Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DO PARANÁ
DEPARTAMENTO DE HIDRÁULICA E SANEAMENTO – DHS
LABORATÓRIO DE ENGENHARIA AMBIENTAL BORSARI NETO
Elaboração e Organização: Carla Cristina Bem e Luiz Fernando Dombroski
Curitiba
2010
MANUAL DE PROCEDIMENTOS
LABORATORIAIS INTEGRA-CLIMASUL
APLICADO AO MONITORAMENTO DE
PARÂMETROS ASSOCIADOS À QUALIDADE
DA ÁGUA EM CORPOS AQUÁTICOS

ÍNDICE
CAPÍTULO 1 - Monitoramento de qualidade da água em corpos aquáticos...........4
CAPÍTULO 2 - Parâmetros de interesse na gestão e monitoramento de qualidade
da água em corpos aquáticos..................................................................................6
CAPÍTULO 3 - Procedimentos para amostragem em campo, conservação e
armazenamento de amostras de água associadas ao monitoramento de qualidade
da água em corpos aquáticos................................................................................12
CAPÍTULO 4 – Considerações gerais sobre coleta, preservação de amostras e
medidas de segurança em laboratório...................................................................15
CAPÍTULO 5 - Procedimentos para limpeza e descontaminação de frascos,
vidrarias e materiais de uso geral utilizados nos ensaios laboratoriais.................20
CAPÍTULO 6 - Procedimentos para amostragem em campo, conservação e
armazenamento de amostras de água associadas ao monitoramento de qualidade
da água em corpos aquáticos................................................................................25
POP 01 - Oxigênio dissolvido.................................................................................30 POP 02 - DBO5.......................................................................................................38 POP 03 - DQO........................................................................................................47 POP 04 - Carbono orgânico dissolvido..................................................................48 POP 05 - Série de sólidos......................................................................................51 POP 06 - UV-Vis.....................................................................................................56 POP 07 a POP 11 - Série de nitrogênio.................................................................58 POP 12 e POP 13 - Série de fósforo......................................................................79 POP 14 - Clorofila-a...............................................................................................90 POP 15 - Metais pesados (Cd, Cr, Cu, Ni, Zn, Pb)................................................93 POP 16 - Alcalinidade total.....................................................................................99 POP 17 - Fluorescência molecular.......................................................................103 POP 18 - Coliformes totais e fecais......................................................................105

LISTA DE SÍMBOLOS
PEAD – polietileno de alta densidade
PP - polipropileno
OD – oxigênio dissolvido
DBO5 – demanda biológica de oxigênio (5 dias de incubação)
DQO – demanda química de oxigênio
COD – carbono orgânico dissolvido
CT – carbono total
CI – carbono inorgânico
UV-Vis – região ultravioleta - visível do espectro eletromagnético
IF – intensidade de fluorescência
N-NO2- – nitrogênio na forma de nitrito
N-NO3- – nitrogênio na forma de nitrato
N-NH3 – nitrogênio amoniacal total
NH4+ – íon amônio (amônia ionizada)
NH3 – amônia não ionizada
NT – nitrogênio total
NOrgT – nitrogênio orgânico total
NINORG – nitrogênio inorgânico total
PT – fósforo total
PTD – fósforo total dissolvido
PTP – fósforo total particulado
PO43- - ortofosfato / fosfato reativo solúvel
POrgD – fósforo orgânico dissolvido
ST – sólidos totais
SF – sólidos totais fixos
SV – sólidos totais voláteis
SST – sólidos suspensos totais
SSF – sólidos suspensos fixos
SSV – sólidos suspensos voláteis
SDT – sólidos dissolvidos totais
– comprimento de onda e – variação do comprimento de onda

4
_____________________________________________________________
Capítulo 1 – Monitoramento de
qualidade das águas em corpos aquáticos ______________________________________________________________
O monitoramento da qualidade ambiental surgiu da necessidade de
mensurar a interferência ou nível de degradação causada por agentes de
natureza antropogênica no ambiente ao longo do tempo (variações temporais) ou
do espaço (variações espaciais), por meio da análise, experimentos e
interpretação de parâmetros físicos, químicos e biológicos.
A freqüência do monitoramento deve ser de acordo com a resposta
requerida e da infraestrutura disponível para realizá-lo. O uso do monitoramento
da qualidade de corpos aquáticos pode ser realizado e aplicado como:
Instrumento de comando e controle e;
Instrumento de gestão do recurso.
O monitoramento de comando e controle consiste em analisar pontos
estratégicos onde possam estar ocorrendo violações nos padrões de qualidade
ambiental estabelecidos por legislação, sendo utilizados, principalmente, pelos
órgãos que possuem poder fiscalizador.
Quando se pretende obter dados de qualidade para dar suporte ao sistema
de gestão, o monitoramento deve ser realizado ao longo de tempo, permitindo
observar as variações temporais da qualidade e, ao longo do espaço,
possibilitando observar plumas de poluição, desde sua origem, dispersão e/ou
degradação completa. Este foco permite a indagação de inúmeras questões
voltadas aos resultados obtidos frente ao monitoramento de parâmetros da
qualidade da água.
Além da importância em se estabelecer uma frequência no monitoramento,
que seja adequada à informação a ser obtida, é fundamental que os dados
produzidos sejam de uma confiabilidade inquestionável (validação dos
procedimentos analíticos) e possam garantir uma verdadeira imagem fotográfica

5
momentânea do que estava ocorrendo no ambiente durante uma determinada
amostragem.
Em relação à qualidade da água, a questão da consistência dos dados de
qualidade e quantidade de água é de grande relevância na gestão dos recursos
hídricos, por estes serem a base principal para a busca pelas ferramentas
necessárias no suporte de tomada de decisão.
Esta confiabilidade dos dados coletados em campo é obtida por meio de
análises físico-químicas realizadas com procedimentos analíticos adequados e
executados com rigor, além da utilização de sensores calibrados, de modo a
produzir resultados que representem com precisão e exatidão as concentrações
existentes na amostra.
A validação dos dados evita que erros sejam propagados e venham a
produzir outros, como no caso da modelagem matemática de cenários futuros,
calibrados com valores de concentrações que possuem distorções devido a
determinação analítica inadequada ou execução do procedimento.
Por fim, é importante notar que esta pequena introdução é um comunicado
de grande importância deste manual, ao analista que venha a desempenhar o
papel fundamental de comunicar aos demais envolvidos do projeto de pesquisa
que os dados obtidos em campo e em laboratório, são de alta qualidade e
veracidade frente ao ambiente aquático estudado.

6
______________________________________________________________
Capítulo 2 – Parâmetros de interesse na
gestão e monitoramento de qualidade da água em corpos aquáticos ______________________________________________________________
As variáveis monitoradas para avaliar a qualidade das águas podem ser
físicas, químicas e biológicas.
As variáveis físicas são:
(a) TEMPERATURA: descrita diretamente como a medida da intensidade de
calor. Em ambientes aquáticos é um parâmetro importante, pois influi em algumas
propriedades da água (densidade, viscosidade, oxigênio dissolvido), com reflexos
sobre a manutenção da vida aquática. A temperatura pode variar em função de
fontes naturais (energia solar, sazonalidade do meio) e fontes antropogênicas
(águas residuárias industriais; águas de resfriamento de máquinas e outros).
(b) TURBIDEZ: parâmetro de qualidade da água relacionado diretamente com
a presença de material em suspensão (sólidos suspensos) na água, como areia,
silte, argila, substâncias orgânicas finamente divididas, organismos microscópicos
e outras partículas.
(c) CONDUTIVIDADE ELÉTRICA: capacidade que a água possui de
conduzir corrente elétrica. Este parâmetro está relacionado com a presença de
íons dissolvidos na água, que são partículas carregadas eletricamente Quanto
maior a quantidade de íons dissolvidos, maior será a condutividade elétrica na
água. Para estudos em corpos aquáticos, os resultados são, normalmente,
mensurados em campo com o uso de sensores (condutivímetro), devidamente
calibrado. O valor se dá através da medida lida pelo sensor e a unidade
apresentada como S.cm-1 ou mS.cm-1.
As variáveis químicas são:

7
POTENCIAL HIDROGENIÔNICO (pH): o pH de uma solução é o logaritmo
decimal negativo da concentração de íons hidrônio (H3O+, em mol.L-1) e avalia o
caráter ácido ou básico da solução. O pH é normalmente determinado por
eletrometria, mas pode ser estimado por titulação ou por papel indicador. A
medição do pH por eletrometria baseia-se na determinação da atividade dos íons
hidrônio pela medição potenciométrica utilizando um eletrodo de vidro associado
a um eletrodo de referência.
ALCALINIDADE: causada por sais alcalinos, principalmente de sódio e cálcio;
mede a capacidade da água de neutralizar os ácidos; em teores elevados, pode
proporcionar sabor desagradável à água, tem influência nos processos de
tratamento da água.
OXIGÊNIO DISSOLVIDO: elemento principal no metabolismo dos
microorganismos aeróbios que habitam as águas naturais ou os reatores para
tratamento biológico de esgotos. A determinação da concentração de oxigênio
dissolvido em águas é também imprescindível para o desenvolvimento da análise
da demanda bioquímica de oxigênio (DBO) e é um dos parâmetros que indicam o
nível de degradação de ambientes aquáticos. Em corpos aquáticos, a
quantificação de OD pode ser feita com o uso de sensores ou por meio de
procedimentos analíticos em laboratório. A unidade de concentração é mg O2.L-1.
DEMANDA BIOQUÍMICA DE OXIGÊNIO: a Demanda Bioquímica de
Oxigênio (DBO) é um teste no qual procedimentos padronizados de laboratório
são usados para determinar a quantidade de oxigênio relativa em águas naturais,
efluentes domésticos e industriais que são consumidos no processo de
estabilização da matéria orgânica presente na amostra durante um período de
tempo, considerando somente a atividade microbiológica.
O teste de DBO é empregado para determinar os níveis de poluição, na
avaliação de cargas poluidoras ou eficiência de um determinado sistema de
tratamento. O teste de DBO mais difundido é o DBO520, no qual, as amostras são
incubadas por 5 dias a 20ºC, mas também há a DBOU (Demanda Bioquímica
Última de Oxigênio). Normalmente é realizado o teste de DBO520 devido ao menor

8
período de incubação e a degradação que ocorre neste tempo equivale a cerca
de 70% da concentração de matéria orgânica presente.
Os valores obtidos no teste de DBO com os obtidos nos testes de DQO
podem ser relacionados. Esta relação DQO/DBO indica a biodegrabilidade da
amostra. Quanto mais elevada for esta relação, menor é a fração biodegradável, e
quando menor a relação, maior a atividade de biodegradação da amostra. O valor
da DBO é expresso em mg O2.L-1.
DEMANDA QUÍMICA DE OXIGÊNIO: a Demanda Química de Oxigênio
(DQO) é definida como a quantidade de oxigênio necessária para oxidar
quimicamente e completamente a matéria orgânica e inorgânica oxidável de uma
determinada amostra, sob condições de análise controladas. O parâmetro DQO é
largamente empregado na avaliação de qualidade das águas naturais e de águas
residuárias industriais e domésticas. Seu valor pode ser correlacionado com
outros parâmetros como, por exemplo, com a Demanda Bioquímica de Oxigênio
(DBO). O valor da concentração é expresso em mg O2.L-1.
SÉRIE DE SÓLIDOS: resíduos ou sólidos são todas as matérias suspensas ou
dissolvidas na água, provenientes de despejos domésticos ou industriais. Pode-se
interpretar o termo sólido a partir de uma definição operacional, como sendo a
matéria que permanece como resíduo após evaporação, secagem ou calcinação,
a uma determinada temperatura padrão e por um tempo fixo de um volume de
amostra conhecido. Os sólidos de uma água podem ser classificados de acordo
com o fluxograma disposto abaixo:
Sólidos Suspensos Totais (SST) Sólidos Dissolvidos Totais (SDT)
Sólidos Voláteis Totais (SVT) Sólidos Fixos Totais (SFT)
Sólidos Totais (ST)
Sólido
Suspensos
Voláteis
(SSV)
Sólido
Suspensos
Fixos
(SSF)
Sólido
Dissolvidos
Voláteis
(SDV)
Sólido
Dissolvidos
Fixos
(SDF)

9
De acordo com o tratamento térmico efetuado na amostra, pode-se, ainda,
fragmentar os sólidos em termos de “fixos” e “voláteis”, sendo que o primeiro é
aplicado ao resíduo total, em suspensão ou dissolvido, após aquecimento e
secagem por um período específico e a uma temperatura específica. A massa
perdida por ignição é chamada de “sólidos voláteis”; a determinação dessas
porções não permite distinguir com precisão a matéria orgânica e inorgânica, uma
vez que a perda por ignição não envolve apenas a matéria orgânica, podendo
ocorrer perdas (pequenas, considerando a temperatura utilizada) na
decomposição ou volatilização de sais minerais, por exemplo.
UV-Vis: a espectrofotometria na região UV-Vis do espectro eletromagnético é
uma das técnicas analíticas mais empregadas, em função de robustez, custo
relativamente baixo e grande número de aplicações desenvolvidas como
exemplo, na caracterização da matéria orgânica. O método consiste na leitura da
amostra a partir de varredura entre faixas de comprimento de onda na região do
UV-Vis, normalmente, entre os comprimentos de onda de 200 a 800nm, para
posterior tratamento dos dados. A resposta do sinal de absorvância será dada em
função dos constituintes orgânicos e/ou inorgânicos que compõe cada amostra,
cada qual com picos de intensidade de sinais em faixas distintas do espectro.
SÉRIE DE NITROGÊNIO: o nitrogênio (N) é um macronutriente essencial ao
metabolismo dos seres vivos pois, após o carbono é o elemento exigido em maior
quantidade pelas células vivas e ao contrário do fósforo (P), é abundante no
ambiente aquático (na grande maioria dos casos, visto que pode atuar também
como nutriente limitante). Sua importância se deve, principalmente, a sua
participação na formação de proteínas e, em baixas concentrações, atuando
como nutriente utilizado na produtividade primária, etapa metabólica dos
ambientes aquáticos. Em ambientes aquáticos, o nitrogênio encontra-se presente
em diferentes espécies: N2 (nitrogênio molecular), NO2- (nitrito), NO3
- (nitrato),
N2O (óxido nitroso), nitrogênio orgânico dissolvido, nitrogênio orgânico
particulado, NH4+ (íon amônio), NH3 (amônia), sendo a soma desses dois últimos
o nitrogênio amoniacal total. Dentre essas espécies, o nitrato e o íon amônio são
de grande importância para os ecossistemas aquáticos, por representarem a
principal fonte de nitrogênio para os produtores primários (Ex.: fitoplâncton).

10
O estado de oxidação dos compostos de nitrogênio em corpos aquáticos
pode indicar a idade e o grau de poluição. Isto significa dizer, por exemplo, que as
formas reduzidas apontam para um foco de poluição próximo, enquanto a
prevalência de NO2- (baixa concentração) e NO3
-, ao contrário, indica que a
influência de atividades antropogênicas, como os lançamentos de esgotos, se
encontram distante, considerando que os estados reduzidos ou oxidados são
função, principalmente, da concentração de oxigênio dissolvido na coluna d’água.
SÉRIE DE FÓSFORO: a grande importância do fósforo deve-se à participação
deste elemento em processos fundamentais do metabolismo dos seres vivos
como macronutriente. Entretanto, quando comparado a outros nutrientes é, em
geral, considerado limitante. Tal fato se deve a tendência de formar compostos
insolúveis associados a argilas, cátions, óxihidróxidos de ferro, material
particulado, os quais acabam incorporando e concentrando uma significativa
quantidade de fósforo nos sedimentos. Em águas naturais, é geralmente
encontrado na forma iônica ou complexado, como o fosfato, por ser a única forma
estável em solução aquosa. Sob esta forma, o fósforo pode ser encontrado como:
PO43-, HPO4
2-, H2PO4-, conhecidas como ortofosfato ou, também, fósforo reativo
solúvel. Estas por sua vez, representam a fração de maior biodisponibilidade e
rápida assimilação pelas algas e plantas aquáticas. As formas dissolvidas ou
particuladas estarão sempre combinadas ou complexadas a outros elementos, a
exemplo do ferro. Ainda, a presença de fósforo nos sedimentos envolve a
contribuição dos minerais do solo da bacia de drenagem ou deposição através da
coluna d’água por meio de processos físicos, químicos ou biológicos.
CLOROFILA-a: monitorada no laboratório como uma variável biológica, a
clorofila-a é um parâmetro de análise realizado normalmente em ambientes
lênticos, mas com possibilidade de casos de florações de algas em ambientes
lóticos. A clorofila-a é o pigmento fotossintetizante primário dos organismos que
produzem oxigênio estando presente em algas, organismos fotossintetizantes e
também em algumas bactérias, sendo o único pigmento que tem a capacidade de
converter energia luminosa em moléculas orgânicas.
O monitoramento da concentração de clorofila-a em sistemas aquáticos
permite avaliar quantitativamente a biomassa fitoplanctônica presente no

11
ambiente, resultado este que pode ser aplicado como indicador de condições
como, por exemplo, o grau de trofia.
METAIS PESADOS: a presença de metais pesados em ambientes aquáticos
se relaciona tanto com fontes naturais, a partir de processos físicos e químicos,
envolvendo, por exemplo, desgate (intemperismo) e lixiviação do material
contindo nos solos e rochas que compôe a bacia de drenagem de rios, lagos,
reservatórios. Neste caso, as concentrações avaliadas para metais são
relacionadas a níveis de “background”, encontrados a partir da ocorrência natural,
considerando a sua variabilidade e disponibilidade geológica entre diferentes
regiões. Por outro lado, atividades de origem antropogênica que repercutem no
impacto sobre a qualidade do ambiente (ar, água, solos e sedimentos,) resultam
em níveis elevados de metais pesados de alto caráter tóxico (ao considerar
valores guia de qualidade para cada metal) como: arsênio (As), mercúrio (Hg),
chumbo (Pb), cádmio (Cd), cromo (Cr), cobre (Cu), níquel (Ni) e zinco (Zn). Estes
metais aportam nos sistemas aquáticos a partir de uma variedade de fontes,
sendo transportados direta ou indiretamente, a exemplo do lançamento de águas
residuárias industriais (distintos ramos industriais) e por meio da deposição
atmosférica (precipitação dos metais, influência das chuvas e dos ventos no
transporte para outras regiões), além da importante contribuição da drenagem
urbana, a qual se caracteriza por níveis relativamente elevados para Cu, Pb e Zn.
No ambiente aquático, a especiação dos metais representa um importante
significado na avaliação da biodisponibilidade e toxicidade, com diferenças
significativas entre diferentes espécies. Neste caso se torna, por exemplo, uma
área específica no estudo das espécies para avaliação dos seus efeitos na biota
aquática. Um exemplo é a verificação das espécies de cromo, a partir do estado
de oxidação quando verificada as formas mais estáveis: Cr (III) e Cr (VI). O Cr (VI)
ou cromo hexavalente é descrito como a forma do elemento mais tóxica, com um
significante impacto em efeitos adversos sobre a qualidade do ambiente aquático.
Neste caso, a necessidade de valores guia de qualidade imposta para as
diferentes espécies é fundamental para obter um melhor entendimento do impacto
causado pelo metal. Outra questã fundamental é a tendência de acúmulo dos
metais em sedimentos de fundo, os quais influenciam diretamente e de forma
dinâmica na disponibilidade para a coluna da água.

12
______________________________________________________________
Capítulo 3 – Procedimentos para
amostragem em campo, conservação e armazenamento de amostras de água associadas ao monitoramento de qualidade da água em corpos aquáticos ______________________________________________________________
A coleta das amostras deve ser realizada de tal forma que seja garantida e
preservada a total integridade de suas características físico-químicas, inicialmente
presentes no ambiente possibilitando obter um “retrato do ponto de amostragem”.
Para tal, os procedimentos de coleta e armazenamento para posterior análise em
laboratório são específicos para cada parâmetro. Estes devem ser aplicados
como metodologia modelo para as linhas de pesquisa relacionadas ao trabalho de
campo, no estudo de ambientes aquáticos.
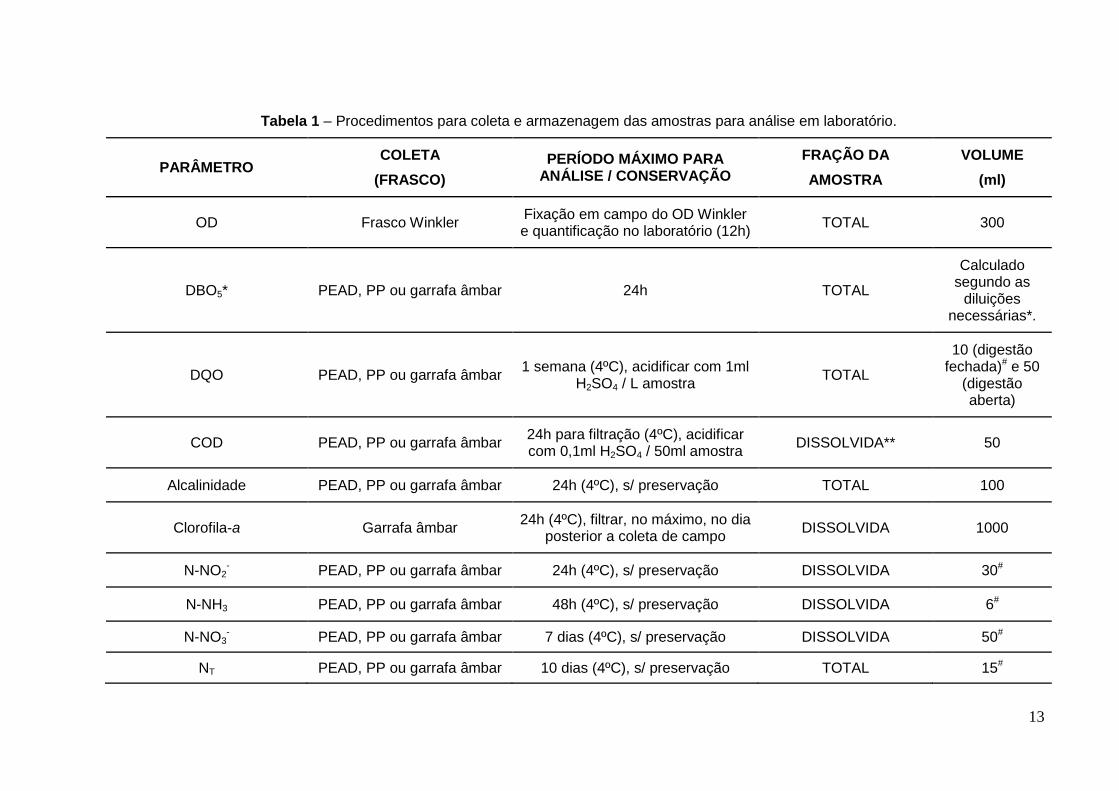
A Tabela 1 apresenta um resumo do procedimento para coleta
considerando: estocagem, período para realização do ensaio, procedimentos para
conservação (quando necessário), volume e fração da amostra utilizada na
determinação dos parâmetros associados ao monitoramento de qualidade da
água de corpos aquáticos.
Os procedimentos para preparo, limpeza e descontaminação dos frascos
para amostragem constam no Capítulo 4 - Procedimentos para limpeza e
descontaminação de frascos, vidrarias e materiais de uso geral utilizados
nos ensaios laboratoriais.
13
Tabela 1 – Procedimentos para coleta e armazenagem das amostras para análise em laboratório.
PARÂMETRO COLETA
(FRASCO)
PERÍODO MÁXIMO PARA ANÁLISE / CONSERVAÇÃO
FRAÇÃO DA
AMOSTRA
VOLUME
(ml)
OD Frasco Winkler Fixação em campo do OD Winkler e quantificação no laboratório (12h)
TOTAL 300
DBO5* PEAD, PP ou garrafa âmbar 24h
TOTAL
Calculado segundo as
diluições necessárias*.
DQO PEAD, PP ou garrafa âmbar 1 semana (4ºC), acidificar com 1ml
H2SO4 / L amostra TOTAL
10 (digestão fechada)# e 50
(digestão aberta)
COD PEAD, PP ou garrafa âmbar 24h para filtração (4ºC), acidificar com 0,1ml H2SO4 / 50ml amostra
DISSOLVIDA** 50
Alcalinidade PEAD, PP ou garrafa âmbar 24h (4ºC), s/ preservação TOTAL 100
Clorofila-a Garrafa âmbar 24h (4ºC), filtrar, no máximo, no dia
posterior a coleta de campo DISSOLVIDA 1000
N-NO2- PEAD, PP ou garrafa âmbar 24h (4ºC), s/ preservação DISSOLVIDA 30#
N-NH3 PEAD, PP ou garrafa âmbar 48h (4ºC), s/ preservação DISSOLVIDA 6#
N-NO3- PEAD, PP ou garrafa âmbar 7 dias (4ºC), s/ preservação DISSOLVIDA 50#
NT PEAD, PP ou garrafa âmbar 10 dias (4ºC), s/ preservação TOTAL 15#
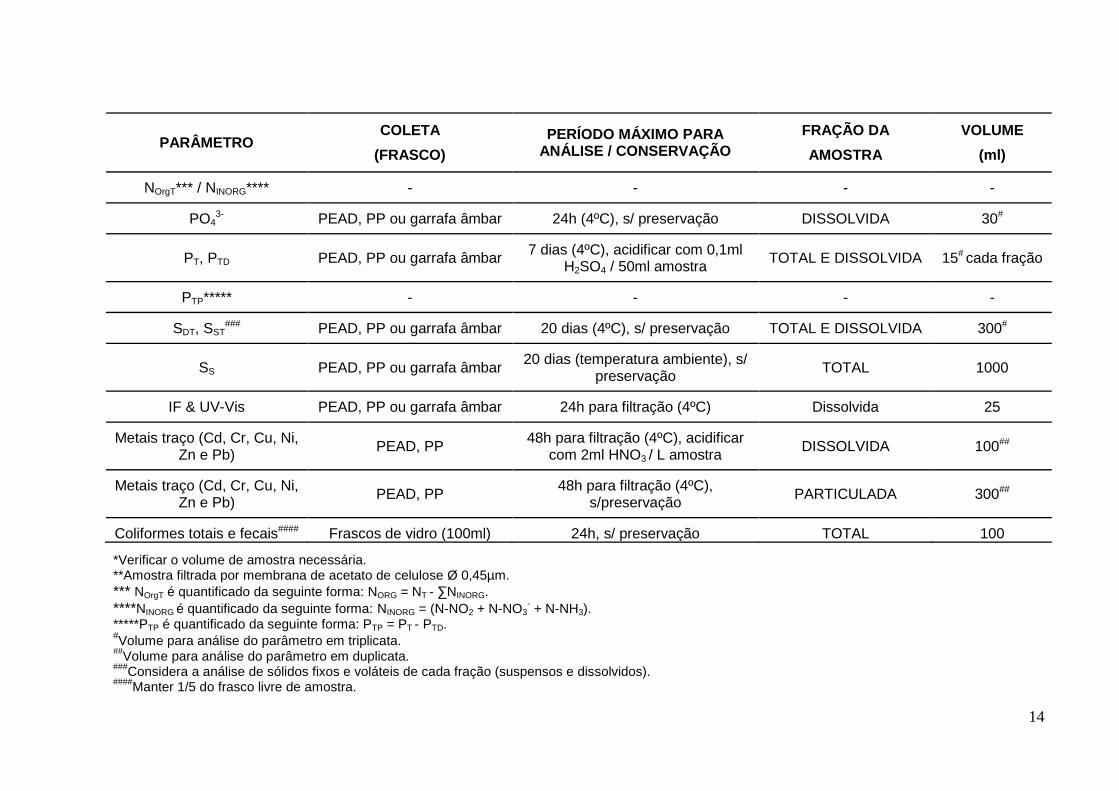
14
PARÂMETRO COLETA
(FRASCO)
PERÍODO MÁXIMO PARA ANÁLISE / CONSERVAÇÃO
FRAÇÃO DA
AMOSTRA
VOLUME
(ml)
NOrgT*** / NINORG**** - - - -
PO43- PEAD, PP ou garrafa âmbar 24h (4ºC), s/ preservação DISSOLVIDA 30#
PT, PTD PEAD, PP ou garrafa âmbar 7 dias (4ºC), acidificar com 0,1ml
H2SO4 / 50ml amostra TOTAL E DISSOLVIDA 15# cada fração
PTP***** - - - -
SDT, SST###
PEAD, PP ou garrafa âmbar 20 dias (4ºC), s/ preservação TOTAL E DISSOLVIDA 300#
SS PEAD, PP ou garrafa âmbar 20 dias (temperatura ambiente), s/
preservação TOTAL 1000
IF & UV-Vis PEAD, PP ou garrafa âmbar 24h para filtração (4ºC) Dissolvida 25
Metais traço (Cd, Cr, Cu, Ni, Zn e Pb)
PEAD, PP 48h para filtração (4ºC), acidificar
com 2ml HNO3 / L amostra DISSOLVIDA 100##
Metais traço (Cd, Cr, Cu, Ni, Zn e Pb)
PEAD, PP 48h para filtração (4ºC),
s/preservação PARTICULADA 300##
Coliformes totais e fecais#### Frascos de vidro (100ml) 24h, s/ preservação TOTAL 100
*Verificar o volume de amostra necessária. **Amostra filtrada por membrana de acetato de celulose Ø 0,45µm.
*** NOrgT é quantificado da seguinte forma: NORG = NT - ∑NINORG.
****NINORG é quantificado da seguinte forma: NINORG = (N-NO2 + N-NO3
- + N-NH3).
*****PTP é quantificado da seguinte forma: PTP = PT - PTD. #Volume para análise do parâmetro em triplicata.
##Volume para análise do parâmetro em duplicata.
###Considera a análise de sólidos fixos e voláteis de cada fração (suspensos e dissolvidos).
####Manter 1/5 do frasco livre de amostra.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
15
______________________________________________________________
Capítulo 4 – Considerações gerais sobre
coleta, preservação de amostras e medidas de segurança em laboratório ______________________________________________________________
O objetivo da amostragem é coletar pequenas porções de um determinado
material, para favorecer o transporte, porém, grandes o suficiente para atenderem
os propósitos da análise, ou seja, representar acuradamente o material
amostrado.
Os métodos de coleta de amostras devem ser realizados de acordo com o
material a ser analisado e com os propósitos da análise, seguindo-se metodologia
específica para cada parâmetro. Porém, algumas regras gerais podem ser
estabelecidas para a realização de um procedimento correto de amostragem.
1. Assegure-se de que todos os equipamentos estão limpos e em boa
qualidade de uso antes de usar;
2. Quando uma determinada substância preservativa for adicionada aos
recipientes de coleta, encher os mesmos sem pré-enxaguar com a amostra
e tomar cuidado para não extravasar no enchimento e não haver formação
de bolhas;
3. Todos os métodos de preservação podem ser inadequados tratando-se de
análises relacionadas com a fração particulada da amostra;
4. Depois de fechado o frasco de coleta, verificar a formação de bolhas por
inversão e leves batidas no frasco. Se houver bolhas de ar, o ideal é
descartar e coletar outra amostra, porém, isto não deve ser feito se ao frasco
tiver sido adicionado algum preservativo antes do enchimento com amostra;
5. Para coleta em rios, no caso de amostra simples, coletar preferencialmente
no meio da seção transversal a meia profundidade (para outros tipos de
coleta, estudar o método mais apropriado);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
16
6. Evite áreas com muita turbulência, pois, pode haver muita perda de
constituintes voláteis e potencial presença de vapores tóxicos mais densos
que o ar;
7. Dependendo do tipo de análise a ser realizada, encha completamente o
recipiente de coleta (p/ a maioria das análises de compostos orgânicos) ou
deixe espaço para aeração, mistura e etc. (análises microbiológicas e
compostos inorgânicos);
8. Evite coletar água da superfície a menos que óleos e graxas sejam
desejáveis ou não interfiram na análise;
9. Exceto para a análise de compostos orgânicos voláteis, deixe um espaço
livre de aproximadamente 1% em todas as amostras para permitir expansão
térmica durante o transporte;
10. Identifique todas as amostras especificando inclusive características físicas e
químicas da água e também características climáticas que possam ser
correlacionas com as características da amostra (utilize caneta com tinta à
prova d’água);
11. Ao acidificar amostras tenha certeza de que a diluição proporcionada pela
acidificação é desprezível ou grande o suficiente para que um fator de
correção possa ser incorporado ao cálculo das concentrações (usar ácido
ultra puro para evitar contaminação);
Alguns fatores importantes que interferem nos resultados das análises são:
a presença de material em suspensão ou turbidez, o método escolhido para
remover a amostra do recipiente de coleta, e as mudanças físicas e químicas
ocorridas devido ao armazenamento ou aeração. Na análise de elementos traço,
procedimentos detalhados para o processamento das amostras são necessários,
especialmente para metais e compostos orgânicos. Deve-se definir e considerar
cuidadosamente no plano de amostragem as técnicas específicas de coleta para
tornar as amostras representativas. Para metais, frequentemente, é apropriado
coletar uma amostra filtrada e outra normal para diferenciar entre metal total,
dissolvido e metal particulado, presente na matriz. Deve-se estar atento para o
fato de que alguns metais podem ser parcialmente adsorvidos ao filtro.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
17
Frequentemente, uma leve turbidez pode ser tolerada se a experiência
mostrar que esta não causará nenhuma interferência em testes gravimétricos ou
volumétricos e que sua influência em testes colorimétricos pode ser corrigida,
pois, é neste teste que se apresentam os maiores efeitos da interferência da
turbidez em amostras.
Frascos de coleta
O tipo de frasco de coleta a ser usado é de grande importância,
principalmente na determinação de elementos traço, por isso, certifique-se de que
todos os recipientes de coleta estão foram descontaminados seguindo
procedimento padrão. Em geral, utilizam-se frascos de plástico ou vidro, a escolha
depende do constituinte a ser analisado, por exemplo, sílica, sódio e boro podem
se desprender de frascos de vidro leve, mas não do plástico, e quantidades traço
de alguns pesticidas e metais podem ser adsorvidos nas paredes de frascos de
vidro. Portanto, frascos de vidro duro (pyrex ou equivalente) são preferíveis. Para
amostras contendo compostos orgânicos, não use frascos de plástico ou somente
aqueles feitos de polímeros fluorados como os de politetrafluoretileno (PTFE).
Sempre que possível, evite plásticos, devido ao seu potencial contaminante por
ésteres de ftalato. Use frascos de vidro para todas as análises de compostos
orgânicos como orgânicos voláteis, orgânicos semi-voláteis, pesticidas, PCBs e
óleos e graxas. Alguns compostos são sensíveis a luz (compostos contendo
bromo, alguns pesticidas, compostos aromáticos polinucleares, etc.), por isso,
utilize nestes casos, frascos de vidro âmbar para minimizar a fotodegradação.
Geralmente, as tampas de frascos de vidro são de plástico, o que pode, em
alguns casos, representar um problema. Não utilize tampas de plástico com forro
de papel. Use forros de PTFE ou lâmina, mas esteja ciente de que forros de metal
podem contaminar amostras para análise de metais e também podem reagir com
amostras ácidas ou alcalinas. Frascos de soro com borracha forrada com PTFE
ou septo de PTFE também podem ser utilizados.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
18
Dicas de segurança
Devido à possibilidade de algum constituinte ser tóxico, deve-se ter muito
cuidado ao manusear as amostras, assim como em todo o processo das análises.
Substâncias tóxicas podem entrar em nosso corpo pela pele, pelos olhos e no
caso de vapores, a partir da respiração, podendo atingir os pulmões. A seguir,
apresenta-se alguns procedimentos de segurança:
1. Use luvas apropriadas ao tipo de método a ser desenvolvido;
2. Sempre use óculos de proteção;
3. Na presença de vapores tóxicos, faça a amostragem em locais bem
ventilados, ou utilize equipamento de respiração adequado;
4. Nunca se alimente dentro de um laboratório ou próximo às amostras e locais
de amostragem;
5. Sempre lave muito bem as mãos antes de se alimentar;
6. Não deixe as amostras próximas de locais com chamas ou muito quentes;
7. No caso de presença de compostos inflamáveis e necessidade de
refrigeração das amostras, utilize apenas refrigeradores especiais à prova de
explosões;
8. Colete as amostras de maneira segura, evitando acidentes;
9. Para coleta de substâncias radioativas, procedimentos de segurança
específicos devem ser adotados;
10. No caso de dúvida quanto ao nível de periculosidade, sempre consulte um
profissional de segurança ou higiene industrial.
Minimização dos resíduos
A minimização dos resíduos de laboratório, além de contribuir com o meio
ambiente, reduz os custos do laboratório. Por isso, algumas práticas devem ser
adotadas em todos os procedimentos de análises químicas. Algumas são:
1. Sempre que possível, compre e utilize quantidades menores de
substâncias químicas. Pode ser mais econômico comprar quantidades

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
19
maiores, porém, deve-se estar atento ao fato de que quantidades maiores
ficam mais sujeitas a ter o seu prazo de validade expirado antes do término
da substância, gerando maior quantidade de resíduo e maior custo para
disposição final e tratamento do mesmo;
2. Utilize primeiro os produtos que estão com seus prazos de validade mais
próximos do vencimento (mais velhos), ou se possível, adquira-os apenas
no momento da análise na quantidade ideal;
3. Produtos químicos vencidos e que não foram abertos, muitas vezes podem
retornar para o fornecedor para serem reciclados ou corretamente
dispostos;
4. De preferência para métodos de análise que utilizem menor quantidade de
reagente, e que estes não sejam perigosos;
5. Evite transformar resíduos não perigosos em perigosos através de mistura
quando descartados no mesmo local;
6. Transfira produtos que estão armazenados à muito tempo para locais onde
outras pessoas possam ter acesso e então utilizar (sempre com a
autorização do responsável pelo laboratório ou produtos).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
20
______________________________________________________________
Capítulo 5 - Procedimentos para limpeza
e descontaminação de frascos, vidrarias e materiais de uso geral utilizados nos ensaios laboratoriais ______________________________________________________________
A inclusão deste capítulo no manual de procedimentos laboratoriais,
voltado às técnicas de limpeza e descontaminação do material utilizado nas
diversas metodologias analíticas, descritas na sequência, no Capítulo 6, é uma
etapa fundamental e de grande importância antes do início de uma metodologia e,
dessa forma, merece especial atenção.
Em muitas situações o procedimento de descontaminação ou a ausência
deste, pode gerar dúvidas preocupantes durante uma determinada análise. Fato
este, provocado pelo equívoco da separação do material, não verificação da
qualidade e validade dos reagentes analíticos, dentre outros.
Muitas vezes, a quantificação do parâmetro pode ser realizada em triplicata
e os valores responderem bem próximos com baixa variabilidade, entretanto para
todas as réplicas, existe a possibilidade de contaminação, originada, por exemplo,
da separação de vidrarias da mesma fonte (Ex.: vidrarias lavadas com detergente
Extran, contendo fosfato na composição, utilizadas para ensaios da série de
fósforo).
Por fim, o presente capítulo apresenta os passos a serem aplicados no
preparo de material para posterior análise dos parâmetros. Os procedimentos
para limpeza dos frascos de amostragem também são apresentados neste
capítulo.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
21
1. PROCEDIMENTOS DE DESCONTAMINAÇÃO
1.1. Oxigênio Dissolvido (OD) e Demanda Bioquímica de Oxigênio (DBO)
Toda a vidraria utilizada na determinação de OD e DBO deve seguir os
passos abaixo:
- Limpeza com escovão para vidraria e detergente Extran. Enxágue diversas
vezes com água normal. Posteriormente, enxágue no mínimo 3 vezes com água
destilada e deionizada. Colocar vidraria para secar em estufa (40±5 ºC);
No caso dos frascos utilizados no preparo dos reagentes analíticos, seguir os
passos abaixo:
- Enxaguar diversas vezes com água normal. Emergir em banho de HCl 10% por
um período mínimo de 12h (ideal: 24h);
- Retirar do banho ácido e enxaguar no mínimo 3 vezes com água destilada e
deionizada. Colocar os frascos para secar em estufa (40±5 ºC);
1.2. Demanda Química de Oxigênio (DQO), Carbono Orgânico
(COT/COD), UV-Vis e Fluorescência (IF)
Toda a vidraria utilizada na quantificação de DQO (refluxo aberto ou refluxo
fechado) e frascos para preparo de reagentes devem seguir os passos abaixo:
- Limpeza com escovão para vidraria e detergente Extran. Enxágue diversas
vezes com água normal;
- Transferir o material para banho de HNO3 10% por um período mínino de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar com água destilada (mínino de 3 vezes);
- Transferir o material para banho de HCl 10% por um período mínimo de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar no mínimo 3 vezes com água destilada e
deionizada. Colocar os frascos para secar em estufa (40±5 ºC);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
22
1.3. Série de Sólidos
O preparo do material para sólidos não apresenta necessidades de
soluções ácidas para lavagem. É necessária apenas uma boa limpeza com
detergente Extran, para remoção do material residual, lavagem no mínimo 3
vezes com água destilada e deionizada e estufa para secar (40±5 ºC).
1.4. Série de Nitrogênio e Clorofila-a
Todo material aplicado aos ensaios laboratoriais da série de nitrogênio,
deve seguir os passos abaixo para descontaminação:
- Limpeza com escovão para vidraria e detergente Extran. Enxágue diversas
vezes com água normal;
- Transferir o material para banho de HCl 10% por um período mínino de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar no mínimo 3 vezes com água destilada e
deionizada. Colocar os frascos para secar em estufa (40±5 ºC);
ATENÇÃO: nunca mantenha ou utilize material que foi imerso em banhos de HNO3 10% para
descontaminação nos ensaios da série de nitrogênio, devido à contaminação pelo ácido na
detecção das formas do elemento.
1.5. Série de Fósforo
Todo material aplicado aos ensaios laboratoriais da série de fósforo, deve
seguir os passos abaixo para descontaminação:
- Após utilizar o material ou para iniciar o ensaio da série de fósforo lavar o
material APENAS com água normal, enxaguando diversas vezes;
- Transferir o material para banho de HNO3 10% por um período mínino de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar com água destilada (mínino de 3 vezes).
Colocar os frascos para secar em estufa (40±5 ºC);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
23
ATENÇÃO: no caso da série de fósforo, se possível, é preferível manter o material separado para
evitar a utilização de vidrarias, frascos, dentre outros que tenham sido lavados com detergente
Extran, visto que é muito utilizado no laboratório. A determinação de fósforo acaba sendo muito
sensível a pequenas contaminações causadas geralmente pela limpeza com detergente. Fato
este, devido às baixas concentrações, quantificadas na maioria dos casos.
1.6. Metais pesados
Todo material aplicado aos ensaios laboratoriais de metais pesados, deve
seguir os passos abaixo para descontaminação:
- Limpeza com escovão para vidraria e detergente Extran. Enxágue diversas
vezes com água normal;
- Transferir o material para banho de HNO3 10% por um período mínino de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar com água destilada (mínino de 3 vezes);
- Transferir o material para banho de HCl 10% por um período mínimo de 12h
(ideal: 24h);
- Retirar do banho ácido e enxaguar no mínimo 3 vezes com água mili-Q (ultra-
pura);
- Para secar o material preparado para análise de metais pesados, não manter o
material exposto ao ar ou estufa de modo a evitar contaminação. Coloque o
material em bandejas e cubra, por exemplo, com papel filme;
ATENÇÃO: durante o manuseio do material e durante o procedimento, utilize de preferência luvas isentas de talco em pó, devido à possibilidade de contaminação, principalmente, por zinco.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
24
______________________________________________________________
Capítulo 6 - Procedimentos e métodos
analíticos para ensaios laboratoriais de parâmetros de qualidade da água ______________________________________________________________
O presente capítulo tem por objetivo, descrever passo a passo os materiais
e métodos analíticos envolvidos na quantificação e validação dos resultados
relacionados aos parâmetros de qualidade da água de corpos aquáticos (rios,
córregos, reservatórios, lagos e outros). Assim, de forma sucinta, cada método
apresenta os seguintes itens: descrição breve da metodologia, materiais
(equipamentos, vidrarias, reagentes analíticos) necessários, procedimentos para
calibração e padronização de métodos, procedimentos para tratamento e análise
da amostra, limites de detecção do método, referências e outros.
De grande importância e apresentado neste capítulo, é a redução do
volume de amostra necessário para as análises de diversos parâmetros,
considerando inclusive a quantificação em triplicata para uma melhor resposta e
confiabilidade da metodologia proposta pelo manual.
Por fim, os seguintes parâmetros são apresentados neste capítulo: OD,
DBO, DQO, UV-Vis, COD, série de sólidos, série de nitrogênio, série de
fósforo, clorofila-a, metais pesados (Cd, Cr, Cu, Ni, Zn e Pb), alcalinidade
total, intensidade de fluorescência molecular (IF) e coliformes (totais e
fecais).
É importante ressaltar que os métodos foram expostos da maneira mais
usualmente aplicada no laboratório de Engenharia Ambiental Borsari Neto nos
projetos que envolvem avaliação da qualidade da água em ambientes aquáticos.
Entretanto, todos os procedimentos analíticos podem ser novamente adaptados e
avaliados quanto à possibilidade de aplicação, dependendo da necessidade do
trabalho, linhas de pesquisa e/ou projetos.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
25
POP 01 – OXIGÊNIO DISSOLVIDO (OD)
1. MÉTODO WINKLER MODIFICADO PELA AZIDA SÓDICA
1.1. MÉTODO WINKLER MODIFICADO PELA AZIDA SÓDICA
Este método consiste em fixar o oxigênio dissolvido da amostra por meio
da adição das soluções de sulfato manganoso (MnSO4) e a solução álcali-iodeto-
azida, que contém hidróxido de sódio (NaOH), iodeto de sódio (NaI) e azida
sódica (NaN3).
A fixação do oxigênio dissolvido ocorre através da formação de óxido de
manganês, segundo a reação 1:
Mn (OH2) + ½O2 → MnO2 + H2O (1)
Nesta etapa ocorre uma intensa floculação da amostra e uma consequente
precipitação do material floculado. A solução de azida sódica é utilizada para a
remoção da interferência de nitritos, representada pelas reações 2 e 3:
NaN3 + H+ → HN3 + Na+ (2)
HN3 + NO2− + H+ → N2 + N2O + H2O (3)
A segunda fase é a liberação de iodo, que ocorre após a adição de ácido
sulfúrico concentrado (H2SO4), provocando a ruptura dos flocos e o
desenvolvimento de uma coloração amarelada, cuja intensidade é proporcional à
concentração de oxigênio dissolvido presente inicialmente na amostra. A reação 4
expressa o procedimento citado acima:
MnO2 + 2I- + 4H+ → Mn+2 + I2 + 2H2O (4)
Note-se que o íon iodeto é oxidado a iodo molecular em proporção
estequiométrica a quantidade de óxido de manganês que, por sua vez, é
proporcional à concentração de oxigênio dissolvido na amostra, conforme
mostrado na reação de fixação (1).
A fase final da análise é a titulação do iodo liberado com solução
padronizada de tiossulfato de sódio (iodometria), representado pela reação 5.
2Na2S2O3 + I2 → Na2S4O6 + 2NaI + 10H2O (5)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
26
O indicador desta reação é uma solução de amido (0,5% - 2,5%), com
ponto de viragem da titulação do azul para incolor.
Os resultados para a concentração de OD presente na amostra são
expressos na unidade de mg O2.L-1.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Não necessita.
Vidrarias
Frascos de DBO winkler de 300ml
Pipetas volumétricas
Béckeres
Erlenmeyer
Bureta de 50ml
Reagentes analíticos
Solução de Sulfato Manganoso (MnSO4): dissolver 480g de
MnSO4.4H2O, ou 400g MnSO4.2H2O ou 364g de MnSO4.H2O em,
aproximadamente, 800ml de água destilada e deionizada, filtrar em papel
filtro Watman nº 40 e diluir em balão volumétrico de 1000ml.
* Estocar em frascos tipo âmbar ou frascos de vidro, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução de Alcali-Iodeto-Azida: dissolver em, aproximadamente, 400ml
de água destilada e deionizada e seguindo a ordem:
- 250g de hidróxido de sódio (NaOH p.a); adicione lentamente devido
à elevada quantidade de NaOH (reação ↑ exotérmica).
- 67,5g de iodeto de sódio (NaI p.a);
Diluir para 500ml em balão volumétrico. Em seguida, transferir 5g de azida
sódica (NaN3 p.a), previamente dissolvida em 20ml de água destilada,
perfazendo um volume final de 520ml.
* Estocar em frascos tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
27
Ácido Sulfúrico concentrado (p.a);
Solução de Dicromato de Potássio (K2Cr2O7) 0,004167 mol.L-1: dissolver
1,226g de dicromato de potássio p.a (K2Cr2O7), previamente seco em
estufa a temperatura de 100ºC por 2h, em 800ml de água destilada.
Transferir para um balão volumétrico de 1000ml, aferindo o volume total
com água destilada.
* Estocar em frascos tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução indicadora de Amido 1%: adicionar em um bécker, 100ml de
água destilada e 1g de amido solúvel (p.a). Levar a mistura para
aquecimento e agitação simultaneamente até a temperatura atingir valores
entre 60 e 70ºC (controle através de um termômetro graduado). Na
sequência, mantenha por, aproximadamente, 15min a solução de amido
nesta faixa de temperatura, sob agitação. Por fim, transfira a solução já
dissolvida ou o sobrenadante para um frasco de reagente.
* Mantenha o frasco em ambiente refrigerado (< 4ºC) para preservação
* Estável por 6 meses (mínimo).
Solução Padrão de Tiossulfato de Sódio (Na2S2O3.5H2O) 0,025 mol.L-1:
dissolver 6,205g de tiossulfato de sódio pentahidratado, juntamente com
1,5ml de hidróxido de sódio 6 mol.L-1 (ou 0,4g de NaOH p.a), em
aproximadamente 800mL de água destilada. Transferir para um balão
volumétrico de 1000ml, aferindo o volume total com água destilada.
* Estocar em frascos tipo âmbar ou frascos de vidro, em temperatura ambiente.
* A utilização da solução é dependente de sua padronização diária (hora do uso) para cálculo
da concentração real.
Padronização: dissolver aproximadamente 2g de iodeto de potássio (KI p.a) em
100ml de água deionizada e destilada em um erlenmeyer de 250ml. Adicionar
10ml de solução H2SO4 1:9 (1ml de H2SO4 e 9ml de água deionizada).
Acrescentar 20ml da solução de dicromato de potássio 0,004167 mol.L-1. Deixar a
solução no escuro por 5min. Em seguida, aferir para 200ml com água destilada.
Titular a solução com o tiossulfato de sódio 0,025 mol.L-1 até a coloração amarelo-
palha, acrescentar então 5 gotas do indicador amido 1% e prosseguir a titulação

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
28
até o ponto de viragem do azul para o incolor. Calcular a concentração real
(mol.L-1) do tiossulfato segundo a equação 1:
)(2
)(1*)()(
322
722722322
OSNav
OCrKvOCrKMOSNaM
(1)
em que:
M (K2Cr2O7) = molaridade da solução de dicromato de potássio (mol.L-1).
v1 (K2Cr2O7) = volume da solução de dicromato de potássio (mL).
v2 (Na2S2O3) = volume da solução de tiossulfato gasto na titulação (mL).
1.3. PROCEDIMENTO ANALÍTICO (Amostra)
1. Transferir a amostra para o frasco Winkler, evitando agitar a amostra e a
formação de bolhas;
2. Adicionar 1ml de sulfato manganoso;
3. Adicionar 1ml de solução alcalina de iodeto azida;
4. Feche o frasco, misture por inversão no mínimo 10 vezes:
Havendo a formação de uma suspensão leitosa, não há oxigênio
dissolvido para ser determinado na amostra;
Havendo a formação de um precipitado de cor marrom, dar
continuidade a análise (ver tópico 5).
* As etapas de 1 a 4 devem ser, preferencialmente, realizadas em campo com a amostragem
direta no frasco Winkler, evitando ao máximo a presença de bolhas.
5. Em laboratório, adicionar ao frasco 1ml de H2SO4 (manuseie
cuidadosamente, com o uso de EPIs – jaleco, luvas e óculos de proteção);
6. Fechar o frasco e misturar por inversão novamente até que o precipitado
seja totalmente dissolvido;
7. Transferir do frasco para um erlenmeyer, 100ml da amostra;
8. Titular com solução padronizada de tiossulfato de sódio 0,025 mol.L-1 (valor
teórico) até o aparecimento de uma coloração amarelo-palha;
9. Acrescentar então, de 3 a 5 gotas do indicador amido 1% e prosseguir a
titulação até o ponto de viragem da cor azul para incolor;
10. Anote o volume gasto na titulação (ml), correspondente a amostra;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
29
1.4. CÁLCULO DA CONCENTRAÇÃO DE OD
fcVgLmgOD *2*)/( (2)
em que:
Vg = volume de tiossulfato de sódio gasto na titulação (mL).
fc= fator de correção do tiossulfato de sódio (Volume prático / Volume teórico).
1.5. LIMITE DE DETECÇÃO
Concentração de oxigênio dissolvido (mg O2.L-1):
Limite máximo: 7 a 9 mg OD.L-1
Limite mínimo: 2 mg OD.L-1
2. DESTINAÇÃO DO RESÍDUO GERADO
Depois de finalizada a etapa de titulação o resíduo gerado na leitura do
OD, pode ser descartado diretamente na pia sem necessidade de um pré-
tratamento.
3. REFERÊNCIA
STANDARD METHODS, methods 4500 - O C & 5210 B (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
30
POP 02 – DEMANDA BIOQUÍMICA DE OXIGÊNIO (DBO5)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO WINKLER MODIFICADO PELA AZIDA SÓDICA
O teste de DBO tem por objetivo, determinar a quantidade de oxigênio
consumido por microorganismos aeróbios presentes na amostra e assim,
relacionar com a quantidade de matéria orgânica de natureza biodegradável.
O método usualmente empregado para a determinação da DBO é o da
diluição, incubação por um período de 5 dias a 20ºC, com a determinação dos
níveis iniciais e finais da concentração de oxigênio através do método da azida
sódica modificada (POP-01).
1.1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Garrafão para água de diluição (compatível com o volume necessário no
ensaio)
Incubadora termo-regulável (20±1ºC)
Vidrarias
Frascos de DBO de 300ml
Pipetas volumétricas
Béckeres
Erlenmeyer
Bureta de 50ml
Reagentes analíticos - Soluções nutrientes
Solução Tampão de Fosfato: dissolver em aproximadamente 600ml de
água destilada:
- 8,5g de fosfato monobásico de potássio (KH2PO4 p.a);
- 21,75g de fosfato dibásico de potássio (K2HPO4 p.a);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
31
- 33,4g de fosfato dibásico de sódio heptahidratado (Na2HPO4.7H2O
p.a);
- 1,7g de cloreto de amônio (NH4Cl p.a)
Transferir para um balão volumétrico de 1000ml e completar o volume com
água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Sulfato de Magnésio (MgSO4): dissolver 22,5g de sulfato de
magnésio heptahidratado (MgSO4.7H2O p.a) em aproximadamente 800ml
de água, transferir para um balão volumétrico de 1000ml e completar o
volume com água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Cloreto de Cálcio (CaCl2): dissolver 27,5g de cloreto de
cálcio anidro (CaCl2 p.a) em aproximadamente 800ml de água, transferir
para um balão volumétrico de 1000ml e completar o volume com água
destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Cloreto Férrico (FeCl3): dissolver 0,25g de cloreto férrico
hexahidratado (FeCl3.6H2O p.a) em aproximadamente 800ml de água,
transferir para um balão volumétrico de 1000ml e completar o volume.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Água de Diluição: adicionar a um garrafão previamente limpo e estéril
água destilada de acordo com a necessidade a ser empregada no teste;
manter o conteúdo desse garrafão em aeração por, no mínimo, 45 min e
máximo de 1h30min; deixar em repouso por aproximadamente 30min; na
sequência, adicionar 1ml de cada uma das soluções nutrientes para cada
litro de água de diluição utilizada.
Reagentes analíticos - Método Winkler modificado pela azida sódica (POP-01)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
32
Os reagentes utilizados na etapa de quantificação do OD (inicial e final),
através da titulação iodométrica, seguem os mesmos procedimentos de preparo e
padronização descritos no POP-01 (procedimento para OD).
1.1.2. PROCEDIMENTO ANALÍTICO (Amostra)
1.1.2.1. Método Winkler sem semente
1. Regular o pH das amostras para 6,8 a 7,2 quando estiverem a temperatura
ambiente. Utilize para o ajuste soluções levemente ácidas ou básicas como
HCl 0,01 mol.L-1 ou NaOH 0,01 mol.L-1;
2. Adicionar ao frasco de DBO identificado, a amostra ou amostra mais a
alíquota da diluição. O Standard recomenda pelo menos 5 diluições. As
diluições devem ser decididas com base na concentração de DQO,
previamente quantificada (ver POP-03);
3. Anotar o nº e volume do frasco e a porcentagem da amostra adicionada em
uma ficha de controle padrão;
4. No caso de diluição da amostra, completar o volume do frasco com água de
diluição evitando a formação de bolhas e turbulências;
5. Preparar, considerando a necessidade de diluição, amostra em réplica de
modo que possibilite a medição do ODinicial e ODfinal;
5. Medir o ODinicial por meio do método modificado pela azida sódica (ver POP-
01);
6. Levar as amostras de DBO5 para a incubação (20±1ºC);
7. Após 5 dias determinar a concentração de ODfinal da amostra por meio do
método modificado pela azida sódica (ver POP-01);
1.1.2.2. Procedimento Winkler com semente
1. Regular o pH das amostras para 6,8 a 7,2 quando estiverem a temperatura
ambiente. Utilize para o ajuste soluções levemente ácidas ou básicas como
HCl 0,01 mol.L-1 ou NaOH 0,01 mol.L-1;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
33
2. Anotar o nº e o volume do frasco ou porcentagem da amostra adicionada
em uma ficha de controle;
3. Adicionar o volume da amostra ao frasco de DBO preparado evitando a
formação de bolhas e turbulências;
4. Em um frasco em separado, incubar a semente, adicionando 2ml da
semente preparada, completando o volume com água de diluição;
5. Completar o volume do frasco com água de diluição evitando a formação
de bolhas e turbulências;
6. Medir o ODinicial por meio do método modificado pela azida sódica (ver
POP-01);
7. Levar as amostras de DBO5 para a incubação (20±1ºC);
8. Após 5 dias determinar a concentração de ODfinal da amostra por meio do
método modificado pela azida sódica (ver POP-01);
Preparo da semente
As sementes utilizadas no teste da DBO podem ser obtidas de amostras in
natura de esgotos, de amostras do próprio efluente coletadas após 3 a 8km a
jusante do ponto de despejo, de forma a obter microorganismos adaptados.
Também podem ser utilizados sólidos suspensos ou serem adquiridas sementes
comerciais para DBO.
CÁLCULO DA DBO SEM SEMENTE
( / ) I FOD ODDBO mg L
P
5
(3)
em que :
ODI = concentração de OD inicial (mg.L-1)
ODF = concentração de OD final (mg.L-1)
P = fração volumétrica da amostra utilizada (ml)
CÁLCULO DA DBO COM SEMENTE
( )*( / ) I F I FOD OD B B f
DBO mg LP
5 (4)
em que :

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
34
ODI = concentração de OD inicial (mg.L-1)
ODF = concentração de OD final (mg.L-1)
BI = concentração de OD no controle da semente inicial (mg.L-1)
BF = concentração de OD no controle da semente final (mg.L-1)
f = razão da semente diluída na amostra em relação à semente de controle
P = fração volumétrica da amostra utilizada (ml)
1.2. DESTINAÇÃO DO RESÍDUO GERADO
Depois de finalizada a etapa de titulação o resíduo gerado nas leituras de
DBO, pode ser descartado diretamente na pia sem necessidade de um pré-
tratamento.
1.3. LIMITE DE DETECÇÃO
Concentração de oxigênio dissolvido (mg.L-):
limite máximo – 7 a 9 mg OD.L-1
limite mínimo – 2 mg OD.L-1
1.4. MÉTODO RESPIROMÉTRICO / MANOMÉTRICO – OXITOP
Baseia-se numa amostra em uma garrafa âmbar sob quantidade suficiente
de microorganismos e nutrientes a temperatura controlada de 201ºC e que por
meio de agitação faz com que o O2 presente na câmara de ar se dissolva no
líquido. Os microorganismos respiram este oxigênio dissolvido na amostra
durante o processo de degradação da matéria orgânica, exalando CO2, que é
absorvido pelos grânulos de NaOH p.a contido em um reservatório de borracha,
produzindo uma diferença de pressão na garrafa, que é medida pelo sensor
Oxitop, cujo sistema realiza este leitura digital e conversão dos valores para mg
O2.L-1.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
35
1.4.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Garrafas de DBO Oxitop
Bandeja de agitação magnética
Agitador magnético
Incubadora termo-regulável (20±1ºC)
Vidrarias
Pipetas volumétricas
Frascos volumétricos padrão
Reagentes analíticos
NaOH (p.a) em pérolas;
Solução Tampão de Fosfato: dissolver em aproximadamente 600ml de
água destilada:
- 8,5g de fosfato monobásico de potássio (KH2PO4 p.a);
- 21,75g de fosfato dibásico de potássio (K2HPO4 p.a);
- 33,4g de fosfato dibásico de sódio heptahidratado (Na2HPO4.7H2O
p.a);
- 1,7g de cloreto de amônio (NH4Cl p.a)
Transferir para um balão volumétrico de 1000ml e completar o volume com
água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Sulfato de Magnésio (MgSO4): dissolver 22,5g de sulfato de
magnésio heptahidratado (MgSO4.7H2O p.a) em aproximadamente 800ml
de água, transferir para um balão volumétrico de 1000ml e completar o
volume com água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Cloreto de Cálcio (CaCl2): dissolver 27,5g de cloreto de
cálcio anidro (CaCl2 p.a) em aproximadamente 800ml de água, transferir

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
36
para um balão volumétrico de 1000ml e completar o volume com água
destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Cloreto Férrico (FeCl3): dissolver 0,25g de cloreto férrico
hexahidratado (FeCl3.6H2O p.a) em aproximadamente 800ml de água,
transferir para um balão volumétrico de 1000ml e completar o volume com
água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de Cloreto de Amônio (NH4Cl): dissolver 38,2g de cloreto de
amônio (NH4Cl p.a) em aproximadamente 800ml de água, neutralize o pH
para 7,0 com KOH, transfira para um balão volumétrico de 1000ml e
completar o volume com água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
1.2.2. PROCEDIMENTO ANALÍTICO OXITOP (Amostra)
1. Conhecer o valor da DQO da amostra ou a faixa de DBO esperada e de
acordo com estes valores observar na Tabela 2, o volume da amostra e
solução nutriente a ser transferido para a garrafa de Oxitop;
Tabela 2. Especificação dos valores de amostra e volume de solução nutriente utilizados
na quantificação de DBO, a partir do procedimento OXITOP segundo as faixas de DBO.
DBO ESPERADA (mg.L-1)
VOLUME DE AMOSTRA (ml)
VOLUME DE SOLUÇÃO NUTRIENTE (ml)
0 – 40 432 1,7 0 – 80 365 1,5 0 – 200 250 1,0 0 – 400 164 0,6 0 – 800 97 0,4
0 – 2000 43,5 0,2
2. Transferir o volume de amostra no frasco de DBO Oxitop;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
37
3. Pipetar o volume da solução nutriente, que é composta por:
a. 6ml da solução de tampão fosfato;
b. 2ml da solução de sulfato de magnésio;
c. 2ml da solução de cloreto férrico;
d. 2ml da solução de cloreto de cálcio;
e. 2ml da solução de cloreto de amônio.
4. Colocar a barra magnética dentro da garrafa;
5. Adicionar de 2 a 4 pastilhas de NaOH (p.a) no reservatório de borracha;
6. Colocar o reservatório com cuidado na boca da garrafa;
7. Fechar a garrafa com o sensor e colocar sobre o sistema de agitação;
8. Ligar o sistema de agitação e verificar os agitadores;
9. Pressionar simultaneamente as teclas M e S até que apareça no visor do
sensor “00”;
1.2.2.1. CÁLCULO DA DBO OXITOP
A concentração final da DBO5 será dada após o término da análise no leitor
do sensor do Oxitop.
1.2.2.2. LIMITE DE DETECÇÃO
0 – 2000 mg O2.L-1
2. DESCARTE DO RESÍDUO GERADO
Depois de finalizada as leituras no equipamento OXITOP, o resíduo
(amostra e mistura de solução nutriente) pode ser descartado diretamente na pia
sem necessidade de um pré-tratamento.
3. REFERÊNCIA
STANDARD METHODS, methods 4500 - O C & 5210 B (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
38
POP 03 – DEMANDA QUÍMICA DE OXIGÊNIO (DQO)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DO REFLUXO ABERTO – TITULOMÉTRICO
A matéria orgânica / inorgânica é oxidada em meio ácido (H2SO4) por um
forte agente oxidante – dicromato de potássio (K2Cr2O7) em excesso, em um
condensador de refluxo do tipo Friedrichs. Toda reação é catalisada por sulfato de
prata (Ag2SO4) e intensa liberação de calor.
Após a digestão, o excesso de dicromato é titulado com solução de sulfato
ferroso amoniacal – SFA – (Fe (SO4)2(NH4)2), previamente padronizado, e assim
determina-se a quantidade de oxidante consumida na reação; tal quantidade será
expressa em termos equivalentes de oxigênio. O tempo ideal para a digestão é de
2 horas.
1.1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Chapa de aquecimento contendo condensadores Friedrichs
Balança analítica (precisão ± 0,0001g)
Vidrarias
Balão de fundo chato de 500ml
Pérolas de vidro para controlar ebulição
Espátulas
Pipetas volumétricas
Balões volumétricos
Dispenser
Proveta
Bureta de 50ml

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
39
Reagentes analíticos
Ácido Sulfúrico com Sulfato de Prata: adicionar lentamente e
cuidadosamente 10g de sulfato de prata (Ag2SO4 p.a) em 1000ml de ácido
sulfúrico concentrado (H2SO4 p.a). Deixar em repouso de um a dois dias
para a dissolução completa.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução de Dicromato de Potássio 0,04167 mol.L-1: dissolver 12,259g
de dicromato de potássio (K2Cr2O7 p.a), previamente seco a 150ºC por 2h,
em aproximadamente 800ml de água destilada, transferir para um balão
volumétrico de 1000ml e aferir com água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo) – padrão primário
Solução de Dicromato de Potássio 0,004167 mol.L-1: diluir 100ml da
solução de dicromato de potássio 0,25 mol.L-1 em um balão volumétrico de
1000ml, completar o volume com água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo) – padrão primário
Solução de Sulfato Ferroso Amoniacal (SFA) 0,25 mol.L-1: dissolver 98g
de SFA (p.a) em aproximadamente 800ml de água destilada, adicionar
lentamente 20ml de ácido sulfúrico concentrado (H2SO4 p.a), aguardar
esfriar e transferir para um balão volumétrico de 1000ml, completar o
volume com água destilada.
* Estocar a solução em frasco tipo âmbar ou frasco de vidro, em temperatura ambiente.
* Estabilidade do reagente é dependente da padronização diária (momento do uso).
Solução de indicador ferroína: dissolver 1,485g de 1,10 – fenantrolina
monohidratada (p.a) e 695mg de sulfato ferroso heptahidratado (p.a) em
100ml de água destilada.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 6 meses (mínimo).
Padronização - SFA:

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
40
1. Adicionar 10 ml de dicromato de potássio 0,04167 mol.L-1 em um
erlenmeyer;
2. Completar o volume do erlenmeyer para 100ml, com água destilada;
3. Adicionar, com um dispenser, 30ml de ácido sulfúrico;
4. Aguardar esfriar;
5. Adicionar de 3 a 6 gotas de indicador ferroína;
6. Titular com a solução de SFA 0,25 mol.L-1 até o ponto de viragem do
indicador (verde para marrom);
7. Calcular a Molaridade da solução de SFA:
2500,0*)(2
)(1)( 722
SFAv
OCrKvSFAM
em que:
M (SFA) = molaridade da solução de sulfato ferroso amoniacal (mol.L-1).
v1 (K2Cr2O7) = volume da solução de dicromato de potássio (mL).
v2 (SFA) = volume da solução de tiossulfato gasto na titulação (mL).
Solução de Sulfato Ferroso Amoniacal (SFA) 0,025 mol.L-1: diluir 100ml
da solução de SFA 0,25 mol.L-1 em um balão volumétrico de 1000ml e
completar o volume com água destilada. Padronizar da mesma forma
descrita para o SFA 0,25 mol.L-1, com uma alteração: utilizar a solução de
dicromato de potássio 0,004167 mol.L-1 no método descrito anteriormente.
Sulfato de Mercúrio (HgSO4 p.a)
1.1.2. PROCEDIMENTO ANALÍTICO (Amostra)
1.1.2.1. Procedimento para DQO > 50 (mg.L-1)
1. Pesar em um balão de fundo chato 0,4g de sulfato de mercúrio;
2. Adicionar ao balão de fundo chato:
a. 20ml da amostra homogeneizada;
b. 10ml da solução 0,04167 mol.L-1 de dicromato de potássio;
c. 5ml de ácido sulfúrico p.a;
d. 3 a 4 pérolas de vidro;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
41
e. 25ml de ácido sulfúrico p.a./sulfato de prata com o auxilio de um
dispenser;
3. Conectar o balão no condensador de refluxo;
4. Deixar em refluxo por 2h; desligar o condensador e deixar esfriar;
5. Adicionar 5 a 6 gotas de solução indicadora de ferroína no balão;
6. Titular a amostra com solução de SFA 0,25 mol.L-1 (padronizada) até
atingir o ponto de viragem do verde azulado para o marrom;
7. Anotar o volume gasto na titulação;
1.1.2.2. Procedimento para DQO < 50 (mg.L-1)
1. Pesar em um balão de fundo chato 0,4g de sulfato de mercúrio;
2. Adicionar ao balão de fundo chato:
a. 50ml da amostra já homogeneizada;
b. 25ml da solução 0,004167 mol.L-1 de dicromato de potássio;
c. 5ml de ácido sulfúrico p.a;
d. 3 a 4 pérolas de vidro;
e. 25ml de ácido sulfúrico p.a./sulfato de prata com o auxilio de um
dispenser;
3. Conectar o balão no condensador de refluxo;
4. Deixar em refluxo por 2h; desligar o condensador e deixar esfriar;
5. Adicionar 5 a 6 gotas de solução indicadora de ferroína no balão;
6. Titular a amostra com solução de SFA 0,025 mol.L-1 (padronizada) até
atingir o ponto de viragem do verde azulado para o marrom;
7. Anotar o volume gasto na titulação.
1.1.3. CÁLCULO DA DQO
V
fcVVLmgODQO ab 8000**1.0*)
)/( 2
(5)
em que :
Vb: volume gasto na titulação do branco (mL)
Va: volume gasto na titulação da amostra (mL)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
42
V: volume da amostra (ml): 20 para DQO > 50 mg.L-1 e 50 para DQO < 50 mg.L-1.
fc: fator de correção do SFA
1.2. MÉTODO DO REFLUXO FECHADO – COLORIMÉTRICO
A matéria orgânica / inorgânica presente na amostra é oxidada por meio do
agente oxidante (K2Cr2O7). O método padrão emprega como reagentes: solução
padrão de hidrogenoftalato de potássio, solução catalítica ácida (Ag2SO4 em
H2SO4 concentrado) e solução digestora (composta de K2Cr2O7, HgSO4 e H2SO4
diluídos em água). O método consiste na redução do cromo (Cr6+ a Cr3+) e
subsequente análise através da modificação da coloração, em um
espectrofotômetro na região do visível. Este procedimento é usualmente
conduzido em um digestor a 150°C, por 2h e as leituras obtidas em
espectrofotômetro de luz visível. Para valores esperados de DQO menores que
90 mg.L-1, a amostra deve ser lida no comprimento de onda de 420nm, se a DQO
estiver entre 100 a 900 mg.L-1, a leitura deve ser realizada no comprimento de
onda de 600nm. Valores de concentração superiores a 900 mg.L-1 podem ser
obtidos através da diluição da amostra, previamente homogeneizada.
1.2.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Aparelho digestor - refluxo fechado
Espectrofotômetro UV-Vis
Balança analítica (precisão ± 0,0001g)
Micropipeta (100 - 1000µl)
Vidrarias
Pipetas graduadas e volumétricas
Frascos de vidro para digestão
Cubeta de vidro
Reagentes analíticos
Ácido sulfúrico (p.a);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
43
Sulfato de mercúrio (p.a);
Ácido Sulfúrico com Sulfato de Prata (solução catalítica): adicionar 10g
de sulfato de prata (Ag2SO4 p.a) em 1000ml de ácido sulfúrico concentrado
(H2SO4 p.a). Deixar um dia para a dissolução total.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução digestora: dissolver 1,0216g de dicromato de potássio (K2Cr2O7
p.a), previamente seco a 150ºC por 2 horas, em aproximadamente 80ml de
água destilada, adicionar lentamente 16,7ml de ácido sulfúrico p.a. e 3,33g
de sulfato de mercúrio. Dissolver, aguardar esfriar e transferir para um
balão volumétrico de 100ml, aferindo com água destilada; O preparo desta
solução considera a leitura em = 600nm (DQO > 100 mg O2.L-1).
Para leitura em = 420nm (DQO < 90 mg O2.L-1), deve ser seguido
o mesmo procedimento de preparo, apenas alterando a quantidade de
dicromato de potássio para 0,1022g, considerando 100ml de solução final.
* Estocar a solução em frasco tipo âmbar, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Hidrogenoftalato de Potássio (solução padrão para calibração):
dissolver 0,0425g de hidrogenoftalato de potássio (KHP), previamente seco
em estufa (110ºC) e dessecador, em água destilada e diluir para 100ml em
balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC) para estabilidade
do reagente (1 mês).
* KHP = 1,176 mg O2 / mg e DQO teórica de 500 mg O2.L-1
.
1.2.2. PROCEDIMENTO ANALÍTICO (Amostra)
1.2.2.1. Calibração do método
A calibração do método é realizada através de curva de calibração padrão,
descrito na seqüência:
1. A partir da solução padrão (KHP), preparar, em balões volumétricos de
50ml, a seguinte curva de calibração (volume final com água destilada):

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
44
Concentração DQO (mg.L-1) Volume (ml) 0 (branco) -
10 1 25 2,5 50 5
150 15 250 25
*Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser preparada
em duplicata ou triplicata.
2. Posteriormente ao preparo das soluções padrões, conforme a tabela
acima, retirar, com auxílio de uma pipeta volumétrica, uma alíquota de 2ml de
cada padrão e reservar nos frascos para digestão;
3. Na sequência, adicionar cuidadosamente (reação exotérmica ↑ calor),
2ml da solução digestora e em seguida, 2ml da solução catalítica;
4. Fechar bem os frascos e misturar lentamente pela parede do mesmo, de
modo a favorecer a mistura dos reagentes adicionados (evitar misturar totalmente
por inversão, pois a presença de H2SO4 pode danificar as tampinhas, podendo
causar vazamento durante a digestão e também, cuidar com o aquecimento dos
frascos);
5. Realizar a digestão em aparelho digestor por um período de 2h e
temperatura de 150ºC;
6. Após completar a digestão, transferir os frascos para um suporte
específico e aguardar o resfriamento;
7. Realizar leitura no espectrofotômetro, ( = 600nm), sem especificação de
tempo máximo para leitura após a digestão;
NOTA: O procedimento descrito acima considera a calibração para leituras em
= 600nm. No caso de calibração para = 420nm os passos metodológicos são os
mesmos, havendo apenas a mudança da solução digestora (K2Cr2O7), (Ver item
1.2.1 – Reagentes).
8. Equação da reta, obtida com a plotagem do gráfico: absorbância (eixo y)
X concentração (eixo x);
9. Verificar coeficiente de linearidade: R > 0,990;
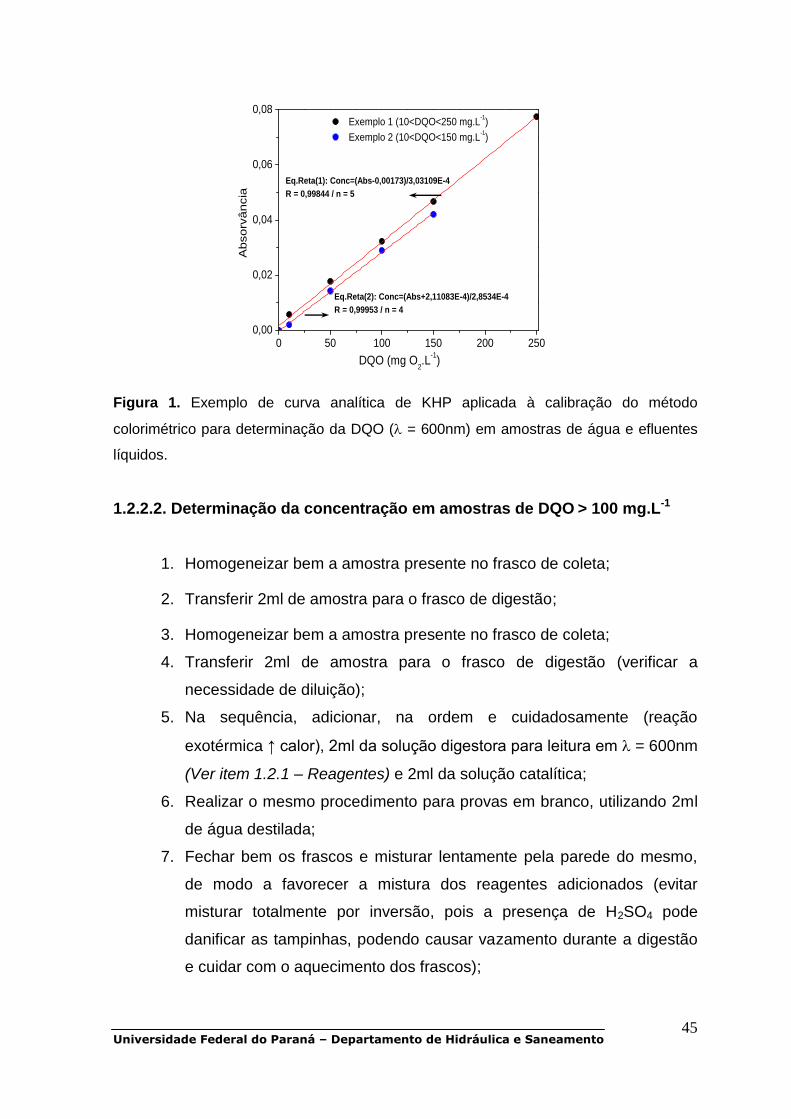
10. Realizar comparação com a curva analítica apresentada abaixo na
Figura 1.
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
45
0 50 100 150 200 2500,00
0,02
0,04
0,06
0,08 Exemplo 1 (10<DQO<250 mg.L
-1)
Exemplo 2 (10<DQO<150 mg.L-1)
Eq.Reta(2): Conc=(Abs+2,11083E-4)/2,8534E-4
R = 0,99953 / n = 4
Eq.Reta(1): Conc=(Abs-0,00173)/3,03109E-4
R = 0,99844 / n = 5
A
bso
rvâ
ncia
DQO (mg O2.L
-1)
Figura 1. Exemplo de curva analítica de KHP aplicada à calibração do método
colorimétrico para determinação da DQO ( = 600nm) em amostras de água e efluentes
líquidos.
1.2.2.2. Determinação da concentração em amostras de DQO > 100 mg.L-1
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir 2ml de amostra para o frasco de digestão;
3. Homogeneizar bem a amostra presente no frasco de coleta;
4. Transferir 2ml de amostra para o frasco de digestão (verificar a
necessidade de diluição);
5. Na sequência, adicionar, na ordem e cuidadosamente (reação
exotérmica ↑ calor), 2ml da solução digestora para leitura em = 600nm
(Ver item 1.2.1 – Reagentes) e 2ml da solução catalítica;
6. Realizar o mesmo procedimento para provas em branco, utilizando 2ml
de água destilada;
7. Fechar bem os frascos e misturar lentamente pela parede do mesmo,
de modo a favorecer a mistura dos reagentes adicionados (evitar
misturar totalmente por inversão, pois a presença de H2SO4 pode
danificar as tampinhas, podendo causar vazamento durante a digestão
e cuidar com o aquecimento dos frascos);

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
46
8. Realizar a digestão em aparelho digestor por um período de 2h e
temperatura de 150ºC;
9. Após completar a digestão, transferir os frascos para um suporte
específico e aguardar o resfriamento;
10. Realizar leitura no espectrofotômetro, ( = 600nm), sem especificação
de tempo máximo para leitura após a digestão;
1.2.2.3. Determinação da concentração em amostras de DQO < 90 mg.L-1
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir 2ml de amostra para o frasco de digestão;
3. Na sequência, adicionar, na ordem e cuidadosamente (reação
exotérmica ↑ calor), 2ml da solução digestora para leitura em = 420nm
(Ver item 1.2.1 – Reagentes) e 2ml da solução catalítica;
4. Realizar o mesmo procedimento para provas em branco, utilizando 2ml
de água destilada;
5. Fechar bem os frascos e misturar lentamente pela parede do mesmo,
de modo a favorecer a mistura dos reagentes adicionados (evitar
misturar totalmente por inversão, pois a presença de H2SO4 pode
danificar as tampinhas, podendo causar vazamento durante a digestão
e cuidar com o aquecimento dos frascos);
6. Realizar a digestão em aparelho digestor por um período de 2h e
temperatura de 150ºC;
7. Após completar a digestão, transferir os frascos para um suporte
específico e aguardar o resfriamento;
8. Realizar leitura no espectrofotômetro, ( = 420nm), sem especificação
de tempo máximo para leitura após a digestão;
1.2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de DQO (mg O2.L-1) através da equação da
reta, obtida na curva analítica (calibração).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
47
1.2.4. LIMITE DE DETECÇÃO
Para leituras no comprimento de onda 420 nm: 0 - 90 mg O2.L-1
Para leituras no comprimento de onda de 600 nm: 100 - 900 mg O2.L-1
2. REFERÊNCIA
Adaptação de STANDARD METHODS, method 5220 D (Apha, 1998)
3. DESCARTE DO RESÍDUO GERADO
Para ambos os métodos (1.1 – refluxo aberto e 1.2 – digestão fechada) o
resíduo gerado apresenta um alto caráter de toxicidade tendo na sua composição:
cromo hexavalente, Cr (VI) e trivalente, Cr (III), mercúrio (Hg2+) e prata (Ag2+) e
um meio fortemente acidificado. Dessa forma, o resíduo deve ser armazenado em
frasco plástico preferencialmente rígido (PEAD) e identificado, inclusive para
novas estocagens. A partir do acúmulo em excesso no laboratório é necessária
requerer junto à Prefeitura da Cidade Universitária uma solicitação para
encaminhamento ao aterro industrial Essencis.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
48
POP 04 – CARBONO ORGÂNICO DISSOLVIDO (COD)
1. MÉTODO PARA DETERMINAÇÃO
A quantificação de COD segue metodologia especificada pelo fabricante do
equipamento. De modo geral, o método consiste na combustão da amostra, sob
alta temperatura, e posterior quantificação através de detecção infravermelha não
dispersiva (NDIR) A determinação da concentração de carbono é realizada pela
combustão á alta temperatura
1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Analisador de carbono orgânico (marca Shimadzu, modelo TOC V-CPH).
Membrana de acetato de celulose Ø 0,45m de porosidade
Bomba de vácuo
Balança analítica (precisão ± 0,0001g)
Frasco plástico (PEAD, PP) para amostra (50ml).
Vidrarias
Conjunto kitassato para filtração
Reagentes analíticos
Ácido clorídrico (HCl) 2 mol.L-1: prepare a solução diluindo uma parte de
HCl concentrado (12 M) em cinco partes de água deionizada.
Soluções Padrões para calibração:
Solução padrão para CT: pesar 0,2125g de biftalato hidrogênio de potássio,
previamente seco a 105-120ºC por 1h e resfriado em dessecador. Transferir para
um balão volumétrico de 100ml e dissolver em água deionizada. Aferir para
100ml. A concentração teórica desta solução é de 1000 mg C.L-1.
Solução padrão para CI: pesar 0,305g de bicarbonato de sódio (p.a), previamente
seco por 2 horas em um dessecador e 0,441g de carbonato de sódio,
previamente seco a 280-290ºC por 1h e resfriado em um dessecador. Transferir

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
49
para um balão volumétrico de 100ml e dissolver em água deionizada. Aferir para
100ml. A concentração teórica desta solução é de 1000 mg C.L-1.
2. PROCEDIMENTO ANALÍTICO
3.1. CALIBRAÇÃO DO MÉTODO
A partir das soluções padrões preparadas de CT e CI, o usuário deve
selecionar uma calibração padrão a partir do software do equipamento, visto que
este realiza automaticamente as diluições, especificando a concentração padrão
inicial (CT e CI de 100 mg C.L-1).
OBS: Não tendo o conhecimento desta etapa de utilização do TOC V-CPH
recorrer aos usuários capacitados para a realização da calibração do aparelho
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE COD EM AMOSTRAS
1. Devido a não adequação deste equipamento com teores elevados de
cloretos nas amostras, este parâmetro deve ser cuidadosamente verificado da
sua presença na amostra, antes de iniciar a leitura;
2. Filtrar, aproximadamente, 50ml de amostra em membrana de acetato de
celulose Ø 0,45µm;
NOTA: a leitura do COD pode ser realizada imediatamente após a realização da
coleta, em um prazo de 2 dias, considerando a refrigeração da amostra (< 4 ºC),
ou você pode transferir a fração filtrada para esta análise para o frasco específico
e congelar para leitura posterior (6 meses).
3. Preparar o equipamento para procedimento de leitura de amostras
(Verificar POP TOC V-CPH);
4. Posteriormente, será solicitado o início das leituras de CT e CI;
5. Inserir o capilar do equipamento analisador de COT no frasco de
amostra e proceder à determinação;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
50
3.3. CÁLCULO DA CONCENTRAÇÃO
O valor do COD (mg C.L-1) se dá automaticamente, através do software do
equipamento, o qual realiza a subtração do CI a partir do CT, para quantificação
da fração orgânica de carbono presente na amostra.
2.4. LIMITE DE DETECÇÃO
O limite de detecção, segundo o manual do aparelho (Shimadzu, 2003), é de
4 µg.L-1, sendo o valor de COD obtido pela subtração do carbono total (CT) pelo
carbono inorgânico (CI), com faixa de detecção igual a: < 25.000 mg.L-1 para CT e
30.000 mg.L-1 para CI.
4. REFERÊNCIA
TOC-VCPH SHIMADZU CORPORATION (2003)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
51
POP 05 – SÉRIE DE SÓLIDOS
1. MÉTODO PARA DETERMINAÇÃO – GRAVIMÉTRICO
O método gravimétrico baseia-se na diferença de peso. Dessa forma, a
determinação das várias formas de sólidos prende-se a diferença entre o peso
seco e úmido, em relação ao volume de amostra utilizado no ensaio.
1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de Vácuo
Balança analítica (precisão ± 0,0001g)
Dessecador
Estufa (105 ± 5ºC)
Mufla (550 ± 5ºC)
Pinça simples metálica ou de madeira e espátula
Porta Filtro
Membrana de filtração de fibra de vidro
Vidrarias
Proveta graduada (50ml e 100ml)
Pipeta graduada e volumétrica
Cápsula de porcelana de 50ml de capacidade
Cápsula de porcelana de 100ml de capacidade
Cadinho de porcelana de 50ml de capacidade
Conjunto kitassato para filtração
Cone Imhoff
Reagentes analíticos
Não é utilizada nenhuma solução, apenas a amostra.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
52
1.2. SÓLIDOS TOTAIS (ST)
1.2.1. PROCEDIMENTO ANALÍTICO (Amostra)
(a) Preparo da Cápsula (NOTA: procedimento explicativo para 1 réplica)
1. Ligar a mufla a 550 ± 5ºC e a estufa a 105 ± 5ºC;
2. Colocar uma cápsula limpa na mufla para aferição por 1h e seguir os
passos abaixo na sequência:
a. Esfriar na estufa por 45min seguido de dessecador por 45min;
b. Pesar a cápsula e anotar o resultado em g (P0);
(b) Determinação de Sólidos Totais (ST)
1. Transferir para a cápsula, uma alíquota homogênea de volume adequado
de amostra, medido em proveta, e manter sob evaporação em banho-maria
até atingir secura completa (MANUSEAR A CÁPSULA SEMPRE COM O AUXÍLIO
DA PINÇA E NÃO DIRETAMENTE COM A MÃO);
2. Levar a cápsula com resíduo em estufa a 105 ± 5ºC por 45min;
3. Deixar a cápsula esfriar no dessecador por 45min;
4. Pesar a cápsula com resíduo e anotar o resultado em g (P1);
5. Expressar o resultado:
)(
000.000.1*01)/(
mLVamostra
PPLmgST
(6)
(c) Determinação de Sólidos Totais Fixos (STF)
1. Após anotar o valor do peso P1, transferir a cápsula para mufla a 550 ± 5ºC
por 1 hora e seguir os passos abaixo na sequência;
2. Esfriar na estufa a 105 ± 5ºC por 30min seguido de dessecador por 45min;
3. Pesar a cápsula e anotar o resultado em g (P2);
4. Expressar o resultado:
)(
000.000.1*02)/(
mLVamostra
PPLmgSTF
(7)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
53
(d) Determinação de Sólidos Totais Voláteis (STV)
1. É obtido através do cálculo da diferença entre os Sólidos totais e os
Sólidos Totais Fixos;
2. Expressar o resultado:
STFSTLmgSTV )/( (8)
1.3. SÓLIDOS SUSPENSOS (SST)
1.3.1. PROCEDIMENTO ANALÍTICO (Amostra)
(a) Preparo do Cadinho (NOTA: procedimento explicativo para 1 réplica)
1. Ligar a mufla a 550 ± 5ºC e a estufa a 105 ± 5ºC;
2. Colocar um cadinho limpo com a membrana de fibra de vidro na mufla para
aferição por 1h;
3. Deixar o cadinho esfriar na estufa por 45min seguido de dessecador por
1h;
4. Pesar o cadinho e anotar o resultado em g (P0);
(b) Determinação de Sólidos Suspensos Totais (SST)
1. Transferir o cadinho contendo o papel filtro de fibra de vidro para um porta
filtro adaptado ao kit kitassato para filtração (MANUSEAR A CÁPSULA SEMPRE
COM O AUXÍLIO DA PINÇA E NÃO DIRETAMENTE COM A MÃO);
2. Filtrar um volume de amostra entre 50 e 200ml (volumes utilizados
rotineiramente), com auxílio de uma proveta graduada, que favoreça a
quantificação de SST na amostra em questão;
3. Retirar o cadinho contendo o papel filtro fibra de vidro e o resíduo sólido
retido no mesmo do kit kitassato;
4. Levar o cadinho com resíduo, para estufa a 105 ± 5ºC por 1h;
5. Em seguida, aguardar esfriar no dessecador por 30min;
6. Pesar o cadinho com resíduo e anotar o resultado em g (P1);
7. Expressar o resultado:

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
54
)(
000.000.1*01)/(
mLVamostra
PPLmgSST
(9)
(c) Determinação de Sólidos Suspensos Fixos (SSF)
1. Após anotar o valor do peso P1, transferir o cadinho para mufla a 550 ±
5ºC por 1h;
2. Deixar a cadinho esfriar na estufa a 105 ± 5ºC por 30min seguido de
dessecado por 45min;
3. Pesar a cadinho e anotar o resultado em g (P2);
4. Expressar o resultado:
)(
000.000.1*02)/(
mLVamostra
PPLmgSSF
(10)
(d) Determinação de Sólidos Suspensos Voláteis (SSV)
1. É obtido pela diferença entre os sólidos suspensos totais e os sólidos
suspensos fixos;
2. Expressar o resultado:
SSFSSTLmgSSV )/( (11)
1.4. SÓLIDOS SEDIMENTÁVEIS (SS)
1.4.1. PROCEDIMENTO ANALÍTICO (Amostra)
2. Homogeneizar vigorosamente a amostra;
3. Transferir a amostra para o cone de Imhoff até a marca de 1000ml;
4. Deixar em repouso por 45min;
5. Passar vagarosamente um bastão de vidro na parede interna do cone, ou
gira-lo suavemente entre as mãos;
6. Deixar em repouso por mais 15min;
7. Determinar o volume, em ml, ocupado pelos sólidos sedimentáveis;
8. Anotar o valor observado;
9. Expressar o resultado em ml.L-1 pela leitura direta no cone Imhoff;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
55
2. REFERÊNCIAS
Adaptação de STANDARD METHODS, method 2540 B (Apha, 1998)
Adaptação de STANDARD METHODS, method 2540 E (Apha, 1998)
STANDARD METHODS, method 2540 F (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
56
POP 06 – UV-Vis
1. MÉTODO PARA DETERMINAÇÃO - ESPECTROFOTOMÉTRICO
Varredura espectrofotométrica na região do UV-Vis. Espécies atômicas ou
moleculares que absorvem energia na região do UV-Vis são detectadas através
do espectro de varredura.
1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Espectrofotômetro UV-Vis
Bomba de vácuo
Membrana de acetato de celulose Ø 0,45m de porosidade
Frasco plástico (PEAD, PP) para amostra (20ml)
Vidrarias
Cubeta de quartzo
Conjunto kitassato para filtração
Reagentes analíticos
Não há uso de soluções para o preparo e quantificação da amostra.
1.2. PROCEDIMENTO ANALÍTICO (Amostra)
1. Filtrar aproximadamente 20ml de amostra em membrana de acetato de
celulose Ø 0,45µm;
2. Transferir a amostra (~ 4ml) para uma cubeta de quartzo;
3. Selecionar no espectrofotômetro UV-Vis a opção espectro e realizar uma
varredura na faixa de comprimentos de onda entre 200 e 700nm (Verificar
POP UV-Vis SHIMADZU);
4. Realizar a análise de UV-Vis para cada amostra;
5. Utilizar água destilada como prova em branco da análise;
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
57
200 250 300 3500,0
0,2
0,4
0,6
Absorv
ância
Comprimento de Onda (nm)
Amostra 1
Amostra 2
Amostra 3
Amostra 4
Amostra 5
Amostra 6
NOTA: a leitura no espectrofotômetro pode ser realizada imediatamente após a
realização da coleta, em um prazo de 2 dias, considerando a refrigeração da
amostra (< 4ºC), ou você pode transferir a fração filtrada para esta análise para o
frasco específico e congelar para leitura posterior (6 meses).
2. EXEMPLO GRÁFICO DO ESPECTRO UV-Vis DE VARREDURA PARA
AMOSTRAS DE ÁGUA
A Figura 2 exemplifica o espectro de varredura obtido após o tratamento
dos dados obtidos a partir do espectrofotômetro de UV-Vis. Este exemplo
considera os valores de absorvância entre os comprimentos de onda de 200 a
350nm.
Figura 2. Espectro de varredura na região do UV-Vis para seis amostras de águas
superficiais coletadas em diferentes pontos de amostragem na região do Alto Iguaçu.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
58
POP 07 – SÉRIE DE NITROGÊNIO – NITRITO (N-NO2-)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DA SULFANILAMIDA / N-1-NAFTIL
A concentração de N-NO2- é determinada através da formação de um azo
corante, de coloração púrpura característica, em faixa de pH entre 2,0 e 2,5,
decorrente da complexação entre sulfanilamida e N-(1-naftil)-etilenodiamina,
conforme esquema abaixo:
Figura 3. Esquema explicativo das etapas de reação de Griess (1879), utilizada como
metodologia analítica na quantificação de N-Nitrito em amostras de água e de efluentes
líquidos.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo
Membranas de acetato de celulose (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Espectrofotômetro UV-Vis

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
59
Micropipeta (100 - 1000µl)
Vidrarias
Balão volumétrico de 50ml
Balão volumétrico de 250ml
Pipetas volumétricas ou graduadas
Cubeta de vidro
Conjunto kitassato para filtração
Reagentes analíticos
Solução estoque padrão de N-NO2-: dissolver 0,308g de nitrito de sódio
(NaNO2 p.a) em água destilada. Aferir para 250ml com água destilada em
balão volumétrico.
* 1 ml = 250µg N ou 250 mg.L-1
* Estável por 6 meses após o preparo.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC)
Solução padrão para calibração de N-NO2-: diluir 0,2ml da solução
estoque padrão em água destilada. Aferir com água destilada para 100ml,
em balão volumétrico.
* 1ml = 0,5 µg N ou 0,5 mg.L-1
* Preparo deve ser realizado apenas para o uso imediato.
Solução reativa para N-NO2-: adicionar 25ml de ácido fosfórico (H3PO4
p.a) 85% em, aproximadamente, 200ml de água destilada. Adicionar 2,5g
de sulfanilamida (C6H8N2O2S p.a). Após dissolução total, adicionar 0,25g
de N-1-naftil etilenodiamina dihidroclórico (C12H4N2.2HCl p.a). Após
dissolução total, aferir com água destilada, para um volume final de 250ml
em balão volumétrico.
* Estável por 1 mês (mínimo).
* Acondicionamento em ambiente refrigerado (4ºC).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
60
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é realizada através de curva de calibração padrão,
descrita na seqüência:
1. A partir da solução padrão para calibração (0,5 mg.L-1), preparar, em
balões volumétricos de 25ml, a seguinte curva de calibração (volume final com
água destilada):
Concentração N-NO2- (µg.L-1) Volume N-NO2
- (ml) 0 (branco) -
5 0,25 10 0,5 25 1,25 50 2,5
100 5 *Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser preparada
em duplicata ou triplicata.
2. Posteriormente ao preparo das soluções padrões, conforme a tabela
acima, retirar, com auxílio de uma pipeta volumétrica, uma alíquota de 10ml de
cada padrão e reservar em outro frasco;
3. Juntamente aos padrões, realizar prova em branco utilizando alíquotas
de 10ml de água destilada;
4. Adicionar 0,4ml da solução reativa para N-NO2- em cada alíquota
(padrão e branco), com auxílio de uma micropipeta e misturar por inversão;
5. Leitura em espectrofotômetro ( = 543 nm) entre 10min e tempo máximo
de 2h;
6. Equação da reta, obtida com a plotagem do gráfico: absorvância (eixo y)
X concentração (eixo x);
7. Verificar coeficiente de linearidade: R > 0,990;
8. Realizar comparação com a curva analítica apresentada na Figura 4a;
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE N-NO2- EM AMOSTRAS
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir 50ml de amostra para o sistema de filtragem de vácuo;
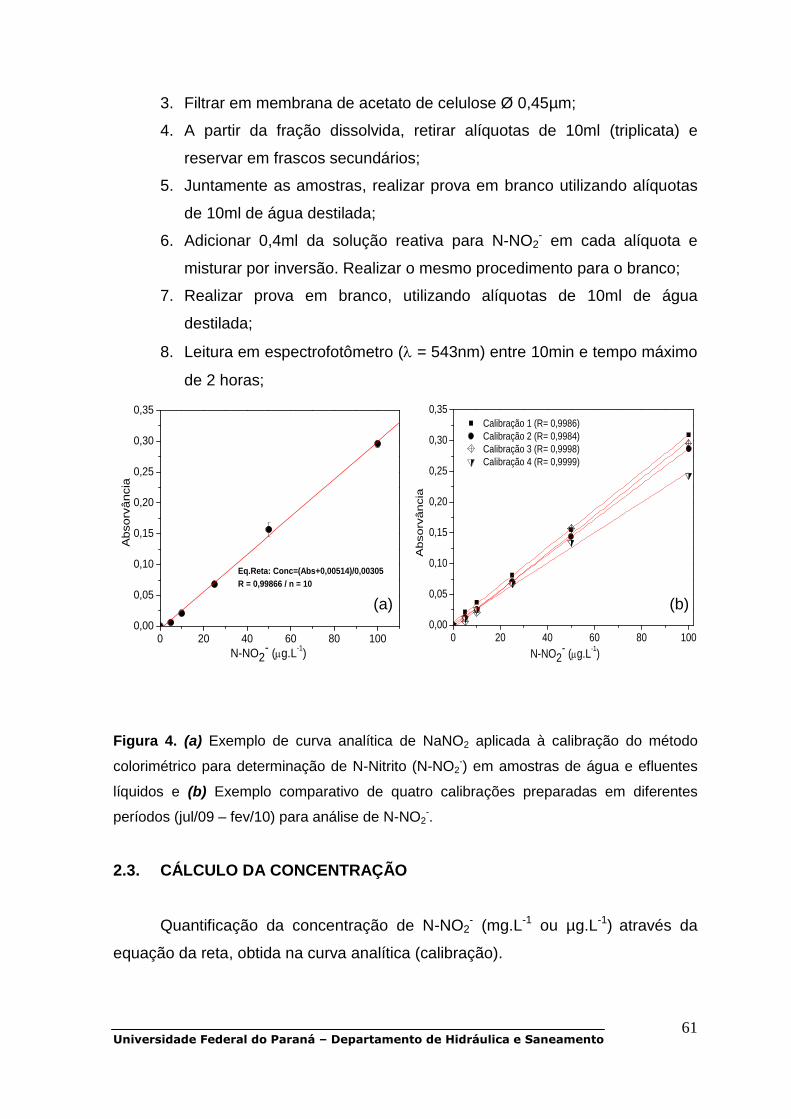
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
61
3. Filtrar em membrana de acetato de celulose Ø 0,45µm;
4. A partir da fração dissolvida, retirar alíquotas de 10ml (triplicata) e
reservar em frascos secundários;
5. Juntamente as amostras, realizar prova em branco utilizando alíquotas
de 10ml de água destilada;
6. Adicionar 0,4ml da solução reativa para N-NO2- em cada alíquota e
misturar por inversão. Realizar o mesmo procedimento para o branco;
7. Realizar prova em branco, utilizando alíquotas de 10ml de água
destilada;
8. Leitura em espectrofotômetro ( = 543nm) entre 10min e tempo máximo
de 2 horas;
Figura 4. (a) Exemplo de curva analítica de NaNO2 aplicada à calibração do método
colorimétrico para determinação de N-Nitrito (N-NO2-) em amostras de água e efluentes
líquidos e (b) Exemplo comparativo de quatro calibrações preparadas em diferentes
períodos (jul/09 – fev/10) para análise de N-NO2-.
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de N-NO2- (mg.L-1 ou µg.L-1) através da
equação da reta, obtida na curva analítica (calibração).
0 20 40 60 80 1000,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
Ab
so
rvâ
ncia
N-NO2- (g.L
-1)
Eq.Reta: Conc=(Abs+0,00514)/0,00305
R = 0,99866 / n = 10
0 20 40 60 80 1000,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35A
bsorv
ância
N-NO2- (g.L
-1)
Calibração 1 (R= 0,9986)
Calibração 2 (R= 0,9984)
Calibração 3 (R= 0,9998)
Calibração 4 (R= 0,9999)
(a) (b)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
62
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de N-NO2- é aplicável para leitura na faixa de
concentração entre 5 e 1000 µg.L-1. Para concentrações superiores, sugere-se a
diluição da amostra e, posteriormente, quantificação através do fator de diluição
fdiluição utilizado.
3. DESCARTE DO RESÍDUO GERADO
O resíduo da análise de nitrito deve, primeiramente, ser neutralizado com
soluções de NaOH (0,01 – 1 mol.L-1) para posterior descarte na pia.
3. REFERÊNCIA
Adaptação de STANDARD METHODS, method 4500- NO2- B (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
63
N
O O (-)
O
(-)
(+) Cd-Cu
N
O O (-) (-)
Cd0Cd
2+
POP 08 – SÉRIE DE NITROGÊNIO – NITRATO (N-NO3-)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DA COLUNA DE Cd-Cu
A concentração de N-NO3- é determinada, quantitativamente, através da
redução para N-NO2-, em presença de grânulos de cádmio (Cd). Posteriormente,
o N-NO2- é quantificado segundo o método da sulfanilamida / N-1-naftil (ver POP
nº. 07 / N-NO2-), como mostra o esquema abaixo:
Figura 5. Esquema explicativo das etapas da redução de N-Nitrato para N-Nitrito através
da coluna de Cd-Cu e reação de Griess (1879), utilizada como metodologia analítica na
quantificação de N-Nitrato em amostras de água e de efluentes líquidos.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo
Membranas de acetato de celulose (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Espectrofotômetro UV-Vis
Micropipeta (100 - 1000l)
Vidrarias
Balão volumétrico de 50ml

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
64
Balão volumétrico de 250ml
Pipetas volumétricas ou graduadas
Cubeta de vidro
Conjunto kitassato para filtração
Tubos de plástico para centrífuga, tipo Falcon (15ml)
Reagentes analíticos
Solução estoque padrão de N-NO3-: dissolver 0,18045g de nitrato de
potássio (KNO3 p.a), previamente seco em estufa (105ºC / 24h) em água
destilada. Aferir para 250ml com água destilada em balão volumétrico.
* 1ml = 100 µg N-NO3- ou 100 mg.L
-1.
* Estável por 6 meses após o preparo.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC)
Solução intermediária padrão de N-NO3-: diluir 25ml da solução estoque
padrão em água destilada. Aferir com água destilada para 250ml, em balão
volumétrico.
* 1ml = 10µg N-NO3- ou 10 mg.L
-1
* Estável por 6 meses após o preparo.
Solução de sulfato de cobre 2% (CuSO4): dissolver 10g de CuSO4 p.a
em aproximadamente 300ml de água destilada. Aferir com água destilada
para 500ml, em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente seco.
Solução estoque de NH4Cl – EDTA: dissolver 13g de cloreto de amônio
(NH4Cl p.a) e 1,7g de EDTA (p.a) em aproximadamente 800ml de água
destilada. Aferir com água destilada para 1000ml, em balão volumétrico.
* Verificar o pH da solução e, se necessário, ajustar para pH 8,5.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 1 ano (mínimo).
Solução diluída de NH4Cl – EDTA: diluir 300ml da solução estoque de
NH4Cl – EDTA para 500ml (volume final), com água destilada, em balão
volumétrico. Solução estoque de NH4Cl – EDTA
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 1 ano (mínimo).
Solução reativa para N-NO2-: ver POP-07, item 1 (N-NO2
-).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
65
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é realizada através de curva de calibração padrão,
descrita na seqüência:
- Partindo-se da solução intermediária padrão preparar, em balões volumétricos
de 50ml, a seguinte curva de calibração (volume final com água destilada):
Concentração N-NO3- (µg.L-1) Volume N-NO3
- (ml) 0 (branco) -
50 0,25 100 0,5 300 1,5 500 2,5 800 4,0
*Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser
preparada em duplicata ou triplicata.
NOTA: após adição do volume respectivo ao padrão apresentando na tabela
acima, adicionar, em cada padrão, 10ml da solução diluída de NH4Cl – EDTA e,
posteriormente, aferir com água destilada;
Após o preparo das soluções padrões e suas respectivas concentrações,
seguir para a próxima etapa: processo de ativação da coluna de Cd-Cu,
descrito na sequência:
1. Passar pela coluna de Cd-Cu uma alíquota de 20 – 25ml de solução de
sulfato de cobre 2%;
2. Na sequência, passar uma alíquota de 50ml da solução diluída de NH4Cl
– EDTA;
3. Após a solução de NH4Cl – EDTA, lavar a coluna com alíquotas de 25 ml
de água destilada;
4. Retirar uma alíquota de 5ml da coluna ( utilizar um tubo de centrífuga
aferido) e adicionar 0,4ml da solução reativa para N-NO2-. Verificar a presença ou
não da coloração púrpura (desenvolvimento da cor indica a presença de N-NO2-);
5. Na presença de cor, lavar a coluna novamente com alíquotas de 25ml de
água destilada e realizar novo teste para verificar a limpeza da coluna;
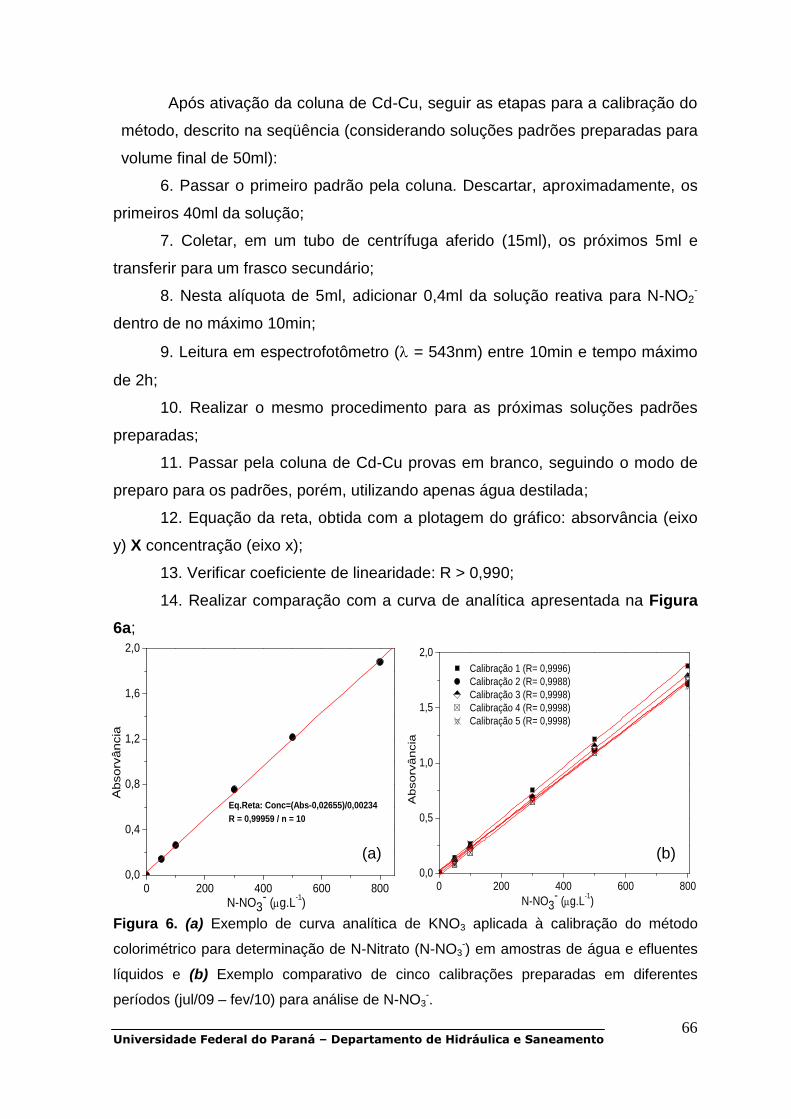
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
66
0 200 400 600 8000,0
0,4
0,8
1,2
1,6
2,0
Ab
so
rvâ
ncia
N-NO3- (g.L
-1)
Eq.Reta: Conc=(Abs-0,02655)/0,00234
R = 0,99959 / n = 10
0 200 400 600 8000,0
0,5
1,0
1,5
2,0
Ab
so
rvâ
ncia
N-NO3- (g.L
-1)
Calibração 1 (R= 0,9996)
Calibração 2 (R= 0,9988)
Calibração 3 (R= 0,9998)
Calibração 4 (R= 0,9998)
Calibração 5 (R= 0,9998)
(a) (b)
Após ativação da coluna de Cd-Cu, seguir as etapas para a calibração do
método, descrito na seqüência (considerando soluções padrões preparadas para
volume final de 50ml):
6. Passar o primeiro padrão pela coluna. Descartar, aproximadamente, os
primeiros 40ml da solução;
7. Coletar, em um tubo de centrífuga aferido (15ml), os próximos 5ml e
transferir para um frasco secundário;
8. Nesta alíquota de 5ml, adicionar 0,4ml da solução reativa para N-NO2-
dentro de no máximo 10min;
9. Leitura em espectrofotômetro ( = 543nm) entre 10min e tempo máximo
de 2h;
10. Realizar o mesmo procedimento para as próximas soluções padrões
preparadas;
11. Passar pela coluna de Cd-Cu provas em branco, seguindo o modo de
preparo para os padrões, porém, utilizando apenas água destilada;
12. Equação da reta, obtida com a plotagem do gráfico: absorvância (eixo
y) X concentração (eixo x);
13. Verificar coeficiente de linearidade: R > 0,990;
14. Realizar comparação com a curva de analítica apresentada na Figura
6a;
Figura 6. (a) Exemplo de curva analítica de KNO3 aplicada à calibração do método
colorimétrico para determinação de N-Nitrato (N-NO3-) em amostras de água e efluentes
líquidos e (b) Exemplo comparativo de cinco calibrações preparadas em diferentes
períodos (jul/09 – fev/10) para análise de N-NO3-.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
67
ATENÇÃO: a leitura das soluções padrões deve manter durante a passagem pela coluna de Cd-
Cu a mesma vazão (7 - 10 mL.min-1
), assim como as amostras a serem quantificadas
posteriormente. Isto se deve ao fato de vazões diferentes ocasionarem alterações na sensibilidade
da coluna, podendo ocasionar alterações na detecção de padrões e amostras.
- A verificação da sensibilidade da coluna de Cd-Cu pode ser realizada da seguinte
maneira: caso você tenha utilizado a mesma solução reativa para N-NO2-, tanto na
calibração de N-NO2- como de N-NO3
-, compare os resultados obtidos para a absorvância
do último padrão de N-NO2- e o primeiro padrão de N-NO3
- (dados a partir das tabelas de
calibração deste manual, indicam a mesma concentração para ambos).
- Considerando a absorvância de N-NO2- (último padrão) igual a 100%, verificar a relação
com o valor encontrado para N-NO3-. Considerando como exemplo, a calibração padrão
para N-NO2- e N-NO3
- presente neste manual, a partir dos dados de absorvância, verifica-
se uma sensibilidade de, aproximadamente 95%, ou seja, indica uma boa eficiência da
coluna de Cd-Cu na redução de N-NO3- para N-NO2
-.
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE N-NO3- EM AMOSTRAS
1. Primeiramente, verificar ativação da coluna de Cd-Cu. Caso, anteriormente
à quantificação das amostras, tenha sido preparado uma calibração
padrão, a coluna não precisa ser ativada novamente;
2. Homogeneizar bem a amostra presente no frasco de coleta;
3. Transferir 50ml de amostra para o sistema de filtragem de vácuo;
4. Filtrar em membrana de acetato de celulose Ø 0,45µm;
5. A partir da fração dissolvida, transferir, com o auxílio de uma pipeta
volumétrica, 5ml da amostra (triplicata) para balão volumétrico de 50ml;
6. Adicionar 10ml da solução diluída de NH4Cl – EDTA;
7. Aferir o balão volumétrico com água destilada;
ATENÇÃO: seguindo este procedimento acima, você estará realizando uma diluição de 10 vezes
de sua amostra, a qual deve ser utilizada posteriormente para correção do valor encontrado
através da curva de calibração. Considerando que uma diluição de 10 vezes pode estourar a
calibração do método ou ficar abaixo da detecção do mesmo, verificar nova diluição para
quantificação de N-NO3-
na amostra, como os exemplos a seguir: 25ml de amostra + 10ml de
solução diluída de NH4Cl - EDTA para volume final de 50ml (diluição de 2 vezes); 1ml de amostra
+ 10ml de solução diluída de NH4Cl - EDTA para volume final de 50ml (diluição de 50 vezes).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
68
8. Dando continuidade ao procedimento analítico, após diluição da amostra,
passar pela coluna de Cd-Cu;
9. Descartar, aproximadamente, os primeiros 40ml da amostra;
10. Coletar, em um tubo de centrífuga aferido (15ml), os próximos 5ml e
transferir para um frasco secundário;
11. Nesta alíquota de 5ml, adicionar 0,4ml da solução reativa para N-NO2-;
12. Leitura em espectrofotômetro ( = 543nm) entre 10min e tempo máximo
de 2h;
13. O mesmo procedimento deve ser realizado para as réplicas da amostra e
para outras amostras;
14. Após a passagem da amostra, seguir o mesmo procedimento para
realização de testes em branco, utilizando água destilada;
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de N-NO3- (mg.L-1 ou µg.L-1) através da
equação da reta, obtida na curva analítica (calibração). É importante ressaltar que
o resultado analisado para N-NO2- deve ser descontando do valor obtido.
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de N-NO3- é aplicável para leitura na faixa de
concentração entre 10 e 1000 µg.L-1. Para concentrações superiores, sugere-se a
diluição da mesma e, posteriormente, quantificação através do fator de diluição
fdiluição utilizado.
3. DESCARTE DO RESÍDUO GERADO
Da mesma forma que apresentado na análise de nitrito, o resíduo da
análise de nitrato deve primeiramente ser neutralizado para descarte em pia.
3. REFERÊNCIA
Adaptação de STANDARD METHODS, method 4500- NO3
- E (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
69
(+)
) N
H H
H
H + OCl- H2NCl + OH-
H2NCl + OH Na2[Fe(CN)5NO]·2H2O
Meio básico O NCl
Na2[Fe(CN)5NO]·2H2O
Meio básico
+ O NCl OH Na2[Fe(CN)5NO]·2H2O
Meio básico Ausência de luz
O N
OH
Azul de indofenol
POP 09 – SÉRIE DE NITROGÊNIO – NITROGÊNIO
AMONIACAL TOTAL (NH3 + NH4+)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO MODIFICADO DO FENATO / AZUL DE INDOFENOL
A presença de um composto de intensa coloração azul-esverdeado
característica, azul de indofenol, é formada através da reação entre a
concentração de N-NH3 presente na amostra, hipoclorito de sódio e fenol em meio
básico, tendo o nitroprussiato de sódio como catalisador da reação, conforme
esquema abaixo:
Figura 7. Esquema explicativo das etapas de reação de Berthelot (1859), utilizada como
metodologia analítica na quantificação de N-Amoniacal em amostras de água e de
efluentes líquidos.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo
Membranas de acetato de celulose (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Espectrofotômetro UV-VIS
Micropipeta (100 - 1000µl)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
70
Papel parafilm®
Vidrarias
Balão volumétrico de 100ml
Balão volumétrico de 500 ml
Pipetas volumétricas e graduadas
Cubeta de vidro
Conjunto kitassato para filtração
Reagentes analíticos
Solução estoque padrão de NH4Cl: dissolver 0,3819g de cloreto de
amônio (NH4Cl p.a), previamente seco em estufa (100ºC/2h), em
aproximadamente 80ml de água destilada. Aferir com água destilada para
100ml, em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 1 mês após o preparo.
* 1ml = 1mg N ou 1ml = 1,22 mg NH3.
Solução intermediária padrão de N-NH3: diluir 5ml da solução estoque
padrão em água destilada. Aferir com água destilada para 50ml (diluição de
10x), em balão volumétrico.
* Concentração final de 100 mg N.L-1
* Preparo deve ser realizado para uso imediato.
Solução de hipoclorito de sódio: solução comercial 2,5%.
Verificar reposição desta solução a cada 6 meses, devido à decomposição após abertura
do frasco.
* Transfira a solução para um frasco âmbar para acondicionamento em ambiente refrigerado
(4ºC).
Solução reativa 1 (Hipoclorito alcalino): adicionar 20ml de hipoclorito de
sódio 2,5% em 80ml de água destilada e deionizada. Na sequência,
adicionar 5g de NaOH p.a. Dissolver e aferir com água destilada para 100
ml, em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (<4ºC).
* Estável por 2 meses após o preparo.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
71
Solução reativa 2 (Fenol + Nitroprussiato de sódio): dissolver 10g de
fenol (p.a) e 0,1g de nitroprussiato de sódio (p.a) em, aproximadamente,
80ml de água destilada. Dissolver e aferir com água destilada para 100ml,
em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 2 meses após o preparo.
* ATENÇÃO: durante o preparo, utilize luvas e óculos de proteção e sempre que for manusear o
fenol (reagente ou solução para N-NH3), trabalhar SEMPRE em capela com ventilação / exaustão;
O fenol é altamente cancerígeno e todos os procedimentos de segurança devem ser adotados na
hora do uso.
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é realizada através de curva de calibração padrão,
descrita na seqüência:
1. Partindo-se da solução intermediária padrão preparar, em balões
volumétricos de 50ml, a seguinte curva de calibração (volume final com água
destilada):
Concentração N-NH3 (µg.L-1) Volume N-NH3 (ml)
0 (branco) - 200 0,1 400 0,2 800 0,4 1200 0,6 2000 1,0
*Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser
preparada em duplicata ou triplicata.
2. Posteriormente ao preparo das soluções padrões, conforme a tabela
acima, retirar, com auxílio de uma pipeta volumétrica, uma alíquota de 2ml de
cada padrão e reservar em outro frasco;
3. Juntamente aos padrões, realizar prova em branco utilizando alíquotas
de 2ml de água destilada;
4. Adicionar nesta ordem: 1ml da solução reativa 1 seguido de 1ml da
solução reativa 2;
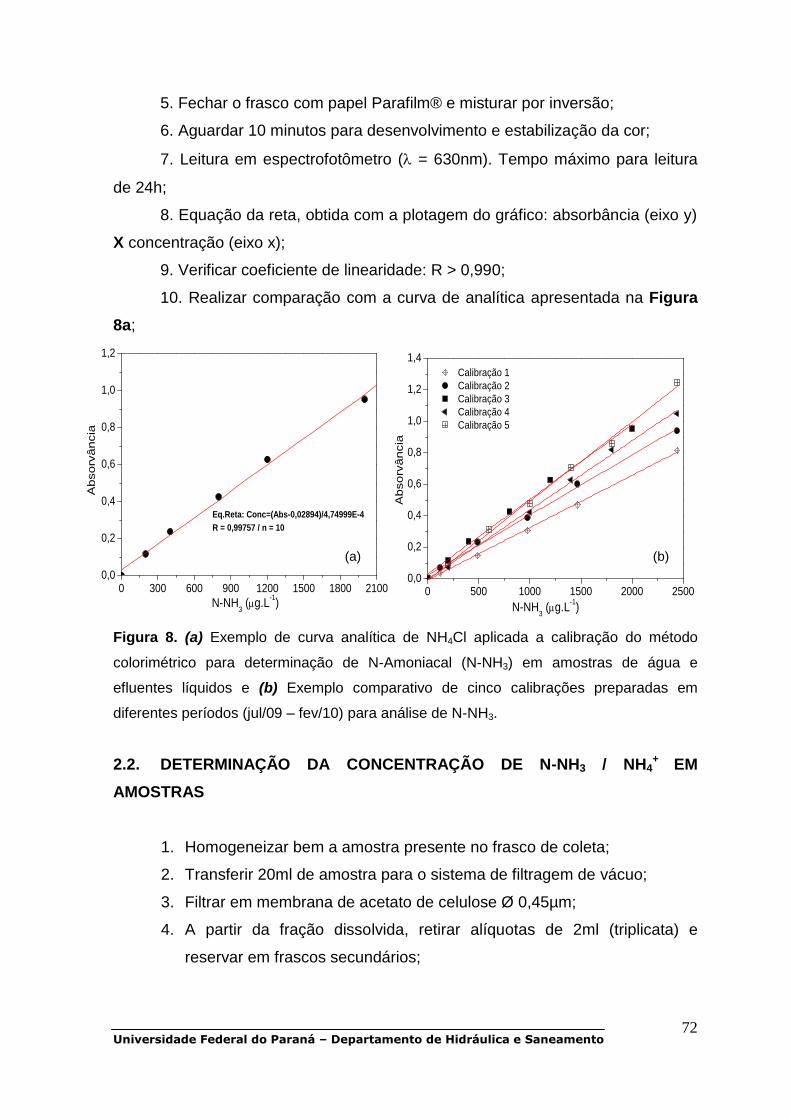
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
72
0 300 600 900 1200 1500 1800 21000,0
0,2
0,4
0,6
0,8
1,0
1,2
Ab
so
rvâ
ncia
N-NH3 (g.L
-1)
Eq.Reta: Conc=(Abs-0,02894)/4,74999E-4
R = 0,99757 / n = 10
0 500 1000 1500 2000 25000,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Ab
so
rvâ
ncia
N-NH3 (g.L
-1)
Calibração 1
Calibração 2
Calibração 3
Calibração 4
Calibração 5
(a) (b)
5. Fechar o frasco com papel Parafilm® e misturar por inversão;
6. Aguardar 10 minutos para desenvolvimento e estabilização da cor;
7. Leitura em espectrofotômetro ( = 630nm). Tempo máximo para leitura
de 24h;
8. Equação da reta, obtida com a plotagem do gráfico: absorbância (eixo y)
X concentração (eixo x);
9. Verificar coeficiente de linearidade: R > 0,990;
10. Realizar comparação com a curva de analítica apresentada na Figura
8a;
Figura 8. (a) Exemplo de curva analítica de NH4Cl aplicada a calibração do método
colorimétrico para determinação de N-Amoniacal (N-NH3) em amostras de água e
efluentes líquidos e (b) Exemplo comparativo de cinco calibrações preparadas em
diferentes períodos (jul/09 – fev/10) para análise de N-NH3.
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE N-NH3 / NH4+ EM
AMOSTRAS
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir 20ml de amostra para o sistema de filtragem de vácuo;
3. Filtrar em membrana de acetato de celulose Ø 0,45µm;
4. A partir da fração dissolvida, retirar alíquotas de 2ml (triplicata) e
reservar em frascos secundários;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
73
5. Juntamente as amostras, realizar prova em branco utilizando alíquotas
de 2ml de água destilada;
6. Adicionar nesta ordem: 1ml da solução reativa 1 seguido de 1ml da
solução reativa 2, para cada alíquota (amostra ou branco);
7. Fechar o frasco com papel Parafilm® e misturar por inversão;
8. Aguardar 10min para desenvolvimento e estabilização da cor;
9. Leitura em espectrofotômetro ( = 630nm). Tempo máximo para leitura
de 24h;
ATENÇÃO: o método do fenato para quantificação de N-NH3 acaba por gerar um resíduo
composto, principalmente, por fenol, devido à existência deste reagente nas etapas de reação do
método. Assim, todo e qualquer material líquido gerado por este método devem ser armazenados
em frascos próprios para o resíduo de fenol / N-NH3 (FRASCOS DE VIDRO PARA DESCARTE).
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de N-NH3 / NH4+ (mg.L-1 ou µg.L-1) através
da equação da reta, obtida na curva analítica (calibração).
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de N-NH3 /NH4+ é aplicável para leitura na
faixa de concentração entre 10 e 2000 µg.L-1. Para concentrações superiores,
sugere-se a diluição da mesma e, posteriormente, quantificação através do fator
de diluição fdiluição utilizado.
3. DESCARTE DO RESÍDUO GERADO
O método proposto no presente manual para análise de nitrogênio
amoniacal utiliza fenol nas etapas de quantificação da amostra. Dessa forma, o
resíduo gerado deve ser, exclusivamente, armazenado em frascos de vidro (≥
1litro). Quando houver um acúmulo excessivo em laboratório deve-se solicitar na
Prefeitura da Cidade Universitária um encaminhamento para transporte e
destinação final no aterro industrial da Essencis.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
74
3. REFERÊNCIAS
WEATHERBURN. Phenol-hypochlorite reaction for determination of ammonia. Anal.
Chem, Vol.8, pp. 130-132, 1962.
Adaptação de STANDARD METHODS, method 4500-NH3 F (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
75
N
O O (-)
O
(-)
(+) N
O O (-) (-)
Cd-Cu
Cd0Cd
2+
NT
K2S2O8 / NaOH
t = 30min T = 120ºC;
N
O O (-)
O
(-)
(+)
POP 10 – SÉRIE DE NITROGÊNIO – NITROGÊNIO TOTAL
(NT)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DA DIGESTÃO POR PERSULFATO DE POTÁSSIO
Quantificação de NT através de uma oxidação alcalina em autoclave (25min
/ 14kg.cm-1) na faixa de temperatura entre 110 - 125ºC, para conversão de todas
as formas de nitrogênio presentes a N-NO3- e posterior redução pela coluna de
Cd-Cu para detecção na forma de N-NO2-, a exemplo do esquema abaixo:
Figura 9. Esquema explicativo das etapas da oxidação por persulfato das formas de
nitrogênio a N-Nitrato, redução de N-Nitrato para N-Nitrito através da coluna de Cd-Cu e
reação de Griess (1879), utilizada como metodologia analítica na quantificação de N-
Total em amostras de água e de efluentes líquidos.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Balança analítica (precisão ± 0,0001g)
Autoclave vertical
Espectrofotômetro UV-Vis

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
76
Vidrarias
Frascos para digestão
Balão volumétrico de 100ml
Balão volumétrico de 500ml
Tubos de plástico para centrífuga, tipo Falcon (15ml)
Pipetas volumétricas e graduadas
Cubeta de vidro
Reagentes analíticos
Solução digestora: dissolver 2,01g de persulfato de potássio (K2S2O8 p.a
< 0,001% N), em aproximadamente 80ml de água destilada. Adicionar
0,300g de hidróxido de sódio (NaOH p.a) e dissolver novamente. Aferir com
água destilada para 100ml, em balão volumétrico.
* Preparo deve ser realizado para uso imediato.
Solução tampão de borato: dissolver 30,9g de ácido bórico (H3BO3 p.a)
em, aproximadamente, 300ml em água destilada. Adicionar 4g de hidróxido
de sódio (NaOH p.a) e dissolver novamente. Aferir com água destilada para
500 mL em balão volumétrico.
* Estável por 1 ano (mínimo).
Solução de sulfato de cobre 2%: ver POP-08, item 1 (N-NO3-).
Solução estoque de NH4Cl – EDTA: ver POP-08, item 1 (N-NO3-).
Solução diluída de NH4Cl – EDTA: ver POP-08, item 1 (N-NO3-).
Solução reativa para N-NO2-: ver POP-07, item 1 (N-NO2
-).
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é mesma utilizada para a determinação do nitrato
(ver POP-08).
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE N-TOTAL EM AMOSTRAS

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
77
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir com o auxílio de uma pipeta volumétrica, 5ml da amostra
para tubos de ensaio com rosca para uso na autoclave;
3. Adicionar ao volume de amostra, 2,5ml da solução digestora;
4. Fechar bem os tubos para digestão e misturar por inversão;
5. Manter na autoclave, por um período de 25min após atingir a
temperatura de 120ºC e pressão de 14 kg.cm-2. Neste momento, mudar
a chave seletora de máximo para médio e controlar a possibilidade de
aumento no manômetro de controle. Neste caso, mudar a chave para
mínimo;
6. Após o término da digestão e resfriamento da autoclave, retirar os tubos
e aguardar que atinjam a temperatura ambiente;
7. Após esfriar, adicionar 1ml da solução tampão de borato e misturar por
inversão;
8. Transferir o volume presente no tubo de ensaio para um balão
volumétrico de 50ml;
9. Adicionar 10ml da solução de NH4Cl-EDTA diluída;
10. Aferir o balão volumétrico com água destilada;
11. Passar a amostra pela coluna de Cd-Cu (ver etapa de ativação da
coluna de Cd-Cu);
12. Descartar, aproximadamente, os primeiros 40ml;
13. Coletar, em um tubo de centrífuga aferido (15ml), os próximos 5ml e
transferir para um frasco secundário;
14. Nesta alíquota de 5ml, adicionar 0,4ml da solução reativa para N-NO2-;
15. Leitura em espectrofotômetro ( = 543nm) entre 10min e tempo máximo
de 2h;
O mesmo procedimento deve ser realizado para as réplicas da amostra e
para outras amostras. Após a passagem da amostra, seguir o mesmo
procedimento para realização das provas em branco, utilizando água destilada.
ATENÇÃO: seguindo este procedimento acima, descrito para NT, você estará fazendo uma
diluição de 10 vezes da amostra, ou seja, foram tomados 5ml iniciais da amostra, passado pela
digestão e antes de passar pela coluna de Cd-Cu foi diluída para 50ml. Para este exemplo, o valor

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
78
obtido para a absorbância deve ser corrigido com um fator de diluição de 10 vezes, de modo a se
obter o valor da concentração real.
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de NT (mg.L-1 ou µg.L-1) através da equação
da reta, obtida na curva de calibração para N-NO3-.
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de NT é aplicável para leitura de
concentrações < 2,9 mg.L-1. Concentrações superiores ao limite podem ser
obtidas através da diluição da amostra e, posteriormente, quantificação através do
fator de diluição fdiluição.
ATENÇÃO: para este método, se a concentração de NT for superior ao limite de detecção,
a diluição da amostra deve ser realizada na etapa 2 deste procedimento. Ao adicionar os 5ml da
amostra, uma diluição de 5x, por exemplo, pode ser preparada a partir de 1ml da amostra e 4ml de
água destilada e, em seguida adição da solução digestora (etapa 3). Ainda, a diluição pode ser
feita separadamente, em balão volumétrico, e posteriormente o volume de 5ml retirado e
transferido para o tubo de digestão. Assim, se houver uma diluição prévia a etapa de digestão,
esta deve ser acrescida a diluição de 10x, aplicada a este procedimento e descrita acima, ou seja,
ambos os fatores de diluição são multiplicados para que a nova diluição seja obtida.
3. DESCARTE DO RESÍDUO GERADO
Idem procedimento apresentado para nitrito e nitrato.
3. REFERÊNCIA
Adaptação de STANDARD METHODS, method 4500- N C (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
79
POP 11 – SÉRIE DE NITROGÊNIO – NITROGÊNIO
ORGÂNICO TOTAL (NOrgT)
1. MÉTODO PARA DETERMINAÇÃO
Aplicando as metodologias descritas para a série de nitrogênio até o
presente momento, quantificamos o N-Orgânico da seguinte forma:
NOrgT = NT - ∑NINORG (12)
em que:
∑NINORG = [N-NH3] + [N-NO2-] + [N-NO3
-]

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
80
POP 12 – SÉRIE DE FÓSFORO – ORTOFOSFATO (PO43-)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DO ÁCIDO ASCÓRBICO
A reação entre molibdato de amônio e antimônio tartarato de potássio em
meio ácido (H2SO4) com ortofosfato, formam o ácido fosfomolibdico, que em
presença de ácido ascórbico é reduzido para fosfomolibdato, composto de
coloração azul intensa característica.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo
Membranas de acetato de celulose (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Espectrofotômetro UV-Vis
Micropipeta (100 - 1000µl)
Vidrarias
Balão volumétrico de 100ml
Balão volumétrico de 250ml
Balão volumétrico de 500ml
Erlenmeyer
Pipetas volumétricas e graduadas
Cubeta de vidro
Conjunto kitassato para filtração
Reagentes analíticos
Solução padrão para fosfato (50 mg.L-1): dissolver 0,02195g de fosfato
de potássio monobásico anidro (KH2PO4 p.a) em, aproximadamente, 80ml

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
81
de água destilada. Aferir com água destilada para 100ml, em balão
volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 1 ano (mínimo).
* Concentração final de 50 mg P.L-1
Solução de ácido sulfúrico 2,5 mol.L-1: adicionar lentamente 70ml de
H2SO4 (p.a) em, aproximadamente, 400ml de água destilada. Aferir com
água destilada para 500ml, em balão volumétrico.
* Armazenar em frascos de vidro normal para reagente, em temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução de tartarato de antimônio e potássio: dissolver 0,6858g de
tartarato de antimônio e potássio (p.a) em, aproximadamente, 200ml de
água destilada. Aferir com água destilada para 250ml, em balão
volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 6 meses (mínimo).
Solução de molibdato de amônio: dissolver 10g de molibdato de amônio
(p.a) em, aproximadamente, 200ml de água destilada. Aferir com água
destilada para 250ml, em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 6 meses (mínimo).
Solução de ácido ascórbico: dissolver 1,76g de ácido ascórbico (p.a) em,
aproximadamente, 80ml de água destilada. Aferir com água destilada para
100ml, em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar em ambiente refrigerado (4ºC).
* Estável por 1 semana (máximo).
Solução reativa combinada (Mix): adicionar com o auxílio de pipetas
volumétricas e/ou graduadas os seguintes reagentes, na ordem e
proporção de volume: 50ml de H2SO4 2,5 mol.L-1 + 5ml de tartarato de
antimônio e potássio + 15ml de molibdato de amônio + 30ml de ácido
ascórbico (volume final de 100ml).
* Antes de promover a misturar, aguardar os reagentes armazenados em refrigerador atingir a
temperatura ambiente.
* Estável por 4 horas após o preparo.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
82
- A cada adição de um reagente, promover a homogeneização da solução.
- Ao final, na adição do ácido ascórbico, uma coloração amarelada bem clara
aparecerá, sendo um indicativo que os reagentes do Mix foram bem preparados,
sem influência de contaminação por fósforo no material utilizado.
- No caso da solução Mix ficar com coloração esverdeada e azulada, indica
contaminação por fósforo, embora possa ser utilizada devido à correção pelo
branco a ser realizado com água destilada;
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é realizada através de curva de calibração padrão,
descrita na seqüência:
1. A partir da solução padrão (50 mg P.L-1) preparar, em balões
volumétricos de 50ml, a seguinte curva de calibração (volume final com água
destilada):
Concentração P-PO4-3 (µg.L-1) Volume P-PO4
-3 (ml) 0 (branco) -
100 0,1 300 0,3 500 0,5 900 0,9 1500 1,5
*Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser
preparada em duplicata ou triplicata.
2. Posteriormente ao preparo das soluções padrões, conforme a tabela
acima, retirar, com auxílio de uma pipeta volumétrica, uma alíquota de 10ml de
cada padrão e reservar em outro frasco;
3. Juntamente aos padrões, realizar prova em branco utilizando alíquotas
de 10ml de água destilada;
4. Adicionar 2ml da solução reativa combinada (MIX) para ortofosfato em
cada alíquota;
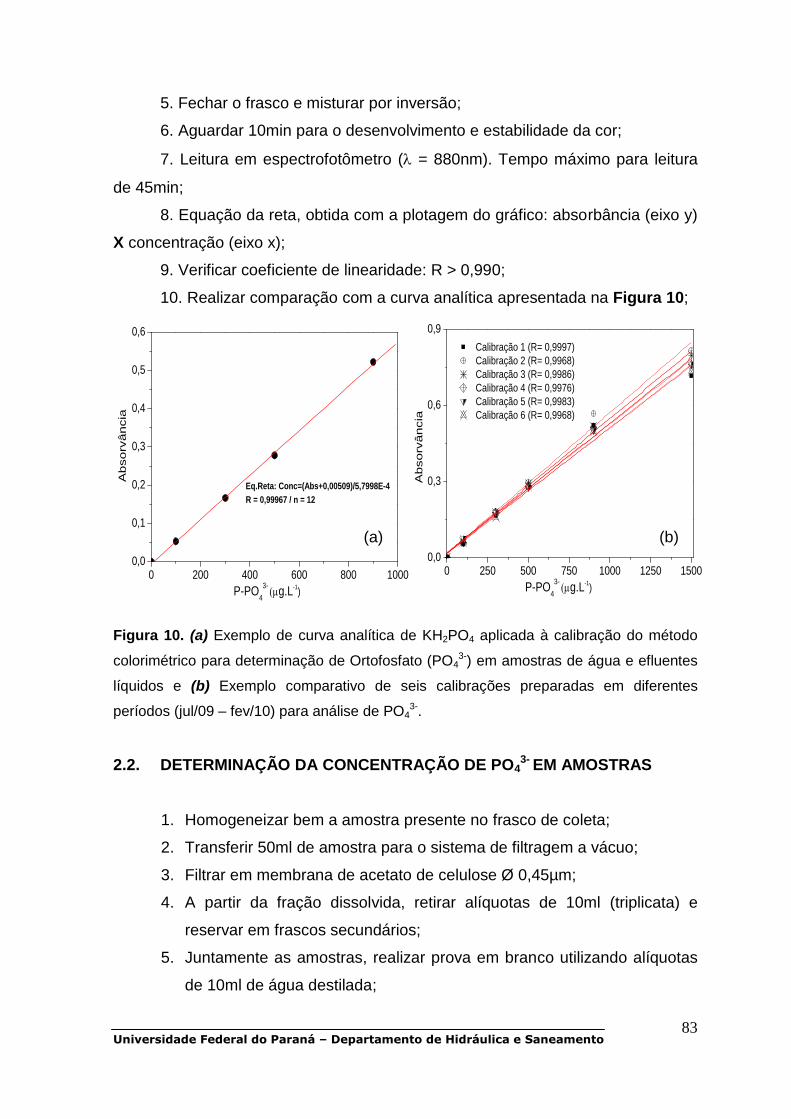
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
83
0 200 400 600 800 10000,0
0,1
0,2
0,3
0,4
0,5
0,6
A
bsorv
ância
P-PO4
3- g.L-1
)
Eq.Reta: Conc=(Abs+0,00509)/5,7998E-4
R = 0,99967 / n = 12
0 250 500 750 1000 1250 15000,0
0,3
0,6
0,9
P-PO4
3- g.L-1
)
Calibração 1 (R= 0,9997)
Calibração 2 (R= 0,9968)
Calibração 3 (R= 0,9986)
Calibração 4 (R= 0,9976)
Calibração 5 (R= 0,9983)
Calibração 6 (R= 0,9968) A
bsorv
ância
(a) (b)
5. Fechar o frasco e misturar por inversão;
6. Aguardar 10min para o desenvolvimento e estabilidade da cor;
7. Leitura em espectrofotômetro ( = 880nm). Tempo máximo para leitura
de 45min;
8. Equação da reta, obtida com a plotagem do gráfico: absorbância (eixo y)
X concentração (eixo x);
9. Verificar coeficiente de linearidade: R > 0,990;
10. Realizar comparação com a curva analítica apresentada na Figura 10;
Figura 10. (a) Exemplo de curva analítica de KH2PO4 aplicada à calibração do método
colorimétrico para determinação de Ortofosfato (PO43-) em amostras de água e efluentes
líquidos e (b) Exemplo comparativo de seis calibrações preparadas em diferentes
períodos (jul/09 – fev/10) para análise de PO43-.
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE PO43- EM AMOSTRAS
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Transferir 50ml de amostra para o sistema de filtragem a vácuo;
3. Filtrar em membrana de acetato de celulose Ø 0,45µm;
4. A partir da fração dissolvida, retirar alíquotas de 10ml (triplicata) e
reservar em frascos secundários;
5. Juntamente as amostras, realizar prova em branco utilizando alíquotas
de 10ml de água destilada;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
84
6. Adicionar 2ml da solução reativa combinada (Mix) em cada alíquota;
7. Fechar o frasco e misturar por inversão;
8. Aguardar 10min para o desenvolvimento e estabilidade da cor;
9. Leitura em espectrofotômetro ( = 880nm). Tempo máximo para leitura
de 45min;
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de PO43- (mg.L-1 ou µg.L-1) através da
equação da reta, obtida na curva analítica (calibração).
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de ortofosfato (PO43-) é aplicável para
concentrações < 1500 µg.L-1 com valor mínimo detectável de 5 µg.L-1.
3. DESCARTE DO RESÍDUO GERADO
A composição do resíduo de fósforo / ortofosfato é fortemente ácida, devido
ao uso de H2SO4 como parcela na mistura do reativo indicador combinado. Dessa
forma, o descarte pode ser realizado na pia, logo após a correção do pH para a
faixa de neutralidade.
O mesmo procedimento deve ser adotado nas análises de PT e PDT,
apresentados no POP-13, na sequência.
3. REFERÊNCIAS
MURPHY, J.; RILEY, J.P. A Modified Single Solution Method for the Determination of
Phosphate in Natural Water. Anal.Chim.Acta, Vol. 27, pp. 31-36, 1962
STANDARD METHODS, method 4500 - P E (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
85
POP 13 – SÉRIE DE FÓSFORO – FÓSFORO TOTAL (PT)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DA DIGESTÃO ALCALINA (K2S2O8 / NaOH) & MÉTODO DO
ÁCIDO ASCÓRBICO
Quantificação de PT através de digestão alcalina utilizando persulfato de
potássio em autoclave (125ºC / 14kg.cm-1 / 25min) para conversão de todas as
formas de fósforo presentes para ortofosfato (PO43-) e posterior quantificação
segundo o método do ácido ascórbico.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo e membranas de acetato de celulose (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Autoclave vertical
Espectrofotômetro UV-Vis
Micropipeta (100 - 1000µl)
Vidrarias
Frascos para digestão (vidro)
Balão volumétrico de 100ml, 250ml e 500ml
Erlenmeyer
Pipetas volumétricas e graduadas
Cubeta de vidro
Reagentes analíticos
Solução digestora alcalina: dissolver 2,01g de persulfato de potássio
(K2S2O8 p.a < 0,001% N), em aproximadamente 80ml de água destilada
Adicionar 0,300g de hidróxido de sódio (NaOH p.a) e dissolver novamente.
Aferir com água destilada para 100ml, em balão volumétrico.
* Preparo deve ser realizado para uso imediato.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
86
Solução reativa combinada (Mix): ver POP-12, item 1 (PO43-).
Solução padrão para fosfato (50 mg.L-1): ver POP-12, item 1 (PO43-).
Solução tampão de borato: dissolver 30,9g de ácido bórico (H3BO3 p.a)
em, aproximadamente, 300ml em água destilada. Adicionar 4g de hidróxido
de sódio (NaOH p.a) e dissolver novamente. Aferir com água destilada para
500 mL em balão volumétrico.
* Estável por 1 ano (mínimo).
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
A calibração do método é realizada através de curva de calibração padrão,
descrita na seqüência:
1. A partir da solução padrão (50 mg P.L-1) (ver POP-12, item 1) preparar,
em balões volumétricos de 50ml a seguinte curva de calibração (volume final com
água destilada):
Concentração P-PO4-3 (µg.L-1) Volume P-PO4
-3 (ml) 0 (branco) -
100 0,1 300 0,3 500 0,5 900 0,9 1500 1,5
*Para uma melhor confiabilidade e verificação da linearidade da curva de calibração, esta pode ser
preparada em duplicata ou triplicata.
2. A partir das soluções padrões preparadas, retirar com o auxílio de uma
pipeta volumétrica, alíquotas de 5ml e transferir para os tubos de ensaio com
rosca para uso em autoclave;
3. Juntamente aos padrões, realizar prova em branco utilizando alíquotas
de 5ml de água destilada;
4. Adicionar a cada tubo, 2,5ml da solução digestora alcalina;
5. Fechar bem os tubos para digestão e misturar por inversão no mínimo
duas vezes;
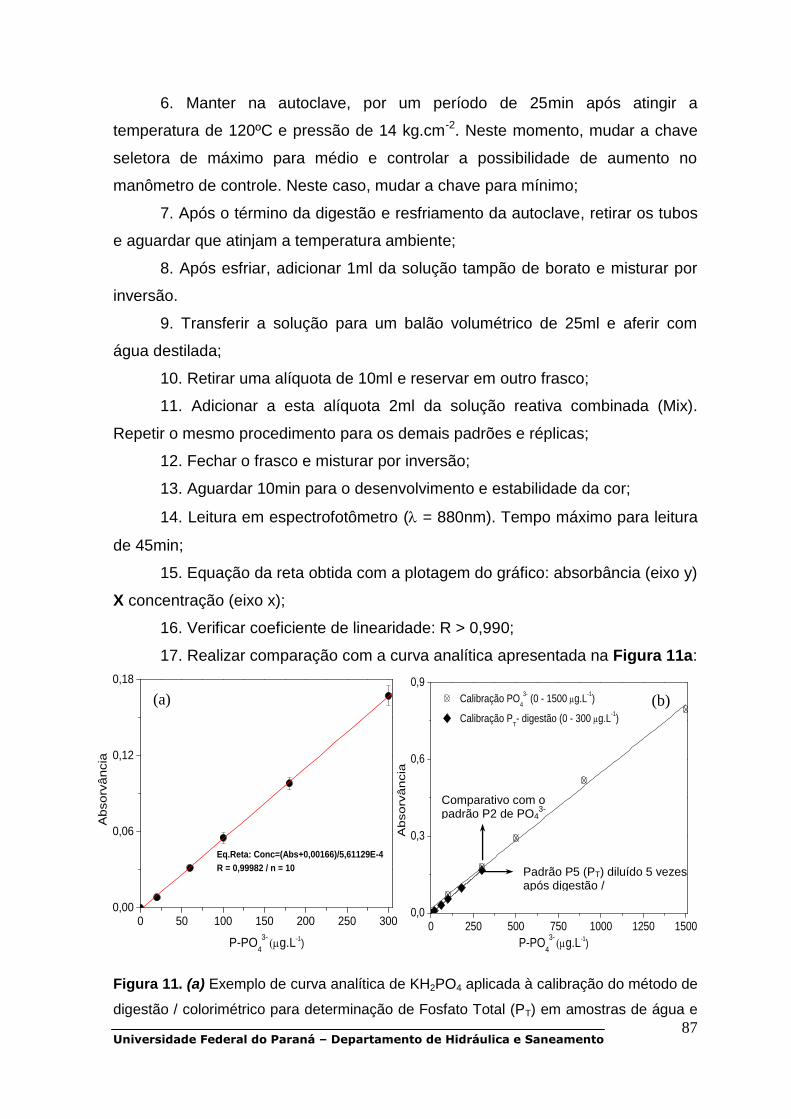
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
87
0 50 100 150 200 250 3000,00
0,06
0,12
0,18
Eq.Reta: Conc=(Abs+0,00166)/5,61129E-4
R = 0,99982 / n = 10
Absorv
ância
P-PO4
3- g.L-1
)
0 250 500 750 1000 1250 15000,0
0,3
0,6
0,9
Calibração PO4
3- (0 - 1500 g.L
-1)
Calibração PT- digestão (0 - 300 g.L
-1)
P-PO4
3- g.L-1
)
Absorv
ância
Padrão P5 (PT) diluído 5 vezes após digestão / tamponamento.
Comparativo com o padrão P2 de PO4
3-
6. Manter na autoclave, por um período de 25min após atingir a
temperatura de 120ºC e pressão de 14 kg.cm-2. Neste momento, mudar a chave
seletora de máximo para médio e controlar a possibilidade de aumento no
manômetro de controle. Neste caso, mudar a chave para mínimo;
7. Após o término da digestão e resfriamento da autoclave, retirar os tubos
e aguardar que atinjam a temperatura ambiente;
8. Após esfriar, adicionar 1ml da solução tampão de borato e misturar por
inversão.
9. Transferir a solução para um balão volumétrico de 25ml e aferir com
água destilada;
10. Retirar uma alíquota de 10ml e reservar em outro frasco;
11. Adicionar a esta alíquota 2ml da solução reativa combinada (Mix).
Repetir o mesmo procedimento para os demais padrões e réplicas;
12. Fechar o frasco e misturar por inversão;
13. Aguardar 10min para o desenvolvimento e estabilidade da cor;
14. Leitura em espectrofotômetro ( = 880nm). Tempo máximo para leitura
de 45min;
15. Equação da reta obtida com a plotagem do gráfico: absorbância (eixo y)
X concentração (eixo x);
16. Verificar coeficiente de linearidade: R > 0,990;
17. Realizar comparação com a curva analítica apresentada na Figura 11a:
Figura 11. (a) Exemplo de curva analítica de KH2PO4 aplicada à calibração do método de
digestão / colorimétrico para determinação de Fosfato Total (PT) em amostras de água e
(a) (b)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
88
de efluentes líquidos e (b) Exemplo comparativo entre a calibração para PO43-, com
ausência da etapa de digestão, e calibração para PT com presença da etapa de digestão /
neutralização.
ATENÇÃO: verifique a seguinte etapa da metodologia: ao adicionar 5ml do padrão para digestão
e após a neutralização, ter aferido para 25ml, você realizou uma diluição de 5 vezes, ou seja, o
padrão deve estar, teoricamente, 5 vezes mais diluído que o valor encontrado para a curva de
calibração de Ortofosfato (POP nº. 12). Um exemplo pode ser observado na Figura 11b.
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE PT EM AMOSTRAS
1. Homogeneizar bem a amostra presente no frasco de coleta;
2. Com o auxílio de uma pipeta volumétrica, transferir 5ml da amostra para
tubos de ensaio com rosca, para digestão;
3. Juntamente aos padrões, realizar prova em branco utilizando alíquotas
de 5ml de água destilada;
4. Adicionar a cada tubo, 2,5ml da solução digestora alcalina;
5. Fechar bem os tubos para digestão e misturar por inversão;
6. Manter na autoclave, por um período de 25min após atingir a
temperatura de 120ºC e pressão de 14 kg.cm-2. Neste momento, mudar
a chave seletora de máximo para médio e controlar a possibilidade de
aumento no manômetro de controle. Neste caso, mudar a chave para
mínimo;
7. Após o término da digestão e resfriamento da autoclave, retirar os tubos
e aguardar que atinjam a temperatura ambiente;
8. Após esfriar, adicionar 1ml da solução tampão de borato e misturar por
inversão;
9. No mesmo frasco, adicionar 1,5ml de água destilada e misturar por
inversão;
ATENÇÃO: verifique a seguinte etapa da metodologia: ao adicionar 5ml do padrão para digestão
e após a adição dos reagentes seguintes (solução digestora, solução tampão e água), a amostra
foi diluída, neste caso, 2x, fator este que deve ser utilizado na correção da concentração de PT
quantificada.
10. Adicionar a esta alíquota 2ml da solução reativa combinada (Mix).
Repetir o mesmo procedimento para as demais amostras e réplicas;
11. Fechar o frasco e misturar por inversão;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
89
12. Aguardar 10min para o desenvolvimento e estabilidade da cor;
13. Leitura em espectrofotômetro ( = 880nm). Tempo máximo para leitura
de 45min;
2.3. CÁLCULO DA CONCENTRAÇÃO
Quantificação da concentração de PT (mg.L-1 ou µg.L-1) através da equação
da reta, obtida na curva analítica (calibração) para PT.
2.4. LIMITE DE DECTEÇÃO
O método para quantificação de PT está relacionado com o método
utilizado na quantificação de ortofosfato (PO43-). Assim, segue a faixa de leitura
descrita para ortofosfato: quantificação para concentrações < 1500 µg.L-1 com
valor mínimo detectável de 5 µg.L-1.
3. DETERMINAÇÃO DA CONCENTRAÇÃO DE FÓSFORO TOTAL
DISSOLVIDO (PTD) EM AMOSTRAS
A quantificação de fosfato total na fração dissolvida (PTD) pode ser
realizada seguindo a metodologia descrita para fosfato total (PT). Entretanto, a
única diferença é uma etapa adicional de pré tratamento da amostra:
A amostra deve, primeiramente, ser filtrada em membrana de acetato de
celulose Ø 0,45µm para obtenção da fração dissolvida e, na sequência, seguir o
método da digestão alcalina (fechada).
4. DETERMINAÇÃO DA CONCENTRAÇÃO DE FÓSFORO TOTAL
PARTICULADO (PTP) EM AMOSTRAS
4.1. MÉTODO PARA DETERMINAÇÃO

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
90
A quantificação da concentração de fosfato total, presente na fração
particulada (PTP) pode ser obtida a partir da subtração da concentração de fosfato
total dissolvido (PTD) pela concentração encontrada para fosfato total (PT):
PTP = PT – PTD (13)
5. DETERMINAÇÃO DA CONCENTRAÇÃO DE FÓSFORO ORGÂNICO
DISSOLVIDO / SOLÚVEL (POrgD) EM AMOSTRAS
5.1. MÉTODO PARA DETERMINAÇÃO
A quantificação da concentração do fósforo orgânico dissolvido (PORG),
presente na amostra pode ser obtida a partir da subtração da concentração de
ortofosfato (PO43-), POP Nº. 12 pelo resultado encontrado para a concentração de
fosfato total dissolvido (PTD):
POrgD = PTD – PO4-3 (14)
6. REFERÊNCIAS
MURPHY, J.; RILEY, J.P. A Modified Single Solution Method for the Determination of
Phosphate in Natural Water. Anal.Chim.Acta, Vol. 27, pp. 31-36, 1962
STANDARD METHODS, method 4500 - P E (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
91
POP 14 – CLOROFILA-a (CLa)
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO DA EXTRAÇÃO COM ACETONA 90%
Quantificação de clorofila-a por meio da extração com acetona 90% e
determinação da concentração pela absorvância medida em espectrofotômetro
UV-Vis.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Bomba de vácuo
Membranas de fibra de vidro (Ø 0,45µm)
Balança analítica (precisão ± 0,0001g)
Tubos para centrífuga graduados com tampa
Gral e pistilo de porcelana
Micropipeta (100 – 1000l)
Centrífuga
Espectrofotômetro UV-Vis
Vidrarias
Pipetas volumétricas e graduadas
Cubeta de vidro
Reagentes analíticos
Solução saturada de carbonato de magnésio: adicione 1,0g de
carbonato de magnésio e dissolva em 100ml de água destilada.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
92
Solução de acetona 9:1: misture 90 partes de solução de acetona
(CH3(CO)CH3) com 10 partes de solução saturada de carbonato de
magnésio.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de HCl 0,1 mol.L-1: adicionar 8,5ml de ácido clorídrico (HCl
p.a) em, aproximadamente, 800ml de água destilada. Aferir para 1000ml
em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 1 ano (mínimo).
2. PROCEDIMENTO ANALÍTICO
2.1. DETERMINAÇÃO DA CONCENTRAÇÃO DE CLa EM AMOSTRAS
1. Homogeneizar bem a amostra presente no frasco de coleta, pelo
menos 10 vezes, fazendo movimento em forma de 8;
2. Após homogeneização da amostra, filtrar em membrana de fibra
de vidro até a saturação da membrana e anotar o volume filtrado;
3. Retirar o filtro e armazená-lo em envelope de papel laminado
identificado. A membrana pode ser congelada por um período de
até 60 dias. A análise para determinação pode ser prosseguida
após a filtração ou posteriormente ao descongelamento da
membrana;
4. Na sequência, a membrana dever ser colocada em um tubo de
centrífuga graduado e, com auxílio de um bastão de vidro,
macere o filtro até que a membrana se desmanche por completo,
adicionando aos poucos os 10ml da solução saturada de acetona;
5. Centrifugar a 4000rpm por 10min. Envolver os tubos, com papel
laminado identificado (ausência de luminosidade) e manter as
amostras armazenadas sob refrigeração (4ºC) por um período de
24h;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
93
6. Após este período proceder à leitura do sobrenadante em
espectrofotômetro UV-Vis, de acordo com a seguinte ordem:
LEITURA 1: com a amostra simples, no comprimento de onda
de 664nm e 750nm, para eliminar a interferência de outros
compostos;
LEITURA 2: adicionar na mesma amostra, diretamente na
cubeta, 300l (6 gotas) de HCl 0,1 mol.L-1. Tampar a cubeta e
misturar por inversão (1 vez). Após 3min da adição do HCl,
realizar novamente a leitura para os comprimentos de onda de
665nm e 750nm;
2.2. CÁLCULO DA CONCENTRAÇÃO
Os valores das concentrações de clorofila-a são obtidos por meio da
seguinte equação:
3 26,7 [664 665 ] 1( / )
2
b ax xVCLa mg m
V xL
(15)
em que:
664b: subtração do sinal obtido em 664nm e 750nm
665a: subtração do sinal obtido em 665nm e 750nm (densidade óptica
após a acidificação)
V1: volume do extrato (acetona, 10ml)
V2: volume filtrado da amostra (L)
L: caminho ótico da cubeta (1cm)
3. REFERÊNCIA
Adaptado de STANDARD METHODS (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
94
POP 15 – METAIS PESADOS (Cd, Cr, Cu, Ni, Zn, Pb)
1. MÉTODO PARA DETERMINAÇÃO
1.1. ESPECTROMETRIA DE ABSORÇÃO ATÔMICA DE CHAMA (EAA-
Chama) / PRÉ CONCENTRAÇÃO DA AMOSTRA (10x)
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos e materiais
Espectrômetro de absorção atômica de chama (EAA-Chama)
Balança analítica (precisão ± 0,0001g)
Micropipeta (100 - 1000µl)
Frasco plástico (PEAD, PP) para soluções padrões (50ml) e tubos de
centrífuga Falcon (15ml) para as amostras
Bomba de vácuo
Membranas de acetato de celulose (Ø 0,45µm)
Chapa de aquecimento
Água ultra pura (mili-Q)
Vidrarias
Balões volumétricos de 100ml
Becker de 50ml
Conjunto kitassato para filtração
Erlenmeyers de 50ml
Reagentes analíticos
Ácido Nítrico concentrado 65% ultra puro (HNO3 p.a 65%)
H2O2 30%
Ácido Nítrico 1:1: adicione, lentamente, 500ml de ácido nítrico
concentrado (HNO3 p.a 65%), com auxílio de pipetas volumétricas de
100ml em 500ml de água destilada e deionizada.
*Armazenar em frasco âmbar e acondicionar a temperatura ambiente.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
95
* Estável por 1 ano (mínimo).
Ácido Clorídrico 1:1: adicione, lentamente, 500ml de ácido clorídrico
concentrado (HCl p.a), com auxílio de pipetas volumétrica de 100ml em
500ml de água destilada e deionizada.
*Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 1 ano (mínimo).
2. PROCEDIMENTO ANALÍTICO
2.1. CALIBRAÇÃO DO MÉTODO
Cada metal a ser quantificado no espectrômetro de absorção atômica
necessita de soluções padrões, preparadas em uma determinada faixa de
concentração do metal para que, antes da leitura das amostras, o equipamento
seja devidamente calibrado e retorne uma resposta adequada frente à
sensibilidade do aparelho para o determinado metal a ser analisado.
Na sequência, são apresentados os procedimentos para o preparo das
soluções padrões para os metais Cd, Cr, Cu, Ni, Zn e Pb.
2.1.1. PREPARO DAS SOLUÇÕES PADRÕES DE METAIS
Os padrões de metais consistem na preparação de cinco soluções de
concentração conhecida, sendo que para cada uma todos os metais (Cd, Cr,
Cu, Ni, Zn e Pb) estão presentes. Por exemplo: o padrão 1 deve conter
igualmente 1 mg.L-1 para Cd, Cr, Cu, Ni, Zn e 2 mg.L-1 para Pb. Retomando, é
necessário adicionar igualmente o mesmo volume para cada metal, ou seja,
6ml totalizando 36ml de solução padrão. Para tal, se deve calcular a
concentração de cada metal que seja 6 vezes superior a concentração final,
visto que, considerando os 6 metais em análise estes devem ser diluídos por
igual. Na Tabela 2 são apresentados os cálculos e procedimento para o
preparo das soluções padrões:
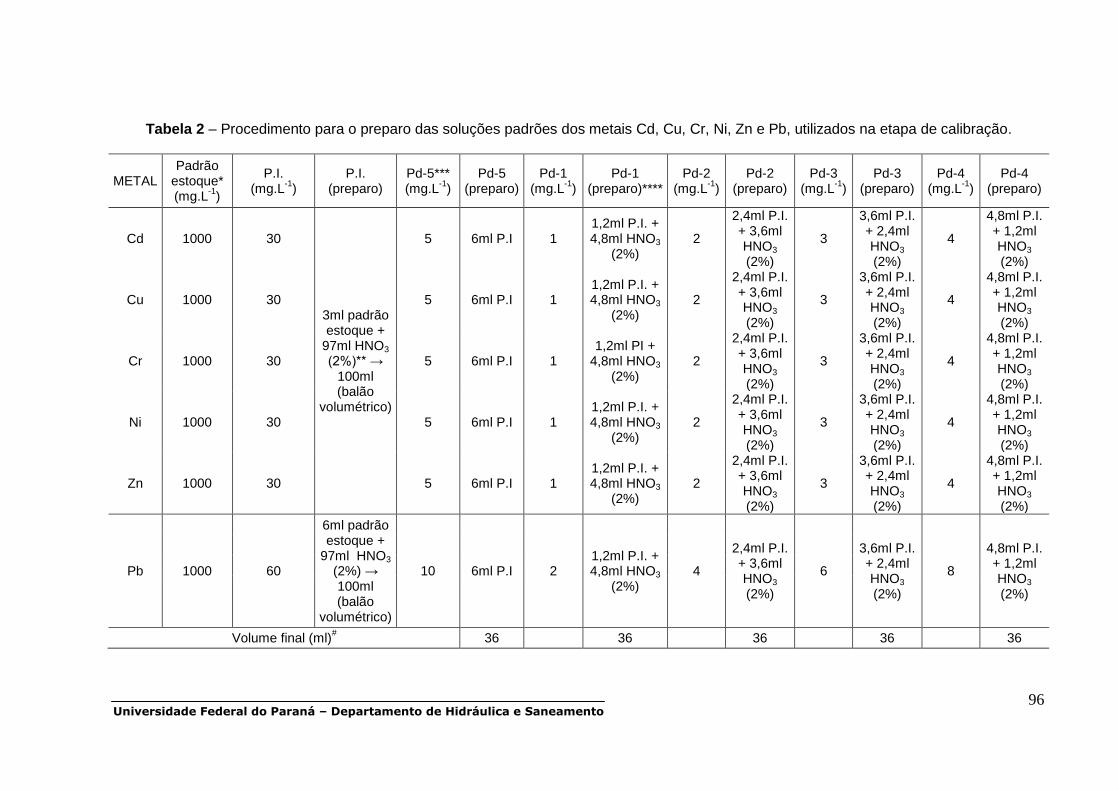
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
96
Tabela 2 – Procedimento para o preparo das soluções padrões dos metais Cd, Cu, Cr, Ni, Zn e Pb, utilizados na etapa de calibração.
METAL Padrão
estoque* (mg.L
-1)
P.I. (mg.L
-1)
P.I. (preparo)
Pd-5*** (mg.L
-1)
Pd-5 (preparo)
Pd-1 (mg.L
-1)
Pd-1 (preparo)****
Pd-2 (mg.L
-1)
Pd-2 (preparo)
Pd-3 (mg.L
-1)
Pd-3 (preparo)
Pd-4 (mg.L
-1)
Pd-4 (preparo)
Cd 1000 30
3ml padrão estoque +
97ml HNO3 (2%)** →
100ml (balão
volumétrico)
5 6ml P.I 1 1,2ml P.I. + 4,8ml HNO3
(2%) 2
2,4ml P.I. + 3,6ml HNO3
(2%)
3
3,6ml P.I. + 2,4ml HNO3
(2%)
4
4,8ml P.I. + 1,2ml HNO3 (2%)
Cu 1000 30 5 6ml P.I 1 1,2ml P.I. + 4,8ml HNO3
(2%) 2
2,4ml P.I. + 3,6ml HNO3 (2%)
3
3,6ml P.I. + 2,4ml HNO3 (2%)
4
4,8ml P.I. + 1,2ml HNO3 (2%)
Cr 1000 30 5 6ml P.I 1 1,2ml PI +
4,8ml HNO3 (2%)
2
2,4ml P.I. + 3,6ml HNO3
(2%)
3
3,6ml P.I. + 2,4ml HNO3 (2%)
4
4,8ml P.I. + 1,2ml HNO3 (2%)
Ni 1000 30 5 6ml P.I 1 1,2ml P.I. + 4,8ml HNO3
(2%) 2
2,4ml P.I. + 3,6ml HNO3 (2%)
3
3,6ml P.I. + 2,4ml HNO3 (2%)
4
4,8ml P.I. + 1,2ml HNO3 (2%)
Zn 1000 30 5 6ml P.I 1 1,2ml P.I. + 4,8ml HNO3
(2%) 2
2,4ml P.I. + 3,6ml HNO3 (2%)
3
3,6ml P.I. + 2,4ml HNO3 (2%)
4
4,8ml P.I. + 1,2ml HNO3 (2%)
Pb 1000 60
6ml padrão estoque +
97ml HNO3
(2%) → 100ml (balão
volumétrico)
10 6ml P.I 2 1,2ml P.I. + 4,8ml HNO3
(2%) 4
2,4ml P.I. + 3,6ml HNO3
(2%)
6
3,6ml P.I. + 2,4ml HNO3 (2%)
8
4,8ml P.I. + 1,2ml HNO3
(2%)
Volume final (ml)#
36
36
36
36
36

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
97
*Padrão certificado, de concentração conhecida. **HNO3 (2%): diluir, em balão volumétrico, 20ml de HNO3 65% em 980ml de água ultra pura. ***Pd: identificação dos padrões na tabela – Pd-5 (padrão 5), Pd-1 (padrão 1)... ****Os padrões Pd-1, Pd-2, Pd-3 e Pd-4 são preparados a partir de diluições de volumes conhecidos dos padrões intermediários (PI) em HNO3 (2%) para um volume final de 6ml . #Representa a soma do volume de solução adicionado para cada metal (6+6+6+6+6+6)ml.
2.2. DETERMINAÇÃO DA CONCENTRAÇÃO DE METAIS PESADOS NA
FRAÇÃO DISSOLVIDA - Pré concentração 10x
1. Homogeneizar bem o frasco contendo a amostra preservada com HNO3;
2. Transferir 100ml de amostra para o sistema de filtragem de vácuo;
3. Filtrar em membrana de acetato de celulose Ø 0,45µm;
4. Transferir 50ml da amostra (dissolvida) com o auxílio de uma pipeta
volumétrica para o erlenmeyer (realizar o procedimento em duplicata ou
triplicata);
5. Adicionar 3ml de ácido nítrico concentrado (HNO3 p.a 65%);
6. Manter os erlenmeyers contendo as amostras em uma chapa de aquecimento
a uma temperatura entre 110 e 130ºC;
7. Manter evaporando a solução até volume inferior a 5ml. Evitar que a solução
evapore completamente;
8. Retirar da chapa e aguardar esfriar. (Mantenha os frascos sempre cobertos
de modo a evitar contaminação);
9. Transferir as amostras para tubos de centrífuga de 15ml e aferir o volume
para 5ml com água ultra pura;
10. Acondicionar em refrigerador (<4ºC) até o momento da leitura;
11. Realizar a quantificação em espectrômetro de absorção atômica de chama;
12. Caso o tempo para leitura seja superior a 30 dias, mantenha as amostras em
congelador;
2.3. DETERMINAÇÃO DA CONCENTRAÇÃO DE METAIS PESADOS NA
FRAÇÃO PARTICULADA - Pré concentração 15x
1. Homogeneizar bem o frasco contendo a amostra preservada com HNO3;
2. Transferir 150ml de amostra para o sistema de filtragem de vácuo;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
98
3. Filtrar em membrana de acetato de celulose Ø 0,45µm. Para cada membrana
utilizada, anotar o valor do seu respectivo peso (P1) e ponto ou réplica
correspondente;
4. Manter as membranas em ambiente livre de contaminação por metais
(temperatura ambiente) para secagem até peso constante (24h);
5. Realizar uma nova verificação do peso e anotar o valor observado (P2);
6. Em erlenmeyers de 50ml, colocar as membranas, seguido de 10ml de HNO3
1:1;
7. Juntamente, realizar brancos analíticos apenas com 5ml de água ultra pura e
10ml de HNO3 1:1 e seguir todos os passos seguintes das amostras;
8. Manter em chapa de aquecimento por um período de 45min, após atingir a
temperatura de 150ºC;
9. Retirar da chapa e aguardar o resfriamento dos frascos, mantendo-os na
capela de exaustão;
10. Adicionar 5ml de HNO3 concentrado (65% p.a) e retornar os frascos para a
chapa de aquecimento, na temperatura de 150ºC, por 2h ou redução do
volume próximo de 3ml;
11. Retirar da chapa e aguardar que os frascos atinjam a temperatura ambiente;
12. Adicionar 2ml de H2O2 (30%) e, novamente, manter sob aquecimento (150ºC);
13. Aguardar a completa efervescência da amostra. Se necessário adicionar
alíquotas de 5ml de água ultra pura, evitando a evaporação total da mesma;
14. Retirar da chapa e aguardar que os frascos atinjam a temperatura ambiente.
Na sequência, adicionar 10ml de HCl 1:1 e retornar para a chapa, mantendo a
uma temperatura de 180ºC por um período de 1h;
15. Verificar um volume aproximado, abaixo de 5ml e retirar os mesmos da
chapa. Manter na capela de exaustão até que atinjam a temperatura
ambiente;
16. Transferir a amostra para o tubo de centrífuga, aferindo com água ultra pura
no volume de 10ml;
17. Centrifugar as amostras para remoção de particulados residuais do
sobrenadante;
18. Acondicionar em refrigerador (<4ºC) até o momento da leitura;
19. Realizar a quantificação em espectrômetro de absorção atômica de chama;

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
99
20. Caso o tempo para leitura seja superior a 30 dias, mantenha as amostras em
congelador;
2.4. CÁLCULO DA CONCENTRAÇÃO
Os resultados da concentração (g.mL-1 ou mg.L-1) são calculados
diretamente pelo software do equipamento, uma vez que antes da leitura das
amostras para cada metal é necessário a calibração com as soluções padrões para
ver a resposta e sensibilidade do aparelho. Ainda, seguindo este procedimento, os
resultados devem ser corrigidos, visto que a amostra foi concentrada 10x (fração
dissolvida) e 15x (fração particulada) para quantificação. Para as amostras da fração
particulada, os resultados também podem ser obtidos em peso seco (ex.: g.g-1)
visto que a quantidade de sólidos pode ser determinada pela diferença do peso da
membrana (g) antes e após a filtração e o respectivo volume filtrado (ml).
2.5. LIMITE DE DETECÇÃO
Os valores de limite de detecção devem ser obtidos juntamente com a leitura
das amostras. Para tal, você deve separar para cada metal, oito alíquotas de água
mili-Q para serem lidas antes das amostras. O cálculo é feito segundo a fórmula: LD
= k.sb/m onde k é o fator de confiança superior (2 ou 3, sendo que o primeiro
apresenta um nível de 92,1% de confiança e o segundo 98%) ao desvio padrão (sb)
das oito réplicas de água mili-Q e a variável m responde quanto à sensibilidade da
calibração (inclinação da curva de calibração para cada metal).
3. DESCARTE DO RESÍDUO GERADO
Para metais, o resíduo necessita de um ajuste de pH prévio, visto que o
procedimento de análise adotado neste manual, tem na etapa ou alíquota final um
meio extremamente ácido, na presença de HNO3. Após a correção do pH, o resíduo
pode ser descartado pela pia. A concentração de metais, inicialmente, não é
considerada, embora, apresente valores inicialmente baixos.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
100
4. REFERÊNCIAS
Method 3050B. Acid Digestion of Sediments, Sludges and Soils. EPA, revision 2, 1996
STANDARD METHODS (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
101
POP 16 – ALCALINIDADE TOTAL
1. MÉTODO PARA DETERMINAÇÃO
1.1. MÉTODO TITULOMÉTRICO
A amostra é titulada com ácido padrão a um pH específico, determinando-se o ponto
final pelo ponto de viragem do indicador aplicado na titulação.
1.2. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Balança analítica (precisão ± 0,0001g)
Agitador magnético
Vidrarias
Erlenmeyer
Bureta
Pipetas volumétricas e graduadas
Reagentes analíticos
Solução indicadora de metilorange: dissolver 0,2g de metilorange
C14H14N3SO3Na (p.a), em água quente e após esfriamento, filtrar se
necessário. Aferir com água destilada / deionizada para 100ml em balão
volumétrico.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução indicadora de fenolftaleína: dissolver 1,0g de fenolftaleína
C6H4COO.C(C6H4OH)2, p.a., em 60 mL de álcool etílico p.a (C2H5OH). Aferir
com água destilada / deionizada para 100 ml em balão volumétrico.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
102
Solução de carbonato de sódio 0,1N: pesar 5,3g de carbonato de sódio,
Na2CO3 (p.a), previamente seco em estufa a 200ºC por 1 hora. Dissolver em
água destilada, transferir para um balão volumétrico e aferir para 1000ml.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).
Diluição da solução de Na2CO3 0,1N para 0,02N: transferir 200ml da
solução de carbonato de sódio 0,1N para um balão volumétrico de 1000 ml e
aferir com água destilada.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 6 meses (mínimo).
Solução de ácido sulfúrico 0,1N: adicionar aproximadamente 500ml de
água destilada em um balão volumétrico de 1000ml. Com o auxílio de uma
pipeta, transferir, lentamente, 2,8ml de ácido sulfúrico concentrado (H2SO4
p.a) para o balão volumétrico aferindo-o posteriormente com água destilada.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 1 ano (mínimo).
Diluição da solução de H2SO4 0,1N para 0,02N: transferir 200ml da solução
de ácido sulfúrico 0,1N para um balão volumétrico de 1000ml e aferir com
água destilada.
* Armazenar em frasco âmbar e acondicionar a temperatura ambiente.
* Estável por 1 ano (mínimo).
Solução de tiossulfato de sódio: dissolver 0,35g de tiossulfato de sódio
pentahidratado, Na2S2O3.5H2O (p.a) em água destilada e aferir para 100ml
em balão volumétrico. Adicionar 1ml desta solução para complexar 2 mg.L-1
de Cloro.
Padronização da solução de H2SO4 0,02N: transferir com auxílio de uma
pipeta volumétrica, 10ml da solução de carbonato de sódio 0,02N para um
elernmeyer. Preparar uma bureta com o agente titulante de ácido sulfúrico
0,02N. Adicionar 4 gotas da solução indicadora de metilorange no elernmeyer
que contém carbonato de sódio e agitar até notar a viragem do indicador
metilorange de amarelo para laranja. Anotar o volume gasto na titulação do
ácido sulfúrico e calcular o fator de correção:

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
103
VtFc=
Vg (16)
em que:
Vt = volume teórico (mL)
Vg = volume gasto na titulação (mL)
2. PROCEDIMENTO ANALÍTICO (amostra)
2.1. DETERMINAÇÃO DA ALCALINIDADE
1. Preencher a bureta com a solução de ácido sulfúrico 0,02N;
2. Colocar 100ml da amostra com pipeta volumétrica em um erlenmeyer e
adicionar de 4 a 5 gostas de solução indicadora de fenolftaleína;
NOTA 1: Não filtrar, diluir ou concentrar, ou alterar a amostra;
NOTA 2: Caso a amostra seja clorada, adicionar algumas gotas de tiossulfato de
sódio;
NOTA 3: A amostra deverá ficar com a coloração rósea após a adição da
fenolftaleína, caso tenha um pH > 8,3 e ficará incolor quando o pH for < 8,3. Para
este último caso não existe alcalinidade à fenolftaleína;
3. Obtendo uma coloração rósea, titular até ficar incolor e anotar o volume
gasto do agente titulante (H2SO4);
4. Adicionar de 4 a 5 gotas da solução de metilorange;
NOTA 4: Caso a amostra tenha um pH > 4,4 ficará com uma coloração amarela e
quando o pH for < 4,4 ficará com uma coloração laranja. Neste último caso, não
existe alcalinidade á metilorange;
5. Obtendo uma coloração amarela, acertar o menisco da bureta, anotar o
ponto de partida da titulação, adicionar o ácido gota a gota agitando a
amostra até o ponto de viragem (coloração laranja) e anotar o volume
do agente titulante (H2SO4) gasto;
6. Anotar o volume total do ácido gasto na titulação (AlcFenolftaleína +
AlcMetilorange);
2.2. CÁLCULO DA CONCENTRAÇÃO

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
104
Alcalinidade à fenolftaleína:
3( / ) 10Alcalinidade mg LCaCO Vg fc (17)
em que:
Vg: volume (ml) de H2SO4 0,02N gasto com a amostra
Fc: fator de correção do H2SO4 0,02N
Alcalinidade total:
3( / ) 10Alcalinidade mg LCaCO Vg fc (18)
em que:
Vg: volume (ml) de H2SO4 0,02N gasto com a amostra
Fc: fator de correção do H2SO4 0,02N
Alcalinidade á metilorange:
Alcalinidade à metilorange (mg/L de CaCO3) = alcalinidade total – alcalinidade à
fenolftaleína
3. REFERÊNCIA
STANDARD METHODS (Apha, 1998)

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
105
POP 17 – FLUORESCÊNCIA MOLECULAR (IF)
1. MÉTODO PARA DETERMINAÇÃO - ESPECTROFLUORÍMETRO
Varredura espectroscópica de fluorescência molecular. Excitação de espécies
atômicas ou moleculares presentes no analito, através de radiações
eletromagnéticas localizadas na região do UV e/ou visível e posterior espectro de
radiações emitidas (quando as espécies retornam ao estado fundamental) em
comprimentos de onda superiores ao valor estabelecido para excitação.
1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Espectrofluorímetro
Bomba de vácuo
Membrana de acetato de celulose Ø 0,45m de porosidade
Vidrarias
Cubeta de quartzo multifacetada
Conjunto kitassato para filtração
Frasco plástico (PEAD, PP) para amostra (20ml)
Reagentes analíticos
Não há uso de soluções para o preparo e quantificação da amostra.
1.2. PROCEDIMENTO ANALÍTICO (Amostra)
1. Filtrar aproximadamente 20ml de amostra em membrana de acetato de
celulose Ø 0,45µm;
2. Transferir a amostra (3ml) para uma cubeta de quartzo multifacetada;
3. Selecionar no espectrofluorímetro a opção desejada: modalidade emissão
(varredura entre 300 e 700nm e comprimento de onda de excitação (Exc) de
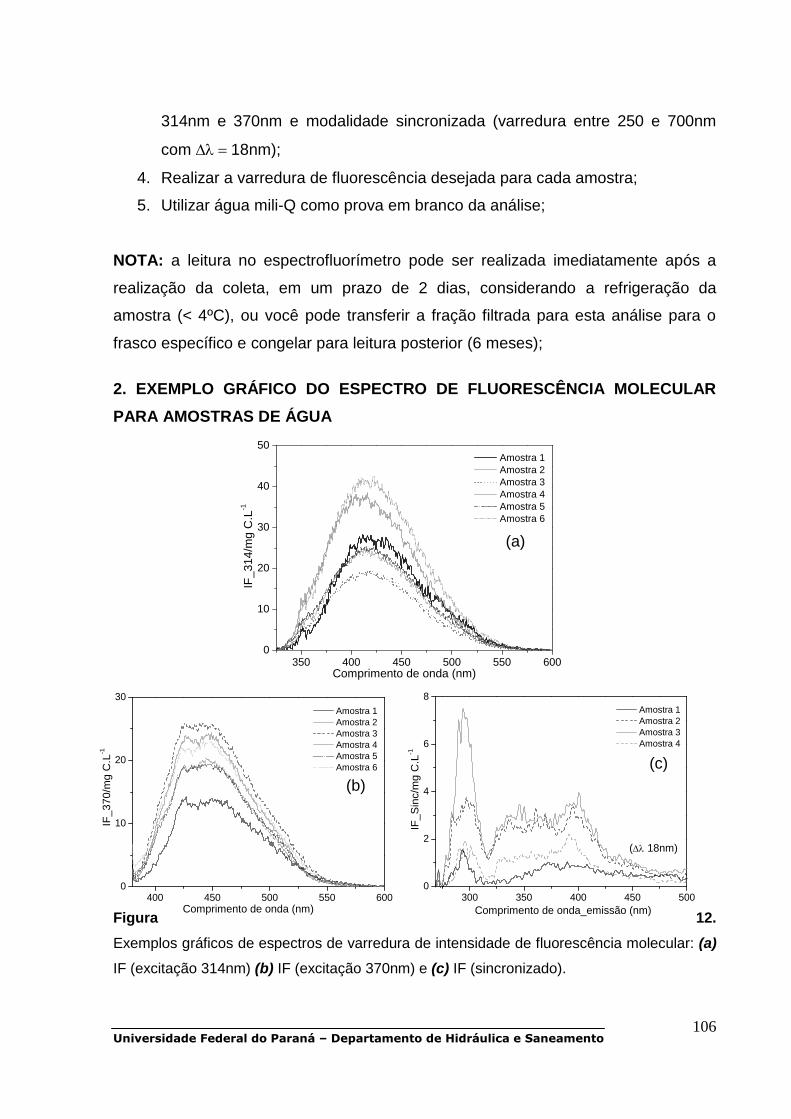
Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
106
350 400 450 500 550 6000
10
20
30
40
50 Amostra 1
Amostra 2
Amostra 3
Amostra 4
Amostra 5
Amostra 6
Comprimento de onda (nm)
IF_
31
4/m
g C
.L-1
400 450 500 550 6000
10
20
30
Amostra 1
Amostra 2
Amostra 3
Amostra 4
Amostra 5
Amostra 6
IF_
37
0/m
g C
.L-1
Comprimento de onda (nm)
300 350 400 450 5000
2
4
6
8
Amostra 1
Amostra 2
Amostra 3
Amostra 4
IF_
Sin
c/m
g C
.L-1
Comprimento de onda_emissão (nm)
( 18nm)
(a)
(b)
(c)
314nm e 370nm e modalidade sincronizada (varredura entre 250 e 700nm
com 18nm);
4. Realizar a varredura de fluorescência desejada para cada amostra;
5. Utilizar água mili-Q como prova em branco da análise;
NOTA: a leitura no espectrofluorímetro pode ser realizada imediatamente após a
realização da coleta, em um prazo de 2 dias, considerando a refrigeração da
amostra (< 4ºC), ou você pode transferir a fração filtrada para esta análise para o
frasco específico e congelar para leitura posterior (6 meses);
2. EXEMPLO GRÁFICO DO ESPECTRO DE FLUORESCÊNCIA MOLECULAR
PARA AMOSTRAS DE ÁGUA
Figura 12.
Exemplos gráficos de espectros de varredura de intensidade de fluorescência molecular: (a)
IF (excitação 314nm) (b) IF (excitação 370nm) e (c) IF (sincronizado).

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
107
POP 18 – COLIFORMES TOTAIS E FECAIS
1. MÉTODO PARA DETERMINAÇÃO – CARTELA COLILERT
O método Colilert consiste na quantificação dos coliformes totais e fecais
presentes em uma dada amostra, através da mistura entre a amostra e o reagente
colilert patenteado, com posterior transferência da solução para uma cartela estéril
(100ml), a qual é selada e mantida incubada a 35±2ºC durante 24h (1º leitura) e 48h
(2º leitura-confirmação). Os resultados são obtidos pela relação de valores positivos
entre os quadrados maiores e menores da cartela, com àqueles verificados na
tabela padrão para o teste colilert.
1.1. EQUIPAMENTOS, VIDRARIAS E REAGENTES
Equipamentos
Seladora para cartelas colilert
Autoclave vertical
Câmara escura equipada de radiação UV
Incubadora termo-regulável (35±2ºC)
Vidrarias
Balão de fundo chato (esterilizado*)
Proveta 100ml (esterilizado*)
* 15 minutos em autoclave, frascos totalmente vedados com tampões (preparados
com gases), papel alumínio e papel kraft (camada dupla)
Reagentes
Reagente específico colilert
1.2. PROCEDIMENTO ANALÍTICO (Amostra)
Primeiramente, é importante avaliar a faixa possível de quantificação para a
análise de coliformes na amostra de interesse, de modo a verificar se existirá ou não

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
108
a necessidade de diluição da mesma. O método utiliza 100ml de amostra, mas em
muitos casos volumes menores são utilizados, devido à grande quantidade de
coliformes presentes. Para estes casos, volumes de 1ml ou 0,1ml da amostra, por
exemplo, são transferidos para 100ml de água destilada, previamente esterilizada, e
o fator de diluição anotado para correção do resultado final. Dessa forma, seguir as
etapas abaixo:
1. Após coleta da amostra, transferir 100ml desta para um balão de fundo chato
estéril ou o volume correspondente para a diluição pretendida, de modo que o
volume final seja de 100ml;
2. Em cada amostra, adicionar uma cartela do reagente colilert e agitar até
dissolução completa;
3. Transferir os 100ml para uma cartela colilert estéril, colocar sobre o suporte
da seladora e selar a mesma;
4. Manter a cartela em incubadora termo-regulável a 35±2ºC;
5. Após 24h na incubadora, anotar os valores positivos nos quadrados grandes
(49 espaços) e pequenos (48 espaços). Os valores positivos são aqueles nos
quais uma coloração amarela forte se desenvolveu;
6. O mesmo procedimento deve ser realizado observando-se as cartelas em
uma câmara escura equipada de luz UV, de modo que, para este caso, os
quadrados grandes e pequenos a serem anotados são aqueles que
desenvolverem uma luminescência azul característica;
7. Os valores anotados pelo item 5 são referentes aos coliformes totais e os
resultados do item 6, os coliformes fecais;
8. Repetir o procedimento dos itens 6 e 7 após 48h para confirmação dos
resultados;
2.1. CÁLCULO DO NÚMERO MAIS PROVÁVEL (NMP/100ml)
Os resultados são obtidos a partir da cartela padrão do método que
correlaciona os valores observados nos quadrados grandes com aqueles
observados nos quadrados pequenos.

Universidade Federal do Paraná – Departamento de Hidráulica e Saneamento
109
Por exemplo, em uma análise que utilizou 100ml da amostra foram
observados 10 quadrados positivos grandes com 15 quadrados positivos pequenos
para coliformes totais e 5 quadrados positivos grandes com 3 quadrados positivos
pequenos para coliformes fecais. Pela tabela, os valores observados são
respectivamente 28 NMP/100ml e 8,2 NMP/100ml. Caso um volume de apenas
0,1ml de amostra tenha sido utilizado e as mesmas observações obtidas, estes
valores acima devem ser multiplicados pelo fator de diluição de 1000x,
correspondendo a 28 000 NMP/100ml para coliformes totais e 8 200 NMP/100ml
para coliformes fecais.
3. REFERÊNCIA
Colilert IDEXX Quanti Tray/200





























