Embed Size (px)
Citation preview

Priority Report
Interleukin-6 Prevents the Initiation but Enhancesthe Progression of Lung CancerZhaoxia Qu1,2, Fan Sun1,2, Jingjiao Zhou1,2, Liwen Li1,2, Steven D. Shapiro1, andGutian Xiao1,2
Abstract
Recent studies suggest that high expression of the proinflam-matory cytokine IL6 is associated with poor survival of lungcancer patients. Accordingly, IL6 has been a target of greatinterest for lung cancer therapy. However, the role of IL6 inlung cancer has not been determined yet. Here, we demonstratethat IL6 plays opposite roles in the initiation and growth of lungcancer in a mouse model of lung cancer induced by the K-Rasoncogene. We find that compared with wild-type mice, IL6-deficient mice developed much more lung tumors after anactivating mutant of K-Ras was induced in the lungs. However,lung tumors developed in IL6-deficient mice were significantlysmaller. Notably, both the lung tumor–suppressing and –pro-
moting functions of IL6 involve its ability in activating thetranscription factor STAT3. IL6/STAT3 signaling suppressed lungcancer initiation through maintaining lung homeostasis, regu-lating lung macrophages, and activating cytotoxic CD8 T cellsunder K-Ras oncogenic stress, whereas it promoted lung cancercell growth through inducing the cell proliferation regulatorcyclin D1. These studies reveal a previously unexplored role ofIL6/STAT3 signaling in maintaining lung homeostasis and sup-pressing lung cancer induction. These studies also significantlyimprove our understanding of lung cancer and provide a molec-ular basis for designing IL6/STAT3-targeted therapies for thisdeadliest human cancer. Cancer Res; 75(16); 3209–15. �2015 AACR.
IntroductionLung cancer is the leading cause of cancer deaths in both
women and men, responsible for roughly 160,000 deaths annu-ally in the United States alone (1). Moreover, approximately 85%of the patients with lung cancer die of the disease within 5 years(1). A better understanding of the mechanisms underlying lungcancer development and progression and therapy resistance isdirely needed to design novel effective therapies for this deadliestcancer. The most predominant risk factor for lung cancer istobacco smoking, which accounts for about 87% of lung cancercases (2). Tobacco smoke induces genetic alterations, particularlyactivatingmutations of the K-Ras oncogene, in lung epithelium toinitiate and promote carcinogenesis (2).
One of the important functions of K-Ras activation is to induceexpression of IL6, a pleiotropic proinflammatory cytokine thathas been suggested to function as a lynchpin between inflamma-tion and cancer in several cancers, such as colon and liver cancers(3, 4). Indeed, IL6 is expressed in over 50% of human lung cancercell lines and primary tissues (5, 6). As a matter of fact, IL6 can be
detected in serum, pleural fluids, bronchioalveolar lavage fluids(BALF), and breath condensate of patients with lung cancer (6–10). More importantly, high IL6 level in tumor tissue, serum,BALF, and breath condensate is associated with lung cancerprogression, resistance to antitumor therapies, and poor survivalof lung cancer patients (6–10). Moreover, high IL6 level is alsoassociated with postoperative complication and postoperativerecurrence of lung cancer (11–13). Mechanistic studies suggestthat IL6 promotes lung cancer cell proliferation and migrationthrough activation of the transcription factor STAT3 (4, 7, 14). Inline with the role of IL6 in STAT3 activation, STAT3 has beenfound to be persistently activated in up to 65% of human lungcancers (4, 14). Also, the constitutive activation of STAT3 isassociated with lung cancer progression, therapy resistance, andpoor survival of lung cancer patients (4, 14). These studies suggesta molecular link between IL6 and lung cancer.
However, it remains unknownwhether andhow IL6 is involvedin the initiation of lung cancer. Current studies on lung cancermainly focus on the role of IL6 in the in vitro growth in cell cultureand in vivo growth in immunodeficient mice of lung cancer celllines (5, 15). Although useful, these studies require validation inendogenously arising lung tumors. They cannot address the roleof IL6 in the early stages of lung tumorigenesis. Furthermore, theycannot determine whether and how the inflammation-regulatoryactivity of IL6 is involved in lung cancer, because the hosts theyused for the in vivo growth of lung cancer cells lack immuneresponses and immunity. Another important issue that stillremains to be determined is the role of IL6 in lung physiologyunder oncogenic stresses. Addressing these issues is of importanceand interest, given the pleiotropic and complex functions of IL6.In particular, using endogenous lung tumorigenesis in immune-competent mice as a model system, we have recently found thatSTAT3 plays opposing roles in the initiation and progression of
1University of Pittsburgh Cancer Institute, Pittsburgh, Pennsylvania.2Department of Microbiology and Molecular Genetics, University ofPittsburgh School of Medicine, Pittsburgh, Pennsylvania.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Authors: Gutian Xiao, University of Pittsburgh Cancer Institute,5117 Centre Ave, HCC 1.19, Pittsburgh, PA 15213. Phone: 412-623-5410; Fax: 412-623-1415; E-mail: [email protected]; or Zhaoxia Qu, Phone: 412-623-1749; Fax:412-623-1415; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-14-3042
�2015 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 3209
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042

lung tumor (16). Accordingly, we also examined the effect ofIL6 deficiency on the initiation and development of endogenouslung tumor in immune-competent mice.
Materials and MethodsAnimals
IL6 knockout (IL6D/D) mice were purchased from The JacksonLaboratory. Lox-Stop-Lox (LSL) K-RasG12D mice were describedpreviously (16). Both IL6D/D mice and LSL-K-RasG12D mice werebackcrossed to FVB/N mice for more than ten generations forpure FVB/N background. IL6D/D FVB/N mice and LSL-K-RasG12D
FVB/N mice were then bred to generate IL6D/D/LSL-K-RasG12D
FVB/N mice. All animals were housed under specific pathogen-free conditions, and all animal experiments were approvedby the Institutional Animal Care and Use Committee of theUniversity of Pittsburgh.
Lung carcinogenesis and tumor enumerationSix- to 8-week-old IL6D/D/LSL-K-RasG12Dmice and IL6wt/wt/LSL-
K-RasG12D mice were intranasally administered 1 � 107 plaque-forming units (pfu) of Cre-expressing adenovirus (adenocre;Gene Transfer Vector Core, University of Iowa, Iowa City, IA) toinduce expression of the K-RasG12D mutant in lungs. Threemonths after Cre induction of K-RasG12D, all mice were sacrificedfor lung tumor examinations. Surface tumors inmouse lungswerecounted by three blinded readers under a dissecting microscope.Tumor diameters were determined by microcalipers.
BALF and immunofluorescence assaysMice were sacrificed, and their lungs were lavaged four times
with PBS. The recovered BALF were centrifuged. Cells from BALFwere visualized byHema 3 staining, and different leukocytes werecounted. Cells from BALF were also subjected to immunofluo-rescence assays as described previously (17). The antibodies usedfor immunofluorescence staining were listed in SupplementaryTable S1.
IHC assaysMouse lungs were excised, fixed in formalin, embedded in
paraffin, and cut into 4-mm-thick sections. Sectionswere subjectedto IHC staining as described previously (16). The antibodies usedfor IHC staining were listed in Supplementary Table S1.
BrdUrd labelingMice were i.p. injected with 50 mg/kg BrdUrd (Sigma-Aldrich)
24hours prior to sacrifice.Mouse lung tissue sectionswere stainedwith anti-BrdUrd (Sigma-Aldrich) according to the vendor'sinstructions.
Real-time PCR analysisMouse lung tissues, lung tumor tissues, BAL cells, or lung
epithelial cells were subjected to RNA extraction, RNA reversetranscription, and real-time PCR as described previously (16).Primer pairs used for real-time PCR were listed in the Supple-mentary Table S2.
Statistical analysisData were reported as mean � SD. The Student t test (two-
tailed) was used to assess significance of differences between two
groups, and P values < 0.05 and 0.01 were considered statisticallysignificant and highly statistically significant, respectively (16).
ResultsIL6D/Dmice are prone to lung tumorigenesis induced bymutantK-Ras
To test the functional significance of IL6 in lung tumorigen-esis, we took advantage of IL6D/D mice and LSL-K-RasG12D mice.After the mutant K-RasG12D transgene is activated in lungsthrough intranasal administration of Cre recombinase, LSL-K-RasG12D mice develop lung cancers. It is worthy to note thatmurine lung cancers driven by oncogenic K-Ras faithfullyrecapitulate human lung cancers, and in particular adenocarci-nomas associated with tobacco smoking (16). They share thesame genetic and molecular changes, as well as morphologyand histology. Moreover, K-Ras–induced lung cancers in mice,like their human counterparts, are associated with pulmonarydamage and immune cell infiltration. Thus, we generatedIL6D/D/LSL-K-RasG12D mice and IL6wt/wt/LSL-K-RasG12D mice bybreeding IL6D/D mice and LSL-K-RasG12D mice. For simplicity,IL6D/D/LSL-K-RasG12D mice and IL6wt/wt/LSL-K-RasG12D miceare hereinafter referred to as IL6D/D mice and IL6wt/wt (or simplyas wild type, WT) mice, respectively.
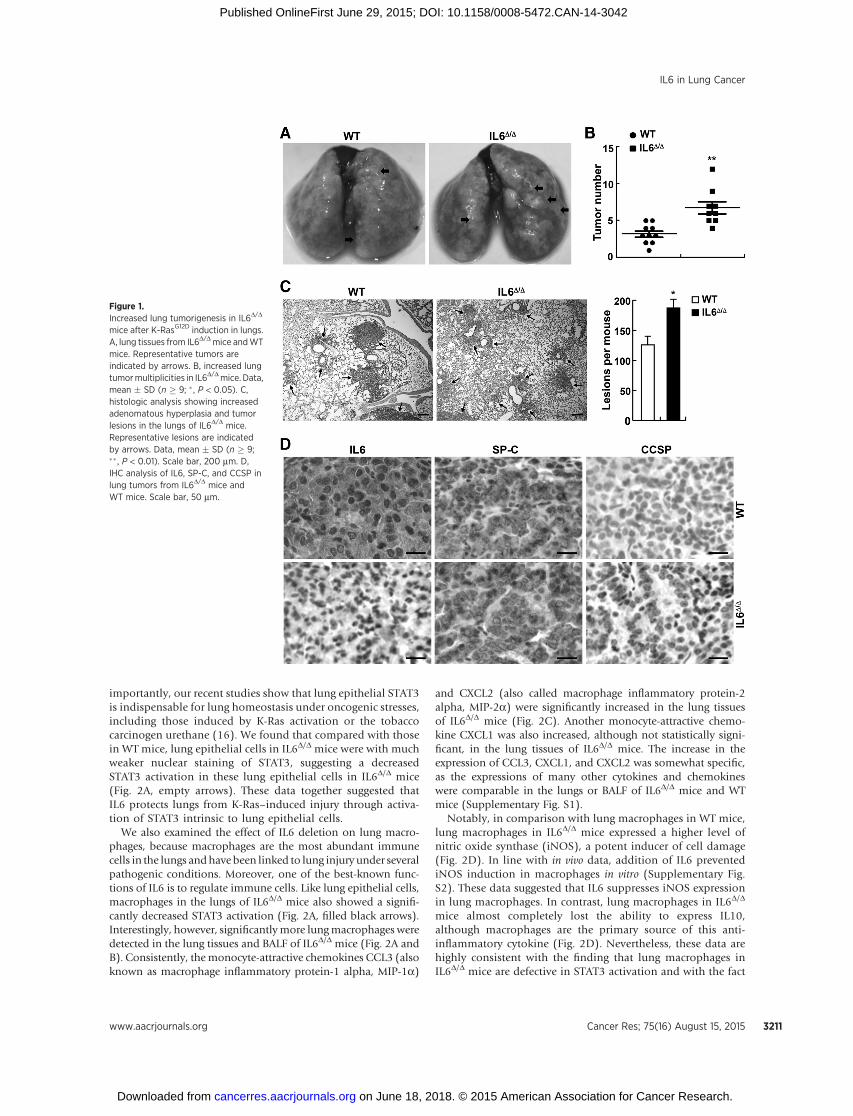
Consistent with previous studies (16), WT mice developedlung tumors after Cre induction of K-RasG12D in the lungs asevidenced by both the surface tumor enumeration and histo-logic assays (Fig. 1A–C). However, IL6D/D mice developed morelung tumors after the same induction of K-RasG12D. Exceptfor their difference in IL6 expression, tumors in IL6D/D miceandWTmice had the same morphologic and histologic features(Fig. 1C and D). In further support of the notion that SP-C–positive alveolar type II epithelial cells and/or BASCs are the cells-of-origin of lung cancers (16), tumors in IL6D/D mice or WT micewere positive for SP-C, while negative for the Clara cell markerCCSP (Clara cell secretory protein; Fig. 1C). These data indicatedthat IL6 suppresses lung tumor initiation induced by K-Ras.
Increased lung tumorigenesis in IL6D/D mice is associated withexacerbated lung damage as well as increased number andtoxicity of lung macrophages
Before K-RasG12D induction, the lungs of IL6D/D mice werenormal and displayed the same morphology and histology asthose of WT mice (data not shown). In agreement with ourrecent findings (16), K-RasG12D expression in the lungs ofWT mice induced mild alveolar congestion and minor impair-ments of alveolar epithelial integrity, indicating mild lungdamage (Fig. 2A, left, black arrowheads). However, the sameK-RasG12D expression caused more significant lung damage inIL6D/D mice, as evidenced by the severe loss of the integrity ofthe alveolar epithelium and the enlarged air space in thelungs of IL6D/D mice (Fig. 2A, right, black arrowheads). Thesedata suggested that IL6 is required for maintaining pulmo-nary homeostasis under K-Ras oncogenic stress, including pul-monary inflammation induced by K-Ras (see the followingsections).
To determine the mechanisms by which IL6 suppressesK-Ras–induced lung damage, we compared the activationstatus of STAT3 in the lung epithelial cells of IL6D/D mice andWT mice. It has been well established that one of themost important functions of IL6 is to activate STAT3. Most
Qu et al.
Cancer Res; 75(16) August 15, 2015 Cancer Research3210
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042
importantly, our recent studies show that lung epithelial STAT3is indispensable for lung homeostasis under oncogenic stresses,including those induced by K-Ras activation or the tobaccocarcinogen urethane (16). We found that compared with thosein WT mice, lung epithelial cells in IL6D/D mice were with muchweaker nuclear staining of STAT3, suggesting a decreasedSTAT3 activation in these lung epithelial cells in IL6D/D mice(Fig. 2A, empty arrows). These data together suggested thatIL6 protects lungs from K-Ras–induced injury through activa-tion of STAT3 intrinsic to lung epithelial cells.
We also examined the effect of IL6 deletion on lung macro-phages, because macrophages are the most abundant immunecells in the lungs andhave been linked to lung injury under severalpathogenic conditions. Moreover, one of the best-known func-tions of IL6 is to regulate immune cells. Like lung epithelial cells,macrophages in the lungs of IL6D/D mice also showed a signifi-cantly decreased STAT3 activation (Fig. 2A, filled black arrows).Interestingly, however, significantlymore lungmacrophages weredetected in the lung tissues and BALF of IL6D/D mice (Fig. 2A andB). Consistently, themonocyte-attractive chemokines CCL3 (alsoknown as macrophage inflammatory protein-1 alpha, MIP-1a)
and CXCL2 (also called macrophage inflammatory protein-2alpha, MIP-2a) were significantly increased in the lung tissuesof IL6D/D mice (Fig. 2C). Another monocyte-attractive chemo-kine CXCL1 was also increased, although not statistically signi-ficant, in the lung tissues of IL6D/D mice. The increase in theexpression of CCL3, CXCL1, and CXCL2 was somewhat specific,as the expressions of many other cytokines and chemokineswere comparable in the lungs or BALF of IL6D/D mice and WTmice (Supplementary Fig. S1).
Notably, in comparison with lung macrophages in WT mice,lung macrophages in IL6D/D mice expressed a higher level ofnitric oxide synthase (iNOS), a potent inducer of cell damage(Fig. 2D). In line with in vivo data, addition of IL6 preventediNOS induction in macrophages in vitro (Supplementary Fig.S2). These data suggested that IL6 suppresses iNOS expressionin lung macrophages. In contrast, lung macrophages in IL6D/D
mice almost completely lost the ability to express IL10,although macrophages are the primary source of this anti-inflammatory cytokine (Fig. 2D). Nevertheless, these data arehighly consistent with the finding that lung macrophages inIL6D/D mice are defective in STAT3 activation and with the fact
Figure 1.Increased lung tumorigenesis in IL6D/D
mice after K-RasG12D induction in lungs.A, lung tissues from IL6D/Dmice andWTmice. Representative tumors areindicated by arrows. B, increased lungtumormultiplicities in IL6D/Dmice. Data,mean � SD (n � 9; � , P < 0.05). C,histologic analysis showing increasedadenomatous hyperplasia and tumorlesions in the lungs of IL6D/D mice.Representative lesions are indicatedby arrows. Data, mean � SD (n � 9;�� , P < 0.01). Scale bar, 200 mm. D,IHC analysis of IL6, SP-C, and CCSP inlung tumors from IL6D/D mice andWT mice. Scale bar, 50 mm.
IL6 in Lung Cancer
www.aacrjournals.org Cancer Res; 75(16) August 15, 2015 3211
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042

that IL10 is a transcriptional target of STAT3. Interestingly, IL6and IL10 induced each other in both macrophages and lungepithelial cells (Supplementary Fig. S3A and S3B), suggesting aparacrine loop of IL6/IL10. More importantly, IL6 and IL10suppressed apoptosis of lung epithelial cells in vitro, whichwas associated with STAT3 activation and induction of cellsurvival genes, such as survivin, Bcl-2, and Bcl-xL (Supplemen-tary Fig. S3C–S3E). Given the abundant expression of IL10receptor (IL-10R) in lung epithelial cells, these data togethersuggested that IL6 and IL10 form a paracrine loop amongmacrophages and lung epithelial cells to activate STAT3, pro-tecting lung epithelial cells from K-Ras–induced injury.
Increased lung tumorigenesis in IL6D/D mice is also associatedwith the decreased expansion and activation of CD8 T cells aswell as the decreased tumor killing
Another important role of IL10 is to activate and expand CD8 Tcells, the lymphocytes that can directly induce apoptosis of tumorcells for tumor suppression. Thus, we hypothesized that IL10-
mediated activation and expansion of cytotoxic CD8 T cells isanother mechanism by which IL6 suppresses lung tumorigenesisinduced by K-Ras. To test the hypothesis, we first examined thetotal numbers of T cells in the BALF of IL6D/D mice and WT mice.We found that compared with WT mice, IL6D/D mice had signif-icantly fewer T cells in their BALF (Fig. 3A). The decrease of T cellsin the lungs of IL6D/D mice was due to the loss of CD8 T cells,because the expression of CD8, but not that of CD4, was muchlower in the BALF of IL6D/Dmice (Fig. 3A). Notably, CD8 T cells inthe lungs of IL6D/D mice had defective tumor-killing ability.Compared with lung CD8 T cells in WT mice, lung CD8 T cells inIL6D/D mice expressed much lower levels of antitumor cytokineIFNg and apoptosis inducers granzyme A and granzyme B(Fig. 3B). Accordingly, lung tumor cells in IL6D/D mice had muchlower apoptosis rate (Fig. 3C). It seems that the defects of the lungCD8T cells in IL6D/Dmicewere largely due to their defect in STAT3activation (Fig. 3D).
To confirm the in vivo data in a simple and direct way, wecompared the in vitro tumor cell killing ability of CD8 T cells
Figure 2.Elevated lung damage and increasedlung macrophages in IL6D/D mice after K-RasG12D induction in lungs. A, histologicand morphologic analysis showingincreased death rate of lung epithelialcells and decreased STAT3 activation inthe lung epithelial cells and lungmacrophages in IL6D/D mice. Lungepithelial cells and infiltratedmacrophages are indicated by emptyarrows and filled black arrows,respectively. Damaged lung epithelialcells are indicated by arrowheads. Scalebar, 50 mm. B, hema 3 staining showingmore macrophages in the BALF fromIL6D/D mice. Data, mean � SD (n � 5;�, P < 0.05). C, real-time PCR assaysshowing increased CCL3, CXCL2, andCXCL1 in the lung tissues of IL6D/D mice.Data, mean � SD (n � 5; � , P < 0.05). D,real-time PCR assays showing increasediNOS but decreased IL10 in the lungmacrophages of IL6D/D mice. Data, mean� SD (n � 5; � , P < 0.05).
Figure 3.Decreased lung CD8 T cells and tumorcell apoptosis in IL6D/Dmice expressingK-RasG12D in their lungs. A, hema 3staining and real-time PCR assaysshowing decreased CD8 T cells but nosignificant change in CD4 T cells in theBALF from IL6D/Dmice.Data,mean� SD(n � 5; � , P < 0.05). B, real-time PCRassays showing decreased expression ofIFNg , granzyme A, and granzyme B inthe lung CD8 T cells of IL6D/Dmice. Data,mean � SD (n � 5; � , P < 0.05). C, IHCassays showing decreased apoptosisof lung tumor cells in IL6D/D mice.Apoptotic tumor cells are indicated byarrows. D, immunofluorescence assaysshowing the lack of STAT3 activation inthe lung CD8 T cells in IL6D/D mice.
Qu et al.
Cancer Res; 75(16) August 15, 2015 Cancer Research3212
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042
isolated from IL6D/D mice and WT mice. As expected, coculturewith CD8 T cells from WT mice led to loss of lung tumor cells(Supplementary Fig. S4A). However, CD8 T cells from IL6D/D
mice significantly lost the tumor killing ability. On the otherhand, addition of IL6 significantly enhanced the tumor killingability of CD8 T cells (Supplementary Fig. S4B). Consistently,we found that addition of IL6 increased expression of cytotoxicmolecules in CD8 cells, such as perforin, granzymes A and B,TNFa, and TRAIL (Supplementary Fig. S4C). Moreover, IL6also induced expression of cell survival genes in CD8 cells,such as Bcl-2, Bcl-xL, survivin, and Mcl-1, and preventedactivation-induced death of CD8 cells in vitro (SupplementaryFig. S4D and S4E). These data altogether clearly indicated thatIL6 also protects and activates cytotoxic CD8 T cells to sup-press K-Ras–induced lung tumorigenesis.
IL6 contributes to the growth of lung cancers induced by K-RasAlthough IL6D/D mice developed more lung tumors than WT
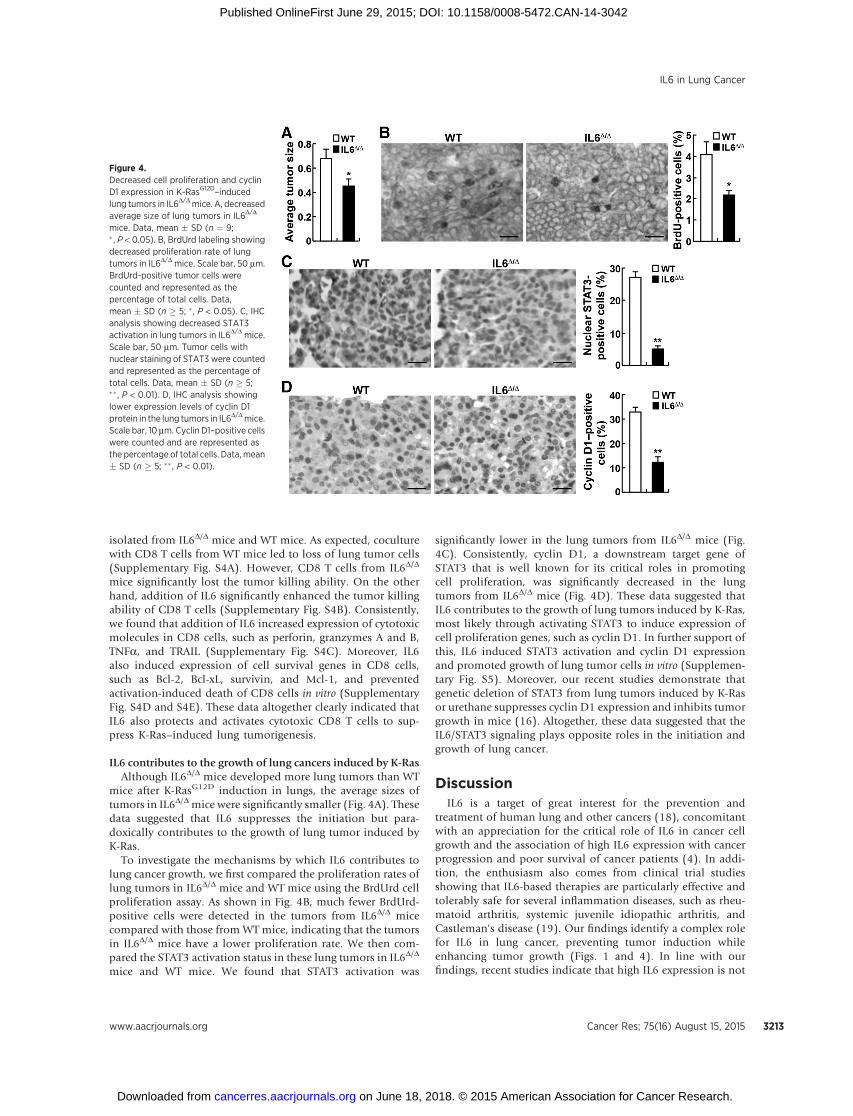
mice after K-RasG12D induction in lungs, the average sizes oftumors in IL6D/Dmice were significantly smaller (Fig. 4A). Thesedata suggested that IL6 suppresses the initiation but para-doxically contributes to the growth of lung tumor induced byK-Ras.
To investigate the mechanisms by which IL6 contributes tolung cancer growth, we first compared the proliferation rates oflung tumors in IL6D/D mice and WT mice using the BrdUrd cellproliferation assay. As shown in Fig. 4B, much fewer BrdUrd-positive cells were detected in the tumors from IL6D/D micecompared with those from WT mice, indicating that the tumorsin IL6D/D mice have a lower proliferation rate. We then com-pared the STAT3 activation status in these lung tumors in IL6D/D
mice and WT mice. We found that STAT3 activation was
significantly lower in the lung tumors from IL6D/D mice (Fig.4C). Consistently, cyclin D1, a downstream target gene ofSTAT3 that is well known for its critical roles in promotingcell proliferation, was significantly decreased in the lungtumors from IL6D/D mice (Fig. 4D). These data suggested thatIL6 contributes to the growth of lung tumors induced by K-Ras,most likely through activating STAT3 to induce expression ofcell proliferation genes, such as cyclin D1. In further support ofthis, IL6 induced STAT3 activation and cyclin D1 expressionand promoted growth of lung tumor cells in vitro (Supplemen-tary Fig. S5). Moreover, our recent studies demonstrate thatgenetic deletion of STAT3 from lung tumors induced by K-Rasor urethane suppresses cyclin D1 expression and inhibits tumorgrowth in mice (16). Altogether, these data suggested that theIL6/STAT3 signaling plays opposite roles in the initiation andgrowth of lung cancer.
DiscussionIL6 is a target of great interest for the prevention and
treatment of human lung and other cancers (18), concomitantwith an appreciation for the critical role of IL6 in cancer cellgrowth and the association of high IL6 expression with cancerprogression and poor survival of cancer patients (4). In addi-tion, the enthusiasm also comes from clinical trial studiesshowing that IL6-based therapies are particularly effective andtolerably safe for several inflammation diseases, such as rheu-matoid arthritis, systemic juvenile idiopathic arthritis, andCastleman's disease (19). Our findings identify a complex rolefor IL6 in lung cancer, preventing tumor induction whileenhancing tumor growth (Figs. 1 and 4). In line with ourfindings, recent studies indicate that high IL6 expression is not
Figure 4.Decreased cell proliferation and cyclinD1 expression in K-RasG12D–inducedlung tumors in IL6D/Dmice. A, decreasedaverage size of lung tumors in IL6D/D
mice. Data, mean � SD (n ¼ 9;� , P < 0.05). B, BrdUrd labeling showingdecreased proliferation rate of lungtumors in IL6D/D mice. Scale bar, 50 mm.BrdUrd-positive tumor cells werecounted and represented as thepercentage of total cells. Data,mean � SD (n � 5; �, P < 0.05). C, IHCanalysis showing decreased STAT3activation in lung tumors in IL6D/D mice.Scale bar, 50 mm. Tumor cells withnuclear staining of STAT3 were countedand represented as the percentage oftotal cells. Data, mean � SD (n � 5;�� , P < 0.01). D, IHC analysis showinglower expression levels of cyclin D1protein in the lung tumors in IL6D/Dmice.Scale bar, 10 mm. Cyclin D1–positive cellswere counted and are represented asthe percentage of total cells. Data, mean� SD (n � 5; �� , P < 0.01).
IL6 in Lung Cancer
www.aacrjournals.org Cancer Res; 75(16) August 15, 2015 3213
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042

associated with lung cancer risk in humans (20), although it isassociated with lung cancer progression and poor survival oflung cancer patients (6–10). It should be pointed out that evencomplete deletion of IL6 can only delay lung cancer growth.Thus, to target IL6 for lung cancer therapy, we need, on onehand, to consider the potential risk in increasing lung damageand tumorigenesis due to long-term IL6 inhibition, and on theother hand, to combine IL6 inhibition with other cancertherapies for efficient clinical outcomes. In this regard, recentphase I and II clinical trials involving 125 lung cancer patientsindicate that IL6-targeted therapy alone has no obvious clinicalbenefits, except for an amelioration of lung cancer–associatedanemia and cachexia in patients (19). Although more carefuland more lung cancer patients–involved clinical trials areneeded to determine the clinical outcomes of IL6-targetedtherapy, it could be speculated that the overall clinical benefitsof IL6 therapy alone might be limited, giving both the tumor-suppressing and -promoting roles of IL6 in lung cancer.
Interestingly, both lung tumor–suppressing and –promotingfunctions of IL6 involve its ability in activating STAT3. IL6suppresses lung cancer induction through maintaining lunghomeostasis and inducing tumor cell killing in STAT3-dependentmanners. In addition to inducing STAT3 activation in lung epi-thelial cells, IL6 activates STAT3 in other cells in lungs, particularlymacrophages, to express IL10, which serves as a paracrine stim-ulus to further enhance lung epithelial STAT3 activation for lunghomeostasis under oncogenic stress (Fig. 2 and SupplementaryFig. S3). IL6 also suppresses lung macrophages to express iNOSand thereby prevents iNOS-induced lung damage (Fig. 2 andSupplementary Fig. S2). IL10 produced by lung macrophages,perhaps together with IL6, also induces STAT3 activation incytotoxic CD8 T cells for their survival and activation, which inturn induce tumor cell apoptosis (Fig. 3 and Supplementary Fig.S4). Thus, it seems that IL6 utilizes multiple related mechanismsto suppress lung cancer initiation. Paradoxically, IL6 contributesto lung cancer growth also through STAT3-dependent mecha-nism. In this case, IL6 activates STAT3 to induce cyclin D1 in lungcancer cells, therefore promoting cancer proliferation (Fig. 4 andSupplementary Fig. S5). In further support of these findings, ourrecent studies indicate that mice selectively deficient in lungepithelial STAT3 show overall similar phenotypes as IL6-deficient
mice in K-Ras–induced lung damage, tumor initiation, and pro-gression (16).
In summary, these data demonstrate that the IL6/STAT3 sig-naling plays overall opposite roles in the initiation and growth oflung cancer—it prevents lung cancer initiation throughmaintain-ing lung homeostasis and inducing cytotoxic CD8 T cells, whereasit contributes to (although it is not absolutely required for) lungcancer growth through inducing expression of key regulators ofcell proliferation. These studies not only greatly improve ourunderstanding of the pathophysiologic actions of IL6/STAT3signaling and the pathogenesis of lung and other cancers associ-ated with IL6/STAT3 signaling, but also provide a mechanisticbasis for targeting IL6/STAT3 signaling to treat IL6- and STAT3-associated cancers.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: Z. Qu, G. XiaoDevelopment of methodology: Z. Qu, F. Sun, G. XiaoAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Z. Qu, F. Sun, J. Zhou, L. Li, G. XiaoAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Z. Qu, F. Sun, J. Zhou, L. Li, G. XiaoWriting, review, and/or revision of the manuscript: Z. Qu, S.D. Shapiro,G. XiaoAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): Z. Qu, F. Sun, G. XiaoStudy supervision: Z. Qu, G. Xiao
AcknowledgmentsThe authors thank other members of the Qu–Xiao collaborative team for
their critical reading and technique supports.
Grant SupportThis study was financially supported in part by the NIH/NCI grants R01
CA172090, R21 CA175252, and P30 CA047904, as well as the American LungAssociation (ALA) Lung Cancer Discovery Award and American Cancer Society(ACS) Fellowship Award RSG-06-066-01-MGO.
Received October 14, 2014; revised May 18, 2015; accepted May 19, 2015;published OnlineFirst June 29, 2015.
References1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin
2014;64:9–29.2. Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and
approaches to prevention. Lancet Oncol 2002;3:461–9.3. Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is
required for tumorigenesis. Genes Dev 2007;21:1714–9.4. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins
between inflammation and cancer. Semin Immunol 2014;26:54–74.5. Yamaji H, Iizasa T, Koh E, Suzuki M, Otsuji M, Chang H, et al. Correlation
between interleukin 6 production and tumor proliferation in non-smallcell lung cancer. Cancer Immunol Immunother 2004;53:786–92.
6. Koh E, Iizasa T, Yamaji H, Sekine Y, Hiroshima K, Yoshino I, et al.Significance of the correlation between the expression of interleukin 6and clinical features in patients with non-small cell lung cancer. Int J SurgPathol 2012;20:233–9.
7. Yeh HH, Lai WW, Chen HH, Liu HS, Su WC. Autocrine IL-6-induced Stat3activation contributes to the pathogenesis of lung adenocarcinoma andmalignant pleural effusion. Oncogene 2006;25:4300–9.
8. Huang F, Wang XL, Geng Y, Li MX. Evaluation of IL-6 level in serum andbronchoalveolar lavage fluid of patients with non-small cell lung cancer(NSCLC). Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2005;21:507–9.
9. Carpagnano GE, Resta O, Foschino-Barbaro MP, Gramiccioni E, Carpag-nano F. Interleukin-6 is increased in breath condensate of patients withnon-small cell lung cancer. Int J Biol Markers 2002;17:141–5.
10. Chang CH, Hsiao CF, Yeh YM, Chang GC, Tsai YH, Chen YM, et al.Circulating interleukin-6 level is a prognostic marker for survival inadvanced nonsmall cell lung cancer patients treated with chemotherapy.Int J Cancer 2013;132:1977–85.
11. Szczesny TJ, Slotwinski R, Stankiewicz A, Szczygiel B, ZaleskaM, KopaczM.Interleukin 6 and interleukin 1 receptor antagonist as early markers ofcomplications after lung cancer surgery. Eur J Cardiothorac Surg 2007;31:719–24.
12. Kita H, Shiraishi Y,Watanabe K, Suda K,Ohtsuka K, Koshiishi Y, et al. Doespostoperative serum interleukin-6 influence early recurrence after curativepulmonary resection of lung cancer? Ann Thorac Cardiovasc Surg 2011;17:454–60.
Qu et al.
Cancer Res; 75(16) August 15, 2015 Cancer Research3214
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042

13. Wang XS, Shi Q, Williams LA, Mao L, Cleeland CS, Komaki RR, et al.Inflammatory cytokines are associated with the development of symptomburden in patients with NSCLC undergoing concurrent chemoradiationtherapy. Brain Behav Immun 2010;24:968–74.
14. Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lungcancer. Cancers (Basel) 2014;6:708–22.
15. Song L, Smith MA, Doshi P, Sasser K, Fulp W, Altiok S, et al. Antitumorefficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mousexenograft models of lung cancer. J Thorac Oncol 2014;9:974–82.
16. Zhou J, Qu Z, Yan S, Sun F, Whitsett JA, Shapiro SD, et al. Differential rolesof STAT3 in the initiation and growth of lung cancer. Oncogene 2014 Oct06. [Epub ahead of print].
17. Yan P, Fu J, Qu Z, Li S, Tanaka T, Grusby MJ, et al. PDLIM2 suppresseshuman T-cell leukemia virus type I Tax-mediated tumorigenesis by target-ing Tax into the nuclear matrix for proteasomal degradation. Blood2009;113:4370–80.
18. Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor incancer: implications for translational therapeutics. Cancer 2007;110:1911–28.
19. Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin BiolTher 2011;11:1663–8.
20. Zhou B, Liu J, Wang ZM, Xi T. C-reactive protein, interleukin 6 and lungcancer risk: a meta-analysis. PLoS One 2012;7:e43075.
www.aacrjournals.org Cancer Res; 75(16) August 15, 2015 3215
IL6 in Lung Cancer
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042

2015;75:3209-3215. Published OnlineFirst June 29, 2015.Cancer Res Zhaoxia Qu, Fan Sun, Jingjiao Zhou, et al. of Lung CancerInterleukin-6 Prevents the Initiation but Enhances the Progression
Updated version
10.1158/0008-5472.CAN-14-3042doi:
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/75/16/3209.full#ref-list-1
This article cites 19 articles, 2 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/75/16/3209.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/75/16/3209To request permission to re-use all or part of this article, use this link
on June 18, 2018. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 29, 2015; DOI: 10.1158/0008-5472.CAN-14-3042





























