Embed Size (px)
Citation preview

Knock-down of PQBP1 impairs anxiety-relatedcognition in mouse
Hikaru Ito1, Natsue Yoshimura1, Masaru Kurosawa2, Shunsuke Ishii3, Nobuyuki Nukina2
and Hitoshi Okazawa1,�
1Department of Neuropathology, Medical Research Institute and 21st Century Center of Excellence Program (COE)
for Brain Integration and Its Disorders, Tokyo Medical and Dental University, Tokyo 113-8510, Japan, 2Laboratory for
Structural Neuropathology, RIKEN Brain Science Institute, Saitama 351-0198, Japan and 3Laboratory for Molecular
Genetics, RIKEN Tsukuba Institute, Ibaraki 305-0074, Japan
Received May 7, 2009; Revised and Accepted August 4, 2009
PQBP1 (polyglutamine tract-binding protein 1) is a causative gene for a relatively frequent X-linked syndromicand non-syndromic mental retardation (MR). To analyze behavioral abnormalities of these patients from molecu-lar basis, we developed a knock-down (KD) mouse model. The KD mice possess a transgene expressing 498 bpdouble-strand RNA that is endogenously cleaved to siRNA suppressing PQBP1 efficiently. After confirming thatPQBP1 is selectively suppressed to nearly 50% of the control mice, we performed behavioral analyses of PQBP1-KD mice. The KD mice possessed normal ability in ordinary memory tests including water-maze test, whereasthey showed abnormal anxiety-related behavior in light/dark exploration test and open-field test and showedobvious declines of anxiety-related cognition in the repetitive elevated plus maze or novel object recognitiontest. Correspondingly, we found c-fos upregulation and histone H3 acetylation after behavior testswere declined in neurons of amygdala, prefrontal cortex and hippocampus. Furthermore, we foundthat 4-phenylbutyric acid, an HDAC inhibitor, efficiently improved expression of these genes and rescued theabnormal phenotypes in adult PQBP1-KD mice. These results suggested that PQBP1 dysfunction inregulating gene expression might underlie the abnormal behavior and cognition of PQBP1-KD mice and thatthe recovery of expression of such PQBP1 target genes might improve the symptoms in adult patients.
INTRODUCTION
Mental retardation (MR) is a highly frequent developmental dis-order that affects nearly 1% of population in advanced countries.The causes are variable, although monogenic MR is a majorgroup of MR in addition to multigenic and environmental factor-induced MR (1). The dominance of male in MR is largely recog-nized (1), and the relevance between X-linked MR genes andevolution of human intelligence is also suspected (2). The fre-quency of MR genes in X chromosome is obviously higherthan that in the other chromosomes, which might be used forevolutional selection of intelligence through males (2–4).
The most frequent X-linked MR is fragile-X syndrome thataffects approximately one in 4000 males and one in 7000females. The following group includes Rett syndrome thataffects one in 6000–10 000 males. Recently, mutations inPQBP1 (polyglutamine tract-binding protein 1) gene located
at Xp11.23 were shown to be causative for syndromic andnon-syndromic MR including Renpenning syndrome,Golabi–Ito–Hall and Sutherland–Haan syndromes (5–9).The frequency of all the PQBP1-linked MR patients seemsto be rather high in western countries and might be equivalentto that of Rett syndrome (9).
PQBP1 was isolated originally as a binding protein to thepolyglutamine (polyQ) tract sequence (10). PQBP1 is impli-cated in neurodegenerative pathologies because it interactswith mutant ataxin-1 (Atx1), the causative gene product of spi-nocerebellar ataxia type-1 (SCA1), and with huntingtin (Htt),the causative gene product of Huntington’s disease (HD).PQBP1 interacts with phosphorylation sites of the C-terminalof RNA polymerase II (Pol II) (11), the core molecule of tran-scription, and with several other proteins such as NpwBP/SIPP/WBP11 (12,13) and U5-15kD (14,15) that are involvedin RNA splicing. PQBP1 and these binding partners are
�To whom correspondence should be addressed. Email: [email protected]
# The Author 2009. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2009, Vol. 18, No. 22 4239–4254doi:10.1093/hmg/ddp378Advance Access published on August 6, 2009
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

identified in the splicing complex also by recent proteome ana-lyses (16,17). Interaction between PQBP1 and mutant polyQproteins might induce dysfunction of PQBP1 in transcriptioncoupled with splicing (11,18). The similar dysfunction in tran-scription/splicing might occur in PQBP1-linked MR.
In this study, we generated a PQBP1-knock-down (KD)mouse model, to investigate the phenotype induced by PQBP1deficiency and the molecular link between phenotype andPQBP1 mutations. We found impairment of anxiety-relatedcognition and memory, and detected abnormal expression ofmemory-related genes that could be induced by dysfunction ofPQBP1-mediated transcription/splicing in this mouse model.Furthermore, we found that an histone deacetylase (HDAC)inhibitor, 4-phenylbutyric acid (PBA), that generally upregu-lates transcription and ameliorates neurodegeneration inanimal models, rescued the abnormal behavior and memory ofthe PQBP1-KD mice.
RESULT
Generation of PQBP1-KD mouse
In this study, we employed the pDECAP system developed byIshii and colleagues (19) to knock-down PQBP1. ThepDECAP plasmid expresses double-strand RNA that isdegraded by endogenous Dicer to siRNA suppressing thetarget mRNA (19). We made three types of plasmids by sub-cloning three different partial PQBP1 cDNAs into pDECAPand compared their suppression efficiencies in P19 cells (Sup-plementary Material, Fig. S1). The most effective suppressionwas obtained by the fragment from 295 to 792 bp, thus wegenerated transgenic mice with pDECAP-PQBP1 (295–792)(Fig. 1A and B).
We obtained two lines of mice with this construct. Theexpression levels of PQBP1 in the two lines were not statisti-cally different (data not shown). Using two pups from line #1and another two from line #2, we tested whether PQBP1 isselectively suppressed in pDECAP-PQBP1 transgenic micein relevance to other control proteins with cerebral cortex iso-lated from pDECAP-PQBP1 transgenic mice and littermatewild-type (WT) mice by western blot analyses. PQBP1expression in the pDECAP-PQBP1 transgenic mice was�50% of that in WT mice (Fig. 1C). There was no apparentchange in the expression levels of GAPDH, a-Tubulin,b-Actin, GFAP and MAP2. In the nucleotide BLAST searchat NCBI (http://blast.ncbi.nlm.nih.gov), no homologousgene sequence was found with the PQBP1 fragment from295 to 792 bp.
PQBP1-KD mice have normal motor function
First, we assessed spontaneous activity and motor function inPQBP1-KD mice. Spontaneous activity monitored under 12 hlight/dark cycle was not changed in PQBP1-KD mice at theage of 1.5, 3 and 6 months (Supplementary Material,Fig. S2A). We also tested their motor function using the stan-dard paradigm for the accelerating rotating rod (rotarod)apparatus, but found no differences between PQBP1-KD andWT mice in the rotarod test (Supplementary Material,Fig. S2B).
PQBP1-KD mice showed abnormal anxiety-relatedbehavior
PQBP1-KD mice behave almost normally in ordinary con-ditions. However, we noticed that they sometimes stayed inthe center of cage. Therefore, we used three behavioral para-digms, the open-field test, the light–dark exploration testand the elevated plus maze test, to evaluate anxiety-related be-havior in PQBP1-KD and WT mice. We performed theseexperiments at three different ages: 1.5 (the earliest timepoint for behavior analysis due to the technical and ethicalregulations), 3 and 6 months.
First, total locomotion activity and anxiety-like responses ofPQBP1-KD and WT mice were measured in a novel open field.In comparison to WT mice, KD mice spent a significantlygreater percentage of time in the center square where anxietyis triggered (P , 0.05) (Fig. 2A). No difference was observedin total locomotion activity between the genotypes (Fig. 2A).Anxiety-related behavior was also tested in the light–darkexploration test as described previously (20). In the light–dark exploration test, PQBP1-KD mice spent more time inlight box when compared with WT mice (P , 0.01) (Fig. 2B).Although the total locomotion distance in the light and darkbox did not differ between genotypes, PQBP1-KD mice tra-veled significantly farther in the light box (P , 0.05)(Fig. 2B). The first latency to enter the dark box and thenumber of transitions between the light and dark boxes didnot differ between the genotypes (data not shown). These abnor-mal behaviors in the open-field and light–dark exploration testswere detected at 1.5 months and resisted at least to 6 months(Fig. 2A and B), suggesting that these phenotypes started inpups. Therefore, neurochemical abnormalities described later(Figs 4–8) might occur during development, whereas, in thisstudy, we focus on the analysis of adult PQBP1-KD mice(1.5–6 months). Developmental analysis of behavior ofnewborn pups will be reported elsewhere in the future.
PQBP1-KD mice showed abnormal anxiety-relatedcognition
We next performed the repetitive elevated plus maze test toevaluate the anxiety-related behavior and memory (21–24).The mice were placed on the same maze twice with a 3 h inter-val. In the first trial, PQBP1-KD mice stayed for a longer timeon the open arms than littermate (WT) mice (Fig. 3A). In thesecond trial, the difference became larger. WT mice hardlystayed on the open arms where the anxiety was induced,whereas PQBP1-KD mice stayed on the open arms for ahalf time of the first trial (Fig. 3A). The difference was statisti-cally significant (Student’s or Welch’s test, P , 0.01). Inaddition, PQBP1-KD mice were more active on the openarms than the WT mice (Fig. 3A1). These results were simi-larly observed from 1.5 to 6 months (Fig. 3A1). In the repeti-tive elevated plus maze test, the improvement of ‘time on openarm’ from trial 1 to trial 2 was not significantly differentbetween WT and PQBP1-KD mice, in contrast to theelongation of ‘time on open arm’ at trial 1 (Fig. 3A2). Itsuggests that abnormal cognition rather than memory inPQBP1-KD mice was unraveled by the elevated plus mazetest.
4240 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

We also examined PQBP1-KD mice with novel object recog-nition test (Fig. 3B), a non-aversive memory task that relies onthe natural exploratory behavior of mice. In this task, mice wereexposed to two identical objects for 10 min during the trainingsession (trials 1 and 2). We then tested their cognition andmemory by replacing one of the objects in trials 3 and 4(Fig. 3B1, 3B2, upper scheme). PQBP1-KD mice spent moretime for exploring in trial 2, suggesting that their explorationis not dependent on the novel object recognition (Fig. 3B1).Since reduction of the exploratory time from trial 1 to trial 2was significantly smaller in PQBP1-KD mice than in WTmice (Supplementary Material, Fig. S3), PQBP1-KD micemight not learn or remember the object in trial 1. In trials 3and 4, the exploring time in Area B was increased from trial 2in WT mice but not in PQBP1-KD mice (Fig. 3B2), suggestingthat KD mice did not distinguish novel and old objects. The ratiobetween trials 2 and 3 was significantly different (Fig. 3B). Thesimilar difference was observed between trials 2 and 4 (Fig. 3B).
Abnormal anxiety-related cognition of PQBP1-KD mice wasobserved in the repetitive elevated plus maze test (Fig. 3A) andthe extent of changes was almost similar in trial 1 and trial2. Meanwhile, in the novel object recognition test, exploringtime to the old object of PQBP1-KD mice was elongated intrial 2, although it was normal in trial 1 (Fig. 3B1). Moreover,
the normal increase of exploring time from trials 2 to 3 orfrom trials 2 to 4 was not found in PQBP1-KD mice(Fig. 3B2). For this increase of exploring time, WT mice remem-bered the old object in trial 1/2 and compared it with a new one intrial 3/4. Therefore, PQBP1 hypofunction might also impair akind of memory related to anxiety.
We evaluated PQBP1-KD mice with the fear-conditioningtest, in which mice were conditioned to a foot shock contextand an auditory cue (the conditioned stimulus) and tested24 h later. There were no significant differences in the fear-conditioning test between the genotypes, althoughPQBP1-KD mice had a tendency to present less freezingresponses compared with WT mice in the contextual testand cued test (Fig. 3C).
In Morris water maze test, PQBP1-KD mice did not showsignificant reduction of the ordinary spatial memory (Sup-plementary Material, Fig. S4), although PQBP1-KD micetended to show lower values than WT mice.
Neurons of PQBP1-KD mice show reduction of c-fosand hypoacetylation of histone H3
We next intended to analyze the molecular mechanismsunderlying the behavioral and cognitive abnormalities in
Figure 1. Generation of PQBP1-KD mice and reduction of PQBP1. (A) The transgene construction. To express double-strand RNA with RNA polymeraseII-mediated transcription, the pDECAP contains cytomegalovirus (CMV) promoter, ribozyme cassette to cut off the cap structure of the transcribed RNA(70), and MAZ site for RNA polymerase II pausing (71). There is no poly(A) additional signal in the construct. Primer sites for checking mice are also indicated.(B) PCR products of mouse genomic DNA. Left and right lanes, a 100 bp ladder DNA marker; Tg, transgenic mouse; WT, wild-type mouse. (C) Western blotanalyses of cortical protein extracts (10 mg) from pDECAP-PQBP1 transgenic (PQBP1-KD) mice and control littermate WT mice at the day of birth (P0)using anti-PQBP1, anti-GAPDH, anti-a-Tubulin, anti-b-Actin, anti-GFAP and anti-MAP2 antibodies. We observed reduction of PQBP1 protein but not theother proteins in PQBP1-KD mice. Right bar graphs show quantitative analysis of western blots in four PQBP1-KD mice and four WT mice. Data are presentedas mean þ SD. Statistical analyses by Student’s t-test: �P , 0.01 versus WT mice.
Human Molecular Genetics, 2009, Vol. 18, No. 22 4241
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

PQBP1-KD mice. First, we examined expression of c-fos animmediate early gene that has been used as a marker of neur-onal activity in anxiety-related behavior and memory (25–27).At 1.5, 3 and 6 months of age, brain samples prepared beforeand 2 h after behavior tests (Fig. 4). We focused on amygdala,prefrontal cortex and hippocampus, because these regions playcritical roles in cognition and emotional memory (reviewed in28). Western blot analysis revealed a clear upregulation ofc-fos protein in response to the behavior tests (repetitive elev-ated plus maze test) in WT mice (age-matched littermates) butnot in PQBP1-KD mice (Fig. 4A). A significant reduction ofc-fos after behavior tests was observed in all the tested brainregions of PQBP1-KD mice (Fig. 4B). In addition, the levelsof c-fos were similarly reduced at all time points from 1.5 to6 months (Fig. 4B). Consistently, immunohistochemistry ofPQBP1-KD mice revealed significant reduction of c-fosprotein in these brain areas after behavior tests (Fig. 4C).Co-staining with neuronal marker NeuN confirmed c-fosreduction occurred in neurons (Fig. 4D and E).
We employed another marker of the memory-related tran-scriptional upregulation, acetylated histone H3, which hasbeen suggested to reflect anxiety-related behavior andmemory (29–33). We compared the signal intensities ofacetylated H3 between PQBP1-KD and age-matched litter-mate (WT) mice in western blot analyses and immunohisto-chemistry (Fig. 5). In this case, acetylated H3 was notsignificantly upregulated by behavior tests in WT mice(Fig. 5A). However, the levels of acetylated H3 were lowerin PQBP1-KD mice both before and after behavior tests(Fig. 5A). The levels of acetylated H3 were similarly
reduced at all time points from 1.5 to 6 months in cortex, hip-pocampus and amygdala when compared with those in WTmice (P , 0.05) (Fig. 5B, Supplementary Material, Fig. S5).Immunohistochemistry performed at 3–6 months of age alsosupported the decrease of acetylated H3 in prefrontal cortex,hippocampus and amygdala (Fig. 5C). Quantitative analysesof the immunostain signals of acetylated histone H3 in NeuN-positive neurons (Fig. 5D) confirmed that acetylated H3 werereduced in neurons of these brain regions (Fig. 5E).
PBA rescues impairment of anxiety-related cognitionin PQBP1-KD mice
Finally, we tested whether transcriptional up-regulation canrescue the phenotype of PQBP1-KD mice at 7 months ofage (Figs 6 and 7). We employed a HDAC inhibitor, PBA,which is known to increase acetylation of histone H3 andH4 in the mouse brain (34). PQBP1-KD and aged-matched lit-termate (WT) mice received daily intra-peritoneal injectionsof PBA or vehicle (phosphate buffered saline, PBS) for4 weeks from the age of 7 months (Fig. 6A). As expected,administration of PBA for 4 weeks increased the level ofacetylated H3 both in the PQBP1-KD and WT mouse brains(Fig. 6B). The level of acetylated H3 in PBA-treatedKD-mice was normalized to that of WT mice without PBAtreatment (Fig. 6B). The similar recovery was observed inimmunohistochemistry (Fig. 6C). Interestingly, PBA-treatedWT mice showed a higher level of acetylated H3 thanthe ordinary level of the PBA-non-treated WT mice (Fig. 6Band C).
Figure 2. PQBP1-KD mice show abnormal anxiety-related behavior. Open-field behavior and light–dark exploration tests were performed with PQBP1-KD andcontrol WT mice at 1.5, 3 and 6 months. (A) Open-field behavior test. Total distance (upper) and percentage of time spent in the center (lower) in a novel openfield during the first 15 min are shown. Trace of movement was shown at right side. In total distance, the variance at 1.5, 3 and 6 months are F(1,7)¼1.130,F(1,21)¼1.905 and F(1,19)¼0.765, respectively. Student’s t-test was employed for comparison, while no significant difference was observed. In %time incenter, the variance at 1.5, 3 and 6 months are F(1,7)¼0.664, F(1,21)¼5.566 and F(1,19)¼1.656, respectively. For statistical analysis, Student’s t-test wasused at 1.5 and 6 months and Welch’s t-test was used at 3 months. PQBP1-KD mice spent a significantly greater percentage of time in the center. �P ,
0.05. (B) In the light–dark exploration test, PQBP1-KD mice spent significantly more time in the light box (upper). PQBP1-KD mice traveled also significantlyfarther in the light box (lower). The variance was not different among groups. �P , 0.05 in Student’s t-test, ��P , 0.01 in Student’s t-test.
4242 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

Figure 3. PQBP1-KD mice show abnormal anxiety-related cognition and memory. (A1) In the repetitive elevated plus maze test, the time spent on open arms(upper), time spent on closed arms (middle) and total distance of locomotion on the elevated plus maze (lower) were analyzed with PQBP1-KD and littermate(WT) mice (upper) at the age of 1.5, 3 and 6 months. In trials 1 and 2, PQBP1-KD mice spent significantly longer time on open arms (upper). �P , 0.05, ��P ,0.01 in Student’s t-test or Welch’s t-test. (A2) Age-dependent changes in the repetitive elevated plus maze test are shown with values in trial 1 (left panels) andthe improvement of values from trial 1 to trial 2 (right panels). �P , 0.05 in Student’s t-test or Welch’s t-test. (B1) The novel object recognition test was per-formed according to the scheme. Trials 1 and 2 were for memory consolidation, and trials 3 and 4 were for recognition of novel objects (upper scheme). Totalexploratory time in each trial is shown at three different ages. No significant difference was observed between genotypes in trial 1, whereas total exploratory timein trial 2 was significantly longer in PQBP1-KD mice at 3 and 6 months. �P , 0.05 in Student’s t-test or Welch’s t-test. (B2) Percentage exploring to a newobject (B/AþB) is shown. Trials 1 and 2 are for learning of old objects. The increase of exploring to a new object from trial 2 to trail 3 or from trial 2 totrial 4 was observed in WT mice but not in PQBP1-KD mice. In trial 3, the relative exploring to a new object was lower in PQBP1-KD mice. The ratios oftrial 2 or 3 to trial 1 were also abnormal in multiple aspects. �P , 0.05 in Student’s t-test or Welch’s t-test. (C) In fear conditioning tests, PQBP1-KD micetended to show less freezing responses in the contextual test and cued test, but statistical difference was not confirmed in Student’s t-test.
Human Molecular Genetics, 2009, Vol. 18, No. 22 4243
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

Figure 4. Upregulation of c-fos in response to learning task was impaired in PQBP1-KD mice. (A) Expression levels of c-fos protein before and after behaviortests were examined by western blot analyses in PQBP1-KD and WT (age-matched littermates) mice at the age of 1.5, 3 and 6 months. Quantitative analysis ofc-fos band intensities (standardized by GAPDH) is shown below. Mean þ SD. Statistical analysis was performed with F-test followed by Student’s t-test: �P ,
0.01. (B) Western blot analyses were performed 2 h after behavior tests with protein extracts from cortex, hippocampus and amygdala using anti-c-fos and anti-GAPDH antibodies at the age of 1.5, 3 and 6 months (upper panels). Quantitative analysis of western blots is shown below (lower panels). Expression level ofc-fos protein is expressed as a ratio to GAPDH (lower). Data are presented as mean þ SD of the results from four animals. Statistical analyses by Student’s t-test:�P , 0.05 versus WT mice. (C) Immunofluorescence images of c-fos at a low magnification show reduction of c-fos in various brain regions of PQBP1-KD andWT mice. (D) NeuN-positive neurons showed a lower level of c-fos in PQBP1-KD mice than in WT mice. Neuorns were stained by antibodies against c-fos(green) and a neuron marker, NeuN (red). Scale bar: 50 mm. (E) Signal intensity of c-fos was quantified in NeuN-labeled neurons in KD or WT mice. Student’st-test indicated a significant difference of c-fos levels between KD and WT mouse brains at each region (P-CTX, Hip, Amyg) (�P , 0.01). Mean þ SEM. Scalebars, 200 mm.
4244 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022
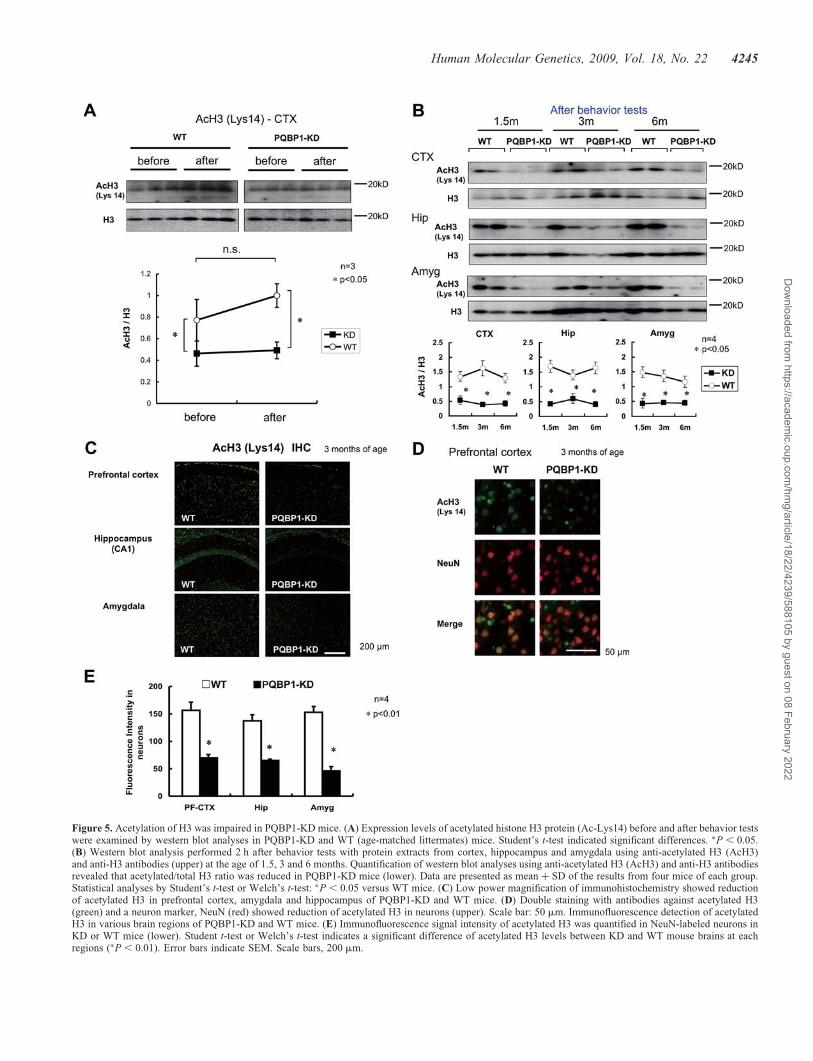
Figure 5. Acetylation of H3 was impaired in PQBP1-KD mice. (A) Expression levels of acetylated histone H3 protein (Ac-Lys14) before and after behavior testswere examined by western blot analyses in PQBP1-KD and WT (age-matched littermates) mice. Student’s t-test indicated significant differences. �P , 0.05.(B) Western blot analysis performed 2 h after behavior tests with protein extracts from cortex, hippocampus and amygdala using anti-acetylated H3 (AcH3)and anti-H3 antibodies (upper) at the age of 1.5, 3 and 6 months. Quantification of western blot analyses using anti-acetylated H3 (AcH3) and anti-H3 antibodiesrevealed that acetylated/total H3 ratio was reduced in PQBP1-KD mice (lower). Data are presented as mean þ SD of the results from four mice of each group.Statistical analyses by Student’s t-test or Welch’s t-test: �P , 0.05 versus WT mice. (C) Low power magnification of immunohistochemistry showed reductionof acetylated H3 in prefrontal cortex, amygdala and hippocampus of PQBP1-KD and WT mice. (D) Double staining with antibodies against acetylated H3(green) and a neuron marker, NeuN (red) showed reduction of acetylated H3 in neurons (upper). Scale bar: 50 mm. Immunofluorescence detection of acetylatedH3 in various brain regions of PQBP1-KD and WT mice. (E) Immunofluorescence signal intensity of acetylated H3 was quantified in NeuN-labeled neurons inKD or WT mice (lower). Student t-test or Welch’s t-test indicates a significant difference of acetylated H3 levels between KD and WT mouse brains at eachregions (�P , 0.01). Error bars indicate SEM. Scale bars, 200 mm.
Human Molecular Genetics, 2009, Vol. 18, No. 22 4245
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

The recovery by PBA was tested also with behavior tests. Inthe novel open-field test, PBA-treated PQBP1-KD mice spentless time in the center square than PBS-treated PQBP1-KDmice (Fig. 7A), indicating that PBA improved theanxiety-reduced behavior. In the light–dark exploration test,PBA-treated PQBP1-KD mice spent less time in light boxwhen compared with PBS-treated PQBP1-KD mice (P ,0.01) (Fig. 7B), indicating the similar improvement ofanxiety deficiency. These results indicate that HDAC inhibitor(PBA) improved the abnormal anxiety-related behaviors ofPQBP1-KD mice. Despite of the super normal level ofacetylated H3 (Fig. 6B–D), the anxiety-related behavior wasnot altered in PBA-injected WT mice (Fig. 7A and B),suggesting that the transcriptional basis for behavior wassaturated.
We further performed the repetitive elevated plus maze to testthe effect of PBA. When compared with PBS-treatedPQBP1-KD mice, PBA-treated PQBP1-KD mice spent lesstime in the open arms (P , 0.01) (Fig. 7C). In this case, notonly the time at trial 1 but also the time reduction from trial 1to 2 was also improved (Fig. 7C). Thus, chronic injection ofPBA recovered anxiety-related cognition in PQBP1-KD mice.
Finally, we confirmed that the HDAC inhibitor also rescuedthe response of c-fos protein to the behavior test (repetitive elev-ated plus maze test) in PQBP1-KD mice (Fig. 7D).
Expression of other anxiety-related genesin PQBP1-KD mice
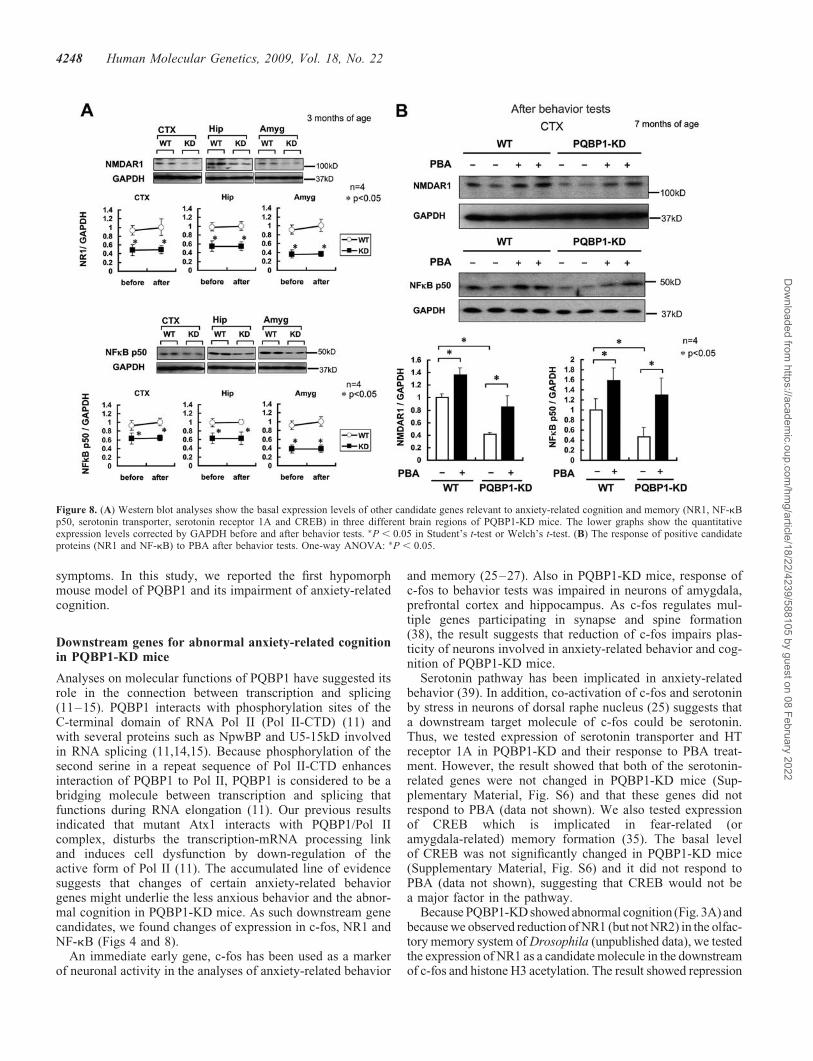
Furthermore, we examined the possibility that expressionchanges of other genes underlie the abnormal anxiety-relatedcognition in PQBP1-KD mice. Serotonin pathway has beenimplicated in anxiety-related behavior. Thus, we decided toanalyze expression of serotonin transporter and HT receptor1A. Cyclic AMP-responsive element binding protein(CREB) was another candidate, because it is implicated infear-related (or amygdala-related) memory formation (35).Because PQBP1-KD showed abnormal cognition (Fig. 3A)and because we observed reduction of NMDA receptorsubunit 1 (NR1) (but not NR2) in the olfactory memorysystem of Drosophila (unpublished data), we also includedNR1 as a candidate. Since NF-kB is implicated inanxiety-related behavior (36) and is known to regulate NR1gene expression (37), we added it to the candidates.
Figure 6. PBA rescues acetylated histone H3 in PQBP1-KD mice. (A) Intraperitoneal injection of PBA was performed every day for 4 weeks with PQBP1-KDand WT mice from the age of 7 months, and immunohistochemistry of acetylated H3 was conducted after behavior test. (B) Upper panels show western blotanalyses of protein extracts from prefrontal cortex of PQBP1-KD and WT mice (n ¼ 3) after treatment of PBA (PBAþ) or PBS (PBA2). Quantitative analysisof the ratios of band intensities between acetylated H3 and total H3. Data are presented as mean þ SD of the results from three animals of each group. One-wayANOVA: �P , 0.05 versus WT mice. (C) Prefrontal cortex of PQBP1-KD and WT mice after PBA treatment were stained by anti-acetylated H3 antibody(green). Scale bar: 200 mm. Immunofluorescent signal intensities of acetylated H3 were measured in prefrontal cortex (lower). Data are presented asmean þ SEM. One-way ANOVA: �P , 0.01 versus WT mice.
4246 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

Among these candidates: serotonin transporter, HT receptor1A, NMDA receptor NR1 subunit, NF-kB p50 and CREB,western blot analyses showed reduction of NF-kB and NR1in the cortex, hippocampus and amygdala of PQBP1-KDmice (Fig. 8A). The reduced level was not significantly differ-ent before and after behavior tests. Serotonin transporter, HTreceptor 1A and CREB did not show such reduction (Sup-plementary Material, Fig. S6). PBA partially recoveredexpression of NR1 and NF-kB in PQBP1-KD mice near tothe levels of non-treated control mice (Fig. 8B).
DISCUSSION
PQBP1 hypofunction induces abnormal anxiety-relatedcognition
Mutations identified so far in human PQBP1-linked MRpatients lead to total or partial deletion of the C-terminaldomain of the PQBP1 protein (5–7). In addition, presumablydue to the RNA decay of the truncated mRNA, the level ofPQBP1 seems to be reduced substantially (5). Therefore,hypofunction of PQBP1 is strongly suggested to cause MR
Figure 7. PBA rescues abnormal anxiety-related behavior, cognition and memory in PQBP1-KD mice. Behaviors in the open field, light–dark exploration, noveland repetitive elevated plus maze test are shown for PBS-treated WT (n ¼ 6), PBA-treated WT (n ¼ 7), PBS-treated PQBP1-KD (n ¼ 6) and PBA-treatedPQBP1-KD (n ¼ 7) mice. (A) The time spent in the center of the open field was quantified for 15 min, and the ratio to the total time was calculated.One-way ANOVA with post hoc Bonferroni/Dunn test: �P , 0.05. (B) Time spent in the light box of light–dark exploration test (10 min) is shown.One-way ANOVA with post hoc Bonferroni/Dunn test: ��P , 0.01. (C) After 4 weeks of treatment with PBA or PBS, the repetitive elevated plus maze testwas performed with PQBP1-KD and WT mice, and the time spent on open arms was analyzed. PBA recovered the abnormal anxiety-related memory. Allresults are presented as mean þ SEM. Statistical analyses by one-way ANOVA with post hoc Bonferroni/Dunn test: �P , 0.05, ��P , 0.01. (D) Westernblot analyses with protein extracts from prefrontal cortex of PQBP1-KD mice after treatment of PBA or PBS (upper). Quantitative analyses of westernblot results are shown as the ratio to GAPDH (lower). Data are presented as mean þ SD of the results from four animals. One-way ANOVA with post hocBonferroni/Dunn test: �P , 0.05.
Human Molecular Genetics, 2009, Vol. 18, No. 22 4247
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022
symptoms. In this study, we reported the first hypomorphmouse model of PQBP1 and its impairment of anxiety-relatedcognition.
Downstream genes for abnormal anxiety-related cognitionin PQBP1-KD mice
Analyses on molecular functions of PQBP1 have suggested itsrole in the connection between transcription and splicing(11–15). PQBP1 interacts with phosphorylation sites of theC-terminal domain of RNA Pol II (Pol II-CTD) (11) andwith several proteins such as NpwBP and U5-15kD involvedin RNA splicing (11,14,15). Because phosphorylation of thesecond serine in a repeat sequence of Pol II-CTD enhancesinteraction of PQBP1 to Pol II, PQBP1 is considered to be abridging molecule between transcription and splicing thatfunctions during RNA elongation (11). Our previous resultsindicated that mutant Atx1 interacts with PQBP1/Pol IIcomplex, disturbs the transcription-mRNA processing linkand induces cell dysfunction by down-regulation of theactive form of Pol II (11). The accumulated line of evidencesuggests that changes of certain anxiety-related behaviorgenes might underlie the less anxious behavior and the abnor-mal cognition in PQBP1-KD mice. As such downstream genecandidates, we found changes of expression in c-fos, NR1 andNF-kB (Figs 4 and 8).
An immediate early gene, c-fos has been used as a markerof neuronal activity in the analyses of anxiety-related behavior
and memory (25–27). Also in PQBP1-KD mice, response ofc-fos to behavior tests was impaired in neurons of amygdala,prefrontal cortex and hippocampus. As c-fos regulates mul-tiple genes participating in synapse and spine formation(38), the result suggests that reduction of c-fos impairs plas-ticity of neurons involved in anxiety-related behavior and cog-nition of PQBP1-KD mice.
Serotonin pathway has been implicated in anxiety-relatedbehavior (39). In addition, co-activation of c-fos and serotoninby stress in neurons of dorsal raphe nucleus (25) suggests thata downstream target molecule of c-fos could be serotonin.Thus, we tested expression of serotonin transporter and HTreceptor 1A in PQBP1-KD and their response to PBA treat-ment. However, the result showed that both of the serotonin-related genes were not changed in PQBP1-KD mice (Sup-plementary Material, Fig. S6) and that these genes did notrespond to PBA (data not shown). We also tested expressionof CREB which is implicated in fear-related (oramygdala-related) memory formation (35). The basal levelof CREB was not significantly changed in PQBP1-KD mice(Supplementary Material, Fig. S6) and it did not respond toPBA (data not shown), suggesting that CREB would not bea major factor in the pathway.
Because PQBP1-KD showed abnormal cognition (Fig. 3A) andbecause we observed reduction of NR1 (but not NR2) in the olfac-tory memory system of Drosophila (unpublished data), we testedthe expression of NR1 as a candidate molecule in the downstreamof c-fos and histone H3 acetylation. The result showed repression
Figure 8. (A) Western blot analyses show the basal expression levels of other candidate genes relevant to anxiety-related cognition and memory (NR1, NF-kBp50, serotonin transporter, serotonin receptor 1A and CREB) in three different brain regions of PQBP1-KD mice. The lower graphs show the quantitativeexpression levels corrected by GAPDH before and after behavior tests. �P , 0.05 in Student’s t-test or Welch’s t-test. (B) The response of positive candidateproteins (NR1 and NF-kB) to PBA after behavior tests. One-way ANOVA: �P , 0.05.
4248 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

of NR1 in PQBP1-KD mice and its recovery by PBA (Fig. 8) inconsistence with the results of behavior tests. As an AP1 site forc-fos binding exists in the upstream of mouse and human NR1genes (http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=frnrec&part=ch5#ch5.r45), c-fos might upregulate NR1 after be-havior tests. Since NR1 expression was reported to be normal inconditional knockout mice of c-fos (40), c-fos seems to regulateonly inducible but not basal gene expression of NR1. Mean-while, hypoacetylation of histone H3 might cause the initialreduction of NR1 before the behavior tests.
On the other hand, histone H3 hypoacetlylation couldinduce reduction of NR1 in collaboration with other transcrip-tion factors like NF-kB that is known to regulate NR1 geneexpression (37). Consistently, we observed mild reduction ofNF-kB p50 subunit in PQBP1-KD mice (Fig. 8A). TheNF-kB p50 subunit KO mice showed reduced anxiety-relatedbehavior (41) similar to PQBP1-KD mice. NMDA receptor isa heterodimer composed of NR1 and NR2. Although the directrelationship between NR1 and anxiety has not been reportedso far, NR2A KO mice exhibited decreased anxiety-related be-havior (42). These notions seem to support our hypothesis.
Connection from PQBP1-KD to anxiety-related genes
Furthermore, we found reduced acetylation of histone H3 inneurons of PQBP1-KD mice (Fig. 5) that might connectfrom PQBP1-KD to the reduced expression of anxiety-relatedgenes. Interestingly, histone acetylation was reported to link tomemory formation (30,32,43). Histone complex interacts withDNA to regulate the higher structure of chromatin and controlthe accessibility of DNA for gene transcription. Generally,acetylation of histone H3 releases genomic DNA fromhistone complex to make it accessible for transcriptionfactors and RNA polymerases, thereby upregulating gene tran-scription (44–46). Our results in study demonstrated thatacetylation of histone H3 in PQBP1-KD mice were reducedin cortex, hippocampus and amygdala. Therefore, it is possibleto imagine that the reduced acetylation of histone H3 wouldmediate the PQBP1’s effect to target genes in the brainregions critical for anxiety-related cognition.
The pathway by which PQBP1-KD leads to histone H3 dea-cetylation has not been identified. However, transcription,RNA splicing and histone modification are closely linked.Multiple factors that mediate elongation, splicing and otherRNA modification interact with Pol II-CTD in aphosphorylation-dependent manner (47,48). As describedabove, PQBP1 is such a molecule connecting splicing andtranscription, and its functional disturbance leads to transcrip-tional repression (11). In the transcriptional cessation undernon-homologous end-joining of DNA double-strand breaks,Sin3p/Rpd3p histone deacetylase complex induces histonedeacetylation around the break point for DNA repair (49).We observed that RNA Pol II inhibition by alpha-amanitininduces DNA damage response (unpublished data), and itwas reported that histone deacetylase is activated in DNAdamage response (50). Regarding the relationship betweenhistone and splicing, H3 methylation is known to facilitatepre-mRNA splicing (51), and inhibition of HAT leads to theblock of splicing at A-like spliceosome (52). Consideringwith these links among histone, RNA Pol II and spliceosome,
abnormal transcription and splicing by PQBP1-KD mightinduce histone deacetylation.
Collectively, these data and considerations might be able toprovide a hypothetical scheme that PQBP1-KD impairsexpression of downstream genes involved anxiety-related be-havior via histone H3 acetylation. However, further analysesusing different types of animal models and human pathologyand extensive investigation on the other signaling pathwaysare needed for complete understanding of the gene pathwaythat links molecular abnormality to patient symptoms.
PBA as a candidate for human therapy
This study also showed that HDAC inhibitor, PBA improvedabnormal anxiety-related behavior and cognition inPQBP1-KD mice. The rescue effect strongly supported ourhypothesis that regulation of certain genes via transcriptionor splicing might underlie the abnormal behavioral phenotypesin PQBP1-KD mice. An intriguing point obtained in this studywas that we could improve such abnormalities in adult mice.Although the results were at the mouse level, the successfulintervention for adult mice is encouraging for treatment ofhuman MR patients. It might suggest that PQBP1-linked MRpatients might be reversible even in adulthood. The use ofHDAC inhibitor should be seriously considered for a groupof MR related to gene expression regulation because asimilar approach was successful also in Rubinstein–Taybisyndrome (29).
It might be possible to imagine that non-specific effect(s) ofPBA benefit mouse and ameliorate the symptoms. PBA actu-ally possesses multiple biological activities like ammoniascavenging in urea cycle dysfunction (53) and regulation ofubiquitin–proteosome or ER stress pathway (54,55).However, PQBP1 is not involved in these biological functions.HDAC inhibitors including PBA are also expected asanti-cancer drugs that inhibit cell proliferation and induceapoptosis (56), whereas these effects would not benefit thecognitive function. Furthermore, PBA has adverse effectslike dyspepsia and fatigue in human patients (57), and theoverdose of sodium butyrate, a homologous molecule, deterio-rates the symptoms in a mouse model of neurodegeneration(58). Collectively, non-specific beneficial effects by PBA aredifficult to consider as the cause for the improvement of cog-nition and memory in our mouse model.
Specific beneficial effects of various HDAC inhibitors werereported in mouse or Drosophila models of polyQ diseases(58–63), Parkinson’s disease (64), amyotrophic lateral scler-osis (65) and Alzheimer’s disease (AD) (66). Among them,PBA was reported to mitigate the phenotype of HD mousemodel via transcriptional improvement (61). Related HDACinhibitors, sodium butyrate and suberoylanilide hydroxamicacid also improve phenotypes of HD and DRPLA modelmice through HDAC inhibition and transcriptional improve-ment (58,62). Interestingly, improved cognition was reportedin the AD mouse model with nicotinamide that inhibits aClass III HDAC Sir2 deacetylase (67,68).
PBA has been already used for human patients of hyperam-monemic encephalopathy due to ornithine transcarbamylasedeficiency (53) or of inherited epilepsy due to glycosyl-phosphatidylinositol deficiency (69). Furthermore, clinical
Human Molecular Genetics, 2009, Vol. 18, No. 22 4249
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

trials of PBA have been performed in various cancersand neurodegenerative diseases (http://clinicaltrials.gov/ct2/results?term=phenylbutyrate). A small part of patientsshowed dyspepsia and fatigue while no major side effectwas reported. As the safety of PBA has been established inthese clinical use and trials, it would be a good candidatefor PQBP1-linked MR and its relevant developmentaldisorders.
PQBP1 in MR and neurodegneration
PQBP1 was isolated originally as a binding protein to thepolyQ tract sequence (10). PQBP1 is implicated in neurode-generative pathologies because it interacts with mutant Atx1,the causative gene product of SCA1, and with Htt, the causa-tive gene product of HD. The interaction with mutant polyQproteins links to dysfunction of PQBP1 in transcriptioncoupled with splicing (11). We have shown in this studythat a homologous dysfunction in transcription/splicing mayoccur in MR. However, a critical difference between MRand neurodegeneration is the onset of dysfunction. PQBP1hypofunction would start from an embryonic stage in theMR pathology, whereas mutant polyQ proteins woulddisturb the PQBP1 function more strongly after the toxic(interacting) form accumulates by overwhelming the cell pro-tection mechanisms such as ubiquitin–proteasome and autho-phagy systems. The chronological difference may beimportant for differentiating the critical period for therapeuticintervention of each symptom of MR or neurodegeneration.
MATERIALS AND METHODS
Construction of PQBP1 double-strand RNAexpression vector
The plasmid to express PQBP1 double-strand RNA(pDECAP-PQBP1) was constructed by subcloning the last498 bp (295–792 fragment) of the mouse PQBP1 codingregion into the pDECAP vector as an inverted repeat with a12 bp spacer (ATCTGCGGTACC). The primers, forward:CCGGAATTCTCAGTCCTGCTGTTTGGTTC and reverse:GCTCTAGAGGTACCGCAGATGCTGAGGACAAGT, wereused to amplify anti-sense sequence with spacer, and theprimers, forward: CGGGGTACCGCTGAGGACAAGTCGGAC and reverse: GCTCTAGATCAGTCCTGCTGTTTGGTTC,were used for amplifying sense sequence. The pDECAP vectorcontains cytomegalovirus promoter, ribozyme cassette andMAZ site for RNA Pol II pausing (70,71). There is nopoly(A) addition signal in the construct.
Generation of pDECAP-PQBP1 transgenic mice
The 2.15 kb BglII–BamHI fragment of pDECAP-PQBP1 wasfreed from background sequences, purified and injected intofertilized mouse oocytes. The fertilized mouse oocytes werederived from matings between C57BL/6 mice. For genotyp-ing, genomic DNA was prepared from tail biopsy and usedfor PCR. Integration of the DNA fragment was confirmed byPCR using three primer pairs: forward-1, 50-GATCTTCAATATTGGCCATTAGC-30, with reverse-1, 50-ACGTAGATGT
ACTGCCAAGTAG-30, forward-2, 50-TGCGGTAGTTTATCACAGTT-30, with reverse-2, 50-GCTCTAGAGGTACCGCAGATGCTGAGGACAAGT-30 and forward-3, 50-CGGGGTACCGCTGAGGACAAGTCGGAC-30, with reverse-3, 50-CACACCTCCCCCTGAACCT-30 (Fig. 1A). Four transgenicfounder mice (F0) were produced and bred twice withC57BL/6J mice to produce F2 hemizygous transgenicanimals. The effective suppression of PQBP1 was obtainedin two lines, which were chosen for further studies.
Tissue preparation and western blotting
Two hours after the last behavior test, mice were deeplyanesthetized and sacrificed by cervical dislocation, andbrains were removed. For western blot sampling, braintissues of PQBP1-KD mice (pDECAP-PQBP1 transgenicmice) and littermate WT mice dissolved in lysis buffer con-taining 62.5 mM Tris/HCl, pH 6.8, 2% (w/v) SDS, 2.5%(v/v) 2-mercaptoethanol and 5% (v/v) glycerol. Protein con-centration was quantified by using the BCA method (MicroBCA Protein Assay Reagent Kit; Pierce Chemical, Rockford,IL, USA). These samples were separated by SDS–PAGE,transferred onto polyvinilydene difluoride membrane FineTraps (Nihon Eido, Tokyo, Japan) through a semidrymethod, blocked by 5% milk in TBS with Tween-20(TBST) (10 mM Tris/Cl, pH 8.0, 150 mM NaCl, 0.05%Tween-20) and incubated with appropriate antibodies asdescribed below. The filters were incubated with eachprimary antibody for over night at 48C, with the correspondinghorseradish peroxidase-conjugated second antibody for 1 h atroom temperature in 5% milk/TBST. Finally, the target mol-ecules were visualized through an enhanced chemilumines-cence Western blotting detection system (AmershamBiosciences, GE Health Care Biosciences, Hong Kong).Immunoblotted bands were quantified using the Image J1.32 software (National Institutes of Health, Bethesda, MD,USA) after densitometric scanning of the films. The dilutionconditions for primary and secondary antibodies were asfollows: goat anti-PQBP1 (C-20, Santa Cruz), 1:1000, mouseanti-GAPDH (Chemicon), 1:5000, mouse anti-b-Actin(AC-74, Sigma), 1:1000, mouse anti-a-Tubulin (Sigma),1:1000, rabbit anti-GFAP (NeoMarkers), 1:1000, mouseanti-MAP2 (Sigma), 1:2000, rabbit anti-acetylated histoneH3 (Upstate), 1:1000, mouse anti-histone H3 (Upstate),1:1000, rabbit anti-c-Fos (K-25, Santa Cruz), 1:1000;anti-NR1 (Abcam), 1:250, anti-NF-kB p50 (Santa Cruz),1:500, anti-serotonin receptor 1A (Santa Cruz), 1:3000, anti-serotonin transporter (Santa Cruz), 1:500, anti-HMGB1(Abcam), 1:2000, anti-CREB1 (Santa Cruz), 1:250, anti-goatIgG (DAKO), anti-mouse IgG (Amersham Biosciences) andanti-rabbit IgG (Amersham Biosciences), 1:3000. Antibodieswere diluted in TBST with 5% skim milk.
For quantitative comparison of western blot band intensi-ties, we performed WB three times for each mouse. Themeans were averaged with the number of mice (indicated infigure or each figure legend) and SEM were calculated.After examining the variance among the groups, statisticalanalysis was performed by Student’s t-test, Welch’s t-test orone-way ANOVA, as described in the method of ‘statisticalanalysis’.
4250 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

Immunohistochemistry and histology
Mouse brains were fixed with 4% paraformaldehyde in 0.1 M
phosphate buffer pH 7.4, embedded in paraffin and sectionedinto 5 mm thick slices. Several sections were stained by hematox-ylin–eosin for routine histology. For immunohistochemistry, theparaffin-embedded mouse sections were deparaffinized, rehy-drated and then microwaved in 10 mM of citrate buffer, pH 6.0,for 15 min. These sections were incubated with primary anti-bodies for overnight at 48C, and finally with Alexa Fluor 488and 594-labeled anti-IgGs (Invitrogen) for 1 h at room tempera-ture. Primary antibodies of anti-NeuN (Millipore), anti-GFAP(NeoMarkers), anti-acetylated histone H3 (Upstate) andanti-c-Fos (Santa Cruz) were diluted at a ratio of 1:100.
Quantification of signal intensity
Signal intensity per cell was calculated by using MetaMorphsoftware (Universal Imaging Corporation, Downingtown).After co-staining of anti-NeuN antibody with anti-c-Fos oranti-acetylated H3 antibody, NeuN-positive neurons wereselected at random, and the fluorescence signal intensitiesper area (mm2) of c-Fos or acetylated H3 were collectedfrom at least three different slides of a mouse and more than100 cells were measured in each slide. To exclude backgroundfluorescence, we measured fluorescence of ten randomlyselected non-cell existing tissue areas of each sample, andtheir mean value was subtracted from the cellular fluorescencesignals on the same slide. The average of the signal intensitiesafter background subtraction were calculated in each mouse,then the mean values from multiple mice (n ¼ 3 or 4) werestatistically analyzed between the groups.
Conditions of testing animals
Mice were segregated by sex, housed two to five to a cage,provided with water and rodent chow and maintained on a12 h light/dark cycle. All experiments were performedduring the light phase (9:00–18:00 h) using PQBP1-KDmale mice and littermate WT male mice between 1.5 and8 months of age and were conducted in accordance with thepolicies of the Institutional Animal Care and Use Committeeof the Tokyo Medical and Dental University. The analyseswere performed in the order of open-field test, novel objectrecognition test, light–dark exploration test, elevated plusmaze test, rotarod test, Morris water maze and fear condition-ing test. After each trial (except the water maze), apparatuseswere wiped and cleaned by 70% alcohol and damp towel.Open-field test, novel object recognition test, light–darkexploration test, elevated plus maze test, Morris water mazetest and fear conditioning test performed using Image XX(O’Hara & Co, Ltd.), a modified software the based on thepublic domain NIH Image program (developed at the USNational Institutes of Health and available on the Internet athttp://rsb.info.nih.gov/nih-image/).
Spontaneous activity
Spontaneous activity was measured in individually caged micein their home cages under 12 h light/dark cycle. Their activity
was monitored for 48 h using SCANET SV-10 (Toyo sangyo,Toyama, Japan).
Open-field test
Mice were placed in the center of an open-field space(50 cm � 50 cm � 40 cm [H]) and allowed to explore for15 min. Light intensity was 70 lux in the center of the field.Distance traveled (cm) and % duration of staying at thecenter area of the field (30% of the field) were adopted asthe indices.
Novel object recognition test
The novel object recognition test was started the day after theopen-field test. During the training session, two novel objectswere placed into the open field, and the spatial position ofobjects was counterbalanced between subjects. During thetraining session, mice were allowed to explore for 10 min oneach of two consecutive days. The time spent exploring eachobject and total approach time was recorded. To test formemory retention, mice were placed back into the same box3 and 24 h after a training session, in which one of the familiarobjects used during the training session was replaced by anovel object and allowed to explore freely for 10 min. A pre-ference index, a ratio of the amount of time spent exploringany one of the two objects (training session) or the novelobject (retention session) over the total time spent exploringboth objects was used to measure recognition memory.
Light–dark exploration test
Light box was made of white plastic (20 cm � 20 cm � 20 cm[H]) and illuminated by LEDs (250 lux at the center of thebox); a CCD camera was equipped on the ceiling. Dark boxwas made of black plastic (20 cm � 20 cm � 20 cm [H])and an infrared camera was equipped on the ceiling. Therewas a tunnel for transition on the center panel between thelight box and dark box (3 cm � 5 cm) with a sliding door.In the L–D box test, mice were individually introduced intothe light box, and the door of the tunnel opened 3 s after theintroduction of a mouse. Then mouse was allowed to movefreely in the L–D box for 10 min. Total distance traveled, dis-tance traveled in the light box, duration staying in the lightbox, numbers of the transition between light and dark boxand the first latency to enter the dark box were measured asindices.
Elevated plus maze test
The elevated plus maze consisted of four 5 � 25 cm orthog-onal arms connected by a 5 � 5 cm central square. The floorof each arm was made of white plastic and the wall ofclosed arms (15 cm) and ridge of open arms (0.3 cm) weremade of clear plastic. Closed arms and open arms werearranged 60 cm above the floor. Light intensity was 70 luxat the central square of the maze. In the elevated plus mazetest, mice were individually put on the central square facingto an open arm, and then mice were allowed to move freelyin the maze for 5 min. To test for memory retention, mice
Human Molecular Genetics, 2009, Vol. 18, No. 22 4251
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

were placed back on the same maze 3 h after the first trial.Duration staying in the open and closed arms, durationstaying in the central square, entries into the open andclosed arms and total distance traveled were measured asindices.
Rotarod test
Mice were placed on a rotating rod (diameter: 3 cm), and therotating speed was linearly increased from 3.5 r.p.m. to35 r.p.m. in 300 s and continued at 35 r.p.m. until 360 s.Mice received nine trials (three trials per day for 3 consecutivedays) with a 30–60 min rest interval between trials. Theamount of time for each mouse to fall from the rod wasrecorded for each trial. The mean latency to fall off therotarod was recorded and used in subsequent analysis.
Morris water maze test
The Morris water maze test was conducted in a circular poolof a 1 m diameter and filled with water at a temperature of22.0+ 18C. Water was colored by white painting in orderthat mice could not see the platform (20 cm high, 10 cm diam-eter; 1 cm below the surface of water) or other cues under thewater. There were some extra-maze landmark cues that werevisible to the mice in the maze. Mice received six trials perday for 5 consecutive days. Each acquisition trial was initiatedby placing an individual mouse into the water facing the outeredge of the maze at one of four designated starting pointquasi-randomly, but the submerged platform remained con-stant for each mouse throughout testing. A trial was terminatedwhen the mouse reached the platform, and the latency and dis-tance swam were measured. Cut-off time of the trial was 60 s,and mice that did not reach the platform within 60 s wereremoved from the water and placed on the platform for 30 sbefore being toweled off and placed back into their homecage. The inter-trial interval was �6 min. After the 5 days’training, a probe test was conducted. In the probe test, the plat-form was taken away, and each mouse was placed into thewater at the point of the opposite position of the target plat-form, and allowed to swim in the maze for 60 s. The distanceswam, the number of crossings the position of the target plat-form and other three platforms, time staying in the quadrantsof the four platforms were measured.
Contextual fear conditioning test
Contextual fear conditioning test was carried out the day afterthe Morris water maze test. This test consisted of three parts: aconditioning trial, a context test trial and a cued test trial. Fearconditioning was carried out on a clear plastic chamberequipped with a stainless steel grid floor (34 cm � 26 cm �30 cm [H]). A CCD camera was equipped on the ceiling ofthe chamber and was connected to a video monitor and com-puter. The grid floor was wired to a shock generator. Whitenoise (80 dB) was supplied from a loudspeaker as an auditorycue (the conditioned stimulus, CS). A continuous 0.4 mA footshock (the unconditioned stimulus, US) for 2 s was adminis-tered at the end of the 30 s CS period. The conditioning trialconsisted of a 2 min exploration period followed by two
CS–US pairings separated by 1 min each. A context testwas performed in the same conditioning chamber for 5 minin the absence of the white noise at 24 h after the conditioningtrial. Further, a cued test was performed in an alternativecontext with distinct cues; the test chamber was differentfrom the conditioning chamber in brightness, floor structure(no grid) and shape. The cued test was conducted 3 h afterthe contextual test and consisted of a 2 min explorationperiod (no CS) to evaluate the non-specific contextual fear fol-lowed by a 3 min CS period (no foot shock) to evaluate theacquired cued fear. Rate of freezing response of mice wasmeasured as an index of fear memory.
Injection of PBA
PBA (Nacalai Tesque, Kyoto, Japan) was dissolved in PBS bytitration with sodium hydroxide to pH 7.4. Mice received dailyintraperitoneal injections of PBA solution (100 mg/kg/day,volume 5 ml/kg) or vehicle (PBS, 5 ml/kg), for 4 weeksfrom 7 months of age. The analyses were performed in theorder of open-field test, light–dark exploration test and elev-ated plus maze test.
Statistical analysis
For statistical analysis of behavioral analyses, we first com-pared the variances among groups with F-test. When the var-iance was similar among groups, we used Student’s t-test orone-way ANOVA. In the case that the variance was notsimilar, we used Welch’s t-test. When a significant effectwas found in one-way ANOVA, Bonferroni/Dunn post hoccomparisons were applied. Average and standard error ofmean (SEM) were presented for each experimental parameterin one group.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENT
We thank Mrs Tayoko Tajima for her excellent technicalsupport and our laboratory members for discussion.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by grants to H.O. from Ministry ofEducation, Culture, Sports, Science and Technology ofJapan (16390249, 16650076, 18390254, 18650097, Researchon Pathomechanisms of Brain Disorders: 17025017,18023014, 20023011), the research grant (19A-8) forNervous and Mental Disorders from Ministry of HealthLabor and Welfare, and grants from Uehara Memorial Foun-dation and The Tokyo Biochemical Research Foundation.
4252 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

REFERENCES
1. Chelly, J. and Mandel, J.L. (2001) Monogenic causes of X-linked mentalretardation. Nat. Rev. Genet., 2, 669–680.
2. Check, E. (2005) Genetics: the X factor. Nature, 434, 266–267.3. Carrel, L. and Willard, H.F. (2005) X-inactivation profile reveals
extensive variability in X-linked gene expression in females. Nature, 434,400–404.
4. Ropers, H.H. and Hamel, B.C. (2005) X-linked mental retardation. Nat.Rev., 6, 46–57.
5. Kalscheuer, V.M., Freude, K., Musante, L., Jensen, L.R., Yntema, H.G.,Gecz, J., Sefiani, A., Hoffmann, K., Moser, B., Haas, S. et al. (2003)Mutations in the polyglutamine binding protein 1 gene cause X-linkedmental retardation. Nat. Genet., 35, 313–315.
6. Kleefstra, T., Franken, C.E., Arens, Y.H., Ramakers, G.J., Yntema, H.G.,Sistermans, E.A., Hulsmans, C.F., Nillesen, W.N., van Bokhoven, H., deVries, B.B. et al. (2004) Genotype–phenotype studies in three familieswith mutations in the polyglutamine-binding protein 1 gene (PQBP1).Clin. Genet., 66, 318–326.
7. Lenski, C., Abidi, F., Meindl, A., Gibson, A., Platzer, M., Frank Kooy, R.,Lubs, H.A., Stevenson, R.E., Ramser, J. and Schwartz, C.E. (2004) Noveltruncating mutations in the polyglutamine tract binding protein 1 gene(PQBP1) cause Renpenning syndrome and X-linked mental retardation inanother family with microcephaly. Am. J. Hum. Genet., 74, 777–780.
8. Stevenson, R.E., Bennett, C.W., Abidi, F., Kleefstra, T., Porteous, M.,Simensen, R.J., Lubs, H.A., Hamel, B.C. and Schwartz, C.E. (2005)Renpenning syndrome comes into focus. Am. J. Med. Genet., 134, 415–421.
9. de Brouwer, A.D., Yntema, H.G., Kleefstra, T., Lugtenberg, D.,Oudakker, A.R., de Vries, B.B., van Bokhoven, H., Van Esch, H., Frints,S.G., Froyen, G. et al. (2007) Mutation frequencies of X-linked mentalretardation genes in families from the EuroMRX consortium. Hum.Mutat., 28, 207–208.
10. Waragai, M., Lammers, C.H., Takeuchi, S., Imafuku, I., Udagawa, Y.,Kanazawa, I., Kawabata, M., Mouradian, M.M. and Okazawa, H. (1999)PQBP-1, a novel polyglutamine tract-binding protein, inhibitstranscription activation by Brn-2 and affects cell survival. Hum. Mol.
Genet., 8, 977–987.11. Okazawa, H., Rich, T., Chang, A., Lin, X., Waragai, M., Kajikawa, M.,
Enokido, Y., Komuro, A., Kato, S., Shibata, M. et al. (2002) Interactionbetween mutant ataxin-1 and PQBP-1 affects transcription and cell death.Neuron, 34, 701–713.
12. Komuro, A., Saeki, M. and Kato, S. (1999) Association of two nuclearproteins, Npw38 and NpwBP, via the interaction between the WWdomain and a novel proline-rich motif containing glycine and arginine.J. Biol. Chem., 274, 36513–36519.
13. Nicolaescu, E., Beullens, M., Lesage, B., Keppens, S., Himpens, B. andBollen, M. (2008) Nature of the nuclear inclusions formed by PQBP1, aprotein linked to neurodegenerative polyglutamine diseases. Eur. J. Cell
Biol., 87, 817–829.14. Waragai, M., Junn, E., Kajikawa, M., Takeuchi, S., Kanazawa, I., Shibata,
M., Mouradian, M.M. and Okazawa, H. (2000) PQBP-1/Npw38, a nuclearprotein binding to the polyglutamine tract, interacts with U5-15kD/dim1pvia the carboxyl-terminal domain. Biochem. Biophys. Res. Commun., 273,592–595.
15. Zhang, Y., Lindblom, T., Chang, A., Sudol, M., Sluder, A.E. and Golemis,E.A. (2000) Evidence that dim1 associates with proteins involved inpre-mRNA splicing, and delineation of residues essential for dim1interactions with hnRNP F and Npw38/PQBP-1. Gene, 257, 33–43.
16. Makarov, E.M., Makarova, O.V., Urlaub, H., Gentzel, M., Will, C.L.,Wilm, M. and Luhrmann, R. (2002) Small nuclear ribonucleoproteinremodeling during catalytic activation of the spliceosome. Science, 298,2205–2208.
17. Makarova, O.V., Makarov, E.M., Urlaub, H., Will, C.L., Gentzel, M.,Wilm, M. and Luhrmann, R. (2004) A subset of human 35S U5 proteins,including Prp19, function prior to catalytic step 1 of splicing. EMBO J.,23, 2381–2391.
18. Okazawa, H., Sudol, M. and Rich, T. (2001) PQBP-1 (Np/PQ): apolyglutamine tract-binding and nuclear inclusion-forming protein. BrainRes. Bull., 56, 273–280.
19. Shinagawa, T. and Ishii, S. (2003) Generation of ski-knockdown mice byexpressing a long double-strand RNA from an RNA polymerase IIpromoter. Genes Dev., 17, 1340–1345.
20. Peier, A.M., Mcllwain, K.L., Kenneson, A., Warren, S.T., Paylor, R. andNelson, D.L. (2000) Over correction of FMR1 deficiency with YACtransgenics: behavioral and physical features. Hum. Mol. Genet., 9, 1145–1159.
21. Itoh, J., Nabeshima, T. and Kameyama, T. (1990) Utility of an elevatedplus-maze for the evaluation of memory in mice: effects of nootropics,scopolamine and electroconvulsive shock. Psychopharmacology (Berl),101, 27–33.
22. Rodgers, R.J., Haller, J., Halasz, J. and Mikics, E. (2003) ‘One-trialsensitization’ to the anxiolytic-like effects of cannabinoid receptorantagonist SR141716A in the mouse elevated plus-maze.Eur. J. Neurosci., 17, 1279–1286.
23. Rodgers, R.J., Johnson, N.J., Cole, J.C., Dewar, C.V., Kidd, G.R. andKimpson, P.H. (1996) Plus-maze retest profile in mice: importance ofinitial stages of trail 1 and response to post-trail cholinergic receptorblockade. Pharmacol. Biochem. Behav., 54, 41–50.
24. Yan, Q.J., Asafo-Adjei, P.K., Arnold, H.M., Brown, R.E. and Bauchwitz,R.P. (2004) A phenotypic and molecular characterization of thefmr1-tm1Cgr fragile X mouse. Genes Brain Behav., 3, 337–359.
25. Amat, J., Baratta, M.V., Paul, E., Bland, S.T., Watkins, L.R. and Maier,S.F. (2005) Medial prefrontal cortex determines how stressorcontrollability affects behavior and dorsal raphe nucleus. Nat. Neurosci.,8, 365–371.
26. Lee, M.D., Somerville, E.M., Kennett, G.A., Dourish, C.T. and Clifton,P.G. (2004) Tonic regulation of satiety by 5-HT receptors in the mouse:converging evidence from behavioural and c-fos immunoreactivitystudies? Eur. J. Neurosci., 19, 3017–3025.
27. Warburton, E.C., Koder, T., Cho, K., Massey, P.V., Duguid, G., Barker,G.R., Aggleton, J.P., Bashir, Z.I. and Brown, M.W. (2003) Cholinergicneurotransmission is essential for perirhinal cortical plasticity andrecognition memory. Neuron, 38, 987–996.
28. LaBar, K.S. and Cabeza, R. (2006) Cognitive neuroscience of emotionalmemory. Nat. Rev. Neurosci., 7, 54–64.
29. Alarcon, J.M., Malleret, G., Touzani, K., Vronskaya, S., Ishii, S., Kandel,E.R. and Barco, A. (2004) Chromatin acetylation, memory, and LTP areimpaired in CBPþ/2 mice: a model for the cognitive deficit inRubinstein–Taybi syndrome and its amelioration. Neuron, 42, 947–959.
30. Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M. and Tsai, L.H. (2007)Recovery of learning and memory is associated with chromatinremodelling. Nature, 447, 178–182.
31. Korzus, E., Rosenfeld, M.G. and Mayford, M. (2004) CBP histoneacetyltransferase activity is a critical component of memoryconsolidation. Neuron, 42, 961–972.
32. Levenson, J.M., O’Riordan, K.J., Brown, K.D., Trinh, M.A., Molfese,D.L. and Sweatt, J.D. (2004) Regulation of histone acetylation duringmemory formation in the hippocampus. J. Biol. Chem., 279, 40545–40559.
33. Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B.,Noebels, J., Armstrong, D., Paylor, R. and Zoghbi, H. (2002) Mice withtruncated MeCP2 recapitulate many Rett syndrome features and displayhyperacetylation of histone H3. Neuron, 35, 243–254.
34. Gardian, G., Browne, S.E., Choi, D.K., Klivenyi, P., Gregorio, J., Kubilus,J.K., Ryu, H., Langley, B., Ratan, R.R., Ferrante, R.J. et al. (2005)Neuroprotective effects of phenylbutyrate in the N171-82Q transgenicmouse model of Huntington’s disease. J. Biol. Chem., 280, 556–563.
35. Kida, S., Josselyn, S.A., Pena de Ortiz, S., Kogan, J.H., Chevere, I.,Masushige, S. and Silva, A.J. (2002) CREB required for the stability ofnew and reactivated fear memories. Nat. Neurosci., 5, 348–355.
36. Kassed, C.A. and Herkenham, M. (2004) NF-kB p50-deficient mice showreduced anxiety-like behaviors in tests of exploratory drive and anxiety.Behav. Brain Res., 154, 577–584.
37. Liu, A., Hoffman, P.W., Lu, W. and Bai, G. (2004) NF-kappaB siteinteracts with Sp factors and up-regulates the NR1 promoter duringneuronal differentiation. J. Biol. Chem., 279, 17449–17458.
38. Cohen, S. and Greenberg, M.E. (2008) Communication between theSynapse and the Nucleus in neuronal development, plasticity, and disease.Annu. Rev. Cell Dev. Biol., 24, 183–209.
39. Canli, T. and Lesch, K.P. (2007) Long story short: the serotonintransporter in emotion regulation and social cognition. Nat. Neurosci., 10,1103–1109.
40. Fleischmann, A., Hvalby, O., Jensen, V., Strekalova, T., Zacher, C.,Layer, L.E., Kvello, A., Reschke, M., Spanagel, R., Sprengel, R., Wagner,E.F. and Gass, P. (2003) Impaired long-term memory and NR2A-type
Human Molecular Genetics, 2009, Vol. 18, No. 22 4253
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022

NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos inthe CNS. J. Neurosci., 23, 9116–9122.
41. Kassed, C.A. and Herkenham, M. (2004) NF-kappaB p50-deficient miceshow reduced anxiety-like behaviors in tests of exploratory drive andanxiety. Behav. Brain Res., 154, 577–584.
42. Boyce-Rustay, J.M. and Holmes, A. (2006) Genetic inactivation of theNMDA receptor NR2A subunit has anxiolytic- and antidepressant-likeeffects in mice. Neuropsychopharmacology, 31, 2405–2414.
43. Tsankova, N., Renthal, W., Kumar, A. and Nestler, E.J. (2007) Epigeneticregulation in psychiatric disorders. Nat. Rev. Neurosci., 8, 355–367.
44. Ho, A.K., Price, D.M., Dukewich, W.G., Steinberg, N., Arnason, T.G. andChik, C.L. (2007) Acetylation of histone H3 and adrenergic-regulatedgene transcription in rat pinealocytes. Endocrinology, 148, 4592–4600.
45. Sterner, D.E. and Berger, S.L. (2000) Acetylation of histones andtranscription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459.
46. Struhl, K. (1998) Histone acetylation and transcriptional regulatorymechanisms. Genes Dev., 12, 599–606.
47. Kornblihtt, A.R., de la Mata, M., Fededa, J.P., Munoz, M.J. and Nogues,G. (2004) Multiple links between transcription and splicing. RNA, 10,1489–1498.
48. Allemand, E., Batsche, E. and Muchardt, C. (2008) Splicing, transcription,and chromatin: a menage a trois. Curr. Opin. Genet. Dev., 18, 145–151.
49. Jazayeri, A., McAinsh, A.D. and Jackson, S.P. (2004) Saccharomyces
cerevisiae Sin3p facilitates DNA double-strand break repair. Proc. Natl
Acad. Sci. USA, 101, 1644–1649.50. Kao, G.D., McKenna, W.G., Guenther, M.G., Muschel, R.J., Lazar, M.A.
and Yen, T.J. (2003) Histone deacetylase 4 interacts with 53BP1 tomediate the DNA damage response. J. Cell Biol., 160, 1017–1027.
51. Sims, R.J. III and Reinberg, D. (2008) Is there a code embedded inproteins that is based on post-translational modifications? Nat. Rev. Mol.
Cell Biol., 9, 815–820.
52. Kuhn, A.N., van Santen, M.A., Schwienhorst, A., Urlaub, H. andLuhrmann, R. (2009) Stalling of spliceosome assembly at distinct stagesby small-molecule inhibitors of protein acetylation and deacetylation.RNA, 15, 153–175.
53. Maestri, N.E., Brusilow, S.W., Clissold, D.B. and Bassett, S.S. (1996)Long-term treatment of girls with ornithine, transcarbamylase deficiency.New Eng. J. Med., 335, 855–859.
54. Gardian, G., Browne, S.E., Choi, D.K., Klivenyi, P., Gregorio, J., Kubilus,J.K., Ryu, H., Langley, B., Ratan, R.R., Ferrante, R.J. and Beal, M.F.(2005) Neuroprotective effects of phenylbutyrate in the N171-82Qtransgenic mouse model of Huntington’s disease. J. Biol. Chem., 280,556–563.
55. Kubota, K., Niinuma, Y., Kaneko, M., Okuma, Y., Sugai, M., Omura, T.,Uesugi, M., Uehara, T., Hosoi, T. and Nomura, Y. (2006) Suppressiveeffects of 4-phenylbutyrate on the aggregation of Pael receptors andendoplasmic reticulum stress. J. Neurochem., 97, 1259–1268.
56. Johnstone, R.W. and Licht, J.D. (2003) Histone deacetylase inhibitors incancer therapy: Is transcription the primary target? Cancer Cell, 4, 13–18.
57. Gilbert, J., Baker, S.D., Bowling, M.K., Grochow, L., Figg, W.D.,Zabelina, Y., Donehower, R.C. and Carducci, M.A. (2001) A phase I doseescalation and bioavailability study of oral sodium phenylbutyrate inpatients with refractory solid tumor malignancies. Clin. Cancer Res., 7,2292–2300.
58. Ferrante, R.J., Kubilus, J.K., Lee, J., Ryu, H., Beesen, A., Zucker, B.,Smith, K., Kowall, N.W., Ratan, R.R., Luthi-Carter, R. and Hersch, S.M.(2003) Histone deacetylase inhibition by sodium butyrate chemotherapyameliorates the neurodegenerative phenotype in Huntington’s diseasemice. J. Neurosci., 23, 9418–9427.
59. Hockly, E., Richon, V.M., Woodman, B., Smith, D.L., Zhou, X., Rosa, E.,Sathasivam, K., Ghazi-Noori, S., Mahal, A., Lowden, P.A. et al. (2003)Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor,ameliorates motor deficits in a mouse model of Huntington’s disease.Proc. Natl Acad. Sci. USA, 100, 2041–2046.
60. Minamiyama, M., Katsuno, M., Adachi, H., Waza, M., Sang, C.,Kobayashi, Y., Tanaka, F., Doyu, M., Inukai, A. and Sobue, G. (2004)Sodium butyrate ameliorates phenotypic expression in a transgenic mousemodel of spinal and bulbar muscular atrophy. Hum. Mol. Genet., 13,1183–1192.
61. Gardian, G., Browne, S.E., Choi, D.-K., Klivenyi, P., Gregono, J.,Kubilus, J.K., Ryu, H., Langley, B., Raton, R.R., Ferrante, R.J. and Beal,M.F. (2005) Neuroprotective effect of phenylbutyrate in the N171-82Qtransgenic mouse model of Huntington’s disease. J. Biol. Chem., 280,556–563.
62. Ying, M., Xu, R., Wu, X., Zhu, H., Zhuang, Y., Han, M. and Xu, T. (2006)Sodium butyrate ameliorates histone hypoacetylation andneurodegenerative phenotypes in a mouse model for DRPLA. J. Biol.Chem., 281, 12580–12586.
63. Steffan, J.S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol,B.L., Kazantsev, A., Schmidt, E., Zhu, Y.Z., Greenwald, M. et al. (2001)Histone deacetylase inhibitors arrest polyglutamine-dependentneurodegeneration in Drosophila. Nature, 413, 739–743.
64. Gardian, G., Yang, L., Cleren, C., Calingasan, N.Y., Klivenyi, P. andBeal, M.F. (2004) Neuroprotective effects of phenylbutyrate againstMPTP neurotoxicity. Neuromolecular Med., 5, 235–241.
65. Ryu, H., Smith, K., Camelo, S.I., Carreras, I., Lee, J., Iglesias, A.H.,Dangond, F., Cormier, K.A., Cudkowicz, M.E., Brown, R.H. Jr andFerrante, R.J. (2005) Sodium phenylbutyrate prolongs survival andregulates expression of anti-apoptotic genes in transgenic amyotrophiclateral sclerosis mice. J. Neurochem., 93, 1087–1098.
66. Green, K.N., Steffan, J.S., Martinez-Coria, H., Sun, X., Schreiber, S.S.,Thompson, L.M. and LaFerla, F.M. (2008) Nicotinamide restorescognition in Alzheimer’s disease transgenic mice via a mechanisminvolving sirtuin inhibition and selective reduction of Thr231-phosphotau.J. Neurosci., 28, 11500–11510.
67. Zhao, K., Harshaw, R., Chai, X. and Marmorstein, R. Structural basis fornicotinamide cleavage and ADP-ribose transfer by NADþ-dependent Sir2histone/protein deacetylases. Proc. Natl Acad. Sci. USA, 101, 8563–8568.
68. Sanders, B.D., Zhao, K., Slama, J.T. and Marmorstein, R. (2007)Structural basis for nicotinamide inhibition and base exchange in Sir2enzymes. Mol. Cell, 25, 463–472.
69. Almeida, A.M., Murakami, Y., Baker, A., Maeda, Y., Roberts, I.A.,Kinoshita, T., Layton, D.M. and Karadimitris, A. (2007) Targeted therapyfor inherited GPI deficiency. New Engl. J. Med., 356, 1641–1647.
70. Huang, Y. and Carmichael, G.G. (1996) Role of polyadenylation innucleocytoplasmic transport of mRNA. Mol. Cell. Biol., 16, 1534–1542.
71. Yonaha, M. and Proudfoot, N.J. (1999) Specific transcriptional pausingactivates polyadenylation in a coupled in vitro system. Mol. Cell, 3,593–600.
4254 Human Molecular Genetics, 2009, Vol. 18, No. 22
Dow
nloaded from https://academ
ic.oup.com/hm
g/article/18/22/4239/588105 by guest on 08 February 2022





























