Embed Size (px)
Citation preview

62
CHAPTER – 2
Terpenes to Ionic Liquids: Synthesis and
Characterization of Citronellal-Based Chiral Ionic
Liquids

63
2.1 Introduction
Ionic liquids are a fascinating class of compounds with unique
properties, which have recently attracted the attention of a wide
number of researchers. Numerous works have been published in the
last couple of years reporting the possibility to perform several organic
reactions and catalyzed processes in these new media. They represent
a new class of non-molecular, ionic solvents with a tremendous
potential to replace conventional solvents. They are also considered
as greener alternatives to conventional organic solvents in chemical
transformations due to their low vapor pressure and high boiling
points.1 Particularly, their property of reusability and tolerance for
flammability has prompted their use in chromatographic separation2
and extraction techniques.3 There has been a flurry of activity on the
synthesis, properties and applications of several ionic liquids in the
last decade culminating in several publications and reviews.4-9
Ionic liquids have the potential to play a significant role in
asymmetric synthesis possibly due to their difference in
thermodynamic and kinetic behavior, selectivity and process
performance as compared to conventional organic solvents. Although
synthesis, characterization and application of ionic liquids have
gained more popularity, literature available on chiral ionic liquids is
very limited.

64
Terpenes are an important part of the “chiral pool” and can be
used in the synthesis of chiral ionic liquids. However, the literature
available on the use of terpenes as a chiral pool in ionic liquids is
limited. Tosoni et al. have reported one of the first chiral ionic liquids
derived from citronellol, 1,3-dicitronellyl-1H- imidazolium bromide,10
Scheme-2.1.
Scheme: 2.1 Synthesis of Citronellyl bromide.
Chiral citronellol has been converted into the corresponding
citronellyl bromide followed by alkylation with 1-alkylimidazoles to
give chiral ionic liquid, Scheme-2.2.
NNR
NNR
Br
40 0C, 5 days
R = CH3, C4H9, C6H13, C12H25, C14H29, C18H37
Br3
4
2
Scheme: 2.2 Synthesis of CILs derived from (3R)-Citronellol.
Similarly alkylation of citronellyl bromide with pyridine gives
chiral pyridinium ionic liquid, Scheme–2.3.

65
Br
40 °C, 5 daysBr
N N
5
6
2
Scheme: 2.3 Synthesis of chiral pyridinium ionic liquids.
Another interesting C2-symmetric 1,3-dicitronellyl-1H-
imidazolium bromide was produced via deprotonation with
tetrabutylammonium hydroxide and then reacted with 2 equivalents
of the citronellyl bromide, Scheme-2.4.
Br
(2 eq.), RT, 3 days
Br
N
NH
1) Bu4NOH
NNH
72 8
Scheme: 2.4 Synthesis of CIL derived from (3R)-Citronellol.
Wang et al. investigated chiral pinene-based ionic liquids using
(1S)-α pinene.11 The cis amino alcohol obtained from α-pinene was
converted into chiral oxazoline, followed by alkylation with suitable
alkyl halide to furnish the chiral ionic liquid, Scheme-2.5. The
counter ion could be exchanged with different counter ions like PF6-
and BF4-.

66
OHO
KMNO4
Acetone
OHN
OHNH2OH
Ethanol
LAH
Ether
OHNH2
H
CH3
O
Cl
Dichloromethane
Saturated Na2CO3
OH
NH
O H
CH3N
OH
CH3
MeSO2Cl
Et3N
Dichloromethane
N
OH
CH3
C3H7
Br15(m.p.= 162 °C )
C3H7Br
N
OH
CH3
C3H7
PF6
HPF6
16(m.p.= 140 °C)
N
OH
CH3
C3H7
BF417(m.p.= 120 °C)
9 10 11 12
1314
Scheme: 2.5 Synthesis of CILs derived from -pinene.
Both the above mentioned examples used imidazolium,
pyridinium, oxazolium moieties to construct chiral ionic liquids.
Chirality in these ionic liquids can arise from the anion or from
the cation counterpart of the ionic liquids. Ionic liquids with chiral
anion counterparts have been reported in the literature.12a However,
application of the same in Diels-Alder reaction did not result in
enantioselectivity. Modifications to the cation counterpart of room
temperature chiral ionic liquids have been reported with ammonium,
pyridinium, oxazolium, thiazolium and imidazolium moieties.12 The

67
following work gives the detailed uses of terpene chiral pool for the
preparation of chiral ionic liquids through simple chemical
transformations.
Citronellal is an easily accessible, less expensive chiral starting
material with an aldehyde functionality, which can be conveniently
used for the several chemical transformations to design the desired
carbon framework. It is extensively used in the field of organic
synthesis and an important chiral precursor in the synthesis of
several natural products. The major advantage of citronellal as chiral
precursor is its commercial accessibility in both the chiral forms. The
low boiling point of citronellal is expected to provide a useful chiral
carbon chain to the ionic liquid under study with low melting points
or offer liquid state at room temperature, thus enabling them to open
new opportunities in the field of asymmetric synthesis.
The chiral ionic liquids can be envisaged from citronellal 18,
using a reductive amination strategy with dimethylamine or
diethylamine followed by treatment with an alkyl halide to give the
corresponding chiral ionic liquids 19a-e and 20a-e respectively, as
mentioned in the Figure 2.1.

68
CHO
H
CHO
H
a. X = I, R = Meb. X = I, R = Etc. X = Br, R = n-Prd. X = Br, R = i-Pre. X = Br, R = n-Bu
(S)-18 (S)-20a-e
(R)-18 (R)-20a-e
H
NR
H
NR
(S)-19a-e
(R)-19a-e
H
NR
H
NR
X X
X X
Figure: 2.1 Retrosynthesis of Citronellal-based chiral ionic liquids.

69
2.2 Results and Discussion
As mentioned in the retro synthetic analysis Figure-2.1, the
citronellal (S)-18 is treated with dimethylamine in the presence of
Raney-nickel in hydrogen environment to give the corresponding
amine (S)-21 in excellent yields. The dimethylamine 21, thus
obtained, is treated with different alkyl halides for eg., methyl iodide,
ethyl iodide, n-propyl bromide, iso-propyl bromide, n-butyl bromide,
etc., to give the corresponding quaternary ammonium salts (S)-19a-e
in good yields. Although the reaction proceeded smoothly with methyl
and ethyl halides, the reactivity with iso-propyl bromide was observed
to be sluggish with longer reaction time. Similarly, the other
commercially available isomer, (R)-citronellal, was also reacted with
dimethylamine in presence of Raney-nickel and hydrogen to give the
corresponding amine (R)-21, which on further treatment with the
alkyl halides furnished the desired chiral ammonium salts (R)-19a-e
in good yields, Scheme-2.6.

70
CHO
H H
N
H
NR X
CHO
H H
N
H
NR X
a. X = I, R = Meb. X = I, R = Etc. X = Br, R = n-Prd. X = Br, R = i-Pre. X = Br, R = n-Bu
(S)-18 (S)-21 (S)-19a-e
(R)-18 (R)-21 (R)-19a-e
a b
ba
a) Dimethylamine, MeOH, Raney-nickel, H2 balloon, RT- overnight.
b) Acetonitrile, R-X, RT- overnight.
Scheme: 2.6 Synthesis of chiral ionic liquids starting from citronellal and dimethylamine as amine source.
The scope of the reaction was extended to diethylamine to
understand the physical properties of the resulting ionic liquids with
citronellal. Thus, citronellal (S)-18 was treated with diethylamine in
the presence of Raney-nickel and hydrogen to give the corresponding
diethylamine (S)-22. The diethylamine derivative (S)-22 was further
treated with different alkyl halides for eg., methyl iodide, ethyl iodide,
n-propyl bromide, iso-propyl bromide, n-butyl bromide, etc., to give
the corresponding quaternary ammonium salts (S)-20a-e in good
yields. Similar observations were experienced in the reaction of iso-
propyl bromide with diethyl substrate, in which the reaction was
observed to be sluggish. Similarly, the chemistry was repeated with
(R)-citronellal and diethylamine in presence of Raney-nickel and

71
hydrogen to give the corresponding amine (R)-22, which on further
treatment with alkyl halides yielded the chiral ammonium salts (R)-
20a-e in good yields, Scheme-2.7. All the chiral ionic liquids prepared
using reductive amination with dimethylamine were found as liquid at
room temperature except (S) and (R)-19a. Similarly, all the new chiral
ionic liquids were prepared using reductive amination with
diethylamine were found to be liquids at room temperature, table-2.1.
CHO
H H
N
CHO
H H
N
a. X = I, R = Meb. X = I, R = Etc. X = Br, R = n-Prd. X = Br, R = i-Pre. X = Br, R = n-Bu
(S)-18 (S)-22 (S)-20a-e
(R)-18 (R)-22 (R)-20a-e
H
NR
H
NR
X
X
a
a
b
b
a) Diethylamine, MeOH, Raney-nickel, H2 balloon, RT- overnight
b) Acetonitrile, R-X, RT-overnight.
Scheme: 2.7 Synthesis of chiral ionic liquids starting from citronellal
and diethylamine as amine source.

72
Table: 2.1
Preparation of compounds (S)-19a-e, (R)-19a-e, (S)-20a-e, and (R)-20a-e
Compound
No. R X
SOR
(C= 1, MeOH) M.P.
(S)-19a Me I- 3.09 Yellow Solid
(S)-19b Et I- 2.96 Liquid
(S)-19c n-Pr Br- 3.96 Liquid
(S)-19d Iso-Pr Br- 3.55 Liquid
(S)-19e n-Bu Br- 3.28 Liquid
(R)-19a Me I- -1.26 Yellow Solid
(R)-19b Et I- -1.34 Liquid
(R)-19c n-Pr Br- -2.27 Liquid
(R)-19d Iso-Pr Br- -2.43 Liquid
(R)-19e n-Bu Br- -2.08 Liquid
(S)-20a Me I- 3.32 Liquid
(S)-20b Et I- 2.94 Liquid
(S)-20c n-Pr Br- 4.38 Liquid
(S)-20d Iso-Pr Br- 2.83 Liquid
(S)-20e n-Bu Br- 3.22 Liquid
(R)-20a Me I- -1.52 Liquid
(R)-20b Et I- -2.15 Liquid
(R)-20c n-Pr Br- -3.0 Liquid
(R)-20d Iso-Pr Br- -2.75 Liquid
(R)-20e n-Bu Br- -3.22 Liquid

73
Reductive amination product dimethylamine (21) was confirmed
from the 1H NMR study, absence of aldehyde proton at δ 9.7 ppm and
appearance of a new peak at δ 2.2 (S, 6H) corresponds to
dimethylamine protons, peak at δ 2.2 (t, 2H) corresponds to N-CH2.
The quaternization of 21 with methyl iodide product (19a) was
confirmed from the 1H NMR peak at δ 3.4-3.5 (S, 9H) which
corresponds to trimethylamine (N-(CH3)3), peak at δ 3.5-3.7 (m, 2H)
corresponds to N-CH2 protons as seen from 1H NMR study.
Reductive amination product diethylamine (22) was confirmed
from 1H NMR study. The aldehyde peak at δ 9.7 ppm disappeared in
the product, and appeared of a new peak at δ 2.5-2.6 (q, 4H)
corresponds to the methylene group of N-(CH2)-CH3)2 and a singlet at
δ 1.7 ppm (6H, 2-CH3 of N-(CH2-CH3)2. The other peak of the product
appeared at δ 2.3-2.4 (m, 2H) of N-CH2. The quaternization of 22 was
carried out using methyl iodide, product 20a was confirmed from 1H
NMR study. The peak at δ 3.5-3.6 (m, 4H) corresponded to N-(CH2-
CH3)2, while the peaks at δ 3.3-3.5 (dd, 2H) of N-CH2-CH2 and δ 3.21
(S, 3H) corresponded to N-CH3, confirming the product.

74
2.3 Conclusion
In summary, several new chiral ionic liquids were prepared
starting from both the chiral isomers of citronellal as the chiral
starting material and dialkylamine as the amine source. The Schiff
base thus obtained was reduced and further reacted with alkyl halides
containing different carbon chains. The strategy could be generalized
with other dialkylamines to design a chiral ionic liquid for any specific
use. Most of the ionic liquids isolated were liquids at room
temperature and amenable for the application in asymmetric
synthesis.

75
2.4 Experimental Section
All the chemicals and reagents of LR grade were used. 1H NMR
spectra were recorded on Varian Gemini 2000 model 400 MHz
instrument using TMS as an internal standard in CDCl3 (Chemical
shifts in ppm) and mass spectrum was recorded on HP 5989A
spectrometer. Optical Rotation of the compounds were recorded on
Polari meter (Jasco) in methanol (c=1).
General procedure for synthesis of compounds (21 & 22) :
To a stirred solution of compound (S) or (R)-18 (1.0 mmol), in
methanol (10 vol) was added appropriate alkylamine (1.5 mmol) and
Raney-nickel (30% w/v) at room temperature. Reaction was left to stir
for 20 hrs at room temperature in hydrogen environment, the catalyst
was filtered and the solvent was evaporated under reduced pressure.
The resulting crude (pale greenish liquid) was subjected to column
chromatography to give pure desired products (21& 22).
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (dimethyl) amine
(21) : To a stirred solution of (3S) or (3R)-3,7-dimethyl-6-octenal (18)
(5.0 g, 32.4 mmol) in methanol (50 mL) was added 40% aqueous
solution of dimethylamine (2.4 mL, 48.6 mmol) and Raney-nickel (30%
w/v) at room temperature. Reaction was left to stir for 20 hrs at room
temperature in hydrogen environment. After completion of reaction,
the catalyst was filtered and the solvent was evaporated under
reduced pressure. The resulting crude (pale greenish liquid) was
subjected to column chromatography using a mixture of ethyl acetate

76
and pet ether as an eluent to yield 4.2 g (70%) of the pure desired
product (3S) or (3R) -3,7-dimethyl-6-octenyl (dimethyl) amine (21).
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 2.2-2.3 (t, J=7.8 Hz,
2H), 2.0 (s, 6H), 1.9-2.1 (m, 2H), 1.7 (s, 3H), 1.6 (s, 3H), 1.4-1.6 (m,
1H), 1.2-1.4 (m, 2H), 1.1-1.2 (m, 2H), 0.8-0.9 (d, J=6.4 Hz, 3H); 13C
NMR (200 MHz, CDCl3) : δ 130.51, 124.59, 57.51 , 45.17, 45.17,
37.01, 34.47, 30.78, 25.37, 25.20, 19.35, 17.27.
Mass: m/z = 183
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (diethyl)amine
(22): To a stirred solution of (3S) or (3R)-3,7-dimethyl-6-octenal (18)
(5.0 g, 32.4 mmol) in methanol (50 mL) was added diethylamine (5.0
mL, 48.6 mmol) and Raney nickel (30% w/v) at room temperature.
The reaction was left to stir for 20 hrs at room temperature in
hydrogen environment. After completion of the reaction, the catalyst
was filtered and the solvent was evaporated under reduced pressure.
The resulting crude (pale greenish liquid) was subjected to column
chromatography using a mixture of ethyl acetate and pet ether as an
eluent to yield 5.1 g (75%) of the pure desired product (3S) or (3R)-3,7-
dimethyl-6-octenyl (diethyl) amine (22).
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 2.5-2.6 (q, 4H), 2.3-2.4
(m, 2H), 1.9-2.1 (m, 2H), 1.6-1.7 (s, 6H), 1.4-1.5 (m, 2H), 1.2-1.4 (m,
2H), 1.1-1.2 (m, 1H), 1.0 (t, J=6.8 Hz, 6H), 0.8 (d, J=6.4 Hz, 3H); 13C
NMR (200 MHz, CDCl3) : δ 130.51, 124.59, 50.43, 46.55, 36.98,
33.47, 30.77, 25.34, 25.21, 19.41, 17.24, 11.29.
Mass: m/z = 212

77
General procedure for synthesis of compounds (S) or (R)-19a-e and
20a-e:
To a stirred solution of compound (S) or (R)-3 (1.0 mmol) in
acetonitrile (10 vol), alkyl halide (1.2 mmol) was added at room
temperature. The reaction was allowed to stir for 12 hrs at room
temperature and the solvent was removed under reduced pressure,
followed by drying under high vacuum to afford the desired product.
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (trimethyl)
ammonium iodide (19a) : To a stirred solution of compound (S) or
(R)-21 (2.0 g, 10.9 mmol) in acetonitrile (20 mL), methyl iodide (2.0
mL,13.1 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure followed by further drying under
high vacuum to afford 3.2 g (90%) of the desired product as a yellow
solid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.5-3.7 (m, 2H), 3.4-3.5
(s, 9H), 1.9-2.1 (m, 2H), 1.7-1.8 (m, 1H), 1.6-1.7 (s, 6H), 1.5-1.6 (m,
2H), 1.2-1.4 (m, 2H), 0.9-1.0 (d, J=6.4 Hz, 3H); 13C NMR (200 MHz,
CDCl3) : δ 131.32, 123.56, 65.51, (3C, 53.43), 36.36, 29.91, 29.39,
25.32, 24.85, 18.99, 17.39.
Mass: m/z =198
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (ethyl) dimethyl
ammonium iodide (19b) : To a stirred solution of compound (S) or
(R)-21 (2.0 g, 10.9 mmol) in acetonitrile (20 mL), ethyl iodide (1.1 mL,
13.1 mmol) was added at room temperature. The reaction was

78
allowed to stir for 12 hrs at room temperature, and the solvent was
removed under reduced pressure followed by drying under high
vacuum to afford 3.3 g (90%) of the desired product as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.7-3.8 (q, 2H), 3.4-3.6
(m, 2H), 3.39 (s, 6H), 1.9-2.1 (m, 2H), 1.7-1.8 (m, 1H), 1.6-1.7 (s, 6H),
1.5-1.6 (m, 2H), 1.4 (t, J=7.2 Hz, 3H), 1.2-1.3 (m, 2H), 1.0 (d, J=6.4
Hz, 3H); 13C NMR (200MHz, CDCl3) : δ 131.19, 123.42, 62.11, 59.20,
50.65, 50.65, 36.19, 29.84, 28.83, 25.18, 24.72, 18.87, 17.26, 8.28.
Mass: m/z = 212
Preparation of (3S or 3R)-3,7-Dimethyl-6-octenyl (dimethyl)
propyl ammonium bromide (19c) : To a stirred solution of compound
(S) or (R)-21 (2.0 g, 10.9 mmol) in acetonitrile (20 mL), n-prppyl
bromide (1.2 mL, 13.1 mmol) was added at room temperature. The
reaction was allowed to stir for 12 hrs at room temperature and the
solvent was evaporated under reduced pressure followed by further
drying under high vacuum yielding 2.3 g (70%) of the desired product
as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.4-3.6 (m, 4H), 3.4 (s,
6H), 1.9-2.1 (m, 2H), 1.7-1.8 (m, 1H), 1.6-1.7 (s, 6H), 1.6 (m, 2H), 1.5-
1.6 (m, 2H), 1.2-1.4 (m, 2H), 1.1 (t, J=7.2 Hz, 3H), 0.9 (d, J=6.4 Hz,
3H); 13C NMR (200MHz, CDCl3) : δ 131.48, 123.63, 64.87, 62.45,
51.14, 36.45, 30.09, 29.10, 25.39, 24.94, 18.96, 17.42, 16.00, 10.40.
Mass: m/z = 226
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (isopropyl)
dimethyl ammonium bromide (19d) : To a stirred solution of

79
compound (S) or (R)-21 (2.0 g, 10.9 mmol) in acetonitrile (20 mL), iso-
propyl bromide (1.6 mL, 13.1 mmol) was added at room temperature.
The reaction was allowed to stir for 12 hrs at room temperature and
the solvent was removed under reduced pressure. The crude product
was purified by column chromatography to yield 1.3 g (40%) of the
desired product as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.7-4.0 (m, 1H), 3.4-3.6
(m, 2H), 3.3-3.4 (s, 6H), 1.9-2.0 (m, 2H), 1.7-1.8 (m, 1H), 1.6-1.7 (s,
6H), 1.4-1.5 (m, 2H), 1.2-1.4 (m, 2H), 1.0-1.1 (m, 6H), 0.9 (d, J=4.8
Hz, 3H); 13C NMR (200MHz, CDCl3) : δ 131.44, 123.63, 64.16, 61.51,
48.04, 36.42, 29.00, 28.87, 25.37, 24.94, 18.93, 17.41, 16.46.
Mass: m/z = 226
Preparation of butyl [(3S or 3R)-3,7-dimethyl-6-octenyl] dimethyl
ammonium bromide (19e) : To a stirred solution of compound (S) or
(R)-21 (2.0 g, 10.9 mmol) in acetonitrile (20 mL), n-butyl bromide (1.4
mL, 13.1 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure. The obtained crude was subjected
to column chromatography to yield 2.1 g (60%) of the desired product
as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.4-3.6 (m, 4H), 3.38 (s,
6H), 1.9-2.1 (m, 2H), 1.8-1.9 (m, 1H), 1.6-1.7 (s, 6H), 1.4-1.5 (m, 4H),
1.39 (m, 2H), 1.2-1.3 (m, 2H), 1.0 (t, J=7.2 Hz, 3H), 0.9 (d, J=6.4 Hz,
3H); 13C NMR (200MHz, CDCl3) : δ 131.67, 123.68, 63.42, 62.43,

80
51.23, 36.57, 30.19, 29.21, 25.50, 25.05, 24.44, 19.39, 19.10, 17.53,
13.52.
Mass: m/z = 240
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (diethyl) methyl
ammonium iodide (20a) : To a stirred solution of compound (S) or
(R)-22 (2.0 g, 9.5 mmol) in acetonitrile (20 mL), methyl iodide (1.8 mL,
11.4 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure followed by drying under high
vacuum to yield 3.0 g (90%) of the desired product as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.5-3.7 (m, 4H), 3.3-3.5
(m, 2H), 3.2 (s, 3H), 1.9-2.1 (m, 2H), 1.7 (m, 2H), 1.6 (s, 6H), 1.4-1.5
(m, 2H), 1.3-1.4 (t, J=7.4 Hz, 6H), 1.2-1.3 (m, 1H), 1.0 (d, J=6.4 Hz,
3H); 13C NMR (200MHz, CDCl3) : δ 130.93, 123.14, 58.73, 56.15,
47.54, 35.89, 29.62, 28.20, 24.93, 24.46, 18.62, 17.00, 7.75.
Mass: m/z = 226
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (triethyl)
ammonium iodide (20b) : To a stirred solution of compound (S) or
(R)-22 (2.0 g, 9.5 mmol) in acetonitrile (20 mL), ethyl iodide (0.9
mL,11.4 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure followed by drying under high
vacuum to yield 2.8 g (80%) of the desired product as a liquid.
1H NMR (400 MHz, CDCl3): δ 5.0-5.2 (m, 1H), 3.4-3.5 (q, 6H), 3.1-3.3
(m, 2H), 1.9-2.1 (m, 2H), 1.7-1.8 (m, 2H), 1.6-1.7 (s, 6H), 1.5-1.6 (m,

81
2H), 1.4 (t, J=7.2 Hz, 9H), 1.2-1.3 (m, 1H), 1.0 (d, J=6.0 Hz, 3H); 13C
NMR (200MHz, CDCl3): δ 129.97, 122.45, 54.62, 52.07, 35.05, 28.84,
27.07, 24.19, 23.71, 17.91, 16.26, 6.80.
Mass: m/z = 240
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (diethyl) propyl
ammonium bromide (20c) : To a stirred solution of compound (S) or
(R)-22 (2.0 g, 9.5 mmol) in acetonitrile (20 mL), n-propyl bromide (1.0
mL, 11.4 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure. The obtained crude was subjected
to column chromatography to yield 1.3 g (50%) of the desired product
as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.5-3.6 (q, 4H), 3.2-3.4
(m, 4H), 1.9-2.1 (m, 2H), 1.8-1.9 (m, 2H), 1.7-1.8 (m, 2H), 1.6-1.7 (s,
6H), 1.5-1.6 (m, 2H), 1.3-1.5 (t, J=7.0 Hz, 6H), 1.2-1.3 (m, 1H), 1.1 (t,
J=7.2 Hz, 3H), 1.0 (d, J=6.8 Hz, 3H); 13C NMR (200MHz, CDCl3) : δ
131.71, 123.59, 59.25, 56.40, 53.86, 36.47, 30.26, 28.47, 25.49,
25.06, 19.14, 17.53, 15.54, 10.71, 7.88.
Mass: m/z = 254
Preparation of (3S or 3R)-3,7-dimethyl-6-octenyl (diethyl)
isopropyl ammonium bromide (20d) : To a stirred solution of
compound (S) or (R)-22 (2.0 g, 9.5 mmol) in acetonitrile (20 mL), iso-
propyl bromide (1.1 mL, 11.4 mmol) was added at room temperature.
The reaction was allowed to stir for 12 hrs at room temperature and
the solvent was removed under reduced pressure. The obtained crude

82
was subjected to column chromatography to yield 0.8 g (25%) of the
desired product as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.5 (q, 4H), 3.2-3.3 (m,
2H), 2.6-2.7 (m, 1H), 1.8-2.2 (m, 6H), 1.6-1.7 (s, 6H), 1.4 (t, J=7.2 Hz,
3H), 1.2 (t, J=7.2 Hz, 3H), 1.0 (d, J=6.4 Hz, 3H), 0.9 (d, J=6.4 Hz, 6H).
Mass: m/z = 254
Preparation of Butyl [(3S or 3R)-3,7-dimethyl-6-octenyl] diethyl
ammonium bromide (20e) : To a stirred solution of compound (S) or
(R)-22 (2.0 g, 9.5 mmol) in acetonitrile (20 mL), n-butyl bromide (1.2
mL, 11.4 mmol) was added at room temperature. The reaction was
allowed to stir for 12 hrs at room temperature and the solvent was
removed under reduced pressure. The obtained crude was subjected
to column chromatography to yield 1.6 g (50%) of the desired product
as a liquid.
1H NMR (400 MHz, CDCl3) : δ 5.0-5.2 (m, 1H), 3.5-3.6 (q, 4H), 3.2-3.4
(m, 2H), 3.1 (t, J=7.2 Hz, 2H), 1.9-2.1 (m, 2H), 1.7-1.8 (m, 6H), 1.6-1.7
(s, 6H), 1.5 (m, 2H), 1.3-1.4 (t, J=7.2 Hz, 6H), 1.2-1.3 (m, 1H), 1.1 (t,
J=7.4 Hz, 3H), 0.9 (d, J=6.8 Hz, 3H); 13C NMR (200MHz, CDCl3) : δ
131.50, 123.45, 57.38, 56.18, 53.64, 36.32, 30.09, 28.28, 25.32,
24.89, 23.59, 19.36, 18.99, 17.36, 13.31, 7.71.
Mass: m/z = 268

83
2.5 Spectral data
1H NMR spectrum (400MHz, CDCl3) of compound 18:
1H NMR spectrum (400MHz, CDCl3) of compound 21:
CHO
H
(R)-18
H
N
(R)-21

84
13C NMR spectrum (400MHz, CDCl3) of compound 21:
Mass spectrum of compound 21:
H
N
(R)-21
H
N
(R)-21

85
1H NMR spectrum (400MHz, CDCl3) of compound 19a:
13C NMR spectrum (400MHz, CDCl3) of compound 19a:

86
DEPT spectrum of compound 19a:
Mass spectrum of compound 19a:

87
1H NMR spectrum (400MHz, CDCl3) of compound 19b:
13C NMR spectrum (400MHz, CDCl3) of compound 19b:

88
DEPT spectrum (400MHz, CDCl3) of compound 19b:
Mass spectrum of compound 19b:

89
1H NMR spectrum (400MHz, CDCl3) of compound 19c:
DEPT spectrum (400MHz, CDCl3) of compound 19c:

90
13C NMR spectrum of compound 19c:
Mass spectrum of compound 19c:

91
1H NMR spectrum (400MHz, CDCl3) of compound 19d:
DEPT spectrum of compound 19d:

92
Mass Spectrum of compound 19d:
1H NMR spectrum of compound 19e:
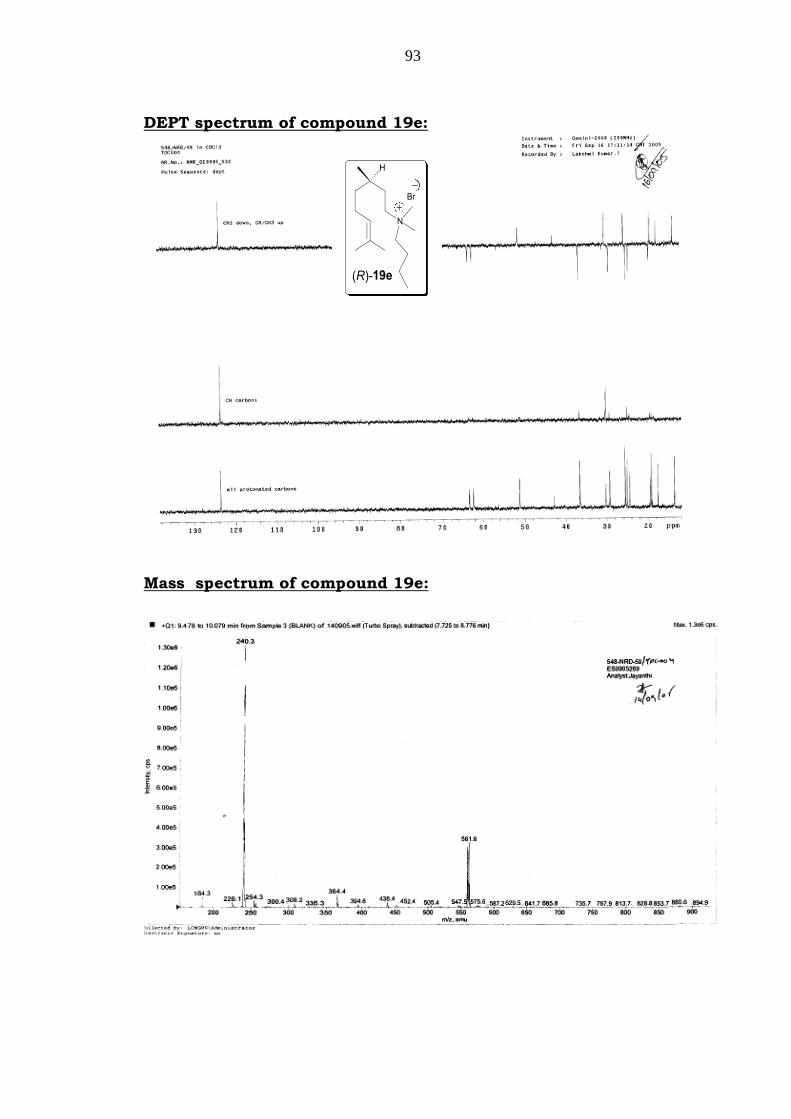
93
DEPT spectrum of compound 19e:
Mass spectrum of compound 19e:

94
13C NMR spectrum of compound 19e:
1H NMR spectrum of compound 22:

95
DEPT spectrum of compound 22:
13C NMR spectrum of compound 22:

96
1H NMR spectrum of compound 20a:
13C NMR spectrum of compound 20a:

97
Mass spectrum of compound 20a:
1H NMR spectrum of compound 20b:

98
DEPT spectrum of compound 20b:
13C NMR spectrum of compound 20b:

99
Mass spectrum of compound 20b:
1H NMR spectrum of compound 20c:
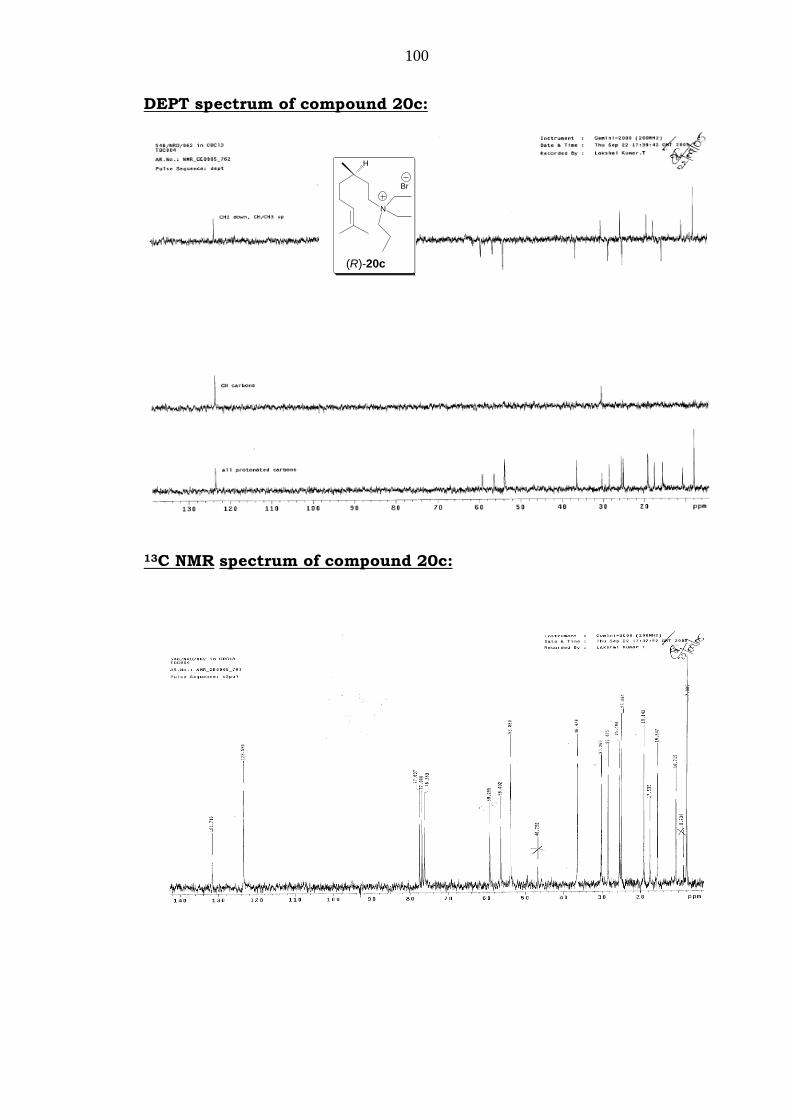
100
DEPT spectrum of compound 20c:
13C NMR spectrum of compound 20c:
H
N
Br
(R)-20c

101
1H NMR spectrum of compound 20d:
Mass spectrum of compound 20d:

102
1H NMR spectrum of compound 20e:
DEPT spectrum of compound 20e:

103
13C NMR spectrum of compound 20e:
Mass spectrum of compound 20e:

104
2.6 References
1. (a) Welton, T. Room Temperature Ionic Liquids Solvents for
Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071-2083; (b)
DuPont, J.; de Souza, R. F.; Suarez, P. A. Z. Ionic Liquid (Molten
Salt) Phase Organometallic Catalysis. Chem. Rev. 2002, 102,
3667-3692; (c) Olivier-Bourbigou, H.; Magna, L. Ionic Liquids
Perspectives for Organic and Catalytic Reactions. J. Mol. Cat. A:
Chem. 2002, 3484, 1-19; (d) Sheldon, R. Catalytic Reactions in
Ionic Liquids. Chem. Commun. 2001, 2399-2407; (e)
Wasserscheid, P.; Keim, W. Ionic Liquids-New Solutions for
Transition Metal Catalysis. Angew. Chem., Int. Ed. Engl. 2000,
39, 3772-3789; (f) Gordon, C. M. New Developments in Catalysis
using Ionic Liquids. Appl. Catal. A: Gen. 2001, 222, 101-117; (g)
Dyson, P. J. pH-Dependent partitioning in room temperature
ionic liquids provides a link to traditional solvent extraction
behavior. Transition Met. Chem. 2002, 27, 353-358; (h)
Freemantle, M. Green Solution for Ionic Liquids. C&E News,
August 24, 1998, 32; May 15, 2000, 37-50; January 1, 1999, 9;
(i) Carmichael, H. Chem. Brit., January 2000, 36; (j) Holbrey J.
D.; Seddon K.R. , Ionic Liquids, Clean Prod. Processes. 1990, 1,
223-226.
2. Visser, A. E.; Swatlowski, R. P.; Rogers, R. D. pH-Dependent
partitioning in room temperature ionic liquids provides a link to

105
traditional solvent extraction behavior. Green Chemistry. 2000, 2
(1), 1.
3. Armstrong, D. W.; He, L.; Liu, Y. S. Examination of Ionic Liquids
and Their Interaction with Molecules, When Used as Stationary
Phases in Gas Chromatography. Anal. Che. 1999, 71, 3873-3876.
4. (a) Adams, C. J.; Earle, M. J.; Roberts, G.; Seddon, K. R. Friedel–
Crafts reactions in room temperature ionic liquids. Chem.
Commun. 1998, 2097-2098; (b) Stark, A.; Maclean, B. L.; Singer,
R. D. 1-Ethyl-3-methylimidazolium halogenoaluminate ionic
liquids as solvents for Friedel–Crafts acylation reactions of
ferrocene. J. Chem. Soc., Dalton Tran. 1999, 63-66.
5. (a) Carmichael, A. J.; Earle, M. J.; Holbrey, J. D.; McCormac, P.
B.; Seddon, K. R. The Heck Reaction in Ionic Liquids: A
Multiphasic Catalyst System. Org. Lett. 1999, 1, 997-1000; (b)
Calo, V.; Nacci, A.; Lopez, L.; Mannarini, N. Heck reaction in ionic
liquids catalyzed by a Pd–benzothiazole carbene complex.
Tetrahedron Lett. 2000, 41, 8973-8976.
6. (a) Fischer, T.; Sethi, A.; Welton, T.; Woolf, J. Diels-Alder
reactions in room-temperature ionic liquids. Tetrahedron Lett.
1999, 40, 793-796; (b) Lee, C. W. Diels-Alder reactions in
chloroaluminate ionic liquids: acceleration and selectivity
enhancement. Tetrahedron Lett. 1999, 40, 2461-2464; (c) Ludley,

106
P.; Karodia, N. Phosphonium tosylates as solvents for the Diels–
Alder reaction. Tetrahedron Lett. 2001, 42, 2011-2014.
7. (a) Ren, R. X.; Zueva, L. D.; Ou, W. Formation of ε-caprolactam
via catalytic Beckmann rearrangement using P2O5 in ionic
liquids. Tetrahedron Lett. 2001, 42, 8441-8443; (b). Peng, J.;
Deng, Y. Catalytic Beckmann rearrangement of ketoximes in ionic
liquids. Tetrahedron Lett. 2001, 42, 403-405.
8. Peng, J.; Deng, Y. Ionic liquids catalyzed Biginelli reaction under
solvent-free conditions. Tetrahedron Lett. 2001, 42, 5917-5919.
9. Potdar, M. K.; Mohile, S. S.; Salunkhe, M. M. Coumarin
syntheses via Pechmann condensation in Lewis acidic
chloroaluminate ionic liquid. Tetrahedron Lett. 2001, 42, 9285-
9287.
10. Tosoni, M.; Laschat, S.; Baro, A. Synthesis of novel chiral ionic
liquids and their phase behavior in mixtures with smectic and
nematic liquid crystals. Helv. Chim. Acta. 2004, 87, 2742 – 2749.
11. Wang, Y. Synthesis and application of novel chiral ionic liquids
derived from a-pinene. M.Sc. thesis. Newark, NJ: New Jersey
Institute of Technology, Department Chemistry and
Environmental Science, 2003.
12. (a) Wasserscheid, P.; Bosmann, A.; Bolm, C. Synthesis and
properties of ionic liquids derived from the chiral pool. Chem.
Commun. 2002, 200-201; (b) Thanh, G. V.; Pegot, B.; Loupy, A.

107
Solvent-Free Microwave-Assisted Preparation of Chiral Ionic
Liquids from (-)-N-Methylephedrine. Eur. J. Org. Chem. 2004,
1112-1116; (c) Pernak, J., Feder-Kubis, J. Synthesis and Properties
of Chiral Ammonium-Based Ionic Liquids. Chem. Eur. J. 2005, 11,
4441-4449; (d) Baudequin, C.; Baudoux, J.; Levillaian, J.; Cahard,
D.; Gaumont, A. C.; Plaquevent, J. C. Ionic liquids and chirality:
opportunities and challenges. Tetrahedron: Asymmetry. 2003, 14,
3081-3093; (e) Haramoto, Y.; Miyashita, T.; Nanasawa, M.; Aoki,
Y.; Nohira, H. Liquid crystal properties of new ionic liquid crystal
compounds having a 1,3-dioxane ring. Liq. Cryst. 2002, 29, 87; (f)
Baudoux, J.; Judeinstein, P.; Cahard, D.; Plaquevent, J. C. Design
and synthesis of novel ionic liquid/liquid crystals (IL2Cs) with axial
chirality. Tetrahedron Lett. 2005, 46, 1137-1140; (g) Levillain, J.;
Dubant, G.; Abrunhosa, I.; Gulea, M.; Gaumont, A. C. Synthesis
and properties of thiazoline based ionic liquids derived from the
chiral pool. Chem. Commun. 2003, 2914-2915; (h) Bukuo Ni, Allan
D Headley, Guigen. Li. Design and Synthesis of C-2 Substituted
Chiral Imidazolium Ionic Liquids from Amino Acid Derivatives. J.
Org. Chem. 2005, 70, 10600-10602.





























