Embed Size (px)
Citation preview

Thèse
Présentée devant
L’UNIVERSITE PAUL SABATIER TOULOUSE III
Ecole doctorale
CHIMIE
En vue de l’obtention du titre de
DOCTEUR DE L’UNIVERSITE PAUL SABATIER
Spécialité
CHIMIE-BIOLOGIE-SANTE
par
Christine TALMARD
Interaction entre le zinc(II) et le peptide
amyloïde bêta lié à la maladie d’Alzheimer
Soutenue le
19 novembre 2007
Rapporteurs : P. DELANGLE Directeur de recherche CEA (DRFMC/LCIB, Grenoble)
A.-M. ALBRECHT-GARY Directeur de recherche CNRS (LPCB, Strasbourg)
Examinateurs : R. MELKI Directeur de recherche CNRS (LEBS, Gif-sur-Yvette)
C. BLONSKI Directeur de recherche CNRS (LSPCMIB, Toulouse)
B. FRANCES Professeur d’Université (UPS, Toulouse) Directeur de thèse : P. FALLER Professeur d’Université (UPS, Toulouse)
Laboratoire de Chimie de Coordination du CNRS 205, route de Narbonne, 31077 Toulouse


1
Table des matières
Liste des abréviations 5
Chapitre 1 9
Le peptide amyloïde bêta, les ions métalliques et la maladie d’Alzheimer I. La maladie d’Alzheimer, vue d’ensemble 11
I.1. Définition clinique 11 I.2. Rappel historique 12 I.3. La maladie d’Alzheimer, priorité de santé publique 13 I.4. Le diagnostic 14 I.5. Facteurs génétiques, biologiques et environnementaux 16
I.5.a. Facteurs génétiques 16 I.5.b. Facteurs biologiques et environnementaux 18 II. Plaques amyloïdes et neurofibrilles 19
II.1. La protéine tau 20 II.2. Le peptide amyloïde bêta 23
II.2.a. Cas général des amyloïdoses 23 II.2.b. La protéolyse de l’APP 27 II.2.c. Le peptide Aβ 29 II.2.d. Structure des fibres amyloïdes 31 II.2.e. Relation entre Aβ et tau 33 II.2.f. Hypothèse de la cascade amyloïde –Toxicité liée à Aβ 35
II.3. Traitements 38 II.3.a. Traitements actuels 38 II.3.b. Voies de recherche 39
II.4. Influence des métaux 45 II.4.a. Introduction : les métaux et l’organisme 45 II.4.b. Modulation des métaux avec l’âge 50 II.4.c. Evolution du stress oxydant avec l’âge 50 II.4.d. Modulation des métaux dans la MA 51 II.4.e. Implication des métaux dans la MA 52 II.4.f. Approche thérapeutique en relation avec le cuivre et le zinc 61 III. Conclusion et contexte de l’étude 64 Références 65

2
Chapitre 2 101
Etude de la liaison du zinc(II) au peptide amyloïde bêta I. Résumé de l’étude 103 II. Publication 104 III. Complément : principe de la calorimétrie de titrage isotherme 128
Chapitre 3 133
Etude de l’oligomérisation du peptide amyloïde bêta durant son agrégation en présence de zinc(II) ou de cuivre(II) I. Contexte bibliographique et étude de l’influence des métaux sur le degré d’agrégation du peptide amyloïde bêta 135 II. Formation d’un complexe monomérique observable 137 III. Matériels et méthodes 142 Références 148
Chapitre 4 151
Etude structurale du mécanisme de promotion de l’agrégation du peptide amyloïde bêta par le zinc(II) I. Introduction 153 II. Résultats et discussion 155 III. Matériels et méthodes 162 Références 166
Chapitre 5 169
Etude de l’interaction entre la métallothionéine-3 et le peptide amyloïde bêta, influence du stress oxydant I. Introduction bibliographique 171

3
II. La MT-3, source potentielle du zinc retrouvé dans les plaques amyloïdes 174 III. Résultats et discussion 175 III.1. Hypothèse 175 III.2. Expériences avec le modèle Cd7-MT-3 176 III.2.a. Interaction entre Cd7-MT-3 et Aβ 176 III.2.b. Interaction entre Cd7-MT-3 et Aβ en présence de H2O2 177 III.2.c. Interaction entre Zn7-MT-3 et Aβ, en présence de H2O2 178
III.3. Agrégation de Aβ en présence de H2O2 et de Zn7-MT-3 180 IV. Matériels et méthodes 182 IV.1. Matériels 182 IV.1.a. Préparation de Cd7-MT-3 et Zn7-MT-3. 182 IV.1.b. Autres solutions 183
IV.2. Méthodes 183 IV.2.a. Analyse d’acides aminés 183 IV.2.b. Spectroscopie d’absorption atomique 184 IV.2.c. Chromatographie d’exclusion stérique 184 IV.2.d. Spectroscopie d’absorption UV-visible 184 Références 185
Chapitre 6 187
Etude de l’interaction du peptide amyloïde bêta avec les membranes, influence du zinc I. Bibliographie 189 II. Résultats et discussion 191 III. Méthode : la BLM 196 Références 201
Conclusion et perspectives 203
Annexes 207 Remerciements 235 Abstract 237

4

Abréviations
5
Liste des abréviations
Aβ Amyloïde bêta
AD Alzheimer’s Disease
ADAM A Disintegrin And Metalloprotease
ADN Acide Désoxyribonucléique
ADRDA Alzheimer's Disease and Related Disorders Association
AICD APP Intracellular Cytosolic Domain
AINS Anti-Inflammatoires Non Stéroïdiens
AMPA α-amino-3-hydroxy-5-méthylisoazol-4-propionate
apoE apolipoprotéine E
Aph-1 Anterior pharynx defective 1
APP Amyloid Precursor Protein
Arc Activity-regulated cytoskeleton-associated protein
ARN(m) Acide Ribonucléique (messager)
BACE1 Beta site Cleaving Enzyme 1
BCA Acide Bicinchoninique
BLM Black Lipid Membrane
BPLED Bipolar Longitudinal Eddy current Delay
CDK Cyclin-Dependent Kinase
CQ Clioquinol
DC Dichroïsme Circulaire
DFOA Desferrioxamine
DMSO Diméthylsulfoxyde
DOPC 1,2-dioleoyl-sn-glycero-3-phosphocoline
EDTA Acide éthylène-diamine-tétraacétique
ESI-MS
Electrospray Ionization Mass Spectrometry (Ionisation par electronébullisation – Spectrométrie de masse)
EXAFS Extended X-Ray Absorption Fine Structure
GIF Growth Inhibitory Factor (MT-3)
GSK Glycogen Synthase Kinase
GTP Guanosine 5’-TriPhosphate
Hepes Acide [(hydroxy-2-éthyl)-4-pipérazinyl-1]-2éthanesulfonique
HPLC High-Performance Liquid Chromatography

Abréviations
6
Hsp Heat shock protein
IDE Insuline Degrading Enzyme (ou insulinase)
IRM Imagerie par Résonance Magnétique
ITC Calorimétrie de titrage isotherme (Isothermal Titration Calorimetry)
IUPAC-IUB
International Union of Pure and Applied Chemistry - International Union of Biochemistry
JIP JNK Interacting Protein
JNK c-Jun N-terminal Kinase
kDa kiloDalton
Kdapp Constante de dissociation apparente
LA-ICPMS
Analyse par couplage de l'ablation laser et de la spectrométrie de masse à source plasma
LCR Liquide Céphalorachidien
MA Maladie d’Alzheimer
Mint Munc18 interacting protein
MMP Matrices MétalloProtéinases
Mr Poids moléculaire
MT Métallothionéine
NADH Forme réduite du nicotinamide adénine dinucléotide
NEP Néprilysine
NG Newport Green
NINCDS
National Institute of Neurological and Communicative Diseases and Stroke (actuellement : National Institute of Neurological Disorders and Stroke)
NMDA N-Méthyl-D-Aspartate
PAQUID Quid des personnes agées
PAR 4-(2-pyridylazo)-resorcinol
Pen-2 Presenilin enhancer 2
PGSE NMR Pulse Gradient Spin-Echo Nuclear Magnetic Resonance
PI-3K PhosphoInositol-3-Kinase
Pin1 Protein interacting with NIMA (never in mitosis A) 1
PPARγ Peroxisome Proliferator-Activated Receptor γ
PP2A Protéine Phosphatase 2A
PS1 Préséniline-1
PS2 Préséniline-2
PSD Post Synaptic Density protein
Rac1 Ras-related C3 botulinum toxin substrate 1

Abréviations
7
Rh Rayon hydrodynamique
RMN Résonance Magnétique Nucléaire
ROS Reactive Oxygen Species
SEC Size Exclusion Chromatography
TACE TNF-Alpha Converting Enzyme
TEP Tomographie par Emission de Positons
TFE 2,2,2-trifluoroéthanol
ThT Thioflavine T
TNF Tumor Necrosis Factor
Tris Tris(Hydroxymethyl)aminométhane
TSH Thyroid Stimulating Hormone
Zi Zincon
ZnT Zinc Transporter

8

9
Chapitre 1
La maladie d’Alzheimer,
introduction bibliographique

10

Chapitre 1 : la maladie d’Alzheimer
11
I. La maladie d’Alzheimer, vue d’ensemble
I.1. Définition clinique
La maladie d'Alzheimer (MA) est une maladie neurodégénérative du système nerveux central.
Elle entraîne progressivement la perte des fonctions mentales, du fait de la détérioration du
tissu cérébral (Figure 1.1), et conduit finalement à un syndrome démentiel. La nature
neurodégénérative de la MA se traduit par des lésions histopathologiques bien précises qui
sont les plaques séniles et les dégénérescences neurofibrillaires.
Figure 1.1. Comparaison de cerveaux de personnes saines et d’individus atteints de la maladie d’Alzheimer (d’après (Mattson 2004)). (a) Volume global du cerveau (rétrécissement marqué du lobe temporal (partie basse) et des lobes frontaux (partie gauche)). (b) Imagerie TEP révélant le glucose : le patient atteint d’Alzheimer montre une forte diminution du métabolisme énergétique dans le cortex
frontal (haut du cerveau) et les lobes temporaux (sur les cotés).
Anatomiquement, elle est caractérisée par des lésions de l’hippocampe (impliqué dans le
processus de mémorisation) et une atrophie corticale diffuse mais souvent à prédominance
pariéto-occipitale, ce qui rend compte de la symptomatologie aphasie-apraxie-agnosie.
Ainsi, le premier symptôme frappant est la perte de la mémoire à court terme (amnésie) ; elle
se manifeste initialement par des distractions mineures qui s'accentuent avec la progression de
la maladie, tandis que les souvenirs plus anciens sont relativement préservés. En même temps
que les désordres progressent, l'affaiblissement cognitif s'étend aux domaines de la langue
(aphasie), de l'adresse des mouvements (apraxie), de la reconnaissance (agnosie), et aux

Chapitre 1 : la maladie d’Alzheimer
12
fonctions exécutives que sont la prise de décision et la planification, rattachées de près aux
lobes frontaux, reflétant l'extension du processus pathologique.
I.2. Rappel historique
Le 4 novembre 1906, lors de la 37ème conférence des psychiatres allemands à Tübingen, Aloïs
Alzheimer, médecin psychiatre à Francfort, décrit un cas de démence d’une femme de 51 ans
Auguste D. (Alzheimer 1907; Alzheimer 1911).
Figure 1.2. Aloïs Alzheimer (1864-1915)
Cette patiente, admise à l’hôpital de Francfort le 25 novembre 1901, décède le 8 avril 1906.
Elle présentait de très nombreux symptômes, allant d’une compréhension et d’une mémoire
réduite jusqu’à l’aphasie, la perte du sens de l’orientation, des comportements imprévisibles,
de la paranoïa, des hallucinations auditives et un délabrement psychosocial avancé.
L’autopsie, à Munich, du cerveau de la patiente révéla des « plaques » formées par une
substance particulière dans le cortex (déjà observé sur un patient épileptique plus âgé par
Blocq et Marinesco)(Blocq and Marinesco 1892) et, pour la première fois, des
enchevêtrements de neurofibrilles (Figure 1.3) (Goedert and Spillantini 2006). Or, en ce
temps-là, un état de démence du sujet âgé etait considéré par la grande majorité des
psychiatres comme normal, lié à l'usure normale du temps, à la fameuse artériosclérose
(maladie artérielle caractérisée par un dépôt de cholestérol et de calcium sur les parois
internes des artères). Cependant, à la suite de cette première observation, Emil Kraepelin, un
des rares psychiatres à croire en l'intérêt de l'étude histologique du cerveau dans les maladies
mentales, parla pour la première fois, dans son traité de psychiatrie en 1912, de la " maladie
d'Alzheimer " définie alors comme une démence du sujet jeune (Auguste D. est décédée à

Chapitre 1 : la maladie d’Alzheimer
13
l’àge de 56 ans seulement), rare et dégénérative, laissant au terme de "démence sénile", les
démences vasculaires du sujet âgé (Kraepelin 1910). C’est pourquoi, jusque dans les années
1960, on supposait que la maladie était rare, mais plus tard on s'aperçut que dans beaucoup de
cas, ce que l'on avait pris pour des aspects normaux de la sénescence relevait en fait de la MA.
Figure 1.3. Section du cortex d’Auguste D. révélée à l’argent de Bielschowsky (a) mettant en évidence la présence de neurofibrilles (b) et de plaques amyloïdes (c) (Goedert et al. 2006).
I.3. La maladie d’Alzheimer priorité de santé publique
La MA est à présent la maladie neurodégénérative la plus répandue dans le monde avec
environ 25 millions de malades. En France, on estime à 860 000 le nombre de personnes
atteintes de la MA (Alzheimer 2006). Si les lésions se développent assez tôt dans la vie, la
maladie ne s’exprime habituellement que tardivement, l’âge étant le premier facteur de risque
(sauf dans le cas de mutations génétiques familiales, cf. section I.5.a). L’affection touche plus
de femmes que d’hommes puisqu’au delà de 75 ans, les proportions sont de 13,2 % pour les
hommes et de 20,5 % pour les femmes. Au-delà de 85 ans, la prévalence s’accroît de manière
exponentielle avec une proportion de 25 % de sujets atteints. L’incidence s’élève à 225 000
nouveaux cas par an en France et l’espérance de vie après le début des symptômes est en
moyenne de 8,5 ans (Tableau 1.1).
Actuellement 40 % des patients sont pris en charge dans une institution, ce qui signifie que
60% des sujets atteints sont à la charge des familles. La dépense moyenne pour la prise en
charge d’un patient est de 22 000 euros par an, soit une charge annuelle de 10 milliards
d’euros (moitié pour l’Etat et moitié pour les familles) qui se répartit en 75 % de dépenses
médico-sociales et 25 % de dépenses médicales. Actuellement, ces dépenses s’élèvent à 0,6 %
du PIB mais, compte tenu du vieillissement de la population, elles passeront à 0,8 % en 2020
(estimation à 1,3 millions de cas) et à 1,8 % en 2040 (2,1 millions). La prise en charge des

Chapitre 1 : la maladie d’Alzheimer
14
personnes atteintes de démence représentera alors, si la tendance actuelle continue, 7 % des
dépenses de santé.
Age Hommes Femmes Total
75-79 4,6 3,7 4,1
80-84 9,6 15,3 13,2
85-89 15,2 23,8 21,0
> 90 21,6 46,5 40,9
Total 9,1 17,2 14,2
Tableau 1.1. Prévalence de la maladie d’Alzheimer en France pour les personnes âgées de 75 ans et plus (données issues de la cohorte PAQUID) (Ramaroson et al. 2003).
Il n’est donc pas étonnant que la MA ait été récemment désignée « grande cause nationale
2007 » par le gouvernement français et que le parlement européen la considère comme un
« fléau » depuis déjà une dizaine d’années.
I.4. Le diagnostic
Aujourd’hui encore, le seul diagnostic définitif de la MA ne peut être établi que post mortem
via l’autopsie du cerveau du malade. Ainsi, dans un premier temps, le diagnostic a reposé sur
des tests cognitifs simples comme les échelles de Blessed (1968), de Pfeiffer (1975) et le
célèbre Mini Mental Status Examination de Folstein (1975). Ces tests ont été conçus comme
des outils de dépistage rapide et des indicateurs de sévérité de la démence, ils ne sont donc
pas spécifiques de la MA.
En 1984, le NINCDS-ADRDA, groupe américain de spécialistes de la MA, propose les
premiers critères diagnostic de la maladie. Peu à peu se dégage un consensus pour
l'élaboration d'outils d'évaluation performants regroupés autour de 4 axes :
- tests cognitifs : Mattis Dementia Rating Scale ; Alzheimer Disease Assessment Scale-
cognitive subscale (ADAS-cog) ; Syndrom Kurztest ;
- évaluation fonctionnelle (activité de la vie quotidienne) : Instrumental Activities of Daily
Living (IADL) ; Physical Self-maintenance Scale ;
- évaluation comportementale (troubles comportementaux et de l'humeur) : ADAS-non
cognitive subscale ; Behavioral pathology in Alzheimer Disease scale ; Neuropsychiatric
Chapitre 1 : la maladie d’Alzheimer
15
Inventory ;
- échelles de classification globale de sévérité : Global Deterioration Scale ; Clinical
Dementia rating Scale.
Actuellement, ces tests sont toujours d’actualité car il n’existe pas encore de marqueur
spécifique de la MA, toutefois ils s’accompagnent de quelques examens complémentaires
plus ou moins routiniers (Sarazin and Dubois 2005).
Le scanner cérébral ou l’IRM sont régulièrement pratiqués mais ces examens ne permettent
pas de mettre en évidence de lésions spécifiques : l'amincissement du cortex (atrophie
corticale ou sous-corticale) se voit dans d'autres maladies de la personne âgée ; ils servent
essentiellement à éliminer d'autres causes : tumeurs, accident vasculaire cérébral, hématome
intra-cérébral ou sous-dural... Des indices sont cependant en cours d'évaluation pour tenter de
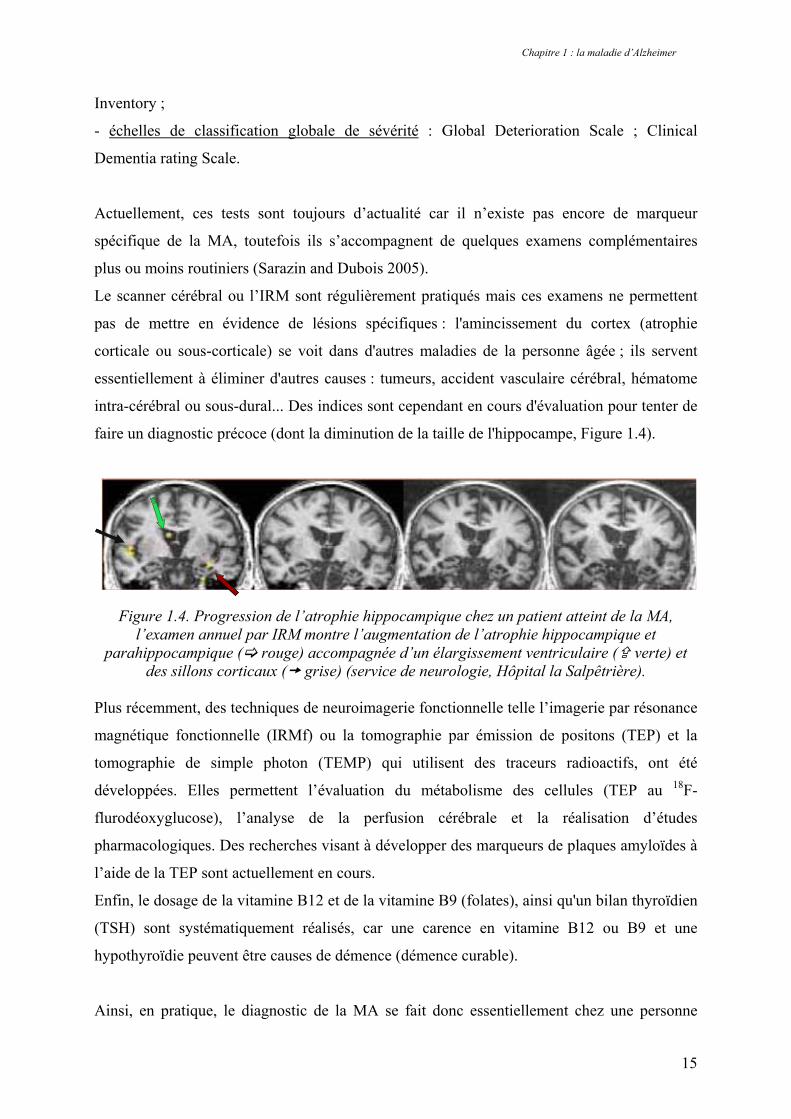
faire un diagnostic précoce (dont la diminution de la taille de l'hippocampe, Figure 1.4).
Figure 1.4. Progression de l’atrophie hippocampique chez un patient atteint de la MA,
l’examen annuel par IRM montre l’augmentation de l’atrophie hippocampique et parahippocampique ( rouge) accompagnée d’un élargissement ventriculaire ( verte) et
des sillons corticaux ( grise) (service de neurologie, Hôpital la Salpêtrière). Plus récemment, des techniques de neuroimagerie fonctionnelle telle l’imagerie par résonance
magnétique fonctionnelle (IRMf) ou la tomographie par émission de positons (TEP) et la
tomographie de simple photon (TEMP) qui utilisent des traceurs radioactifs, ont été
développées. Elles permettent l’évaluation du métabolisme des cellules (TEP au 18F-
flurodéoxyglucose), l’analyse de la perfusion cérébrale et la réalisation d’études
pharmacologiques. Des recherches visant à développer des marqueurs de plaques amyloïdes à
l’aide de la TEP sont actuellement en cours.
Enfin, le dosage de la vitamine B12 et de la vitamine B9 (folates), ainsi qu'un bilan thyroïdien
(TSH) sont systématiquement réalisés, car une carence en vitamine B12 ou B9 et une
hypothyroïdie peuvent être causes de démence (démence curable).
Ainsi, en pratique, le diagnostic de la MA se fait donc essentiellement chez une personne

Chapitre 1 : la maladie d’Alzheimer
16
présentant des signes de démence d'apparition progressive et pour lesquelles les autres causes
ont été éliminées. Un des enjeux majeurs de la recherche sur la maladie d’Alzheimer concerne
donc la découverte de marqueurs prédictifs fiables permettant de faire un diagnostic sûr et le
plus précoce possible, avant l’apparition de la maladie, c’est-à-dire lors de l’étape dite
asymptomatique qui peut durer plusieurs années. Ce dépistage précoce permettrait une
meilleure prise en charge et un allongement de la durée de vie du patient. La mise au point de
marqueurs spécifiques permettrait également un suivi et une évaluation plus efficaces des
traitements pharmacologiques.
I.5. Facteurs génétiques, biologiques et environnementaux
I.5.a. Facteurs génétiques
La MA est une affection polyfactorielle qui résulte de l’interaction entre un terrain génétique
et des facteurs environnementaux. L’implication des gènes dans la maladie d’Alzheimer est
double (Tableau 1.2) : d’une part, il existe des formes monogéniques exceptionnelles,
caractérisées par un début précoce (inférieur à 60 ans) et par l’atteinte d’un sujet sur deux à
chaque génération (formes autosomiques dominantes) ; d’autre part, dans les formes, de loin
les plus courantes, dites sporadiques de la maladie, sont impliqués des facteurs de risque
génétique (polymorphismes de l’ADN) qui constituent simplement un terrain génétique
favorable.
Chromosome Protéine Age moyen de survenue Conséquence biologique Conséquence
clinique
21 APP 50aine Augmentation de la production d’Aβ totale et Aβ42
MA familiale et/ou angiopathie amyloïde
14 Préséniline-1 40aine 50aine
Augmentation de la production d’Aβ42 MA familiale
1 Préséniline-2 50aine Augmentation de la production d’Aβ42 MA familiale
19 Apo E4 60aine et plus
Augmentation de la densité des plaques d’Aβ et des dépôts vasculaires
Risque supérieur de MA
Tableau 1.2. Principales mutations génétiques corrélées à la MA et leurs conséquences

Chapitre 1 : la maladie d’Alzheimer
17
Les formes familiales et héréditaires sont relativement rares (inférieure à 9%) et
correspondent à plus de 150 mutations réparties sur 3 gènes : un gène du chromosome 21
codant pour la protéine APP (amyloid precursor protein), précurseur du peptide amyloïde bêta
(Aβ) principal composant des plaques séniles ; un gène du chromosome 14 et du chromosome
1 codant respectivement pour le préséniline 1 (PS1) et 2 (PS2), les mutations sur ces derniers
favorisent la libération de la forme longue Aβ42 (versus Aβ40), moins soluble.
Quant aux facteurs de susceptibilité génétique, la principale mutation concerne un gène situé
sur le chromosome 19 qui code pour l’apolipoprotéine E (apoE) impliquée dans le transport
du cholestérol (Corder et al. 1993; Strittmatter et al. 1993). En effet, les individus porteur
d’un ou deux allèles ε4 (15 % de la population) ont plus de chance de développer la MA (40 à
65 % des cas sporadiques et familiales), contrairement aux allèles ε3, le plus courant, et ε2,
protecteur (Farrer et al. 1997). L’allèle ε4 est également un facteur de risque pour d’autres
maladies comme les démences vasculaires, la maladie de Creutzfeldt-Jacob et autres
démences. Les mécanismes biologiques qui sous-tendent le rôle de l’apoE sont multiples et
encore mal connus. Plusieurs hypothèses ont été émises : l’apoE E4 peut se lier au peptide Aβ
et pourrait ainsi favoriser son agrégation (Stratman et al. 2005) ; elle augmenterait la
production d’APP (Ye et al. 2005) ; l’apoE E4 est également la moins performante dans la
restauration des membranes lésées via l’apport des lipides constitutifs nécessaires (Mahley et
al. 2006) ; enfin elle pourrait, de manière indépendante au mécanisme amyloïde, avoir des
effets toxiques sur les mitochondries via le clivage de sa partie C-terminale bioactive(Huang
et al. 2001; Chang et al. 2005; Mahley and Huang 2006; Mahley et al. 2006).
Par ailleurs, deux gènes du chromosome 12 codant pour l’α2-macroglobuline et un récepteur
des lipoprotéines de faible densité (tous deux impliqués dans la dégradation de Aβ) ont été
identifiés comme facteurs de risque (Pericak-Vance et al. 1997; Myers and Goate 2001).
Enfin la présence d’un ou plusieurs loci sur le chromosome 10 pourrait également être un
facteur de risque pour la MA (Majores et al. 2000; Pericak-Vance et al. 2000). Cette région
pourrait correspondre à l’enzyme de dégradation de l’insuline (qui jouerait un rôle dans la
dégradation d’Aβ (Vekrellis et al. 2000) ; mais également dans l’augmentation du taux
d’Aβ42 (Ertekin-Taner et al. 2000; Myers et al. 2001)).

Chapitre 1 : la maladie d’Alzheimer
18
I.5.b. Facteurs biologiques et environnementaux
Nous avons déjà vu que l’âge est un facteur prédominant pour l’apparition de la MA (cf.
I.1.c). Le tableau 1.1 montre également que les femmes sont plus touchées que les hommes
du fait de leur espérance de vie plus longue mais également à cause de la baisse en
oestrogènes à la suite de la ménopause (Alberca et al. 2002; Candore et al. 2006). En effet les
oestrogènes interviennent dans de nombreux processus au sein du cerveau (apoptose, stress
oxydant, plasticité synaptique, effets sur les neurotransmetteurs, transport du glucose, réaction
inflammatoire…) dont certains ont une influence dans le développement de la MA
(Henderson 2000), cependant les mécanismes sous-jacents sont complexes et encore à
élucider. En effet, paradoxalement, les femmes de plus de 65 ans ayant une supplémentation
en œstrogène ont plus de chance de développer des démences (Henderson 2006).
De plus, un faible niveau d’éducation, ce qui est souvent le cas chez les femmes âgées, a
également une incidence négative et favorise l’apparition de la MA.
De nombreuses études ont également démontré qu’un taux élevé de cholestérol est un facteur
de risque pour la MA (Michikawa 2003). Des hypothèses, parfois contradictoires, ont
également été avancées concernant l’influence des radeaux lipidiques lors de la coupure de
l’APP (Riddell et al. 2001; Wahrle et al. 2002; Cordy et al. 2003; Ledesma et al. 2003; Chen
et al. 2006). Mais il faut savoir que le taux de cholestérol dans le sang est largement
indépendant de celui du système nerveux central (SNC), en effet ce dernier en synthétise la
majeur partie de novo, le lien serait donc plutôt indirect. D’autant plus que les traitements à
base de statines (visant à diminuer le taux cholestérol sanguin chez les patients atteint de
problèmes cardiovasculaires) ont été supposés diminuer l’incidence de la MA (Jick et al. 2000;
Wolozin et al. 2000), indépendamment de leur propriétés lipophiles et donc de leur capacité à
passer la barrière hémato-méningée (Canevari and Clark 2007).
De manière générale, une carence alimentaire en vitamines antioxydantes (notamment C et
E), fréquentes chez les personnes âgées alors que le stress oxydant est important, est un
facteur aggravant de la MA (Morris et al. 2006).
L’hypertension artérielle, qui est connue pour être un facteur aggravant de la MA, pourrait
également être un facteur de risque. De plus, des études ont montré que des personnes traitées

Chapitre 1 : la maladie d’Alzheimer
19
contre l’hypertension artérielle avaient un risque moindre de développer la MA, reste à savoir
si ce résultat est lié à l'effet direct des molécules ou uniquement au contrôle de la tension
(Birkenhager and Staessen 2004). Paradoxalement, il a été rapporté qu’une faible pression
diastolique (<65 mmHg) était également un facteur de risque pour la MA (Qiu et al. 2003).
La MA s’accompagne généralement de mécanismes inflammatoires (probablement dus au
stress oxydant), ainsi on note un rôle protecteur des anti-inflammatoires non stéroïdiens
(AINS) comme l’aspirine et l’ibuprofène. Mais les effets secondaires de ces médicaments ne
permettent pas d’envisager une prévention systématique. Cependant, de récentes et
prometteuses recherches ont démontré que certains de ces AINS, dont l’ibuprofène, ont la
propriété étonnante de diminuer le taux de peptides Aβ42 (forme longue plus agrégeante), via
peut-être une modulation de l’activité des γ-secrétases (Czirr and Weggen 2006).
Enfin on peut citer comme derniers facteurs de risque : les traumatismes crâniens (Fleminger
et al. 2003), l’exposition aux pesticides (Baldi et al. 2003), les dépressions nerveuses (Jorm et
al. 1991; Alexopoulos et al. 1993; Devanand et al. 1996). Il semblerait finalement qu’une
consommation modérée de vin ou d’alcool diminue le risque de démence et de MA
(Letenneur 2004).
II. Plaques amyloïdes et neurofibrilles
La maladie d'Alzheimer est caractérisée par la rencontre de deux processus dégénératifs
différents, qui semblent se conjuguer pour provoquer la dégénérescence des cellules
nerveuses : l'agrégation de la protéine tau sous forme de filaments dans les cellules nerveuses
(processus de dégénérescence neurofibrillaire ou pathologie tau) et l’agrégation du peptide
amyloïde bêta (Aβ) sous forme de plaques amyloïdes à l’extérieur des neurones. L’état actuel
des connaissances sur l’agrégation de la protéine tau sera présenté succinctement, puis nous
nous intéresserons plus longuement au peptide Aβ sur lequel porte cette thèse.
II.1. La protéine tau
Les protéines tau forment une famille de protéines dont la masse moléculaire s'échelonne de

Chapitre 1 : la maladie d’Alzheimer
20
45 à 62 kilodaltons. Elles sont codées par un seul gène contenant 16 exons et situé sur le
chromosome 17. L'épissage alternatif de ce gène génère la formation de six isoformes (de 48 à
67 kDa), co-existant dans les neurones dans certains rapports (Wischik et al. 1988; Goedert et
al. 1989; Goedert et al. 1989).
Ces protéines comportent des régions répétitives du côté C-terminal (en noir sur la figure 1.5)
qui permettent l'accrochage aux microtubules1 (Lee et al. 1989). En effet, le rôle des protéines
tau est de réguler l’assemblage des sous-unités de tubuline en microtubules : les protéines
phosphorylées induisent la dépolymérisation des microtubules alors que les protéines
déphosphorylées les stabilisent.
Figure 1.5. Représentation schématique des six isoformes de la protéine tau exprimées dans le cerveau humain adulte. Les régions communes à tous les isoformes sont représentées en
bleu, en noir les régions s’associant aux microtubules.
Dans la MA, la dégénérescence neurofibrillaire (Figure 1.6) résulte de l’agrégation
intraneuronale de protéines tau anormalement phosphorylées sous la forme de paires de
filaments appariés en hélice (Kidd 1963; Grundke-Iqbal et al. 1986; Ihara et al. 1986; Goedert
et al. 1988; Wischik et al. 1988; Wischik et al. 1988; Goedert et al. 1992). Ces filaments ont
un diamètre de 10 nm et un pas d'hélice de 80 nm, ils s'accumulent dans les corps cellulaires
des neurones ainsi que dans leurs prolongements neuritiques.
1 Les microtubules, fibres constitutives du cytosquelette, sont des tubes dont la paroi est constituée de plusieurs protofilaments de tubuline. Très nombreux dans les neurones, en particulier dans les dendrites et les axones, ils permettent d'acheminer divers composants vers leurs extrémités.

Chapitre 1 : la maladie d’Alzheimer
21
Figure 1.6. exemples de dégénérescences neurofibrillaires ou neurofibrilles (a : localisation
himmunohistochimique avec anticorps spécifiques de l’état de phosphorylation de tau (Goedert et al. 2006) ; b : section du cortex cérébral d’Auguste D. marqué à l’argent de
Bielscowsky (Graeber et al. 1998)).
Plusieurs observations et hypothèses ont été faites pour tenter d’expliquer la formation de ces
neurofibrilles à partir de protéines tau non mutées et donc a priori fonctionnelles.
• Les protéines tau hyperphosphorylées ont également été décrites comme étant
anormalement glycosylées, ce qui induirait une stabilisation l’hélicité des paires de
filaments (Wang et al. 1996). Cette glycosylation précède et favoriserait
l’hyperphosphorylation et ainsi la formation des filaments (à noter que la
glycosylation seule n’a pas d’effet sur la capacité d’assemblage des microtubules) (Liu
et al. 2002; Liu et al. 2004).
• L’état de phosphorylation d’une phosphoprotéine est fonction d’un équilibre entre
l’activité de protéines kinases et de protéines phosphatase. Or il se trouve que
l’activité d’une de ces dernières, la protéine phosphatase 2A (PP2A), est diminuée
d’environ 20 % dans le cerveau de personnes atteintes de la MA (Gong et al. 1993).
Cette diminution influe sur l’hyperphosphorylation de le protéine tau (Gong et al.
1995) et ainsi sur sa capacité d’assemblage des microtubules (Gong et al. 1994; Gong
et al. 2000; Bennecib et al. 2001). La diminution de PP2A est elle-même
probablement due à l’augmentation (d’environ 20 %) d’une de ces deux protéines
inhibitrices IPP2A 1 (protéine cytosolique de 30 kDa) (Li et al. 1995).
• La protéine tau est également plus facilement phosphorylée quand elle est libre que
liée aux microtubules.
• La séquence peptidique joue aussi un rôle, la vitesse et le taux de phosphorylation
a

Chapitre 1 : la maladie d’Alzheimer
22
variant avec le nombre de domaines de liaison aux microtubules (3 ou 4, en noir sur la
figure 1.5) et d’inserts N-terminaux (0, 1 ou 2, en rose et vert) (Singh et al. 1997).
Mais quelles sont les conséquences de cette hyperphosphorylation en terme de toxicité ? La
quantité globale en protéine tau est équivalente dans un cerveau sain et dans celui atteint de la
MA. Cependant la concentration en protéine tau hyperphosphorylée est environ 8 fois
supérieure dans ce dernier (Khatoon et al. 1992; Khatoon et al. 1994). Or
l’hyperphosphorylation diminue la capacité de la protéine à s’apparier avec les microtubules
(Lindwall and Cole 1984; Bramblett et al. 1993; Yoshida and Ihara 1993; Tanaka et al. 1995).
Pire encore, ces protéines tau anormalement phosphorylées désassemblent les MT déjà formés
en séquestrant les protéines tau normales (Alonso et al. 1994; Santacruz et al. 2005) La perte
en microtubules induit vraisemblablement une inhibition du transport axoplasmique suivie
d’une dégénérescence synaptique et neuronale.
Tauopathies
Maladie d’Alzheimer Sclérose latérale amyotrophique ou maladie de Charcot Maladie des grains argyrophiles Maladie de Parkinson précoce autosomique dominante Parkinsonisme autosomique dominant Maladie de Parkinson juvénile autosomique récessive Dégénérescence cortico-basale Dementia pugilistica (démence des boxeurs) Démence avec dégénerescence neurofibrillaire diffuse et calcifications intracrâniennes Syndrome de Down (trisomie 21) Démence britannique familiale Démence frontotemporale avec parkinsonisme liée au chromosome 17 Syndrome de Gerstmann-Sträussler-Scheinker Parkinsonisme atypique de Guadeloupe Maladie de Hallervorden-Spatz Dystrophie myotonique (maladie de Steinert) Maladie de niemann-pick de type C Maladie de Pick Parkinsonisme post-encéphalitique Angiopathie amyloïde cérébrale à protéine prion Gliose sous-corticale progressive Paralysie supranucléaire progressive Panencéphalite sclérosante subaiguë Démence à dégénérescence neurofibrillaire seule
Tableau 1.3. Maladies dans lesquelles la présence de dépôts de protéine tau a été décrite.

Chapitre 1 : la maladie d’Alzheimer
23
La formation de dépôts de protéines tau n’est pas spécifique à la MA. Ce processus
dégénératif, qui n’est pas forcément doublée de la présence de plaques amyloïdes (Spillantini
et al. 1996; Spillantini et al. 1998; Crowther and Goedert 2000), est en effet le plus répandu
parmi les pathologies démentielles et neurodégénératives. Une grande partie de ces maladies,
nommées tauopathies, est listée dans le tableau 1.3. Les sites de phosphorylation sont
similaires, avec des différences mineures entre les maladies. Cependant la distribution des
différents isoformes dans la composition des filaments varie. Ainsi la MA et le complexe
‘‘sclérose latérale amyotrophique / syndrome parkinsonien’’ de l’île de Guam sont
caractérisés par l’hyperphosphorylation des isoformes de 60, 64 et 68 kDa (présence
également, mais minoritaire, de l’isoforme de 72 kDa). En comparaison, l’isoforme de 60 kDa
n’apparaît pas dans les filaments correspondant à la paralysie supranucléaire progressive et la
dégénération corticobasale (Flament et al. 1991; Ksiezak-Reding et al. 1994), mais est
majoritaire, avec l’isoforme à 64 kDa, dans la maladie de Pick.
L’identification dans les années 90, de mutations dans le gène de l’APP et de la préséniline
dans les cas héréditaires de la MA (environ 1% des personnes atteintes) (Goate et al. 1991;
Murrell et al. 1991; Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995) et le
fait que les dépôts de filaments de protéines tau soient présents dans différentes conditions
sans relations apparentes, a conduit une partie de la communauté scientifique à considérer
cette agrégation comme un épiphénomène de peu d’importance. A l’heure actuelle, le débat
reste ouvert.
II.2. Le peptide amyloïde bêta
II.2.a. Cas général des amyloïdoses
Lors de la synthèse de protéines au sein d’une cellule, il y a toujours un risque qu’elles
s’agrègent. Mais la formation d’agrégats est normalement inhibée par les molécules
chaperons et les processus de dégradation, de plus ils sont défavorisés par les séquences
d’acides aminés choisies par l’évolution. Il arrive pourtant que certaines protéines, du fait de
mutations ou de facteurs environnementaux défavorables, s’agrègent ; dans le cas où les
agrégats sont de type fibrillaire (à distinguer des agrégats amorphes), il s’agit d’une
amyloïdose (ou amylose). Il existe ainsi de nombreuses maladies humaines associées à la
formation de fibres amyloïdes extracellulaires ou d’inclusions intracellulaires de type

Chapitre 1 : la maladie d’Alzheimer
24
amyloïde, à partir de protéines normalement solubles. Quelques exemples d’amyloses sont
donnés dans le tableau 1.4 où elles sont classées en trois groupes distincts : les maladies
neurodégénératives, comme la MA, dans lesquelles l’agrégation a lieu dans le cerveau ; les
amyloses non neuropathiques localisées pour lesquelles l’agrégation a lieu dans un type de
tissu précis autre que le cerveau et enfin les amyloses non neuropathiques systémiques dont
les dépôts sont retrouvés dans différents tissus de l’organisme (Chiti and Dobson 2006;
Gillmore and Hawkins 2006).
Maladie Protéine / peptide impliqué Nombre de résidus
Structure native
Maladies neurodégénératives
Maladie d’Alzheimer a Peptide amyloïde bêta 40-42 Non structuré
Encephalopathie spongiforme a, c
Prion 253 Résidus 1-120 non structuré Résidus 120-230 hélice alpha
Maladie de Parkinson a Α-synucléine 140 Non structuré
Maladie d’Huntington Huntingtine – poly Q 3144 Principalement non structuré
Démence britannique familiale b
Abri 23 Non structuré
Démence familiale danoise b
ADan 23 Non structuré
Amyloses non neuropathiques localisées
Diabète du type II a Amyline 37 Non structuré
Dystrophie héréditaire de la cornée b
kérato-épithéline (fragment C-terminal principalement)
50-200 Inconnu
cataracte a Cristalline γ variable Inconnu
myosite à inclusions (myopathie) a
Peptide bêta amyloïde 40-42 Non structuré
Amyloses non neuropathiques systémiques
Amylose AL (primaire) a
Immunoglobuline (chaîne légère) ~90 Feuillet bêta
Amylose AA (inflammatoire) a
Sérum amyloïde A (fragments) 76-104 Hélice alpha

Chapitre 1 : la maladie d’Alzheimer
25
Amylose ApoAI a, II a ou IV b
Fragment N-terminal de l’apolipoprotéine AI, II ou IV
98, ~70 ou 71
Non structurée
l’amylose de la lysozyme b
Lysozyme muté 130 Hélice alpha + feuillet bêta
l’amylose du fibrinogène b
Fibrinogène (chaîne α) 27-81 Inconnu
Amylose systémique sénile a
Transthyrétine normale 127 Feuillet bêta
Polyneuropathie amyloïde familiale b
Transthyrétine mutée 127 Feuillet bêta
a majoriatirement sporadique b majoritairement héréditaire c 5% de cas par transmission
Figure 1.4. Exemples et classification des amyloses (ou amyloïdoses) (Chiti et al. 2006; Gillmore et al. 2006).
Certaines de ces amyloses sont majoritairement sporadiques, c’est le cas de la MA et de la
maladie de Parkinson, bien que des formes héréditaires soient également présentes (Baltasar-
Rodriguez et al. 2006). D’autres, comme l’amylose de la lysozyme ou celle du fibrinogène,
sont dues à des mutations spécifiques et sont héréditaires. A cela faut-il ajouter
l’encéphalopathie spongiforme qui peut être transmissible (5 % des cas contre 85 % de formes
sporadiques et 10 % héréditaires).
De façon générale, les dépôts amyloïdes extracellulaires sont majoritairement constitués de la
protéine incriminée, qui forme le cœur du dépôt, associée à diverses molécules
(glycosaminoglycanes, apolipoprotéine E, collagène, ions métalliques,… et bien d’autre). Les
fibres qui composent en partie ces dépôts, sont généralement constituées de 2 à 6
protofilaments de 2 à 5 nm de diamètre chacun (Serpell et al. 2000) (à ne pas confondre avec
les protofibrilles, agrégats sphériques de diamètre similaire constitués d’une vingtaine de
protéines en feuillets bêta) (Harper et al. 1997; Harper et al. 1997; Walsh et al. 1997; Walsh et
al. 1999). Une description plus précise de la structure des fibres amyloïdes sera donnée pour
le cas de la MA dans la section II.2.d.
Ces différentes protéines susceptibles de former des dépôts amyloïdes n’ont paradoxalement
pas de séquence peptidique commune ou d’homologie structurale évidente. En effet, autant du
point de vue des structures secondaires que des longueurs / compositions des chaînes

Chapitre 1 : la maladie d’Alzheimer
26
peptidiques, l’hétérogénéité est de mise. Cependant, un nombre élevé de résidus hydrophobes,
l’absence de charge nette locale ou globale élevée, et une propension à former des structures
en feuillets bêta, semblent être les trois dénominateurs communs de ces protéines (Broome
and Hecht 2000; Otzen et al. 2000; Schwartz et al. 2001; Chiti et al. 2002; Uversky 2002;
Wurth et al. 2002).
Enfin, il faut noter que, dans quelques cas relativement rares, la formation de fibres amyloïdes
a été conservée par l’évolution pour générer des nanostructures naturelles exploitées par
l’organisme. La plupart de ces amyloses naturelles connues sont développées par des
bactéries, champignons ou levures (Coustou et al. 1997; Eaglestone et al. 1999; True and
Lindquist 2000; Mackay et al. 2001; Chapman et al. 2002; Claessen et al. 2003; Chien et al.
2004; True et al. 2004). Mais il en existe aussi par exemple chez l’araignée : les fibres de
spidroïne forment la soie de la toile de la Nephila Edulis (Kenney et al. 2002). Chez l’être
humain, un fragment de la glycoprotéine Pmel17 polymérise en fibres amyloïdes afin de
séquestrer la mélanine, durant la biogenèse des mélanosomes. Cette fibrillation est régulée via
la coupure de Pmel17 par la furine, une convertase de proprotéine, ce qui n’est pas sans
rappeler la coupure de l’APP (Amyloid Precurseur Protein) par les γ et β-sécrétases
impliquées dans la MA (cf. section II.2.b). Le mécanisme de ces amyloses naturelles
contrôlées pourrait ainsi inspirer l’élaboration de traitement permettant de réguler les
amyloses pathologiques.
II.2.b. La protéolyse de l’APP
Description et fonction de l’APP
L’APP (amyloid precursor protein) est une protéine transmembranaire de type 1 du système
nerveux central, d’environ 700 acides aminés. Elle est constituée d’un large domaine
extracellulaire (environ 88% du poids total, pour l’isoforme majoritaire), d’une unique région
transmembranaire et d’une petite partie cytosolique. Son rôle exact n’est pas encore élucidé,
mais il est clair qu’elle est impliquée dans la synaptogénèse et la plasticité synaptique (Gralle
and Ferreira 2007). Il a été montré que l’APP interagissait avec de nombreuses protéines : la
protéine Go (protéine liant le GTP et intervenant donc dans la signalisation intracellulaire)
(Nishimoto et al. 1993), des protéines adaptatrices comme, par exemple, Fe65 (impliquée
dans la mobilité cellulaire, elle stimulerait la production d’APP/Aβ et interagirait également

Chapitre 1 : la maladie d’Alzheimer
27
avec la protéine tau) (Sabo et al. 2001; Sabo et al. 2003; Barbato et al. 2005; Yoon et al.
2005), X11/Mint (impliquée, entre autres, dans la formation des synapses, diminuerait la
production d’APP/Aβ) (Lau et al. 2000; Mueller et al. 2000) et JIP (dont l’interaction avec
APP déboucherait sur une dégradation du signal axonal via la protéine kinésine-1) (Matsuda
et al. 2003).
Génération du peptide Aβ
La région Aβ se situe au niveau de la partie membranaire de la protéine APP. Elle est
composée des 28 acides aminés jouxtant le domaine transmembranaire et des 12 à 14 résidus
voisins faisant partie du domaine transmembranaire. L’Aβ résulte d’un clivage séquentiel de
l’APP par deux enzymes protéolytiques (Figure 1.7) : la β- et la γ-sécrétase (Wolfe 2006).
Dans un premier temps, la β-sécrétase, une protéase aspartique aussi nommée BACE1 (pour
Beta site APP Cleaving Enzyme 1, également appelée memapsin 2) et dont l’activité
enzymatique n’est possible qu’en milieu subcellulaire (à pH acide), clive l’APP au niveau du
site bêta, libérant dans le milieu extracellulaire un large ectodomaine soluble (sAPPβ) (Vassar
et al. 1999). Puis le fragment transmembranaire C-terminal restant, C99, est clivé au niveau
de la bicouche lipidique près de la frontière cytosolique par la γ-sécrétase, générant ainsi les
peptide Aβ40 ou Aβ42 et le fragment AICD (APP Intracellular Cytosolic Domain) libéré dans
le cytosol.
L’APP peut également, et plus couramment, être clivé par l’α-sécrétase plutôt que la β-
sécrétase, c’est la voie non amyloïdogénique. L’α-sécrétase est une protéine de la famille
ADAM (A Disintegrin And Metalloprotease, protéines intervenant dans l’adhésion, la
migration cellulaire et le clivage protéolytique de cytokines et facteurs de croissance), la
littérature propose 3 candidates : ADAM-10, ADAM-17 (TACE) et ADAM-9 (Kojro and
Fahrenholz 2005). Le site alpha se trouvant au milieu de la région Aβ, le clivage
correspondant libère un large ectodomaine soluble (sAPPα), puis l’action de la γ-sécrétase sur
le fragment C-terminal restant C83 génère le peptide p3 non pathogène.

Chapitre 1 : la maladie d’Alzheimer
28
Figure 1.7. Représentation schématique du clivage de l’APP libérant le peptide Aβ (Pearson
and Peers 2006).
BACE1 est une protéine membranaire de type 1 caractérisée par un large domaine
extracellulaire comprenant deux résidus aspartiques impliqués dans l’activité β-sécrétase
(Hussain et al. 1999). Elle semble être particulièrement active dans les endosomes (son
activité requérant un pH acide), ce qui pourrait indiquer que le clivage de l’APP se fait
préférentiellement durant son internalisation (Koo and Squazzo 1994; Koo et al. 1996; Perez
et al. 1999). Au niveau de la membrane, elle est localisée dans les radeaux lipidiques
(microdomaines de la membrane biologique enrichis notamment en cholestérol) (Riddell et al.
2001; Cordy et al. 2003; Ehehalt et al. 2003; Abad-Rodriguez et al. 2004), ces derniers
renforçant son activité (Kalvodova et al. 2005). De récentes études ont également avancé que
BACE1 pourrait se dimériser, acquérant ainsi une spécificité catalytique envers l’APP, en
particulier si celui-ci est sous forme d’homodimères ou tétramères (Multhaup 2006).
La γ-sécrétase, qui ne coupe que les liaisons peptidiques situées à l’intérieur de la bicouche
lipidique (Weihofen and Martoglio 2003; Wolfe and Kopan 2004), est un complexe composé
de 4 protéines : la préséniline (PS1 et PS2), la nicastrine, Aph-1 et Pen-2 (De Strooper et al.
1998; Yu et al. 2000; Francis et al. 2002; Goutte et al. 2002). La stoechiométrie et la fonction
de chacune de ces sous-unités au sein du complexe restent à déterminer, on sait cependant que
l’activité catalytique du complexe est très probablement portée par PS1 (Herreman et al. 2000;
Zhang et al. 2000). Il est à noter que la préséniline joue également un rôle essentiel dans la
voie de signalisation Notch (mis-en jeu dans la différenciation cellulaire) (Artavanis-Tsakonas
et al. 1995; De Strooper et al. 1999; Radtke et al. 2004), limitant ainsi les applications

Chapitre 1 : la maladie d’Alzheimer
29
thérapeutiques la ciblant. La majorité des cas héréditaires de la MA proviennent de mutations
au niveau des gènes codant pour PS1 ou PS2.
La phosphorylation de APP sur certains résidus sérine et thréonine joue également un rôle
important dans la production de Aβ (Lee et al. 2003; Lu et al. 2003). Plus précisément, il a été
montré que la phosphorylation du motif Thr668-Pro induit un changement conformationnel
(trans → cis, passage de ~0 à ~10% de formes cis) favorisant la voie amyloïdogénique
(clivage par la β-sécrétase au lieu de l’α-sécrétase) (Ramelot and Nicholson 2001). Pour
rétablir l’équilibre initial, la peptidyl-prolyl isomérase Pin1 catalyse le changement cis →
trans, favorisant ainsi à nouveau la voie non-amyloïdogénique (Figure 1.8).
Figure 1.8. Diminution de la production de Aβ sous l’effet de la protéine Pin1, catalyseur de
l’isomérisation du motif pThr 668-Pro de l’APP (Pastorino et al. 2006).
Une dérégulation de la protéine Pin1 et/ou son inhibition par oxydation pourrait donc
favoriser le développement de la MA (Pastorino et al. 2006).
Pin1 pourrait également avoir un effet protecteur dans les tauopathies (cf. section II.1) et le
cancer (Lu 2003; Lu et al. 2006).
II.2.c. Le peptide Aβ
Description

Chapitre 1 : la maladie d’Alzheimer
30
Le clivage de l’APP par la β- et la γ-sécrétase conduit principalement à un peptide Aβ de 40
acides aminés noté Aβ40. Cependant, d’autres formes plus courtes ou plus longues sont
également produites (entre 39 et 43 acides aminés), en particulier la forme Aβ42, dont la
séquence peptidique est indiquée sur la figure 1.9, représente environ 10 % des espèces d’Aβ.
Les deux peptides Aβ40 et Aβ42 sont normalement présents en tant qu’espèces solubles dans
les fluides biologiques de tous les individus. Cependant l’Aβ42, plus hydrophobe et principal
constituant des plaques séniles, est supposé jouer un rôle plus important qu’Aβ40 dans la
pathologie.
Asp1-Ala-Glu-Phe-Arg-His6-Asp-Ser-Gly-Tyr10-Glu11-Val-His13-His14-Gln-
Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-
Gly-Leu-Met35-Val-Gly-Gly-Val-Val40-Ile41-Ala42
Figure 1.9. Séquence peptidique du peptide Aβ42, écrite du N- vers le C-terminal. La partie en gras correspond au domaine intramembranaire. En bleu sont indiqués les acides aminés correspondant uniquement à Aβ42, en rouge ceux ayant été évoqués comme participant à la chélation des ions métalliques (dans le site de plus forte affinité) (Clippingdale et al. 2001).
Dans le cadre des études portant sur l’interaction du peptide avec les métaux, les formes
tronquées Aβ16 et Aβ28 (c’est-à-dire comprenant les acides aminés respectivement 1 à 16 et
1 à 28) sont couramment utilisées. En effet, elles contiennent les acides aminés correspondant
au principal site de fixation des métaux et ont l’avantage d’être entièrement (Aβ16) ou
partiellement (Aβ28) solubles, ce qui peut dans certains cas faciliter l’expérimentation mais
ne permet généralement pas l’étude à l’état agrégé.
Rôle du peptide Aβ chez le sujet sain
Le fait que le peptide Aβ soit généré tout au long de la vie sans forcément induire une
pathologie, permet de penser qu’il a peut-être une fonction physiologique. Cependant cette
dernière est encore largement méconnue : une activité neurotrophique et un effet stimulant sur
la viabilité neuronale ont été évoqués (Whitson et al. 1990; Yankner et al. 1990), notamment
en contrôlant un relargage excessif de glutamate dans la synapse (Lesne and Kotilinek 2005;
Pearson et al. 2006), l’hypothèse d’une régulation au niveau de canaux ioniques et dans
l’homéostasie du calcium a également été avancée (Abramov et al. 2004; Plant et al. 2006). Il

Chapitre 1 : la maladie d’Alzheimer
31
a également été montré qu’Aβ42 pouvait réguler négativement le taux de sphingomyéline et
Aβ40 la synthèse de cholestérol (Grimm et al. 2005). Il faut cependant être prudent dans
l’assignation d’un rôle physiologique à Aβ, sa séquence peptidique étant très peu conservée
chez les humains et rongeurs (bien que ce ne soit pas le cas chez d’autres mammifères), il est
possible que ce peptide n’ait pas de fonction physiologique essentielle.
II.2.d. Structure des fibres amyloïdes
Le processus de formation de fibres et de plaques amyloïdes à partir du peptide monomérique
soluble, sujet d’intenses recherches, n’a pas encore été élucidé. Mais il est admis que ce
processus passe par l'association de petits agrégats ordonnés ou unités de nucléation, dont la
croissance va déboucher sur de longs protofilaments qui s'enroulent pour former des fibres
amyloïdes matures (figure 1.10).
Figure 1.10. Représentation schématique du processus de formation des fibres amyloïdes.
Cependant, la structure de fibres formées in vitro est, elle, bien décrite dans la littérature.
Cette description a été élaborée à partir de données expérimentales obtenues principalement
par RMN du solide (en accord avec d’autres techniques RMN, diffraction des rayons X et
microscopie électronique) (Malinchik et al. 1998; Antzutkin et al. 2000; Goldsbury et al. 2000;
Antzutkin et al. 2002; Balbach et al. 2002; Petkova et al. 2002; Antzutkin et al. 2003).

Chapitre 1 : la maladie d’Alzheimer
32
Figure 1.11. Modèle structural des protofilaments d’Aβ40 (Tycko 2003),(a) représentation en ruban, (b) représentation atomique (en vert les résidus hydrophobes, en magenta les résidus
polaires, en bleu ceux chargés positivement, en rouge ceux chargés négativement).
Dans le modèle structural des protofilaments obtenu par RMN du solide, figure 1.11, les
peptides sont agencés en couches de deux feuillets bêta, perpendiculairement à l’axe de la
fibre (structure dite « cross bêta »). Les résidus 12-24 et 30-40 sont impliqués dans les
feuillets bêta, 25-29 dans le coude, les 10 premiers acides aminés ne sont pas structurés. Les
liaisons C=O et N-H du squelette sont approximativement perpendiculaires à la page. Le cœur
du protofilament est majoritairement hydrophobe (interactions hydrophobes entre brins), à
l’exception des résidus chargés D23 et K28 qui forment un pont salin. La diversité de
morphologie des fibres observées en microscopie électronique reflète les différences
d’association latérale des protofilaments (Goldsbury et al. 1997; Jimenez et al. 2002).
Les protofilaments formés par le peptide Aβ42 ont vraisemblablement une structure très
similaire, leur plus grande vitesse de formation serait d’avantage due à la moindre solubilité
d’Aβ42 par rapport Aβ40 plutôt qu’à une différence structurale (Antzutkin et al. 2002; Torok
et al. 2002; Tycko 2003).

Chapitre 1 : la maladie d’Alzheimer
33
Les caractéristiques fondamentales des fibres amyloïdes sont une coloration au rouge Congo
(associée à une biréfringence en lumière polarisée), une augmentation/déplacement de la
fluorescence de la Thioflavine T et leur résistance aux protéases.
II.2.e. Relation entre Aβ et tau
Bien que les deux caractéristiques de la MA, les plaques amyloïdes et les neurofibrilles, aient
été identifiées depuis maintenant plus de cent ans, les relations existant entre Aβ et tau sont
encore mal définies. Actuellement, les dernières recherches tendent à montrer qu’Aβ pourrait
directement ou indirectement interagir avec tau pour accélérer la formation de neurofibrilles
(Gotz et al. 2001; Lewis et al. 2001; Oddo et al. 2003), confirmant ainsi l’hypothèse de la
cascade amyloïde. A titre d’exemple, l’accumulation de plaques amyloïdes précède de
quelques mois le dévelopement de neurofibrilles chez des souris triplement mutées (sur l’APP,
tau et PS1) (Oddo et al. 2003). Il semble donc qu’Aβ, via des mécanismes cellulaires et
moléculaires distincts, facilite la phosphorylation, l’agrégation, une mauvaise localisation et
l’accumulation de tau, aboutissant sur l’apparition de neurofibrilles (Blurton-Jones and
Laferla 2006).
Il a été montré qu’un grand nombre de kinases, en particulier GSK3β et CDK5 (impliquées
dans la phosphorylation de tau et donc indirectement dans la formation de neurofibrilles)
(Cruz et al. 2003; Necula and Kuret 2004; Necula and Kuret 2005), voient leur expression
augmentée dans la MA (Pei et al. 2001; Ferrer et al. 2002; Swatton et al. 2004; Derkinderen et
al. 2005; Griffin et al. 2005; Ho et al. 2005; Stoothoff and Johnson 2005). Or des expériences
in vitro ont montré que la présence d’Aβ dans les cultures cellulaires entraîne une activation
de GSK3β et une augmentation de la phosphorylation de tau (Takashima et al. 1993;
Busciglio et al. 1995; Alvarez et al. 1999). De plus, certaines études semblent indiquer que la
toxicité induite par Aβ est dépendante de tau. Par exemple, le blocage in vitro de GSK3β et
CDK5 protège les neurones contre Aβ (Michaelis et al. 1998; Rapoport et al. 2002). De même,
in vivo, des souris transgéniques porteuses du gène muté de l’APP exhibent une augmentation
de l’activation de kinases de tau (Hwang et al. 2004; Puig et al. 2004). Plus récemment, une
réduction in vivo de la quantité d’oligomères solubles d’Aβ (mais pas des insolubles) a été
corrélée avec une réduction de l’activité de GSK3β et de la phosphorylation de tau (Ma et al.
2006). Il faut cependant rappeler que les souris transgéniques porteuses du gène muté de

Chapitre 1 : la maladie d’Alzheimer
34
l’APP ne développent pas la pathologie correspondante à l’hyperphosphorylation de tau et à
la formation de neurofibrilles (excepté une très faible quantité au niveau des plaques
neuritiques extracellulaires). En dehors de l’espérance de vie limitée des rongeurs, il est
possible que la différence d’espèce implique l’absence d’une kinase nécessaire au lien entre
Aβ et tau.
Il est établi que les cerveaux atteints de la MA sont particulièrement sujets à des réactions
inflammatoires (celles-ci étant à la fois bénéfiques – phagocytose d’Aβ - et préjudiciables -
activation des cytokines proinflammatoires) (Duffy et al. 1980; Sheng et al. 1995; Akiyama et
al. 2000; Wyss-Coray and Mucke 2002; Koenigsknecht-Talboo and Landreth 2005). De plus,
des études in vitro ont également montré qu’Aβ était capable d’induire directement une
réponse inflammatoire (Galimberti et al. 1999; Tan et al. 2000; Combs et al. 2001). Or
certaines cytokines proinflammatoires induisent, in vitro et parfois in vivo, une accélération
de l’hyper phosphorylation de tau et de la formation de neurofibrilles (Sheng et al. 2000; Li et
al. 2003; Quintanilla et al. 2004). Ce dernier mécanisme semble impliquer CDK5 (Cruz et al.
2003; Kitazawa et al. 2005).
Plusieurs études ont également montré que la voie ubiquitine-protéasome, mécanisme
principal de dégradation des protéines, était déficient chez les patients atteints de la MA
(réduction de 48 % de l’activité du protéasome dans l’hippocampe) (Keller et al. 2000) et que
cette déficience pourrait contribuer à la formation d’agrégats de tau. En effet de récentes
études semblent indiquer qu’Aβ est impliqué directement ou indirectement dans le
dysfonctionnement du protéasome (Gregori et al. 1997; Lopez Salon et al. 2003; Oh et al.
2005), tandis qu’il existe de nombreux indices attestant d’un lien fort entre la déficience du
protéasome et l’accumulation de tau, même s’il n’est pas encore bien établi si le phénomène
est en aval ou en amont dans la cascade moléculaire et cellulaire (Mori et al. 1987; Perry et al.
1987; Keller et al. 2000; Shimura et al. 2004; Sahara et al. 2005; Qian et al. 2006).
Enfin, une déficience dans le transport neuronal pourrait aussi jouer un rôle important dans la
MA, ainsi que d’autres maladies neurodégénératives (Roy et al. 2005). En effet, des études
montrent une interaction entre l’APP, Aβ et le transport axonal, mais ces interactions sont
complexes et il n’est pas encore clair à partir duquel, du dysfonctionnement du transport
axonal ou de l’augmentation de la production d’Aβ, le mécanisme débute (Koo et al. 1990;
Kamal et al. 2000; Kamal et al. 2001; Pigino et al. 2003; Lazarov et al. 2005; Stokin et al.

Chapitre 1 : la maladie d’Alzheimer
35
2005). Parallèlement, certaines données indiquent que l’augmentation de la production ou la
diminution de la dégradation de tau conduirait à une déficience du transport axonal (Ebneth et
al. 1998; Stamer et al. 2002; Mandelkow et al. 2004). De plus, la protéine tau et son ARNm
sont eux-mêmes transportés le long des axones, ainsi un déficit du transport axonal (du fait
par exemple d’une phosphorylation des kinésines, protéines motrices des microtubules, induit
par Aβ) pourrait conduire à une localisation erronée de tau et de son ARNm (cf. revue
(Blurton-Jones et al. 2006)).
L’ensemble de ces données bibliographiques met clairement en évidence une interaction plus
ou moins directe, via des mécanismes distincts, entre Aβ et tau. Il est à noter que la majorité
des ces données pointe plutôt en faveur de l’hypothèse de la cascade amyloïde : la pathologie
amyloïde induirait la formation de neurofibrilles et serait ainsi la cause première de la MA.
II.2.f. Hypothèse de la cascade amyloïde –Toxicité liée à Aβ
L’hypothèse de la cascade amyloïde (Figure 1.12) considère l’agrégation d’Aβ comme
l’initiateur de la MA, plaçant les pathologies associées à tau, ainsi que d’autres modifications
dégénératives, en aval (Hardy and Higgins 1992; Hardy and Selkoe 2002). Cette hypothèse
provient de l’observation des plaques amyloïdes dans le cerveau des patients atteints de la
MA qui est caractéristique de la pathologie, ce qui n’est pas le cas des dégénérescences
neurofibrillaires (cf le tableau 1.3 des tauopathies) ; mais elle a surtout été renforcée par la
découverte des mutations génétiques responsables de la forme familiale autosomale
dominante de la MA sur les gènes codant pour l’APP, PS1 ou PS2 (Hardy et al. 2002). Ainsi,
d’après cette hypothèse, l’agrégation anormale d’Aβ entraînerait une série de déséquilibres et
de processus neurotoxiques débouchant sur la mort neuronale et la démence.
Cependant, l’hypothèse de la cascade amyloïde, même si elle est considérée par une grande
partie de la communauté scientifique comme une base pour la compréhension du processus de
développement de la MA, est en constante évolution. En effet, la corrélation entre plaques
séniles et pertes cognitives n’était pas satisfaisante (« [plaques appear] at the wrong time and
in the wrong places » (Naslund et al. 2000)). Depuis, un grand nombre d’articles ont attesté
d’une meilleure corrélation et d’une toxicité comparativement bien plus importante pour les
oligomères de faible poids moléculaire (protofilaments, petits oligomères, monomères,
agrégats sphériques…mais la difficulté d’isoler ces différents types d’agrégats n’a pas permis

Chapitre 1 : la maladie d’Alzheimer
36
d’apporter une réponse définitive à ce jour) (Lambert et al. 1998; Haass and Steiner 2001;
Walsh et al. 2002; Kayed et al. 2003; Stefani and Dobson 2003; Klein et al. 2004).
Figure 1.12. Hypothèse classique de la cascade amyloïde pour les formes familiales de la
maladie d’Alzheimer (d’après (Hardy et al. 2002)). Pour les formes sporadiques, les causes de l’augmentation de la production et de l’accumulation d’Aβ sont moins bien définies.
D’un point de vue de la toxicité, ces espèces de faibles poids moléculaire auraient la capacité
d’induire une dépolarisation et une perméabilisation des membranes (celles-ci facilitant
probablement l’étape de nucléation), altérant ainsi l’activité neuronale et débouchant
finalement sur la mort cellulaire (Hartley et al. 1999; Stefani 2006). Plus précisément, un
déséquilibre au niveau du calcium intracellulaire a été plusieurs fois observé (voir revue
(Stefani et al. 2003)), ce déséquilibre pourrait affecter le bon fonctionnement des
mitochondries (perméabilité, libération de cytochrome C) et/ou directement activer les
caspases, conduisant ainsi à l’apoptose (Orrenius et al. 2003). D’autres voies de toxicité ont
également été avancées : déséquilibre dans l’homéostasie des lipides (Janciauskiene and
Ahren 2000), interactions avec des récepteurs membranaires en quantité croissante (du fait de

Chapitre 1 : la maladie d’Alzheimer
37
l’accumulation de produits terminaux de glycation avancée) (Mruthinti et al. 2003; Mruthinti
et al. 2006), liaisons à des intégrines (protéines d’adhésion) activant des voies de signalisation
intracellulaires (Caltagarone et al. 2007), formation de pores au niveau des membranes
(Arispe et al. 1993; Yip and McLaurin 2001; Ji et al. 2002; Quist et al. 2005) (cf. chapitre 5),
accumulation d’Aβ dans les mitochondries couplée à des interactions enzymatiques
perturbant son fonctionnement (Chen and Yan 2006).
Les premiers symptômes de la maladie, comme les défauts de stockage mnésique, pourraient
s’expliquer par une localisation préférentiellement synaptique des oligomères d’Aβ (en
particulier au niveau des clusters de protéines PSD-95) (Allison et al. 2000), associée à la
surexpression de protéines Arc (activity-regulated cytoskeletal-associated protein) inhibitrices
du processus de mémorisation à long terme (Guzowski 2002).
La formation d’un complexe entre Aβ et l’hème pourrait également être source de production
d’espèces oxydantes par les mitochondries, le complexe se comportant comme une
peroxydase (Atamna 2006).
La toxicité liée à l’interaction avec les métaux sera abordée dans la section II.4. de ce chapitre.
Cependant, en marge des courants tauistes (qui place les neurofibrilles à l’origine du
développement de la MA) et βAPPtistes (soutenant l’hypothèse de la cascade amyloïde), une
partie de la communauté scientifique s’interroge encore sur le bien-fondé de ces deux axes de
recherche, argumentant que les deux types de lésions observés chez les patients atteints de la
MA sont pathognomoniques (caractéristiques de la maladie) mais pas forcément
pathologiques, pointant le manque de corrélation entre neurofibrilles / dépôts amyloïdes et
pertes cognitives / mort neuronale (Knowles et al. 1998; Andorfer et al. 2005; Santacruz et al.
2005; Nunomura et al. 2006). En se basant sur (1) l’observation d’une diminution des lésions
représentatives de stress oxydant au voisinage des dépôts amyloïdes (Nunomura et al. 1999;
Nunomura et al. 2000; Nunomura et al. 2004), (2) sur le fait que l’apparition de stress oxydant
semble précéder les dépôts amyloïdes (Misonou et al. 2000; Nunomura et al. 2000; Paola et al.
2000; Pratico et al. 2001; Bayer et al. 2003; Drake et al. 2003; Li et al. 2004; Sung et al.
2004), (3) un effet protecteur d’Aβ40 et 42 sur le stress oxydant induit par le fer et le cuivre
(Zou et al. 2002; Bishop and Robinson 2003) (probablement par chélation des ions
métalliques), ils suggèrent que la production d’Aβ serait une voie de défense contre le stress
oxydant, faisant ainsi de la conséquence (dans l’hypothèse de la cascade amyloïde), la cause.
De même, en attribuant à tau des propriétés anti-oxydantes, la formation de neurofibrilles peut

Chapitre 1 : la maladie d’Alzheimer
38
être vu comme une conséquence du stress oxydant (Nunomura et al. 2001; Wataya et al. 2002;
Gomez-Ramos et al. 2003; Nakashima et al. 2004). Dans le cas où cette théorie s’avèrerait
exacte, il faudra alors rechercher les causes de ce stress oxydant, décrit comme relativement
faible (insuffisant pour entraîner une mort cellulaire rapide) mais chronique (Keyse and
Tyrrell 1989; Rushmore et al. 1990; Davies et al. 1995; Wiese et al. 1995) et débouchant sur
des modifications compensatoires, réversibles dans un premier temps, puis permanentes
(incluant entre autre la formation de neurofilaments, de plaques séniles et, en parallèle, une
dérégulation mitotique) (Zhu et al. 2007).
Ainsi, malgré d’intenses recherches menées en ce sens, l’étiologie de la MA est encore loin
d’être élucidée, retardant d’autant la découverte de traitements curatifs, faute d’axes de
stratégie thérapeutique clairs.
II.3. Traitements
Actuellement, il n’existe aucun traitement thérapeutique curatif et/ou étiologique pour la MA,
seuls sont disponibles des traitements symptomatiques (et, en dernier lieu, palliatifs), qui ont
pour but de retarder le développement de la maladie et d’améliorer les conditions de vie des
malades.
II.3.a.Traitements actuels
Différents traitements sont actuellement utilisés pour améliorer les conditions de vie des
patients atteints de la MA, mais aucun d’entre eux n’est curatifs.
Concernant les troubles cognitifs, les inhibiteurs de cholinestérase (donepezil (Shintani and
Uchida 1997; Rogers et al. 1998; Rogers et al. 1998; Burns et al. 1999), tacrine (Davis et al.
1992; Knapp et al. 1994; Samuels and Davis 1997; Gracon et al. 1998), galantamine (Bickel
et al. 1991; Bores et al. 1996), rivastigmine (Sramek et al. 1996; Rosler et al. 1999)) sont les
plus couramment utilisés, aux différents stades de la maladie. Ils inhibent les enzymes
responsables de l’hydrolyse de l’acétylcholine, compensant ainsi la déficience de ce
neurotransmetteur.
A un stade avancé de la MA, la mémantine, un antagoniste non compétitif des récepteurs
glutamatergiques NMDA (N-methyl-D-aspartate), permettrait de contrebalancer le taux
excessif de glutamate en partie responsable de l’augmentation des troubles mnésiques

Chapitre 1 : la maladie d’Alzheimer
39
(Fleischhacker et al. 1986; Robinson and Keating 2006; Cosman et al. 2007; Schmitt et al.
2007).
D’autres traitements thérapeutiques sont évoqués dans la littérature, mais leur efficacité reste
encore à confirmer, les résultats des précédentes études réalisées étant parfois contradictoires.
Les traitements à base d’œstrogène pour les femmes en fin de ménopause semblent retarder
de quelques années l’apparition de la maladie, mais aucun résultat positif n’a été vérifié avec
les personnes plus âgées atteintes de la MA (Henderson et al. 1994; Paganini-Hill and
Henderson 1996; Schmidt et al. 1996; Asthana et al. 1999).
Des études rétrospectives ont montré que les anti-inflammatoires non stéroïdiens pouvaient
ralentir la progression de la maladie (via l’inhibition des cyclooxygénases, enzymes de la
biosynthèse des dérivés de type prostaglandine intervenant dans les processus inflammatoires)
(Breitner et al. 1994; Stewart et al. 1997), mais ces résultats n’ont malheureusement pas pu
être reproduits dans des études plus récentes (Aisen et al. 2000; Reines et al. 2004). Certains
d’entre eux, cependant, agiraient selon un mécanisme différent et prometteur, ils seront
évoqués dans la section suivante.
La prise d’anti-oxydant (vitamine E, sélégiline qui stimule la production de superoxyde
dismutase) devrait permettre de compenser la baisse d’enzyme anti-oxydante associée à
l’augmentation d’espèces radicalaires, mais là encore les différences observées entre le
placebo et la molécule active ne sont pas significatives (Pratico and Delanty 2000; Ames and
Ritchie 2007).
Quant aux troubles comportementaux (dépression, psychose), ils sont généralement traités à
l’aide d’anti-dépresseur et d’anti-psychotique jugés compatibles avec la maladie (Schachter et
al. 2000).
En dehors des pharmacopées, l’environnement psychosocial joue un rôle primordial. Une
alimentation saine, des exercices physiques et intellectuels réguliers et variés, une bonne
hygiène permettent de retarder significativement l’avancé de la maladie. Un environnement
social apaisant (soutien familial, maintien au domicile quand c’est possible) est également
nécessaire pour ces personnes fragilisées, plus enclins aux insomnies, syndromes d’anxiété et
dépressions (Mittelman et al. 1996; Stern et al. 1997).
II.3.b.Voies de recherche
Puisque la MA est caractérisée par la formation de plaques amyloïdes, l’inhibition de

Chapitre 1 : la maladie d’Alzheimer
40
l’agrégation d’Aβ est apparue comme un axe de recherche logique pour combattre cette
maladie. Dans cette optique, deux groupes de molécules thérapeutiques potentielles ont été
élaborées : les peptides et les « petites » molécules organiques. Dans les deux cas, l’idée est
de concevoir une entité capable de se lier au peptide de façon à empêcher la formation ou
l’accolement de feuillets bêta. Concernant les peptides, de nombreux candidats ont été
avancés : fractions de peptide Aβ sans (Sciarretta et al. 2006) ou avec modifications (Findeis
et al. 1999; Gordon and Meredith 2003), peptides à base d’acides aminés dextrogyres
(Chalifour et al. 2003), ou contenant des résidus prolines (Yamashita et al. 2003) comme la
colostrinine (dérivé du colostrum de mouton) (Gladkevich et al. 2007), certaines protéines
comme la neuroserpine (un inhibiteurs de. protéases à sérine, responsable, dans sa forme
mutée, d’encéphalopathie avec corps d'inclusion de neuroserpine) (Kinghorn et al. 2006).
Même si certains résultats sont encourageants, ce type de molécules a peu de chance de
devenir, en l’état, un médicament efficace, du fait de leur fort potentiel immunogénique
(protéolyse probable avant d’atteindre la cible) et leur faible capacité à passer la barrière
hémato-méningée (Sciarretta et al. 2006). Néanmoins, l’un de ces composés est actuellement
en phase II de tests cliniques : AL-108, développé par Allon, est un petit fragment d’une sous-
unité d’une protéine neuroprotectrice à activité-dépendante (Melnikova 2007).
Une immense panoplie de molécules organiques (non peptidiques) a donc également été
testée, certaines d’entre-elles se sont révélées efficaces dans l’inhibition de l’agrégation d’Aβ :
la DAPH (4,5-dianilinophthalimide) qui désagrège également les fibres préformées
(Blanchard et al. 2004), des dérivés de la triazine (Kim et al. 2006), le scyllo-
cyclohexanehexol (AZD-103, développé par Elan) actuellement en phase II (Melnikova 2007),
la Tramiprosate (considéré comme un glycosaminoglycane-mimétique, développé par
Neurochem) qui est le composé le plus avancé dans les tests cliniques (fin de phase III, cf.
www.neurochem.com/PR209FR.htm) (Melnikova 2007).
Les résultats des tests cliniques devraient permettre d’y voir plus clair quant à l’efficacité de
ce type de traitement. En effet, la réduction des plaques amyloïdes (mesurée in vitro) ne doit
pas impliquer l’augmentation de petits oligomères toxiques (plus difficiles à mesurer). De
plus, comme il a été vu en début de chapitre, il existe dans les mélanocytes (et peut-être
ailleurs) des processus amyloïdiques naturels et contrôlés, nécessaire au bon fonctionnement
de l’organisme. Le manque de spécificité de la plupart de ces composés pourrait donc être
source d’effets secondaires nuisibles (Cohen and Kelly 2003).

Chapitre 1 : la maladie d’Alzheimer
41
Une autre possibilité pour réduire la quantité des plaques séniles serait de stimuler la
dégradation du peptide Aβ et/ou de ses agrégats. On peut envisager d’activer des protéines
chaperons (Hsp20, Hsp27 , Hsp70, Hsp90, Hsp40, αB-cristalline) (Evans et al. 2006;
Wilhelmus et al. 2006), toutefois leur effet est limité par leur localisation intracellulaire. Il
existe également des enzymes connues pour dégrader le peptide Aβ qui sont : la néprilysine
(NEP), glycoprotéine membranaire impliquée dans de nombreux processus physiologiques en
différents endroits de l’organisme (Kenny and Stephenson 1988; Letarte et al. 1988; Kumar et
al. 1990; Turner and Tanzawa 1997; Broccolini et al. 2006) ; son homologue l’endothéline-
convertase ECE-1 (Eckman et al. 2001; Eckman et al. 2003) ; l’insulinase (ou IDE pour
insulin-degrading enzyme, elle dégrade de multiple substrats dont l’amyline) dont l’altération
génétique pourrait être un facteur de risque (Vekrellis et al. 2000; Vepsalainen et al. 2007) ;
l’enzyme de conversion de l’angiotensine (ECA, exopeptidase qui catalyse la conversion de
l'angiotensine I en angiotensine II, un puissant vasoconstricteur) dont l’action est controversée
(Hu et al. 2001; Eckman et al. 2006) ; la plasmine (ou fibrinolysine, hydrolyse la fibrine et
d’autres facteurs de coagulation) (Van Nostrand and Porter 1999; Tucker et al. 2000) ; les
MMP (matrix metalloprotease, enzymes de dégradation de la matrice extracellulaire) (Sung et
al. 2005; Yan et al. 2006). On peut remarquer qu’AICD, le fragment d’APP cytosolique libéré
lors du clivage de la γ-sécrétase, semble réguler l’expression de NEP (Hass and Yankner 2005;
Pardossi-Piquard et al. 2005; Hebert et al. 2006; Pardossi-Piquard et al. 2006). Cette stratégie
thérapeutique est relativement nouvelle, mais les premiers résultats obtenus en laboratoire,
que ce soit via une thérapie génique ou une approche pharmacologique, semblent prometteurs
(Levites et al. 2003; Marr et al. 2003; Iwata et al. 2004; Turner et al. 2004; Saito et al. 2005).
Enfin il est également possible de stimuler le système immunitaire contre le peptide via
l’immunisation active ou passive. Cette dernière alternative est sujette à controverse, en effet
les premiers résultats obtenus en laboratoire étaient très encourageants (réduction des plaques,
amélioration de l’état cognitif chez les animaux) (Schenk et al. 1999; Schenk et al. 2004),
malheureusement les tests cliniques (consistant en l’injection d’anticorps monoclonaux anti
Aβ) durent être arrêtés précipitamment en phase II, du fait de la survenue de cas de méningo-
encéphalite (4 sur 360) (Orgogozo et al. 2003). Les bénéfices de l’immunothérapie semblaient
pourtant (mis à part ces 4 cas) réels (Hock et al. 2003), mais d’autres rapports ont depuis revu
ces bénéfices à la baisse (Fox et al. 2005; Gandy and Heppner 2005; Gilman et al. 2005). Il
faut également considérer le risque que le système immunitaire ainsi activé cible l’APP,
protéine très probablement nécessaire au bon fonctionnement des neurones (cf. section II.2.b).

Chapitre 1 : la maladie d’Alzheimer
42
Néanmoins une nouvelle génération de vaccins (ACC-001, Elan) ne requerrant pas
l’activation du système immunitaire du patient, est actuellement en phase I de test (cf.
www.elan.com/research_development/Alzheimers) (Melnikova 2007). D’autres anticorps
sont aussi en cours de développement : bapineuzumab (Elan/Wyeth) et LY2062430 (Eli Lilly)
en phase II, RN1219 (Pfizer) en phase I (Melnikova 2007).
Le second grand axe de recherche thérapeutique basé sur l’hypothèse amyloïde est la
régulation de la production d’Aβ à partir de l’APP : soit en inhibant la β- et/ou la γ-
sécrétase, soit en activant l’α-sécrétase (voie non-amyloïdogénique).
Malgré la complexité de la γ-sécrétase, protéine composée de plusieurs sous-unités, et le
manque d’information sur sa structure, de nombreux composés inhibant ou modulant son
activité enzymatique ont vu le jour et certains sont déjà entrés en phase de tests cliniques. Le
problème majeur posé par cette sécrétase est qu’elle agit également dans la voie de
signalisation Notch (clivage en S3 du récepteur Notch (De Strooper et al. 1999)), vitale pour
l’organisme et qui est impliquée dans divers processus de différentiation cellulaire durant
l’embryogenèse et chez l’adulte (Artavanis-Tsakonas et al. 1995; Radtke et al. 2004). De plus,
à l’heure actuelle, plus de 30 substrats de la γ-sécrétase ont été rapportés (Nyborg et al. 2006),
ce qui montre clairement la toxicité potentielle que peut entraîner l’utilisation d’inhibiteur de
cette enzyme. La réussite de tels composés repose donc sur la spécificité de la modulation. La
plupart des inhibiteurs actuellement à l’essai reposent sur le motif sulfonamide/sulfone ou
benzodiazepine/benzolactame (Harrison et al. 2004) : le LY-411575, bien que diminuant le
taux de Aβ40 et 42, s’est révélé toxique pour le développement lymphocitaire et le système
gastro-intestinal (attribuable à un disfonctionnement du signal Notch) (Wong et al. 2004) ; les
composés BMS-2999897 et SCH 697466 semblent, d’après les premiers essais effectués sur
des souris transgéniques, pouvoir atteindre un niveau thérapeutique convenable sans toxicité,
cela reste à confirmer (Hyde et al. 2006) ; le LY-450139, même si apparemment non toxique,
ne paraît pas capable, aux doses administrées, de diminuer significativement la production
d’Aβ (tests cliniques en phase II) (Siemers et al. 2006); des premiers résultats positifs ont été
obtenus avec le composé de Merck MK-0752 actuellement en phase II (résultats exposés à la
« 10th international conference on Alzheimer’s disease and related disorders », 2006). Au vu
des difficultés de spécificité rencontrées dans les tentatives d’inhibition, la modulation de
l’activité de la γ-sécrétase pourrait être une bonne alternative. Une nouvelle classe de
médicaments potentiels a récemment attiré l’attention en ce sens : en effet certains anti-

Chapitre 1 : la maladie d’Alzheimer
43
inflammatoires non stéroïdiens (AINS) (ibuprofèn, indométhacine, sulindac sulphide) ont
montré qu’ils pouvaient réduire la sécrétion d’Aβ42 sans perturber la voie de signalisation
Notch, un effet indépendant de leur habilité à inhiber l’activité des cyclooxygénases (Weggen
et al. 2001). Le Flurizan (myriad Genetics), l’énantiomère R de l’AINS flurbiprofène, a
montré des résultats positifs en phase II et si la phase III l’est autant (résultats fin 2008), le
médicament pourrait être commercialisé d’ici 2009/2010 (Lundkvist and Naslund 2007;
Melnikova 2007). Le mécanisme d’action de ces AINS sur la γ-sécrétase est encore largement
méconnu, cependant une étude a montré qu’il pourrait consister en une altération de la
conformation de PS1 et de l’interaction entre PS1 et APP (Lleo et al. 2004). Une autre série
de composés modulateurs de la γ-sécrétase, NGX 83232, structurellement différents des AINS,
sont également à l’étude (SL Wagner, 7th International Conference AD/PD, 2005).
Dans le cadre d’études précliniques, un nouveau composé, EHT 1864, a permis une
diminution significative de la quantité d’Aβ40 et 42 intra et extra cellulaire, sans inhiber la
voie de signalisation Notch (Desire et al. 2005). La raison réside dans le mode d’action
indirect de ce composé. Celui-ci en inhibant la protéine Rac1, une petite protéine G,
déstabiliserait les radeaux lipidiques (siège probable des interaction APP / γ-sécrétase mais
pas du clivage Notch), perturbant ainsi le clivage du site γ (Field et al. 2000; Rozelle et al.
2000; Gianni et al. 2003; Maillet et al. 2003; Zambrano et al. 2004; Desire et al. 2005;
Vetrivel et al. 2005).
La deuxième sécrétase impliquée dans la production de Aβ, BACE1, est une cible attractive.
En effet des souris transgéniques porteuses d’un gène inactif pour cette protéine ne révèlent
aucune aberration phénotypique apparente (Luo et al. 2001; Roberds et al. 2001; Wolfe 2002;
Hamaguchi et al. 2006). De plus la structure du domaine protéase de la BACE1 est connu, ce
qui facilite la conception d’inhibiteur (Hong et al. 2000). Cependant d’autres études ont
également montré que BACE1 modulerait la plasticité synaptique et la myélinisation (Laird et
al. 2005; Hu et al. 2006; Willem et al. 2006), la prudence est donc de mise. Le nombre de
candidats potentiels correspondant à cette stratégie thérapeutique est actuellement en pleine
expansion, en voici quelques exemples. Une série de composés contenant le dipeptide Leu-
Ala a donné de premiers bons résultats in vitro (Ghosh et al. 2006), d’autres composés
peptidomimétiques et une série d’acylguanidines ont également montré une inhibition de
BACE1 in vitro (Hanessian et al. 2005; Cole et al. 2006; Freskos et al. 2007). Afin de palier à
la faible pénétration au travers de la barrière hémato-méningée de ces composés à caractère

Chapitre 1 : la maladie d’Alzheimer
44
peptidique, diverses stratégies consistant à attacher de façon covalente le composé actif à un
« transporteur » ou diminuer la flexibilité conformationelle via une macrocyclisation, ont été
élaborées (Chang et al. 2004; Stachel et al. 2006). Récemment, un inhibiteur non peptidique,
GSK188909, développé par GlaxoSmithKline, s’est également montré satisfaisant in vivo
(tests précliniques)(Hussain et al. 2007) et ATG-Z1, développé par CoMentis, devrait entrer
en phase I de test clinique d’ici peu (annoncé sur www.athenagen.com) (Melnikova 2007).
Une approche potentiellement plus tolérable pour l’organisme serait d’associer les deux types
d’inhibiteur (γ et β), enfin de diminuer la toxicité relatives aux deux médicaments tout en
additionnant leur effet étiologique. De plus, il pourrait être dangereux d’utiliser exclusivement
un type d’inhibiteur, par exemple inhiber uniquement le clivage du site γ pourrait conduire à
l’accumulation de fragments C-terminaux de l’APP, phénomène lié à un déficit mnésique
(Saura et al. 2005). Il semble que les AINS aillent dans le sens de cette « bithérapie », une
récente étude a montré que, en plus de leur capacité à inhiber la γ-sécrétase, ils diminueraient
l’expression de BACE1 en agissant sur le récepteur au facteur activé de prolifération des
peroxysomes γ (Peroxisome proliferator-activated receptor γ, PPARγ), un répresseur de la
transcription du gène de BACE1 (Sastre et al. 2006). Les AINS seraient donc d’excellents
candidats à la thérapie anti-amyloïde pour la MA.
D’autre recherche se sont orientées vers la stimulation de l’α-sécrétase en défaveur de la
voie amyloïdogénique. De plus, certains fragments issus de la protéolyse non-
amyloïdogenique de l’APP ont des propriétés neuroprotectives, rendant cette approche
d’autant plus attractive (Bowes et al. 1994; Mattson 1994; Smith-Swintosky et al. 1994). Or il
a été montré que la stimulation des récepteurs cholinergiques augmentait la production de
sAPPα, diminuant d’autant la production d’Aβ (Rossner et al. 1998). Ainsi, des agonistes
cholinergiques tels que l’acétylcholine, la nicotine ou le carbacol pourrait jouer ce rôle (Seo et
al. 2001; Suh and Checler 2002). Plusieurs composés sont actuellement à l’étude, mais il est
intéressant de remarquer que le donepezyl, un des médicaments actuellement sur le marché
pour les patients atteint de la MA, pourrait avoir, en plus de son effet symptomatique, un effet
étiologique via la stimulation de l’α-sécrétase (Borroni et al. 2001), ce n’est pas le cas de la
tacrine qui aurait tendance à inhiber la voie non-amyloïdogénique (Lahiri et al. 1994; Chong
and Suh 1996; Lahiri et al. 2000).
Au vue des difficultés rencontrées pour traiter les causes de la MA, d’autres voies

Chapitre 1 : la maladie d’Alzheimer
45
thérapeutiques s’attèlent à palier les conséquences (dans l’hypothèse amyloïde) des dépôts
amyloïdes avec le développement : d’anti-inflammatoires (dont, indirectement, des thérapies
induisant une réponse des cellules régulatrices T) (Butovsky et al. 2006; Sela 2006) ; d’anti-
oxydants (Ono et al. 2003) comme, par exemple, les polyphénols du thé vert (Weinreb et al.
2004), le resveratrol (trans-3,4',5-trihydroxystilbene, présent dans le vin rouge) et qui, de plus,
activerait la dégradation des plaques amyloïdes via le protéasome (Dore 2005; Marambaud et
al. 2005; Anekonda 2006), la curcumine capable de réduire les plaques amyloïdes (Yang et al.
2005; Garcia-Alloza et al. 2007), la mélatonine (voir revue (Wang and Wang 2006)), l’acetyl-
L-carnitine (stimule la production de glutathion) (Abdul et al. 2006) ; de composés
neuroprotecteurs (la mémantine, antagoniste des récepteurs NMDA (N-methyl-D-aspartic
acid), agit sur l’excitotoxicité) (Lipton 2007), thérapies stimulant la production de
nicotinamide adénine dinucléotide (agit sur l’axonopathie, dégénérescence de l’axone)
(Belenky et al. 2007; Khan et al. 2007), prévention de l’induction du cycle cellulaire chez les
neurones post-mitotiques (Chong et al. 2005), modulation des récepteurs glutamates
métabotropiques (Riederer and Hoyer 2006; Bernareggi et al. 2007), actions sur les cytokines
et facteurs trophiques (Chong et al. 2005), traitement au lithium(II) (Chuang and Manji 2007).
Cependant, considérant que, en l’état actuel des recherches, il ne semble pas y avoir de voie
unique ou privilégiée conduisant à la mort cellulaire dans la MA, les avis sont mitigés quant à
l’efficacité de ces différentes approches thérapeutiques (Shen et al. 2006).
Les pistes de recherche thérapeutiques sont donc très nombreuses et l’on espère qu’au moins
une d’entres elles aboutisse. Il existe cependant une dernière piste : la thérapie par chélation
des métaux (cf. section II.4.f.). Elle repose sur l’hypothèse d’une influence néfaste des métaux
(cuivre, zinc et fer, en particulier) qui seraient à l’origine de l’agrégation et de la toxicité du
peptide Aβ.
II.4. Influence des métaux
II.4.a. Introduction : les métaux et l’organisme
Un des processus clef qui pourrait expliquer le développement de la MA, est l’altération de
l’homéostasie des métaux, incluant la dérégulation de la neurochimie des métalloprotéines.
C’est cette hypothèse que nous allons traiter dans ce manuscrit, et en particulier le rôle du
zinc dans la MA.

Chapitre 1 : la maladie d’Alzheimer
46
Les métaux sont largement présents dans les systèmes biologiques et peuvent être
grossièrement classés en deux catégories : les métaux essentiels, ayant une fonction
physiologique au sein de l’organisme (Zn Fe, Cu, Mn, Mb, Ca…), et les métaux non
essentiels ou toxiques (Cd, Hg, Pb…). A fortes concentrations, les métaux essentiels peuvent
devenir toxiques d’où l’intérêt pour les organismes de les séquestrer pour ensuite les éliminer
ou les transformer au sein d’organites de stockage spécifiques sous forme inerte ou, au moins,
non toxique (Figure 1.13).
Figure 1.13. Effet des métaux sur le métabolisme cellulaire.
Les métaux non essentiels ne possèdent pas de rôles biologiques connus et peuvent présenter
une haute toxicité pour les organismes. Les êtres vivants doivent donc mettre en oeuvre des
processus qui permettent de limiter l’accumulation de ces métaux ou de les stocker sous
forme non toxique (Figure 1.13).
Le zinc dans l’organisme
Le zinc est un élément indispensable à la survie des êtres vivants (Outten and O'Halloran
2001). Dans les micro-organismes, les plantes et les animaux, le zinc est impliqué dans la
fonction de plus de 300 métalloenzymes, où il remplit trois rôles principaux : catalytique,
cocatalytique et structural (Vallee and Auld 1992; Vallee and Auld 1992; Vallee and Falchuk
1993; Takeda 2000). Ces métalloenzymes sont impliquées dans une grande variété de
fonctions, comme la régulation de l’expression des gènes, l’adressage des protéines ou la

Chapitre 1 : la maladie d’Alzheimer
47
réponse métal-spécifique au stress (O'Halloran 1993). En tant qu’élément catalytique ou co-
catalytique, le zinc est indispensable au fonctionnement d’enzymes telles que les
carboxypeptidases pancréatiques, la phosphatase alcaline, diverses déshydrogénases ou
encore la superoxyde-dismutase. Chez les mammifères, le zinc est absorbé au niveau de la
bordure en brosse de la muqueuse intestinale. Il est ensuite transporté par le sang vers les
tissus où il est utilisé. Une alimentation appauvrie en zinc cause des perturbations chez
l'animal et chez l’homme, et peut affecter le développement du système nerveux central (SNC)
(O'Dell 1993; Sandstead 1994; Prasad 1995). Certaines affections pourraient également
résulter d’une carence en zinc : acrodermatite entéropathique (ou syndrome de Danbolt et
Closs), troubles de la cicatrisation, de la spermatogenèse ou du comportement alimentaire
(diminution de l’appétit, altération du goût), dermites séborrhéiques, eczéma, psoriasis,
alopécie, certains problèmes immunologiques, des surinfections mycosiques et bactériennes,
des inflammations de la commissure des lèvres, etc. Par ailleurs, des troubles
comportementaux comme la léthargie et la dépression pourraient aussi être liés à une
déficience en zinc (Harrison and Gibbons 1994). En outre, des apports excessifs de zinc
(ouvriers travaillant en zinguerie) ou certaines maladies héréditaires (l’hyperzincémie
familiale ou la maladie de Pick) conduisent également à des désordres du SNC et à des
anémies.
La quantité de zinc dans l'organisme varie entre 2 et 3 g et sa concentration dans le sérum et
dans le liquide extracellulaire est estimé à 0,15 μM (Takeda 2000). À l’exception des îlots de
Langerhans (cellules endocrines du pancréas), le SNC est l'organe le plus riche en zinc. La
quantité du zinc dans le SNC est de 10 μg par gramme de tissu (Frederickson 1989). La teneur
moyenne en zinc total des neurones est estimée à environ 150 μmol/L (Wallwork 1987;
Harrison et al. 1994; Takeda 2000) ; la plus grande partie du zinc dans le cytosol n’est pas
libre, mais liée à des métallothionéines et à d’autres protéines (Frederickson 1989). En
utilisant des sondes fluorescentes, la quantité du zinc libre dans le cytosol a été estimée à 1
nM (Canzoniero et al. 1997; Thompson et al. 2002). La distribution du zinc dans le système
nerveux central (SNC) n'est pas uniforme : il est plus concentré dans la substance grise que
dans la substance blanche, et certaines régions du SNC, comme l'hippocampe, en sont
particulièrement riches (cf. figure 1.14). Entre 5 et 15 % de la quantité du zinc total se trouve
dans des neurones glutamatergiques (Haug 1967; Perez-Clausell and Danscher 1985), où il se
localise à l'intérieur des vésicules synaptiques (Frederickson and Moncrieff 1994).
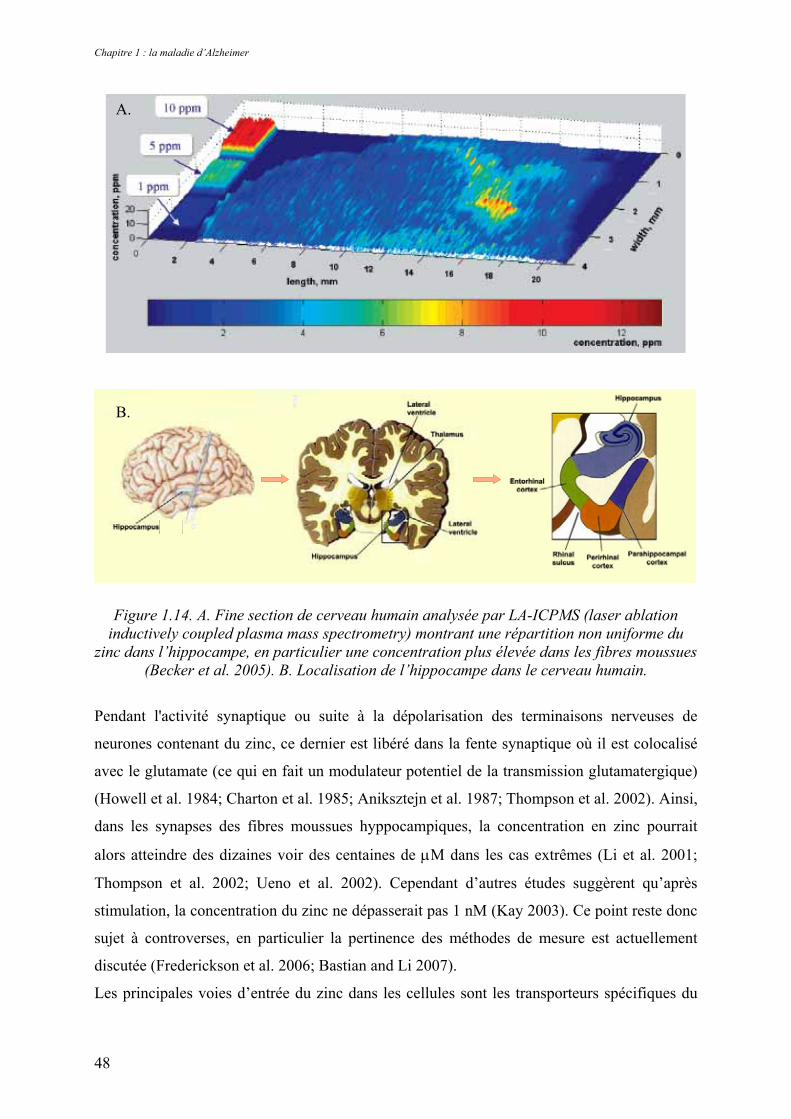
Chapitre 1 : la maladie d’Alzheimer
48
Figure 1.14. A. Fine section de cerveau humain analysée par LA-ICPMS (laser ablation inductively coupled plasma mass spectrometry) montrant une répartition non uniforme du
zinc dans l’hippocampe, en particulier une concentration plus élevée dans les fibres moussues (Becker et al. 2005). B. Localisation de l’hippocampe dans le cerveau humain.
Pendant l'activité synaptique ou suite à la dépolarisation des terminaisons nerveuses de
neurones contenant du zinc, ce dernier est libéré dans la fente synaptique où il est colocalisé
avec le glutamate (ce qui en fait un modulateur potentiel de la transmission glutamatergique)
(Howell et al. 1984; Charton et al. 1985; Aniksztejn et al. 1987; Thompson et al. 2002). Ainsi,
dans les synapses des fibres moussues hyppocampiques, la concentration en zinc pourrait
alors atteindre des dizaines voir des centaines de μM dans les cas extrêmes (Li et al. 2001;
Thompson et al. 2002; Ueno et al. 2002). Cependant d’autres études suggèrent qu’après
stimulation, la concentration du zinc ne dépasserait pas 1 nM (Kay 2003). Ce point reste donc
sujet à controverses, en particulier la pertinence des méthodes de mesure est actuellement
discutée (Frederickson et al. 2006; Bastian and Li 2007).
Les principales voies d’entrée du zinc dans les cellules sont les transporteurs spécifiques du
A.
B.

Chapitre 1 : la maladie d’Alzheimer
49
zinc et différents canaux ioniques (activés par le voltage ou des ligands) qui, sans être
spécifiques au zinc, sont cependant perméables à cet ion. Parmi ces canaux, ont été identifiés
les canaux calciques dépendants du voltage, les récepteurs NMDA et les récepteurs AMPA
perméables au calcium (Weiss et al. 1993; Sheline et al. 2002). Les transporteurs spécifiques
du zinc se classent en plusieurs groupes appelés ZnT 1-7 (cf. revue (Colvin et al. 2003)), mais
seul les ZnT 1, 3, 4 et 6 sont présents dans le cerveau. De nombreuses observations
expérimentales indiquent que ZnT3 serait le transporteur responsable de l’accumulation de
zinc dans les vésicules synaptiques des terminaisons glutamatergiques (Palmiter et al. 1996;
Wenzel et al. 1997; Cole et al. 1999).
Figure 1.15. Transport du zinc synaptique. Les vésicules ornées de ZnT3 sont assemblées dans l’appareil de Golgi du neurone glutamatergique (1) et transportées le long de l’axone
(2).Une fois dans la terminaison présynatique (3), les vésicules contiennent à la fois du zinc et du glutamate qui vont être libérés dans la fente synaptique par exocytose calcium- et
impulsion-dépendante (4). Le zinc module de nombreux canaux, transporteurs et récepteurs localement et diffuse (peut-être) jusqu’à ceux de cellules gliales et d’autres neurones (6-10).
Tous les canaux à calcium sont perméables au zinc (5, 9) le zinc étant ensuite chaperonné par les MT (11). Oxydation et nitrosylation des thiols de la MT libèrent le zinc, ce qui pourrait
avoir des conséquences dans la MA (12) (Frederickson et al. 2005).
Un grand rôle dans l’homéostasie du zinc (Figure 1.15) est également attribué aux
métallothionéines (MT), un groupe de protéines de faible poids moléculaire, riches en résidus
cystéines, qui présentent de nombreux sites de liaison pour le zinc et le cuivre. On a identifié

Chapitre 1 : la maladie d’Alzheimer
50
plusieurs isoformes de ces protéines, appelées MT-1, MT-2, MT-3 et MT-4 (Ebadi et al.
1995). Dans le cerveau, les formes MT-1 et MT-2 ont été trouvées dans les astrocytes
(Hidalgo and Carrasco 1998) tandis que la MT-3 est présente dans les cellules neuronales et
notamment dans les neurones glutamatergiques (Palmiter et al. 1992; Masters et al. 1994).
Certaines études indiquent une déficience en MT-3 chez les personnes atteintes de la maladie
d’Alzheimer (Tsuji et al. 1992; Uchida 1994) mais son implication dans le développement de
cette affection reste controversée (cf. chapitre 5).
II.4.b. Modulation des métaux avec l’âge
Chez la souris, une augmentation des concentrations en cuivre, fer et cobalt avec l’âge a été
observée, tandis que celle du zinc et du manganèse restaient constantes (Maynard et al. 2002).
Similairement, dans le cerveau humain, la concentration en cuivre dans le plasma augmente
régulièrement avec l’âge, tandis qu’elle reste constante pour le zinc jusqu’à ~75 ans pour
diminuer ensuite (McMaster et al. 1992; Madaric et al. 1994; Monget et al. 1996).
L’augmentation en cuivre peut être due à une élévation avec l’âge de ceruloplasmine (ou
ferroxidase, une alpha 2-globuline fixant 90 % du cuivre plasmatique) (Connor et al. 1993;
Milne and Johnson 1993). On observe également une augmentation avec l’âge de clusters de
cuivre et d’ions fer dans le cerveau (Wender et al. 1992). Il faut cependant noter que la plupart
des métaux étudiés (Na, K, P, Ca, Mg, Si, Cr, Cu, Ni, Zn, Fe, Al, Cd, Pb et As) ont des
concentrations différentes en fonction de la localisation dans le cerveau, des altérations dans
cette distribution pourraient également jouer un grand rôle dans le développement de la MA
(Rajan et al. 1997).
Dans le cas du zinc, on dénote aussi chez les sujets âgés une surproduction en
métallothionéine (particulièrement MT-1 et MT-2, les résultats obtenus pour la MT-3 étant
controversés) et α-2 macroglobuline, apparemment en réponse à l’élévation du stress oxydant
(Mocchegiani et al. 2000). Du fait de la forte affinité des ces protéines pour le zinc, cette
surexpression entraînerait une diminution du zinc « accessible » aux autres protéines (en
particulier la thymuline, d’où un déclin du système immunitaire) (Mocchegiani et al. 2006).
II.4.c. Evolution du stress oxydant avec l’âge
La production d’espèce réactives de l’oxygène (ROS) est une des principales hypothèses pour
expliquer le processus de vieillissement (Harman 1956; Barja 2004). C’est également une

Chapitre 1 : la maladie d’Alzheimer
51
hypothèse pour l’apparition de la MA. Ce phénomène est en effet d’une ampleur considérable
puisqu’il semble qu’environ la moitié des protéines d’un animal sénescent soient oxydées
(Gafni 1997). Les ROS sont dérivés de la réduction univalente de l’oxygène et incluent les
espèces telles que l’anion superoxyde, le peroxyde d’hydrogène et les radicaux hydroxyles.
Les ROS peuvent être produits au sein de l’organisme via une grande variété de mécanismes,
cependant la réaction de l’oxygène avec des métaux à activité redox, lié ou non à des
protéines, est la principale source de ROS (Halliwell and Gutteridge 1984). Une petite
quantité de ROS peut être bénéfique à la cellule en jouant le rôle de messager, régulateur de
gènes ou médiateur de l’activation cellulaire. Cependant une augmentation de leur
concentration peut entraîner de nombreux effets néfastes : peroxydation des lipides (Gupta et
al. 1991; Cini and Moretti 1995; Zhu et al. 2006), inactivation d’enzymes, altération des
structures tertiaires de protéines (Forster et al. 1996; Poon et al. 2006) et oxydation de l’ADN
(dont l’ADN mitochondriale) (Hamilton et al. 2001). Parallèlement, on observe une
diminution, avec l’âge, de l’ensemble des mécanismes de défense contre les ROS
(métalloenzymes comme la catalase, glutathion, vitamine E/C, …) (Gracy et al. 1985;
Rosenberger 1991).
Dans les cerveaux de personnes atteintes de la MA, le stress oxydant semble encore plus
important : un taux élevé en lipides peroxydés et la présence de nombreux produits
d’oxydation de protéines et d’ADN ont été observés (Butterfield and Lauderback 2002; Keller
et al. 2005; Sultana et al. 2006).
II.4.d. Modulation des métaux dans la MA
De nombreuses études ont montré la présence d’un déséquilibre de l’homéostasie des métaux
de transition dans le cerveau (hippocampe, amygdale, néocortex et bulbe olfactif) des
personnes atteintes de la MA (Adlard and Bush 2006). En effet, on remarque une
augmentation (facteur 3 à 5) en zinc, cuivre et fer dans les noyaux cortico-médians et baso-
latéraux de l’amygdale, mais les plus grandes concentrations se situent au niveau des plaques
amyloïdes : 390 μM de cuivre, 1050 μM de zinc, 940 μM de fer, à comparés aux
concentrations de 70, 350 et 340 μM respectivement chez le sujet sain (Lovell et al. 1998).
Des analyses histochimiques ont également montré une concentration élevée en zinc au
niveau des neurofilaments et des dépôts amyloïdes caractéristiques de l’angiopathie amyloïde
(Suh et al. 2000). Dans le sérum et le liquide céphalorachidien (LCR), les résultats diffèrent
suivant les études, par exemple : diminution du taux de fer et zinc et une élévation du taux de

Chapitre 1 : la maladie d’Alzheimer
52
cuivre (Basun et al. 1991), ou diminution en zinc dans le LCR (mais pas en fer, cuivre ni
manganèse) et pas de changement notable dans le sérum (Molina et al. 1998). Les principaux
transporteurs de ces métaux (céruloplasmine, protéine de transport du zinc ZnT1,
métallothionéines, protéines liées au fer) semblent également connaître des altérations régio-
spécifiques lors de la MA (Uchida et al. 1991; Connor et al. 1993; Adlard et al. 1998; Smith
et al. 1998; Yu et al. 2001; Bishop et al. 2002; Lovell et al. 2005; Moreira et al. 2005). Ainsi il
apparaît assez clairement une perturbation significative de l’homéostasie du cuivre, fer et zinc
chez les personnes atteintes de la MA. Compte tenu des rôles multiples des ces ions
métalliques au sein de l’organisme et en particulier du système nerveux central (SNC)
(maintient des fonctions enzymatiques et mitochondriales (Yamaguchi et al. 1982; Rossi et al.
2004), immunité (Percival 1998), myélination (Hara and Taketomi 1986; Wegner 2000),
apprentissage et mémoire (Youdim and Yehuda 2000; Bhatnagar and Taneja 2001; Takeda
2001), etc…) leur dérégulation participe inévitablement au spectre des altérations
caractéristiques de la MA. Cependant, l’interaction de ces métaux avec le peptide Aβ suggère
qu’une dérégulation de leur homéostasie pourrait être fondamentalement impliquée dans la
MA en tant que cause et pas seulement en tant que conséquence.
II.4.e.Implication des métaux dans la MA
Implication de métaux toxiques
Dans le cas de métaux particulièrement toxiques comme le plomb, le mercure et le cadmium,
la démonstration de leur implication dans différents processus contribuant à la
neurodégénération est largement établie (Weisskopf et al. 2004; Bleecker et al. 2005;
Lanphear et al. 2005). Leur lien éventuel avec la MA est moins clair. Une surexposition au
plomb en début de vie pourrait favoriser ensuite l’élévation d’APP et Aβ (Basha et al. 2005).
Une théorie concernant l’aluminium est également présente dans la littérature. Il a ainsi été
impliqué dans la formation des neurofilaments, dans l’agrégation et la toxicité d’Aβ (cf.
revues (Gupta et al. 2005; Kawahara 2005)). Ce métal, sans activité redox intrinsèque,
pourrait également promouvoir le stress oxydant via la formation du radical AlO22+ facilitant
les oxydations biologiques dues à l’ion superoxyde (oxydation du coenzyme NADH,
accélération de l’oxydation due au fer, peroxydation des lipides) (Exley 2004). Cependant,
l’aluminium, qui n’a pas de fonction biologique, a des propriétés toxiques, son implication
dans la MA demeure donc controversée (Forbes and Hill 1998; Munoz 1998).

Chapitre 1 : la maladie d’Alzheimer
53
D’un point de vue thérapeutique, la desferrioxamine (DFOA), utilisée depuis 1963 comme
chélateur du fer puis de l’aluminium en 1980 (Keberle 1964; Ackrill et al. 1980; Arze et al.
1981; Swartz 1985; Domingo 1989), semble ralentir le développement de la maladie
(McLachlan 1991), mais l’existence d’effets secondaires ne permet pas un traitement sur la
durée (Kruck et al. 1990; McLachlan 1991; Domingo 1996). De plus, l’administration de
DFOA n’empêche pas la formation de neurofibrilles comme escompté, son action est donc
probablement anti-oxydante et anti-inflammatoire non spécifique (Savory et al. 1994). Des
résultats plus encourageants ont cependant été décrits, en laboratoire, avec des composés
comme Feralex-G (Shin et al. 2003), mais à l’heure actuelle aucun traitement ciblant
l’aluminium dans la MA n’a été rapporté être en phase de tests cliniques. Cependant la
recherche de chélateurs de l’aluminium ayant moins d’effet secondaire que DFOA est
toujours d’actualité, ne serait-ce que pour les cas d’intoxication à ce métal (Missel et al. 2005).
Implication du Fer...
...avec Tau
Le fer semble jouer un rôle important dans la toxicité et l’agrégation de tau : la présence de
fer dans les neurofilaments pourrait exercer une activité pro-oxydante (Sayre et al. 2000) et le
fer (III) (mais pas le fer(II)) peut induire l’agrégation de tau hyperphosphorylé (agrégation
réversible si le fer (III) est réduit en fer(II)) (Yamamoto et al. 2002).
L’homéostasie du fer est également liée indirectement à tau, via le bon fonctionnement des
microtubules. En effet, il a été montré que les microtubules jouent un rôle important dans la
régulation de le ferritine, une protéine de stockage du fer (Hasan et al. 2005). La ferritine a
également été identifiée dans les filaments de tau dans le cas de paralysie supranucléaire
progressive (ou maladie de Steele, Richardson et Olszewski) (Perez et al. 1998) et il semble
que son dysfonctionnement dans la MA soit en partie responsable de l’augmentation du stress
oxydant (Quintana et al. 2006).
… avec l’APP
Le fer semble réguler l’expression de l’APP, comme suggéré par des expériences utilisant le
gène rapporteur codant la luciférase, accolé à la région 5' non traduite de l’ARNm de l’APP.
La biosynthèse de l’APP est donc ainsi sous contrôle post-transcriptionnel « fer-dépendant »,
un taux élevé de fer intracellulaire augmente ainsi la production d’APP (Rogers et al. 2002;
Venti et al. 2004; Reznichenko et al. 2006).

Chapitre 1 : la maladie d’Alzheimer
54
...avec Aβ
La concentration physiologique du fer ionique libre (Fe3+, Fe2+) est de l’ordre du pico- / nano-
molaire. Or la constante de dissociation du fer(II) serait de l’ordre du micromolaire (la
littérature étant très réduite dans ce domaine, cet ordre de grandeur reste à confirmer)
(Garzon-Rodriguez et al. 1999), ce qui n’explique pas la présence massive de fer dans les
plaques amyloïdes (Beauchemin and Kisilevsky 1998). Une dérégulation de l’homéostasie de
ce métal pourrait donc en être l’origine. Il semble par exemple qu’un dysfonctionnement de la
ferritine soit en partie à l’origine de l’accumulation de fer dans les plaques séniles, de plus
elle a été retrouvée agrégée autour des plaques amyloïdes (Quintana et al. 2006).
...relativement à la toxicité
La littérature concernant le rôle du fer dans la MA est beaucoup moins importante que pour le
cuivre et le zinc. Cependant il semble que, de la même manière que pour le cuivre, le peptide
Aβ ait la capacité de catalyser la réduction de Fe3+ en Fe2+ (éventuellement en présence
d’agents réducteurs tels que l’ascorbate ou le gluthathion), favorisant ainsi la production de
ROS (Smith et al. 1997; Huang et al. 1999). On peut envisager le mécanisme suivant, avec
Meox-Aβ représentant Fe3+-Aβ ou Cu2+-Aβ :
Meox-Aβ + e- (agent réducteur) Mered-Aβ (1)
Mered-Aβ + O2 Meox-Aβ + O2•- (2)
O2•- + O2
•- + 2 H+ H2O2 + O2 (3)
Mered-Aβ + H2O2 Meox-Aβ + HO- + HO• (4)
La réaction (3) est catalysée par la superoxyde dismutase mais on peut aussi envisager la
réaction suivante : Mered-Aβ + O2•- + 2 H+ Meox-Aβ + H2O2 (3’)
Concernant l’agrégation d’Aβ, la présence de fer ne semble ni promouvoir, ni retarder le
processus (Yoshiike et al. 2001; House et al. 2004). Par contre, d’un point de vue structural,
l’agrégation d’Aβ en présence de fer(III) conduit à des structures en feuillets bêta, il pourrait
donc directement participer à la formation des plaques amyloïdes (House et al. 2004).
... en corrélation avec une approche thérapeutique
Le principal polyphénol contenu dans le thé vert, l’épigallocatechine-3-gallate, pourrait
chélater le fer intracellulaire, diminuant ainsi la production de l’APP (l’étape post-
traductionnelle étant dépendante du fer à cause de la luciférase) (Reznichenko et al. 2006).
Des études ont également été menées avec la desferrioxamine (DFOA), utilisée depuis une
quarantaine d’années dans le cas de surcharge en fer dans l’organisme (Chaston and
Richardson 2003; Kalinowski and Richardson 2005). Cependant la DFOA, même si elle

Chapitre 1 : la maladie d’Alzheimer
55
donne de bons résultats in vitro (inhibition post-transcriptionnelle de l’APP) n’a pas la
capacité de passer la barrière hémato-méningée. D’autres chélateurs du fer, capables de
franchir la barrière hémato-méningée, sont donc à l’étude (Bandyopadhyay et al. 2006;
Mandel et al. 2007). Ainsi un composé nommé M-30, qui active la différentiation neuronale, a
un effet neuroprotecteur et permet la réduction du taux d’APP et d’Aβ, dans les cultures
cellulaires (Avramovich-Tirosh et al. 2007).
Implication du cuivre…
...avec Tau
Le cuivre peut se lier à des peptides correspondant à la deuxième (R2) et troisième (R3)
région répétitive d’accrochage aux microtubules de la protéine tau, induisant ainsi un
changement conformationnel. Ce dernier correspond, pour le peptide R2, à la formation d’une
structure en hélice alpha, ce qui pourrait favoriser la formation de filaments en paires
d’hélices (Ma et al. 2005; Ma et al. 2006). D’autres études ont également avancé que la
présence de cuivre dans les neurofilaments pourrait exercer une activité pro-oxydante (Sayre
et al. 2000).
Il faut enfin noter que le cuivre perturbe la polymérisation des tubulines et donc l’assemblage
en microtubules, ainsi une augmentation du taux en cuivre pourrait avoir des conséquences
néfastes sur les neurones.
… avec l’APP
L’APP comporte un site de fixation du cuivre composé des ligands His-147, His-151, Tyr-168,
Met-170 (Hesse et al. 1994; Atwood et al. 2000; Wegner 2000; Simons et al. 2002; Barnham
et al. 2003). De plus, son expression semble réguler le taux de cuivre cytosolique (Davis et al.
2000; Armendariz et al. 2004; Bellingham et al. 2004) : la surexpression de l’APP induit une
baisse du taux de cuivre cytosolique et inversement, en accord avec son rôle potentiel de
transporteur du cuivre de l’intérieur vers l’extérieur de la cellule (White et al. 1999; Maynard
et al. 2002; Bayer et al. 2003; Phinney et al. 2003; Bellingham et al. 2004).
Le cuivre pourrait également jouer un rôle dans la protéolyse de l’APP, en effet la β-sécrétase
BACE1 peut lier un atome de cuivre dans son domaine C-terminal (via des résidus cystéines),
or le domaine cytoplasmique de BACE1 interagit avec la protéine chaperon consacrée au
cuivre de la superoxyde dismutase 1 (SOD1) et l’expression de BACE1 réduit l’activité de
SOD1 (Angeletti et al. 2005; Dingwall 2007).

Chapitre 1 : la maladie d’Alzheimer
56
…sur la concentration d’Aβ
Paradoxalement, des études ont montré qu’un taux en cuivre élevé diminue la production
d’Aβ, ce qui implique une régulation au niveau de la protéolyse de l’APP, la production de ce
dernier étant sensée diminuer pour compenser l’élévation du taux de cuivre (Maynard et al.
2002; Bayer et al. 2003; Phinney et al. 2003). En effet le cuivre semble favoriser la voie non-
amyloïdogénique puisqu’une augmentation de la concentration en fragments p3 non-
amyloïdogéniques a été observée (Borchardt et al. 1999).
Cependant dans le cas d’un régime à haute teneur en cuivre et en cholestérol, on observe
l’effet inverse, c’est-à-dire une augmentation de la production d’Aβ et l’apparition de plaques
séniles (Sparks and Schreurs 2003). Ceci dénote la complexité de l’action du cuivre dans la
MA et son caractère potentiellement pléiotropique (probablement dépendant de sa localisation
extra- ou intracellulaire). De plus, suivant la technique utilisée pour révéler la présence de
cuivre et en fonction de la localisation dans le cerveau (amygdale, néocortex ; tissus, plaques
amyloïdes), les résultats peuvent parfois différer (Deibel et al. 1996; Loeffler et al. 1996;
Lovell et al. 1998).
...avec Aβ
In vitro, il n’est pas clair si le cuivre accélère ou non l’agrégation du peptide Aβ, les deux
types de résultats pouvant être trouvés dans la littérature (dépendant probablement du pH,
tampon et de la technique de détection) (Atwood et al. 1998; Zou et al. 2001; Raman et al.
2005; Ha et al. 2007). Cependant, certaines études indiquent que les agrégats formés en
présence de cuivre sont majoritairement amorphes et ne contiendraient donc peu ou pas de
fibres (Exley 2004).
Le site de fixation du cuivre, de géométrie plan carrée, est vraisemblablement constitué d’au
moins 3 azotes provenant du cycle imidazole des histidines 6, 13 et 14 (Curtain et al. 2001;
Schoneich and Williams 2002; Kowalik-Jankowska et al. 2003; Lim and Vachet 2003;
Kowalik-Jankowska et al. 2004; Syme et al. 2004; Karr et al. 2005; Guilloreau et al. 2007).
Le 4ème ligand n’a pas encore été clairement identifié et serait dépendant du pH : oxygène à
bas pH, azote à haut pH (hétérogène à pH physiologique) (Kowalik-Jankowska et al. 2003;
Syme et al. 2004; Guilloreau et al. 2007). Ainsi, en fonction des conditions expérimentales,
différents ligands ont été proposés : N-terminal, carboxylate de l’aspartate 1 ou du Glutamate
11, azote du squelette carboné (Kowalik-Jankowska et al. 2003; Syme et al. 2004; Karr et al.
2005; Hou and Zagorski 2006). Un équilibre entre le N-term et l’oxygène de l’aspartate 1 est
très probable (spectre de dichroïsme circlaire modifié quand le peptide est acétylé, perte du

Chapitre 1 : la maladie d’Alzheimer
57
ligand oxygène par délétion des trois premiers acides aminés, effet paramagnétique important
sur Asp1 en RMN du proton). La présence ainsi que l’absence d’effet de l’eau marquée ont
été rapportés, ce qui ne permet pas de statuer sur la présence de ligand H2O axial labile, qui
dépend probablement des conditions expérimentales (Karr et al. 2005; Guilloreau et al. 2007).
Bush et al. ont montré qu’Aβ (et ses formes tronquées) était capable de fixer 2 équivalents de
cuivre(II) avec des constantes de dissociation différentes, faisant ainsi apparaître un site de
forte affinité et un site de fixation plus faible. Garzon-Rodriguez et al. ont mesuré les
constantes d’affinité du cuivre(II) pour le peptide Aβ40 et 42 sur le site de plus forte affinité
en suivant la fluorescence due à la Tyrosine (en position 10) (Garzon-Rodriguez et al. 1999).
Les expériences ont révélé des constantes de dissociation de l’ordre de 2μM dans le tampon
Tris. Des expériences similaires, dans le même tampon, ont été menées par Karr et al.,
révélant des constantes de dissociation de 11 ; 28 ; 47 μM respectivement pour les peptides
Aβ40, Aβ28 et Aβ16 (Karr et al. 2005). Cependant ces constantes sont des constantes
apparentes car les expériences ont été réalisées dans un tampon qui lui aussi possède une
affinité pour le cuivre. Il y a donc compétition entre le tampon et le peptide. Pour s’affranchir
de ce paramètre, Syme et al. ont effectué des mesures par compétition avec deux ligands du
cuivre (Histidine et Glycine) (Syme et al. 2004). Ils ont ainsi encadré le Kd de l’Aβ28 entre
1,5 et 500 nM. Si on tient compte du tampon utilisé (et de son affinité pour le cuivre), les Kd
sont similaires. Le peptide Aβ soluble et les peptides modèles plus courts présentent à pH
physiologique un Kd apparent de l’ordre du nanomolaire. Les dernières études réalisées par
calorimétrie de titrage isotherme et différentes méthodes spectroscopiques confirment ces
valeurs : Kd apparent de l’ordre 100 nM dans l’Hepes à pH 7,4 (10 μM pour le site de faible
affinité) (Guilloreau et al. 2006).
...relativement à la toxicité
D’un point de vue de l’agrégation, des études in vivo, ont montré une augmentation de
plaques séniles liée à un déclin cognitif, lors d’un régime riche en cuivre et cholestérol
(Sparks et al. 2003; Morris et al. 2006). De plus, l’administration de clioquinol à des souris
transgéniques induit une diminution des plaques amyloïdes de près de 50% (Cherny et al.
2001). In vitro, à pH 7,4, des traces de cuivre(II) (concentration inférieure à 0,1 μM) suffisent
à induire la précipitation du peptide (Atwood et al. 2000). Ce phénomène est amplifié à pH
acide (pH = 6,6), ce qui est également le cas de fer(III) (Atwood et al. 1998). Or le pH au sein
du cerveau d’un patient atteint de la MA a tendance à s’acidifier légèrement suite à un
phénomène d’inflammation, favorisant ainsi la précipitation d’Aβ par le cuivre et le fer.
Or il existe une relation évidente entre les espèces réactives de l’oxygène (ROS) et la toxicité

Chapitre 1 : la maladie d’Alzheimer
58
d’Aβ pour les neurones (Bush et al. 2003). De nombreuses études ont montré qu’Aβ est
toxique in vitro et in vivo en relation avec la présence d’ions métalliques redox actifs trouvés
associés aux lésions amyloïdes dans des cerveaux post-mortem (Smith et al. 1997; Sayre et al.
2000; Tabner et al. 2002; Cuajungco et al. 2005). La présence de peptide Aβ oxydé sur la
méthionine 35, retrouvé associé aux métaux cuivre(II) et zinc(II) dans les plaques amyloïdes
post mortem, traduit également un environnement pro-oxydant au niveau de ces plaques
(Dong et al. 2003). En effet, le peptide Aβ (en particulier les formes longues), en présence
d’ions métalliques FeIII ou CuII et de réducteurs biologiques extérieurs tels que l’ascorbate, la
dopamine ou le cholestérol, est capable de générer du peroxyde d’hydrogène H2O2 (Huang et
al. 1999a; Opazo et al. 2002). Une récente étude a confirmé que le peptide Aβ42 était le plus
toxique vis-à-vis de la production de ROS (produisant notamment 5 fois plus de HO• que
Aβ40), tout en indiquant que la quantité de HO• générée par les complexes CuII-Aβ était
modéré (se situant entre celles de CuII-GHK et CuII-DAHK, 2 complexes physiologiques)
(Guilloreau et al. 2007).
Implication du zinc…
...avec Tau
Plusieurs des kinases phosphorylant tau (notamment p70 S6) peuvent être activées par le zinc
(Harris et al. 2004; Bjorkdahl et al. 2005; Pei et al. 2006). Or il a été montré que le taux de
zinc dans le néocortex augmentait significativement chez la personne atteinte de la MA
(Danscher et al. 1997; Lovell et al. 1998; Opazo et al. 2006), ce qui pourrait favoriser
l’hyperphosphorylation de tau.
Plus généralement, le zinc (ainsi que le magnésium) est également impliqué dans la
polymérisation des tubulines, en effet une carence alimentaire maternelle en zinc peut altérer
le développement du cerveau du fait de perturbations dans la formation des microtubules
(Gaskin and Kress 1977; Oteiza et al. 1990; Oteiza et al. 1990; Yoshiike et al. 2001).
… avec l’APP
L’APP comprend un site de fixation du zinc (Bush et al. 1993; Bush et al. 1994; Bush et al.
1994). Il est compris dans la région 170-188 de la protéine et est constitué de deux résidus
cystéines 186 et 187 et d’autres ligands potentiels (Cys-174, Met-170, Asp-177, Glu-184). La
fixation de zinc pourrait favoriser la formation d’un dimère in vivo (Scheuermann et al. 2001;
Ciuculescu et al. 2005).
Le zinc joue également un rôle important sur la protéolyse de l’APP et donc la production

Chapitre 1 : la maladie d’Alzheimer
59
d’Aβ. Tout d’abord le domaine de fixation du zinc dans la région Aβ de l’APP s’étend sur le
domaine de clivage de l’α-sécrétase, le métal pourrait donc potentiellement moduler le
clivage ainsi que la dégradation du peptide (Hoke et al. 2005). Ensuite, la préséniline, une
sous-unité de la γ-sécrétase, est également sensible au taux de zinc (la production de PS1
augmentant avec l’administration exogène de zinc) (Park et al. 2001).
Une des α-sécrétases potentielles, la TACE, possède un ion zinc dans son domaine
catalytique. La liaison cystéine–zinc régule (parmi d’autres interactions) l’activité catalytique
de la sécrétase (Cross et al. 2002; Gonzales et al. 2004; Buckley et al. 2005). Similairement,
ADAM-10, une autre candidate pour le clivage en α de l’APP, possède également un site de
fixation du zinc indispensable à son activité catalytique (Lammich et al. 1999).
…sur la concentration d’Aβ
Environ 15 % du zinc cérébral est stocké sous forme ionique dans des vésicules pour être
ensuite transitoirement libéré dans la synapse. Des études ont montré que le zinc synaptique
des fibres moussues de l’hippocampe semble avoir un effet important sur la production d’Aβ,
en effet, une ablation du gène codant pour la protéine transportant le zinc dans les vésicules
synaptiques (ZnT3) induit une importante réduction des plaques amyloïdes chez les souris
transgéniques surexprimant l’APP (Lee et al. 2002).
...avec Aβ
La présence de zinc induit la précipitation du peptide dans une large gamme de pH (5,5-7,5)
(Bush et al. 1994; Cherny et al. 1999; Yoshiike et al. 2001). Mais la nature fibrillaire des
agrégats induits par le zinc est controversée (Exley 2006; Ha et al. 2007). Le mécanisme
d’agrégation d’Aβ en présence de zinc sera l’objet des chapitres 3 et 4 de cette thèse.
La fixation du zinc à Aβ correspond, d’après la littérature, à un Kd apparent de l’ordre du
micromolaire (pour plus de détails, se reporter à la partie introduction du chapitre 2)
(Clements et al. 1996; Mekmouche et al. 2005), le peptide fixe donc moins bien le zinc que le
cuivre. De plus, comme pour le fer, les concentrations en zinc libre sont généralement trop
faibles pour permettre à Aβ de se lier au métal et l’affinité du zinc pour les protéines à zinc du
cerveau, telles la MT-3, est beaucoup plus forte (environ 6.1010 M-1) (Hasler et al. 2000; Xiao
et al. 2004), pointant là encore l’hypothèse d’une dérégulation de l’homéostasie du zinc pour
expliquer la présence importante de zinc au sein des plaques amyloïdes. Il existe cependant
une exception au niveau des synapses où les concentrations en zinc peuvent être
transitoirement très importantes (cf paragraphe II.4.a), en particulier dans le cas d’ischémie ou
de traumatisme cérébral.

Chapitre 1 : la maladie d’Alzheimer
60
Le site de fixation du zinc n’est pas encore parfaitement défini, Comme pour le cuivre, la
fixation via les trois histidines 6 et 13 et 14 a largement été décrite, mais le 4ème ligand est
sujet à controverse (Liu et al. 1999; Yang et al. 2000; Kozin et al. 2001; Mekmouche et al.
2005; Syme and Viles 2006). Les dernière études suggèrent respectivement le carboxylate du
glutamate 11 (à pH 6,5 et 7,5, basé sur des données RMN du peptide Aβ16 acétylé,
empêchant ainsi une interaction N-term et altèrant peut-être celle avec l’aspartate 1) (Zirah et
al. 2006) et le N-terminal (Figure 1.16 ; RMN du peptide Aβ40 à pH 7,0-7,3) (Danielsson et
al. 2007), similairement au cuivre. Elles excluent également définitivement la Tyr10 comme
ligand potentiel. Zirah et al. indiquent que la fixation du zinc favoriserait une structuration en
hélice alpha, en accord avec la formation d’agrégats majoritairement amorphes en présence de
zinc (Huang et al. 1997; Curtain et al. 2001). Cependant, il est possible que l’utilisation d’un
peptide acétylé modifie le site de fixation du zinc (Glu11 plutôt que Asp1/N-term) et donc la
structure secondaire du complexe.
Un deuxième site de plus faible affinité (avec un rôle physiologique probablement assez
limité) serait situé au niveau des résidus Asp23, Val24, Asn26 et Lys28 du peptide Aβ
(Danielsson et al. 2007).
Figure 1.16. Représentation schématique du site de fixation du zinc dans le peptide Aβ40 non
acétylé à pH physiologique (Danielsson et al. 2007).

Chapitre 1 : la maladie d’Alzheimer
61
...dans la dégradation d’Aβ
IDE et NEP (cf. paragraphe sur la stimulation de la dégradation d’Aβ, section II.3.b.) font
parties de la famille des métalloprotéines à zinc, lesquelles comprennent généralement une
séquence du type HEXXH où les deux histidines jouent le rôle de ligand pour le zinc
catalytique (Gomis-Ruth 2003). Ainsi ces deux enzymes peuvent potentiellement être
influencées par un dysfonctionnement dans l’homéostasie de zinc. De plus elles peuvent être
oxydées dans le cas de la MA et perdre ainsi une partie de leur activité (Wang et al. 2003;
Caccamo et al. 2005; Shinall et al. 2005). La plasmine semble également liée indirectement
au zinc ; l’activité des matrices métalloprotéinases (MMP) est elle aussi dépendante de l’ion
zinc (cf. revue (Yong et al. 1998)).
...relativement à la toxicité
Contrairement au cuivre et au fer, le zinc, dont les orbitales atomiques d sont saturées, n’a pas
d’activité redox et n’induit pas directement de production de ROS lorsqu’il est lié à Aβ. Ainsi,
en prenant la place du cuivre, le zinc aurait plutôt un effet protecteur (Cuajungco et al. 2000;
Yoshiike et al. 2001). Cependant, la fixation du zinc accélère l’agrégation du peptide, ce qui,
dans la première hypothèse de la toxicité des plaques amyloïdes, aurait un effet toxique. Or,
actuellement, les formes oligomériques solubles semblent plus toxiques. De plus, le zinc ne
semble pas favoriser la formation de feuillet β et de fibres amyloïdes. Des études ont
cependant montré que l’injection dans le cortex cérébral de rats de peptides Aβ42 en présence
de zinc était plus toxique que l’injection du peptide seul (Bishop and Robinson 2004). Mais il
est possible que ce résultat soit uniquement du à l’excitotoxicité du zinc(II) libre (processus
pathologique de destruction neuronale par hyperactivation des récepteurs excitateurs
neuronaux) et pas au complexe ZnII-Aβ42. Certaines études émettent également l’hypothèse
qu’Aβ pourrait former des pores dans la membrane lipidique et entraîner ainsi une
dégénération neuronale. Le zinc bloquerait ces canaux, protégeant ainsi les cellules (Lin et al.
1999; Lin et al. 2001) (cf. chapitre 6). Le rôle direct, toxique ou protecteur, du zinc dans la
MA est très controversé et requiert donc des recherches complémentaires.
II.4.f. Approche thérapeutique en relation avec le cuivre et le zinc
L’utilisation de chélateurs du cuivre et du zinc pour le traitement de la MA repose sur deux
hypothèses principales : 1) chélater le cuivre lié à Aβ (aux différents stades d’agrégation)
permettrait de réduire la production de ROS, 2) chélater le zinc permettrait de réduire la

Chapitre 1 : la maladie d’Alzheimer
62
formation de plaques amyloïdes et de dissoudre celles préexistantes.
Les résultats obtenus en laboratoire (in vitro et in vivo), en particulier avec le clioquinol (5-
chloro-7-iodo-8-hydroxyquinoline, CQ, chélateur du zinc(II) et du cuivre(II)), tendent à
conforter cette théorie puisqu’une forte diminution des plaques amyloïdes et une amélioration
de l’état cognitif ont été observées (Cherny et al. 2001; Regland et al. 2001; Ritchie et al.
2003; Bellingham et al. 2004). Cependant des effets secondaires non négligeables, peut-être
dus à une dérégulation trop importante de l’homéostasie de ces deux métaux associée à une
déficience en vitamine B12, ont eu pour conséquences l’arrêt des tests cliniques (Yassin et al.
2000; Tabira 2001).
Le mécanisme étiologique supposé du clioquinol vis-à-vis du peptide Aβ in vivo n’a
cependant pas été démontré et de récentes études suggèrent une voie d’action bien différente
de l’hypothèse de départ (White et al. 2006). En effet, ce chélateur semble stimuler la
dégradation du peptide Aβ via l’activation des matrices métalloprotéinases (MMP), elle-
même due à l’activation de la phosphoinositol-3-kinase (PI-3K) et des JNK (c-Jun N-terminal
Kinase) du fait de l’augmentation de la concentration en métaux (libérés des plaques
amyloïdes). Cette réduction massive des plaques chez les rongeurs a été observée lors de
l’administration conjointe de CQ (ou d’autres chélateurs) et de Zn2+ ou Cu2+ (sans qu’une
augmentation importante en ROS soit identifiée) (Caragounis et al. 2007). Différents types de
chélateurs ont été testés avec succès sur des cultures cellulaires (Figure 1.17) : 8-
hydroxyquinoline, dérivés de la phénanthroline et un composé sulfuré la pyrrolidine
dithiocarbamate. La diversité de ces chélateurs pointe plutôt vers un rôle de transport des
métaux plutôt qu’une interaction direct avec un récepteur. Généralement, leur capacité à
diminuer le taux de Aβ est corrélée à leur lipophilie et à leur capacité à augmenter les taux de
cuivre et zinc cellulaires.
Cette récente hypothèse demande évidemment à être confirmée d’un point de vue du
mécanisme et de la toxicité à long terme (Minogue et al. 2003), mais elle ouvre le champ à de
nouvelles approches quant à la conception de chélateurs de métaux pour la MA.

Chapitre 1 : la maladie d’Alzheimer
63
Dérivés de la 8-hydroxyquinoline (clioquinol : R1 = Cl et R2 = I)
Dérivés de la phénantroline Pyrrolidine dithiocarbamate
Desferrioxamine (DFOA)
Figure 1.17. Exemples de chélateurs du cuivre et du zinc (d’après (Caragounis et al. 2007))
De manière générale, au vue de la diversité du rôle des différents métaux évoqués dans les
différentes étapes supposées du développement de la MA, il y a de fortes chances que l’action
des chélateurs de métaux soit multifactorielles, la difficulté étant d’obtenir un maximum
d’effet curatif en évitant une trop grande dérégulation de l’homéostasie des métaux, néfaste
pour l’organisme (par exemple dans le cas du CQ, l’élévation du taux de zinc et de cuivre
aurait conduit à une diminution en vitamine B12) (Wang et al. 2006; Caragounis et al. 2007;
Prodan et al. 2007). Cette voie de recherche reste évidemment très prometteuse et requiert une
meilleure compréhension du rôle joué par les métaux de transition, tels le zinc et le cuivre,
dans le développement de la MA.

Chapitre 1 : la maladie d’Alzheimer
64
III. Conclusion et contexte de l’étude
Ce chapitre bibliographique illustre bien la complexité des mécanismes pathogéniques de la
maladie d’Alzheimer. Les difficultés rencontrées par la communauté scientifique pour
élucider l’origine étiologique, probablement multifactorielle, de la maladie a conduit à un
élargissement, voir à une dispersion, de l’éventail des stratégies thérapeutiques. Une meilleure
compréhension des mécanismes de développement de la maladie est donc nécessaire pour
mieux cibler la recherche de traitements curatifs.
Dans cette thèse, nous nous sommes basé sur l’hypothèse principale de la cascade amyloïde.
L’ensemble des études présentées s’articule donc autour du peptide amyloïde bêta. D’autre
part, il est important de comprendre le rôle joué par les ions métalliques (en particulier zinc et
cuivre) dans la maladie d’Alzheimer. En effet, leur implication semble essentielle puisqu’ils
prennent part à l’agrégation et probablement à la toxicité d’Aβ. Ainsi l’étude de l’affinité du
peptide amyloïde bêta pour ces métaux (chapitre 2) s’inscrit logiquement dans cette optique.
Elle est de plus nécessaire à l’élaboration de stratégies thérapeutiques par chélation des
métaux.
Nous nous sommes ensuite focalisés sur le zinc, dont l’interaction avec Aβ a été moins
étudiée que le cuivre. Le zinc a été rapporté induire l’agrégation d’Aβ. Afin d’expliquer cette
influence et de saisir sa pertinence vis-à-vis de la formation des fibres amyloïdes, il nous a
semblé important de rechercher l’existence d’éventuelles espèces intermédiaires (chapitre 3)
et de déterminer leur structure secondaire (chapitre 4).
La provenance des quantités élevées de zinc dans les plaques amyloïdes reste encore à
élucider. Ainsi, l’hypothèse d’une interaction avec la métallothionéine-3, protéine à zinc très
répandue dans le cerveau, a été explorée au chapitre 5.
Enfin, d’un point de vue de la toxicité, une des voies envisagées dans la littérature consistant
en la formation de pores d’Aβ dans les membranes cellulaires, a été analysée au chapitre 6 en
prenant compte de l’influence du zinc.

Chapitre 1 : la maladie d’Alzheimer
65
Références
J. Abad-Rodriguez, M. D. Ledesma, K. Craessaerts, S. Perga, M. Medina, et al. (2004). "Neuronal membrane cholesterol loss enhances amyloid peptide generation." J Cell Biol, 167(5), 953-60.
H. M. Abdul, V. Calabrese, M. Calvani and D. A. Butterfield (2006). "Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1-42-mediated oxidative stress and neurotoxicity: implications for Alzheimer's disease." J Neurosci Res, 84(2), 398-408.
A. Y. Abramov, L. Canevari and M. R. Duchen (2004). "Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture." Biochim Biophys Acta, 1742(1-3), 81-7.
P. Ackrill, A. J. Ralston, J. P. Day and K. C. Hodge (1980). "Successful removal of aluminium from patient with dialysis encephalopathy." Lancet, 2(8196), 692-3.
P. A. Adlard, A. K. West and J. C. Vickers (1998). "Increased density of metallothionein I/II-immunopositive cortical glial cells in the early stages of Alzheimer's disease." Neurobiol Dis, 5(5), 349-56.
P. A. Adlard and A. I. Bush (2006). "Metals and Alzheimer's disease." J Alzheimers Dis, 10(2-3), 145-63.
P. S. Aisen, K. L. Davis, J. D. Berg, K. Schafer, K. Campbell, et al. (2000). "A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study." Neurology, 54(3), 588-93.
H. Akiyama, S. Barger, S. Barnum, B. Bradt, J. Bauer, et al. (2000). "Inflammation and Alzheimer's disease." Neurobiol Aging, 21(3), 383-421.
R. Alberca, E. Montes-Latorre, E. Gil-Neciga, P. Mir-Rivera and P. Lozano-San Martin (2002). "[Alzheimer's disease and women]." Rev Neurol, 35(6), 571-9.
G. S. Alexopoulos, B. S. Meyers, R. C. Young, S. Mattis and T. Kakuma (1993). "The course of geriatric depression with "reversible dementia": a controlled study." Am J Psychiatry, 150(11), 1693-9.
D. W. Allison, A. S. Chervin, V. I. Gelfand and A. M. Craig (2000). "Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules." J Neurosci, 20(12), 4545-54.
A. C. Alonso, T. Zaidi, I. Grundke-Iqbal and K. Iqbal (1994). "Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease." Proc Natl Acad Sci U S A, 91(12), 5562-6.
A. Alvarez, R. Toro, A. Caceres and R. B. Maccioni (1999). "Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death." FEBS Lett, 459(3), 421-6.
A. Alzheimer (1907). Allg. Z. Psychiatr, 64146. A. Alzheimer (1911). Z. Ges. Neurol. Pschiat., 4356. A. F. Alzheimer (2006). "le livre vert de la maladie d'Alzheimer. Etat des lieux et
perspectives." Presses du Louvre. D. Ames and C. Ritchie (2007). "Antioxidants and Alzheimer's disease: time to stop feeding
vitamin E to dementia patients?" Int Psychogeriatr, 19(1), 1-8. C. Andorfer, C. M. Acker, Y. Kress, P. R. Hof, K. Duff, et al. (2005). "Cell-cycle reentry and
cell death in transgenic mice expressing nonmutant human tau isoforms." J Neurosci, 25(22), 5446-54.
T. S. Anekonda (2006). "Resveratrol--a boon for treating Alzheimer's disease?" Brain Res Rev,

Chapitre 1 : la maladie d’Alzheimer
66
52(2), 316-26. B. Angeletti, K. J. Waldron, K. B. Freeman, H. Bawagan, I. Hussain, et al. (2005). "BACE1
cytoplasmic domain interacts with the copper chaperone for superoxide dismutase-1 and binds copper." J Biol Chem, 280(18), 17930-7.
L. Aniksztejn, G. Charton and Y. Ben-Ari (1987). "Selective release of endogenous zinc from the hippocampal mossy fibers in situ." Brain Res, 404(1-2), 58-64.
O. N. Antzutkin, J. J. Balbach, R. D. Leapman, N. W. Rizzo, J. Reed, et al. (2000). "Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta-sheets in Alzheimer's beta-amyloid fibrils." Proc Natl Acad Sci U S A, 97(24), 13045-50.
O. N. Antzutkin, R. D. Leapman, J. J. Balbach and R. Tycko (2002). "Supramolecular structural constraints on Alzheimer's beta-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance." Biochemistry, 41(51), 15436-50.
O. N. Antzutkin, J. J. Balbach and R. Tycko (2003). "Site-specific identification of non-beta-strand conformations in Alzheimer's beta-amyloid fibrils by solid-state NMR." Biophys J, 84(5), 3326-35.
N. Arispe, H. B. Pollard and E. Rojas (1993). "Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1-40)] in bilayer membranes." Proc. Natl. Acad. Sci. U.S.A., 9010573-10577.
A. D. Armendariz, M. Gonzalez, A. V. Loguinov and C. D. Vulpe (2004). "Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App." Physiol Genomics, 20(1), 45-54.
S. Artavanis-Tsakonas, K. Matsuno and M. E. Fortini (1995). "Notch signaling." Science, 268(5208), 225-32.
R. S. Arze, I. S. Parkinson, N. E. Cartlidge, P. Britton and M. K. Ward (1981). "Reversal of aluminium dialysis encephalopathy after desferrioxamine treatment." Lancet, 2(8255), 1116.
S. Asthana, S. Craft, L. D. Baker, M. A. Raskind, R. S. Birnbaum, et al. (1999). "Cognitive and neuroendocrine response to transdermal estrogen in postmenopausal women with Alzheimer's disease: results of a placebo-controlled, double-blind, pilot study." Psychoneuroendocrinology, 24(6), 657-77.
H. Atamna (2006). "Heme binding to Amyloid-beta peptide: mechanistic role in Alzheimer's disease." J Alzheimers Dis, 10(2-3), 255-66.
C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, et al. (1998). "Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis." J Biol Chem, 273(21), 12817-26.
C. S. Atwood, R. C. Scarpa, X. Huang, R. D. Moir, W. D. Jones, et al. (2000). "Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42." J Neurochem, 75(3), 1219-33.
Y. Avramovich-Tirosh, T. Amit, O. Bar-Am, H. Zheng, M. Fridkin, et al. (2007). "Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer's disease." J Neurochem, 100(2), 490-502.
J. J. Balbach, A. T. Petkova, N. A. Oyler, O. N. Antzutkin, D. J. Gordon, et al. (2002). "Supramolecular structure in full-length Alzheimer's beta-amyloid fibrils: evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance." Biophys J, 83(2), 1205-16.
I. Baldi, P. Lebailly, B. Mohammed-Brahim, L. Letenneur, J. F. Dartigues, et al. (2003). "Neurodegenerative diseases and exposure to pesticides in the elderly." Am J

Chapitre 1 : la maladie d’Alzheimer
67
Epidemiol, 157(5), 409-14. L. M. Baltasar-Rodriguez, R. O. Millan-Guerrero, R. Aceves-Themsel, S. Isais-Millan and I.
Delgado-Enciso (2006). "[Longitudinal study of three families with familial Parkinson's disease]." Gac Med Mex, 142(5), 387-91.
S. Bandyopadhyay, X. Huang, H. Cho, N. H. Greig, M. B. Youdim, et al. (2006). "Metal specificity of an iron-responsive element in Alzheimer's APP mRNA 5'untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator." J Neural Transm Suppl, (71), 237-47.
C. Barbato, N. Canu, N. Zambrano, A. Serafino, G. Minopoli, et al. (2005). "Interaction of Tau with Fe65 links tau to APP." Neurobiol Dis, 18(2), 399-408.
G. Barja (2004). "Free radicals and aging." Trends Neurosci, 27(10), 595-600. K. J. Barnham, W. J. McKinstry, G. Multhaup, D. Galatis, C. J. Morton, et al. (2003).
"Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis." J Biol Chem, 278(19), 17401-7.
M. R. Basha, W. Wei, S. A. Bakheet, N. Benitez, H. K. Siddiqi, et al. (2005). "The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain." J Neurosci, 25(4), 823-9.
C. Bastian and Y. V. Li (2007). "Fluorescence imaging study of extracellular zinc at the hippocampal mossy fiber synapse." Neurosci Lett, 419(2), 119-24.
H. Basun, L. G. Forssell, L. Wetterberg and B. Winblad (1991). "Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer's disease." J Neural Transm Park Dis Dement Sect, 3(4), 231-58.
T. A. Bayer, S. Schafer, A. Simons, A. Kemmling, T. Kamer, et al. (2003). "Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice." Proc Natl Acad Sci U S A, 100(24), 14187-92.
D. Beauchemin and R. Kisilevsky (1998). "A method based on ICP-MS for the analysis of Alzheimer's amyloid plaques." Anal Chem, 70(5), 1026-9.
J. S. Becker, M. Zoriy, C. Pickhardt, E. Damoc, G. Juhacz, et al. (2005). "Determination of phosphorus-, copper-, and zinc-containing human brain proteins by LA-ICPMS and MALDI-FTICR-MS." Anal Chem, 77(18), 5851-60.
P. Belenky, K. L. Bogan and C. Brenner (2007). "NAD+ metabolism in health and disease." Trends Biochem Sci, 32(1), 12-9.
S. A. Bellingham, D. K. Lahiri, B. Maloney, S. La Fontaine, G. Multhaup, et al. (2004). "Copper depletion down-regulates expression of the Alzheimer's disease amyloid-beta precursor protein gene." J Biol Chem, 279(19), 20378-86.
M. Bennecib, C. X. Gong, I. Grundke-Iqbal and K. Iqbal (2001). "Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356." FEBS Lett, 490(1-2), 15-22.
A. Bernareggi, Z. Duenas, J. M. Reyes-Ruiz, F. Ruzzier and R. Miledi (2007). "Properties of glutamate receptors of Alzheimer's disease brain transplanted to frog oocytes." Proc Natl Acad Sci U S A, 104(8), 2956-60.
S. Bhatnagar and S. Taneja (2001). "Zinc and cognitive development." Br J Nutr, 85 Suppl 2S139-45.
U. Bickel, T. Thomsen, W. Weber, J. P. Fischer, R. Bachus, et al. (1991). "Pharmacokinetics of galanthamine in humans and corresponding cholinesterase inhibition." Clin Pharmacol Ther, 50(4), 420-8.
W. H. Birkenhager and J. A. Staessen (2004). "Blood pressure and dementia." Panminerva Med, 46(4), 227-37.
G. M. Bishop, S. R. Robinson, Q. Liu, G. Perry, C. S. Atwood, et al. (2002). "Iron: a pathological mediator of Alzheimer disease?" Dev Neurosci, 24(2-3), 184-7.

Chapitre 1 : la maladie d’Alzheimer
68
G. M. Bishop and S. R. Robinson (2003). "Human Abeta1-42 reduces iron-induced toxicity in rat cerebral cortex." J Neurosci Res, 73(3), 316-23.
G. M. Bishop and S. R. Robinson (2004). "The amyloid paradox: amyloid-beta-metal complexes can be neurotoxic and neuroprotective." Brain Pathol, 14(4), 448-52.
C. Bjorkdahl, M. J. Sjogren, B. Winblad and J. J. Pei (2005). "Zinc induces neurofilament phosphorylation independent of p70 S6 kinase in N2a cells." Neuroreport, 16(6), 591-5.
B. J. Blanchard, A. Chen, L. M. Rozeboom, K. A. Stafford, P. Weigele, et al. (2004). "Efficient reversal of Alzheimer's disease fibril formation and elimination of neurotoxicity by a small molecule." Proc. Natl. Acad. Sci. U.S.A., 101(40), 14326-14332.
M. L. Bleecker, D. P. Ford, K. N. Lindgren, V. M. Hoese, K. S. Walsh, et al. (2005). "Differential effects of lead exposure on components of verbal memory." Occup Environ Med, 62(3), 181-7.
P. Blocq and G. Marinesco (1892). Sem. Med., 12445. M. Blurton-Jones and F. M. Laferla (2006). "Pathways by which Abeta facilitates tau
pathology." Curr Alzheimer Res, 3(5), 437-48. T. Borchardt, J. Camakaris, R. Cappai, C. L. Masters, K. Beyreuther, et al. (1999). "Copper
inhibits beta-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion." Biochem J, 344 Pt 2461-7.
G. M. Bores, F. P. Huger, W. Petko, A. E. Mutlib, F. Camacho, et al. (1996). "Pharmacological evaluation of novel Alzheimer's disease therapeutics: acetylcholinesterase inhibitors related to galanthamine." J Pharmacol Exp Ther, 277(2), 728-38.
B. Borroni, F. Colciaghi, L. Pastorino, C. Pettenati, E. Cottini, et al. (2001). "Amyloid precursor protein in platelets of patients with Alzheimer disease: effect of acetylcholinesterase inhibitor treatment." Arch Neurol, 58(3), 442-6.
M. P. Bowes, E. Masliah, D. A. Otero, J. A. Zivin and T. Saitoh (1994). "Reduction of neurological damage by a peptide segment of the amyloid beta/A4 protein precursor in a rabbit spinal cord ischemia model." Exp Neurol, 129(1), 112-9.
G. T. Bramblett, M. Goedert, R. Jakes, S. E. Merrick, J. Q. Trojanowski, et al. (1993). "Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding." Neuron, 10(6), 1089-99.
J. C. Breitner, B. A. Gau, K. A. Welsh, B. L. Plassman, W. M. McDonald, et al. (1994). "Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study." Neurology, 44(2), 227-32.
A. Broccolini, T. Gidaro, R. Morosetti, C. Gliubizzi, T. Servidei, et al. (2006). "Neprilysin participates in skeletal muscle regeneration and is accumulated in abnormal muscle fibres of inclusion body myositis." J Neurochem, 96(3), 777-89.
B. M. Broome and M. H. Hecht (2000). "Nature disfavors sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis." J Mol Biol, 296(4), 961-8.
C. A. Buckley, F. N. Rouhani, M. Kaler, B. Adamik, F. I. Hawari, et al. (2005). "Amino-terminal TACE prodomain attenuates TNFR2 cleavage independently of the cysteine switch." Am J Physiol Lung Cell Mol Physiol, 288(6), L1132-8.
A. Burns, M. Rossor, J. Hecker, S. Gauthier, H. Petit, et al. (1999). "The effects of donepezil in Alzheimer's disease - results from a multinational trial." Dement Geriatr Cogn Disord, 10(3), 237-44.
J. Busciglio, A. Lorenzo, J. Yeh and B. A. Yankner (1995). "beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding." Neuron, 14(4), 879-88.
A. I. Bush, G. Multhaup, R. D. Moir, T. G. Williamson, D. H. Small, et al. (1993). "A novel

Chapitre 1 : la maladie d’Alzheimer
69
zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer's disease." J Biol Chem, 268(22), 16109-12.
A. I. Bush, W. H. Pettingell, Jr., M. de Paradis, R. E. Tanzi and W. Wasco (1994). "The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily." J Biol Chem, 269(43), 26618-21.
A. I. Bush, W. H. Pettingell, G. Multhaup, M. d Paradis, J. P. Vonsattel, et al. (1994). "Rapid induction of Alzheimer A beta amyloid formation by zinc." Science, 265(5177), 1464-7.
A. I. Bush, W. H. Pettingell, G. Multhaup, M. Paradis, J. P. Vonsattel, et al. (1994). "Rapid induction of Alzheimer Abeta amyloid formation by zinc." Science, 265(5177), 1464-1467.
A. I. Bush, C. L. Masters and R. E. Tanzi (2003). "Copper, β-amyloid, and Alzheimer's disease: tapping a sensitive connection." Proc. Natl. Acad. Sci. U.S.A., 100(20), 11193-11194.
O. Butovsky, M. Koronyo-Hamaoui, G. Kunis, E. Ophir, G. Landa, et al. (2006). "Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1." Proc Natl Acad Sci U S A, 103(31), 11784-9.
D. A. Butterfield and C. M. Lauderback (2002). "Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress." Free Radic. Biol. Med., 32(11), 1050-1060.
A. Caccamo, S. Oddo, M. C. Sugarman, Y. Akbari and F. M. LaFerla (2005). "Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders." Neurobiol Aging, 26(5), 645-54.
J. Caltagarone, Z. Jing and R. Bowser (2007). "Focal adhesions regulate Abeta signaling and cell death in Alzheimer's disease." Biochim Biophys Acta, 1772(4), 438-45.
G. Candore, C. R. Balistreri, M. P. Grimaldi, S. Vasto, F. Listi, et al. (2006). "Age-related inflammatory diseases: role of genetics and gender in the pathophysiology of Alzheimer's disease." Ann N Y Acad Sci, 1089472-86.
L. Canevari and J. B. Clark (2007). "Alzheimer's disease and cholesterol: the fat connection." Neurochem Res, 32(4-5), 739-50.
L. M. Canzoniero, S. L. Sensi and D. W. Choi (1997). "Measurement of intracellular free zinc in living neurons." Neurobiol Dis, 4(3-4), 275-9.
A. Caragounis, T. Du, G. Filiz, K. M. Laughton, I. Volitakis, et al. (2007). "Differential modulation of Alzheimer's disease amyloid beta peptide accumulation by diverse classes of metal ligands." Biochem J, 407(3), 435-450.
R. J. Chalifour, R. W. McLaughlin, L. Lavoie, C. Morissette, N. Tremblay, et al. (2003). "Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide." J Biol Chem, 278(37), 34874-81.
S. Chang, T. ran Ma, R. D. Miranda, M. E. Balestra, R. W. Mahley, et al. (2005). "Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity." Proc Natl Acad Sci U S A, 102(51), 18694-9.
W. P. Chang, G. Koelsch, S. Wong, D. Downs, H. Da, et al. (2004). "In vivo inhibition of Abeta production by memapsin 2 (beta-secretase) inhibitors." J Neurochem, 89(6), 1409-16.
M. R. Chapman, L. S. Robinson, J. S. Pinkner, R. Roth, J. Heuser, et al. (2002). "Role of Escherichia coli curli operons in directing amyloid fiber formation." Science, 295(5556), 851-5.
G. Charton, C. Rovira, Y. Ben-Ari and V. Leviel (1985). "Spontaneous and evoked release of

Chapitre 1 : la maladie d’Alzheimer
70
endogenous Zn2+ in the hippocampal mossy fiber zone of the rat in situ." Exp Brain Res, 58(1), 202-5.
T. B. Chaston and D. R. Richardson (2003). "Iron chelators for the treatment of iron overload disease: relationship between structure, redox activity, and toxicity." Am J Hematol, 73(3), 200-10.
T. Y. Chen, P. H. Liu, C. T. Ruan, L. Chiu and F. L. Kung (2006). "The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein." Biochem Biophys Res Commun, 342(1), 266-72.
X. Chen and S. D. Yan (2006). "Mitochondrial Abeta: a potential cause of metabolic dysfunction in Alzheimer's disease." IUBMB Life, 58(12), 686-94.
R. A. Cherny, J. T. Legg, C. A. McLean, D. P. Fairlie, X. Huang, et al. (1999). "Aqueous dissolution of Alzheimer's disease Abeta amyloid deposits by biometal depletion." J. Biol. Chem., 274(33), 23223-23228.
R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, et al. (2001). "Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice." Neuron, 30(3), 665-76.
P. Chien, J. S. Weissman and A. H. DePace (2004). "Emerging principles of conformation-based prion inheritance." Annu Rev Biochem, 73617-56.
F. Chiti, N. Taddei, F. Baroni, C. Capanni, M. Stefani, et al. (2002). "Kinetic partitioning of protein folding and aggregation." Nat Struct Biol, 9(2), 137-43.
F. Chiti and C. M. Dobson (2006). "Protein misfolding, functional amyloid, and human disease." Annu Rev Biochem, 75333-66.
Y. H. Chong and Y. H. Suh (1996). "Amyloidogenic processing of Alzheimer's amyloid precursor protein in vitro and its modulation by metal ions and tacrine." Life Sci, 59(7), 545-57.
Z. Z. Chong, F. Li and K. Maiese (2005). "Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer's disease." Brain Res Brain Res Rev, 49(1), 1-21.
D. M. Chuang and H. K. Manji (2007). "In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked?" Biol Psychiatry, 62(1), 4-6.
M. Cini and A. Moretti (1995). "Studies on lipid peroxidation and protein oxidation in the aging brain." Neurobiol Aging, 16(1), 53-7.
E.-D. Ciuculescu, Y. Mekmouche and P. Faller (2005). "Metal-binding properties of the peptide APP170-188: A model of the ZnII-binding site of Amyloid Precursor Protein (APP)." Chem. Eur. J., 11(3), 903-909.
D. Claessen, R. Rink, W. de Jong, J. Siebring, P. de Vreugd, et al. (2003). "A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils." Genes Dev, 17(14), 1714-26.
A. Clements, D. Allsop, D. M. Walsh and C. H. Williams (1996). "Aggregation and metal-binding properties of mutant forms of the amyloid Aβ peptide of Alzheimer's disease." J. Neurochem., 66(2), 740-747.
A. B. Clippingdale, J. D. Wade and C. J. Barrow (2001). "The amyloid-beta peptide and its role in Alzheimer's disease." J Pept Sci, 7(5), 227-49.
F. E. Cohen and J. W. Kelly (2003). "Therapeutic approaches to protein-misfolding diseases." Nature, 426(6968), 905-9.
D. C. Cole, E. S. Manas, J. R. Stock, J. S. Condon, L. D. Jennings, et al. (2006). "Acylguanidines as small-molecule beta-secretase inhibitors." J Med Chem, 49(21), 6158-61.
T. B. Cole, H. J. Wenzel, K. E. Kafer, P. A. Schwartzkroin and R. D. Palmiter (1999). "Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of

Chapitre 1 : la maladie d’Alzheimer
71
the ZnT3 gene." Proc Natl Acad Sci U S A, 96(4), 1716-21. R. A. Colvin, C. P. Fontaine, M. Laskowski and D. Thomas (2003). "Zn2+ transporters and
Zn2+ homeostasis in neurons." Eur J Pharmacol, 479(1-3), 171-85. C. K. Combs, J. C. Karlo, S. C. Kao and G. E. Landreth (2001). "beta-Amyloid stimulation of
microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis." J Neurosci, 21(4), 1179-88.
J. R. Connor, P. Tucker, M. Johnson and B. Snyder (1993). "Ceruloplasmin levels in the human superior temporal gyrus in aging and Alzheimer's disease." Neurosci Lett, 159(1-2), 88-90.
E. H. Corder, A. M. Saunders, W. J. Strittmatter, D. E. Schmechel, P. C. Gaskell, et al. (1993). "Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families." Science, 261(5123), 921-3.
J. M. Cordy, I. Hussain, C. Dingwall, N. M. Hooper and A. J. Turner (2003). "Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein." Proc Natl Acad Sci U S A, 100(20), 11735-40.
K. M. Cosman, L. L. Boyle and A. P. Porsteinsson (2007). "Memantine in the treatment of mild-to-moderate Alzheimer's disease." Expert Opin Pharmacother, 8(2), 203-14.
V. Coustou, C. Deleu, S. Saupe and J. Begueret (1997). "The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog." Proc Natl Acad Sci U S A, 94(18), 9773-8.
J. B. Cross, J. S. Duca, J. J. Kaminski and V. S. Madison (2002). "The active site of a zinc-dependent metalloproteinase influences the computed pK(a) of ligands coordinated to the catalytic zinc ion." J Am Chem Soc, 124(37), 11004-7.
R. A. Crowther and M. Goedert (2000). "Abnormal tau-containing filaments in neurodegenerative diseases." J Struct Biol, 130(2-3), 271-9.
J. C. Cruz, H. C. Tseng, J. A. Goldman, H. Shih and L. H. Tsai (2003). "Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles." Neuron, 40(3), 471-83.
M. P. Cuajungco, L. E. Goldstein, A. Nunomura, M. A. Smith, J. T. Lim, et al. (2000). "Evidence that the beta -amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of Abeta by zinc." J. Biol. Chem., 275(26), 19439-19442.
M. P. Cuajungco, C. J. Frederickson and A. I. Bush (2005). "Amyloid-β metal interaction and metal chelation." Sub-Cellular Biochem., 38235-254.
C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, et al. (2001). "Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits." J Biol Chem, 276(23), 20466-73.
E. Czirr and S. Weggen (2006). "Gamma-secretase modulation with Abeta42-lowering nonsteroidal anti-inflammatory drugs and derived compounds." Neurodegener Dis, 3(4-5), 298-304.
J. Danielsson, R. Pierattelli, L. Banci and A. Graslund (2007). "High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid beta-peptide." Febs J, 274(1), 46-59.
G. Danscher, K. B. Jensen, C. J. Frederickson, K. Kemp, A. Andreasen, et al. (1997). "Increased amount of zinc in the hippocampus and amygdala of Alzheimer's diseased brains: a proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material." J Neurosci Methods, 76(1), 53-9.
J. M. Davies, C. V. Lowry and K. J. Davies (1995). "Transient adaptation to oxidative stress in yeast." Arch Biochem Biophys, 317(1), 1-6.

Chapitre 1 : la maladie d’Alzheimer
72
C. D. Davis, D. B. Milne and F. H. Nielsen (2000). "Changes in dietary zinc and copper affect zinc-status indicators of postmenopausal women, notably, extracellular superoxide dismutase and amyloid precursor proteins." Am J Clin Nutr, 71(3), 781-8.
K. L. Davis, L. J. Thal, E. R. Gamzu, C. S. Davis, R. F. Woolson, et al. (1992). "A double-blind, placebo-controlled multicenter study of tacrine for Alzheimer's disease. The Tacrine Collaborative Study Group." N Engl J Med, 327(18), 1253-9.
B. De Strooper, P. Saftig, K. Craessaerts, H. Vanderstichele, G. Guhde, et al. (1998). "Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein." Nature, 391(6665), 387-90.
B. De Strooper, W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, et al. (1999). "A presenilin-1-dependant-γ-secretase-like protease mediates release of Notch intracellular domain." Nature, 398518-522.
B. De Strooper, W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, et al. (1999). "A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain." Nature, 398(6727), 518-22.
M. A. Deibel, W. D. Ehmann and W. R. Markesbery (1996). "Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: possible relation to oxidative stress." J Neurol Sci, 143(1-2), 137-42.
P. Derkinderen, T. M. Scales, D. P. Hanger, K. Y. Leung, H. L. Byers, et al. (2005). "Tyrosine 394 is phosphorylated in Alzheimer's paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase." J Neurosci, 25(28), 6584-93.
L. Desire, J. Bourdin, N. Loiseau, H. Peillon, V. Picard, et al. (2005). "RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo." J Biol Chem, 280(45), 37516-25.
D. P. Devanand, M. Sano, M. X. Tang, S. Taylor, B. J. Gurland, et al. (1996). "Depressed mood and the incidence of Alzheimer's disease in the elderly living in the community." Arch Gen Psychiatry, 53(2), 175-82.
C. Dingwall (2007). "A copper-binding site in the cytoplasmic domain of BACE1 identifies a possible link to metal homoeostasis and oxidative stress in Alzheimer's disease." Biochem Soc Trans, 35(Pt 3), 571-3.
J. L. Domingo (1989). "The use of chelating agents in the treatment of aluminum overload." J Toxicol Clin Toxicol, 27(6), 355-67.
J. L. Domingo (1996). "Adverse effects of aluminium-chelating compounds for clinical use." Adverse Drug React Toxicol Rev, 15(3), 145-65.
J. Dong, C. S. Atwood, V. E. Anderson, S. L. Siedlak, M. A. Smith, et al. (2003). "Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence." Biochemistry, 42(10), 2768-73.
S. Dore (2005). "Unique properties of polyphenol stilbenes in the brain: more than direct antioxidant actions; gene/protein regulatory activity." Neurosignals, 14(1-2), 61-70.
J. Drake, C. D. Link and D. A. Butterfield (2003). "Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model." Neurobiol Aging, 24(3), 415-20.
P. E. Duffy, M. Rapport and L. Graf (1980). "Glial fibrillary acidic protein and Alzheimer-type senile dementia." Neurology, 30(7 Pt 1), 778-82.
S. S. Eaglestone, B. S. Cox and M. F. Tuite (1999). "Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism." Embo J, 18(7), 1974-81.
M. Ebadi, P. L. Iversen, R. Hao, D. R. Cerutis, P. Rojas, et al. (1995). "Expression and regulation of brain metallothionein." Neurochem Int, 27(1), 1-22.
A. Ebneth, R. Godemann, K. Stamer, S. Illenberger, B. Trinczek, et al. (1998).

Chapitre 1 : la maladie d’Alzheimer
73
"Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease." J Cell Biol, 143(3), 777-94.
E. A. Eckman, D. K. Reed and C. B. Eckman (2001). "Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme." J Biol Chem, 276(27), 24540-8.
E. A. Eckman, M. Watson, L. Marlow, K. Sambamurti and C. B. Eckman (2003). "Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme." J Biol Chem, 278(4), 2081-4.
E. A. Eckman, S. K. Adams, F. J. Troendle, B. A. Stodola, M. A. Kahn, et al. (2006). "Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme." J Biol Chem, 281(41), 30471-8.
R. Ehehalt, P. Keller, C. Haass, C. Thiele and K. Simons (2003). "Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts." J Cell Biol, 160(1), 113-23.
N. Ertekin-Taner, N. Graff-Radford, L. H. Younkin, C. Eckman, M. Baker, et al. (2000). "Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees." Science, 290(5500), 2303-4.
C. G. Evans, S. Wisen and J. E. Gestwicki (2006). "Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro." J Biol Chem, 281(44), 33182-91.
C. Exley (2004). "The pro-oxidant activity of aluminum." Free Radic Biol Med, 36(3), 380-7. C. Exley (2006). "Aluminium and iron, but neither copper nor zinc, are key to the
precipitation of beta-sheets of Abeta_{42} in senile plaque cores in Alzheimer's disease." J Alzheimers Dis, 10(2-3), 173-7.
L. A. Farrer, L. A. Cupples, J. L. Haines, B. Hyman, W. A. Kukull, et al. (1997). "Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium." Jama, 278(16), 1349-56.
I. Ferrer, M. Barrachina and B. Puig (2002). "Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration." Acta Neuropathol (Berl), 104(6), 583-91.
K. A. Field, J. R. Apgar, E. Hong-Geller, R. P. Siraganian, B. Baird, et al. (2000). "Mutant RBL mast cells defective in Fc epsilon RI signaling and lipid raft biosynthesis are reconstituted by activated Rho-family GTPases." Mol Biol Cell, 11(10), 3661-73.
M. A. Findeis, G. M. Musso, C. C. Arico-Muendel, H. W. Benjamin, A. M. Hundal, et al. (1999). "Modified-peptide inhibitors of amyloid β-peptide polymerization." Biochemistry, 38(21), 6791-6800.
S. Flament, A. Delacourte, M. Verny, J. J. Hauw and F. Javoy-Agid (1991). "Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type." Acta Neuropathol (Berl), 81(6), 591-6.
W. W. Fleischhacker, A. Buchgeher and H. Schubert (1986). "Memantine in the treatment of senile dementia of the Alzheimer type." Prog Neuropsychopharmacol Biol Psychiatry, 10(1), 87-93.
S. Fleminger, D. L. Oliver, S. Lovestone, S. Rabe-Hesketh and A. Giora (2003). "Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication." J Neurol Neurosurg Psychiatry, 74(7), 857-62.

Chapitre 1 : la maladie d’Alzheimer
74
W. F. Forbes and G. B. Hill (1998). "Is exposure to aluminum a risk factor for the development of Alzheimer disease?--Yes." Arch Neurol, 55(5), 740-1.
M. J. Forster, A. Dubey, K. M. Dawson, W. A. Stutts, H. Lal, et al. (1996). "Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain." Proc Natl Acad Sci U S A, 93(10), 4765-9.
N. C. Fox, R. S. Black, S. Gilman, M. N. Rossor, S. G. Griffith, et al. (2005). "Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease." Neurology, 64(9), 1563-72.
R. Francis, G. McGrath, J. Zhang, D. A. Ruddy, M. Sym, et al. (2002). "aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation." Dev Cell, 3(1), 85-97.
C. J. Frederickson (1989). "Neurobiology of zinc and zinc-containing neurons." Int Rev Neurobiol, 31145-238.
C. J. Frederickson and D. W. Moncrieff (1994). "Zinc-containing neurons." Biol Signals, 3(3), 127-39.
C. J. Frederickson, J. Y. Koh and A. I. Bush (2005). "The neurobiology of zinc in health and disease." Nat Rev Neurosci, 6(6), 449-62.
C. J. Frederickson, L. J. Giblin, 3rd, R. V. Balaji, R. Masalha, Y. Zeng, et al. (2006). "Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration." J Neurosci Methods, 154(1-2), 19-29.
J. N. Freskos, Y. M. Fobian, T. E. Benson, M. J. Bienkowski, D. L. Brown, et al. (2007). "Design of potent inhibitors of human beta-secretase. Part 1." Bioorg Med Chem Lett, 17(1), 73-7.
A. Gafni (1997). "Structural modifications of proteins during aging." J Am Geriatr Soc, 45(7), 871-80.
D. Galimberti, P. Baron, L. Meda, E. Prat, E. Scarpini, et al. (1999). "Alpha-MSH peptides inhibit production of nitric oxide and tumor necrosis factor-alpha by microglial cells activated with beta-amyloid and interferon gamma." Biochem Biophys Res Commun, 263(1), 251-6.
S. Gandy and F. L. Heppner (2005). "Alzheimer's amyloid immunotherapy: quo vadis?" Lancet Neurol, 4(8), 452-3.
M. Garcia-Alloza, L. A. Borrelli, A. Rozkalne, B. T. Hyman and B. J. Bacskai (2007). "Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model." J Neurochem, 102(4), 1095-104.
W. Garzon-Rodriguez, A. K. Yatsimirsky and C. G. Glabe (1999). "Binding of Zn(II), Cu(II), and Fe(II) ions to alzheimer's Aβ peptide studied by fluorescence." Bioorg. Med. Chem. Lett., 9(15), 2243.
F. Gaskin and Y. Kress (1977). "Zinc ion-induced assembly of tubulin." J Biol Chem, 252(19), 6918-24.
A. K. Ghosh, N. Kumaragurubaran, L. Hong, H. Lei, K. A. Hussain, et al. (2006). "Design, synthesis and X-ray structure of protein-ligand complexes: important insight into selectivity of memapsin 2 (beta-secretase) inhibitors." J Am Chem Soc, 128(16), 5310-1.
D. Gianni, N. Zambrano, M. Bimonte, G. Minopoli, L. Mercken, et al. (2003). "Platelet-derived growth factor induces the beta-gamma-secretase-mediated cleavage of Alzheimer's amyloid precursor protein through a Src-Rac-dependent pathway." J Biol Chem, 278(11), 9290-7.
J. D. Gillmore and P. N. Hawkins (2006). "Drug Insight: emerging therapies for amyloidosis." Nat Clin Pract Nephrol, 2(5), 263-70.

Chapitre 1 : la maladie d’Alzheimer
75
S. Gilman, M. Koller, R. S. Black, L. Jenkins, S. G. Griffith, et al. (2005). "Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial." Neurology, 64(9), 1553-62.
A. Gladkevich, F. Bosker, J. Korf, K. Yenkoyan, H. Vahradyan, et al. (2007). "Proline-rich polypeptides in Alzheimer's disease and neurodegenerative disorders - Therapeutic potential or a mirage?" Prog Neuropsychopharmacol Biol Psychiatry, 31(7), 1347-55.
A. Goate, M. C. Chartier-Harlin, M. Mullan, J. Brown, F. Crawford, et al. (1991). "Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease." Nature, 349(6311), 704-6.
M. Goedert, C. M. Wischik, R. A. Crowther, J. E. Walker and A. Klug (1988). "Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease." Proc Natl Acad Sci U S A, 854051-4055.
M. Goedert, M. G. Spillantini, R. Jakes, D. Rutherford and R. A. Crowther (1989). "Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease." Neuron, 3(4), 519-26.
M. Goedert, M. G. Spillantini, M. C. Potier, J. Ulrich and R. A. Crowther (1989). "Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain." Embo J, 8(2), 393-9.
M. Goedert, M. G. Spillantini, N. J. Cairns and R. A. Crowther (1992). "Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms." Neuron, 8(1), 159-68.
M. Goedert, A. Klug and R. A. Crowther (2006). "Tau protein, the paired helical filament and Alzheimer's disease." J Alzheimers Dis, 9(3 Suppl), 195-207.
M. Goedert and M. G. Spillantini (2006). "A century of Alzheimer's disease." Science, 314(5800), 777-81.
C. Goldsbury, K. Goldie, J. Pellaud, J. Seelig, P. Frey, et al. (2000). "Amyloid fibril formation from full-length and fragments of amylin." J Struct Biol, 130(2-3), 352-62.
C. S. Goldsbury, G. J. Cooper, K. N. Goldie, S. A. Muller, E. L. Saafi, et al. (1997). "Polymorphic fibrillar assembly of human amylin." J Struct Biol, 119(1), 17-27.
A. Gomez-Ramos, J. Diaz-Nido, M. A. Smith, G. Perry and J. Avila (2003). "Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells." J Neurosci Res, 71(6), 863-70.
F. X. Gomis-Ruth (2003). "Structural aspects of the metzincin clan of metalloendopeptidases." Mol Biotechnol, 24(2), 157-202.
C. X. Gong, T. J. Singh, I. Grundke-Iqbal and K. Iqbal (1993). "Phosphoprotein phosphatase activities in Alzheimer disease brain." J Neurochem, 61(3), 921-7.
C. X. Gong, I. Grundke-Iqbal and K. Iqbal (1994). "Dephosphorylation of Alzheimer's disease abnormally phosphorylated tau by protein phosphatase-2A." Neuroscience, 61(4), 765-72.
C. X. Gong, S. Shaikh, J. Z. Wang, T. Zaidi, I. Grundke-Iqbal, et al. (1995). "Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain." J Neurochem, 65(2), 732-8.
C. X. Gong, T. Lidsky, J. Wegiel, L. Zuck, I. Grundke-Iqbal, et al. (2000). "Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease." J Biol Chem, 275(8), 5535-44.
P. E. Gonzales, A. Solomon, A. B. Miller, M. A. Leesnitzer, I. Sagi, et al. (2004). "Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain." J Biol Chem, 279(30), 31638-45.

Chapitre 1 : la maladie d’Alzheimer
76
D. J. Gordon and S. C. Meredith (2003). "Probing the role of backbone hydrogen bonding in beta-amyloid fibrils with inhibitor peptides containing ester bonds at alternate positions." Biochemistry, 42(2), 475-85.
J. Gotz, F. Chen, J. van Dorpe and R. M. Nitsch (2001). "Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils." Science, 293(5534), 1491-5.
C. Goutte, M. Tsunozaki, V. A. Hale and J. R. Priess (2002). "APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos." Proc Natl Acad Sci U S A, 99(2), 775-9.
S. I. Gracon, M. J. Knapp, W. G. Berghoff, M. Pierce, R. DeJong, et al. (1998). "Safety of tacrine: clinical trials, treatment IND, and postmarketing experience." Alzheimer Dis Assoc Disord, 12(2), 93-101.
R. W. Gracy, M. L. Chapman, J. K. Cini, M. Jahani, T. O. Tollefsbol, et al. (1985). "Molecular basis of the accumulation of abnormal proteins in progeria and aging fibroblasts." Basic Life Sci, 35427-42.
M. B. Graeber, S. Kosel, E. Grasbon-Frodl, H. J. Moller and P. Mehraein (1998). "Histopathology and APOE genotype of the first Alzheimer disease patient, Auguste D." Neurogenetics, 1(3), 223-8.
M. Gralle and S. T. Ferreira (2007). "Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts." Prog Neurobiol, 82(1), 11-32.
L. Gregori, J. F. Hainfeld, M. N. Simon and D. Goldgaber (1997). "Binding of amyloid beta protein to the 20 S proteasome." J Biol Chem, 272(1), 58-62.
R. J. Griffin, A. Moloney, M. Kelliher, J. A. Johnston, R. Ravid, et al. (2005). "Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology." J Neurochem, 93(1), 105-17.
M. O. Grimm, H. S. Grimm, A. J. Patzold, E. G. Zinser, R. Halonen, et al. (2005). "Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin." Nat Cell Biol, 7(11), 1118-23.
I. Grundke-Iqbal, K. Iqbal, Y.-C. Tung, M. Quinlan, H. M. Wisniewski, et al. (1986). "Abnormal phosphorylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology." Proc Natl Acad Sci U S A, 834913-4917.
L. Guilloreau, L. Damian, Y. Coppel, H. Mazarguil, M. Winterhalter, et al. (2006). "Structural and thermodynamical properties of CuII amyloid-beta16/28 complexes associated with Alzheimer's disease." J Biol Inorg Chem, 11(8), 1024-38.
L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller (2007). "Redox chemistry of copper-amyloid-beta: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state." Chembiochem, 8(11), 1317-25.
A. Gupta, M. Hasan, R. Chander and N. K. Kapoor (1991). "Age-related elevation of lipid peroxidation products: diminution of superoxide dismutase activity in the central nervous system of rats." Gerontology, 37(6), 305-9.
V. B. Gupta, S. Anitha, M. L. Hegde, L. Zecca, R. M. Garruto, et al. (2005). "Aluminium in Alzheimer's disease: are we still at a crossroad?" Cell Mol Life Sci, 62(2), 143-58.
J. F. Guzowski (2002). "Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fluorescent imaging approaches." Hippocampus, 12(1), 86-104.
C. Ha, J. Ryu and C. B. Park (2007). "Metal ions differentially influence the aggregation and deposition of Alzheimer's beta-amyloid on a solid template." Biochemistry, 46(20), 6118-25.
C. Haass and H. Steiner (2001). "Protofibrils, the unifying toxic molecule of

Chapitre 1 : la maladie d’Alzheimer
77
neurodegenerative disorders?" Nat Neurosci, 4(9), 859-60. B. Halliwell and J. M. Gutteridge (1984). "Oxygen toxicity, oxygen radicals, transition metals
and disease." Biochem J, 219(1), 1-14. T. Hamaguchi, K. Ono and M. Yamada (2006). "Anti-amyloidogenic therapies: strategies for
prevention and treatment of Alzheimer's disease." Cell Mol Life Sci, 63(13), 1538-52. M. L. Hamilton, H. Van Remmen, J. A. Drake, H. Yang, Z. M. Guo, et al. (2001). "Does
oxidative damage to DNA increase with age?" Proc Natl Acad Sci U S A, 98(18), 10469-74.
S. Hanessian, H. Yun, Y. Hou, G. Yang, M. Bayrakdarian, et al. (2005). "Structure-based design, synthesis, and memapsin 2 (BACE) inhibitory activity of carbocyclic and heterocyclic peptidomimetics." J Med Chem, 48(16), 5175-90.
A. Hara and T. Taketomi (1986). "Cerebral lipid and protein abnormalities in Menkes' steely-hair disease." Jpn J Exp Med, 56(6), 277-84.
J. Hardy and G. A. Higgins (1992). "Alzheimer's disease: the amyloid cascade hypothesis." Science, 256184-185.
J. Hardy and D. J. Selkoe (2002). "The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics." Science, 297(5580), 353-6.
D. Harman (1956). "Aging: a theory based on free radical and radiation chemistry." J Gerontol, 11(3), 298-300.
J. D. Harper, C. M. Lieber and P. T. Lansbury, Jr. (1997). "Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein." Chem Biol, 4(12), 951-9.
J. D. Harper, S. S. Wong, C. M. Lieber and P. T. Lansbury (1997). "Observation of metastable Abeta amyloid protofibrils by atomic force microscopy." Chem Biol, 4(2), 119-25.
F. M. Harris, W. J. Brecht, Q. Xu, R. W. Mahley and Y. Huang (2004). "Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc." J Biol Chem, 279(43), 44795-801.
N. L. Harrison and S. J. Gibbons (1994). "Zn2+: an endogenous modulator of ligand- and voltage-gated ion channels." Neuropharmacology, 33(8), 935-52.
T. Harrison, I. Churcher and D. Beher (2004). "gamma-Secretase as a target for drug intervention in Alzheimer's disease." Curr Opin Drug Discov Devel, 7(5), 709-19.
D. M. Hartley, D. M. Walsh, C. P. Ye, T. Diehl, S. Vasquez, et al. (1999). "Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons." J. Neurosci., 198876-8884.
M. R. Hasan, D. Morishima, K. Tomita, M. Katsuki and S. Kotani (2005). "Identification of a 250 kDa putative microtubule-associated protein as bovine ferritin. Evidence for a ferritin-microtubule interaction." Febs J, 272(3), 822-31.
D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vasak (2000). "Effect of the two conserved prolines of human growth inhibitory factor (metallothionein-3) on its biological activity and structure fluctuation: comparison with a mutant protein." Biochemistry, 39(47), 14567-75.
M. R. Hass and B. A. Yankner (2005). "A {gamma}-secretase-independent mechanism of signal transduction by the amyloid precursor protein." J Biol Chem, 280(44), 36895-904.
F. M. Haug (1967). "Electron microscopical localization of the zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure." Histochemie, 8(4), 355-68.
S. S. Hebert, L. Serneels, A. Tolia, K. Craessaerts, C. Derks, et al. (2006). "Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression

Chapitre 1 : la maladie d’Alzheimer
78
of putative target genes." EMBO Rep, 7(7), 739-45. V. W. Henderson, A. Paganini-Hill, C. K. Emanuel, M. E. Dunn and J. G. Buckwalter (1994).
"Estrogen replacement therapy in older women. Comparisons between Alzheimer's disease cases and nondemented control subjects." Arch Neurol, 51(9), 896-900.
V. W. Henderson (2000). "Hormone therapy and the brain: a clinical perspective on the role of estrogen ". New York, Parthenon Publishing.
V. W. Henderson (2006). "Estrogen-containing hormone therapy and Alzheimer's disease risk: understanding discrepant inferences from observational and experimental research." Neuroscience, 138(3), 1031-9.
A. Herreman, L. Serneels, W. Annaert, D. Collen, L. Schoonjans, et al. (2000). "Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells." Nat Cell Biol, 2(7), 461-2.
L. Hesse, D. Beher, C. L. Masters and G. Multhaup (1994). "The βA4 amyloid precursor protein binding to copper." FEBS Lett., 349109-116.
J. Hidalgo and J. Carrasco (1998). "Regulation of the synthesis of brain metallothioneins." Neurotoxicology, 19(4-5), 661-6.
G. J. Ho, M. Hashimoto, A. Adame, M. Izu, M. F. Alford, et al. (2005). "Altered p59Fyn kinase expression accompanies disease progression in Alzheimer's disease: implications for its functional role." Neurobiol Aging, 26(5), 625-35.
C. Hock, U. Konietzko, J. R. Streffer, J. Tracy, A. Signorell, et al. (2003). "Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease." Neuron, 38(4), 547-54.
D. E. Hoke, J. L. Tan, N. T. Ilaya, J. G. Culvenor, S. J. Smith, et al. (2005). "In vitro gamma-secretase cleavage of the Alzheimer's amyloid precursor protein correlates to a subset of presenilin complexes and is inhibited by zinc." Febs J, 272(21), 5544-57.
L. Hong, G. Koelsch, X. Lin, S. Wu, S. Terzyan, et al. (2000). "Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor." Science, 290(5489), 150-3.
L. Hou and M. G. Zagorski (2006). "NMR reveals anomalous copper(II) binding to the amyloid Abeta peptide of Alzheimer's disease." J Am Chem Soc, 128(29), 9260-1.
E. House, J. Collingwood, A. Khan, O. Korchazkina, G. Berthon, et al. (2004). "Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease." J Alzheimers Dis, 6(3), 291-301.
G. A. Howell, M. G. Welch and C. J. Frederickson (1984). "Stimulation-induced uptake and release of zinc in hippocampal slices." Nature, 308(5961), 736-8.
J. Hu, A. Igarashi, M. Kamata and H. Nakagawa (2001). "Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity." J Biol Chem, 276(51), 47863-8.
X. Hu, C. W. Hicks, W. He, P. Wong, W. B. Macklin, et al. (2006). "Bace1 modulates myelination in the central and peripheral nervous system." Nat Neurosci, 9(12), 1520-5.
X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, et al. (1997). "Zinc-induced Alzheimer's Abeta1-40 aggregation is mediated by conformational factors." J Biol Chem, 272(42), 26464-70.
X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, et al. (1999). "The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction." Biochemistry, 38(24), 7609-16.
X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. A. Tyndall, et al. (1999a). "Cu(II) potentiation of Alzheimer Abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction." J. Biol. Chem., 274(52), 37111-

Chapitre 1 : la maladie d’Alzheimer
79
37116. Y. Huang, X. Q. Liu, T. Wyss-Coray, W. J. Brecht, D. A. Sanan, et al. (2001).
"Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons." Proc Natl Acad Sci U S A, 98(15), 8838-43.
I. Hussain, D. Powell, D. R. Howlett, D. G. Tew, T. D. Meek, et al. (1999). "Identification of a novel aspartic protease (Asp 2) as beta-secretase." Mol Cell Neurosci, 14(6), 419-27.
I. Hussain, J. Hawkins, D. Harrison, C. Hille, G. Wayne, et al. (2007). "Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor protein and amyloid-beta production in vivo." J Neurochem, 100(3), 802-9.
D. Y. Hwang, J. S. Cho, S. H. Lee, K. R. Chae, H. J. Lim, et al. (2004). "Aberrant expressions of pathogenic phenotype in Alzheimer's diseased transgenic mice carrying NSE-controlled APPsw." Exp Neurol, 186(1), 20-32.
L. A. Hyde, Q. Zhang, N. A. McHugh, J. Chen, R. A. Del Vecchio, et al. (2006). In vivo characterization of a novel gama-secretase inhibitor in CRND8 transgenic mice: efficacy and side effects. S. f. N. t. A. Meeting. Atlanta.
Y. Ihara, N. Nukina, R. Miura and M. Ogawara (1986). "Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease." J Biochem (Tokyo), 99(6), 1807-10.
N. Iwata, H. Mizukami, K. Shirotani, Y. Takaki, S. Muramatsu, et al. (2004). "Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain." J Neurosci, 24(4), 991-8.
S. Janciauskiene and B. Ahren (2000). "Fibrillar islet amyloid polypeptide differentially affects oxidative mechanisms and lipoprotein uptake in correlation with cytotoxicity in two insulin-producing cell lines." Biochem Biophys Res Commun, 267(2), 619-25.
S. R. Ji, Y. Wu and S. F. Sui (2002). "Cholesterol is an important factor affecting the membrane insertion of beta-amyloid peptide (A beta 1-40), which may potentially inhibit the fibril formation." J Biol Chem, 277(8), 6273-9.
H. Jick, G. L. Zornberg, S. S. Jick, S. Seshadri and D. A. Drachman (2000). "Statins and the risk of dementia." Lancet, 356(9242), 1627-31.
J. L. Jimenez, E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson, et al. (2002). "The protofilament structure of insulin amyloid fibrils." Proc Natl Acad Sci U S A, 99(14), 9196-201.
A. F. Jorm, C. M. van Duijn, V. Chandra, L. Fratiglioni, A. B. Graves, et al. (1991). "Psychiatric history and related exposures as risk factors for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group." Int J Epidemiol, 20 Suppl 2S43-7.
D. S. Kalinowski and D. R. Richardson (2005). "The evolution of iron chelators for the treatment of iron overload disease and cancer." Pharmacol Rev, 57(4), 547-83.
L. Kalvodova, N. Kahya, P. Schwille, R. Ehehalt, P. Verkade, et al. (2005). "Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro." J Biol Chem, 280(44), 36815-23.
A. Kamal, G. B. Stokin, Z. Yang, C. H. Xia and L. S. Goldstein (2000). "Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I." Neuron, 28(2), 449-59.
A. Kamal, A. Almenar-Queralt, J. F. LeBlanc, E. A. Roberts and L. S. Goldstein (2001). "Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP." Nature, 414(6864), 643-8.

Chapitre 1 : la maladie d’Alzheimer
80
J. W. Karr, H. Akintoye, L. J. Kaupp and V. A. Szalai (2005). "N-Terminal deletions modify the Cu2+ binding site in amyloid-beta." Biochemistry, 44(14), 5478-87.
M. Kawahara (2005). "Effects of aluminum on the nervous system and its possible link with neurodegenerative diseases." J Alzheimers Dis, 8(2), 171-82; discussion 209-15.
A. R. Kay (2003). "Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn." J Neurosci, 23(17), 6847-55.
R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, et al. (2003). "Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis." Science, 300(5618), 486-9.
H. Keberle (1964). "The Biochemistry of Desferrioxamine and Its Relation to Iron Metabolism." Ann N Y Acad Sci, 119758-68.
J. N. Keller, K. B. Hanni and W. R. Markesbery (2000). "Impaired proteasome function in Alzheimer's disease." J Neurochem, 75(1), 436-9.
J. N. Keller, F. A. Schmitt, S. W. Scheff, Q. Ding, Q. Chen, et al. (2005). "Evidence of increased oxidative damage in subjects with mild cognitive impairment." Neurology, 64(7), 1152-6.
J. M. Kenney, D. Knight, M. J. Wise and F. Vollrath (2002). "Amyloidogenic nature of spider silk." Eur J Biochem, 269(16), 4159-63.
A. J. Kenny and S. L. Stephenson (1988). "Role of endopeptidase-24.11 in the inactivation of atrial natriuretic peptide." FEBS Lett, 232(1), 1-8.
S. M. Keyse and R. M. Tyrrell (1989). "Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite." Proc Natl Acad Sci U S A, 86(1), 99-103.
J. A. Khan, F. Forouhar, X. Tao and L. Tong (2007). "Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery." Expert Opin Ther Targets, 11(5), 695-705.
S. Khatoon, I. Grundke-Iqbal and K. Iqbal (1992). "Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein." J Neurochem, 59(2), 750-3.
S. Khatoon, I. Grundke-Iqbal and K. Iqbal (1994). "Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains." FEBS Lett, 351(1), 80-4.
M. Kidd (1963). Nature, 197192-193. W. Kim, Y. Kim, J. Min, D. J. Kim, Y. T. Chang, et al. (2006). "A high-throughput screen for
compounds that inhibit aggregation of the Alzheimer's peptide." ACS Chem Biol, 1(7), 461-9.
K. J. Kinghorn, D. C. Crowther, L. K. Sharp, C. Nerelius, R. L. Davis, et al. (2006). "Neuroserpin binds Abeta and is a neuroprotective component of amyloid plaques in Alzheimer disease." J Biol Chem, 281(39), 29268-77.
M. Kitazawa, S. Oddo, T. R. Yamasaki, K. N. Green and F. M. LaFerla (2005). "Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease." J Neurosci, 25(39), 8843-53.
W. L. Klein, W. B. Stine, Jr. and D. B. Teplow (2004). "Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease." Neurobiol Aging, 25(5), 569-80.
M. J. Knapp, D. S. Knopman, P. R. Solomon, W. W. Pendlebury, C. S. Davis, et al. (1994). "A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer's disease. The Tacrine Study Group." Jama, 271(13), 985-91.
R. B. Knowles, T. Gomez-Isla and B. T. Hyman (1998). "Abeta associated neuropil changes:

Chapitre 1 : la maladie d’Alzheimer
81
correlation with neuronal loss and dementia." J Neuropathol Exp Neurol, 57(12), 1122-30.
J. Koenigsknecht-Talboo and G. E. Landreth (2005). "Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines." J Neurosci, 25(36), 8240-9.
E. Kojro and F. Fahrenholz (2005). "The non-amyloidogenic pathway: structure and function of alpha-secretases." Subcell Biochem, 38105-27.
E. H. Koo, S. S. Sisodia, D. R. Archer, L. J. Martin, A. Weidemann, et al. (1990). "Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport." Proc Natl Acad Sci U S A, 87(4), 1561-5.
E. H. Koo and S. L. Squazzo (1994). "Evidence that production and release of amyloid beta-protein involves the endocytic pathway." J Biol Chem, 269(26), 17386-9.
E. H. Koo, S. L. Squazzo, D. J. Selkoe and C. H. Koo (1996). "Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody." J Cell Sci, 109 (Pt 5)991-8.
T. Kowalik-Jankowska, M. Ruta, K. Wisniewska and L. Lankiewicz (2003). "Coordination abilities of the 1-16 and 1-28 fragments of β-amyloid peptide towards copper(II) ions: a combined potentiometric and spectroscopic study." J. Inorg. Biochem., 95270-282.
T. Kowalik-Jankowska, M. Ruta, K. Wisniewska, L. Lankiewicz and M. Dyba (2004). "Products of Cu(II)-catalyzed oxidation in the presence of hydrogen peroxide of the 1-10, 1-16 fragments of human and mouse beta-amyloid peptide." J Inorg Biochem, 98(6), 940-50.
S. A. Kozin, S. Zirah, S. Rebuffat, G. H. Hoa and P. Debey (2001). "Zinc binding to Alzheimer's Abeta(1-16) peptide results in stable soluble complex." Biochem Biophys Res Commun, 285(4), 959-64.
E. Kraepelin (1910). "Psychiatrie. Ein Lehrbuch für Studierende und Arzte". Leipzig, Barth Verlag.
T. P. Kruck, E. A. Fisher and D. R. McLachlan (1990). "Suppression of deferoxamine mesylate treatment-induced side effects by coadministration of isoniazid in a patient with Alzheimer's disease subject to aluminum removal by ionspecific chelation." Clin Pharmacol Ther, 48(4), 439-46.
H. Ksiezak-Reding, K. Morgan, L. A. Mattiace, P. Davies, W. K. Liu, et al. (1994). "Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration." Am J Pathol, 145(6), 1496-508.
G. K. Kumar, M. Runold, R. D. Ghai, N. S. Cherniack and N. R. Prabhakar (1990). "Occurrence of neutral endopeptidase activity in the cat carotid body and its significance in chemoreception." Brain Res, 517(1-2), 341-3.
D. K. Lahiri, S. Lewis and M. R. Farlow (1994). "Tacrine alters the secretion of the beta-amyloid precursor protein in cell lines." J Neurosci Res, 37(6), 777-87.
D. K. Lahiri, M. R. Farlow, N. Hintz, T. Utsuki and N. H. Greig (2000). "Cholinesterase inhibitors, beta-amyloid precursor protein and amyloid beta-peptides in Alzheimer's disease." Acta Neurol Scand Suppl, 17660-7.
F. M. Laird, H. Cai, A. V. Savonenko, M. H. Farah, K. He, et al. (2005). "BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions." J Neurosci, 25(50), 11693-709.
M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, et al. (1998). "Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins." Proc Natl Acad Sci U S A, 95(11), 6448-53.
S. Lammich, E. Kojro, R. Postina, S. Gilbert, R. Pfeiffer, et al. (1999). "Constitutive and

Chapitre 1 : la maladie d’Alzheimer
82
regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease." Proc Natl Acad Sci U S A, 96(7), 3922-7.
B. P. Lanphear, R. Hornung, J. Khoury, K. Yolton, P. Baghurst, et al. (2005). "Low-level environmental lead exposure and children's intellectual function: an international pooled analysis." Environ Health Perspect, 113(7), 894-9.
K. F. Lau, D. M. McLoughlin, C. L. Standen, N. G. Irving and C. C. Miller (2000). "Fe65 and X11beta co-localize with and compete for binding to the amyloid precursor protein." Neuroreport, 11(16), 3607-10.
O. Lazarov, G. A. Morfini, E. B. Lee, M. H. Farah, A. Szodorai, et al. (2005). "Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited." J Neurosci, 25(9), 2386-95.
M. D. Ledesma, J. Abad-Rodriguez, C. Galvan, E. Biondi, P. Navarro, et al. (2003). "Raft disorganization leads to reduced plasmin activity in Alzheimer's disease brains." EMBO Rep, 4(12), 1190-6.
G. Lee, R. L. Neve and K. S. Kosik (1989). "The microtubule binding domain of tau protein." Neuron, 2(6), 1615-24.
J. Y. Lee, T. B. Cole, R. D. Palmiter, S. W. Suh and J. Y. Koh (2002). "Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice." Proc Natl Acad Sci U S A, 99(11), 7705-10.
M. S. Lee, S. C. Kao, C. A. Lemere, W. Xia, H. C. Tseng, et al. (2003). "APP processing is regulated by cytoplasmic phosphorylation." J Cell Biol, 163(1), 83-95.
S. Lesne and L. Kotilinek (2005). "Amyloid plaques and amyloid-beta oligomers: an ongoing debate." J Neurosci, 25(41), 9319-20.
M. Letarte, S. Vera, R. Tran, J. B. Addis, R. J. Onizuka, et al. (1988). "Common acute lymphocytic leukemia antigen is identical to neutral endopeptidase." J Exp Med, 168(4), 1247-53.
L. Letenneur (2004). "Risk of dementia and alcohol and wine consumption: a review of recent results." Biol Res, 37(2), 189-93.
Y. Levites, T. Amit, S. Mandel and M. B. Youdim (2003). "Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate." Faseb J, 17(8), 952-4.
E. Levy-Lahad, W. Wasco, P. Poorkaj, D. M. Romano, J. Oshima, et al. (1995). "Candidate gene for the chromosome 1 familial Alzheimer's disease locus." Science, 269(5226), 973-7.
J. Lewis, D. W. Dickson, W. L. Lin, L. Chisholm, A. Corral, et al. (2001). "Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP." Science, 293(5534), 1487-91.
F. Li, N. Y. Calingasan, F. Yu, W. M. Mauck, M. Toidze, et al. (2004). "Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice." J Neurochem, 89(5), 1308-12.
M. Li, H. Guo and Z. Damuni (1995). "Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney." Biochemistry, 34(6), 1988-96.
Y. Li, C. J. Hough, S. W. Suh, J. M. Sarvey and C. J. Frederickson (2001). "Rapid translocation of Zn(2+) from presynaptic terminals into postsynaptic hippocampal neurons after physiological stimulation." J Neurophysiol, 86(5), 2597-604.
Y. Li, L. Liu, S. W. Barger and W. S. Griffin (2003). "Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway." J Neurosci, 23(5), 1605-11.

Chapitre 1 : la maladie d’Alzheimer
83
J. Lim and R. W. Vachet (2003). "Development of a methodology based on metal-catalyzed oxidation reactions and mass spectrometry to determine the metal binding sites in copper metalloproteins." Anal Chem, 75(5), 1164-72.
H. Lin, Y. J. Zhu and R. Lal (1999). "Amyloid beta protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles." Biochemistry, 38(34), 11189-96.
H. Lin, R. Bhatia and R. Lal (2001). "Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology." Faseb J, 15(13), 2433-44.
G. Lindwall and R. D. Cole (1984). "Phosphorylation affects the ability of tau protein to promote microtubule assembly." J Biol Chem, 259(8), 5301-5.
S. A. Lipton (2007). "Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation." Curr Drug Targets, 8(5), 621-32.
F. Liu, T. Zaidi, K. Iqbal, I. Grundke-Iqbal, R. K. Merkle, et al. (2002). "Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease." FEBS Lett, 512(1-3), 101-6.
F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart and C. X. Gong (2004). "O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease." Proc Natl Acad Sci U S A, 101(29), 10804-9.
S. T. Liu, G. Howlett and C. J. Barrow (1999). "Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A beta peptide of Alzheimer's disease." Biochemistry, 38(29), 9373-8.
A. Lleo, O. Berezovska, L. Herl, S. Raju, A. Deng, et al. (2004). "Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation." Nat Med, 10(10), 1065-6.
D. A. Loeffler, P. A. LeWitt, P. L. Juneau, A. A. Sima, H. U. Nguyen, et al. (1996). "Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders." Brain Res, 738(2), 265-74.
M. Lopez Salon, L. Pasquini, M. Besio Moreno, J. M. Pasquini and E. Soto (2003). "Relationship between beta-amyloid degradation and the 26S proteasome in neural cells." Exp Neurol, 180(2), 131-43.
M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery (1998). "Copper, iron and zinc in Alzheimer's disease senile plaques." Journal of the Neurological Sciences, 158(1), 47.
M. A. Lovell, J. L. Smith, S. Xiong and W. R. Markesbery (2005). "Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer's disease." Neurotox Res, 7(4), 265-71.
K. P. Lu (2003). "Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics." Cancer Cell, 4(3), 175-80.
K. P. Lu, Y. C. Liou and I. Vincent (2003). "Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer's Disease." Bioessays, 25(2), 174-81.
K. P. Lu, F. Suizu, X. Z. Zhou, G. Finn, P. Lam, et al. (2006). "Targeting carcinogenesis: a role for the prolyl isomerase Pin1?" Mol Carcinog, 45(6), 397-402.
J. Lundkvist and J. Naslund (2007). "Gamma-secretase: a complex target for Alzheimer's disease." Curr Opin Pharmacol, 7(1), 112-8.
Y. Luo, B. Bolon, S. Kahn, B. D. Bennett, S. Babu-Khan, et al. (2001). "Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation." Nat Neurosci, 4(3), 231-2.
Q. Ma, Y. Li, J. Du, H. Liu, K. Kanazawa, et al. (2006). "Copper binding properties of a tau

Chapitre 1 : la maladie d’Alzheimer
84
peptide associated with Alzheimer's disease studied by CD, NMR, and MALDI-TOF MS." Peptides, 27(4), 841-9.
Q. F. Ma, Y. M. Li, J. T. Du, K. Kanazawa, T. Nemoto, et al. (2005). "Binding of copper (II) ion to an Alzheimer's tau peptide as revealed by MALDI-TOF MS, CD, and NMR." Biopolymers, 79(2), 74-85.
Q. L. Ma, G. P. Lim, M. E. Harris-White, F. Yang, S. S. Ambegaokar, et al. (2006). "Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro." J Neurosci Res, 83(3), 374-84.
J. P. Mackay, J. M. Matthews, R. D. Winefield, L. G. Mackay, R. G. Haverkamp, et al. (2001). "The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures." Structure, 9(2), 83-91.
A. Madaric, E. Ginter and J. Kadrabova (1994). "Serum copper, zinc and copper/zinc ratio in males: influence of aging." Physiol Res, 43(2), 107-11.
R. W. Mahley and Y. Huang (2006). "Apolipoprotein (apo) E4 and Alzheimer's disease: unique conformational and biophysical properties of apoE4 can modulate neuropathology." Acta Neurol Scand Suppl, 1858-14.
R. W. Mahley, K. H. Weisgraber and Y. Huang (2006). "Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease." Proc Natl Acad Sci U S A, 103(15), 5644-51.
M. Maillet, S. J. Robert, M. Cacquevel, M. Gastineau, D. Vivien, et al. (2003). "Crosstalk between Rap1 and Rac regulates secretion of sAPPalpha." Nat Cell Biol, 5(7), 633-9.
M. Majores, M. Bagli, A. Papassotiropoulos, S. G. Schwab, F. Jessen, et al. (2000). "Allelic association between the D10S1423 marker and Alzheimer's disease in a German population." Neurosci Lett, 289(3), 224-6.
S. B. Malinchik, H. Inouye, K. E. Szumowski and D. A. Kirschner (1998). "Structural analysis of Alzheimer's beta(1-40) amyloid: protofilament assembly of tubular fibrils." Biophys J, 74(1), 537-45.
S. Mandel, T. Amit, O. Bar-Am and M. B. Youdim (2007). "Iron dysregulation in Alzheimer's disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents." Prog Neurobiol, 82(6), 348-60.
E. M. Mandelkow, E. Thies, B. Trinczek, J. Biernat and E. Mandelkow (2004). "MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons." J Cell Biol, 167(1), 99-110.
P. Marambaud, H. Zhao and P. Davies (2005). "Resveratrol promotes clearance of Alzheimer's disease amyloid-beta peptides." J Biol Chem, 280(45), 37377-82.
R. A. Marr, E. Rockenstein, A. Mukherjee, M. S. Kindy, L. B. Hersh, et al. (2003). "Neprilysin gene transfer reduces human amyloid pathology in transgenic mice." J Neurosci, 23(6), 1992-6.
B. A. Masters, C. J. Quaife, J. C. Erickson, E. J. Kelly, G. J. Froelick, et al. (1994). "Metallothionein III is expressed in neurons that sequester zinc in synaptic vesicles." J Neurosci, 14(10), 5844-57.
S. Matsuda, Y. Matsuda and L. D'Adamio (2003). "Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1." J Biol Chem, 278(40), 38601-6.
M. P. Mattson (1994). "Secreted forms of beta-amyloid precursor protein modulate dendrite outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons." J Neurobiol, 25(4), 439-50.
M. P. Mattson (2004). "Pathways towards and away from Alzheimer's disease." Nature,

Chapitre 1 : la maladie d’Alzheimer
85
430631-639. C. J. Maynard, R. Cappai, I. Volitakis, R. A. Cherny, A. R. White, et al. (2002).
"Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron." J Biol Chem, 277(47), 44670-6.
J. C. McLachlan (1991). "Transcutaneous electrical nerve stimulation." Lancet, 337(8743), 742.
D. McMaster, E. McCrum, C. C. Patterson, M. M. Kerr, D. O'Reilly, et al. (1992). "Serum copper and zinc in random samples of the population of Northern Ireland." Am J Clin Nutr, 56(2), 440-6.
Y. Mekmouche, Y. Coppel, K. Hochgrafe, L. Guilloreau, C. Talmard, et al. (2005). "Characterization of the ZnII binding to the peptide amyloid-beta1-16 linked to Alzheimer's disease." Chembiochem, 6(9), 1663-71.
I. Melnikova (2007). "Therapies for Alzheimer's disease." Nat Rev Drug Discov, 6(5), 341-2. M. L. Michaelis, N. Ranciat, Y. Chen, M. Bechtel, R. Ragan, et al. (1998). "Protection against
beta-amyloid toxicity in primary neurons by paclitaxel (Taxol)." J Neurochem, 70(4), 1623-7.
M. Michikawa (2003). "Cholesterol paradox: is high total or low HDL cholesterol level a risk for Alzheimer's disease?" J Neurosci Res, 72(2), 141-6.
D. B. Milne and P. E. Johnson (1993). "Assessment of copper status: effect of age and gender on reference ranges in healthy adults." Clin Chem, 39(5), 883-7.
A. M. Minogue, A. W. Schmid, M. P. Fogarty, A. C. Moore, V. A. Campbell, et al. (2003). "Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta?" J Biol Chem, 278(30), 27971-80.
H. Misonou, M. Morishima-Kawashima and Y. Ihara (2000). "Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells." Biochemistry, 39(23), 6951-9.
J. R. Missel, M. R. Schetinger, C. R. Gioda, D. N. Bohrer, I. L. Pacholski, et al. (2005). "Chelating effect of novel pyrimidines in a model of aluminum intoxication." J Inorg Biochem, 99(9), 1853-7.
M. S. Mittelman, S. H. Ferris, E. Shulman, G. Steinberg and B. Levin (1996). "A family intervention to delay nursing home placement of patients with Alzheimer disease. A randomized controlled trial." Jama, 276(21), 1725-31.
E. Mocchegiani, M. Muzzioli and R. Giacconi (2000). "Zinc and immunoresistance to infection in aging: new biological tools." Trends Pharmacol Sci, 21(6), 205-8.
E. Mocchegiani, L. Costarelli, R. Giacconi, C. Cipriano, E. Muti, et al. (2006). "Zinc-binding proteins (metallothionein and alpha-2 macroglobulin) and immunosenescence." Exp Gerontol, 41(11), 1094-107.
J. A. Molina, F. J. Jimenez-Jimenez, M. V. Aguilar, I. Meseguer, C. J. Mateos-Vega, et al. (1998). "Cerebrospinal fluid levels of transition metals in patients with Alzheimer's disease." J Neural Transm, 105(4-5), 479-88.
A. L. Monget, P. Galan, P. Preziosi, H. Keller, C. Bourgeois, et al. (1996). "Micronutrient status in elderly people. Geriatrie/Min. Vit. Aux Network." Int J Vitam Nutr Res, 66(1), 71-6.
P. I. Moreira, S. L. Siedlak, G. Aliev, X. Zhu, A. D. Cash, et al. (2005). "Oxidative stress mechanisms and potential therapeutics in Alzheimer disease." J. Neural. Transm., 112921-932.
H. Mori, J. Kondo and Y. Ihara (1987). "Ubiquitin is a component of paired helical filaments in Alzheimer's disease." Science, 235(4796), 1641-4.
M. C. Morris, J. A. Schneider and C. C. Tangney (2006). "Thoughts on B-vitamins and

Chapitre 1 : la maladie d’Alzheimer
86
dementia." J Alzheimers Dis, 9(4), 429-33. S. Mruthinti, W. D. Hill, S. Swamy-Mruthinti and J. J. Buccafusco (2003). "Relationship
between the induction of RAGE cell-surface antigen and the expression of amyloid binding sites." J Mol Neurosci, 20(3), 223-32.
S. Mruthinti, A. Sood, C. L. Humphrey, S. Swamy-Mruthinti and J. J. Buccafusco (2006). "The induction of surface beta-amyloid binding proteins and enhanced cytotoxicity in cultured PC-12 and IMR-32 cells by advanced glycation end products." Neuroscience, 142(2), 463-73.
H. T. Mueller, J. P. Borg, B. Margolis and R. S. Turner (2000). "Modulation of amyloid precursor protein metabolism by X11alpha /Mint-1. A deletion analysis of protein-protein interaction domains." J Biol Chem, 275(50), 39302-6.
G. Multhaup (2006). "Amyloid precursor protein and BACE function as oligomers." Neurodegener Dis, 3(4-5), 270-4.
D. G. Munoz (1998). "Is exposure to aluminum a risk factor for the development of Alzheimer disease?--No." Arch Neurol, 55(5), 737-9.
J. Murrell, M. Farlow, B. Ghetti and M. D. Benson (1991). "A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease." Science, 254(5028), 97-9.
A. J. Myers and A. M. Goate (2001). "The genetics of late-onset Alzheimer's disease." Curr Opin Neurol, 14(4), 433-40.
H. Nakashima, T. Ishihara, O. Yokota, S. Terada, J. Q. Trojanowski, et al. (2004). "Effects of alpha-tocopherol on an animal model of tauopathies." Free Radic Biol Med, 37(2), 176-86.
J. Naslund, V. Haroutunian, R. Mohs, K. L. Davis, P. Davies, et al. (2000). "Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline." Jama, 283(12), 1571-7.
M. Necula and J. Kuret (2004). "Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro." J Biol Chem, 279(48), 49694-703.
M. Necula and J. Kuret (2005). "Site-specific pseudophosphorylation modulates the rate of tau filament dissociation." FEBS Lett, 579(6), 1453-7.
I. Nishimoto, T. Okamoto, Y. Matsuura, S. Takahashi, T. Okamoto, et al. (1993). "Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o)." Nature, 362(6415), 75-9.
A. Nunomura, G. Perry, M. A. Pappolla, R. Wade, K. Hirai, et al. (1999). "RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease." J Neurosci, 19(6), 1959-64.
A. Nunomura, G. Perry, M. A. Pappolla, R. P. Friedland, K. Hirai, et al. (2000). "Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome." J Neuropathol Exp Neurol, 59(11), 1011-7.
A. Nunomura, G. Perry, G. Aliev, K. Hirai, A. Takeda, et al. (2001). "Oxidative damage is the earliest event in Alzheimer disease." J Neuropathol Exp Neurol, 60(8), 759-67.
A. Nunomura, S. Chiba, C. F. Lippa, P. Cras, R. N. Kalaria, et al. (2004). "Neuronal RNA oxidation is a prominent feature of familial Alzheimer's disease." Neurobiol Dis, 17(1), 108-13.
A. Nunomura, R. J. Castellani, H. G. Lee, P. I. Moreira, X. Zhu, et al. (2006). "Neuropathology in Alzheimer's disease: awaking from a hundred-year-old dream." Sci Aging Knowledge Environ, 2006(8), pe10.
A. C. Nyborg, T. B. Ladd, C. W. Zwizinski, J. J. Lah and T. E. Golde (2006). "Sortilin, SorCS1b, and SorLA Vps10p sorting receptors, are novel gamma-secretase substrates." Mol Neurodegener, 13.

Chapitre 1 : la maladie d’Alzheimer
87
B. L. O'Dell (1993). "Roles of zinc and copper in the nervous system." Prog Clin Biol Res, 380147-62.
T. V. O'Halloran (1993). "Transition metals in control of gene expression." Science, 261(5122), 715-25.
S. Oddo, A. Caccamo, J. D. Shepherd, M. P. Murphy, T. E. Golde, et al. (2003). "Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction." Neuron, 39(3), 409-21.
S. Oddo, A. Caccamo, J. D. Shepherd, M. P. Murphy, T. E. Golde, et al. (2003). "Triple-transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction." Neuron, 39409-421.
S. Oh, H. S. Hong, E. Hwang, H. J. Sim, W. Lee, et al. (2005). "Amyloid peptide attenuates the proteasome activity in neuronal cells." Mech Ageing Dev, 126(12), 1292-9.
K. Ono, Y. Yoshiike, A. Takashima, K. Hasegawa, H. Naiki, et al. (2003). "Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer's disease." J. Neurochem., 87172-181.
C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, et al. (2002). "Metalloenzyme-like activity of Alzheimer's disease β-amyloid." J. Biol. Chem., 277(43), 40302-40308.
C. Opazo, S. Luza, V. L. Villemagne, I. Volitakis, C. Rowe, et al. (2006). "Radioiodinated clioquinol as a biomarker for beta-amyloid: Zn complexes in Alzheimer's disease." Aging Cell., 569-79.
J. M. Orgogozo, S. Gilman, J. F. Dartigues, B. Laurent, M. Puel, et al. (2003). "Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization." Neurology, 61(1), 46-54.
S. Orrenius, B. Zhivotovsky and P. Nicotera (2003). "Regulation of cell death: the calcium-apoptosis link." Nat Rev Mol Cell Biol, 4(7), 552-65.
P. I. Oteiza, S. Cuellar, B. Lonnerdal, L. S. Hurley and C. L. Keen (1990). "Influence of maternal dietary zinc intake on in vitro tubulin polymerization in fetal rat brain." Teratology, 41(1), 97-104.
P. I. Oteiza, L. S. Hurley, B. Lonnerdal and C. L. Keen (1990). "Effects of marginal zinc deficiency on microtubule polymerization in the developing rat brain." Biol Trace Elem Res, 24(1), 13-23.
D. E. Otzen, O. Kristensen and M. Oliveberg (2000). "Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: a structural clue to amyloid assembly." Proc Natl Acad Sci U S A, 97(18), 9907-12.
C. E. Outten and T. V. O'Halloran (2001). "Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis." Science, 292(5526), 2488-92.
A. Paganini-Hill and V. W. Henderson (1996). "Estrogen replacement therapy and risk of Alzheimer disease." Arch Intern Med, 156(19), 2213-7.
R. D. Palmiter, S. D. Findley, T. E. Whitmore and D. M. Durnam (1992). "MT-III, a brain-specific member of the metallothionein gene family." Proc Natl Acad Sci U S A, 89(14), 6333-7.
R. D. Palmiter, T. B. Cole, C. J. Quaife and S. D. Findley (1996). "ZnT-3, a putative transporter of zinc into synaptic vesicles." Proc Natl Acad Sci U S A, 93(25), 14934-9.
D. Paola, C. Domenicotti, M. Nitti, A. Vitali, R. Borghi, et al. (2000). "Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells." Biochem Biophys Res Commun, 268(2), 642-6.
R. Pardossi-Piquard, A. Petit, T. Kawarai, C. Sunyach, C. Alves da Costa, et al. (2005). "Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP." Neuron, 46(4), 541-54.

Chapitre 1 : la maladie d’Alzheimer
88
R. Pardossi-Piquard, J. Dunys, G. Yu, P. St George-Hyslop, C. Alves da Costa, et al. (2006). "Neprilysin activity and expression are controlled by nicastrin." J Neurochem, 97(4), 1052-6.
I. H. Park, M. W. Jung, H. Mori and I. Mook-Jung (2001). "Zinc enhances synthesis of presenilin 1 in mouse primary cortical culture." Biochem Biophys Res Commun, 285(3), 680-8.
L. Pastorino, A. Sun, P. J. Lu, X. Z. Zhou, M. Balastik, et al. (2006). "The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production." Nature, 440(7083), 528-34.
H. A. Pearson and C. Peers (2006). "Physiological roles for amyloid beta peptides." J Physiol, 575(Pt 1), 5-10.
J. J. Pei, E. Braak, H. Braak, I. Grundke-Iqbal, K. Iqbal, et al. (2001). "Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration." J Alzheimers Dis, 3(1), 41-48.
J. J. Pei, W. L. An, X. W. Zhou, T. Nishimura, J. Norberg, et al. (2006). "P70 S6 kinase mediates tau phosphorylation and synthesis." FEBS Lett, 580(1), 107-14.
S. S. Percival (1998). "Copper and immunity." Am J Clin Nutr, 67(5 Suppl), 1064S-1068S. J. Perez-Clausell and G. Danscher (1985). "Intravesicular localization of zinc in rat
telencephalic boutons. A histochemical study." Brain Res, 337(1), 91-8. M. Perez, J. M. Valpuesta, E. M. de Garcini, C. Quintana, M. Arrasate, et al. (1998). "Ferritin
is associated with the aberrant tau filaments present in progressive supranuclear palsy." Am J Pathol, 152(6), 1531-9.
R. G. Perez, S. Soriano, J. D. Hayes, B. Ostaszewski, W. Xia, et al. (1999). "Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42." J Biol Chem, 274(27), 18851-6.
M. A. Pericak-Vance, M. P. Bass, L. H. Yamaoka, P. C. Gaskell, W. K. Scott, et al. (1997). "Complete genomic screen in late-onset familial Alzheimer disease. Evidence for a new locus on chromosome 12." Jama, 278(15), 1237-41.
M. A. Pericak-Vance, J. Grubber, L. R. Bailey, D. Hedges, S. West, et al. (2000). "Identification of novel genes in late-onset Alzheimer's disease." Exp Gerontol, 35(9-10), 1343-52.
G. Perry, R. Friedman, G. Shaw and V. Chau (1987). "Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains." Proc Natl Acad Sci U S A, 84(9), 3033-6.
A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, et al. (2002). "A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR." Proc Natl Acad Sci U S A, 99(26), 16742-7.
A. L. Phinney, B. Drisaldi, S. D. Schmidt, S. Lugowski, V. Coronado, et al. (2003). "In vivo reduction of amyloid-beta by a mutant copper transporter." Proc Natl Acad Sci U S A, 100(24), 14193-8.
G. Pigino, G. Morfini, A. Pelsman, M. P. Mattson, S. T. Brady, et al. (2003). "Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport." J Neurosci, 23(11), 4499-508.
L. D. Plant, N. J. Webster, J. P. Boyle, M. Ramsden, D. B. Freir, et al. (2006). "Amyloid beta peptide as a physiological modulator of neuronal 'A'-type K+ current." Neurobiol Aging, 27(11), 1673-83.
H. F. Poon, R. A. Vaishnav, T. V. Getchell, M. L. Getchell and D. A. Butterfield (2006). "Quantitative proteomics analysis of differential protein expression and oxidative modification of specific proteins in the brains of old mice." Neurobiol Aging, 27(7),

Chapitre 1 : la maladie d’Alzheimer
89
1010-9. A. S. Prasad (1995). "Zinc: an overview." Nutrition, 11(1 Suppl), 93-9. D. Pratico and N. Delanty (2000). "Oxidative injury in diseases of the central nervous system:
focus on Alzheimer's disease." Am J Med, 109(7), 577-85. D. Pratico, K. Uryu, S. Leight, J. Q. Trojanoswki and V. M. Lee (2001). "Increased lipid
peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis." J Neurosci, 21(12), 4183-7.
C. I. Prodan, S. S. Bottomley, A. S. Vincent, L. D. Cowan, N. R. Holland, et al. (2007). "Hypocupremia associated with prior vitamin B12 deficiency." Am J Hematol, 82(4), 288-90.
B. Puig, T. Gomez-Isla, E. Ribe, M. Cuadrado, B. Torrejon-Escribano, et al. (2004). "Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice." Neuropathol Appl Neurobiol, 30(5), 491-502.
S. B. Qian, H. McDonough, F. Boellmann, D. M. Cyr and C. Patterson (2006). "CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70." Nature, 440(7083), 551-5.
C. Qiu, E. von Strauss, J. Fastbom, B. Winblad and L. Fratiglioni (2003). "Low blood pressure and risk of dementia in the Kungsholmen project: a 6-year follow-up study." Arch Neurol, 60(2), 223-8.
C. Quintana, S. Bellefqih, J. Y. Laval, J. L. Guerquin-Kern, T. D. Wu, et al. (2006). "Study of the localization of iron, ferritin, and hemosiderin in Alzheimer's disease hippocampus by analytical microscopy at the subcellular level." J Struct Biol, 153(1), 42-54.
R. A. Quintanilla, D. I. Orellana, C. Gonzalez-Billault and R. B. Maccioni (2004). "Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway." Exp Cell Res, 295(1), 245-57.
A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, et al. (2005). "Amyloid ion channels: a common structural link for protein-misfolding disease." Proc Natl Acad Sci U S A, 102(30), 10427-32.
F. Radtke, A. Wilson, S. J. Mancini and H. R. MacDonald (2004). "Notch regulation of lymphocyte development and function." Nat Immunol, 5(3), 247-53.
M. T. Rajan, K. S. Jagannatha Rao, B. M. Mamatha, R. V. Rao, P. Shanmugavelu, et al. (1997). "Quantification of trace elements in normal human brain by inductively coupled plasma atomic emission spectrometry." J Neurol Sci, 146(2), 153-66.
B. Raman, T. Ban, K. Yamaguchi, M. Sakai, T. Kawai, et al. (2005). "Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide." J Biol Chem, 280(16), 16157-62.
H. Ramaroson, C. Helmer, P. Barberger-Gateau, L. Letenneur and J. F. Dartigues (2003). "[Prevalence of dementia and Alzheimer's disease among subjects aged 75 years or over: updated results of the PAQUID cohort]." Rev Neurol (Paris), 159(4), 405-11.
T. A. Ramelot and L. K. Nicholson (2001). "Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR." J Mol Biol, 307(3), 871-84.
M. Rapoport, H. N. Dawson, L. I. Binder, M. P. Vitek and A. Ferreira (2002). "Tau is essential to beta -amyloid-induced neurotoxicity." Proc Natl Acad Sci U S A, 99(9), 6364-9.
B. Regland, W. Lehmann, I. Abedini, K. Blennow, M. Jonsson, et al. (2001). "Traetment of Alzheimer's disease with Clioquinol." Dement. Geriatr. Cogn. Disord., 12408-414.
S. A. Reines, G. A. Block, J. C. Morris, G. Liu, M. L. Nessly, et al. (2004). "Rofecoxib - No effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study."

Chapitre 1 : la maladie d’Alzheimer
90
Neurology, 6266-71. L. Reznichenko, T. Amit, H. Zheng, Y. Avramovich-Tirosh, M. B. Youdim, et al. (2006).
"Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease." J Neurochem, 97(2), 527-36.
D. R. Riddell, G. Christie, I. Hussain and C. Dingwall (2001). "Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts." Curr Biol, 11(16), 1288-93.
P. Riederer and S. Hoyer (2006). "From benefit to damage. Glutamate and advanced glycation end products in Alzheimer brain." J Neural Transm, 113(11), 1671-7.
C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, et al. (2003). "Metal-protein attenuation with Iodochlorohydroxyquin (Clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease." Arch. Neurol., 601685-1691.
S. L. Roberds, J. Anderson, G. Basi, M. J. Bienkowski, D. G. Branstetter, et al. (2001). "BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics." Hum Mol Genet, 10(12), 1317-24.
D. M. Robinson and G. M. Keating (2006). "Memantine: a review of its use in Alzheimer's disease." Drugs, 66(11), 1515-34.
E. I. Rogaev, R. Sherrington, E. A. Rogaeva, G. Levesque, M. Ikeda, et al. (1995). "Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene." Nature, 376(6543), 775-8.
J. T. Rogers, J. D. Randall, C. M. Cahill, P. S. Eder, X. Huang, et al. (2002). "An iron-responsive element type II in the 5'-untranslated region of the Alzheimer's amyloid precursor protein transcript." J Biol Chem, 277(47), 45518-28.
S. L. Rogers, R. S. Doody, R. C. Mohs and L. T. Friedhoff (1998). "Donepezil improves cognition and global function in Alzheimer disease: a 15-week, double-blind, placebo-controlled study. Donepezil Study Group." Arch Intern Med, 158(9), 1021-31.
S. L. Rogers, M. R. Farlow, R. S. Doody, R. Mohs and L. T. Friedhoff (1998). "A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer's disease. Donepezil Study Group." Neurology, 50(1), 136-45.
R. F. Rosenberger (1991). "Senescence and the accumulation of abnormal proteins." Mutat Res, 256(2-6), 255-62.
M. Rosler, R. Anand, A. Cicin-Sain, S. Gauthier, Y. Agid, et al. (1999). "Efficacy and safety of rivastigmine in patients with Alzheimer's disease: international randomised controlled trial." Bmj, 318(7184), 633-8.
L. Rossi, M. F. Lombardo, M. R. Ciriolo and G. Rotilio (2004). "Mitochondrial dysfunction in neurodegenerative diseases associated with copper imbalance." Neurochem Res, 29(3), 493-504.
S. Rossner, U. Ueberham, R. Schliebs, J. R. Perez-Polo and V. Bigl (1998). "The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling." Prog Neurobiol, 56(5), 541-69.
S. Roy, B. Zhang, V. M. Lee and J. Q. Trojanowski (2005). "Axonal transport defects: a common theme in neurodegenerative diseases." Acta Neuropathol (Berl), 109(1), 5-13.
A. L. Rozelle, L. M. Machesky, M. Yamamoto, M. H. Driessens, R. H. Insall, et al. (2000). "Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3." Curr Biol, 10(6), 311-20.
T. H. Rushmore, R. G. King, K. E. Paulson and C. B. Pickett (1990). "Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic

Chapitre 1 : la maladie d’Alzheimer
91
compounds." Proc Natl Acad Sci U S A, 87(10), 3826-30. S. L. Sabo, A. F. Ikin, J. D. Buxbaum and P. Greengard (2001). "The Alzheimer amyloid
precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement." J Cell Biol, 153(7), 1403-14.
S. L. Sabo, A. F. Ikin, J. D. Buxbaum and P. Greengard (2003). "The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo." J Neurosci, 23(13), 5407-15.
N. Sahara, M. Murayama, T. Mizoroki, M. Urushitani, Y. Imai, et al. (2005). "In vivo evidence of CHIP up-regulation attenuating tau aggregation." J Neurochem, 94(5), 1254-63.
T. Saito, N. Iwata, S. Tsubuki, Y. Takaki, J. Takano, et al. (2005). "Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation." Nat Med, 11(4), 434-9.
S. C. Samuels and K. L. Davis (1997). "A risk-benefit assessment of tacrine in the treatment of Alzheimer's disease." Drug Saf, 16(1), 66-77.
H. H. Sandstead (1994). "Understanding zinc: recent observations and interpretations." J Lab Clin Med, 124(3), 322-7.
K. Santacruz, J. Lewis, T. Spires, J. Paulson, L. Kotilinek, et al. (2005). "Tau suppression in a neurodegenerative mouse model improves memory function." Science, 309(5733), 476-81.
M. Sarazin and B. Dubois (2005). "[A guide to diagnosis of Alzheimer's disease]." Rev Prat, 55(17), 1879-90.
M. Sastre, I. Dewachter, S. Rossner, N. Bogdanovic, E. Rosen, et al. (2006). "Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma." Proc Natl Acad Sci U S A, 103(2), 443-8.
C. A. Saura, G. Chen, S. Malkani, S. Y. Choi, R. H. Takahashi, et al. (2005). "Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice." J Neurosci, 25(29), 6755-64.
J. Savory, M. M. Herman, R. T. Erasmus, J. C. Boyd and M. R. Wills (1994). "Partial reversal of aluminium-induced neurofibrillary degeneration by desferrioxamine in adult male rabbits." Neuropathol Appl Neurobiol, 20(1), 31-7.
L. M. Sayre, G. Perry, P. L. Harris, Y. Liu, K. A. Schubert, et al. (2000). "In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals." J Neurochem, 74(1), 270-9.
A. S. Schachter, K. L. Davis, S. Lovestone, L. S. Schneider, A. Burns, et al. (2000). "Alzheimer's Disease." Dialogues Clin Neurosci, 2(2),
D. Schenk, R. Barbour, W. Dunn, G. Gordon, H. Grajeda, et al. (1999). "Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse." Nature, 400(6740), 173-7.
D. Schenk, M. Hagen and P. Seubert (2004). "Current progress in beta-amyloid immunotherapy." Curr Opin Immunol, 16(5), 599-606.
S. Scheuermann, B. Hambsch, L. Hesse, J. Stumm, C. Schmidt, et al. (2001). "Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease." J Biol Chem, 276(36), 33923-9.
R. Schmidt, F. Fazekas, B. Reinhart, P. Kapeller, G. Fazekas, et al. (1996). "Estrogen replacement therapy in older women: a neuropsychological and brain MRI study." J Am Geriatr Soc, 44(11), 1307-13.
F. Schmitt, M. Ryan and G. Cooper (2007). "A brief review of the pharmacologic and therapeutic aspects of memantine in Alzheimer's disease." Expert Opin Drug Metab

Chapitre 1 : la maladie d’Alzheimer
92
Toxicol, 3(1), 135-41. C. Schoneich and T. D. Williams (2002). "Cu(II)-catalyzed oxidation of beta-amyloid peptide
targets His13 and His14 over His6: Detection of 2-Oxo-histidine by HPLC-MS/MS." Chem Res Toxicol, 15(5), 717-22.
R. Schwartz, S. Istrail and J. King (2001). "Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues." Protein Sci, 10(5), 1023-31.
K. L. Sciarretta, A. Boire, D. J. Gordon and S. C. Meredith (2006). "Spatial separation of beta-sheet domains of beta-amyloid: disruption of each beta-sheet by N-methyl amino acids." Biochemistry, 45(31), 9485-95.
K. L. Sciarretta, D. J. Gordon and S. C. Meredith (2006). "Peptide-based inhibitors of amyloid assembly." Methods Enzymol, 413273-312.
M. Sela (2006). "Immunomodulatory vaccines against autoimmune diseases." Rejuvenation Res, 9(1), 126-33.
J. Seo, S. Kim, H. Kim, C. H. Park, S. Jeong, et al. (2001). "Effects of nicotine on APP secretion and Abeta- or CT(105)-induced toxicity." Biol Psychiatry, 49(3), 240-7.
L. C. Serpell, M. Sunde, M. D. Benson, G. A. Tennent, M. B. Pepys, et al. (2000). "The protofilament substructure of amyloid fibrils." J Mol Biol, 300(5), 1033-9.
C. T. Sheline, H. S. Ying, C. S. Ling, L. M. Canzoniero and D. W. Choi (2002). "Depolarization-induced 65zinc influx into cultured cortical neurons." Neurobiol Dis, 10(1), 41-53.
Y. Shen, P. He, Z. Zhong, C. McAllister and K. Lindholm (2006). "Distinct destructive signal pathways of neuronal death in Alzheimer's disease." Trends Mol Med, 12(12), 574-9.
J. G. Sheng, R. E. Mrak and W. S. Griffin (1995). "Microglial interleukin-1 alpha expression in brain regions in Alzheimer's disease: correlation with neuritic plaque distribution." Neuropathol Appl Neurobiol, 21(4), 290-301.
J. G. Sheng, S. G. Zhu, R. A. Jones, W. S. Griffin and R. E. Mrak (2000). "Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo." Exp Neurol, 163(2), 388-91.
R. Sherrington, E. I. Rogaev, Y. Liang, E. A. Rogaeva, G. Levesque, et al. (1995). "Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease." Nature, 375(6534), 754-60.
H. Shimura, D. Schwartz, S. P. Gygi and K. S. Kosik (2004). "CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival." J Biol Chem, 279(6), 4869-76.
R. W. Shin, T. P. Kruck, H. Murayama and T. Kitamoto (2003). "A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer's disease." Brain Res, 961(1), 139-46.
H. Shinall, E. S. Song and L. B. Hersh (2005). "Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral." Biochemistry, 44(46), 15345-50.
E. Y. Shintani and K. M. Uchida (1997). "Donepezil: an anticholinesterase inhibitor for Alzheimer's disease." Am J Health Syst Pharm, 54(24), 2805-10.
E. R. Siemers, J. F. Quinn, J. Kaye, M. R. Farlow, A. Porsteinsson, et al. (2006). "Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease." Neurology, 66(4), 602-4.
A. Simons, T. Ruppert, C. Schmidt, A. Schlicksupp, R. Pipkorn, et al. (2002). "Evidence for a copper-binding superfamily of the amyloid precursor protein." Biochemistry, 41(30), 9310-20.
T. J. Singh, I. Grundke-Iqbal, W. Q. Wu, V. Chauhan, M. Novak, et al. (1997). "Protein

Chapitre 1 : la maladie d’Alzheimer
93
kinase C and calcium/calmodulin-dependent protein kinase II phosphorylate three-repeat and four-repeat tau isoforms at different rates." Mol Cell Biochem, 168(1-2), 141-8.
V. L. Smith-Swintosky, L. C. Pettigrew, S. D. Craddock, A. R. Culwell, R. E. Rydel, et al. (1994). "Secreted forms of beta-amyloid precursor protein protect against ischemic brain injury." J Neurochem, 63(2), 781-4.
M. A. Smith, P. L. Harris, L. M. Sayre and G. Perry (1997). "Iron accumulation in Alzheimer disease is a source of redox-generated free radicals." Proc Natl Acad Sci U S A, 94(18), 9866-8.
M. A. Smith, K. Wehr, P. L. Harris, S. L. Siedlak, J. R. Connor, et al. (1998). "Abnormal localization of iron regulatory protein in Alzheimer's disease." Brain Res, 788(1-2), 232-6.
D. L. Sparks and B. G. Schreurs (2003). "Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease." Proc Natl Acad Sci U S A, 100(19), 11065-9.
M. G. Spillantini, R. A. Crowther and M. Goedert (1996). "Comparison of the neurofibrillary pathology in Alzheimer's disease and familial presenile dementia with tangles." Acta Neuropathol (Berl), 92(1), 42-8.
M. G. Spillantini, T. D. Bird and B. Ghetti (1998). "Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies." Brain Pathol, 8(2), 387-402.
J. J. Sramek, R. Anand, T. S. Wardle, P. Irwin, R. D. Hartman, et al. (1996). "Safety/tolerability trial of SDZ ENA 713 in patients with probable Alzheimer's disease." Life Sci, 58(15), 1201-7.
S. J. Stachel, C. A. Coburn, S. Sankaranarayanan, E. A. Price, G. Wu, et al. (2006). "Macrocyclic inhibitors of beta-secretase: functional activity in an animal model." J Med Chem, 49(21), 6147-50.
K. Stamer, R. Vogel, E. Thies, E. Mandelkow and E. M. Mandelkow (2002). "Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress." J Cell Biol, 156(6), 1051-63.
M. Stefani and C. M. Dobson (2003). "Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution." J Mol Med, 81(11), 678-99.
M. Stefani (2006). "Biological surfaces as catalysts of amyloid aggregate nucleation and primary sites of amyloid toxicity." Ital J Biochem, 55(3-4), 194-204.
Y. Stern, M. X. Tang, M. S. Albert, J. Brandt, D. M. Jacobs, et al. (1997). "Predicting time to nursing home care and death in individuals with Alzheimer disease." Jama, 277(10), 806-12.
W. F. Stewart, C. Kawas, M. Corrada and E. J. Metter (1997). "Risk of Alzheimer's disease and duration of NSAID use." Neurology, 48(3), 626-32.
G. B. Stokin, C. Lillo, T. L. Falzone, R. G. Brusch, E. Rockenstein, et al. (2005). "Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease." Science, 307(5713), 1282-8.
W. H. Stoothoff and G. V. Johnson (2005). "Tau phosphorylation: physiological and pathological consequences." Biochim Biophys Acta, 1739(2-3), 280-97.
N. C. Stratman, C. K. Castle, B. M. Taylor, D. E. Epps, G. W. Melchior, et al. (2005). "Isoform-specific interactions of human apolipoprotein E to an intermediate conformation of human Alzheimer amyloid-beta peptide." Chem Phys Lipids, 137(1-2), 52-61.
W. J. Strittmatter, A. M. Saunders, D. Schmechel, M. Pericak-Vance, J. Enghild, et al. (1993).

Chapitre 1 : la maladie d’Alzheimer
94
"Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease." Proc Natl Acad Sci U S A, 90(5), 1977-81.
S. W. Suh, K. B. Jensen, M. S. Jensen, D. S. Silva, P. J. Kesslak, et al. (2000). "Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains." Brain Res, 852(2), 274-8.
Y.-H. Suh and F. Checler (2002). "Amyloid Precursor Protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease." Pharmacol. Rev., 54(3), 469-525.
R. Sultana, M. Perluigi and D. A. Butterfield (2006). "Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics." Antioxid Redox Signal, 8(11-12), 2021-37.
J. Y. Sung, S. M. Park, C. H. Lee, J. W. Um, H. J. Lee, et al. (2005). "Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases." J Biol Chem, 280(26), 25216-24.
S. Sung, Y. Yao, K. Uryu, H. Yang, V. M. Lee, et al. (2004). "Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease." Faseb J, 18(2), 323-5.
R. D. Swartz (1985). "Deferoxamine and aluminum removal." Am J Kidney Dis, 6(5), 358-64. J. E. Swatton, L. A. Sellers, R. L. Faull, A. Holland, S. Iritani, et al. (2004). "Increased MAP
kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain." Eur J Neurosci, 19(10), 2711-9.
C. D. Syme, R. C. Nadal, S. E. Rigby and J. H. Viles (2004). "Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer's disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques." J Biol Chem, 279(18), 18169-77.
C. D. Syme and J. H. Viles (2006). "Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer's disease." Biochim Biophys Acta, 1764(2), 246-56.
T. Tabira (2001). "Clioquinol's return: cautions from Japan." Science, 292(5525), 2251-2. B. J. Tabner, S. Turnbull, O. M. El-Agnaf and D. Allsop (2002). "Formation of hydrogen
peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer's disease and Parkinson's disease." Free Radic Biol Med, 32(11), 1076-83.
A. Takashima, K. Noguchi, K. Sato, T. Hoshino and K. Imahori (1993). "Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity." Proc Natl Acad Sci U S A, 90(16), 7789-93.
A. Takeda (2000). "Movement of zinc and its functional significance in the brain." Brain Res Brain Res Rev, 34(3), 137-48.
A. Takeda (2001). "Zinc homeostasis and functions of zinc in the brain." Biometals, 14(3-4), 343-51.
J. Tan, T. Town, T. Mori, Y. Wu, M. Saxe, et al. (2000). "CD45 opposes beta-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase." J Neurosci, 20(20), 7587-94.
T. Tanaka, K. Iqbal, E. Trenkner, D. J. Liu and I. Grundke-Iqbal (1995). "Abnormally phosphorylated tau in SY5Y human neuroblastoma cells." FEBS Lett, 360(1), 5-9.
R. B. Thompson, D. Peterson, W. Mahoney, M. Cramer, B. P. Maliwal, et al. (2002). "Fluorescent zinc indicators for neurobiology." J Neurosci Methods, 118(1), 63-75.
M. Torok, S. Milton, R. Kayed, P. Wu, T. McIntire, et al. (2002). "Structural and dynamic features of Alzheimer's Abeta peptide in amyloid fibrils studied by site-directed spin

Chapitre 1 : la maladie d’Alzheimer
95
labeling." J Biol Chem, 277(43), 40810-5. H. L. True and S. L. Lindquist (2000). "A yeast prion provides a mechanism for genetic
variation and phenotypic diversity." Nature, 407(6803), 477-83. H. L. True, I. Berlin and S. L. Lindquist (2004). "Epigenetic regulation of translation reveals
hidden genetic variation to produce complex traits." Nature, 431(7005), 184-7. S. Tsuji, H. Kobayashi, Y. Uchida, Y. Ihara and T. Miyatake (1992). "Molecular cloning of
human growth inhibitory factor cDNA and its down-regulation in Alzheimer's disease." Embo J, 11(13), 4843-50.
H. M. Tucker, M. Kihiko, J. N. Caldwell, S. Wright, T. Kawarabayashi, et al. (2000). "The plasmin system is induced by and degrades amyloid-beta aggregates." J Neurosci, 20(11), 3937-46.
A. J. Turner and K. Tanzawa (1997). "Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX." Faseb J, 11(5), 355-64.
A. J. Turner, L. Fisk and N. N. Nalivaeva (2004). "Targeting amyloid-degrading enzymes as therapeutic strategies in neurodegeneration." Ann N Y Acad Sci, 10351-20.
R. Tycko (2003). "Insights into the amyloid folding problems for solide-state NMR." Biochemistry, 42(11), 3151-3159.
Y. Uchida, K. Takio, K. Titani, Y. Ihara and M. Tomonaga (1991). "The growth inhibitory factor that is deficient in the Alzheimer's disease brain is a 68 amino acid metallothionein-like protein." Neuron, 7(2), 337-47.
Y. Uchida (1994). "Growth-inhibitory factor, metallothionein-like protein, and neurodegenerative diseases." Biol Signals, 3(4), 211-5.
S. Ueno, M. Tsukamoto, T. Hirano, K. Kikuchi, M. K. Yamada, et al. (2002). "Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits." J Cell Biol, 158(2), 215-20.
V. N. Uversky (2002). "Natively unfolded proteins: a point where biology waits for physics." Protein Sci, 11(4), 739-56.
B. L. Vallee and D. S. Auld (1992). "Functional zinc-binding motifs in enzymes and DNA-binding proteins." Faraday Discuss, (93), 47-65.
B. L. Vallee and D. S. Auld (1992). "Active zinc binding sites of zinc metalloenzymes." Matrix Suppl, 15-19.
B. L. Vallee and K. H. Falchuk (1993). "The biochemical basis of zinc physiology." Physiol Rev, 73(1), 79-118.
W. E. Van Nostrand and M. Porter (1999). "Plasmin cleavage of the amyloid beta-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity." Biochemistry, 38(35), 11570-6.
R. Vassar, B. D. Bennett, S. Babu-Khan, S. Kahn, E. A. Mendiaz, et al. (1999). "Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE." Science, 286(5440), 735-41.
K. Vekrellis, Z. Ye, W. Q. Qiu, D. Walsh, D. Hartley, et al. (2000). "Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme." J Neurosci, 20(5), 1657-65.
A. Venti, T. Giordano, P. Eder, A. I. Bush, D. K. Lahiri, et al. (2004). "The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5'-untranslated region." Ann N Y Acad Sci, 103534-48.
S. Vepsalainen, M. Parkinson, S. Helisalmi, A. Mannermaa, H. Soininen, et al. (2007). "Insulin-degrading enzyme is genetically associated with Alzheimer's disease in the Finnish population." J Med Genet, 44(9), 606-8.
K. S. Vetrivel, H. Cheng, S. H. Kim, Y. Chen, N. Y. Barnes, et al. (2005). "Spatial segregation of gamma-secretase and substrates in distinct membrane domains." J Biol

Chapitre 1 : la maladie d’Alzheimer
96
Chem, 280(27), 25892-900. S. Wahrle, P. Das, A. C. Nyborg, C. McLendon, M. Shoji, et al. (2002). "Cholesterol-
dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains." Neurobiol Dis, 9(1), 11-23.
J. C. Wallwork (1987). "Zinc and the central nervous system." Prog Food Nutr Sci, 11(2), 203-47.
D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow (1997). "Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate." J Biol Chem, 272(35), 22364-72.
D. M. Walsh, D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. M. Condron, et al. (1999). "Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates." J Biol Chem, 274(36), 25945-52.
D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, et al. (2002). "Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo." Nature, 416(6880), 535.
D. S. Wang, N. Iwata, E. Hama, T. C. Saido and D. W. Dickson (2003). "Oxidized neprilysin in aging and Alzheimer's disease brains." Biochem Biophys Res Commun, 310(1), 236-41.
J. Z. Wang, I. Grundke-Iqbal and K. Iqbal (1996). "Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease." Nat Med, 2(8), 871-5.
J. Z. Wang and Z. F. Wang (2006). "Role of melatonin in Alzheimer-like neurodegeneration." Acta Pharmacol Sin, 27(1), 41-9.
R. Wang, X. Zhu, F. Guo, W. Zhang and Z. Jia (2006). "Influence of different dietary levels of zinc on performance, vitamin B12, and blood parameters in lambs." Int J Vitam Nutr Res, 76(6), 353-8.
T. Wataya, A. Nunomura, M. A. Smith, S. L. Siedlak, P. L. Harris, et al. (2002). "High molecular weight neurofilament proteins are physiological substrates of adduction by the lipid peroxidation product hydroxynonenal." J Biol Chem, 277(7), 4644-8.
S. Weggen, J. L. Eriksen, P. Das, S. A. Sagi, R. Wang, et al. (2001). "A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity." Nature, 414(6860), 212-6.
M. Wegner (2000). "Transcriptional control in myelinating glia: flavors and spices." Glia, 31(1), 1-14.
A. Weihofen and B. Martoglio (2003). "Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides." Trends Cell Biol, 13(2), 71-8.
O. Weinreb, S. Mandel, T. Amit and M. B. Youdim (2004). "Neurological mechanisms of green tea polyphenols in Alzheimer's and Parkinson's diseases." J Nutr Biochem, 15(9), 506-16.
J. H. Weiss, D. M. Hartley, J. Y. Koh and D. W. Choi (1993). "AMPA receptor activation potentiates zinc neurotoxicity." Neuron, 10(1), 43-9.
M. G. Weisskopf, R. O. Wright, J. Schwartz, A. Spiro, 3rd, D. Sparrow, et al. (2004). "Cumulative lead exposure and prospective change in cognition among elderly men: the VA Normative Aging Study." Am J Epidemiol, 160(12), 1184-93.
M. Wender, J. Szczech, S. Hoffmann and W. Hilczer (1992). "Electron paramagnetic resonance analysis of heavy metals in the aging human brain." Neuropatol Pol, 30(1), 65-72.
H. J. Wenzel, T. B. Cole, D. E. Born, P. A. Schwartzkroin and R. D. Palmiter (1997). "Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey."

Chapitre 1 : la maladie d’Alzheimer
97
Proc Natl Acad Sci U S A, 94(23), 12676-81. A. R. White, R. Reyes, J. F. Mercer, J. Camakaris, H. Zheng, et al. (1999). "Copper levels are
increased in the cerebral cortex and liver of APP and APLP2 knockout mice." Brain Res, 842(2), 439-44.
A. R. White, T. Du, K. M. Laughton, I. Volitakis, R. A. Sharples, et al. (2006). "Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity." J Biol Chem, 281(26), 17670-80.
J. S. Whitson, C. G. Glabe, E. Shintani, A. Abcar and C. W. Cotman (1990). "Beta-amyloid protein promotes neuritic branching in hippocampal cultures." Neurosci Lett, 110(3), 319-24.
A. G. Wiese, R. E. Pacifici and K. J. Davies (1995). "Transient adaptation of oxidative stress in mammalian cells." Arch Biochem Biophys, 318(1), 231-40.
M. M. Wilhelmus, W. C. Boelens, I. Otte-Holler, B. Kamps, R. M. de Waal, et al. (2006). "Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity." Brain Res, 1089(1), 67-78.
M. Willem, A. N. Garratt, B. Novak, M. Citron, S. Kaufmann, et al. (2006). "Control of peripheral nerve myelination by the beta-secretase BACE1." Science, 314(5799), 664-6.
C. M. Wischik, M. Novak, P. C. Edwards, A. Klug, W. Tichelaar, et al. (1988). "Structural characterization of the core of the paired helical filament of Alzheimer disease." Proc Natl Acad Sci U S A, 85(13), 4884-8.
C. M. Wischik, M. Novak, H. C. Thogersen, P. C. Edwards, M. J. Runswick, et al. (1988). "Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease." Proc Natl Acad Sci U S A, 85(12), 4506-10.
M. S. Wolfe (2002). "APP, Notch, and presenilin: molecular pieces in the puzzle of Alzheimer's disease." Int Immunopharmacol, 2(13-14), 1919-29.
M. S. Wolfe and R. Kopan (2004). "Intramembrane proteolysis: theme and variations." Science, 305(5687), 1119-23.
M. S. Wolfe (2006). "The gamma-secretase complex: membrane-embedded proteolytic ensemble." Biochemistry, 45(26), 7931-9.
B. Wolozin, W. Kellman, P. Ruosseau, G. G. Celesia and G. Siegel (2000). "Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors." Arch Neurol, 57(10), 1439-43.
G. T. Wong, D. Manfra, F. M. Poulet, Q. Zhang, H. Josien, et al. (2004). "Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation." J Biol Chem, 279(13), 12876-82.
C. Wurth, N. K. Guimard and M. H. Hecht (2002). "Mutations that reduce aggregation of the Alzheimer's Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis." J Mol Biol, 319(5), 1279-90.
T. Wyss-Coray and L. Mucke (2002). "Inflammation in neurodegenerative disease--a double-edged sword." Neuron, 35(3), 419-32.
Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd (2004). "C-terminal domain of the membrane copper transporter Ctr1 from Saccharomyces cerevisiae binds four Cu(I) ions as a cuprous-thiolate polynuclear cluster: sub-femtomolar Cu(I) affinity of three proteins involved in copper trafficking." J Am Chem Soc, 126(10), 3081-90.
M. Yamaguchi, M. Kura and S. Okada (1982). "Role of zinc as an activator of mitochondrial function in rat liver." Biochem Pharmacol, 31(7), 1289-93.
A. Yamamoto, R. W. Shin, K. Hasegawa, H. Naiki, H. Sato, et al. (2002). "Iron (III) induces

Chapitre 1 : la maladie d’Alzheimer
98
aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer's disease." J Neurochem, 82(5), 1137-47.
T. Yamashita, Y. Takahashi, T. Takahashi and H. Mihara (2003). "Inhibition of peptide amyloid formation by cationic peptides with homologous sequences." Bioorg Med Chem Lett, 13(22), 4051-4.
P. Yan, X. Hu, H. Song, K. Yin, R. J. Bateman, et al. (2006). "Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ." J Biol Chem, 281(34), 24566-74.
D. S. Yang, J. McLaurin, K. Qin, D. Westaway and P. E. Fraser (2000). "Examining the zinc binding site of the amyloid-beta peptide." Eur J Biochem, 267(22), 6692-8.
F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, et al. (2005). "Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo." J Biol Chem, 280(7), 5892-901.
B. A. Yankner, L. K. Duffy and D. A. Kirschner (1990). "Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides." Science, 250(4978), 279-82.
M. S. Yassin, J. Ekblom, M. Xilinas, C. G. Gottfries and L. Oreland (2000). "Changes in uptake of vitamin B(12) and trace metals in brains of mice treated with clioquinol." J Neurol Sci, 173(1), 40-4.
S. Ye, Y. Huang, K. Mullendorff, L. Dong, G. Giedt, et al. (2005). "Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target." Proc Natl Acad Sci U S A, 102(51), 18700-5.
C. M. Yip and J. McLaurin (2001). "Amyloid-beta peptide assembly: a critical step in fibrillogenesis and membrane disruption." Biophys J, 80(3), 1359-71.
V. W. Yong, C. A. Krekoski, P. A. Forsyth, R. Bell and D. R. Edwards (1998). "Matrix metalloproteinases and diseases of the CNS." Trends Neurosci, 21(2), 75-80.
I. S. Yoon, C. U. Pietrzik, D. E. Kang and E. H. Koo (2005). "Sequences from the low density lipoprotein receptor-related protein (LRP) cytoplasmic domain enhance amyloid beta protein production via the beta-secretase pathway without altering amyloid precursor protein/LRP nuclear signaling." J Biol Chem, 280(20), 20140-7.
H. Yoshida and Y. Ihara (1993). "Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau." J Neurochem, 61(3), 1183-6.
Y. Yoshiike, K. Tanemura, O. Murayama, T. Akagi, M. Murayama, et al. (2001). "New insights on how metals disrupt amyloid beta-aggregation and their effects on amyloid-beta cytotoxicity." J Biol Chem, 276(34), 32293-9.
M. B. Youdim and S. Yehuda (2000). "The neurochemical basis of cognitive deficits induced by brain iron deficiency: involvement of dopamine-opiate system." Cell Mol Biol (Noisy-le-grand), 46(3), 491-500.
G. Yu, M. Nishimura, S. Arawaka, D. Levitan, L. Zhang, et al. (2000). "Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing." Nature, 407(6800), 48-54.
W. H. Yu, W. J. Lukiw, C. Bergeron, H. B. Niznik and P. E. Fraser (2001). "Metallothionein III is reduced in Alzheimer's disease." Brain Res, 894(1), 37-45.
N. Zambrano, D. Gianni, P. Bruni, F. Passaro, F. Telese, et al. (2004). "Fe65 is not involved in the platelet-derived growth factor-induced processing of Alzheimer's amyloid precursor protein, which activates its caspase-directed cleavage." J Biol Chem, 279(16), 16161-9.
Z. Zhang, P. Nadeau, W. Song, D. Donoviel, M. Yuan, et al. (2000). "Presenilins are required

Chapitre 1 : la maladie d’Alzheimer
99
for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1." Nat Cell Biol, 2(7), 463-5.
P. Zhu, S. H. Lee, S. Wehrli and I. A. Blair (2006). "Characterization of a lipid hydroperoxide-derived RNA adduct in rat intestinal epithelial cells." Chem Res Toxicol, 19(6), 809-17.
X. Zhu, H. G. Lee, G. Perry and M. A. Smith (2007). "Alzheimer disease, the two-hit hypothesis: an update." Biochim Biophys Acta, 1772(4), 494-502.
S. Zirah, S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, et al. (2006). "Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging." J Biol Chem, 281(4), 2151-61.
J. Zou, K. Kajita and N. Sugimoto (2001). "Cu(2+) Inhibits the Aggregation of Amyloid beta-Peptide(1-42) in vitro." Angew Chem Int Ed Engl, 40(12), 2274-2277.
K. Zou, J. S. Gong, K. Yanagisawa and M. Michikawa (2002). "A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage." J Neurosci, 22(12), 4833-41.

Chapitre 1 : la maladie d’Alzheimer
100

101
Chapitre 2
Etude de la liaison du zinc(II)
au peptide amyloïde bêta

102

Chapitre 2 : la liaison zinc(II) - Aβ
103
I. Résumé de l’étude
Ce chapitre a fait l’objet d’un article dans Biochemistry (sous presse). N’ayant pas d’élément
nouveau à ajouter à cette étude, la publication constitue le corps de ce chapitre. A titre
informatif, un complément sur le fonctionnement et principe de la calorimétrie de titrage
isotherme a été ajouté en dernière partie. Pour plus de clarté, le format des références choisi
par l’éditeur a été conservé dans le résumé de l’article.
Comme exposé dans le chapitre 1, l’agrégation du peptide Aβ est considérée comme la cause
principale de la MA dans l’hypothèse largement acceptée de la cascade amyloïde.(1-4)
Cependant les formes oligomériques solubles semblent être plus toxiques que les fibres ou
protofibrilles.(2) Dans ce contexte, l’étude des facteurs influençant l’agrégation est donc d’un
grand intérêt.(5) Or de nombreuses études ont montré le rôle important joué par les métaux, et
en particulier le zinc, dans l’agrégation d’Aβ.(1, 4, 6, 7) Ce dernier possède en effet un site de
fixation du zinc localisé sur la partie N-terminale du peptide, constitué assez
vraisemblablement des histidines 6, 13 et 14 et d’un quatrième ligand dont l’identification est
encore sujet à discussion.(19, 24-34) La détermination de l’affinité du zinc(II) pour Aβ est
également d’une grande importance pour l’élucidation des sources de zinc et les applications
thérapeutiques par chélation des métaux. Cependant, les valeurs de constante de dissociation
données dans la littérature diffèrent en fonction des conditions et techniques utilisées.(17, 28,
34-37)
Dans cette étude, l’interaction du zinc avec Aβ40 et 42, mais également les formes tronquées
(moins agrégeantes mais possédant le site de fixation du métal) Aβ16 et Aβ28, a été étudiée
par calorimétrie de titrage isotherme (ITC) et réaction de compétition avec différents
chélateurs du zinc (zincon, Newport green, et zinquin).
Les constantes de dissociation apparentes (Kdapp) entre Aβ et le zinc(II) (tampon Hepes 20
mM, NaCl 100 mM, pH 7,4) ainsi déterminées se situent toutes dans le bas micromolaire
(environ 1 à 20 μM), même s’il semble que les peptides les plus longs aient une affinité
légèrement plus forte pour le métal (Aβ40-42 > Aβ28 > Aβ16). L’état d’agrégation du
peptide ne semble pas influer sur le Kdapp, les résultats obtenus avec le peptide à l’état soluble,
sous forme de fibres ou préincubé en présence de zinc, ne diffèrent pas significativement. Il
apparaît également que le zinc présent dans les formes solubles et agrégées de ZnII-Aβ est
facilement accessible par les chélateurs de ce métal, ce qui indique que le développement de

Chapitre 2 : la liaison zinc(II) - Aβ
104
thérapie de chélation des métaux spécifique aux formes solubles de Zn-Aβ semble très
difficile.
Les expériences d’ITC ont également permis d’estimer qu’environ 0,9 protons par peptide
sont libérés lors de la fixation du zinc(II) à pH 7,4. Ce résultat est en accord avec la présence
de 3 ligands histidines et d’un quatrième non déprotoné lors de la fixation. Dans ce contexte,
le groupement carboxylate de l’aspartate 1 (mais pas le N-term) serait un bon candidat.
II. Publication
Zinc Binding to Amyloid-β: Isothermal Titration Calorimetry (ITC)
and Zn-Competition Experiments with Zn-Sensors§
Christine Talmard, Anaïs Bouzan, Peter Faller
Laboratoire de Chimie de Coordination, CNRS UPR 8241 ; associated with University
Toulouse III, 205, route de Narbonne, 31077 Toulouse Cedex 4 France
§ This work was supported by the grant from the French ministry: ACI-INTERFACE PCB
(DRAB). C.T. was supported by a grant from ESF.
Abbreviations: Aβ16: amyloid-β1-16 Aβ28: amyloid-β1-28 Aβ40: amyloid-β1-40 Aβ42:
amyloid-β1-42: Hepes: {2-(4-(2-Hydroxyethyl)-1-piperazinyl)ethanesulfonic acid; ITC:
isothermal titration calorimetry; Kdapp: apparent dissociation constant; Tris:
Tris(Hydroxymethyl)aminomethane.
Abstract
Aggregation of the peptide beta-amyloid (Aβ) to amyloid plaques is a key event in
Alzheimer’s disease (AD). According to the amyloid cascade hypothesis, Aβ-aggregates are
toxic to neurons via the production of reactive oxygen species and are hence directly involved in
the cause of the disease. Zinc ions play an important role, because they are able to bind to Aβ
and influence the aggregation properties. In the present work isothermal titration calorimetry
and Zn-sensors (Zincon, Newport green and zinquin) were used to investigate the interaction
of Zn with the full length Aβ1-40 and Aβ1-42, as well as the truncated Aβ1-16 and Aβ1-28.

Chapitre 2 : la liaison zinc(II) - Aβ
105
The results suggest that Zn binding to Aβ induces a release of ~0.9 protons by the peptide.
This correspond to the expected value upon Zn-binding to the three histidines and indicates
that further ligands are not deprotonated upon Zn-binding. Such behaviour is expected for
carboxylates, but not the N-terminus. Moreover, the apparent dissociation constant (Kdapp) of
Zn-binding to all forms of Aβ is in the low µM range (1-20µM) and rather independent on the
aggregation state including soluble Aβ, Aβ-fibrils or Zn-induced Aβ aggregates. Finally, Zn
in the soluble or aggregated Zn-Aβ form is relatively well accessible for Zn-chelators. The
potential repercussions on the metal chelation therapy are discussed.
Introduction
The aggregation of the peptide amyloid-β (Aβ) into fibrils is considered to be a key
event in Alzheimer’s disease (AD).(1-4) In vivo, the most prevalent forms of Aβ consist of 40
(Aβ40) and 42 amino acids (Aβ42). Aβ is cleaved off from the membrane protein amyloid
precursor protein (APP). The longer form Aβ42 is more prone to aggregation and more toxic
to neurons than Aβ40. This is in agreement with the widely accepted amyloid cascade
hypothesis, which proposes that an increased Aβ accumulation and aggregation lead to the
formation of fibrils in amyloid plaques and to neuronal disfunction and later on dementia.(2)
However, it is thought that soluble, oligomeric forms of Aβ are the most toxic species, rather
than more aggregated fibrils or protofibrils.(2) In this context, factors influencing this
aggregation are of high interest.(5) Studies in vitro, in cell cultures and AD model mice
indicate an important role for metals (Zn, Cu, and Fe) in this respect.(1, 4, 6, 7)
In the case of Zn, a large body of evidence has been accumulated supporting an
important role of this metal ion in the metabolism of Aβ linked to Alzheimer’s disease. Zn has
been found in the amyloid plaques at high concentrations (~mM) and treatment with chelators
partially solubilized the plaques.(8) APP possesses a high affinity Zn-binding site, which is
located outside of the Aβ-region.(9, 10) Transgenic mice, which express human APP, develop
amyloid plaque pathology and then can serve as models for AD. In such mice the lacking of
the Zn-transporter ZnT3 (transports Zn into synaptic vesicles) reduced the plaque load. Hence,
it was concluded that endogenous Zn contributed to the amyloid deposition in transgenic
mice.(11) Chelators, such as clioquinol, DP109 and cyclo-phen-type, known to bind Zn (and
Cu) reduced successfully the amyloid plaques burden in transgenic mice.(7, 12-15)
In vitro studies revealed that Zn binds to Aβ and promotes its aggregation.(5, 16, 17)

Chapitre 2 : la liaison zinc(II) - Aβ
106
Zn binds to Aβ in a monomeric and stoichiometric Zn-Aβ complex, which is transiently stable
prior to aggregation.(18) Moreover, Zn-binding influences also the morphology of the Aβ
aggregates. Generally their structures were described to be more amorphous, i.e. containing
no or less fibrils. Zn-induced aggregates are less reactive with the marker thioflavin T than
aggregates formed in the absence of Zn.(19-23)
The first Zn-binding site of Aβ is located in the N-terminal part. Several studies on
soluble Aβ40 or truncated forms containing the Zn-binding site (i.e. Aβ16 and Aβ28) tried to
identify the ligands of Zn in Aβ.(19, 24-34) Although there is some contradiction, the
involvement of the histidines 6, 13 and 14 has been suggested by most articles. Less
agreement is found concerning further ligands. Mostly suggested was the aspartate at position
1, either by the N-terminus or carboxylate side chain.(28, 29, 34) In the case of an acetylated
N-terminus, a NMR structure of the Zn-Aβ1-16 was solved and showed that Zn is bound to
the three His and Glu11.(31) Less is known for the Zn-binding site in Zn-induced aggregates
of Aβ. Raman spectroscopy studies indicated that it is provided by 3 His similar to soluble
Aβ.(25) In contrast EXAFS measurements detected differences between soluble and
aggregated Aβ, among others the number of His involved.(32) It has been also suggested that
the peptide aggregates through intermolecular His(N(tau))-Zn(II)-His(N(tau)) bridges.(25)
Concerning Zn-binding to Aβ fibrils formed in the absence Zn, to our best knowledge no
information is available.
A key parameter in the interaction of Zn with Aβ is the affinity for the metal. The
knowledge of the dissociation constant allows the comparison with other biological and non-
biological Zn-ligands. This can give insights into the possible sources of Zn ions able to be
chelated by Aβ, as well as which ligands (proteins or others) would be able to withdraw Zn
from Aβ. This is of particular interest concerning the metal chelation therapy. In such a
therapy, chelators should have an intermediate affinity, capable of disrupting low affinity but
pathologically relevant metal-Aβ, without disrupting higher affinity essential metal-
protein/peptide interactions. This allows specific, rather than systemic, chelation of excess
metals in the brain of AD patients.
The dissociation constant of Zn-Aβ found in the literature differed dependent on the
conditions and methods used. Initially a substoichimetric binding of Zn to Aβ40 with a Kd of
~ 100 nM and a second site of 5 µM by using a displacement assay with radioactive and cold
Zn binding to blotted Aβ40 has been reported.(17) In a subsequent study using also blotted
peptide, Clements et al. found no evidence for a sub µM binding but confirmed a Kd of ~ 5
µM for Aβ40 by their value of 3.2 µM.(35) Similar values in the low µM range (i.e. 5-10 µM)

Chapitre 2 : la liaison zinc(II) - Aβ
107
have been also reported for truncated Aβ (Aβ16) by competition with the Zn-sensor zincon
(28) and for Aβ28 by competition with Cu binding followed by fluorescence (~7µM).(34) By
using fluorescence enhancement of the single Tyr10 due to Zn-binding to Aβ different results
have been reported (although under very similar conditions, Tris-buffer, pH 7.4, 100-150 mM
NaCl). Ricchelli et al. 2005 agreed with a Kd in the low µM range for Aβ16/40/42,(36) but
Garzon et al. deduced a higher Kd of 300 µM for Aβ40 and 57 µM for Aβ42.(37) Although
generally the apparent Kd of Aβ and its truncated forms Aβ16/28 is in the low µM range, not
much is known of the influence of the aggregation state on the Kdapp, a parameter which could
explain some of the discrepancy reported.
In the present study we investigated the interaction of Zn with Aβ40 and Aβ42 as well
as the truncated forms Aβ16 and Aβ28. Isothermal titration calorimetry and competition
experiments with Zn-sensors were applied to study this interaction. In particular, the apparent
dissociation constants (Kdapp) were compared between soluble, aggregated Zn-Aβ40 and
Aβ40 fibrils. This could have importance concerning the metal chelation therapy, which is
discussed.
Material and Methods
Material: The peptides Aβ16, Aβ28, Aβ40 and Aβ42 were either purchased from EZBiolab
(Westfield, IN, USA), Geneshere Biotechnologies (Paris, France) or Activotec (Cambridge,
UK). The peptide concentrations were determined by absorption spectroscopy, by using the
well established extinction coefficient of the Tyr at 275 nm ε = 1410 cm-1 M-1. Aβ40/42
which are prone to aggregation, were monomerized according by a short incubation at pH 11-
12.(38, 39)
Aggregating concentration: In order to determine at which concentration the different Aβ
peptides aggregate in the presence of Zn, turbidity measurements at 400 nm were performed
at different concentrations over 3h (maximal time of an ITC experiment). The results showed
that Zn-Aβ16 is mainly soluble up to 60 µM, Zn-Aβ28 up to 20 µM and Aβ40 up to 10 µM.
Isothermal Titration Calorimetry (ITC). ITC experiments were performed at 298.0 ±0.1 K on
a Microcal VP-ITC calorimeter (Microcal, Northampton, MA). Ligand and receptor solutions
were prepared with the same buffer stock solution 20 mM {2-(4-(2-Hydroxyethyl)-1-
piperazinyl)ethanesulfonic acid (Hepes), Tris(Hydroxymethyl)aminomethane (Tris) or
cacodylate buffer with 100 mM NaCl at pH 7.4. All samples were degassed by stirring under

Chapitre 2 : la liaison zinc(II) - Aβ
108
vacuum before use. In the cell, the peptide solution has a concentration of 10 to 140 µM. In
each ITC experiment, about thirty injections of 7-8 µL of 150 to 800 µM zinc solution were
made into the cell containing the peptide. The syringe speed was set at 300 rpm. Control
experiments (heat dilution measurement) were performed by injected zinc solution into the
buffer, suggesting that the heat dilution was negligible for Tris buffer (∆HTris ~-0.15 kcal.mol-
1). In the case of Hepes, and cacodylate the ∆H of dilution was lower, but compared to the
measured ∆H not insignificant (However, fits of the curve showed similar Kdapp independent
if the dilution curve was subtracted or not). Thus the ITC data were corrected for the heat of
dilution of the titrant by subtracting the excess heats at high molar ratios of zinc to peptide.
Some inverse titrations (100 µM Aβ16 solution injected in 500 µM zinc solution) confirmed
the range of the reaction enthalpy values. Data were analysed with Microcal Origin 7.0, the
fitting curves were calculated according to one set of binding site model. The number of
protons nH+ released (> 0) or taken up (< 0) by the buffer during Zn-Aβ complexation is
expressed by the following equation :
ΔHbinding = ΔHreaction + nH+ ΔHionization
where ΔHbinding is the measured enthalpy of binding and ΔHreaction the intrinsic enthalpy of
reaction.(40)
Apparent dissociation constant (Kdapp): The apparent Zn-Aβ dissociation constant was
estimated by competition assay with the colorimetric Zn-chelator zincon (2-carboxy-2’-
hydroxy-5’-(sulfoformazyl)benzene), from Acros organics. It has been reported in the
literature that zincon forms a complex with ZnII in a 1 to 1 stoichiometry (ZnII– zincon),
showing a distinct absorption band at 620 nm (ε = 23200 cm-1M-1) at pH 7.4 and having an
apparent binding constant (Kapp) of 7.9×104, i.e. Kdapp of 12.6 µM (41) We verified this
parameters by titration of ZnSO4 with different concentrations of zincon (10–20 μm) under
our conditions (pH 7.4, 20 mM HEPES, 100 mM NaCl). Slighlty higher affinity was found
(Kdapp of 5.8±1.5 µM) and molecular extinction coefficient was lower than reported (15000 ±
1500 cm-1 M-1).
The binding equilibrium of ZnII between Aβ and zincon can be expressed as Equation (1):
ZnII-Aβ + zincon Aβ + ZnII-zincon (1)
The apparent binding constant of ZnII–Aβ can be calculated by resolving Equation (Zi =
zincon) (2):
(2)

Chapitre 2 : la liaison zinc(II) - Aβ
109
In the case of titration of Aβ-Zn complex with zincon, the absorption band at 620 nm is due to
the Zn–zincon complex, which increases upon addition of zincon. The tailing from the
absorption band at 470 nm due to apo-zincon was subtracted from the Zn-zincon complex
signal. Assuming that all the zinc is bound, to either zincon or Aβ, it can be deduced that, at
half maximal absorption (see Figure 4), [zincon-Zn] = [Aβ-Zn] = [Zn]total /2. [zincon] and [Aβ]
could be calculated by subtracting the Zn-bound fraction from the initial concentration (i.e.,
[zincon]= [zincon]total-[Zn–zincon] and [Aβ]=[Aβ]total-[Zn–Aβ]). Then, using the binding
constant of ZnII–zincon obtained from the titration realized the same day, the apparent binding
constant of ZnII–Aβ could be calculated.(28) In order to make sure that most Zn was bound to
the ligand, an excess of Aβ over Zn (2:1 ratio) was used as a starting point of the titration. An
analogous approach was used for the inverse titration: that is, the addition of Aβ to Zn–zincon.
Both approaches yielded similar apparent binding constants of ZnII–Aβ, indicating that
reaction 1 had reached equilibrium.
Similar approach has been used with the fluorescent sensor Newport Green DCF dipotassium
salt (NG), from Invitrogen (λex = 485 nm, λem = 530 nm).(42) Its dissociation constant with
zinc has been reported to be around 1μM,(43) which was confirmed by our experiments
(0.9±0.5×106 M-1). A concentration of 3 µM has been used in the titration experiment since
the enhancement of fluorescence of the NG-Zn complex has been found to be linear with its
concentration only up to 5 μM.
Zinquin (2-methyl-. 8-[(4-methylphenyl)sulfanylamino]-6(carbowymethyloxy)quinoline, was
purchased from Sigma-Aldrich. Excitation and emission wavelength was 370 nm and 490 nm
respectively. Zinquin was first solubilized in DMSO then in water. The concentration has
been determined using the molecular extinction coefficient of about 9000 M cm-1 at 340 nm
calculated from reference (44).
Fibrils preparation. Solution of 200 μM soluble Aβ40 in either Tris or Hepes 20 mM, NaCl
100 mM, 0.01% NaN3 at pH 7.4, have been placed at 30°C under gentle agitation for 3 weeks.
Presence of fibrils were confirmed by negative stain transmission electron microscopy and
ThT fluorescence.(45)
Results
Isothermal Titration Calorimetry (ITC): Isothermal titration calorimetry experiments can be
used to study the binding between two molecules (often called ligand and receptor).(46-48)

Chapitre 2 : la liaison zinc(II) - Aβ
110
Direct indications of three parameters can be obtained. These are the stoichiometry between
ligand and receptor, the binding constant (K) and the enthalpy (∆H). Based on the binding
constant and the enthalpy, the entropy (∆S) can be calculated. However, it is important to note
that the heat measured, and as a consequence the thermodynamic parameters (K and ∆H),
stem from the entire reaction (i.e.; from starting reagents to final product) and are thus
apparent (for more detailed discussion of ITC measurement of metal-peptide/protein
references 49-54).
As the Kd of Zn-Aβ16 has been reported to be in the low µM range, the first ITC
measurements were conducted with the recommended 10 to 100 times higher concentration.
An ITC measurement of Zn added to 140 µM Aβ16 or Aβ28 in 100 mM Tris/HCl at pH 7.4 is
shown in Figure 1.
Figure 1: ITC: Zn titration to Aβ16 (■) and Aβ28 (●) at 140 µM in 20 mM Tris buffer, 100
mM NaCl, pH 7.4.
The best fits at this concentrations revealed apparent Kd of 61 ± 4 μM and 30 ± 4 μM for
Aβ16 and 28, respectively. But they also revealed a stoichiometry around 1.5 which is above
the integer stoichiometry of the Zn-binding sites as suggested by spectroscopic data.(28, 29,

Chapitre 2 : la liaison zinc(II) - Aβ
111
33, 34) Moreover, parallel turbidity measurement revealed that under this concentration Aβ28
and also partially Aβ16 aggregate in the presence of Zn over the time period of 3h (time for
ITC experiment; for details see Materials and Methods). This means that the measured heat is
the sum of two events, i.e. sum of Zn-binding to is the peptide and the aggregation. In order to
obtain only the contribution of the Zn-binding, ITC measurements at lower concentrations of
10-20 µM Aβ16/28 were conducted (Figure 2).
Figure 2: ITC of Aβ16 (■), 28 (●) and 40 (♦) at low concentration (10 - 20 µM) in 20 mM
Tris buffer, 100 mM NaCl, pH 7.4.
Turbidity and centrifugation measurements showed that at this concentration Zn-Aβ16/28 did
not aggregate significantly. The best fit of Zn titrations to Aβ16 and 28 at lower concentration
revealed generally an integer stoichiometry, i.e. one Zn per Aβ16/28. The Kdapp obtained for
Aβ16 and Aβ 28 were 22 ± 15 and 10 ± 8µM, respectively (Table 1). However at lower
concentration the titration curve is only partially measured and hence the value obtained for
ΔH is less accurate. As shown below, we confirmed the ΔH by inverse titration, where it is
much better defined.

Chapitre 2 : la liaison zinc(II) - Aβ
112
Table 1 :Apparent Kd deduced from ITC a
Aβ16 Aβ28 Aβ40
Low concentration of Zn-Aβ (10-20 µM)
22 ± 15 µM 10 ± 8 µM 7 ± 3 µM
High concentration of Zn-Aβ (70-150 µM)
61 ± 5 µM b 30 ± 4 µM b 3 ± 2 µM
Preformed fibrils of Aβ (without Zn)
9 ± 4 µM
a The variance of the Kdapp values indicated derived from the variance of the Kdapp obtained by fitting several experiments under the same conditions. b the best fit yielded a stoichiometry of about 1.5
After these experiments with the truncated peptides Aβ16 and 28, measurements were
extended to the full length Aβ40. ITC experiments with Aβ40 were conducted at higher
(70µM) and lower (10µM) concentration, i.e. under conditions where aggregation with Zn
occurs or is almost absent. In either case the best fits exhibited an about one to one
stoichiometry. The Kdapp were in the same range: 20 ± 14 and 7 ± 3µM at high and low
concentration, respectively (Table 1). Taken together the ITC experiments indicate that the
Kdapp of Zn to Aβ40 and truncated peptides Aβ16/28 are in the low µM range. It seems that
the longer peptides bind Zn slightly stronger than the shorter, i.e. Aβ40 > Aβ28 > Aβ16.
Moreover, Zn-binding under aggregating and non-aggregating concentrations did not show
large difference, indicating that the aggregation does not contribute much to the measured
reaction heat.
The same experiments have been repeated in the presence of HEPES and cacodylate buffer at
pH 7.4 (100 mM NaCl). The results in HEPES confirmed the about one to one stoichiometry
and a Kdapp in the low µM range obtained in the Tris buffer. In the cacodylate buffer the
signal was positive (ΔH > 0) but very small. The best fits revealed Kdapp in the same range as
in the other buffers Tris and HEPES. However the stoichiometry given by the best fit was
beyond a one to one complex.
The ITC measurement of Zn titration to Aβ in different buffers allows the estimation of the
number of protons displaced by the binding of ZnII to Aβ. The displaced proton will bind to
the buffer, which have different ∆H for proton binding. A difference in ∆H of two titrations,
which differ only in their buffer, should be only due to the proton binding to the buffer. Since

Chapitre 2 : la liaison zinc(II) - Aβ
113
the ∆H of Tris, HEPES and cacodylate are known (11.51, 5.0, and -0.56 kcal/mol,
respectively),(55) the number of protons displaced by the Zn-binding has been calculated to
be 0.76 ± 0.37 from the ITC measurements (Figure 3).
-10
-8
-6
-4
-2
0
2
4
-2 0 2 4 6 8 10 12
∆H bi
ndin
g(k
cal.m
ol-1
)
∆H ionization (kcal.mol-1)
(a)
(b)
(c)
Figure 3: Enthalpy change of binding of Aβ16 to zinc at pH 7.4 as a function of the ionization
enthalpy of the reaction buffer (a: cacodylate, b: Hepes, c: Tris). The absolute value of the
slope of the linear regression yields the number of protons released by the buffer when one
zinc binds to one Aβ16. The circles correspond to the values obtained when Zn was added
(0.76 ± 0.37 proton released) and the squares when the peptide was added (0.89 ± 0.07 proton
released).
Due to the relatively low concentrations of Aβ in the ITC measurements, the ∆H deduced
from the binding isotherm is not that well defined. This is due to the fact that when Zn is
added at low Aβ concentration, Zn is in equilibrium between free and bound. To get a good
estimate of the ∆H of Zn-binding to Aβ, conditions are needed where Zn completely binds to
Aβ. This is achieved by inverse experiment, when clearly substoichiometric amounts of Aβ
are added to Zn in the buffer. Under this conditions all Aβ added will bind to Zn and the ∆H
is better defined. Such experiments were conducted in Tris, HEPES and cacodylate and
revealed ∆H of -8.9, -2.6, +1.8 kcal/mol respectively. This corresponds to a replacement of
0.89 ± 0.07 protons and hence confirming, with more accuracy, the previous value of 0.8

Chapitre 2 : la liaison zinc(II) - Aβ
114
protons displaced by the Zn-binding to Aβ. Moreover, the values for ∆H obtained by the
inverse experiment in Tris and Hepes buffers, are very similar to the ∆H obtained by the fit of
the titration curve from titration of Zn to the peptides, and hence corroborate the other
parameters of the fit, i.e. Kdapp and stoichiometry. (see above)
Determination of Kdapp of Zn-binding to soluble Aβ by competition assay followed by
absorption and fluorescence spectroscopy:In order to confirm the above measured Kdapp of
Zn-Aβ and to have the possibility to measure at lower peptide concentration and thus under
non-aggregating condition, the Kdapp was also measured by a competition assay. Apparent
binding constants can be estimated by competition with a chelator, which binding constant is
known and in the same range. The Zn-chelator zincon has been shown to be appropriate for
Zn-protein complexes (41, 56, 57) and was already successfully applied for the soluble
Aβ16.(28) Thus the same methodology was applied to measure the Kdapp of Aβ28, 40 and 42,
at a concentration where no significant aggregation was observed by turbidity for Aβ28 and
Aβ40.(i.e at 10 µM). Figure 4 showed the titration experiments followed by absorption at 620
nm. The upper panel showed the absorption at 620 nm of the Zn-zincon complex by adding
increasing amounts of Aβ40. The decrease of the absorption at 620 nm reflected the transfer
of Zn from zincon to Aβ40. About 13 µM Aβ40 were necessary to decrease the band at 620
nm by half, indicating that half of the 5 µM Zn bound to zincon was transferred to Aβ40.
Thus globally the binding constants were very similar, whereas zincon was slightly stronger
than Aβ40. The calculations lead to a Kdapp of about 6.3 µM for Zn-Aβ40. The lower panel
showed the inverse experiment, in which zincon was added to Zn-Aβ40. Here half of the 5
µM Zn was bound to zincon after adding 6 µM of zincon (thus, Kdapp.~ 9.6 µM) This
confirmed the above experiment which showed that globally the binding constants were in the
same range, zincon being a little stronger then Aβ40. The fact that the two approaches yielded
the same Kdapp (in experimental limits) indicated that the reaction of Zn exchange reached
equilibrium, a prerequisite for the calculation of Kdapp.

Chapitre 2 : la liaison zinc(II) - Aβ
115
0
20
40
60
80
100
0 10 20 30 40
% A
bs 6
20 n
m
[Zi]t (μM)
0
20
40
60
80
100
0 10 20 30 40 50
% A
bs 6
20 n
m
[Aβ40]t (μM)
Figure 4: Estimation of the Zn(II)-binding constant by competition with the chelator zincon:
Upper panel: Absorption at 620 nm of the Zn-Zincon complex upon addition of increasing
amount Aβ1-40. Conditions: zincon 10 µM; Zn 5µM; pH 7.4, 20 mM HEPES, 100 mM NaCl.
Lower panel: To the complex of Zn-Aβ1-40 increasing concentrations of zincon were added
and the absorption at 620 nm of the Zn-zincon was followed. Conditions: Aβ1-40 10 µM; Zn
50µM; pH 7.4, 20 mM HEPES, 100 mM NaCl. In ordinate is the percent of absorption at 620
nm with 100 % standing for the maximum absorption obtained (at t0 and t∞ respectively).
Analogue experiments were conducted with Aβ42, Aβ28 and Aβ16 (Table 2). In general no
large difference between the entire and truncated peptide was observed at low concentration.
The Kdapp were in the low µM range 1-20 µM, with a tendency to a higher affinity by the
longer peptides. Similar experiments were performed with the Zn-sensitive chelator Newport
green. Newport green has an apparent Kd of 1 µM.(42, 43, 58) This value was confirmed by
Zn-titration to Newport green under our conditions (20 mM Hepes buffer pH 7.4, 100mM
NaCl), which revealed a Kdapp of about 0.9 µM. In general the competition experiments
confirmed a Kdapp in the low µM range for Zn-Aβ. Also the tendency that the longer Aβ-

Chapitre 2 : la liaison zinc(II) - Aβ
116
peptides bind Zn slightly stronger was observed again.
Table 2: apparent Kd from Zincon (Zi added, [Aβ]/[Zn]=2))
Peptide Kdapp (μM)
Aβ16 14 ± 5
Aβ28 12 ± 5
Aβ40 and Aβ42
non aggregated
7 ± 3
Aβ40 Fibrils 9 ± 6
(Aβ40 or 42 + Zn) aggregated 3 hours 3 ± 2
Another widely used fluorescent Zn-chelator is zinquin. Zinquin has a apparent binding
affinity at physiological pH in the pM to low nM range, i.e. much stronger than soluble Zn-
Aβ.(44) Titration experiment of zinquin to Zn-Aβ showed that zinquin withdraws Zn
quantitatively from Aβ. In contrast, Aβ was not able to compete significantly for Zn-binding
with zinquin, even at 10-times excess of Aβ compared to zinquin. These results are in line
with the at least 100 times stronger affinity of zinquin compared to Aβ.
Zn-binding to preformed fibrils of Aβ: Next, the Zn binding to Aβ40 fibrils (incubated over 3
weeks in the absence of Zn) was measured. The ITC measurement is shown in Figure 5. The
best fits revealed an Kdapp of 9 ± 4 µM, i.e. not significantly different of the Zn-binding to
soluble Aβ (Table 1). Also the ΔH did not differ significantly. However, a change was
observed concerning the stoichiometry of the Zn-binding site, i.e. a substoichiometric binding
of ~0.2 equivalents Zn per Aβ-peptide (when using 60 μM of fibrils). This suggests that Zn
binds to only a portion of the binding-sites with a similar Kdapp. This could indicate that the
binding site is perhaps the same. The rest of the binding sites is either not accessible to Zn-
binding because they are buried in the fibril structure (ii) or did change due to structural
modifications induced by aggregation. Such changes could lead to a lower affinity not
detectable by ITC or to a Zn-binding with a very small ΔH not detectable by ITC. In order to
confirm these results the Zn-binding to the fibrils was also analysed by the competition assay

Chapitre 2 : la liaison zinc(II) - Aβ
117
with zincon, which were in agreement with a substoichiometric Zn-binding with a Kdapp in the
low µM range (not shown) (Table 2).
Figure 5: ITC of Zn titration to soluble Aβ40 (70 μM) (♦) and to fibrils of Aβ40 (60 μM) (•)
in 20 mM Tris buffer, 100 mM NaCl, pH 7.4.
Zn-binding in Zn-induced aggregates of Aβ: Zn-binding to Aβ induces rapid aggregation and
formation of Zn-Aβ aggregates. In order to measure the apparent binding affinity of the Zn in
these aggregates, they were titrated with the Zn-sensors zinquin and zincon 2 . First, the
question was addressed if the Zn in the Zn-Aβ aggregates is readily accessible to chelator-
binding. As zinquin has a much stronger (at least 100 times) apparent Zn-binding affinity than
the soluble Zn-Aβ, all Zn should be transferred to zinquin. Indeed, Figure 6 shows that
zinquin immediately bind free Zn and Zn from the soluble Zn-Aβ16 quantitatively after
mixing (i.e; in a couple of second). Addition of zinquin to Zn-Aβ40 aggregates yielded alo a
complete Zn-transfer from the Zn-Aβ aggregates to zinquin. However, in this case the transfer
was only completed after 5 minutes. This indicates that Zn transfer to zinquin is slower in Zn-
2 The opposite titration, i.e. add Zn-Aβ aggregates to the zinc sensor was avoided, because in such an experiment Zn-Aβ aggregates would be highly diluted in to the sensor solution, with the risk to disaggregate the Zn-Aβ aggregates.

Chapitre 2 : la liaison zinc(II) - Aβ
118
Aβ aggregates than in soluble Zn-Aβ.
0
10
20
30
40
50
60
70
0 5 10 15 20
% I
at 4
90 n
m
Time (min)
Figure 6: Fluorescence relative intensity of a solution of 2.1 equivalent of zinquin with 60μM
of zinc (◊), {Aβ16-Zn} (□) or {Aβ40-Zn, incubated overnight} (▲).
Moreover it also shows that zinquin has a stronger affinity than Zn-Aβ aggregates, which
limits the Kdapp of Zn in Zn-Aβ aggregates to a value in the nM range or above. To get a
better estimate of the binding affinity of Zn-Aβ aggregates, the chelator zincon and Newport
green were used. Competition experiments with zincon or Newport green suggested a small
but reproducible 2-3 fold stronger Zn-binding to the Zn-induced aggregates of Aβ28 and
Aβ40 compared to soluble Aβ28/ Aβ40.3
3 The observation that capacity of zincon or Newport green to withdraw Zn from Zn-Aβ aggregates is smaller than from soluble Zn-Aβ is assigned to a higher affinity of Zn in the aggregates (thermodynamics) and not kinetics, because i) the experiments with zinquin (see above) showed that Zn transfer is finished after a couple of minutes and ii) the reaction of addition of zincon and Newport green to Zn-Aβ was followed over 10 min and no increase in the absorption band at 620 nm (reflecting the Zn-Zincon complex) or fluorescence of Zn-Newport green was observed (such experiments are limited to the minutes time scale since the Zn-zincon and –Newport green complexes are degrading slowly with time).

Chapitre 2 : la liaison zinc(II) - Aβ
119
Discussion
The present results indicate the Kdapp of Zn-binding (pH 7.4, 0.1 M NaCl) of different Aβ-
forms are relatively similar, i.e. all in the low µM range (3-15 µM). No indication was found
for the initially reported high affinity binding site (Kd of 104 nM).(17) But the present Kdapp
agrees with values reported for truncated, soluble Aβ16/28.(28, 29, 34) No significant
difference was observed at non-aggregating and aggregating concentration. This includes Zn-
induced Aβ40 aggregates as well as Zn-binding to fibrils formed in the absence of Zn. This
suggests that the affinity of the Zn-binding site is similar in the soluble and the different
aggregated forms. Thus it is well possible that the Zn-binding site is the same in aggregated
and non-aggregated Aβ including the same ligands (3His, Asp1 or Glu11).4
Although no significant difference in Zn affinity between soluble and aggregated Aβ could be
observed, other parameters showed differences. First in the case of the Zn-induced Aβ
aggregates, the kinetics of Zn-transfer from the aggregate to the zinc-sensor was substantially
slower. Two scenarios could explain this slower kinetics i) The Zn-sites are buried in the
aggregate and hence it takes more time to transfer zinc to the sensor ii) as the aggregates are
normally in equilibrium with soluble Aβ,(59, 60) the sensor withdraw Zn rapidly from the
soluble fraction but slower or not from the aggregates. This provokes resolubilisation of Zn-
Aβ from the aggregates to reach equilibrium of soluble and aggregated Zn-Aβ. In this case the
kinetics would reflect the resolubilisation of the Zn-Aβ aggregates.
The second remarkable difference was observed between Zn-binding to soluble Aβ and
preformed Aβ fibrils. Soluble Aβ bound about one equivalent of Zn, the preformed fibrils
bound only a substoichiometric (i.e. ~0.2) amount of Zn, indicating that not all binding sites
were accessible (or existing).
The finding that the Kdapp of Zn to Aβ is in the low µM range independent of its aggregation
state has implication for the so called metal-chelation therapy. The concept of this therapy
consists of chelating the metal ions bound to Aβ and favour the disaggregation from toxic
aggregates forms (e. g. oligomers) to non-toxic monomers. Several chelators of this type have
been developed or have entered clinical trials, like clioquinol,(12) DP-109,(13) bifunctional
metal chelator.(61) Such chelators should have an intermediate affinity, capable of disrupting
4 Supposing that the Zn-binding site is the same in the soluble monomeric and aggregated Aβ would more point to a mechanism where Zn binds to Aβ as a monomeric complex and induces a conformational change, which favours the aggregation by pure peptide-peptide contacts. It points less to a mechanism where Zn-Aβ is first a soluble monomeric complex and aggregation is induced by Zn forming a bridge between two molecules of Aβ, because this would include a change in the coordination (for discussion of the two mechanisms see Talmard et al. 2007).(18)

Chapitre 2 : la liaison zinc(II) - Aβ
120
low affinity but pathologically relevant metal-Aβ, but do not withdraw higher affinity
essential metal-protein/peptide interactions. This allows specific, rather than systemic,
chelation of excess metals in the brain of AD patients. The Kd of Clioquinol at pH 7.4 has
been determined.(62) An apparent Kd value of 1.4 10-9 M has been reported at concentrations
of 1µM Zn and 200 µM Clioquinol. However, since clioquinol forms a 2:1 complex with
Zn,(63) the apparent Kd is dependent on the clioquinol concentration, for which 200µM is
rather high for physiological conditions. A Kd of 4.10-8 M (pH 7.0) was determined for Zn-
DP-109 (Jonathan Friedman, personal communication). Our results indicate that concerning
Zn, a Kdapp of about 1µM is enough to withdraw the majority of the Zn from Aβ, independent
of the aggregation state. Moreover, a chelator with a Kd in this range is unlikely to withdraw
Zn from proteins and enzymes as they have generally a stronger Zn affinity.(64-66)
ITC proton displacement: ITC measurement indicated that Zn-binding to Aβ leads to the
release of 0.8-0.9 protons. It is relatively well determined from the literature that Zn binds to
the 3 His. As a fourth ligand, aspartate at position 1, either by the N-terminus or carboxylate
side chain, is the most reported.(28, 29, 34) However, the carboxylate of Glu11 has also been
suggested (31). As Asp and Glu are normally deprotonated at pH 7.4, Zn-binding does not
lead to a proton release. In contrast, the pKa of the N-terminus of a peptide is normally
around 7.8, a value confirmed by 8.0 deduced from potentiometric measurements.(67) Thus
Zn-binding to the N-terminus would lead to a release of 0.8 protons at pH 7.4. This would in
principle allows the distinction between Zn-binding to a carboxylate and to the N-terminus, if
the pKa of the 3 His are known as well. Unfortunately, two quite different sets of data are
available for pKa of His 6, 13 and 14. NMR measurements on acetylated Aβ16 determined
pKa of 7.0, 6,9 and 6.8 respectively. Potentiometric measurement suggested pKa of 7.0, 6.5,
and 5.7 for non-acetylated Aβ16. On the basis of the NMR data, the calculation gives 0.7 ±
0.3 protons released by Zn-binding to the three His in agreement with our measurement.
When N-terminal binding is added, 1.5± 0.4 protons are calculated to be released, which is
not in agreement with the data. For the potentiometric set, Zn-binding to only 3 His gives 0.4
± 0.3 protons replaced, when including N-terminus as ligand, 1.2 ± 0.4 protons. So these pKa
deduced from potentiometry are more in favour of Zn-binding to the N-terminus. However, in
our previous publication,(28) we found chemical shifts of the three His at different pH for the
non-acetylated Aβ16 almost identical to those reported for the acetylated Aβ16, indicating
that under our conditions the pKa of the three His correspond to 6.9 ± 0.1. Thus it seems that

Chapitre 2 : la liaison zinc(II) - Aβ
121
the measured release of 0.89 protons by Zn-binding to Aβ is entirely due to the binding to the
3 His. It suggests that no other proton is released, which is in favour of binding to a
carboxylate rather than the N-terminus (or tyrosine). 5
ITC comparison between Cu and Zn: We previously reported ITC measurement on the
interaction of Cu2+ with Aβ16 and Aβ28.(53) Cu is supposed to bind to the same three His as
Zn in a square planar geometry. The fourth ligand is likely to be Asp1 (either by N-terminus
and carboxylate). Cu binding was stronger than Zn binding and it was more dependent on the
buffer (see table 1). In Hepes Kdapp of Cu was ~0.1 µM compared to ~10 µM for Zn, whereas
in Tris Kdapp of Cu was only ~1µM. Not only the Kdapp were different, so also did ΔH and ΔS
(Table 3).
Table 3: Comparison of the thermodynamic parameters, obtained by ITC, of the Zn-Aβ16 and
Cu-Aβ16 interaction.
metal Kd (μM) ∆HTris (kcal.mol-1)
∆STris (cal.K-1.mol-1)
∆HHepes (kcal.mol-1)
∆SHepes (cal.K-1.mol-1)
Zn(II) 22 ± 15 -8.4 ± 4.0 -8 ± 15 -2.3 ± 0.2 18 ± 7
Cu(II) 0.1 -3 12 -0.5 31
In the case of Zn-binding ΔS, even if not very accurate, was clearly smaller than for Cu-
binding. The difference of ΔH was less striking, but Zn-binding had a little more negative ΔH.
This means that for Zn-binding the enthalpic contribution is slightly higher and the entropic
lower compared to Cu-binding. Or in other words, it is principally the higher entropic
contribution which is responsible for the higher affinity of Aβ towards Cu (compared to Zn).
The higher entropic contribution could be due to (i) release of more water molecules from the
hydrated CuII upon CuII-Aβ formation. (ii) release of more water molecules from the peptide
hydration or (iii) conformational changes, i.e. less degree of freedom upon binding of CuII or
(iv) entropic changes due to metal-buffer interaction (before complex formation). In particular,
5 The suggestion that Zn is not binding predominantly to the N-terminus is not in contradiction with the NMR measurements, which showed a line-broadening of the resonances of the N-terminal Asp upon Zn-binding. Transiently and partial Zn-binding could be sufficient to explain the line-broadening in NMR, but would not lead to substantial proton release in ITC.

Chapitre 2 : la liaison zinc(II) - Aβ
122
assumption (i) is likely to contribute to the higher entropic contribution of Cu versus Zn-
binding. Hydrated Cu is likely to loose all its water upon binding to Aβ, since no indication
for a labile ligand to the square planar Cu in Cu-Aβ was found.(53) In contrast, Zn, which is
most likely bound to the same four amino acids, prefers penta- and hexacoordination and is
thus likely to keep one or two water molecules upon binding to Aβ. This is supported by the
~13-20 cal mol-1K-1 higher value of ΔS for Cu than Zn-binding (see table 3), and by the fact
that a value of 9.5 calmol-1K-1 per water released from a metal ion has been reported.(52)
Conclusions In conclusion, the present work shows evidence that the apparent Zn-binding affinity to Aβ40
and Aβ42 is in the low µM range and is not much dependent on the aggregation state of
Aβ, i.e. soluble Aβ, Aβ-fibrils or Zn-induced Aβ. Moreover, Zn in the soluble or aggregated
Zn-Aβ form is relatively well accessible for Zn-chelators. This suggests that chelators, i.e. in
the metal chelation therapy, withdraw Zn concomitantly from soluble and aggregated Zn-Aβ,
and specific chelation only from soluble Zn-Aβ seems very difficult.
Acknowledgements:
We wish to thank Dr. D. Fournier (IPBS, Toulouse) for access to the microcalorimeter.

Chapitre 2 : la liaison zinc(II) - Aβ
123
References (1) Suh, Y. H., and Checler, F. (2002) Amyloid precursor protein, presenilins, and alpha-
synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev 54, 469-525.
(2) Hardy, J., and Selkoe, D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353-356.
(3) Klein, W. L., Stine, W. B., Jr., and Teplow, D. B. (2004) Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging 25, 569-580.
(4) Multhaup, G., and Masters, C. L. (1999) Metal binding and radical generation of proteins in human neurological diseases and aging. Met Ions Biol Syst 36, 365-387.
(5) Bush, A. I., Pettingell, W. H., Multhaup, G., d Paradis, M., Vonsattel, J. P., Gusella, J. F., Beyreuther, K., Masters, C. L., and Tanzi, R. E. (1994) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265, 1464-1467.
(6) Selkoe, D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81, 741-766.
(7) Bush, A. I. (2003) The metallobiology of Alzheimer's disease. Trends Neurosci 26, 207-214.
(8) Cherny, R. A., Legg, J. T., McLean, C. A., Fairlie, D. P., Huang, X., Atwood, C. S., Beyreuther, K., Tanzi, R. E., Masters, C. L., and Bush, A. I. (1999) Aqueous dissolution of Alzheimer's disease Abeta amyloid deposits by biometal depletion. J Biol Chem 274, 23223-23228.
(9) Bush, A. I., Multhaup, G., Moir, R. D., Williamson, T. G., Small, D. H., Rumble, B., Pollwein, P., Beyreuther, K., and Masters, C. L. (1993) A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer's disease. J Biol Chem 268, 16109-16112.
(10) Ciuculescu, E. D., Mekmouche, Y., and Faller, P. (2005) Metal-binding properties of the peptide APP170-188: a model of the ZnII-binding site of amyloid precursor protein (APP). Chemistry 11, 903-909.
(11) Lee, J. Y., Cole, T. B., Palmiter, R. D., Suh, S. W., and Koh, J. Y. (2002) Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A 99, 7705-7710.
(12) Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., McLean, C. A., Barnham, K. J., Volitakis, I., Fraser, F. W., Kim, Y., Huang, X., Goldstein, L. E., Moir, R. D., Lim, J. T., Beyreuther, K., Zheng, H., Tanzi, R. E., Masters, C. L., and Bush, A. I. (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665-676.
(13) Lee, J. Y., Friedman, J. E., Angel, I., Kozak, A., and Koh, J. Y. (2004) The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol Aging 25, 1315-1321.
(14) Adlard, P. A., and Bush, A. I. (2006) Metals and Alzheimer's disease. J Alzheimers Dis 10, 145-163.
(15) Boldron, C., Van der Auwera, I., Deraeve, C., Gornitzka, H., Wera, S., Pitie, M., Van Leuven, F., and Meunier, B. (2005) Preparation of cyclo-phen-type ligands: chelators of metal ions as potential therapeutic agents in the treatment of neurodegenerative diseases. Chembiochem 6, 1976-1980.
(16) Mantyh, P. W., Ghilardi, J. R., Rogers, S., DeMaster, E., Allen, C. J., Stimson, E. R., and Maggio, J. E. (1993) Aluminum, iron, and zinc ions promote aggregation of

Chapitre 2 : la liaison zinc(II) - Aβ
124
physiological concentrations of beta-amyloid peptide. J Neurochem 61, 1171-1174. (17) Bush, A. I., Pettingell, W. H., Jr., Paradis, M. D., and Tanzi, R. E. (1994) Modulation
of A beta adhesiveness and secretase site cleavage by zinc. J Biol Chem 269, 12152-12158.
(18) Talmard, C., Guilloreau, L., Coppel, Y., Mazarguil, H., and Faller, P. (2007) Amyloid-beta peptide forms monomeric complexes with Cu(II) and Zn(II) prior to aggregation. Chembiochem 8, 163-165.
(19) Yang, D. S., McLaurin, J., Qin, K., Westaway, D., and Fraser, P. E. (2000) Examining the zinc binding site of the amyloid-beta peptide. Eur J Biochem 267, 6692-6698.
(20) Raman, B., Ban, T., Yamaguchi, K., Sakai, M., Kawai, T., Naiki, H., and Goto, Y. (2005) Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide. J Biol Chem 280, 16157-16162.
(21) Klug, G. M., Losic, D., Subasinghe, S. S., Aguilar, M. I., Martin, L. L., and Small, D. H. (2003) Beta-amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous ageing at neutral pH. Eur J Biochem 270, 4282-4293.
(22) Yoshiike, Y., Tanemura, K., Murayama, O., Akagi, T., Murayama, M., Sato, S., Sun, X., Tanaka, N., and Takashima, A. (2001) New insights on how metals disrupt amyloid beta-aggregation and their effects on amyloid-beta cytotoxicity. J Biol Chem 276, 32293-32299.
(23) House, E., Collingwood, J., Khan, A., Korchazkina, O., Berthon, G., and Exley, C. (2004) Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J Alzheimers Dis 6, 291-301.
(24) Liu, S. T., Howlett, G., and Barrow, C. J. (1999) Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A beta peptide of Alzheimer's disease. Biochemistry 38, 9373-9378.
(25) Miura, T., Suzuki, K., Kohata, N., and Takeuchi, H. (2000) Metal binding modes of Alzheimer's amyloid beta-peptide in insoluble aggregates and soluble complexes. Biochemistry 39, 7024-7031.
(26) Curtain, C. C., Ali, F., Volitakis, I., Cherny, R. A., Norton, R. S., Beyreuther, K., Barrow, C. J., Masters, C. L., Bush, A. I., and Barnham, K. J. (2001) Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 276, 20466-20473.
(27) Kozin, S. A., Zirah, S., Rebuffat, S., Hoa, G. H., and Debey, P. (2001) Zinc binding to Alzheimer's Abeta(1-16) peptide results in stable soluble complex. Biochem Biophys Res Commun 285, 959-964.
(28) Mekmouche, Y., Coppel, Y., Hochgrafe, K., Guilloreau, L., Talmard, C., Mazarguil, H., and Faller, P. (2005) Characterization of the ZnII binding to the peptide amyloid-beta1-16 linked to Alzheimer's disease. Chembiochem 6, 1663-1671.
(29) Syme, C. D., and Viles, J. H. (2006) Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer's disease. Biochim Biophys Acta 1764, 246-256.
(30) Zirah, S., Rebuffat, S., Kozin, S. A., Debey, P., Fournier, F., Lesage, D., and Tabet, J.-C. (2003) Zinc binding properties of the amyloid fragment Aβ(1-16) studied by electrospray-ionization mass spectrometry. International Journal of Mass Spectrometry 228, 999-1016.
(31) Zirah, S., Kozin, S. A., Mazur, A. K., Blond, A., Cheminant, M., Segalas-Milazzo, I., Debey, P., and Rebuffat, S. (2006) Structural changes of region 1-16 of the Alzheimer

Chapitre 2 : la liaison zinc(II) - Aβ
125
disease amyloid beta-peptide upon zinc binding and in vitro aging. J Biol Chem 281, 2151-2161.
(32) Stellato, F., Menestrina, G., Serra, M. D., Potrich, C., Tomazzolli, R., Meyer-Klaucke, W., and Morante, S. (2006) Metal binding in amyloid beta-peptides shows intra- and inter-peptide coordination modes. Eur Biophys J 35, 340-351.
(33) Hou, L., and Zagorski, M. G. (2006) NMR reveals anomalous copper(II) binding to the amyloid Abeta peptide of Alzheimer's disease. J Am Chem Soc 128, 9260-9261.
(34) Danielsson, J., Pierattelli, R., Banci, L., and Graslund, A. (2007) High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid beta-peptide. Febs J 274, 46-59.
(35) Clements, A., Allsop, D., Walsh, D. M., and Williams, C. H. (1996) Aggregation and metal-binding properties of mutant forms of the amyloid A beta peptide of Alzheimer's disease. J Neurochem 66, 740-747.
(36) Ricchelli, F., Drago, D., Filippi, B., Tognon, G., and Zatta, P. (2005) Aluminum-triggered structural modifications and aggregation of beta-amyloids. Cell Mol Life Sci 62, 1724-1733.
(37) Garzon-Rodriguez, W., Yatsimirsky, A. K., and Glabe, C. G. (1999) Binding of Zn(II), Cu(II), and Fe(II) ions to Alzheimer's A beta peptide studied by fluorescence. Bioorg Med Chem Lett 9, 2243-2248.
(38) Hou, L., Shao, H., Zhang, Y., Li, H., Menon, N. K., Neuhaus, E. B., Brewer, J. M., Byeon, I. J., Ray, D. G., Vitek, M. P., Iwashita, T., Makula, R. A., Przybyla, A. B., and Zagorski, M. G. (2004) Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 126, 1992-2005.
(39) Fezoui, Y., Hartley, D. M., Harper, J. D., Khurana, R., Walsh, D. M., Condron, M. M., Selkoe, D. J., Lansbury, P. T., Jr., Fink, A. L., and Teplow, D. B. (2000) An improved method of preparing the amyloid beta-protein for fibrillogenesis and neurotoxicity experiments. Amyloid 7, 166-178.
(40) Pierce, M. M., Raman, C. S., and Nall, B. T. (1999) Isothermal titration calorimetry of protein-protein interactions. Methods 19, 213-221.
(41) Shaw, C. F., Laib, J. E., Sevas, M. M., and Petering, D. H. (1990) Inorg. Chem. 29, 403-408.
(42) Lukowiak, B., Vandewalle, B., Riachy, R., Kerr-Conte, J., Gmyr, V., Belaich, S., Lefebvre, J., and Pattou, F. (2001) Identification and purification of functional human beta-cells by a new specific zinc-fluorescent probe. J Histochem Cytochem 49, 519-528.
(43) Jiang, P., and Gui, Z. (2004) Florescent detection of zinc in biological systems: recent develpoment on the design of chemosensors and biosensors. Coord. Chem. Rev. 248, 205-229.
(44) fahrni, C. J., and O'Halloran. (1999) Aqueous Coordination Chemistry of Quinoline-Based Fluorescence Probes for the Biological Chemistry of Zinc. J. Am. Chem. Soc. 121, 11448-11458.
(45) Nilsson, M. R. (2004) Techniques to study amyloid fibril formation in vitro. Methods 34, 151-160.
(46) Ababou, A., and Ladbury, J. E. (2007) Survey of the year 2005: literature on applications of isothermal titration calorimetry. J Mol Recognit 20, 4-14.
(47) Privalov, P. L., and Dragan, A. I. (2007) Microcalorimetry of biological macromolecules. Biophys Chem 126, 16-24.
(48) Ladbury, J. E. (2004) Application of isothermal titration calorimetry in the biological sciences: things are heating up! Biotechniques 37, 885-887.

Chapitre 2 : la liaison zinc(II) - Aβ
126
(49) Atwood, C. S., Scarpa, R. C., Huang, X., Moir, R. D., Jones, W. D., Fairlie, D. P., Tanzi, R. E., and Bush, A. I. (2000) Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neurochem 75, 1219-1233.
(50) Zhang, Y., and Wilcox, D. E. (2002) Thermodynamic and spectroscopic study of Cu(II) and Ni(II) binding to bovine serum albumin. J Biol Inorg Chem 7, 327-337.
(51) Blasie, C. A., and Berg, J. M. (2002) Structure-based thermodynamic analysis of a coupled metal binding-protein folding reaction involving a zinc finger peptide. Biochemistry 41, 15068-15073.
(52) Blasie, C. A., and Berg, J. M. (2003) Kinetics and thermodynamics of copper(II) binding to apoazurin. J Am Chem Soc 125, 6866-6867.
(53) Guilloreau, L., Damian, L., Coppel, Y., Mazarguil, H., Winterhalter, M., and Faller, P. (2006) Structural and thermodynamical properties of CuII amyloid-beta16/28 complexes associated with Alzheimer's disease. J Biol Inorg Chem 11, 1024-1038.
(54) Zhang, Y., Akilesh, S., and Wilcox, D. E. (2000) Isothermal titration calorimetry measurements of Ni(II) and Cu(II) binding to His, GlyGlyHis, HisGlyHis, and bovine serum albumin: a critical evaluation. Inorg Chem 39, 3057-3064.
(55) Bradshaw, J. M., and Waksman, G. (1998) Calorimetric investigation of proton linkage by monitoring both the enthalpy and association constant of binding: application to the interaction of the Src SH2 domain with a high-affinity tyrosyl phosphopeptide. Biochemistry 37, 15400-15407.
(56) Huang, M., Krepkiy, D., Hu, W., and Petering, D. H. (2004) Zn-, Cd-, and Pb-transcription factor IIIA: properties, DNA binding, and comparison with TFIIIA-finger 3 metal complexes. J Inorg Biochem 98, 775-785.
(57) Armas, A., Sonois, V., Mothes, E., Mazarguil, H., and Faller, P. (2006) Zinc(II) binds to the neuroprotective peptide humanin. J Inorg Biochem 100, 1672-1678.
(58) Thompson, R. B., Peterson, D., Mahoney, W., Cramer, M., Maliwal, B. P., Suh, S. W., Frederickson, C., Fierke, C., and Herman, P. (2002) Fluorescent zinc indicators for neurobiology. J Neurosci Methods 118, 63-75.
(59) Wetzel, R. (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39, 671-679.
(60) Sengupta, P., Garai, K., Sahoo, B., Shi, Y., Callaway, D. J., and Maiti, S. (2003) The amyloid beta peptide (Abeta(1-40)) is thermodynamically soluble at physiological concentrations. Biochemistry 42, 10506-10513.
(61) Dedeoglu, A., Cormier, K., Payton, S., Tseitlin, K. A., Kremsky, J. N., Lai, L., Li, X., Moir, R. D., Tanzi, R. E., Bush, A. I., Kowall, N. W., Rogers, J. T., and Huang, X. (2004) Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer's amyloidogenesis. Exp Gerontol 39, 1641-1649.
(62) Ferrada, E., Arancibia, V., Loeb, B., Norambuena, E., Olea-Azar, C., and Huidobro-Toro, J. P. (2007) Stoichiometry and conditional stability constants of Cu(II) or Zn(II) clioquinol complexes; implications for Alzheimer's and Huntington's disease therapy. Neurotoxicology 28, 445-449.
(63) Di Vaira, M., Bazzicalupi, C., Orioli, P., Messori, L., Bruni, B., and Zatta, P. (2004) Clioquinol, a drug for Alzheimer's disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg Chem 43, 3795-3797.
(64) Blasie, C. A., and Berg, J. M. (2004) Entropy-enthalpy compensation in ionic interactions probed in a zinc finger peptide. Biochemistry 43, 10600-10604.
(65) Krezel, A., and Maret, W. (2006) Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem 11, 1049-1062.

Chapitre 2 : la liaison zinc(II) - Aβ
127
(66) Walkup, G. K., and imperiali, B. (1997) Fluorescent Chemosensors for Divalent Zinc Based on Zinc Finger Domains. Enhanced Oxidative Stability, Metal Binding Affinity, and Structural and Functional Characterization. J. Am. Chem. Soc. 119, 3443-3450.
(67) Kowalik-Jankowska, T., Ruta, M., Wisniewska, K., and Lankiewicz, L. (2003) Coordination abilities of the 1-16 and 1-28 fragments of beta-amyloid peptide towards copper(II) ions: a combined potentiometric and spectroscopic study. J Inorg Biochem 95, 270-282.

Chapitre 2 : la liaison zinc(II) - Aβ
128
III. Complément : principe de la calorimétrie de titrage isotherme
Lorsque deux molécules interagissent (dans le contexte de l’ITC, on les nomme généralement
ligand et récepteur) la réaction s'accompagne d'une libération ou d'une absorption de chaleur
selon le signe de l'enthalpie standard ΔH° de la fixation. Les microcalorimètres modernes
peuvent mesurer de très faibles variations de chaleur et en déduire, dans des conditions
adaptées, les paramètres thermodynamiques de la réaction. Ainsi la calorimétrie de titrage
isotherme (Isothermal Titration Calorimetry, ITC) permet d’étudier l’affinité d’un ligand pour
un récepteur de manière précise. Le principe est le suivant : le récepteur est placé dans la
cellule appropriée (sample cell sur la figure 2.1) situé dans l’enceinte adiabatique, le ligand
placé dans la seringue est ajouté par petits volumes successifs, entre chaque injection
l'appareil détermine la chaleur dégagée ou absorbée puis la compense pour maintenir la
température constante.
Figure 2.1. Schéma constitutif du microcalorimètre de titrage isotherme VP-ITC (Microcal).
Cette méthode non destructive permet, si l’enthalpie de réaction globale dans le tampon utilisé
Système d’injection et d’agitation
Enceinte adiabatique contenant le système
de mesure

Chapitre 2 : la liaison zinc(II) - Aβ
129
est non nulle, de déterminer des constantes d’association comprises entre 102 et 109 M-1.
Le principe des mesures est le suivant : soit [M] la concentration en macromolécule, [X] en
ligand et [MX] en complexe, à l’équilibre. On considère une réaction de stœchiométrie 1 :1,
où Ka est la constante d’équilibre de formation du complexe.
M + X MX ]].[[
][XM
MXKa =
La constante d’équilibre est exprimée en fonction des concentrations totales Mtot et Xtot qui
sont les seules connues.
][][ MMXMtot +=
][][ XMXX tot += d’où ( )( )][.][][
MXMMXXMXK
tottoa −−=
Après réarrangement, on obtient l’équation du second degré suivante :
0.].[1][ 2 =+⎟⎟⎠
⎞⎜⎜⎝
⎛++− tottot
atottot XMMX
KXMMX
Dont les deux racines sont :
2
.411
][
2
totXM
KXM
KXM
MXtot
atottot
atottot −⎟⎟
⎠
⎞⎜⎜⎝
⎛++±++
=
En dérivant la racine positive par rapport à Xtot :
2
2
2
.11
.112
22.11
1
21][
⎟⎟⎠
⎞⎜⎜⎝
⎛++⎟⎟
⎠
⎞⎜⎜⎝
⎛−−
−+
−+=
totatotatot
tot
tot
tot
tot
tottota
tot
MKMKMX
MX
MXMK
dXMXd (*)

Chapitre 2 : la liaison zinc(II) - Aβ
130
Or la quantité de chaleur Q dégagée ou absorbée à la saturation pendant une expérience ITC
peut être exprimée ainsi (soit V0 volume utile de la cellule et ΔH° enthalpie standard de
réaction en J.mol-1) :
De manière générale :
dH = dU + d(PV) = δQ + δW + d(PV) = δQ + VdP
Mais la réaction est isobare donc on peut simplifier :
dH = δQ d’où ΔH = Q
Or, dans les expériences ITC, Q mesuré est en joules et ΔH° doit être obtenue en
joules par mole de réaction (c’est-à-dire par mole de complexe formé) donc :
Q = ΔH°.[MX].V0
Soit, en prenant l’expression dérivée, avec ΔH° etV0 constants :
dQ = d(ΔH°.[MX].V0) = ΔH°.V0.d[MX]
En remplaçant d[MX] par son expression dans (*), il vient :
⎥⎥⎥⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢⎢⎢⎢
⎣
⎡
⎟⎟⎠
⎞⎜⎜⎝
⎛++⎟⎟
⎠
⎞⎜⎜⎝
⎛−−
−+
−+°Δ=
2
2
20
.11
.112
22.11
1
21.1
totatotatot
tot
tot
tot
tot
tottota
tot
MKMKMX
MX
MXMK
HdXdQ
V
Le paramètre expérimental mesuré en ITC est la dérivée de la chaleur par rapport à la
concentration totale de ligand. Ce paramètre dépend de la valeur de Mtot en relation avec Ka et
Xtot.6
Le terme Xtot / Mtot représente le rapport molaire qui augmente au cours de la titration, tandis
que KaMtot est défini comme le paramètre c, sans dimension, qui détermine l’allure de la
courbe de titration 1/ V0 (dQ/dXtot) en fonction du rapport molaire Xtot / Mtot (Figure 2.2).
Cette courbe porte le nom d’isotherme de réaction et a l’allure d’une sigmoïde. Le point
d’inflexion de la courbe indique la valeur du coefficient stoechiométrique n, la différence
entre les deux plateaux permet la détermination de l’enthalpie globale de la réaction ΔH° et la
constante d’affinité est fonction de la pente de la sigmoïde. 6 T. Wiseman, S. Williston, J. F. Brandts, L. N. Lin (1989) Anal Biochem.179(1):131-7.

Chapitre 2 : la liaison zinc(II) - Aβ
131
Figure 2.2. Allures des isothermes de réaction obtenues lors d’une expérience ITC, en
fonction de la valeur du paramètre c. Ainsi, pour une concentration donnée, plus l’affinité est forte, plus la courbe de titrage sera marquée. Une valeur de c élevée (>100) permet une
définition précise de ΔH°, tandis que la détermination de la constante d’affinité requiert une valeur de c intermédiaire (idéalement autour de 40).
Le problème majeur qui s’est posé lors de la détermination des paramètres thermodynamiques
relatifs à l’interaction Aβ-Zn a été l’agrégation importante de ce complexe, empêchant ainsi
l’utilisation des concentrations appropriées pour l’obtention d’une courbe de titrage nette (c
compris entre 5 et 20 suivant les peptides). Il en résulte une incertitude plus grande que celle
généralement obtenue avec l’ITC, en particulier dans l’estimation de l’enthalpie standard
globale de réaction. Ce phénomène étant d’autant plus marqué que la valeur absolue de cette
grandeur est faible : |ΔH°Tris| > |ΔH°Hepes| > |ΔH°Cacodyl| (en effet la libération de protons par le
peptide lors de l’interaction Aβ-Zn implique la présence d’un terme d’enthalpie d’ionisation
du solvant dans l’expression de l’enthalpie globale de la réaction).
Cette propriété de l’association a d’ailleurs permis, en utilisant des tampons avec des
enthalpies d’ionisation différentes, de déterminer le nombre de protons échangés par mole de
réaction. En effet, le nombre de protons libérés (>0) ou captés (<0) par le tampon durant la
complexation du zinc au peptide peut être exprimé suivant l’équation suivante :
ΔHbinding = ΔHreaction + nH+ ΔHionization
où ΔHbinding est l’enthalpie mesuré lors de la réaction et ΔHreaction l’enthalpie intrinsèque de la

Chapitre 2 : la liaison zinc(II) - Aβ
132
complexation.7
D’un point de vue pratique, les expériences ont été réalisées à 298,0 ±0,1 K sur un calorimètre
VP-ITC de Microcal (Nortampton, MA). Les solutions de peptides et métaux ont été
préparées à partir de la même solution stock de tampon (20 mM acide [(hydroxy-2-éthyl)-4-
pipérazinyl-1]-2éthanesulfonique (Hepes, Flucka), Tris(Hydroxymethyl)aminométhane (Tris,
Fluka) ou diméthylarseniate de sodium trihydraté (cacodylate, Acros), avec 100 mM NaCl à
pH 7.4, Figure 2.3).
Figure 2.3. Formules des tampons Hepes, Tris et cacodylate de sodium.
Tous les échantillons ont été préalablement dégazés avant utilisation. La cellule de référence
contenait toujours de l’eau ultra pure changée au moins une fois par semaine. Dans la cellule
de mesure, le peptide a une concentration comprise entre 10 et 140 μM. Une expérience ITC
englobe une quarantaine d’injections de ligand (7-8 μL de 150 à 1000 μM). La vitesse
d’agitation a toujours été fixée à 300 rpm. Des expériences de contrôle consistant à injecter le
ligand dans du tampon montrent que la chaleur de dilution est négligeable dans le cas du Tris
(∆HTris ~-0.15 kcal.mol-1). Pour les deux autres tampons la chaleur de dilution, bien
qu’inférieure, n’est pas négligeable comparée aux enthalpies de réaction. Cependant le fit des
courbes donne des valeurs de constante d’association similaires avec et sans soustraction de la
courbe de dilution. Ainsi les courbes ont été traitées en soustrayant à tous les points de la
courbe la chaleur de dilution (valeur correspondante à l’enthalpie mesurée ou extrapolée pour
une grande valeur du rapport [métal]/[peptide]). Quelques titrages inverses ont également été
réalisés (100 μM Aβ16 injecté dans 500 μM de solution de zinc(II)) et confirment l’ordre de
grandeur des enthalpies de réaction obtenues par titrage direct. Les données ont été analysées
avec le logiciel fourni avec l’appareil (Microcal Origin 7.0) et les courbes expérimentales ont
été lissées suivant le modèle à un site de fixation.
7 M. M. Pierce, C. S. Raman, and B. T. Nall (1999) Methods 19, 213-21.

133
Chapitre 3
Etude de l’oligomérisation du peptide
amyloïde bêta durant son agrégation en
présence de zinc(II) ou de cuivre(II)

134

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
135
I. Contexte bibliographique et étude de l’influence des métaux sur le degré d’agrégation
du peptide amyloïde bêta
Comme énoncé précédemment dans le premier chapitre, l’étude des mécanismes d’agrégation
d’Aβ est primordiale pour l’élucidation des processus de développement de la MA (Multhaup
and Masters 1999; Hardy and Selkoe 2002; Suh and Checler 2002; Klein, Stine et al. 2004;
Maynard, Bush et al. 2005). D’autant plus que les dysfonctionnements neuronaux semblent
être majoritairement le fruit d’agrégats intermédiaires plutôt que des fibres matures.
Cependant, autant les fibres que les petits oligomères ont été décrits comme étant
neurotoxiques (Hardy and Selkoe 2002). Plusieurs intermédiaires de l’agrégation d’Aβ ont été
observés, allant du dimère à de larges agrégats d’environ 10 nm de diamètres (Klein, Stine et
al. 2004). L’étude des conditions influençant l’agrégation et la formation d’intermédiaires est
donc d’un grand intérêt et, en ce sens, nous nous sommes intéressés à l’influence du zinc et du
cuivre, retrouvés in vivo particulièrement concentrés dans les produits finaux de l’agrégation,
les plaques amyloïdes (Multhaup and Masters 1999; Maynard, Bush et al. 2005).
En effet, ces métaux, qui se lient à la partie N-terminale du peptide (acides aminés 1-16),
influent sur l’agrégation d’Aβ : le zinc (II) semble clairement l’accélérer, tandis que le cuivre
(II) a été rapporté tantôt accélérer, tantôt ralentir l’agrégation, probablement en raison des
différentes conditions utilisées (Atwood, Moir et al. 1998; Zou, Kajita et al. 2001; Raman,
Ban et al. 2005).
Aβ40 +2 eq Cu
Aβ40 + DFOA
Aβ40 +1 eq Cu
Aβ40 +1 eq Zn
apo-Aβ40
100
80
60
40
20
0
% a
grég
atio
n
Aβ40 +2 eq Cu
Aβ40 + DFOA
Aβ40 +1 eq Cu
Aβ40 +1 eq Zn
apo-Aβ40
100
80
60
40
20
0
% a
grég
atio
n
Figure 3.1. Effet du zinc (II) et cuivre (II) sur l’agrégation d’Aβ40. Mesure du taux
d’agrégation à pH 7,4 après 3 heures d’incubation à 37°C (concentration initiale en Aβ40 soluble de 50 μM).

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
136
Sous nos conditions (50 mM Hepes, 100 mM NaCl, pH 7,4, Aβ40 à 50 μM, incubation de 3h
à 37 °C) l’ordre de propension à l’agrégation est le suivant (Figure 3.1) : (ZnII-Aβ40) > (CuII2-
Aβ40) > (apo-Aβ40) ≈ (CuII1-Aβ40) > (apo-Aβ40 en présence de desferrioxamine (DFOA)).
L’agrégation d’une protéine ou d’un peptide induite par des métaux peut être modélisée selon
deux mécanismes distincts : le métal se lie à des ligands provenant de deux protéines et forme
un pont (Figure 3.2.A) ; le métal se lie à une protéine en un complexe monomérique et induit
un changement de conformation qui va favoriser l’interaction entre les protéines (Figure
3.2.B). Des mécanismes intermédiaires peuvent également être envisagés, tel que la formation
de dimères induits par le métal qui ensuite agrègent via l’interaction entre dimères (Figure
3.2.C).
…
…
A)
B)
C) …2
…
…
A)
B)
C) …2
Figure 3.2. Mécanismes théoriques potentiels illustrant l’induction de l’agrégation de protéines ou peptides par des métaux.
L’objectif principal a donc été la recherche d’un éventuel dimère d’Aβ induit par la présence
de métaux. En effet, l’existence d’un tel dimère aurait une importance certaine car :
1. cela apporterait une explication plausible quant à l’accélération de l’agrégation d’Aβ
en présence cuivre (II) (> 1 équivalent) et de zinc (II) (Schmechel, Zentgraf et al.
2003) ;
2. cela expliquerait pourquoi une concentration sous stœchiométrique en métaux est
suffisante pour induire l’agrégation (particulièrement le zinc) et pourquoi ces deux
métaux ont été retrouvés en proportion sous stœchiométrique dans les plaques
amyloïdes (Huang, Atwood et al. 2004) ;
3. la dimérisation de protéines induite par des métaux est un schéma classique et peut
être directement reliée à une fonction (Frankel, Bredt et al. 1988; Hopfner, Craig et al.
2002; Ciuculescu, Mekmouche et al. 2005) ;

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
137
4. un dimère d’Aβ a été isolé à partir de tissu de cerveau humain et s’est révélé être
neurotoxique (Klein, Stine et al. 2004) ;
5. basé sur des mesures de RPE à basse température, il a été suggéré que Cu(II) se lie à
Aβ en formant des complexes dimériques de type CuII2-Aβ2 (Curtain, Ali et al. 2001).
II. Formation d’un complexe monomérique observable
Le passage de l’apo-, CuII-, ZnII-Aβ16, 28 et 40 en chromatographie d’exclusion stérique
conduit à la détection d’un seul pic d’élution correspondant à une espèce de faible poids
moléculaire (Figure 3.3). Pour le cas des complexes de métaux, l’analyse des fractions a
confirmé la présence quantitative de cuivre et de zinc (présent, pour ce dernier, dans le
tampon d’élution, cf. matériels & méthodes).
dimère monomère
Figure 3.3. Chromatogramme qualitatif (la hauteur des pics d’absorption ayant été modifiée pour une meilleure lecture) de l’apo- (rouge), CuII- (bleu) et ZnII-Aβ28 (vert). Conditions :
Hepes 20 mM, NaCl 100 mM, pH 7,4. Les flèches indiquent le volume d’élution attendu pour le monomère et le dimère du peptide Aβ28 non structuré (calculée d’après la référence
(Danielsson, Jarvet et al. 2002)).
Il ne semble donc y avoir, dans la gamme de poids moléculaires étudiée (jusqu’à environ 75
kDa), qu’une seule espèce détectable. En comparant les volumes d’élutions vis-à-vis des
poids moléculaires pour apo-Aβ, CuII-Aβ, ZnII-Aβ, les protéines de calibration (globulaires)
et deux oligonucléotides (structure plus allongée), plusieurs constatations peuvent être faîtes

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
138
(Figure 3.4) :
1. les différents segments sont parallèles ce qui confirme que Aβ16, 28 et 40 se comporte
de façon similaire en terme de ratio Vélution / log (Mr) ;
2. pour un poids moléculaire donné, le rayon hydrodynamique de Aβ et de ses
complexes métalliques se situe entre celui des oligonucléotides et celui des protéines
globulaires ;
3. les complexes CuII0,5-Aβ, CuII
1-Aβ et CuII2-Aβ sont élués aux même volumes (pour
chaque longueur de peptide) que les formes apo ;
4. les complexes de zinc sont élués plus tard que les formes apo.
Figure 3.4. Chromatographie d’exclusion stérique d’Aβ ( ) et de ces complexes métalliques (CuII et ZnII ), des molécules de calibration (---) et des oligonucléotides ( ) de 6 et 12
bases. Conditions : Hepes 20 mM, NaCl 100 mM, pH 7,4. Le graphe montre le comportement du volume d’élution en fonction du logarithme du poids moléculaire des molécules et
complexes élués (seule une partie de la régression linéaire correspondant aux molécules de calibration apparaît sur le graphe principal, la totalité du segment est représenté sur le petit
graphe).
Si l’on se base sur les protéines de calibration pour calculer la masse moléculaire apparente
d’Aβ et des complexes métalliques, on obtient des poids moléculaires 2 à 3 fois supérieurs
aux valeurs réelles. Cela a conduit certains auteurs à proposer que l’apo-Aβ40/42 existe à
l’état de di-, tri-, et tétramères stables (cf. par exemple référence (Garzon-Rodriguez,
Sepulveda-Becerra et al. 1997)), bien que d’autres méthodes (RMN à écho de spin avec

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
139
gradient de champ magnétique pulsé, ultracentrifugation analytique) aient suggéré un état
monomérique. Cette erreur provient du fait que les protéines de calibration ont une structure
globulaire ce qui n’est pas le cas d’Aβ, qui est majoritairement random coil (conformation en
pelote statistique non périodique) (Tseng, Esler et al. 1999; Danielsson, Jarvet et al. 2002;
Hou, Shao et al. 2004). Or la chromatographie d’exclusion sépare les molécules suivant leur
rayon hydrodynamique, c’est-à-dire à la fois suivant leur poids moléculaire et leur structure.
Par exemple, les protéines globulaires exhibent un rayon hydrodynamique 2 à 4 fois supérieur
après dénaturation. De même, pour une masse moléculaire donnée, l’oligonucléotide est élué
bien plus tôt que la protéine globulaire (Figure 3.4), du fait de leur structure allongée. Il n’est
donc pas exclu, au contraire, que le volume d’élution des peptides Aβ et de leurs complexes
métalliques correspondent à des monomères (ou complexes monomériques).
De plus, Danielsson et al. (Danielsson, Jarvet et al. 2002) ont montré que l’apo-Aβ40 et 28
étaient monomériques et se comportaient de façon très similaire aux protéines dénaturées.
Pour ces dernières, la formule empirique Rh (en nm) = 0,02 Mr0,5 donne un très bon fit sur une
large gamme de poids moléculaires. Cette relation découle d’une autre formule empirique
développée pour les protéines hautement dénaturées Rh (en Å) = 2,21 N0,57 avec N le nombre
d’acides aminés. Les Rh calculés d’après cette première formule correspondent très bien aux
Rh mesurés des apo- et MeIIx-Aβ (calculés d’après la régression linéaire obtenue avec les
protéines de calibration de Rh connus) si l’on considère qu’Aβ est sous forme monomérique,
ce qui n’est pas le cas avec les formes dimériques (Tableau 3.1). A cela s’ajoute le fait qu’en
RMN du proton, on n’observe qu’un shift modéré de quelques signaux durant la liaison au
métal. Ceci indique que le complexe soluble MeIIx-Aβ n’est pas plus structuré que la forme
apo et justifie l’emploi de la formule empirique pour les complexes métalliques (Syme, Nadal
et al. 2004; Mekmouche, Coppel et al. 2005; Syme and Viles 2005; Hou and Zagorski 2006).
Dans le cas de ZnII-Aβ cependant, le rayon hydrodynamique mesuré en SEC est plus petit que
celui de apo- et CuIIx-Aβ (Figure 3.3 et 3.4, Tableau 3.1). Cela est peut-être dû à une
conformation légèrement plus compacte et structurée comme suggérée par la structure RMN
de ZnII-Aβ16 (Zirah, Kozin et al. 2006). Mais une légère interaction du zinc et donc de ZnII-
Aβ avec la résine de la colonne ne peut pas être exclue (cf. Matériels & méthodes). Afin de
confirmer cette tendance l’apo-Aβ16, -28, -40 et leur complexe avec 0,2 - 0,5 et 1 équivalent
de zinc(II) ont été analysés en RMN à écho de spin avec gradient de champ magnétique pulsé
(PGSE NMR). L’ensemble des Rh déduits de ces expériences correspondent assez bien à ceux
obtenus par SEC (Tableau 3.1). De plus, pour toutes les longueurs d’Aβ mesurées, une

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
140
tendance à la diminution du Rh sous l’effet de la liaison au zinc a été observée, en accord avec
la SEC et l’existence d’un complexe monomérique.
Il est intéressant de noter qu’aucune dimérisation substantielle n’a été observée, même en
présence d’un demi équivalent de zinc(II) ou de cuivre(II) par Aβ, c’est-à-dire dans des
conditions favorisant la formation de complexes par métal pontant (voir Figure 3.2.C). Ce
constat est important d’un point de vue biologique, car les ions métalliques sont supposés être
en proportion sous stoechiométrique dans les plaques amyloïdes et qu’une quantité sous
stoechiométrique en ions métalliques est suffisante pour induire l’agrégation d’Aβ (Tseng,
Esler et al. 1999; Danielsson, Jarvet et al. 2002; Hou, Shao et al. 2004).
Rh (nm) expérimental déterminé par Rh (nm) calculé d’après Mr a SEC b RMN de diffusion Aβ
monomère dimère apo Cu Zn apo Zn
16 1,19 1,69 1,37 1,40 1,14 1,16 ± 0,05 1,14 ± 0,05
28 1,54 2,18 1,69 1,64 1,41 1,5 ± 0,2 1,4 ± 0,1
40 1,78 2,52 1,85 1,86 1,59 1,7 ± 0,2 1,5 ± 0,2
42 1,81 2,57 1,85 1,86 - - - a Calculée d’après la référence (Danielsson, Jarvet et al. 2002). b erreur expérimentale inférieure à 0,1 nm dans les données SEC.
Tableau 3.1. Rayons hydrodynamiques d’Aβ et de ses complexes de cuivre(II) et de zinc(II).
La quantification des expériences de SEC a montré que CuII-Aβ42, ZnII-Aβ28 et ZnII-Aβ40
agrégeaient partiellement à toutes les concentrations utilisées (10-800 μM), avec des pertes
allant jusqu’à environ 80 % pour ZnII-Aβ40. CuII-Aβ40 et ZnII-Aβ16 agrègent seulement à
des concentrations supérieures, tandis que CuII-Aβ16 et 28 n’agrègent pas significativement.
Cependant, les chromatogrammes de l’ensemble de ces complexes exhibent un unique pic
correspondant au monomère. Ces résultats montrent que, entre le monomère et les agrégats
trop grand pour traverser la matrice de la colonne, aucun intermédiaire stable (di-, tri-,
jusqu’au 20-mère) n’a été formé en quantité suffisante pour être détecté.
L’élution du complexe ZnII-Aβ42 n’a pas été possible, même à basse concentration, du fait
d’une agrégation importante et rapide, cependant l’élution du complexe CuII-Aβ42 a mis en

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
141
évidence un pic supplémentaire au niveau du volume mort (Figure 3.5), ce qui n’est pas le cas
pour CuII-Aβ40, indiquant un processus d’agrégation potentiellement différent. Un pic
similaire a été décrit avec apo-Aβ40 et assigné à des protofibrilles (Walsh, Lomakin et al.
1997).
Figure 3.5. Chromatogramme du complexe CuII
1-Aβ42. Les flèches indiquent le volume d’élution attendu pour le monomère et le dimère du peptide Aβ42 non structuré (calculée
d’après la référence (Danielsson, Jarvet et al. 2002)).
Ces résultats montrent que le cuivre(II) et le zinc(II) forment un complexe monomérique avec
Aβ40 ou 42 qui est formé transitoirement, mais qui est assez stable pour être observé. Durant
l’agrégation, aucun oligomère stable de masse apparente inférieure à 75 kDa n’a pu être
observé, indiquant que la formation du dimère est l’étape cinétiquement déterminante en
début d’agrégation (voir Figure 3.6).
Me
Oligomères >75 kDa
ProtofibrillesAβ
Figure 3.6. Model de l’agrégation d’Aβ induite par le zinc(II) ou le cuivre(II).
Conclusion
Il est clair que l’agrégation passe par la formation d’un dimère, en présence ou absence de
métal, cependant ce dimère est très instable et agrége rapidement. La liaison au zinc(II) ou au
cuivre(II) n’induit pas la formation de quantité signifiante de dimère, même dans des
conditions favorables. Il semble donc qu’Aβ40 et 42 n’ont pas d’aptitude à former des

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
142
dimères du fait de la liaison au métal, pointant ainsi vers un mécanisme impliquant un
changement de conformation (Figure 3.2.B) plutôt qu’un ion pontant (Figure 3.2.A).
III. Matériels et méthodes
Préparation des solutions de peptides Aβ.
Les peptides Aβ16, Aβ28, Aβ40, Aβ42 ont soit été synthétisés par processus standard Fmoc
et purifiés par HPLC avec une colonne C8 par Honoré Mazarguil (IPBS, Toulouse), soit
achetés à EZBiolab (Westfield, IN, USA), Geneshere Biotechnologies (Paris, France) ou
Activotec (Cambridge, UK). L’ESI-MS des peptides Aβ16, 28, 40 et 42 montre des masses,
pour les espèces monochargées, correspondantes aux masses calculées à moins de 0,3 unités
près.
Les solutions stocks de peptides sont préparées en dissolvant les peptides dans l’eau ou
directement dans le tampon de travail à pH 9-10 pour faciliter leur dissolution et s’assurer
qu’ils sont sous forme monomérique. Le pH est ensuite ajusté. Les solutions sont conservées à
-20 °C. Dans ces conditions, elles sont stables plusieurs semaines. Les spectres RMN 1H des
peptides Aβ16, Aβ28, Aβ40 ont montré qu’il n’y avait aucune dégradation ni agrégation des
peptides dans ce laps de temps.
La concentration des peptides est déterminée par spectroscopie d’absorption UV-Vis en
utilisant le coefficient d’absorption de la Tyr à λ = 276 nm, ε = 1410 cm-1.M-1 (Gill and von
Hippel 1989). La structure des peptides est principalement “random coil” et la Tyr10 se
comporte donc comme une Tyr libre. Il est donc raisonnable de considérer qu’elle a le même
coefficient d’absorption molaire qu’une Tyr libre. A partir de la Loi de Beer-Lambert, A =
ε.l.c (où l = 1 cm), un simple spectre d’absorption permet de déduire la concentration du
peptide.
Préparation des solutions de métaux.
Les solutions de métaux sont préparées par pesée d’une masse de sulfate de zinc heptahydraté
(ZnSO4, 7 H2O, Alfa Aesar), chlorure de cuivre dihydraté (CuCl2, 2 H2O, Sigma) ou chlorure
de cadmium (CdCl2, Aldrich) diluée dans l’eau. Ces solutions mères stocks sont conservées à
-20°C. Elles sont ensuite diluées dans le tampon approprié jusqu’à 1 mM, 500 ou 100 μM.
L’ajout d’HCl avant dilution est généralement nécessaire afin d’éviter la formation de

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
143
précipité d’hydroxydes Métal(OH)2.
Spectroscopie d’absorption UV-visible
Les spectres d’absorption UV/Visible ont été réalisés à température ambiante avec les
spectromètres Cary 2300 et Agilent 8453 dans une cuve en quartz de 1 cm de large.
Etude de la diffusion par RMN à écho de spin avec gradient de champ magnétique pulsé
(Pulse Gradient Spin-Echo Nuclear Magnetic Resonnance, PGSE NMR)
La mesure de la diffusion d’une particule permet d’en déduire son rayon hydrodynamique et
ainsi sa taille. Soit kB la constante de Boltzmann, T la température, f le coefficient de friction :
La formule du coefficient de friction donnée ici correspond au cas d’une particule sphérique,
où η est la viscosité du liquide et r le rayon de Stockes de la particule diffusante.
La séquence de base du gradient de champ pulsé est représentée sur la figure 3.7. A l’état
initial, l’ensemble des spins des noyaux des atomes d’hydrogènes est en équilibre thermique
et orienté suivant l’axe z, c’est-à-dire parallèle au champ magnétique statique. A t0 on
applique, pendant un cours instant, un champ de π/2 suivant l’axe des x afin d’orienter les
spins dans le plan perpendiculaire au champ magnétique statique. Ensuite un premier gradient
de champ pulsé est appliqué de manière à induire un déphasage des spins en fonction de leur
position suivant z. A la fin de la première période τ, un champ pulsé π selon l’axe y permet
d’inverser le signe de la phase. Ainsi, lorsque l’échantillon reçoit le 2ème gradient de champ
pulsé identique au premier, les particules ne s’étant pas déplacées suivant z voient le
déphasage de leur spins nucléaires entièrement compensé, les autres restent déphasées
proportionnellement à leur déplacement suivant z entre l’application des deux champs de
gradient. Si toutes les particules ne se sont déplacées uniformément, on obtient une
Mesure de la diffusion
Taille de la molécule
Loi de Stokes-Einstein: D = kB.T/f où f = 6π.η.rs

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
144
distribution de phase et une atténuation du signal RMN. On associe enfin à chaque signal du
spectre RMN 1H de la solution un coefficient de diffusion.
Figure 3.7. Représentation schématique d’une séquence RMN à gradient de champ pulsé
(Price 1997).
Les expériences de RMN ont été effectuées avec un spectromètre Bruker Avance500 équipé
d’une sonde 5mm triple résonance inversée selon z. Toutes les mesures ont été réalisées selon
la séquence pulsée BPLED (Wu, Chen et al. 1995) avec un retard de 5 ms (Te) des courants de
Foucault. La température a été fixée à 291 K. Le délai de recyclage a été ajusté à 10 secondes
afin de permettre une complète relaxation. La forme du gradient était rectangulaire avec une
longueur de 2,6 ms pour Aβ40 et 2,4 ms pour Aβ28 et Aβ16, la force variant selon 16 in

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
145
créments (2-95 %) selon la rampe du gradient créé par le logiciel de Bruker DOSY (Diffusion
Ordered Spectroscopy). La force du gradient a été calibrée par mesure de l’auto-diffusion du
signal résiduel HDO dans un échantillon à 100% D2O à 298 K (1,90.10-9 m2.s-1). Le temps de
diffusion Δτ a été pris égal à 180 ms pour Aβ40 et 150 ms pour Aβ28 et Aβ16. Le signal
résiduel de l’eau a été supprimé par une séquence WATERGATE. Les échantillons n’ont pas
été mis en rotation pour éviter une surévaluation du coefficient de diffusion (remarquée
expérimentalement). Tous les échantillons ont été préparés dans D2O pur et le pH a été ajusté
à environ 7,4. Les coefficients de diffusion ont été déterminés en fittant avec la méthode des
moindres carrées les points obtenus par l’équation de Stejskal-Tanner en utilisant
l’algorythme Simfit du logiciel de Bruker T1/T2.
Le rayon hydrodynamique a été calculé d’après les coefficients de diffusion obtenus
expérimentalement selon la relation de Stockes-Einstein (He and Niemeyer 2003).
rTkD B
..6.ηπ
=
avec D le coefficient de diffusion (cm2.s-1), T = 291 K, ηD2O = 1,3.10-3 kg.m-1.s-1 et r le rayon
hydrodynamique (m). Les valeurs de coefficients de diffusion mesurées pour l’apo-Aβ sont
respectivement de 1,39 ; 1,06 et 0,93.10-10 m2.s-1 pour Aβ16, Aβ28 et Aβ40 ; et 1,42 ; 1,19 et
1,08.10-10 m2.s-1 pour les complexes ZnII-Aβ.
Chromatographie d’exclusion stérique (SEC)
L’analyse séparative des mélanges d’oligomères d’Aβ en présence et absence de métaux
(Cu(II) et Zn(II)) est réalisée par chromatographique d’exclusion stérique couplée à une FPLC.
Elle est effectuée à température ambiante sur une colonne superdex 75 10/300 GL avec une
instrumentation AKTA (Amersham Pharmacia Biotech) sous des conditions isocratiques.
L’éluant utilisé est une solution tampon de 20 mM de Hepes à pH 7,4 contenant 100mM de
NaCl. Son débit est généralement de 0,4 ml.min-1. L’absorption a été enregistrée aux 3
longueurs d’ondes : 220 nm, 276 nm et 600 nm. Le système est calibré avec les molécules
globulaires de rayons hydrodynamiques connus suivantes: l’albumine (35,5 Å, 67kDa), la
ribonucléase A (17,5Å ; 13,7 kDa), le cytochrome C (17 Å ; 12,3 kDa), l’aprotinine (13,5 Å;
6,5 kDa), la vitamine B12 (8,5 Å ; 1,35 kDa). Les oligonucléotides (de séquences 5’-
CGTACG et 5’-TCCATATCACAT) ont été donnés par G. Pratviel et B. Meunier (LCC,
Toulouse). Après équilibration de la colonne avec élution du tampon 20 mM Hepes, 100 mM
NaCl, pH 7.4 (2 volumes de colonne), 100 μl d’échantillons de protéines (concentration

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
146
comprise entre 10 et 200 μM) sont injectés. Le pic d’Aβ est collecté puis quantifié par
absorption.
Sous ces conditions, le complexe Aβ-Zn perd partiellement le zinc. Des expériences
similaires ont donc été effectuées en présence de 50 ou 100 μM de ZnCl2 dans le tampon
d’élution afin de maintenir le zinc fixé au peptide (Kd de l’ordre du micromolaire). Les
concentrations de zinc ont été mesurées avec le 4-(2-pyridylazo)-resorcinol (PAR)
(Ciuculescu, Mekmouche et al. 2005). Le PAR, de plus forte affinité que Aβ pour le zinc,
forme des complexes 1:1 et 2:1 avec Zn(II), ainsi l’utilisation d’un large excès de PAR permet
à plus de 99 % du zinc(II) présent de former un complexe 2:1 avec le PAR : Zn(PAR)2. Celui-
ci est caractérisé par un maximum d’absorbance à 490 nm (ε = 66000 M-1.cm-1) (Shaw, Laib
et al. 1990; Walkup and Imperiali 1997).
La quantité de cuivre présente dans la fraction a été estimée par l’absorbance à 600 nm (ε =
60 M-1.cm-1) et par test colorimétrique test avec la bathocuproïne (Poillon and Dawson 1963).
Dans ce test, le Cu(II) est d’abord réduit en Cu(I) par addition d’un excès d’acide ascorbique
(30 équivalents). Le Cu(I) est ensuite complexé par la bathocuproïne, qui exhibe alors une
bande de transition à 480 nm (ε = 13500 M-1.cm-1). L’acide bathocuproïne disulfonique
(Acros) est 10 fois en excès.
Etude de l’agrégation
Des solutions de 50 μM d’Aβ40 monomérique (tampon Hepes 50 mM, NaCl 100 mM, pH 7,4)
ont été incubés pendant 3 heures à 37°C, sous agitation modérée, en présence ou non de métal.
La déféroxamine (achetée chez Sigma, figure 3.8) a été utilisée pour chélater d’éventuelles
traces de métaux. Les échantillons ont ensuite été centrifugés 30 min à 21000g à 4°C. Les
fractions d’Aβ40 soluble et agrégé ont été déterminées en mesurant le contenu en peptide des
culots et surnageants grâce au test colorimétrique de détection de protéines BCA (Pierce,
Rockville CA).
Figure 3.8. Molécule de déféroxamine libre ou liée à du fer(III).

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
147
Le test BCA est étalonné à partir de solutions d’albumine de sérum bovin de concentration
connue (Smith, Krohn et al. 1985). Le réactif de détection est composé d’acide
bicinchoninique (BCA) et de sulfate de cuivre (II) en présence d’hydroxyde de potassium. Le
principe du test repose sur la réaction de réduction des ions cuivriques en ions cuivreux par
les liaisons peptidiques en milieu très alcalin. Les ions Cu+ sont ensuite captés spécifiquement
par le BCA pour former un complexe 2:1 exhibant une bande d’absorption à 562 nm (Figure
3.9).
Figure 3.9. Réactions conduisant à la détection colorimétrique des protéines par le test BCA.
Ce test colorimétrique possède plusieurs avantages, puisqu'il est :
- quantitatif, sensible (détection à partir de 0,02 μM d’Aβ) et linéaire dans la gamme
de concentration de protéine étudiée (jusqu'à 6 μM d’Aβ),
- relativement peu dépendant de la séquence de la protéine puisque ce sont les
liaisons peptidiques qui réagissent lors du dosage et non les résidus des acides
aminés (comme par exemple le test de Bradford basé sur la coloration des
arginines).
- - compatible avec l'utilisation de CuII et de chélateurs d'ions métalliques aux
concentrations de l'étude.
Il est toutefois préférable d’utiliser la protéine ou le peptide étudié plutôt que la BSA lors de
l’étalonnage. En effet, des différences parfois significatives ont été trouvées entre la
concentration initiale réelle et celle déduite des tests BCA.

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
148
Références C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, et al. (1998). "Dramatic
aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis." J Biol Chem, 273(21), 12817-26.
E. D. Ciuculescu, Y. Mekmouche and P. Faller (2005). "Metal-binding properties of the peptide APP(170-188): a model of the Zn(II)-binding site of amyloid precursor protein (APP)." Chemistry, 11(3), 903-9.
C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, et al. (2001). "Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits." J Biol Chem, 276(23), 20466-73.
J. Danielsson, J. Jarvet, P. Damberg and A. Gräslund (2002). "Translational diffusion measured by PFG-NMR on full length and fragments of the Alzheimer Ab(1-40) peptide. Determination of hydrodynamic radii of random coil peptides of varying length." Magnetic Resonance in Chemistry, 40(13), S89-S97.
A. D. Frankel, D. S. Bredt and C. O. Pabo (1988). "Tat protein from human immunodeficiency virus forms a metal-linked dimer." Science, 240(4848), 70-3.
W. Garzon-Rodriguez, M. Sepulveda-Becerra, S. Milton and C. G. Glabe (1997). "Soluble amyloid Abeta-(1-40) exists as a stable dimer at low concentrations." J Biol Chem, 272(34), 21037-44.
S. C. Gill and P. H. von Hippel (1989). "Calculation of protein extinction coefficients from amino acid sequence data." Anal Biochem, 182(2), 319-26.
J. Hardy and D. J. Selkoe (2002). "The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics." Science, 297(5580), 353-6.
L. He and B. Niemeyer (2003). "A novel correlation for protein diffusion coefficients based on molecular weight and radius of gyration." Biotechnol Prog, 19(2), 544-8.
K. P. Hopfner, L. Craig, G. Moncalian, R. A. Zinkel, T. Usui, et al. (2002). "The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair." Nature, 418(6897), 562-6.
L. Hou, H. Shao, Y. Zhang, H. Li, N. K. Menon, et al. (2004). "Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation." J Am Chem Soc, 126(7), 1992-2005.
L. Hou and M. G. Zagorski (2006). "NMR reveals anomalous copper(II) binding to the amyloid Abeta peptide of Alzheimer's disease." J Am Chem Soc, 128(29), 9260-1.
X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, R. E. Tanzi, et al. (2004). "Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Abeta peptides." J Biol Inorg Chem, 9(8), 954-60.
W. L. Klein, W. B. Stine, Jr. and D. B. Teplow (2004). "Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease." Neurobiol Aging, 25(5), 569-80.
C. J. Maynard, A. I. Bush, C. L. Masters, R. Cappai and Q. X. Li (2005). "Metals and amyloid-beta in Alzheimer's disease." Int J Exp Pathol, 86(3), 147-59.
Y. Mekmouche, Y. Coppel, K. Hochgrafe, L. Guilloreau, C. Talmard, et al. (2005). "Characterization of the Zn(II) Binding to the Peptide Amyloid-beta(1-16) linked to Alzheimer's Disease." Chembiochem, 6(9), 1663-1671.
G. Multhaup and C. L. Masters (1999). "Metal binding and radical generation of proteins in human neurological diseases and aging." Met Ions Biol Syst, 36365-87.
W. N. Poillon and C. R. Dawson (1963). "On The Nature Of Copper In Ascorbate Oxidase. Ii. The Role Of Prosthetic Copper In The Enzyme's Function." Biochim Biophys Acta,

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
149
7737-46. W. S. Price (1997). "Pulsed-field gradient nuclear magnetic resonance as a tool for studying
translational diffusion: Part 1. Basic theory." Concepts in Magnetic Resonnance, 9(5), 299-336.
B. Raman, T. Ban, K. Yamaguchi, M. Sakai, T. Kawai, et al. (2005). "Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide." J Biol Chem, 280(16), 16157-62.
A. Schmechel, H. Zentgraf, S. Scheuermann, G. Fritz, R. Pipkorn, et al. (2003). "Alzheimer beta-amyloid homodimers facilitate A beta fibrillization and the generation of conformational antibodies." J Biol Chem, 278(37), 35317-24.
C. F. Shaw, J. E. Laib, M. M. Savas and D. H. Petering (1990). "Biphasic Kinetics of aurothionein formation from gold sodium thiomalate: a novel metallochromic technique to probe Zn2+ and Cd2+ displacement from metallothionein." Inorg. Chem., 29403-408.
P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, et al. (1985). "Measurement of protein using bicinchoninic acid." Anal Biochem, 150(1), 76-85.
Y. H. Suh and F. Checler (2002). "Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease." Pharmacol Rev, 54(3), 469-525.
C. D. Syme, R. C. Nadal, S. E. Rigby and J. H. Viles (2004). "Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer's disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques." J Biol Chem, 279(18), 18169-77.
C. D. Syme and J. H. Viles (2005). "Solution (1)H NMR investigation of Zn(2+) and Cd(2+) binding to amyloid-beta peptide (Abeta) of Alzheimer's disease." Biochim Biophys Acta,
B. P. Tseng, W. P. Esler, C. B. Clish, E. R. Stimson, J. R. Ghilardi, et al. (1999). "Deposition of monomeric, not oligomeric, Abeta mediates growth of Alzheimer's disease amyloid plaques in human brain preparations." Biochemistry, 38(32), 10424-31.
G. K. Walkup and B. Imperiali (1997). "Fluorescent chemosensors for divalent zinc based in zinc finger domains. Enhanced oxidative stability, metal binding affinity, and structural and functional characterization." J Am Chem Soc, 1193443-3450.
D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow (1997). "Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate." J Biol Chem, 272(35), 22364-72.
D. Wu, A. Chen and C. S. Johnson (1995). "An Improved Diffusion Ordered Spectroscopy Experiment Incorporating Bipolar Gradient Pulses." J. Magn. Reson., A115260-264.
S. Zirah, S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, et al. (2006). "Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging." J Biol Chem, 281(4), 2151-61.
J. Zou, K. Kajita and N. Sugimoto (2001). "Cu(2+) Inhibits the Aggregation of Amyloid beta-Peptide(1-42) in vitro." Angew Chem Int Ed Engl, 40(12), 2274-2277.

Chapitre 3 : étude de l’oligomérisation des complexes cuivre(II)- et zinc(II) - Aβ
150

151
Chapitre 4
Etude structurale du mécanisme de
promotion de l’agrégation du peptide
amyloïde bêta par le zinc(II)

152

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
153
I. Introduction
Les amyloïdoses sont caractérisées par un repliement et un assemblage anormal d’une
protéine, conduisant à la déposition de fibres de protéines insolubles (amyloïdes), provoquant
ensuite des dysfonctionnements cellulaires suivis de la mort des cellules (revues : (Sacchettini
and Kelly 2002; Chiti and Dobson 2006)). Il faut noter cependant, comme énoncé dans le
chapitre 1, que toutes les amyloïdoses ne sont pas pathologiques, des amyloïdoses
fonctionnelles ont récemment été découvertes dans un certains nombres d’organisme vivants,
dont l’homme (Fowler et al. 2007). Plus de 20 protéines ou peptides différents ont été
rapportés former des dépôts amyloïdes in vivo. Bien que ces protéines n’aient pas de lien
évident entre elles, tant au niveau de leur séquence peptidique que de leur fonction, elles
polymérisent en des fibres similaires d’un diamètre d’environ 10 nm (Pepys 2006), organisées
en structure dite « cross bêta » et pouvant être révélées par des marqueurs amyloïdophiles tels
la Thioflavine T (ThT) ou le rouge Congo.
Ces protéines peuvent ou non avoir une structure native définie. Quand c’est le cas, comme
pour le lysozyme et la transthyrétine, le changement conformationnel nécessaire à la
formation de structures amyloïdes, passe par une déstabilisation de la structure native du fait
de mutations ou de facteurs environnementaux défavorables (Ohnishi and Takano 2004; Jahn
and Radford 2005). Dans le cas de protéines ou peptides non structurées, tel que Aβ ou
l’alpha-synucléine, la structure du monomère initiateur de l’arrangement en feuillets bêta
n’est pas connu. Teplow et ses collaborateurs (Walsh et al. 1997; Walsh et al. 1999;
Kirkitadze et al. 2001) ont rapporté des indications en faveur de l’existence d’un intermédiaire
de structure partiellement hélice alpha dans la formation de fibres amyloïdes à partir de
peptide Aβ majoritairement non structuré. Cette hypothèse est également confirmée par le fait
que, en présence d’environ 20 % de 2,2,2-trifluoroéthanol (TFE) connu pour promouvoir les
structures en hélice, la formation de fibres est favorisée (Fezoui and Teplow 2002). Cette effet
cesse à des concentrations supérieures en TFE pour lesquelles le taux d’hélices alpha
augmente, soulignant ainsi l’importance d’une conversion partielle en hélice alpha.
Un des facteurs environnementaux physiologiques capable d’affecter la conformation et ainsi
les propriétés d’agrégation des protéines et peptides amyloïdogéniques, est la présence d’ions
métalliques tels le zinc(II) et le cuivre(II). Le zinc a été rapporté se lier à diverses protéines et
peptides comme Aβ, le prion, l’alpha-synucléine, la transthyrétine Abri, etc (Bush et al. 1994;
Atwood et al. 1998; Khan et al. 2004; Gaggelli et al. 2005; Binolfi et al. 2006; Wilkinson-

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
154
White and Easterbrook-Smith 2007). Généralement le zinc favorise l’agrégation de la protéine
ou du peptide amyloïdogénique. Dans le cas de Aβ, dont l’interaction avec le zinc est une des
mieux décrite, la structure des agrégats a été décrite comme étant majoritairement amorphe
(dépendant des conditions expériementales et temps d’incubation). En effet les agrégats
induits par le zinc semblent moins réagir avec la ThT que ceux formés en l’absence de zinc
(Yang et al. 2000; Yoshiike et al. 2001; Klug et al. 2003; House et al. 2004; Raman et al.
2005).
Comme indiqué dans le premier chapitre, le site de fixation du zinc est localisé sur la partie
N-terminale de Aβ (acide aminés 1 à 16). Plusieurs études utilisant Aβ40 ou les formes
courtes de Aβ (Aβ16 et 28) ont tenté d’identifier les ligands du zinc (Liu et al. 1999; Miura et
al. 2000; Yang et al. 2000; Curtain et al. 2001; Kozin et al. 2001; Zirah et al. 2003;
Mekmouche et al. 2005; Hou and Zagorski 2006; Stellato et al. 2006; Syme and Viles 2006;
Zirah et al. 2006; Danielsson et al. 2007). Bien qu’il y ait quelques contradictions,
l’implication des histidines 6, 13 et 14 sont suggérés par la plupart des publications. Les
conclusions concernant d’autres ligands concordent moins, le plus fréquemment évoqué étant
l’aspartate en position 1, via la fonction amine terminale ou du groupement carboxyle du
résidu (Zirah et al. 2003; Mekmouche et al. 2005; Danielsson et al. 2007). Dans le cas d’un
N-terminus acétylé, la structure RMN du complexe ZnII-Aβ16 a été résolue et indique que le
zinc est lié aux trois histidines et au glutamate 11 (Zirah et al. 2006).
Cependant, le mécanisme de promotion de l’agrégation par le zinc est mal connu. Le but de ce
chapitre est donc d’essayer de comprendre l’effet du zinc sur l’agrégation de Aβ en présence
de TFE, ainsi que le rôle de l’intermédiaire partiellement structuré en hélice alpha. Cette étude
a été menée avec le peptide Aβ28, contenant le site de fixation du zinc, qui a l’avantage
d’agréger beaucoup plus lentement que Aβ40 et 42, rendant ainsi possible les études
spectroscopiques avec un suivi dans le temps, tout en ayant la capacité à former des fibres
amyloïdes classiques (Kirschner et al. 1987).8
8 Le zinc(II) a été choisi préférentiellement au cuivre(II) car sa liaison aux histidines ou carboxylates n’absorbe pas dans la région correspondante aux liaisons peptidiques (190-240 nm). Ainsi les changements observés lors de l’ajout du métal dans le spectre de dichroïsme circulaire ne sont dus qu’à des modifications dans la structure secondaires du peptide. Ce n’est pas le cas du cuivre qui absorbe dans cette région via la liaison aux histidines.

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
155
II. Résultats et discussion
Le degré d’agrégation de Aβ28 en présence ou non de zinc(II) et en fonction de la teneur en
TFE est donné sur la figure 4.1. Différentes méthodes ont été utilisées pour mesurer l’état
d’agrégation. Premièrement, après 30 minutes d’incubation puis centrifugation, les quantités
de peptide contenues dans le surnageant et le culot ont été mesurées avec le test BCA (voir
Matériels et méthodes). Cette analyse montre qu’en l’absence de zinc, l’agrégation de l’apo-
Aβ28 est faible quelque soit la teneur en TFE de la solution (0 à 40 %). Par contre, la
présence d’un équivalent de zinc(II) induit une précipitation dont l’importance dépend du
pourcentage de TFE : très faible en l'absence de TFE (~10 %), elle est quasi-totale à 10 % de
TFE, puis diminue progressivement jusqu’à retrouver des valeurs très faibles pour une teneur
en TFE de 40 %.
0
20
40
60
80
100
0 10 20 30 40 50 60
% TFE
% a
grég
atio
n et
inte
nsité
de
flore
scen
ce re
lativ
e
Figure 4.1. Pourcentage d’agrégation (déterminé avec le test BCA et par mesure de
l’absorption du surnageant ) ou intensité de fluorescence relative du à la fixation de ThT , après 30 minutes d’incubation à 37°C, en fonction de la teneur en TFE de la solution, en absence (en pointillé) ou en présence (lignes pleines) d’un équivalent (50 μM) de zinc(II).
Les agrégats ont également été analysés par coloration à la ThT. Celle-ci se fixe
spécifiquement aux structures en feuillet bêta en modifiant son émission de fluorescence
(émission à 480 nm pour une excitation à 445 nm) et est donc logiquement utilisée depuis de
nombreuses années pour révéler les structures amyloïdes riches en feuillet bêta. Les résultats

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
156
obtenus avec la ThT corrèlent tout à fait avec ceux obtenus par centrifugation : la fluorescence
mesurée en absence de zinc est très faible, alors qu’en présence de zinc(II) on retrouve la
même dépendance à la teneur en TFE, c’est-à-dire une fluorescence importante à 10-20 % de
TFE et beaucoup moins en deçà et au-delà. Cette corrélation indique que les agrégats
précédemment observés contiennent des structures en feuillet bêta.
Afin de mieux comprendre le phénomène d’induction de l’agrégation par le zinc, celle-ci a été
suivie dans le temps par turbidimétrie et dichroïsme circulaire à 10 % de TFE (Figure 4.2 et
4.4).
0
0.03
0.06
0.09
0.12
0.15
0.18
0 1 2 3 4 5 6 7 8t (h)
Abs
orpt
ion
à 40
0 nm
Figure 4.2. Mesure de la turbidimétrie d’une solution à 50 μM d’Aβ28 à température
ambiante : seul ( rose), en présence d’un équivalent de ZnII ( orange), en présence de 10 % TFE ( bleu), en présence d’un équivalent de ZnII et de 10 % TFE ( vert).
En présence d’un équivalent de zinc(II) mais sans TFE, Aβ28 exhibe une augmentation
modérée de la turbidité, atteignant un plateau après environ 8 heures d’incubation (pas
d’augmentation significative entre 8 et 30 heures). En revanche, la présence de 10% de TFE,
associée au zinc, accélère et augmente très significativement l’agrégation qui atteint un
plateau au moins trois fois supérieur en à peine 3 heures. Le temps nécessaire pour atteindre
la moitié du maximum de turbidimétrie est de 4 heures en absence de TFE et environ 10
minutes à 10 % TFE.
En absence de zinc, l’augmentation de turbidité est très limitée. Cependant la présence de 10
% de TFE induit au bout de 8 heures, une légère agrégation. Des expériences
complémentaires, sans zinc et à 0 et 10 % de TFE, réalisées sur une semaine, n’ont pas mis en

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
157
évidence une augmentation supplémentaire de la turbidité. De même, une coloration par ThT
a montré une très faible et relativement constante émission de fluorescence sur l’ensemble des
échantillons de 1 à 7 jours en présence de 10 % de TFE. Sans TFE, aucune émission de
fluorescence significative n’a pu être détectée même après 10 jours. Il semble donc que la
présence de 10 % de TFE favorise légèrement l’agrégation, avec une structure en feuillet bêta,
de l’apo-Aβ28. Cet effet peut être attribué à la stabilisation d’un intermédiaire partiellement
structuré en hélice alpha, favorable à la formation d’agrégats de type amyloïde (Figure 4.3).
On peut également émettre l’hypothèse qu’une quantité importante de TFE stabiliserait trop
fortement l’intermédiaire en hélice qui ne pourrait ainsi plus basculer vers une structure en
feuillet bêta.
Majoritairementrandom coil
Favorisée par 10% TFE
1ère étape 2ème étape
Partiellement hélice α
Majoritairement feuillet β
Majoritairementrandom coil
Favorisée par 10% TFE
1ère étape 2ème étape
Partiellement hélice α
Majoritairement feuillet β
Figure 4.3. Modèle d’agrégation d’Aβ via un intermédiaire partiellement hélice alpha.
Le spectre de dichroïsme circulaire (DC) de ZnII-Aβ28 dans 10 % TFE indique une cinétique
similaire à celle mesurée en turbidimétrie (Figure 4.4) : la moitié du temps nécessaire à la
stabilisation de la structure secondaire est également de l’ordre de 10 minutes. Ainsi
l’agrégation s’accompagne d’un changement de structure d’abord principalement random coil
(avec environ 20 % d’hélice alpha) puis finalement majoritairement feuillet bêta (un peu plus
de 50 %, avec seulement ~3-4 % d’hélice alpha).

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
158
-13
-11
-9
-7
-5
-3
-1
1
195 205 215 225 235
Wavelength (nm)
[teta
] (kd
eg c
m2
dmol
-1)
0
20
40
60
80
0 50 100 150 200t (min)
%
Longueur d’onde (nm)
[Θ] (
kdeg
.cm
2 .dm
ol-1
)
-13
-11
-9
-7
-5
-3
-1
1
195 205 215 225 235
Wavelength (nm)
[teta
] (kd
eg c
m2
dmol
-1)
0
20
40
60
80
0 50 100 150 200t (min)
%
-13
-11
-9
-7
-5
-3
-1
1
195 205 215 225 235
Wavelength (nm)
[teta
] (kd
eg c
m2
dmol
-1)
0
20
40
60
80
0 50 100 150 200t (min)
%
-13
-11
-9
-7
-5
-3
-1
1
195 205 215 225 235
Wavelength (nm)
[teta
] (kd
eg c
m2
dmol
-1)
0
20
40
60
80
0 50 100 150 200t (min)
%
Longueur d’onde (nm)
[Θ] (
kdeg
.cm
2 .dm
ol-1
)
Figure 4.4. Dépendance dans le temps du spectre de dichroïsme circulaire d’une solution à
50 μM de ZnII-Aβ28 contenant 10 % de TFE (déconvolution avec le logiciel Dichroprot, base de donnée Fasman : random coil, feuillet β, hélice α). Mesures à 0, 25 sec, 4, 8, 11,
15, 23, 31, 48, 150, 202 et 228 min (Greenfield and Fasman 1969; Deleage and Geourjon 1993).
Les spectres DC d’apo- et ZnII-Aβ28 à 0 et 40 % de TFE n’exhibent que peu, voire pas, de
changement dans cet intervalle de temps, parallèlement à leur faible degré d’agrégation. Ces
résultats sont également confirmés par les mesures par coloration à la ThT, qui indiquent
également que les agrégats contiennent des structures en feuillet bêta. Ces données concordent
donc bien avec l’idée que les agrégats formés en présence de zinc sont, au moins en partie, du
type amyloïde. Au vu de l’ensemble de ces résultats, il est clair que l’association d’un
équivalent de zinc et la présence de 10-20 % de TFE a un effet synergique sur l’agrégation
d’Aβ, en accélérant et augmentant la formation d’agrégats contenant des structures en
feuillets bêta.
Ainsi, en se référant au modèle précédent (Figure 4.3), le zinc pourrait accélérer soit la
formation de l’intermédiaire en hélice alpha (1ère étape), soit la transition de cet intermédiaire
vers une structure en feuillet de type amyloïde (2ème étape). Autrement dit, le zinc soit favorise,
soit déstabilise l’hélice alpha. Afin d’évaluer cette incidence, l’effet du zinc sur la proportion
en hélice alpha a été déterminée par spectroscopie DC. Les figures 4.5 et 4.6 comparent
l’intensité du signal à 208 nm, dont la valeur est principalement conditionnée par le taux

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
159
d’hélice alpha, pour ZnII- et apo-Aβ (respectivement 28 et 16) pour différente teneur en TFE.
A 208 nm, le signal de l’hélice alpha est négatif, la liaison du zinc à Aβ28 induit donc une
diminution de la teneur en hélice alpha en présence de TFE (10-40 %) avec un effet maximal
à 10 et 20 % TFE. Par contre, en absence de TFE, la liaison du zinc semble induire une
augmentation de structure en hélice alpha. Ainsi, dans les conditions favorisant l’agrégation
d’Aβ (10-20 % TFE), le zinc a tendance à déstabiliser l’hélice alpha, pointant donc plutôt vers
une promotion de la 2ème étape dans le modèle précédent.
-16
-14
-12
-10
-8
-6
-4
0 10 20 30 40% TFE
[O] (
kdeg
.cm
2.dm
ol-1
)[Θ
] (kd
eg.c
m2 .d
mol
-1)
-16
-14
-12
-10
-8
-6
-4
0 10 20 30 40% TFE
[O] (
kdeg
.cm
2.dm
ol-1
)[Θ
] (kd
eg.c
m2 .d
mol
-1)
Figure 4.5. Ellipticité molaire à 208 nm, en fonction du pourcentage de TFE, pour des
solutions à 50 μM d’Aβ28, en présence ( vert) ou absence ( bleu) d’un équivalent de zinc(II).
Cependant, la présence de zinc(II) induisant l’agrégation de Aβ28, il est difficile de séparer le
phénomène de formation d’hélice alpha de l’agrégation, bien qu’à la longueur d’onde choisie
les autres structures contribue peu (mais une perte global de signal ne peut être exclue). Le
comportement du peptide modèle Aβ16 ont donc été étudié, en effet il possède le site de
fixation du zinc mais il reste soluble dans les conditions présentes (Mekmouche et al. 2005;
Talmard et al. 2007). Les spectres de dichroïsme obtenus avec Aβ16 sont similaires à ceux
obtenus avec Aβ28. On retrouve la même diminution, en valeur absolue, du signal à 208 nm
du fait de l’ajout de zinc(II) en présence TFE (mais sans l’effet maximal à 10-20% TFE), et
une augmentation an absence de TFE.

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
160
[Θ] (
kdeg
.cm
2 .dm
ol-1
)
-16
-14
-12
-10
-8
-6
-4
0 20 40 60 80% TFE
[Θ] (
kdeg
.cm
2 .dm
ol-1
)
-16
-14
-12
-10
-8
-6
-4
0 20 40 60 80% TFE
Figure 4.6. Ellipticité molaire à 208 nm, en fonction du pourcentage de TFE, pour des solutions à 50 μM d’Aβ16, en présence ( vert) ou absence( bleu) d’un équivalent de
zinc(II).
Néanmoins, il est clair qu’à 10 % TFE la liaison du zinc(II) à Ab16 induit une diminution du
taux de structures en hélice alpha, indiquant que c’est également le cas pour Ab28.
Conclusion
Il a été montré dans la littérature que la présence de 10-20 % de TFE induit la formation
partielle d’hélices alpha et, parallèlement, favorise la formation d’agrégats d’Aβ de type
amyloïde (riches en feuillets bêta). L’hypothèse a donc été émise qu’une structure
partiellement hélice alpha constitue un intermédiaire dans la formation des fibres amyloïdes.
Cependant cette dernière est inhibée par un taux trop important d’hélices alpha (du fait
probablement d’une trop grande stabilité). Dans ce chapitre, l’influence du zinc sur
l’agrégation du peptide modèle amyloïdogénique Aβ28 a été étudié, en fonction de la teneur
en TFE. Le zinc accentue l’effet du TFE, en effet, à 10-20 % de TFE, il accélère la formation
d’agrégats d’Aβ28 de type amyloïde. Etant donné que, à cette concentration de TFE, la
présence de zinc diminue le taux d’hélice alpha, on peut supposer que la liaison au zinc
favorise la transformation de l’intermédiaire partiellement hélice alpha vers une structure plus
encline à former des fibres riches en feuillets bêta. Cette hypothèse est résumée sur la figure
4.7. Ainsi, dans une première étape, la présence de 10-20 % de TFE stabiliserait intermédiaire
de structure partiellement hélice alpha et, dans une deuxième étape, le zinc accélèrerait la
transformation de cet intermédiaire vers les structures caractéristiques des dépôts amyloïdes,
via une légère déstabilisation des hélice alpha, mais sans pour autant favoriser le retour vers

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
161
l’état majoritairement non structuré initial. Cette hypothèse est également en accord avec
l’effet synergique observé du TFE et du zinc : séparément les deux facteurs favorisent
l’agrégation, mais conjointement l’effet promoteur est plus que doublé.
Majoritairement random coil
Favorisée par 10%
TFEPartiellement
hélice αMajoritairement
feuillet β
2ème étape1ère étape
Favorisée par ZnII
Majoritairement random coil
Favorisée par 10%
TFEPartiellement
hélice αMajoritairement
feuillet β
2ème étape1ère étape
Favorisée par ZnII
Figure 4.7. Modèle de l’effet synergique du zinc(II) et du TFE (à 10-20 %) sur la formation
d’agrégats d’Aβ28 de type amyloïde : la faible teneur en TFE stabilise l’intermédiaire partiellement en hélice alpha et la liaison du zinc déstabilise cet intermédiaire en faveur de
structure amyloïde en feuillet bêta.
Il est possible que le mécanisme de promotion de la formation de fibres amyloïdes par le
zinc(II) soit le même en l’absence de TFE. En effet, dans ce cas, les intermédiaires
partiellement hélice alpha sont moins nombreux et l’agrégation plus lente. Néanmoins la
transformation de l’intermédiaire vers une structure amyloïde en feuillet bêta peut encore être
accélérée par le zinc. Il faut cependant noter que, l’intermédiaire en hélice ayant été observé
(Walsh et al. 1997; Walsh et al. 1999), il ne peut pas être le nucleus, et une accélération en
aval (après cet intermédiaire) est possible. On peut également supposer que ce mécanisme de
promotion de fibres amyloïdes peut être applicable à d’autres peptide ou protéines
(préférentiellement de structure native aléatoire) incluant le peptide Aβ40/42.
Il est aussi intéressant de remarquer qu’une récente publication (Garai et al. 2007) rapporte la
capacité du zinc à faire accélérer sélectivement l’agrégation de certains intermédiaires d’Aβ40.
Les intermédiaires concernés ont une taille d’environ 10-300 nm (mesurée par spectroscopie
de corrélation de fluorescence). Ils ont montré que le zinc n’avait pas d’effet sur la transition
du monomère vers l’intermédiaire, mais favorisait l’agrégation de ces intermédiaires vers des
agrégats non fibrillaires. Ainsi, en supposant que l’intermédiaire partiellement hélice alpha
observé par Teplow et coll. et l’intermédiaire de Garai et al. correspondent à la même espèce,
alors le modèle présenté dans ce chapitre concorde avec leurs observations.

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
162
III. Matériels et méthodes
Remarque : pour la préparation des solutions de peptides Aβ et de métaux, se reporter au
paragraphe matériels et méthodes du chapitre 3.
Coloration à la Thioflavine-T (ThT)
La ThT est un produit commercial dont le spectre caractéristique de fluorescence présente
avec un pic d’émission à 445 nm obtenu avec une excitation à 385 nm. Lorsque la ThT
interagit avec les structures en feuillet bêta commune aux structures amyloïdes, il en résulte
un nouveau maximum d’émission à 482 nm pour une excitation vers 450 nm (LeVine 1993).
Une solution de ThT à 2 μM est préparée par dilution successive dans le tampon, à laquelle
sont ajoutés de petits volumes de la solution à analyser (correspondant à 0.1 jusqu’à 2
équivalents de peptide par ThT). Une régression linéaire (hors saturation) permet une
quantification, relative à l’ensemble des échantillons analysés pour une date et une solution de
ThT données, précise.
Figure 4.8. Formule chimique de la ThT composée d’une extrémité hydrophobe (groupe
diméthylamine attaché à un groupe phényl) lié à un groupe benzothiazole plus polaire (via le souffre et l’azote chargé)
Il a été suggéré que la ThT se lie aux fibres amyloïdes selon les sillons formés par
l’interaction entre les feuillets bêta (Krebs et al. 2005). Mais cette hypothèse est controversée,
du fait que la ThT se lie également aux acides nucléiques (Canete et al. 1987) mais pas aux
fibres formées à partir de peptides portant une charge importante. Il a donc été également
proposé que la fixation de la ThT aux fibres d’Aβ implique des interactions à la fois
électrostatiques et hydrophobes (Figure 4.8) (Khurana et al. 2005).
Spectroscopie de fluorescence
Les spectres de fluorescence ont été collectés avec spectrophotomètre de fluorescence Cirad.
Les échantillons sont placés dans une cuve en quartz et les données sont enregistrées à

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
163
température ambiante. Pour la ThT, l’excitation se fait à 440 nm (bande passante : 4), et les
données sont enregistrées entre 460 – 550 nm (bande passante : 3, photomultiplicateur réglé à
1100 V).
Dichroïsme circulaire.
Les macromolécules biologiques, du fait de leur chiralité, sont optiquement actives. Par
conséquent, lorsqu’elles sont placées dans un faisceau de lumière polarisée rectilignement,
leurs chromophores asymétriques interagissent différemment avec la composante circulaire
droite et gauche de la lumière. Ceci donne lieu à deux phénomènes : 1) une rotation du plan
de polarisation de la lumière (ou pouvoir rotatoire) résultant de la biréfringence du milieu
optiquement actif (l’indice de réfraction est différent suivant la polarisation de la lumière), 2)
une absorption différente des deux composantes de la lumière (coefficients d’extinction
différents). Ce dernier phénomène, appelé dichroïsme circulaire, est très sensible à
l’agencement des chromophores entre eux et notamment, pour les protéines, des liaisons
peptidiques (fonction amide des liaisons peptidiques, étude dans l’UV lointain 180-250 nm).
Ainsi il est directement corrélé à la structure secondaire de la protéine, ce qui fait du
dichroïsme circulaire une méthode spectroscopique de choix pour l’étude de leur repliement.
Par comparaison avec les spectres de protéines de structure secondaire connue, il est possible
de déterminer relativement précisément les proportions d’hélices alpha, feuillets bêta
(parallèles ou antiparallèles) et structures désordonnées (random coil) au sein d’une protéine
(Figure 4.9).
Elli
ptic
ité
190 210 230 250Longueur d’onde (nm)
Elli
ptic
ité
190 210 230 250Longueur d’onde (nm)
Figure 4.9. Modèle de spectre de dichroïsme circulaire de protéines de structure 100% hélice
alpha ( ⎯ ), feuillet bêta (---) ou aléatoire ( ).

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
164
L’appareil de dichroïsme circulaire, ou spectropolarimètre, décompose la lumière en ses deux
composantes circulaires droite et gauche de mêmes intensité et amplitude. Après passage dans
l’échantillon optiquement actif, les deux composantes mesurées, absorbées différemment,
n’ont plus la même amplitude, leur résultante décrit une ellipse (figure 4.10).
Figure 4.10. Phénomène de dichroïsme circulaire. Un rayon de lumière polarisée rectiligne
monochromatique peut être en deux composantes circulaires gauche et droite de même amplitude (figure de gauche). Après passage au travers d’un échantillon optiquement actif,
les deux composantes sont absorbées différemment, le rayon polarisé résultant est elliptique.
Historiquement, l’ellipticité, exprimée en millidegrés et définie comme le rapport entre le
petit axe et le grand axe de l’ellipse, était le paramètre mesuré en dichroïsme circulaire. Cette
unité a perduré en dépit du fait que la grandeur mesurée par les instruments actuels est la
différence d’absorption, paramètre qui obéit à la loi de Beer-Lambert. Les deux unités sont
toutefois reliées. Soit Δε le dichroïsme circulaire c’est-à-dire la différence de coefficient
molaire d’absorption, εg et εd les coefficients d’extinction molaire des ondes polarisées
circulairement respectivement à gauche et à droite, ag et ad les absorptions correspondantes, l
longueur du trajet optique et c concentration molaire de l’espèce considérée :
claa dg
dg .−
=−=Δ εεε (M-1.cm-1)
Pour relier le dichroïsme circulaire à l’ellipticité, on fait l’hypothèse que l’effet de ce
dichroïsme est faible, autrement dit Δa << 1. Sous cette approximation, l’ellipticité molaire [θ]
est proportionnelle à Δε :

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
165
εΔ=Θ .3298][ (deg.cm2.dmol-1)
Une fois le spectre obtenu, il est possible d’utiliser l’un des nombreux logiciels de
déconvolution disponibles afin de déterminer le pourcentage de chacune des différentes
conformations (les données exposées dans ce chapitre ont été déconvoluées avec le logiciel
DichoProt) (Greenfield et al. 1969; Andrade et al. 1993; Deleage et al. 1993; Sreerama and
Woody 2000).
Les données décrites dans cet ouvrage ont été obtenues sur un spectropolarimètre Jobin &
Yvon CD6 (Division d’Instrumentation S.A., Longjumeau, France), à 298 K, avec des
cellules en quartz de 1 mm d’épaisseur hermétiquement bouchées pour éviter l’évaporation en
particulier du TFE (solution commerciale à 99%, Aldrich).
Etude de l’agrégation par test BCA.
Les différents échantillons ont été incubés 30 minutes à 37°C puis centrifugés 30 minutes à
20000 g à 4°C. Les fractions d’Aβ28 soluble et agrégé ont été déterminées en mesurant le
contenu en peptide des culots et surnageants grâce au test colorimétrique de détection de
protéines BCA (pour plus de précision quant au principe de ce test, se reporter au paragraphe
Matériels et Méthodes du chapitre 3).

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
166
Références M. A. Andrade, P. Chacon, J. J. Merelo and F. Moran (1993). "Evaluation of secondary
structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network." Protein Eng, 6(4), 383-90.
C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, et al. (1998). "Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis." J Biol Chem, 273(21), 12817-26.
A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, et al. (2006). "Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement." J Am Chem Soc, 128(30), 9893-901.
A. I. Bush, W. H. Pettingell, G. Multhaup, M. d Paradis, J. P. Vonsattel, et al. (1994). "Rapid induction of Alzheimer A beta amyloid formation by zinc." Science, 265(5177), 1464-7.
M. Canete, A. Villanueva, A. Juarranz and J. C. Stockert (1987). "A study of interaction of thioflavine T with DNA: evidence for intercalation." Cell Mol Biol, 33(2), 191-9.
F. Chiti and C. M. Dobson (2006). "Protein misfolding, functional amyloid, and human disease." Annu Rev Biochem, 75333-66.
C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, et al. (2001). "Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits." J Biol Chem, 276(23), 20466-73.
J. Danielsson, R. Pierattelli, L. Banci and A. Graslund (2007). "High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid beta-peptide." Febs J, 274(1), 46-59.
G. Deleage and C. Geourjon (1993). "An interactive graphic program for calculating the secondary structure content of proteins from circular dichroism spectrum." Comput Appl Biosci, 9(2), 197-9.
Y. Fezoui and D. B. Teplow (2002). "Kinetic studies of amyloid beta-protein fibril assembly. Differential effects of alpha-helix stabilization." J Biol Chem, 277(40), 36948-54.
D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly (2007). "Functional amyloid--from bacteria to humans." Trends Biochem Sci, 32(5), 217-24.
E. Gaggelli, F. Bernardi, E. Molteni, R. Pogni, D. Valensin, et al. (2005). "Interaction of the human prion PrP(106-126) sequence with copper(II), manganese(II), and zinc(II): NMR and EPR studies." J Am Chem Soc, 127(3), 996-1006.
K. Garai, B. Sahoo, S. K. Kaushalya, R. Desai and S. Maiti (2007). "Zinc Lowers Amyloid-beta Toxicity by Selectively Precipitating Aggregation Intermediates." Biochemistry, 46(37), 10655-63.
N. Greenfield and G. D. Fasman (1969). "Computed circular dichroism spectra for the evaluation of protein conformation." Biochemistry, 8(10), 4108-16.
L. Hou and M. G. Zagorski (2006). "NMR reveals anomalous copper(II) binding to the amyloid Abeta peptide of Alzheimer's disease." J Am Chem Soc, 128(29), 9260-1.
E. House, J. Collingwood, A. Khan, O. Korchazkina, G. Berthon, et al. (2004). "Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease." J Alzheimers Dis, 6(3), 291-301.
T. R. Jahn and S. E. Radford (2005). "The Yin and Yang of protein folding." Febs J, 272(23), 5962-70.
A. Khan, A. E. Ashcroft, O. V. Korchazhkina and C. Exley (2004). "Metal-mediated

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
167
formation of fibrillar ABri amyloid." J Inorg Biochem, 98(12), 2006-10. R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, et al. (2005).
"Mechanism of thioflavin T binding to amyloid fibrils." J Struct Biol, 151(3), 229-38. M. D. Kirkitadze, M. M. Condron and D. B. Teplow (2001). "Identification and
characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis." J Mol Biol, 312(5), 1103-19.
D. A. Kirschner, H. Inouye, L. K. Duffy, A. Sinclair, M. Lind, et al. (1987). "Synthetic peptide homologous to beta protein from Alzheimer disease forms amyloid-like fibrils in vitro." Proc Natl Acad Sci U S A, 84(19), 6953-7.
G. M. Klug, D. Losic, S. S. Subasinghe, M. I. Aguilar, L. L. Martin, et al. (2003). "Beta-amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous ageing at neutral pH." Eur J Biochem, 270(21), 4282-93.
S. A. Kozin, S. Zirah, S. Rebuffat, G. H. Hoa and P. Debey (2001). "Zinc binding to Alzheimer's Abeta(1-16) peptide results in stable soluble complex." Biochem Biophys Res Commun, 285(4), 959-64.
M. R. Krebs, E. H. Bromley and A. M. Donald (2005). "The binding of thioflavin-T to amyloid fibrils: localisation and implications." J Struct Biol, 149(1), 30-7.
H. LeVine, 3rd (1993). "Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution." Protein Sci, 2(3), 404-10.
S. T. Liu, G. Howlett and C. J. Barrow (1999). "Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A beta peptide of Alzheimer's disease." Biochemistry, 38(29), 9373-8.
Y. Mekmouche, Y. Coppel, K. Hochgrafe, L. Guilloreau, C. Talmard, et al. (2005). "Characterization of the ZnII binding to the peptide amyloid-beta1-16 linked to Alzheimer's disease." Chembiochem, 6(9), 1663-71.
T. Miura, K. Suzuki, N. Kohata and H. Takeuchi (2000). "Metal binding modes of Alzheimer's amyloid beta-peptide in insoluble aggregates and soluble complexes." Biochemistry, 39(23), 7024-31.
S. Ohnishi and K. Takano (2004). "Amyloid fibrils from the viewpoint of protein folding." Cell Mol Life Sci, 61(5), 511-24.
M. B. Pepys (2006). "Amyloidosis." Annu Rev Med, 57223-41. B. Raman, T. Ban, K. Yamaguchi, M. Sakai, T. Kawai, et al. (2005). "Metal ion-dependent
effects of clioquinol on the fibril growth of an amyloid {beta} peptide." J Biol Chem, 280(16), 16157-62.
J. C. Sacchettini and J. W. Kelly (2002). "Therapeutic strategies for human amyloid diseases." Nat Rev Drug Discov, 1(4), 267-75.
N. Sreerama and R. W. Woody (2000). "Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set." Anal Biochem, 287(2), 252-60.
F. Stellato, G. Menestrina, M. D. Serra, C. Potrich, R. Tomazzolli, et al. (2006). "Metal binding in amyloid beta-peptides shows intra- and inter-peptide coordination modes." Eur Biophys J, 35(4), 340-51.
C. D. Syme and J. H. Viles (2006). "Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer's disease." Biochim Biophys Acta, 1764(2), 246-56.
C. Talmard, L. Guilloreau, Y. Coppel, H. Mazarguil and P. Faller (2007). "Amyloid-beta peptide forms monomeric complexes with Cu(II) and Zn(II) prior to aggregation." Chembiochem, 8(2), 163-5.

Chapitre 4 : mécanisme de promotion de l’agrégation d’Aβ par le zinc(II)
168
D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow (1997). "Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate." J Biol Chem, 272(35), 22364-72.
D. M. Walsh, D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. M. Condron, et al. (1999). "Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates." J Biol Chem, 274(36), 25945-52.
L. E. Wilkinson-White and S. B. Easterbrook-Smith (2007). "Characterization of the binding of Cu(II) and Zn(II) to transthyretin: effects on amyloid formation." Biochemistry, 46(31), 9123-32.
D. S. Yang, J. McLaurin, K. Qin, D. Westaway and P. E. Fraser (2000). "Examining the zinc binding site of the amyloid-beta peptide." Eur J Biochem, 267(22), 6692-8.
Y. Yoshiike, K. Tanemura, O. Murayama, T. Akagi, M. Murayama, et al. (2001). "New insights on how metals disrupt amyloid beta-aggregation and their effects on amyloid-beta cytotoxicity." J Biol Chem, 276(34), 32293-9.
S. Zirah, S. Rebuffat, S. A. Kozin, P. debey, F. Fournier, et al. (2003). "Zinc binding properties of the amyloid fragment Ab(1-16) studied by electrospray-ionization mass spectrometry." International Journal of Mass Spectrometry, 228999-1016.
S. Zirah, S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, et al. (2006). "Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging." J Biol Chem, 281(4), 2151-61.

169
Chapitre 5
Etude de l’interaction entre la
métallothionéine-3 et
le peptide amyloïde bêta,
influence du stress oxydant

170

Chapitre 5 : l’interaction MT-3 / Aβ
171
I. Introduction bibliographique
Les métallothionéines (MT) sont une famille de protéines de faible poids moléculaire (6-7
kDa, 60 à 68 acides aminés), riches en cystéines (25 à 30%, Figure 5.1) et qui ne contiennent
généralement pas d’acide aminé aromatique et d’histidine (Vasto et al. 2007). Les MT sont
ubiquitaires dans le règne animal et ont également été décrites chez les végétaux et chez les
procaryotes (Zhou and Goldsbrough 1994; Zhou and Goldsbrough 1995). Du fait de leur
richesse en cystéines, elles possèdent la capacité de fixer, par des interactions non covalentes,
des ions métalliques des groupes Ib et IIb de configuration électronique d10 (en particulier le
zinc(II) et le cuivre(I)) : 7 ions divalents et 8 à 14 ions monovalents par MT. Ainsi, elles sont
supposées jouer un rôle majeur dans le transport et/ou le stockage du zinc intracellulaire mais
sont également impliquées dans l’homéostasie du cuivre et dans la séquestration de métaux
toxiques tels le cadmium et le mercure (affinité relative vis-à-vis du groupement thiol :
Hg(II)>> Cu(I), Ag(I), Bi(III) >> Cd(II) > Pb(II) > Zn(II) > Co(II) > Fe(II) (Vasak 1991).
Dans les conditions naturelles, les métallothionéines ne sont généralement pas complexées par
un métal unique, le contenu en métaux de MT purifiées est variable et dépend de l’organisme,
du tissu, et de l’histoire de l’exposition aux métaux (Hamer 1986). Les métaux peuvent être
libérés des MT lorsque l’on abaisse le pH (Daniels et al. 1998).
Chez les mammifères, on retrouve 4 isoformes : MT-1, 2, 3 et 4 (Vasak and Hasler 2000).
MT-1 et 2 sont présentes dans tous les organes, MT-3 est principalement exprimée dans le
cerveau et MT-4 est limitée aux épithéliums cellulaires tels que celui de la peau ou de la
langue (Quaife et al. 1994; Moffatt and Denizeau 1997; Vasak et al. 2000).
MT-1 MDPN·CSCSTGGSCTCTSSCACKNCKCTSCKKSCCSCCPVGCSKCAQGCVCKGA······ADKCTCCA
MDPN·CSCATDGSCSCAGSCKCKQCKCTSCKKSCCSCCPVGCAKCSQGCICKEA······SDKCSCCAMT-2domaine β domaine α
1 10 20 30 40 50 60 68¦ ¦ ¦ ¦ ¦ ¦ ¦ ¦MDPETCPCPSGGSCTCADSCKCEGCKCTSCKKSCCSCCPAECEKCAKDCVCKGGEAAEAEAEKCSCCQMT-3 / GIF
MT-1 MDPN·CSCSTGGSCTCTSSCACKNCKCTSCKKSCCSCCPVGCSKCAQGCVCKGA······ADKCTCCA
MDPN·CSCATDGSCSCAGSCKCKQCKCTSCKKSCCSCCPVGCAKCSQGCICKEA······SDKCSCCAMT-2domaine β domaine α
1 10 20 30 40 50 60 68¦ ¦ ¦ ¦ ¦ ¦ ¦ ¦MDPETCPCPSGGSCTCADSCKCEGCKCTSCKKSCCSCCPAECEKCAKDCVCKGGEAAEAEAEKCSCCQMT-3 / GIF
Figure 5.1. Séquences peptidiques des MT-3, MT-1 et MT-2
En plus de la régulation de l’homéostasie des métaux, les MT jouent également un rôle anti-
oxydant, anti-apoptotique, protecteur lors de lésions cérébrales (MT-3), régulateur de
croissance neuronale (MT-3) (Moffatt et al. 1997; Hidalgo et al. 2001).

Chapitre 5 : l’interaction MT-3 / Aβ
172
La cristallographie et la résonance magnétique nucléaire (RMN) ont permis la détermination
structurale de quelques MT, mettant ainsi en évidence les caractéristiques communes à cette
famille de protéines. Les MT possèdent 2 domaines de fixation comprenant généralement
chacun un cluster de 3 (domaine β, N-terminal, Zn3(CysS)9) ou 4 (domaine α, C-terminal,
Zn4(CysS)11) métaux (cf. figure 5.2). Dans ces clusters, les métaux sont coordonnés dans une
géométrie tétraédrique formée par les fonctions thiolates des cystéines, pontants ou terminaux
(Figure 5.3). La fixation des ions sur les deux domaines n’est pas figée et leur déplacement
d’un site de fixation à un autre est toujours possible (Otvos et al. 1993). Lorsque les MT ne
sont pas complexées avec des ions métalliques, l’absence de ponts disulfures au sein de la
chaîne confère à ces molécules une structure tridimensionnelle hautement désordonnée
caractéristique d’une chaîne d’acides aminés aléatoire. Au contraire, la structure
tridimensionnelle de la protéine complexée est entièrement dominée par l’organisation de la
chaîne d’acides aminés autour des métaux (Stillman et al. 2000).
Figure 5.2. Structure tridimensionnelle de la (Cd5,Zn2)-MT-2 de rat (les sphères représentent
les ions métalliques insérés entre les résidus cystéines) (Romero-Isart and Vasak 2002).(

Chapitre 5 : l’interaction MT-3 / Aβ
173
Figure 5.3. Représentations schématiques des clusters Zn-thiolate dans les domaines de fixation N- (à gauche) et C-terminal (à droite) des MT (Maret 2006).
L’affinité des MT pour le zinc est élevée, généralement entre 1010 et 1014 M-1 (Kaji et al. 1993;
Jacob et al. 1998), et vers 6,2.1010 pour la MT-3 humaine (Hasler et al. 2000; Romero-Isart et
al. 2002). Les sites de fixation semblent cinétiquement labiles, permettant ainsi des transferts
intramoléculaires (Otvos et al. 1993; Romero-Isart et al. 2002).
Nous nous intéresserons ici plus particulièrement à la MT-3, anciennement appelée growth
inhibitory factor (GIF) du fait de sa propriété à inhiber la croissance neuronale. La MT-3 se
retrouve principalement et logiquement dans les neurones riches en zinc, tels que ceux de
l’hippocampe et para-hippocampe (Kojima et al. 1999). Bien que de nombreuses études
montrent une élévation de la production de MT-1 et 2 avec l’âge, les résultats concernant la
MT-3 sont plus controversés, diminution et élévation de la concentration de MT-3 ont été
décrites (Uchida et al. 1991; Miyazaki et al. 2002; Gomi et al. 2005). De même, chez les
patients atteints de la MA, le taux de MT-3 a été rapporté être, soit nettement diminué
(Uchida et al. 1991; Tsuji et al. 1992; Yu et al. 2001), soit stable (Erickson et al. 1994;
Amoureux et al. 1997). Il est possible que les méthodes de détection utilisées influent sur ces
résultats (utilisation d’anticorps spécifiques de la protéine, mesure de la quantité d’ARN).
Son rôle est encore mal connu, elle est probablement impliquée dans la régulation de
l’homéostasie des métaux dans le cerveau et aurait des fonctions protectrices contre les
métaux toxiques. Elle semble également interagir avec de nombreuses protéines (la protéine
de choc thermique 84 (HSP84) et 70 (HSP70), la protéine reliée à la dihydropyrimidinase 2,
la créatine kinase et la ß-actine) (El Ghazi et al. 2006), ce qui pourrait lui conférer de
nouvelles fonctions qui restent à déterminer. De plus, sous l’influence de stress oxydant, sa
concentration intra- et extracellulaire augmente, reflétant ses propriétés anti-oxydantes et son
probable rôle de messager chimique dans les processus anti-inflammatoire (Chen et al. 2006;
Lynes et al. 2006).
Il a été montré que la MT-3 (mais ni MT-1 ou 2) protégeait les neurones (en culture cellulaire)

Chapitre 5 : l’interaction MT-3 / Aβ
174
des effets toxiques d’Aβ (Irie and Keung 2001), en particulier à faible concentration (Irie and
Keung 2003). Mais le mécanisme sous-tendant cette protection et sa spécificité n’a pas encore
été élucidé. Récemment, une étude a mis-à-jour la capacité de la Zn7MT-3 à lier les ions Cu2+,
via la libération de trois ions zinc(II), pour former un cluster de 4 cuivre(I) réduits
particulièrement stable, dans le domaine N-terminal (Meloni et al. 2007). Ainsi, sur ce modèle,
la MT-3 pourrait prendre et ainsi stabiliser le cuivre, libre (dû à la dérégulation de
l’homéostasie de ce métal dans la MA) ou lié au peptide Aβ (affinité plus faible), supprimant
ainsi la principale source de production de ROS (Barnham et al. 2004; Kitzberger et al. 2005;
Gaggelli et al. 2006).
II. La MT3, source potentielle du zinc retrouvé dans les plaques amyloïdes
Les plaques amyloïdes contiennent une quantité anormalement élevée de métaux de transition
comme le cuivre et le zinc, mais leur provenance est actuellement largement méconnue. Dans
le cas du zinc, il est possible qu’une petite partie du zinc(II) libéré dans la synapse lors de la
transmission synaptique (10 nM à 300μM), se lie à Aβ (Assaf and Chung 1984; Howell et al.
1984). Cependant, la présence de cette forte concentration en zinc libre est très limitée dans le
temps du fait de sa liaison rapide aux ligands et récepteurs de plus forte affinité pour le zinc
qu’Aβ, présents dans l’espace intersynaptique.
Une autre alternative serait de s’intéresser aux protéines de transport ou de stockage du zinc,
or, dans le cerveau la MT-3 en est une des principales. Les études portant sur la variation de
son expression avec l’âge et chez les personnes atteintes de la MA sont contradictoires.
Cependant, dans le cas de la MA, la concentration de MT-3 extracellulaire semble augmenter,
alors qu’elle est plutôt intracellulaire chez le sujet sain (Chen et al. 2006; Lynes et al. 2006). Il
a également était remarqué une nette élévation du stress oxydant avec l’âge et encore plus
chez le sujet atteint de la MA, qui pourrait être la cause de la présence marquée de MT-3
extracellulaire (celle-ci se comportant alors comme senseur du stress oxydant). Ainsi la MT-3
pourrait agir en tant que piégeur d’espèces oxydantes, via la formation d’un pont cystéine et la
libération de Zn2+ (cf. Figure 5.4) (Maret and Vallee 1998). Le zinc libéré, en particulier si le
relarguage a lieu dans le cytosol, pourrait alors être capté par les peptides Aβ.

Chapitre 5 : l’interaction MT-3 / Aβ
175
Figure 5.4. L’oxydation des soufres des cystéines est accompagnée des la libération d’ions
Zn2+ et de la formation d’un pont disulfure (Maret 2006).
Afin de confirmer cette hypothèse, l’interaction entre la MT-3 et le peptide Aβ a été étudiée
en collaboration avec l’équipe du Professeur Vasak de l’université de Zurich.
III. Résultats et Discussion
III.1. Hypothèse
Nous avons précédemment remarqué que la présence d’ions zinc(II) accélère l’agrégation d’
Aβ. Ainsi, à partir d’une certaine concentration en peptide, dépendante de la longueur de ce
dernier, la précipitation peut être relativement rapide (c > 10 μM pour Aβ40 et > 30 μM pour
Aβ28 dans nos conditions). Ainsi, si la MT-3 cède bien le zinc(II) qu’elle contient en
présence de stress oxydant (représenté dans l’ensemble des expériences par du peroxyde
d’hydrogène), on peut formuler l’hypothèse suivante qui va permettre de caractériser les
échanges de zinc entre MT et Aβ (Figure 5.5).
R-S-Zn-S-R’ + H2O2 + 2 H+ R-S-S-R’ + 2 H2O + Zn2+
(Zn7-MT-3) pont disulfure
Agrégation du complexe ZnII-Aβ Aβ
R-S-Zn-S-R’ + H2O2 + 2 H+ R-S-S-R’ + 2 H2O + Zn2+
(Zn7-MT-3) pont disulfure
Agrégation du complexe ZnII-Aβ AβAβ
Figure 5.5. Représentation schématique de l’hypothèse de travail (pour une représentation
correcte du site de fixation de ZnII dans les MT, voir figure 5.3).
En effet, le dosage du culot (Aβ-Zn) et du surnageant (Zn7-x-MT-3, Aβ restant), devrait nous

Chapitre 5 : l’interaction MT-3 / Aβ
176
permettre de quantifier le relarguage du zinc vers le peptide Aβ en présence de stress oxydant.
III.2. Expériences avec le modèle Cd7-MT-3
III.2.a. Interaction entre Cd7-MT-3 et Aβ
Dans un premier temps, il est important de savoir si le peptide Aβ est capable de capter le zinc
lié à la MT-3 en l’absence de peroxyde d’hydrogène. Cela peut sembler peu probable compte
tenu des constantes d’affinité rapportées pour la MT-3, très supérieures à celle d’Aβ.
Cependant il s’agit de constantes globales, c’est-à-dire une moyenne sur les sept ZnII, il est
donc possible qu’un zinc soit moins fortement lié. Une méthode simple consiste à regarder la
perte en liaison métal-Cys en spectroscopie UV, cependant, dans le cas du zinc, la bande
d’absorption de la liaison Zn-Cys (235 nm) est trop proche de celle des liaisons peptidiques.
On utilise donc le la MT-3 chargée en cadmium (bande Cd-Cys à 250 nm) dont la réactivité
est similaire à celle du zinc (sans toutefois être identique, ainsi sa constante d’affinité pour la
MT-3 est supérieure, de l’ordre de 1014 M-1 pour l’ensemble des CdII) (Romero-Isart et al.
2002).
Ainsi des solutions de MT-3 avec 2 et jusqu’à 10 équivalents d’Aβ40 ont été réalisées (en
tampon Hepes 10 mM, NaCl 20 mM, pH 7,4) et suivis pendant plus de 4 heures. La figure 5.6
montre que même pour 10 équivalents d’Aβ (160 μM contre 16 de MT-3), la perte en
cadmium de la MT-3 n’est pas significative. Des résultats similaires ont été obtenus pour des
ratios Aβ / MT-3 inférieurs.

Chapitre 5 : l’interaction MT-3 / Aβ
177
0
1
2
220 240 260 280 300 320
Longueur d'onde (nm)
Abs
orba
nce
0 min40 min1h102h504h20
Figure 5.6. Spectres UV d’une solution d’Aβ40 (160 μM), Cd7-MT-3 (16 μM) (tampon Hepes
10 mM, NaCl 20 mM, pH 7,4) laissée à température ambiante. L’épaulement à 250 nm correspond à la liaison Cd-Cys.
Il apparaît donc que, en l’absence de peroxyde d’hydrogène, le peptide Aβ ne soit pas capable
de capter le cadmium de la MT-3.
III.2.b. Interaction entre Cd7-MT-3 et Aβ en présence de H2O2
Apres avoir montré que la présence d’Aβ n’induit pas de relarguage de cadmium par la MT-3,
l’expérience est répétée en ajoutant du peroxyde d’hydrogène, afin d’évaluer le rôle du stress
oxydant. On observe alors une diminution significative de la bande de transfert de charge
(Cd-Cys) à 250 nm, vraisemblablement due à une oxydation des cystéines et un relarguage de
cadmium. La figure 5.7, représentant la quantité de cadmium relargué (estimée d’après la
diminution du de l’absorption à 250 nm), indique une dépendance au temps et à la quantité de
H2O2 introduite. La réaction est relativement lente (s’étale sur plusieurs heures) et 10µM d’
H2O2 sont suffisants pour relarguer une quantité significative de Cd(II). L’utilisation de
concentrations supérieures en peroxyde d’hydrogène induit une augmentation de la quantité
de cadmium libéré par la MT-3.

Chapitre 5 : l’interaction MT-3 / Aβ
178
0
5
10
15
20
25
30
0 1 2 3 4 5 6t (h)
[CdII ]
rela
rgué
(μM
)0
0.51
1.52
220 250 280 310λ (nm)
Abs
100 μM H2O2
00.5
11.5
2
220 250 280 310λ (nm)
Abs
100 μM H2O2
0
5
10
15
20
25
30
0 1 2 3 4 5 6t (h)
[CdII ]
rela
rgué
(μM
)
0
5
10
15
20
25
30
0 1 2 3 4 5 6t (h)
[CdII ]
rela
rgué
(μM
)0
0.51
1.52
220 250 280 310λ (nm)
Abs
100 μM H2O2
00.5
11.5
2
220 250 280 310λ (nm)
Abs
100 μM H2O2
Figure 5.7. Suivi dans le temps du relarguage de cadmium par 20 μM de Cd7-MT-3 en
présence de 20 μM d’Aβ40 et 0 ( rouge), 10 ( vert), 20 ( orange) ou 100 μM ( bleu) de H2O2 dans le tampon habituel. Les valeurs des concentrations de cadmium relargué ont
été calculées à partir des spectres d’absorption (en insert : diminution dans le temps du signal Cd-Cys à 250 nm, en présence de 20 μM d’ Aβ40 et 100 μM d’ H2O2).
III.2.c. Interaction entre Zn7-MT-3 et Aβ, en présence de H2O2
L’on sait à présent que la présence de H2O2 induit un relarguage du métal pour la Cd7-MT-3,
mais en est-il de même pour la forme native chargée en zinc. Comme indiqué précédemment,
la bande de transfert de charge de Zn-MT-3 se situe vers 235 nm, elle est donc beaucoup plus
difficile à suivre que celle de la MT-3 chargée en cadmium (à 250 nm). Le choix de la
méthode d’analyse pour suivre la réaction entre la Zn7-MT-3 et H2O2 s’est donc porté sur la
chromatographie d’exclusion stérique (SEC). Ainsi des solutions de 20 μM de Zn7-MT-3
(dans le tampon Hepes 10 mM, NaCl 20 mM à pH 7,4) contenant différentes concentrations
de H2O2 ont été éluées sur chromatographie d’exclusion stérique (SEC) après 30 minutes à
température ambiante (Figure 5.8).

Chapitre 5 : l’interaction MT-3 / Aβ
179
0
25
50
75
100
125
150
175
200
225
250
5 6 7 8 9 10 11 12 13 14
Volume d'élution (mL)
Abs
220
nm
0 uM H2O25 uM H2O210 uM H2O220 uM H2O2100 uM H2O2200 uM H2O2500 uM H2O2
monomères
dimèrestrimères
0
25
50
75
100
125
150
175
200
225
250
5 6 7 8 9 10 11 12 13 14
Volume d'élution (mL)
Abs
220
nm
0 uM H2O25 uM H2O210 uM H2O220 uM H2O2100 uM H2O2200 uM H2O2500 uM H2O2
monomères
dimèrestrimères
Figure 5.8. Chromatogramme de solutions à 20 μM en Zn-MT-3 contenant 0, 5, 10, 20, 100, 200 ou 500 μM d’eau oxygénée (détection de l’absorption à 220 nm, tampon 10 mM Hepes,
20 mM Na Cl, pH 7,4).
Le coefficient d’absorption à 220 nm étant affecté par le relarguage du zinc (baisse du signal
Zn-Cys à 235 nm), cette étude est strictement qualitative, cependant on peut constater que
l’ajout de H2O2 provoque une diminution de l’absorbance totale à 220 nm que l’on peut
attribuer au relarguage du zinc. Mais on ne peut pas exclure une perte de matériel due à la
formation d’agrégats trop grands pour s’insérer dans la matrice de la colonne (cependant les
résultats décrits sur la figure 5.10 tendent à invalider cette dernière hypothèse). On peut
également remarquer que l’ajout d’H2O2 entraîne : 1) l’apparition d’espèces oligomériques
qui témoigne de la création de pont disulfures intermoléculaires ; 2) un déplacement du pic
des espèces monomériques attestant d’une diminution du rayon hydrodynamique engendrée
par la formation de pont disulfures intramoléculaires. L’ensemble de ces phénomènes indique
la libération d’ions Zn2+ par la MT-3. L’hypothèse de départ est donc en partie vérifiée
(Figure 5.5) : la présence de peroxyde d’hydrogène provoque un relarguage partiel mais
significatif du zinc de la MT-3.

Chapitre 5 : l’interaction MT-3 / Aβ
180
III.3. Agrégation d’Aβ en présence de H2O2 et de Zn7-MT-3
Afin de vérifier si le zinc (ou du moins une partie des clusters stabilisé par les cystéines)
libéré par la MT-3 est capable de se fixer sur l’Aβ et de provoquer son agrégation, un suivi de
la turbidimétrie d’un mélange de 50 μM d’Aβ40 et 500 μM de H2O2 a été réalisé (le choix,
dans un premier temps, d’une forte concentration en peroxyde d’hydrogène devant permettre
de mettre en évidence le phénomène). La figure 5.9 montre clairement que l’ajout d’un
équivalent de zinc induit rapidement l’agrégation d’une grande partie du peptide Aβ présent
dans la solution. La présence de MT-3 chargée en zinc et de H2O2 n’ont pas d’effet immédiat
mais semble après un certain temps (un peu plus d’une heure) déboucher sur la même
situation, l’agrégation massive du peptide. Cette agrégation ne peut pas être due directement à
l’eau oxygénée puisque cette dernière n’induit quasiment aucune augmentation de
l’absorption et est donc vraisemblablement le fait de la libération de zinc par la MT-3. La
cinétique de l’agrégation d’Aβ est du même ordre de grandeur que celle du relarguage du
cadmium sous les mêmes conditions, donc, en supposant que le zinc se comporte de façon
similaire au cadmium, cela implique que c’est bien le zinc relargué par la MT-3 qui induit
l’agrégation du peptide Aβ.
0
0.05
0.1
0.15
0.2
0.25
0.3
0 10 20 30 40 50 60 70 80
Evol
utio
n de
l'ab
sorp
tion
à 35
0 nm
t (min)
Figure 5.9. Evolution de la turbidité d’échantillons à 50 μM d’Aβ40 (tampon habituel) contenant : ♦ (bleu) un équivalent de zinc(II), ● (rouge) 500 μM de H2O2 et 20 μM de Zn7-
MT-3, ■ (vert) uniquement 500 μM de H2O2.

Chapitre 5 : l’interaction MT-3 / Aβ
181
Afin de corroborer les données obtenues en turbidimétrie par le suivi de l’agrégation d’Aβ
induit par le zinc relargué de la MT-3 en présence de H2O2, il a été choisi de suivre la réaction
par centrifugation et dosage du zinc, Aβ et MT-3 dans le culot et le surnageant. La
composition en acides aminés d’Aβ et MT-3 étant assez différente (par exemple, présence de
cystéines dans la MT-3 et pas dans Aβ, etc.), il est possible de doser leur mélange par analyse
d’acide amine. Ainsi, en considérant l’hypothèse exprimée précédemment, des solutions
contenant Aβ28 (50 μM), Zn7MT-3 (20 μM) et différentes concentrations d’eau oxygénée (0
à 500 μM) ont été laissées 3 heures à 25 °C sous faible agitation, puis centrifugées. Les culots
et surnageants ont ensuite été analysés par spectroscopie d’absorption atomique et analyse
d’acides aminés. Les résultats sont exposés sur la figure 5.10.
0
10
20
30
40
50
60
70
Con
cent
ratio
n ag
régé
e (u
M)
0 5 20 100 500
[H2O2] (uM)
Figure 5.10. Concentrations d’Aβ28 (bleu), MT-3 (violet) et zinc (jaune) ayant agrégées, en 3h, en fonction de la concentration de H2O2 présente dans le milieu (tampon Hepes 10 mM, NaCl 20 mM, pH 7,4 ; pourcentage d’erreur jusqu’à 15%). Concentrations initiales sont 50
μM Aβ28, 20 μM Zn7-MT-3.
Les résultats montrent que les concentrations en Aβ et zinc agrégés augmentent avec la
quantité de peroxyde d’hydrogène introduit initialement. Par contre l’agrégation de la MT-3
reste marginal voir non significative (< 2 μM, c’est-à-dire très proche de la limite de détection
(0,75 μM). Ceci est donc en accord avec l’hypothèse que le H2O2 provoque la relarguage du
zinc, qui se fixe sur l’Aβ et entraîne ainsi l’agrégation du complexe ZnII-Aβ. Cependant,
malgré cette confirmation qualitative des résultats, il faut noter que la présence de zinc dans
les agrégats est largement sur-stoechiométrique par rapport à Aβ28. Cet effet aurait pu être

Chapitre 5 : l’interaction MT-3 / Aβ
182
imputé à la présence de MT-3 qui même agrégée peut encore contenir quelques équivalents de
zinc, mais on ne la retrouve qu’en très petite quantité dans les agrégats. De plus le phénomène
a été reproduit même en absence de MT-3, il s’agit donc probablement d’un problème de bruit
de fond.
Conclusion
Le présent chapitre est principalement constitué de résultats préliminaires, en effet une
majeure partie des manipulations présentées n’ont pas pu être répétées, faute de temps. Ainsi
au vu de ces premiers résultats qui restent à confirmer, il apparaît que si le peptide Aβ et la
MT-3 (majoritairement chargée en zinc dans le cerveau) coexistent dans un milieu où règne
un stress oxydant prolongé (quelques heures), la MT-3 soit capable de céder une partie de son
zinc à Aβ. Le stress oxydant augmentant nettement avec l’âge, de même, d’après la littérature,
que la localisation extracellulaire de la MT-3, il est possible que cette situation favorise la
formation de plaques amyloïdes possédant une grande quantité de zinc.
IV. Matériels et méthodes
Remarque : pour la préparation des solutions de peptides Aβ et de métaux, se reporter au
paragraphe matériels et méthodes du chapitre 3.
IV.1. Matériels
IV.1.a. Préparation de Cd7-MT-3 et Zn7-MT-3.
La MT-3 a été fournie aimablement par le groupe de Milan Vasak de l’université de Zurich.
La MT-3 humaine est surexprimée dans E. coli puis purifiée. Elle est conservée en milieu
réducteur en présence de mercaptoéthanol (en effet, il semble que la ZnxCuy-MT-3 ait
tendance à s’oxyder à l’air, formant ainsi des ponts disulfures pour finalement libérer du zinc).
Les solutions stocks de MT-3 sont préparées en solubilisant le peptide (~ 1mg) dans l’eau
désionisée (150 μl), auxquelles sont rajoutés 8μl de mercaptoéthanol à 144 mM. La séparation
de la MT-3 et du mercaptoéthanol est réalisée juste avant son utilisation sur une colonne
(pipette pasteur) de résine d’exclusion stérique séphadex G-15 avec pour éluant une solution

Chapitre 5 : l’interaction MT-3 / Aβ
183
tampon de 20mM Hepes, 100mM NaCl, pH 7,4. La concentration des échantillons est
déterminée par un dosage des cystéines. Ce dosage est basé sur la réaction de la dithiopyridine
(en excès) avec les thiols de la MT-3 pour donner le 2- pyridinethiol, qui présente une bande
d’absorption caractéristique à 343nm (ε = 7600 M-1.cm-1). Sachant que la MT-3 contient 20
fonctions thiols, la concentration peut être déterminée suivant la formule suivante : [MT] =
DO343/(7060x20) (Roschitzki and Vasak 2002).
Homométallation de la MT-3 :
Dans un premier temps l’apo-MT-3 est préparé, à partir de Znx,Cdy-MT-3 obtenue
précédemment, par chromatographie d’exclusion stérique (Sephadex G-50) à pH 2 (Vasak et
al. 1987). La fraction correspondant au pic monomérique est alors collectée et lyophilisée
après avoir ajuster le pH à 2,5 par dilution. Ensuite, afin de charger la protéine en cadmium ou
zinc, 20 mg d’apo-MT-3 diluée dans 50 mM HCl est mélangé à 8 équivalents de CdCl2 ou
ZnCl2 au même pH. Pour que le métal se lie, le pH est élevé jusqu’à 8,6 avec une solution à
0,5 M Tris. L’ensemble de cette opération se déroule sous atmosphère inerte et les solutions
sont préalablement dégazées. Une petite portion de Chelex 100 est alors ajoutée à la solution,
puis éliminée par centrifugation, le surnageant est concentré par ultrafiltration. Ce dernier est
ensuite passé sur colonne d’exclusion (Sephadex G-50) équilibrée avec 20 mM Tris-HCl / 10
mM NaCl à pH 8,6. Le rapport métal/protéine est vérifié par analyse d’un aliquot en
spectroscopie d’absorption atomique et par spectroscopie d’absorption UV-visible à pH acide.
IV.1.b. Autres solutions
Les solutions diluées d’eau oxygénée utilisées ont été obtenues à partir d’une solution
commerciale à 30 % d’H2O2 (Fluka).
IV.2. Méthodes
IV.2.a. Analyse d’acides aminés
Les échantillons ont été centrifugés 30 minutes à 15000 g et le surnageant entièrement pipeté
pour être remplacé par 200 μL d’acide trifluoroacétique à 0,1 %. Les solutions ont ensuite été
placées dans les tubes adaptés pour l’analyse d’acides aminés et lyophilisés sur la nuit. Les
échantillons ont été analysés par le groupe d’analyse protéique (service commun) de
l’université de Zurich.

Chapitre 5 : l’interaction MT-3 / Aβ
184
IV.2.b. Spectroscopie d’absorption atomique
Les échantillons de 300 μL ont été centrifugés 30 minutes à 15000 g puis le culot et le
surnageant ont été séparés (150 μL chacun). Leur concentration en zinc a été déterminée à
l’aide d’un spectromètre d’absorption atomique monofaisceau SpectrAA-110 de Varian Inc.,
équipé d’une lampe à cathode creuse de zinc. Une flamme air-acétylène permet l’atomisation
des gouttelettes.
L’appareil a été calibré avec les étalons appropriés avant passage des échantillons.
IV.2.c. Chromatographie d’exclusion stérique
L’analyse séparative des mélanges d’oligomères de MT-3 en présence de différentes
concentrations de H2O2 a été réalisée par chromatographique d’exclusion stérique couplée à
une FPLC. Elle est effectuée à température ambiante sur une colonne superdex 75 10/300 GL
avec une instrumentation AKTA (Amersham Pharmacia Biotech) sous des conditions
isocratiques. L’éluant utilisé est une solution tampon de 20 mM de Hepes à pH 7,4 contenant
100mM de NaCl. La présence de MT-3 en sortie de colonne a été détectée par absorption à
220 nm. Les solutions ont toutes été préalablement centrifugées 30 minutes à 10000 g afin de
n’éluer que les espèces solubles.
IV.2.d. Spectroscopie d’absorption UV-visible
Les spectres d’absorption UV/Visible ont été réalisés à température ambiante avec les
spectromètres Cary 2300 et Agilent 8453 dans une cuve en quartz de 1 cm de large.

Chapitre 5 : l’interaction MT-3 / Aβ
185
Références M. C. Amoureux, D. Van Gool, M. T. Herrero, R. Dom, F. C. Colpaert, et al. (1997).
"Regulation of metallothionein-III (GIF) mRNA in the brain of patients with Alzheimer disease is not impaired." Mol Chem Neuropathol, 32(1-3), 101-21.
S. Y. Assaf and S. H. Chung (1984). "Release of endogenous Zn2+ from brain tissue during activity." Nature, 308(5961), 734-6.
K. J. Barnham, C. L. Masters and A. I. Bush (2004). "Neurodegenerative diseases and oxidative stress." Nat Rev Drug Discov, 3(3), 205-14.
W. Q. Chen, Y. Y. Cheng, X. L. Zhao, S. T. Li, Y. Hou, et al. (2006). "Effects of zinc on the induction of metallothionein isoforms in hippocampus in stress rats." Exp Biol Med (Maywood), 231(9), 1564-8.
M. J. Daniels, J. S. Turner-Cavet, R. Selkirk, H. Sun, J. A. Parkinson, et al. (1998). "Coordination of Zn2+ (and Cd2+) by prokaryotic metallothionein. Involvement of his-imidazole." J Biol Chem, 273(36), 22957-61.
I. El Ghazi, B. L. Martin and I. M. Armitage (2006). "Metallothionein-3 is a component of a multiprotein complex in the mouse brain." Exp Biol Med (Maywood), 231(9), 1500-6.
J. C. Erickson, A. K. Sewell, L. T. Jensen, D. R. Winge and R. D. Palmiter (1994). "Enhanced neurotrophic activity in Alzheimer's disease cortex is not associated with down-regulation of metallothionein-III (GIF)." Brain Res, 649(1-2), 297-304.
E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin (2006). "Copper homeostasis and neurodegenerative disorders (Alzheimer's, prion, and Parkinson's diseases and amyotrophic lateral sclerosis)." Chem Rev, 106(6), 1995-2044.
F. Gomi, S. Nakano and Y. Uchida (2005). "Tolerance of aged rat brains to mild hyperoxia: possible involvement of higher GIF content." Brain Res, 1033(1), 113-6.
D. Hamer (1986). "Metallothionein." Annu Rev Biochem, (55), 913-951. D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vasak (2000). "Effect of the two
conserved prolines of human growth inhibitory factor (metallothionein-3) on its biological activity and structure fluctuation: comparison with a mutant protein." Biochemistry, 39(47), 14567-75.
J. Hidalgo, M. Aschner, P. Zatta and M. Vasak (2001). "Roles of the metallothionein family of proteins in the central nervous system." Brain Res Bull, 55(2), 133-45.
G. A. Howell, M. G. Welch and C. J. Frederickson (1984). "Stimulation-induced uptake and release of zinc in hippocampal slices." Nature, 308(5961), 736-8.
Y. Irie and W. M. Keung (2001). "Metallothionein-III antagonizes the neurotoxic and neurotrophic effects of amyloid beta peptides." Biochem Biophys Res Commun, 282(2), 416-20.
Y. Irie and W. M. Keung (2003). "Anti-amyloid beta activity of metallothionein-III is different from its neuronal growth inhibitory activity: structure-activity studies." Brain Res, 960(1-2), 228-34.
C. Jacob, W. Maret and B. L. Vallee (1998). "Control of zinc transfer between thionein, metallothionein, and zinc proteins." Proc Natl Acad Sci U S A, 95(7), 3489-94.
T. Kaji, C. Yamamoto, S. Tsubaki, S. Ohkawara, M. Sakamoto, et al. (1993). "Metallothionein induction by cadmium, cytokines, thrombin and endothelin-1 in cultured vascular endothelial cells." Life Sci, 53(15), 1185-91.
R. Kitzberger, C. Madl and P. Ferenci (2005). "Wilson disease." Metab Brain Dis, 20(4), 295-302.
S. Kojima, A. Shimada, T. Morita, Y. Yamano and T. Umemura (1999). "Localization of metallothioneins-I & -II and -III in the brain of aged dog." J Vet Med Sci, 61(4), 343-9.

Chapitre 5 : l’interaction MT-3 / Aβ
186
M. A. Lynes, K. Zaffuto, D. W. Unfricht, G. Marusov, J. S. Samson, et al. (2006). "The physiological roles of extracellular metallothionein." Exp Biol Med (Maywood), 231(9), 1548-54.
W. Maret and B. L. Vallee (1998). "Thiolate ligands in metallothionein confer redox activity on zinc clusters." Proc Natl Acad Sci U S A, 95(7), 3478-82.
W. Maret (2006). "Zinc coordination environments in proteins as redox sensors and signal transducers." Antioxid Redox Signal, 8(9-10), 1419-41.
G. Meloni, P. Faller and M. Vasak (2007). "Redox silencing of copper in metal-linked neurodegenerative disorders: reaction of Zn7metallothionein-3 with Cu2+ ions." J Biol Chem, 282(22), 16068-78.
I. Miyazaki, M. Asanuma, Y. Higashi, C. A. Sogawa, K. Tanaka, et al. (2002). "Age-related changes in expression of metallothionein-III in rat brain." Neurosci Res, 43(4), 323-33.
P. Moffatt and F. Denizeau (1997). "Metallothionein in physiological and physiopathological processes." Drug Metab Rev, 29(1-2), 261-307.
J. D. Otvos, X. Q. Liu, H. Li, G. Shen and M. Basti, Eds. (1993). Dynamic Aspects of Metallothionein Structure. Metallothionein III.
C. J. Quaife, S. D. Findley, J. C. Erickson, G. J. Froelick, E. J. Kelly, et al. (1994). "Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia." Biochemistry, 33(23), 7250-9.
N. Romero-Isart and M. Vasak (2002). "Advances in the structure and chemistry of metallothioneins." J Inorg Biochem, 88(3-4), 388-96.
B. Roschitzki and M. Vasak (2002). "A distinct Cu(4)-thiolate cluster of human metallothionein-3 is located in the N-terminal domain." J Biol Inorg Chem, 7(6), 611-6.
M. J. Stillman, D. Thomas, C. Trevithick, X. Guo and M. Siu (2000). "Circular dichroism, kinetic and mass spectrometric studies of copper(I) and mercury(II) binding to metallothionein." J Inorg Biochem, 79(1-4), 11-9.
S. Tsuji, H. Kobayashi, Y. Uchida, Y. Ihara and T. Miyatake (1992). "Molecular cloning of human growth inhibitory factor cDNA and its down-regulation in Alzheimer's disease." Embo J, 11(13), 4843-50.
Y. Uchida, K. Takio, K. Titani, Y. Ihara and M. Tomonaga (1991). "The growth inhibitory factor that is deficient in the Alzheimer's disease brain is a 68 amino acid metallothionein-like protein." Neuron, 7(2), 337-47.
M. Vasak, E. Worgotter, G. Wagner, J. H. Kagi and K. Wuthrich (1987). "Metal co-ordination in rat liver metallothionein-2 prepared with or without reconstitution of the metal clusters, and comparison with rabbit liver metallothionein-2." J Mol Biol, 196(3), 711-9.
M. Vasak (1991). "Metal removal and substitution in vertebrate and invertebrate metallothioneins." Methods Enzymol, 205452-8.
M. Vasak and D. W. Hasler (2000). "Metallothioneins: new functional and structural insights." Curr Opin Chem Biol, 4(2), 177-83.
S. Vasto, E. Mocchegiani, M. Malavolta, I. Cuppari, F. Listi, et al. (2007). "Zinc and inflammatory/immune response in aging." Ann N Y Acad Sci, 1100111-22.
W. H. Yu, W. J. Lukiw, C. Bergeron, H. B. Niznik and P. E. Fraser (2001). "Metallothionein III is reduced in Alzheimer's disease." Brain Res, 894(1), 37-45.
J. Zhou and P. B. Goldsbrough (1994). "Functional homologs of fungal metallothionein genes from Arabidopsis." Plant Cell, 6(6), 875-84.
J. Zhou and P. B. Goldsbrough (1995). "Structure, organization and expression of the metallothionein gene family in Arabidopsis." Mol Gen Genet, 248(3), 318-28.

187
Chapitre 6
Etude de l’interaction du peptide amyloïde
bêta avec les membranes, influence du zinc
Etude réalisée à l’université de Würzburg,
dans l’équipe du Prof. R. Benz

188

Chapitre 6 : Aβ et les membranes, influence du zinc
189
I. Bibliographie
De nombreuses études ont tenté de comprendre le mécanisme par lequel l’agrégation de
certaines protéines est toxique pour les cellules (comme c’est le cas pour la grande majorité
des amyloïdoses). Il a été conclu que cette toxicité s’exerçait suivant les mêmes modifications
de propriétés biochimiques spécifiques de la cellule, indépendamment de la nature du peptide
étudié (Bucciantini et al. 2002). De plus, certaines amyloïdoses sont intracellulaires quand
d’autres résident dans le cytosol (Bodles et al. 2000; Kayed et al. 2003). Ces observations font
des membranes cellulaires des cibles potentielles privilégiées pour la pathogénie amyloïde. En
effet, une altération des membranes cellulaires, via des lésions non spécifiques (Green et al.
2004; Kayed et al. 2004) ou la formation de canaux ioniques (Arispe et al. 1993; Rhee et al.
1998; Lin et al. 1999; Kawahara et al. 2000; Hirakura et al. 2002; Kourie and Henry 2002),
peut engendrer une dishoméostasie ioniques et entraîner ensuite la mort cellulaire (Cecchi et
al. 2005).
Le peptide Aβ est amphipatique, les 28 résidus N-terminaux, correspondant à une portion du
domaine extracellulaire de l’APP, sont hydrophiles, tandis que la partie C-terminale restante,
transmembranaire dans l’APP, est hydrophobe. Du fait de sa provenance, Aβ pourrait donc
potentiellement interagir de lui-même avec les membranes cellulaires, par exemple en se
réinsérant sous forme d’hélice alpha via sa partie hydrophobe (favorisant ainsi plutôt la
formation de pores), ou par le biais d’interactions électrostatiques qui accélèreraient
l’agrégation du peptide en favorisant les structures en feuillet bêta et en jouant le rôle de site
de nucléation (Figure 6.1) (Jarrett and Lansbury 1993; Bokvist et al. 2004). Ces interactions
entraîneraient une diminution de la fluidité des membranes (Kremer et al. 2000) induisant une
altération de leurs propriétés, comme leur dépolarisation (Hartley et al. 1999) ou
l’augmentation de leur perméabilité (Arispe et al. 1996).

Chapitre 6 : Aβ et les membranes, influence du zinc
190
Figure 6.1. Représentation schématique de différents types d’interaction Aβ-membranes :
adsorption électrostatique avec accélération de la formation d’agrégat en structure feuillets bêta ; insertion par la partie hydrophobe d’Aβ. Une élévation du potentiel membranaire
favoriserait les deux processus (adsorption facilitée / insertion plus importante du peptide) (Bokvist et al. 2004).
D’un point de vue de la toxicité, le peptide Aβ a été rapporté induire une dégénération
neuritique (Aβ42 à 50 nM), une mort cellulaire (Aβ42 à 10 μM) et une altération de
l’homéostasie du calcium, phénomènes qui découleraient de la formation de canaux ioniques
(Lin et al. 2001). Confirmant cette hypothèse, des images de pores membranaires composés
de plusieurs (généralement 4 ou 6) sous-unités d’Aβ42 ont été publiées (Figure 6.2) (Lin et al.
2001; Quist et al. 2005). Les structures de ces canaux semblent toutefois assez hétérogènes.
Figure 6.2. Pores formés par Aβ40 dans une bicouche lipidique (DOPC, 1,2-dioleoyl-sn-
glycero-3-phosphocoline) visualisé par microscopie à force atomique (taille de l’image : 25 nm) (Quist et al. 2005).
Une augmentation de la conductance des bicouches lipidiques (DOPC), en présence d’Aβ
(10μM Aβ42 et 21μM Aβ40, 100 mM KCl 10 mM Hepes, pH 7,4) a également été observée

Chapitre 6 : Aβ et les membranes, influence du zinc
191
(Figure 6.3) (Lin et al. 2001; Quist et al. 2005), en particulier, avec les oligomères solubles
(Kayed et al. 2004).
Figure 6.3. Visualisation du courant traversant un canal constitué de peptides Aβ (solution
tamponnée à pH 7,4, 100 mM KCl, Aβ à 21 μg.L-1, bicouche plane de phospholipides, d’après (Quist et al. 2005)).
De plus, Aβ interagit chimiquement et biochimiquement avec les lipides : les produits
d’oxydation des lipides modifient Aβ, augmentant ainsi son agrégation (Bieschke et al. 2006) ;
inversement, Aβ peut oxyder les lipides via la production de ROS (en présence de cuivre et de
fer) (Murray et al. 2005; Lau et al. 2007).
II. Résultats et discussion
Dans ce chapitre, on s’intéressera aux effets du zinc(II) sur les « pores » formés par Aβ.
L’ensemble des expériences a été réalisé, sauf mention contraire, dans le tampon Hepes 20
mM KCl 1 M à pH 7, avec une membrane bicouche de cholestérol oxydé et en utilisant le
peptide Aβ42. Des essais ont également été réalisés avec le lipide L-α-Phosphatidylcholine
(Avanti Polar Lipids) plus couramment utilisé, mais l’insertion d’Aβ42 n’y est pas détectable,
il faut y ajouter un minimum de 30 % en cholestérol oxydé pour visualiser un début
d’insertion. Ce groupe de lipides est pourtant plus couramment utilisé dans ce type d’étude, en
effet il fait partie de la famille des phosphatidylcholines (ou lécithine, Figure 6.4), sous-
groupe des phospholipides (ou diacylphosphoglycérides, classe la plus abondante des lipides
des cellules animales) (IUPAC-IUB 1978). En comparaison, le cholestérol (de la famille des
stérols) est beaucoup plus compact et apolaire, mais il constitue tout de même environ 30 %

Chapitre 6 : Aβ et les membranes, influence du zinc
192
de l’ensemble des lipides et des membranes plasmiques. La difficulté d’insertion du peptide
dans une bicouche classique de phospholipides par rapport à la celle constituée de cholestérol
oxydé, semble indiquer que: 1) Aβ ne s’insère pas dans les membranes aussi aisément qu’une
protéine membranaire classique (qui n’a aucune difficulté à s’insérer rapidement dans les
bicouches phospholipidiques) ; 2) Aβ pourrait s’insérer préférentiellement dans les radeaux
lipidiques riches en cholestérol.
Figure 6.4. Haut : structure générale des lipides de la classe des phophatidylcholines,
constitué de l’association des groupements choline, phosphate, glycérol et d’acides gras (nomenclature : palmitoyl-oleyl-sn-phosphatidyl-choline). Bas : formule de la L-α-
Phosphatidylcholine (lécithine de soja).
La concentration en sel semble également influer puisque l’insertion d’Aβ42 se fait moins
aisément à 100 mM de KCl qu’à 1M. Il est possible que l’interaction entre les parties
hydrophobes du peptide et des lipides soit favorisée par l’ajout de sel, ce dernier perturbant la
solvatation des résidus C-term d’Aβ42.
Quant aux peptides Aβ16 et Aβ28, principalement utilisés pour étudier l’interaction avec les
métaux et dépourvu de la partie hydrophobe transmembranaire dans l’APP, leur insertion est
extrêmement faible (même en introduisant 10 μM de peptide dans chaque compartiment et en
augmentant la tension aux électrodes jusqu’à 70 mV). On constate une très légère
augmentation de la conductivité (maximum de 22 nS avec Aβ28) mais celle-ci n’est pas
stable. Il est probable qu’il ne s’agisse que d’une légère déstabilisation de la membrane du fait
de l’interaction du peptide avec la partie hydrophile (groupement alcool ou fonctions oxydées
carbonyles ou carboxyles) des molécules de cholestérol. La partie hydrophobe d’Aβ apparaît
donc indispensable à son insertion dans les membranes.

Chapitre 6 : Aβ et les membranes, influence du zinc
193
Aβ42 s’insère dans les membranes de cholestérol à partir de 0,7 μM de peptide dans les deux
compartiments, l’initiation de l’insertion du peptide peut parfois prendre quelques minutes
voire nécessiter une destruction (un léger choc suffit) puis reformation de la membrane,
comme c’est le cas dans l’expérience illustrée à la figure 6.5 (indiqué par une flèche bleue).
Ensuite la conductivité augmente progressivement jusqu’à une valeur limite, atteinte
généralement en quelques minutes. Cette phase d’insertion nous renseigne sur l’homogénéité
des pores formés : sur la figure 6.5, on n’observe pas de paliers lors de l’insertion des peptides
dans la membrane ce qui indique que les pores formés n’ont pas de conductivité définie, ils
sont hétérogènes. La conductivité à saturation résultant de la présence de 0,7 μM d’Aβ42 est
variable, entre 30 et 100 nS suivant les expériences, confirmant l’hétérogénéité des pores
d’Aβ.
Figure 6.5. Conductivité d’Aβ42 0,7 μM (tampon 20 mM Hepes, 1 M KCl, pH 7) inséré dans une bicouche lipidique cholestérol oxydé (échelle : 1 cm en abscisse, 5 nS en ordonnées), la flèche bleue indique la destruction/reformation volontaire de la membrane, initiatrice dans
cet exemple de l’insertion du peptide).
L’influence du zinc a également été mise en évidence : l’ajout d’ions Zn2+ après stabilisation
d’Aβ42 dans la membrane entraîne une diminution de la conductivité, c’est-à-dire la
fermeture des pores (Figure 6.6). Cette fermeture est réversible, en effet l’ajout d’un chélateur
(EDTA, de plus grande affinité pour le zinc qu’Aβ) à la fin du titrage induit une élévation de
la conductivité.

Chapitre 6 : Aβ et les membranes, influence du zinc
194
Figure 6.6. Suivi de la conductance d’une membrane de cholestérol. 1ère partie :
augmentation du signal avec l’insertion d’Aβ42 (0,7 μM dans chaque compartiment) dans la membrane. 2ème partie : diminution de la conductance durant le titrage par Zn2+. 3ème partie :
réouverture des pores via l’ajout d’un équivalent (1,77 mM) d’EDTA, chélateur du zinc.
On peut déjà constater, en observant la figure 6.6, que l’ajout de 10 équivalents de zinc ne
suffit pas à bloquer les pores (malgré le champ électrique présent dans la solution qui a
tendance à conduire les ions positifs vers la membrane pour l’un des deux compartiments). En
effet, les constantes de dissociation obtenues, dans l’hypothèse d’une stoechiométrie 1:1, sont
beaucoup plus élevées que celles calculées au chapitre 2 : entre 270 et 350 μM, soit presque
cent fois supérieures. Plusieurs hypothèses peuvent être avancées pour expliquer ce
phénomène : 1) l’insertion du peptide dans la membrane induit un changement
conformationnel qui défavorise la liaison au métal ; 2) l’interaction de plusieurs zincs par
peptide est nécessaire pour induire une baisse de la conductivité, mettant en jeu des sites de
plus faible affinité (incluant des ligands appartenant à Aβ et éventuellement au cholestérol
oxydé).
Remarque 1 : afin de mettre en valeur un éventuel renforcement de la toxicité de (ZnII-Aβ)

Chapitre 6 : Aβ et les membranes, influence du zinc
195
sur les membranes lorsqu’il se trouve dans un état agrégé, une tentative d’insertion de (ZnII-
Aβ42) préincubé durant 5 jours (solution à 50 μM dans le tampon habituel, à température
ambiante) a été réalisée. L’étude a montré un temps de latence avant insertion
comparativement très supérieur à l’apo-Aβ42 monomérique. Il est probable que, vu la faible
concentration utilisée (0,3 μM), les agrégats se soient partiellement solubilisés et les
complexes ZnII-Aβ également partiellement rompus. Le faible signal finalement obtenu
(maximum à 20 nS) correspond finalement aux valeurs précédentes ramené à 0,3 μM d’Aβ42
(le zinc n’étant pas assez concentré pour abaisser la conductivité), confirmant la dissolution
des agrégats et leur incapacité à s’insérer directement dans la membrane. Ce résultat devrait
bien sûr être reproduit et confirmé pour être pris en compte.
Remarque 2 : des expériences antérieures réalisées dans le laboratoire (université de
Würzburg, équipe du Prof. Benz) montraient également une baisse de la conductivité lors de
l’ajout d’ions Cu2+ après stabilisation d’Aβ42 dans la membrane. Ces métaux sont donc
protecteurs vis-à-vis de la perméabilité membranaire induite par Aβ. Cependant, la présence
au niveau des membranes de cuivre (II), qui peut conduire à la production de ROS (même lié
à Aβ), est potentiellement toxique pour les cellules.
Remarque 3 : plusieurs tentatives ayant pour but de faire apparaître un éventuel effet
protecteur du peptide humanine (séquence : MAPRGFSCLLLLTSEIDLPVKRRA,
(Hashimoto et al. 2001; Niikura et al. 2004; Benaki et al. 2005; Armas et al. 2006)) via la
fermeture des pores d’Aβ (en présence ou non de zinc) n’ont pas abouti du fait de la
destruction quasi immédiate de la membrane après ajout de l’humanine (Figure 6.7). Cette
forte déstabilisation a été observée indifféremment en présence et en absence d’Aβ, la
reconstruction de la membrane n’étant ensuite plus possible. Au vu de sa séquence peptidique,
il est possible que l’humanine agisse comme un détergent qui interagirait avec la membrane
en favorisant la formation de micelles.

Chapitre 6 : Aβ et les membranes, influence du zinc
196
Figure 6.7. Stabilisation de l’insertion du peptide Aβ42 suivie de la rupture de la membrane
de cholestérol suite à l’ajout de peptide humanine (flèche verte).
Conclusion
On constate une légère déstabilisation des membranes riches en cholestérol lors de l’ajout du
peptide Aβ42, dont la partie hydrophobe semble jouer un rôle essentiel. Les « pores » formés
sont cependant très hétérogènes puisque la conductivité augmente de façon plutôt linéaire
avant de se stabiliser. Le zinc (qui n’induit pas la formation de ROS contrairement au cuivre)
pourrait jouer un rôle protecteur en se liant au peptide et ainsi fermer les canaux. Cependant
des concentrations importantes en zinc libre sont nécessaires pour induire une baisse
significative de la conductivité membranaire et cette situation est peu probable in vivo (ou
alors transitoirement). Toutefois, la déstabilisation de la bicouche lipidique par Aβ reste faible
et dépendante d’un certain nombre de facteurs (haute teneur en cholestérol, forte
concentration en sel, destruction/reformation de la membrane souvent nécessaire). Ainsi, au
vue de ces expériences et en accord avec certaines publications (Gonzalez-Montalban et al.
2007), il ne semble pas que la perméabilité des membranes cellulaires induite par le peptide
Aβ soit la cause principale de toxicité de la maladie d’Alzheimer, il est en effet probable que
plusieurs voies de toxicité coexistent dans la MA. Il faut cependant garder à l’esprit que les
membranes de cholestérol oxydé utilisées (très ordonnées) ne sont que des modèles et que le
comportement du peptide vis-à-vis de membranes biologiques peut différer.
III. Méthode : la BLM
Remarque : pour la préparation des solutions de peptides Aβ et de métaux, se reporter au

Chapitre 6 : Aβ et les membranes, influence du zinc
197
paragraphe matériels et méthodes du chapitre 3.
La membrane noire ou BLM (Black Lipid Membrane) est constituée d’une bicouche lipidique
plate séparant deux solutions aqueuses dans chacune desquelles trempe une électrode (Figure
6.7). Une différence de potentiel est appliquée entre les deux électrodes et l’on mesure le
passage du courant (porté par les ions de la solution, en général KCl) au travers de la
membrane, ce qui permet d’en déduire sa conductance c’est-à-dire sa perméabilité aux ions.
En absence de protéine et d’impureté, le courant entre les deux compartiments est constant et
proche de zéro. La cuve et les électrodes sont placées dans une cage de Faraday pour les isoler
de nuisances électriques et électromagnétiques extérieures et l’ensemble de l’appareil est
placé sur un marbre amortissant les tremblements susceptibles de rompre la membrane.
Figure 6.7. Représentation schématique du matériel de BLM. Les deux compartiments en
téflon sont séparés par une ouverture de 0,1 mm dans laquelle est formée la bicouche lipidique. Les protéines ajoutées ensuite dans un des compartiments, peuvent s’insérer dans
la membrane, laissant ainsi passé des ions et donc du courant. Lorsque l’on étudie des protéines membranaires, qui forment des canaux stables, l’augmentation de courant se fait
par paliers, chaque palier correspondant à la formation d’un pore.

Chapitre 6 : Aβ et les membranes, influence du zinc
198
Les compartiments en téflon sont remplis respectivement de 5 mL de solution tampon (1 M
KCl, 20 mM Hepes, pH 7). Un système d’agitation magnétique permet de travailler avec des
solutions homogènes. Dans l’ensemble des expériences réalisées, sauf mention contraire, les
membranes sont constituées de cholestérol oxydé (Figure 6.8). Les lipides sont conservés
dans le chloroforme à -20°C. Le solvant est évaporé avant utilisation et remplacé par un
mélange de n-décane et butanol, ce dernier stabilisant la membrane.
Figure 6.8. Le cholestérol et ses formes oxydées. La présence d’une double liaison peut
conduire, in vivo, à une oxydation ménagée (aldéhyde) ou complète (acide carboxylique).
Cette solution à 1 % de cholestérol oxydé est appliquée à l’aide d’une boucle sur l’ouverture
de 0,1 mm séparant les deux compartiments (Figure 6.7 et 6.9) (Benz and Gisin 1978). La
qualité de la membrane est vérifiée à l’aide d’un microscope placé devant l’ouverture réalisée
dans l’un des compartiments, la membrane ne doit contenir aucun reflet ou bulle et être
entièrement « noire », signe de la formation d’un film homogène d’épaisseur bimoléculaire.
L’ensemble du matériel utilisé doit être soigneusement lavé et séché entre chaque expérience.
Un temps d’attente d’une heure minimum doit être observé, sous agitation, entre la formation
de la membrane et l’ajout du peptide, afin de vérifier l’absence d’impureté. Toutes les
expériences ont été effectuées à température ambiante.

Chapitre 6 : Aβ et les membranes, influence du zinc
199
Figure 6.9. Photographies de l’appareil et de ses différents éléments : 1- boucle en téflon, 2-
barreaux aimantés, 3- générateur de tension, 4- amplificateur de tension, 5- cage de Faraday, 6- microscope et lampe, 7- marbre, 8- enregistreur à papier déroulant, 9- cuvette en téflon.
Mesure de la conductivité
Les membranes de lipide sont quasiment imperméables aux ions de la solution électrolytique.
C’est pourquoi, lors de l’application d’une tension de l’ordre de 20 mV, aucun courant n’est
décelé. Après ajout de la protéine étudiée, l’interaction avec la membrane se traduit par le
passage d’ions (formation de pore ou simple altération de la membrane induisant une certaine
perméabilité) et l’intensité mesurée augmente. Si la protéine introduite forme des pores
stables (protéines membranaires), on observera un signal en escalier, chaque palier
correspondant à la formation d’un pore. Ceci à condition bien sûr que les pores ne soient pas
spécifiques d’un ion particulier ou que celui-ci fasse partie des électrolytes du tampon.
La conductivité d’une protéine est obtenue d’après les expressions suivantes :
eUIG =
Avec G conductivité en Siemens (S), I intensité (A) et Ue la tension appliquée entres les
électrodes (entre 20 et 70 mV).
L’intensité mesurée est amplifiée d’un facteur Vf (généralement égal à 109 V.A-1) et convertie
en tension (Ua, en V).

Chapitre 6 : Aβ et les membranes, influence du zinc
200
f
a
VUI =
En remplaçant dans l’expression précédente, il vient :
ef
a
UVUG.
=
L’échelle Uv de l’enregistreur à papier déroulant est choisie par rapport à la valeur de Ua.
Ainsi la hauteur du papier déroulant, correspondant à Uv, contient 10 graduations. Une simple
règle de 3 permet d’obtenir la conductivité par graduation Gg, indiquée sur les spectres.
ef
vg UV
UG..10
=
La méthode de titrage
Une fois le peptide inséré et le signal stabilisé, une molécule (métal, protéine,…) peut être
ajoutée dans l’un des compartiments. Si cette molécule, en se liant au canal, est capable de le
bloquer, il est alors possible d’effectuer un titrage et d’en déduire la constante d’association
entre la macromolécule introduite dans la membrane et son ligand. De petits ajouts successifs
de ligand vont ainsi diminuer la conductivité mesurée jusqu’à une valeur maximale
correspondant à la saturation de tous les pores. On associe ensuite la constante de dissociation
à la valeur de la concentration en métal à la moitié de la saturation. Dans le cas du peptide Aβ,
on fait l’hypothèse d’une stœchiométrie 1:1 entre Aβ et le métal puisque le signal n’est pas
assez net pour connaître le nombre de pores et le nombre de peptide par pore (qui semble de
toute façon hétérogène d’après la littérature). De plus même après saturation en métal, il reste
une conductivité résiduelle (10 à 30 % de la conductivité maximale avant titrage)
probablement due à une fermeture incomplète de l’ensemble des pores. On ne peut cependant
pas exclure qu’une fraction des pores aient une structure telle que le zinc ne puisse pas les
fermer. Les constantes de dissociation ont été calculées graphiquement par lecture de la
concentration de zinc totale (quasi identique à la concentration en zinc libre dans l’hypothèse
d’une stœchiométrie 1 :1) à la moitié de la diminution maximale de conductivité (k1/2). Les
valeurs obtenues pour les constantes de dissociation sont donc assez approximatives et
doivent être considérées comme des ordres de grandeur.

Chapitre 6 : Aβ et les membranes, influence du zinc
201
Références
N. Arispe, H. B. Pollard and E. Rojas (1993). "Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes." Proc Natl Acad Sci U S A, 90(22), 10573-7.
N. Arispe, H. B. Pollard and E. Rojas (1996). "Zn2+ interaction with Alzheimer amyloid beta protein calcium channels." Proc Natl Acad Sci U S A, 93(4), 1710-5.
A. Armas, V. Sonois, E. Mothes, H. Mazarguil and P. Faller (2006). "Zinc(II) binds to the neuroprotective peptide humanin." J Inorg Biochem, 100(10), 1672-8.
D. Benaki, C. Zikos, A. Evangelou, E. Livaniou, M. Vlassi, et al. (2005). "Solution structure of humanin, a peptide against Alzheimer's disease-related neurotoxicity." Biochem Biophys Res Commun, 329(1), 152-60.
R. Benz and B. F. Gisin (1978). "Influence of membrane structure on ion transport through lipid bilayer membranes." J Membr Biol, 40(4), 293-314.
J. Bieschke, Q. Zhang, D. A. Bosco, R. A. Lerner, E. T. Powers, et al. (2006). "Small molecule oxidation products trigger disease-associated protein misfolding." Acc Chem Res, 39(9), 611-9.
A. M. Bodles, D. J. Guthrie, P. Harriott, P. Campbell and G. B. Irvine (2000). "Toxicity of non-abeta component of Alzheimer's disease amyloid, and N-terminal fragments thereof, correlates to formation of beta-sheet structure and fibrils." Eur J Biochem, 267(8), 2186-94.
M. Bokvist, F. Lindstrom, A. Watts and G. Grobner (2004). "Two types of Alzheimer's beta-amyloid (1-40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation." J Mol Biol, 335(4), 1039-49.
M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, et al. (2002). "Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases." Nature, 416(6880), 507-11.
C. Cecchi, S. Baglioni, C. Fiorillo, A. Pensalfini, G. Liguri, et al. (2005). "Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates." J Cell Sci, 118(Pt 15), 3459-70.
N. Gonzalez-Montalban, A. Villaverde and A. Aris (2007). "Amyloid-linked cellular toxicity triggered by bacterial inclusion bodies." Biochem Biophys Res Commun, 355(3), 637-42.
J. D. Green, L. Kreplak, C. Goldsbury, X. Li Blatter, M. Stolz, et al. (2004). "Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide." J Mol Biol, 342(3), 877-87.
D. M. Hartley, D. M. Walsh, C. P. Ye, T. Diehl, S. Vasquez, et al. (1999). "Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons." J Neurosci, 19(20), 8876-84.
Y. Hashimoto, T. Niikura, Y. Ito, H. Sudo, M. Hata, et al. (2001). "Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer's disease-relevant insults." J Neurosci, 21(23), 9235-45.
Y. Hirakura, I. Carreras, J. D. Sipe and B. L. Kagan (2002). "Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense." Amyloid, 9(1), 13-23.
IUPAC-IUB (1978). "The nomenclature of lipids (Recommendations 1976) IUPAC-IUB Commission on Biochemical Nomenclature." Biochem J, 171(1), 21-35.
J. T. Jarrett and P. T. Lansbury, Jr. (1993). "Seeding "one-dimensional crystallization" of

Chapitre 6 : Aβ et les membranes, influence du zinc
202
amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie?" Cell, 73(6), 1055-8.
M. Kawahara, Y. Kuroda, N. Arispe and E. Rojas (2000). "Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line." J Biol Chem, 275(19), 14077-83.
R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, et al. (2003). "Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis." Science, 300(5618), 486-9.
R. Kayed, Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, et al. (2004). "Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases." J Biol Chem, 279(45), 46363-6.
J. I. Kourie and C. L. Henry (2002). "Ion channel formation and membrane-linked pathologies of misfolded hydrophobic proteins: the role of dangerous unchaperoned molecules." Clin Exp Pharmacol Physiol, 29(9), 741-53.
J. J. Kremer, M. M. Pallitto, D. J. Sklansky and R. M. Murphy (2000). "Correlation of beta-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes." Biochemistry, 39(33), 10309-18.
T. L. Lau, J. D. Gehman, J. D. Wade, K. Perez, C. L. Masters, et al. (2007). "Membrane interactions and the effect of metal ions of the amyloidogenic fragment Abeta(25-35) in comparison to Abeta(1-42)." Biochim Biophys Acta,
H. Lin, Y. J. Zhu and R. Lal (1999). "Amyloid beta protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles." Biochemistry, 38(34), 11189-96.
H. Lin, R. Bhatia and R. Lal (2001). "Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology." Faseb J, 15(13), 2433-44.
I. V. Murray, M. E. Sindoni and P. H. Axelsen (2005). "Promotion of oxidative lipid membrane damage by amyloid beta proteins." Biochemistry, 44(37), 12606-13.
T. Niikura, T. Chiba, S. Aiso, M. Matsuoka and I. Nishimoto (2004). "Humanin: after the discovery." Mol Neurobiol, 30(3), 327-40.
A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, et al. (2005). "Amyloid ion channels: a common structural link for protein-misfolding disease." Proc Natl Acad Sci U S A, 102(30), 10427-32.
S. K. Rhee, A. P. Quist and R. Lal (1998). "Amyloid beta protein-(1-42) forms calcium-permeable, Zn2+-sensitive channel." J Biol Chem, 273(22), 13379-82.

205
Conclusion
et
perspectives

206

Conclusion et perspectives
207
Conclusion et perspectives
Ces travaux nous ont principalement permis de caractériser l’interaction entre le peptide Aβ,
impliqué dans le maladie d’Alzheimer, et le zinc(II), retrouvé en grande quantité dans les
plaques amyloïdes.
Dans un premier temps, l’affinité du zinc pour le peptide a été étudiée par calorimétrie de
titrage isotherme et réactions de compétition avec différents chélateurs. Il en ressort que la
constante de dissociation apparente du complexe ZnII-Aβ se situe dans le bas micromolaire,
quelle que soit sa longueur (16, 28, 40 ou 42 acides aminés), mais surtout quel que soit l’état
d’agrégation. Il pourrait d’ailleurs être intéressant de vérifier ce résultat avec des plaques
amyloïdes formées in vivo puis isolées. D’après cette étude in vitro, il semble qu’une thérapie
par chélation des métaux (principalement le zinc et le cuivre) visant préférentiellement les
espèces solubles à priori plus toxiques, soit difficile à mettre en œuvre, le risque étant de
solubiliser les plaques amyloïdes (moins toxiques d’après la littérature). Cependant, le
mécanisme d’action des chélateurs de métaux n’étant pas encore élucidé (possible activation
des voies de dégradation d’Aβ), cet inconvénient n’entravera pas forcément les progrès dans
cette stratégie thérapeutique. Des études in vivo seront finalement nécessaires pour définir le
bénéfice des chélateurs de métaux dans la maladie d’Alzheimer.
L’utilisation de différents tampons d’enthalpies d’ionisation bien distinctes avec la
calorimétrie de titrage isotherme a également permis la détermination du nombre de protons
échangés lors de la fixation du zinc(II) sur Aβ à pH 7,4. Ainsi, dans l’hypothèse qui semble la
plus probable, où le premier acide aminé serait le quatrième ligand du zinc (avec les histidines
6, 13 et 14), ce résultat privilégie le groupement carboxylate de l’aspartate 1 par rapport au N-
terminal. Il serait intéressant de faire des expériences similaires à des pH légèrement acides ou
basiques, en effet le site de fixation du cuivre(II), apparemment similaire à celui du zinc(II), a
été rapporté être dépendant du pH, il en est donc peut-être de même pour le zinc(II).
Ensuite, après avoir remarqué que le zinc(II) accélère fortement l’agrégation d’Aβ, nous
avons voulu vérifier l’explication qui semblait la plus plausible pour expliquer ce phénomène :
la formation de dimère par l’intermédiaire de métal pontant. Cependant, aucun oligomère
(jusqu’à 75 kDa) n’a pu être détecté par chromatographie d’exclusion stérique et RMN de
diffusion, au contraire du monomère ZnII-Aβ qui est apparu assez stable. Il semble donc que
l’agrégation d’Aβ induite par le zinc passe par un changement de conformation qui favorise

Conclusion et perspectives
208
l’agrégation par contact entre peptides.
L’agrégation de ZnII-Aβ a donc finalement été quantifiée (par turbidimétrie et test BCA) et
suivie par dichroïsme circulaire en présence de différentes concentrations de TFE qui favorise
les structures en hélice alpha. Cette étude a permis d’établir un modèle de l’agrégation d’Aβ.
Ainsi , dans l’hypothèse où le mécanisme serait le même en absence et en présence de métal,
une première étape, favorisée par la présence de 10-20 % de TFE, constituerait en une
structuration partielle en hélice alpha, puis dans une deuxième étape favorisée par la liaison au
zinc(II), l’hélice alpha serait déstabilisée vers une conformation principalement feuillet bêta.
Il serait cependant intéressant de vérifier la concordance entre agrégation en présence et en
absence de métaux en caractérisant les agrégats de (ZnII-Aβ) en microscopie électronique et
en RMN du solide.
Un mécanisme de toxicité potentielle a également était étudié. En effet, au vu des précédents
résultats, rien n’indique clairement que l’interaction du zinc avec Aβ est source de toxicité, au
contraire, ce métal qui n’a pas d’activité redox, ne semble pas favoriser les oligomères
solubles, les plus toxiques (des études in vivo impliquant des agrégats formés en présence de
zinc seraient cependant nécessaires pour confirmer cette hypothèse). Le peptide Aβ a donc
ainsi été inséré dans des bicouches lipidiques modèles dont on a suivi la conductance. Le zinc
s’est alors révélé protecteur vis-à-vis de cette forme de toxicité, puisqu’il permet, en bouchant
partiellement les pores d’Aβ, de diminuer significativement la conductance de la membrane.
Il serait cependant intéressant d’utiliser des bicouches lipidiques de composition plus proche
des membranes biologiques afin de mieux estimer la toxicité d’Aβ.
Enfin, une hypothèse sur la provenance du zinc retrouvé dans les plaques amyloïdes a été
explorée. En effet, l’affinité du zinc pour Aβ étant relativement faible par rapport aux
principales protéines à zinc présentes dans le cerveau, le stress oxydant pourrait favoriser le
transfert du zinc vers Aβ par l’intermédiaire de la formation de ponts disulfures dans la
métallothionéine-3 qui libèrerait ainsi une partie de son zinc. Les premiers résultats obtenus
dans cette étude semblent confirmer cette hypothèse, cependant les expériences doivent être
répétées. Ce modèle in vitro a toutefois des limites, ainsi il n’est pas certain que la
concentration physiologique en stress oxydant soit suffisante, d’autant que la cinétique avec le
peroxyde d’hydrogène est relativement lente. Ainsi l’utilisation d’autres modèles de stress
oxydant est à envisager afin de comparer les vitesses de réaction. L’influence de réducteurs
comme le glutathion ou la vitamine C serait également intéressant à étudier.

209
Annexes

210

211

212

213

214

215

216

217

218

219

220

221

222

223

224

225

226

227

228

229
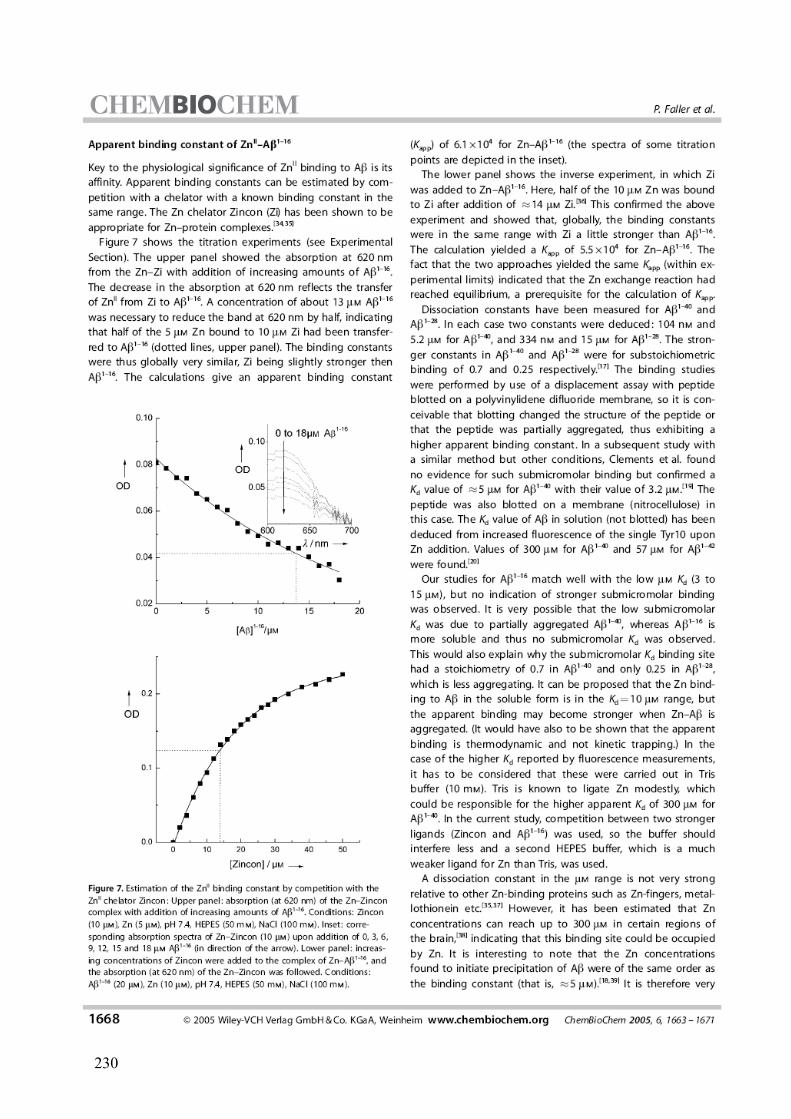
230

231

232

233

234

235
Enfin, pour conclure brièvement ce long manuscrit, je voudrais remercier très
chaleureusement l’ensemble des personnes avec qui j’ai eu le plaisir et la
chance de travailler, en France et à l’étranger, durant ces trois années de thèse.
Last but not least, I would like to express my most sincere acknowledgements to
all people whom I had the pleasure and opportunity to work with during this last
three years.

236
Ce manuscrit est dédicacé : Aux chercheurs d’aiguilles. Aux géniaux ingénieurs. Aux experts techniciens. Aux Fantastiques sponsors.
Ce manuscrit n’est pas dédicacé : Aux faxeurs ineptes. Aux pauseurs grincheux. Aux formations venteuses.
D’une façon générale, ce manuscrit est dédicacé au monde de la recherche avec lequel mes rapports furent aussi divers qu’enrichissants.

237
Interaction between zinc(II) and the amyloid beta peptide in Alzheimer’s disease
Abstract :
Alzheimer’s disease (AD) is a neurodegenerative disorder. Brain regions involved in memory
and learning are diminished in size due to death of neurons and synaptic degeneration. The
morphological hallmarks of AD are the extracellular amyloid plaques and the intracellular
neurofibrillary tangles. The formers are mainly constituted by the amyloid beta peptide (Aβ),
a cleaving product of a membrane protein called amyloid-precursor-protein (APP). According
to the widely held amyloid cascade hypothesis, increased Aβ production and accumulation
leads to its aggregation. The generated oligomers are supposed to provoke neuronal
dysfunction which will later leads to dementia. A large body of evidence indicates that the
essential metal ions zinc, copper and iron are involved in Alzheimer’s disease (AD): they are
found in the amyloid plaques at high concentrations (~mM) and metal-binding to Aβ and APP
has been demonstrated. In vitro and in vivo studies showed that these metal ions influence the
amyloid cascade and thus the neurotoxicity. This thesis deals with the bioinorganic aspects of
the zinc-binding to Aβ. Particular attention has been paid on the binding affinity and the
influence of metal-binding on protein structure and aggregation. The question of whether
metallothionein could provide Aβ with zinc ions has also been investigated. The toxicity
caused by amyloid pores and the influence of metals in this respect is also discussed.
Key words : Alzheimer, amyloid beta peptide, aggregation, zinc, bioinorganic chemistry,
calorimetry, metallothionein.

UNIVERSITE TOULOUSE III - PAUL SABATIER
Thèse d’Université, école doctorale de chimie, spécialité Chimie-Biologie-Santé
Soutenue le 19 novembre 2007 à Toulouse
Christine TALMARD
Directeur de thèse : Prof. Peter FALLER
Interaction entre le zinc(II) et le peptide amyloïde bêta lié à la maladie d’Alzheimer
Résumé : La nature dégénérative de la maladie d’Alzheimer se traduit par des lésions
histopathologiques intra- et extracellulaires, au niveau des neurones. Ces dernières, les
plaques amyloïdes, résultent de l’agrégation en fibres amyloïdes du peptide amyloïde bêta.
L’hypothèse communément admise de la cascade amyloïde place même l’oligomérisation de
ce peptide à l’origine de la maladie. Dans ce contexte, des expériences in vivo et in vitro
attestent d’une influence des ions métalliques (zinc, cuivre et fer principalement) sur
l’agrégation du peptide et sa toxicité. Nous nous sommes donc attaché à étudier l’équilibre de
fixation de l’ion zinc(II) sur le peptide amyloïde bêta (détermination des constantes
thermodynamiques) et l’influence de l’ion métallique sur l’agrégation et la structure
secondaire du peptide. La question de la toxicité causée par la formation de pores dans les
membranes cellulaires, ainsi qu’une hypothèse concernant la provenance du zinc ont
également été abordées.
Mots clés : Alzheimer, peptide amyloïde bêta, agrégation, zinc, chimie bioinorganique,
calorimétrie, métallothionéine.
Laboratoire de Chimie de Coordination, 205 route de Narbonne 31077 Toulouse, France
Interaction between zinc(II) and the amyloid beta peptide in Alzheimer’s disease
Abstract : Alzheimer’s disease (AD) is a neurodegenerative disorder. Brain regions involved
in memory and learning are diminished in size due to death of neurons and synaptic
degeneration. The morphological hallmarks of AD are the extracellular amyloid plaques and
the intracellular neurofibrillary tangles. The formers are mainly constituted by the amyloid
beta peptide (Aβ), a cleaving product of a membrane protein called amyloid-precursor-protein
(APP). According to the widely held amyloid cascade hypothesis, increased Aβ production
and accumulation leads to its aggregation. The generated oligomers are supposed to provoke
neuronal dysfunction which will later leads to dementia. A large body of evidence indicates
that the essential metal ions zinc, copper and iron are involved in Alzheimer’s disease (AD):
they are found in the amyloid plaques at high concentrations (~mM) and metal-binding to Aβ
and APP has been demonstrated. In vitro and in vivo studies showed that these metal ions
influence the amyloid cascade and thus the neurotoxicity. This thesis deals with the
bioinorganic aspects of the zinc-binding to Aβ. Particular attention has been paid on the
binding affinity and the influence of metal-binding on protein structure and aggregation. The
question of whether metallothionein could provide Aβ with zinc ions has also been
investigated. The toxicity caused by amyloid pores and the influence of metals in this respect
is also discussed.
Key words : Alzheimer, amyloid beta peptide, aggregation, zinc, bioinorganic chemistry,
calorimetry, metallothionein.





























