Embed Size (px)
Citation preview

22 January 1999
Ž .Chemical Physics Letters 299 1999 630–636
Interaction of Cr and Crq with NO: a density functional study
Ana Martınez a,), Patrizia Calaminici b, Andreas M. Koster b, Steve A. Mitchell c´ ¨a Departamento de Quımica, DiÕision de Ciencias Basicas e Ingenierıa, UniÕersidad Autonoma Metropolitana-Iztapalapa, A.P.55-534,´ ´ ´ ´ ´
Mexico D.F. 09340, Mexico´b Theoretische Chemie, UniÕersitat HannoÕer, Am Kleinen Felde 30, D-30167 HannoÕer, Germany¨
c Steacie Institute for Molecular Sciences, National Research Council of Canada, Ottawa, Ont. K1A 0R6, Canada
Received 29 July 1998; in final form 9 October 1998
Abstract
The interaction of Cr and Crq with NO was investigated with the all-electron linear-combination-of-Gaussian-type-orbitalsKohn–Sham density functional theory. Several local minima on different potential energy surfaces were found. The lowestminima for CrNO and CrNOq are doublet bent and triplet linear structures with binding energies of 61.9 and 43.2 kcalrmol,respectively. Correlation diagrams of the Kohn–Sham orbitals for the initial stages of the reaction are given. For themechanisms of the reactions, charge transfer processes are suggested. The rate coefficient for the reaction CrqNO obtainedfrom the calculated molecular data is in good agreement with the observed value. q 1999 Elsevier Science B.V. All rightsreserved.
1. Introduction
w xParnis and co-workers 1 have reported an exper-imental study dealing with the reaction of transitionmetal atoms with small molecules. They used thetechnique of gas-phase metal atom formation byvisible multiphoton dissociation of a volatileorganometallic compound to study metal atom reac-
w xtions with a variety of simple molecules 1–5 Withthis technique, they examined the reactivity of
Ž 7 .ground-state a S Cr atoms with small molecules.They found that the dominant feature of Cr atomchemistry is the formation of association complexeswith the stable free radicals O and NO.2
w xIn previous works, Martınez et al. 6,7 studied´the reactivity of Cr and Mo atoms with N and O ,2 2
) Corresponding author.
in order to obtain an explanation for the differentw xreactivities observed 1,8 . For CrO and MoO , the2 2
results indicate that the O molecule is completely2
dissociated after two spin flips, and that metal–oxygen bonds are formed. For the CrN and MoN2 2
system, the thermodynamically unstable side-oncomplex was found as the product of the reaction. Inall these reactions, the metal atom always acted, asthe electron donor.
In order to investigate the reaction of Cr and Crq
with NO, we have performed density functional the-Ž .ory DFT calculations. Bond distances, equilibrium
geometries, binding energies, electronic states, har-monic frequencies, net atomic charges from a Mul-liken population analysis and Kohn–Sham molecularorbital correlation diagrams are presented. A consis-tent interpretation of the kinetic and computationalresults is also given.
0009-2614r99r$ - see front matter q 1999 Elsevier Science B.V. All rights reserved.Ž .PII: S0009-2614 98 01305-0

( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´ 631
2. Computational details
All calculations have been carried out using thew xdensity functional program ALLCHEM 9,10 . ALLCHEM
works with Gaussian basis sets and with a set ofGaussian-type auxiliary functions in order to avoidfour-center Coulomb integrals. A new efficient inte-gral algorithm for the calculation of three-center
w xCoulomb integrals was applied 9 , which exploitssome special features of the structure of the auxiliaryfunction sets used. Recently, an adaptive numerical
w xintegrator was successfully implemented 10 , whichallows an efficient optimization of the grid for anindividual molecular system.
In the DFT code ALLCHEM, both the linear combi-nation of Gaussian-type orbital local spin densityŽ .LCGTO-LSD and the corresponding generalized
Ž .gradient approximation LCGTO-GGA , methods areimplemented. In the case of local density functionalcalculations, the exchange-correlation contributions
Ž . w xproposed by Vosko, Wilk and Nusair VWN 11have been used. In the case of generalized gradient
Ž .approximated GGA spin density functional calcula-w xtions, the functional of Perdew and Wang 12,13 for
w xthe exchange and of Perdew 14 for the correlationcontribution were used.
The basis sets employed were the double-zetaŽ . w xbasis sets DZVP 15 . The contraction pattern, in
Ž .the Huzinaga notation, is 63321r531r41) for theŽ .chromium atom and 621r41r1) for the nitrogen
and oxygen atoms. The exchange-correlation poten-tial was numerically integrated on the adaptive griddescribed above. A grid accuracy of 10y5 was usedfor all calculations. The Coulomb energy was calcu-lated by the variational fitting procedure proposed by
w xDunlap et al. 16,17 . For the fitting of the densityw xthe auxiliary function set A2 15 was used in all
calculations.In order to localize different minima on the poten-
Ž .tial energy surface PES , all molecular structureswere fully optimized, without symmetry constraints,starting from different initial structures using the
w xVWN functional 11 , analytic energy gradients andw xthe Berny quasi-Newton update 18 . A SCF energy
convergence criterion of 10y6 a.u. was used in allthe optimizations. The structure optimization conver-gence was based on the gradient and displacementvectors with a threshold of 10y4 and 10y3 a.u.,
respectively. GGA energies were calculated with thegeometries optimized at the local level. Therefore,the GGA potential was included self-consistently forthe final energy evaluation. According to our experi-ences, the results obtained from this procedure arealmost independent from the particular chosen GGAfunctional.
ŽFor the most stable structures of CrNO neutral.and cationic , a vibrational analysis was performed
in order to discriminate between minima and transi-tion states on the PES. The second derivatives were
Žcalculated by numerical differentiation two-point.finite difference of the analytic energy gradients
using a displacement of 0.002 a.u. from the opti-mized geometry for all 3N coordinates. The energyconvergence threshold for the vibrational analysiscalculations was set to 10y9. The harmonic frequen-cies were obtained by diagonalizing the mass-weighted cartesian force constant matrix. Since thegeometries were optimized at the local VWN level,the vibrations were also calculated in the LSD ap-proximation.
3. Results and discussion
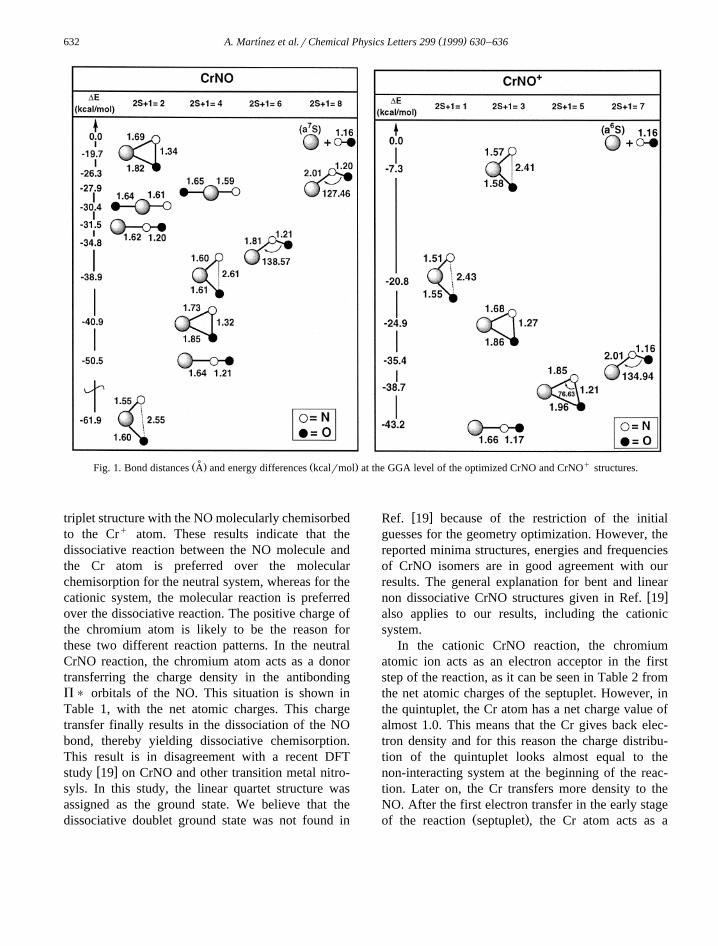
The optimized geometries of CrNO and CrNOq
are summarized in Fig. 1. We started from severalinitial geometries and multiplicities and we foundseveral critical points on the different PES. Bonddistances at the LSD level and energy differences atthe GGA level with respect to the dissociation limitwŽ Ž7 . . Ž qŽ6 . .xCr S qNO and Cr S qNO for the neutraland cationic systems, respectively, are also reported.
In Fig. 1, it can be seen that many thermodynami-cally stable local minima were found. On the octu-
Ž .plet and sextuplet PES for CrNO and on the septu-Ž q.plet PES for CrNO we found minima, which
present a relative weak interaction between the metalatom and the N atom. For CrNOq one bent mini-mum on the quintet PES was found. This structurereflects a weaker interaction between the Crq andthe O atom and a stronger interaction between theCrq and the N atom. In all these structures, the N–Omolecule is not dissociated. For CrNO, the groundstate is a doublet bent structure with the NO moleculedissociatively chemisorbed to the Cr atom. For thecationic system, the most stable structure is a linear
( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´632
˚ qŽ . Ž .Fig. 1. Bond distances A and energy differences kcalrmol at the GGA level of the optimized CrNO and CrNO structures.
triplet structure with the NO molecularly chemisorbedto the Crq atom. These results indicate that thedissociative reaction between the NO molecule andthe Cr atom is preferred over the molecularchemisorption for the neutral system, whereas for thecationic system, the molecular reaction is preferredover the dissociative reaction. The positive charge ofthe chromium atom is likely to be the reason forthese two different reaction patterns. In the neutralCrNO reaction, the chromium atom acts as a donortransferring the charge density in the antibondingP) orbitals of the NO. This situation is shown inTable 1, with the net atomic charges. This chargetransfer finally results in the dissociation of the NObond, thereby yielding dissociative chemisorption.This result is in disagreement with a recent DFT
w xstudy 19 on CrNO and other transition metal nitro-syls. In this study, the linear quartet structure wasassigned as the ground state. We believe that thedissociative doublet ground state was not found in
w xRef. 19 because of the restriction of the initialguesses for the geometry optimization. However, thereported minima structures, energies and frequenciesof CrNO isomers are in good agreement with ourresults. The general explanation for bent and linear
w xnon dissociative CrNO structures given in Ref. 19also applies to our results, including the cationicsystem.
In the cationic CrNO reaction, the chromiumatomic ion acts as an electron acceptor in the firststep of the reaction, as it can be seen in Table 2 fromthe net atomic charges of the septuplet. However, inthe quintuplet, the Cr atom has a net charge value ofalmost 1.0. This means that the Cr gives back elec-tron density and for this reason the charge distribu-tion of the quintuplet looks almost equal to thenon-interacting system at the beginning of the reac-tion. Later on, the Cr transfers more density to theNO. After the first electron transfer in the early stage
Ž .of the reaction septuplet , the Cr atom acts as a
( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´ 633
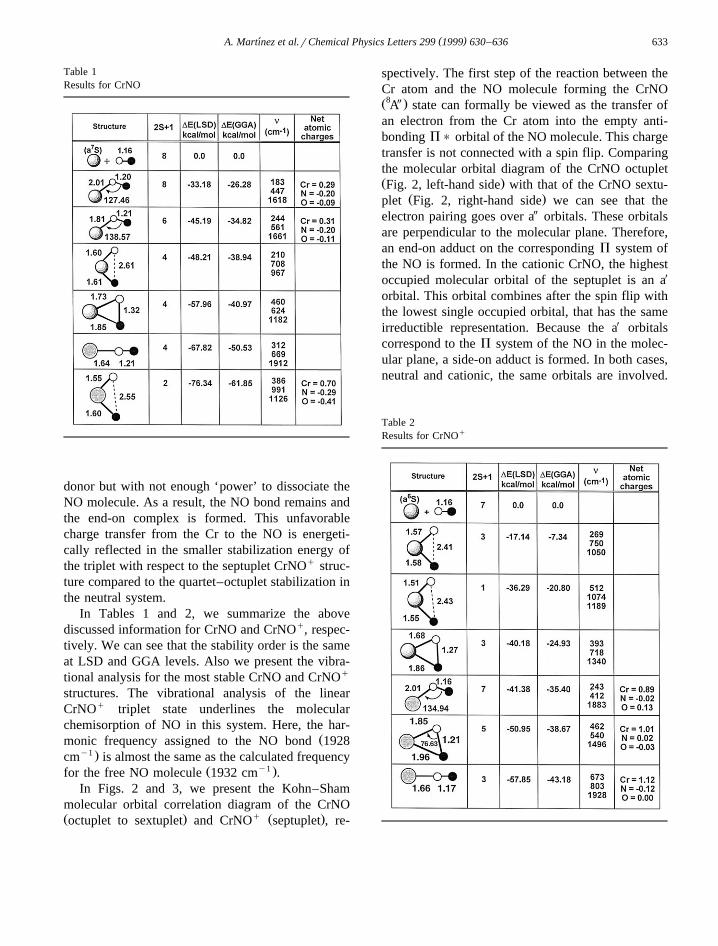
Table 1Results for CrNO
donor but with not enough ‘power’ to dissociate theNO molecule. As a result, the NO bond remains andthe end-on complex is formed. This unfavorablecharge transfer from the Cr to the NO is energeti-cally reflected in the smaller stabilization energy ofthe triplet with respect to the septuplet CrNOq struc-ture compared to the quartet–octuplet stabilization inthe neutral system.
In Tables 1 and 2, we summarize the abovediscussed information for CrNO and CrNOq, respec-tively. We can see that the stability order is the sameat LSD and GGA levels. Also we present the vibra-tional analysis for the most stable CrNO and CrNOq
structures. The vibrational analysis of the linearCrNOq triplet state underlines the molecularchemisorption of NO in this system. Here, the har-
Žmonic frequency assigned to the NO bond 1928y1 .cm is almost the same as the calculated frequency
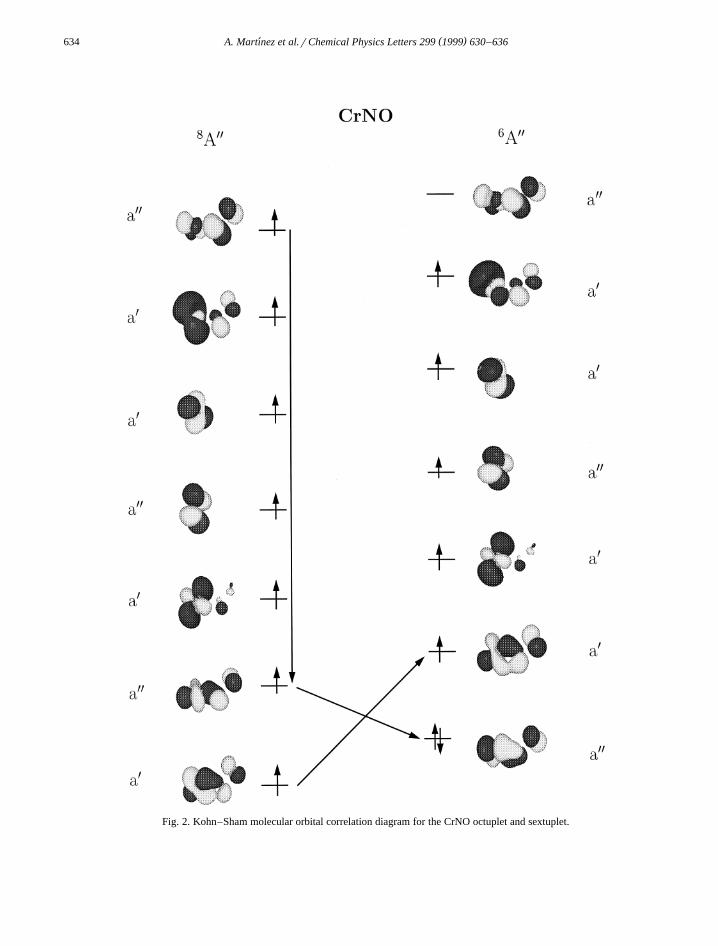
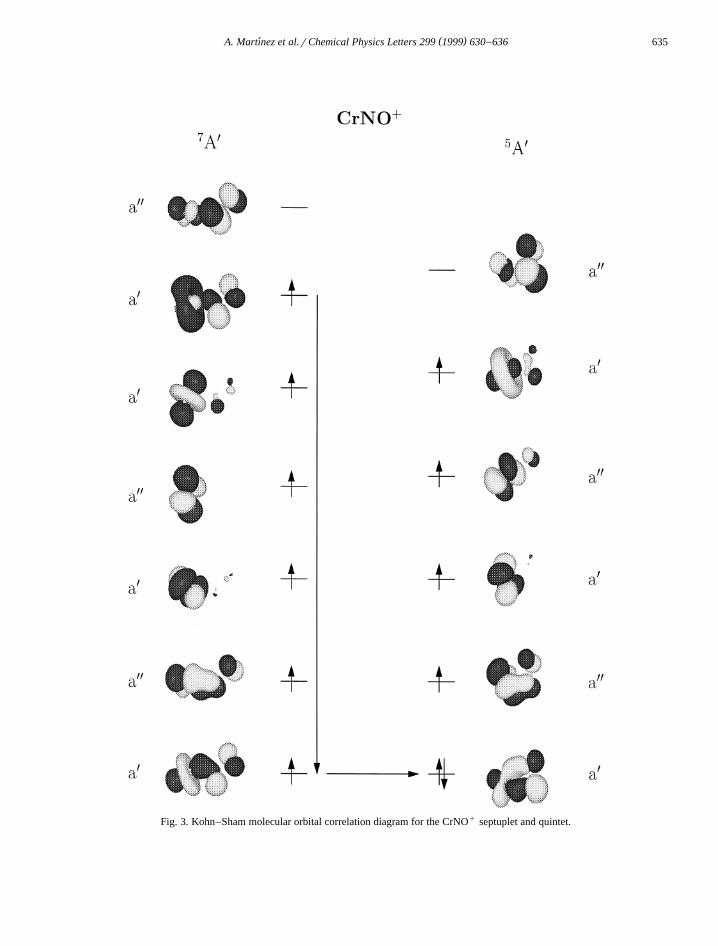
Ž y1 .for the free NO molecule 1932 cm .In Figs. 2 and 3, we present the Kohn–Sham
molecular orbital correlation diagram of the CrNOŽ . q Ž .octuplet to sextuplet and CrNO septuplet , re-
spectively. The first step of the reaction between theCr atom and the NO molecule forming the CrNOŽ8 Y .A state can formally be viewed as the transfer ofan electron from the Cr atom into the empty anti-bonding P) orbital of the NO molecule. This chargetransfer is not connected with a spin flip. Comparingthe molecular orbital diagram of the CrNO octupletŽ .Fig. 2, left-hand side with that of the CrNO sextu-
Ž .plet Fig. 2, right-hand side we can see that theelectron pairing goes over aY orbitals. These orbitalsare perpendicular to the molecular plane. Therefore,an end-on adduct on the corresponding P system ofthe NO is formed. In the cationic CrNO, the highestoccupied molecular orbital of the septuplet is an aX
orbital. This orbital combines after the spin flip withthe lowest single occupied orbital, that has the sameirreductible representation. Because the aX orbitalscorrespond to the P system of the NO in the molec-ular plane, a side-on adduct is formed. In both cases,neutral and cationic, the same orbitals are involved.
Table 2Results for CrNOq
( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´634
Fig. 2. Kohn–Sham molecular orbital correlation diagram for the CrNO octuplet and sextuplet.
( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´ 635
Fig. 3. Kohn–Sham molecular orbital correlation diagram for the CrNOq septuplet and quintet.

( )A. Martınez et al.rChemical Physics Letters 299 1999 630–636´636
Due to the different symmetries of the highest occu-pied orbitals, end-on and side-on adducts are formed.
Simplified RRKM calculations using the formula-w xtion of Troe 20 were carried out to make a connec-
tion between the present computational results andw xpreviously reported experimental measurements 1
of the rate coefficient for the association reaction ofw xCr with NO at 298 K. As discussed elsewhere 1,21
one expects a correlation between the binding energyand the rate coefficient because the latter is con-trolled by the density of vibrational states at thedissociation limit, and this is a steep function of thedissociation energy. The rate coefficient thus carriesinformation on the binding energy, which can beextracted by using either simplified or more elabo-rate RRKM calculations in conjunction with calcu-lated or estimated molecular properties includingvibrational frequencies and moments of inertia. Therelative values of the rate coefficients for the Crq
w xNO and CrqO reactions 1 strongly indicate that2
the products of the CrqNO reaction are relativelyweakly bound. A consistent interpretation of theexperimental kinetic data and the presented computa-tional results is obtained by assuming that only the6AY and 8AY states of CrNO were formed under thereported reaction conditions at room temperature. Byusing the molecular data obtained from our DFTcalculations, we estimate the strong collision ratecoefficient at 300 K to be 1.9=10y29 cm6 sy1.Since a typical value for the collision efficiency ofAr buffer gas is 0.1, this predicts an observed ratecoefficient of 1.9=10y30 cm6 sy1, which compareswell with the experimental value 1.2=10y30 cm6
y1 w xs 1 . Therefore, we conclude that the elementaryreaction that controls the removal rate of Cr by NOat room temperature involves the formation of rela-tively weakly bound sextuplet and octuplet com-plexes.
Acknowledgements
The authors would like to thank J. Mark Parnisfor helpful discussions. We acknowledge LSVP at
Ž .UAM-Iztapalapa Mexico for providing computing´time on the Silicon Graphics Power Challenge com-puter.
References
w x1 J.M. Parnis, S.A. Mitchell, P.A. Hackett, J. Phys. Chem. 94Ž .1990 8152.
w x2 J.M. Parnis, S.A. Mitchell, M. Rayner, P.A. Hackett, J. Phys.Ž .Chem. 92 1988 3869.
w x3 J.M. Parnis, S.A. Mitchell, P.A. Hackett, Chem. Phys. Lett.Ž .151 1988 485.
w x4 S.A. Mitchell, B. Simard, D.M. Rayner, P.A. Hackett, J.Ž .Phys. Chem. 92 1988 1655.
w x5 S.A. Mitchell, P.A. Hackett, D.M. Rayner, M. Cantin, J.Ž .Phys. Chem. 90 1986 6148.
w x6 A. Martınez, A.M. Koster, D.R. Salahub, J. Phys. Chem. 101´ ¨Ž .1997 1532.
w x Ž .7 A. Martınez, J. Phys. Chem. 102 1998 1381.´w x8 L. Lian, S.A. Mitchell, D.M. Rayner, J. Phys. Chem. 98
Ž .1994 11637.w x Ž .9 A.M. Koster, J. Chem. Phys. 104 1996 4114.¨
w x Ž .10 M. Krack, A.M. Koster, J. Chem. Phys. 108 1998 3226.¨w x Ž .11 S.H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58 1980
1200.w x Ž .12 J.P. Perdew, Y. Wang, Phys. Rev. B 33 1986 8800.w x Ž .13 J.P. Perdew, Y. Wang, Phys. Rev. B 34 1986 7406E.w x Ž .14 J.P. Perdew, Phys. Rev. B 33 1986 8822.w x15 N. Godbout, D.R. Salahub, J. Andzelm, E. Wimmer, Can. J.
Ž .Phys. 70 1992 560.w x16 B.I. Dunlap, J.W.D. Connolly, J.R. Sabin, J. Chem. Phys. 71
Ž .1979 4993.w x Ž .17 J.W. Mintmire, B.I. Dunlap, Phys. Rev. A 25 1982 88.w x Ž .18 H.B. Schlegel, J. Comput. Chem. 3 1982 214.w x19 C. Blanchet, H.A. Duarte, D.R. Salahub, J. Chem. Phys. 106
Ž .1997 8778.w x Ž .20 J. Troe, J. Chem. Phys. 66 1977 4758.w x21 S.A. Mitchell, M.A. Blitz, R. Fournier, Can. J. Chem. 72
Ž .1994 587.








![Contour plots of electron density 2D PIC in units of [n |e|] cr wake wave breaking accelerating field laser pulse Blue:electron density green: laser](https://img.pdfslide.us/doc/110x75/56649cb75503460f9497c852/contour-plots-of-electron-density-2d-pic-in-units-of-n-e-cr-wake-wave.jpg)




















