Embed Size (px)
DESCRIPTION
Â
Citation preview

Volume 8 Issue 3I Journal for Clinical Studies
What Do You Need to Know to Ensure Shipping Success To North Africa
Empowering Real World Evidence Analysis Through Risk-based De-identification
Bringing Family Medicine To Clinical Research
Integrating the Patient Perspective into Clinical Research Study DesignsCase Studies and Recommendations
PEER REVIEWED
JOURNAL FOR
Your Resource for Multisite Studies & Emerging MarketsCLINICAL STUDIESU
Volume 8 - Issue 3

Clinical documents arethe visible part of
your clinical programme
Successful documents don’thappen by chance
Tel: +49 69 138 2528-0
Fax: +49 69 138 2528-55
email: [email protected]
www.trilogywriting.com
Trilogy makes surethey do.
Are the regulatorsseeing what yourdocuments have
to say?
TRI026 TRILOGY ICEBERG AD UPDATE.pdf 1 24/07/2014 16:06

Journal for Clinical Studies 1www.jforcs.com
Contents
06 FOREWORD
WATCH PAGES
08 The Real World of Medicine: Regulatory Perspectives on the Pragmatic Data to Support the Approval of New Drug Products
Regina Ballinger, Senior Manager of Regulatory Intelligence with Thomson Reuters, shares her views on what is known as FDA’s “Statute.” A pillar of the FDA’s mission, the statute speaks to requirements of substantial evidence of efficacy as the bar for approving new drug products. When speaking on the approval of medical devices, this becomes “reasonable assurance of safety and efficacy.” Semantics aside, the FDA is looking to tap sources of valid scientific data to support the approval of medical products in the US.
10 Benefiting eCOA through the Right App StrategyChris Watson, Director of Product Strategy for Exco InTouch, explains how electronic clinical outcomes assessment (eCOA) providers are facing heightened demand for user-friendly, accessible solutions that facilitate hassle-free participation in clinical trials. This has sparked debate on how to deliver apps across the different operating platforms of iOS, Android and Windows in order to provide the best user experience. In particular – and for good reason – the use of native apps is becoming an increasingly popular choice.
12 Cardiovascular Medical Devices Watch ColumnThis cardiovascular medical devices watch column was established at the beginning of this year. However, owing to various changes in his job responsibilities, the original writer will no longer be able to provide regular updates. Therefore, Dr Rick Turner, PhD, DSc, FASH, a member of Quintiles’ Cardiac Safety and Cardiovascular Centers of Excellence, stepped in to do so. Dr Turner also wrote cardiovascular safety watch columns discussing biopharmaceutical drugs for this journal for several years, and he will now be writing about cardiovascular medical devices.
14 What do you Need to Know to Ensure Shipping Success to North Africa?
Whilst obviously part of the African continent, North Africa is very definitely its own distinct region, very much divided from sub-Saharan Africa, and most definitely not part of the Middle East. North Africa has been up till now a much under-used area when it comes to clinical trials. Sue Lee of World Courier explains why, across the whole region, preplanning and attention to detail are key for success in shipping.
REGULATORY
16 Empowering Real-world Evidence Analysis Through Risk- based De-identification
Clinical trials produce vast amounts of incredibly valuable data that, outside the initial research objectives, is not being sufficiently leveraged, according to Pamela Neely Buffone, Director of Product Management for Privacy Analytics. We are just starting to see the wealth of patient-level data delivering ground-breaking insights - particularly from real-world evidence (RWE) analysis. For years, companies have used real-world data (RWD) to conduct ongoing pharmacovigilance and run comparative analyses of their products. RWE is becoming a valuable tool for decision-making at multiple stages of the drug development process.
MANAGING DIRECTOR Martin Wright
PUBLISHERMark A. Barker
EDITOR Orsolya Balogh
PUBLICATION MANAGERVictoria [email protected]
EDITORIAL ASSISTANTVeronica [email protected]
DESIGNER Fiona Cleland
RESEARCH & CIRCULATION MANAGEREvelyn [email protected]
ADMINISTRATOR Barbara Lasco
FRONT COVER © istockphoto
PUBLISHED BY Pharma PublicationsUnit J413, The Biscuit Factory Tower Bridge Business Complex 100 Clements Road, London SE16 4DGTel: +44 0207 237 2036Fax: +0014802475316Email: [email protected]
Journal for Clinical Studies – ISSN 1758-5678 is published bi-monthly by PHARMAPUBS.
The opinions and views expressed by the authors in this magazine are not necessarily those of the Editor or the Publisher. Please note that although care is taken in preparation of this publication, the Editor and the Publisher are not responsible for opinions, views and inaccuracies in the articles. Great care is taken with regards to artwork supplied, the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
Volume 8 Issue 3 June 2016PHARMA PUBLICATIONS
JOURNAL FOR
Your Resource for Multisite Studies & Emerging Markets CLINICAL STUDIES U

Volume 8 Issue 32 Journal for Clinical Studies
Contents
20 Demand for Data Puts Safety at Centre in Decision-makingWhere once pharmacovigilance was regarded as a necessary burden, pharmaceutical companies are starting to recognise that safety in fact plays an integral role in business strategy, such as in decisions regarding mergers and acquisitions (M&A). Marco Anelli, MD, head of pharmacovigilance and medical affairs advisory services at ProductLife Group explores how the changing environment is affecting what is required of pharmacovigilance leaders, as well as how safety data is gathered, managed, and turned into critical reports and documents..
24 Medical Management in Clinical Trials: A Roadmap to Operational Excellence (Part 2 – Study Setup)
The more robustly and transparently the medical procedures are defined and documented, the more streamlined it gets during the study conduct and close-out phases. Further, the ground rules that structure the safety standards of the study and the integrity of the patient data collected are laid out during this phase. Mohamed El Malt, Chief Medical Consultant at Europital. & Vijayanand Rajendran, Clinical Research Physician at Europital, discusses in details the responsibilities and activities of a clinical research physician, the study MM, during the study setup phase – starting from clinical trial application till the recruitment of the first patient.
MARKET REPORT
28 Clinical Trial Transparency: New Data Anonymisation Requirements
Faced with new European Medicines Agency (EMA) guidance on the anonymisation of clinical trial data, drug manufacturers targeting the vast EU market have two choices – do what it takes to meet the November deadline – which prioritises clinical study report content - or embrace a smarter and more sustainable strategy that starts with the patient-level data. Chris Olinger, CTO of d-Wise, advises against taking shortcuts.
32 Bringing Family Medicine to Clinical ResearchThe World Organization of Family Doctors (WONCA) reports that 600,000 family doctors in 150 countries provide 3 billion consultations each year. General practice/family medicine (GP/FM) is the core discipline of primary medical care and the cornerstone of many healthcare systems in Europe. The vast majority of European citizens have a general practitioner (GP), who is a first-contact physician in case of health problems. Unfortunately, very few of them are involved in clinical research. Janusz Kabata MD, and Bohdan Tillack MSc, MBA at MedConsult with Paweł Kabata MD, PhD at MedSource Polska share their thoughts on how to bring family medicines to clinical research.
THERAPEUTICS
36 Improving Development of Antiepileptic Drugs for Rare Forms of Epilepsy
Jayne Abraham, PhD, Director of Medical and Scientific Affairs for Neuroscience at Worldwide Clinical Trials Therapeutic and Idil Çavuş, MD PhD is a Senior Medical Director of Medical and Scientific Affairs for Neuroscience at Worldwide Clinical Trials explain how rare diseases cover a broad range of diseases and patients, with about 50% of those affected being children. Many have a genetic component, while others arise from exposure to infections or toxins, from faulty immune responses, or occasionally from trauma or injury (e.g. traumatic brain injury (TBI)).
40 Considerations for Performing Sweat Testing In a Cystic Fibrosis Clinical Trial
Sweat testing was initially standardised in 1959 and it remains the gold standard for diagnosing cystic fibrosis (CF). Guidelines from the US Cystic Fibrosis Foundation and European Cystic Fibrosis Society (ECFS) both note that the sweat test is the preferred method to diagnose CF. Sweat is produced using a technique called iontophoresis, in which an electrical current

Journal for Clinical Studies 3www.jforcs.com
DORA WIRTH (LANGUAGES) LIMITED26-28 HAMMERSMITH GROVELONDONW6 7BA
www.dwlanguages.com
E-mail: [email protected].: +44 (0)20 7229 4552
@DWLanguages
Need global life science solutions?Rely on us. Problem solved.
Yourre
liabl
epa
rtne
rfo
rgl
obal
life
scie
nce
solu
tionsOver 50 years’ experience in providing
expertise, global translation solutionsand language consultancy in thefollowing specialist areas:
n Regulatory Affairs
n Clinical Research
n Medical Devices
n Manufacturing
n Legal
n Medical Research
n Medical Publishing
n Marketing Communications
DWL_Ad_05_2014_DWL Ad Doc 06/05/2014 13:00 Page 1

Volume 8 Issue 34 Journal for Clinical Studies
Contents
is used to simulate sweat production and chloride levels are subsequently measured. A sweat chloride level greater than 60 millimoles per litre (mmol/L) is considered diagnostic of CF. In this white paper, Vikki Brandi, Executive Director, Respiratory and Allergy Clinical Development, INC Research considers the performing of sweat testing in a cystic fibrosis clinical trial.
44 Why is Measuring Cognition Important?Susan McGoldrick at QCTR answers the question: Why is measuring cognition important? Cognition is the hallmark of the diagnosis of dementia, of which the most common form is Alzheimer’s disease (AD). It has proven to be a difficult concept to pin down and yet it is used to assist in diagnosis. There is no definitive diagnostic test for AD and biomarkers are not yet able to definitively diagnose AD – rather, its diagnosis is based on clinical criteria. Depending on how conclusive these criteria are will lead to a diagnosis of ‘probable’ or ‘possible’ AD. It is a sobering thought that the definitive AD diagnosis is only possible pathophysiologically at post mortem. The diagnoses of ‘probable’ or ‘possible’ AD all include a worsening of cognition in their definition.
TECHNOLOGY
48 The Changing Organisation and Data Management Roles Resource Roles Revisited for New Optimisation (Part 1)
Anyone who has been involved in clinical development and data management (DM), in particular for a long period of time, will recognise how the organisation of staff to perform the wide range of data collection and management tasks has changed to better align with advancements in technology. Joette Keen, Head of Biometrics & Clinical Data Execution Systems at KCR, discusses the pre-electronic data capture (EDC) era, when all trials, regardless of size, collected data on paper case report forms (CRFs). This method had a long lifespan, and as such had quite established practices which also drove operational organisation.
50 Beyond Diagnostics – The Added Value of Digital Body Imaging
From the advent of x-rays in 1895 to the latest developments in magnetic resonance imaging (MRI) and computed tomography (CT) technology, imaging has played a vitally important role in improving global public health. When considering more modern techniques, we see how in the past 5-10 years this area has evolved to provide a clearer picture of body composition. Olof Dahlqvist Leinhard, Chief Technology Officer at AMRA, discusses the future of digital body imaging, not only in terms of the development of new techniques and software, but also in the new ways that these can be applied for the benefit of patients..
LOGISTICS
54 Prioritising patient-centricity in your clinical trialsTim Roberts of PCI Pharma Services and Erem Latif of WestRock Healthcare examine how changes in healthcare legislation, alongside unprecedented merger and acquisition activity in the pharmaceutical industry, have increased the stakes in the clinical trials process – including increased outsourcing of clinical trials. Within a context of rising costs – from medication non-adherence and the need for ever wider global footprints – a patient-led approach is essential to help ensure compliance and, ultimately, clinical trial success.
SPECIAL FEATURE
58 Integrating the Patient Perspective into Clinical Research Study Designs: Case Studies and Recommendations
One need not conduct a literature review to readily identify treatments in popular culture that have had a tremendous impact on societal health: for example, penicillin and the polio vaccine come immediately to mind. Indeed, the evolution of clinical research from its early beginnings to the sophisticated machine we see today has been extraordinary, generating vast amounts of data, conclusions, and even more research questions. David Cameron, at Quintiles, shares his case studies and recommendations on patient perspective and clinical research study designs.

Journal for Clinical Studies 5www.jforcs.com
MEDICAL EXPERTISE
EXPERIENCE
GLOBALTRANSPARENCY
24/7
Oncology
Infectious Diseases
Immunology
Gastroenterology
Inflammatory Diseases
Ophtalmology
Hematology
Cardiology
Respiratory Diseases
Medical Management of Clinical Trials
Medical and Safety Review
24/7 Medical Monitoring
A team of Clinical Research Physicians with Prominent Scientific Skills and Exemplary Professional Standards.
Dedicated Medics with In-depth Knowledge, Proactivity and Unparalleled Commitment.
Flexible end-to-end Medical Services with Strong Work Ethics and Sense of Shared Ownership.
WHO WE ARE
OUR EXPERIENCE
MEDICAL SERVICES
ContactTel: +32 9 331 60 30Fax: +32 9 329 55 48Email: [email protected]: europital.com
Medical Monitoring for your next Clinical Trial
46_JCS_January2016.indd 65 03/02/2016 12:31:53

Volume 8 Issue 36 Journal for Clinical Studies
Foreword
Editorial Advisory Board
Art Gertel, VP, Clinical Services, Regulatory & Medical writing, Beardsworth Consulting Group Inc.
Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
Bakhyt Sarymsakova - Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Cellia K. Habita, President & CEO, Arianne Corporation
Chris Tierney, Business Development Manager, EMEA Business Development, DHL Exel Supply Chain, DHL Global
Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
Deborah A. Komlos, Senior Medical & Regulatory Writer, Thomson Reuters
Elizabeth Moench, President and CEO of MediciGlobal
Eileen Harvey, Senior VP/General Partner, PRA International
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet Development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organization (WHO) Expert in ethics
Georg Mathis, Founder and Managing Director, Appletree AG
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Hermann Schulz, MD, CEO, INTERLAB central lab services – worldwide GmbH
Janet Jones, Senior Director, ICON Clinical Research
Jerry Boxall, Managing Director, ACM Global Central Laboratory
Jeffrey Litwin, MD, F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Vice President, ClinArt International, Chair of the GCC Chapter of the ACRP
Nermeen Varawala, President & CEO, ECCRO – The Pan Emerging Country Contract Research Organisation
Patrice Hugo, Chief Scientific Officer,
Clearstone Central Laboratories
Rabinder Buttar – President & Chief Executive Officer of ClinTec International
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Sanjiv Kanwar, Managing Director, Polaris BioPharma Consulting
Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
Stefan Astrom, Founder and CEO of Astrom Research International HB
Steve Heath, Head of EMEA - Medidata Solutions, Inc
T S Jaishankar, Managing Director, QUEST Life Sciences
Clinical studies are being conducted in emerging markets as part of global drug development programmes in order to access large pools of eligible patients and to benefit
from a cost-effective structure. However, over the last few years, the definition of “emerging markets” has been under review, especially from a regulatory perspective.
Multiple factors are considered for placement of clinical studies, such as adherence to good clinical practice (GCP), medical infrastructure & standard of care, number of
eligible patients, etc. We also have to look at other quantitative factors, such as country’s GDP, patent applications, healthcare expenditure, healthcare infrastructure,
corruption, innovation, etc. These different factors and indexes are correlated to the number of clinical studies ongoing in the “emerging markets”. R&D, healthcare
expenditure, technology infrastructure, transparency, and level of innovation show a significant correlation with the number of clinical trials being conducted in these
countries. In recent years there has been an increasing trend across oncology brands for data showing clinical trials conducted in emerging markets. In 2013, more than
55,000 clinical trials were conducted in emerging nations in Asia, Latin America, Eastern Europe and Africa − representing almost 40% of all studies. The shift from more established markets
(US, UK, Western Europe) to emerging markets (India, China, Japan, Eastern Europe) brings to mind a number of questions:
1. Where is this shift coming from? What is driving it? Is it purely based on cost?
2. How are physicians viewing data coming from emerging markets? Are they viewing it the same as if it had come from an established market like the UK, US or the ‘EU4’?
3. What is the impact of this switch on the perception of pharmaceutical brands?
Concerns for data from clinical trials in emerging markets are coming from multiple angles, but the market does seem to be heading increasingly in that direction. So how can pharma
brands address these lingering concerns to make the data out of these markets better accepted? In addition to education on regulations and ethical requirements in the emerging markets,
other options available to pharma brands are to conduct a smaller trial to duplicate the results in an established market, provide a robust number of patients, publish results in a peer-
reviewed journal, or speak at global conferences.
In this issue of JCS, we have some interesting topics being discussed.
Regina Ballinger, Senior Manager of Regulatory Intelligence with Thomson Reuters, shares her views on what is known as the FDA’s “Statute”. A pillar of the FDA’s mission, the statute
speaks to requirements of substantial evidence of efficacy as the bar for approving new drug products. We have re-established the Cardiovascular Medical Device Watch column. Dr Rick
Turner, PhD, DSc, FASH, a member of Quintiles’ Cardiac Safety and Cardiovascular Centers of Excellence, has stepped in to produce this column. Sue Lee of World Courier explains
why, across the whole region of North Africa, preplanning and attention to detail are key for success in shipping. In the Regulatory section, Pamela Neely Buffone, Director of Product
Management for Privacy Analytics, discusses Empowering Real-world Evidence Analysis Through Risk-based De-identification, and Marco Anelli, MD, head of pharmacovigilance and
medical affairs advisory services at ProductLife Group explores how the changing environment is affecting what is required of pharmacovigilance leaders, as well as how safety data is
gathered, managed, and turned into critical reports and documents.
In the Market Report section, Janusz Kabata MD and Bohdan Tillack MSc, MBA at MedConsult, with Paweł Kabata MD, PhD at MedSource Polska, share their thoughts on how to bring
family medicines to clinical research. In the Therapeutic section, Vikki Brandi, Executive Director, Respiratory and Allergy Clinical Development, INC Research considers the performing
of sweat testing in a cystic fibrosis clinical trial, and Susan McGoldrick at QCTR answers the question: Why is measuring cognition important? Cognition is the hallmark of the diagnosis of
dementia, of which the most common form is Alzheimer’s disease. We have a special feature in this issue. David Cameron, at Quintiles, shares his case studies and recommendations on
patient perspective and clinical research study designs.
I hope you enjoy this edition of JCS. My team and I look forward to bringing you more interesting articles in the July issue.
Orsolya Balogh
Editor

Journal for Clinical Studies 7www.jforcs.com
Accelerating Enrollment Digital Marketing Locating Lost Patients
THE PATIENT RECRUITMENT-RETENTION REVOLUTION
“Over MediciGroup’s 24 year history, the last 4 years has witnessed more change than the previous 20 years. The speed of change is unprecedented as more than 85% of patients and families obtain health and clinical trial information online. Medici is proud to be at the forefront of this digital revolution, spearheading more than 30 global online patient communities, and leading in the delivery of successful patient recruitment through digital and social media on a global scale.”
Liz Moench, President & CEO MediciGroup® Inc.
WE’RE SOARING TO NEW HEIGHTS
End to End Services for Patient Recruitment and Retention

Volume 8 Issue 38 Journal for Clinical Studies
Watch Pages
The Real World of Medicine: Regulatory Perspectives on the Pragmatic Data to Support the Approval of New Drug ProductsWhen the US Food and Drug Administration (FDA) considers the approval of new products, the standard lies within what is known as “The Statute.” A pillar of the FDA’s mission, the statute speaks to requirements of substantial evidence of efficacy as the bar for approving new drug products. When speaking on the approval of medical devices, this becomes “reasonable assurance of safety and efficacy.” Semantics aside, the FDA is looking to tap sources of valid scientific data to support the approval of medical products in the US.
The newest child of substantial evidence could very well be “real-world evidence.” Real-world evidence (RWE) can be defined and used in a variety of ways. When applied to medicine, it would reflect data gathered from an actual use environment that is used as evidence to support a medical practice or therapy. Real-world data (RWD) is that information collected from any real environments. RWD from a variety of sources is merged to form RWE. The National Pharmaceutical Council notes that “the focus has shifted from looking simply at what should be compared to how comparative studies should be designed to answer the practical questions about ‘effectiveness,’ particularly in real-world settings.” GlaxoSmithKline has published a position paper that points to improvements in data infrastructure that are allowing new ways to fill gaps in knowledge left by traditional approaches to medical product approval.1
According to the FDA, RWE is already being incorporated into medical product applications, mainly in the areas of rare diseases and medical device approvals. The FDA has stated that it sees additional uses of RWE in supplemental indications, especially in instances where diseases are similar and/or their natural histories are well-characterised. There are many discussions surrounding the potential uses of RWE, including incorporating the data into a regulatory framework that could be used to guide decisions. Pilot programmes are cited as a way to gain experience with the use of RWE, allowing for testing grounds to learn where RWE would be most applicable. Potential uses and benefits include:
• Reducing approval time• Accelerating new/approved therapies• Improving amount and quality of information
available to all physicians• Changes to FDA-approved labelling • Benefit-risk assessment and impact on patient
decisions
There are a host of concerns related to the use of RWE to support approvals that must be addressed. “RWE is not always reliable,” says FDA senior policy-maker Janet Woodcock, MD, the head of the Center for Drug Evaluation and Research (CDER) at FDA. For example in
cancer, if medical records are the source of the data for a trial and the stage of the disease is not correct, assigned treatment would not be accurate. Robert Temple, MD, equates “pragmatic data” (another name for RWE) as similar to historical data in many instances. Dr Temple is the FDA’s Deputy Center Director for Clinical Science and Acting Deputy Director of the Office of Drug Evaluation I in CDER. He makes note of several problems attributed to RWE in the current environment, including:
• Reliability of data• Informed consent complications/challenges• Adapting current methodologies or designing new
ones to fit the data gathered in real-world settings• Linking type of data collection sources together
(e.g., death records with claims)
The FDA is speaking out on this topic in a variety of ways. Drs Woodcock and Temple made their comments as panel members at a public meeting on RWE on March 3-4, 2016, sponsored by the Duke-Margolis Center for Health Policy. In the December 10, 2015 issue of FDA Voice, the FDA describes the revolution of big data and its impact on RWE for medical product approvals in the US. RWE is integrated into discussions.2

Journal for Clinical Studies 9www.jforcs.com
Despite any potential problems posed by RWE, there is a definite trend toward its use with the concept of pragmatic and adaptive clinical trials becoming more readily and openly discussed by policy-makers, pharmaceutical companies, and third-party payers in the US. Patients are another major player — they must agree to enroll in trials that are taking place throughout their everyday life. Privacy issues are at the heart of many discussions on using this type of evidence.
“RWE promises to transform patient outcomes, but it also threatens to upend long-established norms in the generation and use of health care evidence.” This comes from a report published in 2015 by the Network for Excellence in Healthcare Innovation.3 The report continues on to state that RWE may enhance data from randomised controlled trials (RCTs), enhancing the accuracy of safety and efficacy profiles of new drugs and medical devices.
RCTs are the gold standard for providing the repository of data used to evaluate new medical products for marketing. Whether RWE enhances or competes with the RCT as evidentiary support remains to be seen. But what is certain is that data from the “real world” is a vast repository of untapped information that could go a long way to improving the health of populations around the world.
References 1. Position Paper: Communicating Effectiveness
Research and Evidence with Population Health Decision Makers. GlaxoSmithKline website: https://u s . g s k . c o m / m e d i a / 4 4 9 5 1 6 / g s k - p u b l i c - p o l i c y -
posit ion-paper_rwe-cer-communication-final .pdf. Accessed March 19, 2016.
2. Califf, RM and Sherman, R. What We Mean When We Talk About Data. US Food and Drug Administration website: http://blogs.fda.gov/fdavoice/index.php/2015/12/what-we-mean-when-we-talk-about-data/. December 10, 2015. Accessed March 19, 2016.
3. Real World Evidence: A New Era for Health Care Innovation. Network of Excellence in Health Innovations website: http://www.nehi.net/writable/publ icat ion_f i les/f i le/ rwe_issue_br ief_f inal .pdf . Accessed March 12, 2016.
Watch Pages
Regina Ballinger is a Senior Manager of Regulatory Intelligence with Thomson Reuters. She currently manages US regulatory content for the Cortellis Regulatory Intelligence database, and is the executive editor of the AdComm Bulletin. Ms Ballinger has specialised experience
in public health, pharmaceutical regulatory affairs, and health communications. She has had numerous articles published on topics related to new drug approvals and drug regulatory issues. Ms Ballinger was educated at the University of Maryland in law and nursing. She holds an MS degree in Health Systems Management.Email: [email protected]

Volume 8 Issue 310 Journal for Clinical Studies
Watch Pages
Benefiting eCOA Through the Right App Strategy
Electronic clinical outcomes assessment (eCOA) providers are facing heightened demand for user-friendly, accessible solutions that facilitate hassle-free participation in clinical trials. This has sparked debate on how to deliver apps across the different operating platforms of iOS, Android and Windows in order to provide the best user experience. In particular – and for good reason – the use of native apps is becoming an increasingly popular choice.
The way providers capture data, be it through native apps or web-based diaries, will be determined by the way mobile technology is accessed – something which can vary markedly across patient populations.
Apps can be presented in three ways, all of which come with an icon on a mobile device’s home screen. However, native apps, web apps and mobile websites all have fundamentally different architecture and therefore features. When evaluating which option would best suit a particular study, sponsors should weigh up the benefits and limitations of each option:
Native apps are a form of downloadable software which is coded to run directly on a mobile device’s operating system. The key advantage of using this type of app is that they can be used without a mobile or internet connection, making them significantly more versatile than other options. They also have the ability to interface directly with the inbuilt functionality of mobile devices, including cameras and Bluetooth®. This allows medical devices such as spirometers and glucometers, as well as wearable technology such as activity trackers, to be easily connected to collect objective data.
Another important benefit is their ability to provide a consistent look and feel across all screens and device types. These capabilities, along with their suitability for use in transcribing patient diaries, make native apps a very comprehensive solution for the market, helping to deliver clinical programmes in a timely, cost-effective manner across all platforms.
Native apps allow displays to be optimised to suit the specific user population, which is great for older patients or patients with disabilities, and they are automatically adapted to the controlled accessibility settings of their personal device. They are similarly ideal for use in geographies with unreliable connectivity, as data can be stored locally in an encrypted form, protecting patient data until connectivity is restored.
Until recently, the most common approach to data capture has been via web apps using HTML5. This type of solution facilitates speed to market – there is one fundamental build which is embedded into a code base
that enables them to run on different mobile operating systems. However, unlike native apps, this does not enable the layout to be optimised for the device in use, meaning natural button configurations cannot be incorporated. This may create a barrier to use for some patients who would need to become familiar with the new user interface.
Although web apps may look and feel like native apps, and while they boast a degree of interactive functionality as opposed to merely capturing information, they are incapable of readily accessing the inbuilt functions of mobile devices.
The third option, mobile websites, meanwhile, can be optimised for use on a mobile phone or tablet, and by their very nature include web-based functions that allow users to exercise a degree of control. Users can also store clinical trial companies’ website URLs as icons on the home screens of their mobile devices. However, much like web apps, their functionality is limited compared to native apps in that patients may find themselves unable to tailor the website interface according to their individual requirements. Other drawbacks include connectivity issues and lack of data storage. For example, if a patient begins completing a diary entry but then loses connectivity during the process, data which had already been inputted would be lost and need re-entering once connectivity is re-established. Similarly, if there is no connectivity at the time a patient wishes to start completing a diary entry, data cannot be collected. This would result in a delay in recording the data and potentially invoking follow-up activities –creating another barrier to patient engagement.
In evaluating these options, sponsors should carefully consider their patient population, what kind of access patients have to mobile devices and, critically, how best to engage each individual patient within the clinical trial process. Although web apps have, to date, been a popular choice, their limitations are clear. It is through the use of native apps that mobile technology can extend the functionality offered by electronic patient-reported outcomes (ePRO), improving patient experience and expanding the opportunity for quality data capture in clinical trials.
Chris Watson has a PhD in behavioural
neuropharmacology and is an experienced product
strategist with over 16 years’ experience in the
delivery of business- and consumer-based solutions,
the last six of which have been focused in the clinical
technology industry. He has an extensive knowledge of
product and software development processes and is responsible for
implementing mobile product strategy at Exco InTouch.
Email: [email protected]

Journal for Clinical Studies 11www.jforcs.com
Clinical Services
Clinical Technologies
Diagnostics & Biomarker Development
API Services & Chemical Development
PharmaceuticalDevelopment
Commercial Services
Analytical Services
WE UNDERSTAND YOUR NEEDS ARE UNIQUE – SO ARE OUR SOLUTIONSPROVIDING AN UNPARALLELED RANGE OF CONTRACTPHARMACEUTICAL DEVELOPMENT AND MANUFACTURING SERVICES GLOBALLY FOR OVER 40 YEARS
GET IN TOUCH
UK (Global Headquarters)
+44 28 3833 2200
US Headquarters +1 215 660 8500
ASIA PACIFIC Headquarters+65 6309 0720
AM6136

Volume 8 Issue 312 Journal for Clinical Studies
Watch Pages
This cardiovascular medical devices watch column was established at the beginning of this year. However, owing to various changes in his job responsibilities, the original writer will no longer be able to provide regular updates. I am therefore thankful to Dr Rick Turner for stepping in to do so. As many of you may remember, Dr Turner wrote cardiovascular safety watch columns discussing biopharmaceutical drugs for this journal for several years, and I am grateful that he will now be writing about cardiovascular medical devices. (The Editor)
The first column in this series1 introduced the Medical Device Epidemiology Network Initiative (MDEpiNet), part of the Epidemiology Research Program at the US Food and Drug Administration’s Center for Devices and Radiological Health. It highlighted an upcoming meeting, entitled “The Role of Endpoint Adjudication in Cardiovascular Device Clinical Trials,” that has since taken place. In due course, a report of the meeting’s discussions will be published in the literature: in the meantime, presentations given at the meeting are available on the Cardiac Safety Research Consortium’s website.2
It can take a year or so to publish such reports since their preparation typically involves a wide range of co-authors from different constituencies represented at meetings, such as academia, industry, and government organizations, including regulatory agencies. However, the gravitas and impact of these reports is enhanced precisely because of the diversity of their authorship: the reports identify areas of current consensus, areas where consensus is currently elusive, and pathways towards gleaning additional information that will facilitate future consensus. As an example, consider the January 2016 publication in the American Heart Journal of a paper reporting discussions at an MDEpiNet Think Tank focused on the PASSION Program that took place in October 2014. As the report noted, “The PASSION program is an MDEpiNet-sponsored program that aims to demonstrate the goals of MDEpiNet by using cardiovascular medical device registries to bridge evidence gaps across the medical device total product life cycle.”3
The PASSION Think Tank meeting focused on four areas of cardiovascular medicine intended to cultivate interest in various MDEpiNet device-specific working groups: coronary intervention, electrophysiology, valvular disease, and peripheral vascular disease. The report in the American Heart Journal summarised discussions at the meeting and future intended directions for the PASSION programme. In due course, I will let readers know when a report from the “Endpoint Adjudication” meeting is published.
Future columns in this series will address multiple
topics within the domain of cardiovascular medical devices. Examples of these topics are provided in the Bibliography.
References
1. Kindman A. Cardiovascular medical devices watch column.
Journal for Clinical Studies. 2016;8(1):20.
2. Cardiac Safety Research Consortium web page. Scientific
Meetings. Available at: http://cardiac-safety.org/?p=2110
(Accessed 26 May 2016)
3. Zeitler EP, Al-Khatib SM, Drozda JP Jr, et al; MDEpiNet.
Predictable and SuStainable Implementation of National
Cardiovascular Registries (PASSION) infrastructure: A think
tank report from Medical Device Epidemiological Network
Initiative (MDEpiNet). Am Heart J. 2016;171:64-72.e1-2.
Bibliography
• Chimhundu C, De Jager K, Douglas TS. Focus areas of
cardiovascular medical device research in South Africa. S Afr
Med J. 2015;106:55-6.
• Farooqi KM, Saeed O, Zaidi A, et al. 3D printing to guide
ventricular assist device placement in adults with congenital
heart disease and heart failure. JACC Heart Fail. 2016;4:301-
11.
• Head SJ, Mylotte D, Mack MJ, et al. Considerations and
recommendations for the introduction of objective performance
criteria for transcatheter aortic heart valve device approval.
Circulation. 2016;133:2086-93.
• Magruder JT, Crawford TC, Grimm JC, Fredi JL, Shah AS.
Managing mitral regurgitation: focus on the MitraClip device.
Med Devices (Auckl). 2016;9:53-60.
• Toth GG, Vanderheyden M, Bartunek J. Novel device-based
interventional strategies for advanced heart failure. Postepy
Kardiol Interwencyjnej. 2016;12:13-6.
• Vahl TP, Kodali SK, Leon MB. Transcatheter Aortic Valve
Replacement 2016: a modern-day “through the looking-glass”
adventure. J Am Coll Cardiol. 2016;67:1472-87.
• Webb MK, Wang J, Riegel MS, et al. Initial experience with the
pediatric Impella device: a feasibility study in a porcine model.
Catheter Cardiovasc Interv. 2016 May 24 [Epub ahead of print]
Cardiovascular Medical Devices Watch Column
J. Rick Turner, PhD, DSc, FASH, is a member of Quintiles’ Cardiac Safety and Cardiovascular Centers of Excellence. He has published over 100 peer-reviewed papers and 15 books. In addition to cardiac safety, his areas of expertise include hypertension, diabetes drug development,
and cardiovascular medical devices.Email: [email protected]

BAP Pharma has an MHRA Wholesale Dealers Licence and a CD Licence (Schedules 1-5) enabling us to operate in a fully licensed manner. In addition, we have an MIA (IMP) licence which allows us to import from outside the EU.
bappharma.com
When you need a leading supplier of comparator drugswith a global reach you’re in safe hands
A global leader in clinical trial comparators, BAP supply pedigree products with exceptional customer service.
To find out more about how BAP Pharma can help ensure your clinical trials go ahead as planned, visit us at bappharma.com or speak to one of our team on +44 (0)1753 696326
Bespoke sourcing We work with our clients in a consultative manner to determine the optimum supply route. Products are then sourced either directly from manufacturer or from one of our reliable, audited wholesaler suppliers.
Regulated shipment Goods are shipped to BAP Pharma in either cold chain (2-8oC) or controlled ambient conditions (15-25oC) by our approved couriers in either Temperature Controlled Vehicles or Credos. We include a temperature data logger with all of our shipments.
Safe storage and distributionBAP Pharma arranges product distribution around the world, wherever you need it.
Products are stored in our warehouse near Heathrow in either controlled ambient conditions or in our cold room.
Reliable documentation For every product supplied to a customer, being able to source key documentation is central to our business model.
BAP Pharma clients may securely access their documents at any time via their own personalised, encrypted portal. To learn more, simply register at bappharma.com for access to our Demonstration Portal.
46_JCS_January2016.indd 2 03/02/2016 12:30:29

Volume 8 Issue 314 Journal for Clinical Studies
Watch Pages
What Do You Need to Know to Ensure Shipping Success to North Africa?
Whilst obviously part of the African continent, North Africa is very definitely its own distinct region, very much divided from sub-Saharan Africa, and most definitely not part of the Middle East (although the population of 423 million represents some 58% of the Arab League which spreads from the Middle East and over the whole of Northern Africa). North Africa has been up till now a much under-used area when it comes to clinical trials.
The Sahara desert to the south is 3.6 million sq. miles, and is the world’s third biggest desert, and the largest hot desert. The Atlas Mountains extend for 1600 miles, separating the Sahara from the Mediterranean coastline, and this feature dominates the region. The area has been inhabited by man for millennia and there are cave paintings at Tassili n’Ajjer in Algeria dating back to between 8000-4000 BCE. The area has been under the control of many nations including French, Spanish, Arab, and Italian, and this is reflected in the many languages spoken across the countries, dominated for the greatest area by French.
Egypt leads the way for clinical trials with (according to clinicaltrials.gov) almost 1200 either having taken place or in progress, which is no mean feat, particularly given the hiatus experienced for new trials during the political uncertainty of the Arab Spring. It is the most populous country in the region, at around 80 million, which has clearly been a heavy influence.
Tunisia and Morocco are at 195 and 80 studies respectively, then Algeria at 75, and the Sudan trails with 22. The remainder of countries, including Libya (and some tiny Spanish territories), have a handful of studies and at the present time are unlikely to be amongst the first-choice destinations for most pharmaceutical companies and their clinical trials. It is estimated that it will take at least 10 years to rebuild Libya’s infrastructure after the 2011 war.
Concentrating on Egypt for the moment, it is clear that the country operates a complex healthcare system, with public, NGO, religious and charitable institutions all providing services. Spend is approximately 5.1% of GDP on health, which is #142 in the world rankings. Control is in the hands of the Ministry of Health, and they run a complex finance mechanism with some state input, some health insurance options, and a further contribution by the patient or their families. Egypt produces over 90% of the pharmaceuticals it consumes, and pharmaceuticals account for just over one-third of all health spending. Publicly produced medicines are heavily subsidised. There is a clear divide between rich and poor and approximately 25% of Egyptian children under age five have chronic malnutrition. Life expectancy is around 71 years for men
and 76.5 for women, which is very much consistent across the region. In 2009, there were 16.04 physicians and 33.80 nurses per 10,000 inhabitants, and 0.5 beds per 1000 population. Applications to run trials must be done through a sequential process (LEC and then the MOH).
Some consideration towards creating systems for importing clinical trial drugs has been made, but the process remains somewhat variable once shipments arrive into Cairo.
Import Paperwork
Certificate of Origin:May be required for certain items – must be checked on a case-by-case basis. Import Licence:Is always required to import pharmaceuticals and only the consignee can apply for the import licence. Officially the consignee can only make the application at the MoH once the shipment has arrived at the airport, and once they have the original invoice, MAWB and HWB, which must be presented for the application. For temperature-controlled shipments, it is often possible to apply for and to obtain the import licence based on a copy of the invoice, i.e. before physical shipping takes place. The import licence takes a minimum of one week, but can be several weeks or longer. In very rare cases, the MoH has refused to issue an import licence. It is strongly recommended to seek advice prior to shipping.

Journal for Clinical Studies 15www.jforcs.com
Invoices:For low-value shipments: two original proforma invoices between the shipper and the consignee. Zero value is not acceptable.
For high-value or commercial shipments: two original commercial invoices from shipper to consignee.
The invoices need to include
• Incoterms. • Consignee company name and address on invoice,
which must be exactly the same as the holder of the import licence and of the name used on the MAWB.
• Shipper information, including company name, full address, phone, fax, email address and website.
• Batch number and detailed product name.• For shipments originating from Non-European
countries; the original invoices must be legalised (with stamp or signature) by Chamber of Commerce at origin.
Non-compliance to any of these requirements will cause delay and a possible fine. Power of AttorneyStatement – Consignee:A copy of the consignee’s company registration certificate is always required to clear non-documents.
When including a temperature recorder in shipments, the MoH requires the product name and batch number (as a minimum) to be saved as customised data, when the temperature recorder is programmed. Information is checked against the customs invoice at the point of import. Failure to comply may result in MoH refusing the use of the product.
In cases where the multiple batches of materials are used inside the same packaging and the numbers cannot all be programmed onto the recorder, then it is possible to use multiple recorders (even if the consignment is just one box) and add different batch numbers to the monitor, or to add the invoice number, MAWB number and the name of consignee to the recorder data instead, and cross-reference on the paperwork. If the batch numbers fit, they should always be used preferentially.
Clearance time (if everything is in order) should be eight hours, and customs operate every day except Friday. It is strongly recommended to add the external temperature requirements on the MAWB or shipments will not be stored correctly. The handling agent is only authorised to follow the instructions on the airline paperwork. The temperature control rooms in Cairo airport have no temperature-recording system. In tests our representative concluded that these areas were not set/working correctly, so extra care should be taken when selecting packaging. Additionally, the refrigerators (applies to all handlers), particularly during the summer,
may have reduced performance. This is due to the facilities being full and the doors often being opened to the heat of the daytime. Shipments on dry ice are accessible for replenishment during clearance pending release.
Tunisia requires an import permit for every shipment, which is issued by the DPM (Director of Pharmacy and Medicine). The consignee must apply with an invoice for each shipment. As French is the main written language used, it is vital to have thorough business and trial-specific language skills and understanding to be able to make the application.
If we compare this with Algeria, they have a longer-term approach to the granting of import licences. The consignee needs to apply for import authorisation with the MoH (which takes approximately 15-20 days). The company must preregister with customs in advance, before any application can be made. Once obtained, the authorisation paperwork must be sent to customs, together with a statement from the consignee. The Head of Customs will then give their approval and inform customs at the airport. This approval is given for a certain period of time, typically a year, and covers all the shipments for a study.
Across the whole region, pre-planning and attention to detail are key for success in shipping. The temperature challenges, which are not limited to the summer, need extra care in instructing the handling of shipments, but relying on temperature-controlled areas is probably rather optimistic. Choosing robust packaging is a sensible method of ensuring temperature compliance.
Watch Pages
Sue Lee has worked for World Courier for 25 years. During this time she has experienced a variety of customer service and operational functions, including the setting up of numerous, multi national, clinical sites for the transportation of biological samples in her capacity as Head of the Major Clinical
Trial Unit. Sue has orchestrated the shipping thousands of shipments with very specific temperature requirements to a host of challenging locations, and each presenting their own obstacles and dilemmas. More recently in her role as Regional Quality Manager, Sue has been auditing and developing procedures and systems for regulatory compliance, package and vehicle testing, as well as temperature control and mapping. Currently, Sue’s role includes delivering pertinent, technical information and updates on latest industry developments via technical presentations, articles and white papers, workshops, association and discussion group involvement and direct links with other industry professionals. This also includes direct involvement delivering and maintaining World Courier’s online presence. Email: [email protected]

Volume 8 Issue 316 Journal for Clinical Studies
Regulatory
Empowering Real-world Evidence Analysis Through Risk-based De-identification
Clinical trials produce vast amounts of incredibly valuable data that, outside the initial research objectives, is not being sufficiently leveraged. We are just starting to see the wealth of patient-level data delivering ground-breaking insights – particularly from real-world evidence (RWE) analysis. For years, companies have used real world data (RWD) to conduct ongoing pharmacovigilance and run comparative analyses of their products. RWE is becoming a valuable tool for decision-making at multiple stages of the drug development process.
With demands from healthcare payers growing, cost pressures from drug development climbing, and maintaining patient privacy paramount, biopharmaceutical companies must explore systematic approaches for RWE.
All of these facts depend on the availability of high-quality de-identified patient data. To properly leverage RWD at the start of clinical trials and through drug development lifecycles, the data must first be de-identified to protect the privacy of patients. Putting a process in place to automate the delivery of high-quality de-identified data on an ongoing basis is critical to the long-term success of RWE initiatives.
Real-world Evidence, SimplifiedRWE uses data gathered from patients in the real world – RWD – to support decision-making around drug use and safety. Hospitalisation data, electronic medical records (EMRs) and claims databases from insurers are some of the sources of information that contain data on millions of patients. Leveraging this individual patient-level data (IPD), biopharmaceutical companies use information gathered across the continuum of care to better understand the long-term impact of a medication.
As with any secondary use of PHI, the use of RWD to answer questions about a drug’s safety, efficacy or value must heed privacy concerns. When a patient’s data is used for a purpose other than primary care, it needs to be de-identified to protect their privacy. While a number of methods can be used to de-identify PHI, a comprehensive de-identification solution is needed, since RWE can use many different data sources. Traditionally, pharmaceutical companies have used RWE to enhance what is learned from clinical trials, but they are finding new and innovative ways that it can be applied in drug development. These ways promise to improve patient outcomes and reduce costs overall.
The Limitations on Learning from Clinical TrialsThere are many reasons for the differences that can occur between a trial and the real world. Patients who
are recruited to participate in a randomised clinical trial (RCT) must often meet precise medical criteria and are monitored closely to ensure they adhere to trial protocol. In the real world, however, patients are more complex. They will have multiple medical conditions that make them tougher to treat or that limit their options. Often, patients are on other medications for chronic conditions like high blood pressure, diabetes or arthritis. In an environment where the average person has 12 prescriptions filled annually and the average senior fills 28 prescriptions each year5, drug-to-drug interactions become a real concern.
Furthermore, patients don’t always follow their prescribed treatment regimen. There are many reasons why they do not take their medications as directed: forgetting, avoiding unpleasant side-effects, and reducing the expense of prescription drug use are just a few. During a clinical trial, the medication is provided to the participant at no cost. Once a drug is on the market, however, financial issues are a significant consideration. Patients use many strategies to reduce their prescription drug costs, including skipping doses or taking less than the prescribed dose.
Since the specifics of care in a clinical trial can differ markedly from what happens in the real world, the benefits of a trial’s results can be difficult to ascertain more broadly. With the bill for prescription drugs rising annually, payers are pressuring drug companies to prove the long-term benefits of their products.
By analyzing EMR and hospital datasets it’s possible for clinical study sponsors to augment the results of a clinical trial with knowledge of what happens in a larger population over a longer timeframe. Not only is this useful to show a drug’s effectiveness over time but, insights may be revealed that can improve patient care and clinical treatments.
Developing a De-identification Pipeline for RWEA data de-identification pipeline that uses risk-based de-identification is the cornerstone for an RWE platform. This method delivers the rich, granular data required to inform clinical development, market research and physician targeting and detailing.
In addition to providing insights about comparative effectiveness and pharmacovigilance, drug-makers are being spurred to consider RWE in a broader context. With the cost of developing a new drug hitting unprecedented levels, companies have started using RWE to cut waste, curb costs and maximise the benefits of their marketing spend.

Journal for Clinical Studies 17www.jforcs.com
Regulatory
By the time a drug achieves market approval, the process has been underway for more than a decade and has cost as much as a billion dollars. A 2014 study from the Tufts Centre for the Study of Drug Development found that it takes, on average, $2.6 billion to develop a new prescription medicine that gains market approval when including out-of-pocket and time costs7. Compared to a similar study from 2003, the cost for developing a prescription drug has increased 145% over the last decade, even after inflation was taken into account.
If organisations can compress clinical development timelines by learning earlier on which drugs are effective on which patient groups, this can reduce the number of drugs abandoned late in the development process. This can save millions of dollars and months of misplaced effort. Drug manufacturers are also discovering that RWE can deliver value at multiple points along the drug development lifecycle. RWE is emerging as a valuable tool to recruit for clinical trials, improve product launches, target the right prescribers and patients, and support ongoing access through creative pricing and reimbursement mechanisms. It has been estimated that by applying RWE in a systematic way, a top-ten pharmaceutical company could realise $1 billion in value8.
An IMS Health report, RWE Market Impact on Medicines, provides more than 100 non-safety case studies where RWE has been used to influence product decisions in the US and other western nations9. Many of these examples are prospective studies where a drug product was able to achieve reimbursement by having the manufacturer compensate payers for patients who did not respond to therapy or did not meet benchmarks. In other cases, studies that used RWE to show a product’s effectiveness allowed for formulary status and premium pricing to be restored. Another report10 noted a case of a biopharmaceutical company that was able to surpass expectations for a new product’s uptake by more than 10%. Despite the presence of a long-established competitor in that market, the company was able to use RWE to focus their detailing efforts on physicians with relevant patients who met the target treatment profile.
Examples like these illustrate the promise of RWE, but it is still early days for its use in widespread decision-making. There are many sources of data across the healthcare landscape. EMRs provide a wealth of information, including patient demographics, medical conditions, vital signs, lab results, and treatment information. Individual claims databases, prescription databases and hospitalisation records add to the overall picture. The real promise of RWE will be realized by having a comprehensive view of the patient experience. This will only happen when data gathered at the various stages of care can be connected.
The linking of disparate datasets can be facilitated by creating de-identified data that retains a high level of granularity or specificity. Data-masking approaches that
broadly redact data are insufficient for this purpose; a risk-based approach to data de-identification is required, including the removal of identifying variables so that a person cannot be re-identified. However, not all forms of de-identification are concerned with the delivery of high-quality information for analysis. To provide value for decision-making, the data used in RWE analysis must retain as much granularity as possible.
Techniques like masking can drastically reduce the quality of data since they will often completely remove important variables, such as dates or zip codes. While masking appears to offer simple-to-use patient-level data for RWE initiatives, biopharmaceutical companies must first obtain the patient’s consent that their data can be shared for secondary purposes; otherwise, they must de-identify the data. While most patients are willing to share their data for use in research, they expect that their privacy will be maintained. In order to allow de-identification to be done in a way that protects individual privacy while maintaining the quality of the data, the use of a risk-based approach to de-identification, like the Expert Determination method described in the HIPAA Privacy Rule, is recommended. With risk-based de-identification, attributes like dates can be preserved with special techniques rather than eliminated11. Risk-based de-identification can allow chronological information and durations to be retained, thus enabling retention of better information for use in subsequent analysis, providing richer and more accurate findings. A defensible de-identification strategy based on peer-reviewed statistical techniques needs to be documented in detail to support the level of data quality required.
Furthermore, because biopharmaceutical companies operate in the global marketplace, it is prudent for them to follow internationally-accepted standards and guidelines that pertain to the use and sharing of healthcare data. Industry associations like the Health Information Trust Alliance (HITRUST), and government bodies like the Institute of Medicine (IOM) in the US and the Canadian Council of Academies, have all endorsed the use of a risk-based methodology to de-identify healthcare data.
Establishing a data de-identification pipeline helps apply risk-based de-identification automatically and consistently to datasets that are being continuously updated. Companies that have established a data warehouse for RWE purposes need to continuously refresh the data within it so that the information remains current. This provides analysts and researchers with timely access to the most recently available data in a de-identified format, a situation that would be nearly impossible using manual processes. A de-identification pipeline pulls in data from the source, an EMR database for example, on a regular basis (e.g., monthly or quarterly). At this point, the automated de-identification engine would perform a series of steps to manipulate the database variables, reducing the risk of re-identification and protecting the patient’s privacy.

Volume 8 Issue 318 Journal for Clinical Studies
As with any risk-based de-identification approach, the first step is to assess the risk to privacy by looking at who will have access to the data and what security and privacy controls are in place to protect it from unauthorised access. Next, the variables in the data that contain keys to an individual’s identity must be classified. While a data warehouse may consist of hundreds of data tables with thousands of variables, only a subset of these are relevant from a privacy perspective. The final step is to map the data to ensure the de-identified data maintains the integrity of the original database. With the work of the de-identification engine complete, the de-identified RWD can be exported to the data warehouse for analysis. The use of the pipeline limits the risk of a successful re-identification attack on the data warehouse, since it only ever accepts de-identified data.
By engaging with experts in the field of data de-identification, a de-identification pipeline can be implemented that complies with applicable laws and regulations such as the HIPAA Privacy Rule. In the event of a data breach, the ability to show compliant data practices can provide organisations with a defensible position in the event of a lawsuit.
ConclusionWith the cost of pharmaceutical health expenditures mounting, leading biopharmaceutical companies are turning to RWE to show healthcare’s payers the return on investment that medications can provide. Beyond pharmacovigilance, RWE is being used to supplement the learning from clinical trials and inform decisions about labelling, pricing and market access. As governments, insurance companies and patients continue to seek the optimal value for the dollars spent, the impact of RWE will proliferate.
While these opportunities still lie in the future, organisations that want to position themselves to maximum advantage cannot get there by continuing to take ad hoc approaches with their data. Organisational readiness can only be reached by building a robust RWE platform; one that automates de-identification using a robust, risk-based approach to de-identification that delivers high-quality, granular data in an ongoing way.
References1. Munro, Dan (2015, January 4). U.S. Healthcare
Spending On Track to Hit $10,000 per Person This Year. Forbes. Available at: http://www.forbes.com/sites/danmunro/2015/01/04/u-s-healthcare-spending-on-track-to-hit -10000-per-person-this -year/print/
2. National Center for Health Statistics (2015). Health, United States, 2014: With Special Feature on Adults Aged 55-64. Centers for Disease Control and Prevention.
3. National Health Expenditures 2014 Highlights. Centers for Medicare & Medicaid Services. Available
at: https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthExpendData/downloads/highlights.pdf
4. Sarasohn-Kahn, Jane (2015, December 3). U.S. National Health Spending Up Due to More People Covered and Higher Drug Costs. Health Populi. Available at: http://healthpopuli.com/2015/12/03/13644/
5. Retail Prescription Drugs Filled at Pharmacies (Annual per Capita by Age). (2014). The Henry J. Kaiser Family Foundation.
6. Cohen, Robin A. and Maria A. Villarroel (2015, January). Strategies Used by Adults to Reduce Their Prescription Drug Costs: United States, 2013. Centers for Disease Control and Prevention.
7. Costs to Develop and Win Marketing Approval for a New Drug is $2.6 Billion (2014, November 18). Tufts Centre for the Study of Drug Development.
8. Hughes et Al. (2014). Breaking New Ground with RWE: How Some Pharmacos are Poised to Realize a $1 Billion Opportunity. IMS Health. Available at: http://www. im shea l t h . c om /f i l es /web /G lob a l /Se r v i ce s /Services TL/rwes_breaking_new_ground_d10.pdf
9. Hughes, Ben and Marla Kessler. (2013). RWE Market Impact on Medicines: A Lens for Pharma. IMS Health.
10. Hughes, Benjamin, Marla Kessler and Amanda McDonnell (2014). Breaking New Ground with RWE: How Some Pharmacos are Poised to Realize a $1 Billion Opportunity. IMS Health.
11. For more on Expert Determination and risk-based de-identification techniques see Privacy Analytics white paper, De-Identification 201: Fundamentals of Data De-Identification. Available at: http://www.privacy-analytics.com/de-id-university/white-papers/de-identification-201/
12. Volker, Dr. Ronicke, Dr. Michael Ruhl and Dr. Thomas Solbach (2015). Revitalizing pharmaceutical R&D: The value of real world evidence. Strategy&.
Regulatory
Pamela Neely Buffone is Director of Product Management for Privacy Analytics, where her mandate is to make the risk-based approach to de-identification more accessible to meet the dual needs of data utility and privacy protection. The focus of her career has been to make analytics more
consumable and easy to use, thereby bringing the value of insight and discovery to more people across industries and domains.

Journal for Clinical Studies 19www.jforcs.com

Volume 8 Issue 320 Journal for Clinical Studies
Regulatory
Demand for Data Puts Safety at Centre in Decision-making
Where once pharmacovigilance was regarded as a necessary burden, pharmaceutical companies are starting to recognise that safety in fact plays an integral role in business strategy, such as in decisions regarding mergers and acquisitions (M&A). Marco Anelli, MD, head of pharmacovigilance and medical affairs advisory services at ProductLife Group, explores how the changing environment is affecting what is required of pharmacovigilance leaders as well as how safety data is gathered, managed, and turned into critical reports and documents.
The LandscapeThe demands on the pharmacovigilance function are escalating. Regulatory authorities expect more and more safety data from a wider and wider network of sources to support the approval of a product application, as well as for assessing the safety of products already on the market. They’re also seeking a more standardised approach to how data get reported.
Pharmaceutical companies must now keep track of safety information — such as adverse-event reporting — from traditional sources — such as healthcare practitioners and the medical literature — as well as from patients through social media and other reporting avenues.
As a result, safety departments are under greater pressure than ever, and the regulatory authorities have acknowledged that that pressure will only increase. At the same time, however, safety is starting to be viewed as a business enabler and no longer the regulatory burden it’s been viewed as in the past.
A New Role for PharmacovigilanceSenior managements at pharmaceutical companies are starting to draw their safety leaders into their inner circles, thereby making sure that those safety leaders not only have support to fulfil their regulated functions, but also play a more central role in decision-making. Therefore, drug safety people are beginning to get involved in strategic decision-making, such as with mergers and acquisitions and pharmacoeconomics.
When a company is trying to get reimbursement for a drug sold in a European country, for instance, the company has to prove the budget impact, which means how much the reimbursement of the adoption of a drug will cost the country’s national health system. If a drug is more expensive but has fewer side-effects than a similar drug, all of those side-effects can be evaluated — provided the company has the data — at an economic level by subtracting the cost of those events from the overall cost
of the drug. Simply put, a safer but more expensive drug may be more economical for a national health system. And the drug safety data used in assessing that budget impact can be supplied only by pharmacovigilance, which underscores the need for clear and consistent data and a coherent approach to managing the data.
Safety information is also important in an M&A or in-licensing scenario. Companies must conduct their due diligence to verify that everything is in order before finalising an agreement, but in so many cases, data and files go missing — particularly with legacy products. Missing safety information could have huge ramifications when registering a product with the regulators, who may ask a company to redemonstrate the product’s risk–benefit ratio. Having senior safety people involved in the negotiation process gives companies insight they might otherwise not have, and if information is missing, this could be factored into an in-licensing or M&A agreement.
Increasingly, a company has to be able to conduct an economic evaluation of a drug’s safety and produce the drug’s tolerability profile. The company must be able to put a price tag on the drug based on both its efficacy and its safety profile; and again, safety data and the expertise of safety leaders will play important roles in setting that figure.
Cross-functional ImplicationsThe more integral business role that pharmacovigilance now plays poses several considerations. The first has to do with the changing skill-set required of safety leaders that would move them away from the conservative mind-set that safety has tended to adopt in the past and instead towards a more entrepreneurial approach that balances risk with benefit.
A second consideration involves cross-functional collaboration and moving towards common ways of working, which has been problematic in the past because such functions as regulatory, medical affairs, and pharmacovigilance have tended to work in silos. Today, however, because regulators expect more volumes of data before they can determine the safety and efficacy of products on the market, data that was once considered to be regulatory or pharmacovigilance or clinical has to become consistent across all documents.
Take, for example, the situation with drug renewals. In the past, some regulatory authorities would simply request confirmation from a reputable expert saying the drug met acceptable standards as far as the risk-benefit ratio was concerned. Today, product renewal is permanent, and regulatory authorities want more information about

Journal for Clinical Studies 21www.jforcs.com
Regulatory
the drug’s safety profile. That information comes from scientific literature, from post-marketing clinical trials, from periodic safety update reports, and from the company’s pharmacovigilance database.
Third, because so much of the data required for one document is also required in others, there’s a strong chance that the relevant authorities will cross-check the data and the documentation. What’s troubling for companies is that the regulators sometimes report that data is inconsistent from one document to another, which could negatively affect how the authorities respond and which could potentially result in an inspection.
Rapid Information RetrievalThe ability to quickly access and analyse safety information about a drug is becoming increasingly critical. Inevitably, companies confront a crisis by way of a referral at some point. There are different types of referrals, but they’re always triggered by concern about a drug’s safety. Sometimes companies are required to once again prove the risk-benefit ratio of their drugs based on data they’ve gathered since marketing approval was granted. And because referrals have tight timelines, companies have to present that data quickly. The question that safety must ask is, Are we ready for a referral?
Ideally, data should be retrievable almost instantly, but instead, typically, gathering information for a referral takes weeks, thereby adding to the organisation’s costs and stress levels. Information that could have a bearing on a referral can come from multiple sources — such as study results, information from social media, and published papers — and so, having a system with the flexibility to search for different types of information could vastly reduce turnaround time.
All companies have many databases for managing their data, but those databases are seldom interconnected. As a result, even all of that data does not translate into knowledge. A more progressive approach would be to shift from data handling to knowledge handling. Indeed, this is the direction the regulators are moving in. The European Medicines Agency has published a document saying the agency wants to see integrated data management systems, wherein all information about drugs — from preclinical through production and life cycle management — is getting handled consistently.
Realistically, however, few companies are in a position to adopt an integrated data management approach to all documentation in a single step. A more feasible way of improving the management of data about a product’s life cycle is to begin with one key element: the company core data sheet (CCDS). Feedback from regulatory inspections demonstrates that the CCDS is usually incomplete or inadequate. But because the CCDS is the basis for information in many documents, if the information in it is poor or incomplete, that will have a serious knock-on
effect on the summary of product characteristics (SmPC) and all promotional activities. The CCDS is the single most important document for promotional purposes because companies can’t make any claims about their products if corroborating information isn’t in the SmPC — and, therefore, the CCDS.
The best approach is to continuously prepare and update the CCDS during all phases of a drug’s life cycle — and with contributions from all functions. But there’s a difficulty: the information needed for populating the CCDS is of both structured and unstructured kinds, which makes it difficult to find and gather the information.
The use of metadata offers a standardised way to describe, connect, and manage data. However, even though metadata as a concept for managing data is now well-defined and solutions are in place, it’s been difficult to apply the solutions to the pharmaceutical environment.
One way to bridge the divide could be to have data stewards take ownership of this transformation. Data stewards are workers who are knowledgeable about the industry and the data. They will have to be assisted by a team of people with various skills and from different functions such as clinical people, data managers, knowledge managers, and information technology people. By bridging the CCDS’s information gaps, it is hoped the increased burden pharmacovigilance faces can at least be alleviated, and the business as a whole can benefit by applying safety intelligence for both regulatory and commercial purposes.
CCDS Implications: Examples from IndustryThe approach companies are taking to their CCDSs differs significantly from one to the other, with some being very proactive and with others largely ignoring the problem and so now having to deal with the consequences.
One large European pharma company has uncovered significant problems with its CCDS process, which has created challenges with partnerships and in promotional campaigns to the extent that some of the campaigns may have to be delayed, scaled back, or potentially even cancelled if there is insufficient information in the CCDS to substantiate marketing claims. Attempting to fix a poorly maintained CCDS in retrospect can be an uphill task, because the person who initially compiled the report may have left the job, and tracking down the links to published evidence or study reports can be nearly impossible.
At the opposite end of the scale, Italian pharmaceutical company Zambon undertook a large global harmonisation project to ensure oversight of all of its products, comprising 240 marketing authorisations worldwide. The company began by harmonising safety information, starting with activities at the corporate level. The first project was to harmonise core safety information so as to create a common basis for defining safety information in the data sheet in preparation for a major overhaul of the

Volume 8 Issue 322 Journal for Clinical Studies
Regulatory
CCDS process.After two and a half years, the company completed the
CCDS for its main products, and at the same time, it began to harmonise its periodic safety update reports so as to eliminate duplication. Zambon also implemented tools that would create a common repository for managing safety information. The safety overhaul also involved redefining the company’s pharmacovigilance system master file — a project that involved many different functions across the company.
Zambon also implemented a tool to record and archive medical information enquiries, and, separately, the company implemented an initiative to manage and trace quality processes, with special focus on corrective action and preventive action. The major driver behind the quality review is to ensure the company stays abreast of all complaints so it can quickly analyse the issues, correct any defects, and prevent further quality issues.
Continuous monitoring of the product risk-benefit profile is integral to Zambon’s approach, with the introduction of standardised systems safety analyses aimed at detecting changes in reporting rates and evaluating signalling, to determine whether any safety signals warrant increased monitoring. As Zambon’s chief scientific officer, Marco Sardina, MD, says, the heightened oversight enables the company to ensure the CCDS is kept up-to-date and to ensure the best oversight of products the company has on the market.
Ultimately, the CCDS has the potential to affect the value of a company’s products, such as requiring a company to withdraw certain claims about a product or potentially even putting the company at risk of losing indications for already marketed products.
From the TopAcross the pharmaceutical industry, with pharmacovigilance playing an increasingly important role in strategic business decision-making and becoming more intricately connected with other areas of each business — with regard to both the data involved and the collaboration required — perhaps the time has come to consider creating a new executive position: chief pharmacovigilance officer (CPVO). Such a pharmacovigilance leader, as a member of the executive suite, would have to have the type of high-level expertise companies require of, say, a chief scientific officer or other head of function. This would presuppose a leader whose expertise is based less on technical know-how and more on possessing a deep understanding of the role that safety plays at the business level. The CPVO would also have to be entrepreneurial and equipped to interact with the CEO when it comes to, say, evaluating a potential M&A or the net value of a drug.
Even if the industry is not quite ready for a CPVO, companies are being pressed to move towards taking a high-level approach to pharmacovigilance — the area in which resources are invested in managing and leveraging safety information to safeguard a product on the market or to provide insight regarding the company’s key commercial decisions.
Marco Anelli, MD, is head of pharmacovigilance and medical affairs advisory services at ProductLife Groupwww.productlifegroup.com
The writer thanks Erick Gaussens, PhD, of ProductLife Group and Marco Sardina, MD,
PhD, of Zambon for their contributions to this article.

Journal for Clinical Studies 23www.jforcs.com

Volume 8 Issue 324 Journal for Clinical Studies
Regulatory
Medical Management in Clinical Trials: A Roadmap to Operational Excellence (Part 2 – Study Setup)
In the previous article of this series, we discussed the role of the research physician/medical monitor (MM) during the study development phase, focusing on study design, development of essential documents and selection of business partners. In this edition, the responsibilities/activities of a clinical research physician, the study MM, during the study setup phase – starting from clinical trial application (CTA) till the recruitment of the first patient – will be detailed. This phase holds significance for the study MM, as the blueprint for medical management and the associated tools to accomplish the project-specific goals and deliverables will be conceived during this period. The more robustly and transparently the medical procedures are defined and documented, the more streamlined it gets later, during the study conduct and close-out phases. Further, the ground rules that structure the safety standards of the study and the integrity of the patient data collected are laid out during this phase.
In other words, the responsible MM should keep in mind that what is planted in this period will be harvested later on during the study.
Regulatory SupportThe selected business partners for regulatory services, together with the sponsor affiliates in the respective countries, play the primary role in regulatory interactions
with the concerned authorities during the study setup phase, starting with the CTA using essential documents prepared during the development phase. All relevant study setup activities go in tandem with the proceedings to obtain regulatory approvals. The MM plays an important role in providing responses to queries or questions raised by competent authorities (CAs) and the corresponding ethics committee or institutional review board (IRB) in the geographical location of the selected study sites. During that period, any required medical/scientific clarification regarding the protocol, study drug or applicable study procedures should be provided by the MM, together with other relevant members of the study team. Close interaction between the MM, regulatory affairs designee and project manager (PM) is essential, to ensure that all the relevant regulatory approvals are in place to trigger the recruitment activities as per the study plan.
Medical Management Documents and ProceduresOne of the key responsibilities of the MM during the setup phase is to lead the development of all documents and operational procedures related to medical management of the study. All relevant documents should be in place prior to recruiting the first subject in the study, to uphold safety of the study subjects and the integrity of the collected data. Whenever required, training should be provided to involved team members to ensure compliance with the established procedures.
Medical Management Plan (MMP)The MMP details the medical monitoring team responsibilities and maps the procedural flow for all medical management tasks for the study. The MMP is developed by the MM depending on the study-specific requirements and it should be finalised before enrolling the first patient in the study. This document outlines the roles of all the medics involved in the project, both the CRO and the sponsor medics. It also defines the various responsibilities of all involved MMs including, when applicable, the lead MM and regional MMs based on the scope of the study. The MMP should also include sufficient information on the expected contribution from different cross-functional team members towards achieving the desired goals of the plan.

Journal for Clinical Studies 25www.jforcs.com
Regulatory
Key Contents of MMP
Eligibility Review: Determining the applicable approach for eligibility review, either prospectively or retrospectively in the study, and defining the
tools to be utilized.
Management of Medical Questions and Guidance Requests: Specifying the communication channels, escalation pathways and applicable timeframes
for managing incoming questions or guidance requests, together with the established procedures for building the project-specific knowledge-base and information-sharing with sites and study team. When an interactive guidance management system (IGMS) is deployed for medical management (1), the relevant information about such a system should be reflected in the MMP.
Medical Review of Study Data: Specifying the various data sets to be used for medical review during study conduct, the frequency of review and, when applicable,
the essential data alerts concerning significant lab abnormalities and/or adverse events of special interest.
Management of Protocol Deviation (PD): Outlining the applicable procedures for management and prevention of protocol deviations, especially major PDs that might
impact safety of participating subjects or scientific integrity of study data, and specifying the applicable methods for identifying, reporting and recording of PDs (2).
Communication and Escalation:The MMP should lay out the established communication methods and escalation pathways with regard to key decision-making
concerning medically-related issues. This is of prime importance in global studies with large medical teams, including various medics from both the sponsor and the involved CRO.
The design/development of all associated forms, trackers, listings and patient profile utilised in the various procedures of medical management should go in tandem with the development of the MMP.
Safety Management Plan (SMP)The safety management plan (SMP) is a key medical document and requires MM inputs during the setup phase of the study. Depending on the applicable procedures, the SMP can be developed as a separate plan or integrated into the MMP. The SMP should be commensurate with the anticipated risks, and the scope and complexity of the study. The primary purpose of SMP is to monitor the progress of the trial from a safety perspective, laying out the mechanism for serious adverse event reporting to the sponsor, ethics committee, applicable regulatory authority (RA), and plans for ensuring data integrity and consistency between the safety database (DB) and the clinical DB. Additionally, the reporting requirements and the applicable procedures concerning reporting of other safety-relevant events such as adverse events of special interest, pregnancy, overdose, etc. should be clearly defined in the SMP. The agreed process flow for safety review, reporting and management must be described elaborately with information on safety database, responsibilities of each person involved in the safety reporting, regulatory reporting procedures and timelines. The MM should ensure that all safety procedures included in the SMP are in compliance with the regulatory landscape and ICH-GCP guidelines, with clearly defined responsibilities and pathway for each involved stakeholder.
Major Protocol Deviations (PD) GuidanceTo facilitate prevention and management of PDs and ensure appropriate and timely identification, reporting and recording of any major PDs, a comprehensive guidance document should be developed based on the final version of protocol. This document is designed to be illustrative of all anticipated medical or procedural significant deviations that could occur during the study, and would also impact the safety of study subjects or scientific integrity of the study data. The document outlines the applicable strategy for corrective and preventive actions (CAPA) associated with PDs. Additionally, the types of deviations are prospectively classified, categorised and the reporting procedure is described (2). Specific instances of potential deviations are listed accordingly as eligibility or study conduct deviation. Together with the guidance document, developing of a study-specific PD report template and a designated tracker at this stage will establish the tools for appropriate PD management during the study conduct phase. Whenever feasible, system-based reporting and evaluation of PDs can be established to allow essential recording and tracking of all PDs in an automated way, thereby introducing operational efficiency and augmenting quality with regard to this important aspect of the clinical trials’ conduct.

Volume 8 Issue 326 Journal for Clinical Studies
Regulatory
Case Report Form (CRF) DesignThe primary objective of CRF designing is to gather complete and accurate data by facilitating transcription of data from source documents onto the CRF and avoiding duplication of information. The CRF should be designed with the primary and secondary endpoints as the main goal of data collection (3). The electronic case report form (eCRF), usually supplied by selected vendors, is customised in context to the study objectives and the indication, so that essential data for regular review and for final analysis is captured completely with quality. The pre-coded answer options are incorporated to all applicable forms, keeping the free text data fields to a minimum. The built-in data checks are placed wherever appropriate to enable clean data entry. Avoiding redundant capture of data and employing cross-checks at critical data fields would filter out errant data entry from the onset. It is increasingly recognised that the design of the CRF is a key quality step in ensuring the data required by the protocol, regulatory compliance and/or safety needs/comments, study-specific hypothesis attributes, site workflow, and cross-checking of data items within a form or across different forms are addressed (4). Standard coded forms of data collection are employed whenever possible to facilitate easier data extract and interpretation during the statistical analysis. Moreover, timely availability of accurate and complete data would facilitate the medical decision-making particularly when safety issues arise concerning any of the study subjects.
Collaborative efforts between the MM and data management (DM) team are essential during the development of the CRF to institute a user affable and dynamic CRF system that can achieve maximal accuracy and completeness in data capture and transfer, thereby facilitating effective study review and analysis. Hence, due diligence towards the development of the CRF by pooling medical and data management expertise, along with clearly-written associated CRF completion guidelines, saves valuable resources throughout the course of study and also elevates the quality of data delivered.
Medical Review of Relevant Documents and PlansIn addition to the development of plans and procedures related to medical management, the medical input and review is instrumental for a wide range of plans, documents and procedures relevant to the diverse activities of the clinical study. We refer here to some of the key other plans and documents that require careful review by the MM.
• The statistical analysis plan (SAP), authored by the study statistician, requires inputs and review from the MM before approval. Contents of the SAP such as endpoint definitions and variables, safety and efficacy analysis criteria, and templates for tables, listings and figures (TLFs) are focused during the review to assess the methods defined to present and evaluate the data-driven parameters that reflect the safety or efficacy objectives of the study.
• The data management plan (DMP) and the CRF completion guidelines (CRFCG), usually authored by the responsible data manager, need to be reviewed by the MM to confirm the data collection practices, review plans and data exchange procedures. The DMP outlines the various procedures used to manage the study data and sets the specifications for collecting, organising, validating, storing, backing-up and dissemination of clinical data throughout the life cycle of a study. The objective of CRFCG is to lay out the study-specific data entry guidelines within the CRF, by defining each data point and the associated instructions to follow by the investigation site personnel while performing data entry during the study.
• The forms/templates used for reporting significant study information, such as SAE report, pregnancy, overdose, and major protocol deviation, and the corresponding trackers, should be reviewed by MM prior implementation in the study to ensure that all relevant information is completely and appropriately captured.
• Whenever local site laboratories are used for study-related laboratory assessments, it is advisable to establish a standardised conversion factor list to harmonise the lab results collected from various sites in a way that facilitates medical review and interpretation of the information. This list can be prepared by the study statistician, followed by review and approval by the MM. When feasible, automatisation of this conversion within the eCRF will save valuable time for different team members, including MM, data manager and biostatistician, both during regular reviews and at the time of DBL.
• When applicable, the MM should also review the charter for the external data and safety monitoring board (DSMB) or data monitoring committee (DMC), to ensure all relevant information with regard to the required data and procedure for decision-making by those committees are reflected in the corresponding charter.
Study Team TrainingThe primary responsibility to train all the clinical study team members on medical-related aspects of the project rests with the study MM. The investigator and site staff training on protocol specifications and various study-related assessments is a key step before activating a site in the study, and is usually delivered by the MM in a face-to-face meeting, either individually or in a global or regional investigators’ meeting. This training serves as the platform to convey all the study-specific expectations and responsibilities of the investigators, along with the ICH-GCP principles while undertaking an investigative trial. The training should also focus on key information in IB and refer to the sections that would be of significant guidance during the study conduct. The investigators are sensitised on the safety profile of the investigative drug, focusing on expected and class adverse events.

Journal for Clinical Studies 27www.jforcs.com
In addition to the operational aspects of the protocol, the MM must also provide therapeutic area and disease indication training to all operational team members during the setup of the study, to ensure clear understanding of the patient population that would be recruited in the study and the type(s) of treatment that would be provided; thereby the specific and unique needs of a study are well communicated in advance. Detail-oriented and targeted training from the MM will drive quality collection, documentation and monitoring from the sites, which serves to uphold patient safety and ensure that data generated is suitable for planned interpretation and analysis.
Closing RemarksClose collaboration between the MM and other study team members from clinical operations, regulatory affairs, project management, data management and statistics is highly critical to build a medical management platform for the complete course of a trial. Guarding subjects’ safety, ensuring compliance with the approved study protocol, and maintaining scientific integrity of the data during study conduct are the key objectives that the MM should be mindful of while setting up the described
procedures for medical management. To summarise, the MM during the set-up phase
• Takes the strategic lead to bring together all required study team expertise for the harmonised conduct of the study
• Take key decisions on methodology, tools to be employed, and procedures to be followed for effective medical management
• Train the investigators, site staff and study team members on the study protocol, study treatment(s) and relevant procedures
In the subsequent articles, we will discuss the role of MM during the study conduct and close-out phases, breaking down the specific tasks and effective ways to achieve desired operational excellence.
References1. Mohamed El Malt, Vijayanand Rajendran:
Transforming Medical management of clinical trials: Search Engine, European Pharmaceutical Contractor, March 2015: p40-43
2. Mohamed El Malt, Vijayanand Rajendran: Prevention and Management of Protocol Violations, Feb 11, 2014, Applied Clinical Trials. http://www.appl iedc l in ica l t r ia l son l ine .com/prevent ion-and-management-protocol-violations
3. Krishnankutty B, Basics of case report form designing in clinical research, Perspect Clin Res. 2014 Oct-Dec; 5(4): 159–166
4. Lu Z, Su J. Clinical data management: Current status, challenges, and future directions from industry perspectives. Open Access J Clin Trials. 2010;2:93–105
Vijayanand Rajendran, MD*Clinical Research Physician, Europital.Qualified physician with over nine years of clinical and research experience. Hands-on experience in safety monitoring of Phase I-IV trials in a variety of therapeutic areas including oncology, haematology,
gastroenterology and the musculo-skeletal system.Mail: [email protected]
Mohamed El Malt, MD, PhD*Chief Medical Consultant, Europital.Oncology surgeon and expert scientific researcher with more than 32 years of experience as a medical doctor, including 17 years of clinical research and drug development experience in academic
medical centres, pharma and CRO as investigator, project leader and medical director, in addition to 15 years of experience as general and oncology surgeon.
Regulatory

Volume 8 Issue 328 Journal for Clinical Studies
Clinical Trial Transparency: New Data Anonymisation Requirements
Faced with new European Medicines Agency (EMA) guidance on the anonymisation of clinical trial data, drug manufacturers targeting the vast EU market have two choices: do what it takes to meet the November deadline – which prioritises clinical study report content – or embrace a smarter and more sustainable strategy that starts with patient-level data. Chris Olinger, CTO of d-Wise,, advises against taking shortcuts.
The growing market expectation for corporate transparency is putting pressure on life sciences to be more open about their clinical trials results. Patients quite rightly want to know the findings of studies they took part in, and other researchers as well as the general public have an understandable interest in monitoring outcomes too. It is a trend that the pharmaceutical industry has seen coming for some time, and at least 20 leading players have already devised their own processes and channels for making their patient-level data more readily available, in anticipation of eventual regulatory intervention.
But now the European Medicines Agency (EMA) has stepped in with its own formal take on the situation. After drafting a new policy for clinical trial reporting (EMA Policy 0070) in 2014, the Agency has now issued a hefty 91 pages of implementation guidance, and life sciences organisations must comply with the new requirements starting from November this year.
While the current marketing authorisation process already requires the submission of clinical study reports (CSRs), the new regulation stipulates that, within 60 days of an authorisation decision (positive or otherwise), the CSRs must be made available in a format that removes any risk of a subject’s identity being breached.
Although the industry hasn’t quite reached a state of panic in response to the new requirements, concern is running high. November will come around soon enough, and the majority of life sciences companies don’t have a plan yet; they are still working out what to do.
The Danger of Cutting CornersSo what do we know? The extensive guidance issued in March prioritises clinical study reports as the primary target for patient anonymisation. This makes it very tempting for organisations to focus any investment and strategy in this area – on sourcing solutions or outsourced services that can take CSR documents and make the requisite changes to anonymise all references to subjects.
This knee-jerk reaction may meet the initial requirements and stave off imminent panic, but it is not
very efficient and is not a sustainable, long-term solution. Although the EMA has started with CSRs as its target, the published policy document indicates it is only a matter of time before all of the patient-level data behind those reports will need to be given the same treatment.
The smartest, most comprehensive and cost-effective way to approach patient anonymisation in clinical trial documents is to start with the patient-level data. Everything else flows from that, so get it right first time and everything from thereon in should be watertight. In the long term, this will also save a lot of time, expense and risk.
Importantly, this is the only way to ensure that patient-level data is given consistent treatment – which is critical in ensuring that study findings retain their scientific meaning and value. If different algorithms are applied to patient anonymisation between documents and data, it becomes increasingly difficult to rejoin the dots if researchers later need to perform further cross-referencing and analysis.
Introducing unnecessary complexity could also create more work for companies down the line, as they find themselves called up to address numerous follow-on questions once clinical trial findings are in the public domain. As willing as they might be to meet growing market expectations around transparency, manufacturers don’t particularly want to invite an open-ended discussion – administrative work that could tie up valued resource. The ideal is that interested parties should be able to serve themselves, finding all the answers they require through a designated portal (it is possible that the EMA will eventually channel all of the data through a central public website).
Why the EMA did not begin with the source data in its clinical trial anonymisation requirements isn’t clear. Possibly, the Agency thought that starting with CSR reports would make lighter work for organisations in the early stages of adapting to the new demands, shielding companies from the need to worry about the technicalities of thousands of data fields that may be associated with clinical trial data anonymisation.
False StartsSo what are companies doing currently? The industry hasn’t had much time to react to the EMA guidance yet, because it is so new. Concerned about the time pressures, some firms have taken the easy way out – engaging external agencies to process CSR documents. Electronic redaction (which is the equivalent of drawing a thick black line through patient information) is not
Market Report

Journal for Clinical Studies 29www.jforcs.com
Market Report
an option, according to the new guidance. So formulae need to be applied to protect patient A’s identity - which could be open to discovery based on the type of study they took part in, their age, race and demographic, and when they attended hospital or clinic, among other bits of information in their clinical data record.
Some early attempts to keep the costs down by using offshore help seem to have backfired, however, creating quality issues and causing some work to need to be redone. In short, it has proved a false economy. Meanwhile, given that a typical application for marketing authorisation may comprise 50 separate studies, keeping track of the different formulae that have been used to protect patients’ identities is creating its own issues.
And what of all the old studies that may still have relevance to drugs being brought to market today? What scale of workload may be required to bring all of this archived content into line?
With just 60 days of marketing authorisation to turn around all relevant CSR content, or suffer some form of penalty, it makes more sense that companies focus on a more holistic strategy for addressing trials’ patient
data, rather than individual manifestations of that data. This promises to be more economical in the long term, is more reliable, and means firms could prepare compliant, anonymised CSRs so that these are ready on the shelves at the time of marketing authorisation submission.
Get it Right First TimeSticking to the letter of the law and focusing only on the initial requirements of the EMA guidance may be understandable, but it is a short-sighted approach which could lead to more work and cost in the long run. This is something some pharma companies are already starting to find, as their current piecemeal approaches to the challenge begin to struggle.
Another sign of this is that some companies are considering to re-run reports – redoing all of their analyses and recreating study reports using compliant, anonymised patient references. The fact that they are even considering a non-trivial undertaking of this nature, which adds no conceivable value for the business, confirms the level of concern yet lack of real strategy across the industry.

Volume 8 Issue 330 Journal for Clinical Studies
Market Report
If companies understood how much simpler and less painful the clinical trial anonymisation process would be if they harnessed the right tools and started in the right place, they could avoid much of this stress. Patient-level data is structured and well-organised. This makes it easy to manage any transformations, because this can be done systematically and comprehensively in a few simple steps.
When they have got the hang of it, companies can expect to process an entire clinical study’s worth of data in just a day. Once the master data has been given the anonymisation treatment, amending the study reports becomes a simple matter of intelligent search-and-replace; the hard work has already been done.
The overall investment isn’t much more to do things this way, but the long-term gains are substantial. Let’s not forget – the EMA will expect fuller data anonymisation before too long; the CSR-only requirement is a temporary step. So there is no avoiding this. And starting with the data is a much more methodical and safe way to go about patient anonymisation; one that simultaneously makes it easier to ultimately anonymise the CSR, while making it less likely that external parties will discover inconsistencies the public reports and data which cause them to get in touch.
Embrace TransparencyExpecting transparency requirements to grow, and building anonymisation options into the original processes, is the best way life sciences organisations
can stay ahead of the market and minimise their risk. Although the FDA is not committing itself to the path the EMA has taken, it is conceivable that this will change down the line, so there is no justification for complacency whichever markets pharma organisations are targeting today.
The requirement to produce lay language summaries to make clinical trial findings more accessible to the general public is a further indication of how important data sharing is becoming. In this age of digital connectivity and growing consumer consciousness, populations are exercising their right to know more about the studies in which they are participating, the products they are buying and the processes behind them. If not the case already, it will soon get to a point that companies that do not share their data risk being cast under a shadow, causing consumers to wonder what they may be trying to hide.
Patient privacy will always be paramount, so the life sciences industry needs to be clever about this. It’s about finding the middle ground – between compliance and patient safeguarding, and the advance and promotion of science. With the right measures in place, pharma should not have to worry about the risk posed by greater transparency. Rather, their concern should be about meeting a ‘reasonable expectation’ of risk management – one that doesn’t sacrifice the science by stripping the value out of the data.
As time ticks by, companies should develop strong resolve – making their goal not simply to meet the minimum current regulatory requirements around CSR anonymisation, but to remember the spirit and wider purpose of the policy. This is to disseminate knowledge, and to empower external communities to find the answers they need more readily – so that the benefits of research and of medicinal advances have the broadest possible reach. That’s what real progress looks like.
Chris Olinger has more than 30 years’ experience delivering software solutions. Most notably, he spent 14 years at SAS where he led the development of the SAS Output Delivery System (ODS). As R&D product manager, Chris led a team of developers charged with creating ODS, now a core component of SAS software
used in nearly all SAS solutions. Chris has authored many technical papers and is a highly respected and much sought after industry speaker. As CTO of d-Wise Chris has a special interest in technology that can help transform the Health and Life Sciences industries.Email: [email protected]: www.d-wise.com

Leave medical translation to professionals
Your reliable medical language partner since 1996
Medical translation, editing, proofreading
Readability testing, user testing
Pre-translation source-text editing
In-country review
Rijnsburgerweg 10 2333 AA Leiden The Netherlands+31 71 568 08 62 www.medilingua.com

Volume 8 Issue 332 Journal for Clinical Studies
Market Report
The World Organization of Family Doctors (WONCA) reports that 600,000 family doctors in 150 countries provide 3 billion consultations each year. General practice/family medicine (GP/FM) is the core discipline of primary medical care and the cornerstone of many healthcare systems in Europe. The vast majority of European citizens have a general practitioner (GP), who is a first contact physician in case of health problems. Unfortunately, very few of them are involved in clinical research.
According to OECD Health Statistics1 in OECD countries in Europe, 31% of all physicians work in primary care (generalist) and every fifth is a specialist in family medicine (Figure 1).
Primary care is often represented as the base of the pyramid of healthcare. The middle layer is secondary care (provided by medical specialists), while tertiary care (provided mostly by referral centres and academic institutions) is situated at the top of the pyramid.
In Europe there are three models of primary care organisation2:
Public model, where primary care has a central place in the healthcare system and is run by the state rather than by professionals. These systems are usually governed by decentralised authorities and consists of multidisciplinary teams, with usually salaried GPs. This organisation is found in Finland, Lithuania, Portugal, Spain and Sweden.
Professional hierarchical gatekeeper model in which GPs are the cornerstone. Primary care professionals are accountable for management of resources used for healthcare. In this model, usually GPs are self-employed and remunerated with capitation and/or fee for service. There are strong professional associations and cooperation with academic institutions. This system is present in Poland, The Netherlands, Denmark, Estonia, Slovenia and the UK.
Free professional non-hierarchical model, where GPs try to organise primary care delivery independently. This model is characterised by the patients’ and professionals’ freedom, meaning the absence of a list system or gatekeeping. GPs are self-employed, and the academic status of general practice is quite low. This system operates in Austria, Belgium, France, Germany and Switzerland.
Not all countries clearly fit into this classification. Usually there are borders between the first two models, with a decentralisation of healthcare responsibilities, strong willingness to organise primary care at a regional level with advanced primary care management strategies, but with self-employed practitioners paid mainly by capitation, a low academic level for general practice and no nurse practitioners, this describes the case of Italy.
Today, family physicians and their GP practices are backed with the newest IT technologies and frequently are using telemedicine as a tool to access advanced diagnostic methods. IT technology and its applications have revolutionised healthcare systems. The adoption and use of information and communication technologies are increasingly seen as support, redesign and improvement tools for healthcare delivery, especially in primary care. GPs are aware of opportunities provided by technological innovations. An analysis of information and communication technologies (ICT) adoption and use by general practitioners in 31 European countries, published in 20153, shows that 95.97% of GP practices use computers in service delivery (Table 1).
The vast majority of countries have achieved a near-universal adoption of a computer in their primary care practices, with only a few countries near or under the boundary of 90%. However, in several countries in southern and central eastern Europe, there is still the need for further efforts to adopt computers for primary care functions.
Family medicine is a relatively new medical speciality, with rapidly growing academic institutions that closely cooperate with family practices providing healthcare to local communities. Such an approach gives GPs access to
Bringing Family Medicine to Clinical Research
ICT Application % of GPs Lowest UsageSearching medical information on the Internet 82% Cyprus – 45,7%
Issuing drug prescriptions 81% Cyprus – 1,4%Sending referral letters to specialists 70.50% Lithuania – 3,1%
Making appointments 51% Cyprus – 4,6%Issuing invoices 42% Greece – 0,5%Keeping records 77.4% Cyprus – 14,3%Storing test results 74% Lithuania – 18,8%Table 1. Use of information and communication technology in primary care
Figure 1. General Practitioners in OECD countries
0 500,000 1,000,000 1,500,000 2,000,000 2,500,000
All Physicians
All Generalists
GPs
All Physicians All Generalists GPsOECD EU 1,657,680 513,034 346,110OECD 2,070,998 941,943 529,981
GP/FM Specialists in OECD Countries
Figure 1. General Practitioners in OECD countries
Table 1. Use of information and communication technology in primary care
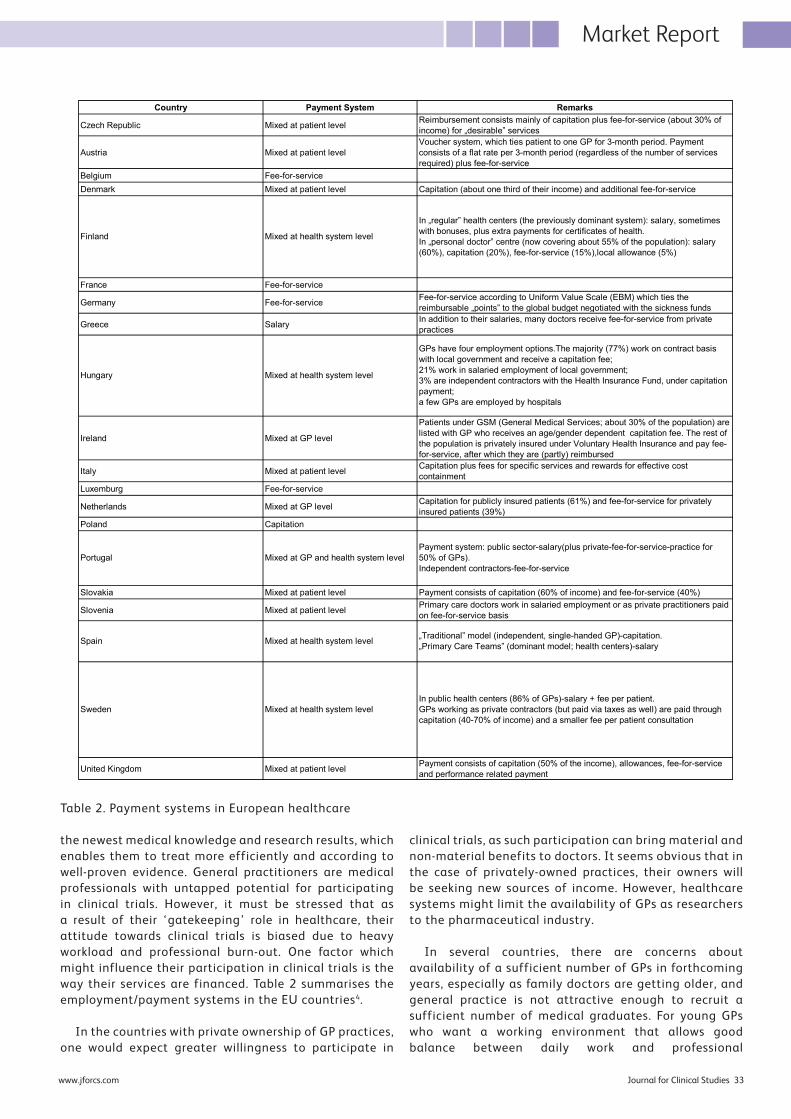
Journal for Clinical Studies 33www.jforcs.com
the newest medical knowledge and research results, which enables them to treat more efficiently and according to well-proven evidence. General practitioners are medical professionals with untapped potential for participating in clinical trials. However, it must be stressed that as a result of their ‘gatekeeping’ role in healthcare, their attitude towards clinical trials is biased due to heavy workload and professional burn-out. One factor which might influence their participation in clinical trials is the way their services are financed. Table 2 summarises the employment/payment systems in the EU countries4.
In the countries with private ownership of GP practices, one would expect greater willingness to participate in
clinical trials, as such participation can bring material and non-material benefits to doctors. It seems obvious that in the case of privately-owned practices, their owners will be seeking new sources of income. However, healthcare systems might limit the availability of GPs as researchers to the pharmaceutical industry.
In several countries, there are concerns about availability of a sufficient number of GPs in forthcoming years, especially as family doctors are getting older, and general practice is not attractive enough to recruit a sufficient number of medical graduates. For young GPs who want a working environment that allows good balance between daily work and professional
Country Payment System Remarks
Czech Republic Mixed at patient level Reimbursement consists mainly of capitation plus fee-for-service (about 30% of income) for „desirable” services
Austria Mixed at patient levelVoucher system, which ties patient to one GP for 3-month period. Payment consists of a flat rate per 3-month period (regardless of the number of services required) plus fee-for-service
Belgium Fee-for-serviceDenmark Mixed at patient level Capitation (about one third of their income) and additional fee-for-service
France Fee-for-service
Germany Fee-for-service Fee-for-service according to Uniform Value Scale (EBM) which ties the reimbursable „points” to the global budget negotiated with the sickness funds
Greece Salary In addition to their salaries, many doctors receive fee-for-service from private practices
Ireland Mixed at GP level
Patients under GSM (General Medical Services; about 30% of the population) are listed with GP who receives an age/gender dependent capitation fee. The rest of the population is privately insured under Voluntary Health Insurance and pay fee-for-service, after which they are (partly) reimbursed
Italy Mixed at patient level Capitation plus fees for specific services and rewards for effective cost containment
Luxemburg Fee-for-service
Netherlands Mixed at GP level Capitation for publicly insured patients (61%) and fee-for-service for privately insured patients (39%)
Poland Capitation
Slovakia Mixed at patient level Payment consists of capitation (60% of income) and fee-for-service (40%)
Slovenia Mixed at patient level Primary care doctors work in salaried employment or as private practitioners paid on fee-for-service basis
United Kingdom Mixed at patient level Payment consists of capitation (50% of the income), allowances, fee-for-service and performance related payment
Table 2. Payment systems in European healthcare
Spain Mixed at health system level
Sweden Mixed at health system level
In „regular” health centers (the previously dominant system): salary, sometimes with bonuses, plus extra payments for certificates of health.In „personal doctor” centre (now covering about 55% of the population): salary (60%), capitation (20%), fee-for-service (15%),local allowance (5%)
GPs have four employment options.The majority (77%) work on contract basis with local government and receive a capitation fee;21% work in salaried employment of local government;3% are independent contractors with the Health Insurance Fund, under capitation payment;a few GPs are employed by hospitals
Payment system: public sector-salary(plus private-fee-for-service-practice for 50% of GPs).Independent contractors-fee-for-service
In public health centers (86% of GPs)-salary + fee per patient.GPs working as private contractors (but paid via taxes as well) are paid through capitation (40-70% of income) and a smaller fee per patient consultation
„Traditional” model (independent, single-handed GP)-capitation.„Primary Care Teams” (dominant model; health centers)-salary
Finland Mixed at health system level
Hungary Mixed at health system level
Portugal Mixed at GP and health system level
Market Report
Table 2. Payment systems in European healthcare
Volume 8 Issue 334 Journal for Clinical Studies
Market Report
development, participation in clinical research may increase the attractiveness of family medicine. A healthcare system with the vast majority of privately-owned practices (Poland, The Netherlands, Denmark, Estonia, Slovenia, Romania) and a capitation-based payment system is ideally suited to the purpose of bringing family medicine to clinical trials. On the opposite end, there are countries with publicly-funded primary care (Greece, Spain, Portugal), where doctors are employed and salaried by the state, which controls the whole system and does not give doctors enough freedom and incentives to participate in clinical studies. Even if the obstacles such as regulations or agreements are taken into consideration, there is always a possibility to introduce family medicine doctors to clinical studies. It should be stressed that GPs’ participation in clinical studies is a win-win(-win) game for doctors, sponsors, and CROs and SMOs (Table 3).
A general practitioner has access to a large and diverse patient population, managing common types of chronic and acute diseases that often present in an early stage of development. Many diseases are treated mostly by GPs and approximately 80% of health problems can be solved in primary care in close proximity to the patient (Table 4)5, 6
According to the clinicaltrials.gov database, there were almost 45 thousand studies conducted in conditions routinely managed by general practitioners. While early phase studies and exploratory studies are the domain of academic investigators, confirmatory and post-registration studies could also be conducted in a primary care setting. Also, in most cases, in contrast to high-volume specialised medical facilities or academic institutions, GPs have access to research-naïve patients, especially in rural or remote areas. Clinicaltrials.gov database searches for terms related to primary care and family medicine are shown in Table 56.
One of the core elements of family medicine is person-centredness, which refers to a style of doctor-patient encounter characterised by responsiveness to patient needs and preferences, using the patient’s informed wishes to guide activity, interaction and information-giving, and shared decision-making7. This type of close relationship might also help the patient to make a
decision to participate in a trial, when presented and led by a trusted and well-known physician. In recent years, many health services and providers aspire to provide person-centred care, including the clinical research industry. The value of general practitioners was highlighted in the Report on General Public and Patient Perceptions of Clinical Research, published by CISCRP in 2013, which shows that for 52% of patients, the primary care physician is the preferred source of clinical research information. In contrast to the results of the survey, up-to-date engagement of family physicians and general practitioners in clinical research is very limited8.
The World Health Organization (WHO) global strategy on integrated people-centred health services (IPCHS) for 2016-2026 is a call for a fundamental paradigm shift in the way health services are funded, managed and delivered9.
Building strong primary care-based systems: strong primary care services are essential for reaching the entire population and guaranteeing universal access to services. It involves ensuring adequate funding, appropriate
Searched Term All Studies Open Studies Industry In Europe
Primary care 28614 7818 1146 403
Family medicine 5023 1117 231 81
General practice 4387 1141 263 134
Family practice 2204 507 71 27Table 5. Clinical Trials in Family Medicine
All Studies III/IV Phase
44.3% Essential (primary) hypertension 546 249 45.6%
25.6% Spondylosis 116 18 15.5%19.8% Atherosclerosis 5001 1519 30.4%
15.8% Type 2 diabetes mellitus 7232 1970 27.2%
12.7% Chronic ischaemic heart disease 583 81 13.9%
11.8% Type 1 diabetes mellitus 6399 617 9.6%
10.3% Hypertensive heart disease 368 37 10.1%
9.2% Polyarthrosis 2 0 0.0%8.3% Heart failure 5213 751 14.4%
5.9% Gonarthrosis [arthrosis of knee] 977 284 29.1%
5.8% Asthma 5009 821 16.4%
5.8% Secondary hypertension 963 30 3.1%
5.2% Angina pectoris 1522 438 28.8%
5.1%Other chronic obstructive pulmonary disease
726 3 0.4%
4.7% Migraine 680 225 33.1%
4.4% Coxarthrosis [arthrosis of hip] 199 59 29.6%
4.4% Gastritis and duodenitis 170 49 28.8%
4.2% Acute myocardial infarction 1489 312 21.0%
3.9%Stroke, not specified as haemorrhage or infarction
5856 584 10.0%
3.8% Osteoporosis without pathological fracture 1409 384 27.3%
44460 8431 19.0%Table 4. Most common medical conditions treated by primary care physicians
Incidence Conditionclinicaltrials.gov Data
Percentage
Total
Table 3. Win-win game to participate in clinical trials
Table 4. Most common medical conditions treated by primary care physicians
Table 5. Clinical Trials in Family Medicine

Journal for Clinical Studies 35www.jforcs.com
training, and connections to other services and sectors. It promotes coordination and continuous care over time for people with complex health problems, facilitating intersectoral action in health. It employs multidisciplinary teams to ensure the provision of comprehensive services for all. It prioritises community and family-oriented models of care as a mainstay of practice. Among other things, strategy implementation will be based on:
• primary care services with a family- and community-based approach,
• multidisciplinary primary care teams,• gatekeeping to access other specialised services,• greater proportion of health expenditure allocated to
primary care.
There is an ongoing discussion on critical need to support patient-centricity in clinical research. Patient-centricity is about engaging the patients in clinical research and bringing trials to their homes10. This is recognised as a key element in patient recruitment and retention. There are several innovative solutions supporting patient-centricity in clinical trials, like direct-to-patient studies, information and communication technologies (ePRO, social media networking), patient advocacy groups, pharmacy-directed outreach, etc.
We believe that the most efficient way to support
patient-centricity is to involve family physicians and their teams in clinical trials. It will improve quality of medical services by providing access to the newest medical solutions and treatment options, especially in areas earlier found to have limited access to those. From an industry perspective, this can improve patient retention and recruitment, enabling trials to be conducted more efficiently.
Therefore, implementing the patient-centred model by connecting the clinical research industry with professionals who are closest to the patients might be a faster, cheaper and easier solution for the many trials, especially those which can be conducted outside of specialist centres.
References
1. OECD iLibrary Health Statistics 2013. http://www.oecd-ilibrary.
org/social-issues-migration-health/data/oecd-health-statistics/
o e c d - h e a l t h - d a t a - h e a l t h - c a r e - r e s o u r c e s _ d a t a - 0 0 5 4 1 -
en?isPartOf=/content/datacollection/health-data-en#, visited
on 17 May 2016.
2. Kringos, D.S. et al. Building primary care in a changing Europe.
World Health Organization (2015).
3. De Rosis, S. & Seghieri, C. Basic ICT adoption and use by
general practitioners: an analysis of primary care systems in
31 European countries. BMC Medical Informatics and Decision
Making 15:70 (2015).
4. Saltman, R.B. et al. Primary Care in the Driver Seat? Open
University Press (2006).
5. Windak, A., Chlabicz, S., Mastalerz-Migas, A.: Family Medicine
– Textbook for Physicians and Students. (in Polish) Termedia
(2015).
6. www.clinicalrials.gov, visited on 10 November 2015.
7. Paparella, G. Person-centred care in Europe: a cross-country
comparison of health system performance strategies and
structures. Policy briefing. Picker Institute Europe (2016).
8. Report on Clinical Trial Information Seekers. The Center for
Information and Study on Clinical Research Participation
(CISCRP) (2013).
9. WHO global strategy on integrated people-centred health
services 2016-2026. Executive Summary. Draft for consultation.
World Health Organization (24/07/2015 version).
10. Kabata, J. & Kabata, P. Family Medicine – Clinical Trials
Enhancement at the Patient’s Level. J. for Clinical Studies, vol
7,1 (2015).
Market Report
Janusz Kabata, MD, PhD, MBA is co-founder and CEO of MedConsult, company that develops European network of family physicians trained to conduct clinical trials at GP practices. Prior to MedConsult , he was co-founder and Chief Medical Officer at GlobalCare Clinical Trials Ltd, and European Director of CRN (now Symphony
Clinical Research), providers of home care services where he developed and managed the network of home care service providers in Europe and Asia. He was associate professor at Gdańsk Medical University. He founded Nova Medical (now Synevo Central Lab), the first central laboratory for clinical trials in Central Eastern Europe. He is specialist in public health and laboratory medicine.
Bohdan Tillack MSc, MBA is co-founder of MedConsult. He is a finance professional and was instrumental in creating banking system in Poland after the political changes of the early 1990s. Bohdan left his successful finance carrer as Vicepresident of Nordea Bank Polska and joined forces with
Dr. Janusz Kabata at MedConsult to develop European Network of Family Physicians trained to conduct clinical trials. Bohdan is responsible for the financial side of the enterprise including value added services offered to the network participants to become investigative site.
Paweł Kabata MD, PhDMedical Director at MedSource Polska. Has 15 years’ experience in clinical research, both as country coordinator of Ambulant Care Services for GlobalCare Clinical Trials, and investigator of academic and industry-sponsored trials. He also works as a surgeon in the Department of Surgical Oncology
at Medical University of Gdańsk, Poland and in family-owned general practice.

Volume 8 Issue 336 Journal for Clinical Studies
Therapeutics
Improving Development of Antiepileptic Drugs for Rare Forms of EpilepsyTherapeutic development in rare diseases involves many challenges such as an incomplete understanding of the disease to inform trial design, requirements for new or more sensitive and specific outcome measures, and difficulties of recruiting a small sample to participation, among others. Rare diseases cover a broad range of diseases and patients, with about 50% of those affected being children. Many have a genetic component, while others arise from exposure to infections or toxins, from faulty immune responses, or occasionally from trauma or injury (e.g. traumatic brain injury (TBI)). For many rare conditions, the causes are frustratingly elusive. Many factors contribute to trial feasibility, but solid understanding of the epidemiology of the targeted condition is necessary to plan successful trials, but due to the small number of potential participants, a standard randomised controlled trial is often not feasible. Indeed this is the case with various forms of epilepsy syndromes. Although epilepsy affects approximately 1 in 100 people, many specific epilepsy syndromes are rare (http://www.ninds.nih.gov/).
Currently there are several incentives in place to encourage the development of new therapies for the rare epilepsy syndromes, and particularly those that do not respond well to the marketed antiepileptic drugs (AEDs). These disorders meet the requirements of orphan indications, for which tax incentives are provided and investments are smaller, with a potentially less demanding path for approval in Europe and the USA. For example, the USA Orphan Drug Act guarantees market exclusivity to the sponsor for seven years, as well as financial and regulatory benefits during development, including tax credits related to clinical trial expenses, and the elimination of fees for users. Another incentive is provided by the American National Institutes for Neurological Disorders and Stroke (NINDS) Anticonvulsant Screening Program (ASP), which provides free screening of antiepileptic compounds for commercial and academic institutions and sophisticated pre-clinical characterisation of promising molecules (Smith et al., 2007). Another opportunity may involve the repurposing of drugs from other therapeutic areas that possess either relevant disease-modifying properties for epilepsy or a novel mechanism of action with substantial synergistic efficacy against refractory epilepsy when combined with an existing AED therapy. This route would markedly reduce the level of investment necessary for discovery and development, and potentially decrease the regulatory data requirements.
As these types of new epilepsy therapies address a major unmet medical need, they also offer a promising incentive for future AED development. Indeed, the urgency to discuss innovative AED drug development was highlighted at the recent ILAE/AES Working Groups
joint meeting in London in which a call for the discovery of disease-modifying treatments that prevent the development of epileptogenesis in at-risk populations and importantly, drug development in specific rare refractory subgroups that would qualify for the much coveted “orphan drug designation” (Wilcox et al., 2013; Simonato et al., 2013). Below we discuss various ways to improve the development of antiepileptic drugs for rare forms of epilepsy.
Innovative Study Designs Epilepsy, by its nature, poses challenges to clinical development, and with the presence of over 20 AEDs on the market and a low success rate for Phase III epilepsy trials, enthusiasm for traditional antiepileptic drug (AED) development has decreased (French, 1997; Simonato et al., 2012; Simonato et al., 2013). However, the need for development of new therapies is as urgent as ever, with ~30% of the epileptic patients continuing to have poorly controlled seizures despite therapy (French, 1997). Given the presence of numerous approved AEDs for seizure control, conducting pure placebo-controlled designs is considered unethical in the epilepsy population, exposing the patients to unnecessary risk. Most AEDs are tested using the traditional Phase III, add-on, placebo-controlled clinical trial design in refractory patients who have frequent partial seizures. This population is usually heterogeneous, with patients maintained at a given dose for a fixed duration (usually 8 – 12 weeks), followed by open-label extension studies. This design has several hurdles for the development of new therapies in rare epilepsy syndromes. They usually require fairly large populations and the presence of the background AEDs can complicate the interpretation of the results. A “superiority” trial design in rare diseases is often not possible, as these trials usually require larger sample sizes. The “pure-placebo” controlled designs are accepted as gold standard in many disease areas, but in diseases such as epilepsy, where a single event can have serious consequences, and there are other therapeutic options, these trials are considered unethical by regulatory agencies, and have faced steep recruitment challenges.
To better understand the role of the investigational drug’s effect as monotherapy, an alternative design that “converts” the patients to monotherapy has also been approved by both FDA and EMA (Sachdeo, 2007; Wilcox et al., 2013). In this design, patients who continue to have seizures despite being maintained on background AEDs are randomised initially to placebo and study drug (often two doses). Following achievement of a maintenance period, the background AEDs are withdrawn, and patients are titrated to monotherapy or placebo. Once patients have an event (which can be pre-defined as one or the nth seizure of specific characteristics), patients are

Journal for Clinical Studies 37www.jforcs.com
Therapeutics
withdrawn from the study and treated with alternative standard of care AEDs. In this time-to-event design, the primary endpoint is retention time in the study following discontinuation of background AED. One other option is to have pseudo-placebo studies, with low and high doses of the study drug that has shown some efficacy in Phase II trials, compared to an active control. This design may be better suited for newly-diagnosed epilepsy patients, who have not yet been tried on any AEDs. Similar to the “conversion” studies above, the study duration in these trials also do not need to be fixed. Rather, these trials can also have the time-to-event design, where the patients receive rescue therapy and transition to an open-label trial following the first or nth seizure (Simonato et al., 2013).
In some very rare diseases, with no prior experience of the drug, companies have been successful securing initial approvals with small open-label studies (specifically if there is a well understood and clinically relevant biomarker for treatment response), or use of historical baseline data (French et al., 2012). For epilepsy, that may necessitate the study of epilepsy syndrome with clearly defined EEG signature that relates to the specific seizures, or another blood/imaging biomarker, and well documented prior seizure frequency/severity (historical control). One can also design longer-term initial studies to understand the natural course of the disease, in order to use as historical controls. This can be done as an observational study, but also can be incorporated as a lead-in arm in a controlled study, where patients are treated with a known AED for several weeks and the seizure frequency and severity are well-documented.
Finally, there are currently no feasible controlled trial designs to study therapies that may prevent the development of epilepsy (epileptogenesis) in at-risk populations (e.g. post-traumatic seizures, those with developmental lesions, febrile seizures etc). These patients are most-often studied only after the seizure pattern has been developed. Development of early-stage disease-modifying or preventative therapies for epileptogenesis will be challenging, but necessary, if we are to get in front of the disease process. Taken together, for new epilepsy therapies, and specifically in the refractory or rare epilepsy syndromes, innovative clinical trial designs are still needed.
Homogeneity of Sub-populationsAnother major problem with current AED study designs is implicit in the patient population included which could lead to many false negatives (Simonato et al., 2012). More specifically, the majority of randomised clinical trials for AEDs include patients with complex partial seizures, with or without secondary generalised seizures. This heterogenous population of epileptic patients includes a wide variety of and diverse epileptogenic and ictogenic mechanisms. Potential compounds tested in a traditional randomised controlled trial (RCT) against any one subset of these seizure types could go unnoticed if it were not effective against any and all the others. As discussed by Simonato and colleagues (2012), these observations raise several important issues related to clinical trial design. First, the patients studied in these trials have medical characteristics that render them treatment-resistant, which may not be relevant to various different target populations. In addition, this concept of a “lump sum enrolment” may lead to ineffective treatment as the

Volume 8 Issue 338 Journal for Clinical Studies
underlying etiology and pathophysiology of the disease is so discrepant from one epilepsy syndrome to another.
Given the nature of rare disease clinical trials, in that these are much smaller studies with only a few patients per study site, it is imperative that the patient population is more homogeneous. One possibility is through target identification based on a specific etiology (e.g. TBI), genetic or other biomarker (e.g. EEG or MRI signature, specific antibody etc). For example, among refractory epilepsy subgroups, MRI can be used to identify subgroups of patients with hippocampal sclerosis or developmental cortical lesions. Many of these patients can achieve freedom from seizures only after a successful respective neurosurgery, and as such there is a clear need for development of effective non-surgical therapies. Among the paediatric epileptic population, approximately 4% suffer from Lennox-Gastaut syndrome (LGS), a severe form of epilepsy that is notoriously difficult to treat (Shields, 2004; Markand, 2003). This population can also be reliably identified based on a well-recognized triad of multiple generalized seizure types, a slow spike-and-wave pattern (less than 2.5 Hz) on EEG and cognitive dysfunction (Markand, 2003). Taken together, further research is needed to more carefully characterise fundamental neuronal mechanisms underlying different types of epilepsy syndromes and ictogenesis that might influence their response to specific antiepileptic compounds.
Future DirectionsDespite the development and availability of more than 20 anti-seizure drugs, current medications still fail to control seizures in 20-30% of patients. However, our understanding of the mechanisms mediating the development of epilepsy and the causes of drug resistance has grown substantially over the past decade, providing opportunities for the discovery and development of more efficacious anti-epileptic and anti-epileptogenic drugs. New strategies for the discovery and development of AEDs that also offer a compelling case for industry investment must be pursued in order to provide new and improved treatment options for patients with epilepsy and, importantly, rare epilepsy syndromes.
References1. h t t p : / / w w w . n i n d s . n i h . g o v / d i s o r d e r s / e p i l e p s y /
epilepsy.htm 2. French JA. Obstacles encountered in designing
antiepileptic drug trials. Epilepsy Res 1993; 10 (suppl): 81–89.
3. French JA. Refractory epilepsy: clinical overview. Epilepsia. 2007;48(Suppl. 1):3–7.
4. French JA, Temkin NR, Shneker BF, Hammer AE, Caldwell PT, Messenheimer JA. Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapeutics. 2012 Jan;9(1):176-84.
5. Markand ON. Lennox-Gastaut syndrome (childhood epileptic encephalopathy.) J Clin Neurophysiol
2003;20: 426–41. 6. Sachdeo R. Monotherapy clinical trial design.
Neurology. Dec 11;69 (2007) (24 Suppl 3):S23-7. 7. Schmidt D, Sillanpaa M. Evidence-based review on the
natural history of the epilepsies. Curr Opin Neurol. 2012;25:159–163
8. Shields WD. Diagnosis of infantile spasms, Lennox-Gastaut syndrome, and progressive myoclonic epilepsy. Epilepsia. 2004;45 Suppl 5:2-4.
9. Simonato M, Löscher W, Cole AJ, Dudek FE, Engel J Jr, Kaminski RM, Loeb JA, Scharfman H, Staley KJ, Velíšek L, Klitgaard H. Finding a better drug for epilepsy: preclinical screening strategies and experimental trial design. Epilepsia. 2012 Nov;53(11):1860-7. Epub 2012 Jun 18.
10. Simonato M, French JA, Galanopoulou AS, O’Brien TJ. Issues for new antiepilepsy drug development. Curr Opin Neurol. 2013 Apr;26(2):195-200. Review.
11. Smith M, Wilcox KS, White HS. Discovery of antiepileptic drugs. Neurotherapeutics 2007; 4: 12–17
12. Wahab, A. Difficulties in Treatment and Management of Epilepsy and Challenges in New Drug Development. Pharmaceuticals (Basel). 2010 Jul; 3(7): 2090–2110.
13. Wilcox KS, Dixon-Salazar T, Sills GJ, Ben-Menachem E, White HS, Porter RJ, Dichter MA, Moshé SL, Noebels JL, Privitera MD, Rogawski MA. Issues related to development of new antiseizure treatments. Epilepsia. 2013 Aug;54 Suppl 4:24-34.
Idil Çavuş, MD PhD is a Senior Medical Director of Medical and Scientific Affairs for Neuroscience at Worldwide Clinical Trials. Dr Cavus has extensive training and experience in clinic, as well as in basic to translational to clinical research in CNS clinical trials in both academia and industry. She has authored several research manuscripts in epilepsy and other disease areas, and has extensive expertise in neuropsychiatric disorders including rare neurological disorders. Email: [email protected].
Jayne Abraham, PhD is a Director of Medical and Scientific Affairs for Neuroscience at Worldwide Clinical Trials. Dr Abraham has been involved in the assessment, treatment and investigation of various CNS drugs and disorders in both industry and academia for the past eight years. Dr Abraham specialises in clinical trials methodology and has advanced training in CNS disorders ranging from cognitive impairment in normal aging to complex epileptogenesis in paediatric patients. Email: [email protected]
Therapeutics

MLM Safeguard Box®MLM Safeguard Box® contributes signifi cantly to the quality of safety data and clearly outmatches conventional packaging methods for blood samples.
MLM Medical Labs® is an innovative central lab exclusively dedicated to clinical trials. We are located in Germany and support studies in more than 30 countries worldwide.
For further information please contact us at: [email protected] visit us under: www.mlm-labs.com
Our logistics innovation that eff iciently protectssamples against thermal stress:
Test data was collected using samples from 3 donors. Samples were held at room temperature for 2 hours, then stressed to extreme temperatures for 3 hours, then kept at room temp. overnight.
TEMPERATURE PROFILE
[°C]
555045403530252015
CardboardSafeguard Box
0 2 4 6 8 10 12 14 16 18 20 22 24
Time [h]
Heat test
WBC
Control Safeguard box
20
15
10
5
0Cardbox
[cel
ls/nl
]
Heat test
GPT (ALT)
Control Safeguard box
80
60
40
20
0Cardbox
[U/l
]
Heat test
INR
Control Safeguard box
1,5
1
0,5
0Cardbox
Not m
easu
rabl
e
MLM Medical Labs GmbHDohrweg 63 41066 MönchengladbachGermany
160425_mlm-anz_safeguardbox_JournalForClinicalStudies_210x297_rz.indd 1 25.04.16 15:30

Volume 8 Issue 340 Journal for Clinical Studies
Therapeutics
Considerations for Performing Sweat Testing in a Cystic Fibrosis Clinical TrialSweat testing was initially standardised in 19591 and it remains the gold standard for diagnosing cystic fibrosis (CF). Guidelines from the US Cystic Fibrosis Foundation and European Cystic Fibrosis Society (ECFS) both note that the sweat test is the preferred method to diagnose CF2, 3. Sweat is produced using a technique called iontophoresis in which an electrical current is used to simulate sweat production and chloride levels are subsequently measured. A sweat chloride level greater than 60 millimoles per litre (mmol/L) is considered diagnostic of CF. Currently, the most frequent method of sweat collection utilises the Westcor Macroduct® method. The Macroduct system uses an external battery in which an iontophoresis device is strapped to the patient’s wrist and his/her sweat is collected via tubing. The technical aspects for collecting sweat can be considered demanding and tedious for both clinical staff and study subjects as it takes a fair amount of preparation and must be performed by trained staff. Despite adequate training, obtaining an insufficient amount of sweat is a common occurrence. The objective of this article is to discuss key considerations when incorporating the sweat test as an assessment in a clinical trial involving patients with CF.
Inclusion of Sweat Testing as an Endpoint Inclusion of sweat testing is generally considered to be a secondary endpoint, however it may be considered as a primary endpoint in early phase proof of concept studies with novel CF transmembrane conductance regulator (CFTR) modulating agents4,5. Ideally, early discussion with regulatory authorities should take place prior to study design. As sweat testing was intended to be a diagnostic test, the acceptability by regulators to be classified as a surrogate biomarker of CFTR activity should be discussed. Also, the clinical relevance of the results, especially when compared to pulmonary function tests such as the forced expiratory volume in one second (i.e., FEV
1), has also been
questioned by clinicians and regulators.
Although obvious, it is important to note that site staff will only receive sweat testing results at screening and not post-randomisation to ensure entry criteria are met, as this may lead to unblinding. Ideally, an unblinded sponsor or CRO team member reviews the results on a regular basis and escalates any issues to the team, such as lack of reproducibility and/or insufficient volume (and not actual results to ensure that blinding is maintained). Additionally, it is important to clarify in the protocol if

GMP Lab Services• immunogenicity testing• biocompatibility testing• drug release testing• stability and formulation testing• hygiene and microbiology for
manufacturing
PK and PD Studies – GLP/GCLP lab services• method development and method
validation• quantification of drug substances
and metabolites• analysis of soluble and cellular
biomarkers
synlab pharma institute
Clinical Trials Phases I to IV• central lab services – worldwide• Phase I lab services – 24/7• pediatric studies – micro-volumes• biomarker identification• visit kits and frozen storage • expertise for medical devices
synlab pharma institutemerger of• INTERLAB lab services• BURECO• Swiss BioAnalyticsand others
www.synlab.com
Wide Range of Lab Services forpharma | biotech | medical devices | novel foods | cosmetics

Volume 8 Issue 342 Journal for Clinical Studies
Therapeutics
other study assessments cannot be performed as each sweat test can take more than 30 minutes.
Equipment Even if sites perform quantitative pilocarpine iontophoresis, the methodology and equipment used may differ from that required for a clinical trial; therefore, it is essential to standardise both across study sites. In addition, sweat testing requires many ancillary supplies, so it is important to provide all items as a kit to ensure standardisation and to reduce the burden on sites. Additional supplies, such as extra 9-volt batteries for the inducer and pilogel disks, should be provided to avoid delays.
Training Training staff is another key aspect and should ideally include more than one person at each study site. It is important to determine if prior certification from other sponsors (and/or patient advocacy groups such as the US CF Foundation) will be acceptable or if study-specific training and re-certification is required. Also important is to verify whether certification will include an analysis of the “mock” results based on volume and reproducibility, and the timeline for these results to be available relative to subject enrolment. It may be prudent for trainees to sign an informed consent as pilocarpine is administered during the training (which may result in localised burning and erythema; extremity hair may also need to be shaved).
Another consideration to keep in mind is outlining a plan for re-training staff. This can include online training and/or a “train the trainer” approach in which other local site staff or a clinical research associate (CRA) provide hands-on training. Generally, the most challenging part of the test is removal of the sample from the coil after collection, and this may require re-training, especially if results are not measurable due to insufficient volume.
The Future of Sweat Testing A new method of sweat testing called the CF Quantum®
Sweat Test (CFQT; PolyChrome Medical, Inc.) is currently under development and results of a multicentre pilot study have been published6,7. The CFQT potentially offers a simpler and quicker way to perform quantitative sweat analysis that does not require an external battery-containing inducer box. The pilot study demonstrated that the sensitivity and specificity of the CFQT in diagnosing CF was respectively 100 per cent and 96 per cent. In addition, 88 per cent of subjects/parents preferred the CFQT test compared to currently approved methods, including the Wescor Macroduct® method. New methods for performing sweat testing that lessen the amount of time and burden for study subjects and research sites are much needed.
SummaryThe conduct of sweat chloride testing in clinical trials involving CF subjects is fairly novel and can be intricate
and detailed. Experience and knowledge of key issues are vital to developing a robust study protocol and database and risk mitigation strategies for operational aspects frequently encountered.
References1. Gibson L, Cooke R. A Test for Concentration of
Electrolytes in Sweat in Cystic Fibrosis of the Pancreas Utilizing Pilocarpine by Iontophoresis. Pediatr. 23, 545-549 (1959).
2. Farrell P, Rosenstein B, White T et al. Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr. 153, S4-S14 (2008).
3. De Boeck K, Derichs N et al. New Clinical Diagnostic Procedures for Cystic Fibrosis in Europe. J Cyst Fibros. 10(2), S53–S66 (2011).
4. Accurso F, Van Goor F et al. Sweat Chloride as a Biomarker of CFTR Activity: Proof of Concept and Ivacaftor Clinical Trial Data. J Cyst Fibros. 13(2); 139-147 (2014).
5. Durmowicz A, Witzmann K, Rosebraugh C, Chowdhury B. Change in Sweat Chloride as a Clinical Endpoint in Cystic Fibrosis Clinical Trials. Chest. 143(1), 14-18 (2013).
6. Rock M, Makholm L, Chatfield B et al. A New Method of Sweat Testing: The CF Quantum® Sweat Test. J Cyst Fibros. 13(5), 520-527 (2014).
7. Rock M, Makholm L, Laguna T et al. Sweat Testing using the CF Quantum® Test System. Pediatr Pulm. 36, 266-267 (2013).
Vikki Brandi, PA, DHSc, Executive Director at INC Research, provides global oversight for INC Research’s Respiratory and Allergy clinical development programmes. Her background includes seven years of clinical practice as a Physician Assistant in addition to more than thirty years in drug
development as an investigator, pharmaceutical and biotech sponsor and clinical research organisation (CRO) professional. She has been involved in CF trials as both a sponsor and CRO professional. Email: [email protected]. Website: https://www.incresearch.com/our-experience/respiratory-clinical-trials.

Could be completed
on time.
So the landmark
trial
Of the drug delayed in customs
Who secured the release
Who called the expert
You are the person
Trust our chain reaction
Clinigen CTS adopts a problem-solving approach to clinical
trial supplies, anticipating hurdles and working swiftly to
overcome them.
Our aim is to deliver a clinical trial experience that is
seamless, secure and hassle free.
Email: [email protected]
Web: www.clinigengroup.com/clinical-trial-services
CTS1697_ITC_Mag_A4_Ad.indd 1 19/06/2015 12:01

Volume 8 Issue 344 Journal for Clinical Studies
Therapeutics
Why is Measuring Cognition Important?
IntroductionDoes the measurement of cognition need to improve before there can be advances in treatments of Alzheimer’s disease and other dementias? This article examines the importance of accurately measuring cognition and the new research harnessing new technologies that offer a completely new approach to this important field.
Why is Measuring Cognition Important?Cognition is the hallmark of the diagnosis of dementia, of which the most common form is Alzheimer’s disease (AD). It has proven to be a difficult concept to pin down and yet it is used to assist in diagnosis. There is no definitive diagnostic test for AD and biomarkers are not yet able to definitively diagnose AD; rather, its diagnosis is based on clinical criteria. Depending on how conclusive these criteria are will lead to a diagnosis of ‘probable’ or ‘possible’ AD. It is a sobering thought that the definitive AD diagnosis is only possible pathophysiologically at post mortem. The diagnosis of ‘probable’ or ‘possible’ AD all include a worsening of cognition in their definition. These range from ‘clear-cut history of worsening of cognition by report or observation’ for probable AD to ‘…. a sudden onset of cognitive impairment …. or objective cognitive documentation of progressive decline’ for possible AD. Worsening cognition is at the very heart of the diagnosis of AD; however, how it should be measured is largely unspecified, although the criteria for All Cause Dementia does note that ‘Cognitive impairment is detected and diagnosed through a combination of (1) history-taking from the patient and a knowledgeable informant and (2) an objective cognitive assessment, either a “bedside” mental status examination or neuropsychological testing.’1 The tests that should be employed, however, are not specified.
The early stages of AD and other dementias, where the majority of drug development companies are now working, offer the most significant challenge for measuring cognition. As discussed below, cognition is a complex concept and is made up of multiple domains. The domains that can be impaired at various stages are mentioned in the diagnosis of probable AD; domains such as impairment in learning and recall of recently learned information, impaired language presentation, visuospatial presentation and executive dysfunction. As the previous diagnostic criteria for AD largely depended on memory impairment and largely ignored cognitive decline in other domains, this was an important step forward.
As well as being the fundamental key to a diagnosis, the accurate measurement of cognition is important in demonstrating efficacy of new treatments. Although global or functional outcomes may be specified
as a primary outcome measure, demonstrating an improvement in cognition or a delay in decline requires an accurate measurement of cognition as an outcome measure.
What is Cognition and How is it MeasuredCognition is a broad umbrella term for a number of component processes, such as our perception of the environment; how we encode incoming information through the senses. Cognitive functions are the mechanisms by which the brain processes information; they are the brain-based skills necessary for carrying out any task and can be categorised into:
Input - our ability to learn and remember information we perceive so that it is available for retrieval at a later date. Processing or ‘thinking’ - the mental organisation and manipulation of information. Output - actions and behaviour, such as how we communicate information (e.g. speech, writing, gestures and expressions).
Cognitive abilities cannot be directly observed, but are instead inferred from how they manifest as behaviour. Cognition is sometimes regarded as a continuous flow of information between input and output, with all stages interacting and overlapping. The ability to create and retrieve memories is fundamental to all aspects of cognition and our ability to function. Attention is also a vital component of cognitive function in that it underlies and maintains the activity of all other cognitive functions.
The components and sub-components of cognition (also referred to as domains and sub-domains) defy simple definition and have been conceptualised and divided in diverse ways by different researchers.2 In one such scheme the individual components or domains that have been subsumed under the umbrella of cognition are: perception, memory, attention, executive function, language/semantic knowledge, visuospatial ability and praxis. In addition to understanding the cognitive domains affected by AD, it is also important to understand and measure how impaired cognition impacts upon individuals’ ability to function in daily life. As previously stated, AD is a heterogeneous syndrome; therefore, no two people will experience AD in the same way. Several factors contribute to the impact and severity of symptoms, even in people with the same apparent degree of impairment and pathology. An individual’s social and health status influence the extent to which impairments impede the ability to function in their everyday environment. So cognition and function are closely related.
In clinical trials a number of cognitive tests can be employed. The Alzheimer’s Disease Assessment Scale-

Journal for Clinical Studies 45www.jforcs.com
Cognitive Subscale (ADAS-Cog)3 is the most widely used assessment of cognition for clinical trials. The ADAS-Cog is simple to administer and is relatively brief (approximately 30-45 minutes), and this makes it attractive as an instrument for clinical trials. It has thus become established as a ‘gold standard’ for assessment of cognition in AD, and has been used as a primary endpoint in over 170 clinical trials. Initially an 11-part test, it has been supplemented with additional tests to overcome criticism of exclusion of tests of executive function, working memory and attention. It does, however, continue to be criticised for its susceptibility to ceiling effects in those with mild AD, e.g. in mild AD, patients don’t show impairment in this test. In addition, an apparent lack of standardised administration and scoring procedures4 causes high inter-rater variability.5 The MMSE is commonly used in clinical settings as a ‘bedside’ cognitive test, however limitations in terms of the domains relevant in the early stages are commonly expressed and subtle early cognitive decline can be missed.6 These are two commonly used cognitive tests, however there are literally hundreds of cognitive tests, covering the range of cognitive domains, posing a challenge for the clinician in selecting a test best suited to aid their diagnosis and to the drug developer to select as a primary endpoint in a clinical trial.
Once a test is selected, there are significant issues due to the need for repeated assessment. The tests employed in clinical trials are typically those borrowed from experimental or neuropsychology which are generally not designed for repeated assessment of the same individual;7 therefore, performance in these measures can change with repeated testing due to practice and learning of testing strategies, rather than as a result of treatment given. To overcome this issue, tests need to have multiple parallel versions available, which most current measures lack. Furthermore, versions would need to demonstrate equivalency to avoid variability of scores due to differences between versions.8 This issue of practice effects can be mitigated by training participants prior to baseline assessment9 but this practice is often not employed.
Historically, cognitive performance has been tested using pencil and paper tasks, administered and scored by trained personnel. However, advances in technology have resulted in the development of computerised tests. Automation of cognitive testing is said to have a number of benefits over the more traditional pencil and paper tests. The ability to record response latency with millisecond precision is an important advantage as psychomotor speed impacts cognitive performance. Computerised testing can also lead to greater standardisation of administration and scoring of tests, resulting in less measurement or operator error.10 It is also claimed that computerised testing allows multiple parallel versions to be created, which would allow for repeated assessment of individuals.11
Despite these advantages, there are potential barriers
to the use of automated testing. A substantial proportion of older individuals have visual and motor impairments which could make computerised assessment challenging. Another potential weakness is they do not record qualitative information about individuals’ approach to the tasks. In addition, validation of computerised batteries is costly and the proprietary nature of these tests may be prohibitive.12 According to Knopman and Caselli (2012) it has yet to be proven that the conceptual advantages of computerised testing translate into actual advantages over traditional methods, whilst Silverberg, Ryan et al. (2011) highlight the need for more information regarding the psychometric properties of computerised tests before they can replace pencil and paper methods in clinical decision-making. That said, new testing paradigms that take advantage of advancing technology are continually being developed, meaning there is great potential in the use of technology to improve cognitive assessment.
A further criticism of traditional cognitive measures is that the tasks individuals are required to carry out seldom resemble the activities they engage in on a daily basis. This has led regulatory authorities to require a co-primary functional measure in order to demonstrate clinical relevance.
Role of BiomarkersAs it currently stands, AD dementia is a clinical diagnosis. The biomarker data is not robust enough to allow a diagnosis of AD without clinical symptoms. The clinical diagnosis is based around the decline in cognition. Thus the measurement of this decline and indeed demonstration of any improvement must be by cognitive testing. Improvement in cognitive testing will help in the development of biomarkers. Biomarkers are much needed in this field if we are to step towards an objective diagnosis and allow the development of treatments of early or pre-symptomatic patients.
New ApproachesAs the awareness of the challenges of measuring cognition grows, and the recognition that this may be holding back the development of new treatments for AD, then scientists are turning to new approaches. This includes the application of new technologies to tackle this challenge.
In January 2014, Akili Interactive Labs Inc. announced an agreement with Pfizer to test the ability of Akili’s mobile video game platform to detect cognitive differences in healthy elderly people at risk of developing AD. Under the agreement, Pfizer will conduct a clinical trial that will evaluate healthy elderly subjects with and without the presence of amyloid in their brains. As stated in the press release, the goal of the trial is to investigate the Akili game as a biomarker or clinical endpoint for potential use in future Alzheimer’s trials. On announcing this deal, Michael Ehlers, Senior Vice President and Chief Scientific Officer of the Neuroscience Research Unit at Pfizer is quoted as saying,
Therapeutics

Volume 8 Issue 346 Journal for Clinical Studies
Therapeutics
“This partnership is another example of Pfizer’s commitment to embracing innovative technologies that have the potential to further research into neuroscience diseases. A tool that enables cognitive monitoring for the selection and assessment of clinical trial patients has the potential to be an important advance in Alzheimer’s research and beyond.”13
More recently, Akili Interactive Labs closed a $30m fundraising to continue the development of these digital tools. Several investors, including JAZZ Venture Partners, Canepa Advanced Healthcare Fund and PureTech Health, participated in the financing.14
The UK has also seen new approaches with new technologies. A new initiative supported by Alzheimer’s UK was announced in May 2016. ‘You can now help scientists tackle dementia by playing on your smartphone. Sea Hero Quest, which launches today (4 May) for Android and iOS devices, will provide data that will help researchers better understand how diseases like Alzheimer’s affect the brain.’
Sea Hero Quest follows the story of a young man who has to sail the ocean recovering his father’s lost memories. Rather than being given points on a map to follow, players must navigate the world themselves using methods that test their memory and orientations skills, such as following landmarks and shooting flares. According to medical research charity Alzheimer’s Research UK, a loss of navigation skills and spatial awareness is one of the earliest detectable symptoms of dementia. However, there’s currently no way for medical professionals to determine whether a person is losing their ability to find their way around due to the disease or through natural ageing. It’s hoped the app will make use of the estimated three billion hours people spend gaming each week collectively; by playing Sea Hero Quest for just two minutes, players will generate the same amount of data that would take scientists five hours to create under lab conditions, its creators claim.15
ConclusionMeasuring cognition is a complex area that, as with many aspects of research into the brain, has been neglected. Current methods for measuring cognition are rooted in the past, heavily relying on pencil and paper tasks, often with an over-reliance on verbal memory tests. The tests offer little comparison to the decline in function that the person is experiencing, and mean that the path to a diagnosis is long and arduous. It also cannot be just a coincidence that there have been no new treatments for AD for over a decade, with hundreds of failed drug trials. The exciting new approaches to this complex and long-standing problem are to be welcomed and offer hope that in the future the diagnosis is definitive and straightforward, and we have more advances in the development of new treatments for AD.
References
1. http://www.alzheimersanddementia.com/article/S1552-5260(11)00101-
4/fulltext#sec2 The diagnosis of dementia due to Alzheimer’s disease:
Recommendations from the National Institute on Aging-Alzheimer’s
Association workgroups on diagnostic guidelines for Alzheimer’s disease
McKhann et al May 2011 Volume 7 Issue 3, Pages 263–269Issue 3, Pages
263–269
2. Glisky, E. L. (2007). Changes in cognitive function in human aging. Brain
aging: models, methods, and mechanisms. D. R. Riddle. USA, CRC Press.
3. Rosen, W. G., Mohs, R. C. and Davis, K. L. (1984). “A new rating scale for
Alzheimer’s disease.” The American Journal of Psychiatry 141(11): 1356-
1364.
4. Connor, D. J. and Sabbagh, M. N. (2008). “Administration and scoring variance
on the ADAS-Cog.” Journal of Alzheimer’s Disease 15(3): 461-464.
5. Schafer, K., De Santi, S. and Schneider, L. S. (2011). “Errors in ADAS-cog
administration and scoring may undermine clinical trials results.” Current
Alzheimer Research 8(4): 373-376.
6. Simard, M. The Mini Mental State Examination Strengths and Weaknesses
of a Clinical Instrument. The Canadian Alzhiemer Disease Review. December
1998 http://stacommunications.com/customcomm/Back-issue_pages/AD_
Review/adPDFs/december1998/10.pdf
7. Harrison, J. and Maruff, P. (2008). “Measuring the mind: assessing cognitive
change in clinical drug trials.” Expert Review of Clinical Pharmacology 1(4):
471-473.
8. Knopman, D. S. and Caselli, R. J. (2012). “Appraisal of cognition in preclinical
Alzheimer’s disease: a conceptual review.” Neurodegenerative disease
management 2(2): 183-195.
9. Harrison, J. and Maruff, P. (2008). “Measuring the mind: assessing cognitive
change in clinical drug trials.” Expert Review of Clinical Pharmacology 1(4):
471-473.
10. Snyder, P. J., Jackson, C. E., Petersen, R. C., Khachaturian, A. S., Kaye, J., Albert,
M. S. and Weintraub, S. (2011). “Assessment of cognition in mild cognitive
impairment: A comparative study.” Alzheimer’s & Dementia 7(3): 338-355.
11. Silverberg, N. B., Ryan, L. M., Carrillo, M. C., Sperling, R., Petersen, R. C.,
Posner, H. B., Snyder, P. J., Hilsabeck, R., Gallagher, M. and Raber, J. (2011).
“Assessment of cognition in early dementia.” Alzheimer’s and Dementia 7(3):
e60-e76
12. Knopman, D. S. and Caselli, R. J. (2012). “Appraisal of cognition in preclinical
Alzheimer’s disease: a conceptual review.” Neurodegenerative disease
management 2(2): 183-195.
13. http://www.prnewswire.com/news-releases/akili-interactive-labs-announces-
partnership-with-pfizer-to-test-video-game-in-people-at-risk-of-alzheimers-
disease-239412291.html
14. http://www.businesswire.com/news/home/20160121006550/en/Digital-
Medicine-Company-Akili-Interactive-Labs-Raises
15. http://www.ibtimes.co.uk/sea-hero-quest-playing-this-smartphone-game-can-
help-fight-dementia-1558195
Susan McGoldrick is an experienced pharmaceutical executive with experience in various roles within the pharma industry, including a small pharma company developing treatments for CNS disorders, and founding and running a CRO specialising in CNS disorders. Susan now
advises The CNS Company on clinical trial issues in CNS disorders.Email: [email protected]

MedConsult services: Feasibility study and site identification
Study start-up
Contracts and budget negotiation
Training and quality assurance
Coordination of study from start to finish
Why Family Medicine?
Many diseases are treated mostly by GPs
Large and diverse patient population
Patient-centred approach
Patients close to the GP practice
Long-term follow-up
Contact us at: www.gp4research.com
Our network of clinical trial-trained GPs will help you reach your study goals on time.
medconsult

Volume 8 Issue 348 Journal for Clinical Studies
Technology
AbstractAnyone who has been involved in clinical development, and data management (DM) in particular, for a long period of time will recognise how the organisation of staff to perform the wide range of data collection and management tasks has changed to better align with advancements in technology.
Consider the pre-electronic data capture (EDC) era, when all trials, regardless of size, collected data on paper case report forms (CRFs). This method had a long lifespan, and as such had quite established practices which also drove operational organisation. The use of paper to record information drove processes, and the processes in turn influenced how staff were organised to perform tasks.
Workflow Depended on MonitoringFor example, it was typical in the paper CRF environment to have clinical research associates (CRAs) perform source data verification (SDV) during site monitoring visits, collect the completed SDV’d CRFs and submit them in a “batch” to a central data management department for entry into an internal system, where data management personnel would perform data review. Therefore, workflow for the data management staff depended on monitoring visit cycles. This created a bolus of work at times, and then a steady decrease until the arrival of the next “batch”. Staff assignments had to deal with this ebb and flow of work. As a result, the typical data manager had to have many assigned study related tasks to make full use of their time.
Classic Staff AssignmentTraditionally, for every trial there was a clinical data manager who performed as a “lead” who would have overall responsibility for the design/development of internal systems to collect data from the paper CRFs, create DM-related documentation, organise and train a team of data reviewers – dependent on the size of the trial and volume of data, perform data review, complete encoding, manage external data integration, facilitate meetings, manage communication with other operational areas, evaluate and escalate risks and manage timelines to ensure delivery of the final quality study data for statistical analysis. This clinical data management (CDM) “lead” role was assigned to the more senior, experienced staff, with junior staff taking on primarily data review, with other tasks gradually added commensurate with their level of experience. Depending on the duration of a trial, the team of individuals, and the CDM lead in particular, would work for many months or even years on a study.
For many years the DM environment and processes remained fairly constant, and the role of a clinical data manager in organisations also remained consistent. However, two major “events” converged that had a major influence on the way clinical data management and supporting roles evolved.
Financial Pressures and Cycle TimeThe pharmaceutical industry came under great pressure as costs in healthcare escalated, and there was an increased focus on the cost of prescription drugs as a contributor. This scrutiny caused pharma to look seriously at opportunities for cost efficiencies, which in turn generated heightened interest in processes; including evaluating what and how people performed tasks. “Lean” became an important goal! And faster execution of tasks meant faster completion to reduce the data review cycle time, allowing studies to potentially complete submissions earlier. Processes were evaluated and changed at a more rapid pace as the industry adopted the mindset of continuous improvement. The way of doing business started changing much more rapidly than in the past as companies tried aggressively to manage costs.
Changing TechnologyWith the introduction of new technical advances, emerging personal computing capability, a technology-savvy population and the internet, the approach for capturing clinical trial data was ripe for change. Opportunistically, EDC software products came to market allowing for entry of clinical trial information into systems at the physician’s site. While this did not eliminate source documentation, it did change the paradigm of recording clinical data on paper CRFs. This technology also held the
The Changing Organisation and Data Management RolesResource Roles Revisited for New Optimisation

Journal for Clinical Studies 49www.jforcs.com
promise of more near-term entry of patient visit data into a “system”, reducing the amount of questions or queries that would have to be generated during data review, and ultimately leading to higher-quality data at the point of capture, as data could be electronically “checked” for accuracy at the point of entry. As such, it was expected that the review work for the clinical data manager would decrease and also the method of review would become more efficient. To achieve these goals, systems had to have more and more sophisticated, integrated features, taking into account all the activities that were previously handled independently. The change to EDC technology required different skill sets for data management staff and new processes and ways of organising resources to perform those tasks.
Consequences to Data Management and Resource Management One could say that the change from paper-based process to EDC processes, and the impact on organisational demands, was seen as a significant breakthrough in time and cost. However, the transition did not happen overnight for pharma or CROs. Once the technology door opened, data management and the roles individuals played faced many challenges. Ongoing studies in one process had to either complete or transition, and staff had to retain knowledge to support the “legacy” work. They also needed to learn the new way of doing business. As a result, staff had to learn and manage multiple processes, including the constant further evolution of technologies. People who were experts suddenly had a brand new learning curve, and if an individual remained on a study for more than a year, they faced another learning curve when assigned to the next study because technology advancements were occurring at a fast pace, and driving process change to keep up as well.
Facing this challenge, data management organisations arrived at what can be termed a “functional resourcing” solution. By taking the data management tasks that previously were managed by one person – who had to be an expert at all – and breaking them into smaller segments for assignment, it became easier for staff to become a master of the task, as well as stay current with the fast pace of technology and process changes, since they encountered them more frequently. These “functions” became production lines for the specific study tasks within data management. The new functional norm broke down personnel organisation by tasks; i.e. start-up activities – which involved system design and testing, conduct – which involved review and query management, and data base lock – which concentrated on bringing the trial to closure on time with clean data. This organisational design also required an overall project manager to be sure that handoffs went smoothly and timelines were met. From the clinical data manager perspective, now instead of supporting one study from beginning to end, depending on their functional assignment, they did their task for the study and moved on to perform the same task for the next study. The goal was operational efficiency due to
repetitive tasks and constant learning and improvement that specialisation would bring.
As with any major shift in conducting work, the influence of technology and the resulting organisation change for data management brought its own NEW set of challenges, so the quest for how to do work most efficiently continued its cycle. While EDC technologies continue to evolve, there has been a push to make the tools more intuitive and diminish the need for specialisation to perform a wide variety of tasks. As a result, today some data management departments are again examining what is the best way to organise tasks and people to deliver quality EDC studies and data efficiently, and to determine whether there is a benefit to do so with a broadly skilled staff who see their work as a more meaningful career path.
References1. Karamehic, J., Ridic, O., Ridic, G., Jukic, T., Coric, J.,
Subasic, D. & Masic, I. (2013). Financial Aspects and the Future of the Pharmaceutical Industry in the United States of America. Materia Socio-Medica, 25(4), 286–290. http://doi.org/10.5455/msm.2013.25.286-290
2. Definition of Lean: Lean Enterprise Institute http://www.lean.org/WhatsLean/
3. Walther, B., Hossin, S., Townend, J., Abernethy, N., Parker, D. & Jeffries, D. (2011). Comparison of Electronic Data Capture (EDC) with the Standard Data Capture Method for Clinical Trial Data. PLoS ONE, 6(9), e25348. http://doi.org/10.1371/journal.pone.0025348
4. Sahoo, U. & Bhatt, A. Electronic data capture (EDC) – A new mantra for clinical trials. Qual Assur. 2003;10:117–21. [PubMed]
5. Krishnankutty, B., Bellary, S., Kumar, N. B. R. & Moodahadu, L. S. (2012). Data management in clinical research: An overview. Indian Journal of Pharmacology, 44(2), 168–172. http://doi.org/10.4103/0253-7613.93842
Joette Keen is Head Biometrics & Clinical Trial Data Execution Systems at KCR, a contract research organisation (CRO). Mrs Keen has more than 30 years of extensive experience in clinical data management, DM systems, as well as international operations management. Over the course of
her career, Mrs Keen was involved in many efforts across various functional areas. Her technical responsibility included developing data capture programmes for paper-based early and late phase drug and vaccine clinical trials, and SAS programmes to transform the entered data for transmission to clinical databases.Email: [email protected]
Technology

Volume 8 Issue 350 Journal for Clinical Studies
Technology
From the advent of x-rays in 1895 to the latest developments in magnetic resonance imaging (MRI) and computed tomography (CT) technology, imaging has played a vitally important role in improving global public health.1 When considering more modern techniques, we see how in the past 5-10 years this area has evolved to provide a clearer picture of body composition. The dual-energy X-ray absorptiometry (DXA) imaging technique, for example, originally developed for bone density measurement, is now being employed for both regional lean tissue volume measurement and regional fat volume measurement. Although a fairly cost-effective technique, DXA imaging does have a strong limitation in that it will always be based on projections through the body, which makes it impossible to assess the actual composition of tissue. To achieve that further step, more advanced imaging technologies are required, such as CT and MRI. These well-established techniques enable 3-D volumetric imaging and provide more specific and accurate measures.
Application in Clinical TrialsMore recently, other uses of imaging have emerged beyond the traditional scope of the hospital or surgical ward, with the pharmaceutical industry increasingly harnessing the power of imaging to support clinical trials. For many reasons, the cost of conducting clinical trials has become greater in recent years – regulations have become more complex, proposed therapies must be compared with the current standard and, if the trial endpoint is survival, it may take longer to reach this endpoint as therapies become more effective.2 Precise and accurate 3-D volumetric imaging offers the opportunity to examine alternative outcome measures that can be observed early after the therapy is given, thereby potentially reducing the cost of conducting clinical trials and accelerating the process of making new therapies available to the public.2
The Emergence of New TechnologiesThe imaging technology solutions developed at AMRA are well-suited to application within clinical trials. Our AMRA® Profiler service is the world’s first CE labelled technology of its kind: a computer-aided, cloud-based body composition measurement service, which uses images from a rapid, six-minute MRI scan to deliver accurate, precise fat and muscle measurements.3,4 This technique adds a new level of accuracy in body composition assessment versus traditional methods such as BMI or waist circumference, not only establishing with accuracy and precision the total amount of fat and lean tissue in the body, but also describing where it is situated – for example, whether the fat in the abdomen is predominantly subcutaneous or visceral, which has differing implications on metabolic health and risk, or whether fat has infiltrated the organs as a sign of atrophy or metabolic disease.
The availability of such detailed body composition measurements and imaging biomarkers can enable pharmaceutical companies conducting clinical trials to select trial subjects that show the highest probability of developing adverse outcomes or have the potential to see the most benefit from treatment within the trial. These insights will also allow us to establish whether if some patients should be placed on more targeted therapies. Additionally, the advanced phenotyping we are now able to conduct allows us to be much more specific about patient inclusion and exclusion criteria for clinical trials. By using these direct measurement techniques as opposed to indirect measures, such as BMI, total weight, or waist circumference, AMRA can offer more accurate patient stratification and establish more homogeneous study populations than previously possible.
Offering Added ValueThe other potential application of this technology is around the longitudinal follow-up of patients and therapies. As AMRA® Profiler has such high precision, it allows for the detailed characterisation of the effects of therapies on individuals. This high precision enables more accurate clinical trial conclusions to be reached based on fewer subjects and also offers investigators the opportunity to examine biomarkers that are more closely related to the physical aspects of metabolic syndrome, such as the distribution of visceral fat, intramuscular fat and liver fat. This information allows us to develop a better understanding of the underlying effects of medical interventions, rather than looking at just weight, which can be confounded by changes in muscle tissue and fat tissue. The speed at which we can complete the scanning examination also means that it can be applied and added to many clinical trial situations – for example, it would be easy to include a scan within a cardiac imaging research trial, which would quickly provide further phenotyping information.
Looking Ahead to the FutureThe focus of AMRA is to further develop our offering to provide more services in one single MRI examination, thereby removing the need to use different modalities to assess different aspects of body composition. We strive to offer one rapid exam that allows the acquisition of relevant information in a short time and at a reasonable cost. The technological progress AMRA has achieved to date has aimed to take this further by streamlining and standardising our delivery, thus ensuring high gains and improved efficiencies.
Earlier this year, AMRA entered into a global co-marketing agreement with GE Healthcare, a development which will mean that our AMRA® Profiler protocol will soon be made available on all GE Healthcare MRI scanners that have
Beyond Diagnostics –
The Added Value of Digital Body Imaging

WE KEEP YOUR
CLINICALTRIALSO N B U D G E T
Blood Pressure Monitoring
Capnography
Centrifuges
ECG Monitoring
Infusion Systems
Laboratory Equipment
Medical Freezersinc. Ultra Low Temperature
Medical Refrigerators
Patient Thermometers
Point of Care
Pulse Oximeter
Spirometer
Temperature Monitoring
Vital Signs Monitoring
Weighing, Measuring & Examination
...we won’t let you down
• Equipment rental/purchase
• Complete worldwide logistics
• Calibration, servicing and technical support available
• Breakdown equipment replaced free of charge
• Global equipment solutions/ outsourced procurement
• No minimum hire/deposit*
Ensuring that your clinical trials arebrought in on budget, we offer full financialmanagement support and a wide range oftailored rental and purchase options.
With over 25 years’ experience wespecialise in global logistics and maintainrigorous cost control across the enterpriseto deliver the most cost-effective solutions.
UK Office:T: +44(0)8456 777001
USA Office:T: 1-800-471-9200
www.woodleyequipment.comFor best value for money visit:
*subject to terms & conditions
JCS-full page-onbudget 26/2/16 14:42 Page 1

Volume 8 Issue 352 Journal for Clinical Studies
Technology
the latest software update, allowing for increased access to our analysis service and providing a further indication of the growing role of body composition measurement in medical imaging.
The Role of Large-scale StudiesInterestingly, it was recently announced that the world’s largest collection of multi-organ scans is now being collated by the UK Biobank, thanks to funding from the Medical Research Council, Wellcome Trust, and the British Heart Foundation.6 One important area that the study will consider is the amount and distribution of fat and muscle mass within the body, measured using MRI, highlighting that there is a growing appetite for this kind of data. Large-scale imaging studies such as this one are very important, as they offer a platform from which we can acquire longitudinal health outcome data to prove the value of this MRI technology.
One such example of this is an ongoing collaboration taking place between AMRA and Pfizer utilising the UK Biobank resource, which is examining and testing the
real-life application of our AMRA® Profiler offering. This unique partnership will assess fat and muscle measurements from magnetic resonance images in up to 6000 subjects who are currently part of the UK Biobank registry.5 The objective of this collaboration is to provide a better understanding of the relationship between body composition and risk for metabolic-related diseases, as well as a broader understanding of conditions linked to body composition. Results from the collaboration will be analysed by Pfizer and AMRA, with the aim of identifying new and more specific biomarkers for chronic conditions where body composition plays an important role.
Evidently, the future of digital body imaging has a great deal to offer, not only in terms of the development of new techniques and software, but also in the new ways that these can be applied for the benefit of patients. Ultimately, our aim is to bring new knowledge to the medical community that supports their needs and the needs of their patients, and assists them in both predicting and preventing disease. At AMRA, we are excited to see the potential role that we can play in advancing the use and broader value of body imaging and composition measurement both in patient trials trials. drug development, and clinical practice.
References
1. http://www.who.int/diagnostic_imaging/en/, visited on 27 April
2016
2. Erickson BJ & Buckner JC. Imaging in Clinical Trials Cancer
Inform. 2007; 4: 13–18.
3. Borga M et al. Validation of a fast method for quantification
of intra-abdominal and subcutaneous adipose tissue for large-
scale human studies. NMR Biomed. 2015, 28:1747-53. doi:
10.1002/nbm.3432
4. Karlsson A et al. Automatic and quantitative assessment
of regional muscle volume by multi-atlas segmentation
using whole-body water–fat MRI. J. Magn. Reson. Imaging.
2015;41:1558–1569.
5. h t t p : / / w w w . u k b i o b a n k . a c . u k / 2 0 1 5 / 0 9 / a m ra - a n d - p f i z e r -
col laborate-to-help- identify-those-at-r isk-from-metabol ic-
disease-and-comorbidities/, visited on 27 April 2016
6. http://www.ukbiobank.ac.uk/2016/04/uk-biobank-launches-
worlds-biggest-imaging-project/, visited on 27 April 2016
Olof Dahlqvist Leinhard, is AMRA’s Chief Technology Officer responsible for technical vision, for leading the execution of technology platforms and partnership strategies, and for overseeing all technology research and product development. Olof is also one of two founders and is a Member
of the Board. Since 2012, Olof has performed as a Junior University Lecturer in Magnetic Resonance (MR) Physics at Linköping University (LiU), within the Department of Medicine and Health (IMH) / Division of Radiological Sciences (RAD).Email: [email protected]
Credit: AMRA (Advanced MR Analytics AB)

Come experience what makes PCI different. We support clients as atrue partner and extension of their business, offering expertise andexperience guiding products to successful outcomes. With over 50 years experience, an integrated supply network to meet the needs of global studies, and an exemplary regulatory profile, we offer adaptable and innovative services to offer our clients real solutions enabling speed-to-market for their lifesaving medicines.
Commercial Services
Clinical Services
Expertise Experience Partnership
Manufacturing | Packaging & Labeling | Global Storage & Distribution
Manufacturing | Packaging | Serialization
www.pciservices.com
Expertise and Care in Clinical Development
PCI Clinical Trial Services
A Trusted Partner
© Copyright 2015 Packaging Coordinators, Inc. All Rights Reserved
PCI_ClinicalAdFINAL03.06.16.indd 1 3/6/16 3:25 PM

Volume 8 Issue 354 Journal for Clinical Studies
Prioritising patient-centricity in your clinical trials
PCI Clinical Services’ Tim Roberts and WestRock Healthcare’s Erem Latif examine how changes in healthcare legislation, alongside unprecedented merger and acquisition activity in the pharmaceutical industry, have increased the stakes in the clinical trials process – including increased outsourcing of clinical trials. Within a context of rising costs – from medication non-adherence and the need for ever wider global footprints – a patient-led approach is essential to help ensure compliance and ultimately clinical trial success.
In 2010, the United States made a significant investment with the introduction of the Affordable Care Act.
Bringing together two separate pieces of legislation — the Patient Protection and Affordable Care Act (P.L. 111-148) and the Health Care and Education Reconciliation Act of 2010 (P.L. 111-152) – the Affordable Care Act provides Americans with better health security by putting in place comprehensive health insurance reforms which purport to lower healthcare costs, guarantee more choice, and ultimately enhance the quality of care for all Americans.
Over the last six years, the Affordable Care Act has had a ripple effect on healthcare, with a different impact on every stakeholder.
Coincidentally over the same period, economic and market forces have driven the rate of mergers and acquisitions (M&A) in the pharmaceutical industry to increase dramatically. Between 2013 and 2014 alone, the rate of M&A activity rose by 170%.
The current global pharmaceutical environment is forcing drug companies to develop better drugs, faster – and at a much lower cost. Despite the increased M&A activity, and resultant downsizing and consolidation, expectations for performance, sales, and research and development remain constant.
In the last ten years, the pharmaceutical industry has invested over $800 million in research and development efforts1. The return on this investment, according to the IMS Institute, has been 252 molecules launched between 2000 and 20112. As a direct result of growing research and development efforts and pharmaceutical consolidation, the global clinical trial service market is expanding. The industry is anticipated to reach more than $64 billion by 2020 (based on a CAGR of 9% between 2015 and 2020).
By 2020, 72% of all global clinical trials will be outsourced to professional clinical research organisations (CROs)3. Demand for clinical trial ‘packaging solutions’ is
Logistics

Journal for Clinical Studies 55www.jforcs.com
You command connections.The in-market resources that come with World
Courier’s unsurpassed knowledge, global reach and
flawless supply chain execution mean that whether
your product is going across town or across the
world, you can trust us with what matters most:
your commitment to improving global health.
Choose confidently. Choose World Courier.
Learn more at worldcourier.com.
195_Journal_Print_Ad_210x297_r1.indd 1 8/7/15 11:48 AM

Volume 8 Issue 356 Journal for Clinical Studies
also forecasted to grow to $830 million (representing 1.2 B units) by 2018 and $1100 million by 20234. Assuming this anticipated growth within the CRO market is achieved, and given that clinical packaging and distribution costs are typically 10-15% of a CRO’s spend, it is estimated that the clinical packaging market share will reach $2.5 billion by 2020.
Medication non-adherence continues to present major economic challenges to global healthcare resources and directly impacts the US economy with losses estimated to be in excess of $350 billion every year. Furthermore, non-adherence also costs the global pharmaceutical industry $564 billion annually and has directly led to a 281% increase in pharma’s spend on adherence programme development. These costs do not include estimated cost impacts relative to clinical investigational studies as a result of poor adherence.
Because of the staggering costs emanating from non-adherence, efforts to better understand the causes have led to many theories. While some patients are impacted by the high cost of medications or side-effects, non-adherence is primarily attributable to patient behaviour. In order to change this, one must understand and apply the Habit Loop theory. The Habit Loop, as proposed by B.J. Fogg, has three stages: the cue or trigger; the behaviour itself; and the reward.
There are three unique phases of medication non-adherence. The first step is initiation, defined by when the patient instigates treatment. Primary medication non-adherence is a common issue within non-adherence and is defined when the patient does not fill the prescription, or does not pick up/initiate his/her prescription.
The second phase of non-adherence is implementation.
This stage is defined by a patient delaying, or not taking, their medication, or taking extra doses.
The final phase of non-adherence includes persistence. This is when the patient makes a conscious decision to discontinue treatment. This can happen before the term of the therapy expires, either because the patient assumes their symptoms have resolved or because they don’t see/feel the impact of the therapeutic regimen.
Let us evaluate the scenario of a recent heart attack victim: There are up to ten concomitant therapies which must be taken to support the body following
an episode, ranging from beta-blockers to blood thinners. The average person may need to take these twice a day. If we then add in another complication such as diabetes (which according to the American Heart Association doubles the likelihood of a heart attack), this can add up to 32 different ‘items’ to the patients’ daily health routine. These may include blood glucose test strips to infusion sets, and multiple medications ranging from tablets to vials.
This regimen can add a significant burden to the patient’s life – even though the repercussions of not taking the prescribed medication in this case can be life-threatening. Once the stress and pressure of hospital admittance are in the past and long forgotten, it is very easy to forget to take medication, or believe that you, as the patient, are once again fit and no longer need it. Therefore it is not surprising that once a patient returns to the formalities of daily life, continued medication adherence can be a real challenge.
Working as a clinical trial professional, it is very easy to lose sight of actual patient requirements while focusing upon the stringent protocol and regulations required by relevant authorities to ensure a drug achieves regulatory approval.
In order to minimise rising costs in research and development budgets, pharmaceutical professionals must – while maintaining safety, control and protocol – look for more innovative and smarter ways to ensure the new chemical entity (NCE) developed for trial is taken correctly and effectively by the patient.
As the global footprint of clinical trials grows and CROs look to recruit patients in remote emerging nations, which have what are considered ‘therapy-naïve’ patients, there
Logistics

Journal for Clinical Studies 57www.jforcs.com
must be a consideration of cost to the pharmaceutical business. Lean Six Sigma and other manufacturing principles are now commonplace across the pharmaceutical arena – doing more with less, while building in quality from the foundations. These are the broader principles which must be applied to the thinking behind clinical trials and research and development in general.
Medication non-adherence in the clinical trial arena has a significant impact on the success of the trial, as well as if the molecule in question will actually reach the marketplace. Patients who do not adhere to the therapy regimen during the clinical trial process are impacted adversely.
This non-adherence may mean that they experience reduced symptom management or increased risk of harm. With inaccurate dose response curves, the pharmacokinetic/pharmacodynamic (PK/PD) dosing data of the molecule in question will be wrong. As a result there is a strong possibility that the dose submitted to the FDA during the new drug application (NDA) process will be the ‘highest safest dose’ rather than the ideal dose for the medication. Furthermore, at the highest safest dose, patients may experience inordinate levels of adverse events.
A comprehensive survey of the pharmaceutical research and development arena reveals that medication non-adherence is the direct cause of many molecules being approved over time with less than accurate therapeutic dosing windows. These molecules are then subject to post-approval regulatory re-evaluations, which can lead to dose adjustments.
Consequently, there is now a priority focus on the patient during the clinical trial process to ensure 100% compliance and improve the success of clinical trials. By improving compliance, and therefore hopefully adherence, clinical trial development decisions will become more accurate and the optimal dosing regimen achieved. This will directly impact cost savings by reducing the chance for protocol adjustments and fewer post-approval dose reductions.
In conclusion, there are many savings to be made by focusing upon the patient first. We live in an age where information-sharing has never been so transparent and data so abundant. Clinical trial patients, like all other consumers, are increasingly able to carry out educated research to pick and choose the trials they enter.
By lowering both the burden on the patient and the R&D costs, a strategic focus on adherence and data collection will yield much better results in future trials, with fewer patients and therefore, reduced costs.
References1. 2015 Biopharmaceutical Research Industry Profile.
PhRMA. http://www.phrma.org/sites/default/files/pdf/2015_phrma_profile.pdf
2. “Restoring innovation as pharma’s center of growth.” IMS Health. https://www.imshealth.c o m / d e p l oye d f i l e s / i m s h e a l t h / G l o b a l / Co n t e n t /Information/Appl ications/Cl inical%20Trials%20Optimization/Restoring%20_Innovation.pdf
3. The New Trends of Global Clinical Development Outsourcing Market. http://www.reuters.com/a r t i c l e / 2 0 1 5 / 0 1 / 3 0 / r e s e a r c h - a n d - m a r k e t s -idUSnBw305621a+100+BSW20150130
4. Pharmaceutical Packaging Products. Freedonia Report. July 2014.
Erem Latif, Global Marketing Director for
Healthcare Adherence, WestRock Healthcare.
Erem Latif serves as the Global Marketing Director
for Healthcare Adherence for WestRock Healthcare
(formerly MWV Healthcare). Previously, she was
with UnitedBioSource Corporation, a subsidiary
of Express Scripts, developing, launching, and
implementing innovative pilots to improve patient engagement and
clinical adherence by leveraging patient-centric recruitment and
retention techniques.
Prior to joining UBC/Express Scripts, Erem held a marketing and
media position with the American Society of Cataract & Refractive
Surgery, supporting ophthalmic pharmaceutical and medical device
product launches. Erem’s career in the healthcare space began with
her time spent at AstraZeneca, supporting the Crestor NDA and
launch. Erem holds an MBA in healthcare from Florida Institute of
Technology as well as an MS in human physiology from Georgetown
University.
Erem is a dynamic, articulate healthcare marketing executive, with
15+ years’ distinguished implementation of strategic initiatives
across three diverse sectors: clinical research; pharmaceutical/
medical device; and payer/PBM and addressing multiple stakeholders’
needs. She is skilled at development and timely launch of healthcare
reform-focused initiatives, including patient engagement strategies
and clinical adherence programmes, leveraging her experience from
across the industry.
Tim Roberts, Director of Sales, NA PCI Clinical
Services.
With over 15 years’ experience working in the
late phase clinical trial industry, Tim Roberts is
responsible for PCI’s North American Clinical
Services Business Development Team and Global
Proposals. Prior to this, Tim was responsible for
PCI’s Clinical Services division across Europe, including project
management, documentation and storage and distribution.
With operational experience of managing clinical trials with patient
populations in over 30 countries, Tim has worked to develop and
implement innovation and experience across his team to ensure PCI
is the number one choice for outsourcing in clinical trials.
With a business degree from Liverpool John Moore’s University,
Tim joined PCI in May 2012 and was previously responsible for
EU business development and marketing. Prior to joining PCI, Tim
worked with Catalent and Aptuit.
Logistics

Volume 8 Issue 358 Journal for Clinical Studies
Integrating the Patient Perspective into Clinical Research Study Designs: Case Studies and Recommendations
Editor’s Note: This article is an abbreviated adaptation of the author’s Master’s Paper written at the University of North Carolina at Chapel Hill in partial fulfilment of the requirements for the degree of Master of Public Health, conferred in December 2015. While the author’s research was therefore conducted in the United States, the topic of integrating patients’ perspectives into study designs is germane to clinical research conducted anywhere in the world. On behalf of the journal and its readers, I thank the author for sharing his work with us.
Introduction One need not conduct a literature review to readily identify treatments in popular culture that have had a tremendous impact on societal health: for example, penicillin and the polio vaccine come immediately to mind. Indeed, the evolution of clinical research from its early beginnings to the sophisticated machine we see today has been extraordinary, generating vast amounts of data, conclusions, and even more research questions. It has spawned an entire industry of conferences and publications that cater to both researchers and stakeholders such as payers, providers, and policy-makers.
Unfortunately, one stakeholder remains all too often outside of the research consumption paradigm: the patient. Simply put, patients are not ‘consumers’ of research, they are ‘subjects’ of research. While ethical and regulatory frameworks exist to legitimately protect patients who participate in research, patients do not typically help define either the inputs (research questions) or outputs (analysis and publications). In contrast, however, patients do retain a growing role in the details of their care, including its financial aspects, and are therefore driven by necessity to understand the current state of the science around that care.
Three main imperatives underlie the growth of the need for a patient role (and, by extension, roles for caregivers, support networks, and communities) in healthcare decision-making and the corresponding need for more and better information to assist in that decision-making: each is discussed here in turn.
FinancialAs consumers are asked to pay more, and also a larger share, market forces will begin to shape decision-making and the need for greater transparency in the linkage between the evidence supporting the cost itemisations patients are receiving more often from providers. This decision-making will require more targeted and more relevant evidence, uniquely suited to the patient. The best way to gather and present this evidence is to involve the patient in study planning and analysis. According to the United States (US) Centers for Medicare & Medicaid Services (CMS), out-of-pocket healthcare expenses (i.e., those costs paid by the insured directly for their care) increased 3.2% between 2012 and 2013, after a 3.6% rise between 2011 and 2012.1 These expenses represent 12% of total healthcare spending.
In addition, health insurance premiums (i.e., those costs paid by the insured to insurance companies) increased 2.8% between
2012 and 2013 after growing 4% between 2011 and 2012. This increase has been greater than the values of 1.7% and 1.5%, respectively, the 12-month inflation rates for January through December, 2012 and 2013.2 Overall, households contributed 28% of all healthcare spending (a percentage that has stayed steady since 2010), the largest share among all stakeholders ultimately responsible for healthcare expenses, a group that includes private businesses and federal, state, and local governments. Thus, as healthcare spending overall increased 3.6% in 2013 over 2012, the dollar amount individual consumers of healthcare expend has increased significantly in both nominal and real terms.
Cultural and TechnologicalThe rise of social media, and specifically the application of social media to the sharing of individual medical information, has allowed patients to connect with each other and to share information, including research, as never before. This sharing, and the convenience and accessibility of the environment that enables it, has created an expectation on the part of the patient that they can, and will, be involved in research. According to its website, PatientsLikeMe® has over 350,000 members sharing information on over 2500 diseases and conditions.3 MediGuardSM, an online community of patients who participate in research and share information on conditions and treatments, has over 2.6 million members.4 Facebook®, moreover, has 164 million daily active users in the US as of June, 2015,5 and it is rare to find a patient advocacy group that does not have its own Facebook page, enabling information-sharing across disease communities for patients.
The rise of social media and its application to patient-centric information-sharing have created both a technological framework for sharing information about diseases and treatments, as well as an expectation that such information is merely a click away with brands that have built a trusted relationship with the member. Further, an important aspect of online patient communities manifests itself in the willingness of their members to participate in clinical research.
Another example of a technological development linking patients to research takes the form of the Apple® ResearchKit for Developers,6 which enables the creation of ‘apps’ for smart phones and other devices that collect data (both user-entered as well as device-generated) for medical research, potentially enabling vast numbers of patients to participate in studies along with the collection of device-captured outcome data (e.g., heart rate) without the explicit need for clinical visits.
While cultural and technological developments such as these might a priori indicate an improvement in the patient-centric design of and accessibility to clinical research; many of these advancements have currently been leveraged only by traditional (i.e., non-patient-centric) research using patients as subjects in traditional research (albeit in novel ways, such as “crowdsourcing” data collection7). However, the distance between that paradigm and true patient-centred research as illustrated in the case
Special Feature

Journal for Clinical Studies 59www.jforcs.com
studies described shortly is not great, and this cultural/technological imperative certainly drives research closer to more comprehensive patient involvement.8
StructuralAn evolution of the basic tenets of the way healthcare is conceived and delivered in the US represents a force necessitating a growth of the patient’s role in research. Physicians themselves have begun to recognise the key part played by patients through not only participation in, but also definition of, research. This recognition is driven in turn by the need to discuss often complex elements of treatment and disease collaboratively with patients when decisions are made. Tinetti and Basch9 observed that, in this context, “To appropriately inform patient-centered decision making, patient involvement is essential for identifying the questions to ask and the outcomes to assess.” This viewpoint goes beyond the traditional call for patients to participate as merely subjects in research, but to contribute substantively to research design itself.
The ultimate indication of the emerging impetus to involve the patient in driving research may be the fact that, in 2010, Congress authorised the Patient-Centered Outcomes Research Institute (PCORI), an independent non-profit, non-governmental organisation charged with improving the quality and relevance of evidence available to various stakeholders. The PCORI approach, detailed in one of the upcoming case studies, incorporates the patient perspective by design, attempting to address two primary weaknesses in current research approaches. The first is that research frequently does not address the specific concerns of patients and caregivers in the real world. Second, existing research is not typically available to those constituencies “in ways they can understand or use most effectively.”10
Three Case StudiesThree case studies of patient-centred research are presented. In each study, patients participated in the design of the research itself.
Case Study 1: Prostate Cancer This study evaluated the utility and feasibility of stakeholder involvement in the protocol design for a cancer trial. The subsequent study report revealed a rigorous process for testing the impact of patient (and other stakeholder) involvement on both the process and the outcome of clinical trial design. The general process followed for this study was as follows:11
1. A Mount Sinai Genitourinary Oncology Research Team initially drafted a complete protocol;
2. A secure, web-based collaboration platform was made available (via electronic communication) to a group of clinical and research experts (through one author’s professional network as well as literature review), and also to prostate cancer support groups (with a request to distribute to members), blogs, and prostate-cancer-related discussion groups on social networking sites;
3. Both open-ended and closed-ended input was enabled for six weeks, after which point responses were categorised, analysed, collated and presented to the Mount Sinai research team who drafted the protocol;
4. The results were evaluated and any modifications to the
protocol were made by consensus of the Mount Sinai team; and,
5. Three independent reviewers assessed the protocol modifications to evaluate the results (and achieved sufficient consensus on the number of changes to validate the results).
To enable this effort, several technologies were leveraged to a considerable extent. These included electronic communication such as email (to enable low-cost, high-volume notification of the study), social media (to reach targeted groups of stakeholders likely to be interested and provide feedback), and a secure, web-based platform for managing collaboration, capturing feedback, and then collating feedback.
The authors agreed up front on two important and fundamental definitions. First, the concept of utility was stratified into the implementation of major and minor changes. Major changes involved those to protocol elements including eligibility, dose, primary end-points, and the statistics plan. Minor changes represented all other changes. The threshold for achieving an acceptable utility of the feedback process required at least one major change or three minor changes to the protocol. In this way, the authors set a standard for the quality of stakeholder engagement. Second, to qualify as crowdsourced, the effort to solicit feedback from stakeholders required at least 20 clinical/research experts and at least 20 patients (or their advocates). Given the wide (and varied) use of the term crowdsourced, the inclusion of this definition indicates that the research team set clear objectives for the quantity of feedback that would be required to result in a useful process. Further, the numbers used indicated consideration by the team for the practicalities of the process: clearly, the ability of a research team to integrate feedback (especially open-ended feedback, which has the potential to provide the greatest insight) is self-limiting.
The researchers defended the limitation of the denominator as (a) representing a significant interest over traditional practices with regard to stakeholder (especially patient) collaboration, and (b) enabling the type of open-ended feedback that results in so-called “gems” which drove protocol modifications (as opposed to simply enabling consensus that, while useful and confirmatory, did not drive process utility). The framework constructed by these two definitions provides a useful way of evaluating the results.11
Results first demonstrated that the process did indeed result in a crowdsourced effort, collecting feedback from 60 clinical/research experts and 42 patients (or their advocates). Second, four major and five minor changes were implemented by the Mount Sinai research team: hence, the process met the utility threshold prospectively agreed upon by the authors. Third, the authors solicited the opinion of the stakeholders with respect to the following statement: “I would participate in a similar clinical trial crowdsourcing effort in the future.” In response, 91% of the clinical/research experts and 76% of the patients (or their advocates) agreed or strongly agreed.
The results of this case study demonstrated the impact of patient stakeholder engagement on clinical design — and the value of this feedback both to the end result (i.e., the utility of
Special Feature

Volume 8 Issue 360 Journal for Clinical Studies
Special Feature
the feedback) as well as stakeholder satisfaction — independently of scale and generalisability. Indeed, it is likely that involving patients in study design at any scale will create a “virtuous cycle” that improves not only clinical research, but, in the vision of our next case study, also patient outcomes.
Case Study 2: Uterine FibroidsThis case study addresses a PCORI-sponsored project entitled: “Comparing Patient-Centered Outcomes after Treatment for Uterine Fibroids.” Part of what makes this case study unique and relevant to the concept of patient involvement in research design can be seen in the evaluation process through which it had to pass to receive funding.
The process for identifying potential PCORI research topics begins with a rigorous evaluation at several levels. These levels included an evaluation by staff, outside experts, and a Science Oversight Committee and Advisory Panel. Examples of criteria considered are the type of research question (comparative effectiveness or not), the impact of the condition on individuals and populations, the likelihood that research findings will improve performance, and the topic’s inclusiveness across multiple populations.12 As a result of this initial process, uterine fibroids was selected as one of several projects.
Once this topic had been identified, the grant application resulting in this case study was evaluated based on a set of strict merit review criteria including the following:13
1. Impact of the specific research question to be studied on the health of individuals and populations
2. Potential for the findings from the study to improve healthcare and outcomes
3. Technical merit4. Patient-centredness5. Patient and stakeholder engagement
Criteria 1 and 2 simply extend the identified strengths of the research topic down to the level of the specific study, while criterion 3 is self-explanatory. Criteria 4 and 5, however, drive much of the specific design elements in this case study and, as such, illustrate the power of patient involvement in study design.Criterion 4, “patient-centredness,” as a research descriptor, has a specific definition according to PCORI14, namely, that such research must answer one or more of the following questions:
1. “Given my personal characteristics, conditions, and preferences, what should I expect will happen to me?”
2. “What are my options, and what are the potential benefits and harms of those options?”
3. “What can I do to improve the outcomes that are most important to me?”
4. “How can clinicians and the care delivery systems they work in help me make the best decisions about my health and healthcare?”
The proposal for this case study15 stated that Question 1 would be addressed as follows:
With regard to the first question, the findings of this study will help women making decisions about different treatments for uterine fibroids understand how long they are likely to
remain symptom-free following treatment and how personal characteristics, such as age and ethnicity, and disease characteristics, such as type of symptoms and severity of disease, are likely to affect their outcome.
Duration of symptom relief was selected based on a literature review of patient preferences, and the need for a follow-up procedure, especially a hysterectomy, was identified as a surrogate for that outcome in the datasets analysed primarily also due to the aversion to such a procedure expressed in the literature among women experiencing uterine fibroids. Thus, documented patient input played a central role in outcome selection.15
The study design proposal also addressed Question 2 “by comparing both hysterectomy and surgical and interventional treatment alternatives.” According to the proposal, a review of the literature as well as prior experience with direct stakeholder engagement led to this choice of comparators. An excellent example of incorporating patient input into study design, that prior experience took the form of both an expert panel and a stakeholder committee represented by multiple stakeholders, including patients, which found primary interest in both the comparative durability of alternatives to hysterectomy and, especially among women, the effects of uterine-sparing alternatives to hysterectomy on long-term symptom relief.15
Therefore, the study was ‘envisioned by design’ to address one weakness of current research described previously, namely identifying research questions of interest to patients (and, by extension, caregivers) through the selection of patient-centred outcomes and comparators. In addition, the study also addressed the second weakness identified by PCORI, i.e., making the results of research available to patients and caregivers in a readily comprehensible manner. The study’s designers therefore specifically sought to engage stakeholders and patients in accordance with criterion 5. To do so, the proposal indicated that a panel of stakeholders, including patients, would be convened to review and provide feedback on the protocol and analytic plan for the study, with specific focus on the following: variable selection, subpopulation selection, data definition and presentation, analytic methods, and the process of interpretation of the analyses.15 This list of elements was primarily driven by the study methodology itself, namely a retrospective observational study using secondary data sources (electronic medical records).
The implementation of this aspect of the study began with the selection of a Stakeholder Partnership Council (SPC) consisting of 17 individuals representing patients/consumers, payers, providers, and policy-makers, selected based upon clinical expertise in uterine fibroid disease, experience supporting research and paying for treatments, and existing participation in patient advocacy activity.16 The end result, currently under review by PCORI, will be a qualitative and semi-qualitative analysis of the engagement process by both investigators and the SPC, with an eye toward evaluating direct engagement measures such as trust, legitimacy, fairness, respect, accountability, and competence.15
Case Study 3: OMERACTIn addition to discrete, study-by-study attempts to involve

Journal for Clinical Studies 61www.jforcs.com
Special Feature
patient feedback in study design, some researchers have endeavoured to create a systematic environment for continuously involving patients in research strategy and design elements. This final case study demonstrates that the results can be cultural as well as scientific.
The Outcome Measures in Rheumatology (OMERACT) group meets biannually to define core outcome measures for rheumatic diseases based on international feedback. The group first included patient participants at the 2002 conference, its sixth, and has included them ever since. Initially, the OMERACT conference represented a collaboration of researchers focused on “the accuracy and responsiveness to change of clinically relevant (to patient and clinician) endpoints” in rheumatoid arthritis (RA) due to a lack of consistency in the application, and the existence of a wide variety, of outcome measures.17 Building on this initial success (the core set of outcome measures agreed upon by consensus at the first conference was endorsed by the World Health Organization), the conference met every other year to continue building similar consensus in other related disease areas. During the fifth conference in 2000, as the researchers explored patient-reported outcomes (PROs), a spontaneous proposal approved unanimously by the attendees was made to invite 11 patients to the next conference in 2002 in order to incorporate their perspectives on the RA core set of outcome measures.17
In 2010, a team attempted to qualitatively assess the benefits of patient involvement in study design (in this case, the outcome measures utilised) and the magnitude of that benefit. The assessment focused on three areas:17
1. The research agenda;2. The development of PROs; and,3. The culture of the OMERACT conference, where research
design occurs.
As explicit patient involvement in research decisions is not always documented in literature, the method for conducting this assessment required innovative approaches. In addition to analysing conference proceedings as published in the Journal of Rheumatology, the authors reviewed less formal correspondence (e.g., session reports, emails, invitations) related to the evolution of patient involvement, including arguments for and against and opinions expressed when such involvement occurred. A coding scheme was developed, which utilised applied 211 individual codes grouped into 27 categories to aggregate data based on descriptive characteristics of the content, and the resulting analysis was utilised in a responsive evaluation in 2010 at the OMERACT conference in Malaysia.17
The responsive evaluation took the form of qualitative interviews with a variety of stakeholders using a hermeneutic approach. This approach seeks to interpret the meaning of social interactions,18 and is frequently utilised employed in the social sciences. According to the study team, applying hermeneutic methods to the evaluation resulted in an ability to respect the plurality of opinions held by the various stakeholders, ensuring all perspectives were incorporated. Further, to reduce bias, two outside experts with no OMERACT affiliation were included in debriefings and methodological discussions.17
Thirty-two semi-structured interviews were held, 16 of which involved patient participants, and topics included:
1. The expected role of patient participants;2. Their selection, preparation and support; and3. The expected or provided contribution to the OMERACT
conference.
To incorporate a broad spectrum of views, interviewees were selected based on a stratified sampling methodology of conference attendees, focusing on four characteristics: stakeholder type, gender, geography of residence, the number of OMERACT conferences attended, and the “opinion about patient involvement” (as assessed by one or more of the authors). The authors attempted to dynamically balance the sample as views were expressed by participants in interviews, and sampling ended when the authors agreed that saturation of views had been reached. Where possible, findings were cross-referenced in multiple sources, such as between documents and interviews, and personal recollections were included to fill any gaps.17
The results of this analysis supported the value of patient involvement in the OMERACT conference. Based on structured interaction with patients at the first workshop in 2002, it became clear that the perspectives of patients and researchers diverged with respect to outcome measures of importance in RA research. As a result, new studies in four topics (e.g., fatigue) were devised that questioned the perspectives of patients and incorporated outcomes of interest, such as foot problems and pain flares, into seven core measure sets, including fibromyalgia and gout. In addition, patient collaboration contributed directly to the creation of four specific new PROs, for example, measuring work productivity and pain flares. Patients also provided valuable input into assessments of the feasibility of outcome instruments and core measure sets.17
The decision to integrate patients directly into the conference was an enlightened one, and one with unforeseen cultural consequences. Conference discussion, by nature, tends to be extremely collaborative and open-ended, with small groups focusing on a particular issue and then reporting back to the conference as a whole, either during the conference or later via the publication of proceedings. As such, the inclusion of patient perspectives in these discussions became part of the ‘DNA’ of the conference implementation, establishing relationships and collaborations that resulted in an ongoing platform for research design and prioritisation. Patients became more than just consultants in OMERACT activities, they were full collaborators and co-authors. In addition, the reliance on patients directly for feedback became the cultural norm for this group of researchers, further cementing the concept and creating synergies that extended beyond OMERACT as participants returned home and introduced patient perspectives into other research endeavours. For example, with input from OMERACT participants, the European League against Rheumatism recommended that scientific projects involve patient representatives. Table 1 presents a selection of quotes from conference participants illustrating this cultural evolution.17
Admittedly, these authors based much of their analysis on subjective criteria open to interpretation, despite the creation of

Volume 8 Issue 362 Journal for Clinical Studies
Special Feature
a well-thought-through systematic framework for document review, a detailed methodology for participant interviews, and specific techniques to mitigate bias. The nature of the social trends being measured certainly do not lend themselves to the same rigorous scientific method as randomised controlled trials, and opinions can be as revealing as fact. However, the results clearly demonstrate the value of patient involvement, both in terms of the “talk” and the “walk” documented throughout the paper. More importantly, however, they describe the evolution of a complex system of stakeholder interaction in the crucible of a conference environment, and demonstrate both second- (i.e., cultural) and third- (i.e., network effects on other non-OMERACT projects) order benefits. Findings and RecommendationsEach of the above case studies tackles patient engagement in slightly different ways, though all present useful lessons for future patient-centred research. Some of these lessons are as follows.
Defining patient involvement in terms of quality and quantity objectives provides a useful yardstick. By attaching numeric definitions and objectives to the size of the feedback cohort, as well as to the definition of feedback, researchers provide a useful and universal metric for comparing the degree of patient involvement in research design, which will further the interests of all stakeholders.
Evaluate not just the outcomes of the process, but the process itself. In addition to measures of participation, utility, and study design improvements, researchers should measure the satisfaction of those involved in providing feedback. As methods for soliciting, collating, evaluating and presenting the results of such feedback evolve, this information will help guide a more collaborative approach.
While this paper focuses primarily on patient involvement, the synergies behind multi-stakeholder insight should not be ignored. The interaction of perspectives, especially when those perspectives are freely shared via a robust technology platform across all collaborators, clinician and patient, creates the potential for new insights. In a very real sense, providing patient access to clinician feedback (and vice versa) can improve the quality of feedback from all.
The relative merits of a curated list of stakeholder-contributors versus a mass audience have not been determined. It is not clear that the use of a curated, finite list of stakeholder-contributors results in a qualitative difference in feedback, though certainly collating, evaluating and presenting feedback from a mass audience presents practical problems that may result in a need to more strictly define feedback (i.e., to only closed-ended questions), thus reducing the quality of insights in favour of purely confirmatory feedback.
Participation can be either direct or indirect. The uterine fibroids case study illustrates this vividly, where study designers incorporated patient feedback indirectly from literature searches and previous, unrelated engagements, as well as through direct solicitation as part of the study. While results on the latter are still incomplete, early indications support the utility and power of indirect participation, especially where specific, detailed,
documented information can be found in the literature. The practicalities of designing, evaluating, and funding research may drive investigators to the former.
The timing of input matters. Qualitatively, input during the conceptualisation of the study likely has a greater impact than that solicited during protocol development and analysis plan finalisation. While input at all phases is useful, the dependencies created during the study concept phase with future deliverables limits flexibility. The uterine fibroids case study supports this finding.
A patient-centric framework for devising and evaluating research increases patient participation. A framework such as the PCORI one described in the uterine fibroids study can play a significant role in systematically building in patient participation. For that case study, consideration of patient engagement and participation in the study design was “baked into” the funding proposal by necessity, resulting in a well-thought-through and multi-faceted approach.
The types of patient engagement should be driven by the study design. For the uterine fibroids case study, the retrospective database analysis drove the types of feedback patients could provide to the investigators (at all phases from conceptualisation to analysis). In other types of studies involving patient communities, it is possible to involve patients in a variety of other aspects of research, including patient-reported outcome instrument selection and data collection frequency.
While traditional statistical analysis may not be appropriate for evaluating the impact of patient involvement in study design, measure selection is important. On occasion, such as the evaluation of the value of engagement by the SPC in the uterine fibroids case study, quantitative analysis may not be called for. However, linking even qualitative analysis to key measures, such as trust and fairness, can significantly bolster the case for such engagement.
Research is a community endeavour, benefitting from standing communities of collaborators, including patients. The entire spectrum of activities involved in defining and conducting research can be seen as a complex system which benefits greatly when all stakeholders, including patients, are integrated into the culture of research, not just the process, via standing communities. Whether the community is virtual (i.e., online) or actual (i.e., a conference), the benefits are the same.
Patients hold different perspectives and priorities with respect to disease than other stakeholders, which can be both a benefit and a challenge. A certain amount of curation is necessary to prepare various stakeholders for collaboration, especially initial collaboration.
Patient contributions can be both subtle and surprising. de Wit et al17 noted in their discussion that patient feedback is valuable when it simply affirms what the researcher already believes, and also when, through dialogue, it introduces new ideas, aspects, or elements that need further exploration. Further, it is sometimes difficult for researchers to distinguish between the two.

Journal for Clinical Studies 63www.jforcs.com
ConclusionThe premises and conclusions of this paper should be understood in the context of the many positive trends in the provision of healthcare in the US.
One important such development is the rapidly evolving area of personalised medicine. According to Chan and Ginsburg,19
personalised medicine is “health care that is informed by each person’s unique clinical, genetic, genomic, and environmental information.” Many equate personalised medicine with so-called “precision medicine,” but while personalised medicine enables precision medicine by the very nature of its personalisation, precision medicine itself is best understood as an objective of personalised medicine. Chan and Ginsburg asserted that a key element of personalised medicine, clinical decision support (CDS), for both patients and clinicians is necessary to reach the goal of personalised medicine, namely “…to optimize medical care and outcomes for each individual, resulting in an unprecedented customization of patient care.”19 Reaching this goal therefore requires better and more accessible evidence for the patient in support of CDS.
The ongoing and intense focus on improving the quality of the US healthcare system, as measured by six proposed aims (i.e., safety, effectiveness, patient-centredness, timeliness, efficiency, and equality) has highlighted the need to focus on the patient. This landmark report, Crossing the Quality Chasm, specifically called for considering and respecting the needs, preferences, and values of patients in clinical decision-making.20 However, as the application of the important principles presented in that report evolve to meet today’s challenges, the ability of patients to understand and express needs, preferences and values in the language of clinical decision-making (i.e., evidence) requires that patients play a larger role in defining the research that supports this decision-making.
Additionally, the Institute of Medicine has extended its focus on quality to also identify ways to improve the value of healthcare. One concept at the heart of their recommendations takes the form of a “continuously learning health system,” where patients and clinicians form partnerships in the provision of care based upon shared science, information, incentives and, ultimately, a culture of continuous improvement.21 The concepts and ideas presented in this paper lend support to and are, in turn, directly supported by, their conclusions.
There are, however, challenges with empowering patients to act beyond the clinical research framework, as detailed by Wicks and Vaughan.22 A well-known study initiated by patients and caregivers using the “Patients Like Me” platform led to a rise in the off-label use of lithium carbonate to treat amyotrophic lateral sclerosis (ALS) after several patients utilised Google Translate to access a paper suggesting it might slow their illness. It was unclear where, if any, ethical oversight lay and lithium carbonate was later found to be ineffective. Further, patients enrolled in clinical trials for two ALS treatments “broke the blind” and shared data online. Not only did this undermine the research, but led inadvertently to a group of unenrolled patients ingesting the industrial cleaner, sodium chlorite, which was suspected to be the active ingredient in one of the drugs in the trial. These patients progressed less well than expected.22
These trends and challenges highlight the dissonance caused by an imbalance in the quantity and quality of information retained by providers when compared to patients. As such, it is necessary not to discard the existing clinical trial framework, but simply to rebalance it so that all stakeholders, including patients, can contribute equally and on the same level playing field using the language of evidence.
In conclusion, the role of the patient in clinical research continues to increase in importance as the complex system that is medical care evolves. A key element of that system, the research agenda and those who maintain and implement it, can certainly benefit from more patient involvement. This collaboration can take many forms and is multi-faceted in the value it provides, including identifying optimal individualised care plans, making better informed decisions, and ultimately improving outcomes. When implemented and measured properly, it encourages participation in research and improves the relevance to both patients and other stakeholders by incorporating the practicalities of the patient’s life and treatment journey into evidence generation. When implemented systematically, it holds the potential to reshape both the culture of research and the relationships between patient, provider, payer and policy-maker, leading to improved clinical outcomes.
Author’s NoteI am grateful to the Editor for allowing me to share this work with the journal’s readership. I have deliberately kept the paper faithful to the content of my Master’s Paper, but, as the topics covered continue to evolve in many and varied ways since initial publication, I would also like to share a few additional references on the general topic of patient engagement in clinical research. Readers who are interested in pursuing this dynamic topic in greater detail will thus have an initial roadmap for further exploration.23-26
References1. Centers for Medicare & Medicaid Services (CMS). National
Health Expenditures 2013 Highlights. Available at: https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthExpendData/Downloads/highlights.pdf (Accessed August 31, 2015).
2. US Inflation Calculator. Table of Inflation Rates (%) by Month and Year (1999-2015). Available at: http://www.usinflationcalculator.com/inflation/current-inflation-rates
Special Feature

Volume 8 Issue 364 Journal for Clinical Studies
(Accessed September 17, 2015). 3. PatientsLikeMe. Home page. Available at: https://www.
patientslikeme.com (Accessed August 31, 2015).4. MediGuard. Home page. Available at: https://www.
mediguard.org (Accessed August 31, 2015).5. Facebook. Facebook Q2 2015 Earnings. Available at: http://
edge.media-server.com/m/p/r62axc3n (Accessed August 31, 2015).
6. Apple. ResearchKit for developers. Available at: https://developer.apple.com/researchkit (Accessed August 31, 2015).
7. Surowiecki J. The wisdom of crowds. New York, NY: Anchor, 2005.
8. Wicks P. Could digital patient communities be the launch pad for patient-centric trial design? Trials. 2014;15:172.
9. Tinetti ME, Basch E. Patients’ responsibility to participate in decision making and research. JAMA. 2013;309:2331-2.
10. Patient-Centered Outcomes Research Institute (PCORI) (2014). Why PCORI Was Created. Available at: http://www.pcori.org/about-us/why-pcori-was-created (Accessed August 31, 2015).
11. Leiter A, Sablinski T, Diefenbach M, et al. Use of crowdsourcing for cancer clinical trial development. J Natl Cancer Inst. 2014;106.pii; dju258.
12. PCORI. Generation and Prioritization of Topics for Funding Announcements. Available at: http://www.pcori.org/research-results/how-we-select-research-topics/generation-and-prioritization-topics-funding-4 (Accessed May 16, 2016).
13. PCORI. Merit Review Criteria. Available at: http://www.pcori.org/funding-opportunities/merit-review-process/merit-review-criteria (Accessed May 16, 2016).
14. PCORI. Patient-Centered Outcomes Research. Available at: http://www.pcori.org/research-results/patient-centered-outcomes-research (Accessed May 16, 2016).
15. Gliklich R. Comparing patient-centered outcomes after treatment for uterine fibroids. 2013. Unpublished PCORI Grant Application.
16. Meyers E, Messner D, Velentgas P. PCORI Uterine Fibroid Project: comparing patient-centered outcomes after treatment for uterine fibroids. 2014. Unpublished Observational Study protocol.
17. de Wit M, Abma T, Koelewijn-van Loon M, Colins S, Kirwan J. Involving patient research partners has a significant impact on outcomes research: a responsive evaluation of the international OMERACT conferences. BMJ Open. 2013;3:e002241.
18. Little D. What is hermeneutic explanation? Available at: h t t p : / / w w w - p e r s o n a l . u m d . u m i c h . e d u / ~ d e l i t t l e /Encyclopedia%20entries/hermeneutic%20explanation.htm (Accessed September 14, 2015).
19. Chan I, Ginsburg G. Personalized medicine: progress and promise. Annu. Rev. Genomics Hum. Genet. 2011;12:217-44.
20. Committee on Quality of Health Care in America, Institute of Medicine. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press, 2001.
21. Smith M, Saunders R, Stuckhardt L, McGinnis JM, Editors. Committee on the Learning Health Care System in America. Institute of Medicine. Best care at lower cost: the path to continuously learning health care in America. Washington,
DC: The National Academies Press, 2013.22. Wicks P, Vaughan T. Subjects no more: what happens when
trial participants realize they hold the power? BMJ. 2014;348:g368.
23. PCORI. Our Programs. Available at: http://www.pcori.org/research-results/our-programs (Accessed May 16, 2016).
24. Sacristán JA, Aguarón A, Avendaño-Solá C, et al. Patient involvement in clinical research: why, when, and how. Patient Prefer Adherence. 2016;10:631-40.
25. Woolf SH, Zimmerman E, Haley A, Krist AH. Authentic engagement of patients and communities can transform research, practice, and policy. Health Aff (Millwood). 2016;35:590-4.
26. Robbins M, Tufte J, Hsu C. Learning to “swim” with the experts: experiences of two patient co-investigators for a project funded by the Patient-Centered Outcomes Research Institute. Perm J. 2016;20(2):85-8.
David Cameron consults with biopharmaceutical organisations, provider associations, and patient advocacy groups on the development of real-world evidence to support quality, safety, and effectiveness. He holds a Bachelor of Science degree in International Economics
from Georgetown’s School of Foreign Service and a Master of Public Health degree from the University of North Carolina at Chapel Hill. David is a frequent writer and speaker, and has given guest lectures at New York University’s Stern School of Business and Dartmouth’s Tuck School of Business.Email: [email protected]
Special Feature


Volume 8 Issue 366 Journal for Clinical Studies
Genomic Data Commons Launches for Data and Clinical Information SharingThe Genomic Data Commons (GDC), a unified data system that promotes sharing of genomic and clinical data between researchers, has launched. An initiative of the National Cancer Institute (NCI), the GDC will be a core component of the National Cancer Moonshot and the President’s Precision Medicine Initiative (PMI), and benefits from $70 million allocated to NCI to lead efforts in cancer genomics as part of PMI for Oncology.The GDC will centralize, standardize and make accessible data from large-scale NCI programs such as The Cancer Genome Atlas (TCGA) and its paediatric equivalent, Therapeutically Applicable Research to Generate Effective Treatments (TARGET). NCI is part of the National Institutes of Health.
Together, TCGA and TARGET represent some of the largest and most comprehensive cancer genomics datasets in the world, comprising more than two petabytes of data (one petabyte is equivalent to 223,000 DVDs filled to capacity with data). In addition, the GDC will accept submissions of cancer genomic and clinical data from researchers around the world who wish to share their data broadly. In so doing, researchers will be able to use the state-of-the-art analytic methods of the GDC, allowing them to compare their findings with other data in the GDC.
Source: CenterWatch.com
Southwest Research, Texas Biomedical Awarded $3.4M for Ebola countermeasureSouthwest Research Institute (SwRI) has announced a one-year, $3.4 million contract award from the Defense Threat Reduction Agency (DTRA) to combine two available medications and test the resulting combination drug therapy against the Ebola virus. SwRI is collaborating with Texas Biomedical Research Institute on this program. The award is a one-year contract with two additional option years.
“We are at the forefront of rapidly developing and fielding new therapeutics,” said Dr. Joe McDonough, director of the Pharmaceuticals and Bioengineering Department at SwRI. “We have a unique approach to repurpose two existing drugs that we believe will more effectively work together to target emerging bio-threats like Ebola.”
Because there are no proven treatments for the Ebola virus currently, outbreaks can cause fatalities as high as 90%. Using its core pharmaceutical capabilities, SwRI will create a more bioavailable, or more easily absorbed formulation of cepharanthine (CEPN). CEPN is a Japanese drug that has been safely used by humans for more than 40 years to treat a wide range of illnesses. In screening for chemical compounds that could potentially fight Ebola virus infection, Texas Biomed scientists discovered CEPN was effective at combating the Ebola virus but required very high doses. This new formulation of CEPN will be combined with chloroquine, a drug used to treat malaria. Texas Biomed will conduct efficacy testing of the formulations in its state-of-the-art Biosafety Level 4 Laboratory.
Source: JCS Staff Reporter
Women Are Being Excluded From Clinical Trials“Complexities of the menstrual cycle” are to blame
Even though sex-based equality has progressed in other fields, women have been routinely excluded from clinical trials, the group of British and American experts says, though medical problems and treatments affect them equally, if not more.
Some studies have shown that women are twice as likely to suffer from adverse reactions to drugs, and 80 percent of drugs are withdrawn from the market due to unacceptable side effects in women.
The group cites a study from 2014, where researchers reviewed nearly 1400 sports and exercise research studies involving six million people over three years. Those researchers found that only 39 percent of those study participants, just slightly more than a third, were women.
Researchers are frequently excluding women from trials due to “complexities of the menstrual cycle,” the scientists say. To conduct studies without possible interactions from fluctuating hormones, many researchers forgo using women participants. Or when they do, they use them in early follicular stages of the menstrual cycle, when hormones are lower and more equivalent to male levels.
This leads to, little understanding of how differing hormone levels would affect experiments, say the researchers. It is unknown if the hormone levels would affect each experiment at all, because they have not been tested. And this does not represent the real world that women live in, where hormones fluctuate cyclically. The scientists say this requires more research.
Source: JCS Staff Reporter
Publishers Announce New Way to Link Clinical Trials with PublicationsA group of publishers today announced a development that will allow clinical trials to be easily linked to related publications, such as the study protocol, statistical analysis plan, and articles reporting trial results. The system will link items using clinical trial numbers (CTNs) and digital object identifiers (DOIs). Publishers will also be able to highlight key information such as funding sources and retractions.
The initiative is the culmination of three years’ collaboration between publishers, led by the open access publisher BioMed Central, and Crossref, a not-for-profit membership organisation for scholarly publishing working to make content easy to find, link, cite, and assess.
Clinical trials can result in a large number of separate publications, from study protocols through to the results articles and secondary analyses, published in many different journals, sometimes years apart.
“Researchers need access to all of these articles if they are to reliably evaluate bias or selective reporting in the study. Identifying and centrally linking all articles related to each
News

Journal for Clinical Studies 67www.jforcs.com
JCS News
individual clinical trial is imperative, and publishers have an important part to play in ensuring researchers can find the information they need easily.
Source: All Trials
Lessons Need to be Learned from Clinical Trial TragedyThe British Journal of Clinical Pharmacology has published an editorial calling for improvements to the safety of clinical trials, following the tragic outcomes of the Bial clinical trial in January 2016, when five volunteers were hospitalised and one subsequently died.
The authors make several recommendations for learning from this tragedy, which include:
• Pre-clinical and clinical study data from this trial should be urgently released to allow lessons to be learnt for ongoing and future studies.
• Reviewers of clinical trials should ask for full pre-clinical study reports and full information on pharmacology, including drug and target interactions.
This is not the first time such calls have been made. In 2006 in the UK six people were left critically ill after taking part in a phase 1 trial of a compound called TGN1412. Following this incident the UK Government’s report said the problem of withholding phase 1 trial data needs to be addressed, as better sharing of such information could help to avert such disasters.
Source: All Trials
Will the EMA’s Guidance Usher in a New Era of Trial Transparency?The European Medicines Agency (EMA) has published new guidance for pharmaceutical companies on how to comply with its policy on the publication of clinical data. The guidelines address how to prepare and submit clinical study reports (CSRs), including how to anonymise data, and how to identify and redact commercially confidential information.
CSRs are very long documents that contain detailed information about the methods and results of clinical trials. They contain crucial information about the effectiveness and safety of treatments that might otherwise go unpublished.
Under its policy the EMA will proactively publish the final redacted versions of CSRs submitted in companies’ applications for approval to market medicines, 60 days after a decision has been made. The policy came into effect on 1st January 2015; publication of the first reports is expected mid-September 2016.
This policy has the potential to make the EMA the most transparent regulator in the world. The policy requires companies to provide release-ready versions of CSRs to the EMA for public consumption. This has the potential to provide unprecedented, proactive access to CSRs and the data they contain. But the devil — as always — lies in the details. If industry takes a liberal view of what information can be redacted and/or the EMA does a poor job of enforcing the definition of ‘commercially
confidential information’ it has set, these release-ready versions of CSRs may prove far less useful than we hope. So we’ll have to say tuned to see how ground breaking this policy really proves to be.
Source: All Trials
Diets That Involve Certain Vegetable Oils Found to Improve Cholesterol and Fat LevelsA recently published experimental study found that particular vegetable oils lead to the reduction of blood sugar and fat levels in diabetic mice.
Researchers from the Animal House of Pharmacology Department at the Kwame Nkrumah University of Science and Technology in Ghana conducted an experimental study with the intention of evaluating the potential benefits of vegetable oil consumption with regards to the management of diabetes in diabetic mice. 48 male mice were divided into groups; some were fed generic rodent food, and 10% palm oil feed, 10% coconut oil feed, 10% groundnut oil feed, or glibenclamide (a drug used in the treatment of diabetes) was added to some rodents’ diets.
The experiment showed that sugar levels decreased in mice fed diets that contained 10% palm oil, groundnut oil, and coconut oil; this effect was also observed in mice that were treated with glibenclamide. Moreover, coconut oil and palm oil did not significantly affect fat levels in the mice. Among the diabetic mice treated with glibenclamide, generic rodent food and palm oil significantly reduced total cholesterol.
Overall, the researchers observed a significant improvement in fat levels among diabetic mice that were fed meals that contained 10% vegetable oils (palm, groundnut or coconut oil). Significant decreases in total cholesterol were only observed in mice that consumed diets consisting of groundnut oil only, and diets with palm oil in addition to glibenclamide.
Source: Kwame Nkrumah University of Science and Technology, Ghana
News

Volume 8 Issue 368 Journal for Clinical Studies
Advertisers Index
I hope this journal guides you progressively, through the maze of activities and changes taking place in these Emerging Countries.
JCS has also launched a Weekly Newsetter. Please visit www.jforcs.com and sign up to receive the very informative weekly news letter.
Page 11 Almac Group
Page 13 BAP Pharma
BC Catalent Pharma Solutions
Page 19 Centre International de Développement Pharmaceutique (CIDP)
Page 43 Clinigen Group plc
Page 3 Dora Wirth Languages Ltd
Page 5 Europital
Page 47 Medconsult
Page 7 MediciGlobal – A Bioclinica Company
Page 31 MediLingua Medical Translations B.V.
Page 23 Synevo Central Lab
Page65 Medtech Summit
Page 39 MLM Medical Labs GmbH
Page 53 Packaging Coordinators Inc.
IBC Sharp Clinical Services
Page 41 Synlab Pharma Institute
IFC Trilogy Writing & Consulting GmbH
Page 51 Woodley Equipment Company Ltd
Page 55 World Courier (An AmerisourceBergen Company)
Volume 8 Issue 3I Journal for Clinical Studies
What Do You Need to Know to Ensure Shipping Success To North Africa
Empowering Real World Evidence Analysis Through Risk-based De-identification
Bringing Family Medicine To Clinical Research
Integrating the Patient Perspective into Clinical Research Study DesignsCase Studies and Recommendations
PEER REVIEWED
JOURNAL FOR
Your Resource for Multisite Studies & Emerging MarketsCLINICAL STUDIESU
Volume 8 - Issue 3
ww
w.jforcs.com
Journal For Clinical Studies Your resource for M
ultisite Studies & Em
erging Markets
Volum
e 8 - Issue 3
Subscribe today at www.jforcs.com or email at [email protected]
JOURNAL FOR
Your Resource for Multisite Studies & Emerging MarketsCLINICAL STUDIESU

Services Include:
• Over-Encapsulation and Placebo Manufacture• Clinical Label Design and Print• Clinical Packaging and Labeling• EU Quali�ed Person (QP) Services• Interactive Response Technologies (IVRS/IWRS)• Comparator Sourcing• Returns and Destruction• Clinical Storage and Distribution• Analytical, Formulation and Manufacturing Services
Sharp Clinical Services (UK) LtdWaller House, Elvicta Business Park, Crickhowell, Powys, NP8 1DF, UKT: +44 (0)1873 812182
Sharp Clinical Services - USA300 Kimberton Road,Phoenixville, PA 19460T: +1-610-422-3200
Sharp Clinical Services is a leading provider of specialist clinical packaging and supply chain services. Our experienced team take pride in delivering a personal service on a global scale, caring for your product as if it were our own.�e business is managed through a fully integrated SAP system which includes bar-coded stock management and tracking of customer projects. Sharp Clinical Services has a proven track record in managing the entire clinical supply chain and has a Global Depot Network of 18 depots supporting over 25 countries.
© 2014 Sharp Clinical Services
1093 Sharp_A4_AW2.pdf 1 15/06/2015 11:34
46_JCS_January2016.indd 53 03/02/2016 12:31:43

Catalent. More products. Better treatments. Reliably supplied.™ us + 1 888 SOLUTION (765-8846) eu + 800 8855 6178 catalentbiologics.com
biologics
25+ YEARS’ CLINICAL SUPPLY EXPERIENCE
1.6M+ CUBIC FEET CLINICAL STORAGE including refrigerated, frozen & cryogenic
60K+ BIOLOGICS SHIPMENTS to 80+ countries annually
˝ 2
016
Cat
alen
t Ph
arm
a So
luti
ons.
All
righ
ts r
eser
ved.
500+ PRE-FILLED SYRINGES PER HOUR with randomized labeling
With 8 GMP clinical packaging facilities and 50+ depots around the world, Catalent’s SMART BIOLOGICS CLINICAL SUPPLY SERVICES provide the global reach, innovative supply models, cold chain, and specialty handling capabilities to reliably supply your clinical trials around the world. As your trusted partner for tailored biologic solutions, including our BIOLOGIC DEVELOPMENT PLATFORM and ANALYTICAL SERVICES, we bring you to clinic faster.
smart biologics clinical supply. specialty handling. faster to clinic.
274-05_Ad2016_BiologicsCSS_JCS.indd 1 4/14/16 3:01 PM