Embed Size (px)
Citation preview

Article No. jmbi.1999.3329 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 294, 1203±1213
Kinetic Analysis of Peptide Binding to the TAPTransport Complex: Evidence for StructuralRearrangements Induced by Substrate Binding
Lars Neumann and Robert TampeÂ*
Institut fuÈ r PhysiologischeChemie, Philipps-UniversitaÈtMarburg, Karl-von-Frisch-Str.1, 35033 Marburg, Germany
E-mail address of the [email protected]
Abbreviations used: ABC, ATP bAEBSF, 4-(2-Aminoethyl) benzenesALD, adrenoleukodystrophy; CFTRtransmembrane conductance regulsecond; ER, endoplasmic reticulumperformance liquid chromatographchain acyl dehydrogenase; MDR, mMHC, major histocompatibility comnucleotide binding domain; PBS, psaline; DTT, 1,4-dithio-DL-threitol;dodecylsulfate; TAP, transporter asprocessing. Single letter code is usesequences.
0022-2836/99/501203±11 $30.00/0
The transporter associated with antigen processing (TAP) plays a keyrole in the class I major histocompatibility complex (MHC) mediatedimmune surveillance. It translocates peptides generated by the protea-some complex into the endoplasmic reticulum (ER) for loading ontoMHC class I molecules. At the cell surface these MHC complexes aremonitored for their antigenic cargo by cytotoxic T-lymphocytes. Peptidebinding to TAP is the essential step for peptide selection and for sub-sequent ATP-dependent translocation into the ER lumen. To examine thepathway of substrate recognition by TAP, we employed peptide epitopes,which were labeled with an environmentally sensitive ¯uorophore. Uponbinding to TAP, a drastic ¯uorescence quenching of the ¯uorescent sub-strate was detected. This allowed us to analyze TAP function in real-timeby using a homogeneous assay. Formation of the peptide-TAP complexis composed of a fast association step followed by a slow isomerizationof the transport complex. Proton donor groups moving in proximity tothe ¯uorescence label cause ¯uorescence quenching. Taken together, thispeptide-induced structural reorganization may re¯ect the crosstalk ofstructural information between the peptide binding site and both nucleo-tide-binding domains within the TAP complex.
# 1999 Academic Press
Keywords: ABC transporter; antigen processing; antigen presentation;binding kinetic; ¯uorescence quenching
*Corresponding authorIntroduction
Cytotoxic T-lymphocytes recognize and elimin-ate virus-infected or malignant cells. This requiresthe presentation of fragments derived from viral ortumor-speci®c proteins on the cell surface in associ-ation with major histocompatibility complex(MHC) class I molecules (Pamer & Cresswell,
ing author:
inding cassette;ulfonyl ¯uoride;, cystic ®brosis
ator; cps, counts per; HPLC, highy; MCAD, medium-ultidrug resistance;plex; NBD,
hosphate-bufferedSDS, sodiumsociated with antigend for peptide
1998). These peptides are generated in the cytosolmainly by proteasomal degradation (Coux et al.,1996; York & Rock, 1996). Subsequently, the MHC-encoded transporter associated with antigen pro-cessing (TAP) translocates these peptides into theendoplasmic reticulum (ER), where chaperone-assisted assembly and loading of MHC class I mol-ecules occurs (Uebel & TampeÂ, 1999). Kineticallystable MHC-peptide complexes can leave the ER tothe cell surface, where they are screened by cyto-toxic T-lymphocytes.
TAP belongs to the family of ATP binding cas-sette (ABC) transporters, which are characterizedby the highly conserved Walker A/B motifs andthe C-loop signature (Higgins, 1992). These trans-port proteins are found in prokaryotes, eukaryotesand archea, where they are responsible for trans-port of a very broad spectrum of substrates. Someimportant members of the ABC-transporter familyare the multidrug resistance P-glycoprotein (MDR),the cystic ®brosis transmembrane conductance reg-ulator (CFTR), the oligopeptide transporter of Sal-monella typhimurium, and the pheromone exporter
# 1999 Academic Press

1204 Kinetics of Peptide Binding to TAP
Ste6 of yeast. TAP is localized in the ER membraneand composed of two subunits TAP1 and TAP2,each containing a transmembrane and nucleotidebinding domain (NBD) (Kelly et al., 1992; Kleijmeeret al., 1992). This heterodimer is responsible forpeptide transport into the ER-lumen ensuring pep-tide loading onto MHC class I molecules(Androlewicz et al., 1993; Meyer et al., 1994; Neefjeset al., 1993; Shepherd et al., 1993). Peptides of 8-16amino acids in length bind to TAP with equal af®-nity (van Endert et al., 1994), whereas peptides of9-12 residues are found to be optimal for transport(Koopmann et al., 1996). TAP accepts peptides withvery large bulky side-chains such as ¯uorophores(Uebel et al., 1995) or branched peptides (GrommeÂet al., 1997), indicating a sterically ¯exible sub-strate-binding site. Using combinatorial peptidelibraries, the recognition principle of TAP was dis-closed in the absence of any sequence context(Uebel et al., 1997; Uebel & TampeÂ, 1997). Peptidesare bound by ®xing the ®rst three N-terminal andlast C-terminal residues via their side-chains andbackbone, while the residues in between do notcontribute to substrate af®nity in a signi®cant man-ner. This binding motif combines maximal diver-sity in a region of the epitope where T-cell receptorcontacts are made with maximal binding af®nity toincrease the ef®ciency of antigen processing (Uebel& TampeÂ, 1999).
The transport mechanism of TAP can be dis-sected in an ATP-independent peptide binding andan ATP-dependent translocation step (Uebel et al.,1995; van Endert et al., 1994). ATP binding doesnot require the presence of peptides (MuÈ ller et al.,1994; Russ et al., 1995). Here we studied the pep-tide binding mechanism, which is the initial step ofpeptide transport using ¯uorescein-labeled pep-tides. The ¯uorescence of these peptides is signi®-cantly quenched upon binding to TAP. Bydeveloping a homogeneous assay, we are able toresolve the pathway of peptide binding to the TAPcomplex in real-time. The kinetic analysis revealedthat a fast peptide association step is followed by aslow conformational change of TAP. The ¯uor-escence quenching is caused by a structuralrearrangement of the TAP-peptide complex mov-ing a proton donor group in close proximity to the¯uorophore.
Results
Fluorescein-labeled peptides are bound andtransported by TAP
The observation that TAP accepts peptides withvery bulky side-chains opens up new alternativeapproaches to study structure and function of theTAP complex. Fluorophores for example can sensesmall changes in pH or polarity even at very lowconcentrations (Ohkuma & Poole, 1978). To adoptthis methodology, we substituted each residue ofthe HLA-B27-restricted epitope RRYQKSTEL bycysteine. Fluorescein was coupled via regiospeci®c
thiol chemistry, yielding to cysteine-acetamido-¯uorescein (f). For each reporter peptide, we ana-lyzed the in¯uence of the label on peptide bindingand transport by TAP. The af®nity of each labeledepitope was studied by saturation-binding assaysas shown for RRYfKSTEL (Figure 1(a)). Theamount of bound peptide can be ®tted using the(1:1 or Langmuir) binding model (equation (5)). Inthe case of RRYfKSTEL, the af®nity constant Kd of93(�9) nM was determined. This value is verysimilar to the Kd of the radiolabeled peptideRRYQKSTEL (Uebel et al., 1995). Comparison of alllabeled peptides revealed that the position of the¯uorophore is critical for peptide recognition byTAP (Figure 1(b)). Fluorescein labeling at theamino or carboxy-terminal residue abolished thebinding to TAP almost completely, whereas coup-ling at the positions two or three reduced the af®-nity only about two- to fourfold in comparison tothe peptide RRYQKSTEL. Labeling of the positionfour to eight did not affect or even increased pep-tide-binding af®nity to TAP by a factor of two tofour. These results imply that the peptide-bindingpocket of TAP is sterically restricted only at pos-itions one, two, three and nine of a bound nona-mer. This con®rms the substrate-binding motif ofTAP identi®ed by combinatorial peptide libraries(Uebel et al., 1997).
Next, we analyzed whether these ¯uorescentpeptides are transported by TAP. In order to trapand detect translocated peptides, an N-glycosyla-tion targeting signal sequence (NST) wasintroduced. To avoid any in¯uence on the N-glyco-sylation machinery the ¯uorophore was shiftedthree residues apart from the asparagine residueyielding the peptide RRYQNSTfL. Glycosylatedand therefore transported peptides were extractedby ConA-Sepharose binding. As shown forRRYQNSTfL, translocation of the ¯uorescent sub-strate is ATP-dependent and peptide-speci®c,demonstrating that the bulky ¯uorophore does nothinder peptide translocation (Figure 1(c)). ATP-dependent transport was observed for all ¯uor-escent peptides except the ones labeled at theamino or carboxy terminus. In addition, the bind-ing and transport of ¯uorescein-labeled peptides isblocked by the TAP-speci®c viral inhibitor ICP47(data not shown) (Ahn et al., 1996; Tomazin et al.,1996). Thus, the ¯uorescein-labeled peptides usedhere are fully accepted as substrates for TAP andpresent a very potent tool to investigate substraterecognition of TAP.
Quenching of fluorescent peptides in thesubstrate binding pocket of TAP
Because ¯uorescein is very sensitive to changesof the surrounding polarity or pH value, we ana-lyzed the ¯uorescence emission during the processof substrate binding and dissociation. After TAP-containing microsomes were added to a solution ofRRYfKSTEL, a striking decrease of the ¯uor-escence signal was observed, reaching equilibrium
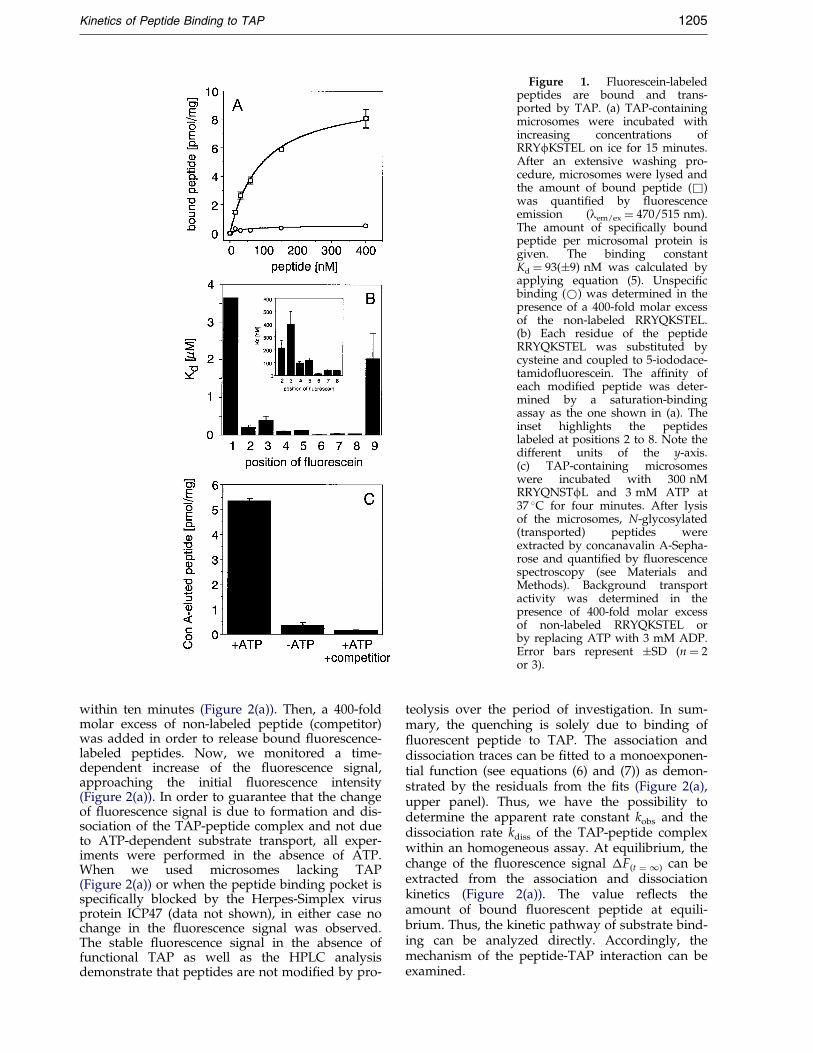
Figure 1. Fluorescein-labeledpeptides are bound and trans-ported by TAP. (a) TAP-containingmicrosomes were incubated withincreasing concentrations ofRRYfKSTEL on ice for 15 minutes.After an extensive washing pro-cedure, microsomes were lysed andthe amount of bound peptide (&)was quanti®ed by ¯uorescenceemission (lem/ex � 470/515 nm).The amount of speci®cally boundpeptide per microsomal protein isgiven. The binding constantKd � 93(�9) nM was calculated byapplying equation (5). Unspeci®cbinding (*) was determined in thepresence of a 400-fold molar excessof the non-labeled RRYQKSTEL.(b) Each residue of the peptideRRYQKSTEL was substituted bycysteine and coupled to 5-iododace-tamido¯uorescein. The af®nity ofeach modi®ed peptide was deter-mined by a saturation-bindingassay as the one shown in (a). Theinset highlights the peptideslabeled at positions 2 to 8. Note thedifferent units of the y-axis.(c) TAP-containing microsomeswere incubated with 300 nMRRYQNSTfL and 3 mM ATP at37 �C for four minutes. After lysisof the microsomes, N-glycosylated(transported) peptides wereextracted by concanavalin A-Sepha-rose and quanti®ed by ¯uorescencespectroscopy (see Materials andMethods). Background transportactivity was determined in thepresence of 400-fold molar excessof non-labeled RRYQKSTEL orby replacing ATP with 3 mM ADP.Error bars represent �SD (n � 2or 3).
Kinetics of Peptide Binding to TAP 1205
within ten minutes (Figure 2(a)). Then, a 400-foldmolar excess of non-labeled peptide (competitor)was added in order to release bound ¯uorescence-labeled peptides. Now, we monitored a time-dependent increase of the ¯uorescence signal,approaching the initial ¯uorescence intensity(Figure 2(a)). In order to guarantee that the changeof ¯uorescence signal is due to formation and dis-sociation of the TAP-peptide complex and not dueto ATP-dependent substrate transport, all exper-iments were performed in the absence of ATP.When we used microsomes lacking TAP(Figure 2(a)) or when the peptide binding pocket isspeci®cally blocked by the Herpes-Simplex virusprotein ICP47 (data not shown), in either case nochange in the ¯uorescence signal was observed.The stable ¯uorescence signal in the absence offunctional TAP as well as the HPLC analysisdemonstrate that peptides are not modi®ed by pro-
teolysis over the period of investigation. In sum-mary, the quenching is solely due to binding of¯uorescent peptide to TAP. The association anddissociation traces can be ®tted to a monoexponen-tial function (see equations (6) and (7)) as demon-strated by the residuals from the ®ts (Figure 2(a),upper panel). Thus, we have the possibility todetermine the apparent rate constant kobs and thedissociation rate kdiss of the TAP-peptide complexwithin an homogeneous assay. At equilibrium, thechange of the ¯uorescence signal �F(t � 1) can beextracted from the association and dissociationkinetics (Figure 2(a)). The value re¯ects theamount of bound ¯uorescent peptide at equili-brium. Thus, the kinetic pathway of substrate bind-ing can be analyzed directly. Accordingly, themechanism of the peptide-TAP interaction can beexamined.

Figure 2. Fluorescence quenching of peptides boundto TAP. (a) The ¯uorescence emission was recorded(lex/em � 470/515 nm) after mixing microsomes contain-ing (grey) or lacking TAP (black) with 40 nM RRYfK-STEL in the absence of ATP at 10 �C (see Materials andMethods). The concentration of TAP and of total proteinwas 2 nM and 100 mg/ml, respectively. Fluorescence-labeled peptide bound to TAP was released by adding a400-fold molar excess of non-labeled RRYQKSTEL. Theassociation and dissociation process were ®tted tomonoexponential functions (see equations (6) and (7))(continuous line). (b) The level of ¯uorescence quench-ing depending on the position of the introduced ¯uor-escein molecule was quanti®ed by comparing the¯uorescence emission (lex/em � 470/515 nm) of 1.2 mM¯uorescent peptide in the presence (Ffree) and absence(Fbound) of a 400-fold molar excess of non-labeled compe-titor peptide at 10 �C. A TAP concentration of 30 nMwas used.
1206 Kinetics of Peptide Binding to TAP
Fluorescence quenching depends on theposition of the fluorophore
Quenching was quanti®ed by the ratio betweenthe ¯uorescent signal measured after reaching
binding equilibrium (Fbound) and after competingbound ¯uorescent peptide with a 400-fold molarexcess of non-labeled peptide (Ffree) (Figure 2(b)).Fluorescent peptides have different af®nities forTAP. In order to ensure the same ratio of boundand free peptides, a peptide concentration of1.2 mM was used. This represents the saturationconcentration of the peptide with lowest af®nity(RRfQKSTEL). The largest quenching effect wasobserved for position two, three, or four of thenonameric peptide (Figure 2(b)). While the level of¯uorescence quenching is equal at these positions,it decreases step-wise if the ¯uorophore is movedfurther to the carboxy terminus. Peptides labeled atposition one and nine could not be analyzedbecause their af®nity for TAP was below the detec-tion limit. In conclusion, upon peptide binding toTAP, the changes in the environment are moredrastic in the amino-terminal region of the peptidethan in the middle and in the C-terminal segment.Because the peptide RRYfKSTEL shows high af®-nity binding to TAP and the strongest quenching,we selected this peptide for further kinetic anal-ysis.
Peptide binding to TAP is a two-step process
In order to extract the molecular steps of pep-tide recognition and binding, we analyzed theassociation and dissociation kinetics by variationof substrate concentrations under pseudo-®rst-order conditions (for details of the kinetic anal-ysis see Appendix). Raising the concentration oflabeled peptide, an increase in association rateand ¯uorescence quenching (�F) was observed(Figure 3(a)). At equilibrium, ¯uorescencequenching (�F(t � 1)) reached saturation at highconcentrations of ¯uorescent peptide. Addition ofa 400-fold excess of non-labeled peptide induceddissociation of the TAP-peptide complex restor-ing the initial ¯uorescence intensity (Figure 3(b)).All association and dissociation reactions couldbe ®tted to a monoexponential function derivedfrom the 1:1 binding model (A � B$AB). Kineticanalysis was performed at 10 �C to avoid theloss of TAP function during prolonged incu-bation in the absence of ATP (L.N. & R.T.,unpublished data; van Endert, 1999). The anal-ysis of the peptide-TAP interaction over a broadtemperature range (6-26 �C) demonstrated thatthe kinetics remain monoexponential. At highertemperatures the association and dissociationreactions become much faster (L.N. & R.T.,unpublished results).
The dependence of kobs and kdiss on the peptideconcentration is summarized in Figure 4(a). Thedissociation rate kdiss of 2.1 � 10ÿ3 sÿ1 is concen-tration-independent. However, the observedassociation rate kobs does not linearly depend onthe peptide concentration as we expected for asimple ligand-receptor interaction. Most strikingly,kobs shows a hyperbolic dependence on the peptideconcentration, reaching a constant value at high

Figure 3. Concentration-depen-dent peptide association and dis-sociation kinetics. (a) Theassociation kinetics at various con-centrations of RRYfKSTEL to TAP-containing microsomes wasmeasured at 10 �C in the absence ofATP. The time-dependent ¯uor-escence quenching �F was plotted.(b) After reaching equilibrium thedissociation of the peptide-TAPcomplex was monitored by addinga 400-fold molar excess of non-labeled peptide.
Kinetics of Peptide Binding to TAP 1207
peptide concentrations. This is diagnostic for atwo-step process composed of a fast bimolecularassociation step followed by slow unimolecularisomerization reaction (Hiromi, 1979) (Scheme 1)(for details see Appendix):
Assuming that k�1 [P] � kÿ14k�2 � kÿ2, the dis-sociation constant of the ®rst step Kÿ1 and the rateconstants for the second step k�2, kÿ2 can be calcu-lated from the observed association rate at variouspeptide concentrations [P] (Hiromi, 1979) (seeAppendix):
kobs � k�2�P�Kÿ1 � �P� � kÿ2; with Kÿ1 � kÿ1
k�1�1�
All kinetic parameters determined from the kin-etic analysis are summarized in Table 1. The strictmonoexponential behavior of the association anddissociation kinetics for all peptide concentrations,as well as the hyperbolic dependence of saturation
Table 1. Rate and equilibrium constants
Rate constants (sÿ1 � 103) Equilibrium constants
k�2 � 7.4 � 0.9 Kÿ1�32.0�3.0 nMkÿ2 � 2.9 � 0.4 K�2�2.6�0.2kdiss � 2.1 � 0.8 Ktotal1�9.1�1.6 nM
Ktotal2�12.0�1.0 nM
The parameter kÿ2, k�2 and Kÿ1 were calculated from thedata shown in Figure 4(a). The dissociation constant kdiss wasdetermined by averaging the rate constants given for differentpeptide concentrations (n � 5) in Figure 4(a). The equilibriumconstant K�2 for the slow second step was calculated usingequation (2). The dissociation constant Ktotal for the total pro-cess was determined by either equation (3) (Ktotal
1 ) or equation(4) (Ktotal
2 ).
behavior of kobs indicate that the second step isexclusively responsible for the observed ¯uor-escence quenching (Hiromi, 1979). kÿ2 extractedfrom equation (1) and kdiss (see Figure 3(b)) appearto be identical in the range of error.
The parameters were used to calculate the equili-brium binding constant of the second step K�2 andthe dissociation constant Ktotal of the overall pro-cess (Table 1):
K�2 � k�2
kÿ2; Ktotal � Kÿ1
1� K�2�2��3�
In an alternative approach and additional cross-check, the equilibrium constant Ktotal of the overallbinding process was analyzed by plotting the equi-librium ¯uorescence quenching (�F(t � 1)) versuspeptide concentration (Figure 4(b)). Applyingequation (4), the equilibrium constant Ktotal andthe maximal equilibrium ¯uorescence quenching�F(t � 1)max was determined:
�F�t�1� � �F�t�1�max�P�Ktotal � �P� �4�
The equilibrium constants Ktotal extracted from thekinetic analysis (9.1 (� 1.6) nM) and from �F(t � 1)
(12.0(� 1.0) nM) are in very good agreement(Table 1).
Fluorescence-labeled peptides sense a protondonating group within the substrate bindingpocket of TAP
The ¯uorescence of ¯uorescein is quenched bychanging the hydrophobicity or by decreasing thepH of the solvent (Ohkuma & Poole, 1978). Protona-tion of the carboxy group of ¯uorescein results in¯uorescence quenching. If the interaction of a ¯uor-escein-labeled ligand with its receptor results in achange of the microenvironment of the ¯uorophore,a decrease in ¯uorescence intensity might beobserved. This was demonstrated for example forthe serotonin receptor 5HT3 and its ¯uorescein-labeled ligand (Tairi et al., 1998). In order to eluci-
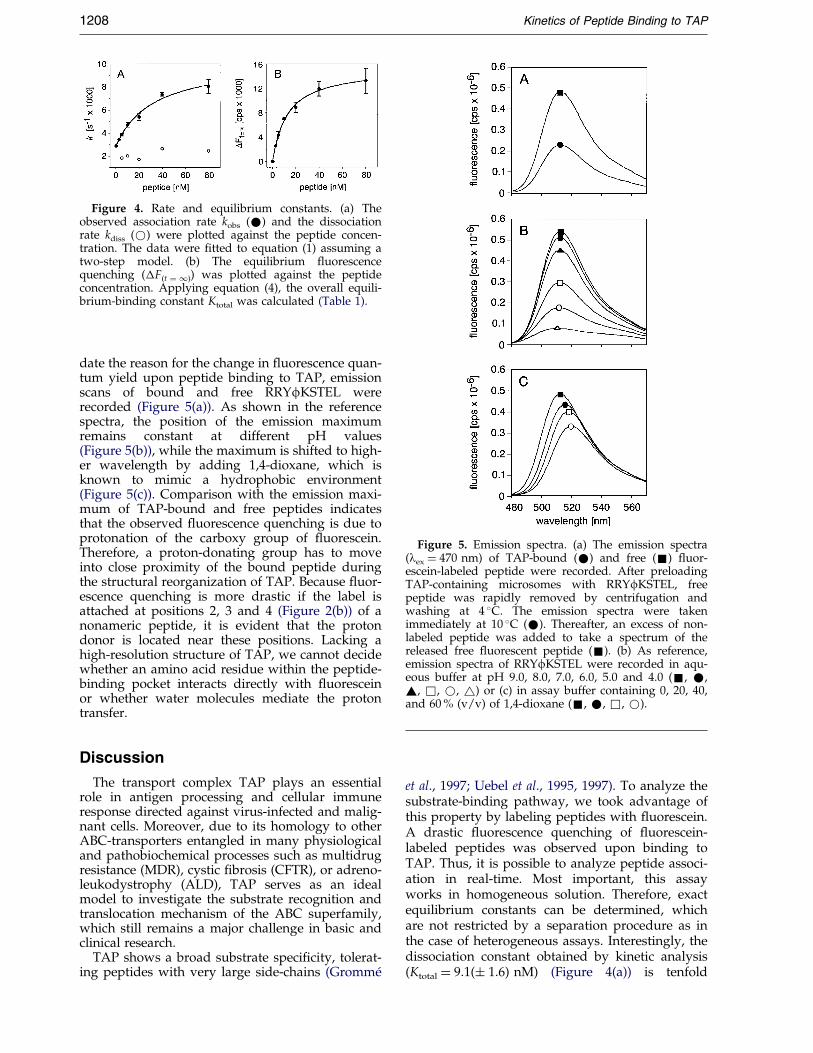
Figure 4. Rate and equilibrium constants. (a) Theobserved association rate kobs (*) and the dissociationrate kdiss (*) were plotted against the peptide concen-tration. The data were ®tted to equation (1) assuming atwo-step model. (b) The equilibrium ¯uorescencequenching (�F(t � 1)) was plotted against the peptideconcentration. Applying equation (4), the overall equili-brium-binding constant Ktotal was calculated (Table 1).
Figure 5. Emission spectra. (a) The emission spectra(lex � 470 nm) of TAP-bound (*) and free (&) ¯uor-escein-labeled peptide were recorded. After preloadingTAP-containing microsomes with RRYfKSTEL, freepeptide was rapidly removed by centrifugation andwashing at 4 �C. The emission spectra were takenimmediately at 10 �C (*). Thereafter, an excess of non-labeled peptide was added to take a spectrum of thereleased free ¯uorescent peptide (&). (b) As reference,emission spectra of RRYfKSTEL were recorded in aqu-eous buffer at pH 9.0, 8.0, 7.0, 6.0, 5.0 and 4.0 (&, *,~, &, *, ~) or (c) in assay buffer containing 0, 20, 40,and 60 % (v/v) of 1,4-dioxane (&, *, &, *).
1208 Kinetics of Peptide Binding to TAP
date the reason for the change in ¯uorescence quan-tum yield upon peptide binding to TAP, emissionscans of bound and free RRYfKSTEL wererecorded (Figure 5(a)). As shown in the referencespectra, the position of the emission maximumremains constant at different pH values(Figure 5(b)), while the maximum is shifted to high-er wavelength by adding 1,4-dioxane, which isknown to mimic a hydrophobic environment(Figure 5(c)). Comparison with the emission maxi-mum of TAP-bound and free peptides indicatesthat the observed ¯uorescence quenching is due toprotonation of the carboxy group of ¯uorescein.Therefore, a proton-donating group has to moveinto close proximity of the bound peptide duringthe structural reorganization of TAP. Because ¯uor-escence quenching is more drastic if the label isattached at positions 2, 3 and 4 (Figure 2(b)) of anonameric peptide, it is evident that the protondonor is located near these positions. Lacking ahigh-resolution structure of TAP, we cannot decidewhether an amino acid residue within the peptide-binding pocket interacts directly with ¯uoresceinor whether water molecules mediate the protontransfer.
Discussion
The transport complex TAP plays an essentialrole in antigen processing and cellular immuneresponse directed against virus-infected and malig-nant cells. Moreover, due to its homology to otherABC-transporters entangled in many physiologicaland pathobiochemical processes such as multidrugresistance (MDR), cystic ®brosis (CFTR), or adreno-leukodystrophy (ALD), TAP serves as an idealmodel to investigate the substrate recognition andtranslocation mechanism of the ABC superfamily,which still remains a major challenge in basic andclinical research.
TAP shows a broad substrate speci®city, tolerat-ing peptides with very large side-chains (GrommeÂ
et al., 1997; Uebel et al., 1995, 1997). To analyze thesubstrate-binding pathway, we took advantage ofthis property by labeling peptides with ¯uorescein.A drastic ¯uorescence quenching of ¯uorescein-labeled peptides was observed upon binding toTAP. Thus, it is possible to analyze peptide associ-ation in real-time. Most important, this assayworks in homogeneous solution. Therefore, exactequilibrium constants can be determined, whichare not restricted by a separation procedure as inthe case of heterogeneous assays. Interestingly, thedissociation constant obtained by kinetic analysis(Ktotal � 9.1(� 1.6) nM) (Figure 4(a)) is tenfold

Kinetics of Peptide Binding to TAP 1209
lower than the one measured by heterogeneous ®l-tra-tion or centrifugation assays (Kd � 93 (� 9) nM)(Figure 1(a)). This difference is probably due to thedilution step before washing and the time lag toseparate free and bound peptides. The higherTAP af®nity may also re¯ect a more ef®cient trans-location of antigenic peptides at low peptide con-centrations expected in vivo.
Most strikingly, analysis of the monoexponentialassociation kinetics revealed a non-linear, hyper-bolic dependence of the association kinetic kobs onpeptide concentration (Figure 4(a)). The data are inagreement with a two-step model composed of afast bimolecular association step followed by aslow unimolecular isomerization step. The strictmonoexponential association kinetic as well as thesaturation behavior of kobs indicates that the secondstep is exclusively responsible for ¯uorescencequenching. Similar two-step interactions includingstructural reorganizations have been identi®ed formany protein-substrate interactions, such as themedium-chain acyl dehydrogenase (MCAD) andits pseudo-substrate 3-indolepropionyl-CoA(Johnson et al., 1992), the muscarinic acetylcholinereceptor and the green mamba toxin (Toomelaet al., 1994), lysozyme and N-acetylglucosamine(Holler et al., 1969), or the Ras-binding domain ofc-Raf-1 and the GTP-bound form of H-Ras (Sydoret al., 1998).
Based on the kinetic analysis, we propose the fol-lowing model of substrate binding to TAP(Figure 6). Formation of the peptide-TAP complexis initiated by a fast binding step. The associationdoes not change the environment in the directvicinity of the ¯uorophore. In the second, slow iso-merization step, TAP-peptide complex undergoes aconformational change, in which a proton donorgroup moves in close proximity of the N-terminalregion of the bound peptide. The effect of ¯uor-escence quenching is most signi®cant if the ¯uoro-phore is attached to positions 2, 3 and 4(Figure 2(b)) of the peptide epitope. There are sev-
Figure 6. Two-step binding model. Peptide binding toTAP is a two-step process composed of a fast bimolecu-lar association step followed by a slow isomerizationreaction, which causes the ¯uorescence quenching of the¯uorescein-labeled peptide in the substrate bindingpocket of TAP.
eral lines of evidence that the structural reorganiza-tion is attributed mostly to TAP and not to thepeptide. First, ¯uorescence quenching is observedfor many peptide epitopes labeled at various pos-itions. Therefore, it seems unlikely that a particularconformation of the peptide causes quenching.Second, the effect on ¯uorescence quenching corre-lates with substrate-binding motif and the stericconstrains of the peptide-binding pocket of TAP(Gubler et al., 1998; Uebel et al., 1997). Third, anextended thermodynamic analysis of the peptideassociation and dissociation process revealed that arather large number of amino acid residues con-tribute to the isomerization, excluding the possi-bility that simply a conformational adaptation ofthe peptide was observed (L.N. & R.T., unpub-lished results). In addition, indirect evidence for amore compact conformation of the TAP hetero-dimer in the presence of peptides was providedby using a crosslinking approach (Lacaille &Androlewicz, 1998).
Green mamba toxin has a high af®nity for themuscarinic acetylcholine receptor (Ktotal � 10 nM)although the dissociation constant for the ®rststep Kÿ1 of the two-step association process wasdetermined to be only 1.4 mM. This is due to anextremely low kÿ2 value relative to its k�2 value(K�2 � k�2/kÿ2 � 140) which causes a trapping ofthe substrate-bound receptor in the rearrangedform (Toomela et al., 1994). Since the dissociationconstant for the overall process of peptide bind-ing to TAP (Ktotal � 12.0(� 1.0) nM) is onlyslightly smaller than the dissociation constant forthe ®rst step (Kÿ1 � 32(� 3) nM), the second stepdoes not signi®cantly contribute to the bindingaf®nity. The isomerization does not seem to lar-gely affect the binding af®nity of TAP. Hence,the structural reorganization of the TAP-peptidecomplex might ful®ll another purpose. Becausethe hydrolysis of ATP is strictly dependent onpeptide binding (S. Gorbulev & R.T., unpub-lished results), it is tempting to speculatewhether the peptide-induced conformationalchange is involved in the communicationbetween the peptide binding site and bothNBDs. The rearranged TAP complex might rep-resent an intermediate within the peptide trans-location cycle, which signals the successfulloading of the peptide-binding site. This signalcould then trigger the ATP-hydrolysis at theNBDs and the translocation of the peptide acrossthe membrane into the ER lumen. The strictcoupling between peptide binding and confor-mational change of the TAP-peptide complexseems highly attractive, because it prevents thespoliation of ATP. The induced ®t of hexokinaseupon binding of glucose was one of the earliestreported examples for a conformational changeavoiding wasteful ATP-hydrolysis (Andersonet al., 1979). Peptide binding and structural reor-ganization of TAP may trigger the crosstalkbetween both NBDs due to a more compact con-formation of TAP enhancing the contact between

1210 Kinetics of Peptide Binding to TAP
both NBDs (Figure 6). This intra/intermolecularcommunication may synchronize the ATP-hydrolysis and the peptide translocation.
Manifold applications for this new ¯uorescencebinding and quenching assay seem to be concei-vable. High-throughput methods under homo-geneous conditions can be developed screeningdifferent TAP-mutants or searching for drugsaffecting TAP-function. Controlling TAP-activityafter solubilization or incorporation into lipo-somes can help to optimize conditions for thereconstitution of TAP. Moreover, the architectureof the peptide-binding site could be mapped.The translocation process of nascent secretoryproteins into the ER and the size of the translo-cation pore was investigated by determining theaccessibility of external quenchers to ¯uoro-phores coupled to de®ned positions within thetranslocated proteins (Crowley et al., 1993, 1994;Hamman et al., 1997). By analogy, ¯uorescencequenching experiments using quencher moleculeswith various size and charge will disclose themicroenvironment of bound peptides. Here, forthe ®rst time, the kinetic pathway of peptidebinding to the TAP complex could be analyzedin real-time using a homogeneous assay. Infuture, related approaches can be applied to fol-low the passage of the substrate through thelocation channel of TAP. It appears to beobvious that besides replacing radioactivemethods ¯uorescence spectroscopy offers numer-ous possibilities to investigate TAP function in avery distinct way.
Materials and Methods
Preparation of TAP-containing microsomes
Coexpression of human TAP1 and TAP2 in Sf9 insectcells has been reported previously (Ahn et al., 1996;Meyer et al., 1994). TAP-containing microsomes were iso-lated by a combination of differential and density gradi-ent centrifugation (Meyer et al., 1994; Uebel et al., 1995).All buffers used for preparation were supplementedwith protease inhibitors (50 mg/ml AEBSF, 1 mg/mlaprotinin, 150 mg/ml benzamidine, 10 mg/ml leupeptin,5 mg/ml pepstatin). Microsomal membranes were resus-pended in PBS, 1 mM DTT (pH 7.4), snap-frozen inliquid nitrogen and stored at ÿ80 �C. Protein concen-tration was analyzed by A280 and MicroBCA (Pierce,Rockford, USA). The concentration of TAP was deter-mined by measuring the maximal amount of speci®callybound peptide (Bmax) using a heterogeneous peptide-binding assay.
Peptide synthesis and fluorescence labeling
All peptides were synthesized by solid phase tech-nique applying conventional Fmoc-chemistry. Peptidescontaining cysteine residues at various positions werecoupled to ¯uorescein by incubating peptides with a 1.2molar excess of 5-iodoacetamido¯uorescein (MolecularProbes, Eugene, USA) in PBS, 20 % (v/v) dimethylforma-mide (pH 6.0) for two hours at room temperature. The
labeled peptides were puri®ed by reversed-phase HPLC.Identity was veri®ed by mass spectrometry.
Heterogeneous peptide binding assays
TAP-containing microsomes (35 mg total protein) wereincubated in the absence of ATP with increasing concen-trations of ¯uorescein-labeled peptide in 150 ml assaybuffer (PBS, 1 mM DTT, 5 mM MgCl2, pH 7.4) for 15minutes at 4 �C. The absence of ATP was ensured by0.03 unit/ml apyrase (Sigma, Deisenhofen, Germany).Subsequently, 350 ml of ice-cold assay buffer were addedand microsomes were pelleted by centrifugation (12,000g, eight minutes, 4 �C). After washing with 500 mlice-cold assay buffer, microsomes were lysed in PBS,1 % (w/v) SDS (pH 7.4). The amount of bound peptidewas quanti®ed by the ¯uorescence emission signal(lex/em � 470/515 nm). All spectra were corrected forlight scattering by subtracting the signal of microsomalsamples lacking ¯uorescent peptides. The amount ofunspeci®c binding was determined by blocking thespeci®c binding sites with a 400-fold molar excess ofnon-labeled peptide. To calculate the dissociation con-stant Kd of the ¯uorescein-labeled peptides, the amountof speci®cally bound peptide B was plotted against thepeptide concentration [P]. Data were ®tted using theequation (5):
B � Bmax�P�Kd � �P� �5�
Peptide transport assay
TAP-containing microsomes (35 mg total protein) wereincubated with 3 mM ATP and 300 nM ¯uorescent pep-tides containing an N-glycosylation consensus sequence(NXS/T) in 100 ml assay buffer. After four minutes at37 �C, adding 400 ml ice-cold assay buffer stopped pep-tide transport. After centrifugation (12,000 g, eight min-utes, 4 �C) and washing with 500 ml cold assay buffer,microsomes were lysed in 700 ml NP-40 lysis buffer(50 mM Tris, 150 mM NaCl, 5 mM MgCl2, 1 % (v/v)NP-40, pH 7.4). N-Glycosylated (transported) peptideswere bound to 30 ml concanavalin A-Sepharose (Sigma,Deisenhofen, Germany) during an 1.5 hour period ofgentle shaking at 4 �C. Thereafter, the beads werewashed three times with 1 ml lysis buffer. Finally, pep-tides were eluted with 200 mM methyl a-D-mannopyra-noside and quanti®ed by ¯uorescence emission (lex/
em � 470/515 nm). Background transport activity wasdetermined by replacing ATP with 3 mM ADP or alter-natively, in the presence of a 400-fold molar excess ofnon-labeled peptide (RRYQKSTEL). All spectra were cor-rected for light scattering by subtracting the signal ofsamples lacking ¯uorescent peptide.
Analysis of the association and dissociation kinetics
Fluorescein-labeled peptides were preincubated in900 ml assay buffer at 10 �C. After reaching a stable ¯uor-escence emission signal (¯uorescence drift <50 cps/min)TAP-containing microsomes in 100 ml assay buffer wereinjected into the cuvette. Prior to use, microsomes wereATP-depleted by pretreatment with 0.03 unit/ml apyrase(Sigma, Deisenhofen, Germany) for ten minutes. The®nal protein and TAP concentration were 100 mg/ml

Kinetics of Peptide Binding to TAP 1211
and 2 nM, respectively. Due to the low concentration ofTAP and the molar excess of ¯uorescent peptide, pseu-do-®rst-order conditions can be applied. After reachingbinding equilibrium indicated by a stable ¯uorescentemission signal (¯uorescence drift <50 cps/min), addinga 400-fold molar excess of non-labeled peptide (RRYQK-STEL) started dissociation of the peptide. The ¯uor-escence emission signal (lex/em � 470/515 nm) wasrecorded using a FLUOLOG-3 spectrometer (InstrumentsS.A., HORIBA Group, Paris, France) equipped with amicro stirring device. Light scattering was reduced bydouble monochromators and narrow slits (3 nm). Inner®lter effects were by-passed by a triangular cuvette,which reduces the pathway of light through the turbidsolution. The time-dependent changes in the ¯uorescencesignal were ®tted to a monoexponential function yield-ing the observed association and dissociation rate con-stants, kobs and kdiss.
association kinetic:
F � �F�t�1�eÿkobst � F�t�1� �6�
dissociation kinetic:
F � �F�t�1��1ÿ eÿkdisst� � F�t�1� �7�
Acknowledgments
We thank Drs Rupert Abele and Lutz Schmitt forhelpful discussion. The work was supported by theDeutsche Forschungsgemeinschaft and the Fonds derChemischen Industrie.
References
Ahn, K., Meyer, T. H., Uebel, S., SempeÂ, P., Djaballah,H., Yang, Y., Peterson, P. A., FruÈ h, K. & TampeÂ, R.(1996). Molecular mechanism and species-speci®cityof TAP inhibition by Herpes-Simplex virus proteinICP47. EMBO J. 15, 3247-3255.
Anderson, C. M., Zucker, F. H. & Steitz, T. A. (1979).Space-®lling models of kinase clefts and confor-mation changes. Science, 204, 375-380.
Androlewicz, M. J., Anderson, K. S. & Cresswell, P.(1993). Evidence that transporter associated withantigen processing translocate a major histocompat-ibility complex class I-binding peptide into theendoplasmic reticulum in an ATP-dependent man-ner. Proc. Natl Acad. Sci. USA, 90, 9130-9134.
Coux, O., Tanaka, K. & Goldberg, A. L. (1996). Structureand functions of the 20 S and 26 S proteasomes.Annu. Rev. Biochem. 65, 801-847.
Crowley, K. S., Reinhart, G. D. & Johnson, A. E. (1993).The signal sequence moves through a ribosomaltunnel into a noncytoplasmic aqueous environmentat the ER membrane early in translocation. Cell, 73,1101-1115.
Crowley, K. S., Liao, S., Worrell, V. E., Reinhart, G. D.& Johnson, A. E. (1994). Secretory proteins movethrough the endoplasmic reticulum membrane viaan aqueous, gated pore. Cell, 78, 461-471.
GrommeÂ, M., van der Valk, R., Sliedregt, K., Vernie, L.,Liskamp, R., HaÈmmerling, G. J., Koopmann, J.-O.,Momburg, F. & Neefjes, J. J. (1997). The rationaldesign of TAP inhibitors using peptide substratemodi®cations and peptidomimetics. Eur. J. Immunol.27, 898-904.
Gubler, B., Daniel, S., Armandola, E. A., Hammer, J.,Caillatzucman, S. & Van Endert, P. M. (1998). Sub-strate selection by transporters associated with anti-gen-processing occurs during peptide binding toTAP. Mol. Immunol. 35, 427-433.
Hamman, B. D., Chen, J. C., Johnson, E. E. & Johnson,A. E. (1997). The aqueous pore through the translo-con has a diameter of 40-60 AÊ during cotransla-tional protein translocation at the ER membrane.Cell, 89, 535-544.
Higgins, C. F. (1992). ABC transporters: from micro-organisms to man. Annu. Rev. Cell Biol. 8, 67-113.
Hiromi, K. (1979). Kinetic of Fast Enzyme Reactions, JohnWiley & Sons, Haltsted Press, New York.
Holler, E., Rupley, J. A. & Hess, G. P. (1969). Kinetics oflysozyme-substrate interactions. Biochem. Biophys.Res. Commun. 37, 423-429.
Johnson, J. K., Wang, Z. X. & Srivastava, D. K. (1992).Mechanistic investigation of medium-chain fattyacyl-CoA dehydrogenase utilizing 3-indolepropio-nyl/acryloyl-CoA as chromophoric substrate ana-logues (published erratum appears in Biochemistry(1993) Sep 21; 32(37):9874). Biochemistry, 31, 10564-10575.
Kelly, A. P., Powis, S. H., Kerr, L.-A., Mockridge, I.,Elliott, T., Bastin, J., Uchanska-Ziegler, B., Ziegler,A., Trowsdale, J. & Townsend, A. (1992). Assemblyand function of the two ABC transporter proteinsencoded in the human major histocompatibilitycomplex. Nature, 355, 641-644.
Kleijmeer, M., Kelly, A., Geuze, H. J., Slot, J. W.,Townsend, A. & Trowsdale, J. (1992). Location ofMHC-encoded transporters in the endoplasmic reti-culum and cis-golgi. Nature, 357, 342-344.
Koopmann, J. O., Post, M., Neefjes, J. J., HaÈmmerling,G. J. & Momburg, F. (1996). Translocation of longpeptides by transporters associated with antigenprocessing (TAP). Eur. J. Immunol. 26, 1720-1728.
Lacaille, V. G. & Androlewicz, M. J. (1998). Herpes-simplex virus inhibitor ICP47 destabilizes thetransporter associated with antigen-processing(TAP) heterodimer. J. Biol. Chem. 273, 17386-17390.
Meyer, T. H., van Endert, P. M., Uebel, S., Ehring, B. &TampeÂ, R. (1994). Functional expression and puri®-cation of the ABC transporter complex-associatedwith antigen-processing (TAP) in insect cells. FEBSLetters, 351, 443-447.
MuÈ ller, K. M., Ebensperger, C. & TampeÂ, R. (1994).Nucleotide binding to the hydrophilic C-terminaldomain of the transporter associated with antigenprocessing (TAP). J. Biol. Chem. 269, 14032-14037.
Neefjes, J. J., Momburg, F. & HaÈmmerling, G. J. (1993).Selective and ATP-dependent translocation of pep-tides by the MHC-encoded transporter. Science, 261,769-771.
Ohkuma, S. & Poole, B. (1978). Fluorescence probemeasurement of the intralysosomal pH in livingcells and the perturbation of pH by various agents.Proc. Natl Acad. Sci. USA, 75, 3327-3331.
Pamer, E. & Cresswell, P. (1998). Mechanisms of MHCclass I-restricted antigen processing. Annu. Rev.Immunol. 16, 323-358.

1212 Kinetics of Peptide Binding to TAP
Russ, G., Esquivel, F., Yewdell, J. W., Cresswell, P.,Spies, T. & Bennick, J. R. (1995). Assembly, intra-cellular localization, and nucleotide binding proper-ties of the human peptide transporters TAP1 andTAP2 expressed by recombinant vaccinia viruses.J. Biol. Chem. 270, 21312-21318.
Shepherd, J. C., Schumacher, T. N., Ashton-Rickardt,P. G., Imaeda, S., Ploegh, H. L., Janeway, C. A. J. &Tonegawa, S. (1993). TAP1-dependent peptidetranslocation in vitro is ATP dependent and peptideselective. Cell, 74, 577-584.
Sydor, J. R., Engelhard, M., Wittinghofer, A., Goody,R. S. & Herrmann, C. (1998). Transient kinetic stu-dies on the interaction of Ras and the Ras-bindingdomain of c-Raf-1 reveal rapid equilibration of thecomplex. Biochemistry, 37, 14292-14299.
Tairi, A. P., Hovius, R., Pick, H., Blasey, H., Bernard, A.,Surprenant, A., Lundstrom, K. & Vogel, H. (1998).Ligand binding to the serotonin 5HT3 receptor stu-died with a novel ¯uorescent ligand. Biochemistry,37, 15850-15864.
Tomazin, R., Hill, A. B., Jugovic, P., York, I., van Endert,P., Ploegh, H. L., Andrews, D. W. & Johnson, D. C.(1996). Stable binding of the Herpes Simplex virusICP47 protein to the peptide binding-site of TAP.EMBO J. 15, 3256-3266.
Toomela, T., Jolkkonen, M., Rinken, A., Jarv, J. &Karlsson, E. (1994). Two-step binding of greenmamba toxin to muscarinic acetylcholine receptor.FEBS Letters, 352, 95-97.
Uebel, S. & TampeÂ, R. (1997). Processing and selectionof antigens by the MHC-encoded peptide transpor-ter TAP. In Symposium in Immunology (Eibl, M. M.,Huber, C., Peter, H. H. & Wahn, U., eds), vol. 5,pp. 155-164, Springer, New York.
Uebel, S. & TampeÂ, R. (1999). Speci®city of the protea-some and the TAP transporter. Curr. Opin. Immunol.11, 203-208.
Uebel, S., Meyer, T. H., Kraas, W., Kienle, S., Jung, G.,WiesmuÈ ller, K. H. & TampeÂ, R. (1995). Require-ments for peptide binding to the human transporterassociated with antigen processing revealed by pep-tide scans and complex peptide libraries. J. Biol.Chem. 270, 18512-18516.
Uebel, S., Kraas, W., Kienle, S., WiesmuÈ ller, K.-H., Jung,G. & TampeÂ, R. (1997). Recognition principle of theTAP-transporter disclosed by combinatorial peptidelibraries. Proc. Natl Acad. Sci. USA, 94, 8976-8981.
van Endert, P. M. (1999). Role of nucleotides and pep-tide substrate for stability and functional state ofthe human ABC family transporters associated withantigen processing. J. Biol. Chem. 274, 14632-14638.
van Endert, P. M., TampeÂ, R., Meyer, T. H., Tisch, R.,Bach, J. F. & McDevitt, H. O. (1994). A sequentialmodel for peptide binding and transport by thetransporters associated with antigen processing.Immunity, 1, 491-500.
York, I. A. & Rock, K. L. (1996). Antigen-processing andpresentation by the class-I major histocompatibilitycomplex. Annu. Rev. Immunol. 14, 369-396.
Appendix
Kinetic analysis is a powerful tool for investi-gating reaction mechanisms. The velocity of a reac-tion can be described by the observed associationrate kobs. This parameter is equal with the exponen-tial coef®cient of the equation which re¯ects the
formation of AB (for example equation (6))(Hiromi, 1979).
Single-step process:
For a single step process (Scheme 2) kobs contains
rate constants for the forward k�1 and the reversereaction kÿ1 as well as the sum of the equilibriumconcentrations of A and B, c0A and c0B (A1):
kobs � k�1�c0A � c0B� � kÿ1 �A1�Thus, the slope of plot kobs against (c0A � c0B) yieldsk�1 while the intercept with the y-axis gives kÿ1.
Two-step process:
Assuming a two-step process composed of abimolecular association step followed by a unimo-lecular isomerization (Scheme 3), one can dis-
tinguish in general between two cases: (a) thebimolecular step is faster, k�1
(c0A � c0B) � kÿ14k�2 � kÿ2; or (b) the unimolecularstep is faster, k�1 (c0A � c0B) � kÿ15k�2 � kÿ2. A biex-ponential kinetic is expected if both steps can berecorded. This is for example possible if both the®rst and the second step cause a ¯uorescencequenching. Hence, two exponential coef®cientskobs1 and kobs2 are obtained. Cases (a) and (b) canbe distinguished from the shape of plots kobs1 andkobs2 against (c0A � c0B) (Hiromi, 1979).
(a) In the case of a fast association reaction and aslow isomerization the observed rate constant forthe ®rst and second step kobs1 and kobs2 is given byequations (A2) and (A3):
kobs1 � k�1�c0A � c0B� � kÿ1 �A2�
kobs2 � k�2�c0A � c0B�Kÿ1 � �c0A � c0B�
� kÿ2 �A3�
Kÿ1 represents the dissociation constant of the ®rststep. As demonstrated in Figure A1(a) and (b) kobs1
shows a linear dependence from (c0A � c0B) whilekobs2 shows saturation behavior. By applyingequations (A2) and (A3) all rate constants and Kÿ1
can be determined.

Figure A1. Fast association followed by a slow isomerization (a) A process composed of a fast association and aslow isomerization shows a linear dependence between the observed association rate kobs1 of the ®rst step and theequilibrium concentrations of the reactants A and B (c0A � c0B). The rate constants can be determined as indicated.(b) The observed rate constant kobs2 for the second step shows saturation behavior. The dissociation constant Kÿ1 forthe ®rst step and the rate constants can be graphically determined as indicated. Slow association followed by a fastisomerization. (c) The observed rate constant kobs1 of a slow association step followed by a fast isomerization shows alinear dependence of the equilibrium concentrations of the reactants A and B (c0A � c0B). The rate constants can bedetermined as indicated. (d) kobs2 describing the rate of the fast isomerization is given by kÿ2 � k�2 and therefore con-centration-independent.
Kinetics of Peptide Binding to TAP 1213
(b) In the case of a slow association rate relativeto the velocity of the unimolecular process kobs1
rises linearly by increasing (c0A � c0B) (equation (A4),Figure A1(c)). kobs2 is independent of (c0A � c0B)(equation (A5), Figure A1(d)):
kobs1 � k�1�c0A � c0B� �kÿ1kÿ2
k�2 � kÿ2�A4�
kobs2 � k�2 � kÿ2 �A5�If only one step causes a detectable change, forexample in ¯uorescence, solely this process is vis-ible. Thus, a single kobs is expected. In such a case atwo-step mechanism can be distinguished from a
single-step mechanism, only if the plot kobs against(c0A � c0B) shows saturation behavior (Figure A1(b))or if it is concentration-independent (Figure A1(d)).Because the initial concentrations of A and B arebetter known than the equilibrium concentrationc0A and c0B one reactant is used usually in largeexcess. So (c0A � c0B) can be replaced by the concen-tration of the reactant used in excess.
References
Hiromi, K. (1979). Kinetic of Fast Enzyme Reac-tions, John Wiley & Sons, Haltsted Press, NewYork.
(Received 19 August 1999; received in revised form 20 October 1999; accepted 20 October 1999)
Edited by W. Baumesiter

![Peptide Nucleic Acids Having Enhanced Binding Affinity and ...[54] PEPTIDE NUCLEIC ACIDS HAVING FOREIGN PATENT DOCUMENTS ENHANCED BINDING AFFINITY AND WO 86/05518 9/1986 WIPO](https://img.pdfslide.us/doc/110x75/5ed9280a6714ca7f4769402c/-peptide-nucleic-acids-having-enhanced-binding-affinity-and-54-peptide-nucleic.jpg)



























