Embed Size (px)
Citation preview

at SciVerse ScienceDirect
European Journal of Medicinal Chemistry 56 (2012) 301e307
Contents lists available
European Journal of Medicinal Chemistry
journal homepage: http: / /www.elsevier .com/locate/ejmech
Original article
Kinetics and docking studies of two potential new inhibitors of the nucleosidehydrolase from Leishmania donovani
Magdalena Nascimento Rennó a,b, Tanos Celmar Costa França a, Dirlei Nico c, Clarisa B. Palatnik-de-Sousa c,Luzineide Wanderley Tinoco d, José Daniel Figueroa-Villar a,*aDepartamento de Química, Instituto Militar de Engenharia, Praça General Tibúrcio 80, 22290-270 Rio de Janeiro, RJ, BrazilbCurso de Farmácia, Universidade Federal do Rio de Janeiro e Campus Macaé, Rua Aluisio da Silva Gomes 50, 27930-560 Macaé, RJ, Brazilc Instituto de Microbiologia Paulo de Góes, Universidade Federal do Rio de Janeiro, Cidade Universitária, 21941-902 Rio de Janeiro, RJ, BrazildNúcleo de Pesquisas de Produtos Naturais, Universidade Federal do Rio de Janeiro, Cidade Universitária, 21941-902 Rio de Janeiro, RJ, Brazil
a r t i c l e i n f o
Article history:Received 13 April 2012Received in revised form27 July 2012Accepted 31 July 2012Available online 10 August 2012
Keywords:Nucleoside hydrolaseLeishmania donovaniLeishmaniasisEnzyme inhibitorNMRMolecular docking
Abbreviations: NH, nucleoside hydrolase; LdNH, nmania donovani; UV, ultraviolet; rLdNH, recombinaLeishmania donovani; rLdNHeMBP, recombinant nucledonovani bound with maltose binding protein; XO,aminophenyliminoribitol; NMR, Nuclear Magnetic Resquare deviation; SDS-polyacrylamide, sodium dodMVD, Molegro Virtual Docker.* Corresponding author. Tel.: þ55 21 2546 7057; fa
E-mail address: [email protected] (J.D. Figuero
0223-5234/$ e see front matter � 2012 Elsevier Mashttp://dx.doi.org/10.1016/j.ejmech.2012.07.052
a b s t r a c t
In this study the recombinant enzyme nucleoside hydrolase of Leishmania donovani (rLdNH) wasexpressed in Escherichia coli in connection with maltose binding protein (MBP). The rLdNHeMBP showedefficient a significant in vitro activity with inosine as substrate. From the coupled reaction with xanthineoxidase (XO) it was possible to determine the kinetic constants of rLdNHeMBP as KM (434 � 109 mM) andVmax (0.20 � 0.02 mM). In addition, two nucleoside analogs (compounds 1 and 2) were tested asprototypes of rLdNH inhibitors. These compounds presented high affinity for the enzyme with Ki valuesof 1.6 � 0.2 and 17.0 � 2.1 mM, respectively, as well as 271 and 26 folds higher than the affinity constantfound for inosine. We also determined the type of enzyme inhibition, using double-reciprocal plot forthese two compounds and the results confirmed a competitive inhibition. Additional docking studiesshowed the binding manner of compounds 1 and 2 inside the active site of LdNH revealing theessential residues for an effective inhibition. These results confirm that compounds 1 and 2 are strongrLdNHeMBP inhibitors.
� 2012 Elsevier Masson SAS. All rights reserved.
1. Introduction
Leishmaniasis is a group of diseases affecting more than 12million people worldwide, which is spread out by the bite of femalephlebotomine sandflies [1,2]. Leishmaniasis is caused by protozoanparasites of the genus Leishmania and represents a complex ofdiseases with an important clinical and epidemiological diversity.One of these diseases is visceral leishmaniasis, also known as kala-azar, which is caused by Leishmania donovani, Leishmania chagasi,and Leishmania infantum and is usually fatal for untreated patients[3]. Many disadvantages on the actual leishmaniasis chemotherapyare related to drug resistance, complex administration mode, high
ucleoside hydrolase of Leish-nt nucleoside hydrolase ofoside hydrolase of Leishmaniaxanthine oxidase; pAPIR, p-sonance; RMSD, root-mean-ecyl sulfate polyacrylamide;
x: þ55 21 2546 7059.a-Villar).
son SAS. All rights reserved.
costs and toxicity. Therefore, the search of new chemotherapytargets and the development of new and more effective drugs fortreatment of leishmaniasis are urgent actions [4]. The DNA differ-ences between protozoan and mammals are being exploited astargets for the development of new and more selective drugs [5].This work is based on the knowledge that these protozoan lack theability of de novo synthesis of purines, which are fundamental forbiosynthesis of their DNA and RNA [5,6]. These parasites use thepurine salvage pathway instead of obtaining nucleosides from theirhost [6]. The most important enzymes involved in this protozoansalvage pathway are the nucleoside hydrolases (NHs) that are ableto catalyze the N-ribosyl hydrolysis of all purine and pyrimidinenucleosides, although differing in their substrate preferences.These enzymes, which have been studied as potential targets forthe development of anti-parasitic drugs, have been found inprokaryotes and eukaryotes, but not in mammals [7e9], makingNHs interesting targets for the development of selective drugs. NHsare strong phylogenic markers of the Leishmania genus [10,11] andthe NH of L. donovani (LdNH) is themain antigen of the first licensedvaccine against canine visceral leishmaniasis [12].
Experimental data suggest that the NHs can be purine-specificor nonspecific enzymes, able to hydrolyze both, purine and

M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307302
pyrimidine nucleosides [2,9,13]. Cui et al. [9] identified a cDNAencoding the LdNH with significant homology to the nonspecificand uridineeinosine-preferring NH [9]. Their studies showed thatthe recombinant enzyme, overexpressed and purified frombacteria, showed significant activity over all naturally occurringpurine and pyrimidine nucleoside substrates [9]. However, thecrystallographic structure of this enzyme has not been reported yetand its available tridimensional structure was obtained byhomology model proposed by França et al. (2008) [14].
The open reading frame encoding the LdNH gene sequence wascloned and inserted downstream from the malE gene of Escherichiacoli, which encodes the maltose-binding protein (MBP), resulting inthe expression of anMBP fusion protein [15]. The recombinant LdNH(rLdNH), is composed by 314 amino acid residues. Using the plasmidpMal-c2-NH36 [15], LdNH was expressed in connection with MBP,and purified by affinity chromatography. This complex rLdNHeMBPdisplays the same enzymatic activity of LdNH and a better stabilityfor this enzyme, thus making unnecessary the cleavage of the MBPby factor XA. The kinetics of rLdNHeMBP with the natural substrateinosine was evaluated in the present work using NMR methods.Further, the enzyme catalytic activity and its potential inhibition bytwo nucleoside analogs (compounds 1 and 2) were tested usingUVevisible spectroscopy. The obtained results showed thatrLdNHeMBP is active for the hydrolysis of inosine, being appropriatefor testing potential LdNH inhibitors. Besides, it was also found thatcompounds 1 and 2 are strong rLdNHeMBP inhibitors.
2. Results and discussion
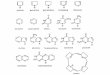
The development of new chemotherapeutic agents againstparasites is still necessary today, especially because the limitedexisting anti-parasitic drugs have many adverse effects and usuallylose their effectiveness over time. There are some reports on studiesinvestigating inhibitors of NH because it is reasonable to assumethat selective inhibition of this enzyme is a particularly attractiveapproach for drug design, especially because NHs have not beenfound yet in mammals [2,7e9,13]. The active site of NH containsa calcium cation, which interacts with the hydroxyl groups ofnucleosides and compounds like the known NH inhibitor p-ami-nophenyl-(1S)-iminoribitol (pAPIR) (Fig. 1) [16]. Accordingly, weselected two riboseequinolone derivatives (1 and 2, Fig. 1) to betested as potential inhibitors of NHs. In this work we used NMR andUV techniques to establish the kinetic parameters with thesubstrate inosine and the capacity of compounds 1 and 2 to inhibitLdNH. In order to understand the interactions of compounds 1 and2with the active site of rLdNHeMBP, the crystallographic structurecoordinates of the pAPIR complex with NH of Crithidia fasciculata(CfNH) (PDB ID 2MAS) [16], were used as reference to perform thedocking studies of these compounds inside the active site of theLdNH model proposed by França et al. [14].
Fig 1. Structures of pAPIR, inosin
2.1. Kinetics parameters
The technique of 1H NMR was used for monitoring the catalyticactivity of the enzyme with the substrate. This technique wasimportant approach for this study because its accuracy providesa direct interpretation of the obtained results without the need ofusing an auxiliary enzyme. The 1H NMR spectra were obtained in5 mm tubes using 20 mM phosphate buffer at pH 8.0 in D2O assolvent. Initially, the spectra of pure inosine (Fig. 2, first spectrum)and hypoxanthine were obtained to acquire information about theintensity and chemical shifts of the nucleobase hydrogen atomsignals, which intensities measured by signal integrationwere usedto monitor the enzyme activity. The values of the nucleobaseproton chemical shifts for inosine (H-2 and H-8, 8.32 and 8.22 ppmin Fig. 2) are different from the respective protons of hypoxanthine(8.17 and 8.19 ppm). The other 1H NMR spectra were obtained afteraddition of the enzyme. Accordingly, the region of the spectrumcorresponding to the nucleobase hydrogen peaks of inosine andhypoxanthine, ranging from 8 to 8.5 ppm, was monitored byobserving the appearance of these signals in the absence andpresence of rLdNHeMBP. The first 1H NMR spectra for monitoringthe enzyme activity was conducted with pure inosine (3.5 mM).The second spectrum was obtained 7 min after addition ofrLdNHeMBP to the NMR tube containing inosine. These 7 minwererequired for the second sample insertion in the NMR magnet, lockand shimming, and was used as the time interval for all the otherspectra. The hydrolysis of the substrate showed a time-increasingintensity of the hypoxanthine protons signals in parallel with theinosine hydrogen signals reduction. The spectra shown in Fig. 2clearly confirm the consumption of substrate and the appearanceof the product during the reaction in a quantitative way.
The KM value for rLdNHeMBP was determined using theUVevisible spectroscopy inosine hydrolysis in the presence of anexcess of xanthine oxidase (XO, 60 milliunits/mL). Since lmax of ino-sine and hypoxanthine are very similar (249 and 250 nm), tomonitorthe hydrolysis of inosine was necessary the presence of XO in excessin order to rapidly transform hypoxanthine to uric acid, which lmax isdifferent (293 nm). The saturation curve (Fig. 3) was obtained byplotting the values of the initial speeds versus substrate concentra-tions, using non-linear regression, to the MichaeliseMenten equa-tion, for determination of the values of Vmax and KM.
The values of KM and Vmax found for rLdNHeMBP, were,respectively, 434�109 mM and 0.20� 0.02 mM/s. The value of KM issimilar to the results reported in literature for rLdNH tailed with sixhistidines [9] and for recombinant Leishmania major NH (rLmNH)[2], which are respectively 329 � 143 mM and 445 � 209 mM. Theseresults confirm that, despite being bound to MBP, rLdNHeMBPmaintained a catalytic activity very similar to the value reportedby Cui et al. [9] for rLdNH (Table 1). Also, the high similarity ofrLdNHeMBP with rLmNH corroborates the findings by França et al.
e and compounds 1 and 2.

Fig 2. Region of the 1H NMR spectra corresponding to the nucleobase hydrogen peaks of inosine and hypoxanthine. The spectra were collected every seven minutes after addition ofrLdNHeMBP. The arrow on the left side indicates the direction of increased reaction time. The signal numbers corresponding to the nucleobase protons are shown for inosine (right)and for hypoxanthine (left). Experiments were performed at pH 8.0, 25 �C at 400 MHz. (For interpretation of the references to color in this figure legend, the reader is referred to theweb version of this article.)
M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307 303
[14], who report that these two enzymes share 100% of identity inthe active site. Furthermore, the presence of MBP confers betterstability and solubility to rLdNH.
2.2. Inhibition assays
2.2.1. Determination of IC50 and Ki
The rLdNHeMBP inhibition effects of compounds 1 and 2 (Fig. 1)were performed by UVevisible spectroscopy tests with the pres-ence of XO, and their IC50 values were determined with fixedconcentrations of rLdNHeMBP (13 nM), inosine (0.3 mM) and XO(60 milliunits/mL) at different concentrations of compounds 1 and2. Before the inhibition assays, a control assay with XO in thepresence of hypoxanthine was performed. The conversion reactionof hypoxanthine to uric acid by XO was monitored in the absenceand presence of compounds 1 and 2, indicating that they do notinterfere with the XO activity. The initial reaction speeds weredetermined based in the variation of uric acid concentration.
The enzymatic rate values were obtained with and withoutcompounds 1 and 2. The doseeresponse data were plotted in orderto determine the effects of compounds 1 and 2 and their concen-tration on rLdNHeMBP enzymatic activity. The inhibitory potency
Fig 3. Saturation curve of rLdNHeMBP obtained by monitoring inosine concentrationon the reaction rate versus substrate concentration. The KM and Vmax were obtained bynonlinear regression. Data represent the mean � standard error of three independentexperiments, obtained by fitting a curve using nonlinear regression.
of these compounds was determined by correlation of theirconcentration with the percentage of enzyme inhibition.
The IC50 values of compounds 1 and 2, which were obtainedfrom non-linear regression of the curves shown in Fig. 4, are2.66 � 0.27 mM and 28.93 � 3.51 mM, respectively.
Our findings demonstrate that, compounds 1 and 2 presentdose-dependent inhibitory effects on the activity of rLdNH, sug-gesting that they are in vivo potential candidate inhibitors of rLdNH.These IC50 values suggest that compound 1 is an rLdNH inhibitorabout 11 times more potent than compound 2.
From the characterization of the inhibitory effects of 1 and 2 onrLdNH it was possible to calculate the values of Ki using the equa-tion of ChengePrussoff [17]. The calculated values of Ki are pre-sented in Table 1, where can be observed that the affinity of 1 forrLdNH is about 270 times greater than inosine.
2.2.2. Determination of the inhibition typeTo determine the inhibition mode of compounds 1 and 2 the
experimental results were analyzed by linear regression, gener-ating the double reciprocal plots of LineweavereBurk [18]. Theseplots are illustrated in Fig. 5. The parameters used in the experi-ments were based on the obtained results of KM for rLdNHeMBPand the experiments were performed from interactive assaysbased on literature [19]. The first experiment was performed in theabsence of the inhibitor in order to determine the parametersthrough the double reciprocal analysis. In this assay we used a fixedconcentration of the enzyme (13 nM) and five different inhibitorconcentrations. The concentrations of compound 2 were greaterthan for compound 1 due to its lower inhibition activity.
The concentration increase of compounds 1 and 2 on the reac-tion catalyzed by rLdNHeMBP significantly increase the KM value(apparent KM), as shown in Fig. 5. These results suggest a compet-itive inhibition profile, probably favored by the structural similarityof compounds 1 and 2 with inosine.
Table 1Ki for compounds 1 and 2 calculated from the IC50 values.
Compound Ki (mM)a IC50 (mM)
1 1.6 � 0.2 2.66 � 0.272 17.0 � 2.1 28.93 � 3.51Inosine 434 � 109b e
a The data corresponds to the mean of three independent experiments(n ¼ 3) � standard error.
b KM for rLdNHeMBP with inosine.

Fig 4. Enzyme activity of rLdNHeMBP using 0.3 mM concentration of inosine (substrate) with different inhibitors concentration on the presence of XO (60 milliunits/mL). Leftpanel: graph of rLdNHeMBP dose-response curve for the compound 1. Right panel: graph of rLdNHeMBP doseeresponse curve for the compound 2. The data correspond to meanvalues of three independent experiments � standard error. The presented data corresponds to the fitting of a standard curve using a four parameter logistic curve.
M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307304
2.3. Docking studies
The RMSD value obtained for the re-docking of pAPIR inside thecrystallographic structure CfNH, was of 0.526 �A. Keeping in mindthat a RMSD value under 2.000�A is acceptable [20e22], this resultwas considered appropriated to validate the used docking protocol.
The ligandeprotein interaction energies (electrostatic and H-bond) were calculated for the best poses of each ligand inside LdNHin order to get a better understanding of the variations betweentheir binding modes and the molecular factors responsible for theiractivity. Table 2 lists the amino acids involved in these interactionsand their distances to the ligands, aswell as the obtained interactionenergy values and the experimental inhibition/affinity constants.
As observed in Table 2 our theoretical results corroborate quali-tatively with the experiments. The inhibitor with the smallest Kivalue (compound 1 in Table 2), experimentally considered as themost promising LdNH inhibitor, corresponds to the inhibitorwith thelowest interacting energy with this enzyme, with greater stabilityinside the active site. It interacts with LdNH active site by means ofseven H-bonds, three more than compound 2 and inosine (Table 2)and also presents smaller distances and better energy values. Thedocking studies of compounds 1 and 2 showed that both displaymore affinity for LdNH active site than inosine, based on their valuesof intermolecular andH-bondenergies (Table 2), being in accordancewith the experimental results. The best poses of inosine andcompounds 1 and 2 inside LdNH are illustrated in Fig. 6, where it ispossible to see that, besides performing various stable interactionswith the active site amino acids, these compounds also show a good
Fig 5. Inhibition kinetics of compounds 1 and 2 with rLdNHeMBP. The experiments were copanel: double reciprocal plot for inosine in the absence and presence of different concentratireciprocal plot for inosine in the absence and presence of different concentrations of compomean of three independent experiments. The presented data corresponds to the fitting of
orientation in the binding sites, reproducing the interactions of theribose portion with Ca2þ and the aspartates of the bottom of theactive site. The halogen groups of compounds 1 and 2, however, didnot establish any direct interaction, but certainly have effect on pepstacking interactions. The absence of direct halogens interactionsprobably happens because these groups are not close enough to anyactive site residue. However, potential interactions with residuesAsn39, Arg76 and Phe167 could happen if a spacer were introducedbetween the Cl atom and the aromatic ring of compound 1 (Fig. 6B).
An additional pep stacking interaction between compound 1and Phe167 can also be observed (Fig. 6A and B). This interaction,together with the additional H-bonds with Asp242 and Glu166,could be contributing to the best experimental results obtained forcompound 1.
3. Conclusions
With the MBP tail it was possible to obtain rLdNH in a solubleform, without inactivating interferents. Besides solubility, the MBPtail also provided stability and a suitable catalytic activity.According to the literature the native form of LdNH have potentialsites of N-glicosilation by post-translational modifications [9] andthis characteristic should contribute to its higher stability. Theexpression in E. coli has the disadvantage of avoiding such modi-fication. Our results suggest that the MBP tail provided the correctfolding, leading to rLdNH in a biologically active conformation.Studies on MBP, glutathione S-transferase and thioredoxin, con-nected to six different proteins, demonstrated that MBP was more
nducted in the absence and presence of increasing concentrations of the inhibitors. Leftons of compound 1. (C) 0 mM; (B) 2 mM; (;) 6 mM and (D) 18 mM. Right panel: doubleund 2. (C) 0 mM; (B) 6 mM; (;) 18 mM and (D) 30 mM. Each point corresponds to thea linear curve.

Table 2Docking results for inosine and compounds 1 and 2 on the active site of LdNH and their experimental values of Ki or KM.
Ligand Residue Dist. (�A) Energy (kcal mol�1) Intermolecular energy (kcal mol�1) H bond energy. (kcal/mol) aKi or KM (mM)
Inosine Asp10 3.13 �2.360 �59.302 �5.538 434 � 109Asp15 3.46 �0.680Glu166 2.82 �2.500Asp242 2.30 �0.001
Compound 1 Asp14 2.62 �2.500 �97.117 �14.286 1.6 � 0.2Thr126 2.93 �2.500Glu166 3.01 �2.420Asn168 3.40 �0.380His241 2.59 �2.450Asp242 2.97 �2.500
Compound 2 Asp14 3.44 �0.800 �92.165 �7.001 17.0 � 2.1Thr126 3.35 �1.230Asn168 2.84 �2.500Asp242 3.11 �2.450
a Experimental data.
M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307 305
effective as a solubilizing agent and can promote the folding of theproteins, favoring their biologically active conformation, suggestingthat that MBP may act as a chaperone [23]. Our results showed thatLdNH has catalytic activity while bounded to MBP, corroboratingthe results reported by Kapust and Waugh [23].
In the XO coupled reaction experiments rLdNHeMBP presenteda KM value similar to the values reported by Cui et al. for LdNH [9]and by Shi et al. for LmNH [2], suggesting similar catalytic proper-ties for these two enzymes as suggested before by França et al. [14]that reported these two enzymes as having a 100% identity on theiractive site. Besides, our spectrometric results also suggest thatcompounds 1 and 2 are new potent LdNH inhibitors. Thesecompounds are analogs to purinic ribonucleosides but presentstructure differences that provide possibilities of better interactionswith the enzyme active site residues.
Our docking results showed that compounds 1 and 2, which arederived of quinolones, have more affinity for the active site of LdNHthan the natural substrate inosine, corroborating the experimentalresults. Also the docking studies showed that compound 1 is able toestablish additional interactions inside the active site that couldexplain its better experimental rLdNHeMBP inhibition resultrelated to compound 2. We also observed that the halogen groupsof compounds 1 and 2 were not able to establish any significantinteraction because they are still far from the residues of the activesite, but that possibly are involved in the pep stacking interactionsof their aromatic rings, with a better influence of Cl in comparisonwith F.
Fig 6. Best poses of inosine (A), compound
In general, our results point rLdNHeMBP as a useful option toperform kinetic studies and for the discovery of new potentialinhibitors of LdNH. The two described inhibitors, compounds 1 and2, are being used as prototypes of new analogs with differentsubstitution patterns in order to achieve more interaction withamino acid residues inside the LdNH active site. Our research groupis actually working on the design and synthesis of new similarinhibitors of LdNH as potential drugs for visceral leishmaniasis.
4. Experimental section
4.1. Materials
Inosine and xanthine oxidase (XO) enzyme were purchasedfrom SigmaeAldrich� Corporation (St Louis, MO, USA) at thehighest purity. Compounds 1 and 2 [24] were donated by theresearch group of Professor Vitor F. Ferreira and Maria Cecília B.V.de Souza from the Fluminense Federal University. All other reagentswere obtained from commercial suppliers.
4.2. Methods
4.2.1. Protein expression and purificationIn order to express the protein, E. coli DH5a cells were trans-
formed with the plasmid pMAL-c2-NH36 [15] and the rLdNH wasobtained in fusionwithMBP. Cells were grown in Luria Bertani brothat 37 �C under agitation, containing ampicillin (100 mg/mL), 1 mM
1 (B) and compound 2 (C) inside LdNH.

M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307306
CaCl2 and glucose (2.0 g/L) until the optical density (OD600nm) of 0.5.The protein expression was induced with 0.3 mM isopropyl-b-D-thiogalactopyranoside for 2 h and harvested by centrifugation at10,000 rpm and 8 �C for 20 min, the supernatant was discarded andthe pellets were kept for 1 h on ice bath. The resuspended pelletswere used for cellular lysis utilizing the cold osmotic shock, 10 mLbuffer (20 mM phosphate buffer pH 8.0), phenyl methylsulfonylfluoride 1 mM, lisozyme 200 mg/mL and sonication in 10 cycles of15 s, pulsed with the sample kept on ice bath. The lysate wascentrifuged at 20,000 rpm for 40min to an efficient clarification andseparation of the soluble fraction, at temperature of 8 �C. The crudeextract was mixed in a 1:5 proportion with the 20 mM phosphatebuffer at pH 8.0, containing 200 mM NaCl. This solution was thencombined with the amylose resin slurry and incubated for 15 min.The fraction containing the rLdNHeMBP was purified by affinitychromatography an amylose resin column (New England Biolabs�
Inc). Theproteinwaselutedwith10mMmaltose solution in the samebuffer and collected in fractions of 3 mL. The rLdNHeMBP proteinwas detected by the Lowry method [25]. The protein purity wasconfirmed by SDS-polyacrylamide with Coomassie brilliant blue asdescribed by Laemmli et al. [26] in gel electrophoresis.
4.2.2. Kinetic assays by 1H Nuclear Magnetic Resonancespectroscopy
The initial kinetic measurements were performed on a BrukerAvance� DRX-400 spectrometer (400MHz) at 25 �C using a 1H pulseangle of 90�, an acquisition time of 1.6 s with 150 scans, with 2 srelaxation delay (d1), and the chemical shifts were reported in partspermillion (ppm) using the HOD signal as reference (4.77 ppm). Theenzymatic substrate transformations and parallel measurementswere carried in 5 mm NMR sample tubes. The inosine solution(3.5mM)was prepared using 20mMphosphate buffer pH 8.0 inD2Oand the solution was lyophilized and dissolved again in the samebuffer solvent. Finally, the sample was prepared in the NMR tubewith 3.5 mM final volume of inosine in 600 mL of the 20 mM phos-phate buffer pH 8.0 in D2O. After acquisition of the pure inosine 1HNMR spectrum the enzyme solution was added to the sample toobtain a final 1 mM enzyme concentration. The kinetics was moni-tored for about 63 min following the integration of the hydrogensignals of hypoxanthine, which have distinct chemical shifts (8.19and 8.17 ppm) in relation with the signals of inosine (8.32 and8.22 ppm). The minimization of the water signal was executed bypresaturation for 1.0 s. All theNMR spectrawere collected, processedand analyzed by the software Spinworks 2.5 fromWindows [27].
4.2.3. Kinetic measurements with UVevis spectrophotometryKinetic measurements were carried out with a UVevisible
spectrophotometer (Biospectro e Model SP-220) using 1 cmpatch length quartz cuvettes. The kinetic parameters ofrLdNHeMBP were determined using a coupled assay using XO fora fast conversion of the inosine hydrolysis product, hypoxanthine(lmax 250 nm), which posses a lmax very similar to inosine (lmax249 nm), to uric acid (lmax 293 nm). The rLdNHeMBP activity wasthenmeasured using the principle that the hypoxanthine producedfrom hydrolysis of inosine by rLdNHeMBP was completely trans-formed into uric acid with an excess of commercial XO (60 milli-units/mL) [28]. Uric acid was monitored spectrophotometrically at293 nm (D 3uric acid ¼ 12.9 mM�1 cm�1) [7,29]. The Michaelisconstants (KM) and maximal velocity (Vmax) for inosine hydrolysiswere determined by reactions started by adding 13 nMrLdNHeMBP, in 20 mM phosphate buffer, pH 8.0 and final 1.0 mLvolume, to the mixtures containing variable concentrations ofinosine. Uric acid formation was monitored at 293 nm, in triplicateat room temperature. The parameters were evaluated by non-linearregression using the MichaeliseMenten equation, Vo ¼ Vmax*[S]/
([S] þ KM), where [S] is the substrate concentration, Vmax is themaximal velocity and KM is the Michaelis constant. In order todeterminate the enzymatic rates there were used several concen-trations of inosine from 0.3 to 3.0 KM, with points above and belowKM, until saturation conditions with the concentration ofrLdNHeMBP kept constant at 13 nM/mL. The saturation curve wasobtained from the values of the initial velocity versus the substrateconcentrations plotted as a nonlinear regression using the equa-tion of MichaeliseMenten for determining the values of Vmax andKM [19].
Values of IC50 were independently determined by ratemeasurements for at least six inhibitor concentrations with fixedconcentrations of enzyme (13 nM) and inosine (0.3 mM). The datawere analyzed based on fits to slope-nonlinear plots. The Ki valuesfor the inhibitors were determined at a substrate concentrationclose to the KM value and using five or six inhibitor concentrations.To calculate Ki from the IC50 values was used the ChengePrussofequation, Ki ¼ [IC50/(1 þ [S]/KM)] [17].
The type of inhibition was determined under the same experi-mental conditions and conducted with five increasing concentra-tions of substrate (0.2, 0.4, 0.5, 0.6, 0.9 mM), and three fixedconcentrations of each inhibitor in each experiment. In parallel trialswere also carried out in the absence of inhibitor. For each concen-tration of inhibitor a new double-reciprocal plot was designed. Todetermine the type of inhibition of compounds 1 and 2 the exper-imental data obtained were analyzed by linear regression, gener-ating the double reciprocal plots of LineweavereBurk [18]. Theparameters usedwere based on results obtained from theKM for thisenzyme and the experiments were conducted from interactivetesting based on literature [19]. The values represent the average ofthree individual experiments. All inhibition patterns were deter-mined at pH 8.0 and provided good fits to slope-linear competitiveinhibition. The data were obtained from the experimental graphs ofbest fit linear regression. The values correspond to themean� standard error of three individual experiments. The kineticsparameters were determined from the collected data employing theSigma Plot 10.0 Enzyme Kinetics Module (Systat Software Inc.).
4.2.4. Docking studiesThe 3D structure of LdNHused for docking studieswas themodel
proposed by França et al. [14], which was built using as templatesthe 3D structures of L. major and C. fasciculataNHs (LmNH and CfNH)obtained from the Protein Data Bank [30] under the codes PDB ID:1EZR [2] and PDB ID: 2MAS respectively [16]. The 3D structure of theinhibitor pAPIR (Fig. 1) and the cofactor Ca2þ co-crystallized withCfNH, were copied into the LdNH active site and used as a referencefor docking of inosine and compounds 1 and 2 (Fig. 1) using theprogramMolegro Virtual Docker (MVD) [31]. The 3D structures of theligands were built using the program PC Spartan Pro� [32] and theirpartial atomic charges calculated by the RM1 semi-empiricalmethod. The compounds were docked in the LdNH binding siteusing MVD [31] according to the same procedure adopted in formerworks with NHs [33]. The binding site was restricted into a spherewith a radius of 6 �A in which all residues were considered flexible.Due to the stochastic nature of the docking algorithm, about 20 runswere performed for each compound with 50 poses (conformationand orientation of the ligand) returned to the analysis of the overlapwith the pAPIR present in the active site of LdNH and theligandeprotein interactions. The best conformation of eachcompound was selected according to its degree of structural simi-larity to pAPIR and the evaluation of the best energy of interactionwith the enzyme.
To validate the docking protocol we first performed the dockingsimulation of pAPIR against the active site of LdNH and compared tothe crystallographic structure of CfNH (re-docking).

M.N. Rennó et al. / European Journal of Medicinal Chemistry 56 (2012) 301e307 307
Acknowledgments
We thank CNPq (The National Council for Scientific andTechnological Development), FAPERJ (Support Research Founda-tion of the State of Rio de Janeiro, CNE fellowship and FAPERJ-Pensa Rio project), CAPES and IMBEB for their financialsupport. We also thank the National Center of Nuclear MagneticResonance Jiri Jonas facilities for the NMR spectra and ProfessorsVitor F. Ferreira and Maria Cecília B.V. de Souza from the Flu-minense Federal University for supplying the samples ofcompounds 1 and 2. We also thank CAPES for the graduatescholarship of M.N. Rennó.
References
[1] R. Duncan, S. Gannavaram, R. Dey, A. Debrabant, I. Lakhal-Naouar,H.L. Nakhasi, Mol. Biol. Int. (2011) 1e10.
[2] W. Shi, W.L. Schramm, S.C. Almo, J. Biol. Chem. 274 (1999) 21114e21120.[3] H.C. Maltezou, J. Biomed. Biotech. (2010) 1e8.[4] M.K. Mittal, S. Misra, M. Owais, N. Goyala, Protein Expr. Purif. 40 (2005)
279e286.[5] D.J. Hammond, W.E. Gutteridge, Mol. Biochem. Parasitol. 13 (1984)
243e261.[6] M. Degano, D.N. Gopaul, G. Scapin, V.L. Schramm, J.C. Sacchettini, Biochemistry
35 (1996) 5971e5980.[7] W. Versées, J. Barlow, J. Steyaert, J. Mol. Biol. 359 (2006) 331e346.[8] W. Versées, J. Steyaert, Curr. Opin. Struct. Biol. 13 (2003) 731e738.[9] L. Cui, G.R. Rajasekariah, S.K. Martin, Gene 280 (2001) 153e162.
[10] J. Lukes, I.L. Mauricio, G. Schönian, J.C. Dujardin, K. Soteriadou, J.P. Dedet,K. Kuhls, W.Q. Tintaya, M. Jirku, E. Chocholova, C. Haralambous, F. Pratlong,M. Oborník, A. Horák, F.J. Ayala, M.A. Miles, Proc. Natl. Acad. Sci. U. S. A. 104(2007) 9375e9380.
[11] I.L. Mauricio, M. Yeo, M. Baghaei, D. Doto, F. Pratlong, E. Zemanova, J.P. Dedet,J. Lukes, M.A. Miles, Int. J. Parasitol. 36 (2006) 757e769.
[12] D. Nico, C. Claser, G.P. Borja-Cabrera, L.R. Travassos, M. Palatnik, I.S. Soares,M.M. Rodrigues, C.B. Palatnik-de-Sousa, PLoS Negl. Trop. Dis. 4 (11) (2010)e866.
[13] B. Giabbai, M. Degano, Structure 12 (2004) 739e749.[14] T.C.C. França, M.R.M. Rocha, B.M. Reboredo, M.N. Rennó, L.W. Tinoco,
J.D. Figueroa-Villar, J. Braz. Chem. Soc. 19 (2008) 64e73.[15] D.M. Santana, G.P. Borja-Cabrera, E.P. Souza, N.R. Sturm, C.P. Sousa,
D.A. Campbell, Mol. Biochem. Parasitol. 120 (2002) 315e319.[16] M. Degano, S.C. Almo, J.C. Sacchettini, V.L. Schramm, Biochemistry 37 (1998)
6277e6285.[17] Y.C. Cheng, W.H. Prussof, Biochem. Pharmacol. 22 (1973) 3099e3108.[18] H. Lineweaver, D. Burk, J. Am. Chem. Soc. 56 (1934) 658e666.[19] I.H. Segel, Enzyme Kinetics Behaviour and Analysis of Rapid Equilibrium and
Steady-State Enzyme System. Wiley Classics Library Edition Published (1993).p. 957.
[20] G.L. Warren, A.C. Webster, A.M. Capelli, B. Clarke, J. LaLonde, M.H. Lambert,M. Lindvall, N. Nevins, S.F. Semus, S. Senger, G. Tedesco, I.D. Wall,J.M. Woolven, C.E. Peishoff, M.S. Head, J. Med. Chem. 49 (2006) 5912e5931.
[21] A.R. Leach, B.K. Shoichet, C.E. Peishoff, J. Med. Chem. 49 (2006) 5851e5855.[22] M. Kontoyanni, L.M. McClellan, G.S. Sokol, J. Med. Chem. 47 (2004) 558e565.[23] R.B. Kapust, D.S. Waugh, Prot. Sci. 8 (1999) 1668e1674.[24] A.D. da Matta, C.V.B. dos Santos, H.S. Pereira, I.C.P.P. Frugulhetti, M.R.P. de
Oliveira, M.C.B.V. de Souza, N. Moussatche, V.F. Ferreira, Heteroat. Chem. 10(1999) 197e202.
[25] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, J. Biol. Chem. 193 (1951)265e275.
[26] U.K. Laemmli, Nature 227 (1970) 680e685.[27] K. Marat, Spinworks 255. Available at: ftp://davinci.chem.umanitoba.ca/pub/
marat/SpinWorks/, (accessed 17.3.08).[28] A. Lewandowicz, E.A. Ringia, L.M. Ting, K. Kim, P.C. Tyler, G.B. Evans, J. Biol.
Chem. 280 (2005) 30320e30328.[29] B.K. Kim, S. Cha, R.E. Parks Jr., J. Biol. Chem. 243 (1968) 1771e1776.[30] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig,
I.N. Shindyalov, P.E. Bourne, Nucleic Acids Res. 28 (2000) 235e242.[31] R. Thomsen, M.H. Christensen, J. Med. Chem. 49 (2006) 3315e3321.[32] W.J. Hehre, B.J. Deppmeier, P.E. Klunzinger, PC SPARTAN Pro, Wavefunction,
Irvine, CA, 1999.[33] A.P. Guimarães, A.A. Oliveira, E.F.F. Cunha, T.C. Ramalho, T.C.C. França,
J. Biomol. Struct. Dynam. 28 (2011) 455e469.