Embed Size (px)
Citation preview

JOHANNES KEPLER
UNIVERSITÄT LINZ
Altenberger Straße 69
4040 Linz, Österreich
www.jku.at
DVR 0093696
Submitted by
Daniel Timelthaler
Submitted at
Institute of Catalysis
Supervisor
DI Dr. Christoph Topf
March 2019
Development of Base
Metal Catalysts for
Green Hydrogenation
Reactions
Master Thesis
To obtain the academic degree of
Diplom-Ingenieur
In the Master’s program
Technical Chemistry – Master Program

Daniel Timelthaler 2/126
STATUTORY DECLARATION
I hereby declare that the thesis submitted is my own unaided work, that I have not used other
than the sources indicated, and that all direct and indirect sources are acknowledged as
references.
This printed thesis is identical with the electronic version submitted.
Linz
Signature

Daniel Timelthaler 3/126
ACKNOWLEDGEMENTS
I would like to express my gratitude towards Univ.-Prof. Dr. Marko Hapke and the entire Institute
of Catalysis for giving me the opportunity to perform the research necessary for the completion of
this thesis. I also want to thank all the members of the Institute for the positive and friendly
atmosphere and supportive working environment.
Special acknowledgements go out to my colleague MSc Kirill Faust for measuring and interpreting
the XRD crystal structures that are included in this thesis. Also, I would like to thank assoz. Univ.-
Prof. Dr. Markus Himmelsbach from the Institute of Analytical Chemistry for performing high
resolution mass spectrometry measurements. Furthermore, I want to thank Fabian Fischer and
Astrid Lehmann at the Leibniz Institute of Catalysis in Rostock (LIKAT) for performing the
elemental analysis.
Last but not least, I want to express special gratitude to DI Dr. Christoph Topf for introducing me
into the topic, his constant support for overcoming difficulties in the practical work and giving
valuable supplementary theoretical inputs. It is unquestioned that this master thesis would not
have been possible without his support.

Daniel Timelthaler 4/126
ABSTRACT
I herein report the development of novel base-metal catalysts for hydrogenation reactions. The
described studies include ligand synthesis as well as different homogeneous and heterogeneous
catalytic systems. Special emphasis has been laid on the optimization of reaction conditions as
well as determining performance dependencies on substrates and additives.
The initial investigations were centered around the synthesis and reduction of
bis(aryl)acenaphthenequinonediimines as representative of the class of α-diimine ligands. The
synthesis was carried out following a literature procedure, which allowed for isolation of BIAN
structures bearing various functional groups. Consecutive experimental work focused on the
non-trivial reduction process of the diimine moiety. The effects of various reducing agents on the
redox-active ligands and selected structural features of the resulting oxidation states were
investigated and a protocol for effective reduction was established.
The synthesized α-diimine ligands were employed in a homogeneous hydrogenation system
featuring a nickel(I)-BIAN hydride complex which was formed in situ by addition of pinacolborane
as an activator. The performance of the complex in catalytic transformations was investigated for
several different functionalities like alkenes, nitriles and ketones. Unfortunately, the system failed
to deliver satisfactory substrate conversions even under high catalyst loadings and harsh reaction
conditions.
Moreover, a cobalt-BIAN complex was evaluated for its catalytic performance in homogeneous
hydrogenation reactions, thereby attempting to utilize zinc metal as a single-electron reductant.
While the alleged homogeneous system performed well in the hydrogenation of nitriles, it was
quickly found to be of heterogeneous nature, featuring in situ generated metallic cobalt particles.
The ligand-free system thus found was successfully optimized by means of catalyst loading,
temperature, H2 pressure, solvents, additives and employed precatalysts. The obtained catalytic
protocol is appealing owing to its convenient setup, allowing all preparations and work-up steps to
be carried out under ambient conditions, as well as the low price and ready availability of the
catalytic components. Employing the optimized protocol, a variety of aliphatic and aromatic nitriles
was converted to their respective primary amines with good yields. The system shows high
functional group tolerance and also succeeds at commercially relevant transformations such as
the hydrogenation of adiponitrile to hexamethylene diamine.

Daniel Timelthaler 5/126
MOLECULE DENOTATION SCHEME
Abbreviation Structure Name
1a
N1,N2-Diphenylacenaphthylene-1,2-
diimine
1a-ZnCl2[a]
(Dichlorido,N1,N2-
diphenylacenaphthylene-1,2-
diimine)zinc(II)
1b
N1,N2-Di-p-tolylacenaphthylene-1,2-
diimine
1c
N1,N2-Dimesitylacenaphthylene-1,2-
diimine
1d
N1,N2-Bis(2,6-
diisopropylphenyl)acenaphthylene-
1,2-diimine
1e
N1,N2-Bis(4-
methoxyphenyl)acenaphthylene-1,2-
diimine
1f
N1,N2-Bis(3,5-
bis(trifluoromethyl)phenyl)-
acenaphthylene-1,2-diimine

Daniel Timelthaler 6/126
1g
N1,N2-Bis(3-(trifluoromethyl)phenyl)-
acenaphthylene-1,2-diimine
1h
N1,N2-Bis(2-(tert-
butyl)phenyl)acenaphthylene-1,2-
diimine
1i
N1,N2-Dicyclopropylacenaphthylene-
1,2-diimine
1j
N1,N2-Bis(4-(trifluoromethyl)phenyl)-
acenaphthylene-1,2-diimine
1k
N1,N2-Bis(2-
chlorophenyl)acenaphthylene-1,2-
diimine
1l
N1,N2-Bis(3-
chlorophenyl)acenaphthylene-1,2-
diimine
1m
N1,N2-Bis(4-
chlorophenyl)acenaphthylene-1,2-
diimine
1n
4,4'-(((Acenaphthylene-1,2-
diylidene)bis(azaneylylidene))-
dibenzonitrile

Daniel Timelthaler 7/126
2a[a]
N1,N2-Diphenylacenaphthylene-1,2-
diamine
3a[a]
N1,N2-Diphenyl-1,2-
dihydroacenaphthylene-1,2-diamine
4a
Bis((hydrido,N1,N2-
Diphenylacenaphthylene-1,2-
diimine)nickel(I))
5a
1-Methylcyclohex-1-ene
6a
Benzonitrile
6b
4-Methylbenzontrile
6c
1-Naphthonitrile
6d
2-Naphthonitrile
6e
4-Methoxybenzonitrile
6f
2-Chlorobenzonitrile

Daniel Timelthaler 8/126
6g
3-Chlorobenzonitrile
6h
4-Chlorobenzonitrile
6i
4-Fluorobenzonitrile
6j
2-Fluoro-6-methoxybenzonitrile
6k
4-(Trifluoromethyl)benzonitrile
6l
3-Pyridinecarbonitrile
6m
N-(4-Cyanophenyl)acetamide
6n
Cinnamonitrile
6o
Cyclohexanecarbonitrile
6p
Pentanenitrile
6q
Acetonitrile
6r
Adiponitrile
6s
(3r,5r,7r)-Adamantane-1-carbonitrile

Daniel Timelthaler 9/126
6t
2-Phenylacetonitrile
6u
4-Formylbenzonitrile
6v
Methyl-4-cyanobenzoate
6w
1,4-Dicyanobenzene
6x
4-Aminobenzonitrile
6y
4-Pyridinecarbonitrile
6z
Furan-2-carbonitrile
6aa
Thiophene-2-carbonitrile
6ab
Thiophene-3-carbonitrile
6ac
4-(1,3-Dioxolan-2-yl)benzonitrile
7a
Acetophenone
7b
2,2,2-Trifluoro-1-phenylethan-1-one

Daniel Timelthaler 10/126
8a
Bis((bromido,N1,N2-
Diphenylacenaphthylene-1,2-
diimine)cobalt(I))
9a
Bis((methoxo,N1,N2-
Diphenylacenaphthylene-1,2-
diimine)cobalt(I))
10a
(Dichlorido,N1,N2-
Diphenylacenaphthylene-1,2-
diimine)cobalt(II))
11a
Bis((chlorido,N1,N2-
Diphenylacenaphthylene-1,2-
diiminoanion)cobalt(II))
12a
trans-Chalcone
12b
3,5,5-Trimethylcyclohex-2-en-1-one
(Isophorone)
13a
But-2-yne-1,4-diole

Daniel Timelthaler 11/126
13b
1,6-Heptadiyne
13c
2-Methylbut-3-byn-2-ol
14a
Cyclohexanecarboxylic acid
14b But-2-enoic acid
14c
3-Aminopropanoic acid
15a[a]
Benzamide
15ad
N-Methylbenzamide
16a
Diphenylmethanimine
16b
N,1-Diphenylmethylimine
16c
N-Methyl-1-phenylmethanimine
17a
Nitrobenzene
18a
Indole
19a
Bromobenzene
20a[a]
Benzylamine

Daniel Timelthaler 12/126
21a[a]
N-Benzyl-1-phenylmethanimine
22a[a]
Dibenzylamine
23a[a]
Benzylalcohol
24a[a]
Methylbenzimidate
25a[a]
Benzylammonium chloride
[a] For this compound, only one form is listed. All compounds encoded by the same number and
foregoing letters consist of the displayed characteristic group on the moiety encoded by the letters
for 1a-n for BIANs, or 6a-ac for nitriles.

Daniel Timelthaler 13/126
TABLE OF CONTENTS
1. Introduction ......................................................................................................................... 14
1.1. Catalytic Hydrogenation Reactions .............................................................................. 14
1.2. Metallic Cobalt as Hydrogenation Catalyst .................................................................. 15
1.3. Heterogeneous Catalytic Hydrogenation of Nitriles ...................................................... 17
1.4. Homogeneous Cobalt and Nickel Catalysts for Hydrogenation .................................... 21
1.5. BIAN Ligands .............................................................................................................. 24
2. Aims and Objectives ........................................................................................................... 26
3. Results and Discussion....................................................................................................... 27
3.1. Synthesis of Ar-BIAN Ligands ..................................................................................... 27
3.2. Reduction of Ar-BIAN Ligands ..................................................................................... 31
3.3. Synthesis of a Ni(I)-BIAN Complex for Hydrogenation ................................................. 36
3.4. Hydrogenation Facilitated by a Cobalt(I)-BIAN Complex ............................................. 40
3.5. Heterogeneous Hydrogenation employing a Co(II)/Zn(0) couple ................................. 47
4. Conclusions ........................................................................................................................ 63
5. Experimental Section .......................................................................................................... 66
5.1. General ....................................................................................................................... 66
5.2. Ar-BIAN Synthesis ....................................................................................................... 67
5.3. Reduction of Ar-BIAN .................................................................................................. 70
5.4. Synthesis of a Ni(I)-BIAN Complex for Hydrogenation ................................................. 71
5.5. Synthesis and Screening of Co(I)-BIAN Complexes .................................................... 72
5.6. Screening of the Heterogeneous Co(II)/Zn(0) System ................................................. 74
6. Used Abbreviations ............................................................................................................. 82
7. References ......................................................................................................................... 83

Daniel Timelthaler 14/126
1. Introduction
1.1. Catalytic Hydrogenation Reactions
Hydrogenation reaction are defined through the addition of two hydrogen atoms onto a substrate
molecule. This catalytic transformation is of utmost importance by virtue of its extensive and broad
use in industrial large-scale processes as well as small-scale synthetic applications. It is part of
processes employed by several of the most relevant branches of chemical industry such as
polymer synthesis, fine chemical production, the manufacture of agrochemicals, and
pharmaceuticals.[1]
No matter the scale or application, hydrogenation processes are defined through their catalysts.
These are required to perform by means of selectivity, stability and activity under the given reaction
conditions.
Catalytic hydrogenation reactions are considered favorable in sustainable synthesis owing to their
intrinsic high atom economy. In traditional synthesis, reductive transformations often rely on the
use of stoichiometric amounts of highly reactive reducing agents such as sodium borohydride or
lithium aluminum hydride. As these agents generate large amounts of inorganic waste during the
course of the reaction, modern catalysis research strives for substituting these reducing agents
for more eco-friendly options such as employing hydrogen gas as the reductant combined with a
potent catalyst.
Hydrogenation catalysts are generally divided into homogeneous and heterogeneous catalysts.
For industrial applications, heterogeneous catalysis takes the spotlight as it has several
advantages in large-scale applications over the homogeneous counterpart. Most importantly,
heterogeneous catalysts are easily separated from the reaction mixture after the catalytic
conversion. While on laboratory scale more complex separation techniques have to be employed,
for industrial scales these are usually connected with high costs, complicated engineering setups
with a concomitant significant decline in overall process efficiency and economic sustainability.
Furthermore, the know-how with respect of engineering and process design is significantly higher
for heterogeneous catalytic processes, thus making it the even more appealing choice.
Commonly employed heterogeneous catalysts consist of an active metal species that is often
immobilized on a support and commonly contains a promoter substance.[1] For several decades,
noble metals such as platinum, rhodium, ruthenium and palladium have been dominating the field.
These metals exhibit excellent activity and stability, often combined with natural or well tunable
selectivity under mild reaction conditions along with low catalyst loadings.

Daniel Timelthaler 15/126
Table 1: Prices of metals for hydrogenation reactions on February 15th 2019[2]
Metal Price (€ / kg)
Rh 80,600
Pd 44,700
Pt 25,100
Ru 8,300
Co 27
Ni 11
Cu 5
However, the high price and limited long-term availability of these metals has drawn recent
research focus towards substitution of noble catalysts for base metals. Amongst these, the most
widely employed metals are cobalt, nickel and copper. A price comparison between metals
commonly employed in hydrogenation reactions is listed in table 1. For most applications, catalysts
based on non-noble elements require higher catalyst loadings, often combined with harsher
reaction conditions. Despite that, base metal catalysts are also well-established in several
industrial processes owing to high selectivity that is otherwise not achieved with noble metals.
One example is the hardening of fats employing a Ni catalyst, which is considered the largest
single application of hydrogenation. In this process, the nickel catalyst plays a vital role in
controlling isomerization reactions of the long carbon chains, while platinum-group metals (PGMs)
fail at these reactions because of the high occurrence of unfavorable side reactions. Copper has
also been proven to exhibit excellent selectivity in the hydrogenation of edible oils. However, a
process utilizing this potential has not yet been commercialized for this application owing to
catalyst separation difficulties.[3]
Apart from this, copper is applied in the production of methanol from syngas and the hydrogenation
of carboxylic acids to alcohols. A general problem with the use of Cu as a hydrogenation catalyst
is its limited activity and the harsh reaction conditions that have to be applied when employing this
metal.[4]
1.2. Metallic Cobalt as Hydrogenation Catalyst
Cobalt catalysts find their most relevant application by volume in the production of alkanes through
the well-known Fischer-Tropsch synthesis.[5] Additionally, this metal shows good performance in
the hydrogenation of nitriles to primary amines as it effectively suppresses side reactions such as
di- and trimerizations. As such, cobalt catalysts find wide-spread application in the reduction of
isophorone nitrile to the corresponding diamine which is a precursor to isophorone diisocyanate,
a crucial monomer for polyurethane synthesis. Very importantly, in this process the cobalt catalyst
does not only catalyze the hydrogenation of the nitrile to its respective amine, but Co also

Daniel Timelthaler 16/126
facilitates the reduction of the imines that are formed as reaction intermediates upon condensation
of the aldehyde group with ammonia. This characteristic increases the viability of cobalt-catalyzed
nitrile hydrogenations as both product yield and selectivity are significantly increased.[6]
In this context, the most prominent type of heterogeneous nickel and cobalt catalysts are the so-
called Raney-type catalysts. Raney nickel was first discovered by US-Chemist Murray Raney in
1927, when he found that treatment of a nickel-aluminum alloy with concentrated sodium
hydroxide solution, leads to selective dissolution of certain intermetallic phases, leaving behind a
highly porous sponge-like material that exhibits high catalytic activity.[7] The sponge is usually
crushed to a fine powder that is storable under anaerobic conditions. This highly active catalyst
continues to be relevant up to now and is still frequently applied. Representative examples of
industrial processes that employ Raney nickel are the hydrogenation of adiponitrile to
hexamethylenediamine for the manufacture of Nylon 6,6 and the heterogeneous hydrogenation of
benzene to cyclohexane.[8]
On the other hand, Raney cobalt is another “skeletal”-type catalyst which is not as popular as its
nickel counterpart and is used to a much lesser extent in industry. Its development by Raney was
patented 6 years after the original discovery of the Raney Catalysts in 1933.[9] Raney cobalt is
usually prepared in a similar fashion as Raney nickel, starting from a cobalt-aluminum alloy
containing 45-50% Co.[10] Literature reports on Raney cobalt are quite sparse owing to its higher
price and generally lower activity compared to Raney nickel.[11] However, in direct comparison
Raney cobalt shows better performance in certain hydrogenolysis reactions such as
hydrodesulfurization (HDS)[12] and oxime cleavage.[13] In what concerns hydrogenation reactions,
Raney cobalt is applicable for selective hydrogenation of C=O bonds as in the catalytic
hydrogenation of citral by Chiang et al.[14] As exemplified in Figure 1, hydrogenation of citral usually
produces a mixture of several different products. Raney cobalt was found to selectively produce
both stereoisomers of I-1b in 45% yield, while Raney nickel preferably catalyzes Michael addition
of H2 to afford product I-1c. However, for both species, the catalyst activity could be successfully
increased by using pertinent metal borides instead of the pure metal catalysts. Raney-Co also
shows certain reactivity towards alkynes, alkenes and arenes.[11] However, the most relevant
application in which cobalt catalysts have decisive advantages over other base metal catalysts
concerns the hydrogenation of nitriles (more on this in chapter 1.3.).
Figure 2: Heterogeneous hydrogenation of citral applying the bulk metal catalyst Raney cobalt.[14]

Daniel Timelthaler 17/126
As effective Raney-Catalysts might be, their most problematic drawback is the high sensitivity
towards oxygen; Raney nickel tends to combust spontaneously upon contact with air. Therefore,
careful handling, usually requiring a glovebox, is indispensable when working with this type of
materials.
Another option of obtaining active metallic cobalt particles was developed by Rieke.[15] In his work,
the active particles are generated by reduction of a dry Co(II) salt with a highly reactive metal,
usually lithium in naphthalene. When performed under well-kept inert conditions, Rieke cobalt
particles form as a dark gray precipitate. Direct use of the thus-produced metal suspension as a
catalyst is often hindered because of the agglomeration tendency of the particles.[16] This method
also suffers from the necessity of lab-technical precautions such as working in a glovebox with
complete exclusion of oxygen and moisture.
A procedure very similar to the synthesis of Rieke cobalt was established by the group of Wolf et
al. On reducing dry CoCl2 with lithium naphtalenide in THF over one day, finely dispersed
catalytically active cobalt nanoparticles were formed. In several experiments, the group
demonstrated the considerable synthetic potential of this benign catalyst. At temperatures as low
as 20 °C, the hydrogenation of alkynes, alkenes, imines and heteroarenes was effected along with
good selectivity and broad substrate scope. Furthermore, the ferromagnetic nature of the catalyst
enabled quantitative catalyst recycling, which guaranteed stability and activity over the course of
several weeks. Furthermore, the group demonstrated that in situ generated Co-nanoparticles gave
almost identical results, hence avoiding laborious catalyst isolation.[16]
With respect to preparing catalysts under ambient conditions, progress could be made with
Urushibara-type catalysts. Urushibara-Co is prepared by rapidly adding a concentrated aqueous
solution of a cobalt chloride to suspension of zinc dust in water at temperatures of approximately
100 °C. The formed catalyst consists of small cobalt particles deposited on Zn dust. For activation
of the catalyst, a change in pH is necessary which is attributed to the dissolution of zinc chloride
deposits on the catalyst surface. Urushibara-type catalysts are highly potent for several catalytic
applications and represent a significantly more convenient catalyst from a preparative point of
view. However, their preparation still suffers from an elongated leaching step and water-wasting
washing procedures.[10]
1.3. Heterogeneous Catalytic Hydrogenation of Nitriles
The catalytic conversion of nitriles into the respective amines has been a widely applied and
industrially relevant process for several decades. It represents an atom economic and efficient
method for the synthesis of amines, which are of pivotal importance in numerous chemical fields,
therefore rendering the abovementioned process highly valuable from an economic point of view.
The applications of the reaction products range from the synthesis of polymer precursors,

Daniel Timelthaler 18/126
surfactants to dyes, and pharmaceuticals. On industrial scale, nitrile hydrogenation is usually
carried out in the liquid phase using slurry bubble columns or packed bed reactors.[17]
The main difficulty of this transformation is the multitude of side products that can evolve during
the catalytic process. Therefore, the primary challenge is the control of product formation by
choosing the catalyst and reaction conditions wisely such that chemoselectivity can be
achieved.[18] Even though literature provides insight into a vast number of experience reports which
allow deducing certain tendencies of catalyst performance, the backgrounds of the observed
effects are often not well understood yet.
The main side reactions of nitrile hydrogenation, as outlined in Figure 2 using benzonitrile BN as
model substrate, include condensation of a semi-reduced benzylimine BI with one equivalent of
fully reduced benzylamine BA. This leads to the formation of dibenzylimine DBI as coupling
product that is further hydrogenated to dibenzylamine DBA. For some catalytic pathways,
elimination of ammonia leading to the formation of toluene T has also been reported.[17] For certain
substrates, this condensation reaction also occurs a third time leading to the formation of the
respective tertiary amine; the mechanism of this reaction was postulated by Braun in 1923.[19]
Common hydrogenation catalysts that are useful for nitrile hydrogenation include PGMs such as
Ru, Rh, Pd and Pt, as well as base metals including Co, Ni and Cu. Each of these metals shows
characteristic tendencies favoring formation of certain reaction products. Amongst the PGMs,
ruthenium catalysts show excellent selectivity for primary amines, while rhodium favors formation
of secondary amines. Palladium and platinum are especially unique as they can be deployed for
the selective production of tertiary amines since these catalysts tend to enhance condensation
remarkably.[20] Concerning base metals, cobalt and nickel exhibit similar properties with cobalt
showing stronger tendencies towards primary amine formation. These characteristics also hold
true for pure metallic catalysts as well as the skeletal Raney-type catalysts.[20] These tendencies
can be explained by comparing the adsorption behavior of the respective metal particles. A higher
adsorption affinity of the primary amine usually results in formation of a higher portion of
condensation products immediately on the catalysts surface.[21]
Figure 2: Possible products of benzonitrile hydrogenation.[17]

Daniel Timelthaler 19/126
The influence of reaction conditions such as temperature, hydrogen pressure and additives turned
out to be difficult to generalize as the exact effects heavily depend on the employed metal and the
catalyst support. In this context, for the vast majority of nitrile hydrogenations aiming at high yields
of primary amines a well-balanced ratio between sufficiently high conversion and satisfactory
selectivity must be found as formation of higher amines is usually enhanced at higher
temperatures. Hydrogen pressure can have mixed effects, but mostly higher H2 pressure
enhances the reaction rate and facilitates the formation of primary amines. With respect to
additives, the most commonly employed reagent is ammonia, which is added to the reaction
mixture either as solution or in gaseous form by virtue of its ability to suppress condensation
reactions.[18][20]
One industrial hydrogenation process of utmost importance is the production of
1,6-hexamethylenediamine HMD from adiponitrile ADN. As HMD is an essential precursor to
Nylon 6,6 the demand of the former will constantly be high, therefore encouraging research into
the development of a more efficient and sustainable process for its production. Commercially,
HMD is either produced by a high-pressure process (90-200 °C, 25–40 MPa) or a low-pressure
route (60-100 °C, 2-5 MPa). The high-pressure process employs a cobalt or iron catalyst that is
prepared by reduction of cobalt oxide or iron oxide in a mixture of hydrogen and ammonia before
the reaction.[22] The low-pressure process, patented by Monsanto, uses Raney nickel as catalyst
and sodium hydroxide instead of ammonia as the additive.[23] For these industrial processes, high
selectivity is crucial as the contamination limits are rather low. Therefore, considerable amounts
of ammonia or sodium hydroxide have to be employed in order to ensure sufficient high selectivity.
The possible side products of adiponitrile hydrogenation are depicted in Figure 3. In the past,
efforts have been made to substitute Raney-Ni in the low-pressure process for Raney-Co. Despite
lower activity, cobalt maintained reactivity even without base addition and showed higher
selectivity towards the formation of HMD than Raney nickel in pilot tests.[17][24]
Owing to its high relevance, research efforts in the field of base metal catalyzed nitrile
hydrogenation have been considerable over the past years. Notably, a significant portion of novel
pertinent catalysts is protected by patent law, indicating the high economic potential of this catalytic
transformation.
Some research efforts focus on the optimization of adiponitrile hydrogenation. One of the most
problematic points of the currently employed processes is the need of recycling the additive,
namely either anhydrous NH3 or NaOH. Therefore, processes utilizing smaller amounts of alkali
metal bases, alkaline earth metal oxides or oxides of lanthanoids as additives for hydrogenation
on a full-contact cobalt catalyst, consisting of at least 90% cobalt are reported. In addition to that,
metals like iron, nickel, PGMs and promotor elements are constituents of the full-fledged catalyst.
The system shows excellent yield on large scale and broad substrate scope.[25]

Daniel Timelthaler 20/126
Figure 3: Products and intermediates during the heterogeneous hydrogenation of adiponitrile.[17]
Recently, the group of Prechtl developed a new approach for hydrogenation employing nickel-
nanoparticles dispersed in an imidazolinium-based ionic liquid. This catalyst was able to perform
the hydrogenation of benzonitrile at catalyst loadings as low as 0.7% at moderate conditions.
Additionally, the group studied the effects of different ionic liquids on the catalytic surface
processes.[26]
Several relevant contributions were made by the group of Beller. In 2016, they developed a potent
catalyst upon immobilization and consecutive pyrolysis of a well-defined 1,10-phenanthroline
cobalt complex. The resulting particles were proven to be useful in the hydrogenation of C≡N and
C=O bonds at catalyst loadings of 4mol%. For nitriles, ammonia was used as an additive to ensure
selective formation of primary amines. Interestingly, the catalyst activity showed strong
dependency on the support material, with Al2O3-supports being superior to the other investigated
materials.[27] A similar system for the semi-hydrogenation of alkynes to alkenes utilizing cobalt in
an N-graphitic matrix was reported.[28] Furthermore, the group documented that an active cobalt
catalyst is obtained through pyrolysis of vitamin B12 on a cerium(IV)-oxide support.[29] For all these
catalyst systems, a significant dependency of catalyst activity on pyrolysis temperature was

Daniel Timelthaler 21/126
observed. This represents a major downside as preparation of the catalysts requires meticulous
monitoring of the catalyst preparation procedure.
An exceptionally simple catalytic protocol employing an in situ generated cobalt catalyst was
recently published by Guan and Dai. In this approach, the reaction vial is directly charged with dry
CoBr2 in THF and the nitrile substrate before 3 eq. of NaBHEt3 are added. Under 20 or 40 bar of
H2 gas, a broad variety of nitriles can be hydrogenated and their amine analogues yielded as
ammonium salts. Outstandingly, the system exhibits good selectivity towards primary amines,
enabling benzylamine yields of 92% in the reduction of benzonitrile, without addition of ammonia
or other additives. It was postulated by the group that the side products of catalyst activation, NaBr
and BEt3, played an important role in controlling the selectivity, most likely due to the Lewis acid
nature of the alkylborane. Interestingly, the reaction selectivity could be reversed by addition of
PNP-ligands, leading to the transformation proceeding via a homogeneous pathway which favored
formation of secondary imine coupling products over complete reduction to primary amines (see
Figure 4). This not only demonstrates the versatility of cobalt catalysis in nitrile hydrogenation, but
also reveals the enormous simplification potential held by in situ systems. However, the reported
process still requires the use of an expensive reducing agent and charging the reaction vessels
under inert conditions in a glove-box.[30]
Figure 4: Co-facilitated nitrile hydrogenation as reported by Guan et al.[30]
1.4. Homogeneous Cobalt and Nickel Catalysts for Hydrogenation
Despite the fact that heterogenous catalysis is usually favored in high-temperature, large-scale
processes, considerable research effort is directed to the development and improvement of
homogeneous catalytic systems. In comparison to its heterogeneous counterpart, homogeneous
catalysis holds certain distinct advantages. Most notably, homogeneous catalysts usually perform
well at much milder conditions, making them favorable for chemical transformation on sensitive
molecules. Also, selectivity of homogeneous catalysts usually outperforms their heterogeneous

Daniel Timelthaler 22/126
congeners, especially when stereoselective transformations are concerned. Additionally, metal
complexes serving as homogeneous catalysts tend to be easier to study with respect to their active
centers and the occurring transformations.
As shown in the previous chapters, cobalt and nickel hold outstanding potential in heterogeneous
catalysis. The knowledge of their hydrogenation capability in homogeneous catalytic systems
however, has been studied to a far less extent.
The first successful hydrogenation of nitriles to primary amines catalyzed by a homogeneous
cobalt complex was reported by the group of Milstein in 2015. They found a PNN-catalyst (see
Figure 5) to fully convert benzonitrile (100%) with a very high selectivity (96%) for primary amines
using a catalyst loading of 4% at 135 °C for 60 hours. The reaction required addition of NaBHEt3
as a hydride source and a strong base for which KHMDS, KOtBu and NaOMe proved applicable.
Although the group was not able to isolate the active form of the complex, it was postulated that it
formed upon deprotonation of the NH-group and the methylene moiety on the nitrogen-bearing
side chain, leading to formation of a monoanionic cobalt(I) hydride complex.[31] Similar catalytic
transformations under milder conditions and shorter reaction times, utilizing a biscarbene-ligand[32]
and transfer hydrogenations using a PNN-ligand[33] were reported as well. A few years before,
successful selective hydrogenations of alkenes, carbonyl compounds and imines using
cobalt-PNP pincer complexes were reported by the group of Hanson.[33][34] A slight alteration of
the ligand substituents was reported by the group of Kempe, that explored the hydrogenation
activity of a cobalt-PNP complex bearing a triazine backbone (see Figure 5). Interestingly, the
resulting catalyst no longer facilitated the hydrogenation of alkenes, selectively reducing C=O
bonds instead.[35] Moving to more challenging substrates, cobalt PNP-pincer were successfully
utilized in the hydrogenation of carboxylic esters. Even though harsh conditions and long reaction
times were required, a broad scope of alkyl esters was reduced to their respective alcohols.
Surprisingly, hydrogenation of aryl esters was not achieved, leading to the assumption that the
reaction proceeds via hydrogenation of the enolate form.[37] Impressively, even hydrogenation of
carboxylic acids was reported by the group of de Bruin under relatively mild conditions (100 °C,
80 bar H2) catalyzed by an insitu generated Co(triphos) assembly (see Figure 5).[38]
Figure 5: Cobalt precatalysts for hydrogenation of C≡N bonds[31] and C=O bonds[35], and activated catalysts for
hydrogenation of carboxylic acids[38] and enamides.[39]

Daniel Timelthaler 23/126
Recent work in the group of Chirik resulted in the development of a catalytically active
bisphosphine-stabilized dimeric Co(I) complex. Notably, reductive single-electron activation was
achieved employing abundant metallic zinc as the reducing agent. The high-spin active complex
(see Figure 5) surprisingly performed best in protic solvents like methanol and exhibited
outstanding selectivity in the enantioselective reduction of enamides (97.0% yield, 98.2% ee).
Furthermore, a Co(0)-complex stabilized by cyclooctadiene was obtained upon a second reduction
of the diphos-ligated Co(I) catalyst, enabling similar degrees of selectivity. The optimized process
represents an attractive alternative to the corresponding rhodium-based equivalent
transformation.[39]
Recent research on homogeneous nickel catalysts has indicated a strong focus on its ability to
effect the hydrogenation of alkenes. The group of Hanson put a simple Ni(II)PNP precatalyst to
use through activation with sodium borohydride. The resulting active complex facilitates
hydrogenation of alkenes such as styrene and 1-octene at 80 °C and 4 bar. Mechanistic studies
indicated that the transformation proceeded via insertion of the alkene into the Ni-H bond, thereby
forming a cationic Ni(II) complex. Product release was found to occur by direct reaction of H2 at
the cationic nickel center.[40] Similarly, employing a bisarylphosphine ligand, hydrogenation and
isomerization of 1-octene were achieved at 50 °C and 50 bar H2.[41] Furthermore, [(dippe)Ni(μ-H)]2
(dippe = 1,2-bis-(diisopropylphosphino)-ethane) was proven to facilitate several transformations
on enones, including simultaneous hydrogenation and reductive amination with primary amines[42]
as well as concerted hydrogenation and alkylation, using methanol as alkylating reagent.[43] The
potential of homogeneous catalysts for enantioselective hydrogenation of C=O bonds was further
demonstrated by the work of Hibino et al.[44]
Certain progress in the field of enantioselective hydrogenation of alkenes was made by the group
of Chirik. In the synthesis of α-unsaturated esters, enantioselective hydrogenation of the adjacent
C=C double bond under complete preservation of the ester group was achieved using a chiral
(S,S)-Me-DuPhos bidentate phosphine ligand with nickel acetate and tetrabutylammonium iodide
(see Figure 6). This transformation proceeded under mild conditions, tolerating a broad variety of
functional groups. The pivotal role of the carboxylate-group could be disclosed, by means of
deuterium labeling studies. This functional group was shown to facilitate the heterolytic cleavage
of the H-H bond.[45]
An alternative to phosphine ligands in homogenous hydrogenation protocols relying on nickel
complexes was again published by the group of Chirik. On treatment of air-stable nickel octanoate
and a DAB α-diimine ligand with an activator, most notably pinacolborane, formation of a dimeric
Ni(I) hydride complex was observed which exhibited catalytic activity in the hydrogenation of
unfunctionalized alkenes (see Figure 6). At exceptionally mild conditions of 50 °C and 4 bar H2
complete conversion and high yields could be realized. The hydride complex was compatible with
most nonpolar, aprotic solvents which gave the best results. Mechanistic studies, employing
deuterium gas and deuterated pinacolborane gave insight into the dual role of the activator. It

Daniel Timelthaler 24/126
facilitated the reduction of Ni(II) to form the active complex on the one hand and increased
hydrogenation activity on the other. Notably, the catalytic protocol demonstrates the possibility of
applying α-diimine ligands as mediators in selective hydrogenation reactions.[46]
Figure 6: Enantioselective hydrogenation of unsaturated esters[45] and the active Ni(I) hydride-complex for hydrogenation of unfunctionalized alkenes.[46]
1.5. BIAN Ligands
Another relevant family of the aforementioned α-diimine ligands is constituted by the
bis(aryl)acenatphtenequinone diimines, usually denoted as BIAN or Ar-BIAN. Ligands of this type
show similarities to 1,4-diaza-1,3-butadiene (DAB) ligands, with the distinct feature of BIAN
ligands being fused with a rigid and plane naphthalene backbone (see Figure 7).
Figure 7: Structural comparison between two commonly employed α-diimine ligands.
Owing to their unique structure, BIAN ligands hold a privileged position amongst diimine-based
catalyst. Their coordinative and electrochemical properties are mostly dominated by three
structural features:[47]
• A comparably high rigidity due to restricted bond rotation provoked by the naphthalene
unit.
• The interplay of the naphthalene and the diimine moiety greatly enhances the ligands
ability to function as an electron acceptor.
• The choice of substituents R situated on the aromatic units significantly influences the
stereo-electronic properties of the coordinating nitrogen atoms.

Daniel Timelthaler 25/126
For the DAB ligand as well as related α-diimine species, the flexible backbone leads to a
preferential s-trans configuration. This steric arrangement allows for minimization of electronic
repulsion between the nitrogen lone pairs and maximization of the orbital overlap between the
nitrogen lone pairs and the C=N π-orbitals. For BIAN ligands however, rotation around the C-C
bond connecting the imine groups is inhibited by the naphthalene backbone. This forces the
coordinating nitrogen atoms into close proximity which has been shown to improve the chelating
effect when acting as a Lewis base in complex formation. Additionally, the naphthalene group
enhances the imine’s resistance against hydrolysis and averts cleavage of the C-C bond
connecting the imine functionalities. This increased stability contributes to the ligand’s applicability
in catalytically active complexes.
When considering the reduction properties of the BIAN ligands, both structural units need to be
considered. Naphthalene has been proven to form useful radical anions upon treatment with alkali
metals. Diimine moieties on the other hand facilitate delocalization of electrons through
antibonding orbitals. The interplay of these effects lays the foundation for the non-innocent
behavior of BIAN ligands, allowing them to accommodate up to four additional electrons.[48]
On discussing the electronic properties of the imine nitrogen atoms, the high σ-donor and π-
acceptor capabilities must be highlighted.[49] This distinguishes BIANs from other ligands in which
the coordinating sp2-hybridized nitrogen is embedded in a cyclic system, such as phenanthroline.
This property greatly influences the coordination ability in metal complexes. The residues on the
aromatic systems adjacent to the nitrogen significantly influence the electron density on the Lewis
base by means of mesomeric and inductive effects. Introduction of bulky groups, especially in the
ortho-position, greatly influence the binding properties as well. Therefore, a judicious choice of the
R groups is pivotal for fine-tuning the BIANs binding properties.[47]
With regard to catalytic applications, BIAN ligands found use in the polymerization of olefins[50] as
well as in the hydrogenation of alkenes.[51] Moreover, coupling reactions were realized to great
success, for example Negishi-[52], Heck-[53] and Suzuki-Miyaura-cross couplings.[54] The most
common metals in catalytic applications of BIAN ligands are nickel, palladium and ruthenium.[47]
However, the full catalytic potential of this ligand class has certainly not been fully explored yet
and there are numerous investigations to be made.

Daniel Timelthaler 26/126
2. Aims and Objectives
The aim of this thesis was the design and development of novel catalytic systems for various
hydrogenation reactions. Special emphasis was put on avoiding the use of noble metals and
employing base metal catalysts for the reduction of selected substance classes. The work included
research on homogeneous and heterogeneous catalysts and the accompanying ligand design.
The detailed goals of the present work included:
• Synthesis and characterization of selected BIAN ligands using literature methods.
• Developing methods for reducing the imine motif in the BIAN framework, therefore
disclosing a novel class of diamine ligands that has only been sparsely reported in the
literature.
• Investigation of Ni(I)-hydride complexes ligated by Ar-BIANs as potential hydrogenation
catalysts formed in situ.
• Employing zinc metal as single electron reductant for cobalt-BIAN complexes and
investigation of the catalytic behavior of the BIAN-metal assemblies thus produced.
• Development and optimization of a convenient heterogeneous hydrogenation system
facilitating selective reduction of nitriles to primary amines by in situ generation of
catalytically active cobalt particles from readily available cobalt(II) salts and zinc metal.
• Elaborating the substrate scope, limitations, additive effects and kinetic properties of the
aforementioned system.

Daniel Timelthaler 27/126
3. Results and Discussion
3.1. Synthesis of Ar-BIAN Ligands
The synthesis of Ar-BIAN ligands was carried out by condensation of acenaphtenequinone with
various aniline derivatives. Simply refluxing the two components in acidic media was discouraged
by literature, thus a two-step synthesis via a Lewis acid complex was chosen. Therefore, the
synthesis followed a literature protocol which employs zinc chloride as Lewis acid (Figure 8).[55]
The reaction was carried out in acetic acid as solvent, and in case fluorinated groups were present
in the aniline, a small portion of toluene (~1/5 of total solvent) was added to ensure quantitative
precipitation of the desired complex. Literature recommended immediate addition of the aniline to
the mixture of acenaphtenequinone and zinc chloride and consecutive refluxing in acetic acid.
However, yields were found to be considerably higher if the sole mixture of acenaphtenequinone
and zinc chloride was stirred at 80 °C for a few minutes before adding the aniline. This process
was accompanied by darkening of the originally yellow suspension, which is most likely attributed
to a certain degree of coordination of quinone to the zinc chloride. In the next step, a small excess
of aniline was added and hereafter the mixture was heated to reflux. Literature recommends
keeping the mixture refluxing for 45 minutes which the authors found to be the point at which the
reversal reaction of imine hydrolysis countervailed the condensation reaction. In my studies
however, I found a certain dependency of the optimal reaction time on the electronic effects of
aniline substituents. While reactions employing electron-poor anilines like
3,5-bis(trifluoromethyl)aniline require long reaction times, electron rich anilines such as
4-methoxyaniline reached the equilibrium after a relatively short reaction time of only 15 minutes.
Figure 8: Synthesis of Ar-BIANs.

Daniel Timelthaler 28/126
The product of the first reaction step, the BIAN-Zn complex shows poor solubility in acetic acid
and precipitates as colorful solid that is readily collected on a frit upon vacuum filtration. Complete
removal of acetic acid from the product was found to be pivotal for satisfying progression of the
next reaction step. The zinc chloride was removed from the BIAN framework by demetallation with
an aqueous solution of potassium oxalate. Therefor, literature recommends a laborious procedure
in which the demetallation is performed by shaking a 2-phase system of BIAN-Zn complex in DCM
and potassium oxalate in water 3 times for 5 minutes each in a separatory funnel. However, this
was soon found to be unnecessary as removal of the zinc atom is achieved by shaking the mixture
once for 1-2 minutes or vigorously stirring it for 2 minutes in a flask. Prolonging the reaction time
for demetallation might actually have a detrimental effect on selectivity, as BIANs that were worked
up following the literature procedure were often found to contain a higher content of the
corresponding hemi-BIAN. Even though BIANs are reported to be exceptionally resistant to
hydrolysis compared to other imines, keeping the product away from water might still prove
favorable.
Employing this novel modified procedure, BIANs derived from several different anilines were
successfully synthesized with yields comparable to the literature. The results of the syntheses are
summarized in table 2. One exception was the preparation of the unsubstituted BIAN 1a which did
not reach satisfying yields if the standard protocol was employed. Investigations including
microwave reactions and procedures without use of Lewis acid ultimately led to the development
of a new procedure that dispenses with the use of acetic acid. Instead, a mixture of
acenaphtenequinone, zinc chloride and a large excess of aniline was simply refluxed in acetonitrile
for 4 hours, resulting in precipitation of the zinc complex in satisfactory yields.
Table 2: Ar-BIAN synthesis results.
Structure Abbreviation Color Yield / %
1a golden 37[a] / 68[b]
1b orange 43[a]
Daniel Timelthaler 29/126
1c bright red 55[a]
1d bright orange 46[a]
1e dark red 49[a]
1f yellow 35[a]
1g yellow 48[a]
[a] Conditions: 2.7 mmol acenapthenequinone, 6.8 mmol ZnCl2, 6 mmol aniline, 10 mL AcOH, 130 °C 15-45 min,
demetallation with K2C2O4 in H2O/DCM. [b] Conditions: 5.5 mmol acenaphtenequinone, 6.4 mmol ZnCl2, 71 mmol
aniline, 30 mL acetonitrile, 170 °C, 4 h
The BIAN-ZnCl2 complexes show slight differences in appearance compared to the demetallized
Ar-BIANs. For example, the pure ligand Mes-BIAN appears as bright red powder, whereas its zinc
complex exhibits a lighter orange color. Figure 9 compares the UV/Vis-spectra of the Mes-BIAN
1c and its zinc complex. It can be clearly seen that, while showing a similar absorption pattern,
the absorption of the zinc complex is slightly shifted to longer wavelengths. Hence, it seems that
coordination induces a bathochromic shift in absorption. The overall lower absorption in the visible
region also seems to be the cause for the observed brighter color. In NMR however, differences
in peak patterns are minute, the one observable difference being a slight upfield shift of the
aromatic signals most likely due to the lowering of electron density induced by the Lewis acidity of
the zinc chloride.

Daniel Timelthaler 30/126
300 400 500 600 700 800
0
1
2
Absorp
tion / a
.u.
Wavelength / nm
Mes-BIAN
Mes-BIAN-ZnCl2
Figure 9: UV/Vis-spectrum of BIAN 1c compared to its zinc complex.
Owing to their high symmetry, BIANs have been found to crystallize easily which allows for simple
purification by recrystallisation. Single crystals of 1c were obtained conveniently by slow
evaporation of dichloromethane from a concentrated solution. The obtained orange crystals were
analyzed by X-Ray diffraction crystallography and the corresponding ORTEP plot of 1c is depicted
in Figure 10. The ligand was found to crystallize in a monoclinic pattern exhibiting complete
C2-symmetry. The mesityl moieties adopt a conformation which minimizes sterical hindrance. The
bond lengths within the naphthalene core were found to be little altered by the adjacent imine
moiety, with an average value of 1.39 Å which closely resembles the mean literature value of
1.40 Å.[56] For the imine bonds, 1.266 Å were found compared to a literature value of 1.279 for
C=N bonds adjacent to an aromatic system. Analysis of the bond lengths and angles within the
five-membered ring reveal interesting information about the extent of aromaticity as can be seen
in Figure 11. In comparison to the aromatic and completely conjugated molecule acenaphthylene,
the bond connecting the two imine bearing carbon atoms (C7-C7a, see appendix) is elongated by
0.122 Å.[57] Also, when compared to average conjugated bonds adjacent to an imine moiety, an
elongation of 0.043 Å is manifested. This observation is in agreement with the literature, which
describes the aromaticity within the BIAN structure to be limited to six-membered rings.[47]
Furthermore, the molecular structure of the Mes-hemi-BIAN was established. In this molecule the
bond connecting the imine to the ketone moiety is stretched even further to 1.546 Å. Moreover, it
is revealed that the asymmetric substitution with heteroatoms inherently leads to a minor unilateral
bond elongation within the naphthalene backbone. The corresponding ORTEP structure of this
hemi-BIAN species is displayed in Figure 12.

Daniel Timelthaler 31/126
Figure 10: ORTEP representation of 1c in the crystal C2/c. Thermal ellipsoids were drawn at the 50% probability level
and 25 °C. Hydrogen atoms were left out for clarity.
Figure 12: ORTEP representation of mesylated hemi-BIAN in the crystal P1. Thermal ellipsoids were drawn at the 50%
probability level and 25 °C. Hydrogen atoms are shown.
Figure 11: Comparison of the degree of aromaticity between unfunctionalized acenaphthylene and Mes-BIAN 1c. The purple bonds exhibit a distinct difference in length which gives information about the extent of the aromatic system,
that is indicated by the red dashed lines.[47][57]
3.2. Reduction of Ar-BIAN Ligands
The next main topic of the present work was the elaboration of a generalized procedure for the
reduction of the imine groups in the BIAN framework, to afford the corresponding bulky secondary
diamine. Though the reduced BIAN has been reported in the literature, its applications as a ligand
in catalysis need to be explored, yet. The transformation itself has only been sparsely reported in
the literature but most often lithium aluminum hydride was used as the reducing agent.[58][59]

Daniel Timelthaler 32/126
The investigations commenced by treating compounds 1c, 1d and 1f with LiAlH4 in different
solvents, at varying conditions. The transformation quickly revealed to be more challenging than
expected. Addition of the reductant usually led to a prompt color change from orange to green.
This intermediate is described in the literature as an adduct of the semi reduced BIAN and lithium
aluminum hydride as highlighted in Figure 13.[60] After completion of the reaction the solution
rapidly readopted the initial colour. This observation is attributed to the fact that BIAN reduction
proceeds over two steps. Initially, uptake of two electrons and two hydrogen atoms generates the
air-sensitive BIANH2 which upon contact with air is reoxidized to the diimine again. Only a second
consecutive reduction step will form the air-stable BIANH4 target compound. These processes are
summarized in Figure 14.
Figure 13: Adduct of BIANH2 and lithium aluminum hydride as described by Sokolov et.al.[60]
As a lack of reactivity of the reductant seemed to be the problem, the temperature and loading of
the reducing agent were increased. However, this led to a rise in formation of decomposition
products. Especially reactions in THF were found to be unfavorable, most likely owing to the
propensity of this solvent to form radicals that hamper the desired chemical transformation.
A variety of reducing agents was then tested for their utility to enable the title reaction. Upon
reaction of then BIAN with sodium borohydride, the transformation again stopped at the
BIANH2-level, an effect that could not be altered on raising temperature, reaction time nor the
mass of reducing agent. If borane in THF was used, the same problem occurred. In contrast to
NaBH4 however, the product of borane reduction did not show significant impurities after being
reoxidized by air rendering it useful for synthesis of BIANH2. Employing NaBHEt3 for the reaction
lead to complete decomposition of the precursor as the solution turned black and NMR revealed
a diverse set of signals.
First success regarding the synthesis of BIANH4 was achieved by directly using the zinc complex
from the BIAN synthesis 1c-ZnCl2 for the reduction. This was accomplished by adding 4
equivalents of solid LiAlH4 to a suspension of the zinc complex in diethyl ether at -80 °C. The
reaction was slowly warmed up to room temperature and stirred for 18 hours (see Figure 15). After
work-up, this reaction was found to produce the fully reduced product 3c in 54% yield, which
outclassed the yields reported in the literature.[58] Employing the same procedure to 1d also gave
the desired product 3d even though the yield dropped to 26%. These experiments clearly indicated

Daniel Timelthaler 33/126
Figure 14: Overview and transformations of the three BIAN stages relevant in the reduction process.
that the presence of ZnCl2 either free in the reaction solution or complexed to the ligand
significantly enhanced reactivity. It can be postulated that the reaction pathway from BIANH2 to
BIANH4 is hindered by the buildup of negative charge at the reduced imines which disfavors further
reaction with hydride ions. The electron pulling effect of the Lewis acid would positively interfere
to counteract this charge build-up and alleviate the next reduction step. In the following studies,
coordinated and carried out by Paul Sunzenauer in our group, this effect was successfully
exploited by combining lithium aluminum hydride with AlCl3 before addition of the BIAN substrate,
forming an alane species which exhibits a strong activating effect as well as strong reducing
power. Employing this protocol, all investigate BIANs could be smoothly reduced with good
yields.[61]
Figure 15: Successful procedure for the BIANH4 synthesis.
The three BIAN reduction stages were further characterized by UV/Vis-spectroscopy owing to their
conspicuous difference in color. The UV/Vis-spectra and a picture of the products in solution are
displayed in Figure 16. The BIAN shows major absorbance in the low wavelength region with weak
absorption being measurable up to wavelengths of 500 nm. This is well in accord with the orange
color of solutions of Mes-BIAN in DCM. Regarding the BIANH2, which appears dark violet as a
solid and violet in liquid, it can be clearly seen that absorption is still present at wavelengths around
650 nm. For Mes-BIANH4 the conjugated system within the molecule is completely disrupted
leading to a hypsochromic shift to shorter wavelengths, i.e. into the UV range.

Daniel Timelthaler 34/126
300 400 500 600 700
0
1
2
3
Absorp
tion / a
.u.
Wavelength / nm
Mes-BIAN
Mes-BIANH2
Mes-BIANH4
Figure 16: UV/Vis-spectra and a photograph of solutions of the three reduction stages of Mes-BIAN. On the picture
from left to right: 1c, 2c and 3c.
In order to gain further insight into the structural implications of the reduction process, crystals of
the BIANH2 2c and BIANH4 3c suitable for XRD measurement were grown. Both compounds were
found to crystallize easily, even though obtaining pure crystals of 2c proved to be more challenging
because of its inherent air sensitivity and co-crystallization of 1c. Compound 2c was obtained by
passing a flow of dry nitrogen over a methanolic solution, achieving slow evaporation under inert
conditions whereas 3c was obtained upon slow evaporation of a solution in diethyl ether under
ambient conditions. 2c was obtained as dark violet crystals that were analyzed by X-Ray
crystallography under oil to avoid air contact. 3c formed clear, colorless crystals that were readily
measured under ambient conditions. ORTEP representations of the obtained structures are shown
in Figure 17 and 18, respectively.
A listing of selected bond lengths and angles is indicated in table 3. Comparison of the carbon-
nitrogen bond length for 1c and 3c shows close resemblance to the literature values for C=N and
C-N bonds of 1.279 Å and 1.469 Å respectively.[56] The bond connecting the two functionalized
carbon atoms C1 and C12 is elongated for 1c as discussed in section 3.1. whereas for 3c a bond
length of 1.567 Å shows similarity to the literature value for tertiary C-C bonds of 1.542 Å. For 2c
however, the C1-C12 bond features strong double bond character which substantiates the way the
molecule is usually drawn in molecular schemes (see Figure 14). The C-N bond length on the
other hand was found to lie in between the measured values for complete sp2-, and complete sp3-
character. Therefore, a certain degree of conjugation between the imine and enamine form can
be assumed. For the bond angles within the five-membered ring, the different conjugation states

Daniel Timelthaler 35/126
account for the difference between 1c and 2c while the sp3-character of the center in 3c shows
influence on the ring structure as well. Very interestingly, for 3c a strong degree of deformation
within the five-membered ring is observed. As visible in the ORTEP representation (see Figure
18), the ring is partly forced out of its planar geometry at the C12-carbon. This is also indicated by
the major difference between the bond angles at the two amine centers of 3c, exhibiting more than
5° difference by virtue of distortion. This deformation is most likely caused by steric hindrance of
the bulky mesityl groups that are present in cis-configuration. It is not clear whether this is the
most stable structure for the molecule or if the cis-structure was forced upon it by coordination of
a Lewis-acid during the course of the reduction.
Table 3: Selected bond lengths, angles and crystal parameters from X-Ray crystallography of the different reduction
states of Mes-BIAN.
Compound C1-N1
bond
length /
Å
C1-C12
bond
length / Å
C2-C1-C12
angle / °
C1-C12-C11
angle / °
Crystal
system
Space
group
Z
1c 1.266 1.517 106.57 106.57 Monoclinic C2/c 4
2c 1.383 1.361 108.60 109.50 Triclinic P1̅ 2
3c 1.484 1.567 102.43 107.52 Monoclinic P21/c 4
Figure 17: ORTEP representation of mesylated BIANH2 2c in the crystal P1̅. Thermal ellipsoids were drawn at the
50% probability level and at 25 °C. Hydrogen atoms are shown to highlight the semi-reduced character of the compound.

Daniel Timelthaler 36/126
Figure 18: ORTEP representation of mesylated BIANH4 3c in the crystal P21/c. Thermal ellipsoids were drawn at the 50% probability level and 25 °C. Hydrogen atoms are left out for clarity except for the amine moiety. The deformation
of the five-membered ring is visible.
3.3. Synthesis of a Ni(I)-BIAN Complex for Hydrogenation
The synthesized and characterized BIAN ligands were employed in synthesis of nickel-BIAN
complexes for application in catalytic hydrogenation. The catalytic activity of nickel(I)-complexes,
ligated by a DAB α-diimine ligand, for hydrogenation of unsubstituted alkenes has been proven
before by the group of Chirik[46] (see section 1.4.). The main goal of my research was to evaluate
whether the more rigid BIAN ligand represents a suitable alternative to the DAB group and which
alterations of catalytic activity and selectivity might result from this substitution.
I commenced my studies by synthesizing a nickel(I)-BIAN complex in a similar fashion as reported
for the DAB based system.[46] Therefor, bis-2,6-(isopropyl)BIAN 1d was suspended in pentane
and mixed with nickel octanoate which resulted in formation of a dark brown solid which
presumably contained the Ni(II)-BIAN complex as displayed in Figure 19. Then pinacolborane was
added as a reductant and hydride source, leading to formation of a blue-black complex which
showed high sensitivity to oxygen. Characterization of the product by NMR did not give clear
results owing to the paramagnetic nature of the complex, but a small peak at -14.5 ppm indicated
the presence of nickel hydrides. The same process was repeated employing
bis(trifluoromethyl)BIAN 1f, giving a dark red product.
For the sake of convenience in application, investigations of the catalytic potential of the complex
focused on the in situ generated species. The screening reactions for optimization of reaction
conditions were performed at a 1 mmol scale with catalyst loadings ranging from 10 to 2mol%.
The standard reaction setup proceeded as follows: A 4 mL glass vial was charged with a nickel
source (nickel octanoate or nickel bromide), 1 equivalent of BIAN ligand and 4 equivalents of
pinacolborane as an activator. The catalytic components were dissolved in 1 mL of solvent and 1
mmol of a substrate was added. The reactions were sealed with a septum-cap, which was

Daniel Timelthaler 37/126
Figure 19: Synthesis of a nickel(I) hydride complex ligated by BIAN chelators.
consecutively penetrated with a needle and hereafter 7 reaction vials were put in an autoclave
inlet. The autoclave was then pressurized with 10 to 50 bar of hydrogen and heated until the end
of the preset reaction time. After completion of the reaction and reaching room temperature, the
pressure was released from the autoclave, the reaction solutions were degassed upon vigorous
stirring on air, and finally an aliquot of the reaction mixture was analyzed by GC-MS.
It is worth mentioning here, that upon addition of pinacolborane as the last reagent, the color of
the solution immediately adopted a dark blue-violet color which was still present after completion
of the reaction. This color change could indicate the successful formation of the Ni(I) hydride
complex.
For substrate screening, four different compounds equipped with diverse functional groups were
chosen (see Figure 20), namely 1-methylcyclohexane (5a) as a trisubstituted alkene, benzonitrile
(6a) as a nitrile, and acetophenone (7a) as well as 2,2,2-trifluoroacetophenone (7b) as ketone
compounds. Results of selected catalytic transformations are listed in table 4, for a more detailed
documentation see table A 5 in the appendix.
Figure 20: Substrates for performance screening of nickel hydride complex ligated by BIAN.
The studies commenced with experiments conducted under relatively mild conditions (50 °C, 10
bar H2). However, as reactions on 5a and 6a did not give the desired results, temperature and
pressure were consequently raised. Screening of the aforementioned functional groups quickly
revealed poor performance of the catalyst in the hydrogenation of alkenes and nitriles. Even with

Daniel Timelthaler 38/126
high catalyst loadings, both conversion and yield usually did not exceed disappointing values
<10%. Apart from the desired hydrogenation of the double bond at 5a, double bond isomerization
was observed. For nitriles, a mixture of the primary amine and various coupling products were
observed (nitrile side products will be discussed in more depth in section 3.5.). The catalyst proved
to be more potent in the transformation of carbonyl compounds. However, conversions still did not
exceed 35%. As the abovementioned nickel complex, which is expected to act as the catalytic
species in this transformation, is sensitive to oxygen and moisture, the screening reactions were
repeated employing dry solvents and anhydrous nickel bromide as metal source. Disappointingly,
this did not improve catalyst performance whatsoever.
Studies of the impact of the nature of the BIAN ligand revealed very similar activity if 1b, 1c, or 1d
were employed, while 1e resulted in a decrease of conversion. As 1b, 1c, and 1d fall into the same
class of BIANs, with the main difference being the degree of steric hindrance, this trend is not
surprising, while 1e underwent cleavage at the C-O bonds making it less favorable for this
application. The high electron density at the ligating nitrogen atoms in 1e could also play a role in
the lower activity as it could result in a lower basicity of the hydride species.
Studies determining the preferential solvent were carried out using 7b as a substrate. It was
quickly found that protic solvents like alcohols were not suitable for this reaction as no conversion
was observed. This is attributed to the intrinsic low stability of hydride species in protic solvents.
Polar, aprotic solvents like 1,4-dioxane and dimethyl formamide gave some conversion but were
outperformed by nonpolar solvents. Among the latter, cyclohexane and n-hexane gave the best
results.
Prolonging the reaction time from 15 to 67 hours did not result in better yields, indicating that the
maximum TON of the system was already reached after 15 hours. Higher temperatures and higher
pressures gave some improvement, but still not satisfactory results. For some experiments,
complete reduction of the nickel to its metallic form was observed at 100 °C and 50 bar hydrogen
pressure. Furthermore, under these harsh conditions, traces of anilines were found in the GC-
chromatograms, indicating partial decomposition of the ligand.
Comparison of the different results of the screening experiments suggests dependence of the
catalyst performance dependency on the concentration of pinacolborane. Lowering the activator
loading from 4 eq. to 2 eq. was accompanied by a sharp decline of reactivity. This is expected
owing to the dual role of the activator as both a reducing agent towards the nickel(II)-species and
a hydride source. To further investigate the system, blank experiments leaving out certain parts of
the catalytic assembly were performed. In these studies, it was proven that pinacolborane without
any additives could almost convert similar amounts of substrate as the mixture of all three catalyst
components could. Even though it is unclear whether all observed conversions can be attributed
solely to the reducing properties of pinacolborane, the poor overall performance of the system did
not encourage and justify any further research into it.

Daniel Timelthaler 39/126
Table 4: Selected screening results for the nickel-BIAN hydride system[a]
Substrate Catalyst
loading /
mol%
Ligand Solvent Temperature /
°C
Conversion /
%[b]
Yield /
%[b]
5a 10 1d THF 100 7 7
5a 5 1d THF 100 1 1
5a 2 1d THF 100 1 0
5a 5 1f THF 100 1 0
5a 5[c] 1f THF 100 5 1
6a 10 1d THF 100 13 0
6a 5 1d THF 100 3 0
6a 2 1d THF 100 0 0
6a 5 1f THF 100 0 0
6a 5[c] 1f THF 100 1 0
7a 10 1b THF 100 13 12
7a 5 1b THF 100 5 5
7a 2 1b THF 100 1 1
7b 10 1b THF 100 33 33
7b 5 1b THF 100 15 15
7b 2 1b THF 100 5 5
7b 5 1c THF 80 13 13
7a 5 1b 1,4-Dioxan 100 6 1
7a 5 1b DMF 100 2 2
7a 5 1b Methanol 100 0 0
7a 5 1b Toluene[d] 80 7 7
7a 5 1b o-Xylene 80 5 5
7a 5 1b Cyclohexane 80 12 12
7a 5 1b n-Hexane[d] 80 11 11
7b 0 - Cyclohexane 80 0 0
7b 5[e] - Cyclohexane 80 0 0
7b 5[f] - Cyclohexane 80 10 10 [a] Reaction conditions: Substrate (1.0 mmol), nickel octanoate (as listed), ligand (equimolar to Ni), pinacolborane (4 eq. to Ni) in 1 mL solvent, temperature as listed, 50 bar H2, reaction time: 16 h. [b] determined by GC/MS. [c] NiBr2 used as Ni source. [d] dry solvent used. [e] only nickel octanoate used. [f] only pinacolborane used.
In summary, the in situ generated nickel(I)-BIAN hydride complex employing pinacolborane did
not live up to the expectations based on the performance of the DAB-ligated system.[46] Formation
of the hydride complex is indicated by a color change even though it is unclear whether this could
also be attributed to a sole change in oxidation state of the BIAN ligand. The system failed to
perform in hydrogenation reactions of alkenes, nitriles and carbonyl compounds, only yielding

Daniel Timelthaler 40/126
small amounts of the desired products. Whether these products were formed by reaction with the
nickel hydride complex or through simple reduction by pinacolborane was not clarified but the lack
of overall reactivity directed our research focus to other base metal systems.
3.4. Hydrogenation Facilitated by a Cobalt(I)-BIAN Complex
As the investigations on hydrogenation featuring nickel-BIAN complexes did not give the desired
results, research efforts were now directed towards the synthesis and utilization of a BIAN-ligated,
catalytically active cobalt complex. The basic idea revolved around employing active
cobalt(I)-species, which have been proven before to be potent catalysts for a variety of
hydrogenation reactions. As cobalt in the oxidation state +I is normally not stable in solution,
stabilizing ligands must be introduced in order to increase the life-time of the active species. In the
literature, this is most commonly achieved using phosphine ligands; most recently the group of
Chirik exploited the reducing power of zinc metal to reduce a phosphine-ligated cobalt(II) center
to the +I oxidation state and employed the reduced complex for the enantioselective hydrogenation
of enamides.[39] Based on the foregoing research on BIAN ligands, their potential in a similar
transformation was evaluated, exploring the principles of single-electron reduction by non-noble
metals in α-diimine complexes.
I commenced my studies by testing three different reactive metals for their ability to form the
respective diamond complex 8b as depicted in Figure 21. For this, a dry cobalt salt was dissolved
in THF in the glovebox and dried reducing agents plus BIAN ligand were added under inert
conditions. The reactions employing manganese and zinc both gave products of similar
appearance (dark, red-brown solid). However, NMR spectroscopy did not reveal any valuable
information owing to the paramagnetic nature of cobalt. ESI-MS-analysis of the zinc-reduced
sample did not point to the formation of 8b. Instead, the corresponding mass signals indicated the
formation of a monomeric cobalt-BIAN complex, coordinated by bromide and a THF-molecule.
The reaction which employed magnesium as a reducing agent gave a very different product as
the solution quickly turned violet upon addition of the Mg metal. Because of its strong reductive
potential, it can be assumed that this change in color is attributed to the formation of a BIANH2
species which is also affirmed by the fact that contact with oxygen led to a rapid color change to
orange. For this compound, ESI-MS also indicated the formation of certain metal complexes, but
the exact nature of these species could not be identified.
In order to render complexes such as 8b applicable for hydrogenation reactions, easy ligand
exchange by hydride ions must be ensured. For this purpose, the present halogens need to be
exchanged for more easily replaceable entities. Two suitable reagents for this purpose are sodium
methanolate and silver tetrafluoroborate. Even though formation of 8b could not be verified,
formation of a Co(I) species could have taken place. Therefore, the products were treated with the
aforementioned reagents. However, the reaction products did not exhibit significant change in

Daniel Timelthaler 41/126
appearance and in the case of sodium methanolate, no corresponding peaks were found in the
NMR spectrum, indicating that the ligand exchange reaction failed. Especially, as purification
proved to be difficult, a different synthesis approach was pursued.
Figure 21: Synthesis of active cobalt-BIAN complexes, route 1.
An alternative protocol centers around the formation of a cobalt(II)-BIAN complex, followed by
reduction with potassium metal. The process is depicted in scheme 23. The synthesis of 10c
proceeded readily by mixing a dry cobalt halide with the BIAN ligand in DCM at room temperature.
The products crystallized as a black solid in case of 10c, whereas a dark red solid was obtained
when 10e was used.
300 400 500 600 700 800
0
1
2
3
4
Absorp
tion / a
.u.
Wavelength / nm
8c
8e
Figure 22: UV/Vis-spectra of cobalt(II)-BIAN complexes 8c and 8e.
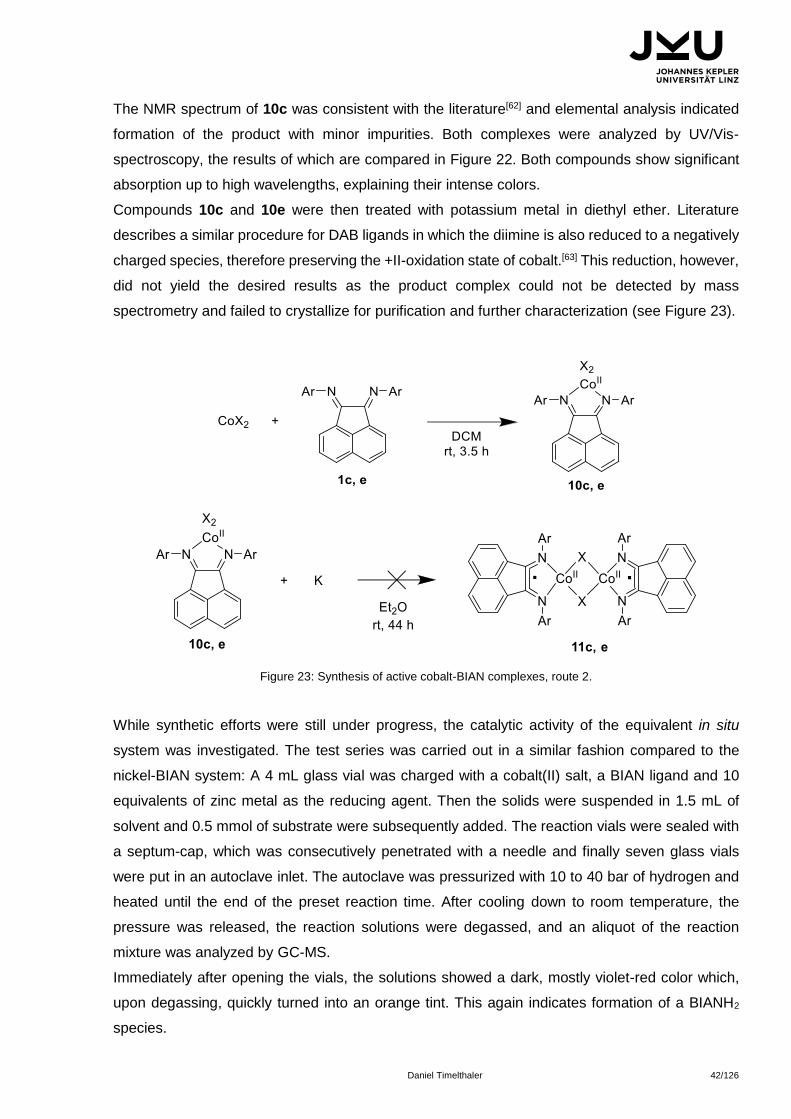
Daniel Timelthaler 42/126
The NMR spectrum of 10c was consistent with the literature[62] and elemental analysis indicated
formation of the product with minor impurities. Both complexes were analyzed by UV/Vis-
spectroscopy, the results of which are compared in Figure 22. Both compounds show significant
absorption up to high wavelengths, explaining their intense colors.
Compounds 10c and 10e were then treated with potassium metal in diethyl ether. Literature
describes a similar procedure for DAB ligands in which the diimine is also reduced to a negatively
charged species, therefore preserving the +II-oxidation state of cobalt.[63] This reduction, however,
did not yield the desired results as the product complex could not be detected by mass
spectrometry and failed to crystallize for purification and further characterization (see Figure 23).
Figure 23: Synthesis of active cobalt-BIAN complexes, route 2.
While synthetic efforts were still under progress, the catalytic activity of the equivalent in situ
system was investigated. The test series was carried out in a similar fashion compared to the
nickel-BIAN system: A 4 mL glass vial was charged with a cobalt(II) salt, a BIAN ligand and 10
equivalents of zinc metal as the reducing agent. Then the solids were suspended in 1.5 mL of
solvent and 0.5 mmol of substrate were subsequently added. The reaction vials were sealed with
a septum-cap, which was consecutively penetrated with a needle and finally seven glass vials
were put in an autoclave inlet. The autoclave was pressurized with 10 to 40 bar of hydrogen and
heated until the end of the preset reaction time. After cooling down to room temperature, the
pressure was released, the reaction solutions were degassed, and an aliquot of the reaction
mixture was analyzed by GC-MS.
Immediately after opening the vials, the solutions showed a dark, mostly violet-red color which,
upon degassing, quickly turned into an orange tint. This again indicates formation of a BIANH2
species.

Daniel Timelthaler 43/126
It is worth noting that in contrast to the foregoing synthesis experiments, the cobalt salt was used
in its hydrated form and the autoclave was charged under ambient conditions to evaluate the
resistance of the system to oxygen and water.
The first screening experiments focused on the determination of functional groups that could be
hydrogenated effectively. Besides the substrates already evaluated in Section 3.3., namely 5a,
6a, 7a, and 7b, two α-unsaturated carbonyl compounds: trans-chalcone 10a and isophorone 10b,
were employed. The structural drawings of the substrates are depicted in Figure 24.
Figure 24: Employed substrates from the class of α-unsaturated carbonyl compounds.
For the first experiments, 100 °C, 40 bar H2 pressure and a reaction time of 21 h were applied.
Compound 1b was selected as ligand since it is more convenient to synthesize than 1a and the
chelator exhibits neutral stereo-electronic properties. Readily available and cheap CoCl2*6H2O
served as cobalt source. To our delight, substrate 12a enabled >99% conversion and complete
selectivity for 1,4-hydrogenation. Also, benzonitrile 6a was fully converted, giving rise to a yield
of 68% of the primary amine. Ketone 7b allowed for complete hydrogenation to the alcohol,
however without the activating effect of the CF3-group, the observed yield of the corresponding
alcohol of 7a was only 48%. Hydrogenation of alkene 5a did not lead to any significant conversion.
The results of the first experiments are summarized in table 5. These first screening experiments
set the path for guiding the ongoing catalysis experiments. Primary focus was drawn to the most
promising functional groups, i.e. nitriles and α-β-unsaturated carbonyl compounds. Therefore,
optimization of the reaction conditions for these substrates was the primary goal in the following
studies. Special interest was paid to the effect of the different BIAN ligands on the reaction
outcome. Screening of a library of available BIAN ligands was carried out, employing benzonitrile
6a and isophorone 12b as model substrates. The results of the studies are summarized in Figures
25 and 26 whereas the corresponding values are listed in table 5, as well as table A 6 in the
appendix. For these studies several BIANs not described in section 3.1. were used. These were
mostly synthesized following the procedure described in section 3.1.[61] The additional employed
ligands are outlined in Figure 27.

Daniel Timelthaler 44/126
Table 5: Initial substrate screening and results for determination of BIAN-influence on the hydrogenation of benzonitrile 6a[a]
Substrate Ligand Conversion / %[b] Yield / %[b][c] Side products / %[b][d]
5a[e] 1b 1 1 0
7b[e] 1b >99 97 2
7a[e] 1b 51 48 3
10a[e] 1b >99 >99 0
6a[e] 1b 100 68 32
6a 1b 84 30 54
6a 1a 92 44 48
6a 1d 79 34 45
6a 1h >99 57 42
6a 1c >99 53 46
6a 1e 33 9 24
6a 1i 50 21 28
6a 1j 9 0 9
6a 1f 50 15 35
6a 1k 29 6 23
3b 1l 43 10 33
3b 1m 17 3 14
3a 1n 3 0 3 [a] Reaction conditions: Substrate (0.5 mmol), CoCl2*6H2O (5mol%), ligand (5mol%), zinc powder (50mol%) in 1.5 mL of methanol, 100 °C, 40 bar H2, reaction time: 14 h. [b] Determined by GC/MS. [c] For substrate 6a, the yield of primary amine is listed. [d] For substrate 6a, the total amount of coupling products is listed. [e] Reaction time was 21 hours.
Figure 27: Further BIAN ligands used for the screening experiments.[61]

Daniel Timelthaler 45/126
1b 1a 1d 1h 1c 1e 1i 1j 1f 1k 1l 1m 1n
0
20
40
60
80
100
Yie
ld / %
Ligand
Adducts
Benzylamine
Benzonitrile
Figure 25: Comparison of product composition in the hydrogenation of benzonitrile as a function of various BIAN
ligands. The corresponding numerical values are listed in table 5.
The results clearly underpin of the influence of the BIAN architecture on the outcome of the
hydrogenation. In general, electron deficient BIANs significantly decreased the catalytic activity of
the system in nitrile hydrogenation. Similar conclusions can be made on the electron- rich methoxy
substituted BIAN 1e. The only ligand from the class of alkyl BIANs 1i did not perform well, either.
The best yields of primary amines and almost full conversion were only achieved by the slightly
electron rich and moderately sterically hindered BIANs 1h and 1c. When considering the need to
form a coordination compound for successful catalysis, it has to be inferred that ligation proceeds
best with ligands displaying moderate sterical hindrance and an average electron density on the
coordinating nitrogen atoms. Moreover, ligands of this type could form complexes which are
intrinsically well suited for the hydrogenation process by means of their steric and electronic
properties.
Interestingly, for selective hydrogenation of the conjugated double bond in isophorone 12b, ligand
1e which did not perform well in hydrogenation of nitriles at all, clearly outperformed the other
employed ligands. For compounds 1a, 1b and 1c, even though complete conversion is realized,
there is a distinct lack in selectivity, as mostly but not exclusively, both unsaturated

Daniel Timelthaler 46/126
1b 1a 1d 1h 1c 1e 1i
0
20
40
60
80
100
Yie
ld /
%
Ligand
ring-opening
1,2-hydrogenation
1,4-hydrogenation
exhaustive hydrogenation, cis
exhaustive hydrogenation, trans
unreacted starting material
Figure 26: Comparison of product composition in the hydrogenation of isophorone as a function of various BIAN
ligands. Exact numerical values are listed in table A 6.
positions were reduced. This could also be attributed to the long reaction time of 64 hours,
although shorter reaction times with these ligands did not lead to the formation of more uniform
product mixtures either.
Despite the fact that reactivity pattern depends on the employed ligand as described above, the
role of the BIAN in the hydrogenation process became more questionable owing to the results
obtained in subsequent experiments. Analysis of the air-sensitive species that was observed in
the reaction solution after completion of the transformation was analyzed through UV/Vis-
spectroscopy. The UV/Vis-analysis of the violet species revealed that the compound of interest
showed identical absorption patterns as the pure BIANH2. This suspicion was further substantiated
by successful growth of crystals from the catalytic reaction solutions which, by X-Ray
crystallographic analysis, were proven to consist of the pure BIANH2, revealing no complexation
of cobalt. The following catalytic experiments, that were performed without the ligand suggest that
the actual catalytically active species had been misidentified: hydrogenation in this system did in
fact not proceed via a homogeneous Co(I)-BIAN complex but instead was catalyzed by
heterogeneous metallic cobalt particles that were formed on in situ reduction of Co(II) by zinc
metal. This was proven by a hot-filtration experiment. The experiment was performed by charging
vials including all catalytically active components and terminating the reaction after a period of 2
hours. Then all vials except a control sample were filtered and the filtrates were subjected to the

Daniel Timelthaler 47/126
reaction conditions again. In all examined reactions, this filtration caused the reaction to cease,
no matter if a BIAN was present or not. This proves the heterogeneous nature of the catalytic
system. At the end of the reaction, the metallic cobalt particles were finely suspended and hardly
visible in the reaction solutions owing to the presence of excess zinc metal and the strong color of
the dissolved BIAN.
The non-innocent nature of the ligand apparently simply led to the reduction of the imine moieties
which were reoxidized upon contact with oxygen. This could also explain why hydrogenations with
electron deficient ligands performed significantly worse than their electron rich counterparts, as
reduction of the cobalt center was effectively hindered by the presence of electropositive BIAN
chelators. Another factor could be incompatibility of the substrate with the present halogenated
anilines even though this assertion remains to be rather speculative.
However, even though the investigated catalytic system is indifferent with respect to the applied
BIAN ligand, it still proved high reactivity and has not been reported in this form, yet. Hence, the
following research efforts focused on exploration, optimization and application of a base metal-
driven heterogeneous hydrogenation reaction facilitated by a ligand-free Co(II)/Zn(0) couple.
3.5. Heterogeneous Hydrogenation employing a Co(II)/Zn(0) couple
The studies on the ligand-free system commenced by evaluating the reactivity towards different
functional groups. In addition to the already mentioned substrates, hydrogenation of alkynes,
carboxylic acids, carboxamides, imines, nitro-functionalities and heterocycles was performed.
Furthermore, the potential of the cobalt-based system for hydrodehalogenation was investigated.
An overview of the tested substrates and the corresponding catalytic results is given in the
appendix (Figures A 5 and A 7).
In summary, the studies revealed the following implications:
• For alkenes (5a) and carbonyl compounds (7a), conversions remained on an equally low
level as compared with the system deploying a BIAN ligand. If ammonia is used as an
additive for carbonyl hydrogenation, reductive amination becomes a dominant side
reaction.
• Nitriles (6a) still proved to be a well-suited substrate for the hydrogenation reaction.
• Unexpectedly, the reactivity towards α,β-unsaturated substrates (12b) decreased
significantly.
• In contrast to alkenes, alkynes (13a, 13b, 13c) proved to be convertible substrates for the
system. The formation of the corresponding alkene product was favored when lowering
the reaction temperature.

Daniel Timelthaler 48/126
• Carboxylic acids (14a, 14b, 14c) could not be hydrogenated, only minute formation of
esters owing to reaction of the acid with the methanol solvent was observed. Therefore, it
seems that hydrogenation of esters is also not facilitated by the system.
• Carboxamides (15a, 15ad) did not show any reaction except for minor ester formation (vide
supra).
• Imines (16a, 16b, 16c) could be hydrogenated well and considering the ease of this
transformation it was explored more in aspiration of understanding the role of imine
intermediates in nitrile hydrogenation rather than for actual substrate screening.
• Nitro-compounds (17a) did not exceed conversions of around 4%.
• Indole (18a), which was used as a heterocyclic substrate could only be hydrogenated in
minute quantities of approximately 3%.
• Hydrodehalogenation of bromobenzene (19a) could be finetuned such that yields of up to
56% were achieved. Especially the water content of the solution had a significant impact
on the performance of the catalytic system. This provided useful information on how to
suppress unwanted dehalogenation events during the course of hydrogenation of
halogenated substrates.
As a result of these studies, nitriles were chosen as the target substrates due to the promising
results and the industrial relevance of the transformation. Further research on hydrogenation of
alkynes will follow in the future.
In the initial experiments a mixture of 5 mol% CoCl2*6H2O and 25 mol% Zn was added to a
methanolic solution of benzonitrile 6a. After 15 h at 100 °C and 40 bar H2 pressure, full conversion
was achieved. However, a well-known and frequently reported problem was the moderate
selectivity of the reaction. In this test run only 63% of the primary amine 20a were formed, with the
rest of the reactant undergoing coupling reactions to form 21a and 22a. The usual way to cope
with this problem is the addition of ammonia to the reaction solution as it disfavors the elimination
of ammonia from the substrate, therefore, inhibiting condensation reactions. For my studies, two
different sources of ammonia and their effects were evaluated, namely a 2 M solution in of NH3 in
methanol (hereafter denoted with 'NH3*MeOH') and an 18 M aqueous solution ('NH3*H2O').
Addition of 1 equivalent of ammonia in methanol to the reaction mixture achieved a boost in
selectivity, yielding 98% of 20a. While it is obvious that the presence of ammonia in the reaction
solution contributes to the higher yields of the desired product, judicious choice of the ammonia
source is a key step to optimize the process: as the results in table 6 indicate, methanolic ammonia
solutions tend to give higher yields of benzylamine 20a after completion after 15 h. It was observed
that reactions employing methanolic ammonia mostly form no side products except for 21a, which
is also effectively reduced by the catalyst, giving an almost pure solution of 20a as a product. If
aqueous ammonia is applied on the other hand, the reaction tends to give side products such as
22a, 23a and 15a as indicated in Figure 28. The final amount of these byproducts usually did not

Daniel Timelthaler 49/126
exceed 1-2%, thus slightly reducing the overall yield of the primary amine. While 23a and 15a are
clearly the products of hydrolysis, the enhanced formation of 22a is a more peculiar phenomenon.
It seems as if reduction of the imine moiety in 21a is promoted by the presence of water, an effect
that was also observed in the hydrogenation of imine substrates 16b and 16c. A possible
explanation is that water interferes by means of nucleophilic catalysis thus activating the imine.
However, even though final yields of reactions that were performed with aqueous NH3 as additive
could not compete with the methanolic alternative in the hydrogenation of benzonitrile 6a, there
are a variety of substrates for which the reaction rate and selectivity by addition of NH3*H2O
outperformed the methanolic system. Therefore, during substrate screening, the aqueous system
was identified to be more versatile with respect to substrate scope.
The required amount of ammonia to achieve decent results without wasting chemicals by
unnecessary overdosing was found to be one molar equivalent with respect to the substrate. Table
6 gives an overview of the results of the optimization experiments. Lower loadings of ammonia led
to a decrease in conversion; e.g. full conversion was no longer reached if 0.5 molar equivalents
of NH3 were added. For higher loadings of ammonia, a decline of selectivity was observed. In the
literature, similar effects are reported which were attributed to a decrease in partial pressure of
hydrogen owing to the high concentration of ammonia in the gas phase.[20] Attempts to tune
selectivity by substituting ammonia with other bases such as triethylamine, diethylamine,
ammonium carbonate and sodium bicarbonate as shown in table 6 failed completely resulting in
a complete drop of conversion. These findings again demonstrated the importance and
distinctiveness of ammonia as an additive.
Figure 28: Observed reaction products in the cobalt-catalyzed hydrogenation of benzonitrile 6a.
The first screening experiments were carried out with metal loadings of 5 mol% Co. As similar
systems reported in recent literature could perform well with lower amounts of catalyst[30],
decreasing the amount of employed cobalt salt to 2mol% was set as a goal. However, simply
performing the reaction with 2mol% of Co(OAc)2*4H2O, 6mol% of Zn and 1 molar equivalent of
methanolic ammonia decreased the selectivity to 94% for 20a. Furthermore, with this setup, yields
were not fully reproducible and tended to decrease to around 80% in control experiments. The
most convenient way to ensure reliable results was addition of a small quantity of Lewis acid.

Daniel Timelthaler 50/126
Complementing the aforementioned system with 2mol% of zinc triflate very high yields of
approximately 99% 20a could be achieved in a reproducible manner. A further decrease of the
Lewis acid-loading to 1mol% however, brought the system to its limits as complete conversion
could no longer be achieved irrespectively of the Lewis acid activation (table 8). As alternatives to
Zn(OTf)2, other Lewis acids, namely ZnCl2 and Al(OTf)3 were tested. However, for the
hydrogenation of 6a, there was no significant enhancement of the catalytic activity of the system.
For more complex substrates on the other hand, certain preferences could be observed as will be
specified later on. By employing 2mol% of Lewis acid, catalyst loadings could be successfully
lowered to 2mol%. But considering the low price of the precursor salt, application of the 5mol%-
protocol without Lewis acid could also be appealing for certain applications to keep the system
more simple and to avoid the use of halogenated compounds.
Table 6: Studies of the influence of different bases and Lewis acid concentration on the heterogeneous Co(II)/Zn(0)
system.[a]
Base[b] Lewis acid Conversion /
%[c]
Yield 20a /
%[c]
Yield 21a /
%[c]
Yield
22a /
%[c]
500% NH3*MeOH none 100 83 17 0
200% NH3*MeOH none 100 87 13 0
100% NH3*MeOH none 100 95 4 1
50% NH3*MeOH none 68 31 37 0
200% NH3*MeOH[d] 2mol% Zn(OTf)2 100 98 2 0
100% NH3*MeOH[d] 2mol% Zn(OTf)2 100 >99 <1 0
50% NH3*MeOH[d] 2mol% Zn(OTf)2 97 51 45 0
100% NH3*H2O[d] 2mol% Zn(OTf)2 100 96 2 1
200% NHEt2 none 0 0 0 0
200% NEt3 none 0 0 0 0
200% NaHCO3[d] 2mol% Zn(OTf)2 0 0 0 0
200% NH4(CO3)2[d] 2mol% Zn(OTf)2 0 0 0 0
[a] Reaction conditions: benzonitrile 6a (0.5 mmol), Co(OAc)2*4H2O (5mol%), zinc powder (15mol %), in 1.5 mL of methanol, 120 °C, 40 bar H2, reaction time: 15 h. [b] Given percentage values refer to molar equivalents of the substrate. [c] Determined by GC/MS. [d] For this reaction, 2mol% of cobalt salt and 6 mol% of zinc were employed.

Daniel Timelthaler 51/126
Investigations into the effect of solvents and their interplay with the ammonia additives were
pursued next. Results thereof are summarized in table 7. The variation of the solvent for the
catalytic hydrogenation identified polarity as a crucial parameter. In order to elucidate the effects
of the reaction medium, experiments were performed at catalyst loadings of 5mol% without the
addition of any NH3. The obtained results clearly revealed the importance of polarity since aprotic,
nonpolar solvents such as toluene and n-heptane did not facilitate any catalytic conversion,
whereas polar solvents allowed for certain conversions. Interestingly, reactions conducted in
methanol performed outstandingly well, reaching full conversion without base addition whereas
polar, aprotic solvents like THF and MTBE or other alcohols such as isopropanol did not exceed
conversions of 30%. A rationale for this was found upon GC/MS analysis of samples taken from
reactions which were stopped before full conversion. In these samples, traces of a methanol
adduct 24a was detected. As this compound apparently formed by nucleophilic addition onto the
triple bond, it can be reasoned that nucleophilic catalysis as facilitated by polar groups in the
solvent, might be crucial to reaction progress.
Addition of ammonia to the different solvents completely change the results of the catalytic
transformations. Especially when NH3*H2O was employed, yields were significantly enhanced for
most solvents. For instance, 100mol% of aqueous ammonia in n-heptane enabled complete
conversion of the nitrile and gave rise to yields of 92% for the primary amine. This substantiates
the theory that a polar solvent could play a catalytic role in the system as the minute quantities
present in these solutions turned out to enhance reactivity in such a distinct manner. This also
allowed for fine-tuning of reactions which turned out to convert certain substrates which could not
be selectively hydrogenated in pure methanol but succeeded in a 2:1 mixture of methanol and
THF. Employing pure water as a solvent led to mediocre results and gave rise to large quantities
of side-products like 15a that are formed upon hydrolysis. To conclude, methanol still proved to
be the best suited solvent for the system and was preferably used for the experiments that
followed.
In the next step, the influence of the cobalt source on the catalytic performance was evaluated.
As the system proved to be well compatible with water, convenient use of hydrated cobalt(II) salt
was encouraged. The fact that this makes laborious charging procedures in the glovebox and use
of expensive equipment obsolete, renders the protocol even more appealing. Of the available
cobalt(II) salt hydrates, three were chosen for screening: CoCl2*6H2O, Co(BF4)2*6H2O and
Co(OAc)2*4H2O. As outlined in table 8, no differences in reactivity were observed when aqueous
ammonia solution was used as additive, whereas cobalt acetate performed best in methanol
solution. Following the principles of green chemistry, which discourages use of halogenated
compounds, cobalt acetate was defined as cobalt source of choice.

Daniel Timelthaler 52/126
Table 7: Results of solvent effects measurements for the heterogeneous Co(II)/Zn(0) system.[a]
Solvent Ammonia
source[b]
Conversion
/ %[b]
Yield
20a /
%[c]
Yield
21a /
%[c]
Yield
22a /
%[c]
Yield
23a /
%[c]
Yield
15a /
%[c]
Yield
24a /
%[c]
MeOH 100% NH3*MeOH 100 >99 <1 0 0 0 0
MeOH 100% NH3*H2O 100 96 2 1 0 0 0
MeOH[d] None 100 62 31 5 0 0 0
EtOH 100% NH3*MeOH 11 2 9 0 0 0 0
EtOH 100% NH3*H2O 100 95 1 3 1 0 0
i-PrOH 100% NH3*MeOH 56 26 30 0 0 0 0
i-PrOH 100% NH3*H2O 100 96 1 3 <1 0 0
i-PrOH[d] None 9 2 7 0 0 0 0
THF 100% NH3*MeOH 49 28 22 0 0 0 0
THF 100% NH3*H2O 30 25 5 0 0 0 0
THF[d] None 28 15 13 0 0 0 0
MTBE 100% NH3*MeOH 100 59 41 0 0 0 0
MTBE 100% NH3*H2O 100 94 4 2 0 0 0
MTBE[d] None 9 0 8 0 0 0 0
H2O 100% NH3*MeOH 89 27 52 0 0 10 0
H2O 100% NH3*H2O 60 16 33 0 0 10 0
toluene 100% NH3*MeOH 87 50 38 0 0 0 0
toluene 100% NH3*H2O 44 41 3 0 0 0 0
toluene[d] None 0 0 0 0 0 0 0
n-heptane 100% NH3*MeOH 20 11 8 0 0 0 0
n-heptane 100% NH3*H2O 100 92 6 2 0 1 0
n-heptane[d] none 0 0 0 0 0 0 0 [a] Reaction conditions: benzonitrile 6a (0.5 mmol), Co(OAc)2*4H2O (2mol%), zinc powder (6mol %), zinc triflate (2mol%) in 1.5 mL of methanol, 120 °C, 40 bar H2, reaction time: 15 h. . [b] Given percentage values refer to molar equivalents of the substrate. [c] Determined by GC/MS. [d] 5mol% of catalyst loading were used but no Lewis acid.

Daniel Timelthaler 53/126
Apart from these cobalt salts in which the metal is present in an oxidation state of +II, I investigated
the potential use of [Co(NH3)6]Cl3 as a precatalyst. It was anticipated that, a larger amount of
reductant would be needed due to its higher oxidation state, hence a higher loading of zinc was
employed. The intriguing fact about this notional precursor is the presence of six built-in
equivalents of base per equivalent of cobalt. Hence, this complex would not only serve as a metal
source but also provide the ammonia molecules that are mandatory for a decent catalytic
performance. Hence, because of this release of the NH3-ligands on reducing the metal, it was
postulated that extra addition of any ammonia solution to the reaction solution could be avoided.
Regrettably, actual experiments quickly discouraged this attempt as the reaction setup has a
surprising dependency on the conditions during the charging procedure. Preparing the reaction
solutions under ambient laboratory atmosphere resulted in formation of completely inactive
catalysts. Accordingly, all reaction vessels and the autoclave must be charged in the glovebox
under an Ar-atmosphere. But, as this tedious handling contradicts the appealing ease of the
system, the [Co(NH3)6]Cl3 approach was not pursued any further.
As can be seen in table 8, variations of the reaction temperature and hydrogen pressure were also
evaluated. It quickly turned out that reaction temperatures of 120 °C enabled better results
compared to experiments that were run at 100 °C. For substrate 6a, good results could be
achieved at 100 °C as well. However, this does not hold true for a variety of other substrates.
Therefore, the reaction temperature was fixed at 120 °C for most transformations. Reduction of
hydrogen pressure to 10 bar was accompanied by a steep decline in catalyst reactivity. The higher
pressure is vital for the hydrogenation process and was therefore set to 40 bar.
Finally, the effect of the reducing metal on the catalyst performance was elaborated. Investigations
started using a 10-fold molar excess of zinc with respect to the cobalt precursor salt. As shown in
table 9 however, there was no difference in reactivity when the loading was reduced to five or
three equivalents. According to the stoichiometric ratio of the electrochemical reduction of
cobalt(II) to its active elemental form, one equivalent of zinc should be sufficient for complete
conversion of the Co(II) salt. In practice however, this could not be verified as conversion of
benzonitrile dramatically dropped in case of only one equivalent of zinc being applied. To ensure
complete reduction in a reliable fashion, three equivalents of reducing agent were used thereafter.
Furthermore, the utility of other reactive metals for the Co(II)-reduction was investigated. Metals
of choice were manganese, magnesium as well as the metalloids boron and silicon. Of these
reagents, only manganese facilitated certain formation of catalytically active particles as
benzonitrile-conversions of 52% were observed. Magnesium showed complete dissolution, but no
hydrogenation of nitrile was achieved applying this reductant. This indicates that the reducing
agent is not only mandatory for providing a low valent oxidations state of the catalytically active
cobalt atoms but might also be engaged in a synergistic process that eventually gives rise to the
hydrogenation activity of the multi-component system. With respect to boron and silicon, neither
dissolution nor benzonitrile-hydrogenation were observed.

Daniel Timelthaler 54/126
Table 8: Studies of the effect of different cobalt sources, reaction temperature and pressure on the performance of the
heterogeneous Co(II)/Zn(0) system[a]
Cobalt source Ammonia
source[b]
Pressure
/ bar
T /
°C
Conversion
/ %[c]
Yield
20a /
%[c]
Yield
21a
/ %[c]
Yield
22a /
%[c]
Co(OAc)2*4H2O 100% NH3*MeOH 40 120 100 99 <1 <1
Co(OAc)2*4H2O 100% NH3*H2O 40 120 100 97 <1 2
CoCl2*6H2O 100% NH3*MeOH 40 120 100 81 11 <1
CoCl2*6H2O 100% NH3*H2O 40 120 100 97 <1 2
Co(BF4)2*6H2O 100% NH3*MeOH 40 120 100 67 1 3
Co(BF4)2*6H2O 100% NH3*H2O 40 120 100 98 <1 1
[Co(NH3)6]Cl3 none[d][e] 40 100 100 87 6 6
[Co(NH3)6]Cl3 none[d][f] 40 100 0 0 0 0
Co(OAc)2*4H2O[g] 100% NH3*MeOH 40 120 94 55 38 0
none 100% NH3*MeOH 40 120 0 0 0 0
Co(OAc)2*4H2O 100%NH3*MeOH 40 100 100 79 21 0
Co(OAc)2*4H2O 100%NH3*MeOH 40 120 100 >99 <1 0
Co(OAc)2*4H2O 200%NH3*MeOH 10 100 55 36 20 0
CoCl2*6H2O 200%NH3*H2O[e] Ar[h] 100 3 0 2 1 [a] Reaction conditions: benzonitrile 6a (0.5 mmol), cobalt salt (2mol%), zinc powder (6mol%), zinc triflate (2mol%) in 1.5 mL of methanol, temperature as indicated, H2 pressure as indicated, reaction time: 15 h. [b] Given percentages refer to molar equivalents of the substrate. [c] Determined by GC/MS. [d] 5mol% of cobalt salt and 25mol% of zinc were used, but no Lewis acid. [e] Charged in the glovebox under inert conditions [f] Charged under ambient conditions. [g] 1 mol% of cobalt salt, 3mol% of zinc and 1mol% of zinc triflate were used. [h] Reaction was carried out in a pressure tube under argon atmosphere.
With the optimized reaction conditions in hand, the reaction progress over time was explored in
more detail to gain further insights into the course of the reaction. For this purpose, 5 solutions
containing different combinations of additives were prepared and their composition was
determined after 1, 2, 4, 8 and 15 h, respectively. The compositions of these solutions were set
as follows:
I 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, 100mol%
NH3*MeOH, Solvent: 1.5 mL MeOH

Daniel Timelthaler 55/126
II 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, 100mol%
NH3*H2O, Solvent: 1.5 mL MeOH
III 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, Solvent:
1.5 mL MeOH
IV 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 100mol% NH3*MeOH,
Solvent: 1.5 mL MeOH
V 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, Solvent: 1.5 mL MeOH
All experiments were carried out at 120 °C, 40 bar H2 and a reaction time of 15 hours. The
cooling-off period before depressurizing the autoclave was 90 minutes. Before degassing, 1mL
of Methanol and 0.5 mmol of n-hexadecane as internal standard were added. Degassing
proceeded by stirring on air under ambient temperature for 15 minutes.
Table 9: Study of the nature of the reducing agent on the performance of the heterogeneous Co(II)/Zn(0) system.[a]
Cobalt
source
Ammonia
source[b]
Reducing
agent
Reducing
agent
loading
/ mol%
Conversion
/ %[c]
Yield
20a /
%[c]
Yield
21a /
%[c]
Yield
22a /
%[c]
Co(OAc)2*4H2O 200% NH3*MeOH Zn 25 100 98 1 1
Co(OAc)2*4H2O
100%
NH3*MeOH Zn 15 100 98 1 1
Co(OAc)2*4H2O
100%
NH3*MeOH Zn 5 53 25 26 0
Co(OAc)2*4H2O
100%
NH3*MeOH Zn 0 0 0 0 0
CoCl2*6H2O
200%
NH3*MeOH Mn 5 52 33 19 0
CoCl2*6H2O
200%
NH3*MeOH Mg 5 0 0 0 0
CoCl2*6H2O
200%
NH3*MeOH B 5 0 0 0 0
CoCl2*6H2O
200%
NH3*MeOH Si 5 0 0 0 0 [a] Reaction conditions: benzonitrile 6a (0.5 mmol), cobalt salt (5mol%), reductant as indicated, in 1.5 mL of methanol, 100 °C, 40 bar H2, reaction time: 15 h. [b] Given percentage values refer to molar equivalents of the substrate. [c] Determined by GC/MS.

Daniel Timelthaler 56/126
It is important to note that for each measurement, the solutions were freshly prepared to exclude
any possible catalyst deactivation by disruption of the reaction. The resulting time/concentration
diagrams are depicted in Figure 29. The corresponding values are listed in the appendix in table
A 8.
Figure 29 a) visualizes the differences described above between reactions employing NH3*MeOH
and NH3*H2O, respectively. The most distinct difference between the systems is the conversion
rate of the starting material. As can be seen, the reaction using ammonia in methanol requires a
total reaction time of 15 hours but shows almost quantitative yield of primary amine after
completion. In contrast to that, the reaction using aqueous NH3 is completed after only 8 hours
and doesn’t change its composition significantly over the remaining 7 hours. The total
concentration of benzylamine 20a in the aqueous reaction II however, is slightly lower than for the
methanolic system. Sample IV with added NH3*MeOH but not containing any Lewis acid, proceeds
in a very similar fashion as sample I but ultimately does not reach full conversion after 15 hours.
Possibly, longer reaction times could also lead to full conversion of this sample. The absence of
ammonia in the reaction mixture as in samples III and V causes almost no reactivity.
A more detailed study of samples I and II is shown in Figure 29 b) and 29 c) respectively.
Interestingly, the reaction seems to start with the accumulation of the imine coupling product 21a,
the concentration of which peaks around 3.5 hours for the aqueous, and 6 hours for the methanolic
system. During the last hours of the reaction, almost all nitrile 6a was already converted and further
0 2 4 6 8 10 12 14
0
20
40
60
80
100
with NH3MeOH, Zn(OTf)2
with NH3H2O, Zn(OTf)2
with Zn(OTf)2
with NH3MeOH
without additiveYie
ld B
enzyla
min
e / %
Time / h
a)
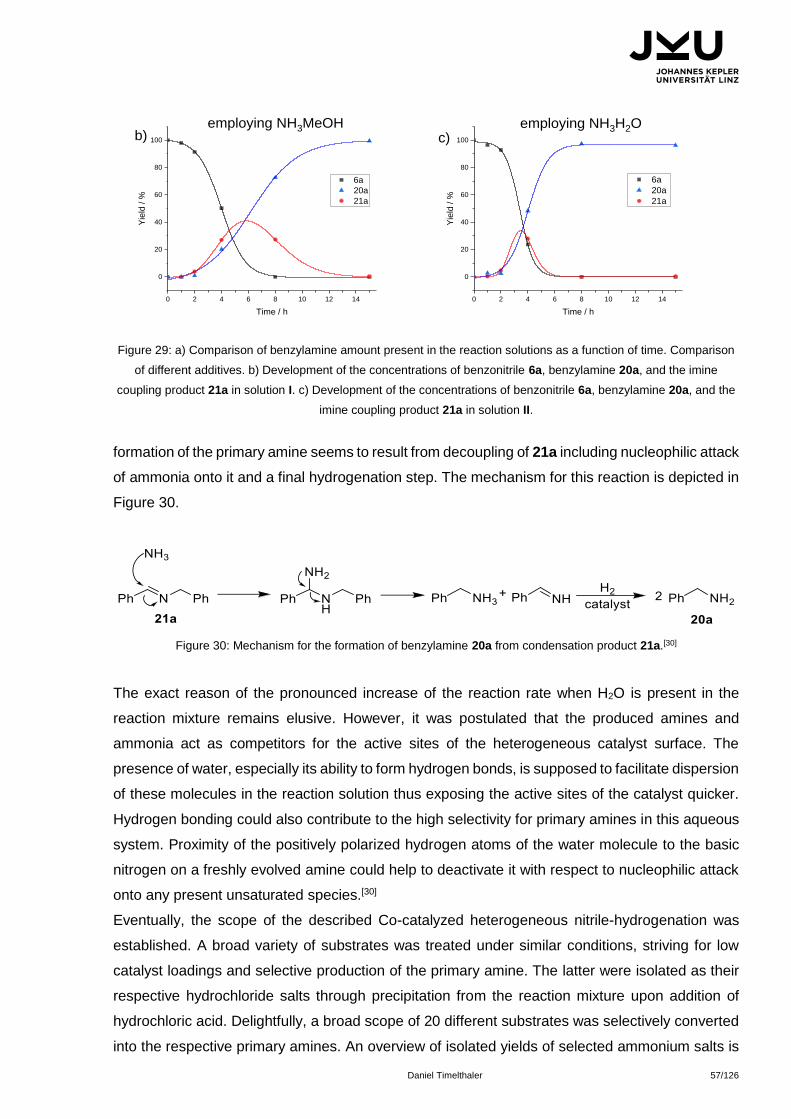
Daniel Timelthaler 57/126
0 2 4 6 8 10 12 14
0
20
40
60
80
100
6a
20a
21a
Yie
ld /
%
Time / h
b)employing NH3MeOH
0 2 4 6 8 10 12 14
0
20
40
60
80
100
6a
20a
21a
Yie
ld /
%
Time / h
c)employing NH3H2O
Figure 29: a) Comparison of benzylamine amount present in the reaction solutions as a function of time. Comparison
of different additives. b) Development of the concentrations of benzonitrile 6a, benzylamine 20a, and the imine
coupling product 21a in solution I. c) Development of the concentrations of benzonitrile 6a, benzylamine 20a, and the
imine coupling product 21a in solution II.
formation of the primary amine seems to result from decoupling of 21a including nucleophilic attack
of ammonia onto it and a final hydrogenation step. The mechanism for this reaction is depicted in
Figure 30.
Figure 30: Mechanism for the formation of benzylamine 20a from condensation product 21a.[30]
The exact reason of the pronounced increase of the reaction rate when H2O is present in the
reaction mixture remains elusive. However, it was postulated that the produced amines and
ammonia act as competitors for the active sites of the heterogeneous catalyst surface. The
presence of water, especially its ability to form hydrogen bonds, is supposed to facilitate dispersion
of these molecules in the reaction solution thus exposing the active sites of the catalyst quicker.
Hydrogen bonding could also contribute to the high selectivity for primary amines in this aqueous
system. Proximity of the positively polarized hydrogen atoms of the water molecule to the basic
nitrogen on a freshly evolved amine could help to deactivate it with respect to nucleophilic attack
onto any present unsaturated species.[30]
Eventually, the scope of the described Co-catalyzed heterogeneous nitrile-hydrogenation was
established. A broad variety of substrates was treated under similar conditions, striving for low
catalyst loadings and selective production of the primary amine. The latter were isolated as their
respective hydrochloride salts through precipitation from the reaction mixture upon addition of
hydrochloric acid. Delightfully, a broad scope of 20 different substrates was selectively converted
into the respective primary amines. An overview of isolated yields of selected ammonium salts is

Daniel Timelthaler 58/126
given in Figure 31, whereas Figure 32 collects substrate that were not amenable to hydrogenation
mediated by the investigated Co-catalyst described herein.
The substrate scope study revealed that the described in situ-prepared heterogeneous Co-catalyst
is of general applicability for the hydrogenation of a variety of nitriles by virtue of its decent group
tolerance. Benzylammonium chloride 25a was isolated in good yields, thus substantiating the
quantification results from GC/MS analysis. The catalyst facilitated hydrogenation of compounds
6f-k, with no indication of hydrodehalogenation side-reactions. Direct comparison of 25f, 25g and
25h showed some interesting effects. While the ortho-substituted chlorobenzonitrile showed very
selective hydrogenation, the meta- and para- substituted analogs could not be converted without
raising the loading of Lewis acid and switching from methanolic to aqueous ammonia as additive.
The reaction product 20m proved to be a special case as it could not be isolated on precipitation
with hydrochloric acid as the amide was cleaved hydrolytically. Alternative work-up by column
chromatography also led to decomposition of the product. Especially interesting was the
hydrogenation of the α-unsaturated cinnamonitrile 6n which gave rise to the reduction of the nitrile
group as well as the adjacent C=C double bond. Aliphatic nitriles were also smoothly
hydrogenated with similar isolated yields compared to their aromatic kindred. The reduction of
acetonitrile 6q to form ethylamine 25q proved to be challenging owing to the high volatility of the
product. The low observed yields are most likely attributed to partial evaporation of the ethylamine
during work-up.
Hydrogenation of adiponitrile 6r proved to be difficult at first. Employing standard reaction
conditions (with catalyst loadings of 5mol% to account for the second nitrile group) a mixture of
the desired hexamethylenediamine, as well as 6-aminohexanenitrile, originating from incomplete
hydrogenation, and ε-caprolactam was formed. Quite counterintuitively, contamination with the
hydrolysis product was avoided by using solely water as solvent. This led to the formation of a
2-phase reactant mixture which converted adiponitrile to hexamethylenediamine (yielded as
ammonium salt 25r) effectively, eliminating all other side-products as confirmed by GC/MS-
analysis. The reason for this is not completely understood. However, we anticipate that the good
solubility of the hydrogenation product 25r in water helps to shift the reaction equilibrium towards
complete hydrogenation, as the product is constantly separated from the reactant on changing the
phase. Moreover, the visual appearance of the reaction solutions in water after completion have
a very different appearance compared to reactions that were conducted in methanol. While in
organic solvents, the catalytically active particles tend to be finely dispersed, they seem to form
larger clusters in water. This might indicate that the formed cobalt particles show a certain degree
of hydrophobicity which might favor catalyst accumulation in the organic reactant layer floating on
top of the water, thus forming a neat reaction system. The constant contact with the phase
boundary is also likely to enhance the aforementioned regeneration of the catalyst surface. This
kind of system has not yet been reported for hydrogenation of nitriles. A similar procedure also
proved to be favorable for the heterogeneous hydrogenation of 25o.
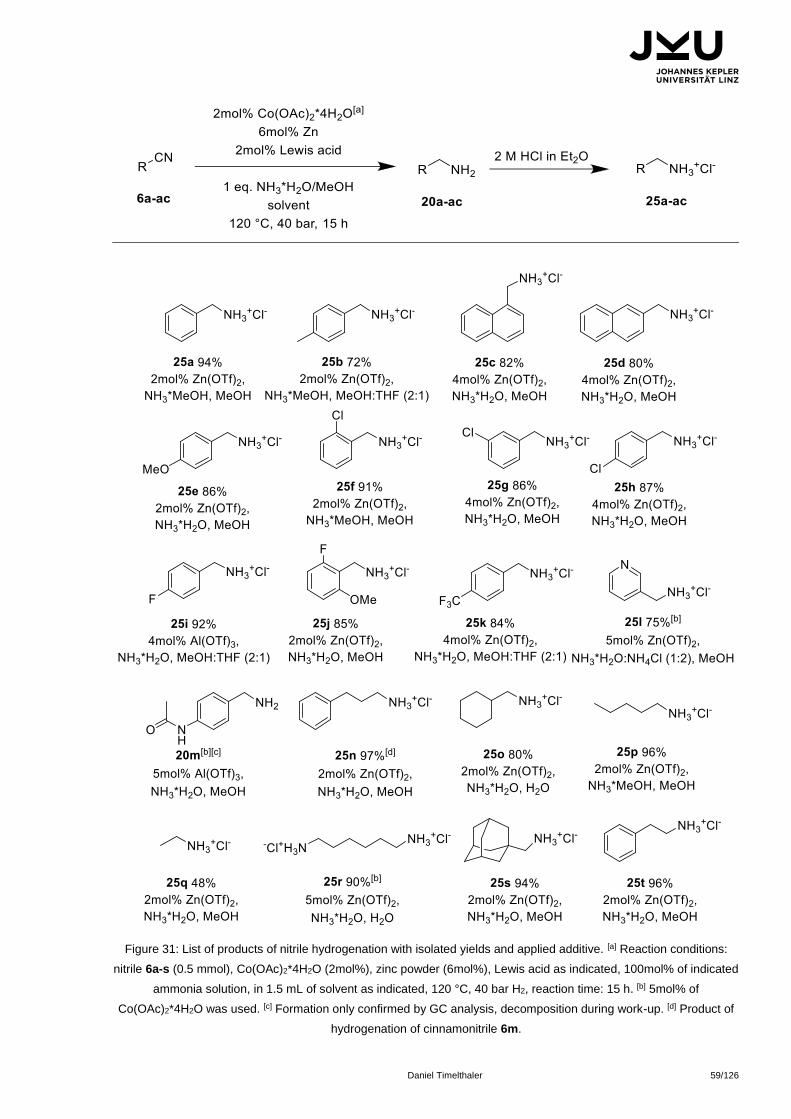
Daniel Timelthaler 59/126
Figure 31: List of products of nitrile hydrogenation with isolated yields and applied additive. [a] Reaction conditions:
nitrile 6a-s (0.5 mmol), Co(OAc)2*4H2O (2mol%), zinc powder (6mol%), Lewis acid as indicated, 100mol% of indicated
ammonia solution, in 1.5 mL of solvent as indicated, 120 °C, 40 bar H2, reaction time: 15 h. [b] 5mol% of
Co(OAc)2*4H2O was used. [c] Formation only confirmed by GC analysis, decomposition during work-up. [d] Product of
hydrogenation of cinnamonitrile 6m.

Daniel Timelthaler 60/126
However, substrate screening experiments revealed a problematic tendency for certain nitriles.
Especially substrates equipped with two functional groups which could act as potential
electrophiles showed a pronounced tendency for oligomerization. The occurrence of these
detrimental reactions was readily identified as a large amount of insoluble, intensely colored solids
was formed in the reaction vials after completion of the hydrogenation reaction. The process was
most likely initiated by multiple attack of nucleophilic amine-nitrogen atoms onto intermediate
electrophilic imine motifs. GC analysis of the reaction solution pertaining to benzonitrile-
hydrogenation allowed to identify a coupling product which gives a clue on how these oligomeric
structures might be formed. The identified structure is depictured in Figure 33. In order to curb the
formation of these undesired oligomers, certain techniques can be applied. Firstly, use of aqueous
ammonia instead of the corresponding methanolic solution proved to be a proper means to
suppress this unwanted side-reaction. Secondly, raising the amount of Lewis acid also counteracts
oligomerization, possibly by decreasing nucleophilicity of certain components present in the
reaction mixture. Lastly, the side-reaction seems to be facilitated in basic pH regimes. However,
the oligomerization was effectively suppressed on using a buffered solution containing a mixture
of NH4Cl/NH3 instead of applying pure ammonia. Using the abovementioned buffer also permitted
access to the hydrogenation of the heterocycle 6l, which was not compatible with the catalytic
protocol relying on NH3 as sole additive. The nucleophilic sp2-nitrogen atom is likely to cause
poisoning of the catalyst upon binding onto the cobalt-surface. However, the protons in the buffer
convert the substrate into a non-nucleophilic pyridinium salt which does not act a catalyst poison
any more.
Among the tested substrates listed in Figure 30, oligomerization occurred in the case of
compounds 6u, 6v, 6w, 6y as well as 6z and no amine was formed in reasonable yields. While 6x
was mostly immune to the catalyst, not showing any significant conversion, the thiophenes 6aa
and 6ab tended to form various condensation and dimerization products including the respective
amides.
Figure 32: Substrates that failed to be hydrogenated to the corresponding primary amine.

Daniel Timelthaler 61/126
Figure 33: Example of an oligomeric byproduct as formed in the cobalt-catalyzed heterogeneous hydrogenation of
benzylamine.
Another strategy for avoiding oligomerization was pursued for substrate 6u, which under usual
reaction conditions always produced various byproducts without any formation of the desired
primary amine. As outlined in Figure 34, this was circumvented by implementing a protecting
group, i.e. 1,3-dioxolane, in order to avoid nucleophilic attack of the formed amine onto the
sensitive aldehyde moiety. To our delight, the protected substrate was neatly hydrogenated to
afford the desired amine. Convenient acidic work-up with simultaneous deprotection and
precipitation furnished the ammonium salt 25u in an overall yield of 80%.
Figure 34: Scheme of circumventing polymerization of 6u by protection with ethylene glycol including pictures of the
reaction solutions.
In summary, the heterogeneous hydrogenation system featuring a Co(II)/Zn(0)-couple shows
excellent activity and selectivity for synthesis of primary amines from nitriles. The most striking
features of this system are its cheap components and the exceptionally simple handling under
ambient atmosphere without the need of inert gas techniques (Schlenk manifold, glovebox). The
catalytic protocol was optimized with respect of the reaction parameters (catalyst loading,
temperature, hydrogen pressure, solvent, additives) and its general applicability was
demonstrated for 20 different substrates, including aliphatic and aromatic nitriles. Although some
compounds were not initially compatible with the given catalytic system owing to the occurrence

Daniel Timelthaler 62/126
of side-reactions, several approaches for eliminating or circumventing this problem were devised,
rendering the catalytic protocol even more practicable.

Daniel Timelthaler 63/126
4. Conclusions
To conclude, I herein reported my advances in developing novel green catalytic hydrogenation
protocols. I performed studies on ligand design, synthesis and characterization of catalytically
active metal complexes and comprehensive studies of in situ systems.
I first conducted research into the synthesis and reduction of bis(aryl)acenaphthenequinone
diimines. The synthesis was readily carried out by condensation of acenaphthenequinone with
various anilines under the presence of zinc chloride (Figure 35). The obtained zinc complex was
subsequently demetallized on extracting with an aqueous solution of potassium oxalate. The
synthesis was convenient to apply and allowed the flexible synthesis of products with a variety of
stereo-electronic properties.
Figure 35: General synthesis of Ar-BIAN ligands.
The consecutive reduction of the ligands extended the knowledge of the redox properties of
sterically rigid α-diimines and their interaction with various reducing agents. While direct treatment
of the BIAN with lithium aluminum hydride did not yield the desired product, the same
transformation employing the corresponding zinc complex as starting material turned out to be a
potent method for preparing the BIANH4 species. These experimental findings gave insight into
the beneficial effects of Lewis acid activation on reduction of this ligand class. The BIANH2 was
found to be synthetically accessible upon reduction with the BH3*THF adduct. Besides preparative
work, studies of the crystal structures of the three BIAN states that evolve during the course of the
pertinent reduction provided information about the stereo-electronic consequences of this
chemical transformation of the BIAN framework (Figure 36).
Figure 36: Overview and interconversion of the three BIAN-reduction stages.

Daniel Timelthaler 64/126
The prepared BIANs were applied in a homogeneous hydrogenation system through coordination
to nickel(II) and subsequently activating the obtained complex with pinacolborane (Figure 37). This
approach was supposed to furnish a dimeric Ni(I) diamond-type hydride complex that mediates
the desired catalytic transformation. The in situ generated system was thoroughly studied in test
reactions with various substrates using molecular hydrogen under pressure. While the system
displayed some activity for certain substrates, especially ketones, the overall reactivity of the
system was found to be low and non-rewarding.
Figure 37: Setup for the Ni(I)-BIAN hydride-catalyzed hydrogenation of acetophenone.
Similarly, a homogeneous system applying the principle of single-electron reduction by zinc metal
to a Co(II)-BIAN complex and the product’s potential in hydrogenation reactions was investigated.
The system performed well for certain substrates such as nitriles and α,β-unsaturated carbonyl
compounds. Studying the effects of ligand substituents on the reaction performance suggested to
apply moderately electron-rich and sterically hindered BIANs. However, studies of the pertinent
catalytic species soon disproved the presumed catalyst concept, identifying the actual active entity
to be heterogeneous cobalt particles formed by in situ reduction with excess zinc metal (Figure
38).
Figure 38: Results of studies on the catalyst nature.

Daniel Timelthaler 65/126
The most thoroughly investigated system of the thesis was the liquid-phase hydrogenation
catalyzed by a Co(II)/Zn(0) couple. Screening the efficiency of the catalyst in the hydrogenation of
various functional groups identified alkynes, imines and nitriles to be reactants that were amenable
to the desired catalytic transformation (Figure 39).
Figure 39: Overview of the performance of the heterogeneous Co(II)/Zn(0) catalytic assembly in hydrogenation reactions of various functionalities. Green color: Good catalyst performance. Yellow color: moderate performance or major problems with side-reactions. Red color: Groups for which the catalyst showed no or very low catalytic activity.
Research efforts were then concentrated on the exploration of nitrile hydrogenation.
Comprehensive screening reactions allowed for optimization of reaction conditions by means of
catalyst loadings, temperature, H2 pressure, solvents, choice of precatalysts and additives. The
most important additive turned out to be ammonia, which enhanced reactivity and led to a dramatic
increase of selectivity regarding the formation of primary amines as main products. As a second
additive, Lewis acids proved to be potent for a further enhancement of the efficacy of the catalyst
assembly. Investigation of the composition of the reaction solution over time gave valuable insights
on the role of coupling products and water as an additive. The catalyst distinguishes itself from
known alternatives by its outstanding practicability, allowing for preparing the reaction setups
under ambient atmosphere and employment of cheap hydrated precursor salts.
Employing the protocol, a scope of over 20 substrates, including aliphatic and aromatic nitriles,
was hydrogenated with good yields. This also included the industrially relevant adiponitrile which
is used on large scale for the production of Nylon-6,6. The system displayed good functional group
tolerance, avoiding side-reactions like hydrodehalogenations. The main limit of the scope was the
occurrence of oligomerization reactions for certain substrates. Rewardingly, these side reactions,
were circumvented in some cases by increased additive loadings and the installation of protecting
groups.

Daniel Timelthaler 66/126
5. Experimental Section
5.1. General
All chemicals were purchased from Merck (including Sigma Aldrich), Acros Organics, Alfa Aesar,
VWR, Roth, TCI, Lancaster Synthesis or Chem Lab and were used as received without further
purification. NMR solvents were purchased from Deutero GmbH. Dry solvents were either
obtained from Acros Organics or received from a MB-SPS-7 solvent system from M. Braun GmbH.
Experiments under inert atmosphere were either carried out in a LABmaster pro glovebox from
Braun, filled with argon (6.0 purity from Linde Gas GmbH.) or on a Schlenk line from Glasbläserei
'Goldmann' connected to a Vacuubrand RZ 2.5 vacuum pump. Column chromatography was
carried out on silica gel-60 40–63 µm from Macherey-Nagel. For fine filtration, Celite 545 from TCI
was applied. Reactions under ultrasonic condition were carried out in a Bandelin Sonorex
ultrasonic bath. Hydrogenation reactions were carried out in a 300 mL autoclave from Parr
Instruments GmbH and the employed hydrogen was purchased from Linde Gas GmbH with a
purity of 5.0. For screening reactions, Braun Sterican 0.45x25mm needles were employed.
GC-MS analysis was carried out on a Shimadzu GC-MS QP-2020 with helium (5.0 purity from
Linde Gas GmbH.) as a carrier gas. UV/VIS absorption spectra were recorded on a Shimadzu UV-
1800 spectrophotometer. High-resolution mass spectrometry was performed on a Thermo Fisher
Scientific LTQ Orbitrap XL or an Agilent Technologies QTOF 6520 with ESI+ ion source for probes
with m/z < 50.
NMR measurements were performed on a Bruker Avance 300 MHz spectrometer. Spectra for
different cores were recorded as follows: 300 MHz for 1H-NMR, 75.5 MHz for 13C and 282.4 MHz
for 19F. Chemical shifts are listed in parts per million (ppm) on the delta scale (δ). Axis calibration
was performed using the residual non-deuterated solvent for 1H- and 13C-NMR as a reference.
Crystallographic measurements were performed on a Bruker Quest D8 Eco diffractometer using
monochromated Mo Kα radiation (λ = 0.71073 Å). Crystals were selected in Fomblin® Y H-VAC
140/13 perfluoropolyether at ambient temperature. Data processing was performed using
APEX3[64], cell refinement and data reduction was done using SAINT V8.37A. Structure solving
was done by intrinsic phasing (XT, Version 2014/15)[65] and refinement was performed by full
matrix least square procedures on F2 using SHELXL, Version 2014/7. For molecular graphics, the
interface Shelxle[66] was used. The material was refined for publication using PublCIF, Diamond
3.2k. All non-hydrogen atoms were assigned anisotropically. All hydrogen atoms were calculated
geometrically, and a riding model was applied in the refinement process.

Daniel Timelthaler 67/126
5.2. Ar-BIAN Synthesis
General method for the synthesis of 1b-1g[55]:
500 mg of acenaphthenequinone (2.7 mmol) and 1 g of anhydrous zinc chloride (7.3 mmol, 2.5
eq.) were placed in a 50 mL round-bottom flask and suspended in 10 mL of acetic acid. For 1f and
1g, a mixture of 7.5 mL of acetic acid and 2 mL of toluene was used instead. The flask was
connected to a condenser and the mixture was heated up to 80 °C and agitated for 30 minutes
until darkening of the suspension was observed. Then the aniline (6.0 mmol, 2.2 eq.) was added,
resulting in a sudden color change. The mixture was heated to 140 °C and kept at reflux for 15-45
minutes. The intensely colored zinc complex 1b-ZnCl2-1g-ZnCl2, was collected on a glass frit
(POR 4), and the solid was subsequently washed with 10 mL of cold acetic acid and 50-100 mL
of diethyl ether. The solid was then placed in a 100 mL separatory funnel and 50 mL of
dichloromethane was added, leading to partial dissolution of the complex. To this suspension, a
saturated solution of potassium oxalate (25 mL) was added and the mixture was shaken for two
minutes, quickly resulting in complete dissolution of the complex. Alternatively, the same mixture
can be vigorously stirred in a 100 mL round-bottom flask for two minutes. The colorful organic
layer was separated, dried with sodium sulfate and the solvent was removed by rotary evaporation,
yielding the crude BIAN as a powder. The crude was purified by recrystallization from a mixture of
THF and ethanol by slowly removing the THF at 55 °C on the rotary evaporator followed by cooling
it to 5 °C, yielding the pure product as colorful crystals.
Alternative method for synthesis of 1a:
1 g of acenaphthenequinone (5.5 mmol) and 0.9 g of anhydrous zinc chloride (6.9 mmol) were
mixed with 6.5 mL of aniline (71.2 mmol) before 30 mL of acetonitrile were added. The mixture
was refluxed for 4 h, resulting in a color change pale yellow to red. The solution was filtered through
a fritted glass filter (POR 4) yielding 1a-ZnCl2 as a yellow solid. After cooling the mother liquor for
a period of five days, additional yellow precipitate was formed which was unified with the initial
product. The solid was then placed in a 100 mL separatory funnel and 50 mL of dichloromethane
was added, leading to partial dissolution of the complex. To this suspension, a saturated solution
of potassium oxalate (25 mL) was added and the mixture was shaken for two minutes, quickly
resulting in complete dissolution of the complex. This process was repeated once, and the
combined organic layers were collected. The solvent was removed by rotary evaporation, leaving
behind a yellow solid. The crude product was recrystallized from 50 mL of methanol, yielding a
crystalline orange-golden solid.

Daniel Timelthaler 68/126
N1,N2-Diphenylacenaphthylene-1,2-diimine (1a)[55]
General method: Synthesized according to the general procedure except that the reaction was
scaled up by a factor of 3. Product was obtained as an orange-golden crystalline solid: 1.039 g
(3.22 mmol, 37% yield)
Alternative method: Golden crystalline solid: 1.252 g (3.77 mmol, 68% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.91 (d, J = 8.28 Hz, 2H), 7.50 (t,
J = 7.50 Hz, 4H), 7.39 (t, J = 7.76 Hz, 2H), 7.29 (t, J = 7.37 Hz, 3H, overlapping), 7.16 (d, J = 7.83
Hz, 4H), 6.86 (d, J = 7.23 Hz, 2H) ppm; 13C NMR (75.5 MHz, CDCl3, 20 °C): δ = 161.3, 151.8,
141.8, 131.2, 129.4, 129.0, 128.5, 127.7, 124.4, 124.0, 118.2 ppm.
N1,N2-Di-p-tolylacenaphthylene-1,2-diimine (1b)[55]
Synthesized according to the general procedure except that the reaction was scaled up by a factor
of 4.
Orange crystalline solid: 1.709 g (4.74 mmol, 43% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.88 (d, J = 8.31 Hz, 2H), 7.38 (t, J =
7.70 Hz, 2H), 7.27 (d, J = 7.17, 4H, overlapping), 7.03 (d, J = 7.53 Hz, 4H), 6.93 (d, J = 7.17 Hz,
2H), 2.45 (s, 6H) ppm; 13C NMR (75.5 MHz, CDCl3, 20 °C): δ = 161.4, 149.3, 141.8, 134.0, 131.3,
130.1, 128.9, 128.8, 127.7, 124.0, 118.3, 21.2 ppm.
N1,N2-Di-mesitylacenaphthylene-1,2-diimine (1c)[55]
Synthesized according to the general procedure except that the reaction was scaled up by a factor
of 4.
Bright red crystalline solid: 2.549 g (6.12 mmol, 55% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.88 (d, J = 8.28 Hz, 2H), 7.39 (t,
J = 7.73 Hz, 2H), 6.97 (s, 4H), 6.77 (d, J = 7.20 Hz, 2H), 2.38 (s, 6H), 2.10 (s, 12H) ppm; 13C NMR
(75.5 MHz, CDCl3, 20 °C): δ = 161.1, 146.9, 140.6 132.9, 131.0, 129.8, 129.0, 128.8, 128.3, 124.7,
122.5, 21.0, 17.8 ppm; UV/VIS (DCM) λmax = 281, 308, 420 nm.

Daniel Timelthaler 69/126
N1,N2-Bis(2,6-diisopropylphenyl)acenaphthylene-1,2-diimine (1d)[55]
Synthesized according to the general procedure except that the reaction was scaled up by a factor
of 2.
Bright orange crystalline solid: 1.365 g (2.73 mmol, 46% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.87 (d, J = 8.28 Hz, 2H), 7.36 (t,
J = 7.77 Hz, 2H), 7.29 – 7.24 (m, 6H, overlapping), 6.63 (d, J = 7.14 Hz, 2H), 2.94 – 3.12 (m, 4H),
1.23 (d, J = 6.84 Hz, 12H), 0.97 (d, J = 6.78 Hz, 12H) ppm; 13C NMR (75.5 MHz, CDCl3, 20 °C):
δ = 161.0, 147.5, 135.4, 131.1, 129.5, 128.8, 127.9, 124.3, 123.5, 123.3, 28.6, 23.4, 23.1 ppm.
N1,N2-Bis(4-methoxyphenyl)acenaphthylene-1,2-diimine (1e)[55]
Synthesized according to the general procedure except that the reaction was scaled up by a factor
of 3.
Dark red crystalline solid: 1.596 g (4.07 mmol, 49% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.89 (d, J = 8.25 Hz, 2H), 7.39 (t, J = 7.74,
2H), 7.14 – 7.06 (m, 4H), 7.06 – 6.98 (m, 6H), 3.90 (s, 6H) ppm; 13C NMR (75.5 MHz, CDCl3, 20
°C): δ = 161.7, 157.0, 145.0, 141.8, 131.4, 129.0, 128.9, 127.7, 123.8, 119.9, 114.8, 55.6 ppm.
N1,N2-Bis(3,5-bis(trifluoromethyl)phenyl)acenaphthylene-1,2-diimine (1f)[55]
Yellow crystalline solid: 0.586 g (0.97 mmol, 35% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 8.04 (d, J = 8.28 Hz, 2H), 7.81 (s, 2H), 7.62
(s, 4H), 7.50 (t, J = 7.80 Hz, 2H), 6.87 (d, J = 7.26 Hz, 2H) ppm; 13C NMR (75.5 MHz, CDCl3, 20
°C): δ = 162.5, 152.4, 142.6, 133.3 (q, J = 33.6 Hz), 131.7, 130.6, 128.3, 127.6, 125.1, 124.1,
121.5, 119.1, 118.4 (t, J = 3.7 Hz) ppm; 19F-NMR (282.4 MHz, CDCl3, 20 °C): δ = -62.9 (s, CF3)
ppm.

Daniel Timelthaler 70/126
N1,N2-Bis(3-(trifluoromethyl)phenyl)acenaphthylene-1,2-diimine (1g)[55]
Yellow crystalline solid: 0.625 g (1.33 mmol, 48% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.96 (d, J = 8.31 Hz, 2H), 7.66 – 7.52 (m,
4H), 7.47 – 7.39 (m, 4H), 7.34 (d, J = 7.62 Hz, 2H), 6.85 (d, J = 7.26 Hz, 2H) ppm; 13C NMR
(75.5 MHz, CDCl3, 20 °C): δ = 161.8, 151.7, 142.1, 132.2, 131.4, 130.1, 129.7, 127.9, 125.8,
124.0, 122.3, 121.8, 121.3, 115.5 ppm; 19F-NMR (282.4 MHz, CDCl3, 20 °C): δ = -62.7 (s, CF3)
ppm.
5.3. Reduction of Ar-BIAN
Method for reduction of 1c-ZnCl2 and 1d-ZnCl2:
The BIAN-ZnCl2 complexes (3.4 mmol) were suspended in 10 mL of diethyl ether and cooled to -
80 °C in a mixture of isopropanol and liquid nitrogen. Then 500 mg of solid lithium aluminum
hydride (13.2 mmol, 3.9 eq.) were added and the mixture was stirred for 18 hours, allowing for
slow acclimatization to room temperature. The resulting green-turquoise suspension was mixed
with 5 mL of acetone to destroy any remaining reductant and was subsequently filtered through a
glass frit (POR 4) twice to remove inorganic residues. Remaining zinc complex was demetallized
by shaking with 10 mL of saturated sodium oxalate solution, and the resulting organic phase was
left on air for slow evaporation, affording the product as large colorless crystals.
N1,N2-dimesityl-1,2-dihydroacenaphthylene-1,2-diamine (3c)
White colorless crystalline solid: 0.779 g (1.85 mmol, 54% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.70 (d, J = 8.22 Hz, 2H), 7.40 (t, J =
7.55 Hz, 2H), 6.98 (d, J = 6.87 Hz, 2H), 6.78 (s, 4H), 5.31 (s, 2H), 3.94 (s, NH), 2.30 (s, 6H), 2.18
(s, 12H) ppm; 13C NMR (75.5 MHz, CD2Cl2, 20 °C): δ = 144.8, 142.7, 135.9, 131.8, 131.1, 130.0,
129.4, 128.4, 124.3, 120.6, 62.5, 20.7, 19.0 ppm; UV/VIS (DCM) λmax = 290 nm.

Daniel Timelthaler 71/126
N1,N2-bis(2,6-diisopropylphenyl)-1,2-dihydroacenaphthylene-1,2-diamine (3d)
Synthesized accordingly to the described method but 0.76 mmol of zinc complex and 6
equivalents of LiAlH4 were used.
White colorless amorphous solid: 0.100 g (0.20 mmol, 26% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.71 (d, J = 8.22 Hz, 2H), 7.37 (t,
J = 7.56 Hz, 2H), 7.25 – 7.19 (m, 6H, overlapping), 6.73 (d, J = 6.90 Hz, 2H), 5.36 (s, 2H), 4.29
(s, NH), 3.30 (sep, J = 6.77 Hz, 4H) 1.23 (dd, J1 = 6.80 Hz, J2 = 15.35 Hz, 24H) ppm.
5.4. Synthesis of a Ni(I)-BIAN Complex for Hydrogenation
Method for synthesis of Ni(I)-BIAN complex 4d and 4f:
The respective BIAN ligand (0.4 mmol) was dried before use and placed in a dry 50 mL Schlenk
flask equipped with a septum. Then 8 mL of dry pentane were added resulting in incomplete
dissolution. After that 308 µL of an 8% solution of Ni octanoate in mineral spirits (0.4 mmol, 1 eq.)
were added and the mixture was stirred. Over the course of 1 hour, dry dichloromethane was
subsequently added to achieve complete dissolution of the ligand. The solution was stirred for 4
h under nitrogen. Then 232 µL of pinacolborane (1.6 mmol, 4 eq.) were added and the mixture
was stirred for another 12 h at room temperature. Removal of the solvent in vacuo resulted in a
colorful solid which was recrystallized from dry pentane.
Bis((hydrido,N1,N2-Dimesitylacenaphthylene-1,2-diimine)nickel(I)) (4d)
Dark-blue crystalline solid, yield not determined.
Bis((hydrido,N1,N2-Bis(3,5-bis(trifluoromethyl)phenyl)acenaphthylene-1,2-diimine)nickel(I)) (4f)
Dark-red amorphous solid, yield not determined.
Protocol for screening of in situ reactions:
A 4 mL glass vial equipped with a magnetic stirring bar was charged with BIAN ligand (0.02 – 0.1
mmol) and 1 mL of solvent was added. To the resulting mixture, an 8% solution of Ni octanoate in
mineral spirits (0.02 – 0.1 mmol) was added. If nickel bromide was used as a nickel source, it was
placed in the vial before the solvent was added. The substrate (1 mmol) was then added, followed

Daniel Timelthaler 72/126
by pinacolborane (0.8 – 0.4 mmol). The dark blue-violet solution was sealed with a septum cap
which was subsequently penetrated with a needle. The thus prepared reaction vials were placed
in an autoclave which was tightly sealed. The autoclave was purged with hydrogen three times
before being pressurized. After that, the autoclave was placed on a heating plate and the content
was stirred for the required reaction time. After completion of the reaction, the heating source was
removed, and the autoclave was allowed to reach room temperature. After that, the pressure was
released, and 1 mL of solvent was added to each of the reaction vials. Thereafter, the solutions
were degassed by stirring on air for 30 minutes. Then, an aliquot of 20 µL was taken from each
vial, mixed with 1 mL of methanol and analyzed by GC-MS. If substrate 7b was employed, the
GC-MS analysis sample was dissolved in THF instead owing to the low stability of the substrate
in methanol.
5.5. Synthesis and Screening of Co(I)-BIAN Complexes
Attempted synthesis of 8b:
In a glovebox filled with argon, 98 mg of dry CoBr2 (0.45 mmol) were placed in a 50mL Schlenk
flask and dissolved in 20 mL of dry THF, resulting in a blue solution. The flask was removed from
the box and connected to a Schlenk line. Then, 16 mg of vacuum-dried zinc metal (0.25 mmol,
0.55 eq.) were added and the solution was refluxed for 24 hours. Upon addition of 160 mg of dry
1b (0.45 mmol, 1 eq.) and further refluxing for 17 h, a brown suspension was obtained. Inert
filtration by cannulation allowed for separation into a brown solid and a green solution. Another
portion of 16 mg of zinc metal (0.25 mmol) and 10 mL of dry THF were added to the solid phase.
After 5 h of refluxing, no dissolution of the additional zinc was observed, and the solvent was
removed in vacuo. The brown product was analyzed by ESI-MS.
Attempted synthesis of 9b:
To the product of the aforementioned synthesis, 27 mg of dry sodium methanolate (0.50 mmol)
were added and the mixture was dissolved in 15 mL of dry acetonitrile. Refluxing for 44 h did not
result in any color change. Inert filtration by cannulation gave a brown solution and a green-black
residue. The solvent was removed in vacuo and the resulting black solid was analyzed by NMR in
benzene-d6.
Synthesis of 10 c, e:[62]
150 mg of dry CoCl2 (1.18 mmol) were dissolved in 20 mL of DCM. Then a solution of 1c or 1e
(1.18 mmol, 1eq.) in 30 mL of DCM was added dropwise over 15 minutes. The solution was stirred
at room temperature for 6 h. The resulting dark solution was filtered through a glass frit (POR 4),
yielding a crystalline solid. Reducing the solvent volume followed by cooling to -40 °C gave

Daniel Timelthaler 73/126
additional product of similar purity. The crude was recrystallized from a mixture of dichloromethane
and heptane yielding the product as dark small crystals.
Dichloro,N1,N2-Dimesitylacenaphthylene-1,2-diimine)cobalt(II) (10c)[62]
Black crystalline powder: 541 mg (0.99 mmol, 84% yield)
Analytical data: 1H NMR (300 MHz, CD2Cl2, 20 °C): paramagnetic, peaks not assignable. δ = 24.78
(s), 16.18 (s), 7.77 (s), 2.16 (d, J = 8.49 Hz), 1.27 (s), 1.12 (s), 0.92 – 0.80 (m) -1.42 (s) ppm;
UV/VIS (DCM) λmax = 262, 315 nm.
Dichloro,N1,N2-Bis(4-methoxyphenyl)acenaphthylene-1,2-diimine)cobalt(II) (10e)[62]
Synthesized in accordance to the described method except that the reaction was scaled down by
a factor of 0.8.
Red amorphous powder: 355 mg (0.68 mmol, 74% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): paramagnetic, peaks not assignable. δ = 11.37
(s), 7.26 (s), 5.32 (s), 3.91 (s), 2.15 (s), 1.68 (s), 1.24 (s), 0.86 – 0.80 (m), -2.08 (s) ppm; UV/VIS
(DCM) λmax = 440 nm.
Attempted synthesis of 11c:
550 mg of compound 10c (1.00 mmol) were dried in vacuo and 40 mL of dry diethyl ether were
added. Stirring for 5 h resulted in incomplete dissolution of the starting material. Then two portions
of 40 mg of potassium metal (1.00 mmol, 1 eq.) each, were freed from oil through washing with n-
heptane and dried in a flow of N2. After adding the first reductant portion, the reaction was stirred
for 14 h at room temperature. After addition of the second portion, the flask was kept in an
ultrasonic bath for 30 minutes to foster dissolution. After another 62 hours the reaction mixture
was separated into a black-brown solid and a red filtrate by inert filtration via cannula. The solvent
was removed in vacuo to obtain a red solid which was characterized by NMR.
Protocol for screening of in situ reactions:
A 4 mL glass vial equipped with a magnetic stir bar was charged with cobalt(II) salt (0.02 – 0.1
mmol), BIAN ligand (0.02 – 0.1 mmol) and zinc metal (0.05 -1 mmol). Then, 1.5 mL of solvent
were added. To the resulting mixture the substrate compound (0.5 mmol) and ammonia in
methanol or water (0.5 – 1.0 mmol) were added. If a solid substrate was tested, it was placed in
the reaction vial before the solvent was added. The resulting mixture was sealed with a with
septum cap which was subsequently penetrated with a needle. Up to 7 reaction vials were placed
in an autoclave which was tightly sealed. The autoclave was purged with hydrogen 3 times before
being pressurized. After that, the autoclave was placed on a heating plate and stirred for the preset

Daniel Timelthaler 74/126
reaction time. After completion, the heating was turned off and the autoclave was allowed to cool
for 1 hour. After that, pressure was released, and 1 mL of solvent was added to each of the reaction
vials. Thereafter, the solutions were degassed by stirring on air for 30 minutes. Then, 20 µL were
withdrawn from each vial, mixed with 1 mL of methanol and analyzed by GC-MS. If substrate 7b
was employed, the GC-MS analysis probe was dissolved in THF instead due to the low stability
of the substrate in methanol.
5.6. Screening of the Heterogeneous Co(II)/Zn(0) System
Protocol for screening of in situ reactions:
A 4 mL glass vial charged with a magnetic stirring bar, cobalt(II) salt (0.02 – 0.05 mmol), Lewis
acid (0.02 – 0.05 mmol) and zinc metal (0.06 - 0.15 mmol). Then, 1.5 mL of solvent were added.
To the resulting mixture, the substrate (0.5 mmol) and ammonia in methanol or water (0.5 – 1.0
mmol) were added. If a solid substrate was tested, it was placed in the vial before the solvent was
added. The resulting mixture was sealed with a septum cap which was subsequently penetrated
with a needle. The reaction vials were placed in a drilled Al-plate that accommodates seven
vessels. The inlet was transferred into the autoclave which was tightly sealed hereafter. The
autoclave was flushed with hydrogen three times before being pressurized. After that, the
autoclave was put on a stirring plate and heated up to the required reaction temperature. On
completion of the catalytic transformation, the autoclave was allowed to reach room temperature.
After that, the H2-pressure was released, and 1 mL of solvent was added to each of the reaction
vials. If the reaction was performed in water, 2 mL of methanol were added to ensure homogeneity.
Thereafter, the solutions were degassed by stirring on air for 30 minutes. Then, an aliquot of 20
µL were taken from each vial, mixed with 1 mL of methanol and analyzed by GC-MS.
Synthesis of 6m:
In a 50 mL Schlenk flask, 575 mg of 4-aminobenzonitrile 6x (4.87 mmol) were dried in vacuo and
dissolved in 20 mL of dry dichloromethane. The solution was then cooled to 0 °C and 567 µL of
acetic anhydride (6.00 mmol, 1.26 eq.) were added in small portions over 15 minutes. The reaction
was allowed to reach room temperature and stirred for 19 h under N2. On completion of the
reaction, the solution was washed with saturated NaHCO3 solution leading to precipitation of a
white solid. The solution was filtered through a glass frit (POR 4) and the solid was washed with
10 mL of DCM. The remaining solvent was removed on the rotary evaporator giving a mixture of
white solid and a yellow oil. The yellow impurity was removed by washing with n-heptane, yielding
additional target compound as a white solid.

Daniel Timelthaler 75/126
N-(4-cyanophenyl)acetamide (6m)
White amorphous powder: 682 mg (4.27 mmol, 88% yield)
Analytical data: 1H NMR (300 MHz, THF-d8, 20 °C): δ = 9.43 (s, NH), 7.78 (d, J = 8.82 Hz, 2H),
7.62 (d, J = 8.78 Hz, 2H), 2.10 (s, 3H) ppm; 13C NMR (75.5 MHz, THF-d8, 20 °C): δ = 168.1, 143.6,
132.7, 118.7, 118.3, 106.1, 23.2 ppm; HR-MS (ESI+): m/z: calcd. for C9H9N2O: 161.0715 [M+H+];
found: 161.0710 [M+H+].
Protection of 6u:
In a 50 mL round-bottom flask, 700 mg of p-cyanobenzaldehyde 6u (5.34 mmol) and 96 mg of
p-toluenesulfonic acid monohydrate (0.50 mmol, 0.095 eq.) were dissolved in 14 mL of toluene.
Then 0.580 µL of ethylene glycol (10.4 mmol, 1.95 eq.) were added and the flask was connected
to a Dean-Stark apparatus. The reaction mixture was heated up to 105 °C and agitated for 15 h
whereas the reaction progress was monitored by GC-MS. After completion of the reaction, the
product solution was washed with 10 mL of saturated aqueous NaHCO3 solution. The aqueous
phase was then extracted three times with toluene in portions of 10 mL. The combined organic
phases were then dried with sodium sulfate and the solvent was remove in vacuo, giving 6ac as
a yellow oil.
4-(1,3-dioxolan-2-yl)benzonitrile (6ac)
Yellow oil: 838 mg (5.34 mmol, 98% yield)
Analytical data: 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.67 (d, J = 7.98 Hz, 2H), 7.59 (d, J =
8.10 Hz, 2H), 5.85 (s, 1H), 4.14 – 4.02 (m, 4H) ppm; 13C NMR (75.5 MHz, CDCl3, 20 °C): δ =
143.1, 132.2, 127.2, 118.6, 112.9, 102.4, 65.4 ppm; HR-MS (ESI+): m/z: calcd. for C10H9NO2:
176.0712 [M+H+]; found: 176.0707 [M+H+].
Procedure for the determination of the isolated yields for the products of nitrile
hydrogenation:
The methanolic product solution was transferred from the reaction vial to a 25 mL round-bottom
flask. The volatiles were removed under reduced pressure and the remains were taken up in 5 mL
of dichloromethane. The flask was washed with dichloromethane several times and the resulting
organic phase was washed with saturated NaHCO3 solution. The aqueous phase was extracted
three times (up to five times for more hydrophilic substrates) and the combined organic layers
were dried with sodium sulfate. Then 3 mL of 2M hydrochloric acid in diethyl ether were added,
resulting in precipitation of a white solid for most compounds. The solid was removed by filtration

Daniel Timelthaler 76/126
through a glass frit (POR 4). The mother liquor was concentrated and cooled to -40 °C to afford
further precipitation. The combined white solids were dried on air and weighed.
Benzylammonium chloride (25a)
Synthesized according to the standard procedure. 0.500 mmol of 6a was used.
White crystalline powder: 67.0 mg (0.468 mmol, 94% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.53 – 7.45 (m, 5H), 4.20 (s, 2H) ppm; 13C
NMR (75.5 MHz, D2O, 20 °C): δ = 132.6, 129.2, 128.8, 43.1 ppm; HR-MS (ESI+): m/z: calcd. for
C7H10N: 108.0808 [M+H+]; found: 108.0805 [M+H+].
4-Methylbenzylammonium chloride (25b)
Synthesized according to the standard procedure. 0.496 mmol of 6b was used.
White crystalline powder: 55.9 mg (0.356 mmol, 72% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.78 (d, J = 8.82 Hz, 2H), 7.62 (d, J = 8.78 Hz,
2H), 4.09 (s, 2H), 2.10 (s, 3H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 139.6, 129.7, 129.5,
128.8, 42.8, 20.2 ppm; HR-MS (ESI+): m/z: calcd. for C9H9N2O: 122.0964 [M+H+]; found: 122.0963
[M+H+].
1-Naphthylmethylammonium chloride (25c)
Synthesized according to the standard procedure. 0.509 mmol of 6c was used.
White crystalline powder: 81.0 mg (0.420 mmol, 82% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.94 – 7.83 (m, 3H), 7.63 – 7.43 (m, 4H),
4.50 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 133.4, 130.3, 129.9, 128.9, 128.1, 127.7,
127.2, 126.5, 125.5, 122.3, 40.1 ppm; HR-MS (ESI+): m/z: calcd. for C11H12N: 158.0964 [M+H+];
found: 158.0965 [M+H+].

Daniel Timelthaler 77/126
2-Naphthylmethylammonium chloride (25d)
Synthesized according to the standard procedure. 0.507 mmol of 6d was used.
White crystalline powder: 77.9 mg (0.403 mmol, 80% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.98 – 7.88 (m, 4H), 7.62 – 7.56 (m, 2H),
7.51 (dd, J1 = 1.41 Hz, J2 = 8.49 Hz, 1H), 4.30 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ =
132.9, 132.8, 130.1, 128.9, 128.2, 127.9, 127.7, 127.0, 126.9, 125.9, 43.2 ppm; HR-MS (ESI+):
m/z: calcd. for C11H12N: 158.0964 [M+H+]; found: 158.0965 [M+H+].
4-Methoxybenzylammonium chloride (25e)
Synthesized according to the standard procedure. 0.497 mmol of 6e was used.
White crystalline powder: 73.6 mg (0.425 mmol, 86% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.43 (d, J = 8.73 Hz, 2H), 7.05 (d, J = 8.73 Hz,
2H), 4.14 (s, 2H), 3.85 (s, 3H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 159.4, 130.6, 125.1,
114.6, 55.4, 42.6 ppm.
2-Chlorobenzylammonium chloride (25f)
Synthesized according to the standard procedure. 0.498 mmol of 6f was used.
White crystalline powder: 80.2 mg (0.453 mmol, 91% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.61 – 7.38 (m, 4H), 4.35 (s, 2H) ppm; 13C
NMR (75.5 MHz, D2O, 20 °C): δ = 133.8, 131.2, 131.1, 130.1, 129.9, 127.8, 40.8 ppm; HR-MS
(ESI+): m/z: calcd. for C7H9ClN: 142.0418 [M+H+]; found: 142.0418 [M+H+].
3-Chlorobenzylammonium chloride (25g)
Synthesized in accordance with the standard procedure. 0.498 mmol of 6g was used.
White crystalline powder: 75.8 mg (0.428 mmol, 86% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.54 – 7.36 (m, 4H), 4.19 (s, 2H) ppm; 13C
NMR (75.5 MHz, D2O, 20 °C): δ = 133.4, 134.1, 130.7, 129.2, 128.7, 127.2, 42.5 ppm; HR-MS
(ESI+): m/z: calcd. for C7H9ClN: 142.0418 [M+H+]; found: 142.0418 [M+H+].

Daniel Timelthaler 78/126
4-Chlorobenzylammonium chloride (25h)
Synthesized in accordance with the standard procedure. 0.502 mmol of 6h was used.
White crystalline powder: 77.4 mg (0.437 mmol, 87% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.66 – 7.30 (m, 4H), 4.19 (s, 2H) ppm; 13C
NMR (75.5 MHz, D2O, 20 °C): δ = 134.5, 131.2, 130.4, 129.2, 42.4 ppm; HR-MS (ESI+): m/z:
calcd. for C7H9ClN: 142.0418 [M+H+]; found: 142.0418 [M+H+].
4-Fluorobenzylammonium chloride (25i)
Synthesized in accordance with the standard procedure. 0.494 mmol of 6i was used.
White crystalline powder: 73.0 mg (0.453 mmol, 92% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.50 (d, J = 5.61 Hz, 1H), 7.48 (d, J = 5.46 Hz,
1H), 7.21 (t, J1 = 8.75, 2H) 4.19 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 162.9 (d, J =
246 Hz, 1C), 131.1 (d, J = 8.77 Hz, 1C), 128.6 (d, J = 3.18 Hz, 1C), 116.0 (d, J = 22.0 Hz, 1C)
42.4 ppm; 19F-NMR (282.4 MHz, D2O, 20 °C): δ = -113.2 (s, Ar-F) ppm; HR-MS (ESI+): m/z: calcd.
for C7H9FN: 126.0714 [M+H+]; found: 126.0713 [M+H+].
2-Fluoro-6-methoxybenzylammonium chloride (25j)
Synthesized according to the standard procedure. 0.498 mmol of 6j was used.
White crystalline powder: 81.3 mg (0.426 mmol, 85% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.45 (q, J = 7.92 Hz, 2H), 6.95 – 6.80 (m,
2H), 4.24 (s, 2H) 3.92 (s, 3H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 161.1 (d, J = 246 Hz,
1C), 158.8 (d, J = 6.86 Hz, 1C), 131.8 (d, J = 10.9 Hz, 1C), 107.8 (d, J = 22.1 Hz, 1C), 107.8,
107.1 (d, J = 2.88 Hz, 1C) 56.1, 31.8 (d, J = 5.98 Hz, 1C) ppm; 19F-NMR (282.4 MHz, D2O, 20 °C):
δ = -117.4 (s, Ar-F) ppm; HR-MS (ESI+): m/z: calcd. for C8H11FNO: 156.0819 [M+H+]; found:
156.0820 [M+H+].

Daniel Timelthaler 79/126
4-(Trifluoromethyl)benzylammonium chloride (25k)
Synthesized according to the standard procedure. 0.491 mmol of 6k used.
White crystalline powder: 87.6 mg (0.415 mmol, 84% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.80 (q, J = 8.04 Hz, 2H), 7.64 (d, J = 8.04
Hz, 2H), 4.29 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 136.6, 130.4 (q, J = 32.3 Hz,
1C), 129.3, 126.0 (q, J = 6.86 Hz, 1C), 124.0 (d, J = 272 Hz, 1C), 42.6 ppm; 19F-NMR (282.4 MHz,
D2O, 20 °C): δ = -62.5 (s, CF3) ppm; HR-MS (ESI+): m/z: calcd. for C8H9F3N: 176.0682 [M+H+];
found: 176.0684 [M+H+].
3-Pyridinmethylammonium chloride (25l)
Synthesized in accordance with the standard procedure. 0.494 mmol of 6l used.
White crystalline powder: 53.7 mg (0.372 mmol, 76% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 9.02 – 8.85 (m, 2H), 8.75 (d, J = 8.22 Hz,
1H), 8.18 (t, J = 6.99 Hz, 1H), 4.51 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 147.4,
142.0, 141.8, 133.0, 127.8, 39.6 ppm; HR-MS (ESI+): m/z: calcd. for C6H9N2: 109.0760 [M+H+];
found: 109.0759 [M+H+].
3-Phenylpropylammonium chloride (25n)
Synthesized according to the standard procedure. 0.500 mmol of 6n was used.
White crystalline powder: 82.7 mg (0.483 mmol, 97% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.40 – 7.23 (m, 5H), 2.97 (t, J = 7.67 Hz, 2H),
2.70 (t, J = 7.67 Hz, 2H), 1.95 (p, J = 7.62, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 140.9,
128.8, 128.4, 126.4, 39.0, 31.8, 28.4 ppm; HR-MS (ESI+): m/z: calcd. for C9H14N: 136.1121
[M+H+]; found: 136.1122 [M+H+].
Cyclohexylmethylammonium chloride (25o)
Synthesized in accordance with the standard procedure. 0.500 mmol of 6o was applied.
White crystalline powder: 60.0 mg (0.402 mmol, 80% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 2.88 (d, J = 6.75 Hz, 2H), 1.83 - 1.61 (m, 6H),
1.38 - 1.15 (m, 3H), 1.10 – 0.94 (m, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 45.2, 35.4,
29.6, 25.6, 25.0 ppm; HR-MS (ESI+): m/z: calcd. for C7H16N: 114.1277 [M+H+]; found: 114.1277
[M+H+].

Daniel Timelthaler 80/126
Pentylammonium chloride (25p)
Synthesized according to the standard procedure. 0.500 mmol of 6p was used.
White crystalline powder: 59.2 mg (0.481 mmol, 90% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 3.01 (t, J = 7.53 Hz, 2H), 1.68 (p, J = 7.29,
2H), 1.42 - 1.30 (m, 4H), 0.91 (t, J = 6.95, 3H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 39.5,
27.7, 26.4, 21.4, 13.1 ppm; HR-MS (ESI+): m/z: calcd. for C5H14N: 88.1121 [M+H+]; found: 88.1119
[M+H+].
Ethylammonium chloride (25q)
Synthesized according to the standard procedure but the autoclave was cooled down to 0 °C
before opening and work-up was performed rapidly using cooled solvents owing to the low boiling
point of ethylamine. 0.500 mmol of 6q was used.
White crystalline powder: 19.3 mg (0.238 mmol, 48 % yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 3.21 (q, J = 7.33 Hz, 2H), 1.29 (t, J = 7.33 Hz,
3H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 46.7, 8.2 ppm; HR-MS (ESI+): m/z: calcd. for
C2H8N: 46.0641 [M+H+]; found: 46.0659 [M+H+].
1-Adamantylammonium chloride (25s)
Synthesized in accordance with the standard procedure. 0.500 mmol of 6s was applied.
White crystalline powder: 94.7 mg (0.471 mmol, 94% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 2.71 (s, 2H), 2.08 – 1.98 (m, 3H), 1.72 (q, J
= 14.37 Hz, 2H), 1.61 – 1.55 (m, 6H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 50.7, 38.8, 35.9,
31.3, 27.6 ppm; HR-MS (ESI+): m/z: calcd. for C11H20N: 166.1590 [M+H+]; found: 166.1594
[M+H+].
2-Phenylethylammonium chloride (25t)
Synthesized according to the standard procedure. 0.500 mmol of 2t was applied.
White crystalline powder: 75.6 mg (0.481 mmol, 96% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 7.49 – 7.33 (m, 5H), 3.30 (t, J = 7.17 Hz, 2H),
3.02 (t, J = 7.28 Hz, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 136.6, 129.1, 128.9, 127.3,
40.6, 32.7 ppm; HR-MS (ESI+): m/z: calcd. for C8H12N: 122.0964 [M+H+]; found: 122.0965 [M+H+].

Daniel Timelthaler 81/126
4-Formylbenzylammonium chloride (25u)
Synthesized according to the standard procedure. 0.500 mmol of 6ac used.
White crystalline powder: 69.5 mg (0.406 mmol, 81% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 9.95 (s, 1H), 8.00 (d, J = 8.10 Hz, 2H), 7.66
(d, J = 8.04 Hz, 2H), 4.31 (s, 2H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 195.8, 139.6, 136.0,
130.6, 129.4, 42.7 ppm; HR-MS (ESI+): m/z: calcd. for C8H10NO: 136.0757 [M+H+]; found:
136.0757 [M+H+].
Procedure for the determination of the isolated yields for 25r:
As the standard procedure led to disappointing results for 25r owing to its high hydrophilicity, a
different approach was pursued: on completion of the reaction, the two-phase reaction solution
(methanol-water) was transferred from the reaction vial to a 25 mL round-bottom flask. Methanol
and ammonia were removed under reduced pressure and the remains were taken up in 5 mL of
ethyl acetate. The flask was washed with water and ethyl acetate several times and the combined
washing phases were filtered through a pad of celite. The pad was thoroughly washed with ethyl
acetate and to the combined layers, 5 mL of hydrochloric acid in diethyl ether were added. The
mixture was shaken vigorously before complete removal of the liquids in vacuo. The product was
isolated as a white solid. The product showed minor impurities from the employed Lewis acid
which were taken in account for the determination of the yield.
1,6-Diammoniumhexyl dichloride (25r)
Synthesized in equivalence to standard procedure. 0.500 mmol of 6r used.
White crystalline powder: 84.7 mg (0.450 mmol, 90% yield)
Analytical data: 1H NMR (300 MHz, D2O, 20 °C): δ = 3.03 (t, J = 7.55 Hz, 4H), 1.76 – 1.65 (m, 4H),
1.48 – 1.40 (m, 4H) ppm; 13C NMR (75.5 MHz, D2O, 20 °C): δ = 39.4, 26.5, 25.1 ppm; HR-MS
(ESI+): m/z: calcd. for C11H20N: 117.1386 [M+H+]; found: 117.1385 [M+H+].
Daniel Timelthaler 82/126
6. Used Abbreviations
Abbreviation
AcOH acetic acid
Ar-BIAN bis(aryl)acenaphthenequinonediimine
Ar-BIANH2 bis(aryl)acenaphthylene-1,2-diamine
Ar-BIANH4 bis(aryl)-1,2-dihydroacenaphthylene-1,2-daimine
DAB Diazabutadiene
DCM Dichloromethane
Dippe 1,2-bis-(diisopropylphosphino)-ethane
DMF dimethylformamide
ESI+ positive mode electrospray ionization
EtOH ethanol
GC-MS gas chromatography with a mass spectrometry detector
Hemi-BIAN 2-(arylimino)acenaphthylene-1-one
HR-MS high-resolution mass spectrometry
i-PrOH 2-propanol
MeOH methanol
Mes-BIAN bis(mesityl)acenaphthenequinonediimine
MTBE Methyl tert-butyl ether
NH3*H2O ammonia (as 18M solution in water)
NH3*MeOH ammonia (as 2M solution in methanol)
NMR nuclear magnetic resonance spectroscopy
ORTEP oak ridge thermal-ellipsoid plot
OTf trifluoromethanesulfonate
THF tetrahydrofuran
UV/VIS ultraviolet-visible spectroscopy

Daniel Timelthaler 83/126
7. References
[1] C.M. Lok, Structure and performance of selective hydrogenation catalysts, in S.D. Jackson,
Hydrogenations (978-3-11.054373-5), Walter de Gruyter GmbH, 2018, p.1-14.
[2] http://www.infomine.com/investment/metal-prices/, accessed on February 17th 2019.
[3] H.B.W. Patterson, The Hydrogenation Reaction in G.R. List, J.W. King, Hydrogenation of
Fats and Oils: Theory and Practice, 2nd Edition, AOCS Press, 1994, p. 5.
[4] G. Onyesták, Period. Polytech. Chem. 2017, 61, 4, 270-277.
[5] G.B. Combes, J.B. Claridge, J.R. Gallagher, M.J. Rosseinsky, 2011, US Patent No.
US9346038B2.
[6] D. Ostgard, M. Berweiler, S. Roder, J. Sauer, B. Jaeger, N. Finke, C. Lettmann, 2000, US
Patent No. US6437186B1.
[7] M. Raney, 1927, US Patent No. 1.628.191.
[8] P. Fouilloux, Appl. Catal. 1983, 1, 1-42.
[9] M. Raney, 1933, US Patent No. 1.915.473.
[10] S. Nishimura, Heterogenous Catalytic Hydrogenation for Organic Synthesis, John Wiley &
Sons Inc., 2001, p. 23-26.
[11] M.G. Banwell, M.T. Jones, T.A. Reekie, B.D. Schwartz, S.H. Tan, L.V. White, Org. Biomol.
Chem. 2014, 12, 38, 7413-7644.
[12] G.M. Badger, N. Kowanko, W.H.F. Sasse, J. Chromatogr. A. 1964, 13, 234.
[13] T.A. Reekie, M.G. Banwell, A.C. Willis, J. Org. Chem. 2012, 77, 10773.
[14] Y.-Z. Chen, B.-J.Liaw, S.-J. Chiang, Appl. Catal. 2005, 284, 97-104.
[15] R.D. Rieke, Chemical Synthesis using Highly Reactive Metals, John Wiley & Sons:
Hoboken, 2017; p 429-441.
[16] P. Büschelberger, E. Reyes-Rodriguez, C. Schöttle, J. Treptow, C. Feldmann, A. Jacobi
von Wangelin, R. Wolf, Catal. Sci. Technol. 2018, 8, 2648-2653.
[17] A.M. Allgeier, S.K. Sengupta, Nitrile hydrogenation, in S.D. Jackson, Hydrogenations,
Walter de Gruyter GmbH, 2018, p.107-147.
[18] S. Nishimura, Heterogenous Catalytic Hydrogenation for Organic Synthesis, John Wiley &
Sons Inc., 2001, p.245-280.
[19] J. von Braun, G. Bleesig, F. Zobel, Ber. Dtsch. Chem. Ges. 1923, 56, 1988.
[20] J. Volf, J. Pasek, Hydrogenation of Nitriles in L. Cerveny, Catalytic Hydrogenations Vol. 5,
Elsevier Science Publishers B.V. 1986, p. 105-141.
[21] M. Kalina, J. Pasek, Kinet. Katal. 1969, 10, 574-580.
[22] D. Bivens, L. Patton, W. Thomas, 1973, US Patent No. US3758584A.
[23] C.E. Cutchens, L.H. Lanier, 1984, US Patent No. US4429159A.
[24] T.A. Koch, K.R. Krause, S.K. Sengupta, 1998, WIPO Patent No. WO1998043940A1.

Daniel Timelthaler 84/126
[25] C.W. Wigbers, W. Mägerlein, T. Krug, J.-P. Melder, T. Heidemann, B. Stein, 2016, WIPO
Patent No. WO2016030383A1.
[26] H. Konnerth, M.H.G. Prechtl, New J. Chem. 2017, 41, 9594-9597.
[27] F. Chen, C. Topf, J. Radnik, C. Kreyenschulte, H. Lund, M. Schneider, A.-E. Surkus, L. He,
K. Junge, M. Beller, J. Am. Chem. Soc. 2016, 138, 8781-8788.
[28] F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A.-E. Surkus, K. Junge, M. Beller, ACS
Catal. 2017, 7, 1526-1532.
[29] R. Ferracioli, D. Borovika, A.-E. Surkus, C. Kreyenschulte, C. Topf, M. Beller, Catal. Sci.
Technol. 2018, 8, 499-507.
[30] H. Dai, H. Guan, ACS Catal. 2018, 8, 9125-9130.
[31] A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David, D. Milstein, J. Am. Chem. Soc.
2015, 137, 8888-8891.
[32] K. Tokmic, B.J. Jackson, A. Salazar, T.J. Woods, A.R. Fout, J. Am. Chem. Soc. 2017, 139,
13554-13561.
[33] Z. Shao, S. Fu, M. Wei, S. Zhou, Q. Liu, Angew. Chem. Int. Ed. 2016, 55, 14653-14657.
[34] G. Zhang, B.L. Scott, S.K. Hanson, Angew. Chem. Int. Ed. 2012, 51, 12102-12106.
[35] G. Zhang, S.K. Hanson, Chem. Commun. 2013, 49, 10151-10153.
[36] S. Rösler, J. Obenauf, R. Kempe, J. Am. Chem. Soc. 2015, 137, 7998-8001.
[37] D. Srimani, A. Mukherjee, A.F.G. Goldberg, G. Leitus, Y. Diskin-Posner, L.J.W. Shimon,
Y.B. David, D. Milstein, Angew. Chem. Int. Ed. 2015, 54, 12357-12360.
[38] T.J. Korstanje, J.I. van der Vlugt, C.J. Elsevier, B. de Bruin, Science 2015, 350, 298-301.
[39] M.R. Friedfeld, H. Zhong, R.T. Ruck, M. Shevlin, P.J. Chirik, Science 2018, 360, 888-893.
[40] K.V. Vasudefan, B.L. Scott, S.K. Hanson, Eur. J. Inorg. Chem. 2012, 30, 4898-4906.
[41] T.J. Mooibroek, E.M.C. Wenker, W. Smit, I. Muitikainen, M. Lutz, E. Bouwman, Inorg.
Chem. 2013, 52, 8190-8201.
[42] N. Castellanos-Blanco, M. Flores-Alamo, J.J. Garcia, Dalton Trans. 2015, 44, 15653-
15663.
[43] N. Castellanos-Blanco, M. Flores-Alamo, J.J. Garcia, Organometallics 2012, 31, 680-686.
[44] T. Hibino, K. Makino, T. Sugiyama, Y. Hamada, Chem. Cat. Chem. 2009, 1, 237-240.
[45] M. Shevlin, M.R. Friedfeld, H. Sheng, N.A. Pierson, J.M Hoyt, L.-C. Campeau, P.J. Chirik,
J. Am. Chem. Soc. 2016, 138, 3562-3569.
[46] N.G. Léonard, P.J. Chirik, ACS Catal. 2018, 8, 342-348.
[47] N.J. Hill, I. Vargas-Baca, A.H. Cowley, Dalton Trans. 2009, 240-253.
[48] M. M. Khusniyarov, K. Harms, O. Burghaus, J. Sundermeyer, Eur. J. Inorg. Chem. 2006,
2985-2996.
[49] R. Asselt, C.J. Elsevier, W.J.J. Smeets, A.L. Spek, R. Benedix, Recl. Trav. Chim. Pays-
Bas 1994, 113, 88-98.

Daniel Timelthaler 85/126
[50] J.M. Rose, A.E. Cherian, G.W. Coates, J. Am. Chem. Soc. 2006, 128, 4186-4187.
[51] R. Asselt, C.J. Elsevier, J. Mol. Catal. 1991, 65, L13-L19.
[52] R. Asselt, C.J. Elsevier, Tetrahedron 1994, 50, 323-334.
[53] G.A. Grasa, R. Singh, E.D. Stevenson, S.P. Nolan, J. Organomet. Chem. 2003, 687, 229-
248.
[54] G.A. Grasa, A.C. Hillier, S.P. Nolan, Org. Lett. 2001, 3, 1077-1080.
[55] M. Gasperini, F. Ragaini, S. Genini, Organometallics 2002, 21, 2950-2957.
[56] F.H. Allen, O. Kennard, D.G. Watson, L. Brammer, A.G. Orpen, R. Taylor, J. Chem. Soc.
1987, Perkin Trans. 2, 12, 1-19.
[57] R.A. Wood, T.R. Welberry, J. Chem. Soc. Perkin Trans. II 1985, 3, 451-456.
[58] M Viganò, F. Feretti, A. Caselli, F. Ragaini, M. Rossi, P. Mussini, P. Macchi, Chem. Eur. J.
2014, 20, 14451-14464.
[59] M Viganò, F. Ragaini, M.G. Buonomenna, R. Lariccia, A. Caselli, E. Gallo, S. Cenini, J.C.
Jansen, E. Drioli, Chem. Cat. Chem. 2010, 2, 1150-1164.
[60] V.G. Sokolov, T.S. Koptseva, M.V. Moskalev, A.V. Piskunov, M.A. Samsunov, I.L.
Fedushkin, Russ. Chem. B+ 2017, 66 (9), 1569-1579.
[61] P. Sunzenauer, A novel class of bidentate amine ligands: Development of a synthetic
strategy and application in coordination chemistry, Master Thesis, JKU Linz, 2018.
[62] V.J.S. Rosa, Synthesis, characterization and reactivity of new cobalt, palladium and
nickel bearing complexes bearing α-diimines and P,O ligands, doctoral thesis,
Universidade Nova de Lisboa, 2008.
[63] X.-J. Yang, X. Fan, Y. Zhao, X. Wang, B. Liu, J.-H. Su, Q. Dong, M. Xu, B. Wu,
Organometallics 2013, 32, 6945-6949.
[64] Bruker (2016), APEX3 v2016.9-0, SAINT V8.37A, SHELXTL-2014, Bruker AXS Inc.:
Madison (WI), USA, 2016.
[65] a) G. M. Sheldrick, SHELXL-2014: Program for the Refinement of Crystal Structures,
University of Göttingen, Germany, 2014. b) G. M. Sheldrick, Acta Crystallogr., Sect. C:
Struct. Chem. 2015, 71, 3−8.
[66] C. B. Hübschle, G. M. Sheldrick, B. Dittrich, J. Appl. Crystallogr. 2011, 44, 1281−1284.

Daniel Timelthaler 86/126
APPENDIX
Table of contents
A1. Crystallographic Data ......................................................................................................... 87
A2. Screening Tables ................................................................................................................ 89
A3. Spectral Data ...................................................................................................................... 95

Daniel Timelthaler 87/126
A1. Crystallographic Data
Compound Mes-BIAN Mes-BIANH2 Mes-BIANH4 Mes-Hemi-BIAN
Empirical formula C30H28N2 C30H30N2 C30H32N2 C42H34N2O2
Formula weight [g/mol] 416.54 418.56 420.57 597.71
Colour orange purple colorless orange
Crystal size [mm] 0.43 × 0.42 × 0.16 0.39 × 0.23 × 0.11 0.54 × 0.39 × 0.17 0.66 × 0.25 × 0.12
Crystal system monoclinic triclinic monoclinic triclinic
Space group C2/c P−1 P21/c P−1
a [Å] 24.0876(7) 10.2487(9) 9.0599(3) 8.097(4)
b [Å] 12.4002(3) 10.3276(9) 18.3903(5) 8.512(5)
c [Å] 7.9888(2) 12.4678(11) 14.0892(5) 13.441(7)
α [°] 90 92.856(4) 90 103.14(4)
β [°] 97.959(1) 108.154(4) 106.549(1) 93.00(3)
γ [°] 90 109.386(4) 90 114.87(3)
V [Å3] 2363.20(11) 1165.45(18) 2409.93(13) 806.9(8)
Z 4 2 4 1
Dcalc [g/cm3] 1.171 1.193 1.159 1.232
μ [mm-1] 0.07 0.07 0.07 0.08
T [K] 296 296 296 296
θ range [°] 3.0-22.7 2.5-23.2 2.6-25.0 2.8-19,5
No. of reflections measured 31884 81227 130173 12770
No. of independent reflections 1703 3361 4261 2310
Obs. Reflections with I > 2σ(I) 1235 2245 3235 969
No. of Parameters
refined/restraints
150/0 298/0 298/0 211/0
Absorption correction multi-scan multi-scan multi-scan multi-scan
Tmin, Tmax 0.88, 0.99 0.87, 0.99 0.89, 0.99 0.83, 0.99
Δρmin/Δρmax [e Å-3] −0.12/0.14 −0.20/0.20 −0.17/0.19 −0.23/0.19
F(000) 888 448 904 316
Rint 0.087 0.193 0.101 0.272
R1 (R[F2 ≥ 2σ(F2))]) 0.048 0.069 0.063 0.085
wR2 (wR(F2)) 0.126 0.152 0.150 0.189
GooF 1.09 1.13 1.15 1.00
CCDC no. N.A. N.A. N.A. N.A.

Daniel Timelthaler 88/126
Figure A 1: Number representation scheme of N1,N2-dimesitylacenaphthylene-1,2-diimine 1c
Table A 1: Selected bond lengths [Å] of 1c
C7-N1 1.266
C7-C7a 1.517
C6-C7 1.471
Figure A 2: Number representation scheme of N1,N2-dimesitylacenaphthylene-1,2-diamine 2c
Table A 2: Selected bond lengths [Å] of 2c
C1-N1 1.383
C1-C12 1.361
C1-C2 1.471
Figure A 3: Number representation scheme of cis-N1,N2-dimesityl-1,2-dihydroacenaphthylene-1,2-diamine 3c

Daniel Timelthaler 89/126
Table A 3: Selected bond lengths [Å] of 3c
C1-N1 1.484
C1-C12 1.567
C1-C2 1.515
Figure A 4: Number representation scheme of 2-(mesitylimino)acenaphthylene-1(2H)-one
Table A 4: Selected bond lengths [Å] of hemi form of 1c
C12-N1 1.268
C1-C12 1.546
C11-C12 1.485
C1-O1 1.208
A2. Screening Tables
Table A 5: Screening results for nickel-BIAN hydride system (cf. page 41)[a]
Substrate Catalyst
loading /
mol%
Ligand Solvent Temperature
/ °C
Conversion
/ %[b]
Yield
/ %[b]
5a 5 1d THF 50[c] 1 <1
5a 5 1d THF 100[c] 2 <1
6a 5 1d THF 50[c] 2 1
6a 5 1d THF 100[c] 1 0
5a 10 1d THF 100 7 7
5a 5 1d THF 100 1 1
5a 2 1d THF 100 1 <1
6a 10 1d THF 100 13 0
6a 5 1d THF 100 3 0
6a 2 1d THF 100 0 0
7b 10 1b THF 100 33 33

Daniel Timelthaler 90/126
7b 5 1b THF 100 15 15
7b 2 1b THF 100 5 5
7b 5 1c THF 80 13 13
7a 10 1b THF 100 13 12
7a 5 1b THF 100 5 5
7a 2 1b THF 100 1 1
5a 5 1e THF 100 1 0
5a 5[d] 1e THF 100 5 1
6a 5 1e THF 100 0 0
6a 5[d] 1e THF 100 1 0
7b 5 1b THF 100 12 4
7a 5 1b THF 100 1 1
7b 5 1b THF 100 3 3
7b 5 1b THF 100 3 3
7b 5 1b THF 100 0 0
7b 0 - Cyclohexane 80 0 0
7b 5[e] 1b Cyclohexane 80 0 0
7b 5[f] - Cyclohexane 80 0 0
7b 5[g] - Cyclohexane 80 10 10
7b 5[e][f] 1b Cyclohexane 80 0 0
7b 5[e][g] 1b Cyclohexane 80 10 10
7b 5[f][g] - Cyclohexane 80 12 12
7a 5 1b THF 100 2 1
7a 5 1b 1,4-Dioxane 100 6 1
7a 5 1b MeOH 100 0 0
7a 5 1b EtOH 100 0 0
7a 5 1b i-PrOH 100 0 0
7a 5 1b Cyclohexane 100 12 12
7a 5 1b DMF 100 2 2
7a 5 1b THF 80 1 1
7a 5 1b Toluene[h] 80 7 7
7a 5 1b o-Xylene 80 5 5
7a 5 1b m-Xylene 80 5 5
7a 5 1b p-Xylene 80 7 7
7a 5 1b Cyclohexane[h] 80 10 10
7a 5 1b n-Hexane[h] 80 11 11 [a] Reaction conditions: substrate (1.0 mmol), nickel octanoate (as listed), ligand (equimolar to Ni), pinacolborane (4 eq. to Ni) in 1 mL solvent, temperature as listed, pressure as listed, reaction time: 16 h. [b] determined by GC-MS. [c] 10 bar of H2 was used. [d] NiBr2 was used as Ni source. [d] only BIAN used. [e] only nickel octanoate was used. [f] only pinacolborane was used. [h] dry solvent was applied.

Daniel Timelthaler 91/126
Table A 6: Results of the determination of the BIAN-influence on the catalytic hydrogenation of isophorone 12b (cf. page 47) [a]
Ligand A / %[b] B / %[b] C / %[b] D/ %[b] E / %[b] Ring-
opening /
%[b]
1b 5 15 3 2 69 6
1a 5 16 2 2 69 7
1d 1 33 2 3 58 3
1h 27 43 2 1 23 6
1c 21 29 2 1 40 6
1e 0 92 2 3 1 2
1i 52 31 2 1 14 1 [a] Reaction conditions: Isophorone (0.5 mmol), CoCl2*6H2O (5mol%), ligand (5mol%), zinc powder (50mol%) in 1.5 mL of methanol, 100 °C, 40 bar H2, reaction time: 64 h. [b] Determined by GC-MS.
Figure A 5: Substrates for application-screening of the heterogeneous Co(II)/Zn(0) system (cf. page 49).

Daniel Timelthaler 92/126
Table A 7: Initial substrate screening for the heterogeneous Co(II)/Zn(0) system (cf. page 49).[a]
Substrate Cobalt
source
Solvent Temperature Conversion
/ %[b]
Yield
/ %[b]
Additives[c]
5a CoCl2*6H2O MeOH 100 1 1 -
6a CoCl2*6H2O MeOH 100 >99 63 -
7a CoCl2*6H2O MeOH 100 61 15
100%
NH3*MeOH
12b CoCl2*6H2O Toluene 100 2 2[d]
100%
NH3*H2O
13a Co(OAc)2*4H2O MeOH 100 >99
60[e] /
40[f]
200%
NH3*MeOH
13a Co(OAc)2*4H2O MeOH 80 >99 >99[e]
200%
NH3*MeOH
13a Co(OAc)2*4H2O MeOH 60 80
14[e] /
66[f]
50%
NH3*MeOH
13b Co(OAc)2*4H2O MeOH 100 >99 96[e]
200%
NH3*MeOH
13b Co(OAc)2*4H2O MeOH 60 89
13[e] /
28[f]
50%
NH3*MeOH
13c Co(OAc)2*4H2O MeOH 100 >99 >99[e]
200%
NH3*MeOH
13c Co(OAc)2*4H2O MeOH 80 >99 >99[e]
200%
NH3*MeOH
13c Co(OAc)2*4H2O MeOH 60 >99 >99[e]
50%
NH3*MeOH
14a Co(BF4)2*6H2O MeOH 100 Unknown[g] 0 -
14b Co(BF4)2*6H2O MeOH 100 Unknown[g] 0 -
14c Co(BF4)2*6H2O MeOH 100 Unknown[g] 0 -
15a CoCl2*6H2O MeOH 120 Unknown[g] 0
200%
NH3*MeOH
15ad CoCl2*6H2O MeOH 120 Unknown[g] 0
200%
NH3*MeOH
16a CoCl2*6H2O MeOH 100 >99 89
100%
NH3*MeOH
16b CoCl2*6H2O MeOH 100 >99 9
100%
NH3*MeOH
16c CoCl2*6H2O MeOH 100 97 71
100%
NH3*MeOH
17a Co(OAc)2*4H2O MeOH 120 4 2[h]
200%
NH3*MeOH
18a Co(OAc)2*4H2O MeOH 120 3 3
200%
NH3*MeOH

Daniel Timelthaler 93/126
Time/Conversion diagram (cf. page 58):
All experiments were carried out at 120 °C, 40 bar H2 applying reaction time of 15 hours. The
cooling-off period amounted to 90 minutes. Before degassing, 1mL of Methanol and 0.5 mmol of
n-hexadecane as internal standard were added. Degassing proceeded by stirring the reaction
vessels on air at ambient temperature for 15 minutes.
Sample compositions:
I 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, 100mol%
NH3*MeOH, Solvent: 1.5 mL MeOH
II 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, 100mol%
NH3*H2O, Solvent: 1.5 mL MeOH
III 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 2mol% Zn(OTf)2, Solvent:
1.5 mL MeOH
IV 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, 100mol% NH3*MeOH,
Solvent: 1.5 mL MeOH
V 0.5 mmol benzonitrile 6a, 2mol% Co(OAc)2*4H2O, 6mol% Zn, Solvent: 1.5 mL MeOH
19a Co(OAc)2*4H2O MeOH 120 21 21[i]
200%
NH3*MeOH
19a Co(OAc)2*4H2O i-PrOH 120 12 12[i]
200%
NH3*MeOH
19a Co(OAc)2*4H2O
MeOH:H2O
(2:1) 120 45 45[i]
200%
NH3*MeOH
19a Co(OAc)2*4H2O
MeOH:H2O
(1:1) 120 56 56[i]
200%
NH3*MeOH
19a Co(OAc)2*4H2O
MeOH:H2O
(1:2) 120 42 58[i]
200%
NH3*MeOH
19a Co(OAc)2*4H2O H2O 120 12 12[i]
200%
NH3*H2O [a] Reaction conditions: Substrate (0.5 mmol), cobalt source (5mol%), zinc powder (50mol%) in 1.5 mL of solvent, temperature as indicated, 40 bar H2, reaction time: 15 h. [b] Determined by GC-MS. [c] Given percentage values refer to molar equivalents of the substrate. [d] Selective Michael-type hydrogenation. [e] Refers to the yield of alkane. [f] Refers to the yield of alkene. [g] Substrate still detectable in GC/MS, conversion unclear as non-volatile products
were formed that could not be analyzed by GC/MS. [h] Refers to the yield of aniline. [i] Refers to yield of toluene.

Daniel Timelthaler 94/126
Table A 8: Results of time/conversion measurements for the heterogeneous Co(II)/Zn(0) system(cf. page 58).[a]
Sample Reaction
time / h
Conversion
/ %[b]
Yield
20a /
%[b]
Yield
21a /
%[b]
Yield
22a /
%[b]
Yield
23a /
%[b]
Yield
15a /
%[b]
Yield
24a /
%[b]
I 1 2 0 0 0 0 0 2
I 2 9 1 4 0 0 0 4
I 4 50 20 27 0 0 0 3
I 8 100 73 27 0 0 0 0
I 15 100 97 <1 0 0 0 0
II 1 3 3 <1 0 0 0 <1
II 2 7 2 5 0 0 0 0
II 4 76 48 28 0 0 0 0
II 8 100 95 0 1 1 0 0
II 15 100 94 <1 2 1 <1 0
III 1 3 0 0 0 0 0 3
III 2 <1 0 0 0 0 0 <1
III 4 4 < 0 0 0 0 4
III 8 5 2 0 0 0 0 3
III 15 5 3 0 0 0 0 2
IV 1 2 <1 <1 0 0 0 2
IV 2 5 0 2 0 0 0 3
IV 4 27 7 16 0 0 0 4
IV 8 84 52 34 0 0 0 1
IV 15 0 79 21 <1 0 0 0
V 1 <1 0 <1 0 0 0 <1
V 2 <1 0 <1 0 0 0 <1
V 4 2 0 1 0 0 0 1
V 8 8 <1 5 0 0 0 2
V 15 25 3 18 0 0 0 2 [a] Reaction conditions as indicated above. [b] Determined by GC-MS using hexadecane as an internal standard for 20a.

Daniel Timelthaler 95/126
A3. Spectral Data
Figure A 6: 1H NMR of 1a.
Figure A 7: 13C NMR of 1a.

Daniel Timelthaler 96/126
Figure A 8: 1H NMR of 1b.
Figure A 9: 13C NMR of 1b.

Daniel Timelthaler 97/126
Figure A 10: 1H NMR of 1c.
Figure A 11: 13C NMR of 1c.

Daniel Timelthaler 98/126
Figure A 12: 1H NMR of 1d *H2O.
Figure A 13: 13C NMR of 1d.
*
Daniel Timelthaler 99/126
Figure A 14: 1H NMR of 1e.
Figure A 15: 13C NMR of 1e.

Daniel Timelthaler 100/126
Figure A 16: 1H NMR of 1f.
Figure A 17: 13C NMR of 1f.
Figure A 18: 19F NMR of 1f.

Daniel Timelthaler 101/126
Figure A 19: 1H NMR of 1g.
Figure A 20: 13C NMR of 1g.
Figure A 21: 19F NMR of 1g.

Daniel Timelthaler 102/126
Figure A 22: 1H NMR of 3c.
Figure A 23: 13C NMR of 3c.
Figure A 23: 1H NMR of 3d.
Daniel Timelthaler 103/126
Figure A 24: 1H NMR of 10c.
Figure A 25: 1H NMR of 10e.

Daniel Timelthaler 104/126
Figure A 26: 1H NMR of 6m.
Figure A 27: 13C NMR of 6m.
Figure A 28: HR-MS of 6m.
DDT22 #2-10 RT: 0.02-0.25 AV: 9 NL: 2.84E4T: FTMS + p ESI Full ms [50.00-2000.00]
158 159 160 161 162 163 164 165 166
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
161.0710
165.0910
158.9640
163.0277164.9843
162.0744158.1902 159.4803 164.0082159.8406
159.1212
160.5049 162.6976 165.4718161.2029 164.7861163.4001
158.8330

Daniel Timelthaler 105/126
Figure A 29: 1H NMR of 6ac.
Figure A 30: 13C NMR of 6ac.
Figure A 31: HR-MS of 6ac.
DDT21 #2-10 RT: 0.02-0.25 AV: 9 NL: 4.00E4T: FTMS + p ESI Full ms [50.00-2000.00]
175 176 177 178 179 180 181 182 183
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
176.0707
182.1176
177.0546
183.1207181.9796179.0701175.5673
175.0582
181.0833177.4421 177.9281176.5569 180.1591 183.7939182.5758

Daniel Timelthaler 106/126
Figure A 32: 1H NMR of 25a.
Figure A 33: 13C NMR of5.
Figure A 34: HR-MS of 25a.
DDT01 #2-10 RT: 0.01-0.24 AV: 9 NL: 4.56E5T: FTMS + p ESI Full ms [70.00-2000.00]
106 107 108 109 110 111 112 113
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
108.0805
109.0838
111.0203107.0699 112.8954109.1900108.2111 110.0872106.6169 111.5987 112.1821106.0225105.6016 107.4566

Daniel Timelthaler 107/126
Figure A 35: 1H NMR of 25b.
Figure A 36: 13C NMR of 25b.
Figure A 37: HR-MS of 25b.
DDT02 #2-10 RT: 0.01-0.24 AV: 9 NL: 2.72E6T: FTMS + p ESI Full ms [70.00-2000.00]
120 121 122 123 124 125 126 127 128
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
122.0963
123.0996
125.9859124.1027122.2530 125.1512120.9784 121.9398 127.9790123.7167 126.9226122.6857
120.6000 124.6304 127.4633

Daniel Timelthaler 108/126
Figure A 38: 1H NMR of 25c.
Figure A 39: 13C NMR of 25c.
Figure A 40: HR-MS of 25c.
DDT03 #2-10 RT: 0.01-0.24 AV: 9 NL: 2.84E6T: FTMS + p ESI Full ms [70.00-2000.00]
156.0 156.5 157.0 157.5 158.0 158.5 159.0 159.5 160.0 160.5 161.0 161.5 162.0 162.5 163.0
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
158.0965
159.0997
158.9638 160.1028158.3275 161.0118156.0804 162.1272157.0834155.6839
162.4688161.4937157.7271 159.4415156.5097

Daniel Timelthaler 109/126
Figure A 41: 1H NMR of 25d.
Figure A 42: 13C NMR of 25d.
Figure A 43: HR-MS of 25d.
DDT04 #2-10 RT: 0.01-0.24 AV: 9 NL: 1.79E6T: FTMS + p ESI Full ms [70.00-2000.00]
155 156 157 158 159 160 161 162
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
158.0965
159.0997
158.9638 160.1030 161.0118
158.3274
157.0832 162.4818159.7382 162.1281156.3939155.7015 157.8662155.1065

Daniel Timelthaler 110/126
Figure A 44: 1H NMR of 25e.
Figure A 45: 13C NMR of 25e.

Daniel Timelthaler 111/126
Figure A 46: 1H NMR of 25f.
Figure A 47: 13C NMR of 25f.
Figure A 48: HR-MS of 25f.
DDT06 #2-10 RT: 0.01-0.24 AV: 9 NL: 2.51E6T: FTMS + p ESI Full ms [70.00-2000.00]
140 141 142 143 144 145 146 147
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
142.0418
144.0387
143.0451
145.0420140.9522 142.2385 146.9735146.0452141.8456 144.2396139.2004 140.0265 143.8530

Daniel Timelthaler 112/126
Figure A 49: 1H NMR of 25g.
Figure A 50: 13C NMR of 25g.
Figure A 51: HR-MS of 25g.
DDT07 #2-10 RT: 0.01-0.24 AV: 9 NL: 1.91E6T: FTMS + p ESI Full ms [70.00-2000.00]
139.5 140.0 140.5 141.0 141.5 142.0 142.5 143.0 143.5 144.0 144.5 145.0 145.5 146.0 146.5
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
142.0418
144.0387
143.0451
145.0420140.9523142.2385141.8456 145.9541140.0254 143.1481 146.7629139.1292 144.2397
143.8449

Daniel Timelthaler 113/126
Figure A 52: 1H NMR of 25h.
Figure A 53: 13C NMR of 25h.
Figure A 54: HR-MS of 25h.
DDT08 #2-10 RT: 0.01-0.24 AV: 9 NL: 1.64E6T: FTMS + p ESI Full ms [70.00-2000.00]
139 140 141 142 143 144 145 146
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
142.0418
144.0388
143.0450
140.9523 145.0420146.9735142.9180
142.2384
140.0260139.6329 144.5022143.7905 145.4881 146.1405141.5906
144.1553141.9316

Daniel Timelthaler 114/126
Figure A 55: 1H NMR of 25i.
Figure A 56: 13C NMR of 25i.
Figure A 57: 19F NMR of 25i.

Daniel Timelthaler 115/126
Figure A 58: HR-MS of 25i.
Figure A 59: 1H NMR of 25j.
Figure A 60: 13C NMR of 25j.
DDT09 #2-10 RT: 0.01-0.24 AV: 9 NL: 1.63E6T: FTMS + p ESI Full ms [70.00-2000.00]
122 123 124 125 126 127 128 129
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
126.0713
127.0745
129.0300125.0150122.0961 128.0778126.2357125.9072123.0057 124.0549 129.9795127.5754123.4118

Daniel Timelthaler 116/126
Figure A 61: 19F NMR of 25j.
Figure A 62: HR-MS of 25j.
Figure A 63: 1H NMR of 25k.
DDT10 #2-10 RT: 0.01-0.24 AV: 9 NL: 3.20E6T: FTMS + p ESI Full ms [70.00-2000.00]
153 154 155 156 157 158 159 160
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
156.0820
157.0852
158.9638158.0882156.3085154.9678 155.8560156.9338
154.0661153.1458 160.7285159.9001157.6683

Daniel Timelthaler 117/126
Figure A 64: 13C NMR of 25k.
Figure A 65: 19F NMR of 25k.
Figure A 66: HR-MS of 25k.
DDT11 #2-10 RT: 0.01-0.24 AV: 9 NL: 1.14E7T: FTMS + p ESI Full ms [70.00-2000.00]
173 174 175 176 177 178 179 180 181
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
176.0684
177.0715
178.0744176.3398175.7976174.9755 179.6231174.0526176.8552
180.8051180.2568178.9953173.6960 177.3452

Daniel Timelthaler 118/126
Figure A 67: 1H NMR of 25l.
Figure A 68: 13C NMR of 25l.
Figure A 69: HR-MS of 25l.
DDT12 #2-10 RT: 0.02-0.25 AV: 9 NL: 6.14E5T: FTMS + p ESI Full ms [50.00-2000.00]
105 106 107 108 109 110 111 112 113
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
109.0759
110.0821
113.0959111.0803106.9569 108.0443 108.9539 109.1917105.0696 112.0834106.1347109.8036
110.5080
Daniel Timelthaler 119/126
Figure A 70: 1H NMR of 25n *DCM.
Figure A 71: 13C NMR of 25n *DCM.
Figure A 72: HR-MS of 25n.
DDT13 #2-10 RT: 0.02-0.25 AV: 9 NL: 4.19E6T: FTMS + p ESI Full ms [50.00-2000.00]
133.5 134.0 134.5 135.0 135.5 136.0 136.5 137.0 137.5 138.0 138.5 139.0 139.5 140.0 140.5
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
136.1122
137.1154
134.0968 138.9831138.1185136.2967134.9584 135.9281136.8060
133.6118 139.9235 140.6991137.2213
*
*

Daniel Timelthaler 120/126
Figure A 73: 1H NMR of 25o.
Figure A 74: 13C NMR of 25o.
Figure A 75: HR-MS of 25o.
DDT14 #2-10 RT: 0.02-0.25 AV: 9 NL: 4.63E6T: FTMS + p ESI Full ms [50.00-2000.00]
110.5 111.0 111.5 112.0 112.5 113.0 113.5 114.0 114.5 115.0 115.5 116.0 116.5 117.0 117.5
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
114.1277
115.1309116.9857
116.1340114.2693111.0204 113.9864112.8526111.8053115.0237
117.1497110.6078 115.3813 116.8742 117.8446
110.1839
112.3223

Daniel Timelthaler 121/126
Figure A 76: 1H NMR of 25p.
Figure A 77: 13C NMR of 25p.
Figure A 78: HR-MS of 25p.
DDT15 #2-10 RT: 0.02-0.25 AV: 9 NL: 2.61E5T: FTMS + p ESI Full ms [50.00-2000.00]
85.5 86.0 86.5 87.0 87.5 88.0 88.5 89.0 89.5 90.0 90.5 91.0 91.5 92.0 92.5
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
88.1119
89.115390.976387.0188 90.627390.182385.3027 87.932686.2564 92.400385.9414 88.6391 91.4055 91.7228
88.398289.6375 92.7608

Daniel Timelthaler 122/126
Figure A 79: 1H NMR of 25q.
Figure A 79: 13C NMR of 25q.
Figure A 80: HR-MS of 25q.

Daniel Timelthaler 123/126
Figure A 81: 1H NMR of 25r.
Figure A 82: 13C NMR of 25r.
Figure A 83: HR-MS of 25r.
DDT17 #2-10 RT: 0.02-0.25 AV: 9 NL: 1.52E6T: FTMS + p ESI Full ms [50.00-2000.00]
114.0 114.5 115.0 115.5 116.0 116.5 117.0 117.5 118.0 118.5 119.0 119.5 120.0 120.5 121.0 121.5
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
117.1385
116.9858
118.1418
117.9890 120.9785118.9787114.1275 115.0238
117.2858
115.6149 116.0760 120.4283 121.3443118.3282 119.9816116.7664

Daniel Timelthaler 124/126
Figure A 84: 1H NMR of 25s.
Figure A 85: 13C NMR of 25s.
Figure A 86: HR-MS of 25s.
DDT18 #2-10 RT: 0.02-0.25 AV: 9 NL: 1.33E7T: FTMS + p ESI Full ms [50.00-2000.00]
163 164 165 166 167 168 169 170 171
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
166.1594
167.1626
168.1656166.4082165.9112164.9846166.8807
163.1330 169.4180167.6664163.7810 164.2391 169.9803169.0494 171.0096

Daniel Timelthaler 125/126
Figure A 87: 1H NMR of 25t.
Figure A 88: 13C NMR of 25t.
Figure A 89: HR-MS of 25t.
DDT19 #2-10 RT: 0.02-0.25 AV: 9 NL: 1.66E6T: FTMS + p ESI Full ms [50.00-2000.00]
119 120 121 122 123 124 125
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
122.0965
123.0998
120.9786 124.1029 125.9860118.9788 122.2533 124.9756121.9402 123.4838118.4175 120.2875122.7372
119.9193

Daniel Timelthaler 126/126
Figure A 90: 1H NMR of 25u, *DCM.
Figure A 91: 13C NMR of 25u, *DCM.
Figure A 92: HR-MS of 25u.
DDT20 #2-10 RT: 0.02-0.25 AV: 9 NL: 2.01E6T: FTMS + p ESI Full ms [50.00-2000.00]
134 135 136 137 138 139 140 141
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
136.0757
137.0790135.0441140.9524138.9831138.0823134.0665 136.9539135.9709
136.2236137.5185 140.2466139.6602133.3512 134.6819
*
*





























