Embed Size (px)
DESCRIPTION
Information theoretical approaches for biological network reconstruction. Farzaneh Farhangmehr (supported by STC) UCSD Presentation#12 July. 30, 2012. Outlines. 1- Introduction: - PowerPoint PPT Presentation
Citation preview

Information theoretical approaches for biological network reconstruction
Farzaneh Farhangmehr (supported by STC)UCSD
Presentation#12 July. 30, 2012

Outlines
1- Introduction: Systems Biology Biological networks Types of biological networks
2- Network reconstruction methods3-Information theoretic approaches
Background Mutual information networks Data Processing Inequality ARACNe Algorithm Time-delay ARACNe algorithm Conditional mutual information
4- Applications in protein-cytokine network reconstructions Background Methods and materials Results
5- Future works: Microarrays Introduction Data Analysis Yeast cell-cycle
References
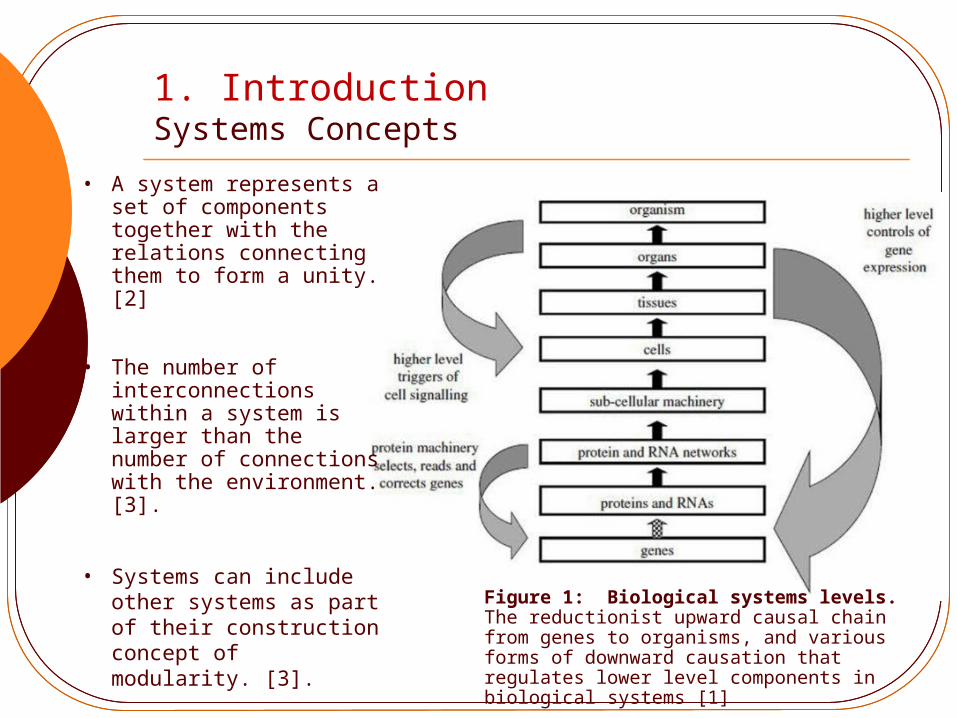
1. IntroductionSystems Concepts
Figure 1: Biological systems levels.The reductionist upward causal chain from genes to organisms, and various forms of downward causation that regulates lower level components in biological systems [1]
• A system represents a set of components together with the relations connecting them to form a unity. [2]
• The number of interconnections within a system is larger than the number of connections with the environment. [3].
• Systems can include other systems as part of their construction concept of modularity. [3].

1. IntroductionSystems Biology
Systems biology defines and analyze the interrelationships of all of the elements in a functioning system in order to understand how the system works [5]:
- To integrate different levels of information to understand how biological systems function.
- To study living cells, tissues, etc. by exploring their components and their interactions.
- To understand the flow of mass, energy and information in living systems.

1. IntroductionBiological Networks
Network is a mathematical structure composed of points connected by lines [6].
A network can be built for any functional system: System vs. Parts = Networks vs. Nodes [7].
By studying network structure and dynamics one can get answers of important biological questions [4]:
- Which interactions and groups of interactions are likely to have equivalent functions across species?
- Based on these similarities, can we predict new functional information
about interactions that are poorly characterized?
- What do these relationships tell us about the evolution of proteins, networks and whole species?

1. IntroductionTypes of Biological Networks
Biological Networks [8],[36]:
- Intra-Cellular Networks:- Protein interaction networks- Metabolic Networks- Signaling Networks- Gene Regulatory Networks- Composite networks- Networks of Modules, Functional Networks Disease networks
- Inter-Cellular Networks
- Neural Networks
- Organ and Tissue Networks
- Ecological Networks
- Evolution Network

2. Biological Network Reconstructions:Reverse Engineering
Reverse engineering of biological networks [17]:
- structural identification: to ascertain network structure or topology.
- identification of dynamics to determine interaction details.
Main approaches:
- Statistical methods- Simulation methods- Optimization methods- Regression techniques- Clustering

2. Network Reconstruction:Statistical methods
Based on the calculation of the correlation for interactions and analyzing their statistical dependencies by using correlation measurements as a metric.
Correlation Measurements:
- Pearson Correlation coefficients - Euclidean distance
- Rank correlation coefficients
- Mutual Information

2. Statistical methods:Pearson Correlation coefficient
Pearson's correlation coefficient between two variables is defined as the covariance of the two variables divided by the product of their standard deviations [18].
Widely used in the sciences as a measure of the strength of linear dependency between two variables.
For two series of n measurements of X and Y written as xi and yi where i = 1, 2, ..., n:
n
ii
n
ii
n
iii
YXyx
yyxxn
yyxxnYX
r
1
2
1
2
1,
)(.)(1
)).((1
.
),cov(
mean samplex
deviation standardX

2. Statistical methods:Euclidean distance
The ordinary distance between two points defined as the square root of the sum of the squares of the differences between the corresponding coordinates of the points.
The Euclidean distance between two genes is the square root of the sum of the squares of the distances between the values in each condition (dimension) [19].
For two series of n measurements of X and Y written as Xi and Yi where i = 1, 2, ..., n, Euclidean distance can be calculated as:
2
1
)(),(
n
iiiEuc yxYXD

2. Statistical methods:Rank Correlation Coefficient
Rank correlation coefficient (RCC) is the Pearson correlation coefficient between the ranked variables [20].
It does not take into account the actual magnitude of the variables, but takes into account the rank of variables.
For two series of n measurements of X and Y written as Xi and Yi where i = 1, 2, ..., n, Xi and Yi are converted to ranks xi and yi and:
n= is the number of conditions (dimension of the profile) di= the difference between ranks of xi and yi at condition i.
)1(61),(
2
2
1
nn
dYX
i
n
i

2. Statistical methodsMutual Information
It gives us a metric that is indicative of how much information from a variable can be obtained to predict the behavior of the other variable [21].
The higher the mutual information, the more similar are the two profiles.
For two discrete random variables of X={x1,..,xn} and Y={y1,…ym}:
p(xi,yj) is the joint probability of xi and yj
P(xi) and p(yj) are marginal probability of xi and yj
m
j
n
i ji
jiji ypxp
yxpyxpYXI
1 1 )()(
),(log),();(

2. Network Reconstruction:Simulation
Key factors: the relevant selection of key characteristics and behaviors; the use of simplifying approximations and assumptions, and validity of the simulation outcomes [37]:
- Boolean networks: Modeled by Boolean variables that represent active and inactive states [38].
- Petri nets: A directed-bipartite graph with two different types of nodes: places and transitions; places represent resources of the system, while transitions correspond to events that can change the state of the resources and arcs connect places with transitions [39].

2. Network Reconstruction:Other approaches
Optimization methods: Minimizing or maximizing a real function by systematically choosing the values of real or integer variables from a feasible set mathematically [40].
Regression analysis includes many techniques for modeling and analyzing several variables, when the focus is on the relationship between a dependent variable and one or more independent variables [41].
Clustering: Partitioning a given set of data points into subgroups, each of which should be as homogeneous as possible [42].

3. Information theoretical approachBackground
Information is any kind of events that affects the state of a system [9].
Hartley’s model of information [1928] [10]:
Information contained in an event has to be defined in terms of some measure of the uncertainty of that event
Less certain events has to contain more information than more certain events.
The information of independent events taken as a single event should be equal to the sum of the information of the independent events.

3. Information theoretical approachShannon theory
Once we agreed to define the information of an event in terms of its probability, the other properties is satisfied if the information of an event is defined as a log function of its probability. [11].
Based on Shannon’s definition (1948), entropy of a random variable is defined in terms of its probability distribution and is a good measure of randomness or uncertainty [12].
Shannon denoted the entropy H of a discrete random variable X with n possible values {xi : i = 1, 2, ..., n} :
where E is the expected value, and I is the self- information content of X
))(log()()()())(()(11
i
n
iii
n
ii xPxPxIxPXIEXH

3. Information theoretical approachShannon theory
Joint Entropy: The joint entropy H(X,Y) of a pair of discrete random variables (X, Y) with a joint distribution p(x, y):
Conditional entropy:- Quantifies the remaining entropy (i.e. uncertainty) of a random variable Y given that the value of another random variable X is known.

3. Information theoretical approachShannon theory
Mutual Information I(X;Y):The reduction in the uncertainty of X due to the knowledge of Y. For two discrete random variables of X={x1,..,xn} and Y={y1,…ym}:
I(X;Y) = H(X) + H(Y) -H(X,Y) =
H(Y) - H(YlX) = H(X) - H(XlY)
m
j
n
i ji
jiji ypxp
yxpyxpYXI
1 1 )()(
),(log),();(

3. Information theoretical approachMutual information networks
X={x1 , …,xi} Y={y1 , …,yj}
The ultimate goal is to find the best model that maps X Y- The general definition: Y= f(X)+U. In linear cases: Y=[A]X+U where [A]
is a matrix defines the linear dependency of inputs and outputs
Information theory maps inputs to outputs (both linear and non-linear models) by using the mutual information:
m
j
n
i ji
jiji ypxp
yxpyxpYXI
1 1 )()(
),(log),();(

3. Information theoretical approachMutual information networks
The entire framework of network reconstruction using information theory has two stages: 1-Mutual information measurements2- The selection of a proper threshold.
Mutual information networks rely on the measurement of the mutual information matrix (MIM). MIM is a square matrix whose elements (MIMij = I(Xi;Yj)) are the mutual information between Xi and Yj.
Choosing a proper threshold is a non-trivial problem. The usual way is to perform permutations of expression of measurements many times and recalculate a distribution of the mutual information for each permutation. Then distributions are averaged and the good choice for the threshold is the largest mutual information value in the averaged permuted distribution.

3. Mutual information networksData Processing Inequality (DPI)
The DPI [21] states that if genes g1 and g3 interact only through a third gene, g2, then:
Checking against the DPI may identify those gene pairs which are not directly dependent even if
)],();,(min[),( 322131 ggIggIggI
)()(),( jiji gpgpggp

3. Mutual information networksARACNE algorithm
Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. “ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context” March 2006, BMC Bioinformatics [25].
ARACNE stands for “Algorithm for the Reconstruction of Accurate Cellular NEtworks”.
ARACNE uses information theory to compute the mutual information between pairs of markers (or genes) in a set of microarray experiments. From these mutual information computations, an interaction network is inferred.
ARACNE identifies candidate interactions by estimating pairwise gene expression profile mutual information, I(gi, gj) and then filter MIs using an appropriate threshold, I0, computed for a specific p-value, p0. In the second step, ARACNe removes the vast majority of indirect connections using the Data Processing Inequality (DPI).
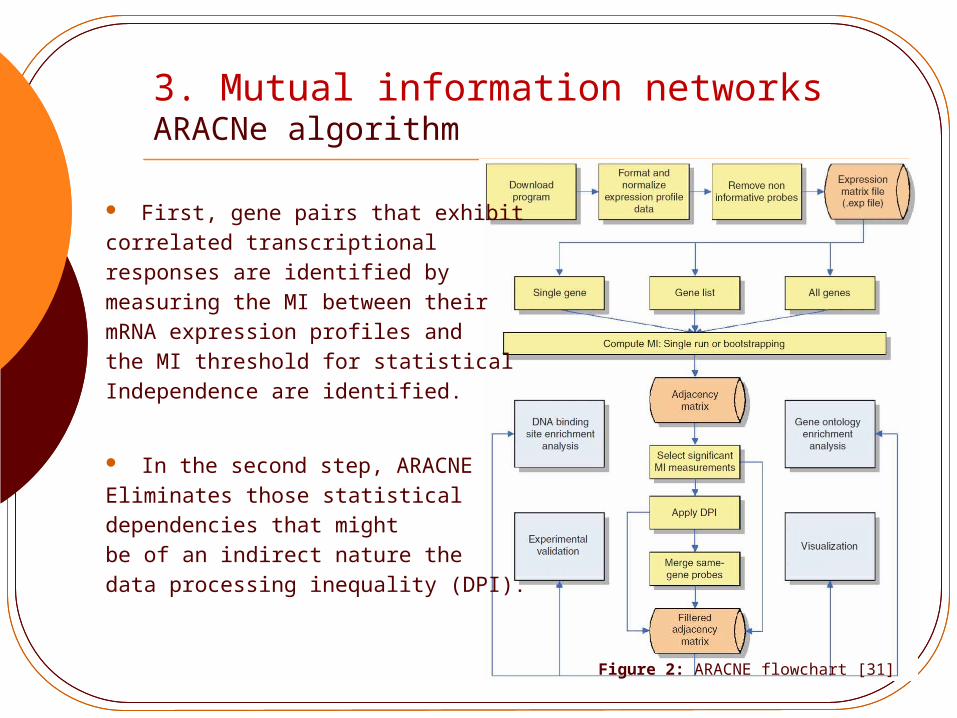
3. Mutual information networksARACNe algorithm
First, gene pairs that exhibit correlated transcriptional responses are identified by measuring the MI between theirmRNA expression profiles andthe MI threshold for statistical Independence are identified.
In the second step, ARACNEEliminates those statistical dependencies that might be of an indirect nature thedata processing inequality (DPI).
Figure 2: ARACNE flowchart [31]

3. Mutual information networksTimeDelay-ARACNE algorithm
An interesting feature of TimeDelay-ARACNE algorithm, is the fact that the time-delayed dependencies can eventually be used for derive the direction of the connections between the nodes of the network, trying to discriminate between regulator gene and regulated genes.
Similar to ARACNE, TimeDelay-ARACNE estimates MI using Gaussian Kernel estimators and performs a selection of the kernel bandwidth, by choosing the bandwidth which (approximately) minimizes the mean integrated squared error (MISE).

3. TimeDelay-ARACNEAlgorithm
Step1:
The first step of the algorithm is aimed at the selection of the initial change expression points in order to flag the possible regulator genes:
If is the sequence of expression of gene ga; and are two thresholds, the initial change of expression (IcE) is defined as:
The thresholds are chosen with :
In all reported experiments, it used = 1.2 and consequently = 0.83.
The quantity IcE(ga) can be used in order to reduce the unnecessary influence relations between genes.
Indeed, a gene ga can eventually influence gene gb only if IcE(ga) ≤ IcE(gb). [33].
,...,...,, 10 taaa ggg
down0j
aup0 /gor /{minarg)( a
jaa
ja ggggIcE
updown
downup
1
up down

3. TimeDelay-ARACNEAlgorithm
Step2:
The basic idea of the proposed algorithm is to detect time-delayed statistical dependencies between the activation of a given gene ga at time t and
another gb at time t + κ with IcE(ga) ≤ IcE(gb).
Time-dependent MIs are calculated for each expression profile obtained by shifting genes by one time step till the defined maximum time delay is reached. Influence is defined as the max time-dependent MIs, Iκ (ga, gb), over all possible delays k:
After the computation of the Infl(ga, gb) estimations, TimeDelay-ARACNE filters them using the threshold, I0.
)IcE( )IcE( with 1,...,2,1:),(max),Infl(
)()(
),(log),(),(
1
aabak
kba
kn
iki
bia
kib
iaki
biaba
k
ggnkggIgg
gPgp
ggPggPggI

3. TimeDelay-ARACNEAlgorithm
Step3:
The last step TimeDelay-ARACNE applies the DPI.

3. TimeDelay-ARACNEApplication: Yeast cell-cycle
Pietro Zoppoli, Sandro Morganella, Michele Ceccarelli: TimeDelay-ARACNE: Reverse engineering of gene networks from time-course data by an information theoretic approach. BMC Bioinformatics 11: 154 (2010) [32].
This study tests the algorithm both on synthetic networks and on microarray expression profiles. The results are compared with the ones of two previously published algorithms: Dynamic Bayesian Networks and systems of ODEs, showing that TimeDelay-ARACNE has good accuracy, recall and F-score for the network reconstruction task.
In order to test TimeDelay-ARACNE performance on Microarray Expression Profiles the time course profiles is a set of 11 genes selected from the yeast, Saccharomyces cerevisiae, cell cycle microarray data [34]. This study selects one of the profiles in which the gene expressions of cell cycle synchronized yeast cultures were collected over 17 time points taken in 10-minute intervals.
In order to test TimeDelay-ARACNE performance on expression profiles, this study selects a set of eight genes network from E. Coli pathway [35].

4. ApplicationProtein-Cytokine Network Reconstruction
• Release of immune-regulatory Cytokines during inflammatory response is medicated by a complex signaling network [45].
• Current knowledge does not provide a complete picture of these signaling components.
• we developed an information theoretic-based model that derives the responses of seven Cytokines from the activation of twenty two signaling Phosphoproteins in RAW 264.7 macrophages.
• This model captured most of known signaling components involved in Cytokine releases and was able to reasonably predict potentially important novel signaling components.

4. Protein-Cytokine NetworkBackground
22 Signaling proteins responsible for cytokine releases:
cAMP, AKT, ERK1, ERK2, Ezr/Rdx, GSK3A, GSK3B, JNK lg, JNK sh, MSN, p38, p40Phox, NFkB p65, PKCd, PKCmu2,RSK, Rps6 , SMAD2, STAT1a, STAT1b, STAT3, STAT5
7 released cytokines (as signal receivers):G-CSF, IL-1a, IL-6, IL-10, MIP-1a, RANTES, TNFa
Using information-theoretic model we want to reconstruct this network from the microarray data and determine what proteins are responsible for each cytokine releases

4. Protein-Cytokine NetworkReleased Cytokines
TNF alpha: Mediates the inflammatory response. Regulates the expression of many genes in many cell types important
for the host response to infection. IL-6:
Interleukin 6 is a pro-inflammatory cytokine and is produced in response to infection and tissue injury. IL-6 exerts its effects on multiple cell types and can act systemically.
Causes T-cell activation IL-10:
Has effect on the production of pro-inflammatory cytokines IL-1a:
Pro-inflammatory mediator produced by monocytes Mediates expression of the gene encoding
MIP-1a: Modulate several aspects of the inflammatory response such as fever
response. Belongs to the group of chemokines

4. Protein-Cytokine NetworkReleased Cytokines
RANTES: Is a chemokine that is predominantly chemotactic for macrophages
G-CSF: Enhances the functional activities of mature neutrophils The expression of its gene encoding is regulated by a combination of
transcriptional and post-transcriptional mechanisms

3. Information theoretical approachesMI Estimation using KDE
Consider two vectors X and Y. A kernel density estimator (KDE) for mutual information is defined as [13]:
Where:
where N is sample size and h is the kernel width. f(x) and f(x,y) represents the kernel density estimators.
i
ii
ii
yfxf
yxf
NYXI
)()(
),(log
1),( ^^
^
)2
)()(exp(
2
1),(
2
22
2
^
i
ii
h
yyxx
NhYXf
ii
h
xx
NhXf )
2
)(exp(
2
1)(
2
2
2
^

3. Information theoretical approachesMI Estimation using KDE
There is not a universal way of choosing h, however the ranking of the MI’s depends only weakly on them [25].
The most common criterion used to select the optimal kernel width is to minimize expected risk function, also known as the mean integrated squared error (MISE) [14].
If Gaussian basis functions are used to approximate univariate data and the underlying density being estimated is Gaussian, then it can be shown that the optimal choice for h is [44]:
Where is the standard deviation of the N samples.
)))()((()( 2^
dXXfXfEhMISEh
5
1
)3
4(N
h

3. Information theoretical approachThreshold Estimation
The probability that zero true mutual information results in an empirical value greater than I0 is: [15]
p ( I>I0 ׀ Ῑ=0)
Where the bar denotes the true MI, N is the sample size and c is a constant. After taking the logarithm of both sides of the above equation:
Log p = a + bI0
Therefore, Log P can be fitted as a linear function of I0 and the slope of b, where b is proportional to the sample size N. For each sample size, the resulting fits are averaged to avoid biased sampling. Using these results, for any given dataset with sample size N and a desired p-value, the corresponding threshold can be obtained.
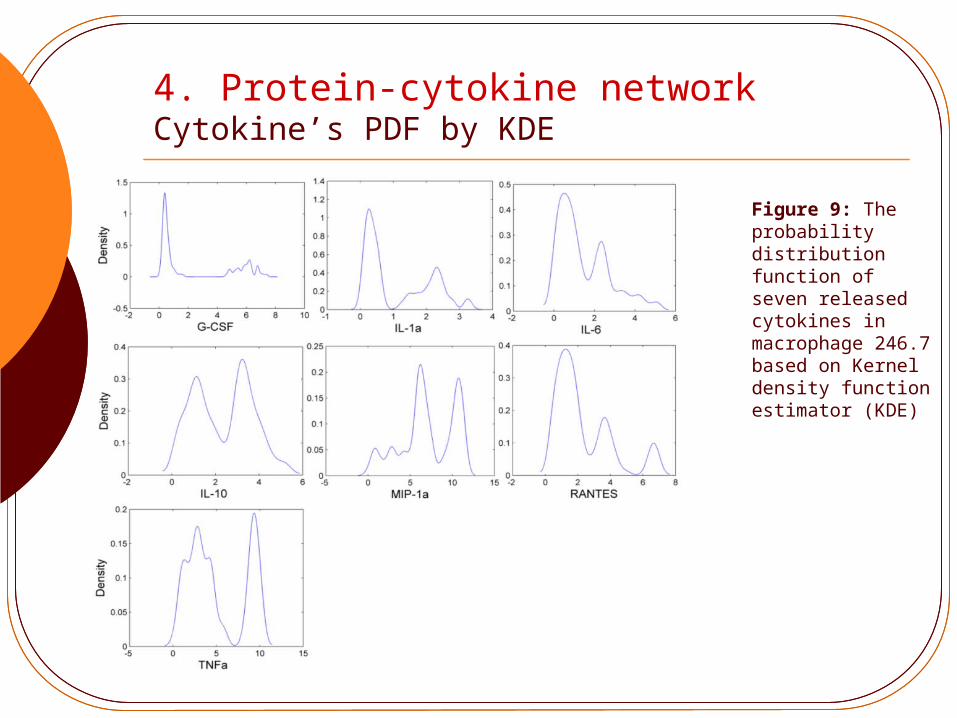
4. Protein-cytokine network Cytokine’s PDF by KDE
Figure 9: The probability distribution function of seven released cytokines in macrophage 246.7 based on Kernel density function estimator (KDE)
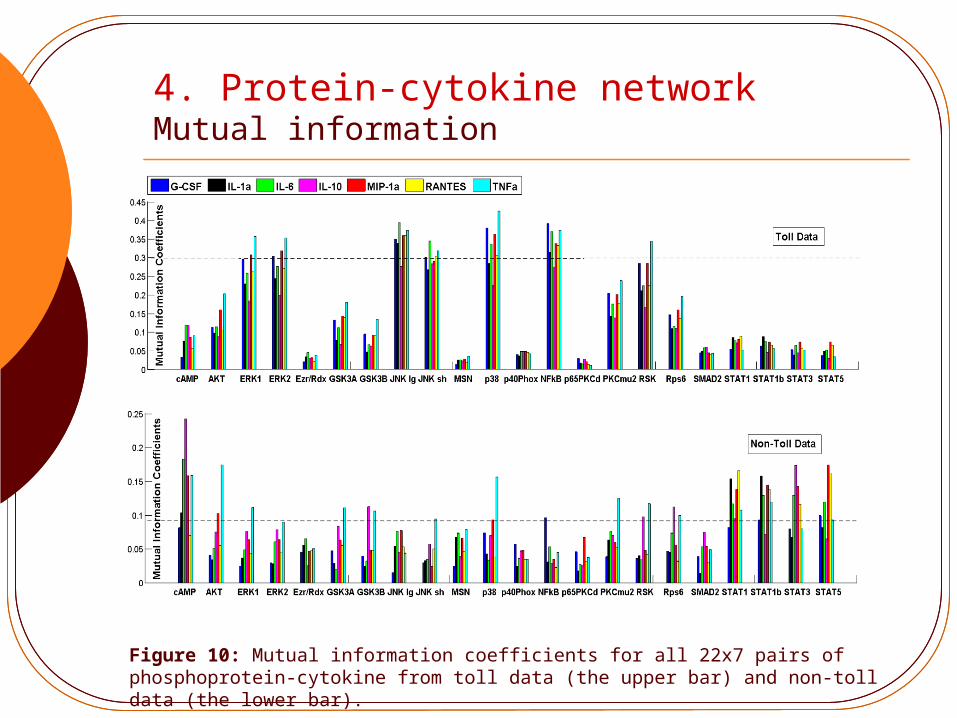
4. Protein-cytokine network Mutual information
Figure 10: Mutual information coefficients for all 22x7 pairs of phosphoprotein-cytokine from toll data (the upper bar) and non-toll data (the lower bar).

4. Protein-cytokine network reconstructionInformation theoretical approach
Figure 11: The phosphoprotein-cytokine network reconstructed from information theoretical approach.
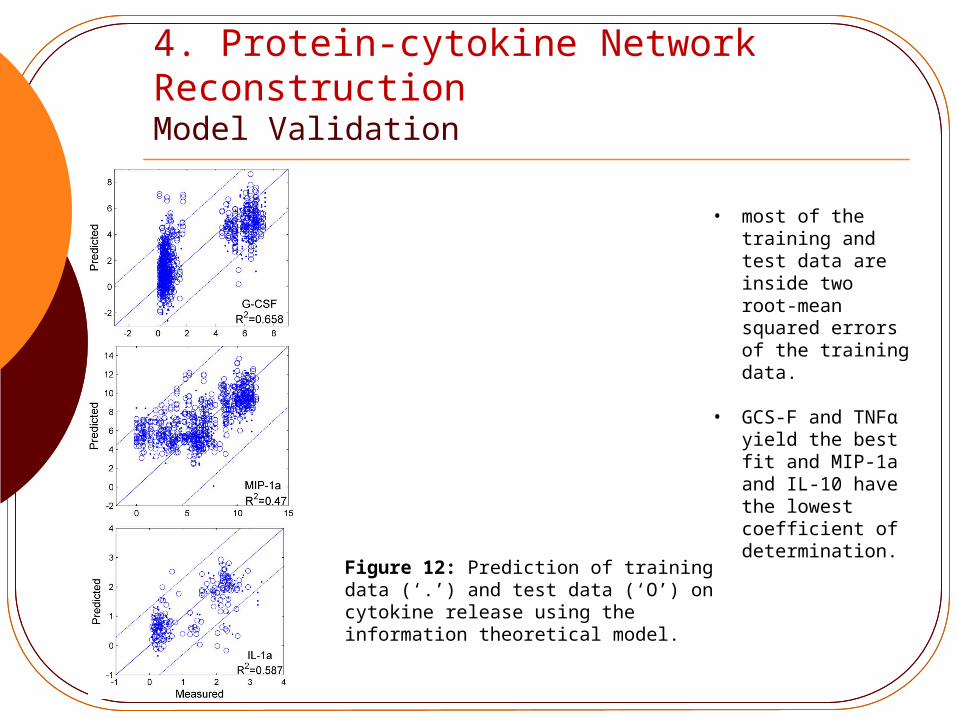
4. Protein-cytokine Network Reconstruction Model Validation
• most of the training and test data are inside two root-mean squared errors of the training data.
• GCS-F and TNFα yield the best fit and MIP-1a and IL-10 have the lowest coefficient of determination.
Figure 12: Prediction of training data (‘.’) and test data (‘O’) on cytokine release using the information theoretical model.

4. Protein-cytokine network modelResults
This model successfully captures known signaling components involved in cytokine releases
It predicts two potentially new signaling components involved in releases of cytokines including: Ribosomal S6 kinas on Tumor Necrosis Factor and Ribosomal Protein S6 on Interleukin-10.
For MIP-1α and IL-10 with low coefficient of determination data that lead to less precise linear the information theoretical model shows advantage over linear methods such as PCR minimal model [16] in capturing all known regulatory components involved in cytokine releases.





























