Embed Size (px)
Citation preview

Introduc)on to Bioinforma)cs of next-‐genera)on sequencing
Sequence acquisi)on and processing; genome mapping and alignment manipula)on
Ruslan Sadreyev
Director of Bioinformatics Department of Molecular Biology, MGH Department of Pathology, MGH, HMS
Next-Generation Sequencing Core and Bioinformatics group at Molecular Biology
5/18/17 NGS analysis 1
Next-‐genera)on sequencing core Website: nextgen.mgh.harvard.edu Email: [email protected]
Bioinforma)cs team Website: molbio.mgh.harvard.edu/department/bioinformaDcs Email: [email protected]
Bring your DNA/RNA!
Bring your data!
2

Founding members of MGH Bioinformatics Consortium
• AnalyDc and TranslaDonal GeneDcs Unit • Biomedical InformaDcs Core • Center for Integrated DiagnosDcs BioinformaDcs Group • BioinformaDcs at the MGH Cancer Center and Department of Pathology • Genomics and Technology Core • ITN/PHS InformaDon Systems/Immune Tolerance Network • BioinformaDcs group at the Department of Molecular Biology • MGH next-‐generaDon sequencing core
5/18/17 NGS analysis 1 [email protected] 3

Missions of Bioinformatics Consortium
Support and collaboration Develop and support the informaDcs component of fundamental, translaDonal, and clinical research projects
Consulting
Plan experimental design and execuDon of data generaDon and analysis; advise on best pracDces
Educational outreach
EducaDon in general BioinformaDcs concepts and methods; helping researchers think about their data in quanDtaDvely rigorous terms
Training Teach hands-‐on computaDonal skills in staDsDcs and BioinformaDcs workflows
5/18/17 NGS analysis 1 [email protected] 4

About this mini-course
5/18/17 NGS analysis 1 [email protected]
You can (a) get oriented in basic NGS BioinformaDcs concepts, approaches, and tools. (b) start asking right quesDons about your data. You cannot get (a) very deep coverage of a specific area/method/applicaDon (b) hands-‐on computaDonal experience
Two seminars: Sequence acquisi)on and processing; genome mapping and alignment manipula)on
Thurs May 18 at 2 pm Specific NGS applica)ons and public datasets
Thurs May 25 at 2 pm
5

I used slides/images/material by …
NGS analysis 1 [email protected]
BF Francis Ouelleae Istvan Albert Mik Black & ChrisDn Print Thomas Keane Illumina
5/18/17 6
NGS analysis 1 [email protected]
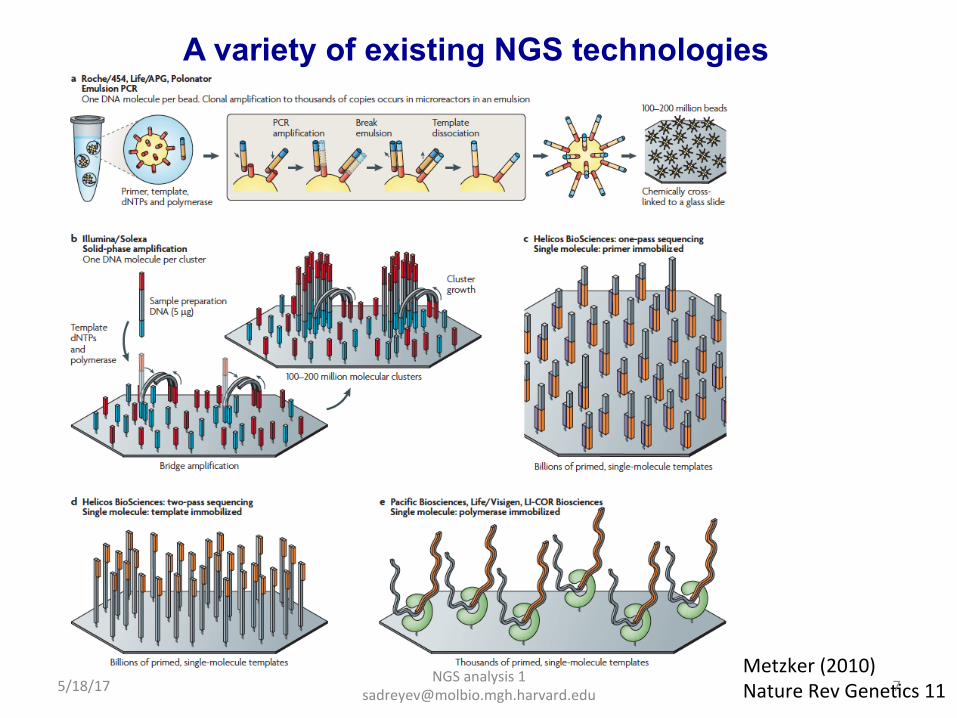
A variety of existing NGS technologies
Metzker (2010) Nature Rev GeneDcs 11 5/18/17 7
NGS analysis 1 [email protected]
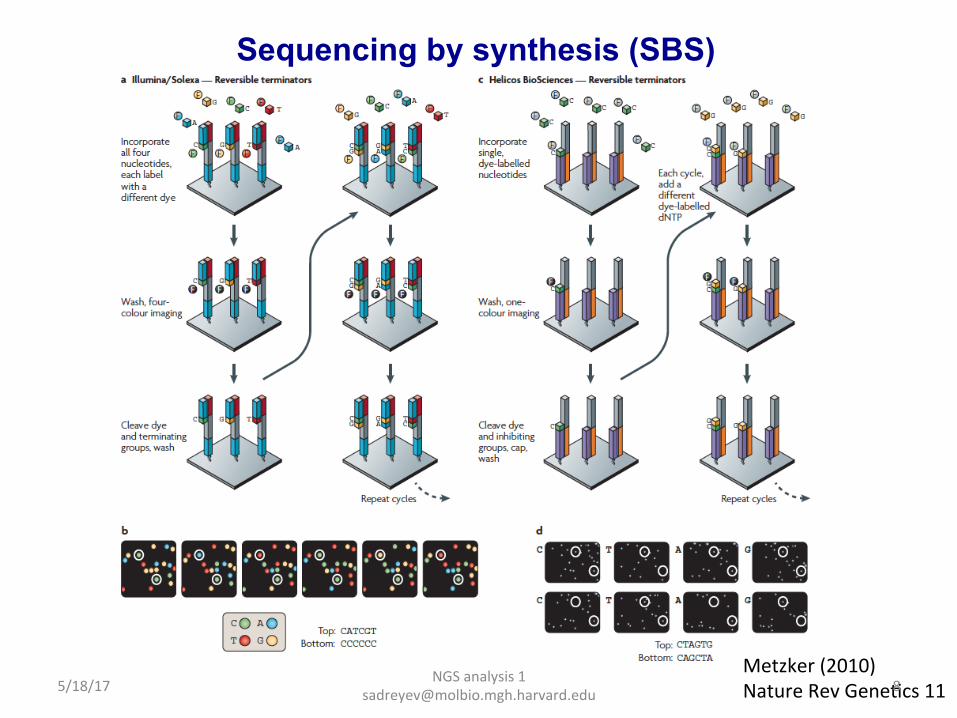
Sequencing by synthesis (SBS)
Metzker (2010) Nature Rev GeneDcs 11 5/18/17 8
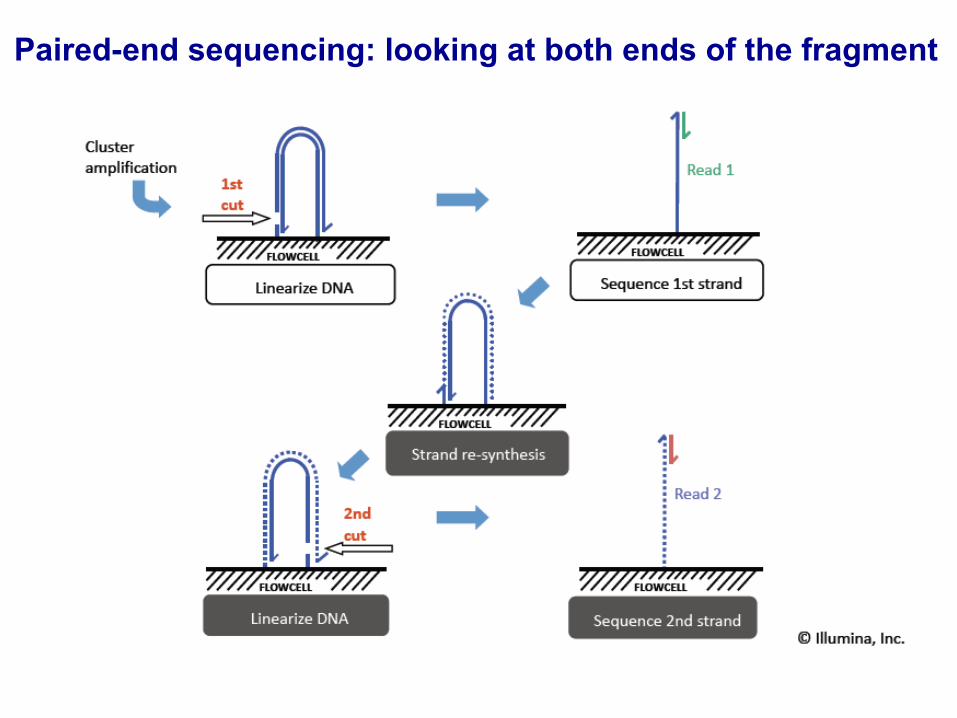
Paired-end sequencing: looking at both ends of the fragment
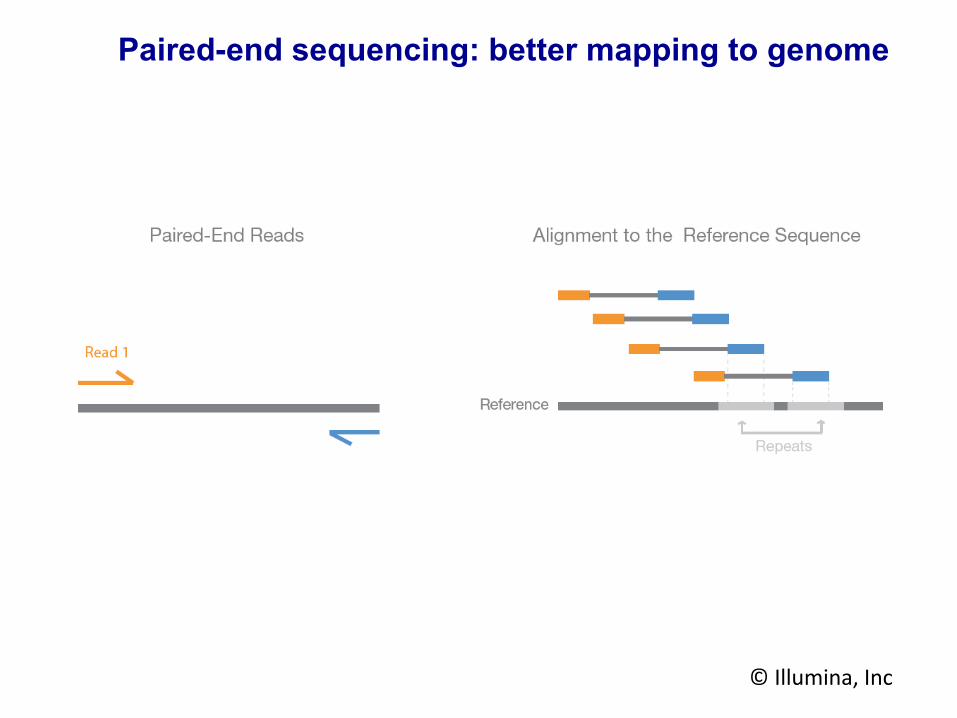
Paired-end sequencing: better mapping to genome
© Illumina, Inc
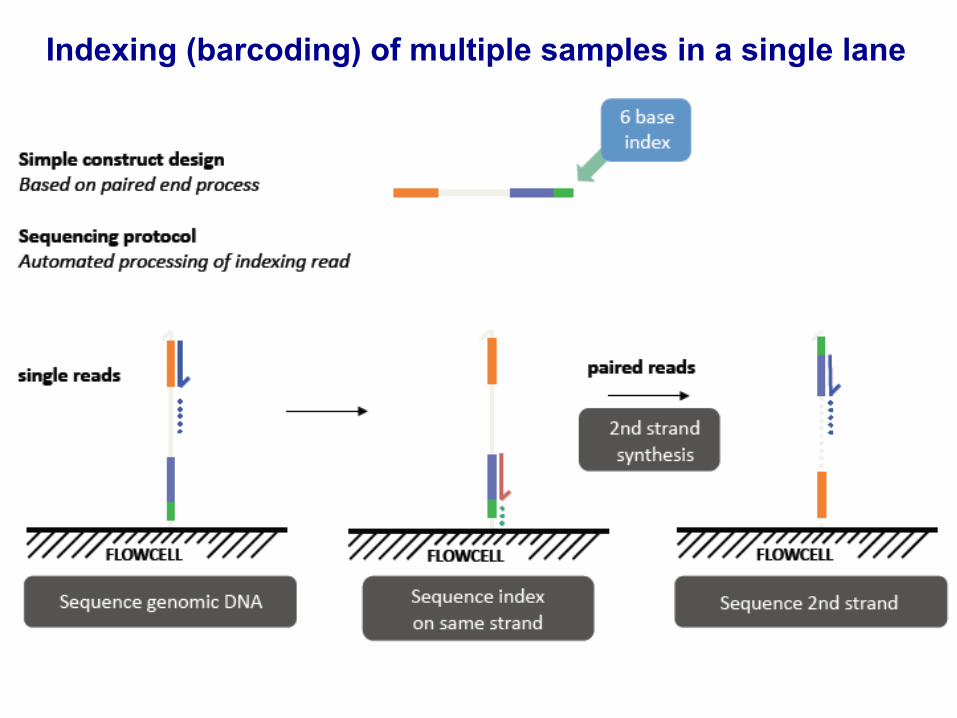
Indexing (barcoding) of multiple samples in a single lane

NGS analysis 1 [email protected]
Major NGS applications: examples
• Whole Genome Shotgun Sequencing (WGS) • Targeted/exome sequencing • RNA-‐seq • ChiP-‐seq • Metagenomics (targeted region/ whole genome sequencing) • MANY more
5/18/17 12
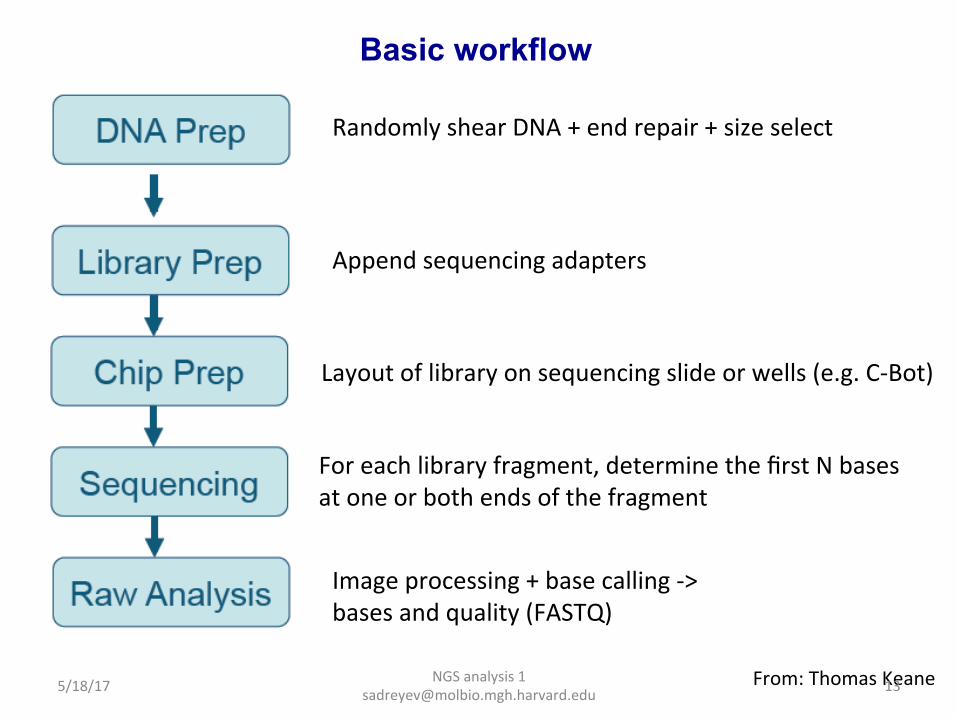
NGS analysis 1 [email protected]
Basic workflow
From: Thomas Keane
Randomly shear DNA + end repair + size select
Append sequencing adapters
Layout of library on sequencing slide or wells (e.g. C-‐Bot)
For each library fragment, determine the first N bases at one or both ends of the fragment
Image processing + base calling -‐> bases and quality (FASTQ)
5/18/17 13

Illumina HiSeq
From hap://www.qbi.uq.edu.au Our current output: 8 lanes, 150-‐200 M reads per lane

Illumina MiSeq: desktop device
Our current output: 1 lane, 10-‐20 M reads per lane
• Fast • Flexible (can do longer reads, up to 500 bp) • ~10x fewer reads than HiSeq • Cheaper per run (but not per read)
ApplicaDons: • Amplicon sequencing • QC before large-‐scale runs • Bacterial genomes • …..
www.illumina.com

NGS analysis 1 [email protected]
Base quality calibration
Phred score: measure of p = Prob(erroneous base call): -‐10log10(p) Q10 = 1 in 10 chance of incorrect base call Q20 = 1 in 100 chance of incorrect base call Q30 = 1 in 1000 chance of incorrect base call Rule of thumb: not useful data If <Q20 Standard assessment: proporDon of bases with score ≥ Q30 Highest Phred scores are typically around Q35-‐40
5/18/17 17

NGS analysis 1 [email protected]
FASTQ format for NGS sequences
hap://en.wikipedia.org/wiki/FASTQ_format 5/18/17 18

NGS analysis 1 [email protected]
Quality control of raw sequences: FASTQC hap://www.bioinformaDcs.babraham.ac.uk/projects/fastqc
5/18/17 19
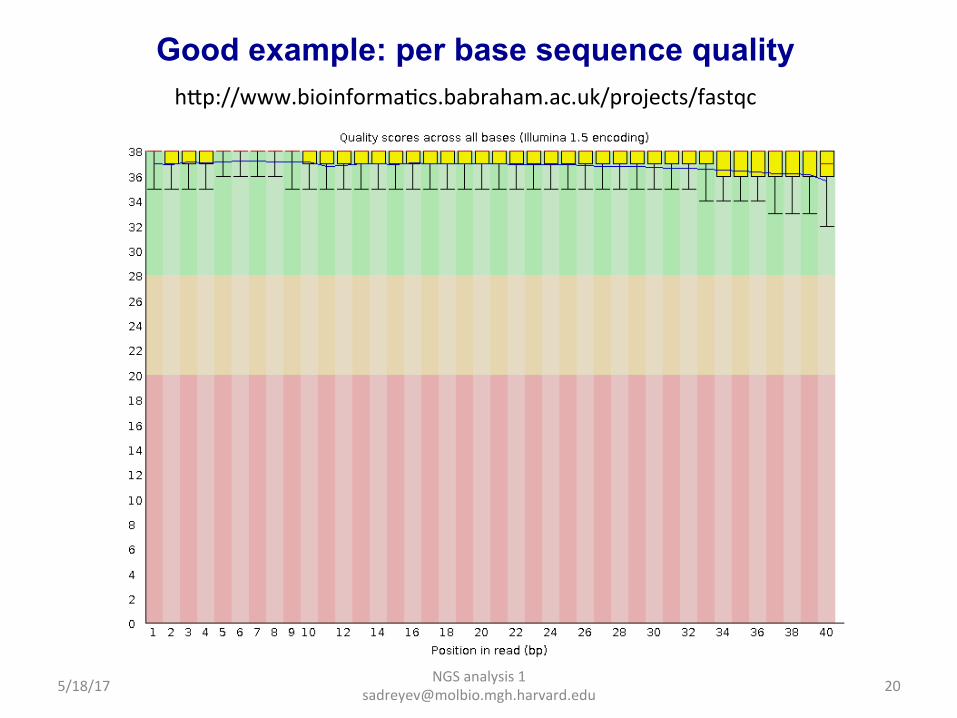
NGS analysis 1 [email protected]
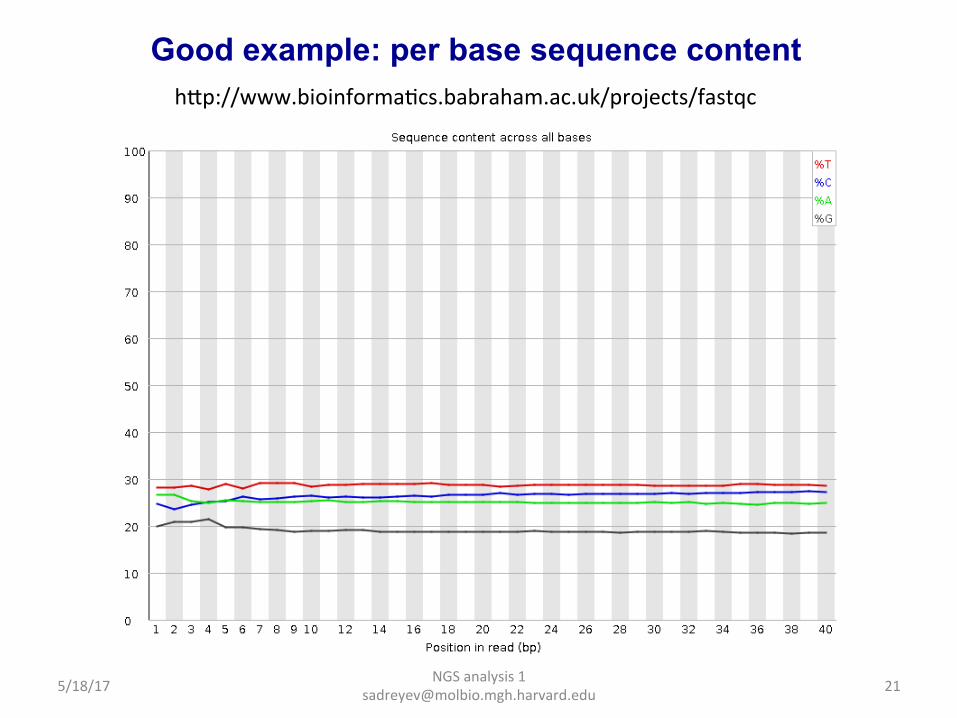
Good example: per base sequence quality hap://www.bioinformaDcs.babraham.ac.uk/projects/fastqc
5/18/17 20
NGS analysis 1 [email protected]
hap://www.bioinformaDcs.babraham.ac.uk/projects/fastqc
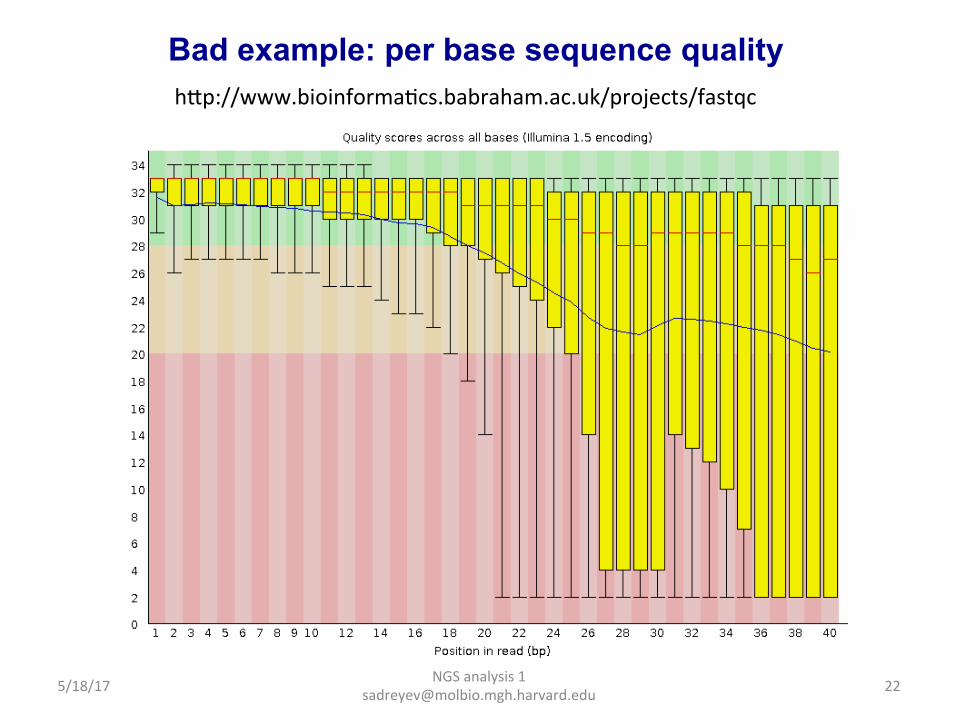
Good example: per base sequence content
5/18/17 21
NGS analysis 1 [email protected]
hap://www.bioinformaDcs.babraham.ac.uk/projects/fastqc
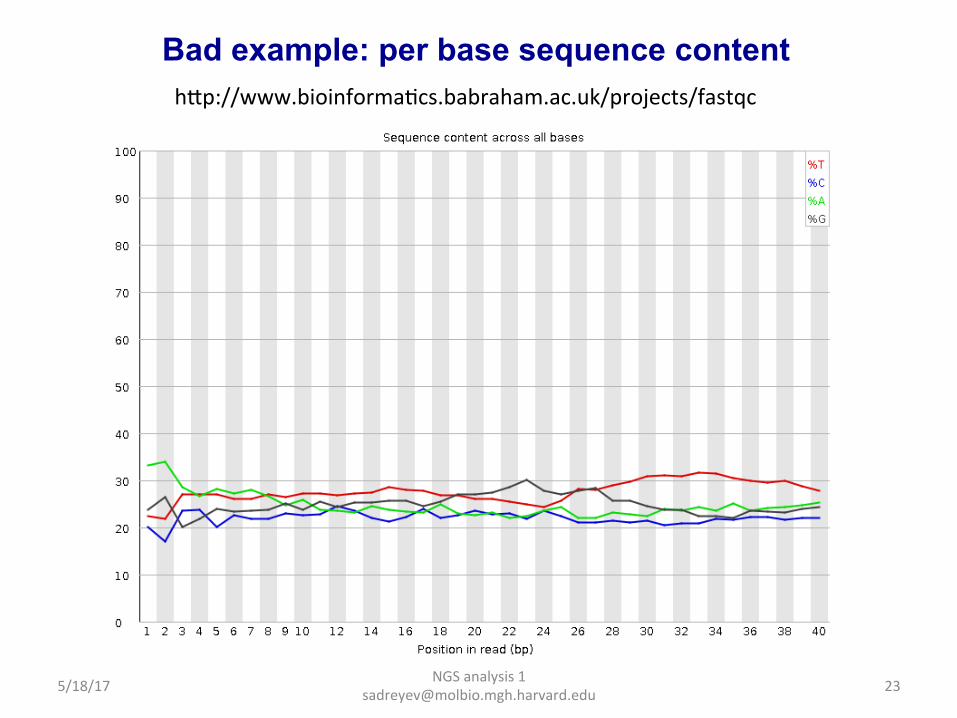
Bad example: per base sequence quality
5/18/17 22
NGS analysis 1 [email protected]
hap://www.bioinformaDcs.babraham.ac.uk/projects/fastqc
Bad example: per base sequence content
5/18/17 23
NGS analysis 1 [email protected]
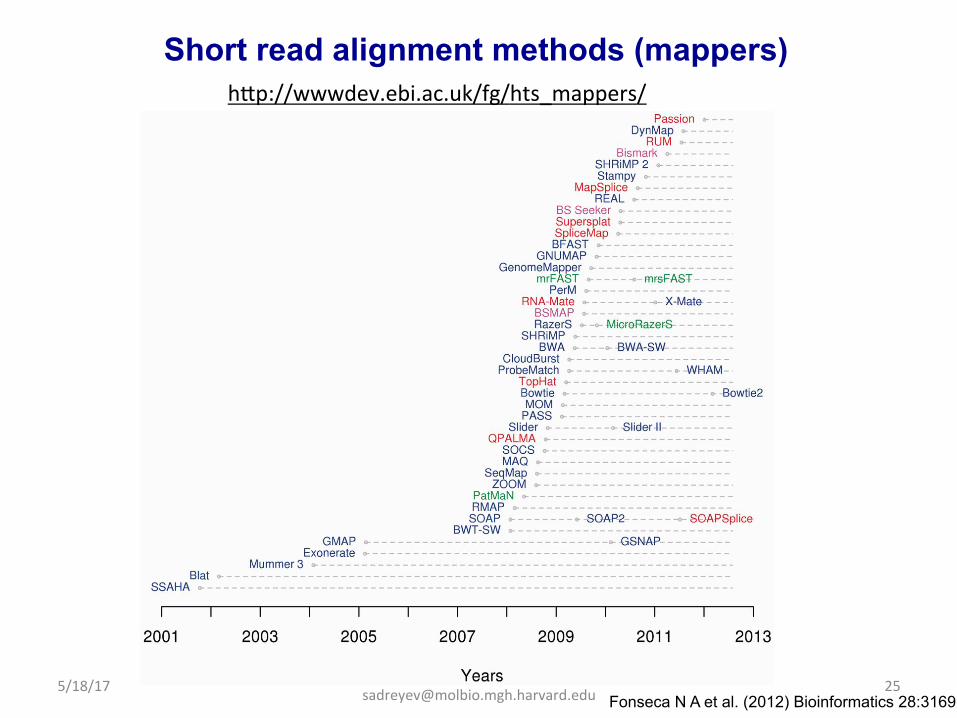
Short read alignment methods (mappers) hap://wwwdev.ebi.ac.uk/fg/hts_mappers/
Fonseca N A et al. (2012) Bioinformatics 28:3169 5/18/17 25

NGS analysis 1 [email protected]
Two popular mappers • BowDe: hap://bowDe-‐bio.sourceforge.net • BWA: hap://bio-‐bwa.sourceforge.net Both are based on Burrows-‐Wheeler Transform (BWT)
Examples of common fast mappers
5/18/17 26
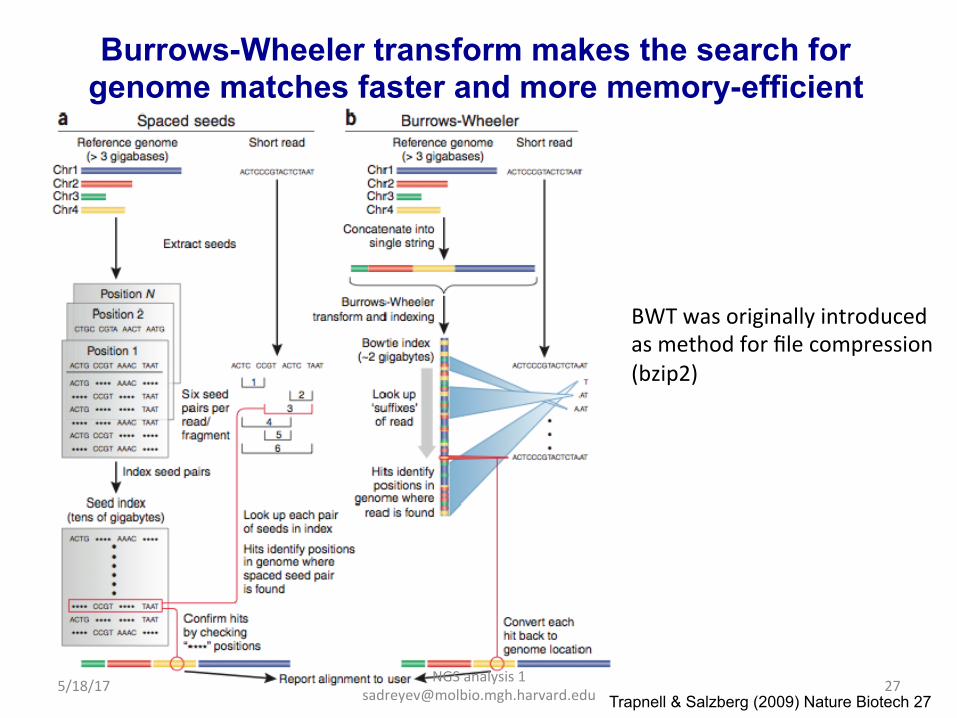
NGS analysis 1 [email protected] Trapnell & Salzberg (2009) Nature Biotech 27
Burrows-Wheeler transform makes the search for genome matches faster and more memory-efficient
BWT was originally introduced as method for file compression (bzip2)
5/18/17 27

NGS analysis 1 [email protected]
Using BWA: example
• Create index for the genome: bwa index [-a bwtsw|div|is] [-c] <in.fasta>• -‐a STR BWT construc)on algorithm: bwtsw or is • bwtsw for human size genome, is for smaller genomes Create index for reads: bwa aln [options] <prefix> <in.fq>• Align each single end fastq file individually • Various op)ons to change the alignment parameters/scoring matrix/seed length
• Using sai files produced by aln step, produce alignment For paired-‐end reads: bwa sampe [options] <prefix> <in1.sai> <in2.sai> <in1.fq> <in2.fq>
For unpaired reads: bwa samse [-n max_occ] <prefix> <in.sai> <in.fq>
5/18/17 28

NGS analysis 1 [email protected]
Not all reads are alignable
Sources of mismatches: 1. Sequencer miscalls 2. Actual differences between sample and reference: (a) Genome variaDon (not the reference genome) ; (b) ContaminaDon (adapters, primers, different biological
species) … Typical good mappability rate > 70%-‐80%
5/18/17 29

NGS analysis 1 [email protected]
SAM/BAM formats
5/18/17 30
• Recent addition: CRAM is a more compact and efficient binary version of SAM

NGS analysis 1 [email protected]
SAM format information at SAMtools website hap://samtools.sourceforge.net
5/18/17 32
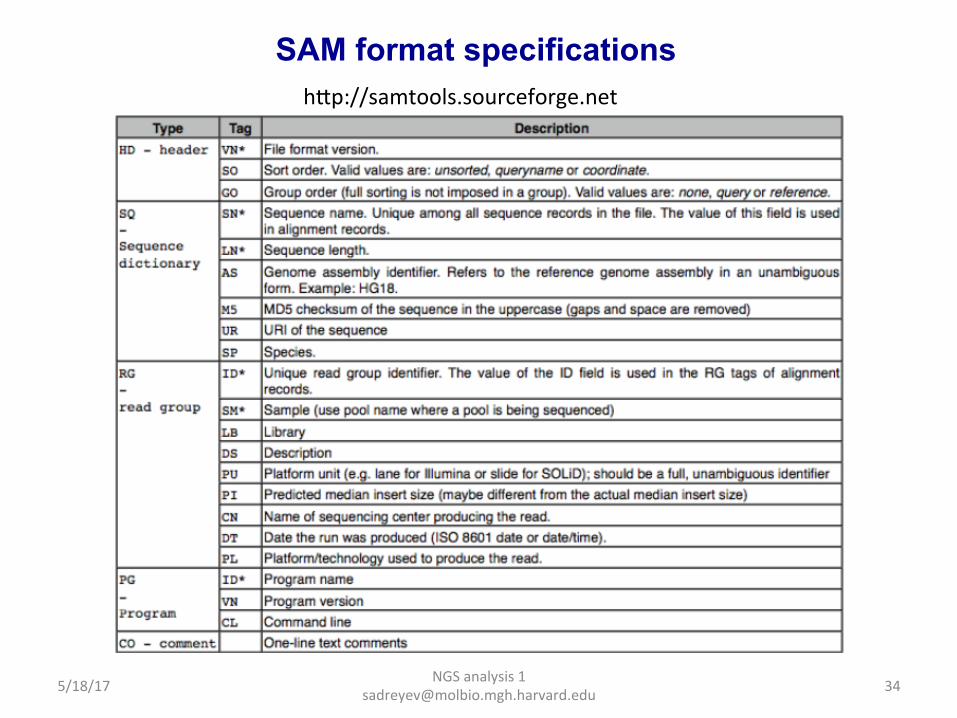
NGS analysis 1 [email protected]
SAM format specifications hap://samtools.sourceforge.net
5/18/17 34

NGS analysis 1 [email protected]
Tools to work with SAM/BAM alignments
Samtools -‐ Sanger/C (hap://samtools.sourceforge.net) Convert SAM <-‐> BAM Sort, index, BAM files Flagstat – summary of the mapping flags Merge mulDple BAM files Rmdup – remove PCR duplicates from the library preparaDon Picard -‐ Broad InsDtute/Java (hap://picard.sourceforge.net) MarkDuplicates, CollectAlignmentSummaryMetrics, CreateSequenceDicDonary, SamToFastq, MeanQualityByCycle, FixMateInformaDon……. Bio-‐SamTool – Perl (hap://search.cpan.org/~lds/Bio-‐SamTools/) Pysam – Python (hap://code.google.com/p/pysam/) …
5/18/17 35

NGS analysis 1 [email protected]
Example: generate and manipulate alignment: commands Unix/Linux/MacOS…
hap://manuals.bioinformaDcs.ucr.edu/home/ ht-‐seq#TOC-‐Rsamtools
# extract specific region of genomesamtools view –b output.sorted.bam chr1:100000-110000 > myregion.bam
5/18/17 37

NGS analysis 1 [email protected]
Viewing data in a rich context on the web: UCSC Genome Browser
5/18/17 39
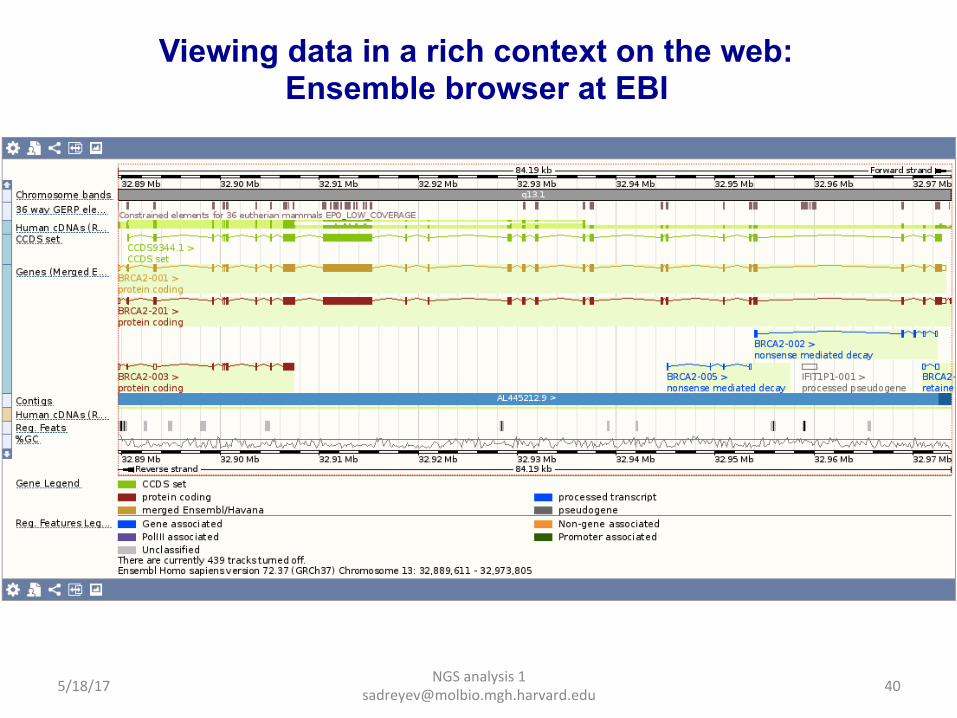
NGS analysis 1 [email protected]
Viewing data in a rich context on the web: Ensemble browser at EBI
5/18/17 40

NGS analysis 1 [email protected]
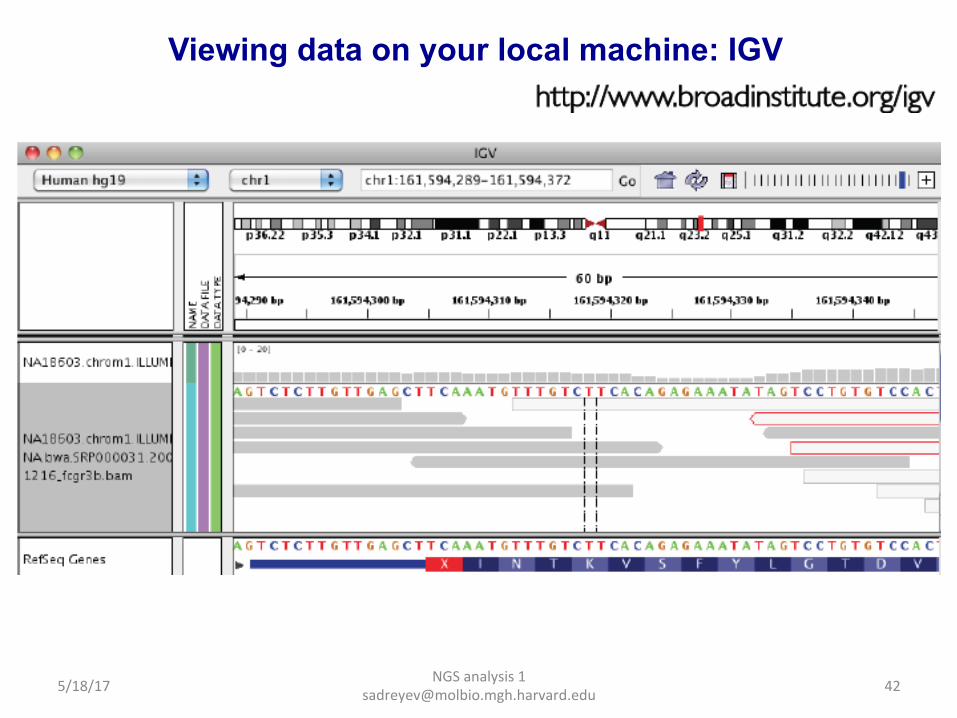
Viewing data on your local machine: IGV (Integrative Genomics Viewer)
5/18/17 41
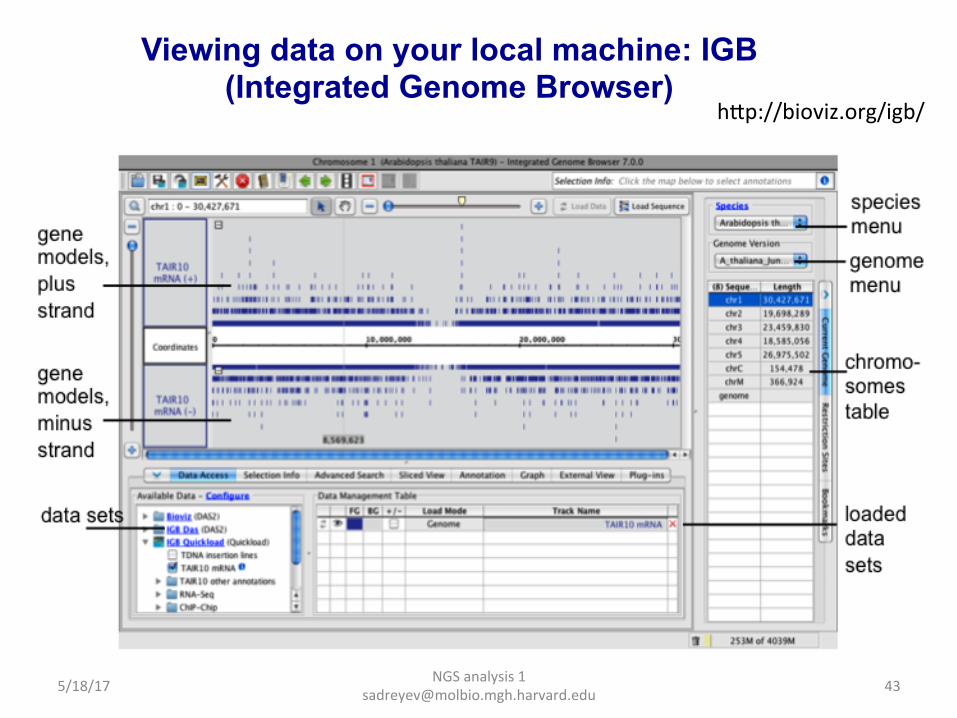
NGS analysis 1 [email protected]
Viewing data on your local machine: IGB (Integrated Genome Browser)
hap://bioviz.org/igb/
5/18/17 43
NGS analysis 1 [email protected]
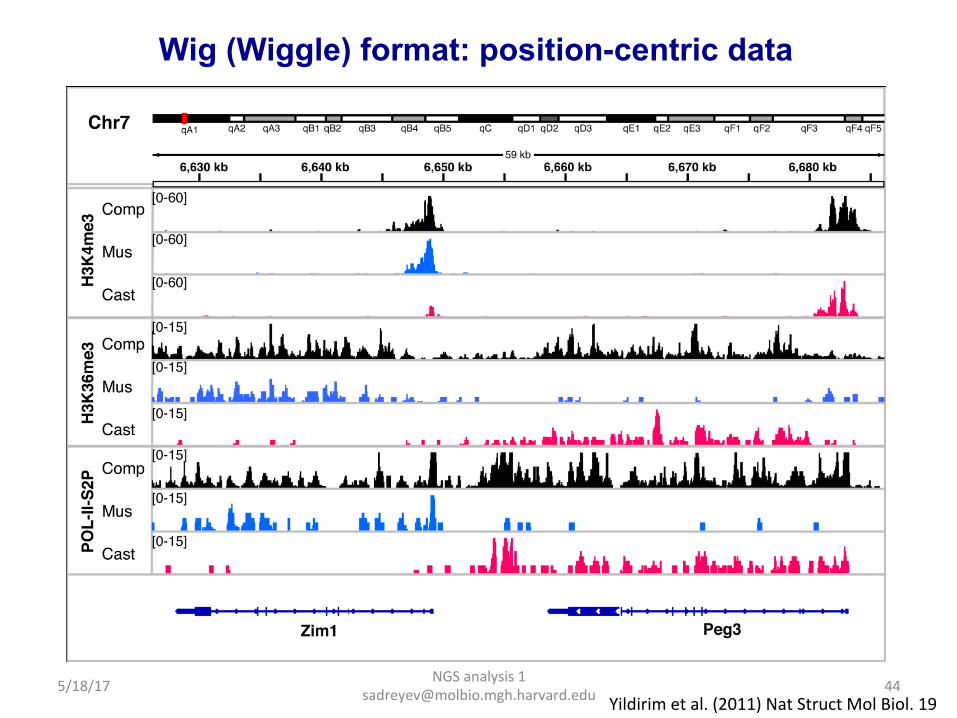
Wig (Wiggle) format: position-centric data
Yildirim et al. (2011) Nat Struct Mol Biol. 19 5/18/17 44
NGS analysis 1 [email protected]
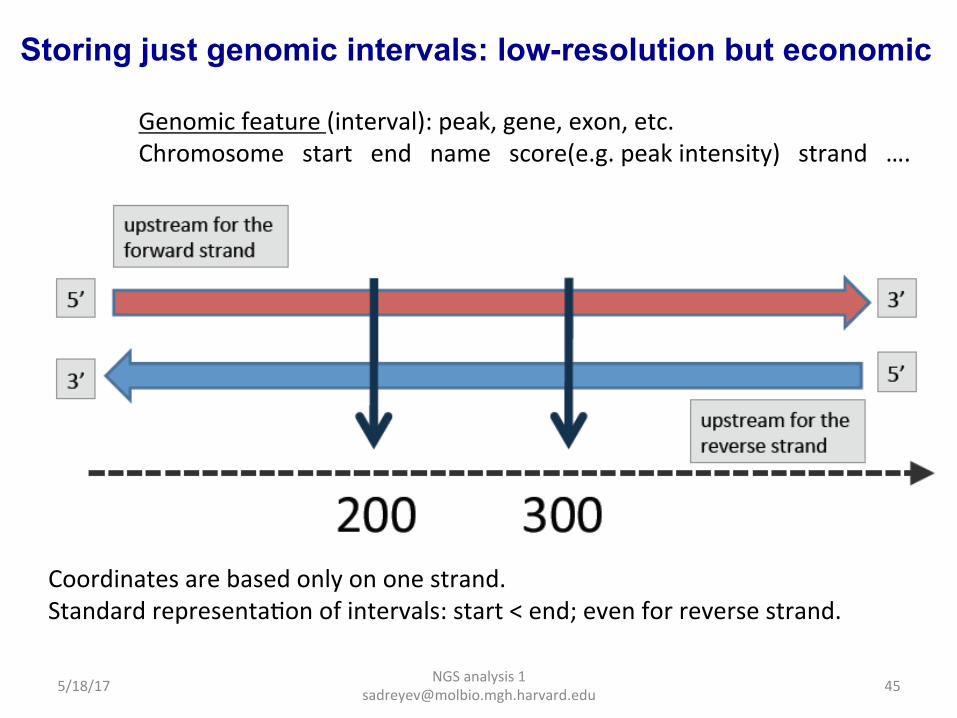
Storing just genomic intervals: low-resolution but economic
Coordinates are based only on one strand. Standard representaDon of intervals: start < end; even for reverse strand.
Genomic feature (interval): peak, gene, exon, etc. Chromosome start end name score(e.g. peak intensity) strand ….
5/18/17 45

NGS analysis 1 [email protected]
Two de facto standards of coordinate system
GFF format (Sanger): BED format (USCS Browser):
5/18/17 46

NGS analysis 1 [email protected]
Working with genomic intervals: BedTools
hap://code.google.com/p/bedtools
High-‐performance package operaDng on genomic intervals in various file formats: BED, GFF, VCF, SAM, BAM
Easy to download, install, and use in Unix/Linux/MAcOS …
5/18/17 47

NGS analysis 1 [email protected]
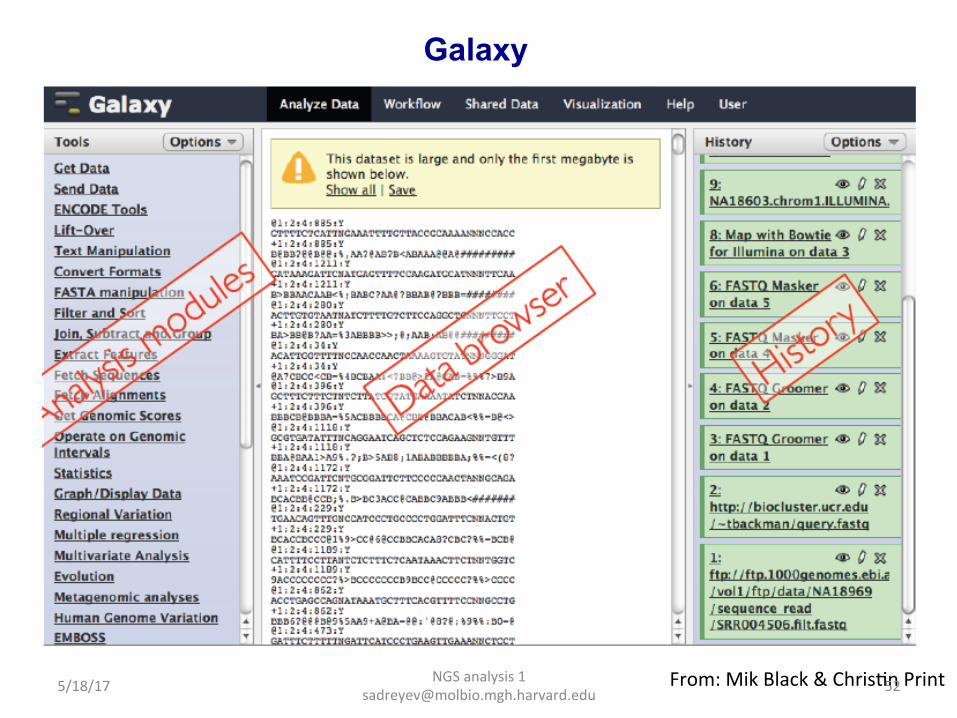
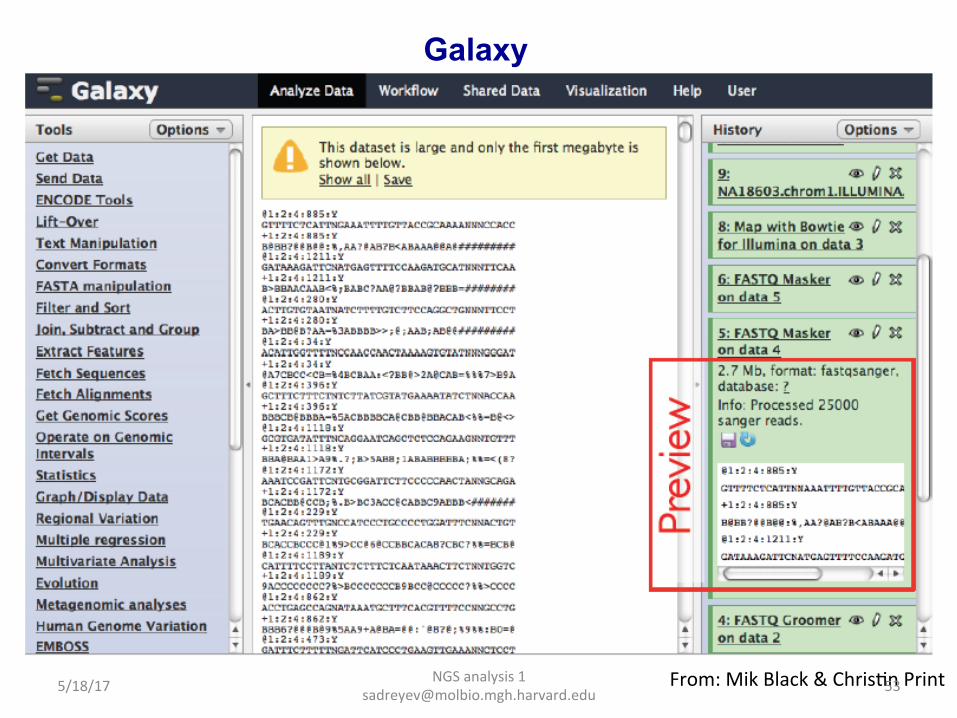
Galaxy hap://galaxy.psu.edu/
Our Galaxy server at Molbio: hap://galaga.mgh.harvard.edu/galaxy (can be accessed inside Partners network)
5/18/17 51

Schedule
5/18/17 NGS analysis 1 [email protected]
Sequence acquisition and processing; genome mapping and alignment manipulation
Thurs May 18 at 2 pm Specific NGS applications and public datasets
Thurs May 25 at 2 pm
56


















![[Thomas Scovel] Psycholinguistics (Oxford Introduc(BookFi.org)](https://img.pdfslide.us/doc/110x75/55cf9856550346d033970ef0/thomas-scovel-psycholinguistics-oxford-introducbookfiorg.jpg)





















