Embed Size (px)
Citation preview

Sequence Assembly&
MASURCA as a hybrid approach
ZEHADY ABDULLAH KHANPhD 1st Year Student,
Department Of Computer Science,Purdue University

DNA Sequencing
The process of determining the precise order of neucleotides in a DNA molecule.






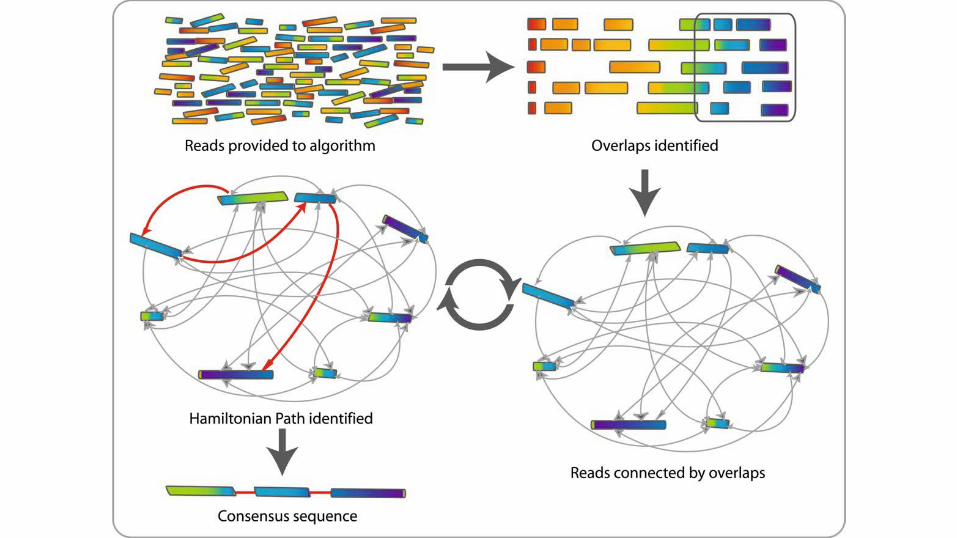
Overlap Layout Consensus● Compute all pairwise overlaps between reads.● Creates a layout
o An alignment of all overlapping reads. ● Extracts a consensus sequence
o by scanning the multiread alignment, column by column. ● Celera Assembler (Miller et al., 2008; Myers et al., 2000), PCAP (Huang, 2003), Arachne
(Batzoglou et al., 2002) and Phusion (Mullikin and Ning, 2003).
● Benefits:o Flexibility with respect to read lengths o Robustness to sequencing errors.
● Problem:
o Exponential computation.
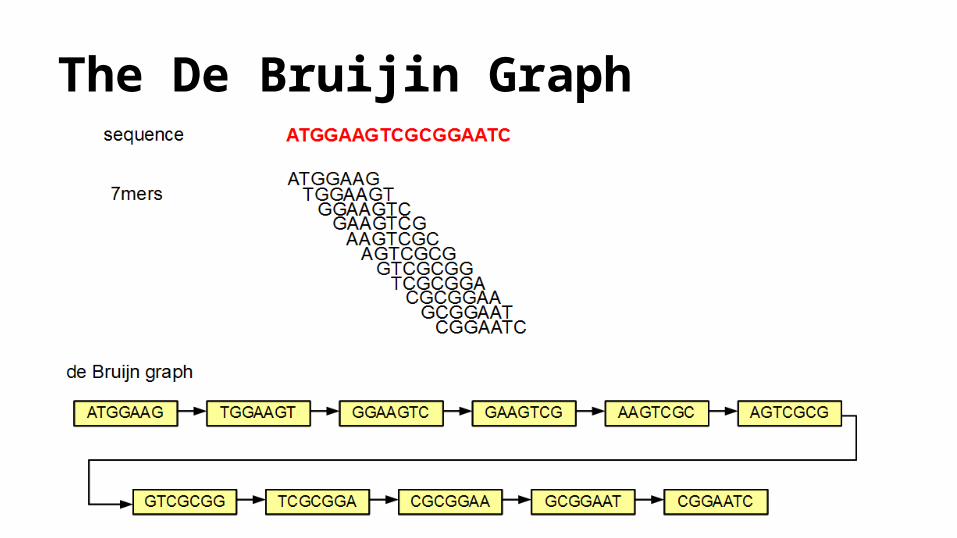
The De Bruijin Graph

The De Bruijin Graph ● Allpaths-LG (Gnerre et al., 2010), SOAPdenovo (Li et al., 2008), Velvet (Zerbino and Birney,
2008), EULER-SR (Chaisson and Pevzner, 2008) and ABySS (Simpson et al., 2009) ● Any path through the graph that visits every edge exactly once, formally known as an Eulerian
path, forms a draft assembly of the read. o Given all reads are perfect, which will match the de Bruijn graph of the genome
● Practically reads are not perfect.● These graphs are complex with many intersecting cycles, and many alternative Eulerian paths
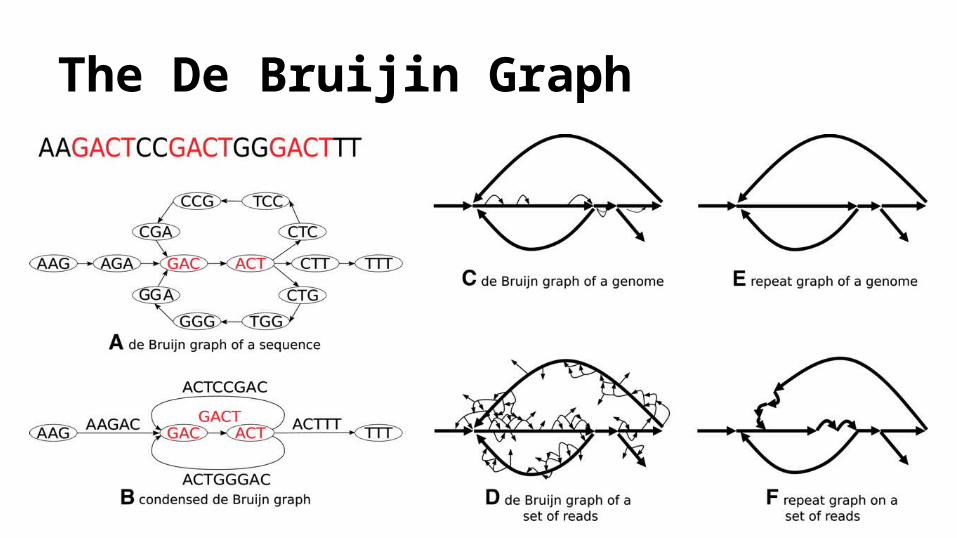
The De Bruijin Graph

What is Masurca?
● A new hybrid methodo Computational efficiency of the De Bruijn graph
method.o Flexibility of the OLC assembly

Super Read● Extend each original read forwards and backwards, base by base, as long as the extension is
unique. ● k-mer count look-up table
o An efficient hash table o Determine quickly how many times each k-mer occurs in our reads
● Given a k-mer found at the end of a read, there are four possible k-mers for the next k-mer.o The strings formed by appending A, C, G or T to the last k-1 bases in the read
● If only one of the four possible k-mers occurs, we say the read has a unique following k-mer and we append that base to the read.

k-Unitig● A k-mer is called a k-mer simple if it has a unique preceding k-mer and a unique following k-
mer. ● A k-unitig is a string of maximal length such that every k-mer in it is simple except for the first
and the last. ● By the construction, no k-mer can belong to more than one k-unitig. ● If a read has a k-mer that occurs in a k-unitig, the read and the k-unitig can be aligned to one
another. ● Use individual reads to merge the k-unitigs that overlap them into a single longer super-read.

Super Reads from paired-reads● If the reads are paired,
o We examine each pair of reads o Map each read to the k-unitigs,o Look for a unique path of k-unitigs connected by k-unitig overlaps that connects the two
reads. o If we find such a path, then we extend both paired-end reads to a new super-read.
Merge the k-unitigs on this unique path

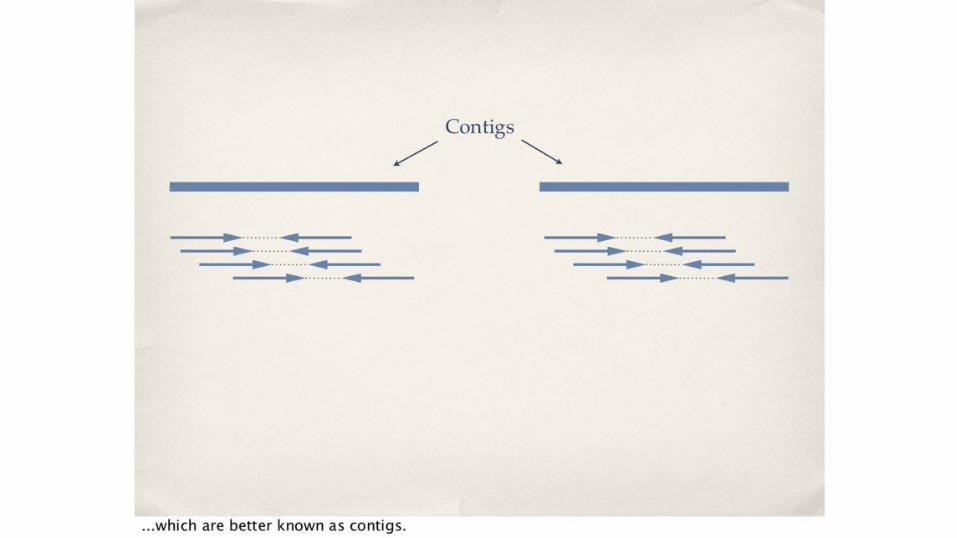
Assembler in Masurca● Modified version of the CABOG assembler ● Only super-reads used are maximal super-reads
o Those that are not exact substrings of another super-reads. ● Use of other data in assembly
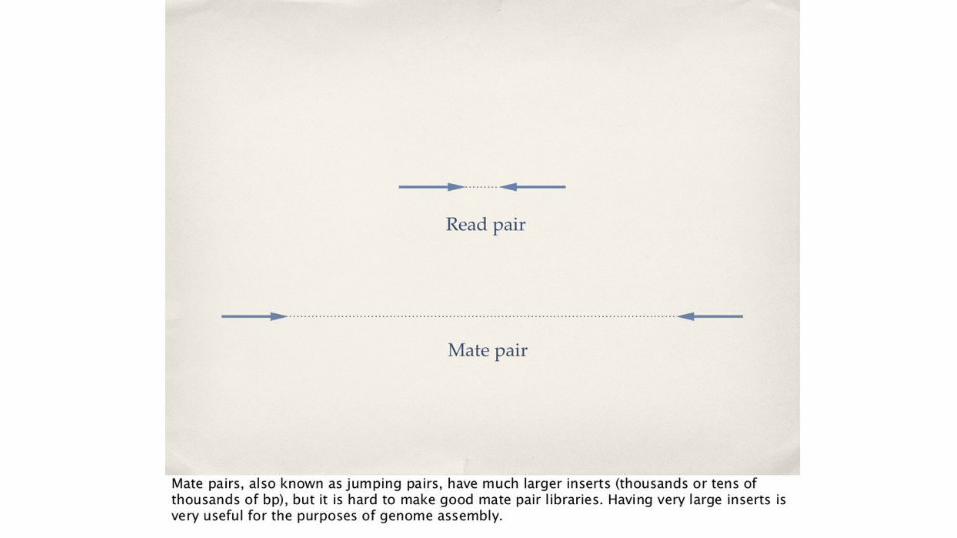
o Jumping libraries o 454 read data o Sanger read datao Mate pairs
● Coverage of the genome by maximal super-reads typically varies from 2–3x o Independent of the raw read coverage
● MaSuRCA automatically chooses the k-mer size for creating super-reads.

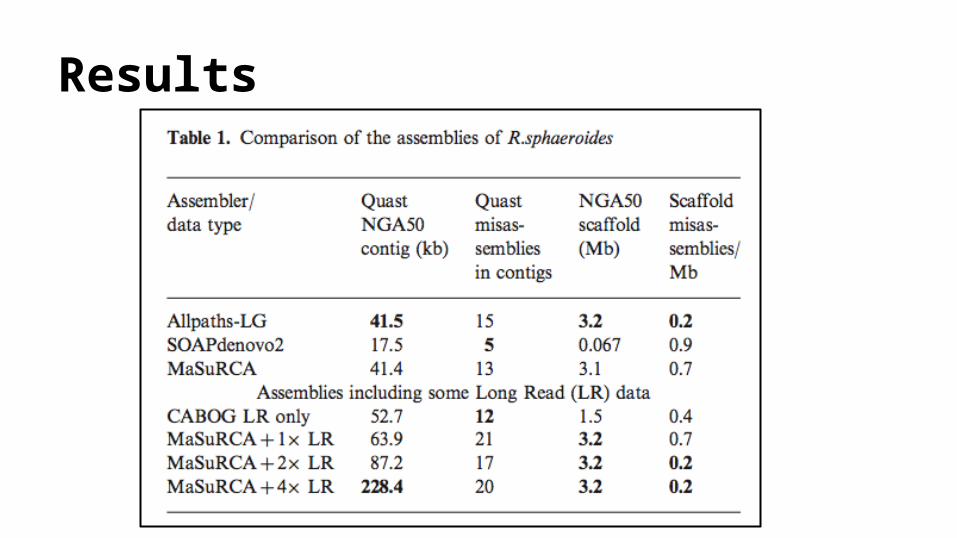
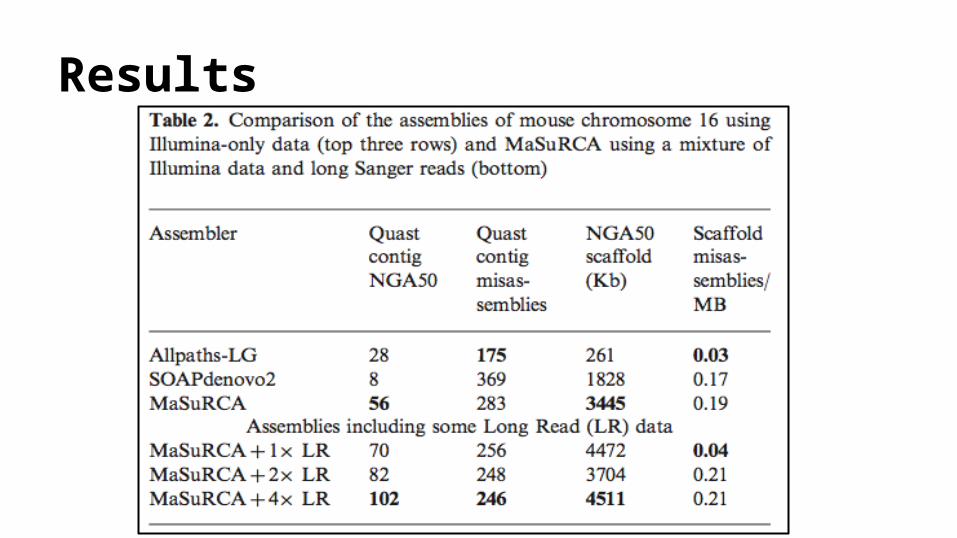
Results: Choice of Genome● Reference Paper:
The MaSuRCA genome assemblerAleksey V. Zimin, Guillaume Marc C ais, Daniela Puiu, Michael Roberts, Steven L. Salzberg and
James A. Yorke
● bacterium R.sphaeroides str. 2.4.1 (Rhodobacter) ● chromosome 16 of M.musculus lineage B6 (mouse)

Metrics of Evaluation● N50 :
o The length for which the collection of all contigs of that length or longer contains at least half of the sum of the lengths of all contigs.
● NGA50: o The value N such that 50% of the finished sequence is contained in contigs whose
alignments to the finished sequence are of size N or larger .
Results
Results

The End











![Aligning reads to a genome - Analysis of Next-Generation … · 2020. 4. 14. · Whydowealign? Whatdowelearn? [Reinertetal.,2015,Pfeifer,2017] LuceSkrabanek (ABC,WCM) Aligningreadstoagenome](https://img.pdfslide.us/doc/110x75/61471cf3f4263007b1359c77/aligning-reads-to-a-genome-analysis-of-next-generation-2020-4-14-whydowealign.jpg)

















