Embed Size (px)
Citation preview

Innovative Enzymatic Approach to Resolve HomogalacturonansBased on their Methylesterification PatternMarie-Christine Ralet,*,† Martin A. K. Williams,‡,§,∥ Abrisham Tanhatan-Nasseri,† David Ropartz,†
Bernard Quemener,† and Estelle Bonnin†
†INRA, UR1268 Biopolymeres Interactions Assemblages, rue de la Geraudiere, BP 71627, F-44300 Nantes, France‡Institute of Fundamental Sciences, Massey University, Palmerston North, New Zealand§MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand∥The Riddet Institute, Palmerston North, New Zealand
ABSTRACT: Three series of model homogalacturonans (HGs) covering a largerange of degree of methylesterification (DM) were prepared by chemical and/orenzymatic means. Randomly demethylesterified HGs, HGs containing a few longdemethylesterified galacturonic acid stretches, and HGs with numerous but shortdemethylesterified blocks were recovered. The analysis of the degradation productsgenerated by the action of a purified pectin lyase allowed the definition of two newparameters, the degree of blockiness, and the absolute degree of blockiness of thehighly methylesterified stretches (DBMe and DBabsMe, respectively). By combining this information with that obtained by themore traditional endopolygalacturonase digestion, the total proportion of degradable zones for a given DM was measured andwas shown to permit a clear differentiation of the three types of HG series over a large range of DM. This double enzymaticapproach will be of interest to discriminate industrial pectin samples exhibiting different functionalities and to evaluate pectin finestructure dynamics in vivo in the plant cell wall, where pectin plays a key mechanical role.
■ INTRODUCTIONPlant cells are encapsulated in a cell wall, whose mostprominent components are polysaccharides. The latter, jointlywith some structural proteins, determine the shape andmechanical properties of plant cells. The fine structure of cellwall polysaccharides governs their functional properties bothafter extraction and in planta. Pectin is an abundantpolysaccharide that is a key component controlling thearchitecture of dicotyledon primary cell walls and is involvedin various cell functions and plant processes.1−4
Pectin molecules are composed of several acidic and neutralstructural domains among which homogalacturonan (HG), alinear partly methylesterified α-(1,4)-linked D-galacturonic acid(GalA) homopolymer, is the most abundant. The degree ofmethylesterification (DM), i.e., the percentage of total GalAresidues methylesterified, and the distribution of thesemethylesters in the HG domains are key features for pectinfunctionality. Indeed, many of the properties, in particular thegelling capabilities, and biological functions of pectin are closelylinked to its ionic binding abilities.1,3,5 The latter are governednot only by the number of methylesters but also by theirdistribution pattern. Two main patterns of methylesterdistribution have classically been described: random orblockwise. Fungal-pectin-methylesterases (PMEs) and basetreatment lead to a random distribution of methylesters.6−9
Plant-PMEs by contrast demethylesterify pectin in a processivemanner, leading to the appearance of demethylesterifiedstretches or blocks.6−13 These enzymes initiate action on amethylesterified carboxyl group adjacent to a nonmethylesteri-
fied one and can then sequentially remove neighboringmethylesters on the HG chain.14,15 Since 8−15 consecutivenonmethylesterified GalA units are required to form a stablecalcium-mediated junction zone between two HG chains,16−18
the starting substrate, the final DM achieved, and thedemethylesterification process implemented will all play arole in determining the ionic sensitivity and ionotropic gelforming properties of modified pectins.6,7,11,19−22
The relationship between the methylester distribution in HGdomains and pectin functionality in the presence of calcium hasrecently been further detailed. It has been shown that plant-PME-demethylesterified pectin samples can produce networkscontaining different types of junction zones, depending on thelength of the nonmethylesterified stretches.23−26 Pectinencompassing small stretches (∼ 10) of consecutive non-methylesterified GalA units gel through rather short dimericcalcium-chelating junction zones, which leads to the formationof a flexible network architecture. For a similar DM, pectinexhibiting longer stretches of consecutive nonmethylesterifiedGalA units gel, in contrast, through the formation of bundlesconsisting of extensively aggregated dimeric junction zones,leading to the formation of a semiflexible network architec-ture.26 It can be hypothesized that within the cell wall, in whichenzymes consistently tailor the amount and distribution ofresident methylesters,5,24 different network architectures ful-
Received: March 1, 2012Revised: April 10, 2012Published: April 20, 2012
Article
pubs.acs.org/Biomac
© 2012 American Chemical Society 1615 dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−1624

filling different structural and mechanical functions might exist.Parameters that can discriminate between pectin samplesexhibiting subtle differences in methylesterification pattern arethus of great interest in order, among other things, to predictthe properties of resulting networks.To date, two main parameters have been introduced in order
to characterize the presence or absence of demethylesterifiedstretches: (i) the degree of blockiness (DB), which is amount ofnonmethylesterified monomers, dimers, and trimers released byincubation with endopolygalacturonase (endo-PG) divided bythe amount of nonmethylesterified GalA present in the pectinsample;8 and (ii) the absolute degree of blockiness (DBabs),which is the amount of nonmethylesterified monomers, dimersand trimers released by endo-PG divided by the total amount ofGalA present in the pectin sample (methylesterified GalAincluded).27 The endo-PG specificity renders the amount anddistribution of its oligogalacturonate digestion productsmethylester-sequence-dependent and simple DB or DBabsmeasurements often allow a clear differentiation betweenpectin substrates demethylesterified in a random or blockwisefashion.21,28−30 Differences in the exact sizes of demethyles-terified blocks however remains extremely difficult to assess.Although differences in demethylesterified blocklength havebeen related to the relative proportion of mono-, di-, andtrigalacturonic acid liberated in endo-PG digests,28,31 sucheffects are of limited applicability and similar proportions ofmono-, di-, and trigalacturonic acid have been observed in suchdigests above a critical DBabs value.30 More recently,13
unmethylesterified blocklengths in pectin substrates havebeen estimated from the lengths of unmethylesterifiedfragments released from very limited endo-PG treatments.With the aid of evaporative light scattering detection, the longeroligomeric unmethylesterified sections excised by limitedtreatments could be identified and quantified and serve as anapproximation to the distribution of blocklengths in thepolymer backbone. Although these studies are a promisingline of enquiry, focusing entirely on demethylesterifiedstretches through the examination of endo-PG hydrolysisproducts gives only a partial view of HG structure. Herein thehighly methylesterified stretches have also been studied throughthe examination of pectin lyase degradation products.32
Although pectin lyase has been used to qualitatively character-ize pectin samples differing in their methylesterification patternpreviously,9,33 in the work described herein clear parameters areintroduced related to the methylesterified stretches in analogyto those defined previously for unmethylesterified stretchesbased on endo-PG digestion. Furthermore it is shown usingmodel HGs with tailored patterns of methylesterification22 thatby examining both endo-PG and pectin lyase digests HGsamples exhibiting only subtle differences in methylesterifica-tion pattern can be resolved over a large range of DM.
■ EXPERIMENTAL SECTIONModel Homogalacturonans. Three series of model homoga-
lacturonans (B-, P-, and BP-series) were prepared, as fully described inTanhatan-Nasseri et al.22 Briefly, demethylesterified citrus pectin washydrolyzed by 0.1 M HCl at 80 °C for 72 h.34 The acid-insolublefraction containing HGs was resuspended in distilled water, the pH ofthe suspension was brought to 7 by tetrabutyl ammonium hydroxide,and the resulting solution (HG0-TBA) was extensively dialyzed againstdistilled water and freeze-dried. Methylation of HG0-TBA wasachieved with methyl iodide in dimethyl sulfoxide.35 The highlymethylesterified HG (HG96) obtained was demethylesterified by 0.2M NaOH or by plant-PME [EC 3.1.1.11] (Sigma P5400; 194 U/mg)
to yield B- and P-series, respectively. A third series quoted BP- wasobtained by demethylesterifying selected NaOH-demethylesterifiedHG samples a further time with plant-PME. HG samples are quotedHGx-By, HGx-Py or HGx-Bx′-Py with B = basic demethylester-ification, P = plant-PME demethylesterification, x = DM of ″mother″HG, x′ = DM achieved after basic demethylesterification and y = finalDM achieved (Figure 1).
Characterization of Model Substrates. GalA measurementswere performed by the automated m-hydroxydiphenyl method.36 DMwas determined by quantifying base-released methanol according toAnthon and Barrett (2004).37 Briefly, 600 μL of HGs solutions (1 mg/mL) were demethylesterified with 600 μL of 0.2 M NaOH for 1 h at 4°C. The solution was neutralized with 600 μL of 0.2 M HCl anddiluted by adding 1.2 mL of H2O. An aliquot of the solution was usedfor GalA determination. A total of 100 μL of 200 mM Tris-HCl bufferpH 7, 400 μL (3 mg/mL) of methylbenzothiazolinone-2-hydrazone(MBTH, Sigma M8006−1G), 50 μL of sample or 40 mM NaCl (as ablank), and 20 μL of alcohol oxidase (E.C. 1.1.3.13, Sigma, A2404;0.02 UI/μL) were mixed in this order to oxidize the releasedmethanol. After addition of the alcohol oxidase, the samples wereincubated for 20 min at 30 °C prior to addition of 200 μL of a solutioncontaining 5 mg/mL each of dodecahydrated ferric ammonium sulfateand sulfamic acid. After 20 min at room temperature, 600 μL of H2Owas added and the tubes were vortexed vigorously. The absorbance at620 nm was then measured and quantification carried out with the useof an external calibration curve (generated using methanol standards inthe range 0 to 20 μg/mL).
DM was calculated as
= ×DMMeOH(mg/g)/32GalA(mg/g)/176
100
Enzymes. Endopolygalacturonase II. Endo-PGII [EC 3.2.1.15,Uniprot P26214] was purified from an Aspergillus niger preparationprovided by Novozymes (Copenhagen, DK).38
Pectin Lyase. PL [EC 4.2.2.10] was purified from the crudepreparation Peclyve of A. niger provided by Lyven (Colombelles, F). Atotal of 20 mL of crude preparation were dialyzed against ultra purewater at 4 °C for 8 h changing water every 2 h and dialysis tubingevery 4 h. The dialyzed enzymatic solution was saturated at 60%ammonium sulfate and centrifuged 15 min at 18000 g at 4 °C. Theammonium sulfate precipitate (AS60) was solubilized in a minimalvolume of ultra pure water, dialyzed against ultra pure water at 4 °Cfor 8 h changing water and dialysis tubing every 2 h, and then dialyzedtwice against 500 mL of 50 mM Na-phosphate buffer pH 6 at 4 °C for90 min. Ammonium sulfate was added to reach a final concentration of1.5 M. Hydrophobic interaction chromatography (HIC) wasperformed on an Akta Purifier 10 system, driven by Unicorn 3.21software (GE Healthcare). AS60 (5 × 1 mL) was loaded onto a
Figure 1. Nomenclature of the homogalacturonan samples.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241616

Resource ether column (GE Healthcare, 0.86 mL) previouslyequilibrated with 50 mM Na-phosphate buffer containing 2 M(NH4)2SO4 pH 6. Elution was performed at a flow rate of 1 mL/minusing the following gradient: 0−7 mL, 100% B; 7−25 mL, lineargradient 100−0% B; 25 mL, 100% A; with A being 50 mM Na-phosphate buffer pH 6 and B being 50 mM Na-phosphate buffer pH 6containing 2 M (NH4)2SO4. Fractions of 500 μL were collected. HIC-excluded fractions were pooled, dialyzed overnight against 1 L of ultrapure water at 4 °C and then dialyzed for 24 h against 50 mM Na-acetate buffer pH 5 at 4 °C. Anion exchange chromatography (AEC)was performed on an Akta Purifier 10 system driven by Unicorn 3.21software (GE Healthcare). HIC excluded fractions (5 × 2 mL) wereloaded onto a MonoQ column (GE Healthcare, 1 mL) previouslyequilibrated with 50 mM Na-acetate buffer pH 5. Elution wasperformed at a flow rate of 1 mL/min using the following gradient: 0−6 mL, 100% A; 6−26 mL, linear gradient 100 to 50% A; 26−37 mL,linear gradient 50 to 0% A; with A being 50 mM Na-acetate buffer pH5 and B being 50 mM Na-acetate buffer pH 5 containing 1 M NaCl.Fractions of 500 μL were collected. Fractions eluting at 0.3 M NaClwere pooled and dialyzed against 50 mM Na-acetate buffer pH 5.The different pools obtained along the purification process were
analyzed for their pectin lyase, pectate lyase, endo-PG and galactanaseactivity and for their protein content using BSA as standards.39 Toassess the activity of pectin and pectate lyase, raw lemon pectin(Grinsted 3450) and polygalacturonic acid were used as substrates,respectively. Activity was followed using 2.5 mg/mL of substrates in 50mM Na-acetate buffer pH5 at 40 °C. The reaction was initiated byaddition of the enzymatic fraction and the activity measured at 20 and30 min. The rate of reaction was determined spectrophotometricallyby measuring the rate of formation of double bond (CC) by itsabsorbance at 235 nm (ε = 5500 M−1 cm−1 as the molar absorptioncoefficient of CC at 235 nm).40 Galactanase and endo-PG activitieswere assessed in 50 mM Na-acetate buffer pH 5 on potato galactan(2.5 mg/mL) and polygalacturonic acid as substrates (2.5 mg/mL),respectively, by measuring the appearance of reducing ends.41 Apurification summary is given in Table 1.Characterization and Quantification of Pectin-Lyase Digest
Products. Preparative Low-Pressure Size-Exclusion Chromatogra-phy. Fifteen mg of HG samples were solubilized in 5 mL of 50 mMNa-acetate buffer pH5. Thirty μL of pectin lyase (57.6 nkat/mL) wereadded prior to incubation for 24 h at 40 °C. Digested samples weresubsequently concentrated to approximately 1 mL under vacuum anddesalted on a Sephadex G10 column (78 × 4.4 cm) eluted withdistilled water at 90 mL/h. The elution was monitored usingrefractometric detection. The resultant desalted enzymatic digestswere concentrated to less than 1 mL under vacuum at 40 °C andinjected to a chromatographic system constituted of a Biogel P6-extrafine (92 × 5 cm) column and a Biogel-P4- fine (86 × 5 cm) column
mounted in series. Elution was performed using a 100 mM Na-acetatebuffer pH 3.6 at a flow rate of 10 mL/h and collected fractions wereanalyzed for their GalA content.36 Appropriate fractions were pooledand either extensively dialyzed against distilled water (polymericpools), or concentrated under vacuum at 40 °C to ∼3 mL, desalted ona Sephadex G10 column (78 × 4.4 cm) and concentrated to reach afinal GalA concentration of ∼150 μg/mL.
In order to prepare large amounts of unsaturated oligogalacturonatestandards a HG-P64 pectin lyase digest was chromatographed asdescribed above. Recovered dp-resolved-fractions were desalted andfurther purified on a Dionex system equipped with a semipreparativeCarbopac PA-100 column (250 × 9 mm). The flow rate of the eluantwas constant at 3 mL/min. The elution was performed using a lineargradient of 250 mM to 500 mM Na-acetate in 100 mM NaOH (0−20min). The column was then re-equilibrated with the starting buffer for30 min. Pulsed amperometry was used for detection. Recoveredfractions were neutralized by 100 mM HCl, desalted on a SephadexG10 column (78 × 4.4 cm) and analyzed for their GalA content.36
High-Performance Anion-Exchange Chromatography. Two mgof the HG sample to be analyzed was solubilized in 2 mL of 50 mMNa-acetate buffer pH 5. Twelve μL of pectin lyase (57.6 nkat/mL) wasadded and the sample incubated for 24 h at 40 °C. Completed digestswere filtered (0.45 μm Millipore) and 20 μL were injected on HPAEC.A Waters system equipped with an analytical Carbopac PA1 column(250 × 2 mm) with pulsed amperometric detection was used. Flowrate was kept constant at 0.25 mL/min. The elution was carried outwith four linear gradient phases of Na-acetate in 100 mM NaOH asdescribed in Tanhatan-Nasseri et al.22 The mobile phases were alldegassed with helium in order to prevent absorption of carbon dioxideand transformation to carbonate. The column was thermostatted at 20°C. Chromeleon Software (Dionex) was used for collecting andprocessing the data. The unsaturated oligogalacturonate standards,whose preparation is described above, were individually injected inorder to calculate the response factors for each degree of polymer-ization that were used to quantify unsaturated oligogalacturonates inHG digests. Analyses were performed in triplicate.
Capillary Electrophoresis. Two mg of the HG sample to beanalyzed were solubilized in 2 mL of 50 mM Na-acetate buffer pH5.Twelve μL of pectin lyase (57.6 nkat/mL) was added and the sampleincubated for 24 h at 40 °C. Completed digests were filtered (0.45 μmMillipore) and freeze-dried. They were subsequently rehydrated in 100μL of Milli-Q water and run in capillary electrophoresis (CE).Experiments carried out in this work used an automated CE system(HP 3D), equipped with a diode array detector. Electrophoresis wascarried out in a fused silica capillary of internal diameter 50 μm and atotal length of 46.5 cm (40 cm from inlet to detector). The capillaryincorporated an extended light-path detection window (150 μm) andwas thermostatically controlled at 25 °C. Phosphate buffer at pH 7.0
Table 1. Summary of Pectin Lyase Purification
fraction activity nkat total activity yield (%) protein yield (%) specific activity (nkat/mg) purification rate
peclyve 100pectin lyase 12921 100 446 1galactanase 1605 100endo-PG 152000 100
AS60 precipitate 12.3pectin lyase 1881 14.6 528 1.2galactanase 52 3.2endo-PG 728 0.5
HIC excluded 2.8pectin lyase 524 4.1 639 1.4galactanase 6 0.4endo-PG 345 0.2
AEC 0.3 M NaCl 0.5pectin lyase 547 4.2 3600 8.1galactanase 0 0endo-PG 0 0
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241617
was used as a CE background electrolyte (BGE) and was prepared bymixing 0.2 M Na2HPO4 and 0.2 M NaH2PO4 in appropriate ratios andsubsequently reducing the ionic strength to that desired (typically 50or 90 mM). Detection was carried out using UV absorbance at 235 nm(reporting on the presence of the unsaturated double bond left at thenonreducing end of all pectin-lyase generated fragments). Sampleswere loaded hydrodynamically (various injection times at 5000 Pa,typically giving injection volumes of the order of 10 nL), and typicallyelectrophoresed across a potential difference of 25 kV. All experimentswere carried out at normal polarity (inlet anodic) unless otherwisestated.Mass Spectrometry. Mass spectrometry studies were carried out on
the dp resolved fractions separated from the pectin-lyase digests inorder to obtain more information on the component oligogalactur-onides. 2,5-Dihydroxybenzoic acid (DHB) was purchased from Sigma-Aldrich Co. (Saint Quentin Fallavier, F). N,N-Dimethylaniline (DMA)was purchased from Fisher Scientific (Fair Lawn, NJ, U.S.A.). Milli-Qwater (Millipore, Bedford, MA, U.S.A.) was used in preparation of allsolutions. All chemical reagents used were HPLC grade. DMA/DHBwas prepared by dissolving 100 mg of DHB in 1 mL of H2O/acetonitrile/DMA (49/49/2). One μL of sample was directly mixedwith 1 μL of matrix solution on a polished steel target plate.Acquisition was performed on an Autoflex III MALDI TOF/TOFmass spectrometer (Bruker Daltonics, Bremen, D) equipped with aSmartbeam laser (355 nm) in positive ion mode with a reflector. Laserpower was adapted for each sample. Mass spectra were automaticallyprocessed by FlexAnalysis software (Bruker Daltonics, Bremen, D).Characterization and Quantification of endo-PG Digest
Products by High-Performance Anion-Exchange Chromatog-raphy. Two mg of samples were solubilized in 2 mL of 50 mM Na-acetate buffer pH4. Twelve μL of endo-PG (35.5 nkat/mL) wereadded prior to incubation for 72 h at 40 °C, 6 μL of fresh enzymebeing added at t = 24 h and 48 h. Hydrolyzates were filtered (0.45 μmMillipore) and analyzed by HPAEC as described above. Analyses wereperformed in triplicate. Monomer, dimer and trimer of GalA were usedas standards. The degree of blockiness (DB) and absolute degree ofblockiness (DBabs) were calculated as follows:
=∑
×DB[oligoGalA ]
[GalA ]100dp3
dp1saturated
unmethylesterified
=∑
×DBGalA
[oligoGalA ]
[ ]100abs
dp3dp1
saturated
total
■ RESULTS AND DISCUSSIONModel HGs. HGs were chemically and/or enzymatically
tailored with the aim of controlling the distribution ofmethylester groups, as described in the Experimental Section(Figure 1) and elsewhere.22 Randomly demethylesterified HGs(B-series) were obtained in the classical manner, by alkalidemethylesterification of a very highly methylesterified“mother” HG (HG96). Manipulating the initial DM ofrandomly methylesterified “mother” HGs prior to enzymaticblockwise-demethylesterification by plant-PME allowed therecovery of two types of blocky HG. Samples containing eithera few long demethylesterified blocks of GalAs (P-series) weregenerated directly from the HG96 mother sample, oralternatively HGs with more numerous but shorter blocks ofdemethylesterified GalA residues (BP-series) were producedfrom “mother” HGs of lower DM (HG96-B ≈ 80). Theselower DM starting substrates offered the processive PME botha larger number of start points (epitopes required for an initialsuccessful binding) and a greater chance, once blockwisedemethylesterification was underway, of encountering alreadynonmethylesterified residues that increase the chances ofdetaching from the chain. HGs of moderate to high DMexhibiting subtle, yet potentially functionally important differ-ences in methylesterification patterns, such as “slightly blocky”versus random, have previously proved difficult to differentiatefrom each other on the basis of their DB and DBabs values, ashave HGs of low DM (<20) that have been obtained by alkalitreatment or plant-PME demethylesterification.22 Therefore, an
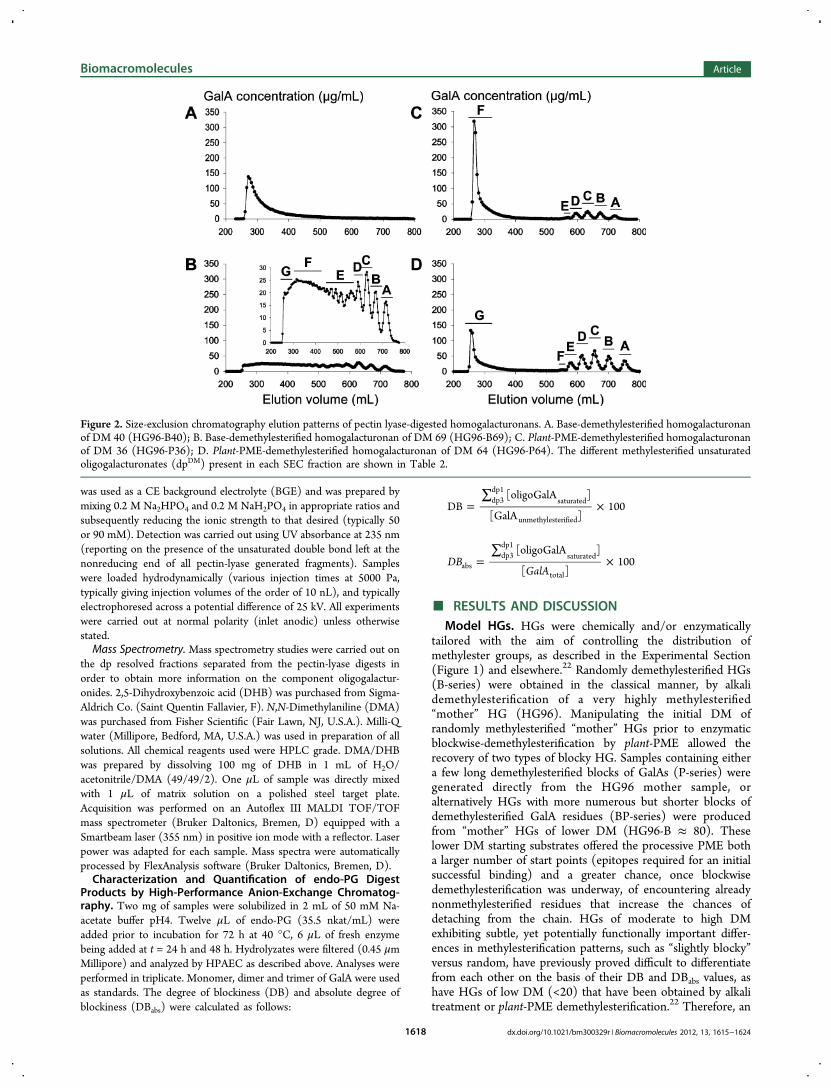
Figure 2. Size-exclusion chromatography elution patterns of pectin lyase-digested homogalacturonans. A. Base-demethylesterified homogalacturonanof DM 40 (HG96-B40); B. Base-demethylesterified homogalacturonan of DM 69 (HG96-B69); C. Plant-PME-demethylesterified homogalacturonanof DM 36 (HG96-P36); D. Plant-PME-demethylesterified homogalacturonan of DM 64 (HG96-P64). The different methylesterified unsaturatedoligogalacturonates (dpDM) present in each SEC fraction are shown in Table 2.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241618

alternative enzymatic degradation approach using pectin lyasehas been investigated for its potential to reveal further insightsinto the distribution of methylesters.Enzymatic Digestion of Model HGs. A pectin lyase was
purified from a crude pectolytic preparation using a three-stepprocedure as described in the Experimental Section. Thepurified sample appeared homogeneous in SDS-PAGE experi-ments using Coomassie blue staining (not shown) and wasdevoid of endo-PG and galactanase activities (Table 1). HGmodel substrates taken from the B-, P-, and BP-series weredegraded by the purified pectin lyase and the digests werestudied: (i) by analytical high-performance anion-exchangechromatography (HPAEC) that permitted the quantification ofreleased unsaturated oligogalacturonides of different dp and (ii)by low-pressure size-exclusion chromatography (SEC) thatpermitted preparative scale production of dp-resolved fractions.These fractions were used to determine HPAEC responsefactors and provide samples for mass spectrometry and capillaryelectrophoresis, which both have the potential to reveal moreinformation about the fine structure variations found in thedifferent dp-resolved species.Preparative SEC of the dp-Resolved Digest Frag-
ments. The results of SEC fractionation, performed asdescribed in the Experimental Section, are shown in Figure 2for the pectin-lyase digests of lowly and highly methylesterifiedHGs with random or blockwise distributions of the freecarboxyl groups, i.e. HG96-B69, HG96-B40, HG96-P64 andHG96-P36. For HG digests from the B-series, chromatogramsdiffered widely depending on the DM of the sample. TheHG96-B40 digest exhibited a sole polymeric peak eluting at thevoid volume indicating that this HG sample is not substantiallydegraded by pectin lyase (Figure 2A). The HG96-B69 digestexhibited a radically different elution pattern with only limitedamounts of material eluting at the void volume and a largerange of peaks eluting between 280 and 750 mL (Figure 2B).Oligomeric fractions that eluted later, labeled A to D, exhibitedhigher DM (DM 77−87) than that of the initial HG sample(DM 69; Table 2). The earlier eluting fractions, labeled E, F,and G, represent nonhydrolyzed or partially hydrolyzedfractions and exhibited a DM close to or slightly lower thanthat of the initial HG sample (Table 2).For HG digests from the P-series, chromatograms were very
much alike whatever the DM of the sample with a polymericfraction eluting at the void volume and 5 to 6 well-resolvedoligomeric peaks eluting between 550 and 780 mL (Figure 2Cand Figure 2D). These P-series digests showed very similaroligomer chain length distributions with an increase in totaloligomer concentration with increasing DM. The pectin lyasecould digest HG96-P36 to some extent demonstrating that,even at this low DM, there are long enough blocks ofmethylesterified GalAs in these plant-PME demethylesterifiedsamples to interact with pectin lyase active site, in goodagreement with the findings of Limberg et al.9 (although itshould be cautioned that the degree to which theintermolecular distribution of DM of the substrate plays arole is unclear.) For both HG96-P36 and HG96-P64 digests,fractions eluting at the void volume exhibited a very low DM(Table 2). These fractions represent long stretches ofnonmethylesterified GalAs that are not suitable for pectinlyase digestion. In both samples, the oligomeric fractionsappeared close to totally methylesterified (Table 2) and arisepredominantly from the digestion of long blocks of consecutive
methylesterified GalAs contained within the fine structure ofthe starting substrate.
Fine Structure of the dp-Resolved Digest Fragments.The oligomeric fractions collected from SEC, that had beenfractionated purely on the grounds of dp, were further analyzedin order to obtain more information on distribution of theirmethylesterification within the samples.
Mass Spectrometry. MALDI-TOF-MS was applied inpositive ion mode and the dp and DM of GalA oligomerspresent in the different SEC fractions was thereby assessedsimultaneously (Table 2). The MS spectrum of the HG96-B69fraction C (Figure 3A) is fully described for clear illustration ofthe observed ions. This MS spectrum was characterized by thepredominance of sodium adducts representative of unsaturatedGalA oligomers of dp5 bearing 2 to 5 methyl groups (m/z 931,945, 959, and 973). Sodiation of the carboxylic functions ofpredominant ions (m/z 945 and 959) was observed (m/z 967,981 for single sodiation of 53 and 54, respectively, and m/z 989for double sodiation of 53). The major species (54, m/z 959)existed also in the saturated form (m/z 977). This ion relates tooligomers arising from the nonreducing end of HG molecules,thereby bearing no unsaturation.32
Mass spectra of the different SEC fractions were similarlyinterpreted and results obtained are summarized in Table 2.Although the same digest-derived oligomers of dp 3 to 6 wereobserved for HG96-B69, HG96-P36 and HG96-P64, theirrelative intensities differed widely. The relative amounts of thedifferent oligomers present in one fraction cannot be assessedby the intensity of the peaks since ionization rates may vary
Table 2. Repartition, DM, and UnsaturatedOligogalacturonates Detected by MALDI-TOF MassSpectrometry in Fractions Recovered after Size-ExclusionChromatographya
sampleSEC
fractionrepartition(% w/w) DM oligomer (dpDM)b
HG96-B69
A 3 81 31, 32, 33
B 5 87 42, 43, 44
C 8 77 52, 53, 54, 55
D 6 81 63, 64, 65, 66
E 19 66 74, 75, 76, 84, 85, 86, 95, 96, 97, 105,106, 107
F 23 61 116, 117, 118, 126, 127, 128, 136,137, 138, 139, 147, 148, 149, 157,158, 159, 1510, 168, 169, 1610,1611
G 36 58 ndHG96-P36
A 2 >95 31, 32, 33
B 5 >95 42, 43, 44
C 11 >95 53, 54, 55
D 7 >95 64, 65, 66
E <1 nd 75, 76, 77
F 75 8 ndHG96-P64
A 9 >95 31, 32, 33
B 13 >95 42, 43, 44
C 15 >95 53, 54, 55
D 13 >95 64, 65, 66
E 6 nd 75, 76, 77
F 1 nd ndG 43 8 nd
and not determined. bbase peak for each dp appears in bold.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241619
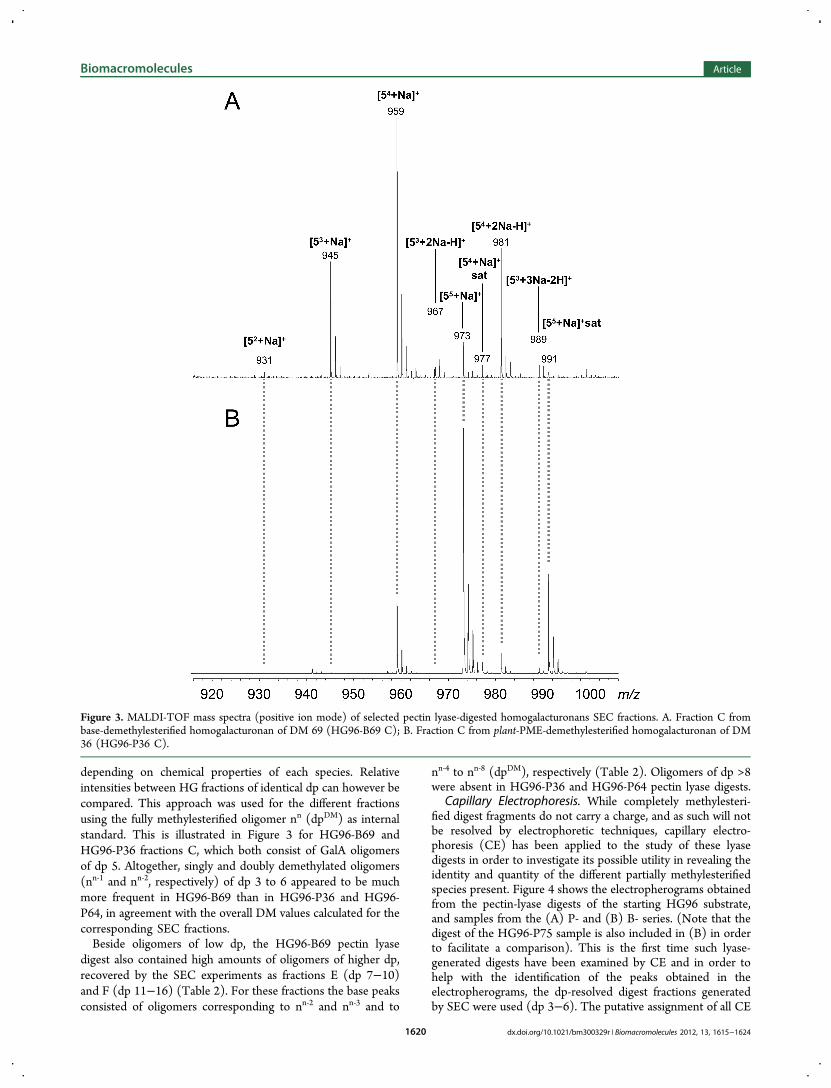
depending on chemical properties of each species. Relativeintensities between HG fractions of identical dp can however becompared. This approach was used for the different fractionsusing the fully methylesterified oligomer nn (dpDM) as internalstandard. This is illustrated in Figure 3 for HG96-B69 andHG96-P36 fractions C, which both consist of GalA oligomersof dp 5. Altogether, singly and doubly demethylated oligomers(nn‑1 and nn‑2, respectively) of dp 3 to 6 appeared to be muchmore frequent in HG96-B69 than in HG96-P36 and HG96-P64, in agreement with the overall DM values calculated for thecorresponding SEC fractions.Beside oligomers of low dp, the HG96-B69 pectin lyase
digest also contained high amounts of oligomers of higher dp,recovered by the SEC experiments as fractions E (dp 7−10)and F (dp 11−16) (Table 2). For these fractions the base peaksconsisted of oligomers corresponding to nn‑2 and nn‑3 and to
nn‑4 to nn‑8 (dpDM), respectively (Table 2). Oligomers of dp >8were absent in HG96-P36 and HG96-P64 pectin lyase digests.
Capillary Electrophoresis. While completely methylesteri-fied digest fragments do not carry a charge, and as such will notbe resolved by electrophoretic techniques, capillary electro-phoresis (CE) has been applied to the study of these lyasedigests in order to investigate its possible utility in revealing theidentity and quantity of the different partially methylesterifiedspecies present. Figure 4 shows the electropherograms obtainedfrom the pectin-lyase digests of the starting HG96 substrate,and samples from the (A) P- and (B) B- series. (Note that thedigest of the HG96-P75 sample is also included in (B) in orderto facilitate a comparison). This is the first time such lyase-generated digests have been examined by CE and in order tohelp with the identification of the peaks obtained in theelectropherograms, the dp-resolved digest fractions generatedby SEC were used (dp 3−6). The putative assignment of all CE
Figure 3. MALDI-TOF mass spectra (positive ion mode) of selected pectin lyase-digested homogalacturonans SEC fractions. A. Fraction C frombase-demethylesterified homogalacturonan of DM 69 (HG96-B69 C); B. Fraction C from plant-PME-demethylesterified homogalacturonan of DM36 (HG96-P36 C).
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241620

peaks observed in digests was complex and involved bothobserving the electrophoretic consequences of further process-ing of these dp-resolved fractions using a fungal-PME and analkali treatment, and running samples against other saturatedand unsaturated standards, and will be reported in detailelsewhere (Manuscript in Preparation). The work reportedhere however clearly shows the potential of the technique forthe investigation of the partially methylesterified unsaturatedfragments released by the lyase digests.As found in the SEC experiments the results obtained from
the P-series substrates show predominantly the same peakspresent in all digests (specifically the first six peaks present inboth series, assigned in the figure captions) with the amount ofthese highly methylesterified species reducing as the DM of thesubstrate is decreased. Although comparisons of the amounts ofdifferent species present and their absolute quantitationrequires accounting for differential absorption coefficients, themeasurement of which is being pursued, the relative amounts ofthe same species present in different digests can be directlyinferred from the comparison of peak areas. All completelymethylesterified species can be observed migrating with theelectroosmotic flow regardless of dp while the partiallymethylesterified species can be resolved in this technique.The speciation of these fragments is also seen to be consistentwith the results obtained from MS analysis and the average DMof the SEC fractions, with the singly demethylesterified speciesoverwhelmingly dominant and a smaller amount of doublydemethylesterified oligomers detectable. The B series resultsare also consistent with the previous results showing an overall
smaller amount of fragments released from substratescompared with the P-series polymers of the same DM, morespecies types liberated, and a greater dependence of thespeciation on the initial substrate DM. CE clearly offers thepotential of quantifying the different partially methylesterifiedspecies, providing extra information that might be useful in themore detailed modeling of the enzyme action and this is beingpursued in further work.Figure 5A shows the results obtained from the lyase digests
of the BP-series compared with that obtained from the mother
sample. The effect of a further round of demethylesterification,this time carried out using the processive PME are clearly seen.While further reducing the still relatively high starting DM(albeit randomly distributed) yields an overall reduction in theamount of low DM digest fragments liberated, the relativeamount of the different species clearly becomes closer to thatfound in the P-series digests, as opposed to those of the B-series, as more blocky stretches are introduced prior to lyasetreatment. Figure 5B shows the resulting digest pattern fromthree substrates with similar DM values, but that have beengenerated from the starting HG96 in quite different ways, onedirectly with plant-PME, one with alkali treatment and one withlimited alkali treatment followed by plant-PME. As expected theP-series sample shows the smallest number of distinct speciesand the largest amount of low-dp material liberated by pectinlyase. The B- and BP- series both show more complex digestpatterns but clearly with the B- series producing less overallfragments, again as expected, and also a decrease in the amountof completely methylesterified species released compared withthose with one or two methylester groups removed.
Figure 4. Electropherograms obtained (235 nm) from the pectin-lyasedigests of the starting HG96 substrate, and samples from the (A) P-and (B) B- series. Based on the investigations involving fractionsseparated from various complete digests on the grounds of dp, theidentity of several key unsaturated species is suggested. The six peaksmarked in (A), left to right are proposed to be (i) completelymethylated oligomers of any dp; (ii) 65; (iii) 54; (iv) 43; (v) 32; and(vi) 53; and in (B) the three additional peaks, again left to right (i) 64;(ii) 53; 42; (iii) 31.
Figure 5. A. Electropherograms obtained (235 nm) from the lyasedigests of the BP-series compared with that obtained from the mothersample. B. Resulting digest patterns from three substrates with verysimilar DM values, but that have been generated from the startingHG96 in quite different ways, one directly with plant-PME, one withalkali treatment and one with limited alkali treatment followed byplant-PME.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241621
Quantification of the dp-Resolved Digest Fragments.HG model substrates from B-, P-, and BP-series were degradedby pectin lyase as described and, in addition to performingpreparative SEC, the digests were analyzed by high-perform-ance anion-exchange chromatography in order to quantify thegenerated oligogalacturonides of different dps. As previouslypointed out,32,33,42 not only the DM of the pectic substrates butalso the distribution pattern of methylesters influences theactivity of pectin lyases. Although the enzyme accepts partiallymethylesterified substrates,32,33 tolerance for carboxyl groups israther limited and the products of dp 3−8, which have beencharacterized here, can be considered as faithful representativesof the highly methylesterified stretches within the HGmolecule.After pectin lyase digestion of an ideal substrate (HG96),
unsaturated GalA oligomers with dp 3−7 were recovered asmajor products with unsaturated GalA oligomers of dp2 anddp8 as very minor ones. The overall recovery (mg of recoveredunsaturated GalA oligomers/100 mg of GalA initially present inthe HG sample) was >92. Saturated GalA oligomers of dp 4and 5, representing altogether around 5 mg/100 mg of GalAinitially present, were also detected. As previously evidenced forHG96-P36 (Figure 3B), those saturated GalA oligomersappeared totally methylesterified by MALDI-TOF-MS analysisand correspond to HG nonreducing ends. The overall recoveryof GalA oligomers was >97%, which demonstrates the validityof the response factors used.Degree of Blockiness and Absolute Degree of
Blockiness of Highly Methylesterified Stretches (DBMeand DBabsMe). Several parameters have been introduced tocharacterize the presence or absence of nonmethylesterifiedblocks among which the degree of blockiness (DB)8 and theabsolute degree of blockiness (DBabs)
27 are the most widelyused. Similarly, in this work, a degree of blockiness and anabsolute degree of blockiness of the highly methylesterifiedstretches (DBMe and DBabsMe, respectively) have beencalculated based upon the results of the pectin lyase digestion
=∑
×DBMe[oligoGalA ]
[GalA ]100dp8
dp2unsaturated
methylesterified
=∑
×DB Me[oligoGalA ]
[GalA ]100abs
dp8dp2
unsaturated
total
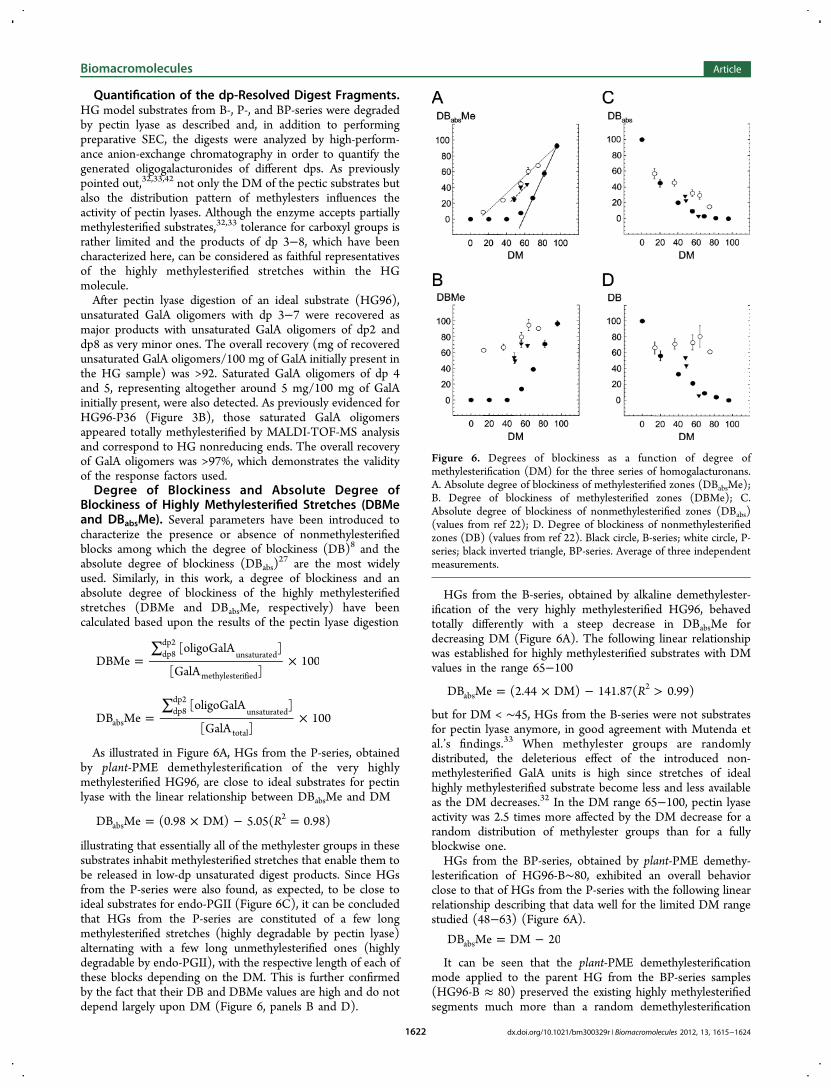
As illustrated in Figure 6A, HGs from the P-series, obtainedby plant-PME demethylesterification of the very highlymethylesterified HG96, are close to ideal substrates for pectinlyase with the linear relationship between DBabsMe and DM
= × − =RDB Me (0.98 DM) 5.05( 0.98)abs2
illustrating that essentially all of the methylester groups in thesesubstrates inhabit methylesterified stretches that enable them tobe released in low-dp unsaturated digest products. Since HGsfrom the P-series were also found, as expected, to be close toideal substrates for endo-PGII (Figure 6C), it can be concludedthat HGs from the P-series are constituted of a few longmethylesterified stretches (highly degradable by pectin lyase)alternating with a few long unmethylesterified ones (highlydegradable by endo-PGII), with the respective length of each ofthese blocks depending on the DM. This is further confirmedby the fact that their DB and DBMe values are high and do notdepend largely upon DM (Figure 6, panels B and D).
HGs from the B-series, obtained by alkaline demethylester-ification of the very highly methylesterified HG96, behavedtotally differently with a steep decrease in DBabsMe fordecreasing DM (Figure 6A). The following linear relationshipwas established for highly methylesterified substrates with DMvalues in the range 65−100
= × − >RDB Me (2.44 DM) 141.87( 0.99)abs2
but for DM < ∼45, HGs from the B-series were not substratesfor pectin lyase anymore, in good agreement with Mutenda etal.’s findings.33 When methylester groups are randomlydistributed, the deleterious effect of the introduced non-methylesterified GalA units is high since stretches of idealhighly methylesterified substrate become less and less availableas the DM decreases.32 In the DM range 65−100, pectin lyaseactivity was 2.5 times more affected by the DM decrease for arandom distribution of methylester groups than for a fullyblockwise one.HGs from the BP-series, obtained by plant-PME demethy-
lesterification of HG96-B∼80, exhibited an overall behaviorclose to that of HGs from the P-series with the following linearrelationship describing that data well for the limited DM rangestudied (48−63) (Figure 6A).
= −DB Me DM 20abs
It can be seen that the plant-PME demethylesterificationmode applied to the parent HG from the BP-series samples(HG96-B ≈ 80) preserved the existing highly methylesterifiedsegments much more than a random demethylesterification
Figure 6. Degrees of blockiness as a function of degree ofmethylesterification (DM) for the three series of homogalacturonans.A. Absolute degree of blockiness of methylesterified zones (DBabsMe);B. Degree of blockiness of methylesterified zones (DBMe); C.Absolute degree of blockiness of nonmethylesterified zones (DBabs)(values from ref 22); D. Degree of blockiness of nonmethylesterifiedzones (DB) (values from ref 22). Black circle, B-series; white circle, P-series; black inverted triangle, BP-series. Average of three independentmeasurements.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241622

would have. HGs from the BP-series however contain onlyrelatively short nonmethylesterified stretches, as evidenced bytheir limited degradation by endo-PGII (Figure 6, panels B andD). These are interspersed with long highly methylesterifiedstretches, as evidenced by their significant degradation bypectin lyase (Figure 6A and Figure 6C).Products obtained after degradation by endo-PGII (dp 1−3)
and pectin lyase (dp 2−8) can be considered as mirror imagesof each other, with endo-PGII degradation products reportingon the blockiness of lowly methylesterified zones of themolecule, and pectin lyase degradation products on the highlymethylesterified ones. It seems reasonable to suggest then thatthe total proportion of zones degradable by endo-PGII and bypectin lyase will be highly sensitive to the intramolecularpattern of DM and may discriminate between pectin sampleshitherto difficult to distinguish, such as the P- and BP- seriesinvestigated herein. Indeed, the total proportion of degradablezones appears radically different depending on the methylesterdistribution (Figure 7).
The following linear relationship between the totalproportion of degradable zones and DM could be establishedfor the data from the digests of the P-series substrates
+ = × + =RDB DB Me (0.35 DM) 59.88( 0.85)abs abs2
demonstrating that a high proportion of HG molecules in theP-series substrates were degradable either by endo-PGII, or bypectin lyase, but with this proportion decreasing slowly withdecreasing DM. This decrease with DM simply reflects the factthat the amount of dp 2−8 unsaturated oligomers released for acompletely methylesterified HG and the amount of dp 1−3saturated oligomers released from a totally nonmethylesterifiedsubstrate of the same length are not the same.In contrast, HGs from the B-series encompassed a low total
proportion of degradable zones, with a mimimum when theDM was close to 50, as expected. Above that DM the amountof lyase-generated fragments increases and the total rises; belowthat, the amount of endo-PG digest products increases andagain the total rises. The following polynomial relationship wasfound to reasonably capture this dependence:
+ = − ×
+ × >R
DB DB Me 99.66 (3.41 DM)
(0.035 DM )( 0.99)abs abs
2 2
With the results from the P- and B- series providingconfidence in the methodology the results from the HGsoriginating from the BP-series were plotted in the samemanner. It is clear that indeed these substrates exhibit anintermediate behavior and in contrast from previouslysuggested discriminators monitoring the sum of DBabs andDBabsMe could clearly differentiate these HGs from the twoother series studied.
■ CONCLUSIONHGs with engineered distributions of methylester groups wereprepared using chemical and enzymatic means.22 As shownschematically in Figure 8, three main series were obtained with
respect to their methylester distributions: (i) a random (B-series), (ii) chains containing long methylesterified stretchesinterspersed with long unmethylesterified ones (P-series), and(iii) HGs with long methylesterified stretches interspersed withshort unmethylesterified ones (BP-series). The amounts ofendo-PGII degradation products (dp 1−3) were similar forrandom or slightly blocky HGs of DM > 55, and so were theamounts of pectin lyase degradation products (dp 2−8)
Figure 7. Sum of absolute degree of blockiness of methylesterifiedzones and absolute degree of blockiness of nonmethylesterified zones(DBabsMe + DBabs) as a function of degree of methylesterification(DM) for the three series of homogalacturonans. Black circle, B-series;white circle, P-series; black inverted triangle, BP-series. Average ofthree independent measurements.
Figure 8. Schematic representation of methylesterified GalA (whitecircle) and nonmethylesterified GalA (black circle) distribution for thethree series of homogalacturonans and the respective “mother”homogalacturonans they arise from.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241623

obtained from slightly blocky and highly blocky HGs of DM <55. However, combining the two enzymatic approaches andthereby quantifying the total proportion of degradable zones,by endo-PGII and by pectin lyase, allowed clear differentiationbetween the three HG series (Figure 8). This double enzymaticapproach will be useful not only for discriminating industrialpectin samples exhibiting different functionalities21 but also forobtaining further insights into pectin fine structure dynamics invivo either at different developmental stages or followingspecific mutations.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank Jacqueline Vigouroux and Marie-Jeanne Crepeau fortheir excellent technical assistance.
■ REFERENCES(1) Ridley, B. L.; O’Neill, M. A.; Mohnen, D. Phytochemistry 2001,57, 929−967.(2) Willats, W. G. T.; McCartney, L.; Mackie, W.; Knox, J. P. PlantMol. Biol. 2001, 47, 9−27.(3) Willats, W. G. T.; Knox, J. P.; Mikkelsen, J. D. Trends Food Sci.Technol. 2006, 17, 97−104.(4) Mohnen, D. Curr. Opin. Plant Biol. 2008, 11, 266−277.(5) Willats, W. G. T.; Orfila, C.; Limberg, G.; Buchholt, H. C.; vanAlebeek, G. J. W. M.; Voragen, A. G. J.; Marcus, S. E.; Christensen, T.M. I. E.; Mikkelsen, J. D.; Murray, B. S.; Knox, J. P. J. Biol. Chem. 2001,76, 19404−19413.(6) Kohn, R.; Markovic, O.; Machova, E. Collect. Czech. Chem.Commun. 1983, 48, 790−797.(7) Thibault, J.-F.; Rinaudo, M. Biopolymers 1985, 24, 2131−2143.(8) Daas, P. J. H.; Meyer-Hansen, K.; Schols, H. A.; De Ruiter, G. A.;Voragen, A. G. J. Carbohydr. Res. 1999, 326, 120−129.(9) Limberg, G.; Korner, R.; Buchholt, H. C.; Christensen, T. M. I.E.; Roepstorff, P.; Mikkelsen, J. D. Carbohydr. Res. 2000, 327, 293−307.(10) Denes, J.-M.; Baron, A.; Renard, C. M. G. C.; Pean, C.; Drilleau,J.-F. Carbohydr. Res. 2000, 327, 385−393.(11) Ralet, M.-C.; Dronnet, V.; Buchholt, H. C.; Thibault, J.-F.Carbohydr. Res. 2001, 336, 117−125.(12) Savary, B. J.; Hotchkiss, A. T.; Cameron, R. G. J. Agric. FoodChem. 2002, 50, 3553−3558.(13) Cameron, R. G.; Luzio, G. A.; Goodner, K.; Williams, M. A. K.Carbohydr. Polym. 2008, 71, 287−299.(14) Sajjaanatakul, T.; Pitifer, L. A. In The Chemistry and Technologyof Pectin; Walter, R. H., Ed.; Academic Press: New York, 1991; pp135−156.(15) Fries, M.; Ihrig, J.; Brocklehurst, K.; Shevchik, V. E.; Pickersgill,R. W. EMBO J. 2007, 26, 3879−3887.(16) Benen, J. A. E.; Kester, H. C. M.; Visser, J. Eur. J. Biochem. 1999,259, 577−585.(17) Chen, E. M. W.; Mort, A. J. Carbohydr. Polym. 1996, 29, 129−136.(18) Luzio, G. A.; Cameron, R. G. Carbohydr. Polym. 2008, 71, 300−309.(19) Lofgren, C.; Guillotin, S.; Evenbratt, H.; Schols, H. A.;Hermansson, A.-M. Biomacromolecules 2005, 6, 646−652.(20) Slavov, A.; Garnier, C.; Crepeau, M.-J.; Durand, S.; Thibault, J.-F.; Bonnin, E. Carbohydr. Polym. 2009, 77, 876−884.(21) Strom, A.; Ribelles, P.; Lundin, L.; Norton, I.; Morris, E. R.;Williams, M. A. K. Biomacromolecules 2007, 8, 2668−2674.
(22) Tanhatan-Nasseri, A.; Crepeau, M.-J.; Thibault, J.-F.; Ralet, M.-C. Carbohydr. Polym. 2011, 86, 1236−1243.(23) Williams, M. A. K.; Vincent, R. R.; Pinder, D. N.; Hemar, Y. J.Non-Newtonian Fluid Mech. 2008, 149, 63−70.(24) Vincent, R. R.; Cucheval, A.; Hemar, Y.; Williams, M. A. K. Eur.Phys. J. E. 2009, 28, 79−87.(25) Vincent, R. R.; Williams, M. A. K. Carbohydr. Res. 2009, 344,1863−1871.(26) Schuster, E.; Cucheval, A.; Lundin, L.; Williams, M. A. K.Biomacromolecules 2011, 12, 2583−2590.(27) Guillotin, S. E.; Bakx, E. J.; Boulenguer, P.; Mazoyer, J.; Schols,H. A.; Voragen, A. G. J. Carbohydr. Polym. 2005, 60, 391−398.(28) Dass, P. J. H.; Voragen, A. G. J.; Schols, H. A. Carbohydr. Res.2000, 326, 120−129.(29) Fraeye, I.; Doungla, E.; Duvetter, T.; Moldenaers, P.; Van Loey,A.; Hendrickx, M. Food Hydrocolloids 2009, 23, 2069−2077.(30) Ngouemazong, D. E.; Tengweh, F. F.; Duvetter, T.; Fraeye, I.;Van Loey, A.; Moldenaers, P.; Hendrickx, M. Food Hydrocolloids 2011,25, 434−443.(31) Daas, P. J. H.; Voragen, A. G. J.; Schols, H. A. Biopolymers 2001,58, 195−203.(32) van Alebeek, G.-J. W. M.; Christensen, T. M. I. E.; Schols, H. A.;Mikkelsen, J. D.; Voragen, A. G. J. J. Biol. Chem. 2002, 277, 25929−25936.(33) Mutenda, K. E.; Korner, R.; Christensen, T. M. I. E.; Mikkelsen,J. D.; Roepstorff, P. Carbohydr. Res. 2002, 337, 1217−1227.(34) Thibault, J.-F.; Renard, C. M. G. C.; Axelos, M. A. V.; Roger, P.;Crepeau, M.-J. Carbohydr. Res. 1993, 238, 271−286.(35) Renard, C. M. G. C.; Jarvis, M. C. Carbohydr. Polym. 1999, 39,201−207.(36) Thibault, J.-F. Lebensm-Wiss.−Technol. 1979, 12, 247−251.(37) Anthon, G. E.; Barrett, D. M. J. Agric. Food. Chem. 2004, 52,3749−3753.(38) Bonnin, E.; Le Goff, A.; Korner, R.; Vigouroux, J.; Roepstorff, P.;Thibault, J.-F. Biochim. Biophys. Acta 2002, 1596, 83−94.(39) Bradford, M. M. Anal. Biochem. 1976, 72, 248−254.(40) Voragen, A. G. J.; Rombouts, F. M.; Pilnik, W. Lebensm-Wiss.−Technol. 1971, 4, 126−128.(41) Nelson, N. J. Biol. Chem. 1944, 153, 375−380.(42) Voragen, A. G. J. Ph.D. thesis; Wageningen, The Netherlands,1972.
Biomacromolecules Article
dx.doi.org/10.1021/bm300329r | Biomacromolecules 2012, 13, 1615−16241624





























