Embed Size (px)
Citation preview

Biol. Chem. 2020; 401(4): 517–531
Julia R. Seiza, Johannes Klinkea, Laura Scharlibbe, Dirk Lohfink, Marisa Heipel, Hendrik Ungefroren, Klaudia Giehlb and Andre Menkeb,*
Different signaling and functionality of Rac1 and Rac1b in the progression of lung adenocarcinomahttps://doi.org/10.1515/hsz-2019-0329Received July 30, 2019; accepted December 2, 2019; previously published online December 7, 2019
Abstract: Rac1 is a ubiquitously expressed Rho GTPase and an important regulator of the actin cytoskeleton. Its splice variant Rac1b exhibits a 19-amino acid (aa) in-frame insertion and is predominantly active. Both proteins were described in tumorigenesis or metastasis. We investigated the contribution of Rac1 and Rac1b to tumor progression of human non-small-cell lung adenocarcinoma (NSCLA). Rac1 protein was present in 8/8 NSCLA cell lines analyzed, whereas Rac1b was expressed in only 6/8. In wound-healing assays, enhanced green fluorescence protein (EGFP)-Rac1 slightly decreased cell migration, whereas proliferation was increased in both, Rac1- and Rac1b-expressing cells. In the in vivo chorioallantoic invasion model, EGFP-Rac1-expressing cells formed more invasive tumors compared to EGFP-Rac1b. This increased invasive-ness correlated with enhanced phosphorylation of p38α, AKT and glycogen synthase kinase 3β (GSK3β), and acti-vation of serum response- and Smad-dependent gene promoters by Rac1. In contrast, Rac1b solely activated the mitogen-activated protein kinase (MAPK) JNK2, together with TCF/LEF1- and nuclear factor kappa B (NFκB)-responsive gene reporters. Rac1b, as Rac1, phosphorylated p38α, AKT and GSK3β. Knockdown of the splicing factor epithelial splicing regulatory protein 1 (ESRP1), which mediates out-splicing of exon 3b from Rac1 pre-messenger
RNA, resulted in increased Rac1b messenger RNA (mRNA) and suppression of the epithelial-mesenchymal transi-tion (EMT)-associated transcription factor ZEB1. Our data demonstrate different signaling and functional activities of Rac1 and Rac1b and an important role for Rac1 in lung cancer metastasis.
Keywords: cell migration; epithelial-to-mesenchymal transition; lung cancer metastasis; NSCLC; Rac1 signal-ing; Rho GTPases; tumor invasion.
IntroductionThe small GTPases of the Rho family, namely Rho, Rac, Cdc42 and its isoforms are well-known regulators of the actin cytoskeleton dynamics and cellular migration, as well as transducers of multiple signals in a variety of onco-genic pathways (Wolpert et al., 2000; Hall, 2005). Typical Rho GTPases cycle between an inactive guanosine diphos-phate (GDP)-bound and an active guanosine triphosphate (GTP)-bound state. In its active GTP-bound form, Rac1 interacts with effector proteins such as PAK1 or IQ motif containing GTPase activating protein 1 (IQGAP) and leads to activation of downstream kinases such as JNK or p38 to regulate many cellular processes, such as formation of lamellipodia, cell-cell adhesion, migration, transforma-tion or cell survival (Nobes and Hall, 1995; Ridley, 2001). In addition, Rac1 influences gene expression and cell prolif-eration by regulating several transcription factors, such as the serum-response factor (SRF) or nuclear factor kappa B (NFκB) (Hill et al., 1995; Minden et al., 1995; Bar-Sagi and Hall, 2000; Gastonguay et al., 2012).
The splice isoform Rac1b, which was first described in 1999 (Jordan et al., 1999; Schnelzer et al., 2000), includes an additional exon 3b immediately behind the switch II region comprising a 19-amino acid (aa) in-frame insertion. This insertion resulted in a strong increase in its nucleotide exchange rate and a delayed GTP hydrolysis (Fiegen et al., 2004). So, in most cells, Rac1b is in its active, GTP-bound state and located at the plasma membrane (Jordan et al., 1999; Schnelzer et al., 2000). Unlike Rac1, Rac1b does not associate with GDP dissociation inhibitors (GDIs), further
aJulia R. Seiz and Johannes Klinke share first authorship.bKlaudia Giehl and Andre Menke share last authorship.*Corresponding author: Andre Menke, Molecular Oncology of Solid Tumors, Science Unit for Basic and Clinical Medicine, Justus Liebig University Giessen, Aulweg 128, D-35392 Giessen, Germany, e-mail: [email protected]. https://orcid.org/0000-0002-3321-9805 Julia R. Seiz, Laura Scharlibbe and Dirk Lohfink: Molecular Oncology of Solid Tumors, Internal Medicine, Science Unit for Basic and Clinical Medicine, Justus Liebig University Giessen, Aulweg 128, D-35392 Giessen, GermanyJohannes Klinke, Marisa Heipel and Klaudia Giehl: Signal Transduction of Cellular Motility, Internal Medicine, Science Unit for Basic and Clinical Medicine, Justus Liebig University Giessen, Aulweg 128, D-35392 Giessen, GermanyHendrik Ungefroren: First Department of Medicine, UKSH, Campus Lübeck, Ratzeburger Allee 160, D-23538 Lübeck, Germany
Open Access. © 2020 Julia R. Seiz et al., published by De Gruyter. This work is licensed under the Creative Commons Attribution 4.0 Public License.

518 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
underscoring the constitutive activity of Rac1b (Matos et al., 2003). Moreover, Rac1b, in contrast to Rac1, is unable to induce lamellipodia formation in the colon adenocarci-noma cell line SW480, and hence may not be a direct regu-lator of the actin cytoskeleton (Matos et al., 2003).
Rac1 and Rac1b are highly homologous splice variants. The process of alternative splicing is mainly regulated by RNA-binding proteins, which function as splice factors (Chen and Manley, 2009). Two of these factors, the epithe-lial splicing regulatory protein 1 (ESRP1) and 2 (ESRP2), are both present in epithelial cells and are involved in the splicing of RNAs derived from a large number of genes involved in epithelial-mesenchymal transition (EMT), cell motility or cytoskeletal dynamics (Warzecha et al., 2010; Dittmar et al., 2012). ESRP1 and ESRP2 are involved in the preservation of Rac1 and epithelial-specific isoforms of transcription factors (Shapiro et al., 2011). However, the role of ESRP1 and ESRP2 in tumor progression seems to be strongly cell type-specific. In colon as well as head and neck squamous cell carcinoma, the downregulation of ESRPs is associated with EMT, but there are also reports that show a correlation between ESRP expression and poor survival of patients especially with breast cancer (Shapiro et al., 2011; Yae et al., 2012; Ishii et al., 2014). Recently, Li et al. demon-strated an impaired expression of ESRP1 in lung adenocar-cinoma and correlated this with alternatively spliced CD44 forms and enhanced EMT (Li et al., 2018).
Lung cancer is the leading cause of cancer-related death with more than 150 000 estimated deaths in the US. Patients with non-small-cell lung cancer (NSCLC) and in particular adenocarcinoma had a 5-year survival rate of only 18% in 2017 (Siegel et al., 2017). A Rac1 dysregula-tion in patients suffering from non-small-cell lung adeno-carcinoma (NSCLA) correlates strongly with a low 5-year survival rate, due to elevated EMT and increased meta-static potential (Zhou et al., 2016). For the splice variant Rac1b, it has been shown that it promotes tumorigenesis of K-Ras-induced lung cancer by increasing proliferation and tumor growth (Zhou et al., 2013).
As little is known about the role of Rac1 and Rac1b in lung cancer, we analyzed Rac1- and Rac1b-specific sign-aling pathways and their effects on cell proliferation and migration in lung adenocarcinoma cell lines. Here, we show that Rac1 and Rac1b, when ectopically expressed in H23 NSCLC cells, activate different signaling pathways and differentially modulate gene expression, cell proliferation and invasion. Rac1 activated mitogen-activated protein kinases (MAPKs), AKT as well as glycogen synthase kinase 3β (GSK3β) and increased cell proliferation and invasion, thereby pointing toward a major role of Rac1 in the devel-opment and progression of lung adenocarcinoma.
ResultsExpression and cellular localization of Rac1 and Rac1b in lung cancer cell lines
The small GTPase Rac1 and its closely related isoform Rac1b have been described as protooncogenes in different types of tumors such as pancreatic carcinoma, colorectal carcinoma or mammary adenocarcinoma (Melzer et al., 2017). To delin-eate the function of both isoforms in lung adenocarcinoma, we determined the total protein levels in different NSCLC cell lines (Figure 1A). Rac1 was present in comparable amounts in all analyzed NSCLC cell lines, with H23 cells exhibiting the lowest Rac1 concentration. The expression of Rac1b, detected with a Rac1b-specific antibody, was much more heterogeneous, with the strongest amounts of protein in the more differentiated H358 and HTB55 cells and barely detect-able levels in A549 and the mesenchymal-appearing H1299 cells. Of note, when using a Rac1 antibody, which detects both isoforms, it becomes evident that Rac1b is expressed at a much lower level than Rac1 (data not shown).
For further studies, we ectopically expressed both Rac1 isoforms as wild-type enhanced green fluorescence protein (EGFP) fusion proteins in H23 cells, which harbor low amounts of endogenous Rac1 and Rac1b proteins, fol-lowed by the selection of individual clones. Most assays were repeated with EGFP-Rac1/1b-transfected HCC44 lung adenocarcinoma cells (Supplementary Figures S1, S2) with results comparable to those obtained with H23 cells. Western blot analyses of the selected H23 clones, stably expressing EGFP-Rac1 (termed D1 and D2) or EGFP-Rac1b (termed B1 and B3), show a comparable, intermediate protein level of the EGFP-Rac proteins. EGFP-expressing cells served as controls (Figure 1B). The cellular localization of the fusion proteins was studied by fluorescence microscopy in living cells. Figure 1C exem-plifies that EGFP-Rac1 was located in the cytoplasm as well as at the plasma membrane of H23 cells, whereas EGFP-Rac1b was localized predominantly at the plasma membrane and in areas of cell-cell contacts (marked by arrows in Figure 1C). The cytoplasmic signals of EGFP-Rac1b were weaker compared to those of EGFP-Rac1. The differential intracellular localization of EGFP-Rac1 and EGFP-Rac1b was supported by results obtained from cell fractionation studies in which the soluble and the par-ticulate protein fractions were separated by high-speed centrifugation. Subsequent western blot analyses demon-strated that EGFP-Rac1 was present in the soluble S100 (cytosolic) and the insoluble P100 (membrane) frac-tion, whereas EGFP-Rac1b was mainly detectable in the P100 membrane protein-containing fraction (Figure 1D).
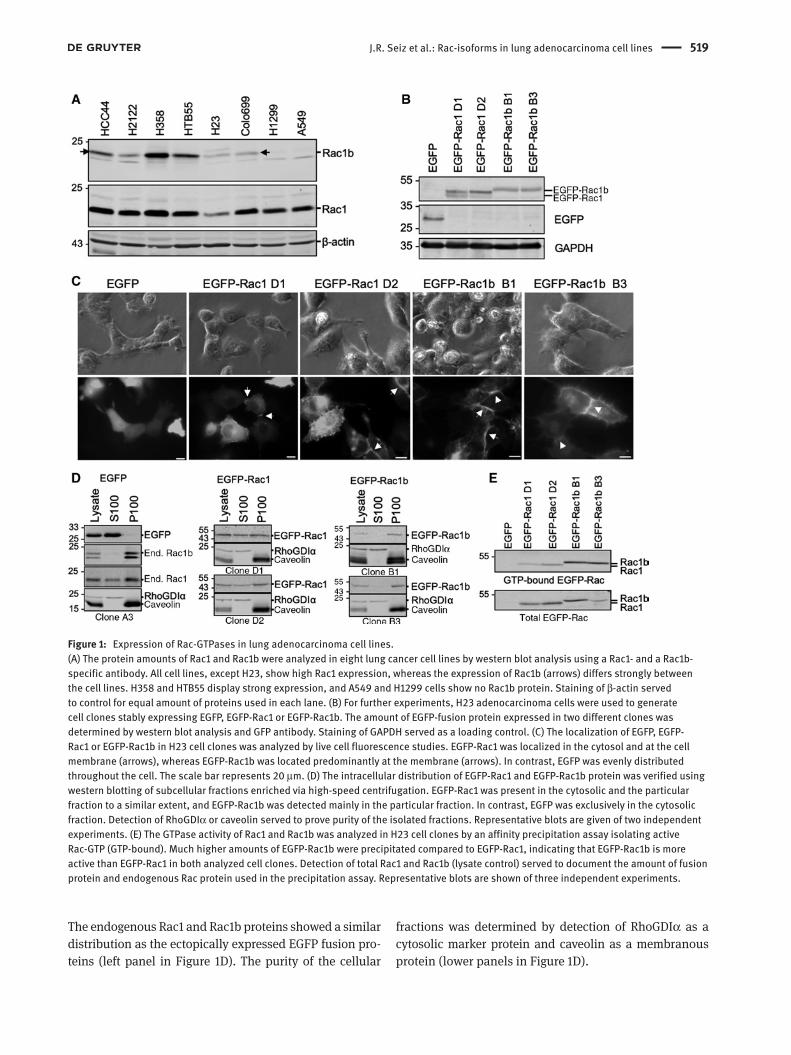
J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 519
The endogenous Rac1 and Rac1b proteins showed a similar distribution as the ectopically expressed EGFP fusion pro-teins (left panel in Figure 1D). The purity of the cellular
fractions was determined by detection of RhoGDIα as a cytosolic marker protein and caveolin as a membranous protein (lower panels in Figure 1D).
Figure 1: Expression of Rac-GTPases in lung adenocarcinoma cell lines.(A) The protein amounts of Rac1 and Rac1b were analyzed in eight lung cancer cell lines by western blot analysis using a Rac1- and a Rac1b-specific antibody. All cell lines, except H23, show high Rac1 expression, whereas the expression of Rac1b (arrows) differs strongly between the cell lines. H358 and HTB55 display strong expression, and A549 and H1299 cells show no Rac1b protein. Staining of β-actin served to control for equal amount of proteins used in each lane. (B) For further experiments, H23 adenocarcinoma cells were used to generate cell clones stably expressing EGFP, EGFP-Rac1 or EGFP-Rac1b. The amount of EGFP-fusion protein expressed in two different clones was determined by western blot analysis and GFP antibody. Staining of GAPDH served as a loading control. (C) The localization of EGFP, EGFP-Rac1 or EGFP-Rac1b in H23 cell clones was analyzed by live cell fluorescence studies. EGFP-Rac1 was localized in the cytosol and at the cell membrane (arrows), whereas EGFP-Rac1b was located predominantly at the membrane (arrows). In contrast, EGFP was evenly distributed throughout the cell. The scale bar represents 20 μm. (D) The intracellular distribution of EGFP-Rac1 and EGFP-Rac1b protein was verified using western blotting of subcellular fractions enriched via high-speed centrifugation. EGFP-Rac1 was present in the cytosolic and the particular fraction to a similar extent, and EGFP-Rac1b was detected mainly in the particular fraction. In contrast, EGFP was exclusively in the cytosolic fraction. Detection of RhoGDIα or caveolin served to prove purity of the isolated fractions. Representative blots are given of two independent experiments. (E) The GTPase activity of Rac1 and Rac1b was analyzed in H23 cell clones by an affinity precipitation assay isolating active Rac-GTP (GTP-bound). Much higher amounts of EGFP-Rac1b were precipitated compared to EGFP-Rac1, indicating that EGFP-Rac1b is more active than EGFP-Rac1 in both analyzed cell clones. Detection of total Rac1 and Rac1b (lysate control) served to document the amount of fusion protein and endogenous Rac protein used in the precipitation assay. Representative blots are shown of three independent experiments.
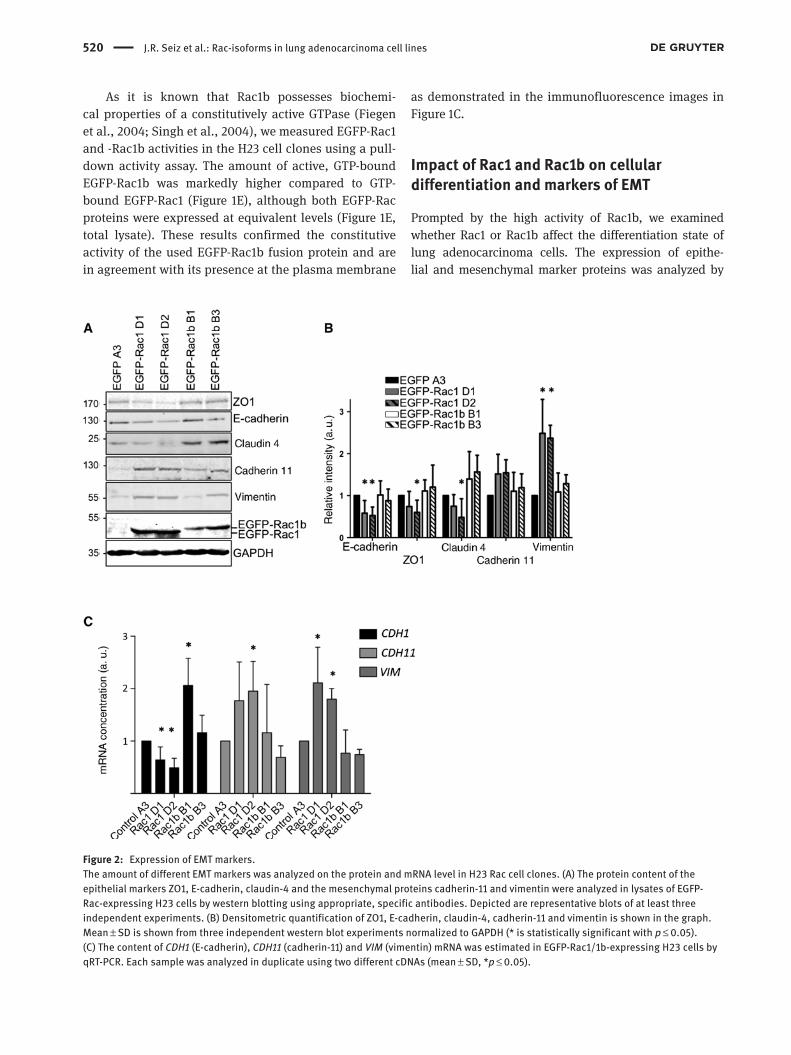
520 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
As it is known that Rac1b possesses biochemi-cal properties of a constitutively active GTPase (Fiegen et al., 2004; Singh et al., 2004), we measured EGFP-Rac1 and -Rac1b activities in the H23 cell clones using a pull-down activity assay. The amount of active, GTP-bound EGFP-Rac1b was markedly higher compared to GTP-bound EGFP-Rac1 (Figure 1E), although both EGFP-Rac proteins were expressed at equivalent levels (Figure 1E, total lysate). These results confirmed the constitutive activity of the used EGFP-Rac1b fusion protein and are in agreement with its presence at the plasma membrane
as demonstrated in the immunofluorescence images in Figure 1C.
Impact of Rac1 and Rac1b on cellular differentiation and markers of EMT
Prompted by the high activity of Rac1b, we examined whether Rac1 or Rac1b affect the differentiation state of lung adenocarcinoma cells. The expression of epithe-lial and mesenchymal marker proteins was analyzed by
Figure 2: Expression of EMT markers.The amount of different EMT markers was analyzed on the protein and mRNA level in H23 Rac cell clones. (A) The protein content of the epithelial markers ZO1, E-cadherin, claudin-4 and the mesenchymal proteins cadherin-11 and vimentin were analyzed in lysates of EGFP-Rac-expressing H23 cells by western blotting using appropriate, specific antibodies. Depicted are representative blots of at least three independent experiments. (B) Densitometric quantification of ZO1, E-cadherin, claudin-4, cadherin-11 and vimentin is shown in the graph. Mean ± SD is shown from three independent western blot experiments normalized to GAPDH (* is statistically significant with p ≤ 0.05). (C) The content of CDH1 (E-cadherin), CDH11 (cadherin-11) and VIM (vimentin) mRNA was estimated in EGFP-Rac1/1b-expressing H23 cells by qRT-PCR. Each sample was analyzed in duplicate using two different cDNAs (mean ± SD, *p ≤ 0.05).
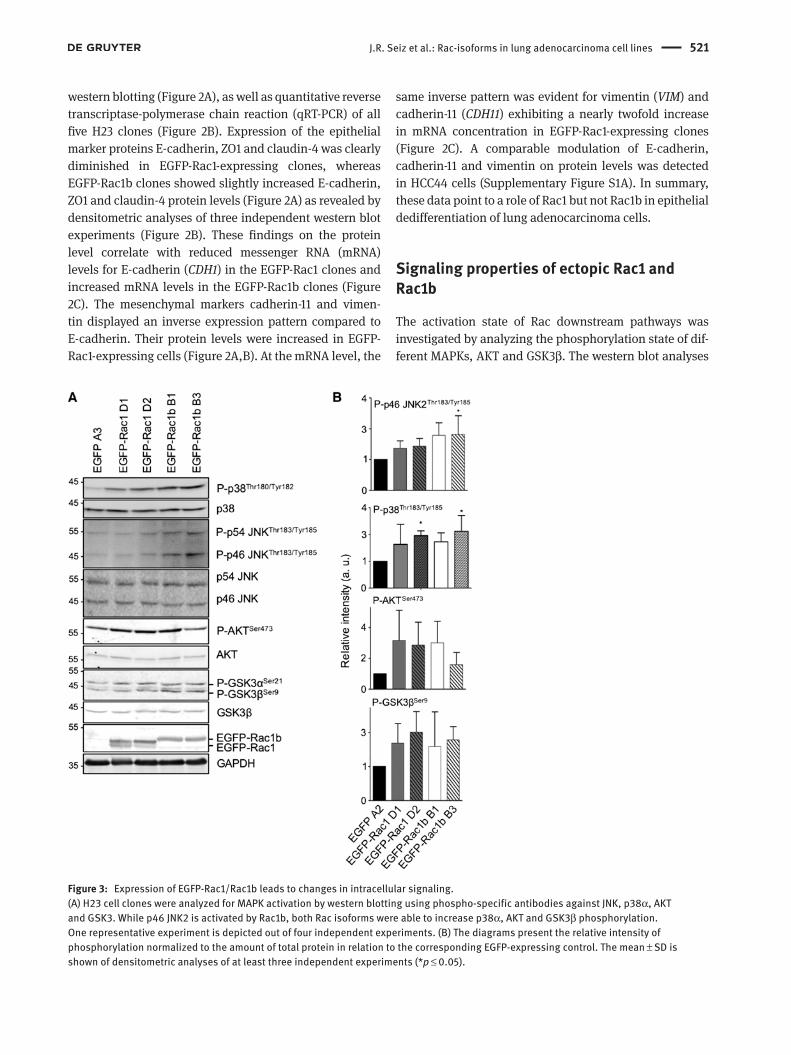
J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 521
western blotting (Figure 2A), as well as quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) of all five H23 clones (Figure 2B). Expression of the epithelial marker proteins E-cadherin, ZO1 and claudin-4 was clearly diminished in EGFP-Rac1-expressing clones, whereas EGFP-Rac1b clones showed slightly increased E-cadherin, ZO1 and claudin-4 protein levels (Figure 2A) as revealed by densitometric analyses of three independent western blot experiments (Figure 2B). These findings on the protein level correlate with reduced messenger RNA (mRNA) levels for E-cadherin (CDH1) in the EGFP-Rac1 clones and increased mRNA levels in the EGFP-Rac1b clones (Figure 2C). The mesenchymal markers cadherin-11 and vimen-tin displayed an inverse expression pattern compared to E-cadherin. Their protein levels were increased in EGFP-Rac1-expressing cells (Figure 2A,B). At the mRNA level, the
same inverse pattern was evident for vimentin (VIM) and cadherin-11 (CDH11) exhibiting a nearly twofold increase in mRNA concentration in EGFP-Rac1-expressing clones (Figure 2C). A comparable modulation of E-cadherin, cadherin-11 and vimentin on protein levels was detected in HCC44 cells (Supplementary Figure S1A). In summary, these data point to a role of Rac1 but not Rac1b in epithelial dedifferentiation of lung adenocarcinoma cells.
Signaling properties of ectopic Rac1 and Rac1b
The activation state of Rac downstream pathways was investigated by analyzing the phosphorylation state of dif-ferent MAPKs, AKT and GSK3β. The western blot analyses
Figure 3: Expression of EGFP-Rac1/Rac1b leads to changes in intracellular signaling.(A) H23 cell clones were analyzed for MAPK activation by western blotting using phospho-specific antibodies against JNK, p38α, AKT and GSK3. While p46 JNK2 is activated by Rac1b, both Rac isoforms were able to increase p38α, AKT and GSK3β phosphorylation. One representative experiment is depicted out of four independent experiments. (B) The diagrams present the relative intensity of phosphorylation normalized to the amount of total protein in relation to the corresponding EGFP-expressing control. The mean ± SD is shown of densitometric analyses of at least three independent experiments (*p ≤ 0.05).
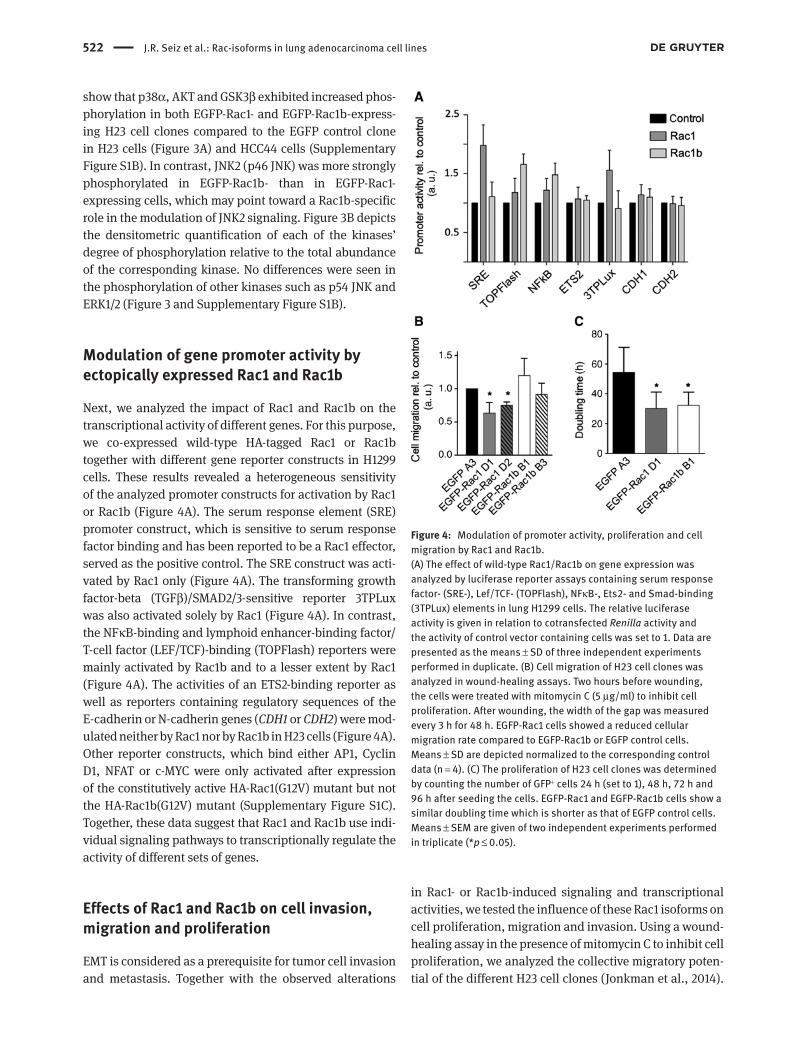
522 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
show that p38α, AKT and GSK3β exhibited increased phos-phorylation in both EGFP-Rac1- and EGFP-Rac1b-express-ing H23 cell clones compared to the EGFP control clone in H23 cells (Figure 3A) and HCC44 cells (Supplementary Figure S1B). In contrast, JNK2 (p46 JNK) was more strongly phosphorylated in EGFP-Rac1b- than in EGFP-Rac1-expressing cells, which may point toward a Rac1b-specific role in the modulation of JNK2 signaling. Figure 3B depicts the densitometric quantification of each of the kinases’ degree of phosphorylation relative to the total abundance of the corresponding kinase. No differences were seen in the phosphorylation of other kinases such as p54 JNK and ERK1/2 (Figure 3 and Supplementary Figure S1B).
Modulation of gene promoter activity by ectopically expressed Rac1 and Rac1b
Next, we analyzed the impact of Rac1 and Rac1b on the transcriptional activity of different genes. For this purpose, we co-expressed wild-type HA-tagged Rac1 or Rac1b together with different gene reporter constructs in H1299 cells. These results revealed a heterogeneous sensitivity of the analyzed promoter constructs for activation by Rac1 or Rac1b (Figure 4A). The serum response element (SRE) promoter construct, which is sensitive to serum response factor binding and has been reported to be a Rac1 effector, served as the positive control. The SRE construct was acti-vated by Rac1 only (Figure 4A). The transforming growth factor-beta (TGFβ)/SMAD2/3-sensitive reporter 3TPLux was also activated solely by Rac1 (Figure 4A). In contrast, the NFκB-binding and lymphoid enhancer-binding factor/ T-cell factor (LEF/TCF)-binding (TOPFlash) reporters were mainly activated by Rac1b and to a lesser extent by Rac1 (Figure 4A). The activities of an ETS2-binding reporter as well as reporters containing regulatory sequences of the E-cadherin or N-cadherin genes (CDH1 or CDH2) were mod-ulated neither by Rac1 nor by Rac1b in H23 cells (Figure 4A). Other reporter constructs, which bind either AP1, Cyclin D1, NFAT or c-MYC were only activated after expression of the constitutively active HA-Rac1(G12V) mutant but not the HA-Rac1b(G12V) mutant (Supplementary Figure S1C). Together, these data suggest that Rac1 and Rac1b use indi-vidual signaling pathways to transcriptionally regulate the activity of different sets of genes.
Effects of Rac1 and Rac1b on cell invasion, migration and proliferation
EMT is considered as a prerequisite for tumor cell invasion and metastasis. Together with the observed alterations
in Rac1- or Rac1b-induced signaling and transcriptional activities, we tested the influence of these Rac1 isoforms on cell proliferation, migration and invasion. Using a wound-healing assay in the presence of mitomycin C to inhibit cell proliferation, we analyzed the collective migratory poten-tial of the different H23 cell clones (Jonkman et al., 2014).
Figure 4: Modulation of promoter activity, proliferation and cell migration by Rac1 and Rac1b.(A) The effect of wild-type Rac1/Rac1b on gene expression was analyzed by luciferase reporter assays containing serum response factor- (SRE-), Lef/TCF- (TOPFlash), NFκB-, Ets2- and Smad-binding (3TPLux) elements in lung H1299 cells. The relative luciferase activity is given in relation to cotransfected Renilla activity and the activity of control vector containing cells was set to 1. Data are presented as the means ± SD of three independent experiments performed in duplicate. (B) Cell migration of H23 cell clones was analyzed in wound-healing assays. Two hours before wounding, the cells were treated with mitomycin C (5 μg/ml) to inhibit cell proliferation. After wounding, the width of the gap was measured every 3 h for 48 h. EGFP-Rac1 cells showed a reduced cellular migration rate compared to EGFP-Rac1b or EGFP control cells. Means ± SD are depicted normalized to the corresponding control data (n = 4). (C) The proliferation of H23 cell clones was determined by counting the number of GFP+ cells 24 h (set to 1), 48 h, 72 h and 96 h after seeding the cells. EGFP-Rac1 and EGFP-Rac1b cells show a similar doubling time which is shorter as that of EGFP control cells. Means ± SEM are given of two independent experiments performed in triplicate (*p ≤ 0.05).

J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 523
As demonstrated in Figure 4B, the two clones expressing EGFP-Rac1 exhibited a decreased cell migration (clone D1 0.63 ± 0.16, clone D2 0.73 ± 0.06) as compared to the EGFP-expressing control (set to 1.0), while the migratory activi-ties of EGFP-Rac1b-expressing cells (clone B1 1.19 ± 0.25, clone B3 0.91 ± 0.17) were not significantly different from the control.
Measurement of the population doubling times of the EGFP-Rac clones by detection of EGFP-positive cells by flow cytometry revealed an increased proliferation rate with a nearly bisected doubling time for Rac1- and Rac1b-expressing cells of 26 h and 28 h, respectively, compared to 54 h for the EGFP-expressing cells. Hence, ectopic EGFP-Rac1 and EGFP-Rac1b expression enhanced the pro-liferation of H23 cells by a factor of ~2 (Figure 4C).
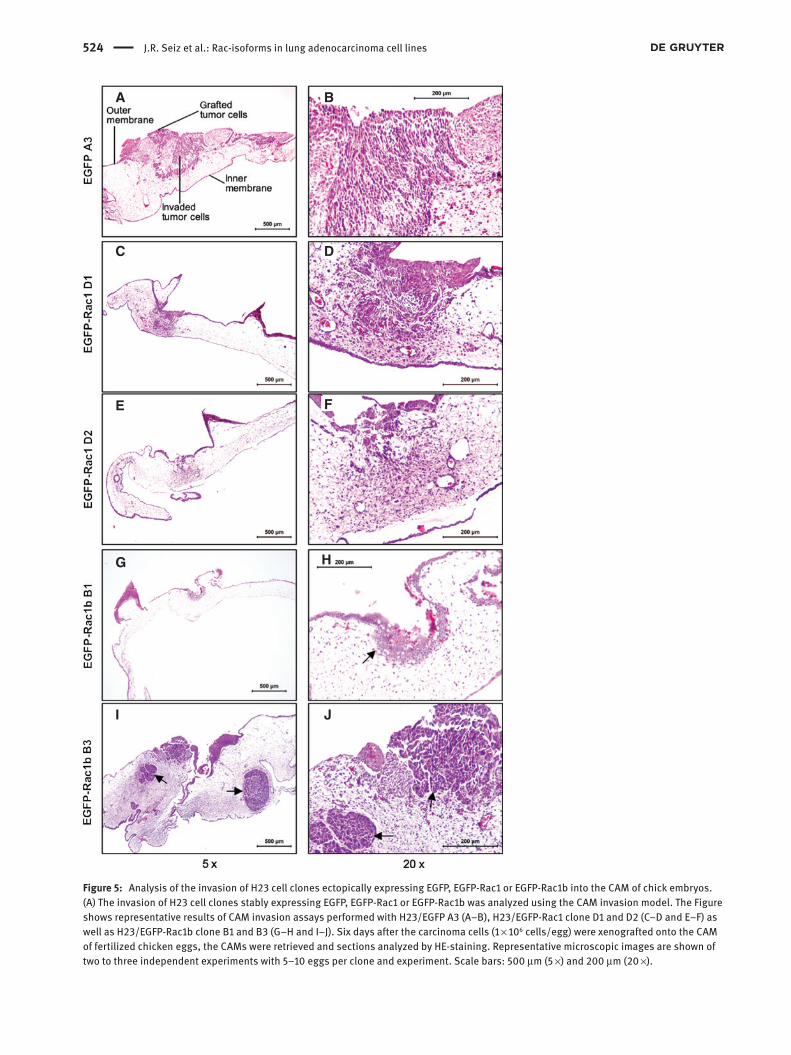
Furthermore, we estimated the invasive capacity of the Rac-expressing H23 cell clones using the chorioal-lantoic membrane (CAM) of fertilized chicken eggs as a model for cell invasion (Hofer et al., 2004; Dreissigacker et al., 2006). EGFP-expressing control, EGFP-Rac1- or EGFP-Rac1b-expressing cells were cultivated for 6 days on the CAM. In all cases, the growth of the tumor cells was evident. However, the degree of invasion through the outer epithelium of the CAM and spreading into the underlying mesenchymal tissue was different between the clones. Although the EGFP-Rac1b-expressing clones were able to penetrate the outer epithelial CAM layer, they invaded the underlying mesenchymal tissue only to a minor extent (clone B1 in 7/13 and clone B3 in 5/8 experi-ments; Figure 5G–J) compared to EGFP-Rac1 cells (clone D1 in 9/10 and clone D2 in 6/6 experiments; Figure 5C–F) and EGFP control cells (in 6/9 experiments) (Figure 5A,B). The histological evaluation revealed for the EGFP-Rac1b-expressing H23 cells more dense, encapsulated tumors (Figure 5H–J, arrows) when compared to EGFP-Rac1 or EGFP control cells.
Regulation of Rac1, Rac1b and differentiation markers by ESPR1 and 2
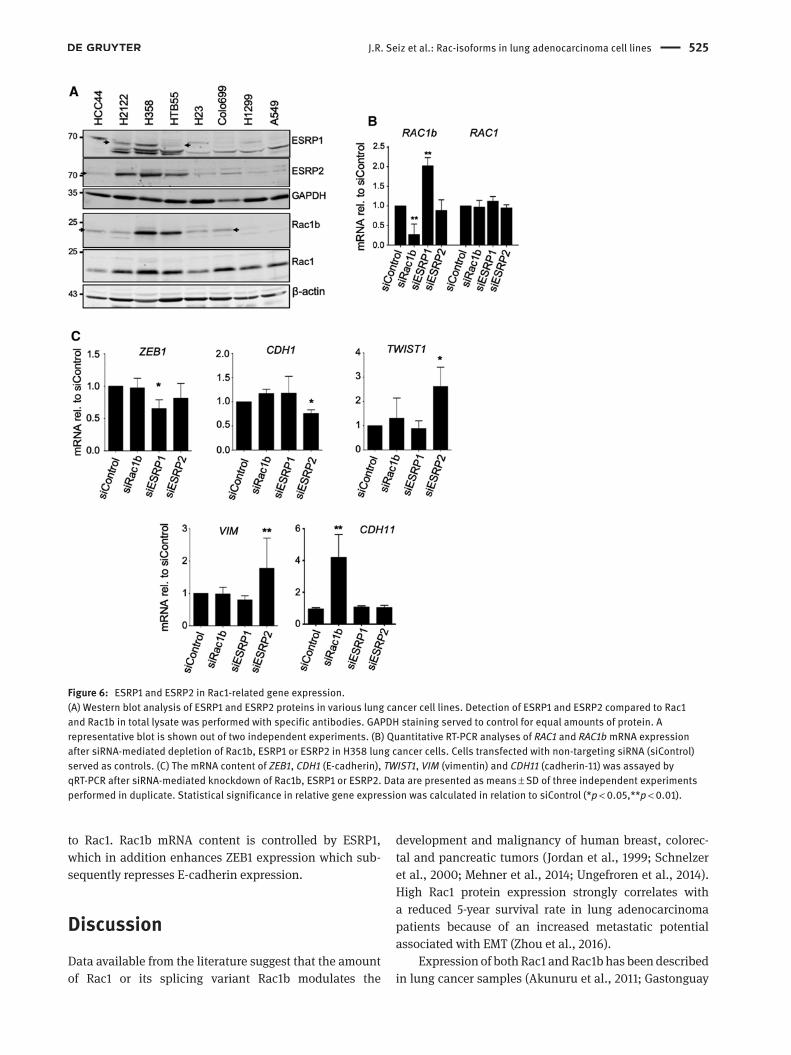
Next, we analyzed the role of the ESRP1 and ESRP2, which have been implicated in the alternative splicing of Rac1 and Rac1b and some EMT-associated transcription factors, such as ZEB, snail or twist in lung cancer cells. First, we evaluated the protein content of both splice factors in different NSCLC cell lines. ESRP1 and 2 showed high protein abundance in cell lines with a more differentiated phenotype and high Rac1b expression such as H358 and H2122, whereas A549 and H1299 cells with low amounts of Rac1b displayed barely detectable amounts of ESRP1 and
2 (Figure 6A). As the ESRP1 antibody detected more than one band in western blots, the specific bands (indicated by arrows) were identified after siRNA-mediated depletion of ESPR1 (Supplementary Figure S2B).
In a next step, we analyzed whether Rac1b, ESRP1 and ESRP2 are involved in regulating the observed EMT markers and cell differentiation by using an siRNA-mediated knock-down approach. Silencing of ESRP1 in H358 lung cancer cells resulted in an increased amount of Rac1b mRNA, whereas a knockdown of ESRP2 did not affect Rac1b mRNA content (Figure 6B). The increase in Rac1b mRNA (Figure 6B) was paralleled by higher Rac1b protein abundance after knockdown of ESRP1 (Supplementary Figure S2B). The successful knockdown of ESRP1 and 2 was demon-strated by qRT-PCR and western blotting in Supplementary Figure S2A,B. This finding supports the notion that ESRP1 is involved in exclusion of exon 3b from the immature Rac RNA also in lung adenocarcinoma (Figure 6B) (Ishii et al., 2014). Moreover, depletion of ESRP1 resulted in a decrease in the mRNA concentration of the Zn-finger transcription factor ZEB1 (Figure 6C). ZEB1 is mainly known as a repres-sive factor in the regulation of different EMT marker genes (Schmalhofer et al., 2009; Larsen et al., 2016). Knockdown of ESRP2 did not alter Rac1b mRNA, but higher levels of twist1 mRNA, another EMT-associated transcription factor, and lower amounts of E-cadherin mRNA were evident. Fur-thermore, the ESRP2 knockdown increased mRNA levels of the mesenchymal marker vimentin (Figure 6C). In contrast, knockdown of either ESRP1, ESRP2 or Rac1b failed to alter the expression of the EMT transcription factors snail or slug in lung adenocarcinoma cells (data not shown). Interest-ingly, silencing of Rac1b resulted in a dramatic upregula-tion of cadherin-11 mRNA, another mesenchymal marker (Figure 6C). Analyses of ESRP1, ZEB1 and twist1 mRNA contents by qRT-PCR confirmed increased amounts of ESRP1 mRNA and enhanced expression of ZEB1 and twist1 in EGFP-Rac1-expressing cells (Supplementary Figure S2C). One target of ZEB1 is CDH1, the E-cadherin gene, which was downregulated in both EGFP-Rac1-expressing clones as shown in Figure 2C. Moreover, detection of ZEB1 in the nuclear fraction of H23 EGFP-Rac clones confirmed that only the EGFP-Rac1-expressing clones exhibited an increased amount of ZEB1 (Supplementary Figure 2D), which correlates with the reduced E-cadherin concentra-tion in these cells (Figure 2A).
Taken together, we were able to demonstrate differen-tial signaling by Rac1 and Rac1b after their ectopic expres-sion in lung adenocarcinoma cells and identified a role for Rac1 in promoting invasion of NSCLA cells. In contrast, Rac1b appeared to be associated with a differentiated epithelial phenotype by modulating signals not related
524 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
A
C D
E F
G H
I J
B
Figure 5: Analysis of the invasion of H23 cell clones ectopically expressing EGFP, EGFP-Rac1 or EGFP-Rac1b into the CAM of chick embryos.(A) The invasion of H23 cell clones stably expressing EGFP, EGFP-Rac1 or EGFP-Rac1b was analyzed using the CAM invasion model. The Figure shows representative results of CAM invasion assays performed with H23/EGFP A3 (A–B), H23/EGFP-Rac1 clone D1 and D2 (C–D and E–F) as well as H23/EGFP-Rac1b clone B1 and B3 (G–H and I–J). Six days after the carcinoma cells (1 × 106 cells/egg) were xenografted onto the CAM of fertilized chicken eggs, the CAMs were retrieved and sections analyzed by HE-staining. Representative microscopic images are shown of two to three independent experiments with 5–10 eggs per clone and experiment. Scale bars: 500 μm (5 ×) and 200 μm (20 ×).
J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 525
to Rac1. Rac1b mRNA content is controlled by ESRP1, which in addition enhances ZEB1 expression which sub-sequently represses E-cadherin expression.
DiscussionData available from the literature suggest that the amount of Rac1 or its splicing variant Rac1b modulates the
development and malignancy of human breast, colorec-tal and pancreatic tumors (Jordan et al., 1999; Schnelzer et al., 2000; Mehner et al., 2014; Ungefroren et al., 2014). High Rac1 protein expression strongly correlates with a reduced 5-year survival rate in lung adenocarcinoma patients because of an increased metastatic potential associated with EMT (Zhou et al., 2016).
Expression of both Rac1 and Rac1b has been described in lung cancer samples (Akunuru et al., 2011; Gastonguay
Figure 6: ESRP1 and ESRP2 in Rac1-related gene expression.(A) Western blot analysis of ESRP1 and ESRP2 proteins in various lung cancer cell lines. Detection of ESRP1 and ESRP2 compared to Rac1 and Rac1b in total lysate was performed with specific antibodies. GAPDH staining served to control for equal amounts of protein. A representative blot is shown out of two independent experiments. (B) Quantitative RT-PCR analyses of RAC1 and RAC1b mRNA expression after siRNA-mediated depletion of Rac1b, ESRP1 or ESRP2 in H358 lung cancer cells. Cells transfected with non-targeting siRNA (siControl) served as controls. (C) The mRNA content of ZEB1, CDH1 (E-cadherin), TWIST1, VIM (vimentin) and CDH11 (cadherin-11) was assayed by qRT-PCR after siRNA-mediated knockdown of Rac1b, ESRP1 or ESRP2. Data are presented as means ± SD of three independent experiments performed in duplicate. Statistical significance in relative gene expression was calculated in relation to siControl (*p < 0.05,**p < 0.01).

526 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
et al., 2012; Mehner et al., 2014; Zhou et al., 2016). In the present study, we provide evidence that enhanced amounts of the GTPase Rac1, in contrast to its closely related isoform Rac1b, contribute to progression of lung adenocarcinoma by increasing tumor invasion, whereas both Rac isoforms led to increased proliferation of H23 and HCC44 lung adenocarcinoma cells. This increased tumor development associated with high Rac1 expres-sion correlated with a partial EMT in lung NSCLA cells, as documented by decreased E-cadherin, ZO1 and claudin-4 concentrations, and increased cadherin-11 and vimen-tin expression. In agreement with the results obtained in pancreatic carcinoma cells (Ungefroren et al., 2014), expression of Rac1b did not induce EMT in lung adeno-carcinoma cell lines. This observation is in line with its expression in NSCLC cell lines with a more differentiated epithelial phenotype such as H358 and HTB55 (Figure 1A). Here, we demonstrate that Rac1b expression correlates with a differentiated epithelial phenotype (Figure 2A,B, Supplementary Figure S1) (Stallings-Mann et al., 2012). In contrast, EGFP-Rac1 expression resulted in a reduc-tion of epithelial marker proteins and an increase in vimentin and cadherin-11. The latter proteins have been described as important markers of mesenchymal differ-entiation in lung cancer, as their expression correlates with increased metastasis and poor prognosis for NSCLC patients (Dauphin et al., 2013). Inhibition of vimentin in lung cancer cells disturbed cell adhesion and diminished cell migration and invasion (Havel et al., 2015). Regarding a possible mechanism, we have reported previously that activated Rac1 destabilizes E-cadherin-mediated cellular adhesion by interacting with the scaffold protein IQGAP, whereas pharmacological inhibition of Rac1 resulted in epithelial differentiation of pancreatic carcinoma cells (Hage et al., 2009).
The Rac1-mediated dedifferentiation of H23 cells described here is in agreement with a more aggressive growth and high invasive activity of Rac1-expressing H23 cells in the CAM invasion model. In contrast, Rac1b-expressing H23 cells, although being able to penetrate the CAM, did not exhibit an increased cellular migration and invasion compared to control H23 cells. Moreover, they produced tumors of a more differentiated type than Rac1-expressing cells, which is in accordance with their more differentiated phenotype.
The tumor-promoting effects of Rac1 were associated with modulation of the transcriptional activation of gene promoters containing SRF-, SMAD- or NFκB-binding sites in NSCLA cells. In contrast to Rac1, Rac1b activated LEF/TCF- and NF-κB-sensitive promoters together with JNK2 in H23 and HCC44 cells, but failed to activate other promoters
containing SRF-, ETS2- or SMAD-responsive sequences. These data are in accordance with results of Singh et al. which demonstrated the inability of Rac1b to induce SRF, NFκB and cyclin D1 promoter activation (Singh et al., 2004). The effect of both Rac1 isoforms on proliferation of H23 cells correlates with the finding that both Rac1 forms increased the activation of different kinases involved in proliferation control such as p38, AKT and GSK3β in the analyzed lung adenocarcinoma cells. A recent publication by Li et al. supports the role of Rac1b in proliferation and inhibition of apoptosis by the activation of JNK2 (Li et al., 2016). However, earlier studies emphasize that Rac1b did not activate p21 protein kinase nor its downstream protein kinase JNK (Matos et al., 2003; Esufali et al., 2007). An interesting difference between Rac1 and Rac1b, not ana-lyzed in this study, has been described regarding their sensitivity toward ubiquitinylation which is involved in regulation of protein stability (Visvikis et al., 2008). Lys147 of Rac1 has been identified to be modified by the ubiquitin E3 ligases HACE1, whereas Rac1b was poorly ubiquitinated and more resistant against proteasomal degradation.
The observed increase in Rac1b expression during tumor development has been associated with alternative splicing events. ESRP1 has been shown to be responsible for exclusion of exon 3b, resulting in Rac1 mRNA. The ESPRP1 knockdown increased the generation of Rac1b mRNA and simultaneously decreased that of ZEB1, a tran-scription factor well known for its role in triggering EMT. In the Rac1-expressing cell clones, ZEB1 was upregulated and E-cadherin was downregulated on the protein and mRNA level (as shown in Figure 2 and Supplementary Figure S2C.) This may explain why elevated levels of Rac1 promote EMT and increase the invasive capacities of lung adenocarcinoma cells. The decrease in E-cadherin protein steady-state level, shown here, may be regulated at the level of mRNA processing or stability, as CDH1 promoter activity remained unaffected by Rac1 in the reporter gene assays (Figure 4A). ESRP2, another member of this splice factor family, inhibited twist1 expression, a bHLH tran-scription factor, which induces mesenchymal differentia-tion during EMT (Zeisberg and Neilson, 2009; Nakashima et al., 2012; Stemmler et al., 2019). The increase in twist1 correlates with increased vimentin mRNA content, sup-porting the mesenchymal differentiation of lung cells after suppression of ESRP2. A function of ESRP1 and 2 in EMT is also supported by data of Ishii et al., demonstrating the plasticity of ESRP expression during metastasis and invasion of head and neck cancer cells. This study shows that cancer cells from primary tumors lost expression of ESRPs, but re-expressed them after reaching tumor stroma

J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 527
or lymph nodes (Ishii et al., 2014). These alterations corre-lated with increased amounts of ZEB after knockdown of ESRP2 in HNSCC cells.
Possible functions of Rac1 and Rac1b in tumorigenesis are controversially discussed in the literature. Although Rac1b has been described to correlate with progression of breast and colorectal cancer, Rac1b alone does not seem to be sufficient for tumor induction (Jordan et al., 1999; Schnelzer et al., 2000). However, in combination with mutationally activated K-Ras, with increased amounts of the metalloproteinase MMP3, or with tumorigenic TGFβ signaling, Rac1b may be able to enhance tumor initiation. Zhou et al. described a strong correlation of Rac1b expres-sion in human lung tumors with accelerated K-Ras(G12D) and elevated cell proliferation in a mouse lung adenocar-cinoma model (Zhou et al., 2013). Our data support these findings, as Rac1b and also Rac1 increased the prolifera-tion of H23 NSCLA cells, which exhibit heterozygously activated K-Ras [(Zondag et al., 2000) and K. Giehl unpub-lished results]. Along the same line of evidence, Li et al. demonstrated that Rac1b is specifically able to activate JNK2/c-jun together with AKT2, thereby improving cell viability and cell cycle progression of colon cancer cells (Li et al., 2016). However, Rac1b did not induce EMT or enhanced cell invasion in our model. Stallings-Mann and coworkers described the induction of Rac1b in a transgenic mouse model expressing MMP3, suggesting that MMP3 promotes Rac1b expression in pancreatic and lung cancer (Mehner et al., 2014; Stallings-Mann et al., 2012). Moreover, the combination of both proteins was needed to induce EMT with reduced E-cadherin protein levels and increased expression of vimentin (Stallings-Mann et al., 2012).
The data presented demonstrate that elevated amounts of Rac1 rather than Rac1b augmented EMT, cell proliferation and invasion in the H23 lung cancer cell model. This is in line with overexpression of Rac1 in 111/153 NSCLC tissues and the corresponding low survival rates of patients (Zhou et al., 2016). Data from Dai et al. support this assumption because upregulation of Rac1b plays a pivotal role in ARHGAP11A-induced EMT, inva-sion and migration (Dai et al., 2018). Thus, the data point to an independent and sometimes opposing role of Rac1 and Rac1b in the development of lung adenocarcinomas. With regard to this opposing role, it has been reported that Rac1 activation [induced by platelet-derived growth factor (PDGF)] can be suppressed by Rac1b in HeLa cells (Nimnual et al., 2010). Altogether, these data provide evi-dence that Rac1 modulates cell migration and invasion in the progression of lung adenocarcinoma. Further support came from an earlier study which showed in the pancre-atic carcinoma cell line Panc1 that Rac1b, in contrast to
Rac1, was able to inhibit TGFβ1-induced cell migration (Ungefroren et al., 2014). In a more recent work, the TGFβ type I receptor ALK5 has been identified as a Rac1b target, the expression of which was suppressed by Rac1b (Unge-froren et al., 2019).
Our data support an important role of increased Rac1 concentrations in induction of EMT followed by enhanced tumor cell invasion of lung adenocarcinoma cells. The closely related isoform Rac1b did not exhibit such an impact on tumor progression. The data point to an inde-pendent and potentially opposing role of Rac1 and Rac1b in the development of lung adenocarcinomas, metastasis and invasive growth.
Materials and methodsAntibodies and plasmids used in this study were commercially available or kindly provided by colleagues: Anti-P-AKTSer473 XP, anti-pan-AKT (40D4), anti-P-ERK1/2Thr202/Tyr204 XP, anti-ERK1/2, anti-P-p38αThr180/Tyr182, anti-p38, anti-P-JNK1/2Thr183/Tyr185, anti-P-GSK3α/βSer21/Ser9, anti-GSK3β, anti-vimentin D21H3, anti-lamin B1 and anti-claudin-4 were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). Anti-caveolin, anti-Rac1, anti-paxillin, anti-N-cadherin and anti-E-cadherin were obtained from BD Bio-sciences (Franklin Lakes, NJ, USA). Anti-ZO1 and anti-β-actin were purchased from Sigma-Aldrich (Taufkirchen, Germany). Anti-JNK2, anti-GFP and anti-RhoGDIα were from Santa Cruz Biotechnologies (Dallas, TX, USA) and anti-Rac1b from EMD Millipore (Darmstadt, Germany). Anti-cadherin-11, anti-ESRP1 and anti-ESRP2 were pur-chased from Invitrogen (Leiden, The Netherlands) and anti-GAPDH from Acris (Herford, Germany).
For luciferase reporter assays, the following vectors were used: pGL3-SRE.L (A. Nordheim, University of Tuebingen, Germany), pTOPflash (Upstate, Lake Placid, NY, USA), p3TP-Lux (Wrana et al., 1992), pGL3-NFAT (A. Koenig, University of Goettingen, Germany), pGL3-N-Cadherin (P.J. Marie, University Paris Diderot, Paris, France), pGL3-FHRE (L.O. Klotz, University of Jena, Germany), pGL3-E-Cad-herin (M. Park, McGill University, Toronto, Canada), pGL-Cyclin D1 (F. McCormick, UCSF, San Francisco, CA, USA). pGL3-Myc (He et al., 1998) and pGL3-ETS2 (Tetsu and McCormick, 1999) were obtained from Addgene (Watertown, MA, USA). pAP1-Luc was obtained from Clontech Laboratories (Mountain View, CA, USA) and pGL3-NFκB (pGL4.32) was obtained from Promega (Mannheim, Germany).
Cell lines and cell culture conditions
H23 (CRL-5800), H1299 (CRL-5803), H358 (CRL-5807), HTB55 (HTB-55) and H2122 (CRL-5985) cells were obtained from ATCC (Manassas, VA, USA). HCC44, A549 and Colo699 cells were from DSMZ (Braunsch-weig, Germany). The cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) (Capricorn, Ebsdorfergrund, Germany), 1% GlutaMAX and 1% non-essential amino acids (both from Gibco-ThermoFisher, Freiburg, Germany).

528 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
Plasmid and siRNA transfection
For Rac1 expression, H23 and HCC44 cells were transfected with pEGFP, pEGFP-Rac1 and pEGFP-Rac1b (Hage et al., 2009) with Lipo-fectamine LTX (ThermoFisher Scientific, Langenselbold, Germany) according to manufacturer’s instructions. Cell lysates were analyzed 24–48 h post transfection. To generate stable EGFP-Rac1-/EGFP-Rac1b-expressing cell clones, transfected cells were selected with 1.5 mg/ml G418 (Capricorn). Resistant cells were subcloned and EGFP/EGFP-Rac1/EGFP-Rac1b expression was confirmed by western blot analyses and fluorescence microscopy (IX81, Olympus, Hamburg, Germany). Two clones per construct were chosen for further experiments.
For siRNA transfection, H23 and H358 cells were transfected with scrambled, non-targeting siRNA (Dharmafect ON-TARGET plus control pool) (Dharmacon, Lafayette, CO, USA), a mixture of Rac1b-si-1A and Rac1b-si-1B or GFP-22 siRNA (all Qiagen, Hilden, Germany), siESRP1_SMARTpool and siESRP2_SMARTpool (both Dharmacon) using Lipofectamin RNAiMax (ThermoFisher Scientific). The cells were transfected twice with siRNAs and lysed 72 h after the second transfection.
Protein analysis
Cultured cells were lysed in NOP buffer (10 mm Tris-HCl pH 7.4, 150 mm NaCl, 1 mm MgCl2, 1 mm CaCl2, 1% nonidet P40, 0.2% Tri-ton X-100) supplemented with protease inhibitors [1 μm aprotinin (Carl Roth, Karlsruhe, Germany), 1 mm pefabloc (Serva, Heidelberg, Germany), 2.5 μm soy bean trypsin inhibitor, 2.1 μm leupeptin (both Sigma-Aldrich)] and with phosphatase inhibitors [10 mm sodium fluo-ride (AppliChem, Darmstadt, Germany), 1 mm sodium orthovanadate, 25 mm β-glycerophosphate and 10 mm sodium pyrophosphate (all Sigma-Aldrich), 750 nm okadaic acid (Cayman Chemical Company, Ann Arbor, MI, USA)]. Fifty micrograms of protein lysates were sepa-rated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Protran BA85, GE Healthcare, Munich, Germany). Proteins were visualized using the Odyssey Sa Infrared Imaging System (LI-COR Bioscience, Lincoln, NE, USA) or enhanced chemiluminescence (ECL, Ther-moFisher Scientific). The quantification of the bands’ intensities was calculated using the software ImageStudio (LI-COR, version 5.02).
Subcellular fractionation
For preparation of cell membrane fractions, the cell clones were lysed and homogenized in HEPES buffer [50 mm HEPES pH 7.6, 8.6% saccharose, 10 mm ethylenediaminetetraacetic acid (EDTA), 10 mm ethylene glycol tetraacetic acid (EGTA)] supplemented with protease inhibitors, as described in Dreissigacker et al. (2006). Briefly, to enrich membranous and cytosolic proteins, the nucleus-free super-natant was centrifuged at 100 000 g for 1 h at 4°C. The pellet (P100 fraction), containing the membranous proteins, was resuspended in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl pH 7.6, 4 mm NaCl, 10 mm EDTA) supplemented with protease inhibi-tors. Fifty micrograms of the supernatant, containing the cytosolic proteins (S100 fraction) and 30 μg of P100 fraction, was separated by SDS-PAGE.
Rac activity assay
Rac activity was estimated by pull-down assays. The assay was per-formed as described in Stähle et al. (2003). Briefly, confluent H23 cell clones were lysed in Rac-RIPA buffer (50 mm Tris-HCl pH 7.2, 150 mm NaCl, 10 mm MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm pefabloc) and homogenized. To pull-down active GTP-bound Rac, GST-PAK-CD fusion protein attached to glutathione sepharose 4B beads (GE Healthcare) was incubated with the H23 cell lysate for 1 h. The beads were washed and the amount of active GTP-Rac was analyzed by immunoblotting.
Quantitative RT-PCR studies
SYBR green-based, qRT-PCRs were performed using QuantiTect primer pairs (Qiagen) for the detection of RAC1, ESRP1, ESRP2, CDH1, ZEB1, SNAI1 and VIM. The primers specific for RAC1B (forward: TGA ATC TGG GCT TAT GGG ATA CA, back: GGT TAT ATC CTT ACC GTA CGT TTC TCC) and RPLP0 (forward: GTC GGA GTC GGA CGA G, back: GGT TAT ATC CTT ACC GTA CGT TTC TCC) mRNAs were purchased from Biomers.net (Ulm, Germany). Each sample was analyzed in duplicate. The experi-ments were carried out using the SensiMixTM SYBR No-ROX PCR mix according to the instruction of the manufacturer (Bioline, Heidelberg, Germany) in the Stratagene Mx3005P real time PCR cycler (Agilent Technologies, Santa Clara, CA, USA). Detection of RPLP0 or ACTB was used as a reference. The amplification, calculated by the 2-ΔΔCt method, and the dissociation curves were calculated using the MxPro qPCR software (Agilent Technologies).
Luciferase reporter gene assay
Cells were co-transfected with the indicated luciferase reporter con-structs, a Renilla luciferase-expressing plasmid pRL-TK (Promega), the control vector pCGN, pCGN-HA-RAC1 or pCGN-HA-RAC1b (wild-type or constitutively active G12V mutants of Rac) using the DNA transfection reagent jetPEI (Polyplus Transfection, Illkirch-Graffen-staden, France). After 24 h, the cells were harvested and the firefly and Renilla luciferase activities were measured using the Dual-Lucif-erase Reporter Assay System (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity to eliminate the effect of different transfection efficiencies. The experiments were performed at least 3 times in duplicate.
Proliferation assay
Cell proliferation of EGFP-, EGFP-Rac1- and EGFP-Rac1b-expressing cells was determined by counting EGFP-positive cells by FACS analy-sis at t = 0, after 24 h, 48 h and 72 h. The volume was normalized to co-counted fluorescent particles of a defined concentration (Spherotech, Lake Forest, IL, USA). Additionally, cell doubling time was recorded in real time using the xCELLigence RTCA-DP biosensor technology (ACEA Bioscience, San Diego, CA, USA) according to manufactur-er’s instructions. Twelve thousand cells were seeded in wells of an

J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 529
E-plate-16-containing micro-electronic sensors. Cells were incubated with growth medium containing 10% FCS. The impedance, which correlates with the cell number, was measured for 72 h and the cell index was calculated using the RTCA software (ACEA Bioscience). Three different biological replicates were analyzed in quadruplicate.
Migration assay
To analyze the migration rate of H23 cell clones, cells of a confluent cell layer were treated with 5 μg/ml mitomycin C (Sigma-Aldrich) for 2 h to inhibit cell proliferation and scratched with a 200-μl pipet tip. The wound size was documented microscopically 2 h after scratch-ing and thereafter every 3 h using the EVOS FL Auto II system (Ther-moFisher Scientific). The migration rate in μm/h was calculated from the reduction of the cell gap, which was measured using the Celleste imaging analysis software (ThermoFisher Scientific). Depicted is the mean ± standard deviation (SD) of four independent experiments per cell clone with six measuring points per scratch and condition.
Chorioallantoic membrane (CAM) invasion assay
The invasive potential of the cells was analyzed using the CAM model previously described in Hofer et al. (2004) with some modifications (Hofer et al., 2004). Briefly, cells (1 × 106 cells in 20 μl DMEM) were seeded into a silicone ring (5 mm diameter) placed on the CAM of fertilized chicken eggs on day 5 of the breeding protocol and incu-bated for another 6 days at 37°C and 60% relative humidity. The chorioallantoic tissue area containing the silicone ring was isolated, fixed with 4% formaldehyde, paraffin-embedded, 5 μm sections were transferred to microscopic slides and stained with hematoxylin and eosin. Microscopic examination was carried out using a DMLA light microscope (Leica Microsystems, Wetzlar, Germany).
Statistics
All depicted values represent the mean ± SD of at least three independ-ent experiments. For statistical comparisons, student’s t-test or one-way analysis of variance (ANOVA) with Tukey’s post-test were performed using GraphPad Prism 6 (Graphpad Software, San Diego, CA, USA).
Acknowledgments: This work was supported by a Research Grant of the University Medical Center Giessen and Marburg (UKGM), Germany, the LOEWE excellence cluster “Universities-Giessen-Marburg-Lung-Center” (UGMLC) of the federal state Hessen, and the Verein zur Förderung der Krebsforschung in Gießen.
ReferencesAkunuru, S., Palumbo, J., Zhai, Q.J., and Zheng, Y. (2011). Rac1 tar-
geting suppresses human non-small cell lung adenocarcinoma cancer stem cell activity. PLoS One 6, e16951.
Bar-Sagi, D. and Hall, A. (2000). Ras and Rho GTPases: a family reunion. Cell 103, 227–238.
Chen, M. and Manley, J.L. (2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741.
Dai, B., Zhang, X., Shang, R., Wang, J., Yang, X., Zhang, H., Liu, Q., Wang, D., Wang, L., and Dou, K. (2018). Blockade of ARHGAP11A reverses malignant progress via inactivating Rac1B in hepato-cellular carcinoma. Cell. Commun. Signal. 16, 99.
Dauphin, M., Barbe, C., Lemaire, S., Nawrocki-Raby, B., Lagonotte, E., Delepine, G., Birembaut, P., Gilles, C., and Polette, M. (2013). Vimentin expression predicts the occurrence of metas-tases in non small cell lung carcinomas. Lung Cancer (Amster-dam) 81, 117–122.
Dittmar, K.A., Jiang, P., Park, J.W., Amirikian, K., Wan, J., Shen, S., Xing, Y., and Carstens, R.P. (2012). Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol. Cell. Biol. 32, 1468–1482.
Dreissigacker, U., Mueller, M.S., Unger, M., Siegert, P., Genze, F., Gierschik, P., and Giehl, K. (2006). Oncogenic K-Ras down-regulates Rac1 and RhoA activity and enhances migration and invasion of pancreatic carcinoma cells through activation of p38. Cell. Signal. 18, 1156–1168.
Esufali, S., Charames, G.S., Pethe, V.V., Buongiorno, P., and Bapat, B. (2007). Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 67, 2469–2479.
Fiegen, D., Haeusler, L.-C., Blumenstein, L., Herbrand, U., Dvorsky, R., Vetter, I.R., and Ahmadian, M.R. (2004). Alternative splic-ing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 279, 4743–4749.
Gastonguay, A., Berg, T., Hauser, A.D., Schuld, N., Lorimer, E., and Williams, C.L. (2012). The role of Rac1 in the regulation of NF-κB activity, cell proliferation, and cell migration in non-small cell lung carcinoma. Cancer Biol. Ther. 13, 647–656.
Hage, B., Meinel, K., Baum, I., Giehl, K., and Menke, A. (2009). Rac1 activation inhibits E-cadherin-mediated adherens junctions via binding to IQGAP1 in pancreatic carcinoma cells. Cell. Commun. Signal. 7, 23.
Hall, A. (2005). Rho GTPases and the control of cell behaviour. J. Biochem. Soc. 7, 891–895.
Havel, L.S., Kline, E.R., Salgueiro, A.M., and Marcus, A.I. (2015). Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 34, 1979–1990.
He, T.C., Sparks, A.B., Rago, C., Hermeking, H., Zawel, L., da, C.L.T., Morin, P.J., Vogelstein, B., and Kinzler, K.W. (1998). Identifi-cation of c-MYC as a target of the APC pathway. Science 281, 1509–1512.
Hill, C.S., Wynne, J., and Treisman, R. (1995). The Rho family GTPases RhoA, Racl, and CDC42Hsregulate transcriptional activation by SRF. Cell 81, 1159–1170.
Hofer, M.D., Menke, A., Genze, F., Gierschik, P., and Giehl, K. (2004). Expression of MTA1 promotes motility and invasiveness of PANC-1 pancreatic carcinoma cells. Br. J. Cancer 90, 455–462.
Ishii, H., Saitoh, M., Sakamoto, K., Kondo, T., Katoh, R., Tanaka, S., Motizuki, M., Masuyama, K., and Miyazawa, K. (2014). Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem. 289, 27386–27399.

530 J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines
Jonkman, J.E.N., Cathcart, J.A., Xu, F., Bartolini, M.E., Amon, J.E., Stevens, K.M., and Colarusso, P. (2014). An introduction to the wound healing assay using live-cell microscopy. Cell Adh. Migr. 8, 440–451.
Jordan, P., Brazao, R., Boavida, M.G., Gespach, C., and Chastre, E. (1999). Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 18, 6835–6839.
Larsen, J.E., Nathan, V., Osborne, J.K., Farrow, R.K., Deb, D., Sullivan, J.P., Dospoy, P.D., Augustyn, A., Hight, S.K., Sato, M., et al. (2016). ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Invest. 126, 3219–3235.
Li, G., Ying, L., Wang, H., Wei, S.S., Chen, J., Chen, Y.H., Xu, W.P., Jie, Q.Q., Zhou, Q., Li, Y.G., et al. (2016). Rac1b enhances cell sur-vival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget 7, 17970–17985.
Li, L., Qi, L., Qu, T., Liu, C., Cao, L., Huang, Q., Song, W., Yang, L., Qi, H., Wang, Y., et al. (2018). Epithelial Splicing Regulatory Protein 1 inhibits the invasion and metastasis of lung adenocarcinoma. Am. J. Pathol. 188, 1882–1894.
Matos, P., Collard, J.G., and Jordan, P. (2003). Tumor-related alterna-tively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J. Biol. Chem. 278, 50442–50448.
Mehner, C., Miller, E., Khauv, D., Nassar, A., Oberg, A.L., Bamlet, W.R., Zhang, L., Waldmann, J., Radisky, E.S., Crawford, H.C., et al. (2014). Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol. Cancer Res. 12, 1430.
Melzer, C., Hass, R., von der Ohe, J., Lehnert, H., and Ungefroren, H. (2017). The role of TGF-β and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell. Commun. Signal. 15, 19.
Minden, A., Lin, A., Claret, F.-X., Abo, A., and Karin, M. (1995). Selec-tive activation of the JNK signaling cascadeand c-Jun transcrip-tional activity by the small GTPases Rac and Cdc42Hs. Cell 81, 1147–1157.
Nakashima, H., Hashimoto, N., Aoyama, D., Kohnoh, T., Saka-moto, K., Kusunose, M., Imaizumi, K., Takeyama, Y., Sato, M., Kawabe, T., et al. (2012). Involvement of the transcription factor twist in phenotype alteration through epithelial-mesenchy-mal transition in lung cancer cells. Mol. Carcinogenesis 51, 400–410.
Nimnual, A.S., Taylor, L.J., Nyako, M., Jeng, H.H., and Bar-Sagi, D. (2010). Perturbation of cytoskeleton dynamics by the opposing effects of Rac1 and Rac1b. Small GTPases 1, 89–97.
Nobes, C.D. and Hall, A. (1995). Rho, rac, and cdc42 GTPases regu-late the assembly of multimolecular focal complexes associ-ated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62.
Ridley, A.J. (2001). Rho GTPases and cell migration. J. Cell Sci. 114, 2713–2722.
Schmalhofer, O., Brabletz, S., and Brabletz, T. (2009). E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 28, 151–166.
Schnelzer, A., Prechtel, D., Knaus, U., Dehne, K., Gerhard, M., Graeff, H., Harbeck, N., Schmitt, M., and Lengyel, E. (2000). Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 19, 3013.
Shapiro, I.M., Cheng, A.W., Flytzanis, N.C., Balsamo, M., Condee-lis, J.S., Oktay, M.H., Burge, C.B., and Gertler, F.B. (2011). An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 7, e1002218.
Siegel, R.L., Miller, K.D., and Jemal, A. (2017). Cancer statistics, 2017. Cancer J. Clinicians 67, 7–30.
Singh, A., Karnoub, A.E., Palmby, T.R., Lengyel, E., Sondek, J., and Der, C.J. (2004). Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 23, 9369–9380.
Stähle, M., Veit, C., Bachfischer, U., Schierling, K., Skripczynski, B., Hall, A., Gierschik, P., and Giehl, K. (2003). Mechanisms in LPA-induced tumor cell migration: critical role of phosphorylated ERK. J. Cell Sci. 116, 3835–3846.
Stallings-Mann, M.L., Waldmann, J., Zhang, Y., Miller, E., Gauthier, M.L., Visscher, D.W., Downey, G.P., Radisky, E.S., Fields, A.P., and Radisky, D.C. (2012). Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci. Transl. Med. 4, 142ra195.
Stemmler, M.P., Eccles, R.L., Brabletz, S., and Brabletz, T. (2019). Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 21, 102–112.
Tetsu, O. and McCormick, F. (1999). Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426.
Ungefroren, H., Sebens, S., Giehl, K., Helm, O., Groth, S., Fändrich, F., Röcken, C., Sipos, B., Lehnert, H., and Gieseler, F. (2014). Rac1b negatively regulates TGF-β1-induced cell motility in pan-creatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 5, 277–290.
Ungefroren, H., Otterbein, H., Fiedler, C., Mihara, K., Hollenberg, D.M., Gieseler, F., Lehnert, H., and Witte, D. (2019). RAC1B suppresses TGF-β1-dependent cell migration in pancreatic carcinoma cells through inhibition of the TGF-β type I receptor ALK5. Cancers 11, 691–701.
Visvikis, O., Lorès, P., Boyer, L., Chardin, P., Lemichez, E., and Gacon, G. (2008). Activated Rac1, but not the tumorigenic vari-ant Rac1b, is ubiquitinated on Lys147 through a JNK-regulated process. FEBS J. 275, 386–396.
Warzecha, C.C., Jiang, P., Amirikian, K., Dittmar, K.A., Lu, H., Shen, S., Guo, W., Xing, Y., and Carstens, R.P. (2010). An ESRP-regu-lated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 29, 3286–3300.
Wolpert, L., Smith, J.C., Hall, A., and Nobes, C.D. (2000). Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Phil. Trans. R. Soc. Lond. B Biol. Sci. 355, 965–970.
Wrana, J.L., Attisano, L., Carcamo, J., Zentella, A., Doody, J., Laiho, M., Wang, X.F., and Massague, J. (1992). TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71, 1003–1014.
Yae, T., Tsuchihashi, K., Ishimoto, T., Motohara, T., Yoshikawa, M., Yoshida, G.J., Wada, T., Masuko, T., Mogushi, K., Tanaka, H., et al. (2012). Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Com-mun. 3, 883.
Zeisberg, M. and Neilson, E.G. (2009). Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437.

J.R. Seiz et al.: Rac-isoforms in lung adenocarcinoma cell lines 531
Zhou, C., Licciulli, S., Avila, J.L., Cho, M., Troutman, S., Jiang, P., Kos-senkov, A.V., Showe, L.C., Liu, Q., Vachani, A., et al. (2013). The Rac1 splice form Rac1b promotes K-ras-induced lung tumori-genesis. Oncogene 32, 903–909.
Zhou, Y., Liao, Q., Han, Y., Chen, J., Liu, Z., Ling, H., Zhang, J., Yang, W., Oyang, L., Xia, L., et al. (2016). Rac1 overexpression is correlated with epithelial mesenchymal transition and predicts poor prognosis in non-small cell lung cancer. J. Cancer 7, 2100–2109.
Zondag, G.C.M., Evers, E.E., ten Klooster, J.P., Janssen, L., van der Kammen, R.A., and Collard, J.G. (2000). Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J. Cell Biol. 149, 775–781.
Supplementary Material: The online version of this article offers supplementary material (https://doi.org/10.1515/hsz-2019-0329).





























