-month time point, TFMPs in the treatedgroup contained cytoprotective heat shock proteins(HSPs) such as HSPA1A, HSPA1B, and HSPA5 (Figure 6).HSPwas not detected in any other group. In addition tothe cytoprotective proteins, pigs in the treated groupcontained proteins required for the maintenance ofcardiac function (ATP1A1 and ATP1A2). There were nounique proteins or functional patterns observed in theinfarcted, untreated animals at this time point.HIGH-SENSITIVITY CARDIAC TnT ASSESSMENT. Serumlevels of TnT were assessed between groups (Figure 7).Area under the curve analyses for the period from
2.5 to 6 months post-MI were conducted. ANOVA washighly statistically significant across all comparisons:MI versus control, p < 0.001; MI versus CRTþBB,p <0.002; and CRTþBB versus control, p < 0.002.
ADRB2, cAMP, AND GRK2 CONCENTRATIONS. Themean concentration of ADRB2 in myocardial tissue inthe control group animals was 0.681 � 0.056 ng/mlcompared to 0.205 � 0.086 ng/ml in the infarcted,untreated group and 0.22 � 0.223 ng/ml in theCRTþBB–treated group (Supplemental Figure 3).Statistically, there were differences in the averageconcentration between the infarcted but untreatedgroup and the control group (p ¼ 0.003) and betweenthe CRTþBB–treated group and the control group
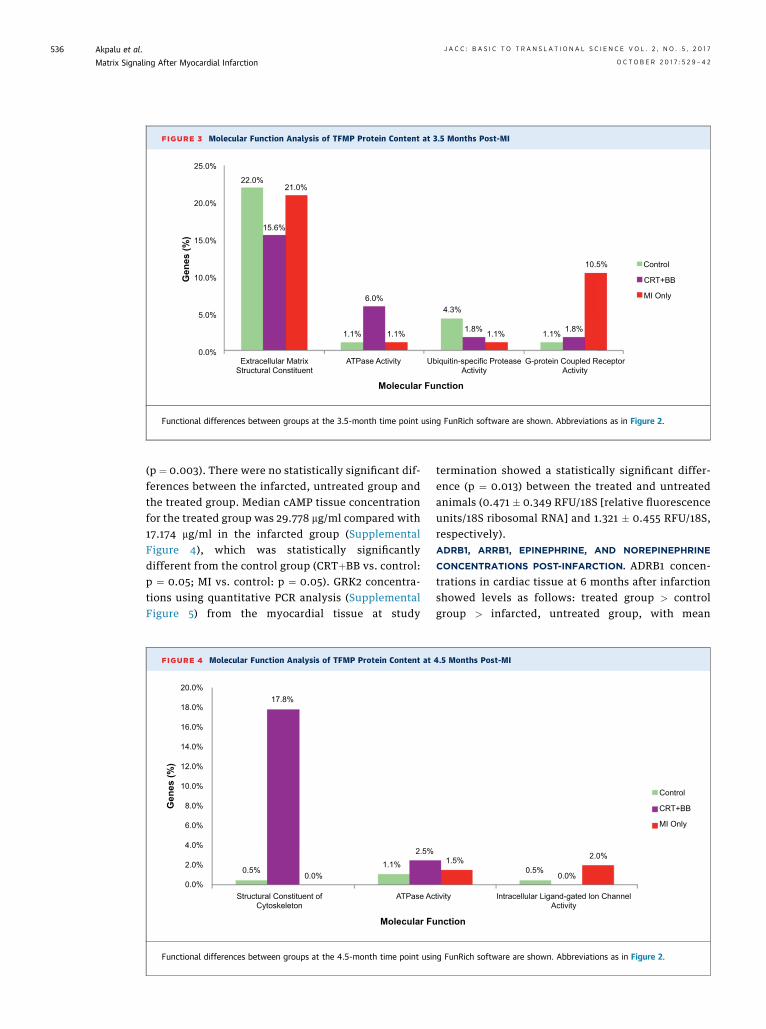
FIGURE 3 Molecular Function Analysis of TFMP Protein Content at 3.5 Months Post-MI
25.0%
20.0%
15.0%
10.0%
5.0%
0.0%
22.0%21.0%
15.6%
1.1%
6.0%
1.1%
4.3%
1.8% 1.8%
10.5% Control
CRT+BB
MI Only
1.1% 1.1%
Gen
es (%
)
Molecular Function
Extracellular MatrixStructural Constituent
ATPase Activity Ubiquitin-specific ProteaseActivity
G-protein Coupled ReceptorActivity
Functional differences between groups at the 3.5-month time point using FunRich software are shown. Abbreviations as in Figure 2.
Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
536
(p ¼ 0.003). There were no statistically significant dif-ferences between the infarcted, untreated group andthe treated group. Median cAMP tissue concentrationfor the treated group was 29.778 mg/ml compared with17.174 mg/ml in the infarcted group (SupplementalFigure 4), which was statistically significantlydifferent from the control group (CRTþBB vs. control:p ¼ 0.05; MI vs. control: p ¼ 0.05). GRK2 concentra-tions using quantitative PCR analysis (SupplementalFigure 5) from the myocardial tissue at study
FIGURE 4 Molecular Function Analysis of TFMP Protein Content at
20.0%
18.0%
16.0%
14.0%
12.0%
10.0%
8.0%
6.0%
4.0%
2.0%
0.0%
Gen
es (%
)
Molecular Fu
0.5%0.0%
1.1%
2.5%
17.8%
ATPase AcStructural Constituent ofCytoskeleton
Functional differences between groups at the 4.5-month time point usin
termination showed a statistically significant differ-ence (p ¼ 0.013) between the treated and untreatedanimals (0.471 � 0.349 RFU/18S [relative fluorescenceunits/18S ribosomal RNA] and 1.321 � 0.455 RFU/18S,respectively).ADRB1, ARRB1, EPINEPHRINE, AND NOREPINEPHRINE
CONCENTRATIONS POST-INFARCTION. ADRB1 concen-trations in cardiac tissue at 6 months after infarctionshowed levels as follows: treated group > controlgroup > infarcted, untreated group, with mean
4.5 Months Post-MI
nction
1.5%0.5%
0.0%
2.0%
tivity Intracellular Ligand-gated lon ChannelActivity
Control
CRT+BB
MI Only
g FunRich software are shown. Abbreviations as in Figure 2.

FIGURE 6 Pathway Studio Analysis of Proteomic Content of TFMPs at 6 Months in the Infarcted Groups Compared With
Control Group Profile
(A) Heat shock proteins (HSPs) associated with post-ischemic conditioning detected in CRTþBB–treated animals only. Metoprolol administration enhanced levels of
HSPA1A. (B) Factors related to cardiac function improvement were detected in treated animals 6 months after infarction, but no such observation made in the
infarcted, untreated animals.
FIGURE 5 Molecular Function Analysis of TFMP Protein Content at 6 Months Post-MI
30.0%
25.0%
20.0%
15.0%
10.0%
5.0%
0.0%
Gen
es (%
)
ATPase ActivityExtracellular MatrixStructural Constituent
Extracellular Ligand-GatedIon Channel Activity
Ubiquitin-Specific ProteaseActivity
Molecular Function
22.0%
27.6%25.8%
1.1%
4.7%3.2%
2.2%
0.0%
6.5%
4.3%
6.5%
0.8%
Control
CRT+BB
MI Only
Functional differences between groups at the 6-month time point using FunRich software are shown. Abbreviations as in Figure 2.
J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
537

FIGURE 7 Serum Troponin T Levels Over the 6-Month Period Post-Infarction
Showing Chronic Myocardial Cell Death in Infarcted Animals Over the
6-Month Study Duration
2.01.81.61.41.21.00.80.60.40.20.0
CRT+ BB
2.5 mth
Con
cent
ratio
n of
Car
diac
Tro
poni
n T
(ng/
mL)
Avg 3.5mth Avg 4.5mth Avg 6 mth Avg
Untreated
Control
The differences in mean values between the infarcted groups became statistically
significant at the 6-month time point (p ¼ 0.008). Avg ¼ average; mth ¼ month; other
abbreviations as in Figure 2.
Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
538
concentrations of 0.133 ng/ml, 0.027 ng/ml, andbelow limits of detection, respectively. There were nosignificant differences in the mean concentrationsof ARRB1, epinephrine, and norepinephrine betweenthe experimental groups in our study; however, therewas the expected elevation in norepinephrine andepinephrine concentrations in the infarcted animalscompared with the control group at all time points.
DISCUSSION
In heart failure patients with severe systolicdysfunction and asynchronous contraction, CRT isan established, effective adjunctive therapy whenpaired with BB. To adequately emulate the humancondition, we used an animal model with decreasedEF and asynchrony. The CRTþBB regimen improvesleft ventricular function and prognosis, whereasmetoprolol reduces infarct size (31,32). What is notfully characterized is the long-term signaling impactof b1-adrenergic blockade on the infarcted heart.
Reports indicate that many of the cellular re-sponses after an MI, such as fibrous tissue deposition,occur within the first few days to weeks of the post-infarction period (33–35). The degree of fibrosis(grade 3 or 4) observed in the septal area, border, andinfarct regions of the hearts of both groups ofinfarcted pigs was in line with the expectation ofstructural changes (fibrous tissue deposition) re-ported previously (36). Additionally, the morpholog-ical changes in cardiac dimensions showed that thisstudy also depicted the expected morphologicalalterations observed in clinical settings (37) and
confirmed that the experimental procedure success-fully induced the MI and mimicked post-MI observa-tions. Furthermore, the hematologic observationsdepicted the functional consequences of infarction,as well as treatment, with respect to time. Collec-tively, the structural, morphological, and functionalterations observed are evidence of pathologicalremodeling in response to infarction. Clinically, theobservation of improved EF and the differences inEDV and ESV between the infarcted animals point to atrend toward better outcomes in our treatment groupanimals than in the infarcted, untreated animals. Thisobservation is in line with general clinical observa-tions of better prognosis with treatment with CRTþBBand also aligns with the observed decline in TnTlevels with treatment in our study. Results from thisstudy therefore confirmed the documented beneficialeffects of b1 blockade in improving outcomes (31,32).More importantly, the proteomic profile of the TFMPsprovided insights into the long-term impact ofmetoprolol usage on active signaling entities of thesurviving cardiomyocytes. First, the inability todifferentiate experimental groups in the long-termpost-MI stage using TFMP numbers confirmed ourview that the more relevant information is the pro-teomic profile of the TFMPs. Second, our findingsdemonstrate unique signatures for each infarcted pigcohort associated with either adaptive or healingchanges, as well as progressive pathological remod-eling or maladaptive signaling leading to worseningheart failure. Additionally, the proteomic profile ofthe TFMPs provided a matrix view of the spatial andtemporal changes and differences in signaling en-tities between the study groups. We infer from ourTFMP proteomic data and Pathway Studio analysesthat the improved prognosis of the CRTþBB–treatedanimals proceeded in a stagewise manner via thefollowing general modalities: 1) cardiomyocyte sur-vival and function enhancement; 2) structural repair;and 3) cytoprotection and post-ischemic conditioning(Figures 8 and 9).
A month after treatment initiation, cardiomyocytesurvival and function enhancing factors were detec-ted as depicted by Supplemental Figure 1A. Collec-tively, the listed proteins confer protective propertiesto the myocardium by promoting chemomechanicaland antiapoptotic properties (38) while attenuatingchronic cardiac remodeling (39–41). The detection ofSERCA proteins in the CRTþBB–treated group isimportant, because metoprolol has been reported torestore cellular levels of SERCA2a (42,43). The detec-tion of cardiomyocyte survival factors perhaps ex-plains the trend toward the decrease in levels of TnTin the treated group compared with the infarcted,

FIGURE 8 Summary Observations Based on Proteomic Profile of TFMPs From CRTþBB–Treated Animals From Treatment
Initiation Until End of Study
Abbreviations as in Figure 2.
J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
539
untreated animals. In the absence of treatment, thedetection of hypoxia-driven factors (SupplementalFigure 1B) predicted worse outcomes for theinfarcted, untreated animals, as confirmed by the re-sults of the currently used biomarkers. At stage2 (2 months after treatment initiation), a wide range ofstructural or repair proteins were present in theTFMPs of the treated group (Supplemental Figure 2).
FIGURE 9 Summary Observations Based on Proteomic Profile of TFM
Abbreviations as in Figure 2.
This was in contrast to the observed narrower array ofstructural proteins detected in the infarcted, un-treated group. Additionally, the differences in thestructural proteins and the late detection of chemo-mechanical proteins could be evidence of delayed andperhaps ineffective cellular signaling in the absenceof treatment. The observed wide range of structuralproteins in the treated animals could be indicative of
Ps From Untreated Animals From 3.5 Months Until End of Study

Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
540
ongoing or incipient repair and healing processestaking effect in the treated group that contributed tothe reported resolution of cardiac cell damage. Sixmonths after infarction, there was an indication ofpost-ischemic conditioning and cytoprotection, asevidenced by the presence of HSP proteins in thetreated group (Figure 6). HSP was not detected in anyother group, and its detection in the treated group is aconsequence of metoprolol administration (44). Thisis relevant because HSP overexpression attenuatesmyocardial apoptosis (45,46), and it confirms theactivation of cytoprotective processes months afterCRTþBB treatment initiation.
The results from the biomarkers used confirm animproved prognosis for the treated animals and sup-port the investigational and interventional opportu-nity offered by TFMP proteomic profiling post-MI.This was in line with a decline in TnT levels in thetreated animals, whereas chronic troponin leakresulting from the death of cardiomyocytes persistedin the infarcted, untreated animals. By having lesscell death characterized as a smaller troponin leak,these data suggest that treatment with CRTþBBgradually attenuated cell death in the treated group.The observed increase in ADRB1 levels in the treatedgroup and its decrease in the infarcted animals trackswith reports of increasing b-AR expression after theadministration of metoprolol (47,48) and providesfurther evidence of the long-term cellular effects ofmetoprolol administration. This observation mightsuggest that chronic b1-blockade is associated withrecirculation of ADRB1 receptors to the car-diomyocyte surface as the levels of ischemic stressorsdecline and the surviving cardiomyocyte recovers,possibly via a negative feedback loop. The lowerlevels of ADRB2 in both infarcted groups are inagreement with published data proposing that mostprotective adrenergic signaling is mediated via b2-receptors and Gi signaling. This explains the observeddecreased b-AR expression along with loweredsensitivity in the aftermath of an MI (49). Further-more, impaired intracellular Ca2þ handling in thefailing heart has been ascribed to either a decreasedexpression of SERCA2a or a shift in the interactionbetween phospholamban and SR Ca2þ-ATPase activity(50,51). Additionally, in the failing heart, there is acorresponding increase in SR Ca2þ ATPase activitywith increasing cAMP concentrations (51). Thisperhaps accounts for the observed cAMP increase inour infarcted animals compared with the control an-imals. Because metoprolol treatment restores cAMP-dependent inotropic effects independent of b-AR(52), this might explain the observed higher levels ofcAMP in the treated animals than in the other groups.
The elevated epinephrine and norepinephrine levelsin our infarcted group animals compared with thecontrol animals are in line with expectations in theacute phase of an MI but highlight the ensuingcellular derangement chronically post-MI. Finally,the observed elevation of GRK2 levels underscoresthe heart’s decreased contractile function by theblunting of procontractile signaling of the b-ARs (53).
The composite of these biomarker evaluations andthe proteomic profiling of the TFMPs showed thattreatment with CRTþBB resulted in improvement inoutcome. Using pathway and functional analyses, theproteomic profile of TFMPs coupled the detection ofdiverse signaling mediators with temporal occurrenceto disease status and allowed the prediction ofoutcome in the post-MI setting. Most importantly, theobserved TFMP proteomic profile provided informa-tion of additive value to what was obtained by anal-ysis of 8 current biomarkers in our chronic ischemiccardiomyopathy model.
This study focused on chronic ischemic cardiomy-opathy in a porcine model of ischemic cardiomyopa-thy and did not examine the utility of the TPMPproteomic profile in the acute phase. Additionally, inour study, MI was induced by use of a collagen plugand therefore might have missed prelude molecularand cellular information that preceded the incidenceof an MI. This, however, does not minimize thedemonstration of the utility of TFMP protein contentas a cellular methodology to be used to further ourunderstanding of signaling after an MI. These resultsmay not be completely generalizable, but the wealthof information obtained from this study suggests thata follow-on translational study examining the utilityof profiling TFMPs in the acute phase is warranted asa next step. Furthermore, given the amount of in-formation obtained from the proteomic profile ofTFMPs, it would be interesting to determine whetherprofiling of the entire MP population would provideadditional information. Another necessary next stepin this line of inquiry must involve quantitativemethods to explore levels of candidate proteins.Furthermore, it would be important to establishrelevant flow cytometry thresholds of TFMP countsthat would yield levels of putative proteins thatwould allow for assessment in clinical settings.
CONCLUSIONS
The composite of the findings of this study is inagreement with previous studies and clinical obser-vations that treatment with CRTþBB after an MIleads to better outcomes. More importantly, ourfindings, while confirming the heterogeneity of TFMP

PERSPECTIVES
COMPETENCY IN MEDICAL KNOWLEDGE: Current models
of post-MI signaling involve small animals with monitoring for a
few hours to days. Usually, only a few proteins are assessed,
which does not adequately depict the pathophysiology in
humans. TFMP proteomic profiling after an MI enables observa-
tion of both spatial and temporal events in multiple signaling
mechanisms in a long-term mini-swine model. Thus, a dynamic
evaluation of active signaling pathways using TFMP proteomic
profiling would be a valuable source of molecular markers to
investigate bAR signaling post-MI.
TRANSLATIONAL OUTLOOK: A chronic animal model,
6 months post-MI, might more closely emulate human disease
and provide greater clinical translation to the human condition of
the long-term consequences of an MI. In addition to spatial
signaling (membrane to the nucleus), this study assessed chronic
changes in signaling longitudinally, rather than at a single time
point, using the proteomic profile of TFMPs. Thus, we demon-
strated that time and intracellular or spatial signaling impact the
observed outward phenotype of pathological remodeling after
an MI within and between groups.
J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7 Akpalu et al.O C T O B E R 2 0 1 7 : 5 2 9 – 4 2 Matrix Signaling After Myocardial Infarction
541
protein content, also demonstrate that proteomicprofiling of TFMPs from both the infarcted and non-infarcted pigs captured the diversity of the proteincontents between and within groups temporally.Changes in the identified proteins within andbetween groups corresponded to relevant changes inmolecular function and reflected the physiologicalstatus of the host. Spatially, the contents of theTFMPs displayed a variety of proteins and providedadditional information on multiple entities supple-mental to what we obtained from assessing 8 of thecurrent cardiac biomarkers. Additionally, for bothinfarcted groups, there was a noticeable time differ-ence in the detected proteins that predictablyreflected active, ongoing cellular activities. There-fore, results of this study support recommendingTFMP protein content profiling prospectively as aviable investigative methodology for chronicischemic cardiomyopathy that could help improveour understanding of bAR signaling after an MI. Theability to dynamically capture active signaling path-ways instead of tracking the presence or absence of asingle molecular entity, as typified by the currentgroup of biomarkers, would provide pertinentanswers to questions about bAR signaling pathwayspost-MI and in heart failure.
ACKNOWLEDGMENTS The authors would like toexpress their gratitude to Jane Chu, Mahfuz Khan,Irena Brandt, and Kayla Jackson for their contribu-tions in sample and data generation for this study.
ADDRESS FOR CORRESPONDENCE: Dr. David S.Feldman, Division of Cardiology, University of CincinnatiMedical Center, 234 Goodman Street, Cincinnati,Ohio 45221. E-mail: [email protected].
RE F E RENCE S
1. Liaudet L, Rosenblatt-Velin N. Role of innateimmunity in cardiac inflammation after myocardialinfarction. Front Biosci (Schol Ed) 2013;5:86–104.
2. Boateng S, Sanborn T. Acute myocardialinfarction. Dis Mon 2013;59:83–96.
3. Vilahur G, Juan-Babot O, Peña E, Oñate B,Casaní L, Badimon L. Molecular and cellularmechanisms involved in cardiac remodeling afteracute myocardial infarction. J Mol Cell Cardiol2011;50:522–33.
4. Maack C, Elter T, Böhm M. Beta-blocker treat-ment of chronic heart failure: comparison of car-vedilol and metoprolol. Congest Heart Fail 2003;9:263–70.
5. Shin J, Johnson JA. Beta-blocker pharmacoge-netics in heart failure. Heart Fail Rev 2010;15:187–96.
6. Lalani GG, Birgersdotter-Green U. Cardiacresynchronisation therapy in patients with chronicheart failure. Heart 2015;101:1008–14.
7. Mookadam F, Moustafa SE. Prevention of latepostmyocardial infarction left ventricular remodel-ing: an update. Curr Heart Fail Rep 2009;6:245–53.
8. Combes V, Simon AC, Grau GE, et al. In vitrogeneration of endothelial microparticles andpossible prothrombotic activity in patients withlupus anticoagulant. J Clin Invest 1999;104:93–102.
9. Martinez MC, Tual-Chalot S, Leonetti D,Andriantsitohaina R. Microparticles: targets andtools in cardiovascular disease. Trends PharmacolSci 2011;32:659–65.
10. Morel O, Toti F, Freyssinet JM. Markers ofthrombotic disease: procoagulant microparticles[in French]. Ann Pharm Fr 2007;65:75–84.
11. Owens AP 3rd, Mackman N. Microparticles inhemostasis and thrombosis. Circ Res 2011;108:1284–97.
12. Martinez MC, Andriantsitohaina R. Microparti-cles in angiogenesis: therapeutic potential. CircRes 2011;109:110–9.
13. Puddu P, Puddu GM, Cravero E, Muscari S,Muscari A. The involvement of circulating mi-croparticles in inflammation, coagulation andcardiovascular diseases. Can J Cardiol 2010;26:140–5.
14. Simpson RJ, Jensen SS, Lim JW. Proteomicprofiling of exosomes: current perspectives. Pro-teomics 2008;8:4083–99.
15. Zwaal RF, Schroit AJ. Pathophysiologic impli-cations of membrane phospholipid asymmetry inblood cells. Blood 1997;89:1121–32.
16. Dadu RT, Nambi V, Ballantyne CM. Developingand assessing cardiovascular biomarkers. TranslRes 2012;159:265–76.
17. Morel OF, Ohlmann PF, Morel NF, et al.Microparticles and cardiovascular disease. ArchMal Coeur Vaiss 2005;98:226–35.
18. Shantsila E, Kamphuisen PW, Lip GY. Circu-lating microparticles in cardiovascular disease:implications for atherogenesis and athero-thrombosis. J Thromb Haemost 2010;8:2358–68.
19. Tushuizen ME, Diamant M, Sturk AF,Nieuwland R. Cell-derived microparticles in thepathogenesis of cardiovascular disease: friend orfoe? Arterioscler Thromb Vasc Biol 2011;31:4–9.
20. Viera AJ, Mooberry M, Key NS, et al. Micropar-ticles in cardiovascular diseasepathophysiologyandoutcomes. J Am Soc Hypertens 2012;6:243–52.

Akpalu et al. J A C C : B A S I C T O T R A N S L A T I O N A L S C I E N C E V O L . 2 , N O . 5 , 2 0 1 7
Matrix Signaling After Myocardial Infarction O C T O B E R 2 0 1 7 : 5 2 9 – 4 2
542
21. Bakouboula BF, Morel OF, Faure AF, et al.Procoagulant membrane microparticles correlatewith the severity of pulmonary arterial hyper-tension. Am J Respir Crit Care Med 2008;177:536–43.
22. Housden BE, Perrimon N. Spatial and temporalorganization of signaling pathways. Trends Bio-chem Sci 2014;39:457–64.
23. Cui J, Li J, Mathison M, et al. A clinicallyrelevant large-animal model for evaluation oftissue-engineered cardiac surgical patch materials.Cardiovasc Revasc Med 2005;6:113–20.
24. Nishijima Y, Sridhar A, Viatchenko-Karpinski S,et al. Chronic cardiac resynchronization therapyand reverse ventricular remodeling in a model ofnonischemic cardiomyopathy. Life Sci 2007;81:1152–9.
25. Wikstrand J. MERIT-HF: description of thetrial. Basic Res Cardiol 2000;95 Suppl 1:I90–7.
26. Pathan M, Keerthikumar S, Ang CS, et al.FunRich: An open access standalone functionalenrichment and interaction network analysis tool.Proteomics 2015;15:2597–601.
27. FunRich: Functional Enrichment Analysis Tool.Available at: http://funrich.org/index.html.Accessed April 1, 2016.
28. Park HJ, Zhang Y, Georgescu SP, Johnson KL,Kong D, Galper JB. Human umbilical vein endo-thelial cells and human dermal microvascularendothelial cells offer new insights into the rela-tionship between lipid metabolism and angiogen-esis. Stem Cell Rev 2006;2:93–102.
29. Hauser S, Jung F, Pietzsch J. Human endo-thelial cell models in biomaterial research. TrendsBiotechnol 2017;35:265–77.
30. Onat D, Brillon D, Colombo PC, Schmidt AM.Human vascular endothelial cells: a modelsystem for studying vascular inflammation indiabetes and atherosclerosis. Curr Diab Rep2011;11:193–202.
31. Barrese V, Taglialatela M. New advances inbeta-blocker therapy in heart failure. Front Physiol2013;4:323.
32. Prakash A, Markham A. Metoprolol: a review ofits use in chronic heart failure. Drugs 2000;60:647–78.
33. Talman V, Ruskoaho H. Cardiac fibrosis inmyocardial infarction-from repair and remodelingto regeneration. Cell Tissue Res 2016;365:563–81.
34. Piek A, de Boer RA, Silljé HH. The fibrosis-celldeath axis in heart failure. Heart Fail Rev 2016;21:199–211.
35. Ghasemi O, Ma Y, Lindsey ML, Jin YF. Usingsystems biology approaches to understand cardiacinflammation and extracellular matrix remodelingin the setting of myocardial infarction. WileyInterdiscip Rev Syst Biol Med 2014;6:77–91.
36. Kong P, Christia P, Frangogiannis NG. Thepathogenesis of cardiac fibrosis. Cell Mol Life Sci2014;71:549–74.
37. Karpinski L, Witkowska M. Postinfarction car-diac remodeling: clinical consequences [in Polish].Przegl Lek 2009;66:380–3.
38. Kumar SR, Masood R, Spannuth WA, et al. Thereceptor tyrosine kinase EphB4 is overexpressed inovarian cancer, provides survival signals and pre-dicts poor outcome. Br J Cancer 2007;96:1083–91.
39. Kishimoto I, Tokudome T, Horio T, Garbers DL,Nakao K, Kangawa K. Natriuretic peptide signalingvia guanylyl cyclase (GC)-A: an endogenous pro-tective mechanism of the heart. Curr Cardiol Rev2009;5:45–51.
40. Li Y, Kishimoto I, Saito Y, et al. Guanylylcyclase-A inhibits angiotensin II type 1A receptor-mediated cardiac remodeling, an endogenousprotective mechanism in the heart. Circulation2002;106:1722–8.
41. Nakanishi M, Saito Y, Kishimoto I, et al. Roleof natriuretic peptide receptor guanylyl cyclase-Ain myocardial infarction evaluated using geneti-cally engineered mice. Hypertension 2005;46:441–7.
42. George I, Sabbah HN, Xu K, Wang N, Wang J.b-adrenergic receptor blockade reduces endo-plasmic reticulum stress and normalizes calciumhandling in a coronary embolization model ofheart failure in canines. Cardiovasc Res 2011;91:447–55.
43. Mao Y, Tokudome T, Otani K, Kishimoto I,Miyazato M, Kangawa K. Excessive sym-pathoactivation and deteriorated heart functionafter myocardial infarction in male ghrelinknockout mice. Endocrinology 2013;154:1854–63.
44. Tsoporis J, Rizos IK, Toumpoulis I, Salpeas V,Izhar S, Parker TG. The induction of Hsp70 by betablockade in the human aorta during coronary ar-tery bypass grafting (CABG) or replacement of anascending thoracic aortic aneurysm (ATAA) is
associated with less apoptosis. Can J Cardiol 2013;Suppl:S141.
45. Nylandsted J, Jäättelä M, Hoffmann EK,Pedersen SF. Heat shock protein 70 inhibitsshrinkage-induced programmed cell death viamechanisms independent of effects on cellvolume-regulatory membrane transport proteins.Pflugers Arch 2004;449:175–85.
46. Suzuki K, Sawa Y, Kagisaki K, et al. Reductionin myocardial apoptosis associated with over-expression of heat shock protein 70. Basic ResCardiol 2000;95:397–403.
47. Sharma V, Parsons H, Allard MF, McNeill JH.Metoprolol increases the expression of beta(3)-adrenoceptors in the diabetic heart: effects onnitric oxide signaling and forkhead transcriptionfactor-3. Eur J Pharmacol 2008;595:44–51.
48. Najafi A, Sequeira V, Kuster DW, van derVelden J. b-adrenergic receptor signalling and itsfunctional consequences in the diseased heart. EurJ Clin Invest 2016;46:362–74.
49. Bernstein D, Fajardo G, Zhao M. The role ofbeta-adrenergic receptors in heart failure: differ-ential regulation of cardiotoxicity and car-dioprotection. Prog Pediatr Cardiol 2011;31:35–8.
50. Roe AT, Frisk M, Louch WE. Targeting car-diomyocyte Ca2þ homeostasis in heart failure. CurrPharm Des 2015;21:431–48.
51. Schmidt U, Hajjar RJ, Kim CS, Lebeche D,Doye AA, Gwathmey JK. Human heart failure:cAMP stimulation of SR Ca(2þ)-ATPase activityand phosphorylation level of phospholamban. AmJ Physiol 1999;277 Pt 2:H474–80.
52. Böhm M, Deutsch HJ, Hartmann D, Rosée KL,Stäblein A. Improvement of postreceptor eventsby metoprolol treatment in patients with chronicheart failure. J Am Coll Cardiol 1997;30:992–6.
53. Cannavo A, Liccardo D, Koch WJ. Targetingcardiac b-adrenergic signaling via GRK2 inhibitionfor heart failure therapy. Front Physiol 2013;4:264.
KEY WORDS bAR signaling, chronicischemic cardiomyopathy, matrix signaling,myocardial infarction, tissue factor-bearingmicroparticles, Yucatan mini swine
APPENDIX For expanded results, please seethe online version of this article.





























